
Identification and characterization of a spontaneously aggregating amyloid-forming variant of human...
14
The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676 Available online at www.sciencedirect.com Identification and characterization of a spontaneously aggregating amyloid-forming variant of human PrP (90–231) through phage-display screening of variants randomized between residues 101 and 112 Archana Verma a,1 , Swati Sharma b,1 , Nirmal Kumar Ganguly a , Siddharta Majumdar a , Purnananda Guptasarma b , Manni Luthra-Guptasarma a,∗ a Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh 160012, India b Institute of Microbial Technology, Sector 39-A, Chandigarh 160036, India Received 7 June 2007; received in revised form 3 September 2007; accepted 5 October 2007 Available online 13 October 2007 Abstract The N-terminal ‘unstructured’ region of the human prion protein [PrP (90–231) ] is believed to play a role in its aggregation because mutations in this region are associated with seeding-independent deposition disorders like Gerstmann–Straussler–Scheinker disease (GSS). One way of examining the effects of such mutations is to search combinatorially derived libraries for sequence variants showing a propensity to aggregate and/or the ability to interact with prion molecules folded into a -sheet-based conformation (i.e., -PrP or PrP Sc ). We created a library of 1.8 × 10 7 variants randomized between positions 101 and 112, displayed it on filamentous bacteriophage, and ‘spiked’ it with a ∼25% population of phages-bearing wild-type prion (wt-PrP). Screening was performed through four rounds of biopanning and amplification against immobilized -PrP, and yielded three -PrP-binding populations: wt- PrP (26% representation) and two non-wt-PrP variants (∼10% and ∼64% representation, respectively). The remarkable enrichment of one non-wt-PrP variant (MutPrP) incorporating residues KPSKPKTNMKHM in place of KGVLTWFSPLWQ, despite its initial representation at a 5 million-fold lower level than wt-PrP, caused us to produce it and discover: (i) that it readily aggregates into thioflavin-T-binding amyloids between pH 6.0 and 9.0, (ii) that it adopts a soluble -sheet based monomeric structure at pH 10.0, (iii) that it is less thermally stable and more compact than wt-PrP, and (iv) that it displays significantly greater resistance to proteolysis than wt-PrP. Our results suggest that sequence variations in the 101–112 region can indeed predispose the prion for aggregation. © 2007 Elsevier Ltd. All rights reserved. Keywords: Prion aggregation; Protein misfolding; Phage display; Sequence variants; Amyloid formation; Conformational transition 1. Introduction In its normal form, the prion protein, PrP c , is a GPI-anchored, metal-binding, cell-surface polypeptide ∗ Corresponding author. Tel.: +91 172 2755196; fax: +91 172 2744401/5078. E-mail address: [email protected] (M. Luthra-Guptasarma). 1 These authors contributed equally to the work. folded into a predominantly -helical conformation (Prusiner et al., 1982). In its abnormal or diseased form, the same polypeptide chain adopts a predominantly -sheet conformation, PrP Sc , which engages in inter- molecular interactions to deposit into fibrillar amyloid aggregates. The switching of conformation from PrP c to PrP Sc is widely believed to occur through a contact- based molecular mechanism in which infective PrP Sc aggregates function as templates directing the structural remodeling of PrP c through direct molecule–molecule 1357-2725/$ – see front matter © 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.biocel.2007.10.009
Transcript of Identification and characterization of a spontaneously aggregating amyloid-forming variant of human...

A
m(s�btPorttt©
K
1
G
f
(
1
The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676
Available online at www.sciencedirect.com
Identification and characterization of a spontaneously aggregatingamyloid-forming variant of human PrP(90–231) through phage-display
screening of variants randomized between residues 101 and 112
Archana Verma a,1, Swati Sharma b,1, Nirmal Kumar Ganguly a, Siddharta Majumdar a,Purnananda Guptasarma b, Manni Luthra-Guptasarma a,∗
a Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh 160012, Indiab Institute of Microbial Technology, Sector 39-A, Chandigarh 160036, India
Received 7 June 2007; received in revised form 3 September 2007; accepted 5 October 2007Available online 13 October 2007
bstract
The N-terminal ‘unstructured’ region of the human prion protein [PrP(90–231)] is believed to play a role in its aggregation becauseutations in this region are associated with seeding-independent deposition disorders like Gerstmann–Straussler–Scheinker disease
GSS). One way of examining the effects of such mutations is to search combinatorially derived libraries for sequence variantshowing a propensity to aggregate and/or the ability to interact with prion molecules folded into a �-sheet-based conformation (i.e.,-PrP or PrPSc). We created a library of 1.8 × 107 variants randomized between positions 101 and 112, displayed it on filamentousacteriophage, and ‘spiked’ it with a ∼25% population of phages-bearing wild-type prion (wt-PrP). Screening was performedhrough four rounds of biopanning and amplification against immobilized �-PrP, and yielded three �-PrP-binding populations: wt-rP (26% representation) and two non-wt-PrP variants (∼10% and ∼64% representation, respectively). The remarkable enrichmentf one non-wt-PrP variant (MutPrP) incorporating residues KPSKPKTNMKHM in place of KGVLTWFSPLWQ, despite its initial
epresentation at a 5 million-fold lower level than wt-PrP, caused us to produce it and discover: (i) that it readily aggregates intohioflavin-T-binding amyloids between pH 6.0 and 9.0, (ii) that it adopts a soluble �-sheet based monomeric structure at pH 10.0, (iii)hat it is less thermally stable and more compact than wt-PrP, and (iv) that it displays significantly greater resistance to proteolysishan wt-PrP. Our results suggest that sequence variations in the 101–112 region can indeed predispose the prion for aggregation.2007 Elsevier Ltd. All rights reserved.
uence v
eywords: Prion aggregation; Protein misfolding; Phage display; Seq. Introduction
In its normal form, the prion protein, PrPc, is aPI-anchored, metal-binding, cell-surface polypeptide
∗ Corresponding author. Tel.: +91 172 2755196;ax: +91 172 2744401/5078.
E-mail address: [email protected]. Luthra-Guptasarma).1 These authors contributed equally to the work.
357-2725/$ – see front matter © 2007 Elsevier Ltd. All rights reserved.doi:10.1016/j.biocel.2007.10.009
ariants; Amyloid formation; Conformational transition
folded into a predominantly �-helical conformation(Prusiner et al., 1982). In its abnormal or diseased form,the same polypeptide chain adopts a predominantly�-sheet conformation, PrPSc, which engages in inter-molecular interactions to deposit into fibrillar amyloidaggregates. The switching of conformation from PrPc
to PrPSc is widely believed to occur through a contact-based molecular mechanism in which infective PrPSc
aggregates function as templates directing the structuralremodeling of PrPc through direct molecule–molecule

f Bioch
association with �-PrP bound to a solid substrate. Alter-
664 A. Verma et al. / The International Journal o
interactions, causing PrPc to be cast in the imageof PrPSc. This conversion is believed to be responsi-ble for the increase in deposited quantities of PrPSc
in the diseased state. PrPSc differs from PrPc in itsphysico-chemical properties. It is insoluble even in milddetergents in contrast to PrPc which is soluble. Fur-thermore, PrPc is sensitive to degradation by proteases,whereas limited proteolysis of PrPSc by proteinaseK results in N-terminally truncated fragments with amolecular mass of 27–30 kDa, designated as PrP 27–30(Safar et al., 2005).
In certain naturally occurring, genetically derivedmutants of the prion polypeptide, the conformationalswitch from PrPc to PrPSc is believed to occurspontaneously, obviating the need for exogenous ‘infec-tion’ by PrPSc particles. In two well-known diseases,Gerstmann–Straussler–Scheinker (GSS) syndrome andCreutzfeldt–Jacob disease (CJD), mutations are linkeddirectly to the spontaneous appearance and amplifica-tion of the PrPSc conformation (Collinge, 1997; Prusiner,Scott, De Armond, & Cohen, 1998). Certain naturallyoccurring mutations in the human population associatedwith these diseases locate in a region in the N-terminalsection of the prion protein between residues 101 and112 (Goldfarb et al., 1992; Huang et al., 1994; Leclercet al., 2001; Riek et al., 1996). It has been proposedthat semi-structured hinges, or loops bridging substruc-tures within the prion could be destabilized by mutationsoccurring in this region, making the prion more prone toundergoing spontaneous misfolding into the PrPSc con-formation. Notably, the entire region between residues90 and 112 is thought to be loosely structured and lacksdefined secondary structure, even though it appears toplay an important role in the switching of conformationsbetween the �-helical and �-sheet states (James et al.,1997); in particular, residues 101, 108, and 111–113,have been shown to participate in structurally stabilizinghydrophobic interactions with the C-terminal sections ofthe protein (Barron et al., 2001; Levy, Hanan, Solomon,& Becker, 2001; Supattapone et al., 2001). It is possi-ble, therefore, that naturally occurring mutations in thisregion destabilize key interactions of the region withother regions and cause it to dissociate away from the C-terminal of the prion molecule in the PrPc conformation,thereby increasing the scope for its structural remodel-ing through contact with PrPSc. According to availableNMR spectroscopic data, recombinant prion protein con-tains a well-structured C-terminal region consisting ofthree �-helices and two very short �-strands, whereas
the N-terminal region of the molecule is highly flexible(Zahn et al., 2000). If perchance the helical structure ofthe C-terminal region were to happen to be dependent,emistry & Cell Biology 40 (2008) 663–676
at least kinetically, on packing interactions with residuesfrom the flexible N-terminal region, it would be conceiv-able for mutations in the N-terminal region to facilitatethe profound molecule-wide helix-to-sheet conversionaccompanying amyloid formation. Thus, regions such asthe stretch between residues 101 and 112 which containnaturally occurring mutations predisposing individualsto prion disease become worthy of further attention.
