
Modeling of glucose regulation and insulin-signaling pathways
Upload
independentCategory
view
1download
0
Hypoxia in High Glucose Followed by Reoxygenation inNormal Glucose Reduces the Viability of CorticalAstrocytes Through Increased Permeability of Connexin43 Hemichannels
JUAN A. ORELLANA,1* DIEGO E. HERN�ANDEZ,1 PASCAL EZAN,2 VICTORIA VELARDE,1
MICHAEL V. L. BENNETT,3 CHRISTIAN GIAUME,2 AND JUAN C. S�AEZ1,4
1Departamento de Ciencias Fisiol�ogicas, Pontificia Universidad Cat�olica de Chile, Santiago, Chile2INSERM, U840, College de France, 11 Place Marcelin Berthelot, 75231 Paris, Cedex 05, France3Department of Neuroscience, Albert Einstein College of Medicine, Bronx, New York4N�ucleo de Inmunolog�ıa e Inmunoterapia, Santiago, Chile
KEY WORDSstroke; diabetes; cell cultures; connexin channels; astrocyticdeath
ABSTRACTBrain ischemia causes more extensive injury in hyperglyce-mic than normoglycemic subjects, and the increased dam-age is to astroglia as well as neurons. In the present work,we found that in cortical astrocytes from rat or mouse,reoxygenation after hypoxia in a medium mimicking inter-stitial fluid during ischemia increases hemichannel activityand decreases cell–cell communication via gap junctions asindicated by dye uptake and dye coupling, respectively.These effects were potentiated by high glucose during thehypoxia in a concentration-dependent manner (and by zeroglucose) and were not observed in connexin 432/2 astro-cytes. The responses were transient and persistent aftershort and long periods of hypoxia, respectively. The persis-tent responses were associated with a progressive reductionin cell viability that was prevented by La31 or peptidesthat block connexin 43 (Cx43) hemichannels or by inhibi-tion of p38 MAP kinase prior to hypoxia–reoxygenation butnot by treatments that block pannexin hemichannels. Blockof Cx43 hemichannels did not affect the reduction in gapjunction mediated dye coupling observed during reoxygena-tion. Cx43 hemichannels may be a novel therapeutic targetto reduce cell death following stroke, particularly in hyper-glycemic conditions. VVC 2009 Wiley-Liss, Inc.
INTRODUCTION
Stroke is a major cause of death in industrializedcountries and results from a transient or permanentreduction in cerebral blood flow produced, in most cases,by arterial occlusion or local thrombosis, i.e., focal ische-mia (Dirnagl et al., 1999). Severe and/or prolongedreduction in cerebral blood flow leads to deprivation ofoxygen and glucose as well as build-up of potentiallytoxic substances. Even a brief period of ischemia inducesdepletion of intracellular ATP paralleled by a progres-sive reduction of electrochemical gradients across theplasma membrane (Silver et al., 1997) followed by meta-bolic changes characteristic of necrosis and/or apoptosis(Benjelloun et al., 2003; Walton et al., 1999).
During and after transient ischemia, death is moreprominent in neurons than in astrocytes primarily dueto the neurotoxic effect of secreted glutamate (Choiet al., 1987). In addition, metabolic differences betweenneurons and astroglia are likely to contribute. The rela-tive insensitivity of astrocytes to oxygen/glucose depriva-tion (OGD) has been ascribed to their ability to switchfrom aerobic to anaerobic metabolism (Peuchen et al.,1996); nevertheless, prolonged ischemia will also impacttheir functioning and eventually kill them (Sugawaraet al., 2002). Many astrocytic functions are impaired bysublethal ischemic insults, and these functions could becritical for neuronal viability (Orellana et al., 2009).Astrocytes provide metabolic and structural support andcontrol the extracellular concentrations of glutamate,K1, and H1 (Fields and Stevens-Graham, 2002).
Many of the functions of astrocytes are facilitated byspatial buffering mediated through gap junctionsbetween astrocytes (Giaume et al., 2007). Gap junctionsare membrane specializations that provide a cytoplasmicpathway between contacting cells that is permeable tomolecules upto a size of �1.4 nm diameter (and may ormay not be charge selective). Generally, gap junctionsare aggregates or plaques that contain a few tens tothousands of cell–cell channels. Each gap junction chan-nel spans two apposed membranes and is formed by ahemichannel or connexon contributed by each mem-brane (S�aez et al., 2003). Each hemichannel is composedof six protein subunits termed connexins (Cxs), a highlyconserved family encoded by 21 genes in human and 20in mouse with orthologs in other mammals (Cruciani
Additional Supporting Information may be found in the online version of thisarticle.
Grant sponsor: CONICYT; Grant number: 24080055 (to J.A.O.); Grant sponsor:FONDECYT; Grant number: 1070591 (to J.C.S.); Grant sponsor: FONDEF; Grantnumber: D07I1086 (to J.C.S.); Grant sponsor: NIH; Grant numbers: NS37402,NS45287 (to M.V.L.B.); Grant sponsor: INSERM/CONICYT (to C.G. and J.C.S.).
*Correspondence to: Juan A. Orellana, Departamento de Ciencias Fisiol�ogicas,Pontificia Universidad Cat�olica de Chile, Alameda 340, Santiago, Chile.E-mail: [email protected]
Received 2 March 2009; Accepted 22 July 2009
DOI 10.1002/glia.20926
Published online 24 August 2009 in Wiley InterScience (www.interscience.wiley.com).
GLIA 58:329–343 (2010)
VVC 2009 Wiley-Liss, Inc.

and Mikalsen, 2005). Connexins are widely expressed inmammalian tissues (S�aez et al., 2003); astrocytesexpress primarily Cx43 with lesser amounts of Cx26 andCx30 (Orellana et al., 2009), and gap junctions betweenthem are permeable to both positive and negative ionsand small molecules, including second messengers suchas Ca21, inositol 1,4,5-trisphosphate and cyclic nucleo-tides, ATP, transmitters, e.g., glutamate, and energy-producing metabolites, e.g., glucose and lactate (Taber-nero et al., 2006).
Recently, the presence of functional connexin hemi-channels in nonjunctional membrane of astrocytes wasdemonstrated by several experimental approaches (S�aezet al., 2005). The use of exogenous expression systemspermitted the study of their electrophysiological and per-meability properties and the mechanisms that controltheir activity. Under appropriate physiological conditions,hemichannels release physiologically relevant quantitiesof signaling molecules (e.g., ATP, glutamate, NAD1, andPGE2) to the extracellular milieu (S�aez et al., 2005).In vitro ischemia-like conditions enhance hemichannelactivity in astrocytes and many other cell types (Orellanaet al., 2009). Opposite to its action on hemichannels,in vitro ischemia-like conditions reduce gap junctionalcommunication between astrocytes (Orellana et al.,2009). Thus, during the last decade, it has become clearthat connexins in astrocytes can play either a protectiveor a deleterious role in neuronal and glial survival duringor after ischemia (Orellana et al., 2009).
It has long been known that hyperglycemia duringacute brain ischemia increases the extent of tissueinjury in animals (Myers and Yamaguchi, 1977) andhumans (Kagansky et al., 2001). Previous studies ruledout increased lactate (Cronberg et al., 2004; Lin et al.,1995) and glutamate concentrations (Cronberg et al.,2003) as crucial determinants of this added injury. Inhippocampal slices, glucose itself or in combination withacidosis mediates the detrimental effects (Cronberget al., 2004), but the mechanisms remain unknown. Theprotocol of chemical ischemia that we used previouslyincreases activity of Cx43 hemichannels, which acceler-ates cell death (Contreras et al., 2003). However, themetabolic inhibitors used were irreversible and for amore physiological procedure we subjected astrocyte cul-tures to oxygen deprivation in the presence of variousconcentrations of glucose in a saline mimicking intersti-tial fluid during ischemia (Bondarenko and Chesler,2001). We then assayed hemichannel activity followingreperfusion with normoxic and normoglycemic solutions.We found that hypoxia did induce opening of hemichan-nels that occurred largely after returning to normoxicand normoglycemic conditions and that the effect wasgreater after hypoxia in hyperglycemic conditions.
MATERIALS AND METHODSReagents and Antibodies
SuperSignal kit for enhanced chemiluminescence(ECL) detection, antirabbit IgG antibody-conjugated to
horseradish peroxidase, Sulfo-NHS-SS-biotin, and im-mobilized NeutrAvidin were purchased from Pierce.Previously described Gap26, Gap27, 10panx1, and E1bpeptides (Evans and Leybaert, 2007; Pelegrin and Sur-prenant, 2006; Wang et al., 2007) were obtained fromNeoMPS, SA (Strasbourg, France). HEPES, H2O(W3500), LaCl3 (La31), ethidium bromide (EtdBr), Luci-fer yellow (LY), 1,4-dithiothreitol (DTT), SB202190, andoATP were purchased from Sigma-Aldrich (St Louis,MO). Minimum Eagle’s medium (MEM), Dulbecco’sMinimum Eagle’s medium (DMEM), HCO2
3 free/F-12medium, fetal bovine serum (FBS), normal goat serum(NGS), penicillin, streptomycin, and trypsin–EDTAwere obtained from GibcoBRL (Grand Island, NY).Hoechst 33342 and Rhodamine B dextran 10 kDa(Rdex) were obtained from Molecular Probes (Eugene,OR). Where applicable, the provider’s protocol wasfollowed.
