
Hypothalamic lipotoxicity and the metabolic syndrome
12
Review Hypothalamic lipotoxicity and the metabolic syndrome Pablo B. Martínez de Morentin a,b , Luis Varela a,b , Johan Fernø a , Rubén Nogueiras a,b , Carlos Diéguez a,b , Miguel López a,b, ⁎ a Department of Physiology, School of Medicine, University of Santiago de Compostela-Instituto de Investigación Sanitaria, S. Francisco s/n, Santiago de Compostela (A Coruña), 15782, Spain b CIBER Fisiopatología de la Obesidad y Nutrición (CIBERobn), Spain abstract article info Article history: Received 29 June 2009 Received in revised form 16 September 2009 Accepted 17 September 2009 Available online 29 September 2009 Keywords: Endoplasmic reticulum (ER) stress Fatty acids Hypothalamus Lipid metabolism Lipid sensing Lipotoxicity Ectopic accumulation of lipids in peripheral tissues, such as pancreatic β cells, liver, heart and skeletal muscle, leads to lipotoxicity, a process that contributes substantially to the pathophysiology of insulin resistance, type 2 diabetes, steatotic liver disease and heart failure. Current evidence has demonstrated that hypothalamic sensing of circulating lipids and modulation of hypothalamic endogenous fatty acid and lipid metabolism are two bona fide mechanisms modulating energy homeostasis at the whole body level. Key enzymes, such as AMP-activated protein kinase (AMPK) and fatty acid synthase (FAS), as well as intermediate metabolites, such as malonyl-CoA and long-chain fatty acids-CoA (LCFAs-CoA), play a major role in this neuronal network, integrating peripheral signals with classical neuropeptide-based mechanisms. However, one key question to be addressed is whether impairment of lipid metabolism and accumulation of specific lipid species in the hypothalamus, leading to lipotoxicity, have deleterious effects on hypothalamic neurons. In this review, we summarize what is known about hypothalamic lipid metabolism with focus on the events associated to lipotoxicity, such as endoplasmic reticulum (ER) stress in the hypothalamus. A better understanding of these molecular mechanisms will help to identify new drug targets for the treatment of obesity and metabolic syndrome. © 2009 Elsevier B.V. All rights reserved. 1. Introduction Obesity and its related metabolic disorders are increasing at epidemic rate in the developed and developing world. The reasons for the increasing prevalence of obesity are multifaceted, but social and environmental factors in combination with genetic predisposition are leading to overall positive energy balance. The imbalanced energy homeostasis that elicits obesity can further lead to insulin resistance and type 2 diabetes, fatty liver and a range of other disorders, generally known as the metabolic syndrome [27,35,37,38,88,104,118], as well as several types of cancer [11]. Thus, controlling obesity should have a beneficial effect on all these harmful complications. For this reason, much effort is being placed on identifying the basic molecular mechanisms controlling energy balance. Although it is obvious that the development of obesity is caused by a positive energy balance, less evident is why the expansion of white adipose tissue (WAT) characteristic of obese individuals is strongly associated with insulin resistance and diabetes [42,140]. There are two main hypotheses, not necessarily exclusive, that attempt to explain this elusive link. On one hand, this may be the result of changes in the repertoire of adipocytokines (i.e., more leptin and resistin and less adiponectin) in a way that promotes insulin resistance [116]. The second hypothesis suggests that obesity leads to a failure of the adipose tissue expandability. Although the WAT displays a high degree of expandability, its storage capacity may become saturated, resulting in that excess fat is redirected to non- Biochimica et Biophysica Acta 1801 (2010) 350–361 Abbreviations: ACC, acetyl-CoA carboxylase; Acetyl-CoA, acetyl-coenzyme A; ACL, ATP citrate lyase; AgRP, agouti-related protein; AICAR, 5-aminoimidazole-4-carbox- yamide-1-β-D-ribofuranoside; AMPK, AMP-activated protein kinase; ARC, arcuate nucleus of the hypothalamus; ATF, activating transcription factor; CaMKK2, Ca2+/ calmodulin-dependent protein kinase kinase 2; CART, cocaine and amphetamine- regulated transcript; CHOP, C/EBP homologous protein; CNS, central nervous system; CPT1, carnitine palmitoyltransferase 1; CPT2, carnitine palmitoyltransferase 2; DMH, dorsomedial nucleus of the hypothalamus; eEF2, elongation factor 2; eIF2α, eukaryotic translation initiation factor 2 α; ER, endoplasmic reticulum; FAS, fatty acid synthase; FFAs, free fatty acids; HFD, high fat diet; ICV, intracerebroventricular; IRE1, inositol- requiring protein-1; LCFA, long-chain fatty acid; LCFACS, long-chain fatty acyl-CoA synthetase; LHA, lateral hypothalamic area; Malonyl-CoA, malonyl-coenzyme A; MCD, malonyl-CoA decarboxylase; MCFAs, medium chain fatty acids; MCH, melanin- concentrating hormone; mTOR, mammalian target of rapamycin; NEFA, non-esterified fatty acids; NPY, neuropeptide Y; NTS, nucleus of the solitary tract; OA, oleic acid; PERK, protein kinase RNA (PKR)-like ER; PGC1α, peroxisome proliferator-activated receptor coactivator 1 alpha; POMC, proopiomelanocortin; PPARα, peroxisomal proliferator- activated receptor alpha; PTP1B, protein tyrosine phosphatase 1B; PVH, paraventricular nucleus of the hypothalamus; RSTN, resistin; SOCS3, suppressor of cytokine signaling 3; SREBP, sterol response element-binding protein; STAT3, signal transducer and activator of transcription 3; TSC1 and TSC2, tuberous sclerosis complex genes 1 and 2; TG, triacylglycerol; TMX, tamoxifen; UCP3, uncoupling protein 3; UPR, unfolded protein response; VMH, ventromedial nucleus of the hypothalamus; WAT, white adipose tissue; Xbp-1, X-box binding protein 1 ⁎ Corresponding author. Department of Physiology, School of Medicine, University of Santiago de Compostela-Instituto de Investigación Sanitaria, S. Francisco s/n, Santiago de Compostela (A Coruña) 15782, Spain and CIBER “Fisiopatología de la Obesidad y Nutrición,” Instituto de Salud Carlos III, Santiago de Compostela, Spain. Tel.: +34 981 582658; fax: +34 981 574145. E-mail address: [email protected] (M. López). 1388-1981/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.bbalip.2009.09.016 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbalip
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Hypothalamic lipotoxicity and the metabolic syndrome

Biochimica et Biophysica Acta 1801 (2010) 350–361
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bba l ip
Review
Hypothalamic lipotoxicity and the metabolic syndrome
Pablo B. Martínez de Morentin a,b, Luis Varela a,b, Johan Fernø a, Rubén Nogueiras a,b,Carlos Diéguez a,b, Miguel López a,b,⁎a Department of Physiology, School of Medicine, University of Santiago de Compostela-Instituto de Investigación Sanitaria, S. Francisco s/n, Santiago de Compostela (A Coruña), 15782, Spainb CIBER Fisiopatología de la Obesidad y Nutrición (CIBERobn), Spain
Abbreviations: ACC, acetyl-CoA carboxylase; Acetyl-ATP citrate lyase; AgRP, agouti-related protein; AICARyamide-1-β-D-ribofuranoside; AMPK, AMP-activatednucleus of the hypothalamus; ATF, activating transcricalmodulin-dependent protein kinase kinase 2; CARTregulated transcript; CHOP, C/EBP homologous proteinCPT1, carnitine palmitoyltransferase 1; CPT2, carnitinedorsomedial nucleus of the hypothalamus; eEF2, elongatranslation initiation factor 2 α; ER, endoplasmic reticuFFAs, free fatty acids; HFD, high fat diet; ICV, intracererequiring protein-1; LCFA, long-chain fatty acid; LCFAsynthetase; LHA, lateral hypothalamic area; Malonyl-Comalonyl-CoA decarboxylase; MCFAs, medium chainconcentrating hormone; mTOR, mammalian target of rafatty acids; NPY, neuropeptide Y; NTS, nucleus of the soliprotein kinase RNA (PKR)-like ER; PGC1α, peroxisomecoactivator 1 alpha; POMC, proopiomelanocortin; PPAactivated receptor alpha; PTP1B, protein tyrosine phosphnucleus of the hypothalamus; RSTN, resistin; SOCS3, supSREBP, sterol response element-binding protein; STAT3,of transcription 3; TSC1 and TSC2, tuberous sclerosistriacylglycerol; TMX, tamoxifen; UCP3, uncoupling proresponse; VMH, ventromedial nucleus of the hypothtissue; Xbp-1, X-box binding protein 1⁎ Corresponding author. Department of Physiology, Sc
Santiago de Compostela-Instituto de Investigación Sanitde Compostela (A Coruña) 15782, Spain and CIBER “FNutrición,” Instituto de Salud Carlos III, Santiago de Com582658; fax: +34 981 574145.
E-mail address: [email protected] (M. López).
1388-1981/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.bbalip.2009.09.016
a b s t r a c t
a r t i c l e i n f oArticle history:Received 29 June 2009Received in revised form 16 September 2009Accepted 17 September 2009Available online 29 September 2009
Keywords:Endoplasmic reticulum (ER) stressFatty acidsHypothalamusLipid metabolismLipid sensingLipotoxicity
Ectopic accumulation of lipids in peripheral tissues, such as pancreatic β cells, liver, heart and skeletalmuscle, leads to lipotoxicity, a process that contributes substantially to the pathophysiology of insulinresistance, type 2 diabetes, steatotic liver disease and heart failure. Current evidence has demonstrated thathypothalamic sensing of circulating lipids and modulation of hypothalamic endogenous fatty acid and lipidmetabolism are two bona fide mechanisms modulating energy homeostasis at the whole body level. Keyenzymes, such as AMP-activated protein kinase (AMPK) and fatty acid synthase (FAS), as well asintermediate metabolites, such as malonyl-CoA and long-chain fatty acids-CoA (LCFAs-CoA), play a majorrole in this neuronal network, integrating peripheral signals with classical neuropeptide-based mechanisms.However, one key question to be addressed is whether impairment of lipid metabolism and accumulation ofspecific lipid species in the hypothalamus, leading to lipotoxicity, have deleterious effects on hypothalamicneurons. In this review, we summarize what is known about hypothalamic lipid metabolism with focus onthe events associated to lipotoxicity, such as endoplasmic reticulum (ER) stress in the hypothalamus. Abetter understanding of these molecular mechanisms will help to identify new drug targets for the treatmentof obesity and metabolic syndrome.
© 2009 Elsevier B.V. All rights reserved.
CoA, acetyl-coenzyme A; ACL,, 5-aminoimidazole-4-carbox-protein kinase; ARC, arcuateption factor; CaMKK2, Ca2+/, cocaine and amphetamine-; CNS, central nervous system;palmitoyltransferase 2; DMH,tion factor 2; eIF2α, eukaryoticlum; FAS, fatty acid synthase;broventricular; IRE1, inositol-CS, long-chain fatty acyl-CoAA, malonyl-coenzyme A; MCD,fatty acids; MCH, melanin-pamycin; NEFA, non-esterifiedtary tract; OA, oleic acid; PERK,proliferator-activated receptorRα, peroxisomal proliferator-atase 1B; PVH, paraventricularpressor of cytokine signaling 3;signal transducer and activatorcomplex genes 1 and 2; TG,tein 3; UPR, unfolded proteinalamus; WAT, white adipose
hool of Medicine, University ofaria, S. Francisco s/n, Santiagoisiopatología de la Obesidad ypostela, Spain. Tel.: +34 981
ll rights reserved.
1. Introduction
Obesity and its related metabolic disorders are increasing atepidemic rate in the developed and developing world. The reasonsfor the increasing prevalence of obesity aremultifaceted, but social andenvironmental factors in combination with genetic predisposition areleading to overall positive energy balance. The imbalanced energyhomeostasis that elicits obesity can further lead to insulin resistanceand type 2 diabetes, fatty liver and a range of other disorders, generallyknown as themetabolic syndrome [27,35,37,38,88,104,118], aswell asseveral types of cancer [11]. Thus, controlling obesity should have abeneficial effect on all these harmful complications. For this reason,much effort is being placed on identifying the basic molecularmechanisms controlling energy balance.
