
Identification and quantification of human DNA repair protein NEIL1 by liquid...
13
Identification and Quantification of Human DNA Repair Protein NEIL1 by Liquid Chromatography/Isotope-Dilution Tandem Mass Spectrometry Prasad T. Reddy, †,# Pawel Jaruga, †,# Gü ldal Kirkali, † Gamze Tuna, †,§ Bryant C. Nelson, † and Miral Dizdaroglu* ,† † Biochemical Science Division, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, United States § Department of Biochemistry, School of Medicine, Dokuz Eylul University, Izmir, Turkey * S Supporting Information ABSTRACT: Accumulated evidence points to DNA repair capacity as an important factor in cancer and other diseases. DNA repair proteins are promising drug targets and are emerging as prognostic and therapeutic biomarkers. Thus, the knowledge of the overexpression or underexpression levels of DNA repair proteins in tissues will be of fundamental importance. In this work, mass spectrometric assays were developed for the measurement in tissues of the human DNA repair protein NEIL1 (hNEIL1), which is involved in base excision and nucleotide excision repair pathways of oxidatively induced DNA damage. Liquid chromatography/isotope-dilution tandem mass spectrometry (LC−MS/MS), in combination with a purified and fully characterized recombinant 15 N- labeled analogue of hNEIL1 ( 15 N-hNEIL1) as an internal standard, was utilized to develop an accurate method for the quantification of hNEIL1. Both hNEIL1 and 15 N- hNEIL1 were hydrolyzed with trypsin, and 18 tryptic peptides from each protein were identified by LC−MS/MS on the basis of their full-scan mass spectra. These peptides matched the theoretical peptides expected from trypsin hydrolysis of hNEIL1 and provided a statistically significant protein score that would unequivocally identify hNEIL1. The product ion spectra of the tryptic peptides from both proteins were recorded, and the characteristic product ions were defined. Selected-reaction monitoring was used to analyze mixtures of hNEIL1 and 15 N- hNEIL1 on the basis of product ions. Additional confirmation of positive identification was demonstrated via separation of the proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and in-gel tryptic digestion followed by LC−MS/MS analysis. These results suggest that the developed assays would be highly suitable for the in vivo positive identification and accurate quantification of hNEIL1 in tissues. KEYWORDS: human DNA repair protein, NEIL1, LC−MS/MS ■ INTRODUCTION DNA repair is an essential life function and protects genomic stability and thus contributes to prevention of disease processes. 1,2 DNA repair deficiencies predispose cells to accumulation of DNA damage and consequently mutations, resulting in genomic instability, which is a major factor in carcinogenesis. 3−8 Germline mutations and polymorphisms in DNA repair genes also contribute to genomic instability and predisposition to cancer. 4,6,8−10 Mounting evidence shows that cancer cells accumulate mutations in their DNA repair proteins, as cancer progresses. 6,8 For example, 60% of cancer cell lines possess mutated DNA repair genes (see http://www.sanger.ac. uk.genetics/CGP). Although some cancer cells can lose their capacity to repair various forms of DNA damage, they acquire alternative mechanisms that can lead to therapeutic resist- ance. 6,8,11 On the other hand, recent findings suggest that some types of malignant tumors possess increased DNA repair capacity that may affect the therapy and outcome of cancer. 8,12−17 Although additional mutations may provide cancer cells with survival advantage, they may also cause cell death late in tumor evolution. However, tumors that over- express DNA repair genes may be favored by natural selection to increase their survival and consequently their DNA repair capacity. Accumulated evidence strongly suggests that DNA repair capacity will be an important factor in predicting patient response to DNA-damaging agents such as chemotherapeutic drugs and ionizing radiation. 8 The determination of the overexpression or underexpression levels of DNA repair proteins in tumors will help develop and guide treatment strategies that will likely lead to the best treatment results for patients. In this context, DNA repair proteins are becoming predictive, prognostic and therapeutic factors in cancer, and also promising drug targets for cancer treatment as DNA repair inhibitors are being developed to increase the efficacy of cancer therapy. 4,6,8 DNA repair consists of various pathways that involve a plethora of enzymes. Oxidatively induced DNA damage caused Received: November 2, 2012 Technical Note pubs.acs.org/jpr © XXXX American Chemical Society A dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXX
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Identification and quantification of human DNA repair protein NEIL1 by liquid...

Identification and Quantification of Human DNA Repair ProteinNEIL1 by Liquid Chromatography/Isotope-Dilution Tandem MassSpectrometryPrasad T. Reddy,†,# Pawel Jaruga,†,# Guldal Kirkali,† Gamze Tuna,†,§ Bryant C. Nelson,†
and Miral Dizdaroglu*,†
†Biochemical Science Division, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, United States§Department of Biochemistry, School of Medicine, Dokuz Eylul University, Izmir, Turkey
*S Supporting Information
ABSTRACT: Accumulated evidence points to DNA repair capacity as an importantfactor in cancer and other diseases. DNA repair proteins are promising drug targets andare emerging as prognostic and therapeutic biomarkers. Thus, the knowledge of theoverexpression or underexpression levels of DNA repair proteins in tissues will be offundamental importance. In this work, mass spectrometric assays were developed for themeasurement in tissues of the human DNA repair protein NEIL1 (hNEIL1), which isinvolved in base excision and nucleotide excision repair pathways of oxidatively inducedDNA damage. Liquid chromatography/isotope-dilution tandem mass spectrometry(LC−MS/MS), in combination with a purified and fully characterized recombinant 15N-labeled analogue of hNEIL1 (15N-hNEIL1) as an internal standard, was utilized todevelop an accurate method for the quantification of hNEIL1. Both hNEIL1 and 15N-hNEIL1 were hydrolyzed with trypsin, and 18 tryptic peptides from each protein wereidentified by LC−MS/MS on the basis of their full-scan mass spectra. These peptidesmatched the theoretical peptides expected from trypsin hydrolysis of hNEIL1 and provided a statistically significant protein scorethat would unequivocally identify hNEIL1. The product ion spectra of the tryptic peptides from both proteins were recorded,and the characteristic product ions were defined. Selected-reaction monitoring was used to analyze mixtures of hNEIL1 and 15N-hNEIL1 on the basis of product ions. Additional confirmation of positive identification was demonstrated via separation of theproteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and in-gel tryptic digestion followed by LC−MS/MSanalysis. These results suggest that the developed assays would be highly suitable for the in vivo positive identification andaccurate quantification of hNEIL1 in tissues.
KEYWORDS: human DNA repair protein, NEIL1, LC−MS/MS
■ INTRODUCTION
DNA repair is an essential life function and protects genomicstability and thus contributes to prevention of diseaseprocesses.1,2 DNA repair deficiencies predispose cells toaccumulation of DNA damage and consequently mutations,resulting in genomic instability, which is a major factor incarcinogenesis.3−8 Germline mutations and polymorphisms inDNA repair genes also contribute to genomic instability andpredisposition to cancer.4,6,8−10 Mounting evidence shows thatcancer cells accumulate mutations in their DNA repair proteins,as cancer progresses.6,8 For example, 60% of cancer cell linespossess mutated DNA repair genes (see http://www.sanger.ac.uk.genetics/CGP). Although some cancer cells can lose theircapacity to repair various forms of DNA damage, they acquirealternative mechanisms that can lead to therapeutic resist-ance.6,8,11 On the other hand, recent findings suggest that sometypes of malignant tumors possess increased DNA repaircapacity that may affect the therapy and outcome ofcancer.8,12−17 Although additional mutations may providecancer cells with survival advantage, they may also cause cell
death late in tumor evolution. However, tumors that over-express DNA repair genes may be favored by natural selectionto increase their survival and consequently their DNA repaircapacity. Accumulated evidence strongly suggests that DNArepair capacity will be an important factor in predicting patientresponse to DNA-damaging agents such as chemotherapeuticdrugs and ionizing radiation.8 The determination of theoverexpression or underexpression levels of DNA repairproteins in tumors will help develop and guide treatmentstrategies that will likely lead to the best treatment results forpatients. In this context, DNA repair proteins are becomingpredictive, prognostic and therapeutic factors in cancer, andalso promising drug targets for cancer treatment as DNA repairinhibitors are being developed to increase the efficacy of cancertherapy.4,6,8
DNA repair consists of various pathways that involve aplethora of enzymes. Oxidatively induced DNA damage caused
Received: November 2, 2012
Technical Note
pubs.acs.org/jpr
© XXXX American Chemical Society A dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXX

by endogenous and exogenous sources such as free radicals inliving cells is mainly repaired by base excision repair (BER) andalso by nucleotide excision repair (NER), albeit to a lesserextent.2,18 Among the DNA glycosylases involved in the firststep of BER, mammalian NEIL1 protein is a bifunctionalenzyme with an additional β,δ-elimination activity.19,20 Itpossesses a unique substrate specificity and specifically removes4,6-diamino-5-formamidopyrimidine (FapyAde) and 2,6-diami-no-4-hydroxy-5-formamidopyrimidine (FapyGua) from DNA;however, it exhibits no significant activity toward 8-hydroxy-guanine (8-OH-Gua).19−28 Some pyrimidine-derived lesionssuch as thymine glycol are also excised albeit to a lesser extent.In addition, there is evidence for the involvement of NEIL1 inNER.29 Since its discovery, accumulated evidence has pointedto a significant role of this unique enzyme in the prevention ofdisease processes including carcinogenesis.21,25,26,28−33 Poly-morphic variants of human NEIL1 (hNEIL1) with reduced orno activity have been discovered, suggesting that individualscarrying neil1 mutations may be at risk for disease develop-ment.25,30,34
The knowledge of the overexpression or underexpressionlevels of DNA repair proteins in tumors will be important, ifthese proteins are to be used as disease biomarkers, as guidesand predictors for development of treatments, and astherapeutic response indicators in patients undergoingantitumor treatments. Our laboratory developed a programfor the accurate and reproducible identification and quantifi-cation of DNA repair proteins by mass spectrometrictechniques such as liquid chromatography-tandem massspectrometry (LC−MS/MS) with isotope-dilution using full-length and fully 15N-labeled analogues of these proteins asinternal standards.35,36 In the present work, we report on thedevelopment of mass spectrometric assays for the measurementby LC−MS/MS of hNEIL1 using full-length and fully 15N-labeled hNEIL1 (15N-hNEIL1) as an internal standard.
