
IB Genetic Polymorphisms and Invasive Pneumococcal Disease
7
IB Genetic Polymorphisms and Invasive Pneumococcal Disease Stephen J. Chapman 1,2 , Chiea C. Khor 1 , Fredrik O. Vannberg 1 , Angela Frodsham 1 , Andrew Walley 1 , Nicholas A. Maskell 2 , Christopher W. H. Davies 3 , Shelley Segal 4 , Catrin E. Moore 4 , Stephen H. Gillespie 5 , Paul Denny 6 , Nicholas P. Day 7 , Derrick W. Crook 8 , Robert J. O. Davies 2 , and Adrian V. S. Hill 1 1 The Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom; 2 Oxford Centre for Respiratory Medicine, Churchill Hospital Site, Oxford Radcliffe Hospital, Oxford, United Kingdom; 3 Department of Respiratory Medicine, Royal Berkshire Hospital, Reading, United Kingdom; 4 Department of Paediatrics, John Radcliffe Hospital, Oxford, United Kingdom; 5 Centre for Medical Microbiology, Department of Infection, University College London, London, United Kingdom; 6 Medical Research Council UK Mouse Genome Centre and Mammalian Genetics Unit, Harwell, Oxon, United Kingdom; 7 Centre for Tropical Diseases, Cho Quan Hospital, Ho Chi Minh City, Vietnam; and 8 Department of Microbiology, John Radcliffe Hospital, Oxford, United Kingdom Rationale: Increasing evidence supports a key role for the transcrip- tion factor nuclear factor (NF)-B in the host response to pneumo- coccal infection. Control of NF-B activity is achieved through inter- actions with the IB family of inhibitors, encoded by the genes NFKBIA, NFKBIB, and NFKBIE. Rare NFKBIA mutations cause immuno- deficiency with severe bacterial infection, raising the possibility that common IB gene polymorphisms confer susceptibility to common bacterial disease. Objectives: To determine whether polymorphisms in NFKBIA, NFKBIB, and NFKBIE associate with susceptibility to invasive pneumococcal disease (IPD) and thoracic empyema. Methods: We studied the frequencies of 62 single-nucleotide poly- morphisms (SNPs) across NFKBIA, NFKBIB, and NFKBIE in individuals with IPD and control subjects (n 1,060). Significantly associated SNPs were then studied in a group of individuals with thoracic empyema and a second control group (n 632). Measurements and Main Results: Two SNPs in the NFKBIA promoter region were associated with protection from IPD in both the initial study group and the pneumococcal empyema subgroup. Significant protection from IPD was observed for carriage of mutant alleles at these two loci on combining the groups (SNP rs3138053: Mantel- Haenszel 2 2 2 13.030, p 0.0003; odds ratio [OR], 0.60; 95% confidence interval [CI], 0.45–0.79; rs2233406: Mantel-Haenszel 2 2 2 18.927, p 0.00001; OR, 0.55; 95% CI, 0.42–0.72). An NFKBIE SNP associated with susceptibility to IPD but not pneumo- coccal empyema. None of the NFKBIB SNPs associated with IPD susceptibility. Conclusions: NFKBIA polymorphisms associate with susceptibility to IPD. Genetic variation in an inhibitor of NF-B therefore not only causes a very rare immunodeficiency state but may also influence the development of common infectious disease. Keywords: genetic polymorphisms; pneumococcal infection; nuclear factor-B Infection with Streptococcus pneumoniae remains a significant global problem, accounting for the deaths of more than 1 million children younger than 5 years worldwide (1). Invasive pneumo- coccal disease (IPD) is defined by the isolation of S. pneumoniae (Received in original form February 1, 2007; accepted in final form April 26, 2007 ) Supported by the Wellcome Trust, UK. Correspondence and requests for reprints should be addressed to Stephen J. Chapman, M.R.C.P., The Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford, OX3 7BN, UK. E-mail: [email protected] This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org Am J Respir Crit Care Med Vol 176. pp 181–187, 2007 Originally Published in Press as DOI: 10.1164/rccm.200702-169OC on April 26, 2007 Internet address: www.atsjournals.org AT A GLANCE COMMENTARY Scientific Knowledge on the Subject The nuclear factor (NF)-B inflammatory pathway plays a key role in the host immune response to pneumococcal infection, and very rare genetic mutations in an NF-B inhibitor cause immunodeficiency with severe bacterial infection. What This Study Adds to the Field This study describes novel associations between common genetic polymorphisms in NF-B inhibitors and susceptibil- ity to invasive pneumococcal disease in humans. from a normally sterile site, such as blood (septicemia), cerebro- spinal fluid (meningitis), or pleural fluid (thoracic empyema). The incidence rate of IPD ranges from 10 to 100 per 100,000 persons per year, and despite advances in medical treatments, the mortality rate of IPD in adults remains at least 10 to 20% (1). Colonization of the nasopharynx by the pneumococcus is widespread, yet only a minority of individuals develop invasive disease (2). An important, and often neglected, factor that influ- ences the development of infectious diseases such as IPD is the host’s genetic profile (3). The ubiquitous transcription factor nuclear factor (NF)-B is central to a diverse array of cellular processes, including host innate and adaptive immune responses (4, 5). Activation of NF-B occurs after stimulation of a variety of immune receptors, including Toll-like receptors (TLRs) and members of the in- terleukin (IL)-1 and tumor necrosis factor receptor superfami- lies. The signaling pathway downstream of the TLR and IL-1 family of receptors is complex and incompletely understood; key mediators include the cytoplasmic adaptor molecules MyD88 and Mal/TIRAP, which activate TRAF6 via IL-1 receptor- associated kinases (IRAK1 and IRAK4) (6, 7). In unstimulated cells, NF-B transcription factors are prevented from binding DNA due to their association with the inhibitors of NF-B (IB) protein family; phosphorylation of the IB inhibitors by the IB kinase complex leads ultimately to their degradation and the release of NF-B, which is then capable of inducing gene transcription (5, 8). The best-studied members of the IB family are IB-,IB-, and IB-, encoded by the genes NFKBIA (Chr14q13.2), NFKBIB (Chr19q13.2), and NFKBIE (Chr6p21.1), respectively (5). The re- cent identification of functional homologs of NF-B and IB in
Transcript of IB Genetic Polymorphisms and Invasive Pneumococcal Disease

