
Identification of a novel mechanism for the removal of glucose residues from high mannose-type...
7
THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1992 by The American Society for Biochemistry and Molecular Biology, Inc Vol. 267, No. 30, Issue of October 25, pp. 21671-21677,1992 Printed in U.S. A. Identification of a Novel Mechanism forthe Removal of Glucose Residues from High Mannose-type Oligosaccharides* (Received for publication, May 22, 1992) Kyungsun SuhS, Christopher A. Gabel$, and John E. Bergmannll From the DeDartment of Anatomy and Cell Biology, Columbia University College of Physicians and Surgeons, New York, New York 10032 ” . The roleof glucosylated oligosaccharides in the bio- genesis of the glycoprotein (G protein) of vesicular stomatitis virus was studied in PhaR2.’, a mouse lym- phoma cell line deficient in glucosidase I1 activity. As expected, the great majority of cell-associated G pro- tein remained glucosylated in PhaR2.7, and the G pro- tein was rapidly deglucosylated in BW5147, the paren- tal cell line. Despite these differences in glucosylation, the rates of G protein trimerization and transport to the cell surface were as rapid and efficient in the PhaR2.7 mutant as in BW6147. Surprisingly, greater than 73% of the oligosaccharides on G proteins re- covered from released virions were complex-type units. The efficient processing of the G protein oligo- saccharides coincided with the efficient removal of glucose residues from its oligosaccharides. Aftertreat- ment with deoxynojirimycin, an inhibitor of endo- plasmic reticulum (ER)glucosidases I and 11, the total percentage of G protein-associated high mannose-type oligosaccharides increased more in the parental cells than in the mutant cells. Furthermore, when the G protein was retained in the ER of PhaR2.7 cells by depletion of the cellular ATP pools with carbonyl cy- anide m-chlorophenylhydrazone, its oligosaccharides remained glucosylated. Under identical conditions, BW5147 cells removed the glucose residues from > 90% of the retained G protein’s oligosaccharides. Thus, PhaR2.7 cells efficiently remove glucose residues from high mannose-type oligosaccharides of selected pro- teins using a deoxynojirimycin-insensitive enzyme lo- cated in a post-ER compartment. The existence of a second mechanism for the deglucosylation of N-linked oligosaccharides provides evidence for the important role of glucose removal in glycoprotein maturation. N-linked glycosylation occurs cotranslationally by the en bloc transfer of a preformed oligosaccharide from a dolichol- linked donor to nascent polypeptides within the rough endo- * These studies were supported by National Institutes of Health Grants GM32617 and GM33342 and career development awards (to J. E. B. and C. A. G.) from the Hirschl-Monique Foundation. The costs of publication of thisarticle were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. $ Present address: Rockefeller University, 1230 York Ave., New York, NY 10021. Present address: Dept. of Immunology and Infectious Diseases, Central Research Division, Pfizer, Inc., Eastern Point Rd., Groton, CT 06340. ll Present address: Cell Culture Research and Development, Cen- tocor, 244 Great Valley Pkwy., Malvern, PA 19355. Tel.: 215-889- 4507; Fax: 215-651-6201. To whom correspondence should besent,. plasmic reticulum. The preformed oligosaccharide is invariant in its structure and contains 3 glucose, 9 mannose, and 2 N- acetylglucosamine residues (1, 2). The addition of only glu- cosylated oligosaccharides to nascent glycoproteins appears to be important as the oligosaccharyltransferase has evolved to preferentially transfer only the glucosylated oligosaccha- rides from the lipid-linked donor to nascent polypeptides (3- 6). The glucose residues, however, are removed subsequently by the action of ER’-associated glucosidases I and 11. Such removal is necessary for an oligosaccharide to be processed to a complex-type unit (7,8). The role of these transient glucose residues in protein biogenesis remains unknown. In a previous study we characterized the asparagine-linked oligosaccharides associated withtwo unrelated mutant forms of the G protein of vesicular stomatitis virus (VSV), ts045 and pdTM12 (9). Structural analysis revealed that most of the high mannose-type oligosaccharides attached to these ER- retained G proteins contained a single glucose residue posi- tioned at a nonreducing terminus. In contrast, high mannose- type oligosaccharides associated with other glycoproteins are rapidly deglucosylated (10-16); removal of glucose residues begins even before the translation of a nascent glycoprotein is finished (11). The mutant G protein-associated glucosylated oligosaccharides were derived, in part, from an unusual post- translational reglucosylation of previously deglucosylated ol- igosaccharides (9). The reglucosylation of these mutant G proteins’ oligosac- charides suggested that the glucose residues “mark”these improperly folded proteins. As previous studies hadsuggested that glucosylated high mannose-type oligosaccharides impair transport of newly synthesized glycoproteins to the Golgi apparatus (17-19) we suggested thatthemarking glucose residue might mediate the prolonged residence of these mu- tant G proteins within the ER (9). To examine the role of glucosylated high mannose-type oligosaccharides in the bio- genesis of the wild-type VSV G protein, we examined the G protein’s post-translational maturation in a mutant mouse lymphoma cell line (PhaR2.7) deficient in glucosidase I1 (20). We demonstrate that despite a marked deficiency of glucosi- dase I1 activity, the maturation of the G protein in these cells is rapid. We further demonstrate that removal of the glucose residues from the G protein’s high mannose-type oligosaccha- rides is mediated by a novel processing activity. ‘The abbreviations used are: ER, endoplasmic reticulum; VSV, vesicular stomatitis virus; MEM, minimal essential medium; Hepes, 4-(2-hydroxyethyl)-l-piperazineethanesulfonic acid; PBS, phos- phate-buffered saline; SDS, sodium dodecyl sulfate; ConA, concana- valin A; HPLC, high performance liquid chromatography; Mes, 4- morpholineethanesulfonic acid; EGTA, [ethylenehis(oxyethyleni- tri1o)ltetraacetic acid dNM, deoxynojirimycin; CCCP, carbonyl cya- nide m-chlorophenylhydrazone; endoH, endoglycosidase H;TBS, Tris-buffered saline. 21671
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Identification of a novel mechanism for the removal of glucose residues from high mannose-type...

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1992 by The American Society for Biochemistry and Molecular Biology, Inc
Vol. 267, No. 30, Issue of October 25, pp. 21671-21677,1992 Printed in U.S. A.
Identification of a Novel Mechanism for the Removal of Glucose Residues from High Mannose-type Oligosaccharides*
(Received for publication, May 22, 1992)
Kyungsun SuhS, Christopher A. Gabel$, and John E. Bergmannll From the DeDartment of Anatomy and Cell Biology, Columbia University College of Physicians and Surgeons, New York, New York 10032
” .