Animals
Newborn (P1–P2) Sprague–Dawley rats were obtainedfrom the Animal Institute of the Pontificia UniversidadCat�olica de Chile, and mice were maintained in the ani-mal facility of the Institut de Biologie at the College deFrance. Mouse astrocyte cultures were made from new-born (P1–P2) animals. Normal cells were obtained fromOF1 mice (Charles River, L’Arbresle, France). Astrocytecultures lacking Cx43 were also prepared from the corti-ces of P0 littermates from Cx431/2 breedings. Eachbrain was treated separately, giving rise to cultures ofCx432/2, Cx431/2, or Cx431/1 astrocytes. Cx43 expres-sion was first characterized by immunocytochemicallabeling and confirmed afterwards by genotyping per-formed as described for Cx432/2 mice (Theis et al.,2001). Genotyping was performed from a tissue sample,using PCR analysis, as described below.
All experimental protocols were approved by the Ethi-cal committee of the Pontificia Universidad Cat�olica deChile or carried out in accordance with the EuropeanCommunity Council Directives, November 24, 1986 (86/609/EEC), and all efforts were made to minimize thenumber of animals used and their suffering.
Cell Cultures
For experiments in Chile, cortical astrocytes wereobtained from neonatal rats or mice. Briefly, meningealtissue was removed, and the neocortex was minced andincubated at 37�C for 30 min in Ca21-free PBS contain-ing trypsin (0.5%) and EDTA (5 mM). Trypsin was thenremoved, and tissue immersed in MEM medium supple-mented with 10% horse serum and containing 1 mg/mLbovine pancreas DNase I, and triturated using a Pasteurpipette. The dissociated cells were centrifuged, and thepellet was resuspended in MEM supplemented with 10%FBS, 100 U/mL penicillin, and 100 lg/mL streptomycinsulfate, and the cells were plated in plastic bottles
330 ORELLANA ET AL.
GLIA

(25 mL, Nunc Clone, Marienfeld, Germany). Confluentcells were replated on 90-mm plastic culture dishes(Nunc Clone), with 5 3 105 cells per plate for biotinyla-tion or 2.5 3 103 cells per glass coverslip (12 mm in di-ameter and 1.3–1.7 mm thick). Cells were grown at37�C in a 5% CO2/95% air atmosphere at nearly 100%relative humidity. All experiments were carried outwhen cells reached �80% confluence. Cells were alwaysfed the day before an experiment.
For experiments in Paris, astrocyte cultures were pre-pared from the cortex of Cx432/2 and Cx431/1 mice.Briefly, cells were seeded (2 3 105 cells per well) onglass coverslips placed inside 16-mm diameter 4-wellplastic plates (NunClon, Nunc) in DMEM, containingglucose (5 mM), penicillin (5 U/mL), and streptomycin (5lg/mL) (Invitrogen, Carlsbad, CA) and 10% FCS(Hyclone, Logan, UT). The medium was changed twice aweek. The mouse genotype was determined by PCRanalysis. Briefly, a mouse tissue sample was digested inbuffer (50 mM KCl; 1.5 mM MgCl2; 10 mM Tris–HCl,pH 5 8.0; IGEPAL CA-630 0.45%; Tween-20 0.45%) con-taining Proteinase K (500 lg/mL; Promega, Madison,WI) at 56�C over night. After digestion, 1 lL of the su-pernatant, containing mouse DNA, was added in 24 lLof primer solution (1:1,000 in pure water). Two sets ofprimers were used: one for the Cx43 wild-type gene, a22 mer forward oligonucleotide and a 25 mer reverse oli-gonucleotide (50-CCCCACTCTCACCTATGTCTCC-30 and50-ACTTTTGCGCCTAGCTAGCTATCCC-30, respectively);the second for the LacZ insert in the Cx43 coding region,a 22 mer forward oligonucleotide and a 22 mer reverseoligonucleotide (50-GGCATACAGACCCTTGGACTCC-30
and 50-TGCGGGCCTCTTCGCTATTACG-30, respec-tively). The PCR reaction was achieved using ‘‘PCRready to go’’ kit (Amersham Biosciences, Saclay, France)with the solution described above, following the instruc-tions of the kit. DNA was firstly annealed at 94�C andthen amplified at 55�C for 40 cycles. The PCR productswere analyzed by electrophoresis in a 2% agarose geland stained with ethidium (Etd). The specific amplifiedsequences were 550 and 850 bp long for the mutantgene and wild-type gene, respectively.
All primary cultures were highly enriched in astro-cytes as determined by the fraction reactive to antiGFAP(�99%) and very small fraction labeled by the microgliamarker isolectin B4 (<1%).
Human cervix carcinoma (HeLa) cells (ATCC, CCL-2,Rockville, MD) transfected with cDNA encoding ratCx43 with EGFP attached to the C terminus (Cx43-EGFP) were grown in DMEM medium containing 100lg/mL streptomycin, 100 U/mL penicillin, and 500 lg/mL G-418 and supplemented with 10% FBS. The me-dium was replaced at 2-day intervals. Subconfluent cellswere routinely harvested by trypsinization and platedon glass coverslips or on 60-mm plastic culture dishes(Nunc Clone). Parental and Cx43–EGFP cells were keptat 37�C in a 5% CO2/95% air atmosphere at nearly 100%relative humidity. Cx43–EGFP cells could be recognizedby their fluorescence emission at 530 nm and excitationat 489 nm.
Hypoxia–Reoxygenation Protocol
Astrocyte cultures were subjected to 3 or 6 h of hy-poxia in an artificial cerebrospinal fluid medium (previ-ously bubbled with 5% CO2/95% N2 for 15 min) thatmimics the interstitial ionic concentrations of ischemicbrain (‘‘ischemic saline’’; Bondarenko and Chesler, 2001)(in mM: 51 NaCl, 65 K-gluconate, 0.13 CaCl2, 1.5MgCl2, 10 HEPES, pH 6.8) containing different glucoseconcentrations (in mM: 0, 5, 12, 17, 22, 27, 32, or 37).Hypoxia was induced by placing the cultures inside anincubator chamber and removing the air with a CO2/N2
(5, 95%) flow for 7 min, after which the chamber wasclosed for 3 or 6 h as previously described (Mart�ınez andS�aez, 2000). Then, reoxygenation was allowed in controlsaline (MEM with 5 mM glucose or different concentra-tions when appropriate and 10% FBS at 37�C in a 5%CO2/95% air atmosphere at nearly 100% relativehumidity).
Dye Coupling
Cells plated on glass coverslips were bathed with re-cording medium (HCO3-free F-12 medium buffered with10 mM HEPES, pH 7.2), and permeability mediated bygap junctions was tested by evaluating the transfer toneighboring cells of LY microinjected into one cell. Thecultures were observed on an inverted microscopeequipped with xenon arc lamp illumination and a NikonB filter (excitation wavelength 450–490 nm; emissionwavelength above 520 nm). LY (115 mM in 150 mMLiCl) was microinjected through a glass microelectrodeby brief overcompensation of the negative capacitancecircuit in the amplifier to cause oscillations until theimpaled cell was brightly fluorescent. Three minutes af-ter dye injection, cells were observed to determinewhether dye transfer occurred. The coupling index wascalculated as the mean number of cells to which the dyespread excluding injected cells from which there was nospread and was expressed as percent of control [Dyecoupling (%)]. In all experiments, dye coupling wastested by injecting a minimum of 10 cells.
Dye Uptake and Time-Lapse FluorescenceImaging
For visualization of Etd dye uptake by ‘‘snapshot,’’control and treated cells were exposed to 5 lM EtdBr for10 min at 37�C. Then, cells were washed with Hank’sbalanced salt solution (in mM: NaCl: 137; KCl: 5.4;Na2HPO4: 0.34; KH2PO4: 0.44, CaCl2:1.2, at pH 7.4),and examined by epifluorescence (518 nm excitation and605 nm emission) on an inverted microscope (Daiphot-Nikon) equipped with a CCD camera (Nikon) associatedwith image analysis software (Lucia-Nikon). Images ofEtd uptake were analyzed with the image J program(NIH software).
331ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA

For time-lapse fluorescence imaging, cells plated onglass coverslips were washed twice in phosphate-buf-fered saline (PBS) solution, pH 7.4. All chemicals weredissolved in ultra pure H2O. Cells were exposed toLocke’s solution (containing 154 mM NaCl, 5.4 mM KCl,2.3 mM CaCl2, 5 mM HEPES, at pH 7.4) with 5 lM ofEtd. Phase-contrast and fluorescence microscopy withtime-lapse imaging were used to record cell appearanceand fluorescence intensity changes in each condition.Changes were monitored using an imaging systemequipped with a Retga 1300I fast-cooled monochromaticdigital camera (12-bit) (Qimaging, Burnaby, BC, Can-ada), monochromator for fluorophore excitation, andMETAFLUOR software (Universal Imaging, Downing-town, PA) for image acquisition and analysis. The cul-tures on glass coverslips were placed in an Olympus BX51W1I upright microscope with water immersion lenses.Regions of interest were placed over random cells andbackground subtracted. Fluorescence was recorded every30 s. To test for changes in slope, regression lines werefitted to points before and after various treatmentsusing the Excel program and mean values of slopeswere compared using GraphPad Prism software.
Western Blot Analysis
Cultures were rinsed twice with a saline solution(Hank’s), pH 7.4, and cells were scraped off the dishwith a rubber policeman and harvested in a solutioncontaining inhibitors of proteases (200 mg/mL soybeantrypsin inhibitor, 1 mg/mL benzamidine, 1 mg/mL e-ami-nocaproic acid, 2 mM PMSF) and phosphatases (20 mMNa4P2O7 and 100 mM NaF). The suspension was centri-fuged (14,000 rpm for 2 min at 4�C), and the pellet wasresuspended in 50 mL of the solution containing prote-ase inhibitors and sonicated (Ultrasonic cell disrupter,Microson). Proteins were measured in aliquots of celllysates with the Bio-Rad protein assay. Samples weremixed with Laemmli buffer and either stored at 280�Cor resolved in 8% SDS/PAGE. After separating the pro-teins by electrophoresis, they were electrotransferred tonitrocellulose sheets. The sheets were incubated in PBS-BLOTTO (5% nonfat milk in PBS) overnight at 4�C toblock nonspecific binding, and then incubated with pri-mary antibody overnight at 4�C, followed by washing inPBS and incubation with secondary goat anti-rabbit IgGantibody conjugated to horseradish peroxidase for 1 h atroom temperature. Immunoreactivity was detected byECL by using the SuperSignal kit. Resulting immuno-blot signals were scanned, and densitometric analysiswas performed with SCION IMAGE software.
