
Identification of the Nuclear Export Signal and STAT-Binding Domains of the Nipah Virus V Protein...
11
10.1128/JVI.78.10.5358-5367.2004. 2004, 78(10):5358. DOI: J. Virol. Jason J. Rodriguez, Cristian D. Cruz and Curt M. Horvath Underlying Interferon Evasion Virus V Protein Reveals Mechanisms and STAT-Binding Domains of the Nipah Identification of the Nuclear Export Signal http://jvi.asm.org/content/78/10/5358 Updated information and services can be found at: These include: REFERENCES http://jvi.asm.org/content/78/10/5358#ref-list-1 at: This article cites 31 articles, 18 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on January 31, 2014 by guest http://jvi.asm.org/ Downloaded from on January 31, 2014 by guest http://jvi.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Identification of the Nuclear Export Signal and STAT-Binding Domains of the Nipah Virus V Protein...

10.1128/JVI.78.10.5358-5367.2004.
2004, 78(10):5358. DOI:J. Virol. Jason J. Rodriguez, Cristian D. Cruz and Curt M. Horvath Underlying Interferon EvasionVirus V Protein Reveals Mechanismsand STAT-Binding Domains of the Nipah Identification of the Nuclear Export Signal
http://jvi.asm.org/content/78/10/5358Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/78/10/5358#ref-list-1at:
This article cites 31 articles, 18 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from
on January 31, 2014 by guest
http://jvi.asm.org/
Dow
nloaded from

JOURNAL OF VIROLOGY, May 2004, p. 5358–5367 Vol. 78, No. 100022-538X/04/$08.00�0 DOI: 10.1128/JVI.78.10.5358–5367.2004Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Identification of the Nuclear Export Signal and STAT-Binding Domainsof the Nipah Virus V Protein Reveals Mechanisms
Underlying Interferon EvasionJason J. Rodriguez, Cristian D. Cruz, and Curt M. Horvath*
Immunobiology Center, The Mount Sinai School of Medicine, New York, New York 10029
Received 8 November 2003/Accepted 19 December 2003
The V proteins of Nipah virus and Hendra virus have been demonstrated to bind to cellular STAT1 andSTAT2 proteins to form high-molecular-weight complexes that inhibit interferon (IFN)-induced antiviraltranscription by preventing STAT nuclear accumulation. Analysis of the Nipah virus V protein has revealed aregion between amino acids 174 and 192 that functions as a CRM1-dependent nuclear export signal (NES).This peptide is sufficient to complement an export-defective human immunodeficiency virus Rev protein, anddeletion and substitution mutagenesis revealed that this peptide is necessary for both V protein shuttling andcytoplasmic retention of STAT1 and STAT2 proteins. However, the NES is not required for V-dependent IFNsignaling inhibition. IFN signaling is blocked primarily by interaction between Nipah virus V residues 100 to160 and STAT1 residues 509 to 712. Interaction with STAT2 requires a larger Nipah virus V segment betweenamino acids 100 and 300, but deletion of residues 230 to 237 greatly reduced STAT2 coprecipitation. Further,V protein interactions with cellular STAT1 is a prerequisite for STAT2 binding, and sequential immunopre-cipitations demonstrate that V, STAT1, and STAT2 can form a tripartite complex. These findings characterizeessential regions for Henipavirus V proteins that represent potential targets for therapeutic intervention.
The biological effects of alpha and beta interferon (IFN-�and -�) are mediated by a transcription factor complex, ISGF3,that is comprised of the signal transducer and activator oftranscription (STAT) proteins, STAT1 and STAT2, in associ-ation with a DNA-binding subunit, an IFN regulatory factor,IRF9 (1, 12). IFN-induced, ISGF3-mediated transcription re-sults in a cellular antiviral state that provides protection againsta broad range of virus types. In response to this negativeselection, most viruses have evolved adaptations that allowthem to evade IFN-induced innate antiviral responses (13, 26).
In the case of the paramyxovirus family of negative-strandRNA viruses, IFN signaling to ISGF3 is disrupted by directtargeting of STAT proteins (4). Current evidence from thestudy of several family members indicates that the commonoutcome of STAT targeting and IFN evasion is achieved byindividual paramyxovirus species through a diverse range ofmolecular mechanisms. For example, the Rubulaviruses, simianvirus 5 (SV5), human parainfluenza virus 2, and mumps virusall encode STAT-directed E3 ubiquitin ligase activities thatfunction in combination with cellular proteins to targetSTAT1, STAT2, or STAT3 for proteasomal degradation (5, 18,19, 28, 29). Distinctly, measles virus, a prototype Morbillivirus,does not induce STAT ubiquitylation and degradation butinstead prevents IFN-induced ISGF3 assembly and STAT pro-tein nuclear translocation (17, 27). These IFN evasion mech-anisms rely on protein-protein interactions between STATsand the paramyxovirus-encoded V protein. The paramyxovirusV protein is characterized by a highly conserved cysteine-richC-terminal domain (CTD), a zinc-binding domain that is
essential for virus replication in IFN-competent systems andfor diverse V protein activities including cell cycle arrest, re-striction of apoptosis, suppression of IRF3 activity, and pro-tein-protein interactions (6, 9, 14, 15, 22, 23, 28). As an IFNresponse modifier, the V protein is a virulence and pathogen-esis-determining factor. In the case of SV5, species-specificrestriction of STAT1 degradation has been shown to be adeterminant of host species tropism, restricting SV5 replica-tion in IFN-competent murine cells (20, 31).
A new genus of recently emerged paramyxoviruses, Henipa-virus, was demonstrated to share V-dependent IFN signalingevasion properties with other paramyxoviruses (21, 24, 25).Encoded by a polycistronic gene, the 405-amino-acid V proteinN terminus is shared with the co-amino-terminal P and Wproteins (3). In addition, a C protein is encoded in an alternateopen reading frame. In comparison to the STAT-degradingRubulavirus V proteins, the V proteins of Nipah virus andHendra virus, the two known Henipavirus species, share ap-proximately 55% amino acid identity within the CTD. Despitethis CTD conservation, it is dispensable for IFN signaling in-hibition (21). The Henipavirus V protein N terminus is entirelyunique compared to other paramyxovirus proteomes, and theV proteins have no obvious homology to any cellular protein.The Henipavirus V proteins are overall 58% identical in aminoacid sequence, with 81% identity between amino acids (aa) 1 to140, 44% identity between aa 141 to 405, and 83% identitywithin the CTD (aa 406 to 456). This sequence conservationaccounts for the functional similarity in the IFN evasion activ-ities of the Nipah virus and Hendra virus V proteins. Both Vproteins have been demonstrated to subvert IFN responses bysequestering STAT1 and STAT2 in high-molecular-weight cy-toplasmic complexes (24, 25). In addition to the ability to bindto both STAT1 and STAT2, the Henipavirus V proteins ex-hibit nuclear-cytoplasmic shuttling behavior that depends on
* Corresponding author. Mailing address: Immunobiology Center,The Mount Sinai School of Medicine, Box 1630, 1 Gustave L. Levy Pl.,New York, NY 10029. Phone: (212) 659-9406. Fax: (212) 849-2525.E-mail: [email protected].