The role of glucosylated oligosaccharides in the bio- genesis of the glycoprotein (G protein) of vesicular stomatitis virus was studied in PhaR2.’, a mouse lym- phoma cell line deficient in glucosidase I1 activity. As expected, the great majority of cell-associated G pro- tein remained glucosylated in PhaR2.7, and the G pro- tein was rapidly deglucosylated in BW5147, the paren- tal cell line. Despite these differences in glucosylation, the rates of G protein trimerization and transport to the cell surface were as rapid and efficient in the PhaR2.7 mutant as in BW6147. Surprisingly, greater than 73% of the oligosaccharides on G proteins re- covered from released virions were complex-type units. The efficient processing of the G protein oligo- saccharides coincided with the efficient removal of glucose residues from its oligosaccharides. After treat- ment with deoxynojirimycin, an inhibitor of endo- plasmic reticulum (ER) glucosidases I and 11, the total percentage of G protein-associated high mannose-type oligosaccharides increased more in the parental cells than in the mutant cells. Furthermore, when the G protein was retained in the ER of PhaR2.7 cells by depletion of the cellular ATP pools with carbonyl cy- anide m-chlorophenylhydrazone, its oligosaccharides remained glucosylated. Under identical conditions, BW5147 cells removed the glucose residues from > 90% of the retained G protein’s oligosaccharides. Thus, PhaR2.7 cells efficiently remove glucose residues from high mannose-type oligosaccharides of selected pro- teins using a deoxynojirimycin-insensitive enzyme lo- cated in a post-ER compartment. The existence of a second mechanism for the deglucosylation of N-linked oligosaccharides provides evidence for the important role of glucose removal in glycoprotein maturation.
N-linked glycosylation occurs cotranslationally by the en bloc transfer of a preformed oligosaccharide from a dolichol- linked donor to nascent polypeptides within the rough endo-
* These studies were supported by National Institutes of Health Grants GM32617 and GM33342 and career development awards (to J. E. B. and C. A. G.) from the Hirschl-Monique Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Present address: Rockefeller University, 1230 York Ave., New York, NY 10021.
Present address: Dept. of Immunology and Infectious Diseases, Central Research Division, Pfizer, Inc., Eastern Point Rd., Groton, CT 06340.
ll Present address: Cell Culture Research and Development, Cen- tocor, 244 Great Valley Pkwy., Malvern, PA 19355. Tel.: 215-889- 4507; Fax: 215-651-6201. To whom correspondence should be sent,.
plasmic reticulum. The preformed oligosaccharide is invariant in its structure and contains 3 glucose, 9 mannose, and 2 N - acetylglucosamine residues (1, 2). The addition of only glu- cosylated oligosaccharides to nascent glycoproteins appears to be important as the oligosaccharyltransferase has evolved to preferentially transfer only the glucosylated oligosaccha- rides from the lipid-linked donor to nascent polypeptides (3- 6). The glucose residues, however, are removed subsequently by the action of ER’-associated glucosidases I and 11. Such removal is necessary for an oligosaccharide to be processed to a complex-type unit (7,8). The role of these transient glucose residues in protein biogenesis remains unknown.
In a previous study we characterized the asparagine-linked oligosaccharides associated with two unrelated mutant forms of the G protein of vesicular stomatitis virus (VSV), ts045 and pdTM12 (9). Structural analysis revealed that most of the high mannose-type oligosaccharides attached to these ER- retained G proteins contained a single glucose residue posi- tioned at a nonreducing terminus. In contrast, high mannose- type oligosaccharides associated with other glycoproteins are rapidly deglucosylated (10-16); removal of glucose residues begins even before the translation of a nascent glycoprotein is finished (11). The mutant G protein-associated glucosylated oligosaccharides were derived, in part, from an unusual post- translational reglucosylation of previously deglucosylated ol- igosaccharides (9).
The reglucosylation of these mutant G proteins’ oligosac- charides suggested that the glucose residues “mark” these improperly folded proteins. As previous studies had suggested that glucosylated high mannose-type oligosaccharides impair transport of newly synthesized glycoproteins to the Golgi apparatus (17-19) we suggested that the marking glucose residue might mediate the prolonged residence of these mu- tant G proteins within the ER (9). To examine the role of glucosylated high mannose-type oligosaccharides in the bio- genesis of the wild-type VSV G protein, we examined the G protein’s post-translational maturation in a mutant mouse lymphoma cell line (PhaR2.7) deficient in glucosidase I1 (20). We demonstrate that despite a marked deficiency of glucosi- dase I1 activity, the maturation of the G protein in these cells is rapid. We further demonstrate that removal of the glucose residues from the G protein’s high mannose-type oligosaccha- rides is mediated by a novel processing activity.
‘The abbreviations used are: ER, endoplasmic reticulum; VSV, vesicular stomatitis virus; MEM, minimal essential medium; Hepes, 4-(2-hydroxyethyl)-l-piperazineethanesulfonic acid; PBS, phos- phate-buffered saline; SDS, sodium dodecyl sulfate; ConA, concana- valin A; HPLC, high performance liquid chromatography; Mes, 4- morpholineethanesulfonic acid; EGTA, [ethylenehis(oxyethyleni- tri1o)ltetraacetic acid dNM, deoxynojirimycin; CCCP, carbonyl cya- nide m-chlorophenylhydrazone; endo H, endoglycosidase H; TBS, Tris-buffered saline.
21671

21672 Deglucosylation of N-Linked Oligosaccharides
MATERIALS AND METHODS
Cell C u l t ~ r e - P h a ~ ~ . ~ cells and BW5147 cells were grown in a- MEM containing 10% fetal bovine serum, 20 mM Hepes, 100 units/
atmosphere of 5% CO, a t 37 "C. ml penicillin, and 100 pg/ml streptomycin and maintained in an
Viral Infection-Cells (1-2 X lo' cells/ml) were mixed with 20 plaque-forming units/cell of wild-type VSV strain WT-2B in growth medium and incubated for 30 min a t 32 "C in an atmosphere contain- ing 5% CO,. To enhance uniform viral adsorption, the cell suspension was shaken every 10 min. The cells were harvested by centrifugation at 1,000 rpm for 5 min, and nonadsorbed virus was discarded. The pellet was resuspended in growth medium at 1-5 X IO6 cells/ml and incubated a t 37 "C in an atmosphere containing 5% CO, for 4 h.
Metabolic Labeling of the G Protein-Virus-infected cells were harvested by centrifugation, and the resulting pellet was washed twice with glucose-free a-MEM. The cell pellet (1-5 X lo7 cells) was resuspended a t lo7 cells/ml in 5 ml of the labeling medium (glucose- free a-MEM, 10% dialyzed fetal bovine serum, and 0.5-1 mCi of [2- "Hlmannose (American Radiolabeled Chemicals Inc., St. Louis, MO; 14 Ci/mmol)) and incubated for 30 min at 37 "C in 5% COz. At the end of the labeling period, the cell suspension was diluted with a- MEM containing 10% fetal bovine serum, 1 mM sodium pyruvate, 10 mM glucose, 10 mM mannose, and 150 p~ cycloheximide. When G protein was to be recovered from the chase medium, the final concen- tration of serum was reduced to 1% to minimize its interference with the immunoprecipitation. At each time point, the labeled cells were harvested by centrifugation, and the resultingpellet was washed twice with 5 ml of PBS at 4 "C. The pelleted cells then were solubilized with 1 ml of lysis buffer containing 1% Triton X-100, 0.4% sodium deoxycholate, and 1 mM phenylmethylsulfonyl fluoride in PBS. Chase media also were centrifuged at 30,000 rpm for 60 min in a Beckman Ti-70.1 rotor to isolate virus-associated G protein. The resultingpellet was resuspended in Laemmli sample buffer (21).
