
Identifying conserved protein complexes between species by constructing interolog networks
16
RESEARCH Open Access Identifying conserved protein complexes between species by constructing interolog networks Phi-Vu Nguyen 1 , Sriganesh Srihari 2 , Hon Wai Leong 1* From Asia Pacific Bioinformatics Network (APBioNet) Twelfth International Conference on Bioinformatics (InCoB2013) Taicang, China. 20-22 September 2013 Abstract Background: Protein complexes conserved across species indicate processes that are core to cellular machinery (e.g. cell-cycle or DNA damage-repair complexes conserved across human and yeast). While numerous computational methods have been devised to identify complexes from the protein interaction (PPI) networks of individual species, these are severely limited by noise and errors (false positives) in currently available datasets. Our analysis using human and yeast PPI networks revealed that these methods missed several important complexes including those conserved between the two species (e.g. the MLH1-MSH2-PMS2-PCNA mismatch-repair complex). Here, we note that much of the functionalities of yeast complexes have been conserved in human complexes not only through sequence conservation of proteins but also of critical functional domains. Therefore, integrating information of domain conservation might throw further light on conservation patterns between yeast and human complexes. Results: We identify conserved complexes by constructing an interolog network (IN) leveraging on the functional conservation of proteins between species through domain conservation (from Ensembl) in addition to sequence similarity. We employ ‘state-of-the-art’ methods to cluster the interolog network, and map these clusters back to the original PPI networks to identify complexes conserved between the species. Evaluation of our IN-based approach (called COCIN) on human and yeast interaction data identifies several additional complexes (76% recall) compared to direct complex detection from the original PINs (54% recall). Our analysis revealed that the IN- construction removes several non-conserved interactions many of which are false positives, thereby improving complex prediction. In fact removing non-conserved interactions from the original PINs also resulted in higher number of conserved complexes, thereby validating our IN-based approach. These complexes included the mismatch repair complex, MLH1-MSH2-PMS2-PCNA, and other important ones namely, RNA polymerase-II, EIF3 and MCM complexes, all of which constitute core cellular processes known to be conserved across the two species. Conclusions: Our method based on integrating domain conservation and sequence similarity to construct interolog networks helps to identify considerably more conserved complexes between the PPI networks from two species compared to direct complex prediction from the PPI networks. We observe from our experiments that protein complexes are not conserved from yeast to human in a straightforward way, that is, it is not the case that a yeast complex is a (proper) sub-set of a human complex with a few additional proteins present in the human complex. Instead complexes have evolved multifold with considerable re-organization of proteins and re- distribution of their functions across complexes. This finding can have significant implications on attempts to extrapolate other kinds of relationships such as synthetic lethality from yeast to human, for example in the identification of novel cancer targets. Availability: http://www.comp.nus.edu.sg/~leonghw/COCIN/. * Correspondence: [email protected] 1 Department of Computer Science, National University of Singapore, Singapore 117590 Full list of author information is available at the end of the article Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8 http://www.biomedcentral.com/1471-2105/14/S16/S8 © 2013 Nguyen et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Identifying conserved protein complexes between species by constructing interolog networks

RESEARCH Open Access
Identifying conserved protein complexes betweenspecies by constructing interolog networksPhi-Vu Nguyen1, Sriganesh Srihari2, Hon Wai Leong1*
From Asia Pacific Bioinformatics Network (APBioNet) Twelfth International Conference on Bioinformatics(InCoB2013)Taicang, China. 20-22 September 2013
Abstract
Background: Protein complexes conserved across species indicate processes that are core to cellular machinery (e.g.cell-cycle or DNA damage-repair complexes conserved across human and yeast). While numerous computationalmethods have been devised to identify complexes from the protein interaction (PPI) networks of individual species,these are severely limited by noise and errors (false positives) in currently available datasets. Our analysis usinghuman and yeast PPI networks revealed that these methods missed several important complexes including thoseconserved between the two species (e.g. the MLH1-MSH2-PMS2-PCNA mismatch-repair complex). Here, we notethat much of the functionalities of yeast complexes have been conserved in human complexes not only throughsequence conservation of proteins but also of critical functional domains. Therefore, integrating information ofdomain conservation might throw further light on conservation patterns between yeast and human complexes.
Results: We identify conserved complexes by constructing an interolog network (IN) leveraging on the functionalconservation of proteins between species through domain conservation (from Ensembl) in addition to sequencesimilarity. We employ ‘state-of-the-art’ methods to cluster the interolog network, and map these clusters back tothe original PPI networks to identify complexes conserved between the species. Evaluation of our IN-basedapproach (called COCIN) on human and yeast interaction data identifies several additional complexes (76% recall)compared to direct complex detection from the original PINs (54% recall). Our analysis revealed that the IN-construction removes several non-conserved interactions many of which are false positives, thereby improvingcomplex prediction. In fact removing non-conserved interactions from the original PINs also resulted in highernumber of conserved complexes, thereby validating our IN-based approach. These complexes included themismatch repair complex, MLH1-MSH2-PMS2-PCNA, and other important ones namely, RNA polymerase-II, EIF3 andMCM complexes, all of which constitute core cellular processes known to be conserved across the two species.
Conclusions: Our method based on integrating domain conservation and sequence similarity to constructinterolog networks helps to identify considerably more conserved complexes between the PPI networks from twospecies compared to direct complex prediction from the PPI networks. We observe from our experiments thatprotein complexes are not conserved from yeast to human in a straightforward way, that is, it is not the case thata yeast complex is a (proper) sub-set of a human complex with a few additional proteins present in the humancomplex. Instead complexes have evolved multifold with considerable re-organization of proteins and re-distribution of their functions across complexes. This finding can have significant implications on attempts toextrapolate other kinds of relationships such as synthetic lethality from yeast to human, for example in theidentification of novel cancer targets. Availability: http://www.comp.nus.edu.sg/~leonghw/COCIN/.
* Correspondence: [email protected] of Computer Science, National University of Singapore,Singapore 117590Full list of author information is available at the end of the article
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
© 2013 Nguyen et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction inany medium, provided the original work is properly cited.