5358
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

CRM1-dependent nuclear export signals (NES). Not only doesthis shuttling affect the steady-state subcellular distribution ofthe V protein, but it also alters the distribution of the latentSTAT1, relocalizing the protein to the cytoplasm (24, 25).
To decipher the molecular mechanisms underlying Heni-pavirus IFN evasion activities, the functional domains of theNipah virus V protein involved in nuclear cytoplasmic distri-bution and STAT protein binding were examined experimen-tally. The NES and minimal interaction domains for bothSTAT1 and STAT2 were identified as discrete regions withinNipah virus V protein aa 100 to 300. The Nipah virus NESmaps within aa 174 to 192, a peptide that is necessary andsufficient for directing protein subcellular distribution. IFNevasion activity is independent of nuclear export but correlatesexactly with the ability of Nipah virus V amino acid residues100 to 160 to interact with STAT1 residues 509 and 712. Cel-lular STAT1 is required for V protein interactions withSTAT2, and therefore, the STAT2 interaction region is con-strained to a larger overlapping region between aa 100 and300, but evidence suggests that contact between STAT2 andNipah virus V requires aa 230 to 237. Moreover, a tripartiteprotein complex between Nipah virus V, STAT1, and STAT2is demonstrated.
MATERIALS AND METHODS
Cell culture. Human 2fTGH, 293T, U3A (STAT1 deficient), and U6A(STAT2 deficient) cells were maintained in Dulbecco’s modified Eagle’s medium(Gibco-BRL) supplemented with 10% cosmic calf serum (HyClone) and 1%penicillin-streptomycin (Gibco-BRL). U3A cells stably expressing either STAT1or STAT1-STAT3 chimeric fusion proteins (15083 and 35141) (8) were main-tained in the above-mentioned medium supplemented with G418 (Calbiochem).
Plasmids and reagents. Expression vectors for FLAG epitope-tagged Nipahvirus and Hendra virus V proteins were obtained as described previously (24, 25).To create truncated Nipah virus V proteins, PCR products were generated withBglII and NotI restriction sites and were ligated into a pEFN expression vectorcontaining an in-frame N-terminal FLAG epitope tag. Truncations were made atamino acid positions 100 (�4, �5), 220 (�3, �6), 300 (�2), and 406 (�1). Togenerate Rev1.4-green fluorescent protein (GFP) Nipah virus V fusion proteins,PCR products flanked by BamHI and AgeI restriction sites were ligated into aRev1.4-GFP expression vector (7) to place the Nipah virus V peptide segment inframe and between the Rev1.4 and GFP. Nipah virus V deletion mutants (�NES,�S2) and Nipah virus V NES point mutants (L174A, V175A, and L186A) weregenerated by a three-step PCR. Final PCR products were ligated into pEFNFLAG epitope expression vector as described above. All constructs were verifiedby DNA sequencing.
Luciferase and Rev1.4 reporter gene assays. Luciferase reporter gene assayswere performed as described previously (24, 25). 2fTGH cells were transfectedwith expression vectors for an ISRE-luciferase reporter gene, empty vector, ordifferent Nipah virus and Hendra virus V expression vectors described above.Cells were treated with 1,000 U of IFN-�/ml for 6 h prior to lysis and luciferaseassays. All conditions represent the average values from triplicate samples, nor-malized to cotransfected Renilla luciferase and expressed as percentages ofIFN-stimulated controls.
Rev1.4-GFP reporter gene assays were performed essentially as described inreference 7. 2fTGH cells were transfected with expression vectors for Rev1.4-GFP, Rev1.4-NES-GFP, or Rev1.4-GFP fused to full-length Nipah virus V orvarious Nipah virus V peptide segments. Five hours prior to fixation, cells weretreated with either 5 �g of actinomycin D (ACTD)/ml or 10 ng of leptomycin B(LMB)/ml. All samples were treated with 20 �g of cycloheximide/ml to preventnew protein synthesis. After 24 h, cells were fixed and permeabilized and GFPfluorescence was visualized by a confocal scanning microscope as describedpreviously (24, 25). Quantitative analysis was done by visually scoring ACTD-treated cells (n � 100) for cytoplasmic GFP fluorescence in a blind comparison.
Immunofluorescence microscopy. Immunofluorescence was carried exactly asdescribed previously (24, 25). Samples were stimulated with either 1,000 U ofIFN-�/ml or 5 ng of IFN-�/ml for 30 min or 10 ng of LMB/ml for 5 h prior tofixation. Fixed and permeabilized cells were stained with antisera to STAT1 (6.6
�g/ml; Santa Cruz), STAT2 (6.6 �g/ml; Santa Cruz), and FLAG epitope tag(24.5 �g/ml; Sigma). Images were visualized and collected with a Leica TCSSPconfocal microscope.
Cell extracts, immunoblotting, and immunoprecipitation. For preparationof cell extracts, cells were lysed in whole-cell extract buffer supplemented with1 mM dithiothreitol, 1 mM NaVO4, and protease inhibitors (Roche) exactly asdescribed previously (24). For immunoblotting, cell extracts were separated bysodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), trans-ferred to nitrocellulose, probed with antisera to STAT1�, STAT1�/�, STAT2,STAT3 (Santa Cruz), and FLAG epitope tag (Sigma), and visualized with chemi-luminescence (NEN Life Sciences). For immunoprecipitation of FLAG epitope-tagged proteins, cell extracts were incubated with M2 affinity gel exactly asdescribed previously (28). Immunoprecipitates were then separated by SDS-PAGE and analyzed by immunoblotting for coprecipitated proteins. For immu-noprecipitation of GFP fusion proteins, lysates (2 mg) were first precleared withprotein G-agarose for 1 h and incubated with antiserum to GFP for 4 h withrocking at 4°C. Immunoprecipitated complexes were then incubated with proteinG-agarose for 12 h, washed five times with whole-cell extract buffer, separated bySDS-PAGE, and analyzed by immunoblotting with specific antisera.
RESULTS
Subcellular distribution of Nipah virus N-terminal frag-ments. Protein shuttling between nucleus and cytoplasm in-volves competitive interactions between nuclear localizationsignals and NES. Many NES sequences have been character-ized as having regularly spaced leucine or hydrophobic resi-dues, similar to that of the Rev protein from human immuno-deficiency virus type 1 (HIV-1). However, export signals oftendo not conform to the Rev paradigm and must be identified byexperimental approaches (11).