Although it is obvious that the development of obesity is caused bya positive energy balance, less evident is why the expansion of whiteadipose tissue (WAT) characteristic of obese individuals is stronglyassociated with insulin resistance and diabetes [42,140]. There aretwo main hypotheses, not necessarily exclusive, that attempt toexplain this elusive link. On one hand, this may be the result ofchanges in the repertoire of adipocytokines (i.e., more leptin andresistin and less adiponectin) in a way that promotes insulinresistance [116]. The second hypothesis suggests that obesity leadsto a failure of the adipose tissue expandability. Although the WATdisplays a high degree of expandability, its storage capacity maybecome saturated, resulting in that excess fat is redirected to non-
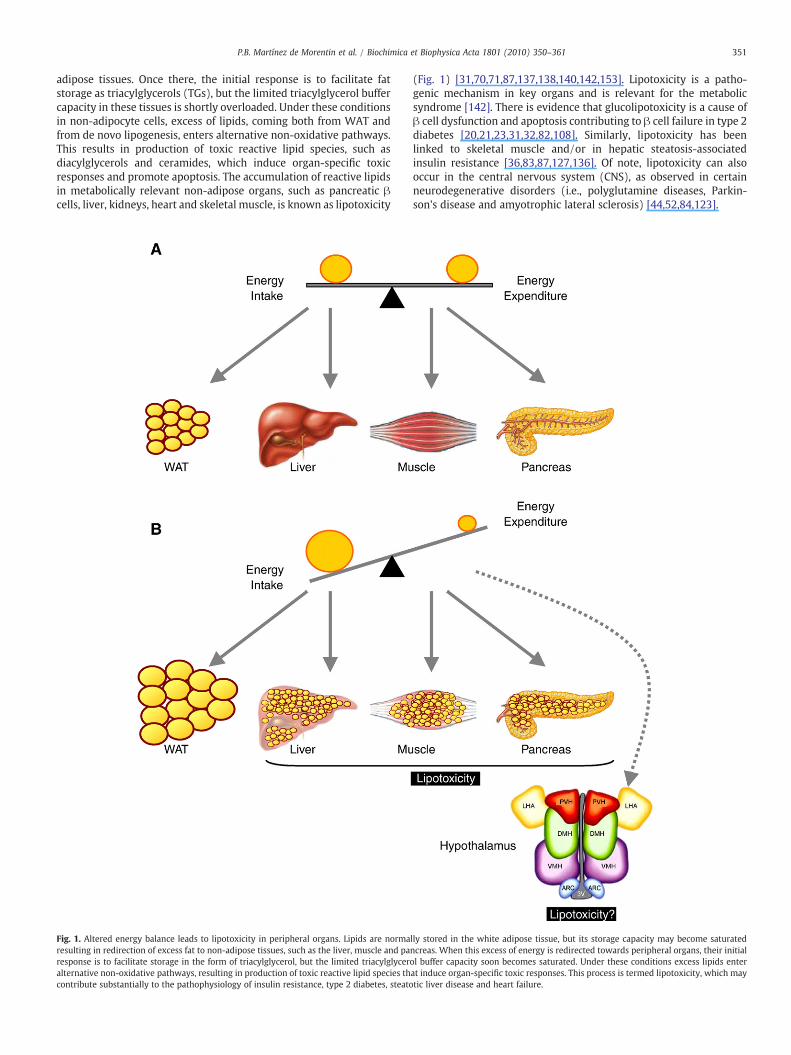
351P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
adipose tissues. Once there, the initial response is to facilitate fatstorage as triacylglycerols (TGs), but the limited triacylglycerol buffercapacity in these tissues is shortly overloaded. Under these conditionsin non-adipocyte cells, excess of lipids, coming both from WAT andfrom de novo lipogenesis, enters alternative non-oxidative pathways.This results in production of toxic reactive lipid species, such asdiacylglycerols and ceramides, which induce organ-specific toxicresponses and promote apoptosis. The accumulation of reactive lipidsin metabolically relevant non-adipose organs, such as pancreatic βcells, liver, kidneys, heart and skeletal muscle, is known as lipotoxicity
Fig. 1. Altered energy balance leads to lipotoxicity in peripheral organs. Lipids are normaresulting in redirection of excess fat to non-adipose tissues, such as the liver, muscle and paresponse is to facilitate storage in the form of triacylglycerol, but the limited triacylglyceralternative non-oxidative pathways, resulting in production of toxic reactive lipid species thcontribute substantially to the pathophysiology of insulin resistance, type 2 diabetes, steato
(Fig. 1) [31,70,71,87,137,138,140,142,153]. Lipotoxicity is a patho-genic mechanism in key organs and is relevant for the metabolicsyndrome [142]. There is evidence that glucolipotoxicity is a cause ofβ cell dysfunction and apoptosis contributing to β cell failure in type 2diabetes [20,21,23,31,32,82,108]. Similarly, lipotoxicity has beenlinked to skeletal muscle and/or in hepatic steatosis-associatedinsulin resistance [36,83,87,127,136]. Of note, lipotoxicity can alsooccur in the central nervous system (CNS), as observed in certainneurodegenerative disorders (i.e., polyglutamine diseases, Parkin-son's disease and amyotrophic lateral sclerosis) [44,52,84,123].
lly stored in the white adipose tissue, but its storage capacity may become saturatedncreas. When this excess of energy is redirected towards peripheral organs, their initialol buffer capacity soon becomes saturated. Under these conditions excess lipids enterat induce organ-specific toxic responses. This process is termed lipotoxicity, which maytic liver disease and heart failure.

352 P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
Bearing in mind that lipotoxicity can occur in the CNS, twoimportant questions arise, which remain to be fully addressed: (1)whether lipotoxicity takes place in the brain areas involved in thecontrol of energy balance, such as the hypothalamus, and, if this is thecase, (2) whether it has any pathophysiological relevance. Historical-ly, the role of lipid metabolism in the brain has been consideredminor. In fact, classically, neuroscience and metabolism books statedthat the brain uses just glucose as a fuel molecule or, alternatively,ketone bodies when energy is scarce, i.e., during starvation. Underthat “orthodox” view, lipids are considered just as structural andsignaling molecules in the CNS, with negligible or totally absent roleas metabolic fuel molecules. Despite this traditional dogma, recentevidence, mainly obtained during the last 10 years, has shown thatperipheral circulating lipids can be sensed by a particular brain area –
the hypothalamus – where lipid metabolism plays a major role in thecontrol of energy balance. Thus, a potential pathophysiological role oflipotoxicity in the hypothalamus is intriguing. The aim of this reviewis to summarize what is known about hypothalamic lipids and theregulation of energy homeostasis. Moreover, we will discuss whetherand why impaired lipid metabolism in the hypothalamus, potentiallyleading to neuronal lipotoxicity, may be a pathophysiologicalmechanism under alterations in energy balance. The information onthis particular field is very scarce, in fact so far there are just four veryrecent and very interesting papers first describing the existence ofendoplasmatic reticulum stress (ER stress, an event usually linked tolipotoxicity) in the hypothalamus [46,101,145,151]. These findingsmay open up a new field, possible identifying new pharmacologicaltargets for the treatment of obesity and associated comorbidities.
2. The hypothalamus plays a major role in the modulation ofenergy balance
The CNS receives multiple inputs of information about the levels ofenergy storage in the body. The hypothalamus is located under thethalamus, including the major portion of the ventral diencephalon.The hypothalamus consists of anatomically-defined neuronal clusters,or nuclei, that integrate both the control of energy expenditure andfeeding. The arcuate nucleus of the hypothalamus (ARC) is believed tobe the “master hypothalamic centre” for energy balance. Two distinctneuronal clusters in the ARC integrate peripheral nutritional/feedingsignals, such as glucose, leptin, ghrelin, adiponectin, resistin (RSTN)and insulin. One set of neurons expresses the orexigenic (feeding-promoting) neuropeptides agouti-related protein (AgRP) and neuro-peptide Y (NPY). These neurons mostly project to “second order”neurons located in other hypothalamic nuclei, such as the paraven-tricular nucleus (PVH). A second ARC population of neurons expressesthe anorexigenic (feeding inhibiting) products of proopiomelanocor-tin (POMC), the precursor of alpha-melanocyte stimulating hormone(α-MSH), and the cocaine and amphetamine-regulated transcript(CART). This set of neurons projects more broadly within the CNS, tosecondary hypothalamic nuclei such as the dorsomedial nucleus(DMH), the PVH and the lateral hypothalamic area (LHA), whichexpresses the orexigenic neuropeptides orexins (OXs) and melanin-concentrating hormone (MCH). Dorsal to the ARC lays the ventro-medial nucleus of the hypothalamus (VMH), which mainly receivesprojections from AgRP/NPY and CART/POMC neurons in the ARC. TheVMH neurons project their axons to the ARC, DMH and LHA, as well asbrainstem regions, such as the nucleus of the solitary tract (NTS) inthe brainstem. Hypothalamic nuclei respond to changes in energystatus by altering the expression of the orexigenic and anorexigenicneuromodulators, which elicits changes in energy intake andexpenditure. When energy intake surpasses expenditure, the expres-sion of orexigenic neuropeptides (such as AgRP, NPY, OXs and MCH)diminishes, and the expression anorexigenic neuropeptides (such asCART and POMC) increases. Reverse changes occur when energyexpenditure exceeds intake. For extensive revision of hypothalamic
mechanism regulating energy homeostasis, see the following reviews[24,25,27,37,39,78,80,93,94,96,104,118].
3. Lipid metabolism in the hypothalamus plays a major role in theregulation of energy balance
Despite the well established role of neuropeptides and peripheralsignals on the regulation of energy balance, current data unequivo-cally show that modulation of hypothalamic lipid metabolism is a keymechanism regulating energy homeostasis. Under lipogenic condi-tions, surplus glucose in the cell is first converted to pyruvate viaglycolysis in the cytoplasm. Pyruvate goes into the mitochondria andis converted to acetyl-CoA and transported as citrate from mitochon-dria into cytoplasm. ATP citrate lyase (ACL) then reconverts citrate toacetyl-CoA. Acetyl-CoA carboxylase (ACC) catalyzes the carboxylationof acetyl-CoA to malonyl-CoA in an ATP-dependent way. Acetyl-CoAand malonyl-CoA are then employed as the substrates for theproduction of palmitate by the seven-enzymatic reactions catalyzedby fatty acid synthase (FAS) (Fig. 2). The resulting saturated fatty acidmolecule can be desaturated to form unsaturated fatty acids, which inturn can be stored as triglyceride molecules or involved in thesynthesis of a range of phospholipids and derivatives for membraneand signaling functions. Alternatively, fatty acids can also be furthermetabolized, depending on cellular energy requirements[55,67,78,81]. Generally, fatty acids are degraded by β-oxidation inthe mitochondria. The fatty acid is first activated in the outermitochondrial membrane in a reaction catalyzed by long-chain fattyacyl-CoA synthetase (LCFACS). Next, it is translocated to themitochondrial matrix, by the action of carnitine palmitoyltransferase1 and 2 (CPT1 and CPT2), where β-oxidation occurs (Fig. 2)[55,67,78,81]. Malonyl-CoA is an intermediate in the biosynthesis offatty acids but also an important regulator of the balance between denovo lipogenesis and fatty acid oxidation. Levels of malonyl-CoAdepend on the equilibrium between the enzyme activities of ACC, FASandMCD,which are regulated by a common upstream kinase, namely,AMP-activated protein kinase (AMPK) (Fig. 2). Activated AMPKphosphorylates and inhibits ACC, whilst activating MCD, therebyreducing malonyl-CoA and the flux of substrates in the fatty acidbiosynthetic pathway [55,63,117]. On the other hand, AMPK regulatesFAS at transcriptional level, through a sterol response element-bindingprotein 1 (SREBP-1) dependent mechanism [76].