BackgroundComplexes of physically interacting proteins form funda-mental units responsible for driving key biological pro-cesses within cells. Even in the simple model organismSaccharomyces cerevisae (budding yeast), these complexesare composed to several protein subunits that work in acoherent fashion to carry out cellular functions. Thereforea faithful reconstruction of the entire set of complexes(the ‘complexosome’) from the set of physical interactions(the ‘interactome’) is essential to understand their organi-sation and functions as well as their roles in diseases [1-4].In spite of the significant progress in computational
identification of protein complexes from protein interac-tion (PPI) networks over the last few years (see the surveys[1,2]), computational methods are severely limited bynoise (false positives) and lack of sufficient interactions(e.g. membrane-protein interactions) in currently availablePPI datasets, particularly from human, to be able to com-pletely reconstruct the complexosome [1,2]. For example,several complexes involved in core cellular processes suchas cell cycle and DNA damage response (DDR) are notpresent in a recent (2012) compendium of human proteincomplexes (http://human.med.utoronto.ca/) assembledsolely by computational identification of complexes fromhigh-throughput PPIs[5]; a web-search (as of Feb 2013) inthis compendium for BRCA1 does not yield any com-plexes even though BRCA1 is known to participate inthree fundamental complexes in DDR viz. BRCA1-A,BRCA1-B and BRCA1-C complexes [6-8]. A possible rea-son for missing these complexes is the lack of sufficientPPI data required for identifying them even using the bestavailable algorithms. But, the authors of this compendiumnote that many human complexes appear to be ancientand slowly evolving - roughly a quarter of the predictedcomplexes overlapped with complexes from yeast and fly,with half of their subunits having clear orthologs [5].Therefore, it is useful to devise effective computationalmethods that look for evidence from evolutionary conser-vation to complement PPI data to reconstruct the full setof complexes.In the attempt to integrate evolutionary information with
PPI networks, Kelley et al. [9] and Sharan et al. [10]devised methods to construct an orthology graph of con-served interactions from two species, which in their experi-ments were yeast (S. cerevisae) and bacteria (H. pylori),using a sequence homology-based (using BLAST E-scoresimilarity) mapping of proteins between the species. Densesub-graphs induced in this orthology graph representedputative complexes conserved between the two species.The complexes so-identified were involved in core cellularprocesses conserved between the two species - e.g. those inprotein translation, DDR and nuclear transport. Van Damand Snel (2008) [11] studied rewiring of protein complexesbetween yeast and human using high-throughput PPI
datasets mapped onto known yeast and human complexes.From their experiments, they concluded that a majority ofco-complexed protein pairs retained their interactionsfrom yeast to human indicating that the evolutionarydynamics of complexes was not due to extensive PPI net-work rewiring within complexes but instead due to gain orloss of protein subunits from yeast to human. Hirsh andSharan [12] developed a protein evolution-based modeland employed it to identify conserved protein complexesbetween yeast and fly, while Zhenping et al. [13] used inte-ger quadratic programming to align and identify conservedregions in molecular networks. Marsh et al. [14] integrateddata on PPI and structure to understand mechanisms ofprotein conservation; they found that during evolutiongene fusion events tend to optimize complex assembly bysimplifying complex topologies, indicating genome-widepathways of complex assembly.
Integrating domain conservationInspired from these works, here we devise a novel com-putational method to identify conserved complexes andapply it to yeast and human datasets. A crucial point wenote on the conservation from yeast to human is thatmany cellular mechanisms, though conserved, have infact evolved many-fold in complexity - for example, cellcycle and DDR. Consequently, while several proteins inthese mechanisms are conserved by sequence similarity(e.g. RAD9 and hRAD9), there are others that are unique(non-conserved) to human (e.g. BRCA1); see Figure 1.These non-conserved proteins perform similar functions(e.g. cell cycle and DDR) as their conserved counterparts,but do not show high sequence similarity to any of theyeast proteins. A deeper examination reveals that theseproteins in fact contain conserved functional domains -for example, the BRCT domain which is present in yeastRAD9 and human hRAD9 is also present in the non-conserved human BRCA1 and 53BP1; all of these playcrucial roles in DDR [15]. Similar structure can be seenin the case of RecQ helicases - several helicase domainsare conserved from the yeast SGS1 to human BLM andWRN, but there are three helicases RECQ1,4,5 which areunique to human that also contain these helicasedomains [16]. Therefore, integrating information onfunctional conservation, mainly through domain conser-vation, can help to identify considerably more (function-ally) conserved complexes than mere sequence similarity,thereby throwing further light on the conservationpatterns of complexes in particular and cellular processesin general.In order to achieve this, simple BLAST-based scores
as used in earlier works [9-13] to measure homologybetween yeast and human proteins do not suffice. Here,we integrate multiple databases including Ensembl [17]and OrthoMCL [18] to build homology relationships
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 2 of 16

among proteins; these databases use a variety of infor-mation to construct orthologous groups among proteinsincluding checking for conserved domains. The integra-tion of these databases generates many-to-many corre-spondence between yeast and human proteins instead ofthe predominantly one-to-one correspondence obtainedby from BLAST-based similarity.
We devise a novel computational method to constructan interolog network using domain information alongwith PPI conservation between human and yeast. Next,we identify dense clusters within the interolog networkusing current ‘state-of-the-art’ PPI-clustering methods(as against traditional clustering methods used in[9,10]). These clusters when mapped back to the PPI
Figure 1 Conservation of complexes between yeast and human. Many proteins in yeast have either ‘split’ into multiple proteins or fusedinto common proteins in human during evolution. This mechanism is a result of selecting optimal protein assemblies [14] thereby resulting inmulti-fold expansion of complexity in human. In order to capture these conservation mechanisms it is necessary to integrate domain along withPPI information.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 3 of 16

networks reveal conserved dense regions, many of whichcorrespond to conserved complexes.Our experiments here reveal that,
(i) integrating domain information generates manyvaluable interactions from the many-to-many ortho-log relationships in the interolog network, therebyenhancing its quality;(ii) interolog network also reduces false-positiveinteractions by accounting for conserved PPIs;(iii) our interolog network construction aids cluster-ing algorithms to identify far more conserved com-plexes than direct clustering of the individual PPInetworks; and(iv) many of these conserved complexes are involvedin core cellular processes such as cell cycle and DDRthrowing further light to the conservation of thesecellular processes.