I�B Genetic Polymorphisms and InvasivePneumococcal DiseaseStephen J. Chapman1,2, Chiea C. Khor1, Fredrik O. Vannberg1, Angela Frodsham1, Andrew Walley1,Nicholas A. Maskell2, Christopher W. H. Davies3, Shelley Segal4, Catrin E. Moore4, Stephen H. Gillespie5,Paul Denny6, Nicholas P. Day7, Derrick W. Crook8, Robert J. O. Davies2, and Adrian V. S. Hill1
1The Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom; 2Oxford Centre for Respiratory Medicine,Churchill Hospital Site, Oxford Radcliffe Hospital, Oxford, United Kingdom; 3Department of Respiratory Medicine, Royal Berkshire Hospital,Reading, United Kingdom; 4Department of Paediatrics, John Radcliffe Hospital, Oxford, United Kingdom; 5Centre for Medical Microbiology,Department of Infection, University College London, London, United Kingdom; 6Medical Research Council UK Mouse Genome Centreand Mammalian Genetics Unit, Harwell, Oxon, United Kingdom; 7Centre for Tropical Diseases, Cho Quan Hospital, Ho Chi Minh City,Vietnam; and 8Department of Microbiology, John Radcliffe Hospital, Oxford, United Kingdom
Rationale: Increasing evidence supports a key role for the transcrip-tion factor nuclear factor (NF)-�B in the host response to pneumo-coccal infection. Control of NF-�B activity is achieved through inter-actions with the I�B family of inhibitors, encoded by the genesNFKBIA, NFKBIB, and NFKBIE. Rare NFKBIA mutations cause immuno-deficiency with severe bacterial infection, raising the possibility thatcommon I�B gene polymorphisms confer susceptibility to commonbacterial disease.Objectives: To determine whether polymorphisms in NFKBIA, NFKBIB,and NFKBIE associate with susceptibility to invasive pneumococcaldisease (IPD) and thoracic empyema.Methods: We studied the frequencies of 62 single-nucleotide poly-morphisms (SNPs) across NFKBIA, NFKBIB, and NFKBIE in individualswith IPD and control subjects (n � 1,060). Significantly associatedSNPs were then studied in a group of individuals with thoracicempyema and a second control group (n � 632).Measurements and Main Results: Two SNPs in the NFKBIA promoterregion were associated with protection from IPD in both the initialstudy group and the pneumococcal empyema subgroup. Significantprotection from IPD was observed for carriage of mutant alleles atthese two loci on combining the groups (SNP rs3138053: Mantel-Haenszel 2 � 2 �2 � 13.030, p � 0.0003; odds ratio [OR], 0.60; 95%confidence interval [CI], 0.45–0.79; rs2233406: Mantel-Haenszel2 � 2 �2 � 18.927, p � 0.00001; OR, 0.55; 95% CI, 0.42–0.72). AnNFKBIE SNP associated with susceptibility to IPD but not pneumo-coccal empyema. None of the NFKBIB SNPs associated with IPDsusceptibility.Conclusions: NFKBIA polymorphisms associate with susceptibility toIPD. Genetic variation in an inhibitor of NF-�B therefore not onlycauses a very rare immunodeficiency state but may also influencethe development of common infectious disease.
Keywords: genetic polymorphisms; pneumococcal infection; nuclearfactor-�B
Infection with Streptococcus pneumoniae remains a significantglobal problem, accounting for the deaths of more than 1 millionchildren younger than 5 years worldwide (1). Invasive pneumo-coccal disease (IPD) is defined by the isolation of S. pneumoniae
(Received in original form February 1, 2007; accepted in final form April 26, 2007 )
Supported by the Wellcome Trust, UK.
Correspondence and requests for reprints should be addressed to Stephen J.Chapman, M.R.C.P., The Wellcome Trust Centre for Human Genetics, RooseveltDrive, Oxford, OX3 7BN, UK. E-mail: [email protected]
This article has an online supplement, which is accessible from this issue’s tableof contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 176. pp 181–187, 2007Originally Published in Press as DOI: 10.1164/rccm.200702-169OC on April 26, 2007Internet address: www.atsjournals.org
AT A GLANCE COMMENTARY
Scientific Knowledge on the Subject
The nuclear factor (NF)-�B inflammatory pathway plays akey role in the host immune response to pneumococcalinfection, and very rare genetic mutations in an NF-�Binhibitor cause immunodeficiency with severe bacterialinfection.
What This Study Adds to the Field
This study describes novel associations between commongenetic polymorphisms in NF-�B inhibitors and susceptibil-ity to invasive pneumococcal disease in humans.
from a normally sterile site, such as blood (septicemia), cerebro-spinal fluid (meningitis), or pleural fluid (thoracic empyema).The incidence rate of IPD ranges from 10 to 100 per 100,000persons per year, and despite advances in medical treatments,the mortality rate of IPD in adults remains at least 10 to 20%(1). Colonization of the nasopharynx by the pneumococcus iswidespread, yet only a minority of individuals develop invasivedisease (2). An important, and often neglected, factor that influ-ences the development of infectious diseases such as IPD is thehost’s genetic profile (3).
The ubiquitous transcription factor nuclear factor (NF)-�Bis central to a diverse array of cellular processes, including hostinnate and adaptive immune responses (4, 5). Activation ofNF-�B occurs after stimulation of a variety of immune receptors,including Toll-like receptors (TLRs) and members of the in-terleukin (IL)-1 and tumor necrosis factor receptor superfami-lies. The signaling pathway downstream of the TLR and IL-1family of receptors is complex and incompletely understood; keymediators include the cytoplasmic adaptor molecules MyD88and Mal/TIRAP, which activate TRAF6 via IL-1 receptor-associated kinases (IRAK1 and IRAK4) (6, 7). In unstimulatedcells, NF-�B transcription factors are prevented from binding DNAdue to their association with the inhibitors of NF-�B (I�B) proteinfamily; phosphorylation of the I�B inhibitors by the I�B kinasecomplex leads ultimately to their degradation and the release ofNF-�B, which is then capable of inducing gene transcription (5,8). The best-studied members of the I�B family are I�B-�, I�B-�,and I�B-�, encoded by the genes NFKBIA (Chr14q13.2), NFKBIB(Chr19q13.2), and NFKBIE (Chr6p21.1), respectively (5). The re-cent identification of functional homologs of NF-�B and I�B in

182 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 176 2007
the horseshoe crab suggests that these components originated morethan 500 million years ago and further underlines the key role ofthis pathway in host defense (9).