Data gleaned in the last decade have demonstrated thathypothalamic fatty acid metabolism plays a major role in themodulation of energy balance, affecting both feeding and energyexpenditure. Anatomical data showed that central enzymes in-volved in lipid metabolism, namely, AMPK, ACC, CPT1, FAS andMCD, are expressed at particularly high levels in all the hypotha-lamic nuclei ARC, DMH, PVH and VMH in rodents and humans[57,67,76,77,90]. Although morphological data suggested that thefatty acid biosynthetic pathway is important in hypothalamicneurons, the interest for a role of hypothalamic fatty acidmetabolism in energy balance did curiously not originate in thefields of neuroscience or metabolism, but in the field of oncology[62]. The finding that several types of tumors expressed elevatedlevels of FAS raised the possibility that inhibition of this enzymemight be a therapeutic target for cancer treatment [62]. Interest-ingly, treatments with FAS inhibitors, such as cerulenin and C75, ordrugs that downregulated FAS expression, such as tamoxifen (TMX),induced a remarkable weight loss and hypophagic effect [48,75,77].The anorectic effect of these drugs, especially C75, (1) requiresthe accumulation of malonyl-CoA in the hypothalamus, which maybe sensed as a signal of nutrient abundance by critical neuronsregulating food intake (“malonyl-CoA hypothesis”) [48,67,77],and (2) decreased expression of orexigenic (AgRP and NPY) andelevated expression of anorexigenic (CART, POMC) neuropeptidesin the ARC [41,67,75,77,125]. The molecular mechanisms of the FAS
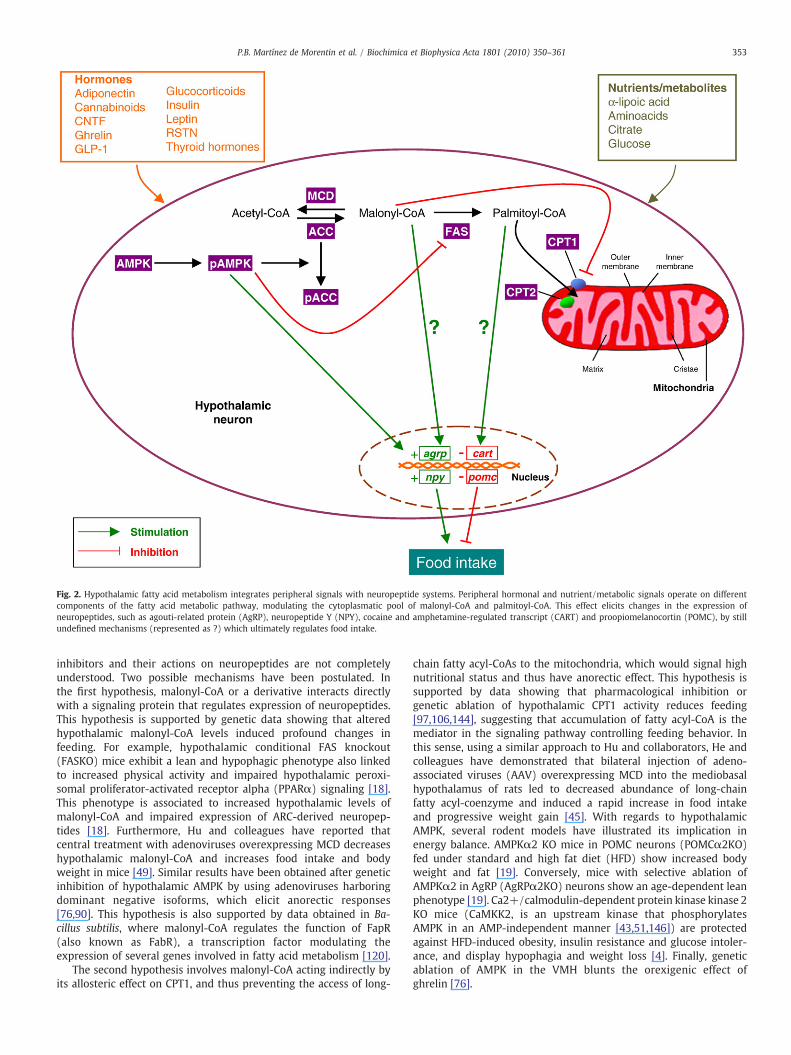
Fig. 2. Hypothalamic fatty acid metabolism integrates peripheral signals with neuropeptide systems. Peripheral hormonal and nutrient/metabolic signals operate on differentcomponents of the fatty acid metabolic pathway, modulating the cytoplasmatic pool of malonyl-CoA and palmitoyl-CoA. This effect elicits changes in the expression ofneuropeptides, such as agouti-related protein (AgRP), neuropeptide Y (NPY), cocaine and amphetamine-regulated transcript (CART) and proopiomelanocortin (POMC), by stillundefined mechanisms (represented as ?) which ultimately regulates food intake.
353P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
inhibitors and their actions on neuropeptides are not completelyunderstood. Two possible mechanisms have been postulated. Inthe first hypothesis, malonyl-CoA or a derivative interacts directlywith a signaling protein that regulates expression of neuropeptides.This hypothesis is supported by genetic data showing that alteredhypothalamic malonyl-CoA levels induced profound changes infeeding. For example, hypothalamic conditional FAS knockout(FASKO) mice exhibit a lean and hypophagic phenotype also linkedto increased physical activity and impaired hypothalamic peroxi-somal proliferator-activated receptor alpha (PPARα) signaling [18].This phenotype is associated to increased hypothalamic levels ofmalonyl-CoA and impaired expression of ARC-derived neuropep-tides [18]. Furthermore, Hu and colleagues have reported thatcentral treatment with adenoviruses overexpressing MCD decreaseshypothalamic malonyl-CoA and increases food intake and bodyweight in mice [49]. Similar results have been obtained after geneticinhibition of hypothalamic AMPK by using adenoviruses harboringdominant negative isoforms, which elicit anorectic responses[76,90]. This hypothesis is also supported by data obtained in Ba-cillus subtilis, where malonyl-CoA regulates the function of FapR(also known as FabR), a transcription factor modulating theexpression of several genes involved in fatty acid metabolism [120].
The second hypothesis involves malonyl-CoA acting indirectly byits allosteric effect on CPT1, and thus preventing the access of long-
chain fatty acyl-CoAs to the mitochondria, which would signal highnutritional status and thus have anorectic effect. This hypothesis issupported by data showing that pharmacological inhibition orgenetic ablation of hypothalamic CPT1 activity reduces feeding[97,106,144], suggesting that accumulation of fatty acyl-CoA is themediator in the signaling pathway controlling feeding behavior. Inthis sense, using a similar approach to Hu and collaborators, He andcolleagues have demonstrated that bilateral injection of adeno-associated viruses (AAV) overexpressing MCD into the mediobasalhypothalamus of rats led to decreased abundance of long-chainfatty acyl-coenzyme and induced a rapid increase in food intakeand progressive weight gain [45]. With regards to hypothalamicAMPK, several rodent models have illustrated its implication inenergy balance. AMPKα2 KO mice in POMC neurons (POMCα2KO)fed under standard and high fat diet (HFD) show increased bodyweight and fat [19]. Conversely, mice with selective ablation ofAMPKα2 in AgRP (AgRPα2KO) neurons show an age-dependent leanphenotype [19]. Ca2+/calmodulin-dependent protein kinase kinase 2KO mice (CaMKK2, is an upstream kinase that phosphorylatesAMPK in an AMP-independent manner [43,51,146]) are protectedagainst HFD-induced obesity, insulin resistance and glucose intoler-ance, and display hypophagia and weight loss [4]. Finally, geneticablation of AMPK in the VMH blunts the orexigenic effect ofghrelin [76].

354 P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
In addition to this pharmacological and genetic evidence,physiological data have shown that regulation of hypothalamic fattyacid metabolism is part of the adaptive changes observed duringphysiological regulation of feeding. Fasting stimulates hypothalamicAMPK and inhibits ACC and FAS activities, whereas refeeding inducesopposite changes [40,76,77,90]. In keeping with these observations,recent data have highlighted the critical physiological importance ofhypothalamic AMPK, ACC and FAS in the control of food intake.Anorectic hormones, such as leptin, insulin, glucagon-like peptide-1(GLP-1), CNTF and melanocortin receptors agonists, such as melano-tan II (MTII), inhibit hypothalamic AMPK activating ACC and/or FAS(Fig. 2) [5,40,90,95,121,129,143]. On the other hand, orexigenicsignals, such as cannabinoids, glucocorticoids, adiponectin, ghrelinand agouti-related protein (AgRP), activate AMPK and inhibit ACC andFAS (Fig. 2) [5,6,53,60,61,76,79,124]. Interestingly, hormonal-inducedalterations in hypothalamic AMPK, ACC and FAS function inducechanges in the hypothalamic concentration of malonyl-CoA, whichcorrelate with changes of neuropeptide expression, mainly in the ARC,such as AgRP, NPY and POMC (Fig. 2) [6,19,56,76,90,124]. Besideshormonal signals and neuropeptides, hypothalamic AMPK and ACCare modulated by nutrients and intermediate metabolites, includingglucose [17,86,90], α-lipoic acid [58], citrate [130] and lactate [15]; allof them inhibit feeding via inhibition of AMPK and activation of ACC.
Interestingly, in addition to food intake control, hypothalamic fattyacid metabolism modulates peripheral energy expenditure. Increasedhypothalamic malonyl-CoA stimulates mitochondrial biogenesis andoxidative gene expression in skeletal muscle via sympathetic nervoussystem [13,14,16,135]. Thus, inhibition of hypothalamic FAS inducesfatty acid oxidation and increases the expression of uncouplingprotein 3 (UCP3) and peroxisome proliferator-activated receptorcoactivator 1 alpha (PGC1α) and also the number of mitochondria inskeletal muscle [13,14,16,135]. Overall, these data indicate thathypothalamic lipid metabolism is a bona fide component of theenergy homeostatic system, integrating peripheral signals with thecentral mechanisms regulating energy balance. Furthermore, inaddition to the important role of endogenous lipid metabolism inthe hypothalamus, very recent and elegant evidence by Rossetti andcolleagues has demonstrated that peripheral lipids exert a keysignaling role on hypothalamic neurons and, very importantly, thatimpairment of this process could be a pathophysiological cause ofobesity.
4. Lipid sensing in the hypothalamus
Circulating nutrients are derived either exogenously (via foodintake) or endogenously (via hepatic production and adipocytelipolysis). Plasma long-chain fatty acids (LCFAs) are mainly boundto albumin and cross the blood–brain barrier (BBB) mostly by simplediffusion of the unbound form. A small proportion of fatty acids enterthe brain via straight uptake of lipoprotein particles mediated bylipoprotein receptor on the luminal surface of the cerebrovascularendothelium [65,110,112]. In general, the rate of entry of fatty acidsinto the brain is relative to its plasma concentration [89,111]. Aftercrossing the BBB, LCFAs are esterified to LCFAs-CoAs, a reactioncatalyzed by the enzyme long-chain fatty acyl-CoA synthetase(LCFACS). This takes place inside neurons and glial cells, where theLCFAs-CoAs are metabolized for use in lipid biosynthesis and, to amuch lesser extent, within oxidative pathways, such as β-oxidationvia CPT1 [65,89].
One of the early macronutrient-related hypotheses attempting toexplain the link between peripheral signals and CNS-controlledregulation of food intake was the lipostatic hypothesis [12,85]. Thismodel proposed that circulating lipids were generated in proportionto body fat stores or feeding status and that these signals could informthe brain about the nutritional status, which would induce adaptivehomeostatic changes in energy intake and expenditure [12]. Even
with the discovery of leptin [152] and its receptors [132], as well as abroad range of other adipocyte-derived hormonal regulators of foodintake, known as adipocytokines [1,59,134], the underlying idea of thelipostatic hypothesis was never fully ruled out. In fact, the hypothesisof CNS-regulated metabolic control via nutritional factors is at thistime enjoying resurgence, complementing and enhancing the studiesthat have been carried out on hormonal factors.