We call our method COCIN (COnserved Complexesfrom Interolog Networks).
MethodsConstructing the interolog networkGiven two PPI networks from two species S1 and S2,and the homology information between proteins of thetwo networks, we construct an interolog network GI asfollows. The two PPI networks are represented as G1
(V1, E1) and G2(V2, E2), and the homology relationshipbetween the proteins is governed by a many-to-manycorrespondence θ: V1 ® V2. The interolog network isdefined as GI(VI, EI), where VI = {vI= {p, q} | pÎV1,qÎV2, and (p, q)Î θ}, and EI= {(vI, v’I) | vI ={p,q}; v’I={r,s}; (p, r)ÎE1 and (q,s)ÎE2}.
Each node in the interolog network represents a pairof homologous proteins, one from each species. Eachedge in the interolog network represents an interactionthat is conserved in both species (interolog). However, ifa protein p Î V1 can be orthologous to multiple pro-teins x Î V2 and x Î V2, then we add two vertices to GI
namely {p, x} and {p, y}, and add an edge between twovertices. Doing so integrates the many-to-many relation-ships obtained due to domain conservation into theinterolog network. Figure 2 below gives a simple exam-ple of this network-construction.Any connected sub-network in this interolog network
can be mapped back to conserved sub-networks in thetwo PPI networks, and this is similar to the orthologygraph method introduced by Kelley et al. [9] and Sharanet al. [10]. However, one unique advantage of our inter-olog network offers is that we can infer a collection ofhomologous complexes between the species. This prop-erty is highly relevant for identifying conserved com-plexes between yeast and human (revisit Figure 1).In order to achieve this, we integrate multiple data-
bases including Ensembl [17] and OrthoMCL [18] tobuild our homology relationships among proteins; thesedatabases use a variety of information to constructorthologous groups among proteins including checkingfor conserved domains.
Clustering the interolog network and detection ofconserved complexesWe identify dense clusters in the interolog network todetect conserved complexes between the two species.To do this, we tested a variety ‘state-of-the-art’ PPI net-work-clustering methods, and found the following three
Figure 2 Construction of the interolog network - a simplified example. Our interolog network constructing integrates PPI and domainconservation information to generate a network that is conducive for clustering algorithms to identify considerably more conserved complexescompared to direct clustering of the original PPI networks from species.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 4 of 16

to perform the best - CMC (Clustering by merging Maxi-mal Cliques) by Liu et al. [19], MCL (Markov Clustering)by van Dongen [20] and HACO (Hierarchical Clusteringwith Overlaps) by Wang et al. [21]. The comparativeassessment of these methods has been confirmed withearlier works [1,2,22-24].CMC operates by first enumerating all maximal cliques
in network, and ranks them in descending order of theweighted interaction density. It then iteratively mergeshighly overlapping cliques to identify dense clusters in thenetwork. MCL simulates a series of random paths (called aflow) and iteratively decomposes the network into a num-ber of dense clusters. HACO performs hierarchical cluster-ing by repeatedly identifying smaller dense clusters andmerging these into larger clusters. HACO has an advantageover the traditional hierarchical clustering because it allowsfor overlaps (protein-sharing) among the clusters.Upon finding dense clusters in the interolog network,
we map back these clusters to sub-networks within thetwo PPI networks to identify conserved complexes.
Building a benchmark dataset for conserved proteincomplexesDue to lack of benchmark datasets of conserved proteincomplexes between human and yeast in the literature,we built our own “gold standard” conserved dataset asfollows. Using currently available datasets of manuallycurated protein complexes of human and yeast, weselected pairs of complexes that shared significant frac-tion of (homologous) proteins.
For measuring the conservation level of a given com-plex pair {C1, C2}, where C1 belongs to species S1 and C2
belongs to species S2, we adopted the following Multi-setJaccard score:Multi-set Jaccard score: Let GC1 and GC2 be the col-
lections of ortholog groups in complexes C1 and C2,respectively. For any group giÎGci(i = 1, 2), let ICi repre-sent the multiplicity of the group gi in complex Ci,which essentially is the number of paralogs within thegroup. Multi-set Jaccard score is then given by:
MSJ(C1, C2) =
∑
gi∈(GC1∪GC2)min(I C1(gi), I C2(gi))
∑
gi∈(GC1∪GC2)max(I C1(gi), I C2(gi))
,
There are often duplication of genes (paralogs)within complexes and clusters. Therefore, MSJ takesinto account the multiplicity of the groups and does amore conservative and accurate estimation of the con-servation between C1 and C2. See Figure 3 for anillustration.We selected pairs of complexes that show MSJ ≥ 50%
(see result section for details).
ResultsPreparation of experimental dataWe combined multiple PPI datasets to enhance the cover-age of our interactome. We collected PPIs from IntAct[25] (version November 13, 2012) and Biogrid [26] (ver-sions 3.2.95 and 3.2.89) databases for yeast; and from
Figure 3 Conservation scores for building benchmark complex datasets. We generate a “gold standard” conserved complexes dataset totest our method. We use two scores here - the Jaccard score for orthologous groups and multi-set Jaccard score.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 5 of 16

Biogrid [26] and HPRD [27] (Release 9, 2010) for human.Table 1 and 2 summarise these datasets.Yeast curated complexes were gathered from Wodak
database (CYC2008) [28] and human curated complexesfrom CORUM (version 09/2009) [29]; these form ourbenchmark complex datasets (details in Table 3). Weused Ensembl [17] and OrthoMCL [18] for the homologymapping between human and yeast proteins.
Criteria for evaluating predicted complexesFor a predicted complex Ci of one species and a manu-ally curated (benchmark) complex Bj, we used Jaccardscore based on collections of complex proteins:
J(Ci, Bj) =|Ci ∩ Bj||Ci ∪ Bj|, which considers Ci a correct predic-
tion for Bj if J(Ci, Bj)≥t, a match threshold. We chose t =0.50 in our experiments as suggested by earlier works[19,22]. Ci is then referred to as a matched prediction ormatched predicted complex, and Bj is referred to as aderived benchmark complex.Based on this, precision is computed as the fraction of
predicted complexes matching benchmark complexes,and the recall is computed as the fraction of benchmarkprotein complexes covered by our predicted complexes.A correctly predicted complex is also checked against our“gold standard” testing dataset to see if it is a conservedcomplex, in which case the derived complex is a derivedconserved complex.