In order to understand the molecular events underly-ing the conversion of PrPc to PrPSc, studies have reliedon heterologous expression systems in order to obtainsufficient quantities of protein for biophysical studies.For most studies, the region of the reading frame span-ning residues 90–231 of human PrP (corresponding toPrP 27–30) has been cloned and expressed in E. coli.This segment encompasses the entire sequence of theprotease-resistant protein found in prion diseased brain,and contains all known point mutations associated withhuman prion disorders, and is sufficient for the propaga-tion of the disease (Leffers et al., 2004).
To further explore the importance of this region,we used the phage-display library-based screeningapproach, developed over a decade ago as a rapidselection method for identification of protein variantswith altered binding characteristics to a known ligand(Smith & Scott, 1993). As is widely acknowledged, thisapproach allows one to simultaneously make qualitativeand semi-quantitative analyses of the binding propertiesof a large number of variants within a single ‘compe-tition’ experiment, to select the best binding variants.Given the difficulties of obtaining and working withPrPSc, we decided to use as the ligand a �-sheet-basedunimolecular form of the prion protein called �-PrPwhich is generated through the refolding of prion proteinunder reducing conditions at acidic pH (Jackson et al.,1999b). We immobilized �-PrP and allowed it to inter-act with a phage-displayed library of prions varying inrespect of their sequences between residue positions 101and 112.
We hoped, specifically, to examine (i) whether theregion between residues 101 and 112 is important forthe conformational switching mechanism, by examin-ing whether sequence changes in this region facilitate,under physiological conditions, the adoption of a �-sheet conformation by prion molecules displayed onphage. Without the attendant complications of aggre-gation, the adoption of the �-sheet-based conformationcould imaginably occur through induction involving
natively, it could also occur through the natural adoptionof a �-sheet-based conformation incidentally capable ofbinding to �-PrP, i.e., capable of interaction with immo-

f Bioch
bawpstPdhiss
drobes2ntapos
rpaiOlmsihnv‘fip
2
2o
wbT
A. Verma et al. / The International Journal o
ilized �-PrP, allowing phage-display library screeningnd identification as a �-PrP-binding variant. In otherords, adoption of the �-sheet structure by a variantrion could constitute the cause, rather than the con-equence, of interactions with �-PrP. We also hopedo examine (ii) whether the prion polypeptide in therPc conformation (i.e., wt-PrP, as a control) itself alsoisplays a tendency to switch conformation from an �-elical form to a �-sheet-based form and bind to �-PrP,n competition with a large number of mutant, potentiallytructurally destabilized forms capable of undergoing theame structural transition.
A phage library of mutant proteins can be pro-uced through either the individual randomization of keyesidue positions, one at a time, or through the simultane-us collective randomization of all positions assumed toe important. The latter approach can offer a much morexhaustive scope for panning of the available ‘sequencepace’ (i.e., sampling of 20n variations instead of the0 × n variations sampled by the former approach; whereindicates the number of positions being varied); in par-
icular, where variations at many neighboring locationsppear to be important and one is seeking a ‘proof-of-rinciple’ demonstration of the importance of a stretchf sequence, rather than the discovery of very specificingle-site mutations.
Therefore, we opted to completely randomize a 12esidue region lying in the N-terminus of the prionolypeptide PrP(90–231) (residues 101–112) by varyingll 12 positions simultaneously, and screening for bind-ng to �-PrP. We report here the isolation of two variants.ne of these variants is shown to be spontaneously amy-
oid forming. We also report that wt-PrP (an �-helicalolecule) appears to bind to �-PrP, a unimolecular �-
heet-based form of the prion, presumably by switchingnto a �-sheet-based conformation itself. Interestingly,owever, wt-PrP appears to show a six orders of mag-itude lower tendency to bind to �-PrP than the twoariants, suggesting that the latter may be some sort ofsuper-prions’, that straightaway deposit into amyloidbers instead of simply possessing an apparent predis-osition to do so.
. Methods
.1. Cloning, expression, purification and refoldingf wt-PrP to α and β forms
The intron-less gene segment encoding PrP(90–231)as amplified from genomic DNA isolated from humanlood using the oligonucleotide, ATG CGG TAC CGCAG CGG ATC CGG TCA AGG AGG TGG CAC
emistry & Cell Biology 40 (2008) 663–676 665
CCA CAG T, as the forward primer, F1 (incorporat-ing BamHI and NheI sites) and the oligonucleotide,TTC AAG CTT ACG CGT TAA GCT TGC TCG ATCCTC TCT GGT AAT AGG C, as the reverse primer,R1 (incorporating HindIII and MluI sites). Using stan-dard protocols, the PCR product was digested withBamHI and HindIII, and ligated into the polylinker siteof the pQE-30 vector (Qiagen) to encode PrP(90–231)in genetic fusion with an N-terminal 6XHis histidine-affinity tag. Following transformation into E. coliJM109, and subcloning into SG13009[pREP4] usingamp/kan screening, the integrity of the construct wasconfirmed through sequencing on an ABI Prism310sequencer. SG13009[pREP4] cells induced to expressPrP(90–231) were lysed in 8 M urea and the protein puri-fied through standard procedures using Ni-NTA agarose(Qiagen). Refolding under oxidizing conditions wasdone by the method of Jackson et al. (1999a). Therefolded product obtained was examined by far-UV cir-cular dichroism (CD) spectroscopy, and electrophoreticevaluation of susceptibility to proteinase K digestion, forassessment of whether it was analogous to PrPc, i.e., the�-helical form of the prion protein (�-PrP). Separately,purified protein was also refolded into the predominantly�-sheet-containing form, �-PrP, under acidic and reduc-ing conditions by the method of Jackson et al. (1999a) atpH 4.0, and checked for conversion to the �-PrP usingCD spectroscopy and proteinase K digestion after trans-fer to pH 8.0. The �-sheet conformation is known to bestable for upto a week after transfer to pH 8.0 (Jacksonet al., 1999b). CD measurements were carried out with�-PrP and �-PrP concentrations of 0.2 and 0.46 mg/ml,respectively.
2.2. Construction of a phage-display library of PrPvariants
Incorporation of randomization in residues 101–112of PrP(90–231) was carried out through two steps. Briefly,(i) custom-synthesis was used to create a degener-ate set of oligonucleotides incorporating a randomizedsequence of 36 bases (corresponding to the 12 residues),flanked by 23 bases on either side corresponding toregions naturally flanking the residues. Using these oli-gos as forward primer, F2 [GTG GCA CCC ACA GTCAGT GGA AC(NNS)12 GCT GGT GCT GCA GCAGCT GGG GC; where N is A, C, G, or T (equimo-lar) and S is G or C (equimolar)], R1 as reverse primer,
and the DNA encoding PrP(90–231) as template (300 ng),the library of F2 primers was incorporated into a con-struct extending upto the 3′ end of the DNA encodingPrP(90–231). Further, (ii) this library (438 bp; 600 ng) was
f Bioch
666 A. Verma et al. / The International Journal oused as template in a reaction in which F1 and R1 wereused as forward and reverse primers for splicing by over-lap extension (SOE), using the 14 base overlap of F1with the template. It may be noted that the product ofStep (i) was used as template in Step (ii) without gelpurification, so that some DNA encoding the originalwt-PrP prion would also exist in the library, for reasonsdetailed in discussion relating to our need to spike thelibrary with wt-PrP-bearing phages. The spliced product(470 bp) from Step (ii) was gel purified, digested withNheI and MluI (500 units of each enzyme per 25 �gof vector), repurified (yield ≈7.6 �g) and cloned intothe phage-display vector, fdtetTA74 (Sampath, Abrol,& Chaudhary, 1997), obtained as a gift from Dr. V.K.Chaudhary (Dept. of Biochem., Univ. of Delhi) through25 separate ligation reactions using a molar ratio of 1:6for vector: insert, with 1 �g vector in each reaction, and 3units of T4 DNA ligase in volumes of 10 �l each, in 4 ◦Covernight ligations. The ligation products were pooled,ethanol-precipitated and electro-transformed into com-petent JM109 E. coli cells that secreted the encodedphages. Amplification of the library of secreted phageswas done by growing transformed cells in media contain-ing tetracycline at 37 ◦C for 12 h, followed by harvestingof phage by PEG/NaCl treatment. Centrifugation at12,000 × g for 20 min resulted in precipitation of phagecomprising the library of PrP variants. Phage titers wereestimated in terms of plaque-forming units (pfu)/ml bystandard methods (Smith & Scott, 1993).