The first evidence showing a role for peripheral lipids in themodulation of energy balance came from the seminal work of Rossettiand colleagues. They demonstrated that central administration ofLCFAs exerted a signaling role within specific hypothalamic energycenters. Indeed, intracerebroventricular (ICV) administration of oleicacid (OA, C18:1) inhibited food intake [91,98], an effect that was notreproduced by medium chain fatty acids (MCFAs), such as octanoicacid (C8) [98]. These data indicate that accumulation of lipids in thehypothalamus is sensed as a signal of energy surplus. Of note, octanoicacid does not require CPT1 for entry in the mitochondria for β-oxidation, indicating the importance of this step in LCFA-mediatedhypothalamic regulation of feeding [98]. Equally important, theesterification of LCFAs to LCFA-CoA is an obligatory step for theanorexigenic action of LCFAs in the hypothalamus [64,65]. Theanorectic effects of OA are mainly exerted in the ARC where AgRPand NPY mRNA expression was shown to be decreased after OAtreatment [91,98] and they are also dependent of glucose levels[69,141]. One of the most interesting conclusions of the work fromRossetti and colleagues is the fact that besides its role on feedingcontrol, hypothalamic lipid sensing also appears to be required forglucose homeostasis in the liver. Elevation in hypothalamic levels ofLCFA, by ICV administration of OA [97] or inhibition of CPT1 [98,107],results in a marked decrease in hepatic glucose production that occursvia decreased glycogenolysis. Similar to the hypothalamic anorexi-genic signal, esterification of hepatic LCFAs to LCFA-CoA is required forthis metabolic effect to take place [64,65]. In addition, intact vagusnerve efferences between hypothalamus and liver and centralactivation of ATP-sensitive potassium channels (KATP) are required[64,105]. The physiological relevance of these data is unclear. It isknown that circulating LCFAs can directly act on hepatic glucoseproduction by stimulating gluconeogenesis [66]. Thus, it has beenpostulated that the central action of LCFAs decreasing glycogenolysismay be required to counteract the direct action of LCFAs stimulatinghepatic gluconeogenesis. This could control hepatic glucose produc-tion in response to an increase in systemic availability of lipids[64,105]
Whether alterations in the lipid sensing mechanism are patho-physiological mechanisms leading to obesity and comorbidities is stillunclear. However, it is worth mentioning that (1) impairment of thiscentral nutrient-sensing pathway is sufficient to disrupt energyhomeostasis and induce obesity and (2) the anorectic response toOA depends on the nutritional state being suppressed by short-termoverfeeding [91,106]. In fact, the hypothalamic responses to LCFAsdisappear in overfed animals, which did not show changes either inAgRP or NPY after OA treatment [91,106]. Bearing in mind these data,it is conceivable that in hyperphagic and obese states there may be aresistance to satiety effects of LCFAs and a desensitization of the AgRPand NPY responses to circulating fatty acids, which contributes tobody weight gain. Interestingly, inhibition of CPT1 activity restoreslipid sensing, normalizing the hypothalamic levels of LCFA-CoAs andmarkedly inhibited feeding behavior and hepatic glucose fluxes inoverfed rats [106]. It is clear that in this model central inhibition oflipid oxidation is sufficient to restore hypothalamic lipid sensing, aswell as glucose and energy homeostasis, and may be an effectiveapproach to the treatment of diet-induced obesity and insulinresistance. Nevertheless, before this can be carried out in a clinicalsetting the molecular underpinnings of this event, which are partiallyunsolved, must be understood. Some lines of evidence suggest thatreactive oxygen species (ROS) could be one of the mitochondrial

355P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
effectors implied in both the hormonal [6] and nutrient sensing at thelevel of the hypothalamus [9]. This is a very interesting idea but itnecessarily implies the presence of mechanisms downstream of β-oxidation. An alternative hypothesis may be that excessive accumu-lation of LCFAs in hypothalamic neurons might be redirected to non-oxidative pathways, producing non-esterified fatty acids (NEFA),which would result in the production of toxic reactive lipid species,which might cause endoplasmic reticulum stress (ER) and neuronallipotoxic effects, affecting the mechanisms of energy homeostasis.
5. Hypothalamic endoplasmic reticulum stress is apathophysiological mechanism leading to impairedenergy balance
One of the mechanisms by which lipids can be toxic is throughthe induction of ER stress. The term ER stress refers to thealterations of the protein-folding functionality of the ER[8,31,31,47,72,100,114,115,149]. In addition to being the site ofsynthesis of secretory proteins and resident proteins, the ER alsoparticipates in diverse functions such as lipid biosynthesis, metab-olism, oligosaccharide synthesis, calcium storage and drug detoxi-
Fig. 3. A model illustrating how prolonged and unresolved ER stress disrupts lipid homephysiological ER stress, cells respond by activating the unfolded protein response (UPR). Teukaryotic translation initiation factor 2α (eIF2α) reduces cellular stress (left side). Howeveexpression and/or phosphorylation of eIF2α can result in increased expression of lipogenicmetabolic changes induced by ER stress can lead to the development of metabolic syndrom
fication [31,31,72,100,114,115]. In eukaryotic cells, most secretedand transmembrane proteins fold and mature in the lumen of theER in a process that requires the unfolded protein response (UPR).Three different classes of UPR transducers have been identified: (1)inositol-requiring protein-1 (IRE1), (2) activating transcriptionfactor-6 (ATF6) and (3) protein kinase RNA (PKR)-like ER kinase(PERK) [31,31,72,100,114,115] (Fig. 3). Activation of these pathwaysas part of the ER stress response (by accumulation of unfoldedproteins in the lumen of the ER) results in a coordinatedtranscriptional response associated to attenuation of proteinsynthesis and upregulation of ER folding machinery (ER chaperonesproteins, such as GRP78, GRP94 and ERp72) and degradation ofirreversibly misfolded proteins [31,31,72,100,114,115]. If this UPRadaptive response is not sufficient to resolve the protein-foldingdefect, ER dysfunction can lead to cell dysfunction and ultimately toapoptotic cell death [31,31,72,114,115].
Interestingly, there is evidence that ER stress occurs underconditions of overnutrition in both peripheral tissues and the CNS[46,101,151]. Free fatty acids (FFAs) and hyperglycemia in β cellspromote activation of the UPR, leading to decreased mRNA insulinexpression and inhibition in the insulin signaling [29,32,73,74]. FFAs
ostasis and results in the development of metabolic syndrome. Under conditions ofhe subsequent increase in the expression of ER chaperones and dephosphorylation ofr, if UPR signaling is impaired (right side), extended C/EBP homologous protein (CHOP)genes, as well as decreased expression of genes required for fatty acid oxidation. Thesee hallmarks, such as hepatic steatosis.

356 P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
and glucose have been involved in PERK-mediated eukaryotictranslation initiation factor 2α (eIF2α) phosphorylation andIRE1α-mediated X-box binding protein 1 (Xbp-1) mRNA splicingin vivo and in isolated rat islets [22,29,32,33,73,74], adipose tissueof obese models in rodents and human biopsies [10,122]. The typeof lipids involved and particularly their degree of saturationseems to be relevant, with saturated fat being particularly proneto promote higher UPR in β cells inducing Xbps, C/EBP homologousprotein (CHOP, also known as GADD153) and GRP78 expression[21,29,32,73,74]. CHOP is a member of the C/EBP family oftranscription factors [100,113] and it is known to inhibit adipocytedifferentiation in response to metabolic stress [131]. Chop geneinduction is preferentially regulated through the PERK/eIF2α/ATF4UPR pathway, although the IRE1α/Xbp-1 and ATF6α pathways havealso been implicated [31,31,72,114,115]. There is compellingevidence showing that impaired CHOP function in peripheral tissuesinduces alterations in energy homeostasis. Female CHOPKO micedevelop obesity; however, no clear mechanisms involving eitherfood intake or thermogenesis have been defined [7]. HFD feedingresulted in obesity in female CHOPKO and heterozygous mice,although caloric intake did not differ from that of wild-type. Fat cellarea increased in mesenteric fat but not in subcutaneous fat inCHOPKO mice fed under a HFD [7]. There is evidence that UPRpromotes CHOP expression in other systems such as promoting βcell apoptosis and diabetes [50,68,99,150]. Further evidence indi-cates that genetic ablation of CHOP provides an advantage to the βcell by preventing oxidative damage and promoting β cell survival[128]. In the context of the CNS, induction of CHOP has been relatedto neuronal toxicity and death in diverse neurodegenerativediseases [103,119,126]. Thus, on the basis of these data, it istempting to speculate that CHOP could be a mediator or ER stress inthe hypothalamus after selective lipid damaging. Further work willbe necessary to address this hypothesis.
Current data also link inflammation to hypothalamic ER stressand leptin resistance. Four recent seminal, challenging and elegantpapers from three independent groups have demonstrated thatobesity and overnutrition elicit ER stress and initiate UPR signalingpathways in the hypothalamus, which in turn led to inhibition ofleptin receptor signaling and leptin resistance (Fig. 4)[46,101,151].Zhang et al. demonstrated that IKKβ/NF-κB, a well-known mediatorof metabolic inflammation, is highly expressed in hypothalamicneurons, although it normally remains inactive [151]. Interestingly,chronic (dietary or genetic obesity) or acute (glucose or oleic acidexposure) overnutrition activates hypothalamic IKKβ/NF-κBthrough, at least in part, elevated hypothalamic ER stress, whichleads to impaired insulin and leptin signaling through a mechanismpartially mediated by suppressor of cytokine signaling (SOCS3), aninhibitor of insulin and leptin signaling. Experimental activation ofthe IKKβ/NF-κB signaling pathway in the hypothalamus, throughvirus-mediated introduction of a constitutively active IKKβ, leads toweight gain and increased food intake alongside remarkablyimpaired insulin and leptin signaling in the hypothalamus in micefed under HFD. Furthermore, the authors demonstrate thatsuppression of IKKβ/NF-κB signaling in the MBH protects againstinsulin and leptin resistance, and obesity [151]. Finally, Zhang et al.demonstrate that obesity-induced ER stress in the hypothalamusdue to diet could also be suppressed by administration oftauroursodeoxycholic acid (TUDCA), a chemical chaperone thatimproves the protein-folding capacity, which resulted in reducedfood intake and decreased body weight [151]. Altogether, these datashow that the hypothalamic IKKβ/NF-κB program is a generalneural mechanism for energy balance underlying obesity andsuggest that suppressing hypothalamic IKKβ/NF-κB may representa strategy to combat obesity and related diseases and maybehypothalamic lipotoxicity. In the other two studies Hosoi et al. andOzcan et al. described that ER stress markedly inhibited leptin-
induced signal transducer and activator of transcription 3 (STAT3)phosphorylation and that ER stress-induced leptin resistance wasmediated through protein tyrosine phosphatase 1B (PTP1B) but,contrary to Zhang et al., not through SOCS3 [46,101]. It isnoteworthy that both studies showed that treatment with chemicalchaperones, such as 4-phenyl butyric acid (PBA) and TUDCA, notonly rescued ER stress in the CNS and reduced body weight but alsoacted as leptin sensitizing agents, dramatically improving ER stress-induced leptin resistance [46,101]. In keeping with these observa-tions, a very recent paper has shown that central administration ofthapsigargin, an ER stress inducer, inhibits the anorexigenic effectsof leptin and insulin [145].