Although polymorphisms within genes encoding the activat-ing TLRs have been associated with a number of disease states(10), the role of genetic variation within downstream compo-nents of the NF-�B pathway in disease development remainslargely unexplored. Exceptions are the associations describedbetween variants in NFKBIA with Crohn’s disease, trachoma,and sarcoidosis (11–13), and the recent association of a functionalpolymorphism in IRAK1 with outcomes from sepsis (14).
There is increasing evidence to support a critical role for theNF-�B pathway in the host immune response to pneumococcalinfection. Many of the immune receptors capable of activatingNF-�B are stimulated during pneumococcal infection, and, inparticular, TLR2 recognizes components of gram-positive bacte-ria such as S. pneumoniae (15–17) and TLR4 recognizes thepneumococcal toxin pneumolysin (18). Activation of NF-�B bypneumococci has been clearly demonstrated both in vitro andin animal models (19–22), and targeted genetic disruption ofthe NF-�B p50 subunit in mice has been shown to increasesusceptibility to overwhelming pneumococcal infection (23). Re-cently, two patients have been described with mutations in theNFKBIA gene leading to impaired NF-�B activation and a pri-mary immunodeficiency syndrome characterized by recurrentsevere bacterial infections in association with the skin conditionanhidrotic ectodermal dysplasia (24, 25). On the basis of thesefindings, we hypothesized that common polymorphisms in the I�Bgenes may be associated with susceptibility to the phenotype ofIPD regularly encountered in clinical practice. To investigatethis further, we studied the frequencies of polymorphisms in thethree major I�B genes NFKBIA, NFKBIB, and NFKBIE ingroups of individuals with IPD and thoracic empyema, as wellas two control groups.
METHODS
Sample Information
The IPD sample collection has been previously described in detail (26).Briefly, blood samples were collected from patients with IPD (definedby the isolation of S. pneumoniae from a normally sterile site) as partof an enhanced active surveillance program in three hospitals inOxfordshire, United Kingdom (John Radcliffe, Horton General, andWycombe General Hospitals). Frequencies of clinical presentation wereas follows: pneumonia, 69%; isolated bacteremia, 15%; meningitis,11%; and other presentations, 5%. This collection was designated theIPD study group (1). Individuals with thoracic empyema (suppurativeinfection of the pleural cavity) were recruited on entry to the U.K.MIST1 (First Multicenter Intrapleural Sepsis Trial) trial, as previouslydescribed (27–29). A bacteriological diagnosis was made in 59% of theavailable samples, with the breakdown of bacterial species within thisgroup as follows: S. pneumoniae, 27%; Streptococcus intermedius-anginosus-constellatus (milleri) group, 24%; other streptococcal species,7%; Staphylococcus aureus, 8%; anaerobes, 16%; gram-negative bacte-ria, 6%; and others, 12%. Individuals with pneumococcal empyemacomprised the IPD study group (2).
Two control groups were available for study. The first control group(control 1) comprised a combination of 180 United Kingdom healthyadult blood donors and 590 cord blood samples, as previously described(26). The second control group (control 2) was collected independentlyand consisted of 370 United Kingdom healthy adult blood donors.Individuals of non-European ancestry were excluded from cases andcontrol subjects. The study was approved by the research ethics commit-tees of the participating hospitals.
Genotyping Techniques
Genotyping was performed using the Sequenom mass-array Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF)primer extension assay (30), as described in the online supplement.
Statistical Analysis
Statistical analysis of genotype associations and logistic regression wasperformed using SPSS version 12.0 (SPSS, Inc., Chicago, IL). Analysisof linkage disequilibrium (LD) and haplotypes was performed usingthe Haploview version 3.2 program (31). Haplotype blocks were definedas regions demonstrating strong evidence of historical recombinationbetween less than 5% of single-nucleotide polymorphism (SNP)–paircomparisons (32). All control genotype distributions were in Hardy-Weinberg equilibrium (0.05 level).
RESULTS
We genotyped a total of 62 SNPs in the initial IPD study group(IPD 1) and control group (control 1). Of these SNPs, 19 wereeither nonpolymorphic or extremely rare (minor allele frequency �0.01) in the population studied, leaving a total of 43 SNPs acrossthe three I�B genes for analysis (Table E2 of the online supple-ment). None of the SNPs genotyped in NFKBIB were associatedwith IPD susceptibility. Of the remaining SNPs, six appeared tobe associated with susceptibility to IPD at the 0.05 significancelevel (Table E2). Within NFKBIA, the associated polymor-phisms were clustered particularly in the promoter region of thegene, where the adjacent SNPs rs2233406 and rs3138053 weremost significantly associated with disease (Table E2). Only asingle SNP in NFKBIE (rs529948) appeared to be associatedwith IPD susceptibility (Table E2). The genotype frequenciesfor the three SNPs with strongest evidence of association withIPD susceptibility are presented in Table 1 (control 1 and IPD1 groups). In each case, conditional logistic regression confirmedthat the pattern of association best fits a dominant, protectiveeffect of the mutant allele. Logistic regression analysis demon-strated no effect of age, comorbid conditions, or sex on I�Bgenotype. Analysis of I�B haplotypes is described in the onlinesupplement.