Cell Surface Biotinylation and Quantitation
Cell cultures (90-mm dishes) were washed three timeswith ice-cold Hank’s saline solution, pH 8.0, and 3 mL ofsulfo-NHS-SS-biotin (0.5 mg/mL) was added to each cellculture followed by incubation for 30 min at 4�C. Cells
were washed three times with ice-cold saline plus 15mM glycine, pH 8.0, to quench unreacted biotin. Then,cells were harvested as for Western blots describedabove. Presence of 1% SDS during this part of the prep-aration did not affect the yield of Cx43. An excess of im-mobilized NeutrAvidin was added (1 mL of NeutrAvidinper 3 mg of biotinylated protein, as recommended by theproviders), and the mixture was incubated for 1 h at4�C. Then, 1 mL of washing buffer (saline solution, pH7.2, plus 0.1% SDS and 1% Nonidet P-40) was added;the mixture was centrifuged for 2 min at 14,000 rpm, at4�C; the supernatant was removed and discarded. Thiswashing procedure was repeated three times. After thefinal wash, the supernatant was removed, and 40 mL ofsaline solution, pH 2.8, plus 0.1 M glycine was added torelease the proteins from the biotin; the pellet wasresuspended, and the mixture was centrifuged at 14,000rpm for 2 min at 4�C. The supernatant was placed in anEppendorf tube (1.5 mL), and the pH was adjusted im-mediately by adding 10 ll of 1 M Tris, pH 7.5. Relativelevels of Cx43 present in each sample were measured byWestern analysis as described above.
Immunofluorescence and Confocal Microscopy
For all immunostaining experiments, cells grown oncoverslips were fixed at room temperature with 2% para-formaldehyde for 30 min, washed three times with PBS,incubated in 0.1 M PBS-glycine three times for 5 mineach and then in 0.1% PBS-Triton X-100 containing 10%NGS for 30 min. To identify astrocytes and microglia,we used anti-GFAP and isolectin B4, respectively. Wefirst incubated cells for 2 h at room temperature withanti-GFAP monoclonal antibody (IgG1, 1:500) diluted in0.1% PBS-Triton X-100 with 2% NGS. After three rinsesin 0.1% PBS-Triton X-100, cells were then incubated for50 min at room temperature with both goat anti-mouseAlexa Fluor 355 (1:1,500) and isolectin GS-IB4 (1:100),diluted in the same solution as the primary antibody. Tolabel Cx43, cells were incubated with the anti-Cx43monoclonal antibody (1:500) for 1 h at room tempera-ture. After three washes, cells were incubated for 50min at room temperature with goat antimouse AlexaFluor 488. After several washes, coverslips weremounted in Fluoromount and examined with an uprightmicroscope equipped with epifluorescence (Eclipse E800,Nikon). To visualize double immunostaining, a confocallaser-scanning microscope (TBCS SP2; Leica, Wetzlar,Germany) was used. Stacks of consecutive confocalimages taken with a 633 objective at 500-nm intervalswere acquired sequentially with two lasers (argon 488nm and helium/neon 543 nm), and Z projections werereconstructed using Leica confocal software. The immu-nofluorescence images were analyzed with ImageJ(NIH). The images were obtained as a stack in whicheach image had an optical thickness of 1 lm; each imagewas put into gray scale and digitized. Afterwards, theImageJ feature for analysis of particles was used, andthe Feret’s diameter was measured to quantify the
332 ORELLANA ET AL.
GLIA

diameter of Cx43 immunoreactive puncta. The Feret’sdiameter is the longest measured distance between anytwo points along the ROI boundary.
Cell Death Quantification
Astrocyte membrane breakdown was evaluated byincorporation of Rhodamin B dextran (Rdex, 10 kDa) af-ter each treatment of interest. Astrocytes cultures wereincubated for 3 min with Rdex (100 lM) to label dam-aged cells and with Hoechst 33342 (1 lM) to label allthe cells. The cultures were rinsed five times in PBSand then the staining was evaluated using an OlympusBX 51W1 microscope.
Data Analysis and Statistics
Data are reported as means 6 SEM. The statisticalanalyses were performed using GraphPad Prism soft-ware. Effects on dye coupling and dye uptake were ana-lyzed by one- or two-way ANOVA, followed by a multiplecomparisons Tukey’s test or the Bonferroni correction,respectively. In all cases, a P value less than 0.05 wasconsidered statistically significant.
RESULTSHypoxia–Reoxygenation Increases Hemichannel
Activity Assessed by Dye Uptake, an EffectPotentiated by High Glucose During the Hypoxia
Previous studies using metabolic inhibition demon-strated an increase in Cx43 hemichannel activity incortical astrocytes (Orellana et al., 2009). The inhibitorsused, antimycin A and iodoacetate, are not readily re-versible, and here we have used a more physiologic pro-cedure of hypoxia–reoxygenation that has the addedadvantage of allowing investigation of an equivalent ofreperfusion (Mart�ınez and S�aez, 2000). With thismethod, astrocyte cultures were deprived of oxygen inthe presence of varying concentrations of glucose andthen returned to normal (control) medium with 5 mMglucose. We used ‘‘ischemic saline’’ during the hypoxicperiod (see Materials and Methods), which better mimicsthe interstitial fluid composition during in vivo ischemiaand results in greater ischemic damage to astrocytes inculture (Bondarenko and Chesler, 2001). We first eval-uated the effect of hypoxia in normal glucose on hemi-channel activity measured as uptake of ethidium ion (5lM Etd, added to the medium as EtdBr). Astrocytesunder control conditions showed a low rate of Etduptake (Fig. 1A,B) as previously demonstrated (Con-treras et al., 2003; Retamal et al., 2006). After 3 h in is-chemic saline with low oxygen and normal glucose (5mM) and 1 h reoxygenation in control saline and 5 mMglucose, uptake relative to control was almostunchanged (Fig. 1A,C,I). With the same protocol buthigh glucose (27 mM) present only during hypoxia,
uptake increased to �150% of control at the time ofreoxygenation, suggesting that the cell permeabilizationresponse began during the hypoxic period. Uptake thenincreased to >300% of control at 1 h before recovering tonear control at 2.5 h. Hypoxia in medium containingzero glucose increased uptake to an intermediate degree(Fig. 1I). When the hypoxic period was 6 h in ischemicsaline with 0, 5, or 27 mM glucose, the effects were simi-lar but persistent throughout the 3 h reoxygenationperiod in 5 mM glucose (Fig. 1E–H,J). Dye uptake waselevated (to �190% of control) at the start of the reoxy-genation after hypoxia in 27 mM glucose and increasedto �430% of control after 1 h reoxygenation in controlsaline with 5 mM glucose (Fig. 1E,J). After 6 h hypoxiain 5 mM glucose and 1 h reoxygenation in 5 mM glu-cose, the Etd uptake rate was increased to �220% (P <0.001) of control (Fig. 1E,J). Uptake after 3 or 6 hhypoxia in different glucose concentrations and after 1 hreoxygenation in 5 mM glucose was dependent on glu-cose concentration during the hypoxia, and was greaterat zero as well as high glucose (Fig. 1K).
The increases in uptake were not observed, if the cellswere maintained in control saline throughout thehypoxia–reoxygenation period independent of the glu-cose concentration (not shown); this result suggests thatthe differences in ionic concentration during ischemiaplay a significant role in the permeability changes con-sistent with earlier observations on damage to neuronsin hippocampal slice cultures (Cronberg et al., 2004;Rytter et al., 2003). The long period of hypoxia requiredto see significant changes in cultured astrocytes as com-pared with just 5 min of ischemia followed by reperfu-sion in hyperglycemic animals (Muranyi et al., 2006)might be explained by changes in astrocyte phenotypeunder the culturing conditions as well as interactionsbetween neurons and glia in vivo.
Increasing osmotic pressure of the 5 mM glucose sa-line by adding sucrose equimolar to the added glucosedid not significantly enhance the hypoxia–-inducedincreases in dye uptake (not shown). Furthermore, thehypoxia-induced decrease in dye coupling to be describedbelow (Fig. 4) was unaffected by the osmotic pressuredifferences.
We found no differences in Etd uptake if cells werebathed in 5 or 27 mM glucose before or after the hypoxicepisode (Fig. 2A); thus, the effects of high and zero glu-cose begin during the hypoxic period. Furthermore,there was no difference between reoxygenation in nor-mal and in ischemic saline (not shown). To determinewhether the astrocyte permeabilization induced by hy-poxia in high glucose was mediated by Cx43 hemichan-nels, we applied various blocking agents during dyeuptake after 3 h hypoxia in 27 mM glucose and 1 h re-oxygenation in 5 mM glucose. We used La31 or specific‘‘connexin mimetic peptides’’ (Gap26 or Gap27), whichhave the same sequence of a region of the first or secondextracellular loops of Cx43, respectively (Evans and Ley-baert, 2007). The application of La31 (200 lM) or Gap26(200 lM) during Etd uptake greatly reduced therate induced by hypoxia–reoxygenation (Fig. 1A,E); also,
333ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA

Gap27 (200 lM), La31 (200 lM), or Gap26 (200 lM)prior to Etd application reduced the percentage oflabeled cells below control values in ‘‘snapshot’’ experi-ments (Fig. 2B).