Despite the fact that these seminal papers have added a newdimension to the complexity of hypothalamic control of energyhomeostasis, several important questions remain to be addressed inthe forthcoming years. Firstly, it would be important to know howthe brain senses metabolic surplus and how this can trigger ERstress response; for this reason, the contribution of other potentialUPR transducers, as well as other inflammatory signaling molecules,needs to be examined. Furthermore, it is critical to know whetherand how endogenous and/or exogenous lipid species can lead tolipotoxicity-induced hypothalamic ER stress. Very recent in vitrodata have demonstrated that exposure of high levels of palmiticacid induces lipotoxicity in rat cortical cells and triggers a strongcell death apoptotic response through a mechanism that includesgeneration of ROS, alterations in the mitochondrial transmembranepotential and increase in the mRNA levels of key cell death/survivalregulatory genes [2]. Secondly, it is necessary to properly dissect theER stress-mediated mechanism under the development of leptinresistance. The observation that chemical chaperones, such as PBAand TUDCA, act as leptin-sensitizing agents, ameliorating leptinresistance and reversing the diet-induced obesity phenotype[46,101,151], may open a novel therapeutic target for obesity.Thirdly, it would be interesting to address whether hypothalamic ERstress is a phenomenon affecting the whole hypothalamus orrestricted to key hypothalamic nuclei/cell clusters. Finally, it wouldbe necessary to understand how ER stress affects hypothalamicnetworking, modulating energy balance. The most straightforwardoption is that ER stress and impaired protein folding elicits thealteration of the synthesis and release of hypothalamic neuropep-tides. However, other options, such as impaired metabolic routes ortransduction cascades, should also be considered. Among these, oneinteresting target could be hypothalamic AMPK [55,63,117]. In thissense, recent work in cardiomyocytes has shown that AMPKactivation contributes to protection of the heart against hypoxicinjury through reduction of ER stress, suggesting that attenuation ofprotein synthesis via eEF2 inactivation may be the mechanism ofcardioprotection by AMPK [133]. Considering the raising knowledgeof hypothalamic AMPK in the modulation of whole body energyhomeostasis [55,63,117], a potential link should be considered.Another interesting candidate that can be affected by ER stresscould be the kinase known as mammalian target of rapamycin(mTOR), which integrates different sensory inputs to regulateprotein synthesis rates in individual cells, and it has recently beenimplicated in the CNS to regulate food intake and body weight[26–28,109,147]. Given that both nutrients and inflammatorysignaling could engage the mTOR pathway, this molecule is also apotential player in obesity-induced ER stress in the CNS. Actually,current results demonstrate that ER stress is a critical component ofthe pathologies associated with dysregulated mTOR activity, and theevidence is as follows. To begin with, in a role distinct from theregulation of energy homeostasis, mTOR modulates protein trans-lation after ER stress induced by human cytomegalovirus (HCMV)[3] or choinic hepatitis B virus (HBV) infections [148]. Data obtainedin recent years have demonstrated that mTOR plays a major role inthe translational control of gene expression during hypoxia- or

Fig. 4. Model of hypothalamic IKKβ/NF-κB and ER stress in metabolic syndrome. Chronic overnutrition activates hypothalamic IKKβ/NF-κB through, at least in part, elevatedhypothalamic ER stress, leading to impaired insulin and leptin signaling. Treatment with chemical chaperones, such as 4-phenyl butyric acid (PBA) and tauroursodeoxycholic acid(TUDCA), reduced ER stress in the CNS, lowered body weight and also acted as leptin sensitizing agents, dramatically improving ER stress-induced leptin resistance. On the otherhand, administration of thapsigargin, an ER stress inducer, inhibits the anorexigenic effects of leptin and insulin. Whether hypothalamic lipotoxicity elicits a similar ER stressresponse will require further research.
357P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
oxidative-induced ER stress, in both normal and cancer cells[34,54,139]. In the CNS, some cues can be found in the tuberoussclerosis complex (TSC), a neurogenetic disorder caused by loss-of-function mutations in either the TSC1 or TSC2 genes that frequentlyresults in prominent CNS-related symptoms, including epilepsy,mental retardation and autism spectrum disorder [30]. Very recentdata have demonstrated that loss TSC1 or TSC2 leads to constitutivemTOR activation in cell lines and mouse or human tumors andcauses ER stress and activation of UPR [102]. In addition,hippocampal Tsc2-deficient neurons and brain lysates from a Tsc1-deficient mice show elevated ER stress, indicated by increased
expression of CHOP; of interest this effect is completely reversed bythe mTOR inhibitor rapamycin both in vitro and in vivo [30]. Theseobservations indicate that ER stress modulates mTOR activity inneurons through the TSC protein complex and that ER stress iselevated in cells lacking this complex. Whether the TSC–mTOR axismediates ER stress in the hypothalamus needs to be clarified. Ofnote, a very recent paper has pointed in this direction, showing thathypothalamic Rip-TSC1KO mice developed hyperphagia and obesity,suggesting that hypothalamic disruption of Tsc1 may dysregulatefeeding circuits via mTOR activation. Indeed, Rip-TSC1KO micedisplayed increased mTOR signaling and enlarged neuron cell size in

358 P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
a number of hypothalamic populations, including POMC neurons.Furthermore, Tsc1 deletion with Pomc/Cre POMC-TSC1KO miceresulted in dysregulation of POMC neurons and hyperphagicobesity. Treatment with the mTOR inhibitor, rapamycin, amelioratedthe hyperphagia, obesity and the altered POMC neuronal morphol-ogy in developing or adult POMC-TSC1KO mice [92]. Thus, thispaper unequivocally demonstrates that mTOR activation in POMCneurons blocks the catabolic function of these cells to promotenutrient intake and increased adiposity. Whether these effects arelinked to specific changes in ER stress tone would need furtherresearch, but considering the link between both facts, this link isquite appealing.
6. Concluding remarks
More than 50 years ago, it was proposed that the CNS sensedcirculating levels of metabolites such as glucose, lipids and amino acidsand that feedingwasmodified according to the levels of thosemolecules.This led to the formulation of the glucostatic, lipostatic and aminostatichypotheses [12,85]. It has taken almost that time to demonstrate thatcirculating LCFAs act as nutrient surplus signals in the hypothalamus[64,69,91,98,105,141]. Pharmacological, genetic and physiological evi-dence has clearly shown that endogenous hypothalamic lipid metab-olism is a bona fide mechanism, modulating energy balance at thewhole body level [5,6,15,17–19,40,45,48,49,53,55,57,58,60,61,75,77–79,86,90,95,97,106,121,124,125,129,130,143,144]. Although these dataclearly support a role for the hypothalamic lipid homeostasis in thecontrol of energy balance, there are two important issues that remainto be addressed. (1) Can alterations in hypothalamic lipid metabolismimpair energy homeostasis and lead to obesity and metabolicsyndrome? (2) Which is the molecular nature of those alterations?Ectopic accumulation of lipids in peripheral tissues, such as pancreaticβ cells, liver, heart and skeletal muscle, leads to lipotoxicity, a processthat contributes substantially to the pathophysiology of insulinresistance, type 2 diabetes, steatotic liver disease and heart failure[31,70,71,87,137,138,140,142,153]. (3) Could hypothalamic lipotoxi-city play a role in the pathophysiology of metabolic syndrome? Somecues point towards that this may the case. For example, the fourseminal and elegant papers previously described above have demon-strated that one of the main mechanisms how lipids can be toxic, theinduction of ER stress, is a general neural mechanism for energyhomeostasis, underlying obesity and, importantly, leptin resistance[46,101,145,151]. However, this potential link between lipotoxicityand ER stress in the hypothalamus needs to be demonstrated.Remarkably, these studies also show that pharmacological manipula-tion of ER stress may be an ideal target for obesity treatment, acting asleptin-sensitizer [46,101,145,151]. This observation can be crucial inthe approaching strategies for drug design and targeting ER stresscould be a useful therapeutic strategy to prevent or even revert obesityand its metabolic complications in humans.
Overall, the presented evidence suggests that hypothalamic ERstress could be an important pathogenic mechanism leading tosome of the diverse manifestations of the metabolic syndrome.Furthermore, these data could represent a connection between ERstress and lipotoxicity. Our challenge for the years to come will beto validate this hypothesis and to identify the molecular effectorslinking hypothalamic lipotoxicity, ER stress and the classical path-ways involved on energy homeostasis. Moreover, when consideringthe concept of hypothalamic lipotoxicity it is necessary toinvestigate whether specific nutrients influence the developmentof ER stress in the hypothalamus and how this can associate withthe severity of metabolic disturbances. We believe that a betterunderstanding of these metabolic networks will help in thedevelopment of more rational and powerful drugs against obesityand related disorders.
Acknowledgments
This work has been supported by grants from Xunta de Galicia(C.D.: PGIDIT06PXIB208063PR and M.L.: GRC2006/66), FondoInvestigationes Sanitarias (M.L.: PI061700), Ministerio de Educaciony Ciencia (C.D.: BFU2005; M.L.: RyC-2007-00211), European Union(R.N., C.D. and M.L.: Health-F2-2008-223713: “Reprobesity”). CIBERde Fisiopatología de la Obesidad y Nutrición is an initiative of ISCIII.
References
[1] R.S. Ahima, J.S. Flier, Adipose tissue as an endocrine organ, Trends Endocrinol.Metab. 11 (2000) 327–332.
[2] F.G. Almaguel, J.W. Liu, F.J. Pacheco, C.A. Casiano, M. De Leon, Activation andreversal of lipotoxicity in PC12 and rat cortical cells following exposure topalmitic acid, J. Neurosci. Res. 87 (2009) 1207–1218.
[3] J.C. Alwine, Modulation of host cell stress responses by human cytomegalovirus,Curr. Top. Microbiol. Immunol. 325 (2008) 263–279.
[4] K.A. Anderson, T.J. Ribar, F. Lin, P.K. Noeldner, M.F. Green, M.J. Muehlbauer, L.A.Witters, B.E. Kemp, A.R. Means, Hypothalamic CaMKK2 contributes to theregulation of energy balance, Cell Metab. 7 (2008) 377–388.
[5] U. Andersson, K. Filipsson, C.R. Abbott, A. Woods, K. Smith, S.R. Bloom, D. Carling,C.J. Small, AMP-activated protein kinase plays a role in the control of food intake,J. Biol. Chem. 279 (2004) 12005–12008.
[6] Z.B. Andrews, Z.W. Liu, N. Walllingford, D.M. Erion, E. Borok, J.M. Friedman, M.H.Tschop, M. Shanabrough, G. Cline, G.I. Shulman, A. Coppola, X.B. Gao, T.L.Horvath, S. Diano, UCP2 mediates ghrelin's action on NPY/AgRP neurons bylowering free radicals, Nature 454 (2008) 846–851.
[7] Y. Ariyama, H. Shimizu, T. Satoh, T. Tsuchiya, S. Okada, S. Oyadomari, M. Mori, M.Mori, Chop-deficient mice showed increased adiposity but no glucose intoler-ance, Obesity 15 (2007) 1647–1656 (Silver. Spring).
[8] S. Basseri, R.C. Austin, ER stress and lipogenesis: a slippery slope toward hepaticsteatosis, Dev. Cell 15 (2008) 795–796.
[9] A. Benani, S. Troy, M.C. Carmona, X. Fioramonti, A. Lorsignol, C. Leloup, L.Casteilla, L. Penicaud, Role for mitochondrial reactive oxygen species in brainlipid sensing: redox regulation of food intake, Diabetes 56 (2007) 152–160.
[10] G. Boden, X. Duan, C. Homko, E.J. Molina, W. Song, O. Perez, P. Cheung, S. Merali,Increase in endoplasmic reticulum stress-related proteins and genes in adiposetissue of obese, insulin-resistant individuals, Diabetes 57 (2008) 2438–2444.
[11] E.E. Calle, R. Kaaks, Overweight, obesity and cancer: epidemiological evidenceand proposed mechanisms, Nat. Rev. Cancer 4 (2004) 579–591.
[12] L.A. Campfield, F.J. Smith, P. Burn, The OB protein (leptin) pathway—a linkbetween adipose tissue mass and central neural networks, Horm. Metab. Res. 28(1996) 619–632.
[13] S.H. Cha, Z. Hu, S. Chohnan, M.D. Lane, Inhibition of hypothalamic fatty acidsynthase triggers rapid activation of fatty acid oxidation in skeletal muscle, Proc.Natl. Acad. Sci. U. S. A. 102 (2005) 14557–14562.
[14] S.H. Cha, Z. Hu, M.D. Lane, Long-term effects of a fatty acid synthase inhibitor onobese mice: food intake, hypothalamic neuropeptides, and UCP3, Biochem.Biophys. Res. Commun. 317 (2004) 301–308.
[15] S.H. Cha, M.D. Lane, Central lactate metabolism suppresses food intake via thehypothalamic AMP kinase/malonyl-CoA signaling pathway, Biochem. Biophys.Res. Commun. 386 (2009) 212–216.
[16] S.H. Cha, J.T. Rodgers, P. Puigserver, S. Chohnan, M.D. Lane, Hypothalamicmalonyl-CoA triggers mitochondrial biogenesis and oxidative gene expression inskeletal muscle: role of PGC-1{alpha}, Proc. Natl. Acad. Sci. U. S. A. 103 (2006)15410–15415.