2.3. Screening of the library of variants for bindingto β-PrP
For coating of 6XHis-tagged �-PrP, His-Sorb (Qia-gen) strips were used. The protein (�-PrP) which hadbeen created through refolding in pH 4.0 buffer underreducing conditions was transferred to pH 7.5 bufferimmediately before coating. There were three reasonsfor doing the coating at pH 7.5: (i) the 6XHis tag onthe �-PrP could be expected to bind to Ni-NTA only atbasic pH (at acidic pH, the nitrogen on the imidazolering becomes protonated causing the histidine residuesin the tag to lose affinity for Ni-NTA); (ii) it is known(Jackson et al., 1999a) that �-PrP retains its �-sheetconformation for several days in pH 7–8 buffer fol-lowing transfer from buffer of pH 4.0, i.e., the �-sheetdominated unimolecular conformation shows sufficientkinetic stability to be subjected to coating and phage-
display biopanning; (iii) it was desirable to performbiopanning with the library of PrP(90–231) variants at a pHclose to physiological pH, so that any spontaneous aggre-gation shown by a prion variant could be considered toemistry & Cell Biology 40 (2008) 663–676
be biologically relevant and feasible. Thus, immediatelyfollowing transfer from pH 4.0 buffer, approximately50 �g of �-PrP in 200 �l of pH 7.5 buffer (10 mM Triscontaining 10 mM sodium acetate) were subjected toovernight incubation in one well of a 96-well microtiterplate, with a control sample (lacking prion, but includingblocking agents) incubated similarly in a different well.Following overnight incubation with �-PrP, and requi-site washing procedures, equivalent amounts of inputlibrary phage were incubated in both wells to effectbiopanning, following which phages were eluted, titeredand amplified using standard procedures (Parmley &Smith, 1988; Smith & Scott, 1993). Four such cycles ofbiopanning and amplification were carried out; in eachcycle the input phage used from the previous round ofamplification was carefully ensured to be 2 × 1011. Fol-lowing each cycle, the eluted phage was titered throughplating of an aliquot mixed with host strain XL1-Blueonto LB/tet/X-gal/IPTG plates and counting of plaques.About 20 plaques were picked up for PCR amplificationusing PrP(90–231) specific primers (as detailed earlier)and sequenced on an ABI Prism 310 sequencer.
2.4. Creation of the recombinant mutant (MutPrP)selected through phage display
Fig. S5 of the supplemental data shows the scheme ofcreation of MutPrP. We first introduced mutations in theregion spanning the residues 101–112 using a custom-synthesized primer (Mut Primer). This primer had anoverlap (with the wild-type PrP gene) of 26 bases on the5′ end and 17 bases on the 3′ end of the region incor-porating the mutations [shown in red color in Fig. S5].The first PCR step was carried out using the Mut Primeras forward primer and Primer 2 as reverse primer. ThisPCR product was shorter than the DNA segment neededto encode the full length PrP(90–231), and so another PCRreaction was carried out using Primer 1 as forward primerand Primer 2 as reverse primer to synthesize the genefor full length MutPrP (spanning residues 90–231 alongwith appropriate 5′ and 3′ extensions for subsequentrestriction digestion and ligation into the vector). Thesequences of the primers are mentioned below:
Primer 1 (forward primer): 5′ ATG CGG TAC CGCTAG CGG ATC CGG TCA AGG AGG TGG CAC CCACAG T 3′.Primer 2 (reverse primer): 5′ TTC AAG CTT ACG
CGT TAA GCT TGC TCG ATC CTC TCT GGT AATAGG C 3′.Mut Primer (forward primer): 5′ GTG GCA CCC ACAGTC AGT GGA ACA AGG GCG TCC TCA CGT
f Bioch
Tat
2
2tcdsratMtbeamamaEast
2
Pawpwslfvs
2
PJm
A. Verma et al. / The International Journal o
GGT TCA GCC CGC TCT GGC AGG CTG GTG CTGCAG CAG C 3′.
he final PCR product was double digested with BamHInd HindIII restriction enzymes and finally cloned intohese very sites in the pQE 30vector.
.5. Protein concentration estimations
Absorbance of protein solutions was measured at80 nm (A280 nm) using a Cary 50 UV–Visible spec-rophotometer. The absorbance was converted to proteinoncentration on the basis of the theoretically pre-icted absorption coefficients calculated by Vector NTIoftware. For wt-PrP, an A280 nm of 1.0 was taken to cor-espond to a concentration of 0.89 mg/ml. For MutPrP,n A280 nm of 1.0 was taken to correspond to a concen-ration of 0.57 mg/ml. The drop in the concentration of
utPrP for an absorbance of 1.0 was due to the introduc-ion of two tryptophan residues in the altered sequenceetween residues 101 and 112. It may be noted thatstimation of protein concentrations based on predictedbsorption coefficients is the next-best method of deter-ining protein concentrations to the use of actual known
bsorption coefficients determined through the primaryeasurement of the amounts of non-hydrolysable amino
cids generated upon acid hydrolysis of peptide bonds.specially for proteins differing profoundly in aromaticmino acid content, this method is preferable to mea-urements based on other indirect colorimetric methodshat tend to be skewed by differential aromatic contents.
.6. Gel filtration chromatography
The gel filtration elution behavior of wt-PrP and Mut-rP was studied on a Pharmacia SMART system. Pumpsnd columns were washed with filtered Millipore Elix-3ater and pre-equilibrated with buffers of appropriateH corresponding to the sample pH. The column usedas the micro-analytical Superdex-75 (Amersham Bio-
ciences; bed volume ∼2.4 ml). The volume of sampleoaded was 50 �l. Samples were loaded onto the columnor either purification or in analytical mode. The elutionolumes were compared with the elution profiles of thetandards run on the same column.
.7. Fluorescence spectroscopy
Fluorescence spectroscopy was carried out on aerkin-Elmer spectrofluorimeter (LS-50B) or on aasco J810 spectropolarimeter fitted with an emissiononochromator. Tryptophan fluorescence spectra: For
emistry & Cell Biology 40 (2008) 663–676 667
fluorescence emission spectra of proteins, the excitationwavelength was set at 280 or 275 nm and the emissionspectrum recorded between 300 and 400 nm. The excita-tion/emission band passes used were either 5/5, 10/10 or5/10 nm. Scan speeds were set at 100 or 120 nm/min andall spectra were averaged over 5 or 10 scans. ThioflavinT (ThT) fluorescence spectra: thioflavin T is a reagentknown to become strongly fluorescent upon binding toamyloid fibrils through intercalation into the cross-�-structure formed by polypeptide chains in these fibrils.ThT-binding studies for detection of amyloids wereperformed by incubating protein solutions or microsus-pensions in 6 �M solution of thioflavin T (in 10 mMTris, pH 8.0) for 1 hr and recording the emission spec-trum (455–550 nm) using an excitation wavelength of444 nm. Excitation and emission slits were set at 7 nmand scan speed was set at 100 nm/min. Control spectrawere collected for buffer containing ThT but no protein,as well as for buffer containing ThT and another protein(lysozyme, 0.2 mg/ml).
2.8. Circular dichroism spectroscopy
Far-UV CD spectra were collected for wt-PrP andMutPrP in buffers of pH 10.0, or pH 8.0 as nec-essary on a Jasco J-810 CD spectropolarimeter incuvettes of 1.0 cm/0.5 cm/0.2 cm path length, withflushing of Iolar grade I nitrogen gas at 6–12 l/min.The wavelength range of data collection was mostly250–190, or 250–200 nm. Data integration time usedwas approximately 1 s/nm. The raw ellipticity obtainedwas converted to mean residue ellipticity [θ] indeg cm2 dmol−1, using the formula: [θ] = {θobs inmillidegrees × 100 × MRW}/{1000 × protein concen-tration (in mg/ml) × path length (in cm)}, where MRW(mean residue weight) was equal to the total molecu-lar weight of the protein/total number of amino acids.For CD data at a single wavelength, e.g. in thermal meltscans, heating was carried out using the Peltier accessoryof the J-810 instrument at 3 ◦C/min for both wt-PrP andMutPrPm using a buffer of pH 8.0 for the former, andpH 10.0 for the latter.
2.9. FTIR spectroscopy
FTIR spectra were collected using a SpectrumBX spectrometer (Perkin-Elmer) between 4000 and600 cm−1 using a resolution of 1 cm−1 and by averag-
ing 32 scans. The solid aggregated sample of MutPrPwas resuspended in water, deposited on the inner surfaceof one CaF2 window of a double window demountableFTIR cell, and allowed to dry completely before the cell
f Bioch
668 A. Verma et al. / The International Journal owas assembled and subjected to scanning. The region ofinterest in the spectrum is between 1700 and 1600 cm−1,where the amide I absorption band for peptide bonds ischaracteristic of the secondary structural content of theprotein.