The three most strongly associated SNPs were then examinedin the thoracic empyema and second control study groups toassess whether the associations were limited to pneumococcaldisease or more generally applicable to other forms of gram-positive invasive respiratory bacterial disease. No associationwas observed between genotype and susceptibility to thoracicempyema overall (cases [n � 262]; for NFKBIArs3138053, 2 �1.177, p � 0.56; for NFKBIArs2233406, 2 � 1.505, p � 0.471;for NFKBIErs529948, 2 � 4.015, p � 0.134). Analysis of thevery small (n � 42) subgroup of individuals with pneumococcalempyema, however, revealed associations between eachNFKBIA genotype and disease susceptibility (Table 1, control2 and IPD 2 groups). For each of the two SNPs in NFKBIA(rs3138053 and rs2233406), protection from pneumococcal em-pyema was associated with carriage of the mutant allele, thesame direction of association as that seen with IPD group 1 andcontrol 1 (Table 1). For NFKBIErs529948, on the other hand,individuals with pneumococcal empyema were overrepresentedin the mutant homozygote group, a different direction of associa-tion from that previously observed for this SNP and IPD (Table 1);indeed, the association with pneumococcal empyema was notsignificant when analyzed using the previous dominant model(p � 0.292, Table 1). In keeping with this, significant heterogene-ity of odds ratios (ORs) for NFKBIErs529948 between the twoIPD-control groups was noted (Tarone’s homogeneity of OR2 � 5.519, p � 0.019). Comparison of ORs for NFKBIArs3138053and NFKBIArs2233406 did not demonstrate any evidence ofheterogeneity between the two IPD-control groups for eitherSNP; after combining and stratifying the study groups, carriageof the mutant allele (mutant homozygotes and heterozygotescombined) was associated with protection from IPD whencompared with the wild-type homozygous state (for rs3138053:

Chapman, Khor, Vannberg, et al.: I�B Genes and Pneumococcal Disease 183
TABLE 1. NFKBIA AND NFKBIE POLYMORPHISM GENOTYPE FREQUENCIES IN INDIVIDUALS WITH INVASIVE PNEUMOCOCCALDISEASE AND CONTROL SUBJECTS
Dominant Model§Genotypic 2 Allelic 2
SNP Status AA (% )* AB (% )* BB (% )* Total (p value)† (p value)‡ OR§ (95% CI ) p Value‡
NFKBIA rs3138053 Control 1 374 (48.8) 322 (42.0) 70 (9.1) 766 8.587 (0.014) 8.204 (0.004) 0.64 (0.48–0.87) 0.003IPD 1 135 (59.7) 77 (34.1) 14 (6.2) 226
Control 2 169 (47.3) 159 (44.5) 29 (8.1) 357 (0.029)|| 4.205 (0.040) 0.42 (0.21–0.83) 0.011IPD 2 28 (68.3) 10 (24.4) 3 (7.3) 41
NFKBIA rs2233406 Control 1 375 (49.2) 315 (41.3) 72 (9.4) 762 14.985 (0.0005) 13.534 (0.0002) 0.57 (0.43–0.76) 0.0001IPD 1 164 (63.1) 79 (30.4) 17 (6.5) 260
Control 2 173 (46.8) 164 (44.3) 33 (8.9) 370 (0.092)|| 3.315 (0.068) 0.47 (0.24–0.93) 0.028IPD 2 26 (65.0) 11 (27.5) 3 (7.5) 40
NFKBIE rs529948 Control 1 534 (70.6) 211 (27.9) 11 (1.5) 756 13.043 (0.001) 6.318 (0.012) 0.59 (0.43–0.83) 0.001IPD 1 231 (80.2) 50 (17.4) 7 (2.4) 288
Control 2 232 (69.9) 96 (28.9) 4 (1.2) 332 (0.043)|| 2.626 (0.105) 1.43 (0.73–2.78) 0.292IPD 2 26 (61.9) 13 (31.0) 3 (7.1) 42
Definition of abbreviations: CI � confidence interval; IPD � invasive pneumococcal disease; OR � odds ratio; SNP � single-nucleotide polymorphism.* Number of individuals (%): AA, wild-type homozygote; AB, heterozygote; BB, mutant homozygote.† 3 � 2 2 comparison, 2 degrees of freedom.‡ 2 � 2 2 comparison, 1 degree of freedom.§ Comparison of mutant allele carriers (BBAB) with noncarriers (AA).|| Fisher-Freeman-Halton exact test.
Mantel-Haenszel 2 � 2 2 � 13.030, p � 0.0003; OR, 0.60;95% confidence interval [CI] for OR, 0.45–0.79; for rs2233406:Mantel-Haenszel 2 � 2 2 � 18.927, p � 0.00001; OR, 0.55; 95%CI, 0.42–0.72).
Outcome data were available for 127 individuals with IPD andfor all of the individuals with pneumococcal empyema; mortalityrates were 10 and 14% in the two groups, respectively. No associ-ation was observed between genotypes and outcome (data notshown), although the number of individuals in the poor outcomegroups was small and a significantly larger study would be re-quired to examine effects of genotype on mortality.
An important question is whether the associations with IPDsusceptibility are truly with polymorphisms in NFKBIA andNFKBIE, or whether these simply represent markers in LD withdisease-associated polymorphisms located in other genes. Toaddress this, we examined the pattern of LD between polymor-phisms (Figures 1–3). The associated SNP in NFKBIE (rs529948)is located within a region of strong LD in this population (Figure3); the lack of associated SNPs outside this region suggests thatthe observed association is localized to NFKBIE, rather thanwith a gene elsewhere along the chromosome. The pattern ofLD across NFKBIA and its flanking regions is more complex(Figure 1). Nevertheless, the disease-associated SNPs are con-tained within the region studied, and the flanking markers geno-typed are in only weak LD with the associated SNPs and do notthemselves demonstrate any association with IPD (Table E2),again indicating that the association is likely to be with NFKBIArather than a neighboring gene. The absence of any predictedgenes in the vicinity of NFKBIA and NFKBIE (Figures 1 and3) further suggests that the observed disease associations arewith these genes, although genotyping of a significantly largernumber of polymorphisms would be required to absolutely ex-clude long-range LD with a variant elsewhere.
Finally, we performed logistic regression analysis to investi-gate possible interactions between the most statistically signifi-cant disease-associated SNPs in NFKBIA (rs2233406) andNFKBIE (rs529948), and between these SNPs and the (nonasso-ciated) polymorphisms in NFKBIB. This analysis was performedusing the IPD 1 and control 1 study groups, because the pneumo-coccal empyema group was too small to assess for interactions.Carriage of a protective allele (either the NFKBIArs2233406T allele or the NFKBIErs529948 A allele) was associated with
an OR for IPD of approximately 0.6 (Table 2). Carriage ofprotective alleles at both loci was associated with an OR fordisease of 0.31 (Table 2). No evidence of epistasis was observedbetween NFKBIArs2233406 and NFKBIErs529948 and poly-morphisms in NFKBIB (data not shown).