Recently, it was demonstrated that cultured astrocytesexpress pannexin1 (Px1) in vitro (Huang et al., 2007).However, in primary astrocytes, Px1 was not found atthe cell surface (Huang et al., 2007). To investigate thepossible contribution of Px1 hemichannels in posthy-poxic dye uptake, we used an extracellular loop peptide(10pnx1) that has been reported to block Px1 hemichan-nels (Pelegrin and Surprenant, 2006), but it was withouteffect on uptake induced in mouse astrocytes by 3 hhypoxia in high glucose and 1 h reoxygenation (Fig. 2B).Since Px1 hemichannels can be activated by extracellu-lar ATP acting on P2X7 receptors (Pelegrin and Surpren-ant, 2006), we also studied the effect of oATP, a P2X7
receptor blocker; again, there was no effect on hypoxia–reoxygenation-induced Etd uptake (Fig. 2B). Similarresults were obtained in rat astrocytes (not shown).Thus, these findings suggest that Cx43, but not Px1
hemichannels, mediate the increase in astrocyte perme-ability induced by hypoxia in high glucose and reoxyge-nation. Further evidence was obtained using astrocytesfrom Cx432/2 mice. Subjecting these cells to 3 h ofhypoxia in 27 mM glucose and 1 h of reoxygenation didnot cause an increase in Etd uptake like the oneobserved in wild-type astrocytes (Fig. 2B), supportingthe inference that astroglial permeabilization occurredspecifically through Cx43 hemichannels.
We also used HeLa cells transfected with Cx43-EGFPto evaluate if hemichannel permeability induced byhypoxia occurs in another cell type and whether theeffect could be reproduced in an exogenous expressionsystem. These cells show basal Etd uptake at the restingpotential and exhibit unitary events with conductanceand pharmacological sensitivity predicted for Cx43 hem-ichannels (Contreras et al., 2003). As reported, cellsexpressing more Cx43-EGFP showed faster Etd uptake(Fig. 3A–D). Already at 1 min of reoxygenation following6 h of hypoxia in 5 and 27 mM glucose, Etd uptake wasincreased to �250 and 520% of control, respectively
Fig. 1. Hypoxia–reoxygenation increases rate of Etd uptake by ratastrocytes, an effect potentiated by high glucose. (A) Time-lapse meas-urements of Etd uptake in rat astrocytes under control conditions (s)and starting at 1 h reoxygenation after 3 h hypoxia in 5 mM ( ) or 27mM glucose (�). Gap 26 (200 lM), a Cx43 hemichannel blocker, wasapplied after 12 min of Etd uptake measurement. (B–D) Fluorescencemicrographs of Etd uptake (10 min exposure to dye after 1 h reoxygena-tion) in astrocytes under control conditions (B) and at 1 h reoxygenationafter 3 h hypoxia in 5 mM (C) or 27 mM glucose (D). (E) Time-lapsemeasurements of Etd uptake in astrocytes under control conditions (s)and starting at 1 h of reoxygenation after 6 h of hypoxia in 5 mM ( ) or27 mM glucose (�). (F–H) Fluorescence micrographs of Etd uptake(10 min exposure to dye) in astrocytes under control conditions (F) andat 1 h reoxygenation after 6 h hypoxia in 5 mM (G) or 27 mM glucose
(H). (I) Averaged data normalized to control (---) of Etd uptake rate byastrocytes measured at the beginning of reoxygenation (time 0) and afterincreasing periods of reoxygenation following 3 h hypoxia in 0 mM (s),5 mM ( ), or 27 mM glucose (�). Zero time is at the beginning of reoxyge-nation. ***P < 0.001, (�) versus ( ); ���P < 0.001, (�) versus (s); £££P <0.001, (s) versus ( ). (J) Etd uptake rate of astrocytes after increasingreoxygenation periods following 6 h hypoxia in 0 mM (s), 5 mM ( ), or27 mM glucose (�). Zero time is at the beginning of reoxygenation. ***P< 0.001, **P < 0.005, *P < 0.05, (�) versus ( ); ���P < 0.001, ��P < 0.005,�P < 0.05, (�) versus (s); £P < 0.05, (s) versus ( ). (K) Etd uptake rateat 1 h reoxygenation following 3 h (h) or 6 h (n) of hypoxia in differentglucose concentrations. ***P < 0.001, *P < 0.05, (n) versus (h). Eachvalue corresponds to mean 6 SE of 20 cells in a representative of fiveexperiments. Bar 5 60 lm.
334 ORELLANA ET AL.
GLIA

(Fig. 3G); these increases almost certainly occurred dur-ing the hypoxic period. In both cases complete recoveryoccurred after 2–3 h reoxygenation (Fig. 3G). Theincrease in Etd uptake was due to Cx43 hemichannels,since it was sensitive to La31 (Fig. 3F) and was directlyrelated to Cx43-EGFP fluorescence in both control cellsand in cells subjected to hypoxia–reoxygenation (P <0.001, r2 5 0.93, and P < 0.001, r2 5 0.94, respectively;
Fig. 3E). In summary, hypoxia-induced increase in hemi-channel-mediated dye uptake occurs in another cell typeexpressing an exogenous connexin, Cx43-EGFP. Theeffects are as large as in astrocytes, but in contrast arereversible. The greater resistance of the HeLa cells maybe related to their tumor origin and selection for sur-vival with poor circulation (Anderson et al., 2006).
Hypoxia–Reoxygenation Reduces Dye CouplingBetween Astrocytes, an Effect Potentiated by
High Glucose During the Hypoxia
In the central nervous system of vertebrates, gap junc-tional coupling of astrocytes is prominent and plays im-portant roles in ionic homeostasis, thus helping toensure the normal functioning of neurons (Simard andNedergaard, 2004). In view of data presented thus farand of previous studies showing that a hypoxic episodecan cause delayed and transient reduction in dye cou-pling of cortical astrocytes in culture (Mart�ınez andS�aez, 2000), we investigated whether astroglial couplingis more sensitive to hypoxia in high glucose.
Since leakage of the microinjected dye (LY) to evaluateintercellular communication via gap junctions mightoccur through hemichannels, particularly in view oftheir increased opening after hypoxia–reoxygenation, wemeasured dye coupling in both the absence and the pres-ence of 200 lM La31 in the extracellular solution. La31
did not significantly modify the intercellular diffusion ofmicroinjected LY in astrocytes under control conditionsor after hypoxia in normal or high glucose followed byreoxygenation in normal glucose (not shown). Threehours of hypoxia in 27 mM glucose and ischemic salinefollowed by reoxygenation reduced dye coupling com-pared with control (Fig. 4A,C,E). The maximal effectinduced by 27 mM glucose occurred at 1–1.5 h of reoxy-genation with �60% reduction in dye coupling comparedwith control (Fig. 4E). In addition, hypoxia in 5 mM glu-cose or in zero glucose reduced dye coupling by �20%and �40% of control, respectively, at 1 h of reoxygena-tion, less than the effect in 27 mM glucose (Fig. 4A–C,E). The inhibitory effects on dye coupling induced by 6h hypoxia in 27 mM glucose, normal glucose, or zeroglucose were more pronounced than those induced by 3h of hypoxia in the same glucose concentrations (Fig.4D,F). Unlike the transient reduction induced by 3 h ofhypoxia, 6 h of hypoxia in 27 mM glucose induced a per-sistent reduction in dye coupling reaching about �90%inhibition at 1 h of reoxygenation and recovering verylittle by 3.5 h reoxygenation (Fig. 4F). Similar to theconcentration dependence of the effect of glucose duringhypoxia on subsequent hemichannel-mediated dyeuptake (Fig. 1K), glucose during 3 or 6 h of hypoxiareduced dye coupling after 1 h reoxygenation in a con-centration-dependent manner with a moderate effect atzero glucose, a smaller effect at 5–12 mM glucose, andthen an effect increasing to a plateau at 27–37 mM glu-cose (Fig. 4G).
Fig. 2. High or zero glucose levels during hypoxia increase astroglialEtd uptake after reoxygenation, an effect independent of high glucosebefore or after the hypoxic period; uptake is mediated exclusively byCx43 hemichannels. (A) Averaged data normalized to control (dashedline) of Etd uptake rate by rat astrocytes after 3 h hypoxia in 0 mM(left), 5 mM (middle), or 27 mM glucose (right) and 1 h reoxygenationand maintained 24 h before hypoxia and during the 1 h reoxygenationwith 5 mM (white bar), 27 mM glucose (dashed white bar), or with thetwo possible combinations of one before and the other after (black andgray bars). *P < 0.001, with respect to control. (B) Percentage of Etd-positive cells in snapshot pictures of mouse astrocytes compared withcontrol (dashed line) after 3 h hypoxia in 27 mM glucose and 1 h reoxy-genation. Cells were then exposed to 5 lM Etd for 20 min and snap-shots were taken. In parallel experiments, cells were simultaneouslyexposed for 20 min to Etd and a connexin hemichannel blocker, La31
(200 lM), Gap26 (200 lM) or Gap27 (200 lM), or a Px1 hemichannelblocker, 10panx1 (200 lM) or a P2X channel blocker, oATP (200 lM).The number of Etd-positive cells in astrocyte cultures of Cx432/2 miceat 1 h of reoxygenation following 3 h hypoxia in 27 mM glucose is alsoshown.
335ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA

Hypoxia–Reoxygenation Increases the Level ofCx43 on the Cell Surface, an Effect Potentiated
by High Glucose
During metabolic inhibition Etd uptake is ascribed toan increase in Cx43 hemichannel activity, largelyaccounted for by elevated levels of surface hemichannelswith little change in open probability (Retamal et al.,2006). To test if reoxygenation after hypoxia in high glu-cose affects the surface levels of Cx43, cell surface pro-teins were biotinylated, isolated with NeutrAvidin beads,and resolved by Western blotting. After 3 h hypoxia and1 h reoxygenation, the levels of surface Cx43 were de-
pendent on the concentration of glucose present duringthe hypoxia period; they were unaffected by 5 mM glu-cose but increased by zero and 22–37 mM glucose [Fig.5A (left panel), B]. The increases in surface Cx43 levelswere delayed and transient; they were significantlyhigher at 1 h than at 40 min reoxygenation and hadreturned to control levels by 3 h of reoxygenation [Fig.5C (left panel), D]. Levels of surface Cx43 were alsoelevated in astrocytes exposed to 1 h reoxygenation after6 h of hypoxia in zero glucose or in 22 mM glucose orhigher but not in 5 mM glucose [Fig. 5E (left panel), F].After 6 h hypoxia in 27 mM glucose, surface Cx43 wasincreased after a shorter period of reoxygenation (20
Fig. 3. Hypoxia–reoxygenation increases Cx43-hemichannel activityin HeLa cells expressing Cx43-EGFP, an effect potentiated by highglucose. Etd uptake was measured in mixed cultures of Cx43-EGFPand parental cells (5 lM Etd). (A) Fluorescence micrograph showingdifferent degrees of EGFP expression. (B) Etd uptake (10 min incuba-tion in 5 lM) after 6 h hypoxia in 27 mM glucose (6 h hypox/27 mM)and 1 h reoxygenation. (C) Merge of EGFP and Etd fluorescence. (D)Time course of Etd uptake in Cx43-EGFP cells (1–4 in A) and parentalHeLa cells (5–7 in A) after 6 h hypoxia in 27 mM glucose and 1 h reox-ygenation. (E) Rate of Etd uptake was proportional to Cx43-EGFP fluo-
rescence for seven cells after 6 h hypoxia in 27 mM glucose and 1 hreoxygenation (�) and for six cells in control conditions (s). (F) Time-lapse measurements of Etd uptake in Cx43-EGFP cells under controlconditions (s) and after 6 h hypoxia in 5 ( ) or 27 mM (�) glucose and1 h reoxygenation. (G) Averaged Etd uptake normalized to control (---)by Cx43-EGFP cells for different reoxygenation periods after 6 h hy-poxia in 5 mM (s) or 27 mM (�) glucose. Each point corresponds to themean 6 SE of 20 cells in a representative experiment of four. Bar 5 10lm.
336 ORELLANA ET AL.
GLIA

min) compared with the increase induced by reoxygena-tion after 3 h hypoxia (40 min) [Fig. 5G (left panel), H].Moreover, after 6 h hypoxia the surface Cx43 levelsremained high for at least 3 h reoxygenation, whereasafter 3 h hypoxia and 3 h reoxygenation, recovery wascomplete.
Under all conditions described above, the total levelsof Cx43 were not affected [Fig. 5A (right panel), C (rightpanel), E (right panel), G (right panel),B,D,F,H]. Fur-thermore, in all Western blot analyses of surface or totalCx43 levels the characteristic phosphorylated P1–P2bands were predominant, and the unphosphorylated NPband was less intense (Fig. 5A,C,E,G), suggesting thatthe treatments did not reduce the expression or increasethe degradation of Cx43 or induce dephosphorylation ofCx43 detectable by this technique.
Hypoxia–Reoxygenation Causes Disappearanceof Large Cx43 Aggregates and Increases GFAP
Expression
To examine further the altered Cx43 distribution inthe cells, we applied indirect immunofluorescence to
astrocytes under conditions that induced the greatestchanges in hemichannel activity and gap junctionalcoupling. In control astrocytes, Cx43 labeling was het-erogeneous including areas of different shape and size;the larger ones likely correspond to gap junction pla-ques or internalized gap junctions, and the smallerones are likely trans Golgi vesicles for Cx43 delivery tothe plasma membrane and small junctional regionsinternalized and targeted for degradation (Fig. 6A andSupp. Info. Figure 1). In astrocytes subjected to 6 h ofhypoxia in 5 mM glucose and 1 h of reoxygenation,Cx43 immunoreactivity was not located in big continu-ous areas, and labeling was as smaller intracellularpuncta, possibly vesicles (Fig. 6B and Supp. Info.Figure 1). The larger areas had likely been internalizedas small vesicles (Gaietta et al., 2002). No obvious dif-ference in Cx43 immunoreactivity was observedbetween astrocytes subjected to 6 h hypoxia in thepresence of 27 mM glucose and those subjected to 6 hhypoxia in 5 mM glucose (1 h reoxygenation, Fig. 6Cand Supp. Info. Figure 1). Cells subjected to 6 h hy-poxia with 5 or 27 mM glucose and 1 h reoxygenationshowed an increase in GFAP immunoreactivity and inthe number of astroglial processes (Fig. 6C), two
Fig. 4. Hypoxia–reoxygenation reduces dye coupling in rat astro-cytes, an effect potentiated by high glucose. (A–D) Fluorescence micro-graphs of LY coupling in astrocytes under control conditions (A), at 1 hof reoxygenation after 3 h hypoxia in 5 mM (B) or 27 mM glucose (C),and at 1 h of reoxygenation after 6 h hypoxia in 27 mM glucose (D).The (*) in panels A–D denotes the cell microinjected with LY. Bar 5 60lm. (E) Average number of astrocytes to which LY spread from aninjected cell after different reoxygenation periods following 3 h of hy-poxia in 5 mM ( ), 27mM (�), or 0 mM (s) glucose [expressed as per-centage of the number in control conditions, (---)]. Zero time is at the
beginning of reoxygenation. **P < 0.005, ***P < 0.001, (�) versus ( ).(F) LY coupling by astrocytes after increasing reoxygenation periods fol-lowing 6 h hypoxia in 5 mM ( ), 27 mM (�), or 0 mM (s) glucose. ***P< 0.001, (�) versus ( ); ���P < 0.001, ��P < 0.005, (�) versus (s); ##P <0.005, #P < 0.05, ( ) versus (s). (G) LY coupling at 1 h reoxygenationafter 3 h (h) or 6 h (n) hypoxia in different glucose concentrations. *P< 0.05, (n) versus (h). For each graph, values are means 6 SE of 10cells in a representative experiment of five; separate cultures wereused for each time point.
337ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA

properties that are characteristic of astroglial activation(Pekny and Nilsson, 2005).
Increase in Dye Uptake Induced byHypoxia–Reoxygenation is Reversed by DTT,
but Decrease in Dye Coupling is not
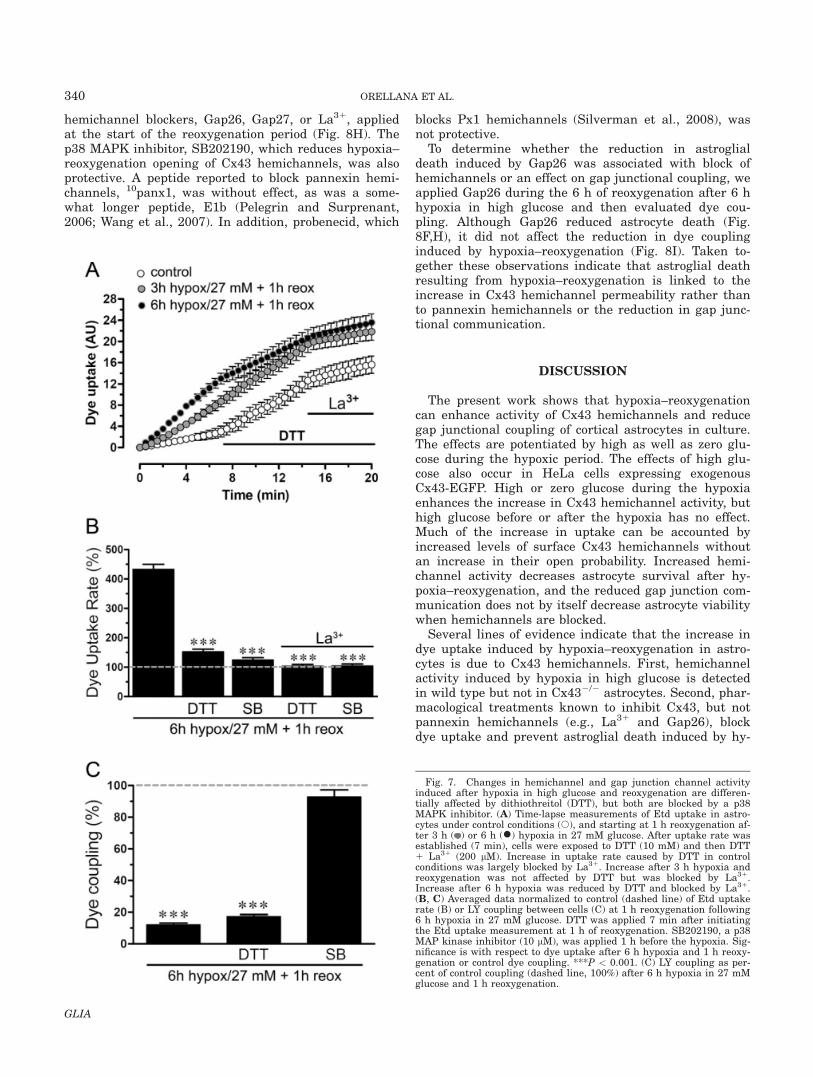
Redox potential modulates the activity of Cx43hemichannels in astrocytes (Retamal et al., 2006,
2007b). Reducing agents reverse the increase in mem-brane permeability through hemichannels induced bymetabolic inhibition or by application of nitric oxidedonors or proinflammatory cytokines (Retamal et al.,2007b). However, reducing agents increase membranepermeability under normoxic conditions (Retamal et al.,2006, 2007b). To evaluate the redox state of Cx43 hemi-channels in astrocytes subjected to hypoxia with normalor high glucose, we measured dye uptake after 3 or 6 hof hypoxia and 1 h reoxygenation and then applied DTT
Fig. 5. Hypoxia–reoxygenation increases the level of surface Cx43in astrocytes, an effect potentiated by high glucose. Cultured astrocyteswere subjected to 3 h (A–D) or 6 h (E–H) hypoxia (hypox) in presenceof different glucose concentrations and then to reoxygenation (reox) fordifferent periods of time. Representative western blots of surface (leftpanels in A, C, E, and G) and total Cx43 (right panels) levels areshown. (A, E) The left panels show levels of surface Cx43 present inastrocytes under control conditions, i.e., not subjected to hypoxia (lane1) or subjected to 1 h of reoxygenation after 3 h (A) or 6 h (E) hypoxiain 0 mM (lane 2), 5 mM (lane 3), 22 mM (lane 4), 27 mM (lane 5), and37 mM (lane 6) glucose. The right panels show the levels of total Cx43under the same conditions along with a-tubulin as a loading control.