2.10. Electron microscopy
Transmission electron microscopy (TEM) of theprotein aggregates was done on a JEOL 1200 EX2 instru-ment using the standard procedure of negative stainingwith phosphotungstic acid (PTA) and uranyl acetate.
2.11. MALDI-TOF mass spectrometry
MALDI-TOF mass spectrometry was performed onan Applied Biosystems Voyager DE-STR instrument.Protein samples were prepared by mixing with equal vol-ume of the matrix, Sinapinic acid. Approximately 1 �lof protein sample was added to 1 �l of the matrix andspotted and dried on sample plates. Samples were set upin duplicates or triplicates. The matrix stock solutions(10 mg/ml) were prepared in 0.3% TFA (trifluoroaceticacid) and 50% acetonitrile.
2.12. N-terminal sequencing
Sequencing of the 14 kDa band was done in the fol-lowing manner. The contents of the entire gel weretransferred through electro-blotting onto a PVDF mem-brane (immobilon PSQ, Sigma Chemical Co.), using theelectro-transfer accessory of a BioRad Mini-Protean IIapparatus. The blot was stained by the dye amido black[0.1% amido black (Sigma Chemical Co.) in 1% aceticacid]. The band on the stained PVDF blot was excisedfrom the dried membrane, cut into small pieces anddestained with 50% methanol. Then it was thoroughlyrinsed with water and allowed to dry. This was thenloaded onto the ABI 476A protein sequencer (AppliedBiosystems).
3. Results
3.1. Cloning, expression, purification and folding ofPrP(90–231) into α-PrP and β-PrP
As described in the Experimental section, the segmentof the intron-lacking gene encoding the prion protein
between residues 90 and 231 [PrP(90–231)] was amplifiedfrom human genomic DNA and cloned into the vectorpQE-30 (Qiagen) to produce a fusion at the N-terminuswith a 6XHis affinity tag (Fig. S1A). The tag was addedemistry & Cell Biology 40 (2008) 663–676
to facilitate ease of purification using Ni-NTA (nickel-nitrilotriacetic acid) conjugated sepharose resin-basedaffinity chromatography, and also for facile immobiliza-tion of PrP(90–231) on Ni-NTA coated His-Sorb plates(Qiagen) after conversion into a �-sheet-based con-formation, for screening for binding to populations ofphage-displayed variants.
Expression levels of the fusion protein amounted toroughly 20% of cellular protein (Fig. 1A), with theprotein present mainly in the form of intracellular inclu-sion bodies. Pure wt-PrP polypeptide (18.2 kDa) wasobtained through affinity chromatography under reduc-ing, denaturing conditions. The polypeptide chain wasthen oxidized and refolded into �-PrP at pH 8.0 toproduce native PrP(90–231), as confirmed by circulardichroism spectroscopy (Fig. S2) and by its suscepti-bility to proteinase K digestion (Fig. S3A). Separately,the PrP(90–231) polypeptide was also refolded into the�-sheet-based conformation (�-PrP) at pH 4.0, underreducing rather than oxidizing conditions, and far-UV CD spectra (Fig. S2) and proteinase K digestion(Fig. S3B) were used to confirm the formation of the rel-atively more protease-resistant �-sheet-based structureunder these conditions.
3.2. Creation and characterization of the library ofprion variants
A schematic of the strategy used to construct thelibrary of PrP(90–231) variants randomized between 101and 112 is shown in Fig. 2. Data relating to the inter-mediate stages of construction of the library (PCR1and PCR2), together with evidence of incorporationinto the phage-display vector fdtetTA74 is shown inFig. S1B and C. The transformation of the phage vec-tor occurred at an efficiency of approximately 1 × 106
clones/�g of DNA. Through sequencing of clones pickedat random, it was discovered that roughly three-fourthsof the clones contained unique sequences differing fromthe native sequence in the region between residues 101and 112, whereas one-fourth sequences contained DNAidentical to that of the original gene [wt-PrP(90–231)].This wt-PrP component of the library originated fromthe amplification of a small amount of the gene encod-ing PrP(90–231) left over as a template in the reactiontube from the first PCR reaction (Step I; see Fig. 2)used to create the library; thus allowing the libraryto contain phages-bearing wt-PrP . Cells trans-
(90–231)formed with phage were grown in media containingtetracycline to amplify the phage. A total of 25 �g ofDNA was transformed. Given the efficiency of transfor-mation and the observed fraction of unique sequences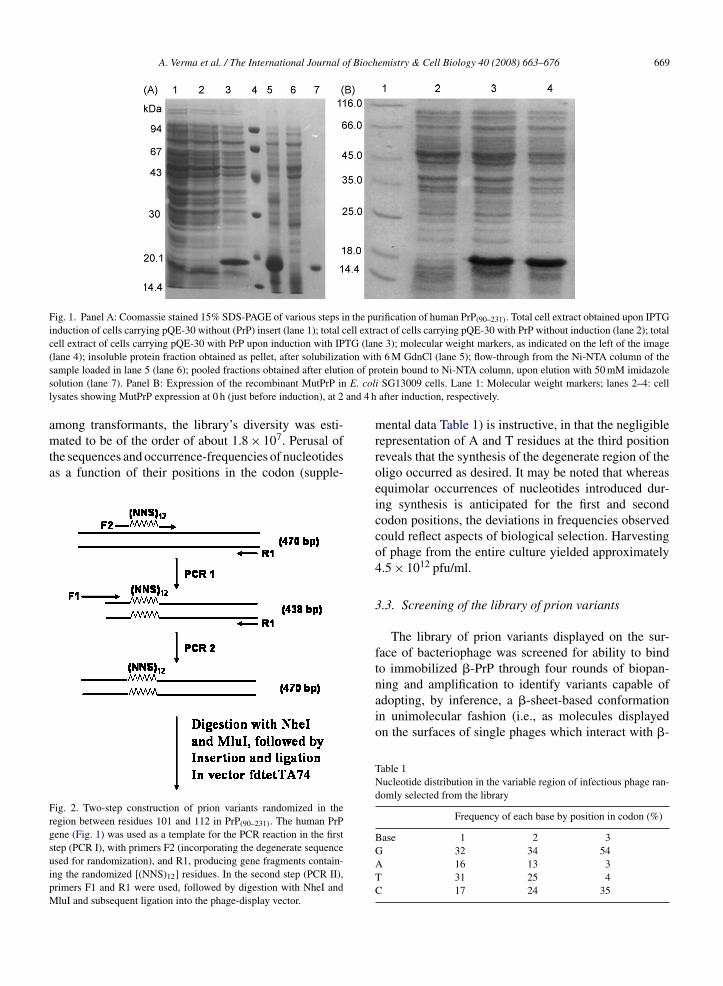
A. Verma et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676 669
Fig. 1. Panel A: Coomassie stained 15% SDS-PAGE of various steps in the purification of human PrP(90–231). Total cell extract obtained upon IPTGinduction of cells carrying pQE-30 without (PrP) insert (lane 1); total cell extract of cells carrying pQE-30 with PrP without induction (lane 2); totalcell extract of cells carrying pQE-30 with PrP upon induction with IPTG (lane 3); molecular weight markers, as indicated on the left of the image( ion wits on of prs E. coll and 4 h
amta
FrgsuipM
lane 4); insoluble protein fraction obtained as pellet, after solubilizatample loaded in lane 5 (lane 6); pooled fractions obtained after elutiolution (lane 7). Panel B: Expression of the recombinant MutPrP inysates showing MutPrP expression at 0 h (just before induction), at 2
mong transformants, the library’s diversity was esti-ated to be of the order of about 1.8 × 107. Perusal of
he sequences and occurrence-frequencies of nucleotidess a function of their positions in the codon (supple-
ig. 2. Two-step construction of prion variants randomized in theegion between residues 101 and 112 in PrP(90–231). The human PrPene (Fig. 1) was used as a template for the PCR reaction in the firsttep (PCR I), with primers F2 (incorporating the degenerate sequencesed for randomization), and R1, producing gene fragments contain-ng the randomized [(NNS)12] residues. In the second step (PCR II),rimers F1 and R1 were used, followed by digestion with NheI andluI and subsequent ligation into the phage-display vector.
h 6 M GdnCl (lane 5); flow-through from the Ni-NTA column of theotein bound to Ni-NTA column, upon elution with 50 mM imidazolei SG13009 cells. Lane 1: Molecular weight markers; lanes 2–4: cellafter induction, respectively.
mental data Table 1) is instructive, in that the negligiblerepresentation of A and T residues at the third positionreveals that the synthesis of the degenerate region of theoligo occurred as desired. It may be noted that whereasequimolar occurrences of nucleotides introduced dur-ing synthesis is anticipated for the first and secondcodon positions, the deviations in frequencies observedcould reflect aspects of biological selection. Harvestingof phage from the entire culture yielded approximately4.5 × 1012 pfu/ml.