■ MATERIALS AND METHODS
Reagents
Trypsin (Proteomics grade), acetonitrile (HPLC-grade), andwater (HPLC-grade) for analysis by LC−MS/MS werepurchased from Sigma (St. Louis, MO). Water purified througha Milli-Q system (Millipore, Bedford, MA) was used for allother applications. Acrylamide, bisacrylamide, and proteaseinhibitor cocktail tablets were obtained from Sigma-Aldrich (St.Louis, MO). 15N-NH4Cl was purchased from CambridgeIsotope Laboratories (Andover, MA). Nickel agarose resinwas from Qiagen (Valencia, CA). 4,6-Diamino-5-formamido-pyrimidine-13C,15N2 (FapyAde-13C,15N2), 2,6-diamino-4-hy-droxy-5-formamidopyrimidine-13C,15N2 (FapyGua-13C,15N2),and 8-hydroxy-2′-deoxyguanosine-15N5 were purchased fromCambridge Isotope Laboratories (Andover, MA). 8-Hydrox-yguanine-15N5 (8-OH-Gua-
15N5) was obtained by hydrolysis of8-hydroxy-2′-deoxyguanosine-15N5 with 60% formic acid at 140°C for 30 min followed by lyophilization. Subsequently, 8-OH-Gua-15N5 was dissolved in 10 mM NaOH before use. TheLaemmli sample buffer, 12.5% Tris-HCl Criterion precast gels,1×Tris/glycine/SDS running buffer and Coomassie BrilliantBlue R-250 Staining Solutions kit were purchased from Bio-RadLaboratories (Hercules, CA). The In-Gel Tryptic Digestion kitwas from Pierce (Rockford, IL). Prestained protein markerswere from New England BioLabs (Ipswich, MA).
Production and Purification of hNEIL1 and 15N-hNEIL1
The human neil1 gene in the expression vector pET22b for His-tagged production of the protein was kindly provided by Dr.Stephen Lloyd (Oregon Health and Science University,Portland, Oregon). The sequence analysis of the gene resultedin the protein sequence with 390 amino acids that matched thepreviously published sequence of human NEIL119,20,37,38 (seealso, http://www.ncbi.nlm.nih.gov/protein/Q96FI4.3). This isthe edited form of NEIL1 with arginine at position 242.38−40
The sequence of the C-terminal His-tag was found to beKLAAALEHHHHHH. E. coli BL21 (DE3) cells harboring theplasmid were grown at 37 °C in LB medium supplementedwith ampicillin and chloramphenicol to a final concentration of100 μg/mL and 40 μg/mL, respectively. At midlogarithmicphase of growth (A600 ∼ 0.6), cells were briefly cooled in iceand hNEIL1 production was induced with 100 μmol IPTG/mLat 25 °C for 16 h. Cells were harvested at 6000g for 20 min andwashed with 25 mM Tris buffer (pH 7.5). The wet weight ofcells obtained in this procedure was 3.5 g/L culture.Minimal medium was prepared as described.41 The
composition of the medium was 6 g of NaH2PO4, 3 g ofK2HPO4, 0.5 g of NaCl, and 1 g of 15N-NH4Cl, 5 g of glucose,246 mg of MgSO4·7H2O per L. Ampicillin and chloramphe-nicol were added to a final concentration of 50 μg/mL and 20μg/mL, respectively. E. coli BL21 (DE3) harboring pET22b/human neil1 recombinant plasmid was grown at 37 °C for 20 hon LB agar plate containing 100 μg of ampicillin and 40 μg ofchloramphenicol/mL. A colony was carefully (without touchinginto the LB medium) transferred to 10 mL minimal mediumcontaining 1 mg of 15N-NH4Cl/mL and 50 μg of ampicillin and20 μg of chloramphenicol/mL. Cells were grown for 16 h at 37°C at 250 rpm in a 50 mL tube. This inoculum was transferredto 90 mL minimal medium, in a 250 mL flask, containing 15N-NH4Cl, ampicillin, and chloramphenicol as above. This culturewas grown at 37 °C for 9 h. Next, this seed culture wastransferred to 900 mL of minimal medium containing 15N-NH4Cl, ampicillin, chloramphenicol in a 2 L flask. This culturewas grown at 37 °C for 4 h. Cells (A600 = 0.45) were brieflycooled in ice, and 15N-hNEIL1 production was induced with100 μmol IPTG/mL at 25 °C for 16 h. Cells were harvested at6000g for 20 min and washed with 25 mM Tris buffer (pH 7.5).The wet weight of cells obtained in this procedure was 2.8 g/Lculture.Cell pellet was suspended at a rate of 1 g/10 mL of 50 mM
Na2HPO4−NaH2PO4 buffer (pH 8.0), 10 mM β-mercaptoe-thanol, 10 mM imidazole, and 300 mM NaCl (lysis buffer).One tablet of protease inhibitors was added to the cellsuspension. Cell suspension was passed through a French Pressat 7 × 104 kPa twice. The cell-free extract was centrifuged at36000g for 30 min. Meanwhile, 1 mL of nickel-agarose slurry(0.5 mL resin) was washed with the lysis buffer in a 30 mL Bio-Rad polypropylene column. The supernatant was added to theresin and mixed on a rocker for 2 h at 4 °C. The flow throughwas collected, and the column was washed successively withthree 10 mL aliquots of the lysis buffer. Next, the resin waswashed successively two times with 10 mL aliquots of the lysisbuffer containing an additional 30 mM imidazole (total 40mM). Next, the resin was washed successively three times with5 mL aliquots of the lysis buffer containing an additional 70mM imidazole (total 80 mM). The first imidazole80 wash didelute about 30% of 15N-hNEIL1 with a number ofcontaminating proteins as judged by SDS-PAGE. The secondand third imidazole80 washes eluted about 30% of nearly
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXB

homogeneous 15N-hNEIL1. Next, 15N-hNEIL1 was elutedsuccessively with three 5 mL aliquots of the lysis buffercontaining 150 mM imidazole. The first 5 mL contained about30% of 15N-hNEIL1. The subsequent two elutions containedthe remaining 15N-hNEIL1. The second and third imidazole80elutions and all the imidazole150 elutions were pooled anddialyzed overnight against 1 L of 50 mM Na2HPO4−NaH2PO4buffer (pH 8.0) and 300 mM NaCl. Slight precipitate ofhNEIL1 appeared after dialysis even in the presence of 300 mMNaCl. The precipitate was centrifuged off from the dialyzedpool, and the clear supernatant was concentrated on NMLW5membrane in an Amicon cell. The above purification proceduredeveloped for cells from 1 L culture was proportionately scaledup to 5 L as and when needed. Protein concentration wasdetermined by the Lowry method using BSA as the standard.42
For the production of hNEIL1, NH4Cl was used instead of15N-
NH4Cl. Otherwise the procedure was the same as that for 15N-hNEIL1. The final yields of 15N-hNEIL1 and hNEIL1 were ∼2mg/L culture.Gas Chromatography/Tandem Mass Spectrometry
The enzymic activities of hNEIL1 and 15N-hNEIL1 weremeasured using gas chromatography/tandem mass spectrom-etry (GC-MS/MS) with isotope-dilution. Calf thymus DNAsamples were prepared, treated with hNEIL1 or 15N-hNEIL1,and then analyzed by GC-MS/MS as described.35,43 The detailsof the sample preparation and GC-MS/MS are given in theSupporting Information.Hydrolysis with Trypsin
An aliquot of 100 μg of hNEIL1 or 15N-hNEIL1 was incubatedwith 2 μg trypsin in 500 μL Tris-HCl buffer (30 mM, pH 8.0)at 37 °C for 2 h. Then, an aliquot of 2 μg trypsin was addedagain. After another 22 h of incubation, the sample was heatedat 95 °C for 5 min to deactivate trypsin prior to analysis byLC−MS/MS.Liquid Chromatography/Mass Spectrometry
Analyses were performed using a liquid chromatograph−massselective detector (1100 Series, Agilent Technologies, Wil-mington, DE) equipped with an automatic injector andatmospheric pressure ionization-electrospray in the positiveionization mode. The flow and temperature of the drying gas(nitrogen) were 10 L/min and 350 °C, respectively. Thenebulizing gas pressure was 308 kPa. The capillary potentialwas 4000 V. The fragmentor and electron multiplier potentialwere 100 and 2600 V, respectively. A Zorbax Extend-C-18,Rapid Resolution HT column (2.1 mm × 100 mm, 1.8 μmparticle size) (Agilent Technologies, Wilmington, DE) with anattached Agilent Eclipse XDB-C8 guard column (2.1 mm ×12.5 mm, 5 μm particle size) was used. The autosampler andcolumn temperature were kept at 5 and 40 °C, respectively.Mobile phase A was water plus 2% acetonitrile and 0.1% formicacid (v/v). Mobile phase B consisted of acetonitrile plus 0.1%formic acid (v/v). A gradient analysis starting from 1% B andlinearly increasing to 51% B in 25 min was used. Afterward, Bwas increased to 90% in 0.1 min and kept at this level for 5 minand then decreased to 1% to equilibrate the column for 20 min.The flow rate was 250 μL/min.Liquid Chromatography/Tandem Mass Spectrometry
LC−MS/MS analyses were performed using an Agilent 1290Infinity LC system coupled to a Thermo-Scientific FinniganTSQ Quantum Ultra AM triple quadrupole MS/MS systemwith an installed heated electrospray-ionization (HESI) source.