To test for functional domains directing cytoplasmic accu-mulation of Nipah virus V, truncated FLAG epitope-taggedNipah virus V protein fragments were constructed. The largestfragment, �1, is truncated at the P/V/W RNA editing site and,hence, contains aa 1 to 406 in common with the P, V, and Wproteins but none of the CTD residues. The remaining con-structs are truncated within lower-homology regions in be-tween highly conserved Henipavirus V protein domains: theC-terminal boundaries of �2, �3, and �4 are residues 300, 220,and 100, respectively, while the �5 and �6 N-terminal bound-aries are residues 100 and 220, respectively (Fig. 1A). Thesefragments were expressed by transfection of human 2fTGHcells, and the distribution of the expressed proteins was ana-lyzed by indirect immunofluorescence with antisera to the N-terminal FLAG epitope tag (Fig. 1B). The steady-state distri-bution of the largest N-terminal fragment, �1, or a shorterpiece, �2, were indistinguishable from that of full-length Nipahvirus V, exhibiting exclusive cytoplasmic accumulation (Fig.1B). Truncation to aa 220 (�3) resulted in a clearly cytoplasmicaccumulation but with a slightly increased nuclear staining, andfurther truncation to residue 100 (�4) produced a greaternuclear accumulation pattern (Fig. 1B). N-terminal truncationof Nipah virus V to residue 100 (�5) had no effect on thecytoplasmic accumulation, whereas further N-terminal trunca-tion to residue 220 (�6) demonstrated a nuclear cytoplasmicdistribution. Like the full-length protein, all of the truncated Vproteins accumulated in the nucleus when exposed to LMB,indicating that nuclear export is driven by a CRM1-dependentmechanism. The nuclear cytoplasmic accumulation patterns ofthese truncated V proteins suggest that signals controlling Vprotein distribution map to the N terminus and that an LMB-sensitive Nipah virus V NES lies between aa 100 and 220.
VOL. 78, 2004 NIPAH VIRUS V PROTEIN FUNCTIONAL DOMAINS 5359
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

Dissection of the Nipah virus V protein NES. Inspection ofthe Nipah virus V protein primary amino acid sequence did notclearly identify a region similar to other well-defined NESpeptides. To experimentally define the NES region, a Revcomplementation strategy devised by Henderson and Elefthe-riou was utilized (7). The HIV-1 Rev protein shuttles betweenthe nucleus and cytoplasm due to a well-characterized NESand nuclear localization signal. Rev import into the nucleusand nucleolus is sensitive to the drug ACTD, whereas exportfrom the nucleus is CRM1 dependent and sensitive to LMB. Inthis system, an export-defective HIV-1 Rev protein mutant,Rev1.4, which contains an inactivated NES, is fused to GFP.Putative Nipah virus V NES sequences were subcloned be-tween Rev1.4 and GFP, expressed as tripartite fusions, andtested for their nuclear export activity.
A series of Rev1.4-Nipah virus V-GFP fusions were con-structed as illustrated in Fig. 2A, and the use of this reporterwith Nipah virus V is demonstrated in Fig. 2B. The Rev1.4mutant remains in the nucleus regardless of drug treatments,but addition of the Rev NES reconstitutes a shuttling protein,
as revealed by cytoplasmic distribution in the presence ofACTD and nuclear accumulation in response to LMB treat-ment (all panels include cycloheximide to prevent new proteinsynthesis that might obscure localization patterns). Similarly,fusion with the full-length Nipah virus V protein produces ashuttling protein, consistent with CRM1-dependent export(24). Treatment with ACTD or LMB confirms the shuttlingbehavior of Nipah virus V in the Rev1.4-GFP fusion system.
The Nipah virus V aa 100 to 300 also complemented Rev1.4to produce a shuttling protein (Fig. 2C), supporting the local-ization of a Nipah virus V NES to this region. Analysis of aseries of subfragments reveals that aa 100 to 220, but not aa220 to 300, carry NES activity. Nuclear export activity was alsoassociated with both aa 130 to 190 and aa 160 to 220 but not aa100 to 160. Quantification of the distribution patterns ofACTD-treated cells confirms that the nuclear export activitymaps clearly to aa 100 to 220. The smaller fragments weresomewhat attenuated in their ability to relocalize Rev1.4 to thecytoplasm, but a statistically significant number of cells exhib-ited a cytoplasmic distribution pattern, suggesting that an NESsufficient for Rev1.4 complementation lies near or within aa160 to 220 (Fig. 2C).
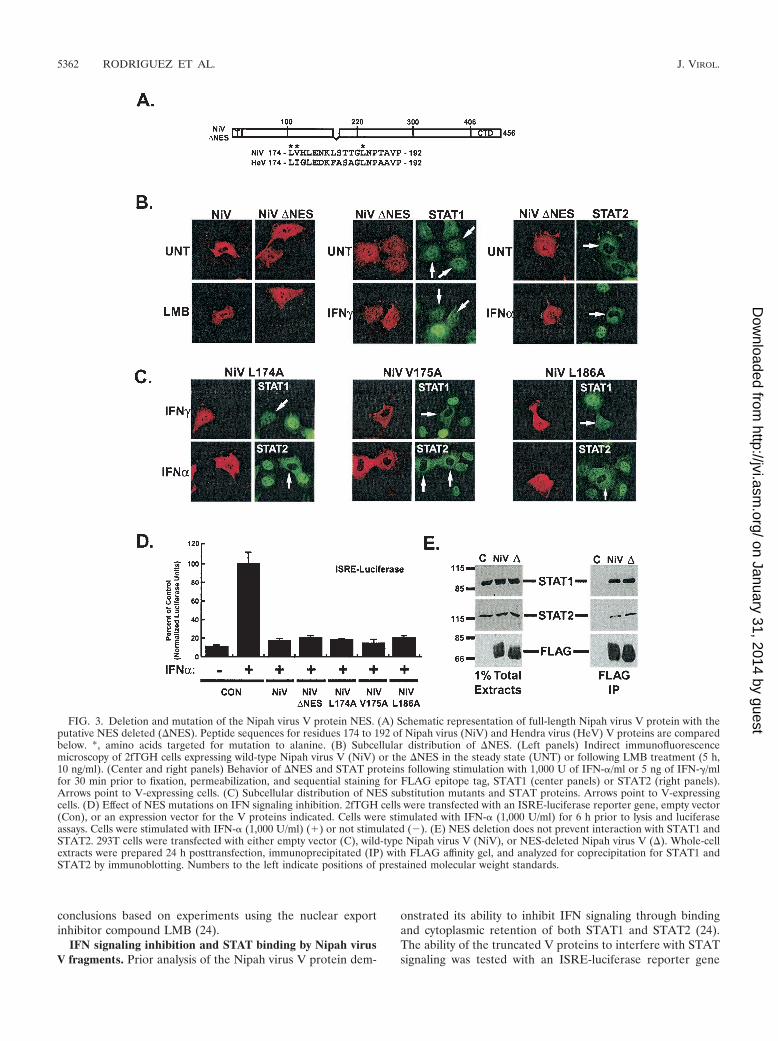
Mutation of the Nipah virus V NES. NES peptides oftencontain regularly spaced leucine residues, although some well-characterized NES peptides do not conform to this archetype(7). Examination of the Nipah virus and Hendra virus V pro-tein sequences within aa 160 to 220 reveals a highly conservedcandidate NES peptide between aa 174 and 192 that containsseveral leucine residues (Fig. 3A). To test if this candidateNES was needed to establish the Nipah virus V protein cyto-plasmic distribution pattern, this peptide was deleted from thefull-length protein to generate a deletion mutant, �NES (Fig.3A). The subcellular distribution of �NES is substantially dif-ferent from that of wild-type Nipah virus V protein, as it lackscytoplasmic accumulation (Fig. 3B). Treatment with LMB re-sulted in redistribution of the wild-type Nipah virus V proteinto the nucleus but did not change the distribution pattern ofthe mutant protein, indicating that the deletion eliminates theCRM1-dependent shuttling.