3.3. Screening of the library of prion variants
The library of prion variants displayed on the sur-face of bacteriophage was screened for ability to bindto immobilized �-PrP through four rounds of biopan-
ning and amplification to identify variants capable ofadopting, by inference, a �-sheet-based conformationin unimolecular fashion (i.e., as molecules displayedon the surfaces of single phages which interact with �-Table 1Nucleotide distribution in the variable region of infectious phage ran-domly selected from the library
Frequency of each base by position in codon (%)
Base 1 2 3G 32 34 54A 16 13 3T 31 25 4C 17 24 35

670 A. Verma et al. / The International Journal of Bioch
Table 2Titer of phage (in pfu/ml) obtained after each of the four rounds ofbiopanning and amplification
Number ofbiopanning cycles
Titer of unamplifiedeluate (pfu/ml)
Titer of amplifiedeluate (pfu/ml)
1st cycle 1.5 × 106 1 × 1013
2nd cycle 3.3 × 1010 4.1 × 1012
with subtilisin, a non-specific protease known to cutany exposed peptide bond (using a 1:300 molar ratioof the protease to the soluble purified wt-PrP). Interest-ingly, subtilisin cleavage yielded a 14 kDa species along
Fig. 3. The entire sequence of the recombinant PrP(90–231) produced
3rd cycle 1.9 × 1010 2.3 × 1013
4th cycle 1.3 × 1010
PrP). Table 2 lists the titer of phage eluted and amplifiedafter each round of biopanning. The input phage in eachround was 2 × 1011. In the control well, in the absenceof �-PrP, with the same amount of input phage used,biopanning resulted in no elution of phages, confirmingthat there was no non-specific binding of phage particleswith the His-Sorb plates [pre-blocked to prevent bind-ing of any substance lacking Ni-NTA-binding function].Note, however, that while the recombinant prion wasproduced in fusion with the His-tag to facilitate purifi-cation, conversion to �-PrP, and immobilization on theHis-Sorb plates, the prion displayed was not producedin fusion with a His-tag; this was to ensure that phage donot bind to the His-Sorb plates at any stage of the screen-ing, even by chance, as an additional precaution overand above the blocking done prior to screening. As seenfrom Table 1, phage eluted in the first round was ampli-fied roughly 107-fold and because the number of inputphages in each cycle was the same, in the second round ofbiopanning there was effectively a much stronger compe-tition among �-PrP binding-competent phages, each ofwhich was now present in sufficient numbers to saturate,if possible, all binding sites individually. This com-petition was made progressively more intense throughsuccessive rounds of biopanning and amplification, eachround amplifying the titers of the best binding variants.After the completion of the fourth round of biopanningseveral of the plaques were individually picked up forsequencing.
3.4. Details of β-PrP-binding variants identifiedselected through screening
The translated sequences of the residues in the regionbetween 101 and 112 found to be encoded by 19plaques picked randomly, and their frequency of occur-rence, were as follows: (i) KGVLTWFSPLWQ (12 out
of 19 sequences); (ii) VRLLDLTSGVSM (2 out of19 sequences); (iii) KPSKPKTNMKHM (5 out of 19sequences). The last sequence which occurred 5 outof 19 times was identical to wt-PrP, indicating thatemistry & Cell Biology 40 (2008) 663–676
PrPc (in the form of wt-PrP) can also bind to �-PrP,although clearly much more poorly because no sig-nificant change occurs in the fraction population ofwt-PrP recovered through four rounds of biopanningand amplification; in contrast, the other two sequenceswould appear to have climbed up through a nearly5 million-fold representational handicap with respectto wt-PrP, to ultimately rival, or exceed, the numbersof wt-PrP present after the fourth round. We decidedto produce and purify the first variant constituting 12out of 19 sequences, or 64% of the sequenced popu-lation, which contained residues ‘KGVLTWFSPLWQ’between positions 101 and 112 instead of the residues‘KPSKPKTNMKHM’. This variant is given the nameMutPrP.
3.5. Characterization of wt-PrP and MutPrPproduced for biophysical studies
Vector-NTI (Invitrogen) based bioinformatic analy-ses of wt-PrP and MutPrP (sequences shown in Fig. 3)predict their molecular masses to be 17.7 and 17.8 kDarespectively. The predicted isoelectric points (pIs) of thetwo proteins, however, differ significantly; the pI of wt-PrP is predicted to be 8.0, while that of MutPrP is 9.06.The aromatic residue contents and absorption coeffi-cients of the two proteins too are predicted to be quitedifferent. An absorbance of 1.0 at 280 nm correspondsto a protein concentration of 0.89 mg/ml for wt-PrP and0.57 mg/ml for MutPrP. The mass of the wt-PrP producedin E. coli (seen in Fig. 1A) was determined to be 17.7 kDaby MALDI-TOF mass spectral analysis (Fig. S4A). Weinvestigated the proteolytic susceptibility of this recom-binant protein through a controlled proteolysis reaction
in E. coli. The wild-type prion (wt-PrP) 90–231 sequence is shownin green with the sequence corresponding to 101–112 of the mutantshown in red. The residues from the vector are represented in black.(For interpretation of the references to color in this figure legend, thereader is referred to the web version of the article.)
A. Verma et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676 671
Fptw
wsaa(pdPtb1
sswrt(
3i
Pp
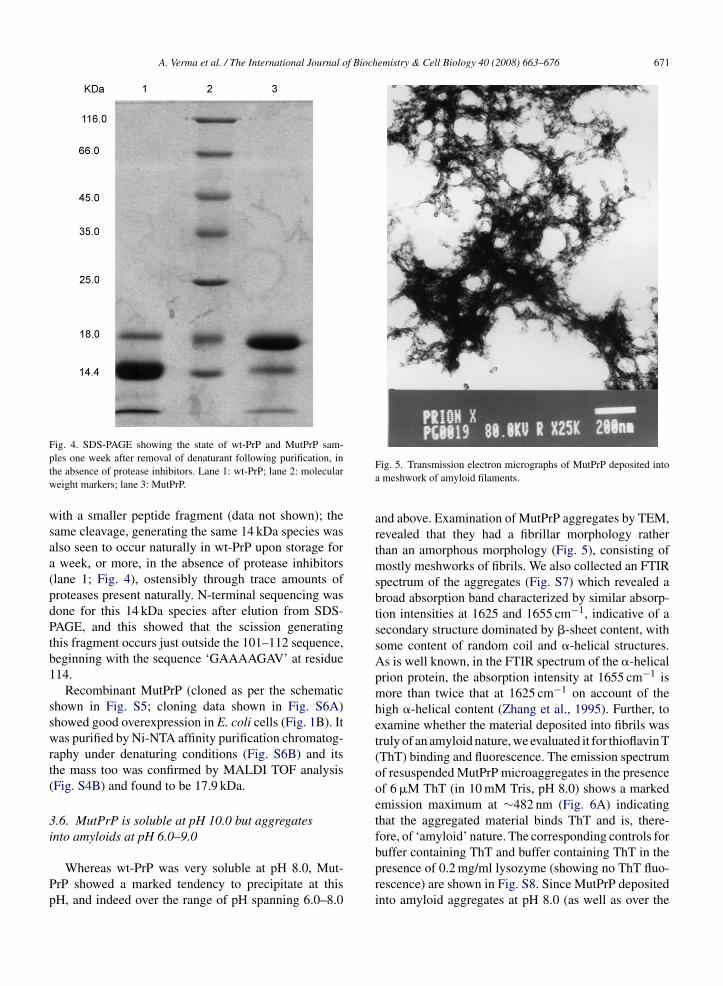
ig. 4. SDS-PAGE showing the state of wt-PrP and MutPrP sam-les one week after removal of denaturant following purification, inhe absence of protease inhibitors. Lane 1: wt-PrP; lane 2: moleculareight markers; lane 3: MutPrP.
ith a smaller peptide fragment (data not shown); theame cleavage, generating the same 14 kDa species waslso seen to occur naturally in wt-PrP upon storage forweek, or more, in the absence of protease inhibitors
lane 1; Fig. 4), ostensibly through trace amounts ofroteases present naturally. N-terminal sequencing wasone for this 14 kDa species after elution from SDS-AGE, and this showed that the scission generatinghis fragment occurs just outside the 101–112 sequence,eginning with the sequence ‘GAAAAGAV’ at residue14.
Recombinant MutPrP (cloned as per the schematichown in Fig. S5; cloning data shown in Fig. S6A)howed good overexpression in E. coli cells (Fig. 1B). Itas purified by Ni-NTA affinity purification chromatog-
aphy under denaturing conditions (Fig. S6B) and itshe mass too was confirmed by MALDI TOF analysisFig. S4B) and found to be 17.9 kDa.