The column, mobile phases, and gradient analysis were as givenabove for LC/MS measurements. The autosampler and columntemperature were kept at 15 and 30 °C, respectively. The flowrate was 300 μL/min. Experimental conditions for theautomated mass calibration and operating parameters of theMS/MS system in the positive ion mode were as previouslydescribed,36 except for sheath gas (nitrogen) pressure and scanwidth being 60 (arbitrary units) and m/z 2, respectively.Sodium Dodecyl Sulfate-Polyacrylamide GelElectrophoresis and In-Gel Tryptic Digestion
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis(SDS-PAGE) was performed as described,44 using the gelelectrophoresis system Criterion Cell (Bio-Rad Laboratories,Hercules, CA). Separations and in-gel trypsin digestions wereperformed according to previously published procedures.45,46
Briefly, the protein samples were denatured with Laemmlisample buffer in a 1:1 ratio. The samples (10 μg each) wereloaded into a 12.5% Tris-HCl Criterion precast gel (13.3 × 8.7cm (W × L), 1 mm thick and run at 180 V in 1× Tris/glycine/SDS running buffer for 90 min). Prestained protein standardswere used as protein markers. The gels were stained anddestained with Coomassie Brilliant Blue R-250 StainingSolutions kit. The in-gel tryptic digestion was carried outusing an In-Gel Tryptic Digestion kit. The proteins bands wereon the order of 2 mm to 4 mm in diameter with the gelthickness of 1 mm. The bands were cut from the gel with asharp scalpel and divided into smaller pieces (1 × 1 mm to 2 ×2 mm). According to the kit protocol, the bands of interestfrom the gel were destained, and then reduced and alkylated.Subsequently, the buffers were exchanged to wash thereduction and alkylation reagents out of the samples. Prior totrypsin hydrolysis, the gel pieces were dehydrated in 100%acetonitrile, completely shrank and were dried by vacuumcentrifugation for 5 min. In the final step, the hydrolysis wasperformed using activated modified trypsin. The samples wereincubated overnight at 30 °C with shaking and thencentrifuged. The supernatant fractions were separated andanalyzed by LC−MS/MS.
■ RESULTS
Production, Purification, and Characterization of hNEIL1and Fully 15N-Labeled hNEIL1
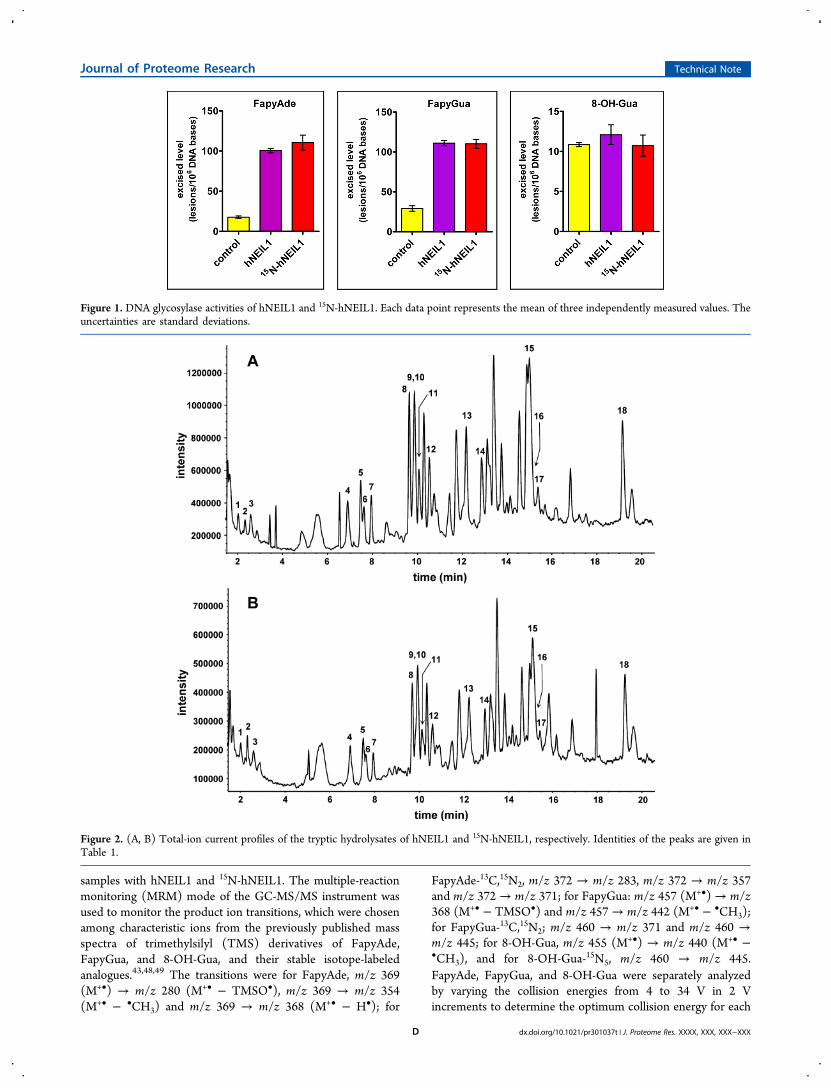
Both hNEIL1 and 15N-hNEIL1 were overproduced in E. coli asdescribed in the experimental procedures. Both proteins werepurified according to the steps shown in Supplemental Figure 1,Supporting Information. First, the enzymic activity of theseproteins was tested. Among other properties, the enzymicactivity of a stable isotope-labeled protein must be identical tothat of its analogous analyte protein. This will ensure that theactive site of the labeled protein was not perturbed by 15N-labeling and other isolation procedures. The glycosylase activityof hNEIL1 is well-known and efficiently removes FapyAde andFapyGua from DNA containing multiple lesions; however, thisenzyme exhibits no significant activity toward 8-OH-Gua.19,22,24−26 The enzymic activities of both hNEIL1 and15N-hNEIL1 were tested using a DNA sample containingmultiple lesions as substrates. The details and applications ofthis approach that is used to accurately determine the substratespecificities of DNA glycosylases have previously beendescribed.47 In the present work, GC-MS/MS with isotope-dilution was used for the measurement of the levels ofFapyAde, FapyGua, and 8-OH-Gua after treatment of DNA
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXC
samples with hNEIL1 and 15N-hNEIL1. The multiple-reactionmonitoring (MRM) mode of the GC-MS/MS instrument wasused to monitor the product ion transitions, which were chosenamong characteristic ions from the previously published massspectra of trimethylsilyl (TMS) derivatives of FapyAde,FapyGua, and 8-OH-Gua, and their stable isotope-labeledanalogues.43,48,49 The transitions were for FapyAde, m/z 369(M+•) → m/z 280 (M+• − TMSO•), m/z 369 → m/z 354(M+• − •CH3) and m/z 369 → m/z 368 (M+• − H•); for
FapyAde-13C,15N2, m/z 372 → m/z 283, m/z 372 → m/z 357and m/z 372 → m/z 371; for FapyGua: m/z 457 (M+•)→ m/z368 (M+• − TMSO•) and m/z 457→ m/z 442 (M+• − •CH3);for FapyGua-13C,15N2; m/z 460 → m/z 371 and m/z 460 →m/z 445; for 8-OH-Gua, m/z 455 (M+•) → m/z 440 (M+• −•CH3), and for 8-OH-Gua-15N5, m/z 460 → m/z 445.FapyAde, FapyGua, and 8-OH-Gua were separately analyzedby varying the collision energies from 4 to 34 V in 2 Vincrements to determine the optimum collision energy for each