Results of complex detection using interolog network (IN)Table 4 summarizes the interolog network constructedfrom yeast and human PPIs. We map back each predicted
cluster from the IN to the original PPI networks to predictconserved complexes between the two species.Firstly, we compared the results of complex detection
from COCIN with direct clustering of the original PPInetworks using CMC, HACO and MCL as shown inTables 5 and 6. Interestingly, we observed that COCIN,which employs CMC, HACO and MCL for clusteringthe interolog network, yielded a better recall than thesemethods on the original PPI networks. Further, becauseIN capitalises on the existence of interactions in bothPPI networks (that is, conservation of interactions), thenumber of noisy dense clusters in COCIN is consider-ably reduced thereby enhancing its precision.Figure 4 compares a predicted complex Ci through
COCIN with two predictions Cy and Chfrom the originalPPI networks; Cy and Chform a pair of orthologouscomplexes, but by direct clustering of the original PPInetworks and matching them and not using COCIN.We noticed that Cy and Chcontained several noisy proteinsand interactions among them which were false positives.These false positives reduced the Jaccard accuracy of thesecomplexes when matched to known benchmark com-plexes. We also note that when we computed the com-plex-derivability index called Component-Edge score (thisindex measures how much of chance a complex canbe detected given the topology of a PPI network) proposedin [24], Ci had a higher CE-score compared to Cy and Ch
in the networks.Figure 5 highlights the improvement of COCIN over
CMC, that is, the additional protein complexes ofhuman and yeast detected by COCIN. As many noisyinteractions are removed in the IN, among the conservedcomplexes that are detected by both CMC and COCIN,COCIN on an average obtained higher Jaccard scores.Some important additional conserved complexes found
Table 1 Properties of yeast physical PPI datasets
Database #proteins
# (non self and duplicated)interactions
IntAct (version Nov 13,2012)
5276 18834
Biogrid (version 3.2.95, Nov30, 2012)
5886 73923
IntAct ∪Biogrid 6332 83777
IntAct∩Biogrid 4620 8930
ICDScore(IntAct ∪ Biogrid) 5239 71636
Table 2 Properties of human physical PPI datasets
Database # proteins #interactions
HPRD (Release 9, 2010) 9617 39184
Biogrid (April 25, 2012) 12515 59027
HPRD ∪ Biogrid 13624 76719
HPRD ∩ Biogrid 8615 21491
ICDScore(HPRD ∪ Biogrid) 8521(EntrezID) 61868
ICDEnrich(HPRD ∪ Biogrid) 9764 (EntrezID) 192053 (EntrezID)
Table 3 Properties of manually curated protein complexdatasets
Databases # complexes
Wodak[28] yeast complexes (CYC 2008) 149 with size>3(36.5%)
Total: 408
CORUM [29] human complexes (September2009)
722 with size>3 (39.1%)
Total: 1843
Table 4 Properties of the interolog network constructedfrom yeast and human PPIs
# Mapped nodes using orthology 2470
# Interologs 6133
Size of biggest connected component 2434 nodes, 6112 edges
#Other connected components 16 (size from 2-3)
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 6 of 16

Table 5 Comparisons of different methods on yeast data.
Method # Predictedcomplexes
# Matchedpredictions
Precision # Gold standard conservedcomplexes
# Detected conservedcomplexes
Recall (of conservedcomplexes)
COCIN 71 36 50.7% 42 32 76.2%
CMC 1202 145 12.1% 42 23 54.8%
HACO 1040 69 6.6% 42 17 40.5%
MCL 387 37 9.6% 42 5 11.9%
Predicted complexes: resulting network clusters
Matched predictions: resulting network clusters that match with benchmarks
Precision = #matched prediction/#predicted complexes
Recall = # detected conserved complexes/# gold standard conserved complexes
Table 6 Comparisons of different methods on human data
Method # Predictedcomplexes
# Matchedpredictions
Precision # Gold standard conservedcomplexes
# Detected conservedcomplexes
Recall (of conservedcomplexes)
COCIN 71 36 50.7% 118 78 66.1%
CMC 1389 156 11.2% 118 66 55.9%
HACO 1290 80 6.2% 118 36 30.5%
MCL 631 45 7.1% 118 24 20.3%
Predicted complexes: resulting network clusters
Matched predictions: resulting network clusters that match with benchmarks
Precision = #matched prediction/#predicted complexes
Recall = # detected conserved complexes/# gold standard conserved complexes
One predicted complex of COCIN can match with many benchmark complexes, this explains for #detected conserved complexes > #matched predictions (asillustrated in Figures 5-8)
Figure 4 An illustration on a predicted complexes from IN. (a) A predicted complex in the IN. (b) The corresponding complex in the humanPPI network. (c) The corresponding complex in the yeast PPI network.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 7 of 16

using COCIN were: RNA Polymerase II, EIF3 complex,MSH2-MLH1-PMS2-PCNA DNA-repair initiation com-plex, MCM complex, MMR complex, Ubiquitin E3 ligase,transcription factor TFIID, DNA replication factor C, 20Sproteasomes (descriptions of these complexes are listedin Tables 7 and 8).
The result of complex detection in the conservedsubnetworksTo further understand the advantage of COCIN on lever-aging conservation for better detection of complexes, weperformed another experiment alternative to the interolognetwork as follows. We predicted complexes from the
Figure 5 COCIN compared to CMC. COCIN over the interolog network identifies significantly more conserved complexes compared to directclustering of the original PPI networks using CMC [19].
