Studies in Infantile Marasmus. IV. Impairment of Immunologic ...

Seminars .m
Arthritis and Rheumatism VOL 25, NO 1 AUGUST 1995
Hormonal and Pregnancy Relationships to Rheumatoid Arthritis: Convergent Effects With Immunologic and
Microvascular Systems
Alfonse T. Masi, Seth L. Feigenbaum, and Robert T. Chatterton
Objective: To review sex hormones and rheumatoid arthrit is (RA) and the interrelationships between hormonal, immunological, and vascular systems. Data Sources: Publications detailing serum sex hormone levels and their HLA interactions, steroidogenesis, pregnancy, and therapeutic uses of sex hormones in RA. Study Selection: Controlled studies of sex hormone levels in RA patients not previously treated wi th glucocorticoids. Data Extraction: Mean (-+SD) serum levels of dehydroepiandrosterone sulfate (DHEAS), testosterone (T), and estradiol (E2). Data Synthesis: Mean (_+SD) levels were collated into tables for women wi th pre- versus postmenopausal onsets of disease and men. Data were also ordered across all study groups by increasing mean levels of the control subjects. Pooled data were summarized statistically, and major sources of variation between the studies were identified. Conclusions: Serum DHEAS, an adrenal androgen, was impressively decreased among women wi th premenopausal onset of RA. One study showed such deficiency years before disease onset. Serum T was somewhat decreased in the premenopausal onset group, but could be explained by decreased peripheral conversion of the lower levels of adrenal androgens. Women wi th postmeno- pausal onset of RA had modestly decreased serum DHEAS levels overall, but no difference in serum T, compared wi th controls. Male RA cases had consistently decreased serum levels of T, but not of DHEAS. Serum E2 was comparable in all RA versus control groups. The complex biology of pregnancy was interpreted as an example of vital interactions between hormonal, immunological, and vascu- lar systems, as they may relate to the physiopathology of RA. The major age, sex, and hereditable determinants of RA were compared wi th in a composite
Alfonse T. Masi, MD, DRPH, FACP, Professor of Medi- cine, University of Illinois College of Medicine at Peoria (UICOM-P); Seth L. Feigenbaum, MPH, MD: Assistant Clinical Professor, Department of Obstetrics, Gynecology and Reproductive Sciences, University of California, San Fran- cisco; Director, Reproductive Endocrinology, Kaiser Perman- ente Medical Center; Robert T. Chatterton, PhD: Professor, Department of Obstetrics~Gynecology, The Medical School, Northwestern University.
Supported in part by grants from Sanofi Winthrop Pharma- ceuticals, HealthCORE Fund of the Methodist Medical Center Foundation, The Proctor Foundation, and The BielfeMt Foun- dation.
Address" reprint requests to Alfonse T. Masi, MD, DRPH, FACP, University of lUinois College of Medicine of Peoria, One Illinois Dr, Peoria, IL 61656.
Copyright © 1995 by W.B. Saunders Company 0049-0177/95/2501-000155.00/0
Seminars in Arthritis and Rheumatism, Vo125, No 1 (August), 1995: pp 1-27 1

2 MASI, FEIGENBAUM, AND CHATTERTON
table of estimated relative risks. Elucidation of the interacting risk factors offers promising avenues of research in this complex disease. Copyright © 1995 by W.B. Saunders Company
INDEX WORDS: DHEAS; immunologic system; microvascular system; preg- nancy; rheumatoid arthritis; sex hormones,
HEUMATOID arthritis (RA) is a pro- foundly complex systemic rheumatic dis-
ease of unknown etiology. 1-3 It is classified as a diffuse connective tissue disease (CTD) in the American Rheumatism Association (ARA; now American College of Rheumatology) nomencla- ture and classification of arthritis and rheuma- tism (1983). 4 An understanding of its biology and pathogenesis involves knowledge of at least the immunomediated and inflammatory pro- cesses in joints ~4-7 and systemically1.8; hereditary and immunogenetic mechanisms9-14; microvascu- lar dynamics in jointsS,15-21; systemic vascular involvements22; and steroid hormone physiol- ogy, both of the glucocorticoids 23-31 and sex hormones.3,9,24,30 -39
This perplexing disorder may be analyzed from different viewpoints. It may be considered as an example of an autoimmune clinical- immunopathologic disease process that is au- tonomously driven, s,~4 Alternatively, it may be viewed as a physiopathogenic illness that is dynamically changing in the affected persons (ie, patients). 3,39 Investigators are expected to pursue different research pathways in search of mechanisms in RA and to derive alternative perspectives of its primary determinants.