DISCUSSION
In this study, we demonstrate that common polymorphisms in theI�B genes NFKBIA and NFKBIE associate with susceptibility toIPD. We consider these associations to be particularly compelingin the setting of evidence from in vitro, animal, and rare human(primary immunodeficiency) models of disease, all of which sup-port an important role for I�B proteins in susceptibility to inva-sive bacterial disease. The increased risk (wild-type homozygote)genotypes for NFKBIArs2233406 and NFKBIErs529948 arepresent in nearly 50 and 70% of the individuals studied, respec-tively (Table 1). As a result, these polymorphisms might be ex-pected to make a significant contribution to the burden of invasivepneumococcal disease in this population. The polymorphismsare located in the upstream regions of the genes and are likelyto exert subtle, regulatory functional effects in comparison tothe NFKBIA mutations responsible for primary immunodefi-ciency (24, 25). These findings raise the possibility that polymor-phisms in other genes associated with rare primary immuno-deficiency states may also influence susceptibility to commoninfectious disease.
An important cause of type I error in genetic associationstudies is the failure to correct significance levels when multipleindependent markers have been examined. Here, we analyzed43 polymorphisms in total, and applying a Bonferroni correctionresults in a threshold significance level of 0.001, rather than 0.05.Even with this corrected significance level, rs2233046 and rs529948remain associated with IPD susceptibility. The Bonferroni cor-rection assumes, however, that markers are independent, whereasmany of the SNPs studied here are in strong or complete LDand are therefore not truly independent from each other. As aresult, the Bonferroni adjustment is likely to markedly overcor-rect in this case. A correction based on the total number of LDblocks and singleton (not part of an LD block) SNPs has beenproposed; such a correction has been demonstrated to generateacceptable type I error rates when using the blocking algorithm

184 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 176 2007
Figure 1. Genomic organization, location of single-nucleotide polymorphisms (SNPs), and linkage disequilibrium (LD) map for NFKBIA. (A ) Genestructure. Exons are shown as rectangles or vertical lines and are numbered. There are no neighboring genes in the region studied. (B ) LD betweenthe polymorphisms studied. Polymorphisms are identified by their dbSNP rs numbers, and their position relative to the gene structure (A ) is markedby a vertical line. Empty squares indicate a high degree of LD (LD coefficient D� � 1) between pairs of markers. Numbers indicate the D� valueexpressed as a percentile. Red squares indicate pairs in strong LD with logarithm of odds (LOD) scores for LD � 2; pink squares, D� � 1 with LOD � 2;white squares, D� � 1.0 and LOD � 2.
described by Gabriel and colleagues across regions of moderateand high LD (33). Using this approach, 18 independent testswere performed in this study, suggesting a threshold p value of0.0027 for statistical significance. It should be noted that eventhis approach is likely to be conservative because it does nottake into account interblock LD.
Whichever approach is used, the observed p values of associa-tion for rs2233406 and rs529948 suggest that these are unlikelyto represent artifacts of multiple testing. Population stratificationis also unlikely to account for the observed association, becauseall cases and control subjects were of white United Kingdomdescent. Further evidence for the association between NFKBIAand IPD is provided by the observation of significant associationsbetween the linked SNPs rs3138053 and rs2233406 and suscepti-bility to pneumococcal empyema in an independently collectedgroup of samples (Table 1). The direction of association in thetwo groups is the same, with carriage of the mutant allele ineach case associated with protection from IPD. It should be
noted that the number of individuals in the pneumococcal empy-ema subgroup is extremely small, however, and this result shouldtherefore be interpreted with some caution. The absence of anassociation between the polymorphisms and susceptibility tothoracic empyema overall suggests that the effect may be specificto invasive pneumococcal disease, although the number of indi-viduals in each of the remaining bacterial groups is also relativelysmall. The lack of clear replication for NFKBIErs529948 withpneumococcal empyema does not necessarily indicate that theinitial association with IPD is false. This result could simplyreflect sampling variation in the context of a small replicationgroup, and attempted replication of the association with rs529948in a larger sample set is required.
Further research is needed to identify the functional effects ofthe associated polymorphisms in each gene. The most statisticallysignificant associated SNPs in NFKBIA are clustered in the up-stream region of the gene, suggesting a possible effect on pro-moter function, although neither rs2233406 nor rs3138053 appear
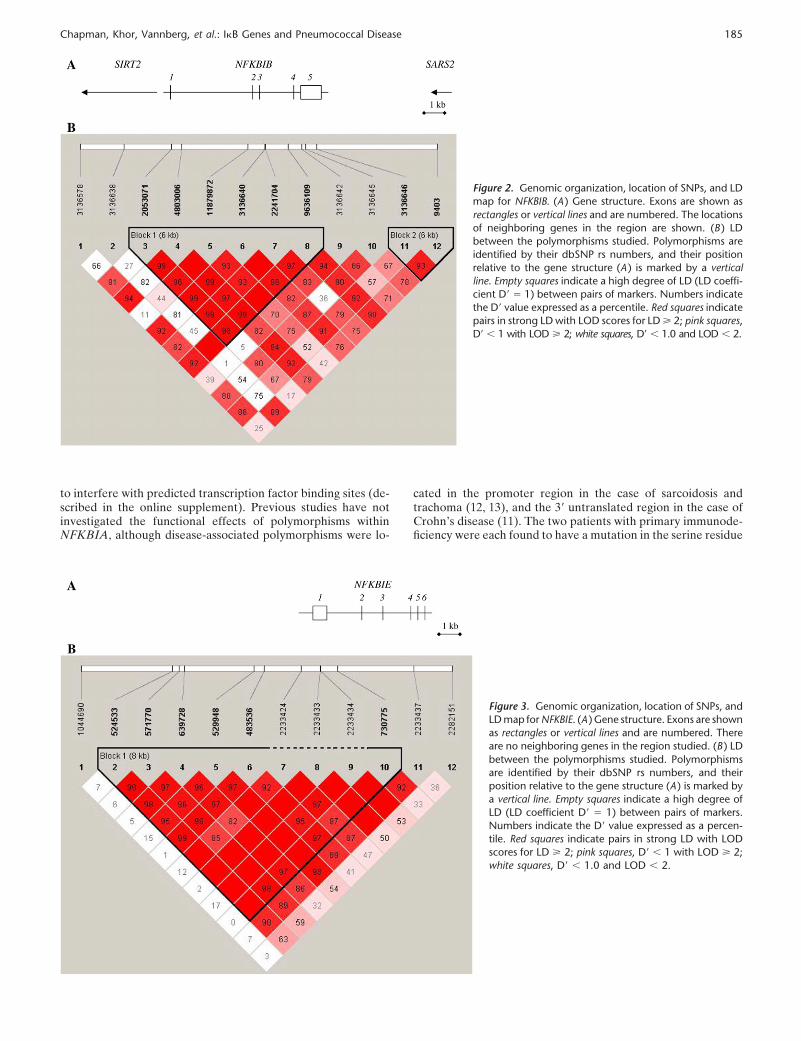
Chapman, Khor, Vannberg, et al.: I�B Genes and Pneumococcal Disease 185
Figure 2. Genomic organization, location of SNPs, and LDmap for NFKBIB. (A ) Gene structure. Exons are shown asrectangles or vertical lines and are numbered. The locationsof neighboring genes in the region are shown. (B ) LDbetween the polymorphisms studied. Polymorphisms areidentified by their dbSNP rs numbers, and their positionrelative to the gene structure (A ) is marked by a verticalline. Empty squares indicate a high degree of LD (LD coeffi-cient D� � 1) between pairs of markers. Numbers indicatethe D� value expressed as a percentile. Red squares indicatepairs in strong LD with LOD scores for LD � 2; pink squares,D� � 1 with LOD � 2; white squares, D� � 1.0 and LOD � 2.