Figure 1. DNA glycosylase activities of hNEIL1 and 15N-hNEIL1. Each data point represents the mean of three independently measured values. Theuncertainties are standard deviations.
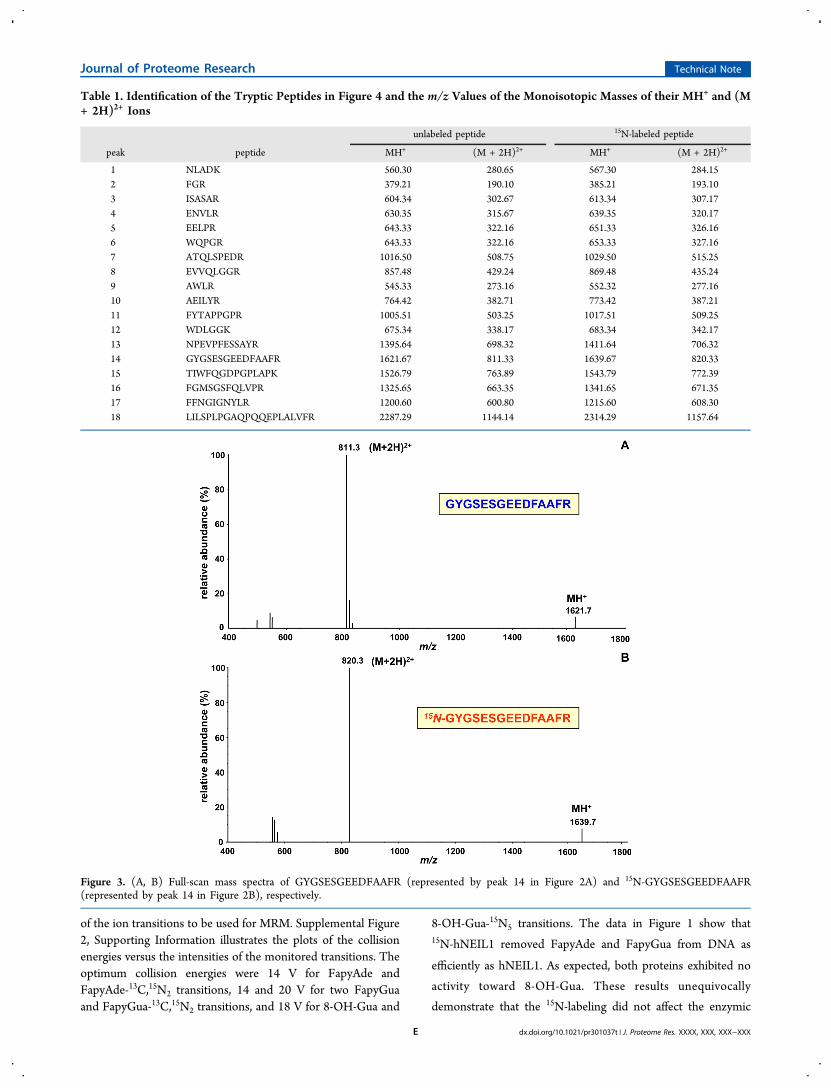
Figure 2. (A, B) Total-ion current profiles of the tryptic hydrolysates of hNEIL1 and 15N-hNEIL1, respectively. Identities of the peaks are given inTable 1.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXD
of the ion transitions to be used for MRM. Supplemental Figure2, Supporting Information illustrates the plots of the collisionenergies versus the intensities of the monitored transitions. Theoptimum collision energies were 14 V for FapyAde andFapyAde-13C,15N2 transitions, 14 and 20 V for two FapyGuaand FapyGua-13C,15N2 transitions, and 18 V for 8-OH-Gua and
8-OH-Gua-15N5 transitions. The data in Figure 1 show that15N-hNEIL1 removed FapyAde and FapyGua from DNA as
efficiently as hNEIL1. As expected, both proteins exhibited no
activity toward 8-OH-Gua. These results unequivocally
demonstrate that the 15N-labeling did not affect the enzymic
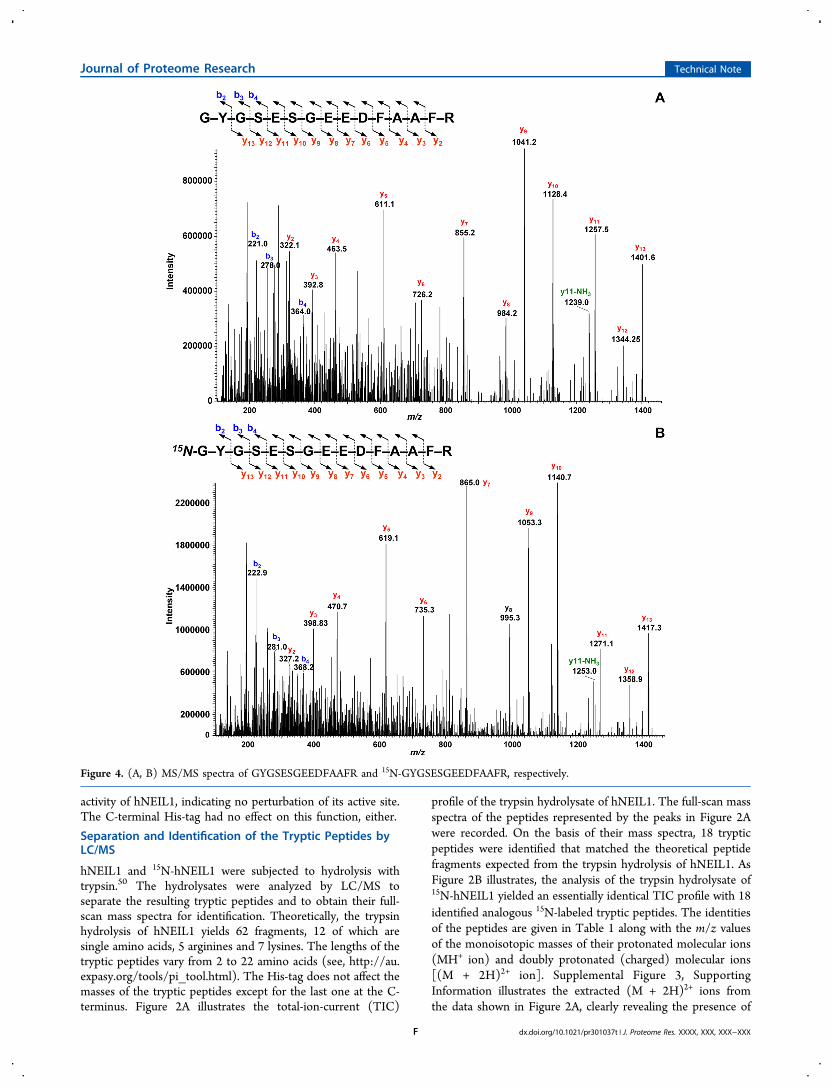
Table 1. Identification of the Tryptic Peptides in Figure 4 and the m/z Values of the Monoisotopic Masses of their MH+ and (M+ 2H)2+ Ions
unlabeled peptide 15N-labeled peptide
peak peptide MH+ (M + 2H)2+ MH+ (M + 2H)2+
1 NLADK 560.30 280.65 567.30 284.152 FGR 379.21 190.10 385.21 193.103 ISASAR 604.34 302.67 613.34 307.174 ENVLR 630.35 315.67 639.35 320.175 EELPR 643.33 322.16 651.33 326.166 WQPGR 643.33 322.16 653.33 327.167 ATQLSPEDR 1016.50 508.75 1029.50 515.258 EVVQLGGR 857.48 429.24 869.48 435.249 AWLR 545.33 273.16 552.32 277.1610 AEILYR 764.42 382.71 773.42 387.2111 FYTAPPGPR 1005.51 503.25 1017.51 509.2512 WDLGGK 675.34 338.17 683.34 342.1713 NPEVPFESSAYR 1395.64 698.32 1411.64 706.3214 GYGSESGEEDFAAFR 1621.67 811.33 1639.67 820.3315 TIWFQGDPGPLAPK 1526.79 763.89 1543.79 772.3916 FGMSGSFQLVPR 1325.65 663.35 1341.65 671.3517 FFNGIGNYLR 1200.60 600.80 1215.60 608.3018 LILSPLPGAQPQQEPLALVFR 2287.29 1144.14 2314.29 1157.64
Figure 3. (A, B) Full-scan mass spectra of GYGSESGEEDFAAFR (represented by peak 14 in Figure 2A) and 15N-GYGSESGEEDFAAFR(represented by peak 14 in Figure 2B), respectively.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXE
activity of hNEIL1, indicating no perturbation of its active site.The C-terminal His-tag had no effect on this function, either.
Separation and Identification of the Tryptic Peptides byLC/MS
hNEIL1 and 15N-hNEIL1 were subjected to hydrolysis withtrypsin.50 The hydrolysates were analyzed by LC/MS toseparate the resulting tryptic peptides and to obtain their full-scan mass spectra for identification. Theoretically, the trypsinhydrolysis of hNEIL1 yields 62 fragments, 12 of which aresingle amino acids, 5 arginines and 7 lysines. The lengths of thetryptic peptides vary from 2 to 22 amino acids (see, http://au.expasy.org/tools/pi_tool.html). The His-tag does not affect themasses of the tryptic peptides except for the last one at the C-terminus. Figure 2A illustrates the total-ion-current (TIC)
profile of the trypsin hydrolysate of hNEIL1. The full-scan massspectra of the peptides represented by the peaks in Figure 2Awere recorded. On the basis of their mass spectra, 18 trypticpeptides were identified that matched the theoretical peptidefragments expected from the trypsin hydrolysis of hNEIL1. AsFigure 2B illustrates, the analysis of the trypsin hydrolysate of15N-hNEIL1 yielded an essentially identical TIC profile with 18identified analogous 15N-labeled tryptic peptides. The identitiesof the peptides are given in Table 1 along with the m/z valuesof the monoisotopic masses of their protonated molecular ions(MH+ ion) and doubly protonated (charged) molecular ions[(M + 2H)2+ ion]. Supplemental Figure 3, SupportingInformation illustrates the extracted (M + 2H)2+ ions fromthe data shown in Figure 2A, clearly revealing the presence of
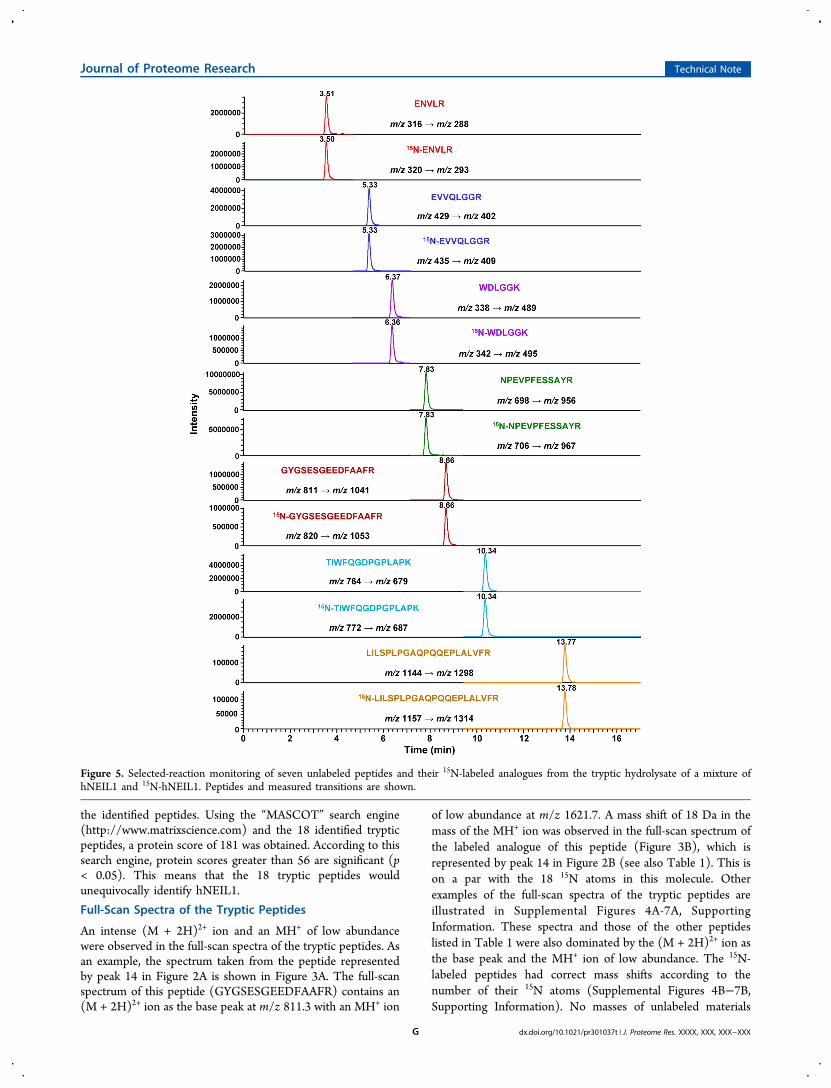
Figure 4. (A, B) MS/MS spectra of GYGSESGEEDFAAFR and 15N-GYGSESGEEDFAAFR, respectively.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXF
the identified peptides. Using the “MASCOT” search engine(http://www.matrixscience.com) and the 18 identified trypticpeptides, a protein score of 181 was obtained. According to thissearch engine, protein scores greater than 56 are significant (p< 0.05). This means that the 18 tryptic peptides wouldunequivocally identify hNEIL1.