The pathogenic mechanisms in RA are cur- rently believed to be immunogenetically deter- mined 14 and immunologically mediated. 1,5-8,21 However, these theories alone are insufficient to explain its considerable heterogeneity in clin- ical expression, a2,14 the significant interactions of age and sex on its characteristic incidence patterns, 38-4° or the complex circadian, men- strual, and pregnancy fluctuations in symptoms and objective manifestations. 14,3°,32 For such reasons, research in hormonal mechanisms has been pursued and indicates that sex hormones are important determinants in e A , 3'24'32"40 possi- bly in conjunction with microvascular system interactions as found during pregnancyY 6,39
Over all ages, females uniformly predominate over males in RA. The female-to-male (F:M)
sex ratio is greater than 1 and is usually 2 or 3 over all ages. 4° However, the ratio of incidence (ie, rate of onset) changes importantly with age, reaching a peak of approximately 5F: 1M during the female reproductive years. This contrasts with sex ratios of 2F:IM or less during juvenile and old ages. 4° The young adult female prepon- derance and exaggerated risk of RA is believed to reflect effects of hypoandrogenicity relative to both normal female and male counter- parts.24.38-40
The incidence of RA rises dramatically over the adult ages, with the exception being men during their third through fifth decades. 4° The ratio of incidence at ages 70+ years versus 20 years of age is approximately eightfold in- creased for women and possibly as high as 20-fold for men. The marked aging effect in RA and its interactions with F:M sex ratios may be contributed by deficiencies of androgenic ana- bolic (AA) steroids. 3,24,37-4° Complex hormone- related mechanisms 3° have also been proposed to influence cyclical fluctuations in activity of RA, under stress and during menstrual cycles.
Autosomal HLA-DRB1 haplotype interac- tions correlate significantly with outcome sever- ity of RA. 13A4 However, these genetic mecha- nisms are not believed to determine onset risks or the complex, heterogeneous course patterns of RA. 12,14 HLA-DRB1 alleles are believed to have a limited pathogenetic role in the overall spectrum of RA. 12,14 Twin studies 12 estimate a maximum genetic effect on concordance of RA of approximately 15%. Postgenomic, somatic cell, and environmental factors, therefore, en- compass the major determinants of causation and pathogenesis of RA, considering the full spectrum of disease. 12 Nevertheless, heredity is important in the risk of acquiring RA. After age and gender adjustments, relative risks of acquir- ing RA were estimated to be 3.4-fold increased for dizygotic (DZ) and 8.6-fold increased for monozygotic (MZ) twin pairs when one mem- ber is affected. 12

SEX HORMONES AND PREGNANCY IN RA 3
The dramatic improvements (in approxi- mately 75%) and even remissions of RA in some women during pregnancy 32,41 have been long-recognized. 32 This fundamental biologic phenomenon in RA, which contrasts with the other CTDs, 42 has not yet been scientifically explained. 41-43 Maternal-fetal disparity in HLA class II alloantigens significantly correlated with the likelihood of remission in one study of pregnant women with RA, suggesting alter- ations in maternal immune responsiveness to paternal HLA antigens. 41
Pregnancy is a complex physiologic state that involves vital hormonal, immunologic, and vas- cular interactions. 3,39,41-45 Chronic exposure to significantly elevated serum levels of free gluco- corticoids and reproductive steroids causes im- portant changes in intravascular protein produc- tion, hormone binding, and microvascular/ tissue permeability, 42 in addition to alterations in immunoreactivity. 41 Therefore, fetoplacental- maternal production of hormones or other fac- tors (see below) not presently identified 42 may also explain the marked pregnancy-ameliorative effects in R A . 42
Within the past decade, fundamental biologic interrelationships have been discovered be- tween the hormonal and immunologic sys- tems. 3'253°,34'35 Reciprocal activation and suppres- sion pathways exist between these dynamic, integrated systems in health and in disease. 3° Also, reciprocal hormonal-vascular 3,44-46 and im- munoIogic -vascu la r 1,3,5,s,21,39,47 interactions have been documented that have potential relevance to e A , 1'3'39 Integrated interaction of the hor- monal-immunologic-vascular systems was re- cently reviewed in relation to RA and its pregnancy-ameliorating phenomenon. 3 Such in- teractions are believed to contribute signifi- cantly to the presently unexplained host and environmental determinants in the onset and course of RA.