to interfere with predicted transcription factor binding sites (de-scribed in the online supplement). Previous studies have notinvestigated the functional effects of polymorphisms withinNFKBIA, although disease-associated polymorphisms were lo-
Figure 3. Genomic organization, location of SNPs, andLD map for NFKBIE. (A ) Gene structure. Exons are shownas rectangles or vertical lines and are numbered. Thereare no neighboring genes in the region studied. (B ) LDbetween the polymorphisms studied. Polymorphismsare identified by their dbSNP rs numbers, and theirposition relative to the gene structure (A ) is marked bya vertical line. Empty squares indicate a high degree ofLD (LD coefficient D� � 1) between pairs of markers.Numbers indicate the D� value expressed as a percen-tile. Red squares indicate pairs in strong LD with LODscores for LD � 2; pink squares, D� � 1 with LOD � 2;white squares, D� � 1.0 and LOD � 2.
cated in the promoter region in the case of sarcoidosis andtrachoma (12, 13), and the 3� untranslated region in the case ofCrohn’s disease (11). The two patients with primary immunode-ficiency were each found to have a mutation in the serine residue

186 AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE VOL 176 2007
TABLE 2. ANALYSIS OF INTERACTION BETWEEN NFKBIARS2233406 AND NFKBIERS529948 INCONFERRING PROTECTION AGAINST INVASIVE PNEUMOCOCCAL DISEASE
NFKBIE Control Subjects, IPD Cases,NFKBIA rs2233406 rs529948 n (% ) n (% ) OR (95% CI )* p Value*
CC GG 238 (33.7) 120 (49.2) 1 —CTTT GG 265 (37.5) 75 (30.7) 0.56 (0.40–0.78) 0.0007CC GAAA 110 (15.6) 34 (13.9) 0.61 (0.39–0.95) 0.03CTTT GAAA 94 (13.3) 15 (6.1) 0.31 (0.17–0.56) 0.0001
Definition of abbreviations: CI � confidence interval; IPD � invasive pneumococcal disease; OR � odds ratio.* Multiple logistic regression analysis using wild-type homozygous (nonprotective) genotypes as reference.
at position 32 of I�B-�, rendering the protein resistant to phos-phorylation and degradation and enhancing its inhibition ofNF-�B (24, 25). This mutation appears to be extremely rare,however, and no common nonsynonymous polymorphisms havebeen identified on sequencing of the NFKBIA exons (11, 34).NFKBIE has not, to our knowledge, been subject to previousdisease-association studies and the functional effects of polymor-phisms in this gene are unknown. Although the location ofrs529948 may be in keeping with an enhancer regulatory effect(see the online supplement), this SNP is within a region of strongLD extending across the gene and therefore may simply be amarker linked to a functional variant within the gene itself.
No associations were observed in our study between polymor-phisms in NFKBIB and susceptibility to IPD. Furthermore, theextensive LD across NFKBIB implies that all possible haplotypiccombinations are likely to have been captured, suggesting thatit is unlikely that a common IPD-associated polymorphism withinthis gene has been missed. Why do polymorphisms in NFKBIAand NFKBIE but not NFKBIB appear to be associated withIPD susceptibility? The precise role of each of these I�B proteinsin NF-�B activation remains unclear, although their degradationand resynthesis kinetics differ (35). After cellular stimulation,both I�B-� and I�B-� are degraded and then resynthesized asa result of activation by NF-�B itself, thereby providing negativefeedback control of NF-�B activity (35–38). The synthesis ofI�B-�, in contrast, is not controlled by NF-�B and consequentlythe degradation of this inhibitor leads to more sustained NF-�Bactivation (35). The temporal control of NF-�B activity appearsin turn to determine the dynamics and pattern of gene expression(35, 38–40). Functional polymorphisms in the I�B genes mayaffect the relative levels of the different inhibitors, potentiallyresulting in different NF-�B activation profiles and differentpatterns of gene expression. Although the presence of disease-associated polymorphisms may simply reflect the historical chanceoccurrence of mutations in functional sites within NFKBIA andNFKBIE but not NFKBIB, it is intriguing that I�B-� and I�B-�are both induced by NF-�B, suggesting that interference withthis negative control pathway may have particularly importantfunctional consequences.
Given the similar functions of I�B-� and I�B-�, it would beunsurprising if polymorphisms within NFKBIA and NFKBIEinteract in determining susceptibility to IPD. The risk of IPDwas found to be reduced to just below 0.6 of expected in thepresence of a protective allele, but further reduced to less than athird of that expected in the presence of both protective alleles(Table 2). This finding suggests that the effect of NFKBIErs529948on susceptibility to IPD is not redundant in the setting of theNFKBIArs223306 effect. The interaction between these poly-morphisms and functional genetic variants in other NF-�B path-way components is worthy of further investigation. Indeed, ina study of wild flies, naturally occurring polymorphisms in cactus(the Drosophila melanogaster homolog of I�B) were not only
themselves associated with susceptibility to infection but alsoexhibited significant epistasis with other intracellular signalingloci in determining immune competence (41).