Immunoprecipitation of G Protein-G protein was isolated from cell lysates as described previously (9). To immunoprecipitate extra- cellular G proteins, media samples were adjusted to 0.1% Triton X- 100, 0.04% deoxycholate, and 0.01% SDS and incubated on ice with 10 pg/ml rabbit anti-G protein. Antigen-antibody complexes were recovered by the addition of 100 p1 of fixed Staphylococcus aureus cells/ml of the culture medium (a 10% suspension from Calbiochem). After a 30-min incubation on ice, the bacteria were collected by centrifugation a t 12,000 X g for 20 min, washed five times with 10 mM Tris, pH 8, 10 mM EDTA, 1% Triton X-100, 0.4% deoxycholate, 0.1% SDS, and washed once with 10 mM Tris, pH 8, 1 mM EDTA.
To immunoprecipitate G protein that had reached the plasma membrane, the cells were harvested by centrifugation, washed twice with 10 ml of PBS at 4 "C, and resuspended in 1 ml of PBS containing 20 pg of affinity-purified rabbit antibody to the VSV G protein. After incubation on ice for 50 min, the cells were harvested by centrifuga- tion, washed twice in PBS containing 2 mg/ml bovine serum albumin, and solubilized in 200 p1 of lysis buffer. Nuclei were removed by centrifugation in an Eppendorf microcentrifuge, and the antigen- antibody complexes were recovered as described above.
To quantify the amount of extracellular, cell surface, and total cell- associated G protein simultaneously, cells were separated from the chase medium by centrifugation. The G protein was recovered from the medium fraction as described above, and the cell fraction was washed twice with ice-cold PBS and split. Half of the cells were suspended in 1 ml of lysis buffer, and total cell-associated G protein was immunoprecipitated. The other half was used for the determi- nation of cell surface G protein as detailed above. All immunoprecip- itates were analyzed by SDS-polyacrylamide gel electrophoresis and fluorography as described previously (9).
Isolation and Analysis of G Protein Oligosaccharides-Regions of the dried gels that contained radiolabeled G protein were excised, and radioactive glycopeptides were prepared by Pronase digestion. The glycopeptides were fractionated on concanavalin A (ConA)-Sepharose (Pharmacia LKB Biotechnology, Inc.) as described previously (22).
To calculate the percentage of each species recovered from ConA- Sepharose, the average number of mannose residues present on the high mannose-type oligosaccharides was estimated using data from HPLC analysis of the a-mannosidase digestion products of high mannose-type oligosaccharides (see below). This analysis yielded essentially the same results for high mannose-type oligosaccharides linked to either virus-associated or newly synthesized intracellular G protein, demonstrating that the normal glucosylated forms of the N- linked oligosaccharides are transferred to nascent G proteins in
PhaRZ.7 cells (not shown). The data on high mannose-type oligosac- charides isolated from extracellular G protein have therefore been presented (see Fig. 5). Assuming that the glucosylated and nonglu- cosylated high mannose-type structures contained the same number of mannose residues, the formula for the average number of mannose residues would be
Man., = 1 + ManF + 3/4*ManB ManAG + 1 /4Man~
where ManF are the cpm in the mannitol peak (fractions 20-25), ManAG are the cpm in the mannose-N-acetylglucosaminitol peak (fractions 30-35), and ManB are the cpm in the Glc2ManrN-acetyl- glucosaminitol peak (fractions 70-90). Based on this formula, the high mannose-type oligosaccharides in BW5147 contained on average 7.3 mannose residues, and those in PhaRZ.' contained 6.4 mannose residues. Further, assuming that complex-type oligosaccharides (peaks I and 11) contained 3 labeled mannose and 1 labeled fucose residue (the 3H is retained when mannose is converted to fucose), one must divide the amount of radioactivity in peak 111 by 1.8 (for BW5147) or 1.6 (for PhaRZ.') before estimating the percentage of the oligosaccharides that were processed to complex-type units.
High mannose-type glycopeptides (peak 111 from ConA-Sepharose) were dissolved in 0.025 ml of citrate-phosphate buffer, pH 5.6, and endoglycosidase H (endo H) was added to a final concentration of 50 milliunits/ml. Digestion of the high mannose-type oligosaccharides with jack bean a-mannosidase was performed as described previously (22). Exoglycosidase digests were analyzed by descending paper chro- matography in ethyl acetate/pyridine/acetic acid/HZO (5/5/1/3). The dried chromatograms were cut into 1-cm strips, and the radioactivity associated with each strip was determined by liquid scintillation counting. HPLC analysis was performed as described previously (9).
MEM) were labeled with [35S]methionine for 3 min and incubated Trirnerization Assay-Virus-infected cells (5 X 10' in 0.5 ml of a-
for 6 min in chase medium (a-MEM containing 10% fetal bovine serum, 10 mM glucose, 10 mM mannose, 1 mM sodium pyruvate, 100 units/ml penicillin, and 100 pg/ml streptomycin) at 37 "C. At the end of the chase, the labeled cell suspension was diluted with an equal volume of lysis buffer (40 mM Mes, 60 mM Tris, 2.5 mM EDTA, 2 mM EGTA, pH 5.5) as described by Doms et al. (23). Nuclei were pelleted by centrifugation in an Eppendorf microcentrifuge for 1 min. 200 p1 of each supernatant was loaded onto a 5-20% linear 5-ml sucrose gradient and subjected to centrifugation in a SW 55 rotor (Beckman Instruments) a t 47,000 rpm for 16 h at 4 "C (23). 350-pl fractions were collected. The VSV proteins were immunoprecipitated from each fraction and analyzed by polyacrylamide gel electrophoresis as described above. The locations of the monomer and trimer peaks were judged from the distribution of N/NS and M proteins on the gradients. Radioactivity in the G protein bands was quantified by Pronase digestion followed by scintillation counting.