This article reviews and integrates recent literature on sex hormones and pregnancy in RA. The objective is to interpret their potential physiopathogenic mechanisms in RA. A broad view of determinants in RA is encouraged to better understand its complex physiopathology. Knowledge of the conjoint effects of hormonal, immunologic, and vascular factors on RA may better explain its onset and course patterns.
Insights from the data reviewed may reveal promising new research avenues into the physio- pathogenesis of this mysterious disease. The ultimate aim is to improve its management and achieve measures of prevention.
SEX HORMONES AND RA
Sex Steroid Synthesis and Physiology
Steroidogenesis (Fig 1) and its physiology are profoundly complex and incompletely under- stood in health and disease. 3 Interactions of these various hormones with the immunologic and vascular systems, as they may relate to RA, were reviewed elsewhere. 3,39 However, mention should be made of essential points. The adrenal cortex is a more important source of androgenic- anabolic (AA) steroids than the gonads in women, unlike men. 3,39 The major circulating AA steroid hormones are androstenedione (A), dehydroepiandrosterone (DHEA), the DHEA sulfate (DHEAS or DHAS), and testosterone (r).
Production and serum levels of adrenal andro- gens vary greatly over life cycles. Levels begin to increase before adolescence (ie, adrenarche), reach their peak in young adult men and women, decline in middle ages, particularly in women (ie, adrenopause), and reach very low levels in senescence. 3 On the contrary, serum cortisol levels are stable during these life cycles. Serum DHEAS is a good indicator of adrenal andro- gen function because it is derived predomi- nantly from that organ, has high concentrations in the blood, and essentially no diurnal varia- tion. 3 On the contrary, serum T is predomi- nantly derived from the gonads in men, but about equally from the adrenals and gonads in women.