[17] S.H. Cha, M. Wolfgang, Y. Tokutake, S. Chohnan, M.D. Lane, Differential effects ofcentral fructose and glucose on hypothalamic malonyl-CoA and food intake,Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 16871–16875.
[18] M.V. Chakravarthy, Y. Zhu, M. López, L. Yin, D.W. Wozniak, T. Coleman, Z. Hu, M.Wolfgang, A. Vidal-Puig, M.D. Lane, C.F. Semenkovich, Brain fatty acid synthaseactivates PPAR-alpha to maintain energy homeostasis, J. Clin. Invest. 117 (2007)2539–2552.
[19] M. Claret, M.A. Smith, R.L. Batterham, C. Selman, A.I. Choudhury, L.G. Fryer, M.Clements, H. Al Qassab, H. Heffron, A.W. Xu, J.R. Speakman, G.S. Barsh, B. Viollet,S. Vaulont, M.L. Ashford, D. Carling, D.J. Withers, AMPK is essential for energyhomeostasis regulation and glucose sensing by POMC and AgRP neurons, J. Clin.Invest. 117 (2007) 2325–2336.
[20] M. Cnop, Fatty acids and glucolipotoxicity in the pathogenesis of type 2 diabetes,Biochem. Soc. Trans. 36 (2008) 348–352.
[21] M. Cnop, J.C. Hannaert, A. Hoorens, D.L. Eizirik, D.G. Pipeleers, Inverserelationship between cytotoxicity of free fatty acids in pancreatic islet cellsand cellular triglyceride accumulation, Diabetes 50 (2001) 1771–1777.
[22] M. Cnop, L. Ladriere, P. Hekerman, F. Ortis, A.K. Cardozo, Z. Dogusan, D. Flamez,M. Boyce, J. Yuan, D.L. Eizirik, Selective inhibition of eukaryotic translationinitiation factor 2 alpha dephosphorylation potentiates fatty acid-inducedendoplasmic reticulum stress and causes pancreatic beta-cell dysfunction andapoptosis, J. Biol. Chem. 282 (2007) 3989–3997.
[23] M. Cnop, N. Welsh, J.C. Jonas, A. Jorns, S. Lenzen, D.L. Eizirik, Mechanisms ofpancreatic beta-cell death in type 1 and type 2 diabetes: many differences, fewsimilarities, Diabetes 54 (Suppl. 2) (2005) S97–S107.

359P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
[24] A.P. Coll, I.S. Farooqi, S. O'Rahilly, The hormonal control of food intake, Cell 129(2007) 251–262.
[25] R. Coppari, G. Ramadori, J.K. Elmquist, The role of transcriptional regulators incentral control of appetite and body weight, Nat. Clin. Pract. Endocrinol. Metab. 5(2009) 160–166.
[26] D. Cota, E.K. Matter, S.C. Woods, R.J. Seeley, The role of hypothalamic mammaliantarget of rapamycin complex 1 signaling in diet-induced obesity, J. Neurosci. 28(2008) 7202–7208.
[27] D. Cota, K. Proulx, R.J. Seeley, The role of CNS fuel sensing in energy and glucoseregulation, Gastroenterology 132 (2007) 2158–2168.
[28] D. Cota, K. Proulx, K.A. Smith, S.C. Kozma, G. Thomas, S.C. Woods, R.J. Seeley,Hypothalamic mTOR signaling regulates food intake, Science 312 (2006)927–930.
[29] D.A. Cunha, P. Hekerman, L. Ladriere, A. Bazarra-Castro, F. Ortis, M.C. Wakeham,F. Moore, J. Rasschaert, A.K. Cardozo, E. Bellomo, L. Overbergh, C. Mathieu, R.Lupi, T. Hai, A. Herchuelz, P. Marchetti, G.A. Rutter, D.L. Eizirik, M. Cnop, Initiationand execution of lipotoxic ER stress in pancreatic beta-cells, J. Cell. Sci. 121(2008) 2308–2318.
[30] A. Di Nardo, I. Kramvis, N. Cho, A. Sadowski, L. Meikle, D.J. Kwiatkowski, M. Sahin,Tuberous sclerosis complex activity is required to control neuronal stressresponses in an mTOR-dependent manner, J. Neurosci. 29 (2009) 5926–5937.
[31] D.L. Eizirik, A.K. Cardozo, M. Cnop, The role for endoplasmic reticulum stress indiabetes mellitus, Endocr. Rev. 29 (2008) 42–61.
[32] W. El Assaad, J. Buteau, M.L. Peyot, C. Nolan, R. Roduit, S. Hardy, E. Joly, G. Dbaibo,L. Rosenberg, M. Prentki, Saturated fatty acids synergize with elevated glucose tocause pancreatic beta-cell death, Endocrinology 144 (2003) 4154–4163.
[33] H. Elouil, M. Bensellam, Y. Guiot, M.D. Vander, S.M. Pascal, F.C. Schuit, J.C. Jonas,Acute nutrient regulation of the unfolded protein response and integrated stressresponse in cultured rat pancreatic islets, Diabetologia 50 (2007) 1442–1452.
[34] H. Endo, K. Murata, M. Mukai, O. Ishikawa, M. Inoue, Activation of insulin-likegrowth factor signaling induces apoptotic cell death under prolonged hypoxia byenhancing endoplasmic reticulum stress response, Cancer Res. 67 (2007)8095–8103.
[35] I.S. Farooqi, S. O'Rahilly, Monogenic obesity in humans, Annu. Rev. Med. 56(2005) 443–458.
[36] G.C. Farrell, C.Z. Larter, Nonalcoholic fatty liver disease: from steatosis tocirrhosis, Hepatology 43 (2006) S99–S112.
[37] J.S. Flier, Obesity wars: molecular progress confronts an expanding epidemic,Cell 116 (2004) 337–350.
[38] G. Fruhbeck, Are we really tackling the obesity epidemic, Obes. Metab. 4 (2008)91–95.
[39] Q. Gao, T.L. Horvath, Neurobiology of feeding and energy expenditure, Annu. Rev.Neurosci. 30 (2007) 367–398.
[40] S. Gao, K.P. Kinzig, S. Aja, K.A. Scott, W. Keung, S. Kelly, K. Strynadka, S. Chohnan,W.W. Smith, K.L. Tamashiro, E.E. Ladenheim, G.V. Ronnett, Y. Tu, M.J. Birnbaum,G.D. Lopaschuk, T.H. Moran, Leptin activates hypothalamic acetyl-CoA carbox-ylase to inhibit food intake, Proc. Natl. Acad. Sci. U. S. A. 104 (2007)17358–17363.
[41] S. Gao, M.D. Lane, Effect of the anorectic fatty acid synthase inhibitor C75 onneuronal activity in the hypothalamus and brainstem, Proc. Natl. Acad. Sci. U. S. A.100 (2003) 5628–5633.
[42] S.M. Haffner, Relationship of metabolic risk factors and development ofcardiovascular disease and diabetes, Obesity 14 (Suppl. 3) (2006) 121S–127S(Silver. Spring).
[43] S.A. Hawley, D.A. Pan, K.J. Mustard, L. Ross, J. Bain, A.M. Edelman, B.G.Frenguelli, D.G. Hardie, Calmodulin-dependent protein kinase kinase-beta isan alternative upstream kinase for AMP-activated protein kinase, Cell Metab. 2(2005) 9–19.
[44] T. Hayashi, A. Saito, S. Okuno, M. Ferrand-Drake, R.L. Dodd, P.H. Chan, Damage tothe endoplasmic reticulum and activation of apoptotic machinery by oxidativestress in ischemic neurons, J. Cereb. Blood Flow Metab. 25 (2005) 41–53.
[45] W. He, T.K. Lam, S. Obici, L. Rossetti, Molecular disruption of hypothalamicnutrient sensing induces obesity, Nat. Neurosci. 9 (2006) 227–233.
[46] T. Hosoi, M. Sasaki, T. Miyahara, C. Hashimoto, S. Matsuo, M. Yoshii, K. Ozawa,Endoplasmic reticulum stress induces leptin resistance, Mol. Pharmacol. 74(2008) 1610–1619.
[47] G.S. Hotamisligil, Inflammation and endoplasmic reticulum stress in obesity anddiabetes, Int. J. Obes. (Lond) 32 (Suppl 7) (2008) S52–S54.
[48] Z. Hu, S.H. Cha, S. Chohnan, M.D. Lane, Hypothalamic malonyl-CoA as a mediatorof feeding behavior, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 12624–12629.
[49] Z. Hu, Y. Dai, M. Prentki, S. Chohnan, M.D. Lane, A role for hypothalamic malonyl-CoA in the control of food intake, J. Biol. Chem. 280 (2005) 39681–39683.
[50] C.J. Huang, C.Y. Lin, L. Haataja, T. Gurlo, A.E. Butler, R.A. Rizza, P.C. Butler, Highexpression rates of human islet amyloid polypeptide induce endoplasmicreticulum stress mediated beta-cell apoptosis, a characteristic of humans withtype 2 but not type 1 diabetes, Diabetes 56 (2007) 2016–2027.
[51] R.L. Hurley, K.A. Anderson, J.M. Franzone, B.E. Kemp, A.R. Means, L.A. Witters, TheCa2+/calmodulin-dependent protein kinase kinases are AMP-activated proteinkinase kinases, J. Biol. Chem. 280 (2005) 29060–29066.
[52] E.V. Ilieva, V. Ayala, M. Jove, E. Dalfo, D. Cacabelos, M. Povedano, M.J. Bellmunt, I.Ferrer, R. Pamplona, M. Portero-Otin, Oxidative and endoplasmic reticulumstress interplay in sporadic amyotrophic lateral sclerosis, Brain 130 (2007)3111–3123.
[53] S. Ishii, J. Kamegai, H. Tamura, T. Shimizu, H. Sugihara, S. Oikawa, Triiodothy-ronine (T3) stimulates food intake via enhanced hypothalamic AMP-activatedkinase activity, Regul. Pept. 151 (2008) 164–169.
[54] H.O. Jin, S.K. Seo, S.H.Woo, E.S. Kim, H.C. Lee, D.H. Yoo, S. An, T.B. Choe, S.J. Lee, S.I.Hong, C.H. Rhee, J.I. Kim, I.C. Park, Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target ofrapamycin via Redd1 expression in response to oxidative and endoplasmicreticulum stress, Free Radic. Biol. Med. 46 (2009) 1158–1167.
[55] B.B. Kahn, T. Alquier, D. Carling, D.G. Hardie, AMP-activated protein kinase:ancient energy gauge provides clues to modern understanding of metabolism,Cell Metab. 1 (2005) 15–25.
[56] E.K. Kim, I. Miller, S. Aja, L.E. Landree, M. Pinn, J. McFadden, F.P. Kuhajda, T.H.Moran, G.V. Ronnett, C75, a fatty acid synthase inhibitor, reduces food intake viahypothalamic AMP-activated protein kinase, J. Biol. Chem. 279 (2004)19970–19976.
[57] E.K. Kim, I. Miller, L.E. Landree, F.F. Borisy-Rudin, P. Brown, T. Tihan, C.A.Townsend, L.A. Witters, T.H. Moran, F.P. Kuhajda, G.V. Ronnett, Expression of FASwithin hypothalamic neurons: a model for decreased food intake after C75treatment, Am. J. Physiol. Endocrinol. Metab. 283 (2002) E867–E879.
[58] M.S. Kim, J.Y. Park, C. Namkoong, P.G. Jang, J.W. Ryu, H.S. Song, J.Y. Yun, I.S.Namgoong, J. Ha, I.S. Park, I.K. Lee, B. Viollet, J.H. Youn, H.K. Lee, K.U. Lee, Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamicAMP-activated protein kinase, Nat. Med. 10 (2004) 727–733.
[59] S. Klaus, Adipose tissue as a regulator of energy balance, Curr. Drug Targets 5(2004) 241–250.