RESULTS
Kinetics of Trimerization and Secretion of G Protein by PhaR2.7 and BW5147 Cells-To determine whether glucosidase I1 deficiency inhibited the transport of wild-type G protein, VSV infected PhaR2.7 and BW5147 cells were pulse labeled with [35S]methionine and chased in the absence of the isotope. The distribution of the 35S-labeled G protein was determined with respect to the cell surface and the total cell-associated fraction by selective immunoprecipitation (see "Materials and Methods"). At the end of the 10-min pulse, no radiolabeled G proteins were recovered from either the medium or the cell surface (Fig. 1, A and B ) . However, 35S-labeled G proteins were detected at the cell surface and in the medium of both the wild-type (Fig. 1A) and the PhaR2.7 mutant (Fig. 1B) cells after 30 min of chase. The amounts of radiolabeled G protein recovered in each of the three fractions was quantified after excision of the radioactive regions of the gel (Fig. 2). The amount of radioactive G protein produced was higher in BW5147 cells than in PhaR2.7 cells (compare the zero time points in Fig. 2, B and D). This difference was seen consist- ently and may be caused by different rates of methionine uptake by the mutant and wild-type cells or differences in their infectibility. The kinetics of G protein transport to the
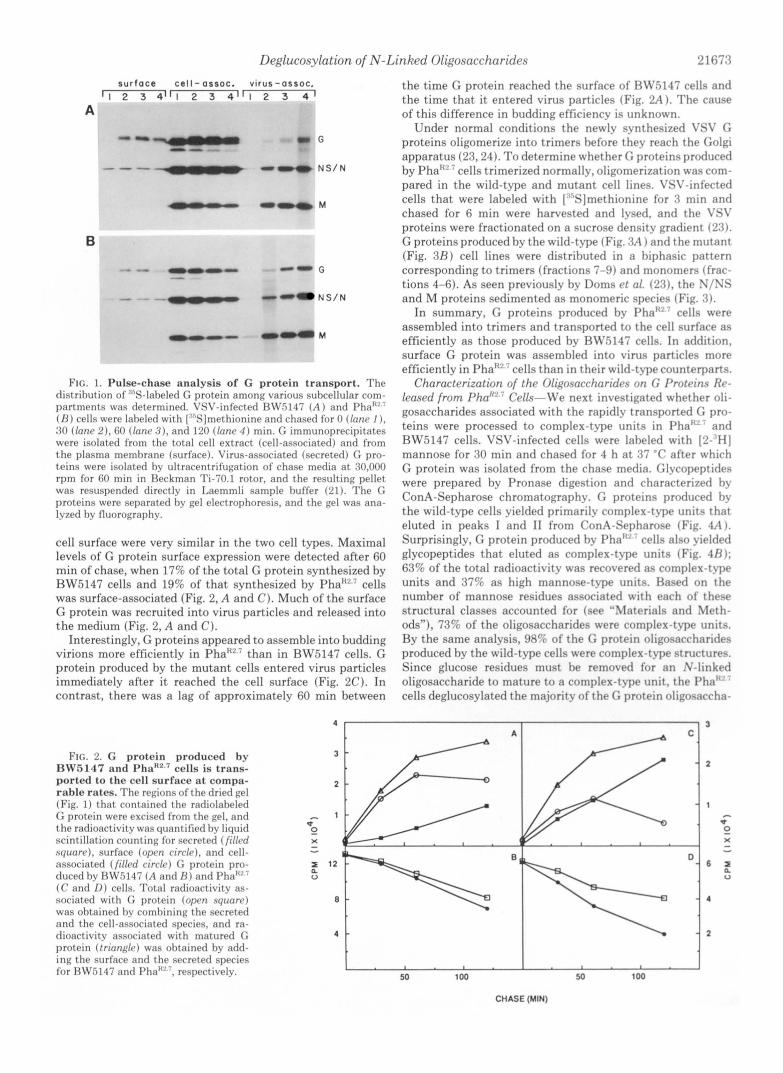
Deglucosylation of N-Linked Oligosaccharides 2 1673
surface ce l l -assoc. v i rus-assoc. ' I 2 3 4 " l 2 3 4 " l 2 3 4 '
A
G
N S / N
N W N
FIG. 1. Pulse-chase analysis of G protein transport. The distrihrltion of .'"S-laheled G protein among various subcellular com- partments was determined. VSV-infected RLV.5147 ( A ) and Pha"" ( R ) cells were Inheled with [:"S]methionine and chased for 0 (lone I ) . 30 (lone 2) . GO (Inn<, : I ) , and 120 (lone 4 ) min. G immunoprecipitates were isolated from the total cell extract (cell-associated) and lrom the plasma memhrane (surface). Vinls-associated (secreted) C pro- teins were isolated hv ultracentrifugation of chase media at 30,000 rpm for 60 min in Heckman Ti-70.1 rotor, and the resulting pellet was resuspended directly in Laemmli sample huffer (21). The G proteins were separated hv gel electrophoresis, and the gel was ana- Ivzed hv fluorography.
cell surface were very similar in the two cell types. Maximal levels of G protein surface expression were detected after 60 min of chase, when 17% of the total G protein synthesized by RW.5147 cells and 19% of that synthesized by Pha":!' cells was surface-associated (Fig. 2, A and C). Much of the surface G protein was recruited into virus particles and released into the medium (Fig. 2, A and C).
Interestingly, G proteins appeared to assemble into budding virions more efficiently in Pha"" than in RW.5147 cells. G protein produced by the mutant cells entered virus particles immediately after it reached the cell surface (Fig. 2C). In contrast, there was a lag of approximately 60 min between
FIG. 2. G protein produced by RWR147 and Pha'c2,' cells is trnns- ported to the cell surface at compa- rnble rates. The regions o f the dried gel (Fig. 1) that contained the radiolaheled (; protein were excised from the gel, and the radioactivitv was quantified hv liquid scintillation counting lnr secreted (filled squorr), surfare (oprn rirrle), and cell- associated (f i l led circle) C, protein pro- duced hv 13W5147 ( A and R ) and I'ha"'' (( ' and 1)) cells. Total radioactivity as- sociated with protein (open nqunrc) was ohtained hy romhining the secreted and the cell-associated species, and ra- dioactivitv associated with matured G protein ( t r innglc) was ohtained hv add- ing the surface and the secreted species lor HW5147 and I'ha""', respectivelv.
the t ime G protein reached the surface of RW5147 cells and the t ime tha t it entered virus particles (Fig. 2A 1. The cause of this difference in budding efficiencv is unknown.
Under normal conditions the newlv synthesized VSV G proteins oligomerize into trimers before thev reach the Golgi apparatus (25,241. To determine whether G proteins produced by Pha""'cel1s trimerized normally, oligomerization was com- pared in the wild-t-ype and mutant cell lines. VSV-infected cells that were labeled with [""Sjmethionine for 3 min and chased for 6 min were harvested and lysed, and the C'SI' proteins were fractionated on a sucrose density gradient ('23). G proteins produced by the wild-t?.pe (Fig. 3A ) and the mutant (Fig. 3 R ) cell lines were distributed in a hiphasic pattern corresponding to trimers (fractions 7-9) and monomers (frar- tions 4-6). As seen previouslv by Doms et 01. ('2.7). the N/NS and M proteins sedimented as monomeric species (Fig. 3) .
In summary, G proteins produced hv J'ha" ' ' cells were assembled into trimers and transported to the cell surface as efficiently as those produced bv H\Z':i147 cells. In addition. surface C protein was assembled into virus particles more efficiently in Pha"" cells than in their wild-type counterparts.
Characterization of the Oligosnccharidcs on G Protcins I+- leased from Pha"" Cells-iYe next investigated whether oli- gosaccharides associated with the rapidlv transported G pro- teins were processed to complex-t?lpe units in Pha""' and RW.5147 cells. VSV-infected cells were labeled with 12- HI mannose for 30 min and chased for 4 h at 37 "C after which G protein was isolated from the chase media. Glycopeptides were prepared bv Pronase digestion and characterized by ConA-Sepharose chromatography. G proteins produced by the wild-t-ype cells yielded primarily complex-t?lpe units that eluted in peaks I and I1 from ConA-Sepharose (Fig. 4A j .