Controlled Serum Sex Steroid Studies in RA
Introductory Caveats'
This review of clinical studies on serum sex hormones and RA includes essentially only controlled reports that were performed on pa- tients who had not previously received glucocor- ticoids or sex hormones (Tables 1 through 4, Figs 2 and 3). Glucocorticoids can particularly depress serum adrenal androgen levels for pro- longed intervals. 3,39,48 Results of these studies must still be strictly interpreted. First, the ef- fects of RA per se on hormonal function must

4 MASI, FEIGENBAUM, AND CHATTERTON
Cholesterol (LDL) (~)@ Z ~ @ ACTH
AS-Pregnenolone • 17-OH-pregnenolone ~'** Dehydroepiandrosterone
1' i, Ir ®
A4-Progesterone • 17-OH-progesterone
11.Deoxycorticosterone 11-Deoxycortisol
k k Corticosterone Cortisol
L®® 1L
• Androstenedione
Estrone @ •
Aldosterone Cortisone
Dehydroepiandrosterone sulphate
Testosterone ~ Dihydrotestosterone
L ® L ® Estradiol 5~-Androstanediol
C21 MineralocorUcoids
(Non-17-hydroxylated)
C21 Glucocorticoids
(17-hydroxylated)
C19 Androgenic and C18 Estrogenic Steroids
(17,20-1yase side chain cleaved)
Fig 1: Simplified schema of the biosynthesis of the major adrenal cortical hormone products showing conventional pathways for mineralocorticoid, glucocorticoid, and sex steroids. Note that ACTH stimulates steroidogenesis by side chain cleavage (P450scc) of free cholesterol to pregnenolone. Subsequently, 17-hydroxylation directs substrate from the mineralocorticoid to glucocorticoid path- ways. The 17-hydroxylase/17,20-1yase enzyme complex, ie, C21 steroid side chain cleavage (P450m7) forms sex steroid precursors. The enzyme complex of 31~-hydroxysteroid dehydrogenase and delta 5, delta 4 isomerase converts steroids with a double bond at the C-5,6 position to the C-4,5 position. Relative facilitation or inhibition at this enzyme level could influence the cortisol DHEA product ratio. Differential activity of the 17-hydroxylase/17,20-1yase complex may possibly also influence this cortisol/DHEA product ratio. Enzymatic steps in steroidogenesis are indicated as follows: (1) 20,22-hydroxylase; (2) 20,22-1yase complex; (3) 3~-hydroxysteroid dehydrogenase; (4) delta 5, delta 4 isomerase; (5) 17-hydroxylase; (6) 21-hydroxylase; (7) 11(~-hydroxylase; (8) 18-hydroxylase; (9) 18-dehydrogenase; (10) 17,20-1yase (delta 5 and delta 4 specificities); (11) 171~-hydroxysteroid dehydrogenase; (12) peripheral 1 ll3-hydroxysteroid dehydrogenase; (13) peripheral aromatization of A ring; (14) sulfokinase; (15) sulfatase; (16) 5~-reductase; and (17) 3~-hydroxysteroid dehydrogenase. (Reproduced with permission of Clinical and Experimental Rheumatology S.A.S.)