.6. MutPrP is soluble at pH 10.0 but aggregatesnto amyloids at pH 6.0–9.0
Whereas wt-PrP was very soluble at pH 8.0, Mut-rP showed a marked tendency to precipitate at thisH, and indeed over the range of pH spanning 6.0–8.0
Fig. 5. Transmission electron micrographs of MutPrP deposited intoa meshwork of amyloid filaments.
and above. Examination of MutPrP aggregates by TEM,revealed that they had a fibrillar morphology ratherthan an amorphous morphology (Fig. 5), consisting ofmostly meshworks of fibrils. We also collected an FTIRspectrum of the aggregates (Fig. S7) which revealed abroad absorption band characterized by similar absorp-tion intensities at 1625 and 1655 cm−1, indicative of asecondary structure dominated by �-sheet content, withsome content of random coil and �-helical structures.As is well known, in the FTIR spectrum of the �-helicalprion protein, the absorption intensity at 1655 cm−1 ismore than twice that at 1625 cm−1 on account of thehigh �-helical content (Zhang et al., 1995). Further, toexamine whether the material deposited into fibrils wastruly of an amyloid nature, we evaluated it for thioflavin T(ThT) binding and fluorescence. The emission spectrumof resuspended MutPrP microaggregates in the presenceof 6 �M ThT (in 10 mM Tris, pH 8.0) shows a markedemission maximum at ∼482 nm (Fig. 6A) indicatingthat the aggregated material binds ThT and is, there-fore, of ‘amyloid’ nature. The corresponding controls for
buffer containing ThT and buffer containing ThT in thepresence of 0.2 mg/ml lysozyme (showing no ThT fluo-rescence) are shown in Fig. S8. Since MutPrP depositedinto amyloid aggregates at pH 8.0 (as well as over the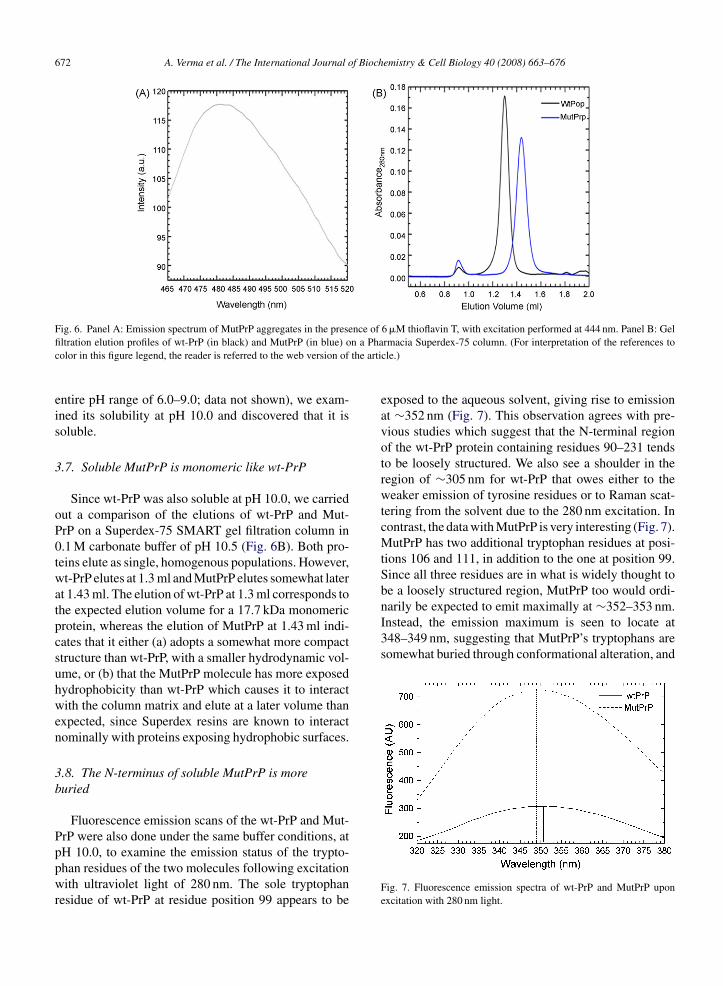
672 A. Verma et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676
nce ofn a Phathe arti
narily be expected to emit maximally at ∼352–353 nm.Instead, the emission maximum is seen to locate at348–349 nm, suggesting that MutPrP’s tryptophans aresomewhat buried through conformational alteration, and
Fig. 6. Panel A: Emission spectrum of MutPrP aggregates in the presefiltration elution profiles of wt-PrP (in black) and MutPrP (in blue) ocolor in this figure legend, the reader is referred to the web version of
entire pH range of 6.0–9.0; data not shown), we exam-ined its solubility at pH 10.0 and discovered that it issoluble.
3.7. Soluble MutPrP is monomeric like wt-PrP
Since wt-PrP was also soluble at pH 10.0, we carriedout a comparison of the elutions of wt-PrP and Mut-PrP on a Superdex-75 SMART gel filtration column in0.1 M carbonate buffer of pH 10.5 (Fig. 6B). Both pro-teins elute as single, homogenous populations. However,wt-PrP elutes at 1.3 ml and MutPrP elutes somewhat laterat 1.43 ml. The elution of wt-PrP at 1.3 ml corresponds tothe expected elution volume for a 17.7 kDa monomericprotein, whereas the elution of MutPrP at 1.43 ml indi-cates that it either (a) adopts a somewhat more compactstructure than wt-PrP, with a smaller hydrodynamic vol-ume, or (b) that the MutPrP molecule has more exposedhydrophobicity than wt-PrP which causes it to interactwith the column matrix and elute at a later volume thanexpected, since Superdex resins are known to interactnominally with proteins exposing hydrophobic surfaces.
3.8. The N-terminus of soluble MutPrP is moreburied
Fluorescence emission scans of the wt-PrP and Mut-PrP were also done under the same buffer conditions, at
pH 10.0, to examine the emission status of the trypto-phan residues of the two molecules following excitationwith ultraviolet light of 280 nm. The sole tryptophanresidue of wt-PrP at residue position 99 appears to be6 �M thioflavin T, with excitation performed at 444 nm. Panel B: Gelrmacia Superdex-75 column. (For interpretation of the references tocle.)
exposed to the aqueous solvent, giving rise to emissionat ∼352 nm (Fig. 7). This observation agrees with pre-vious studies which suggest that the N-terminal regionof the wt-PrP protein containing residues 90–231 tendsto be loosely structured. We also see a shoulder in theregion of ∼305 nm for wt-PrP that owes either to theweaker emission of tyrosine residues or to Raman scat-tering from the solvent due to the 280 nm excitation. Incontrast, the data with MutPrP is very interesting (Fig. 7).MutPrP has two additional tryptophan residues at posi-tions 106 and 111, in addition to the one at position 99.Since all three residues are in what is widely thought tobe a loosely structured region, MutPrP too would ordi-
Fig. 7. Fluorescence emission spectra of wt-PrP and MutPrP uponexcitation with 280 nm light.
A. Verma et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676 673
Fa
tu
3w
wttihaasscRsPaewuu
taoakssM
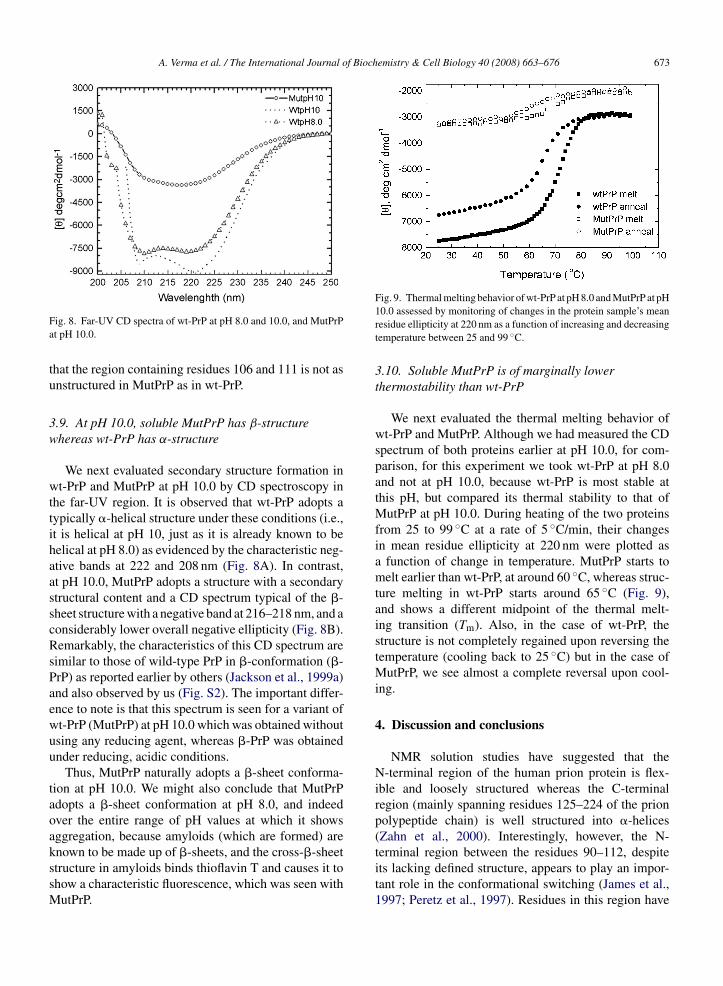
Fig. 9. Thermal melting behavior of wt-PrP at pH 8.0 and MutPrP at pH
ig. 8. Far-UV CD spectra of wt-PrP at pH 8.0 and 10.0, and MutPrPt pH 10.0.
hat the region containing residues 106 and 111 is not asnstructured in MutPrP as in wt-PrP.