Surprisingly, G protein produced hy Pha" . cells also yielded glycopeptides that eluted as complex-type units (Fig. 4R): 63% of the total radioactivity was recovered as complex-type units and 37% as high mannose-type units. Haserl on the number of mannose residues associated with each of these structural classes accounted for (see "\herials and Meth- ods"), 7.1% of the oligosaccharides were complex-t>pe units. Ry the same analvsis, 98'; of the G protein oligosaccharides produced bv the wild-type cells were complex-type structures. Since glucose residues must be removed for an .\'-linked oligosaccharide to mature to a complex-tvpe unit, the I'ha'!' '
cells deglucosylated the majority of the C, protein oligosaccha-
A C
I I I 50 100 50 100
CHASE (MIN)
21674 Deglucosylation of N-Linked Oligosaccharides
A 1 2 3 4 5 6 7 8 9 101112
"0"" G
L " N S / N
B
" G
" N S / N
- " M
TOP BOTTOM
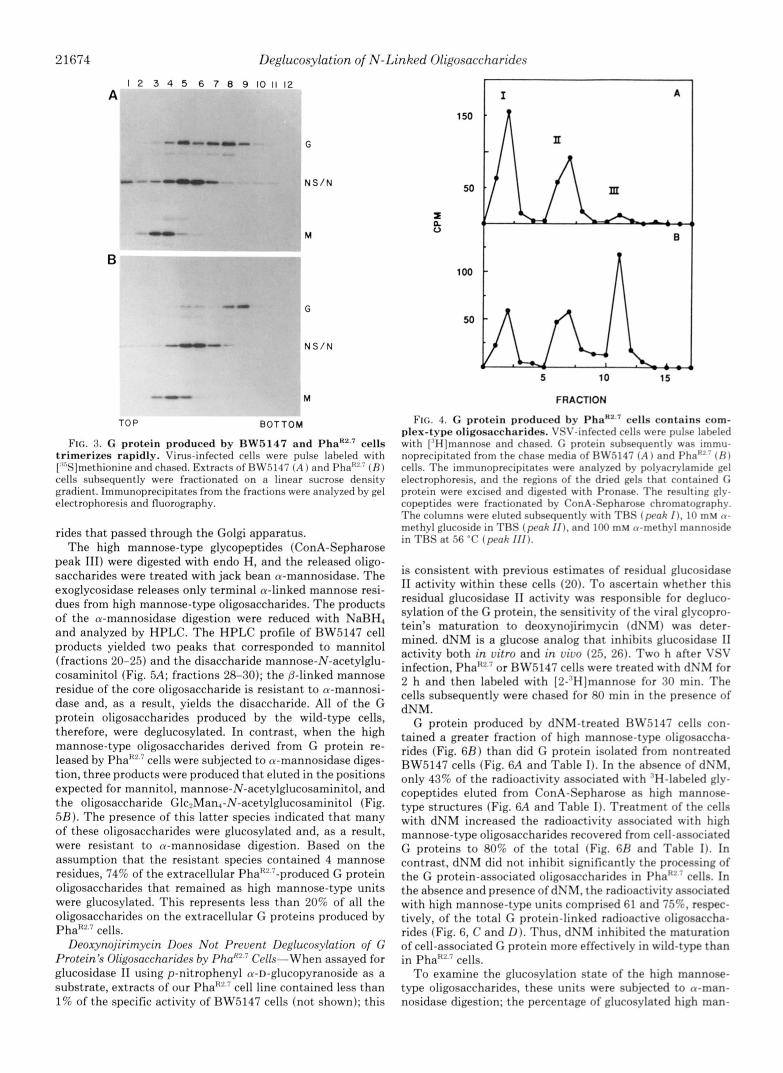
FIG. 3. G protein produced by BW5147 and PhaRZ.' cells trimerizes rapidly. Virus-infected cells were pulse labeled with [""Sjmethionine and chased. Extracts of HW5147 ( A ) and Pha"'.7 ( H ) cells subsequently were fractionated on a linear sucrose density gradient. Immunoprecipitates from the fractions were analyzed by gel electrophoresis and fluorography.
rides that passed through the Golgi apparatus. The high mannose-type glycopeptides (ConA-Sepharose
peak 111) were digested with endo H, and the released oligo- saccharides were treated with jack bean a-mannosidase. The exoglycosidase releases only terminal a-linked mannose resi- dues from high mannose-type oligosaccharides. The products of the a-mannosidase digestion were reduced with NaBH, and analyzed by HPLC. The HPLC profile of RW5147 cell products yielded two peaks that corresponded to mannitol (fractions 20-25) and the disaccharide mannose-N-acetylglu- cosaminitol (Fig. 5A; fractions 28-30); the P-linked mannose residue of the core oligosaccharide is resistant to a-mannosi- dase and, as a result, yields the disaccharide. All of the G protein oligosaccharides produced by the wild-type cells, therefore, were deglucosylated. In contrast, when the high mannose-type oligosaccharides derived from G protein re- leased by Pha"'.' cells were subjected to a-mannosidase diges- tion, three products were produced that eluted in the positions expected for mannitol, mannose-N-acetylglucosaminitol, and the oligosaccharide GlcpMan,-N-acetylglucosaminitol (Fig. 5 R ) . The presence of this latter species indicated that many of these oligosaccharides were glucosylated and, as a result, were resistant to n-mannosidase digestion. Based on the assumption that the resistant species contained 4 mannose residues, 74% of the extracellular PhaR'.'-produced G protein oligosaccharides that remained as high mannose-type units were glucosylated. This represents less than 20% of all the oligosaccharides on the extracellular G proteins produced by
Deoxynojirirnycin Does Not Prevent Deglucosylation of G Protein's Oligosaccharides by Phaf''.7 Cells-When assayed for glucosidase I1 using p-nitrophenyl a-D-ghcopyranoside as a substrate, extracts of our Pha"'.' cell line contained less than 1% of the specific activity of BW5147 cells (not shown); this
PhalC2.7 cells.
150
50
aE n 0
100
56
l 1 A
I B
FRACTION
FIG. 4. G protein produced by PhaR2.' cella contains com- plex-type oligosaccharides. VSV-infected cells were pulse laheled with ["Hjmannose and chased. C protein suhsequentlv was immu- noprecipitated from the chase media of HW.5147 ( A ) and I'ha"' ( H I cells. The immunoprecipitates were analyzed hv polyacrylamide gel electrophoresis, and the regions of the dried gels that contained C protein were excised and digested with Pronase. The resulting glv- copeptides were fractionated hy ConA-Sepharose chromatographv. The columns were eluted subsequently with TRS ( p r a k I ) , 10 m u n- methyl glucoside in THS (prah / I ) , and 100 m u a-methyl mannoside in TRS at 56 "C (peak 111).
is consistent with previous estimates of residual glucosidase I1 activity within these cells (20). To ascertain whether this residual glucosidase I1 activity was responsible for degluco- sylation of the G protein, the sensitivity of the viral glycopro- tein's maturation to deoxynojirimycin (dNM) was deter- mined. dNM is a glucose analog that inhibits glucosidase I1 activity both in vitro and in vivo (25, 26). Two h after VSV infection, or BW5147 cells were treated with dNM for 2 h and then labeled with [2-'H]mannose for 30 min. The cells subsequently were chased for 80 min in the presence of dNM.