be considered in an interpretation of these findings. 3,24,3°,39 Additional study limitations in- clude inadequate comparison subjects, various patient selection factors, small numbers of sub- jects in most studies, and possible influences of medications other than glucocorticoids.
Serum Estrogen Studies
In premenopausal (Table 1) and postmeno- pausal (Table 2) women, most controlled stud-
ies of sex steroids reported since 1985 have compared serum estradiol (E2) levels among RA versus comparison (CN) subjects. No signifi- cant difference in this hormone was found in any study. Premenopausal RA and CN subjects (Table 1) had expectedly higher mean E2 levels (pmol/L) than the postmenopausal groups (Tables 2 and 4). In males (Table 4), the single study that reported E2 levels 61 also showed similar concentrations in 14 RA (72.0 pmol/L)
SEX HORMONES AND PREGNANCY IN RA
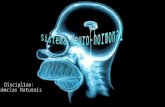
Table 1 : Concentrations of Serum T, E2, and DHEAS in Premenopausal Female RA Patients Not Treated With Glucocorticoids or ACTH Compared With Control Subjects
Study and No. DHEAS and Type o f T ( n m o l / L ) E2 ( p m o l / L ) ( l~mol /L )
Subjects RA Control RA Control RA Control
D o u g a d o s et a148
10 R A v 13 NL 0.87 (0.4) 1.14 (0.5)
10 R A v 11 CD 0.87 (0.3)
Feh6r and Feh~r 49
8 R A v 1 4 N L Feh~r et al 5s *
29 RA v 29 NL (T) 0.8 (0.6) 1.6 (0.7)1"
29 R A v 68 NL (DHEAS)
Cuto lo et a151
7 R A v 7 0 A 0.66 (0.5) 0.59 (0.3)
Spec tor et a152
1 2 R A v 8 O A 1.55(0.4) 1.45(0.4)
Arna l ich et a153
26 RA v 20 CL 0.66 (0.3) 0.52 (0.1)
1.86 (1.1) 2,58 (1,2)
2.18 (1,0)
2.61 (1.9) 4.1 (1.5):1:
2.17 (1.3) 4.2 (1.8)1
182 (82) 238 (159) 4.89 (4.0) 4.23 (2.1)
3.27 (1.7) 2.93 (2.1)
198 (44) 206 (25) 0,34 (0.1) 0.44 (0.1)
NOTE. Data are means (-+SD). Abbreviations: T, testosterone; E2, estradiol; DHEAS, dehydroepiandrosterone; CN, control; CD, chronic disease; CL, chronic lumbalgia; NL, normal; OA, osteoarthritis; RA, rheumatoid arthritis. *Values extrapolated from figures; see text of Feh~r et aP ° for details: Subjects studied for serum T were 18 to 68 years old (ie, 77 RAv 44 NL), but were not divided by pro- v postmenopausar status. Therefore, for serum T analyses, the RA cases were divided as specified for DHEAS, ie, 29 premenopausal, with a matching number of controls, and 15 postmenopausal, with 15 controls, as was specified for DHEAS. Mean data for serum T shown in Tables 1 and 2 are the same, with numbers apportioned as specified. I"P < .01 compared with RA. :~P < .05 compared with RA.