.9. At pH 10.0, soluble MutPrP has β-structurehereas wt-PrP has α-structure
We next evaluated secondary structure formation int-PrP and MutPrP at pH 10.0 by CD spectroscopy in
he far-UV region. It is observed that wt-PrP adopts aypically �-helical structure under these conditions (i.e.,t is helical at pH 10, just as it is already known to beelical at pH 8.0) as evidenced by the characteristic neg-tive bands at 222 and 208 nm (Fig. 8A). In contrast,t pH 10.0, MutPrP adopts a structure with a secondarytructural content and a CD spectrum typical of the �-heet structure with a negative band at 216–218 nm, and aonsiderably lower overall negative ellipticity (Fig. 8B).emarkably, the characteristics of this CD spectrum are
imilar to those of wild-type PrP in �-conformation (�-rP) as reported earlier by others (Jackson et al., 1999a)nd also observed by us (Fig. S2). The important differ-nce to note is that this spectrum is seen for a variant oft-PrP (MutPrP) at pH 10.0 which was obtained withoutsing any reducing agent, whereas �-PrP was obtainednder reducing, acidic conditions.
Thus, MutPrP naturally adopts a �-sheet conforma-ion at pH 10.0. We might also conclude that MutPrPdopts a �-sheet conformation at pH 8.0, and indeedver the entire range of pH values at which it showsggregation, because amyloids (which are formed) are
nown to be made up of �-sheets, and the cross-�-sheettructure in amyloids binds thioflavin T and causes it tohow a characteristic fluorescence, which was seen withutPrP.
10.0 assessed by monitoring of changes in the protein sample’s meanresidue ellipticity at 220 nm as a function of increasing and decreasingtemperature between 25 and 99 ◦C.
3.10. Soluble MutPrP is of marginally lowerthermostability than wt-PrP
We next evaluated the thermal melting behavior ofwt-PrP and MutPrP. Although we had measured the CDspectrum of both proteins earlier at pH 10.0, for com-parison, for this experiment we took wt-PrP at pH 8.0and not at pH 10.0, because wt-PrP is most stable atthis pH, but compared its thermal stability to that ofMutPrP at pH 10.0. During heating of the two proteinsfrom 25 to 99 ◦C at a rate of 5 ◦C/min, their changesin mean residue ellipticity at 220 nm were plotted asa function of change in temperature. MutPrP starts tomelt earlier than wt-PrP, at around 60 ◦C, whereas struc-ture melting in wt-PrP starts around 65 ◦C (Fig. 9),and shows a different midpoint of the thermal melt-ing transition (Tm). Also, in the case of wt-PrP, thestructure is not completely regained upon reversing thetemperature (cooling back to 25 ◦C) but in the case ofMutPrP, we see almost a complete reversal upon cool-ing.
4. Discussion and conclusions
NMR solution studies have suggested that theN-terminal region of the human prion protein is flex-ible and loosely structured whereas the C-terminalregion (mainly spanning residues 125–224 of the prionpolypeptide chain) is well structured into �-helices(Zahn et al., 2000). Interestingly, however, the N-
terminal region between the residues 90–112, despiteits lacking defined structure, appears to play an impor-tant role in the conformational switching (James et al.,1997; Peretz et al., 1997). Residues in this region have
f Bioch
674 A. Verma et al. / The International Journal obeen shown to participate in key structurally stabilizinghydrophobic interactions with the C-terminal sectionsof the protein (Barron et al., 2001; Levy et al., 2001;Supattapone et al., 2001). Also, as already mentioned, innumerous instances it has been observed that mutationsleading to disease locate in this N-terminal region andit may be that semi-structured hinges, or loops, bridgingsubstructures within the prion could be destabilized bysuch mutations, making the prion more prone to spon-taneous misfolding to the PrPSc conformation (Goldfarbet al., 1992; Huang et al., 1994; Leclerc et al., 2001; Rieket al., 1996). It has been suggested that a structural con-version from the �-helical structure to the �-sheet richform might involve a monomeric species rich in �-sheetstructure. One group (Jackson et al., 1999b) has shownthat in acidic reducing conditions, it is possible to causePrPc to switch from its �-helical conformation to a com-pact, highly soluble, monomeric form rich in �-structure(�-PrP).
Given the above background, we decided to exam-ine the importance of the N-terminal region of theprion protein (indicated by the location of natu-rally occurring mutations in the region and also bystructural–biochemical studies), through the route ofphage-display library creation and screening of sequencevariants covering the region 101–112 for binding to �-PrP. To increase competition for �-PrP binding betweenvariants and to check whether wt-PrP prion also binds to�-PrP, the phage library was created in such a manner asto allow wt-PrP to be represented at a very much higherlevel (six orders of magnitude higher) than that at whichany of the other individual sequence variants was repre-sented. While this difference existed in the input phagelibrary used for the first biopanning step, it remainedconceivable for phages possessing the ability to switchconformation and bind to �-PrP to overcome this lowerrepresentation if they possessed a tendency to bind to�-PrP that was several orders of magnitude higher thanthat of wt-PrP.
Our study enabled us to confirm that wt prion in the �-PrP conformation is capable of switching conformationto �-PrP and binding to the �-PrP presented on His-Sorbplates through simple bi-molecular interactions, at somebasal level. What was especially satisfying was that thiswas achieved without the attendant complications usu-ally accompanying unregulated aggregation and withoutour having to reckon for, and control, sizes of aggre-gates (note: this would have become necessary if PrPSc,
instead of �-PrP, had been used for presentation, sinceit would be difficult to maintain uniformity of coatingthrough successive rounds of biopanning with amyloidfibrillar aggregates).emistry & Cell Biology 40 (2008) 663–676
Our use of such a library containing 25% wt-PrP-bearing phages helped us to identify variants capableof binding to �-PrP in the face of strong competi-tion from wt prion. Both kinetic and thermodynamicconsiderations could be of importance, in this regardfor a sequence variant could compete with wt prioneither by (a) displaying a faster kinetics of switchingto a �-PrP binding-competent conformation (becauseof its possessing a different sequence), or (b) throughhaving already adopted a �-PrP binding-competent con-formation, due to the sequence alterations, and throughpossession of a greater affinity for �-PrP than conforma-tionally switched wt-PrP.
Our results with the recombinant MutPrP indicatethe latter of the above scenarios to be likely to be thecase. Most residues in the prion protein have a prefer-ence for �-conformation though the protein itself hasa predominantly �-helical fold (Jackson et al., 1999b).Both variant prions obtained in this study show residuereplacements with increased propensity for adoption ofa �-sheet conformation (Table 3). The nature of residuereplacements suggests that they could very well be muchmore prionogenic than wt prion, or at the very leastmore capable of binding to �-PrP through bimolecu-lar reactions. The residue replacements also alter thehydrophobicity of the 101–112 region, and it is relevantto mention here that it has been suggested (Leffers et al.,2004) that the interactions of the N-terminal region withthe rest of the prion polypeptide are largely hydropho-bic.
Our study shows that the �-sheet-dominated confor-mation of wt PrP which can be created through refoldingof the prion protein in acidic buffer (pH 4.0) underreducing conditions can also be naturally formed, undernon-reducing conditions and at the alkaline pH of 10.0,by MutPrP which was selected through biopanning ofa phage-displayed library of N-terminally randomizedvariants. The much greater propensity of this vari-ant to form a �-sheet-rich structure, and to depositinto amyloid aggregates at near-neutral pH, appear tobe completely in keeping with the fact that it wasselected and amplified through four rounds of biopan-ning and amplification during screening for binding to�-PrP.
There is a �-sheet composed of two short strandsin the structural core of the C-terminal domain of theprion protein. In the sequence, one of these strandsis near the junction of the N- and C-terminal regions.