The Cx43 phosphorylated (P1–P2) and nonphosphorylated (NP) formsare indicated on the left of each gel. (B, F) Quantitation of data exem-plified by A and E; surface and total Cx43 after 3 h (B) or 6 h (F)hypoxia in the indicated glucose concentrations followed by 1 h reoxy-genation in 5 mM glucose. (C, G) Surface and total Cx43 after 3 h (C)or 6 h (G) hypoxia in 27 mM glucose and at the indicated times ofreoxygenation. (D, H) Quantitation of data exemplified by C and G; sur-face and total Cx43 after 3 h (D) or 6 h (H) hypoxia in 27 mM glucoseand at the indicated times of reoxygenation. In D,F and H total levelswere normalized according to the levels of a-tubulin detected in eachlane.
338 ORELLANA ET AL.
GLIA

(10 mM) during time lapse measurement of Etd uptake.In control conditions, the application of 10 mM DTTincreased Etd uptake to �400% of control (Fig. 7A,B).However, in cells that were exposed to 3 h of hypoxia in27 mM glucose and 1 h reoxygenation, DTT applied dur-ing Etd uptake did not affect the uptake rate, whereasfollowing 6 h of hypoxia in 27 mM glucose and 1 h reox-ygenation, DTT reduced the rate, suggesting that thehemichannels had been oxidized (Fig. 7A,B). La31
largely blocked the increases in uptake rate, whetherthey were induced by DTT or by hypoxia–reoxygenation.The reduction in dye coupling induced by 6 h hypoxia in27 mM glucose and 1 h reoxygenation (Fig. 4D,F) wasnot reversed by DTT (Fig. 7C). These observations sug-gest that 6 h hypoxia in high glucose and 1 h reoxygena-tion oxidizes at least some of the hemichannels or anassociated molecule and that the processes involved inthe regulation of Cx43 hemichannels and gap junctionchannels during hypoxia–reoxygenation are different.
Changes in Hemichannel and Gap JunctionChannel Activity Induced by Hypoxia–
Reoxygenation are p38 MAPK Dependent
Because Cx43 hemichannels and gap junction chan-nels in astrocytes treated with proinflammatory cyto-kines are modulated through a p38 MAPK-dependentpathway (Retamal et al., 2007a), and hypoxia–reoxyge-nation is a proinflammatory condition, we examinedwhether the changes in hemichannels and gap junctionsinduced by hypoxia–reoxygenation in high glucose aremediated by a p38 MAPK pathway. The addition ofSB202190, an inhibitor of p38 MAPK, 1 h before the hy-poxia largely prevented the Etd uptake increase inducedby 6 h hypoxia in 27 mM glucose and 1 h reoxygenation(Fig. 7B). Moreover, the same treatment with SB202190prevented the decrease in dye coupling induced by thesame hypoxia–reoxygenation protocol (Fig. 7C).
The Increased Hemichannel Activity Induced byHypoxia–Reoxygenation, Which is Potentiated by
High Glucose, Promotes Astroglial Death
To examine if the rise in hemichannel activity inducedby hypoxia–reoxygenation is deleterious, we evaluatedastroglial viability measured by exclusion of Rdex at dif-ferent reoxygenation periods after 3 or 6 h hypoxia innormal or high glucose. Because of its large size, Rdex istaken up only by cells with disrupted membranes(Kondo et al., 2000). Under control conditions and after3 h hypoxia in 5–27 mM glucose and 6 h reoxygenation,few astrocytes took up Rdex (Fig. 8A,G). However, after6 h of hypoxia in 5 mM glucose and 6 h reoxygenation,�20% of the astrocytes took up Rdex and were, by thismeasure, dead (Fig. 8D,G), and after 6 h hypoxia in 27mM glucose and 3–6 h reoxygenation, �40–45% of astro-cytes were dead (Fig. 8E,G). Cell death induced by 6 hof hypoxia in high glucose was greatly reduced by the
Fig. 6. Hypoxia–reoxygenation causes disappearance of the largerCx43 immunoreactive areas found in control cells and increases astro-glial activation during reoxygenation. Representative confocal imagesdepicting Cx43 (green) and GFAP (red) immunolabeling of astrocytesunder control conditions (A) or at 1 h reoxygenation after 6 h hypoxiain 5 mM (B) or 27 mM (C) glucose. The larger Cx43 labeled areas in(A) may represent gap junction plaques (arrows); Cx43 labeling in (B)and (C) is only of smaller areas (arrow heads). Increased GFAP labelingin (B) and (C) indicates increased activation, which appears more pro-nounced for hypoxia in 27 mM glucose. Insets: 23 magnification of theindicated area of each panel. Bar 5 50 lm.
339ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA
hemichannel blockers, Gap26, Gap27, or La31, appliedat the start of the reoxygenation period (Fig. 8H). Thep38 MAPK inhibitor, SB202190, which reduces hypoxia–reoxygenation opening of Cx43 hemichannels, was alsoprotective. A peptide reported to block pannexin hemi-channels, 10panx1, was without effect, as was a some-what longer peptide, E1b (Pelegrin and Surprenant,2006; Wang et al., 2007). In addition, probenecid, which
blocks Px1 hemichannels (Silverman et al., 2008), wasnot protective.
To determine whether the reduction in astroglialdeath induced by Gap26 was associated with block ofhemichannels or an effect on gap junctional coupling, weapplied Gap26 during the 6 h of reoxygenation after 6 hhypoxia in high glucose and then evaluated dye cou-pling. Although Gap26 reduced astrocyte death (Fig.8F,H), it did not affect the reduction in dye couplinginduced by hypoxia–reoxygenation (Fig. 8I). Taken to-gether these observations indicate that astroglial deathresulting from hypoxia–reoxygenation is linked to theincrease in Cx43 hemichannel permeability rather thanto pannexin hemichannels or the reduction in gap junc-tional communication.
DISCUSSION
The present work shows that hypoxia–reoxygenationcan enhance activity of Cx43 hemichannels and reducegap junctional coupling of cortical astrocytes in culture.The effects are potentiated by high as well as zero glu-cose during the hypoxic period. The effects of high glu-cose also occur in HeLa cells expressing exogenousCx43-EGFP. High or zero glucose during the hypoxiaenhances the increase in Cx43 hemichannel activity, buthigh glucose before or after the hypoxia has no effect.Much of the increase in uptake can be accounted byincreased levels of surface Cx43 hemichannels withoutan increase in their open probability. Increased hemi-channel activity decreases astrocyte survival after hy-poxia–reoxygenation, and the reduced gap junction com-munication does not by itself decrease astrocyte viabilitywhen hemichannels are blocked.
Several lines of evidence indicate that the increase indye uptake induced by hypoxia–reoxygenation in astro-cytes is due to Cx43 hemichannels. First, hemichannelactivity induced by hypoxia in high glucose is detectedin wild type but not in Cx432/2 astrocytes. Second, phar-macological treatments known to inhibit Cx43, but notpannexin hemichannels (e.g., La31 and Gap26), blockdye uptake and prevent astroglial death induced by hy-
Fig. 7. Changes in hemichannel and gap junction channel activityinduced after hypoxia in high glucose and reoxygenation are differen-tially affected by dithiothreitol (DTT), but both are blocked by a p38MAPK inhibitor. (A) Time-lapse measurements of Etd uptake in astro-cytes under control conditions (s), and starting at 1 h reoxygenation af-ter 3 h ( ) or 6 h (�) hypoxia in 27 mM glucose. After uptake rate wasestablished (7 min), cells were exposed to DTT (10 mM) and then DTT1 La31 (200 lM). Increase in uptake rate caused by DTT in controlconditions was largely blocked by La31. Increase after 3 h hypoxia andreoxygenation was not affected by DTT but was blocked by La31.Increase after 6 h hypoxia was reduced by DTT and blocked by La31.(B, C) Averaged data normalized to control (dashed line) of Etd uptakerate (B) or LY coupling between cells (C) at 1 h reoxygenation following6 h hypoxia in 27 mM glucose. DTT was applied 7 min after initiatingthe Etd uptake measurement at 1 h of reoxygenation. SB202190, a p38MAP kinase inhibitor (10 lM), was applied 1 h before the hypoxia. Sig-nificance is with respect to dye uptake after 6 h hypoxia and 1 h reoxy-genation or control dye coupling. ***P < 0.001. (C) LY coupling as per-cent of control coupling (dashed line, 100%) after 6 h hypoxia in 27 mMglucose and 1 h reoxygenation.