versus 8 osteoarthritis (OA; 81.1 pmol/L) sub- jects. Summation of the pooled data from the available studies of serum E2 concentrations among the premenopausal and postmenopausal RA and (CN) women showed no significant difference (Table 4).
Serum Androgen Studies in Premenopausal Women
The mean serum concentrations of DHEAS and total T are shown in Figs 2 and 3, respec- tively, for each study in order of increasing levels reported for the controls.
Among premenopausal women (Table 1), the earliest reported study of androgens was from Paris, France, 48 which showed a nonsignificant gradient in mean serum DHEAS and T concen- trations among study subjects. Mean serum DHEAS levels increased from 1.86 ~mol /L in 10 RA patients to 2.18 ~xmol/L in 11 chronic disease (CD) controls to 2.58 p, mol /L in 13 normal (NL) women.
Two later studies from Budapest, Hun- gary, 49,5° reported significantly lower serum
DHEAS levels in RA versus normal control women. The larger study 5° included serum T levels, which were also significantly lower among 29 premenopausal RA versus 29 normal con- trols. Two subsequent smaller studies from Genoa, Italy, 51 and London, UK, 52 showed essen- tially no difference in serum T or DHEAS between RA and OA patients (Table 1, Fig 2).
The most recent study from Madrid, Spain, 53 also showed no difference in serum T or DHEAS concentrations between 26 RA and 20 chronic back pain patients. However, their controls had an inordinately low mean serum DHEAS level (0.44 txmol/L), which is inexplicably below the weighted mean results for premenopausal con- trol women among the preceding studies (3.75 Ixmol/L; Table 1; Fig 2). Their control mean is also far below the reported lower normal limit (1 to 2 ixmol/L) for premenopausal women 3 or the mean for CN postmenopausal women (1.63 ~xmol/L) found in this review (Tables 2 and 4; Fig 2). These same data 53 were subsequently published in another language. 57 More recent studies 58 indicated low sulfo-conjugated steroid
MASI, FEIGENBAUM, AND CHATTERTON
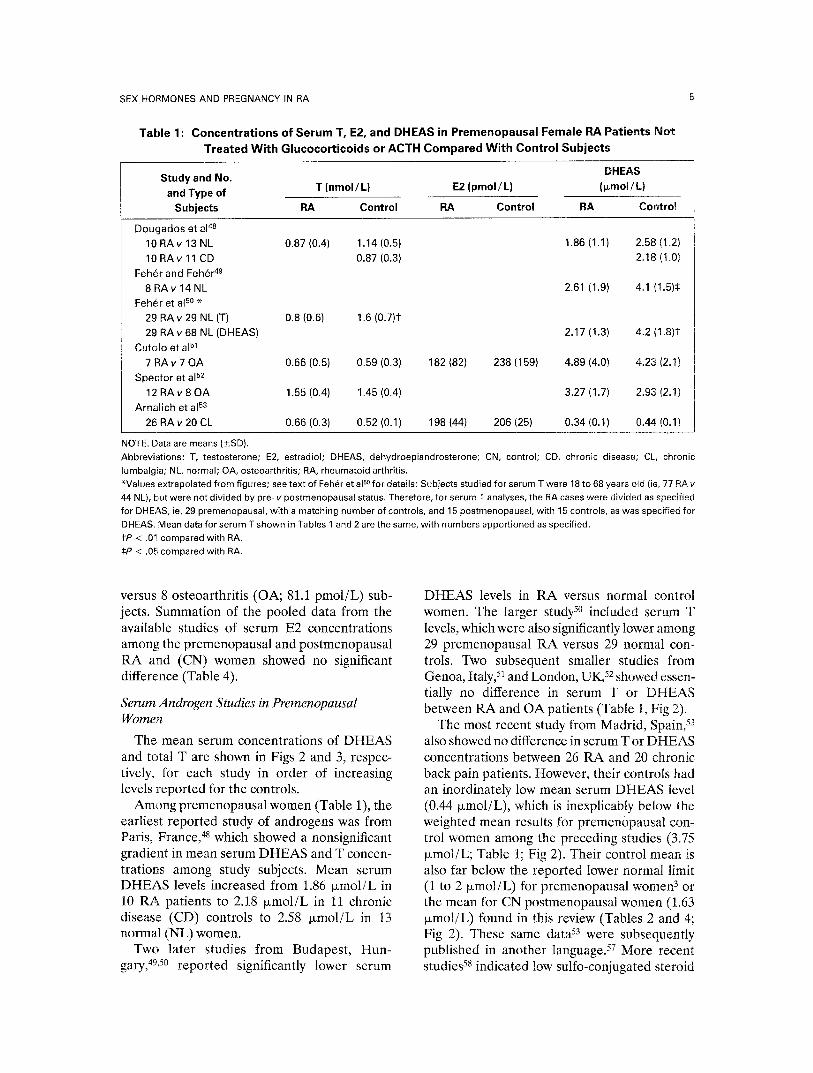
Table 2: Concentrations of Serum T, E2, and DHEAS in Postmenopausal Female RA Patients Not Treated With Glucocorticoids or ACTH Compared With CN Subjects