G protein produced by dNM-treated RW.5147 cells con- tained a greater fraction of high mannose-t-ype oligosaccha- rides (Fig. 6 R ) than did G protein isolated from nontreated BW5147 cells (Fig. 6A and Table I). In the absence of dNM, only 43% of the radioactivity associated with "H-labeled gly- copeptides eluted from ConA-Sepharose as high mannose- type structures (Fig. 6A and Table I ) . Treatment of the cells with dNM increased the radioactivity associated with high mannose-type oligosaccharides recovered from cell-associated G proteins to 80% of the total (Fig. 6R and Table I) . In contrast, dNM did not inhibit significantly the processing of the G protein-associated oligosaccharides in Pha"" cells. In the absence and presence of dNM, the radioactivity associated with high mannose-type units comprised 61 and 7S%, respec- tively, of the total G protein-linked radioactive oligosaccha- rides (Fig. 6, C and D). Thus, dNM inhibited the maturation of cell-associated G protein more effectively in wild-type than
To examine the glucosylation state of the high mannose- type oligosaccharides, these units were subjected to n-man- nosidase digestion; the percentage of glucosylated hiEh man-
in phal<2.7 cells.

Deglucosylation of N-Linked Oligosaccharides 21675
In summary, dNM treatment greatly inhibited the deglu- cosylation of the high mannose-type oligosaccharides at- tached to the G protein produced by BW5147 cells. However, dNM was less effective in reducing the processing of oligosac- charides in PhaR2.7 cells despite the fact that the specific activity of glucosidase I1 was less than 1% that present in the parental cells. The PhaR2.? cells, therefore, must possess ele- vated levels of a compensatory dNM-insensitive deglucosylat- ing activity.
PhaR2.7 Cells Remove the Glucose Residues from the G Pro- tein in a Post-ER Compartment-To determine the intracel- lular location of this compensatory deglucosylating activity, the extent of deglucosylation of G proteins that were retained in the ER was assessed. PhaR2.? and BW5147 cells were infected with VSV and labeled with [ 2 - 3 H ] m a n n ~ ~ e for 15 min. At the end of the labeling period, the cells either were harvested and lysed immediately or were chased for 90 min in the presence or absence of CCCP. CCCP is an uncoupler of oxidative phosphorylation that lowers cellular ATP levels sufficiently to prevent export of proteins from the ER (27, 28). Cycloheximide also was added to the chase medium to
1200
800
400
400
200
FRACTION
FIG. 5. Virus-associated VSV G protein produced by PhaR2.7 cells contains glucosylated high mannose-type oligosaccha- rides. High mannose-type glycopeptides derived from G proteins released by BW5147 ( A ) or PhaRZ-7 ( B ) cells were digested with endo H. The oligosaccharides in turn were digested with a-mannosidase, reduced with NaBH,, and analyzed by HPLC. The peaks of radioac- tivity eluted in the positions expected for mannitol (fractions 20-25, 2,330 cpm in panel A and 1,198 cpm in panel B ) , mannose-N- acetylglucosaminitol (fractions 30-35, 358 cpm, panel A and 98 cpm, panel B ) , and the oligosaccharide Glc2Man4N-acetylglucosaminitol (fractions 70-90,98 cpm, panel A and 1,097 cpm, panel B ) .
nose-type species was estimated as described above. G protein isolated from the dNM-treated BW5147 cells contained 3- fold more glucosylated units than when isolated from un- treated cells (Table I). Thus, the failure of the G protein to obtain complex-type units in dNM-treated BW5147 cells correlated with an inhibition of glucosidase 11. G protein produced by PhaR2.? cells contained the same number of glucosylated high mannose-type units in the presence and absence of dNM (Table I). Thus, dNM treatment of PhaR2.7 cells neither prevented complex-type oligosaccharide forma- tion (Fig. 6) nor caused the accumulation of glucosylated oligosaccharides.
block subsequent protein synthesis and, thus, improve the effectiveness of the chase. G protein recovered from both cell types immediately after the labeling period contained only high mannose-type oligosaccharides (Table 11). When BW5147 cells were chased for 90 min in the absence of CCCP, the total G protein-associated radioactivity declined; this loss reflects processing of the N-linked oligosaccharides, budding of G protein containing virions into the medium, and degra- dation. As expected, many of the oligosaccharides were proc- essed to complex-type units during the chase; only 52% of the radioactivity associated with the G protein was recovered as high mannose-type oligosaccharides (Table 11). When BW5147 cells were chased in the presence of CCCP, the G protein oligosaccharides remained primarily as high mannose- type structures (Table 11). Again, the total radioactivity de- creased, suggesting that G protein was degraded within the ER of the transport-impaired cells (29). The total amount of radiolabeled G protein isolated from PhaR2.7 cells also declined during the chase in the presence and absence of CCCP (Table 11). In the absence of the ionophore the fraction of G protein oligosaccharides that were high mannose-type structures de- clined, but CCCP prevented the processing of the high man- nose-type oligosaccharides to complex-type units.
FIG. 6. Deglucosylation of the G protein's high mannose-type oligo- 200
sitive to dNM. VSV-infected BW5147 saccharides in PhaR2.7 cells is insen-
( A and B ) and PhaR2.7 (C and D) cells were treated with ( B and D) or without ( A and C) dNM for 2 h (at 2 h postin- fection), labeled with [3H]mannose for 30 min, and chased in the presence or absence of dNM for 80 min. G proteins = were immunoprecipitated from extracts 0
n
of the radiolabeled cells and analyzed by polyacrylamide gel electrophoresis. The regions of the dried gels containing G protein were excised, and the radioactiv- ity was solubilized by Pronase digestion. The resulting glycopeptides were applied 300 t o ConA-Sepharose, and the columns were eluted with TBS (peak I ) , 10 mM tu-methyl glucoside in TBS (peak I I ) , and 100 mM a-methyl mannoside in
100
800
TBS a t 56 "C (peak 111).