Full-Scan Spectra of the Tryptic Peptides
An intense (M + 2H)2+ ion and an MH+ of low abundancewere observed in the full-scan spectra of the tryptic peptides. Asan example, the spectrum taken from the peptide representedby peak 14 in Figure 2A is shown in Figure 3A. The full-scanspectrum of this peptide (GYGSESGEEDFAAFR) contains an(M + 2H)2+ ion as the base peak at m/z 811.3 with an MH+ ion
of low abundance at m/z 1621.7. A mass shift of 18 Da in themass of the MH+ ion was observed in the full-scan spectrum ofthe labeled analogue of this peptide (Figure 3B), which isrepresented by peak 14 in Figure 2B (see also Table 1). This ison a par with the 18 15N atoms in this molecule. Otherexamples of the full-scan spectra of the tryptic peptides areillustrated in Supplemental Figures 4A-7A, SupportingInformation. These spectra and those of the other peptideslisted in Table 1 were also dominated by the (M + 2H)2+ ion asthe base peak and the MH+ ion of low abundance. The 15N-labeled peptides had correct mass shifts according to thenumber of their 15N atoms (Supplemental Figures 4B−7B,Supporting Information). No masses of unlabeled materials
Figure 5. Selected-reaction monitoring of seven unlabeled peptides and their 15N-labeled analogues from the tryptic hydrolysate of a mixture ofhNEIL1 and 15N-hNEIL1. Peptides and measured transitions are shown.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXG
were observed in the spectra of all 15N-labeled peptides,confirming the full 15N-labeling of hNEIL1.
Product-Ion Spectra of the Tryptic Peptides
Trypsin hydrolysates of hNEIL1 and 15N-hNEIL1 wereanalyzed by LC−MS/MS. The full-scan mass spectra of thetryptic peptides and their 15N-labeled analogues were essentiallyidentical to those obtained using LC/MS. Next, the product-ion spectra (MS/MS spectra) were obtained. We calculated thetheoretical masses of the typical b- and y-series ions46 as theproduct ions that are expected to result from the collision-induced fragmentation of these peptides (Supplemental Tables1 and 2, Supporting Information). As an example, thecalculation of the masses of the b- and y-ions of the peptideGYGSESGEEDFAAFR and its 15N-labeled analogue is shownin Supplemental Table 3, Supporting Information. The valuesof the b- and y-ions for the unlabeled peptides were inagreement with those calculated using the “ProteinProspector”database of the University of California (http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msproduct). Inorder to obtain the MS/MS spectra of the tryptic peptides,optimum collision energies for the fragmentation of thesepeptides must be measured experimentally. However, therelationship between the collision energies and the m/z valuescan also be determined empirically as was previously describedfor (M + 2H)2+ ions in a tandem quadrupole instrument.46 Thecollision energy for each identified tryptic peptide of hNEIL1was initially chosen according to this empirically determinedrelationship using the m/z value of their (M + 2H)2+ ions.Subsequently, several experiments were performed to deter-mine the optimum collision energy. This was followed by therecording of the MS/MS spectra at the chosen collision energyfor each peptide.
Figure 4A illustrates the MS/MS spectrum of GYGSES-GEEDFAAFR (represented by peak 14 in Figure 2A; see alsoTable 1). The spectrum was dominated by the y-ion series(Supplemental Table 1, Supporting Information) from the y2-ion to the y11-ion with high intensities. In the typical b-ionseries (Supplemental Table 2, Supporting Information), the b2-,b3-, and b4-ions only were observed with some discernibleintensity. The 15N-labeled analogue of this peptide gave anessentially identical MS/MS spectrum (Figure 4B) with massshifts according to the 15N-content of the fragments(Supplemental Tables 1 and 2, Supporting Information). Asfurther examples, the MS/MS spectra of EVVQLGGR,FYTAPPGPR, NPEVPFESSAYR, and TIWFQGDPGPLAPK,and their 15N-labeled analogues (represented by peaks 8, 11, 13and 15, respectively, in Figure 2A,B) are given in SupplementalFigures 8−11, respectively. These MS/MS spectra weredominated by the y-ion series, whereas only a few ions of theb-ion series were observed. The shifts in the masses of thefragments in the spectra of the 15N-labeled analogues were infull agreement with the 15N-contents of the fragments(Supplemental Figures 8B−11B, Supporting Information). Insome cases, the ions resulting from loss of NH3 from y-ions (y-NH3 ion) were also observed as indicated in the spectra.
Selected-Reaction Monitoring for Identification andQuantification
For the positive identification and accurate quantification ofpeptides at low concentrations in a complex mixture such astrypsin hydrolysates of proteins, the mode of selected-reactionmonitoring (SRM) [also called multiple-reaction monitoring(MRM)] of an LC−MS/MS instrument is used.46,51 For thispurpose, it is necessary to choose the (M + 2H)2+ ions or theMH+ ions of the tryptic peptides of a given protein as theprecursor ions and some of their product ions of high
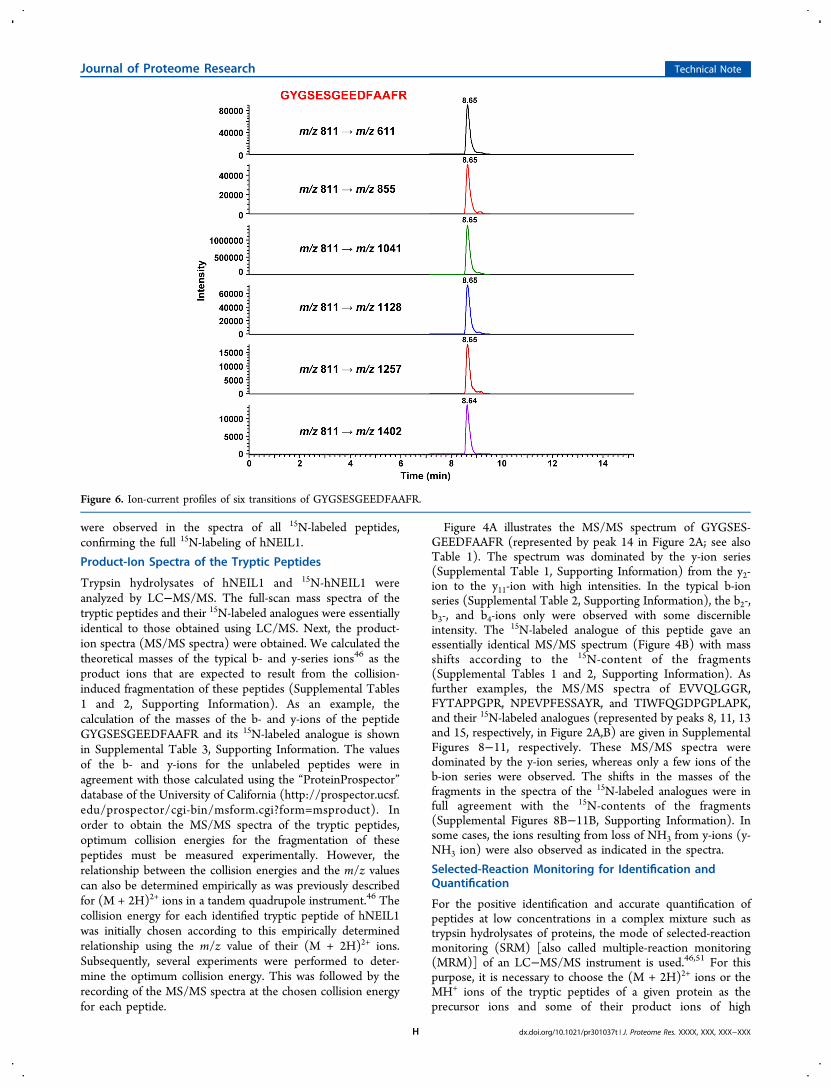
Figure 6. Ion-current profiles of six transitions of GYGSESGEEDFAAFR.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXH
abundance. Subsequently, the transitions of each precursor ionto its chosen product ions are monitored. For quantification,this process is performed for the corresponding transitions ofthe 15N-labeled analogues of the tryptic peptides. Themonitored transitions along with the typical retention time ofthe tryptic peptides enable a positive identification and accuratequantification of these peptides and consequently thecorresponding protein.46,51 For this purpose, however, theknowledge of the full-scan mass spectra and MS/MS spectra ofthe tryptic peptides of an analyte protein is necessary todetermine the appropriate transitions. Furthermore, theretention time of a given peptide on the LC column must beknown under the experimental conditions used. In order toachieve the optimum sensitivity and selectivity, the transitionsof a targeted peptide are recorded within a time window aroundits known retention time.Having identified the tryptic peptides of hNEIL1 at their
respective retention times and determined their product ions asdescribed above, the trypsin hydrolysates of the mixtures ofhNEIL1 and 15N-hNEIL1 were analyzed using SRM in order tocheck the suitability of this technique for the identification andquantification of hNEIL1 using 15N-hNEIL1 as an internalstandard. The (M + 2H)2+ ion was chosen as the precursor ionfor transitions, because this ion generally represents the highestcharge state of the tryptic peptides.46 In addition, the (M +2H)2+ ion was more prominent than the MH+ ion in the full-scan spectra (Figure 3, and Supplemental Figures 4−7,Supporting Information). The ion-current profiles of thetransitions of seven tryptic peptides from the trypsin hydro-lysate of a mixture of hNEIL1 and 15N-hNEIL1 are illustratedin Figure 5. A baseline separation of the monitored peptideswas achieved with excellent peak shapes of the transitionprofiles. As expected, each unlabeled peptide and its respective15N-labeled analogue exactly coeluted at the same retentiontime. In addition to characteristic transitions, the retention timeof a peptide under defined experimental conditions is anabsolutely essential criterion for a positive identification. Itshould be pointed out that, if there is any doubt about thespecificity of a monitored product ion, the identity of thetargeted peptide can be confirmed by monitoring multipletransitions that are characteristic for the targeted peptide.51 Anexample of this approach is shown in Figure 6 in the case of thepeptide GYGSESGEEDFAAFR. The ion-current profiles of sixcharacteristic transitions for this peptide are lined up at itsappropriate retention time, confirming its identity. In addition,the intensities of the transitions agree with those of the productions in the MS/MS spectrum of this peptide (Figure 4). Twoother examples of this approach are given in SupplementalFigure 12, Supporting Information, which illustrates fourtransitions of each of the peptides EVVQLGGR andFYTAPPGPR with retention times only 0.4 min apart. Ifnecessary, additional validation would be provided bymonitoring the corresponding transitions for the coeluting15N-labeled analogue of the targeted peptide.