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 8 of 16

Table 7 Additional conserved complexes found in yeast
ID Complex name Size Jaccardscore
Functionalcategory
Functional description
96 eIF3 complex 7 0.63 Translation Eukaryotic translation initiation factor
247 Transcription factorTFIID complex
15 0.73 Transcription mRNA synthesis
27 DNA-directed RNApolymerase II complex
12 0.69 Transcription mRNA synthesis
45 DNA replication factorC complex (Rad24p)
5 0.67 DNA processing DNA synthesis and replication
152 DNA replication factorC complex (Rcf1p)
5 0.67 DNA processing DNA synthesis and replication
294 Mcm2-7 complex 6 0.6 DNA processing Chromosome maintainance, DNA synthesis and replication
268 SF3b complex 6 0.57 RNA processing mRNA splicing
65 U6 snRNP complex 8 0.5 RNA processing This complex combines with other snRNPs, unmodified pre-mRNA, and variousother proteins to assemble a spliceosome, a large RNA-protein molecularcomplex upon which splicing of pre-mRNA occurs.
375 AP-3 adaptor complex 4 0.67 Cellular transport,vesicular transport
This complex is responsible for protein trafficking to lysosomes and otherrelated organelles.
25 20S proteasome 14 0.5 Cell cycle, proteinfate
Proteasomal degradation (ubiquitin/proteasomal pathway), protein processing(proteolytic)
137 Chaperonin-containingT-complex
8 0.67 Protein fate A multisubunit ring-shaped complex that mediates protein folding in thecytosol without a cofactor.
Table 8 Additional conserved complexes found in human
ID Complex name Size Jaccardscore
Functionalcategory
Function description
4392 EIF3 complex (EIF3A, EIF3B,EIF3G, EIF3I, EIF3C)
5 0.57 Translation Translation initiation
4403 EIF3 complex (EIF3A, EIF3B,EIF3G, EIF3I, EIF3J)
5 0.57 Translation Translation initiation
104 RNA polymerase II core complex 12 0.69 Transcription mRNA synthesis
2685 RNA polymerase II 17 0.59 Transcription mRNA synthesis
2686 BRCA1-core RNA polymerase IIcomplex
13 0.64 Transcription mRNA synthesis
471 PCAF complex 10 0.6 Transcription, DNAprocessing
DNA conformation modification (e.g. chromatin), modification byacetylation, deacetylation, organization of chromosome structure.
2200 RFC2-5 subcomplex 4 0.5 DNA processing DNA synthesis and replication
387 MCM complex 6 0.6 DNA processing Chromosome maintainance, DNA synthesis and replication
369 MMR complex 2 4 0.67 DNA processing DNA damage repair
290 MSH2-MLH1-PMS2-PCNA DNA-repair initiation complex
4 0.67 DNA processing DNA damage repair initiation
1169 SNARE complex 4 0.6 Cellular transport,vesicular transport
Vesicle fusion, synaptic vesicle exocytosis
562 LSm2-8 complex 7 0.67 RNA processing mRNA splicing
561 LSm1-7 complex 7 0.67 RNA processing Control of mRNA stability during splicing
3036 Ubiquitin E3 ligase (SKP1A, SKP2,CUL1, CKS1B, RBX1)
5 0.5 Cell cycle, proteinfate
Mitotic cell cycle and cell cycle control, modification byubiquitination, deubiquitination
2188 Ubiquitin E3 ligase (CDC34,NEDD8, BTRC, CUL1, SKP1A,RBX1)
5 0.5 Cell cycle, proteinfate
Mitotic cell cycle and cell cycle control, modification byubiquitination, deubiquitination
2189 Ubiquitin E3 ligase (SMAD3,BTRC, CUL1, SKP1A, RBX1)
5 0.5 Cell cycle, proteinfate
Mitotic cell cycle and cell cycle control, modification byubiquitination, deubiquitination
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 9 of 16

subset of protein interactions of the first species that areconserved in the second (we call this the conserved subnet-work in the first species). However, this can only find com-plexes of one species at a time, so we map these predictedcomplexes onto the PPI network of the other species toidentify the corresponding conserved complexes. Weemployed CMC to do clustering on the conservedsubnetworks.Complex prediction from conserved subnetworks
showed similar result as COCIN -16 additional con-served complexes in human and 9 additional conservedcomplexes in yeast are found. This supported the pur-pose of IN - to leverage conserved interactions forimproving complex prediction.Figure 6 shows two other examples that explain why
additional conserved complexes are found by COCIN butmissed by CMC. We see from this picture that the pre-dicted human complex from IN (the leftmost figure) andthe corresponding predicted complex from the conservedsubnetwork (the center figure) were contained in a largerCMC-predicted complex (the rightmost figure) from theoriginal PPI networks. This larger complex included sev-eral noisy proteins that reduce the accuracy of the com-plex, thereby causing the complex to be missed.
Comparisons with other complex detection methods inPPI networksSimilar results were obtained using the other two meth-ods HACO and MCL as well, thereby supporting theeffectiveness of COCIN in identifying conserved proteincomplexes. Tables 5 and 6 present these comparisons inmore details, while Figures 7 and 8 highlight furthersubstantiate these results.
Integrating domain information significantly enhancesinterolog constructionFinally, Table 9 summarizes the quality of our testingdataset for conserved protein complexes between yeastand human. We compared the number of benchmarkconserved complexes found in both human and yeastusing mappings from Ensembl and OrthoMCL undermultiple conservation score thresholds (Figure 9). Notethat Ensembl contains homology information based onboth sequence similarity as well as domain conservation,while OrthoMCL is predominantly based on sequencesimilarity. We noticed that using Ensembl homologyinformation can yield more conserved complexes atall conservation score thresholds. Further, Figure 10shows that there exist 1-to-many and many-to-many
Figure 6 Some examples of additional conserved complexes found in IN. The clusters detected from the original PPI networks includeseveral noisy proteins and noisy interactions (false positives), thereby reducing their Jaccard accuracies.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 10 of 16

relationships of conservation between human and yeastcomplexes.Sharan et al. used whole-sequence similarity to con-
struct the interolog network. Here, we used OrthoMCL
as a substitute for the whole-sequence similarity due totechnical difficulties of running BLAST for a large num-ber of proteins. We compared the performance of usingOrthoMCL against using Ensembl, which uses domain
Figure 7 COCIN compared to HACO. COCIN over the interolog network identifies significantly more conserved complexes compared to directclustering of the original PPI networks using HACO [20].
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 11 of 16
conservation along with sequence similarity to deter-mine orthology. Table 10 and Figure 11 show that weobtain an overall improvement in terms of the numberof mapped protein pairs, interologs, as well as con-served protein complexes in both human and yeast byincorporating domain information (through Ensembl).This substantiates the improved performance ofCOCIN over traditional sequence-similarity basedmethods.