To determine the ability of �NES to alter STAT subcellulardistribution, the STAT1 and STAT2 accumulation patternswere also analyzed in �NES-expressing cells (Fig. 3B). STAT1was found in both the nucleus and cytoplasm of �NES-express-ing cells, and there was no relocalization of STAT1 to thecytoplasm as observed with wild-type Nipah virus V (24) (Fig.4C). However, STAT1 did not relocalize to the nucleus inIFN-stimulated �NES-expressing cells, and it retained the dis-tribution pattern typical of unstimulated cells. Similarly, IFN-dependent redistribution of STAT2 was prevented by �NES.These findings indicate that NES activity and STAT interfer-ence are not strictly linked.
To determine the amino acid residues within the NES regionrequired for nuclear export, conserved leucines at positions174 and 186 or a nonconserved valine at position 175 wasindividually mutated to alanine, and the subcellular distribu-tion patterns of the mutant proteins were analyzed (Fig. 3C).Mutant V175A was indistinguishable from the wild type andhad no effect on the cytoplasmic accumulation of Nipah virusV. In contrast, both L174A and L186A mutants failed to ac-
FIG. 1. Subcellular distribution of Nipah virus V protein frag-ments. (A) Schematic diagram of truncated Nipah V proteins. Boxesillustrate positions of the CTD and FLAG epitope tag (T). (B) 2fTGHcells transfected with expression plasmids for FLAG-tagged Nipahvirus V (NiV) protein and truncations (�1 to �6) were either untreated(UNT) or treated with 10 ng of LMB/ml for 5 h prior to fixation andpermeabilization. Fixed cells were stained with antiserum to the FLAGepitope tag and visualized with a confocal scanning microscope.
5360 RODRIGUEZ ET AL. J. VIROL.
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

cumulate in the cytoplasm. These two leucine residues are nec-essary elements of the Nipah virus V protein NES.
The effect of the mutant V proteins on STAT distributionwas also analyzed by immunofluorescence. Like wild-type Nipahvirus V, mutant V175A caused a redistribution of STAT1 tothe cytoplasm of unstimulated cells (data not shown) and alsoprevented IFN-induced nuclear redistribution of STAT1 andSTAT2 (Fig. 3C), indicating that this mutation does not alterthe activity of Nipah virus V protein. Analysis of Nipah virusV mutants L174A and L186A revealed a different STAT lo-calization phenotype. Neither leucine mutant induced relocal-ization of latent STAT1 to the cytoplasm (data not shown),consistent with a loss of shuttling activity. Both leucine substi-tutions prevented IFN-induced STAT1 and STAT2 nuclearredistribution, resulting in a pattern similar to that of cells notstimulated with IFN (Fig. 3C). Together, these data indicate
that within the NES, amino acid residues 174 and 186 arerequired for the export of Nipah virus V and basal STAT1redistribution. Since the export-deficient Nipah virus V pro-teins prevented IFN-induced STAT nuclear accumulation,export activity is not strictly required for IFN signaling in-hibition.
To directly test whether the NES has a role in IFN signalingevasion, assays for both IFN inhibition and STAT proteininteractions were carried out. The �NES protein and leucinesubstitution mutants retained the ability to disengage IFN-dependent gene regulation (Fig. 3D). Deletion of the NESdoes not disrupt the interaction with STAT1 and STAT2 (Fig.3E). These functional assays confirm that the Nipah virus VNES mutants are not grossly malfolded and suggest that thecellular distribution of the V protein is not a formal determi-nant of IFN evasion activity, a result consistent with earlier
FIG. 2. Localization of a CRM1-dependent NES in the Nipah virus V (NiV) protein. (A) Illustration of the Rev1.4-GFP fusions analyzed.(B) Use of Rev1.4-Nipah V-GFP fusions. (Top) 2fTGH cells were transfected with expression vectors for Rev1.4-GFP, Rev1.4-NES-GFP, andfull-length Nipah virus V (NiV) fused to Rev1.4-GFP and fixed and permeabilized 24 h later for confocal microscopy. CHX, cycloheximide; ACTD,actinomycin D; LMB, leptomycin B. (Bottom) Quantitation of NES activity. ACTD-treated cells were scored for cytoplasmic GFP (n � 100).Images were inverted in grayscale to better visualize cytoplasmic fluorescence. (C, Top) Localization of Nipah virus V NES. Same is in panel Bbut using Nipah virus V fragments fused to Rev1.4-GFP. (Bottom) Quantitation of NES activity (n � 100).
VOL. 78, 2004 NIPAH VIRUS V PROTEIN FUNCTIONAL DOMAINS 5361
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from
conclusions based on experiments using the nuclear exportinhibitor compound LMB (24).
IFN signaling inhibition and STAT binding by Nipah virusV fragments. Prior analysis of the Nipah virus V protein dem-
onstrated its ability to inhibit IFN signaling through bindingand cytoplasmic retention of both STAT1 and STAT2 (24).The ability of the truncated V proteins to interfere with STATsignaling was tested with an ISRE-luciferase reporter gene
FIG. 3. Deletion and mutation of the Nipah virus V protein NES. (A) Schematic representation of full-length Nipah virus V protein with theputative NES deleted (�NES). Peptide sequences for residues 174 to 192 of Nipah virus (NiV) and Hendra virus (HeV) V proteins are comparedbelow. *, amino acids targeted for mutation to alanine. (B) Subcellular distribution of �NES. (Left panels) Indirect immunofluorescencemicroscopy of 2fTGH cells expressing wild-type Nipah virus V (NiV) or the �NES in the steady state (UNT) or following LMB treatment (5 h,10 ng/ml). (Center and right panels) Behavior of �NES and STAT proteins following stimulation with 1,000 U of IFN-�/ml or 5 ng of IFN-�/mlfor 30 min prior to fixation, permeabilization, and sequential staining for FLAG epitope tag, STAT1 (center panels) or STAT2 (right panels).Arrows point to V-expressing cells. (C) Subcellular distribution of NES substitution mutants and STAT proteins. Arrows point to V-expressingcells. (D) Effect of NES mutations on IFN signaling inhibition. 2fTGH cells were transfected with an ISRE-luciferase reporter gene, empty vector(Con), or an expression vector for the V proteins indicated. Cells were stimulated with IFN-� (1,000 U/ml) for 6 h prior to lysis and luciferaseassays. Cells were stimulated with IFN-� (1,000 U/ml) (�) or not stimulated (�). (E) NES deletion does not prevent interaction with STAT1 andSTAT2. 293T cells were transfected with either empty vector (C), wild-type Nipah virus V (NiV), or NES-deleted Nipah virus V (�). Whole-cellextracts were prepared 24 h posttransfection, immunoprecipitated (IP) with FLAG affinity gel, and analyzed for coprecipitation for STAT1 andSTAT2 by immunoblotting. Numbers to the left indicate positions of prestained molecular weight standards.