Study and No. DHEAS T (nmol /L) E2 (pmol /L) (l~mol/L) and Type of
Subjects RA CN RA CN RA CN
Feh~r and Feh~r 49
8 RA v 8 NL 1.59 (0.7) 1.8 (0.7) Bijlsma et a154
10 RAv 10 NL 0.70 (0.1) 0.85 (0.1) 76 (40) 104 (33) [4.95] (1.1) [10.89] (2.1)11 Feh~r et al so *
47 RAv 15 NL 0.8 (0.6) 1.6 (0.7)¶ 1.15 (1.1) 2.89 (2.0)¶ Cutolo et a151
14 RAv 12 OA 0.69 (0.3) 0.45 (0.2)11 90 (59) 55 (40) 2.60 (1.6) 1.02 (1.0)¶ Spector et a152
19 RAv 22 OA 1.28 (0.4) 1.12 (0.5) 2.22 (1.8) 1.83 (1.7) Sambrook et a155 §
49 RAv 49 NL 0.6 (0.4) 0.95 (0.3)# 10 (20) 10 (25) 0.3 (0.3) 2.0 (1.0)#
27 RA (no steroids) 0.8 (0.3) 0.95 (0.3) 10 (20) 10 (25) 0.6 (0.5) 2.0 (1.0)# Aroso Dias et al 5e i" . . . . .
21 RAv 15 OA 0.27 (0.2) 0.34 (0.2) 38 (44) 43 (79) 0.47 (0.3) 1.22 (0.8)¶ RA (no steroids) 0.30 (0.3) 27 (24) 0.64 (0.1)
Arnalich et a153
19 RA v 20 OA 1.04 (0.3) 0.42 (0.1)¶ 95 (29) 77 (12) 0.26 (0.05) 0.13 (0.03)¶
NOTE. Data are means (_+SD). Abbreviations: T, testosterone; E2, estradiol; DHEAS, dehydroepiandrosterone; RA, rheumatoid arthritis; CN, control; NL, normal; OA, osteoarthritis. *Values extrapolated from figures; see Feh~r et al so for details and Table 1 note. 1See Aroso Dias et a156 for details. ~=Values in brackets are DHEA in nmol/L (not DHEAS in ~mol/L); subjects were both premenopausal and postmenopausal; RA aged 35 to 65 years; healthy controls matched for age and premenopausal or postmenopausal state. §Underscored values are medians; 22 RA patients received glucocorticoids (mean prednisolone dose, 8 mg four times per day), 27 received none. lIP < .05 compared with RA. ¶P < .01 compared with RA. #P < .001 compared with RA.
(DHEAS) levels in blood and synovial fluids in RA, but statistics were not performed on age- adjusted comparison patients.
Serum Androgen Studies in Postmenopausal Women
Of the three earliest studies (ie, from Buda- pest, Hungary, 49,5° and Utrecht, The Nether- landsS4), two 5°,54 showed significantly lower mean serum DHEAS 5° or DHEA 54 levels in RA than control subjects. The largest study, 5° including 47 postmenopausal RA patients and 15 normal controls, also showed significantly lower serum T among the RA patients than controls.
Interestingly, the Genoa study 5t showed the reverse results; ie, significantly higher serum T and DHEAS levels among 14 RA versus 12 OA
patients. A later study from London, UK, 52 showed no significant difference in serum andro- gens between patient groups. Subsequent inves- tigations from Sydney, Australia, 55 included 22 RA patients on low-dose prednisolone (mean, 8.2 mg/d) therapy, 27 RA patients not taking corticosteroids, and 49 healthy volunteers. Se- rum DHEAS was highly (P < .001) significantly lower among the RA women versus normals (2.0 ixmol/L), whether they were taking corticos- teroids (0.3 txmol/L) or not (0.6 txmol/L). Serum T was significantly lower only in the RA patient group taking steroids compared with the controls. A study from Oporto, Portugal, 56 also included a combination of RA patients taking (N = 12) and not taking (N = 9) low-dose gluco- corticoids, with a mean age of 50.9 -+ 6.8 years

SEX HORMONES AND PREGNANCY IN RA
Table 3: Serum Concentrations of T, Free T, [Derived], LH, and DHEAS in Male RA Patients Not Treated With Glucocorticoids or ACTH and CN Subjects
Study and No. DHEAS T (nmol /L) LH (mlU/L) (~rnol/L) and Type of
Subjects RA CN RA CN RA CN
Cutolo et a159
7 R A v 6 O A Gordon et al 6° 1"
31 RAv 95 NL 31 RAv 33 AS
31 RAv 95 NL