The NMR structure of the human prion protein (Zahnet al., 2000) shows that both �-strands of the core �-sheet have interactions with the helices comprising thebulk of the C-terminal domain. It is possible that the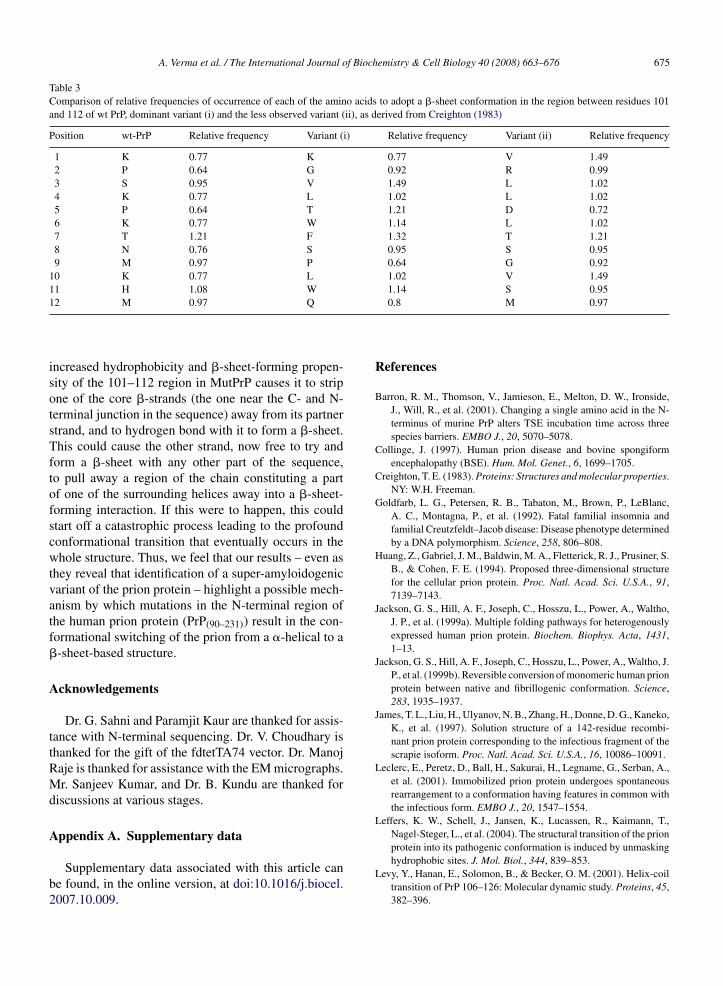
A. Verma et al. / The International Journal of Biochemistry & Cell Biology 40 (2008) 663–676 675
Table 3Comparison of relative frequencies of occurrence of each of the amino acids to adopt a �-sheet conformation in the region between residues 101and 112 of wt PrP, dominant variant (i) and the less observed variant (ii), as derived from Creighton (1983)
Position wt-PrP Relative frequency Variant (i) Relative frequency Variant (ii) Relative frequency
1 K 0.77 K 0.77 V 1.492 P 0.64 G 0.92 R 0.993 S 0.95 V 1.49 L 1.024 K 0.77 L 1.02 L 1.025 P 0.64 T 1.21 D 0.726 K 0.77 W 1.14 L 1.027 T 1.21 F 1.32 T 1.218 N 0.76 S 0.95 S 0.959 M 0.97 P 0.64 G 0.92
10 K 0.77 L 1.02 V 1.4911 H 1.08 W 1.14 S 0.951
isotsTftofscwtvatf�
A
ttRMd
A
b2
2 M 0.97 Q
ncreased hydrophobicity and �-sheet-forming propen-ity of the 101–112 region in MutPrP causes it to stripne of the core �-strands (the one near the C- and N-erminal junction in the sequence) away from its partnertrand, and to hydrogen bond with it to form a �-sheet.his could cause the other strand, now free to try and
orm a �-sheet with any other part of the sequence,o pull away a region of the chain constituting a partf one of the surrounding helices away into a �-sheet-orming interaction. If this were to happen, this couldtart off a catastrophic process leading to the profoundonformational transition that eventually occurs in thehole structure. Thus, we feel that our results – even as
hey reveal that identification of a super-amyloidogenicariant of the prion protein – highlight a possible mech-nism by which mutations in the N-terminal region ofhe human prion protein (PrP(90–231)) result in the con-ormational switching of the prion from a �-helical to a-sheet-based structure.
cknowledgements
Dr. G. Sahni and Paramjit Kaur are thanked for assis-ance with N-terminal sequencing. Dr. V. Choudhary ishanked for the gift of the fdtetTA74 vector. Dr. Manojaje is thanked for assistance with the EM micrographs.r. Sanjeev Kumar, and Dr. B. Kundu are thanked for
iscussions at various stages.
ppendix A. Supplementary data
Supplementary data associated with this article cane found, in the online version, at doi:10.1016/j.biocel.007.10.009.
0.8 M 0.97
References
Barron, R. M., Thomson, V., Jamieson, E., Melton, D. W., Ironside,J., Will, R., et al. (2001). Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across threespecies barriers. EMBO J., 20, 5070–5078.
Collinge, J. (1997). Human prion disease and bovine spongiformencephalopathy (BSE). Hum. Mol. Genet., 6, 1699–1705.
Creighton, T. E. (1983). Proteins: Structures and molecular properties.NY: W.H. Freeman.
Goldfarb, L. G., Petersen, R. B., Tabaton, M., Brown, P., LeBlanc,A. C., Montagna, P., et al. (1992). Fatal familial insomnia andfamilial Creutzfeldt–Jacob disease: Disease phenotype determinedby a DNA polymorphism. Science, 258, 806–808.
Huang, Z., Gabriel, J. M., Baldwin, M. A., Fletterick, R. J., Prusiner, S.B., & Cohen, F. E. (1994). Proposed three-dimensional structurefor the cellular prion protein. Proc. Natl. Acad. Sci. U.S.A., 91,7139–7143.
Jackson, G. S., Hill, A. F., Joseph, C., Hosszu, L., Power, A., Waltho,J. P., et al. (1999a). Multiple folding pathways for heterogenouslyexpressed human prion protein. Biochem. Biophys. Acta, 1431,1–13.
Jackson, G. S., Hill, A. F., Joseph, C., Hosszu, L., Power, A., Waltho, J.P., et al. (1999b). Reversible conversion of monomeric human prionprotein between native and fibrillogenic conformation. Science,283, 1935–1937.
James, T. L., Liu, H., Ulyanov, N. B., Zhang, H., Donne, D. G., Kaneko,K., et al. (1997). Solution structure of a 142-residue recombi-nant prion protein corresponding to the infectious fragment of thescrapie isoform. Proc. Natl. Acad. Sci. U.S.A., 16, 10086–10091.
Leclerc, E., Peretz, D., Ball, H., Sakurai, H., Legname, G., Serban, A.,et al. (2001). Immobilized prion protein undergoes spontaneousrearrangement to a conformation having features in common withthe infectious form. EMBO J., 20, 1547–1554.
Leffers, K. W., Schell, J., Jansen, K., Lucassen, R., Kaimann, T.,Nagel-Steger, L., et al. (2004). The structural transition of the prion
protein into its pathogenic conformation is induced by unmaskinghydrophobic sites. J. Mol. Biol., 344, 839–853.Levy, Y., Hanan, E., Solomon, B., & Becker, O. M. (2001). Helix-coiltransition of PrP 106–126: Molecular dynamic study. Proteins, 45,382–396.

f Bioch
676 A. Verma et al. / The International Journal oParmley, S. F., & Smith, G. P. (1988). Antibody-selectable filamentousfd phage vectors: Affinity purification of target genes. Gene, 73,305–318.
Peretz, D., Williamson, R. A., Matsunaga, Y., Serban, H., Pinilla, C.,Bastidas, R. B., et al. (1997). A conformational transition at the Nterminus of the prion protein features in formation of the scrapieisoform. J. Mol. Biol., 273, 614–622.
Prusiner, S. B., Bolton, D. C., Groth, D. F., Bowman, K. A., Cochran,S. P., & McKinley, M. P. (1982). Further purification and charac-terization of scrapie prions. Biochemistry, 21, 6942–6950.
Prusiner, S. B., Scott, M. R., De Armond, S. J., & Cohen, F. E. (1998).Prion protein biology. Cell, 93, 337–345.
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., &
Wuthrich, K. (1996). NMR structure of the mouse prion proteindomain PrP (121–231). Nature, 382, 180–182.Safar, J. G., Geschwind, M. D., Deering, C., Didorenko, S., Sattavat,M., Sanchez, H., et al. (2005). Diagnosis of human prion disease.Proc. Natl. Acad. Sci. U.S.A., 102, 3501–3506.
emistry & Cell Biology 40 (2008) 663–676
Sampath, A., Abrol, S., & Chaudhary, V. K. (1997). Versatile vectorsfor direct cloning and ligation-independent cloning of PCR ampli-fied fragments for surface display on filamentous bacteriophagegene. Gene, 190, 5–10.
Smith, G. P., & Scott, J. K. (1993). Libraries of peptides and pro-teins displayed on filamentous phage. Methods Enzymol., 217,228–257.
Supattapone, S., Muramoto, T., Legname, G., Mehlhorn, I., Cohen,F. E., DeArmond, S. J., et al. (2001). Identification of two prionprotein regions that modify scrapie incubation time. J. Virol., 75,1408–1413.
Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C., Lopez Garcia,F., et al. (2000). NMR solution structure of the human prion protein.
Proc. Natl. Acad. Sci. U.S.A., 97, 145–150.Zhang, H., Kaneko, K., Nguyen, J. T., Livshits, T. L., Baldwin, M. A.,Cohen, F. E., et al. (1995). Conformational transitions in peptidescontaining two putative alpha-helices of the prion protein. J. Mol.Biol., 250, 514–526.




