5362 RODRIGUEZ ET AL. J. VIROL.
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

assay. All of the truncated V proteins effectively preventedinduction of the ISRE reporter gene except �4 (aa 1 to 100)and �6 (aa 220 to 456), indicating that a peptide segmentbetween aa 100 to 220, the same region containing the NES, isimportant for inhibition of IFN signaling (Fig. 4A). To analyzewhether Nipah virus V protein fragments interact with STAT1and STAT2, FLAG-tagged V protein fragments were ex-pressed and immunoprecipitated, and then the immune com-
plexes were probed for STAT1, STAT2, and STAT3 (Fig. 4B).Full-length Nipah virus V, the two largest N-terminal frag-ments (�1 and �2), and the larger C-terminal fragment (�5)exhibited similar levels of association with STAT1 and STAT2.In contrast, fragment �3 (aa 1 to 220) coprecipitated STAT1but not STAT2. Fragment �4 (aa 1 to 100) bound to neitherSTAT protein. N-terminal truncation to amino acid residue220 (�6) lost the ability to bind STAT1 but retained a weak,
FIG. 4. Localization of Nipah virus V regions for IFN signaling inhibition and STAT binding. (A) 2fTGH cells were transfected with anISRE-luciferase reporter gene and the Nipah virus V expression vectors indicated and subjected to luciferase assays. Cells were stimulated withIFN-� (1,000 U/ml) (�) or not stimulated (�). (B) Interaction of Nipah virus V (NiV) fragments with STAT proteins. Whole-cell extracts fromtransfected cells were prepared 24 h posttransfection and immunoprecipitated (IP) with FLAG affinity gel (Sigma). Total extracts (left panels) andimmunoprecipitates (right panels) were separated by SDS-PAGE and analyzed by immunoblotting with antisera to STAT1, STAT2, STAT3, orFLAG epitope tag. Numbers to the left indicate positions of prestained molecular weight standards. (C) Effect of Nipah virus V fragments onSTAT subcellular distribution. Expression vectors for Nipah virus V and truncations were transfected into 2fTGH cells and processed for indirectimmunofluorescence. Arrows point to V-expressing cells.
VOL. 78, 2004 NIPAH VIRUS V PROTEIN FUNCTIONAL DOMAINS 5363
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

but reproducible, capacity to coprecipitate STAT2. The ob-served 20% IFN signaling inhibition by �6 (Fig. 4A) mightbe a result of this weak interaction with STAT2. None of thefragments interacted with STAT3. These binding results dem-onstrate that STAT1 interacts with Nipah virus V aa 100 to 220but that optimal STAT2 interaction requires residues 100 to300.
The effect of Nipah virus V protein fragments on STATdistribution was tested by indirect immunofluorescence mi-croscopy. All of the Nipah virus V fragments except �4 and�6 redistributed STAT1 to the cytoplasm of unstimulated cells(data not shown) and prevented IFN-induced STAT1 andSTAT2 nuclear accumulation (Fig. 4C). Fragment �6 (aa 220to 456) was slightly capable of preventing STAT2 nuclear ac-cumulation in response to IFN-�, but the phenotype was notcompletely penetrant, possibly the result of the observed weakinteraction with STAT2. These data indicate that the region ofNipah virus V protein specifying STAT interference lies be-tween aa 100 and 220.
Localization of STAT protein binding domains. To furtherdelineate the site(s) of STAT association, the set of Rev1.4-GFP fusion proteins was used for coimmunoprecipitation ex-periments. Lysates from transfected cells were immunoprecipi-tated with GFP-specific antiserum, and the immune complexeswere tested for the presence of STAT1 or STAT2 by immu-noblotting. The Rev1.4-GFP protein did not interact with ei-ther STAT protein, but both STAT1 and STAT2 were presentin immune complexes containing the full-length Nipah virusV protein or aa 100 to 300 (Fig. 5A), but smaller fragmentsdifferentially coprecipitated the two STATs. STAT1 was co-precipitated by aa 100 to 220 and aa 100 to 160, mappingSTAT1 interactions to a 60-aa peptide. In contrast, none of theshorter fragments coprecipitated STAT2. In conjunction withdifferential STAT binding of truncated Nipah virus V proteins,these data indicate separate but overlapping binding sites forSTAT1 and STAT2.
To verify that these tripartite fusion proteins could imitateIFN evasion by Nipah virus V protein, the Rev1.4-GFP serieswere tested for IFN-� signaling inhibition. All of the fragmentsencompassing aa 100 to 160 were capable of preventing IFN-responsive transcription (Fig. 5B). These data demonstrate thatinteraction with STAT1 is the primary determinant of Nipahvirus V IFN evasion and is mediated by amino acid residues100 to 160.
Because �6 retained weak STAT2 binding in the absence ofa STAT1 interaction site, the presence of a STAT2 interactionsite downstream of aa 220 was inferred. In this region, a pep-tide that is highly conserved between Nipah virus and Hendravirus V proteins was identified between amino acid residues230 and 271. Deletion of 7 aa from the N terminus of thisconserved peptide generated a Nipah virus V protein (�S2)that was capable of coprecipitating STAT1 (Fig. 5C) but defi-cient for STAT2 interaction. This finding verifies the presenceof individual STAT1 and STAT2 binding sites on the Nipahvirus V protein.
FIG. 5. Localization of STAT1 and STAT2 binding sites on theNipah virus V (NiV) protein. (A) Selective STAT binding by Nipahvirus V fragments. 293T cells were transfected with the Rev1.4-GFPfusion vectors indicated and lysates immunoprecipitated with anti-serum to GFP. Immune complexes were probed with antisera forSTAT1 or STAT2. Immunoblotting for GFP demonstrates similarexpression in total extracts. (B) IFN signaling inhibition correlates withSTAT1 binding. Cells were transfected with an ISRE-luciferase re-porter gene and the Rev1.4-Nipah V-GFP fusion proteins indicated.Cells were stimulated with IFN-� (1,000 U/ml) (�) or not stimulated(�). (C) STAT2 binding requires Nipah virus V residues 230 to 237.Whole-cell extracts from cells expressing Nipah virus V (N) or deletionof residues 230 to 237 (�S2) were immunoprecipitated (IP) withFLAG affinity gel. Total extracts (left panels) and immunoprecipitates(right panels) were analyzed by immunoblotting with antisera toSTAT1�/�, STAT2, or FLAG epitope tag. (D) Nipah virus V bindsSTAT1 residues 509 to 712. STAT1-deficient U3A cells stably express-ing complementary STAT1-STAT3 fusion proteins 13F and 31F de-scribed in the text were transfected with either empty vector (C) orHA-tagged Nipah virus V (N), and extracts were immunoprecipitatedwith FLAG affinity gel and processed by immunoblotting for HAepitope tag, STAT1�, or STAT3. Numbers to the left of immunoblots
in panels A, C, and D indicate positions of prestained molecular weightstandards.