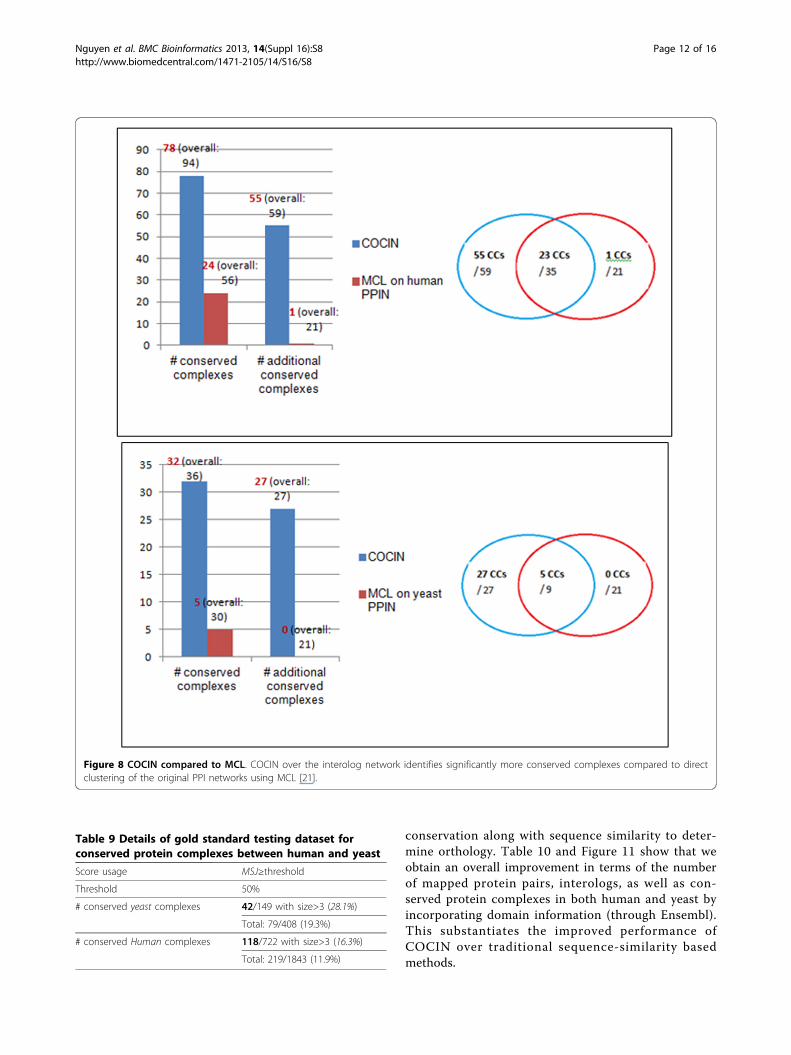
Figure 8 COCIN compared to MCL. COCIN over the interolog network identifies significantly more conserved complexes compared to directclustering of the original PPI networks using MCL [21].
Table 9 Details of gold standard testing dataset forconserved protein complexes between human and yeast
Score usage MSJ≥threshold
Threshold 50%
# conserved yeast complexes 42/149 with size>3 (28.1%)
Total: 79/408 (19.3%)
# conserved Human complexes 118/722 with size>3 (16.3%)
Total: 219/1843 (11.9%)
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 12 of 16
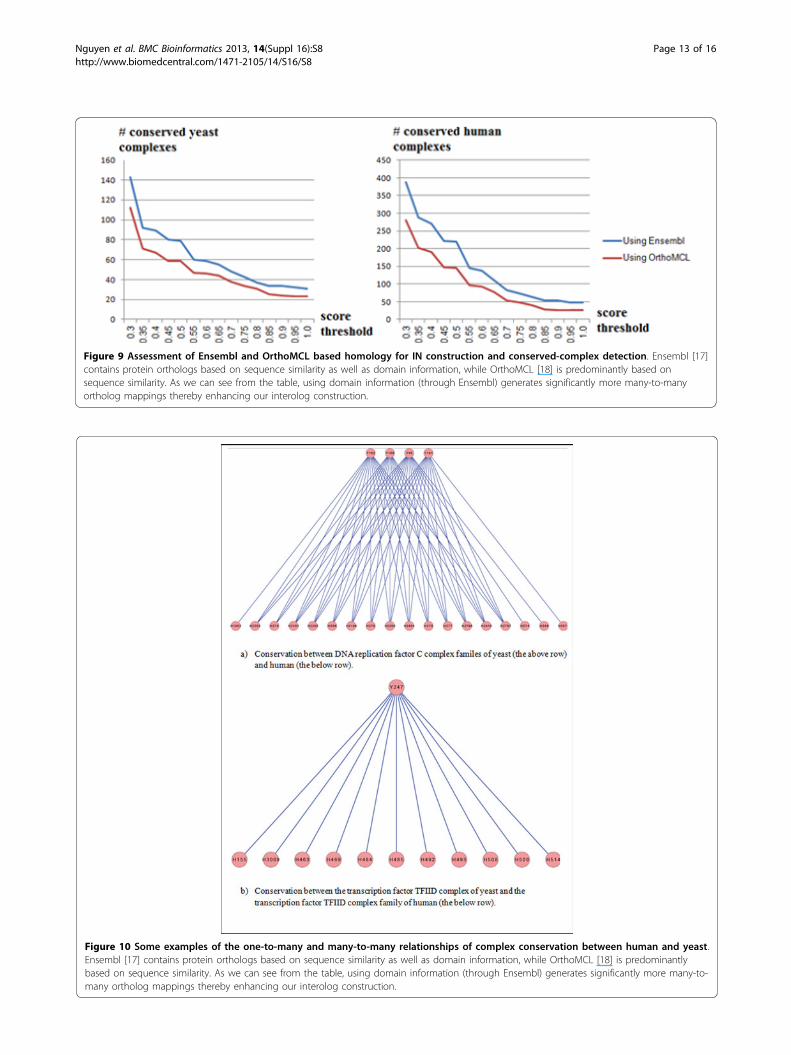
Figure 9 Assessment of Ensembl and OrthoMCL based homology for IN construction and conserved-complex detection. Ensembl [17]contains protein orthologs based on sequence similarity as well as domain information, while OrthoMCL [18] is predominantly based onsequence similarity. As we can see from the table, using domain information (through Ensembl) generates significantly more many-to-manyortholog mappings thereby enhancing our interolog construction.