340 ORELLANA ET AL.
GLIA

poxia in high glucose. Third, the increase in dye uptakeinduced by hypoxia is not observed in Cx432/2 astro-cytes. Fourth, two pannexin mimetic peptides, 10panx1and E1b, as well as probenecid, which are presumptivepannexin hemichannel blockers, do not affect dyeuptake.
To examine the generality of the Cx43 response, wetested whether Cx43-EGFP hemichannels expressed inHeLa cells were similarly sensitive to hypoxia–reoxyge-nation in high glucose. Reoxygenation after hypoxiaincreases the activity of Cx43-EGFP hemichannels, andthe response is potentiated by high glucose concentra-tions present during the hypoxic episode. Therefore, thisaspect of the response to hypoxia and high glucose con-ditions may be cell type independent.
Differences in intensity, duration, and/or ‘‘quality’’ (ab-sence of or high glucose) of an ischemic episode deter-mine whether there is no lasting response, induction of‘‘ischemic tolerance,’’ also termed ischemic precondition-ing, or damage, which may appear with some delay(Gidday, 2006). In our experiments, 6 h hypoxia inducedgreater effects on hemichannels and gap junctional com-munication than 3 h hypoxia, and the effects were morepronounced at high and zero glucose during the hypoxiaperiod (difference in quality) and could be irreversible inthe case of hemichannel activity after 6 h hypoxia and6 h of reoxygenation. Previous studies showed that
metabolic inhibition with iodoacetate and antimycin A,an in vitro ischemia model, increases Cx43 hemichannelopening and reduces gap junctional coupling of corticalastrocytes (Orellana et al., 2009), but in this model thereis no analog of reperfusion, and all changes occur in nor-moxia. The present study demonstrates that hypoxia ina saline that mimics the interstitial ionic concentrationsof ischemic brain (Bondarenko and Chesler, 2001) fol-lowed by reoxygenation in control saline causes changesin Cx43-based channels similar to those induced by met-abolic inhibition, but largely delayed until the period ofreoxygenation. The hypoxia–reoxygenation procedure ismore physiologic and allows the temporal dissection ofchanges occurring upon reoxygenation from those duringthe absence of oxygen. In our experiments, most of thereduction in gap junctional communication, like theincrease in hemichannel activity, occurred during re-oxygenation, which is in agreement with previousreports on gap junctions using similar conditions (Orel-lana et al., 2009).
The opposite changes in hemichannel opening and gapjunction communication are unlikely to be directlyrelated. The current view is that gap junctions aredegraded by internalization of small regions of theentire junction thickness and then transported to lyso-somes (Gaietta et al., 2002). Internalization of large pla-ques into one or the other of the coupled cells is also
Fig. 8. Connexin hemichannel activity induced by hypoxia–reoxyge-nation leads to astroglial death. (A–F) Fluorescence micrographs ofRdex (red) uptake and Hoechst 33342 nuclear staining (blue) in ratastrocytes under control conditions (A), at 6 h of reoxygenation after 3h hypoxia in 5 mM (B, 3 h hypox/5 mM) or 27 mM glucose (C, 3 hhypox/27 mM) or at 6 h reoxygenation after 6 h hypoxia in 5 mM glu-cose (D, 6 h hypox/5 mM) or 27 mM glucose (E, 6 h hypox/27 mM). (F)Gap26 (300 lM) applied at the start of reoxygenation after 6 h of hy-poxia in 27 mM glucose prevented uptake of Rdex at 1 h reoxygenation.(G) Quantitation of cell death measured by Rdex staining as percentage
of total cells identified with Hoechst 33342 at 0, 1, 3, and 6 h reoxyge-nation following 3 or 6 h hypoxia in 5 or 27 mM glucose. *P < 0.001,compared with control. (H) Effect of Gap26 (200 lM), Gap27 (200 lM),La31 (200 lM), 10panx1 (200 lM), E1b (200 lM), probenecid (2 mMProb), and SB202190 (10 lM SB) on cell death induced by 6 h reoxyge-nation following 6 h hypoxia in 27 mM glucose. ***P < 0.001, comparedwith the effect induced by 6 h hypoxia/27 mM 1 6 h reox. (I) Gap26had no effect on the reduction in dye coupling induced by 6 h reoxyge-nation following 6 h hypoxia in 27 mM glucose. Each bar correspondsto the mean 6 SE of four experiments. Bar in F: 50 lm.
341ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA

reported (Piehl et al., 2007). There is no indication thatdocked hemichannels can be separated from each otherand left in the membrane where they might open in theabsence of a docked hemichannel in an apposed mem-brane. However, if junction formation is reduced, theremay be more hemichannels in the membrane (VanSlykeand Musil, 2005), and an indirect relation betweenincreased hemichannel opening and reduction in cou-pling is possible.
Astrocytes bathed with saline containing normal glu-cose (5 mM) did not show enhanced dye uptake orreduced coupling at the onset of reoxygenation after 3 hhypoxia, and by these measures hypoxia in normal glu-cose is less deleterious than hypoxia at zero or a highglucose concentration in agreement with work of othersperformed on neurons (Cronberg et al., 2004; Goldbergand Choi, 1993; Pringle et al., 1997). Delayed changeswere also greater when the hypoxia was in zero or highglucose. Hypoxia in high glucose, during but not beforeor after the hypoxia, induced similar changes in Cx43-based channels. The effect of zero glucose might beexplained by loss of the normal glucose effect mentionedabove. The effect of high glucose concentration showed aconcentration-dependent relationship, presumablycaused by glucose itself and not hypertonicity, sinceequimolar sucrose concentrations failed to enhance thehemichannel activity.
Several mechanisms could contribute to increasedhemichannel activity measured as dye uptake: increasedopening of hemichannels already present in the plasmamembrane, increase in number of hemichannels in themembrane by increased insertion or decreased internal-ization, and increase in the permeability of open hemi-channels. Glucose during the hypoxia increased the sur-face levels of Cx43 after 1 h reoxygenation in a concen-tration-dependent manner, perhaps enough to accountfor the increase in permeability considering the differen-ces in the techniques of measurement.
The reduction in dye coupling induced by hypoxia–reoxygenation cannot be attributed to a reduction inCx43 levels or altered phosphorylation state detectableby changes in electrophoretic mobility, because at differ-ent periods of reoxygenation, the Cx43 total levels andpattern of immunoreactive bands were similar to thoseof control cells. However, immunofluorescence labelingshowed a marked decrease in structures compatiblewith gap junction plaques after 6 h hypoxia in normalor high glucose and 1 h reoxygenation, suggesting thatinternalization and degradation of gap junctions mightexplain the reduction in dye coupling.
Hypoxia–reoxygenation is a proinflammatory condi-tion, and high glucose enhances the effects on hemichan-nels and gap junction channels. Furthermore, these find-ings parallel recent reports showing that gap junctionchannels and hemichannels are oppositely regulated incell lines transfected with Cx43 (Orellana et al., 2009).In addition, we investigated the signaling pathwaysinvolved in the opposite modulation of Cx43-based chan-nels by hypoxia–reoxygenation and found that p38MAPK inhibition reduced the effect of hypoxia–reoxyge-
nation on both Cx43 hemichannels and gap junctions.This observation is in agreement with previous reportsshowing that the opposite regulation of the two types ofconnexin channel in astrocytes treated with TNF-a plusIL-1b, another proinflammatory condition, depends onthe p38 MAP kinase pathway (Retamal et al., 2007a).p38 MAP kinase activity is needed to induce increasedactivity of Cx43 HCs (Schalper et al., 2008), possiblyusing a Ca21-dependent pathway (Mu et al., 2008) or asconsequence of oxidant stress (Guyton et al., 1996).
Furthermore, hemichannel-mediated dye uptakeinduced by metabolic inhibition or treatment with NOdonors in astrocytes is blocked by DTT without changingCx43 hemichannel phosphorylation as indicated by theelectrophoretic mobility (Retamal et al., 2006). In con-trast, under normoxic conditions DTT increases the openprobability of hemichannels in astrocytes and in Cx43-transfected HeLa cells, and a gradual transition betweenthe two responses was observed during metabolic inhibi-tion (Retamal et al., 2007b). Here, we found that hemi-channel-mediated Etd uptake induced by 3 h of hypoxiain high glucose and 1 h of reoxygenation was insensitiveto DTT, but after 6 h of hypoxia and 1 h reoxygenation,DTT reduced Etd uptake. A possible explanation forthese different DDT sensitivities of hemichannel-medi-ated Etd uptake is that 3 h of hypoxia followed by reoxy-genation do not induce a drastic unbalance between lev-els of free radical scavengers (i.e., reduced glutathione)and generation of NO, but after 6 h hypoxia and reoxy-genation there is depletion of free radical scavengersand persistent generation of NO.
The irreversible increase in hemichannel activityinduced by 6 h hypoxia in high glucose might be expectedto increase astroglial death, and hemichannel blockersdelay membrane breakdown following irreversible meta-bolic inhibition (Contreras et al., 2003). We examined theincorporation of Rdex, indicative of loss of membrane in-tegrity, at different periods of reoxygenation following 3or 6 h of hypoxia in high glucose. By this measure weobserved cell death that was greater after longer periodsof hypoxia at higher glucose concentrations.
Although these results were obtained from cultures ofcortical glial cells, they provide a new insight into therole of astroglial connexins in neuronal survival. Loss ofneuronal protection by astrocytes could result from dys-regulation of their gap junctions and hemichannels. Areduction in gap junctional communication impairs thespatial buffering capacity of astroglial networks, and anincrease in hemichannel activity could in turn affect theviability of other cells through a paracrine mechanism.In agreement, ischemic saline in high glucose enhancesthe release of ATP in retinal cell cultures (Costa et al.,2008). Thus, excessive release or deficient uptake of sig-naling molecules such as ATP and other hemichannelpermeant molecules, e.g., glutamate, arachidonic acidbyproducts, could enhance neuronal excitotoxicity. Cx43gap junction channels and hemichannels are regulatedoppositely by hypoxia in high glucose and reperfusion,and both kinds of modulation may be relevant in relatedpathologies, such as diabetes and stroke.