Determination of the Optimum Collision Energies forQuantification
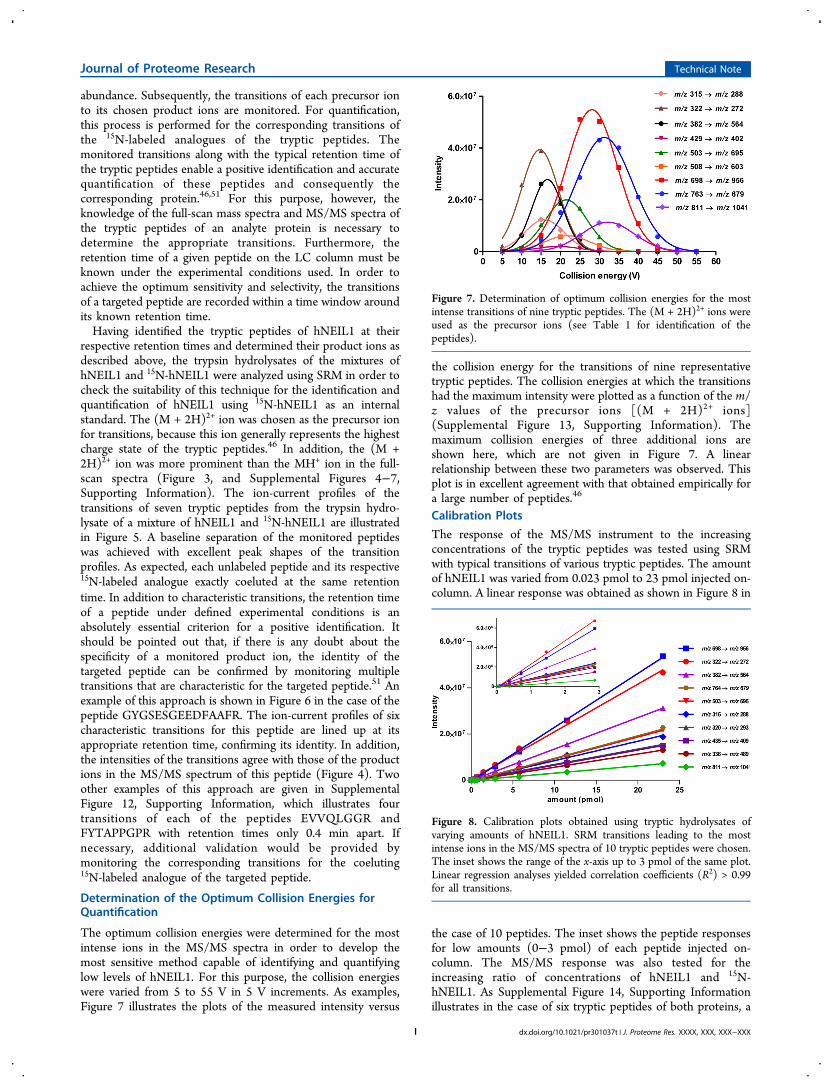
The optimum collision energies were determined for the mostintense ions in the MS/MS spectra in order to develop themost sensitive method capable of identifying and quantifyinglow levels of hNEIL1. For this purpose, the collision energieswere varied from 5 to 55 V in 5 V increments. As examples,Figure 7 illustrates the plots of the measured intensity versus
the collision energy for the transitions of nine representativetryptic peptides. The collision energies at which the transitionshad the maximum intensity were plotted as a function of the m/z values of the precursor ions [(M + 2H)2+ ions](Supplemental Figure 13, Supporting Information). Themaximum collision energies of three additional ions areshown here, which are not given in Figure 7. A linearrelationship between these two parameters was observed. Thisplot is in excellent agreement with that obtained empirically fora large number of peptides.46
Calibration Plots
The response of the MS/MS instrument to the increasingconcentrations of the tryptic peptides was tested using SRMwith typical transitions of various tryptic peptides. The amountof hNEIL1 was varied from 0.023 pmol to 23 pmol injected on-column. A linear response was obtained as shown in Figure 8 in
the case of 10 peptides. The inset shows the peptide responsesfor low amounts (0−3 pmol) of each peptide injected on-column. The MS/MS response was also tested for theincreasing ratio of concentrations of hNEIL1 and 15N-hNEIL1. As Supplemental Figure 14, Supporting Informationillustrates in the case of six tryptic peptides of both proteins, a
Figure 7. Determination of optimum collision energies for the mostintense transitions of nine tryptic peptides. The (M + 2H)2+ ions wereused as the precursor ions (see Table 1 for identification of thepeptides).
Figure 8. Calibration plots obtained using tryptic hydrolysates ofvarying amounts of hNEIL1. SRM transitions leading to the mostintense ions in the MS/MS spectra of 10 tryptic peptides were chosen.The inset shows the range of the x-axis up to 3 pmol of the same plot.Linear regression analyses yielded correlation coefficients (R2) > 0.99for all transitions.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXI

linear response was obtained within a concentration ratioranging from 0.1 to 4.0.Analytical Sensitivity of LC−MS/MS for Tryptic Peptides
The analytical sensitivity of the instrument was examined byanalyzing a tryptic hydrolysate of hNEIL1, the amount of whichranged from 5 fmol to 100 fmol injected on-column. The limitof detection (LOD) was approximately 10 fmol with a signal-to-noise ratio (S/N) of at least 3. The limit of quantification(LOQ) was approximately 30 fmol with an S/N of 10 (data notshown). However, it should be mentioned that the sensitivitywill depend on the monitored transition of a peptide and thusmay vary from transition to transition of each peptide and frompeptide to peptide.SDS-PAGE Analysis and in-Gel Tryptic Digestion
The analysis of both hNEIL1 and 15N-hNEIL1 was alsoperformed using isolation by SDS-PAGE, and subsequent in-geltryptic digestion and LC−MS/MS. Supplemental Figure 15,Supporting Information shows the SDS gel where the proteinswere separated. E. coli Fpg and human OGG1, and their 15N-labeled analogues were also separated for comparison. Theisolation of 15N-labeled E. coli Fpg and 15N-labeled humanOGG1 has been reported in our previous work.35,36 The band(lane 6) containing the mixture of hNEIL1 and 15N-hNEIL1was excised and subjected to in-gel tryptic digestion. Thehydrolysate was analyzed by LC−MS/MS. SupplementalFigure 16, Supporting Information illustrates the ion-currentprofiles of several transitions of the unlabeled and labeledpeptides, revealing a successful isolation from the SDS gel andin-gel tryptic digestion of both proteins.
■ DISCUSSIONSince its discovery, isolation and characterization,19,20,23
hNEIL1 has become the focus of intense studies. This is dueto its unique substrate specificity as a bifunctional DNAglycosylase in BER, the evidence for its critical role inmaintaining the genomic stability and in prevention of diseasessuch as cancer and metabolic syndrome-associated diseases, andits involvement in replication-associated repair (reviewed in ref52). Moreover, the known mutagenic effects and otherproperties of its substrates added a new dimension to theimportance of this protein. hNEIL1 increases during the Sphase indicating its involvement in replication repair and isvariably expressed in human tissues with the highest expressionobserved in the liver, pancreas, and thymus followed bymoderate expression in the brain, spleen, prostate, and ovaryand by low expression in the testis and leukocytes.19,53 Thus far,all evidence accumulated in the past decade strongly points to acritical role of hNEIL1 in the DNA repair capacity of a livinghuman cell. Knowledge of the expression level of this proteinmay provide a greater understanding of the DNA repaircapacity of a given tissue and may be a critical prerequisite fordetermining whether DNA repair pathways are altered incancer (or any other type of disease) tissues. DNA repairalterations may in turn serve as predictive cancer biomarkers.To understand its role in disease processes and its use as areliable biomarker, hNEIL1 must be positively identified andaccurately quantified in relevant human tissues by properchemical and physical techniques.LC−MS/MS with isotope-dilution is the choice of technique
for protein measurements.46,54−60 In order to achieve highprecision and accuracy for the quantitative measurement of aprotein by this technique, a stable isotope-labeled analogue of
this protein must be used as an internal standard. A fully stableisotope-labeled analogue would possess identical chemical andphysical properties of the analyte protein and thus compensatefor eventual losses and drifts during all stages of analysis. Theuse of such a labeled internal standard may also overcomeeventual problems with fluctuations in signal intensity. Anotherimportant reason for this approach is that trypsin hydrolysis canoften be inefficient leading to incomplete yields of trypticpeptides and thus to measurement bias. Since the labeledanalogue of the analyte protein would undergo the same trypsinhydrolysis in a given sample, its use as an internal standardwould minimize the adverse effect of the possible inefficienthydrolysis. Moreover, the labeled analogue of a protein as aninternal standard would be absolutely essential for its accuratequantification when one- or two-dimensional gel electro-phoresis and “in-gel tryptic digestion” are used prior to LC−MS/MS analysis.Previously, full-length expressed proteins containing 13C-
and/or 15N-labeled lysine and arginine have been used asinternal standards for quantification of proteins by massspectrometry.56−60 In our laboratory, full-length and fully15N-labeled proteins were used as internal standards for thequantitative measurement of E. coli Fpg and human OGG1proteins by LC−MS/MS.35,36 In the present work, 15N-hNEIL1 was overexpressed, purified, and characterized as aninternal standard. Its DNA glycosylase activity was essentiallyidentical to that of the unlabeled protein, indicating noperturbation of its active site during isolation and purification.The identification of 18 tryptic peptides of hNEIL1 and 15N-hNEIL1 was achieved. The full-scan mass spectra of thesepeptides contained intense (M + 2H)2+ and MH+ ions. Theshifts in the masses of these ions observed in the full-scan massspectra of the labeled peptides were in full agreement with thenumber of the N-atoms in each peptide. No discernible ionscorresponding to unlabeled peptides were observed in the full-scan mass spectra of labeled peptides, evidencing that a full 15N-labeling of hNEIL1 was achieved. This meets an essentialrequirement for an ideal stable isotope-labeled internalstandard. The MS/MS spectra of the tryptic peptides providedthe ions of the typical b- and y-ion series. The masses of theseions matched those expected from the sequences of thepeptides. The mass shifts observed in the MS/MS spectra ofthe labeled peptides were in full agreement with the 15N-content of each fragment ion. Again, no ion corresponding toan unlabeled fragment was observed, confirming the resultsobtained with the full-scan spectra of the tryptic peptides of15N-hNEIL1. The isolation of hNEIL1 and 15N-hNEIL1 wasalso achieved from gels after SDS-PAGE. This is a crucial stepprior to LC−MS/MS for the identification and quantification ofhNEIL1 in complex tissue samples.The neil1 gene has been sequenced by the National Institute
of Environmental Health Sciences Environmental GenomeProject at the University of Washington in a variety ofindividuals. Variants have been identified that predictedchanges in the amino acid sequence of hNEIL1, i.e., Ser82Cys,Gly83Asp, Cys136Arg, Asp252Asn, and I182M. The variants ofhNEIL1 with Cys82, Asp83, Arg136, and Asn252 have beencloned, expressed in E. coli, and purified.25 hNEIL1-Met182 wasnot isolated. Two of the variants, hNEIL1-Cys82 and hNEIL1-Asn252, exhibited wild type activity, whereas the other twowere devoid of glycosylase activity. The positions 82 (Ser) and83 (Gly) are located in the tryptic peptide FGMSGSFQLVPR,whereas the tryptic peptides AEILYR and GYGSESGEED-
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXJ

FAAFR contain the positions 182 (Ile) and 252 (Asp),respectively. These three peptides were identified in thepresent work. On the other hand, the theoretical peptidewith the position 136 (Cys) (GPCVLQEYQQFR) was notfound. It should be pointed out that, when Cys is replaced byArg, this peptide is expected to produce two peptides upontrypsin hydrolysis because of the cleavage of the peptide bondbetween Arg and the next amino acid. Inactivating neil1mutations reduced hNEIL1 expression, and three rare poly-morphisms in the neil1 promoter region have been identified inhuman gastric cancers.30,34 Such polymorphisms or othermutations may lead to differences in the expression level ofhNEIL1,34 as there is evidence for nucleotide changes causingdifferences in protein expression levels.61 One of the identifiedmutations in hNEIL1 in gastric cancer is Gly245Arg.30 Thetryptic peptide GYGSESGEEDFAAFR identified in the presentwork contains this position. Because of the replacement ofGly245 by Arg in the mutated hNEIL1, this peptide wouldundergo the hydrolysis of the peptide bond between Arg andthe next amino acid, leading to the peptides GYR andSESGEEDFAAFR. The methodology developed in the presentwork would readily identify the disappearance of GYGSES-GEEDFAAFR and perhaps the subsequent appearance of GYRand SESGEEDFAAFR in the trypsin hydrolysate of themutated hNEIL1, proving the presence of the said mutation.For the reasons outlined here, it is fair to assume that, inaddition to the identification and quantification of wild typehNEIL1, the methodology developed in this work may permitthe identification of the known variants of hNEIL1 and helpdiscover unknown variants containing amino acid replacementsin one of the 18 peptides identified in this work. In such cases,15N-hNEIL1 may serve not only as the ideal internal standardfor accurate quantification but also help validate the identity oftryptic peptides with amino acid replacements.In a relevant context, NEIL1 mRNA may be altered via RNA-
editing, which may modulate the sequence and activity ofNEIL1.40 hNEIL1 used in this work was the edited form witharginine at position 242 as originally established.19,20,37,38 Theunedited form of hNEIL1 with lysine at position 242 has beenobserved in untreated U87 cells, and it has been overexpressedand purified.40 The unedited and edited forms of hNEIL1exhibited different glycosylase activities and lesion specificitytoward a number of substrates in different sequence contexts.These two forms may be present in mammalian cells undercertain conditions. The methodology described in this workmay permit the distinction of these two proteins from eachother. The tryptic peptide of EVVQLGGR with arginine atposition 242 was identified in this work (see Table 1; peak 8 inFigure 2; Supplemental Figures 4 and 8). Replacing argininewith lysine at position 242 would change the mass of the MH+
ion from 857.48 to 829.47 Da and also the masses of all the y-ions, but not those of the b-ions. Therefore, it would bepossible to identify the unedited form of hNEIL1 by themethodology described here. The use of the 15N-labeled editedform of hNEIL1 will also help identify and quantify theunedited form. The identification and quantification of boththese forms of hNEIL1 may provide important insight into theextent that editing occurs in mammalian cells.In conclusion, mass spectrometric assays were developed for
the identification and quantification of hNEIL1. The full-scanand MS/MS spectra of a large number of tryptic peptides ofthis protein and its 15N-labeled analogue were described. Theuse of the fully 15N-labeled hNEIL1 as an internal standard will
be a great advantage for accurate quantification when complextissue samples are used. The assays described in this work arelikely to be highly suitable for the positive identification andaccurate quantification of hNEIL1 in vivo. The accurate andreliable measurement of expression levels of DNA repairproteins in human tissues will be of fundamental importancefor understanding of their role in disease processes, fordevelopment of DNA repair inhibitors, and for their use asdisease biomarkers.
■ ASSOCIATED CONTENT*S Supporting Information
Supplemental Tables 1−3 and Supplemental Figures 1−16.This material is available free of charge via the Internet athttp://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author
*Address: 100 Bureau Drive, MS 8311, Gaithersburg, MD20899. Tel: 301-975-2581. Fax: 301-975-8505. E-mail: [email protected] Contributions#P.T.R. and P.J. contributed equally to this work.Notes
The authors declare no competing financial interest.
■ ACKNOWLEDGMENTSCertain commercial equipment or materials are identified inthis paper in order to specify adequately the experimentalprocedure. Such identification does not imply recommendationor endorsement by the National Institute of Standards andTechnology, nor does it imply that the materials or equipmentidentified are necessarily the best available for the purpose.
■ REFERENCES(1) Hoeijmakers, J. H. Genome maintenance mechanisms forpreventing cancer. Nature 2001, 411, 366−374.(2) Friedberg, E. C.; Walker, G. C.; Siede, W.; Wood, R. D.; Schultz,R. A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASMPress: Washington, DC, 2005.(3) Beckman, R. A.; Loeb, L. A. Genetic instability in cancer: theoryand experiment. Semin. Cancer Biol. 2005, 15, 423−435.(4) Madhusudan, S.; Middleton, M. R. The emerging role of DNArepair proteins as predictive, prognostic and therapeutic targets incancer. Cancer Treat. Rev. 2005, 31, 603−617.(5) Liu, R.; Yin, L. H.; Pu, Y. P. Reduced expression of human DNArepair genes in esophageal squamous-cell carcinoma in china. J.Toxicol. Environ. Health A 2007, 70, 956−963.(6) Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer2008, 8, 193−204.(7) Loeb, L. A. Human cancers express mutator phenotypes: origin,consequences and targeting. Nature Rev. Cancer 2011, 11, 450−457.(8) Kelley, M. R. Future directions with DNA repair inhibitors: Aroadmap for disruptive approaches to cancer therapy. In DNA Repairin Cancer Therapy: Molecular Targets and Clinical Applications; Kelley,M. R., Ed.; Academic Press: Amsterdam, 2012; pp 301−310.(9) Mohrenweiser, H. W.; Jones, I. M. Variation in DNA repair is afactor in cancer susceptibility: a paradigm for the promises and perilsof individual and population risk estimation? Mutat. Res. 1998, 400,15−24.(10) Loeb, L. A. Mutator phenotype in cancer: origin andconsequences. Semin. Cancer Biol. 2010, 20, 279−280.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXK

(11) Rowe, B. P.; Glazer, P. M. Emergence of rationally designedtherapeutic strategies for breast cancer targeting DNA repairmechanisms. Breast Cancer Res. 2010, 12, 203.(12) Bosken, C. H.; Wei, Q.; Amos, C. I.; Spitz, M. R. An analysis ofDNA repair as a determinant of survival in patients with non-small-celllung cancer. J. Natl. Cancer Inst. 2002, 94, 1091−1099.(13) Rosell, R.; Taron, M.; Barnadas, A.; Scagliotti, G.; Sarries, C.;Roig, B. Nucleotide excision repair pathways involved in Cisplatinresistance in non-small-cell lung cancer. Cancer Control 2002, 10, 297−305.(14) Mambo, E.; Chatterjee, A.; de Souza-Pinto, N. C.; Mayard, S.;Hogue, B. A.; Hoque, M. O.; Dizdaroglu, M.; Bohr, V. A.; Sidransky,D. Oxidized guanine lesions and hOgg1 activity in lung cancer.Oncogene 2005, 24, 4496−4508.(15) Schmid, K.; Nair, J.; Winde, G.; Velic, I.; Bartsch, H. Increasedlevels of promutagenic etheno-DNA adducts in colonic polyps of FAPpatients. Int. J. Cancer 2000, 87, 1−4.(16) Obtulowicz, T.; Swoboda, M.; Speina, E.; Gackowski, D.;Rozalski, R.; Siomek, A.; Janik, J.; Janowska, B.; Ciesla, J. M.; Jawien,A.; Banaszkiewicz, Z.; Guz, J.; Dziaman, T.; Szpila, A.; Olinski, R.;Tudek, B. Oxidative stress and 8-oxoguanine repair are enhanced incolon adenoma and carcinoma patients. Mutagenesis 2010, 25, 463−471.(17) Kirkali, G.; Keles, D.; Canda, A. E.; Terzi, C.; Reddy, P. T.;Jaruga, P.; Dizdaroglu, M.; Oktay, G. Evidence for upregulated repairof oxidatively induced DNA damage in human colorectal cancer. DNARepair (Amst) 2011, 10, 1114−1120.(18) Evans, M. D.; Dizdaroglu, M.; Cooke, M. S. Oxidative DNAdamage and disease: induction, repair and significance. Mutat. Res.2004, 567, 1−61.(19) Hazra, T. K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y. W.;Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterizationof a human DNA glycosylase for repair of modified bases in oxidativelydamaged DNA. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 3523−3528.(20) Bandaru, V.; Sunkara, S.; Wallace, S. S.; Bond, J. P. A novelhuman DNA glycosylase that removes oxidative DNA damage and ishomologous to Escherichia coli endonuclease VIII. DNA Repair(Amst) 2002, 1, 517−529.(21) Rosenquist, T. A.; Zaika, E.; Fernandes, A. S.; Zharkov, D. O.;Miller, H.; Grollman, A. P. The novel DNA glycosylase, NEIL1,protects mammalian cells from radiation-mediated cell death. DNARepair (Amst) 2003, 2, 581−591.(22) Jaruga, P.; Birincioglu, M.; Rosenquist, T. A.; Dizdaroglu, M.Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimi-dine from oxidatively damaged DNA. Biochemistry 2004, 43, 15909−15914.(23) Doublie, S.; Bandaru, V.; Bond, J. P.; Wallace, S. S. The crystalstructure of human endonuclease VIII-like 1 (NEIL1) reveals azincless finger motif required for glycosylase activity. Proc. Natl. Acad.Sci. U. S. A. 2004, 101, 10284−10289.(24) Hu, J.; de Souza-Pinto, N. C.; Haraguchi, K.; Hogue, B. A.;Jaruga, P.; Greenberg, M. M.; Dizdaroglu, M.; Bohr, V. A. Repair offormamidopyrimidines in DNA involves different glycosylases: role ofthe OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280,40544−40551.(25) Roy, L. M.; Jaruga, P.; Wood, T. G.; McCullough, A. K.;Dizdaroglu, M.; Lloyd, R. S. Human polymorphic variants of theNEIL1 DNA glycosylase. J. Biol. Chem. 2007, 282, 15790−15798.(26) Chan, M. K.; Ocampo-Hafalla, M. T.; Vartanian, V.; Jaruga, P.;Kirkali, G.; Koenig, K. L.; Brown, S.; Lloyd, R. S.; Dizdaroglu, M.;Teebor, G. W. Targeted deletion of the genes encoding NTH1 andNEIL1 DNA N-glycosylases reveals the existence of novel carcinogenicoxidative damage to DNA. DNA Repair (Amst) 2009, 8, 786−794.(27) Muftuoglu, M.; de Souza-Pinto, N. C.; Dogan, A.; Aamann, M.;Stevnsner, T.; Rybanska, I.; Kirkali, G.; Dizdaroglu, M.; Bohr, V. A.Cockayne syndrome group B protein stimulates repair of formamido-pyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 2009, 284,9270−9279.