Figure 10 Some examples of the one-to-many and many-to-many relationships of complex conservation between human and yeast.Ensembl [17] contains protein orthologs based on sequence similarity as well as domain information, while OrthoMCL [18] is predominantlybased on sequence similarity. As we can see from the table, using domain information (through Ensembl) generates significantly more many-to-many ortholog mappings thereby enhancing our interolog construction.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 13 of 16

DiscussionFigure 1 paints a very complicated picture for the con-servation pattern of protein complexes from yeast tohuman. We believe that this picture reflects the actualsituation, and it overrides the belief that a yeast complexis essentially a (proper) subset of a human complex withonly a few new proteins added to the human complex.In other words, the conservation pattern from yeast tohuman is highly intricate involving dispersion andre-distribution of proteins and their functions acrosscomplexes along with addition of several new proteins
in human. At a very high level, though core cellularmechanisms such as cell cycle and DDR are indeedconserved from yeast to human, within these mechan-isms, considerable re-arrangements have occurred.This finding can have implications on studies attempt-ing to extrapolate relationships such as synthetic leth-ality (SL) from yeast to human. In particular, we believethat many of the SL relationships may not be conservedfrom yeast to human, or even if conserved, may not beidentifiable by simple BLAST-based sequence-similaritymappings.
Table 10 Homology data: Ensembl and OrthoMCL
Ensembl database OrthoMCL database
# Ortholog groups: # 1-to-1 groups 1096 1153
# 1-Yeast-to-many groups 756 434
# 1-Human-to-many groups 116 116
# many-to-many groups 197 167
Total: 2165 (5503 pairs) 1870
# Human paralog groups: 2573 2435
# Yeast paralog groups: 426 393
Total # homolog groups: 5164 4698
Ensembl [17] contains protein orthologs based on sequence similarity as well as domain information, while OrthoMCL [18] is predominantly based on sequencesimilarity. As we can see from the table, using domain information (through Ensembl) generates significantly more many-to-many ortholog mappings therebyenhancing our interolog construction.
Figure 11 Comparison between using Ensembl and OrthoMCL in constructing the interolog network. Ensembl [17] contains proteinorthologs based on sequence similarity as well as domain information, while OrthoMCL [18] is predominantly based on sequence similarity. Aswe can see from the table, using domain information (through Ensembl) generates significantly more many-to-many ortholog mappings therebyenhancing our interolog construction.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 14 of 16

ConclusionsIdentifying conserved complexes between species is afundamental step towards identification of conservedmechanisms from model organisms to higher levelorganisms. Current methods based on clustering PPInetworks do not work well in identifying conservedcomplexes, and they are severely limited by lack of trueinteractions and presence of large amounts of falseinteractions in existing PPI datasets. Here, we presenteda method COCIN based on building interolog networksfrom the PPI networks of species to identify conservedcomplexes. Our experiments on yeast and human data-sets revealed that our method can identify considerablymore conserved complexes that plain clustering of theoriginal PPI networks. Further, we demonstrated thatintegrating domain information generates many-to-manyortholog relationships which significantly enhances inter-olog quality and throws further light on conservation ofmechanisms between yeast and human.
AvailabilityOur COCIN software and the datasets used in this work arefreely available at: http://www.comp.nus.edu.sg/~leonghw/COCIN/ or alternately at: https://sites.google.com/site/mclcaw/. The preliminary version of this work appeared asa poster paper (abstract) at RECOMB 2013 [30].
Competing interestsThe authors declare that they have no competing interests.
Authors’ contributionsAll the authors have equal contributions to the ideas of the work. PVNperformed the experiments and analysis, software development. SS and PVNtook part in writing the manuscript. HWL supervised the project andreviewed the manuscript. All the authors have read and approved themanuscript.
AcknowledgementsPVN wants to thank the RAS Group at School of Computing, NationalUniversity of Singapore for all the helpful discussions he had during thiswork; he also thanks Professor Hoai-Bac Le and the University of Science,Vietnam National University at Ho Chi Minh City for the moral supportduring his overseas study.
DeclarationsPublication of this article was funded by an NUS Singapore ARF grant R252-000-461-112, and SS was supported by an Australian NHMRC grant 1028742to Dr Peter T. Simpson and Professor Mark A. Ragan.This article has been published as part of BMC Bioinformatics Volume 14Supplement 16, 2013: Twelfth International Conference on Bioinformatics(InCoB2013): Bioinformatics. The full contents of the supplement areavailable online at http://www.biomedcentral.com/bmcbioinformatics/supplements/14/S16.
Authors’ details1Department of Computer Science, National University of Singapore,Singapore 117590. 2Institute for Molecular Bioscience, The University ofQueensland, St. Lucia, QLD 4072, Australia.
Published: 22 October 2013
References1. Srihari S, Leong HW: A survey of computational methods for protein
complex prediction from protein interaction networks. Journal ofBioinformatics and Computational Biology 2013, 11(2):1230002.
2. Li X, Min Wu, Ng SK: Computational approaches for detecting proteincomplexes from protein interaction networks: a survey. BMC Genomics2010, 11(Suppl 1):S3.
3. Srihari S, Ragan MA: Systematic tracking of dysregulated modulesidentifies novel genes in cancer. Bioinformatics 2013, 29(12):1553-1561.
4. Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R: Associating genes andprotein complexes with disease via network propagation. PLoSComputational Biology 2010, 6(1):e1000641.
5. Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI,Boultz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, Wan C, Vlashblom J,Dar VU, Bezginov A, Clark GW, Wu GC, Wodak SJ, Tillier ER, Paccanaro A,Marcotte EM, Emili A: A consensus of human soluble protein complexes.Cell 2012, 150(5):1068-1081.
6. Khanna KK, Jackson SP: DNA double-strand breaks: signalling, repair andthe cancer connection. Nature Genetics 2001, 27:247-254.