5364 RODRIGUEZ ET AL. J. VIROL.
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

To determine the STAT protein region that interacts withNipah virus V protein, U3A cell lines stably expressing FLAG-tagged complementary STAT1-STAT3 fusion proteins wereused (8, 19). Expression vectors for these fusions, 13F (aa 1 to508 of STAT1 fused to aa 514 to 770 of STAT3) and 31F (aa1 to 514 of STAT3 fused to aa 509 to 750 of STAT1) weretransfected with hemagglutinin (HA)-tagged Nipah virus Vexpression vector, and lysates were subjected to FLAG im-munoprecipitation (Fig. 5D). Immunoblotting demonstratescoprecipitation of Nipah virus V protein only in the cellsexpressing the C-terminal region (aa 509 to 750) of STAT1, afragment that encompasses the STAT protein linker domain,SH2 domain, and transcriptional activation domain. STAT1�,a splice variant lacking the STAT1 C-terminal 38 aa, also wasfound to coprecipitate with Nipah virus V (Fig. 5C), narrowingthe Nipah virus V contact region to STAT1 residues 509 to712.
STAT2 binding requires STAT1. Since the minimal interac-tion sites for STAT1 and STAT2 overlap, the ability of theSTATs to independently bind to Henipavirus V protein wastested (Fig. 6A). Parental 2fTGH cells, STAT1-deficient U3Acells, and STAT2-deficient U6A cells were transfected withexpression vectors for FLAG tagged Nipah virus and Hendravirus V proteins and tested for copurification of STATs. Inintact cells (2fTGH cells), the V proteins coprecipitated bothSTAT1 and STAT2, and in the absence of STAT2 (U6A cells)the V proteins coprecipitated STAT1, confirming independent
interaction between V and STAT1. In contrast, neither V pro-tein coprecipitated STAT2 in the absence of STAT1 (U3Acells). Complementation of the U3A cells by stable expressionof a STAT1 cDNA rescued coprecipitation of STAT2, indi-cating that cellular STAT1 is required for the Henipavirus Vproteins to bind STAT2.
The ability of Henipavirus V proteins to form a trimericcomplex with STAT1 and STAT2 was also tested. FLAG-tagged Henipavirus V proteins or FLAG-tagged GFP wereaffinity purified from transfected cells, and interacting proteinswere eluted with a FLAG tripeptide. Eluates were then sub-ject to a second immunoprecipitation with antisera to eitherSTAT1 or STAT2 (Fig. 6B), and the immune complexes werethen tested for the presence of STAT1 and STAT2 by immu-noblotting. In cells expressing control GFP, no interactionbetween STAT1 and STAT2 was detected. Reimmunoprecipi-tation of V protein eluates with antiserum to either STATprotein revealed an interaction with the confederate STATprotein. These data indicate that Henipavirus V proteins forma trimeric protein complex containing both STAT1 and STAT2.
DISCUSSION
The Henipavirus V protein encodes an intrinsic nuclear ex-port activity and is able to mediate IFN signaling evasionthrough interaction with cellular STAT proteins. In this study,discrete V protein sequences mediating nuclear export and
FIG. 6. Henipavirus V proteins can form tripartite complexes but require STAT1 to interact with STAT2. (A) STAT2 interaction requiresSTAT1. Parental 2fTGH cells, STAT2-deficient U6A cells, STAT1-deficient U3A cells, and U3A cells stably complemented with STAT1 weretransfected with either empty vector (C) or expression vectors for FLAG epitope-tagged Nipah virus V (N) or Hendra virus V (H). After 24 hposttransfection, cells were lysed and extracts were immunoprecipitated with FLAG affinity gel. Coprecipitation of STAT1 and STAT2 wasanalyzed by immunoblotting. (B) Henipavirus V proteins can interact with STAT1 and STAT2 in the same protein complex. Cell extracts expressingFLAG-tagged GFP (G) or Nipah virus (N) or Hendra virus (H) V proteins were affinity purified with FLAG M2 affinity gel (left panel), andpeptide-eluted protein complexes were immunoprecipitated (IP) a second time (right panels) with antisera for STAT1 or STAT2. Immunecomplexes were separated by SDS-PAGE and tested for the presence of STAT1 and STAT2 by immunoblotting with specific antisera. Numbersto the left of immunoblots in panels A and B indicate positions of prestained molecular weight standards. (C) Sequence alignment of amino acids100 to 300 of Nipah virus (NiV) and Hendra virus (HeV) V proteins. The STAT binding sites and NES are indicated by overlining. (D) Schematicdiagram summarizing the functional domains and protein interactions mapped in this study.
VOL. 78, 2004 NIPAH VIRUS V PROTEIN FUNCTIONAL DOMAINS 5365
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

STAT interaction were delineated. Results indicate that all ofthese activities map within Nipah virus V protein amino acids100 to 300.
The Nipah virus V protein NES that is required for itsCRM1-dependent cytoplasmic accumulation was identified.This NES encompasses aa 174 to 192, a peptide that is highlyconserved between Nipah virus and Hendra virus not only inthe V protein but also in the P and W proteins. NES peptidesfrequently contain 10 to 30 aa with indispensable leucine res-idues at irregularly spaced intervals (7). Sequence comparisonsindicate that the Henipavirus NES is distinct from other knowncellular and viral NES peptides that mediate CRM1-depen-dent export. This NES was demonstrated to be required for theobserved V-dependent redistribution and retention of latentSTAT1 from the nucleus to the cytoplasm. However, in spite ofits essential role in controlling V protein shuttling behavior,the NES is dispensable for interaction with either STAT1 orSTAT2 and for the execution of V-dependent IFN signalinginhibition. In fact, results from deletion and substitution mu-tagenesis indicate that the NES and IFN evasion activities areseparable functions of a similar protein region encompassingamino acid residues 100 to 300.
STAT1 and STAT2 are bound by distinct regions of theNipah virus V protein. STAT1 contacts Nipah virus V throughinteraction with a �60-aa peptide between Nipah virus V res-idues 100 and 160 and STAT1 residues 509 to 712 (Fig. 6D).This STAT1 interaction site expressed in cells as a heterolo-gous Rev1.4-GFP fusion protein is sufficient to mediate IFNsignaling inhibition, suggesting that contact with STAT1 issufficient to block the antiviral response. The domain structureof STAT1 has been well characterized, and the V proteinsinteract with the SH2 and Linker domains of the STAT1 pro-tein core (2). The SH2 domain is the primary receptor recog-nition motif and also an essential component of phosphoty-rosine-mediated STAT dimerization. The function of thelinker domain is not well understood, but sequence perturba-tions alter STAT1 DNA binding affinity (30). Apparently, theHenipavirus V protein has evolved to contact these domains toprevent STAT activation, dimerization, and transcriptional ac-tivity. Furthermore, it was observed that Nipah virus V seg-ments lacking this STAT1-binding peptide are also unable toefficiently bind to STAT2. The STAT2 interaction region ofNipah virus V maps to a larger segment, requiring aa 100 to300. Deletion of aa 230 to 237 greatly impaired STAT2 copre-cipitation, but it is important to note that this is merely theextreme N terminus of a longer peptide (aa 230 to 271) that ishighly conserved (67% identical) between Nipah virus andHendra virus.