Our findings raise the interesting question of why the protec-tive alleles are not more common in the population studied,given the considerable historical selective pressure exerted byIPD. One possible explanation is that these alleles confer in-creased susceptibility to another major cause of mortality, withthe observed allele frequencies reflecting a balance betweenthese opposing selective pressures. This theory is consistent withincreasing evidence that “optimal” levels of NF-�B pathwayactivity may reflect a balance between the deleterious conse-quences of impaired or excessive pathway activation. Although,on one hand, impaired NF-�B activation is associated with immu-nodeficiency (24, 25), there is also considerable evidence for acentral role of NF-�B activation in the pathogenesis of inflam-matory disease (42, 43), and increased levels of NF-�B activationare, for example, associated with a worse outcome from sepsis(14, 44, 45). Greater understanding of the genetic control of thismajor inflammatory pathway is of particular relevance in thesetting of growing interest in the modulation of NF-�B activityas a treatment for inflammatory disease (43, 46). Further studiesinvestigating the role of common genetic polymorphisms inNF-�B pathway components in susceptibility to other infectiousand inflammatory diseases may shed light on this issue.
Conflict of Interest Statement : S.J.C. does not have a financial relationship witha commercial entity that has an interest in the subject of this manuscript. C.C.K.does not have a financial relationship with a commercial entity that has an interestin the subject of this manuscript. F.O.V. does not have a financial relationshipwith a commercial entity that has an interest in the subject of this manuscript.A.F. does not have a financial relationship with a commercial entity that has aninterest in the subject of this manuscript. A.W. does not have a financial relation-ship with a commercial entity that has an interest in the subject of this manuscript.N.A.M. does not have a financial relationship with a commercial entity that hasan interest in the subject of this manuscript. C.W.H.D. does not have a financialrelationship with a commercial entity that has an interest in the subject of thismanuscript. S.S. does not have a financial relationship with a commercial entitythat has an interest in the subject of this manuscript. C.E.M. does not have afinancial relationship with a commercial entity that has an interest in the subjectof this manuscript. S.H.G.’s research group has been in receipt of a research grantfrom Wyeth Pharmaceuticals supporting the development of a pneumococcaldiagnostic test. P.D. does not have a financial relationship with a commercialentity that has an interest in the subject of this manuscript. N.P.D. does not havea financial relationship with a commercial entity that has an interest in the subjectof this manuscript. D.W.C. received a grant from Wyeth worth £200,000 from2005–2007 for invasive pneumococcal surveillance. R.J.O.D. does not have afinancial relationship with a commercial entity that has an interest in the subjectof this manuscript. A.V.S.H. does not have a financial relationship with a commer-cial entity that has an interest in the subject of this manuscript.
Acknowledgment : S.J.C. is a Wellcome Trust Clinical Research Fellow; A.V.S.H. isa Wellcome Trust Principal Fellow. CCK is a scholar of the Agency for Science,Technology, and Research (A-STAR), Singapore, and member of the MBBS-PhDprogramme, Faculty of Medicine, National University of Singapore.
References
1. World Health Organization. Pneumococcal vaccines. Wkly EpidemiolRec 2003;14:110–119.

Chapman, Khor, Vannberg, et al.: I�B Genes and Pneumococcal Disease 187
2. Bogaert D, de Groot R, Hermans PWM. Streptococcus pneumoniaecolonisation: the key to pneumococcal disease. Lancet Infect Dis2004;4:144–154.
3. Cooke GC, Hill AVS. Genetics of susceptibility to human infectiousdisease. Nat Rev Genet 2001;2:967–977.
4. Chen F, Castranova V, Shi X, Demers LM. New insights into the roleof nuclear factor-�B, a ubiquitous transcription factor in the initiationof diseases. Clin Chem 1999;45:7–17.
5. Baldwin AS. The NF-�B and I�B proteins: new discoveries and insights.Annu Rev Immunol 1996;14:649–681.
6. Sabroe I, Read RC, Whyte MKB, Dockrell DH, Vogel SN, Dower SK.Toll-like receptors in health and disease: complex questions remain.J Immunol 2003;171:1630–1635.
7. Janssens S, Beyaert R. Functional diversity and regulation of differentinterleukin-1 receptor-associated kinase (IRAK) family members. MolCell 2003;11:293–302.
8. Tergaonkar V, Correa RG, Ikawa M, Verma IM. Distinct roles of I�Bproteins in regulating constitutive NF-�B activity. Nat Cell Biol2005;7:921–923.
9. Wang XW, Tan NS, Ho B, Ding JL. Evidence for the ancient origin ofthe NF-�B/I�B cascade: its archaic role in pathogen infection andimmunity. Proc Natl Acad Sci USA 2006;103:4204–4209.
10. Schroder NWJ, Schumann RR. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet InfectDis 2005;5:156–164.
11. Klein W, Tromm A, Folwaczny C, Hagedorn M, Duerig N, Epplen JT,Schmiegel WH, Griga T. A polymorphism of the NFKBIA gene isassociated with Crohn’s disease patients lacking a predisposing alleleof the CARD15 gene. Int J Colorectal Dis 2004;19:153–156.
12. Mozzato-Chamay N, Corbett EL, Bailey RL, Mabey DCW, Raynes J,Conway DJ. Polymorphisms in the I�B-� promoter region and riskof diseases involving inflammation and fibrosis. Genes Immun2001;2:153–155.
13. Abdallah A, Sato H, Grutters JC, Veeraraghavan S, Lympany PA, RuvenHJ, van den Bosch JM, Wells AU, du Bois RM, Welsh KI. Inhibitorkappa B-alpha (I�B-�) promoter polymorphisms in UK and Dutchsarcoidosis. Genes Immun 2003;4:450–454.
14. Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR,Abraham E. Variant IRAK-1 haplotype is associated with increasednuclear factor-�B activation and worse outcomes in sepsis. Am J RespirCrit Care Med 2006;173:1335–1341.
15. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, GolenbockD. Recognition of gram-positive bacterial cell wall components by theinnate immune system occurs via Toll-like receptor 2. J Immunol 1999;163:1–5.
16. Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, ZahringerU, Gobel UB, Weber JR, Schumann RR. Lipoteichoic acid (LTA)of Streptococcus pneumoniae and Staphylococcus aureus activatesimmune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are notinvolved. J Biol Chem 2003;278:15587–15594.