7. Xu B, Seong-tae K, Kastan MB: Involvement of BRCA1 in S-phase and G2-phase checkpoints after ionizing irradiation. Molecular Cell Biology 2001,21:3445-3450.
8. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J: BASC, a supercomplex of BRCA1-associated proteins involved in the recognition andrepair of aberrant DNA structures. Genes and Development 2000,14:927-939.
9. Kelley BP, Sharan R, Karp RM, Sittler T, Root DE, Stockwell BR, Ideker T:Conserved pathways within bacteria and yeast as revealed by globalprotein network alignment. Proceedings of the National Academy ofSciences USA 2003, 100:11394-11399.
10. Sharan R, Ideker T, Kelley B, Shamir R: Identification of protein complexesby comparative analysis of yeast and bacterial protein interaction data.Journal of Computational Biology 2005, 12(6):835-846.
11. van Dam JP, Snel B: Protein complex evolution does not involve extensivenetwork rewiring. PLoS Computational Biology 2008, 4(7):e1000132.
12. Hirsh E, Sharan R: Identification of conserved protein complexes basedon a model of protein network evolution. Bioinformatics 2007, 23(2):e170-e176.
13. Zhenping L, Zhang S, Wang Y, Zhang XS, Chen L: Alignment of molecularnetworks by integer quadratic programming. Bioinformatics 2007,23(13):1631-1639.
14. Marsh JA, Hernandenz H, Hall Z, Ahnhert SE, Perica T, Robinson CV,Teichmann SA: Protein complexes are under evolutionary selection toassemble via ordered pathways. Cell 2011, 153(2):461-470.
15. Bork P, Hoffman K, Bucher P, Neuwald AF, Alstchul SF, Koonin EV: Asuperfamily of conserved domains in DNA damage-responsive cell cyclecheckpoint proteins. FASEB Journal 1997, 11(1):68-76.
16. Larsen NB, Hickson ID: RecQ helicases: conserved gaurdians of genomicintegrity. DNA Helicases and DNA Motor Proteins: Advances in ExperimentalMedicine and Biology (Springer New York) 2013, 973:161-184.
17. Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P,Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, Hendrix M, Hourlier T,Johnson N, Kähäri AK, Keefe D, Keenan S, Kinsella R, Komorowska M,Koscielny G, Kulesha E, Larsson P, Longden I, McLaren W, Muffato M,Overduin B, Pignatelli M, Pritchard B, Riat HS, Ritchie GR, Ruffier M,Schuster M, Sobral D, Tang YA, Taylor K, Trevanion S, Vandrovcova J,White S, Wilson M, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I,Durbin R, Fernández-Suarez XM, Harrow J, Herrero J, Hubbard TJ, Parker A,Proctor G, Spudich G, Vogel J, Yates A, Zadissa A, Searle SM: Ensembl 2012.Nucleic Acids Research 2012, 40:D84-D90.
18. Li L, Stoeckert CJ, Ross DS: OrthoMCL: Identification of ortholog groupsfor eukaryotic genomes. Genome Research 2003, 13:2178-2189.
19. Liu G, Wong L, Chua HN: Complex discovery from weighted PPInetworks. Bioinformatics 2009, 25(15):1891-1897.
20. van Dongen SM: Graph clustering by flow simulation. PhD thesisUniversity of Utrecht; 2000.
21. Wang H, Kakaradov B, Collins SR, Karotki L, Fiedler D, Shales M, Shokat KM,Walther TC, Krogan NJ, Koller D: A complex-based reconstruction of theSaccharomyces cerevisiae interactome. Molecular and Cellular Proteomics2009, 8(6):1361-1381.
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 15 of 16

22. Srihari S, Leong HW: MCL-CAw: a refinement of MCL for detecting yeastcomplexes from weighted PPI networks by incorporating core-attachment structure. BMC Bioinformatics 2010, 11:504.
23. Srihari S, Leong HW: Temporal dynamics of protein complexes in PPInetworks: a case study using yeast cell cycle dynamics. BMCBioinformatics 2012, 17(S16).
24. Srihari S, Leong HW: Employing functional interactions forcharacterization and detection of sparse complexes from yeast PPInetworks. International Journal of Bioinformatics Research and Applications2012, 8:286-304, Nos. ¾, September.
25. Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C,Dimmer E, Feuermann M, Friedrichsen A, Huntley R, Kohler C, Khadake J,Leroy C, Liban A, Lieftink C, Montecchi-Palazzi L, Orchard S, Risse J,Robbe K, Roechert B, Thorneycroft D, Zhang Y, Apweiler R, Hermjakob H:IntAct - open source resource for molecular interaction data. NucleicAcids Research 2007, 35:D561-D565.
26. Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R,Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, Reguly T, Rust JM,Winter A, Dolinski K, Tyers M: The BioGrid Interaction Database: 2011Update. Nucleic Acids Research 2011, 39(Suppl 1):D698-D704.
27. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S,Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A,Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A,Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK,Mohmood R, Ramachandra YL, Krishna V, Rahiman BA, Mohan S,Ranganathan P, Ramabadran S, Chaerkady R, Pandey A: Human ProteinReference Database - 2009 Update. Nucleic Acids Research 2009, 37:D767-D772.
28. Pu S, Wong J, Turner B, Cho E, Wodak SJ: Up-to-date catalogue of yeastprotein complexes. Nucleic Acids Research 2009, 37(3):825-831.
29. Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G,Frishman G, Montrone C, Mewes HW: CORUM: the comprehensiveresource of mammalian protein complexes. Nucleic Acids Research 2008,36(Database):D646-650.
30. Nguyen PV, Srihari S, Leong HW: Identifying conserved protein complexesbetween species by constructing interolog interaction networks, (posterpaper). 17th International Conference on Research in ComputationalMolecular Biology (RECOMB 2013) 2013, April.
doi:10.1186/1471-2105-14-S16-S8Cite this article as: Nguyen et al.: Identifying conserved proteincomplexes between species by constructing interolog networks. BMCBioinformatics 2013 14(Suppl 16):S8.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Nguyen et al. BMC Bioinformatics 2013, 14(Suppl 16):S8http://www.biomedcentral.com/1471-2105/14/S16/S8
Page 16 of 16




