It was also observed that cellular expression of STAT1 isabsolutely required for Henipavirus V proteins to bind toSTAT2, as precipitation of V proteins from cells deficient inSTAT1 did not copurify STAT2. This phenotype was comple-mented by STAT1 expression, indicating it is a direct conse-quence of STAT1 availability and not the result of additionalunidentified alterations in the STAT1-deficient cells. In con-trast, STAT1 was readily detected in V protein precipitationsfrom STAT2-deficient cells, in agreement with previously re-ported Nipah virus V-induced IFN-� signaling inhibition in theabsence of cellular STAT2 (24). The requirement for STAT1to efficiently bind STAT2 in part explains the observation that
STAT2 requires a larger interaction site; it reflects the need toaccommodate the STAT1-binding region. This codependentrelationship between STAT1 and STAT2 is somewhat similarto the requirement for accessory STAT proteins in IFN signal-ing evasion by the Rubulavirus V protein-dependent ubiquitinligases. Both SV5 and mumps virus V proteins strictly requirecellular STAT2 expression to target STAT1, whereas humanparainfluenza virus 2 requires cellular STAT1 for STAT2 tar-geting (19, 29). In fact, for SV5, the requirement for STAT2provides the molecular basis for host range restriction, as themurine STAT2 orthologue fails to support STAT1 degradationand IFN evasion (20). The exact role for this STAT codepen-dence is currently unknown, but available evidence suggeststhat V proteins form multisubunit targeting complexes thatutilize both cellular and viral partners, and the confederateSTAT may provide contact sites for the recruitment of essen-tial components (28).
Sequential immunoprecipitation demonstrates that in intactcells, the Henipavirus V proteins can form tripartite complexeswith both STAT1 and STAT2. It is likely that independentSTAT1-V complexes are also formed, supported by the higherabundance of STAT1 found in STAT1 immune complexescompared to STAT2 immune complexes (Fig. 6B). Based onthe known molecular mass of each protein, the predicted massof this tripartite complex would be in the range of 300 kDa.This figure is in agreement with earlier gel filtration estimatesof 300 to 500 kDa (24), but it is reasonable to assume thatadditional cellular proteins may also be associated with theexpressed Henipavirus V proteins, as this has been demon-strated for several other paramyxovirus V proteins.
The results of this study indicate that the highly conservedcysteine-rich CTD of the Henipavirus V proteins is entirelydispensable for all aspects of STAT-directed IFN signalinginhibition. Instead, the minimal peptide region capable ofSTAT1 binding and IFN evasion maps to aa 100 to 160 andprovides a mechanistic explanation for the observed rescue ofan IFN-sensitive reporter virus and IFN signaling suppressionconferred by the common N-termini of V and W proteins (21).The Henipavirus proteins, P and W, which are translated frompartially overlapping open reading frames, share the same406-aa N terminus prior to the editing site. Clearly, all proteinsencoded by this reading frame include the STAT binding sitesand NES defined in this report. It has yet to be demonstratedwhether the Henipavirus P protein is capable of attenuatingIFN responses, but considering that the P protein shares the Nterminus, it is predicted that, if accessible to STAT proteins,the P protein would in principle block IFN signal transduction.The W protein consists of the same N terminus linked to adifferent C terminus, and its ability to block IFN signaling hasbeen demonstrated (21). The presence of an NES in P and Wproteins also predicts that they might have an intrinsic abilityto shuttle between cytoplasm and nucleus. Interestingly, exam-ination of the primary amino acid sequence of the W proteinC-terminal extension reveals two pairs of basic residues thatare suggestive of a bipartite nuclear localization signal. Theroles for shuttling proteins in Henipavirus physiology is yet tobe determined.
Given that the only homology between the Henipavirus Vproteins and other paramyxovirus V proteins is within theconserved CTD, a significant question remains regarding
5366 RODRIGUEZ ET AL. J. VIROL.
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from

the function of the zinc-binding region. For some paramyxo-viruses, the CTD has been demonstrated to be required for Vprotein function in IFN antagonism (6, 9, 16). Furthermore,for several paramyxoviruses, expression of the isolated CTDalone has been reported to evade IFN responses (10, 16, 21).However, expression of the CTD alone is not sufficient for IFNevasion by all species tested (e.g., compare results for New-castle disease virus and Nipah virus V in reference 21) andnotably fails to copurify proteins important for Rubulavirus Vprotein-dependent ubiquitin ligase complexes (28). Therefore,this domain may play a role in additional aspects of IFN eva-sion, including the prevention of de novo IFN biosynthesis (6,23). It is also likely that Henipavirus V proteins have evolved touse different peptide regions to counteract IFN signaling, whilethe conservation of the CTD is reflective of a broader functionin the context of virus infection. It may be relevant that otherparamyxoviruses have been demonstrated to form intracyto-plasmic inclusions that contain viral proteins, nucleic acids,IRF3, and STATs (6, 17, 29), but more detailed analysis of Vprotein functions in the context of intact viruses will be re-quired to shed light on the precise role of the CTD.
Further studies encompassing Henipavirus infection must becarried out to examine the potential for a common role for theN termini of V, W, and P proteins in the inhibition of IFNsignaling and viral pathogenesis. Although the precise contri-butions of all protein products generated from the polycis-tronic V/P/W/C locus towards Henipavirus replication andpathogenesis are yet to be determined, the functional domainsdefined here represent logical targets for the development ofspecific Henipavirus antiviral compounds.
ACKNOWLEDGMENTS
We are grateful to Beric Henderson for providing the Rev1.4-GFPreagents, Christina Ulane for assistance with FLAG purification meth-ods, and members of the Horvath laboratory for helpful discussionsand comments on the manuscript.
This study was supported by research grants to C.M.H. from theAmerican Cancer Society (Research Scholar Grant 103079) and theNIH (AI-48722, AI-50707, and AI-55733). J.J.R. is a trainee of theintegrated training program in pharmacological sciences (GM-62754).
REFERENCES
1. Aaronson, D. S., and C. M. Horvath. 2002. A road map for those who don’tknow JAK-STAT. Science 296:1653–1655.
2. Chen, X., U. Vinkemeier, Y. Zhao, D. Jeruzalmi, J. E. Darnell, Jr., and J.Kuriyan. 1998. Crystal structure of a tyrosine phosphorylated STAT-1 dimerbound to DNA. Cell 93:827–839.
3. Chua, K. B., W. J. Bellini, P. A. Rota, B. H. Harcourt, A. Tamin, S. K. Lam,T. G. Ksiazek, P. E. Rollin, S. R. Zaki, W. Shieh, C. S. Goldsmith, D. J.Gubler, J. T. Roehrig, B. Eaton, A. R. Gould, J. Olson, H. Field, P. Daniels,A. E. Ling, C. J. Peters, L. J. Anderson, and B. W. Mahy. 2000. Nipah virus:a recently emergent deadly paramyxovirus. Science 288:1432–1435.
4. Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. Sendaivirus and simian virus 5 block activation of interferon-responsive genes:importance for virus pathogenesis. J. Virol. 73:3125–3133.
5. Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. The Vprotein of simian virus 5 inhibits interferon signalling by targeting STAT1 forproteasome-mediated degradation. J. Virol. 73:9928–9933.
6. He, B., R. G. Paterson, N. Stock, J. E. Durbin, R. K. Durbin, S. Goodbourn,R. E. Randall, and R. A. Lamb. 2002. Recovery of paramyxovirus simianvirus 5 with a V protein lacking the conserved cysteine-rich domain: themultifunctional V protein blocks both interferon-beta induction and inter-feron signaling. Virology 303:15–32.
7. Henderson, B. R., and A. Eleftheriou. 2000. A comparison of the activity,sequence specificity, and CRM1-dependence of different nuclear export sig-nals. Exp. Cell Res. 256:213–224.
8. Horvath, C. M., Z. Wen, and J. E. Darnell, Jr. 1995. A STAT protein domainthat determines DNA sequence recognition suggests a novel DNA-bindingdomain. Genes Dev. 9:984–994.
9. Kawano, M., M. Kaito, Y. Kozuka, H. Komada, N. Noda, K. Nanba, M.Tsurudome, M. Ito, M. Nishio, and Y. Ito. 2001. Recovery of infectioushuman parainfluenza type 2 virus from cDNA clones and properties of thedefective virus without V-specific cysteine-rich domain. Virology 284:99–112.
10. Kubota, T., N. Yokosawa, S. Yokota, and N. Fujii. 2001. C terminal CYS-RICH region of mumps virus structural V protein correlates with block ofinterferon alpha and gamma signal transduction pathway through decreaseof STAT 1-alpha. Biochem. Biophys. Res. Commun. 283:255–259.
11. la Cour, T., R. Gupta, K. Rapacki, K. Skriver, F. M. Poulsen, and S. Brunak.2003. NESbase version 1.0: a database of nuclear export signals. NucleicAcids Res. 31:393–396.
12. Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control andbiological impact. Nat. Rev. Mol. Cell Biol. 3:651–662.
13. Levy, D. E., and A. Garcia-Sastre. 2001. The virus battles: IFN induction ofthe antiviral state and mechanisms of viral evasion. Cytokine Growth FactorRev. 12:143–156.
14. Lin, G. Y., and R. A. Lamb. 2000. The paramyxovirus simian virus 5 Vprotein slows progression of the cell cycle. J. Virol. 74:9152–9166.
15. Lin, G. Y., R. G. Paterson, C. D. Richardson, and R. A. Lamb. 1998. The Vprotein of the paramyxovirus SV5 interacts with damage-specific DNA bind-ing protein. Virology 249:189–200.
16. Nishio, M., M. Tsurudome, M. Ito, M. Kawano, H. Komada, and Y. Ito. 2001.High resistance of human parainfluenza type 2 virus protein-expressing cellsto the antiviral and anti-cell proliferative activities of alpha/beta interferons:cysteine-rich V-specific domain is required for high resistance to the inter-ferons. J. Virol. 75:9165–9176.
17. Palosaari, H., J. P. Parisien, J. J. Rodriguez, C. M. Ulane, and C. M.Horvath. 2003. STAT protein interference and suppression of cytokine sig-nal transduction by measles virus V protein. J. Virol. 77:7635–7644.
18. Parisien, J.-P., J. F. Lau, J. J. Rodriguez, B. M. Sullivan, A. Moscona, G. D.Parks, R. A. Lamb, and C. M. Horvath. 2001. The V protein of humanparainfluenza virus 2 antagonizes type I interferon responses by destabilizingsignal transducer and activator of transcription 2. Virology 283:230–239.
19. Parisien, J.-P., J. F. Lau, J. J. Rodriguez, C. M. Ulane, and C. M. Horvath.2002. Selective STAT protein degradation induced by paramyxoviruses re-quires both STAT1 and STAT2, but is independent of alpha/beta interferonsignal transduction. J. Virol. 76:4190–4198.
20. Parisien, J. P., J. F. Lau, and C. M. Horvath. 2002. STAT2 acts as a hostrange determinant for species-specific paramyxovirus interferon antagonismand simian virus 5 replication. J. Virol. 76:6435–6441.
21. Park, M. S., M. L. Shaw, J. Munoz-Jordan, J. F. Cros, T. Nakaya, N.Bouvier, P. Palese, A. Garcia-Sastre, and C. F. Basler. 2003. Newcastledisease virus (NDV)-based assay demonstrates interferon-antagonist activityfor the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.77:1501–1511.
22. Paterson, R. G., G. P. Leser, M. A. Shaughnessy, and R. A. Lamb. 1995. Theparamyxovirus SV5 V protein binds two atoms of zinc and is a structuralcomponent of virions. Virology 208:121–131.
23. Poole, E., B. He, R. A. Lamb, R. E. Randall, and S. Goodbourn. 2002. TheV proteins of simian virus 5 and other paramyxoviruses inhibit induction ofinterferon-beta. Virology 303:33–46.
24. Rodriguez, J. J., J. P. Parisien, and C. M. Horvath. 2002. Nipah virus Vprotein evades alpha and gamma interferons by preventing STAT1 andSTAT2 activation and nuclear accumulation. J. Virol. 76:11476–11483.
25. Rodriguez, J. J., L. F. Wang, and C. M. Horvath. 2003. Hendra virus Vprotein inhibits interferon signaling by preventing STAT1 and STAT2 nu-clear accumulation. J. Virol. 77:11842–11845.
26. Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev.14:778–809.
27. Takeuchi, K., S. I. Kadota, M. Takeda, N. Miyajima, and K. Nagata. 2003.Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBSLett. 545:177–182.
28. Ulane, C. M., and C. M. Horvath. 2002. Paramyxoviruses SV5 and HPIV2assemble STAT protein ubiquitin ligase complexes from cellular compo-nents. Virology 304:160–166.
29. Ulane, C. M., J. J. Rodriguez, J. P. Parisien, and C. M. Horvath. 2003.STAT3 ubiquitylation and degradation by mumps virus suppress cytokineand oncogene signaling. J. Virol. 77:6385–6393.
30. Yang, E., M. A. Henriksen, O. Schaefer, N. Zakharova, and J. E. Darnell, Jr.2002. Dissociation time from DNA determines transcriptional function in aSTAT1 linker mutant. J. Biol. Chem. 277:13455–13462.
31. Young, D. F., N. Chatziandreou, B. He, S. Goodbourn, R. A. Lamb, and R. E.Randall. 2001. Single amino acid substitution in the V protein of simian virus5 differentiates its ability to block interferon signaling in human and murinecells. J. Virol. 75:3363–3370.
VOL. 78, 2004 NIPAH VIRUS V PROTEIN FUNCTIONAL DOMAINS 5367
on January 31, 2014 by guesthttp://jvi.asm
.org/D
ownloaded from




