[60] B. Kola, E. Hubina, S.A. Tucci, T.C. Kirkham, E.A. Garcia, S.E. Mitchell, L.M.Williams, S.A. Hawley, D.G. Hardie, A.B. Grossman, M. Korbonits, Cannabinoidsand ghrelin have both central and peripheral metabolic and cardiac effects viaAMP-activated protein kinase, J. Biol. Chem. 280 (2005) 25196–25201.
[61] N. Kubota, W. Yano, T. Kubota, T. Yamauchi, S. Itoh, H. Kumagai, H. Kozono, I.Takamoto, S. Okamoto, T. Shiuchi, R. Suzuki, H. Satoh, A. Tsuchida, M. Moroi, K.Sugi, T. Noda, H. Ebinuma, Y. Ueta, T. Kondo, E. Araki, O. Ezaki, R. Nagai, K. Tobe, Y.Terauchi, K. Ueki, Y. Minokoshi, T. Kadowaki, Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake, CellMetab. 6 (2007) 55–68.
[62] F.P. Kuhajda, K. Jenner, F.D. Wood, R.A. Hennigar, L.B. Jacobs, J.D. Dick, G.R.Pasternack, Fatty acid synthesis: a potential selective target for antineoplastictherapy, Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 6379–6383.
[63] R. Lage, C. Diéguez, A. Vidal-Puig, M. López, AMPK: a metabolic gauge regulatingwhole-body energy homeostasis, Trends Mol. Med. 14 (2008) 539–549.
[64] T.K. Lam, A. Pocai, R. Gutierrez-Juarez, S. Obici, J. Bryan, L. Aguilar-Bryan, G.J.Schwartz, L. Rossetti, Hypothalamic sensing of circulating fatty acids is requiredfor glucose homeostasis, Nat. Med. 11 (2005) 320–327.
[65] T.K. Lam, G.J. Schwartz, L. Rossetti, Hypothalamic sensing of fatty acids, Nat.Neurosci. 8 (2005) 579–584.
[66] T.K. Lam, W.G. Van de, A. Giacca, Free fatty acids increase basal hepatic glucoseproduction and induce hepatic insulin resistance at different sites, Am. J. Physiol.Endocrinol. Metab. 284 (2003) E281–E290.
[67] M.D. Lane, M. Wolfgang, S.H. Cha, Y. Dai, Regulation of food intake and energyexpenditure by hypothalamic malonyl-CoA, Int. J. Obes. (Lond) 32 (Suppl 4)(2008) S49–S54.
[68] D.R. Laybutt, A.M. Preston, M.C. Akerfeldt, J.G. Kench, A.K. Busch, A.V. Biankin, T.J.Biden, Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2diabetes, Diabetologia 50 (2007) 752–763.
[69] C. Le Foll, B.G. Irani, C. Magnan, A.A. Dunn-Meynell, B.E. Levin, Characteristics andmechanisms of hypothalamic neuronal fatty acid sensing, Am. J. Physiol. Regul.Integr. Comp. Physiol. 297 (2009) R655–R664.
[70] Y. Lee, H. Hirose, M. Ohneda, J.H. Johnson, J.D. McGarry, R.H. Unger, Beta-celllipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus ofobese rats: impairment in adipocyte–beta-cell relationships, Proc. Natl. Acad. Sci.U. S. A. 91 (1994) 10878–10882.
[71] C. Lelliott, A.J. Vidal-Puig, Lipotoxicity, an imbalance between lipogenesis denovo and fatty acid oxidation, Int. J. Obes. Relat. Metab. Disord. 28 (Suppl. 4)(2004) S22–S28.
[72] J.H. Lin, P.Walter, T.S. Yen, Endoplasmic reticulum stress in disease pathogenesis,Annu. Rev. Pathol. 3 (2008) 399–425.
[73] K.L. Lipson, S.G. Fonseca, S. Ishigaki, L.X. Nguyen, E. Foss, R. Bortell, A.A. Rossini, F.Urano, Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmicreticulum-resident protein kinase IRE1, Cell Metab. 4 (2006) 245–254.
[74] K.L. Lipson, R. Ghosh, F. Urano, The role of IRE1alpha in the degradation of insulinmRNA in pancreatic beta-cells, PLoS ONE 3 (2008) e1648.
[75] T.M. Loftus, D.E. Jaworsky, G.L. Frehywot, C.A. Townsend, G.V. Ronnett, M.D. Lane,F.P. Kuhajda, Reduced food intake and body weight in mice treated with fattyacid synthase inhibitors, Science 288 (2000) 2379–2381.
[76] M. López, R. Lage, A.K. Saha, D. Pérez-Tilve, M.J. Vàzquez, L. Varela, S. Sangiao-Alvarellos, S. Tovar, K. Raghay, S. Rodríguez-Cuenca, R.M. Deoliveira, T.Castañeda, R. Datta, J.Z. Dong, M. Culler, M.W. Sleeman, C.V. Álvarez, R. Gallego,C.J. Lelliott, D. Carling, M.H. Tschop, C. Diéguez, A. Vidal-Puig, Hypothalamic fattyacid metabolism mediates the orexigenic action of ghrelin, Cell Metab. 7 (2008)389–399.
[77] M. López, C.J. Lelliott, S. Tovar, W. Kimber, R. Gallego, S. Virtue, M. Blount, M.J.Vázquez, N. Finer, T. Powles, S. O'Rahilly, A.K. Saha, C. Diéguez, A.J. Vidal-Puig,Tamoxifen-induced anorexia is associated with fatty acid synthase inhibition inthe ventromedial nucleus of the hypothalamus and accumulation of malonyl-CoA, Diabetes 55 (2006) 1327–1336.
[78] M. López, C.J. Lelliott, A. Vidal-Puig, Hypothalamic fatty acid metabolism: ahousekeeping pathway that regulates food intake, Bioessays 29 (2007) 248–261.
[79] M. López, A.K. Saha, C. Diéguez, A. Vidal-Puig, The AMPK–malonyl-CoA–CPT1axis in the control of hypothalamic neuronal function, Cell Metab. 8 (2008) 176.

360 P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
[80] M. López, S. Tovar, M.J. Vázquez, L.M. Williams, C. Diéguez, Peripheral tissue–brain interactions in the regulation of food intake, Proc. Nutr. Soc. 66 (2007)131–155.
[81] M. López, A. Vidal-Puig, Brain lipogenesis and regulation of energy metabolism,Curr. Opin. Clin. Nutr. Metab. Care 11 (2008) 483–490.
[82] K. Maedler, G.A. Spinas, D. Dyntar, W. Moritz, N. Kaiser, M.Y. Donath, Distincteffects of saturated and monounsaturated fatty acids on beta-cell turnover andfunction, Diabetes 50 (2001) 69–76.
[83] F. Marra, A. Gastaldelli, B.G. Svegliati, G. Tell, C. Tiribelli, Molecular basis andmechanisms of progression of non-alcoholic steatohepatitis, Trends Mol. Med.14 (2008) 72–81.
[84] S. Matus, F. Lisbona, M. Torres, C. Leon, P. Thielen, C. Hetz, The stress rheostat: aninterplay between the unfolded protein response (UPR) and autophagy inneurodegeneration, Curr. Mol. Med. 8 (2008) 157–172.
[85] J. Mayer, Regulation of energy intake and the body weight: the glucostatic theoryand the lipostatic hypothesis, Ann. N.Y. Acad. Sci. 63 (1955) 15–43.
[86] R.J. McCrimmon, X. Fan, Y. Ding, W. Zhu, R.J. Jacob, R.S. Sherwin, Potential role forAMP-activated protein kinase in hypoglycemia sensing in the ventromedialhypothalamus, Diabetes 53 (2004) 1953–1958.
[87] G. Medina-Gomez, S. Gray, A. Vidal-Puig, Adipogenesis and lipotoxicity: role ofperoxisome proliferator-activated receptor gamma (PPARgamma) and PPAR-gammacoactivator-1 (PGC1), Public Health Nutr 10 (2007) 1132–1137.
[88] G. Medina-Gomez, A. Vidal-Puig, Gateway to the metabolic syndrome, Nat. Med.11 (2005) 602–603.
[89] J.C. Miller, J.M. Gnaedinger, S.I. Rapoport, Utilization of plasma fatty acid in ratbrain: distribution of [14C]palmitate between oxidative and synthetic pathways,J. Neurochem. 49 (1987) 1507–1514.
[90] Y. Minokoshi, T. Alquier, N. Furukawa, Y.B. Kim, A. Lee, B. Xue, J. Mu, F. Foufelle, P.Ferre, M.J. Birnbaum, B.J. Stuck, B.B. Kahn, AMP-kinase regulates food intake byresponding to hormonal and nutrient signals in the hypothalamus, Nature 428(2004) 569–574.
[91] K. Morgan, S. Obici, L. Rossetti, Hypothalamic responses to long-chain fatty acidsare nutritionally regulated, J. Biol. Chem. 279 (2004) 31139–31148.
[92] H. Mori, K. Inoki, H. Munzberg, D. Opland, M. Faouzi, E.C. Villanueva, T. Ikenoue,D. Kwiatkowski, O.A. MacDougald, M.G. Myers Jr., K.L. Guan, Critical role forhypothalamic mTOR activity in energy balance, Cell Metab. 9 (2009) 362–374.
[93] G.J. Morton, D.E. Cummings, D.G. Baskin, G.S. Barsh, M.W. Schwartz, Centralnervous system control of food intake and body weight, Nature 443 (2006)289–295.
[94] M.G. Myers Jr., H. Munzberg, G.M. Leinninger, R.L. Leshan, The geometry of leptinaction in the brain: more complicated than a simple ARC, Cell Metab. 9 (2009)117–123.
[95] C. Namkoong, M.S. Kim, P.G. Jang, S.M. Han, H.S. Park, E.H. Koh, W.J. Lee, J.Y. Kim,I.S. Park, J.Y. Park, K.U. Lee, Enhanced hypothalamic AMP-activated proteinkinase activity contributes to hyperphagia in diabetic rats, Diabetes 54 (2005)63–68.
[96] R. Nogueiras, M.H. Tschop, J.M. Zigman, Central nervous system regulation ofenergy metabolism: ghrelin versus leptin, Ann. N.Y. Acad. Sci. 1126 (2008)14–19.
[97] S. Obici, Z. Feng, A. Arduini, R. Conti, L. Rossetti, Inhibition of hypothalamiccarnitine palmitoyltransferase-1 decreases food intake and glucose production,Nat. Med. 9 (2003) 756–761.
[98] S. Obici, Z. Feng, K. Morgan, D. Stein, G. Karkanias, L. Rossetti, Centraladministration of oleic acid inhibits glucose production and food intake,Diabetes 51 (2002) 271–275.
[99] S. Oyadomari, A. Koizumi, K. Takeda, T. Gotoh, S. Akira, E. Araki, M.Mori, Targeteddisruption of the Chop gene delays endoplasmic reticulum stress-mediateddiabetes, J. Clin. Invest 109 (2002) 525–532.
[100] S. Oyadomari, M. Mori, Roles of CHOP/GADD153 in endoplasmic reticulumstress, Cell Death Differ. 11 (2004) 381–389.
[101] L. Ozcan, A.S. Ergin, A. Lu, J. Chung, S. Sarkar, D. Nie, M.G. Myers Jr., U. Ozcan,Endoplasmic reticulum stress plays a central role in development of leptinresistance, Cell Metab. 9 (2009) 35–51.
[102] U. Ozcan, L. Ozcan, E. Yilmaz, K. Duvel, M. Sahin, B.D. Manning, G.S. Hotamisligil,Loss of the tuberous sclerosis complex tumor suppressors triggers the unfoldedprotein response to regulate insulin signaling and apoptosis, Mol. Cell 29 (2008)541–551.
[103] M. Pennuto, E. Tinelli, M. Malaguti, U. Del Carro, M. D'Antonio, D. Ron, A.Quattrini, M.L. Feltri, L. Wrabetz, Ablation of the UPR-mediator CHOP restoresmotor function and reduces demyelination in Charcot–Marie–Tooth 1B mice,Neuron 57 (2008) 393–405.
[104] L. Plum, B.F. Belgardt, J.C. Bruning, Central insulin action in energy and glucosehomeostasis, J. Clin. Invest 116 (2006) 1761–1766.