17. Knapp S, Wieland CW, van’t Veer C, Takeuchi O, Akira S, Florquin S,van der Poll T. Toll-like receptor 2 plays a role in the early inflamma-tory response to murine pneumococcal pneumonia but does not con-tribute to antibacterial defense. J Immunol 2004;172:3132–3138.
18. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, ThompsonCM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recogni-tion of pneumolysin by Toll-like receptor 4 confers resistance to pneu-mococcal infection. Proc Natl Acad Sci USA 2003;100:1966–1971.
19. Spellerberg B, Rosenow C, Sha W, Tuomanen EI. Pneumococcal cell wallactivates NF-�B in human monocytes: aspects distinct from endotoxin.Microb Pathog 1996;20:309–317.
20. Schmeck B, Zahlten J, Moog K, van Laak V, Huber S, Hocke AC, OpitzB, Hoffmann E, Kracht M, Zerrahn J, et al. Streptococcus pneumoniae-induced p38 MAPK-dependent phosphorylation of RelA at theinterleukin-8 promoter. J Biol Chem 2004;279:53241–53247.
21. Amory-Rivier CF, Mohler J, Bedos JP, Azoulay-Dupuis E, Henin D,Muffat-Joly M, Carbon C, Moine P. Nuclear factor-kappaB activationin mouse lung lavage cells in response to Streptococcus pneumoniaepulmonary infection. Crit Care Med 2000;28:3249–3256.
22. Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-�Bactivation and neutrophils recruitment require IL-1 and TNF receptorsignaling during pneumococcal pneumonia. J Immunol 2005;175:7530–7535.
23. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption ofthe p50 subunit of NF-kappa B leads to multifocal defects in immuneresponses. Cell 1995;80:321–330.
24. Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, BonnetM, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, et al. A hyper-morphic I�B� mutation is associated with autosomal dominant anhi-drotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest2003;112:1108–1115.
25. Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M,van Dongen J, van de Vosse E, van Tol M, Bredius R, Ottenhoff TH,et al. The same I�B� mutation in two related individuals leads tocompletely different clinical syndromes. J Exp Med 2004;200:559–568.
26. Roy S, Knox K, Segal S, Griffiths D, Moore CE, Welsh KI, SmarasonA, Day NP, McPheat WL, Crook DW, et al.; Oxford PneumococcalSurveillance Group. MBL genotype and risk of invasive pneumococcaldisease: a case-control study. Lancet 2002;359:1569–1573.
27. Maskell NA, Davies CW, Nunn AJ, Hedley EL, Gleeson FV, Miller R,Gabe R, Rees GL, Peto TE, Woodhead MA, et al.; First MulticenterIntrapleural Sepsis Trial. (MIST1) Group. UK controlled trial of in-trapleural streptokinase for pleural infection. N Engl J Med 2005;352:865–874.
28. Chapman SJ, Khor CC, Vannberg FO, Maskell NA, Davies CWH, Hed-ley EL, Segal S, Moore CE, Knox K, Day NP, et al. PTPN22 andinvasive bacterial disease. Nat Genet 2006;38:499–500.
29. Maskell NA, Batt S, Hedley EL, Davies CW, Gillespie SH, Davies RJO.The bacteriology of pleural infection by genetic and standard methodsand its mortality significance. Am J Respir Crit Care Med 2006;174:817–823.
30. Jurinke C, van den Boom D, Cantor CR, Koster H. The use ofMassARRAY technology for high throughput genotyping. AdvBiochem Eng Biotechnol 2002;77:57–74.
31. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualiza-tion of LD and haplotype maps. Bioinformatics 2005;21:263–265.
32. Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B,Higgins J, DeFelice M, Lochner A, Faggart M, et al. The structure ofhaplotype blocks in the human genome. Science 2002;296:2225–2229.
33. Nicodemus KK, Liu W, Chase GA, Tsai Y-Y, Fallin MD. Comparisonof type I error for multiple test corrections in large single-nucleotidepolymorphism studies using principal components versus haplotypeblocking algorithms. BMC Genet 2005;6:S78.
34. Miterski B, Bohringer S, Klein W, Sindern E, Haupts M, Schimrigk S,Epplen JT. Inhibitors in the NF-�B cascade comprise prime candidategenes predisposing to multiple sclerosis, especially in selected combi-nations. Genes Immun 2002;3:211–219.
35. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The I�B-NF-�Bsignalling module: temporal control and selective gene activation.Science 2002;298:1241–1245.
36. Whiteside ST, Epinat J-C, Rice NR, Israel A. I kappa B epsilon, a novelmember of the I�B family, controls RelA and cRel NF-�B activity.EMBO J 1997;16:1413–1426.
37. Lee S-H, Hannink M. Characterization of the nuclear import and exportfunctions of I�B�. J Biol Chem 2002;277:23358–23366.
38. Nelson DE, Ihekwaba AEC, Elliott M, Johnson JR, Gibney CA, ForemanBE, Nelson G, See V, Horton CA, Spiller DG, et al. Oscillations inNF-�B signaling control the dynamics of gene expression. Science2004;306:704–708.
39. Kearns JD, Basak S, Werner SL, Huang CS, Hoffmann A. I�B� providesnegative feedback to control NF-�B oscillations, signaling dynamics,and inflammatory gene expression. J Cell Biol 2006;173:659–664.
40. Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expres-sion programs determined by temporal control of IKK activity. Science2005;309:1857–1861.
41. Lazzaro BP, Sceurman BK, Clark AG. Genetic basis of natural variationin D. melanogaster antibacterial immunity. Science 2004;303:1873–1876.
42. Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor-�Bin pulmonary diseases. Chest 2000;117:1482–1487.
43. Tak PP, Firestein GS. NF-�B: a key role in inflammatory diseases. J ClinInvest 2001;107:7–11.
44. Bohrer H, Qiu F, Zimmermann T, Zhang Y, Jllmer T, Mannel D, BottigerBW, Stern DM, Waldherr R, Saeger HD, et al. Role of NF�B in themortality of sepsis. J Clin Invest 1997;100:972–985.
45. Abraham E. Alterations in cell signaling in sepsis. Clin Infect Dis2005;41:S459–S464.
46. Karin M, Yamamoto Y, Wang QM. The IKK NF-�B system: a treasuretrove for drug development. Nat Rev Drug Discov 2003;3:17–26.




