342 ORELLANA ET AL.
GLIA

REFERENCES
Anderson KM, Tsui P, Guinan P, Rubenstein M. 2006. The proliferativeresponse of hela cells to 2-deoxy-D-glucose under hypoxic or anoxicconditions: An analogue for studying some properties of in vivo solidcancers. Anticancer Res 26:4155–4162.
Benjelloun N, Joly LM, Palmier B, Plotkine M, Charriaut-MarlangueC. 2003. Apoptotic mitochondrial pathway in neurones and astrocytesafter neonatal hypoxia–ischaemia in the rat brain. Neuropathol ApplNeurobiol 29:350–360.
Bondarenko A, Chesler M. 2001. Rapid astrocyte death induced bytransient hypoxia, acidosis, and extracellular ion shifts. Glia 34:134–142.
Contreras JE, S�aez JC, Bukauskas FF, Bennett MV. 2003. Gating andregulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad SciUSA 100:11388–11393.
Costa G, Pereira T, Neto AM, Cristovao AJ, Ambrosio AF, Santos PF.2008. High glucose changes extracellular adenosine triphosphate lev-els in rat retinal cultures. J Neurosci Res 87:1375–1380.
Cronberg T, Rytter A, Asztely F, Soder A, Wieloch T. 2004. Glucose butnot lactate in combination with acidosis aggravates ischemic neuro-nal death in vitro. Stroke 35:753–757.
Cruciani V, Mikalsen SO. 2005. The connexin gene family in mammals.Biol Chem 386:325–332.
Choi DW, Maulucci-Gedde M, Kriegstein AR. 1987. Glutamate neuro-toxicity in cortical cell culture. J Neurosci 7:357–368.
Dirnagl U, Iadecola C, Moskowitz MA. 1999. Pathobiology of ischaemicstroke: An integrated view. Trends Neurosci 22:391–397.
Evans WH, Leybaert L. 2007. Mimetic peptides as blockers of connexinchannel-facilitated intercellular communication. Cell Commun Adhes14:265–273.
Fields RD, Stevens-Graham B. 2002. New insights into neuron-gliacommunication. Science 298:556–562.
Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW,Sosinsky GE, Tsien RY, Ellisman MH. 2002. Multicolor and electronmicroscopic imaging of connexin trafficking. Science 296:503–507.
Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A.2007. Glia: The fulcrum of brain diseases. Cell Death Differ 14:1324–1335.
Gidday JM. 2006. Cerebral preconditioning and ischaemic tolerance.Nat Rev Neurosci 7:437–448.
Goldberg MP, Choi DW. 1993. Combined oxygen and glucose depriva-tion in cortical cell culture: Calcium-dependent and calcium-inde-pendent mechanisms of neuronal injury. J Neurosci 13:3510–3524.
Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. 1996. Activation ofmitogen-activated protein kinase by H2O2. Role in cell survival fol-lowing oxidant injury. J Biol Chem 271:4138–4142.
Huang Y, Grinspan JB, Abrams CK, Scherer SS. 2007. Pannexin1 isexpressed by neurons and glia but does not form functional gap junc-tions. Glia 55:46–56.
Kagansky N, Levy S, Knobler H. 2001. The role of hyperglycemia inacute stroke. Arch Neurol 58:1209–1212.
Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. 2000. Metabolicinhibition activates a non-selective current through connexin hemi-channels in isolated ventricular myocytes. J Mol Cell Cardiol 32:1859–1872.
Lin B, Busto R, Globus MY, Martinez E, Ginsberg MD. 1995. Braintemperature modulations during global ischemia fail to influenceextracellular lactate levels in rats. Stroke 26:1634–1638.
Mart�ınez AD, S�aez JC. 2000. Regulation of astrocyte gap junctions byhypoxia–reoxygenation. Brain Res Brain Res Rev 32:250–258.
Mu D, Zhang W, Chu D, Liu T, Xie Y, Fu E, Jin F. 2008. The role ofcalcium, P38 MAPK in dihydroartemisinin-induced apoptosis of lungcancer PC-14 cells Cancer Chemother Pharmacol 61:639–645.
Muranyi M, Ding C, He Q, Lin Y, Li PA. 2006. Streptozotocin-induceddiabetes causes astrocyte death after ischemia and reperfusion injury.Diabetes 55:349–355.
Myers RE, Yamaguchi S. 1977. Nervous system effects of cardiac arrestin monkeys. Preservation of vision. Arch Neurol 34:65–74.
Orellana JA, S�aez PJ, Shoji KF, Schalper KA, Palacios-Prado N,Velarde V, Giaume C, Bennett MV, S�aez JC. 2009. Modulation of
brain hemichannels and gap junction channels by pro-inflammatoryagents and their possible role in neurodegeneration. Antioxid RedoxSignal 11:369–399.
Pekny M, Nilsson M. 2005. Astrocyte activation and reactive gliosis.Glia 50:427–434.
Pelegrin P, Surprenant A. 2006. Pannexin-1 mediates large pore forma-tion and interleukin-1beta release by the ATP-gated P2X7 receptor.EMBO J 25:5071–5082.
Peuchen S, Duchen MR, Clark JB. 1996. Energy metabolism of adultastrocytes in vitro. Neuroscience 71:855–870.
Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM.2007. Internalization of large double-membrane intercellular vesiclesby a clathrin-dependent endocytic process. Mol Biol Cell 18:337–347.
Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE.1997. Neuroprotection by both NMDA and non-NMDA receptorantagonists in in vitro ischemia. Brain Res 755:36–46.
Retamal MA, Cort�es CJ, Reuss L, Bennett MV, S�aez JC. 2006. S-nitro-sylation and permeation through connexin 43 hemichannels in astro-cytes: Induction by oxidant stress and reversal by reducing agents.Proc Natl Acad Sci USA 103:4475–4480.
Retamal MA, Froger N, Palacios-Prado N, Ezan P, S�aez PJ, S�aez JC,Giaume C. 2007a. Cx43 hemichannels and gap junction channels inastrocytes are regulated oppositely by proinflammatory cytokinesreleased from activated microglia. J Neurosci 27:13781–1392.
Retamal MA, Schalper KA, Shoji KF, Bennett MV, S�aez JC. 2007b.Opening of connexin 43 hemichannels is increased by lowering intra-cellular redox potential. Proc Natl Acad Sci USA 104:8322–8327.
Rytter A, Cronberg T, Asztely F, Nemali S, Wieloch T. 2003. Mouse hip-pocampal organotypic tissue cultures exposed to in vitro ‘‘ischemia’’show selective and delayed CA1 damage that is aggravated by glu-cose. J Cereb Blood Flow Metab 23:23–33.
S�aez JC, Berthoud VM, Bra~nes MC, Mart�ınez AD, Beyer EC. 2003.Plasma membrane channels formed by connexins: Their regulationand functions. Physiol Rev 83:1359–1400.
S�aez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. 2005.Connexin-based gap junction hemichannels: Gating mechanisms. Bio-chim Biophys Acta 1711:215–224.
Schalper KA, Palacios-Prado N, Retamal MA, Shoji KF, Mart�ınez AD,S�aez JC. 2008. Connexin hemichannel composition determines theFGF-1-induced membrane permeability and free [Ca21]i responses.Mol Biol Cell 19:3501–3513.
Silver IA, Deas J, Erecinska M. 1997. Ion homeostasis in brain cells:Differences in intracellular ion responses to energy limitationbetween cultured neurons and glial cells. Neuroscience 78:589–601.
Silverman W, Locovei S, Dahl GP. 2008. Probenecid, a gout remedy,inhibits pannexin 1 channels. Am J Physiol Cell Physiol 295:C761–C767.
Simard M, Nedergaard M. 2004. The neurobiology of glia in the contextof water and ion homeostasis. Neuroscience 129:877–896.
Sugawara T, Lewen A, Noshita N, Gasche Y, Chan PH. 2002. Effects ofglobal ischemia duration on neuronal, astroglial, oligodendroglial,and microglial reactions in the vulnerable hippocampal CA1 subre-gion in rats. J Neurotrauma 19:85–98.
Tabernero A, Medina JM, Giaume C. 2006. Glucose metabolism andproliferation in glia: Role of astrocytic gap junctions. J Neurochem99:1049–1061.
Theis M, Mas C, Doring B, Kruger O, Herrera P, Meda P, Willecke K.2001. General and conditional replacement of connexin43-codingDNA by a lacZ reporter gene for cell-autonomous analysis of expres-sion. Cell Commun Adhes 8:383–386.
VanSlyke JK, Musil LS. 2005. Cytosolic stress reduces degradation ofconnexin43 internalized from the cell surface and enhances gap junc-tion formation and function. Mol Biol Cell 16:5247–5257.
Walton M, Connor B, Lawlor P, Young D, Sirimanne E, Gluckman P,Cole G, Dragunow M. 1999. Neuronal death and survival in two mod-els of hypoxic–ischemic brain damage. Brain Res Brain Res Rev29:137–168.
Wang J, Ma M, Locovei S, Keane RW, Dahl G. 2007. Modulation ofmembrane channel currents by gap junction protein mimetic pepti-des: Size matters. Am J Physiol Cell Physiol 293:C1112–C1119.
343ISCHEMIA AND HEMICHANNELS IN ASTROGLIAL DEATH
GLIA
Copyright © 2022 FDOKUMEN




