(28) Liu, M.; Bandaru, V.; Bond, J. P.; Jaruga, P.; Zhao, X.; Christov,P. P.; Burrows, C. J.; Rizzo, C. J.; Dizdaroglu, M.; Wallace, S. S. Themouse ortholog of NEIL3 is a functional DNA glycosylase in vitro andin vivo. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 4925−4930.(29) Jaruga, P.; Xiao, Y.; Vartanian, V.; Lloyd, R. S.; Dizdaroglu, M.Evidence for the involvement of DNA repair enzyme NEIL1 innucleotide excision repair of (5′R)- and (5′S)-8,5′-cyclo-2′-deoxy-adenosines. Biochemistry 2010, 49, 1053−1055.(30) Shinmura, K.; Tao, H.; Goto, M.; Igarashi, H.; Taniguchi, T.;Maekawa, M.; Takezaki, T.; Sugimura, H. Inactivating mutations of thehuman base excision repair gene NEIL1 in gastric cancer. Carcino-genesis 2004, 25, 2311−2317.(31) Das, A.; Hazra, T. K.; Boldogh, I.; Mitra, S.; Bhakat, K. K.Induction of the human oxidized base-specific DNA glycosylaseNEIL1 by reactive oxygen species. J. Biol. Chem. 2005, 280, 35272−35280.(32) Vartanian, V.; Lowell, B.; Minko, I. G.; Wood, T. G.; Ceci, J. D.;George, S.; Ballinger, S. W.; Corless, C. L.; McCullough, A. K.; Lloyd,R. S. The metabolic syndrome resulting from a knockout of the NEIL1DNA glycosylase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1864−1869.(33) Maiti, A. K.; Boldogh, I.; Spratt, H.; Mitra, S.; Hazra, T. K.Mutator phenotype of mammalian cells due to deficiency of NEIL1DNA glycosylase, an oxidized base-specific repair enzyme. DNA Repair(Amst) 2008, 7, 1213−1220.(34) Goto, M.; Shinmura, K.; Tao, H.; Tsugane, S.; Sugimura, H.Three novel NEIL1 promoter polymorphisms in gastric cancerpatients. World J. Gastrointest. Oncol. 2010, 2, 117−120.(35) Reddy, P. T.; Jaruga, P.; Nelson, B. C.; Lowenthal, M.;Dizdaroglu, M. Stable isotope-labeling of DNA repair proteins, andtheir purification and characterization. Protein Expr. Purif. 2011, 78,94−101.(36) Dizdaroglu, M.; Reddy, P. T.; Jaruga, P. Identification andquantification of DNA repair proteins by liquid chromatography/isotope-dilution tandem mass spectrometry using their fully 15N-labeled analogues as internal standards. J. Proteome Res. 2011, 10,3802−3813.(37) Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.;Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutMsuperfamily: an alternative pathway for the repair of 8-oxoguanine andother oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926−4936.(38) Takao, M.; Kanno, S.; Kobayashi, K.; Zhang, Q. M.; Yonei, S.;van der Horst, G. T.; Yasui, A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J. Biol.Chem. 2002, 277, 42205−42213.(39) Hegde, M. L.; Theriot, C. A.; Das, A.; Hegde, P. M.; Guo, Z.;Gary, R. K.; Hazra, T. K.; Shen, B.; Mitra, S. Physical and functionalinteraction between human oxidized base-specific DNA glycosylaseNEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028−27037.(40) Yeo, J.; Goodman, R. A.; Schirle, N. T.; David, S. S.; Beal, P. A.RNA editing changes the lesion specificity for the DNA repair enzymeNEIL1. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20715−20719.(41) Davis, R. W.; Botstein, D.; Roth, J. R. A Manual for GeneticEngineering: Advanced Bacterial Genetics; Cold Spring HarborLaboratory; Cold Spring Harbor, NY, 1980.(42) Lowry, O. H.; Rosebrough, N. J.; Farr, A. L.; Randall, R. J.Protein measurement with the Folin phenol reagent. J. Biol. Chem.1951, 193, 265−275.(43) Jaruga, P.; Kirkali, G.; Dizdaroglu, M. Measurement offormamidopyrimidines in DNA. Free Radic. Biol. Med. 2008, 45,1601−1609.(44) Laemmli, U. K. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature 1970, 227, 680−685.(45) Rosenfeld, J.; Capdevielle, J.; Guillemot, J. C.; Ferrara, P. In-geldigestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 1992, 203, 173−179.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXL

(46) Kinter, M.; Sherman, N. E. Protein Sequencing and IdentificationUsing Tandem Mass Spectrometry; Wiley-Interscience: New York, 2000.(47) Dizdaroglu, M. Base-excision repair of oxidative DNA damageby DNA glycosylases. Mutat. Res. 2005, 591, 45−59.(48) Dizdaroglu, M. Application of capillary gas chromatography-mass spectrometry to chemical characterization of radiation-inducedbase damage of DNA; implications for assessing DNA repair processes.Anal. Biochem. 1985, 144, 593−603.(49) Dizdaroglu, M. Quantitative determination of oxidative basedamage in DNA by stable isotope-dilution mass spectrometry. FEBSLett. 1993, 315, 1−6.(50) Eidhammer, I.; Flikka, K.; Martens, L.; Mikalsen, S.-O.Computational Methods for Mass Spectrometric Proteomics; John Wiley& Sons, Ltd.: West Sussex, England, 2007.(51) Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selectedreaction monitoring for quantitative proteomics: a tutorial. Mol. Syst.Biol. 2008, 4, 222.(52) Dizdaroglu, M. Oxidatively induced DNA damage: Mechanisms,repair and disease. Cancer Lett. 2012, 327, 26−47.(53) Theriot, C. A.; Hegde, M. L.; Hazra, T. K.; Mitra, S. RPAphysically interacts with the human DNA glycosylase NEIL1 toregulate excision of oxidative DNA base damage in primer-templatestructures. DNA Repair (Amst) 2010, 9, 643−652.(54) Ong, S. E.; Mann, M. Mass spectrometry-based proteomicsturns quantitative. Nat. Chem. Biol. 2005, 1, 252−262.(55) Kuster, B.; Schirle, M.; Mallick, P.; Aebersold, R. Scoringproteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol.2005, 6, 577−583.(56) Brun, V.; Dupuis, A.; Adrait, A.; Marcellin, M.; Thomas, D.;Court, M.; Vandenesch, F.; Garin, J. Isotope-labeled protein standards:toward absolute quantitative proteomics. Mol. Cell Proteomics. 2007, 6,2139−2149.(57) Janecki, D. J.; Bemis, K. G.; Tegeler, T. J.; Sanghani, P. C.; Zhai,L.; Hurley, T. D.; Bosron, W. F.; Wang, M. A multiple reactionmonitoring method for absolute quantification of the human liveralcohol dehydrogenase ADH1C1 isoenzyme. Anal. Biochem. 2007,369, 18−26.(58) Brun, V.; Masselon, C.; Garin, J.; Dupuis, A. Isotope dilutionstrategies for absolute quantitative proteomics. J. Proteomics. 2009, 72,740−749.(59) Singh, S.; Springer, M.; Steen, J.; Kirschner, M. W.; Steen, H.FLEXIQuant: a novel tool for the absolute quantification of proteins,and the simultaneous identification and quantification of potentiallymodified peptides. J. Proteome Res. 2009, 8, 2201−2210.(60) Zinn, N.; Winter, D.; Lehmann, W. D. Recombinant isotopelabeled and selenium quantified proteins for absolute proteinquantification. Anal. Chem. 2010, 82, 2334−2340.(61) Guillemette, C.; Millikan, R. C.; Newman, B.; Housman, D. E.Genetic polymorphisms in uridine diphospho-glucuronosyltransferase1A1 and association with breast cancer among African Americans.Cancer Res. 2000, 60, 950−956.
Journal of Proteome Research Technical Note
dx.doi.org/10.1021/pr301037t | J. Proteome Res. XXXX, XXX, XXX−XXXM




