I 0 a
5 10 15 5 10 15
FRACTION

21676 Deglucosylation of N-Linked Oligosaccharides
TABLE I Differential effect of d N M on G protein oligosaccharide maturation The quantity of the various glycopeptide species recovered after
ConA-Sepharose chromatography (Fig. 6) is indicated as percentage of total cpm present in the oligosaccharides eluting in ConA-Sepha- rose peak 111. High mannose-type glycopeptides (ConA-Sepharose peak 111 of Fig. 6) were digested with endo H and n-mannosidase. The digestion products were analyzed by descending paper chroma- tography, and the percentage of glucosylated oligosaccharides was calculated based on the amount of radioactivity recovered in the n- mannosidase-resistant fraction ( R ) and with the disaccharide man- nose-N-acetylglucosamine peak ( S ) using the formula:
% glucosylated = - 100*(H/4) ( R / 4 + S ) ‘
cpm in ConA w h cpm X peak Total
peak 1 peak 11 peak 111 111 sylated ated Cells in peak 111 gluco- gi:c:s;
BW.5147 -dNM 24,600 34,100 43,800 42.73 69 30,222 +dNM 11,800 14,600 105,500 79.98 88 29,480
pha!?!.7 -dNM 9,600 11,800 33,500 61.02 88 29,480 +dNM 6,300 6,800 38,800 74.76 93 36,084
TABLE I1 Deglucosylation of G protein oligosaccharides by Phax” cells i n a
post-ER compartment VSV-infectedcells were pulse labeled with [“Hjmannose for 15 min
and chased for 90 min in the presence (+) or absence (-) of CCCP. Glycopeptides were prepared from the immunoprecipitated G protein, and these were treated with endo H and n-mannosidase; the digestion products were analyzed by descending paper chromatography. The percentage of the high mannose-type units that were glucosylated was determined as detailed in Table I.
Cells Total 96 cpm in high X high Total cpm
cpm (ConA peak 111) glucosylated mannose mannose glucosylated
BW5147 0 chase 58,800 98 48 27,659 - CCCP
90-min chase 12,800 52 44 2,929 - CCCP
90-min chase 22,400 90 9 1,814 + CCCP
phaH2.7 0 chase 22,400 97 72 15,644 - CCCP
90-min chase 6,800 58 70 2,761 - CCCP
90-min chase 16,200 98 79 12,542 + CCCP
~~~ ~~ ~ ~~
The extent of glucosylation of the high mannose-type oli- gosaccharides again was determined using the a-mannosidase sensitivity assay. Immediately after labeling, 48% of the high mannose-type oligosaccharides on G protein produced by BW5147 cells were glucosylated, as were 72% of those re- covered from PhaR2.7 cells (Table 11). G protein oligosaccha- rides that remained high mannose-type units during the 90- min chase in the absence of CCCP were comparably glucosy- lated. Treatment of the BW5147 cells with CCCP resulted in the accumulation of deglucosylated high mannose-type oli- gosaccharides; only 9% of the G protein’s high mannose-type oligosaccharides remained glucosylated. In contrast, the G protein oligosaccharides that remained in the ER of PhaR2.7 cells were highly glucosylated. An alternate way of analyzing these data is to estimate the total radioactivity associated
with glucosylated oligosaccharides. When BW5147 cells were chased for 90 min in the presence or absence of CCCP, there was a dramatic reduction in labeled glucosylated oligosaccha- rides associated with G protein (from 27,659 cpm to 1,814 cpm or 2,929 cpm, respectively). Similarly, when PhaR2.7 cells were chased in the absence of CCCP, radioactivity associated with these structures declined from 15,644 to 2,761 cpm. After treatment of PhaR2.7 cells with CCCP, however, 12,542 cpm still were associated with these structures. Thus, despite the prolonged residence within the ER of PhaR2.7 cells, the G proteins were not deglucosylated. Therefore, the dNM-insen- sitive deglucosylating activity must reside in a post-ER com- partment.
DISCUSSION
High mannose-type oligosaccharides attached to several mutant forms of the VSV G protein remain glucosylated within the ER. In part, these glucosylated structures are formed by the post-translational reglucosylation of the deglu- cosylated precursor oligosaccharide (9). The function of this post-translational modification is unknown. However, the persistence of the glucosylated high mannose-type oligosac- charides on the aberrant G proteins in vivo coupled with the recent demonstration that denatured thyroglobulin is a better substrate than the native protein for reglucosylation i n vitro (30) suggested that glucosylation serves as a component of a cellular proofreading machinery.
In this study, we found the wild-type G protein reaches the plasma membrane with normal kinetics in PhaR2.7 cells (Fig. 2). Moreover, the majority of the oligosaccharides attached to the extracellular G protein were complex-type units (Fig. 4). Since removal of glucose residues from the precursor high mannose-type oligosaccharide is required for the formation of complex-type structures, G proteins produced by the mutant cells must be deglucosylated efficiently. This was unexpected since our PhaR2.7 cells possess less than 1% of the glucosidase I1 activity of BW5147 cells (not shown), the wild-type cell line from which PhaR2.7 was derived. Previous studies indi- cated that PhaR2.7 cells contain a much higher level of gluco- sylated high mannose-type oligosaccharides on endogenous proteins than do BW5147 cells (20). The VSV G protein, therefore, must be an especially good substrate for the residual glucosidase I1 activity or for a novel enzyme that is present within these mutant cells.
We have presented two lines of evidence which indicate that the mutant cells contain a novel deglucosylating activity. First, removal of the glucose residues from the G protein required normal cellular ATP levels. When ATP synthesis was inhibited with CCCP, an uncoupler of oxidative phos- phorylation, glucose residues were not removed from the high mannose-type oligosaccharides of G protein produced by PhaR2.7 cells. Glucosidase I1 does not require ATP in vitro (32), and CCCP-treated BW5147 cells continued to remove glucose residues efficiently from the G protein in vivo. Al- though the enzyme responsible for the removal of the glucose residues from the G protein in PhaR2.7 cells may require ATP for its activity, we believe that it is more likely that the deglucosylating enzyme resides within a post-ER compart- ment and that ATP is required for the G protein to reach this compartment. Second, glucosidase I1 is inhibited by the glu- cose analog dNM, and treatment of infected BW5147 cells with dNM increased the number of glucosylated high man- nose-type oligosaccharides on G protein 2.8-fold. Similarly, Schlessinger et al. (31) showed that dNM prevents the deglu- cosylation of G protein in normal chick embryo fibroblasts. If residual glucosidase I1 were responsible for deglucosylation of

Deglucosylation of N-Linked Oligosaccharides 21677
the G protein, then these cells would be expected to be more sensitive to dNM treatment than BW5147 cells. However, treatment with dNM only slightly impaired the removal of glucose residues from the G protein oligosaccharides produced by PhaR2.7 cells.
Thus, our data suggest that PhaR2.7 cells contain a dNM- insensitive enzyme that deglucosylates the G protein’s oligo- saccharides in a post-ER compartment. Since the dNM- treated PhaR2.7 cells processed G proteins more efficiently than dNM-treated BW5147 cells, the mutant cells may have up-regulated this alternative pathway to compensate for their deficiency in glucosidase I1 activity. A possible candidate for this alternate activity is the endomannosidase described by Lubas and Spiro (33, 34). Both its intracellular location and its insensitivity to dNM make this enzyme an attractive candidate for the PhaRZ.? processing activity (33, 34). Using the in vitro conditions described by Lubas and Spiro, however, we were unable to demonstrate a difference in endomannosi- dase activity between PhaR2.7 and BW5147 cells (not shown). Thus, PhaR2.7 cells may use yet another activity to remove the glucose residues from the G protein oligosaccharides.