[105] A. Pocai, T.K. Lam, R. Gutierrez-Juarez, S. Obici, G.J. Schwartz, J. Bryan, L. Aguilar-Bryan, L. Rossetti, Hypothalamic K(ATP) channels control hepatic glucoseproduction, Nature 434 (2005) 1026–1031.
[106] A. Pocai, T.K. Lam, S. Obici, R. Gutierrez-Juarez, E.D. Muse, A. Arduini, L. Rossetti,Restoration of hypothalamic lipid sensing normalizes energy and glucosehomeostasis in overfed rats, J. Clin. Invest. 116 (2006) 1081–1091.
[107] A. Pocai, S. Obici, G.J. Schwartz, L. Rossetti, A brain–liver circuit regulates glucosehomeostasis, Cell Metab. 1 (2005) 53–61.
[108] D. Porte Jr., S.E. Kahn, Beta-cell dysfunction and failure in type 2 diabetes:potential mechanisms, Diabetes 50 (Suppl. 1) (2001) S160–S163.
[109] K. Proulx, D. Cota, S.C. Woods, R.J. Seeley, Fatty acid synthase inhibitors modulateenergy balance via mammalian target of rapamycin complex 1 signaling in thecentral nervous system, Diabetes 57 (2008) 3231–3238.
[110] K. Qi, M. Hall, R.J. Deckelbaum, Long-chain polyunsaturated fatty acid accretionin brain, Curr. Opin. Clin. Nutr. Metab. Care 5 (2002) 133–138.
[111] S.I. Rapoport, In vivo labeling of brain phospholipids by long-chain fatty acids:relation to turnover and function, Lipids 31 (Suppl.) (1996) S97–S101.
[112] S.I. Rapoport, In vivo fatty acid incorporation into brain phosholipids in relationto plasma availability, signal transduction and membrane remodeling, J. Mol.Neurosci. 16 (2001) 243–261.
[113] D. Ron, J.F. Habener, CHOP, a novel developmentally regulated nuclear proteinthat dimerizes with transcription factors C/EBP and LAP and functions as adominant-negative inhibitor of gene transcription, Genes Dev. 6 (1992)439–453.
[114] D. Ron, S.R. Hubbard, How IRE1 reacts to ER stress, Cell 132 (2008) 24–26.[115] D. Ron, P. Walter, Signal integration in the endoplasmic reticulum unfolded
protein response, Nat. Rev. Mol. Cell. Biol. 8 (2007) 519–529.[116] E.D. Rosen, B.M. Spiegelman, Adipocytes as regulators of energy balance and
glucose homeostasis, Nature 444 (2006) 847–853.[117] N.B. Ruderman, A.K. Saha, E.W. Kraegen, Minireview: malonyl CoA, AMP-
activated protein kinase, and adiposity, Endocrinology 144 (2003) 5166–5171.[118] P.A. Sarafidis, P.M. Nilsson, The metabolic syndrome: a glance at its history, J.
Hypertens. 24 (2006) 621–626.[119] J. Schapansky, K. Olson, P.R. Van Der, G. Glazner, NF-kappaB activated by ER
calcium release inhibits Abeta-mediated expression of CHOP protein: enhance-ment by AD-linked mutant presenilin 1, Exp. Neurol. 208 (2007) 169–176.
[120] G.E. Schujman, L. Paoletti, A.D. Grossman, D. de Mendoza, FapR, a bacterialtranscription factor involved in global regulation of membrane lipid biosynthe-sis, Dev. Cell 4 (2003) 663–672.
[121] S. Seo, S. Ju, H. Chung, D. Lee, S. Park, Acute effects of glucagon-like peptide-1 onhypothalamic neuropeptide and AMP activated kinase expression in fasted rats,Endocr. J. 55 (2008) 867–874.
[122] N.K. Sharma, S.K. Das, A.K. Mondal, O.G. Hackney, W.S. Chu, P.A. Kern, N. Rasouli,H.J. Spencer, A. Yao-Borengasser, S.C. Elbein, Endoplasmic reticulum stressmarkers are associated with obesity in nondiabetic subjects, J. Clin. Endocrinol.Metab. 93 (2008) 4532–4541.
[123] M. Shimazawa, Y. Inokuchi, Y. Ito, H. Murata, M. Aihara, M. Miura, M. Araie, H.Hara, Involvement of ER stress in retinal cell death, Mol. Vis. 13 (2007) 578–587.
[124] H. Shimizu, H. Arima, M. Watanabe, M. Goto, R. Banno, I. Sato, N. Ozaki, H.Nagasaki, Y. Oiso, Glucocorticoids increase neuropeptide Y and agouti-relatedpeptide gene expression via AMP-activated protein kinase signaling in thearcuate nucleus of rats, Endocrinology 149 (2008) 4544–4553.
[125] T. Shimokawa, M.V. Kumar, M.D. Lane, Effect of a fatty acid synthase inhibitor onfood intake and expression of hypothalamic neuropeptides, Proc. Natl. Acad. Sci.U. S. A. 99 (2002) 66–71.
[126] R.M. Silva, V. Ries, T.F. Oo, O. Yarygina, V. Jackson-Lewis, E.J. Ryu, P.D. Lu, S.J.Marciniak, D. Ron, S. Przedborski, N. Kholodilov, L.A. Greene, R.E. Burke, CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neuronsin an in vivo neurotoxin model of parkinsonism, J. Neurochem. 95 (2005)974–986.
[127] M. Slawik, A.J. Vidal-Puig, Lipotoxicity, overnutrition and energy metabolism inaging, Ageing Res. Rev. 5 (2006) 144–164.
[128] B. Song, D. Scheuner, D. Ron, S. Pennathur, R.J. Kaufman, Chop deletion reducesoxidative stress, improves beta cell function, and promotes cell survival inmultiple mouse models of diabetes, J. Clin. Invest 118 (2008) 3378–3389.
[129] G.R. Steinberg, M.J. Watt, B.C. Fam, J. Proietto, S. Andrikopoulos, A.M. Allen, M.A.Febbraio, B.E. Kemp, Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice, Endocrinology 147 (2006)3906–3914.
[130] G.R. Stoppa, M. Cesquini, E.A. Roman, P.O. Prada, A.S. Torsoni, T. Romanatto, M.J.Saad, L.A. Velloso, M.A. Torsoni, Intracerebroventricular injection of citrateinhibits hypothalamic AMPK and modulates feeding behavior and peripheralinsulin signaling, J. Endocrinol. 198 (2008) 157–168.
[131] Q.Q. Tang, M.D. Lane, Role of C/EBP homologous protein (CHOP-10) in theprogrammed activation of CCAAT/enhancer-binding protein-beta during adi-pogenesis, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 12446–12450.
[132] L.A. Tartaglia, M. Dembski, X. Weng, N. Deng, J. Culpepper, R. Devos, G.J. Richards,L.A. Campfield, F.T. Clark, J. Deeds, Identification and expression cloning of aleptin receptor, OB-R, Cell 83 (1995) 1263–1271.
[133] K. Terai, Y. Hiramoto, M. Masaki, S. Sugiyama, T. Kuroda, M. Hori, I. Kawase, H.Hirota, AMP-activated protein kinase protects cardiomyocytes against hypoxicinjury through attenuation of endoplasmic reticulum stress, Mol. Cell Biol. 25(2005) 9554–9575.
[134] P. Trayhurn, C. Bing, I.S. Wood, Adipose tissue and adipokines—energy regulationfrom the human perspective, J. Nutr. 136 (2006) 1935S–1939S.
[135] Y. Tu, J.N. Thupari, E.K. Kim, M.L. Pinn, T.H. Moran, G.V. Ronnett, F.P. Kuhajda, C75alters central and peripheral gene expression to reduce food intake and increaseenergy expenditure, Endocrinology 146 (2004) 486–493.
[136] R.H. Unger, Lipotoxic diseases, Annu. Rev. Med. 53 (2002) 319–336.[137] R.H. Unger, Longevity, lipotoxicity and leptin: the adipocyte defense against
feasting and famine, Biochimie 87 (2005) 57–64.[138] R.H. Unger, L. Orci, Lipotoxic diseases of nonadipose tissues in obesity, Int. J.
Obes. Relat. Metab. Disord. 24 (Suppl. 4) (2000) S28–S32.[139] B.T. van den, M. Koritzinsky, B.G. Wouters, Translational control of gene
expression during hypoxia, Cancer Biol. Ther. 5 (2006) 749–755.[140] S. Virtue, A. Vidal-Puig, It's not how fat you are, it's what you do with it that
counts, PLoS Biol. 6 (2008) e237.[141] R. Wang, C. Cruciani-Guglielmacci, S. Migrenne, C. Magnan, V.E. Cotero, V.H.
Routh, Effects of oleic acid on distinct populations of neurons in the

361P.B. Martínez de Morentin et al. / Biochimica et Biophysica Acta 1801 (2010) 350–361
hypothalamic arcuate nucleus are dependent on extracellular glucose levels, J.Neurophysiol. 95 (2006) 1491–1498.
[142] J.P. Wilding, The importance of free fatty acids in the development of type 2diabetes, Diabet. Med. 24 (2007) 934–945.
[143] M.J. Wolfgang, S.H. Cha, A. Sidhaye, S. Chohnan, G. Cline, G.I. Shulman, M.D. Lane,Regulation of hypothalamic malonyl-CoA by central glucose and leptin, Proc.Natl. Acad. Sci. U. S. A. 104 (2007) 19285–19290.
[144] M.J. Wolfgang, T. Kurama, Y. Dai, A. Suwa, M. Asaumi, S. Matsumoto, S.H. Cha, T.Shimokawa, M.D. Lane, The brain-specific carnitine palmitoyltransferase-1cregulates energy homeostasis, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 7282–7287.
[145] J.C. Won, P.G. Jang, C. Namkoong, E.H. Koh, S.K. Kim, J.Y. Park, K.U. Lee, M.S. Kim,Central administration of an endoplasmic reticulum stress inducer inhibits theanorexigenic effects of leptin and insulin, Obesity (2009) 1861–1865 (Silver. Spring).
[146] A.Woods, K. Dickerson, R.Heath, S.P. Hong,M.Momcilovic, S.R. Johnstone,M. Carlson,D. Carling, Ca(2+)/calmodulin-dependent protein kinase kinase-beta acts upstreamof AMP-activated protein kinase in mammalian cells, Cell Metab. 2 (2005) 21–33.
[147] S.C. Woods, R.J. Seeley, D. Cota, Regulation of food intake through hypothalamicsignaling networks involving mTOR, Annu. Rev. Nutr. 28 (2008) 295–311.
[148] J.C. Yang, C.F. Teng, H.C. Wu, H.W. Tsai, H.C. Chuang, T.F. Tsai, Y.H. Hsu, W. Huang,L.W. Wu, I.J. Su, Enhanced expression of vascular endothelial growth factor-A inground glass hepatocytes and its implication in hepatitis B virus hepatocarci-nogenesis, Hepatology 49 (2009) 1962–1971.
[149] L. Yang, G.S. Hotamisligil, Stressing the brain, fattening the body, Cell 135 (2008)20–22.
[150] B.Yusta, L.L. Baggio, J.L. Estall, J.A.Koehler,D.P.Holland,H. Li,D. Pipeleers, Z. Ling,D.J.Drucker, GLP-1 receptor activation improves beta cell function and survivalfollowing induction of endoplasmic reticulumstress, CellMetab. 4 (2006)391–406.
[151] X. Zhang, G. Zhang, H. Zhang, M. Karin, H. Bai, D. Cai, Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity, Cell135 (2008) 61–73.
[152] Y. Zhang, R. Proenca, M. Maffei, M. Barone, L. Leopold, J.M. Friedman, Positionalcloning of the mouse obese gene and its human homologue, Nature 372 (1994)425–432.
[153] Y.T. Zhou, P. Grayburn, A. Karim, M. Shimabukuro, M. Higa, D. Baetens, L. Orci, R.H.Unger, Lipotoxic heart disease in obese rats: implications for human obesity, Proc.Natl. Acad. Sci. U. S. A. 97 (2000) 1784–1789.




