The ubiquitous presence of glucose residues on the oligo- saccharides initially transferred to glycoproteins suggests an important role for these residues in glycoprotein metabolism. Our earlier demonstration of the post-translational addition of a single glucose residue to a class of mutant G proteins suggested that glucosylation might serve to tag molecules that were not properly folded (9). Lodish and Kong (17) showed that treatment of HepG2 cells with dNM blocked the secre- tion of cqantitrypsin and cYlantichymotrypsin and demon- strated that the block occurred prior to the exit of these proteins from the ER. Other glycoproteins such as C3 and transferrin were secreted normally. Parent et al. (18) extended these findings by showing that the glycoproteins secreted in the presence of dNM were processed in the Golgi apparatus to endo H-resistant forms. These authors concluded that the glycoproteins secreted in the presence of dNM were processed by the residual glucosidase activity within the ER.
In general, our results are consistent with those presented previously. Wild-type G protein appears to fall into the class of proteins whose rate of transport from the ER to the Golgi apparatus is affected minimally by the absence of ER gluco- sidase activity. As observed with hepatic secretory proteins in the presence of dNM (18), the G proteins released from PhaR2.7 cells contained primarily complex-type N-linked oligosaccha- rides that necessarily were deglucosylated. However, as dis- cussed above, our finding that CCCP significantly impaired the deglucosylation reaction suggests that “escape” is not caused by residual ER glucosidase activity. Rather, the G protein oligosaccharides are deglucosylated in a post-ER com- partment. Since the G protein was transported to the cell surface with normal kinetics in PhaR2.7 cells, we conclude that
glucose residues do not hinder the transport of the G protein to this post-ER compartment. An intriguing possibility sug- gested by recent work with GRP78/BiP (35) is that glucosy- lated proteins may migrate to an intermediate compartment between the ER and the Golgi apparatus but recycle to the ER unless the glucose residues are removed.
In conclusion, our data indicate that PhaR2.7 cells have compensated for a deficiency in glucosidase I1 activity by upregulating a second system capable of efficiently degluco- sylating glycoproteins in a post-ER compartment. These ob- servations suggest that the removal of the glucose residues is of great importance to proper cell function.
Acknowledgments-We thank Linda Friedman and Edith Abreu for the photographic and graphic artwork.
REFERENCES
2. Struck, D. K., and Lennarz, W. J. (1980) in The Biochemistty of Glycopro- 1. Li, E., Tabas, I., and Kornfeld, S. (1978) J. Biol. Chem. 2 5 3 , 7762-7770
teins and Proteoglycam (Lennarz, W. J., ed) pp. 35-83, Plenum Press,
8 7
9
10, 11 12
13.
14. 15.
16
17. 18.
19.
20.
21.
23. 22.
24. 25.
26. 27.
28. 29.
30.
31.
32.
33. 34. 35.
New York
7668-7674
CJ. S. A. 74,4411-4414
105 , 275-278
11892-11895
~~
3. Spiro, M. J., Spiro, R. G., and Bhoyroo, V. D. (1979) J. Biol. Chem. 254 ,
4. Turco, S. J., Stetson, B., and Robbins, P. W. (1977) Proc. Natl. Acad. Sci.
5. Staneloni, R. J., Ugalde, R. A., and Leloir, L. F. (1980) Eur. J . Biochem.
6. Trimble, R. B., Byrd, J. C., and Maley, F. (1980) J. Biol. Chem. 255 ,
Hubbard,S.C., and Ivatt, R. J. (1981) Annu. Reo. Biochem. 50,555-583 Kornfeld, R., and Kornfeld, S. (1985) Annu. Reu. Biochem. 54,631-64 Suh, K., Bergmann, J. E., and Gabel, C. A. (1989) J. Cell Biol. 108 , 811-
Kabcenell, A. K., and Atkinson, P. H. (1985) J. Cell Biol. 101, 1270-1280 Atkinson, P. H., and Lee, J. T. (1984) J. Cell Biol. 9 8 , 2245-2249 Hickman, S., Theodorakis, J. L., Greco, J. M., and Brown, P. H. (1984) J.
Brands, R., Snider, M. D., Hino, Y., Park, S. S., Gelboin, H. V., and
Hubbard, S. C., and P. W. Robbins. (1979) J. Biol. Chem. 254,4568-4576 McElduff, A,, Watkinson, A,, Hedo, J. A,, and Gorden, P. (1986) Biochem.
Rosenfeld, M. G., Marcantonio E. E., Hakimi, J., Ort, V. M. Atkinson, P.
Lodish, H. F., and Kong, N. (1984) J. Cell Blol. 9 8 , 1720-1729 Parent, J. B., Yeo, T. K., Yeo, K. T., and Olden, K. (1986) Mol. Cell.
820
Cell Biol. 9 8 , 407-416
Rothman, J. E. (1985) J. Cell Biol. 101 , 1724-1732
J. 239,679-683
H.,Sabatini, D., and Kreibicd, G. (1984) J , Cell Biol. 9 9 , 1676-1082
Biochem. 72.21-33 Lemansky, P., Gieselmann, V., Hasilik, A., and von Figura, K. (1984) J.
Reitman, M. L., Trowbridge, I . S., and Kornfeld, S. (1982) J. Biol. Chem. Biol. Chem. 2 5 9 , 10129-10135
257.10357-10363 Laemmli, U. K. (1970) Nature 227,680-685
Doms, R. W., Keller, D. S., Helenius. A., and Balch. W. E. (1987) J. Cell Gabel, C. A,, and Bergmann, J. E. (1985) J. Cell Biol. 101,460-469
Biol. 106 . 1957-1969 Kreis, T E.,’and Lodish, H. F. (1986) Cell 4 6 , 929-937 Saunier, B., Kilker, R. D., Jr., Tkacz, J. S., Quaroni, A,, and Herscovics, A.
Hettkamp, H., Bause, E., and Legler, G. (1982) Biosci. Rep. 2, 899-906 Tartakoff, A. M., Vassalli, P., and Detraz, M. (1977) J. Exp. Med. 146,
(1982) J. Biol. Chem. 2 5 7 , 14155-14161
1332-1345 Datema, R., and Schwarz, R. T. (1981) J. Biol. Chem. 256 , 11191-11198 Li pincott-Schwartz, J., Bonifacino, J. S., Yuan, L. C., and Klausner, R.
Trombetta, S. E., Bosch, M., and Parodi, A. (1989) Biochemistry 28,8108- 6. (1988) Cell 54,209-220
R l l 6 Schlesinger, S., Malfer, C., and Schlesinger, M. J. (1984) J . Biol. Chem.
Michael, J. M., and Kornfeld, S. (1980) Arch. Biochen. Biophys. 199, 249-
””
2 5 9 , 7597-7601
9.58 Lubai, W. A., and Spiro, R. G. (1988) J. Bid. Chem. 263,3990-3998 Lubas, W. A., and S iro R. G. (1987) J. Biol. Chem. 2 6 2 , 3775-3781 Pelham, H. R. (19887 E h B O J. 7,913-918




