31 RAv 33 AS Cutolo et a161
1 4 R A v 8 O A
8 R A v 8 N L Specter et a162
25 RAv 27 OA
Specter et a163 1:1:
87 RAv 141 NL 87 RAv 48 AS
87 RAv 141 NL 87 RAv 48 AS
Oilier et a164 §
15 R A v 2 6 NL (HLA-B15+)
56 RAv 112 NL (HLA-B15-) Cutolo et a165 I/
7 R A v 8 O A
7.7 {4.5) 16.6 (8.6)1[
19,5 22.8¶ 3.04 2.161[
23.61[ 2.101[ [0.35] [0.41]#
[0.42]#
9.0 (3.8) 10.8 (3.6)
15.3 (4.6) [0.32] (0.1)
12.5 (4.7)
[0.15]
18.4 (6.9)# 8.8 (5.0) 7.7 (2.9) 23.6 (6.9)*#
17.6 (5.8) 6.96 (3.8) 7.97 (8.8)
[0.46] (0.3)1[
15.8 (5.6)**
16.9 (4.6)**
[0.52] (0.5)** [0.27] (0.4)**
10.61[ 13,8 13.11[ 16.2
1.74 (1.7) 4.20 (1.7)1[
2.85 (2.0) 4.53 (2.6)
4.31 {2.3) 4.15 (3.4)
13.2 (4.2) 18.4 (6.9)'[1 12.1 (2.9) 7.7 (2.9)I[ 1.71 (0.8) 4.53 (2.6)1[
NOTE. Data are means (+-SD). Abbreviations: T, testosterone; LH, luteinizing hormone; DHEAS, dehydroepiandrosterone sulfate; RA, rheumatoid arthritis; CN, control; AS, ankylosing spondylitis; NL, normal; OA, osteoarthritis. *Subset analysis on 8 of original 14 RA patients v 8 NL (see Cutolo et aP 1 for details). tT, free T, and LH mean values not adjusted for age differences. $RA patients who received glucocorticoids could not be accurately excluded. §P values of HLA-B15+ v HLA-B15- : .03 in RA, .07 in NL (see text). !lSame OA control data as reported in Cutolo et al61; values extrapolated from Fig 2. lIP < .05 compared with RA, adjusted for age differences. #P < .01 compared with RA, adjusted for age differences. **P < .001 compared with RA, adjusted for age differences.
(six pre- and 15 postmenopausal patients). The investigators stated that "low plasma androgens were present in corticosteroid-free patients."
The reported series from Madrid, 53 like that from Genoa, 51 also showed significantly higher mean serum T and DHEAS levels (P < .01 for both) in their 19 postmenopausal RA versus 20 OA control patients. Again, the mean DHEAS level among the control subjects (0.13 ~xmol/L) was anomalously and inordinately low (Tables 2 and 4; Fig 2). Furthermore, among their RA patients, the mean serum T level was higher in the 19 postmenopausal (1.04 nmol/L) than the
26 premenopausai (0.66 nmol/L) women. This unexpected trend of higher serum T levels in postmenopausal versus premenopausal women contrasts with that of their OA controls (0.42 v 0.52 nmol/L, respectively) or the total control series summarized in Table 4 (0.86 v 1.10 nmol/L, respectively) and shown in Fig 3.
Serum Androgen Studies in Men
Serum total T or derived free T levels were reported in seven studies of male RA patients versus controls; ie; OA patients, ankylosing spondylitis (AS) subjects, or normals (NL)

8 MASI, FEIGENBAUM, AND CHATTERTON
Table 4: Summary of Mean Serum Concentrations of DHEAS, Total T, E2, and LH in Premenopausal and Postmenopausal Women and in Men With RA Not Treated With Glucocorticoids or ACTH and CN
Subjects: Calculation of P Values on Patient Groups Using a Summation t Test for the Respective Studies
DHEAS Subjects Studied (l~mol/L) T (nmol/L) E2 (pmol/L) LH (mlU/L)
and Parameters RA CN RA CN RA CN RA CN
Premenopausal N 92 141 84 88 33 27
Mean levels 2.01 3.28 0.86 1.10 194.7 214.0
(CN - RA)/CN (%)* 39 22 9
ttest, Pvalue 5.99, <.0011 3.38, <.001~ 1.0, NS
Postmenopausal N 140 141 142 143 76 106
Mean levels 1.32 1.63 0.86 0.86 56.0 41.3
(CN - RA)/CN (%) 19 0 - 3 6
t test, P 2.35, < .02 - - 0.03, NS
Men N 53 41 179 374 14 8
Mean levels 3.24 4.23 13.6 19.0 72.0 81.1
(CN - RA)/CN (%) 23 28 11
t test, P 1.82, .07 10.4, < .001 0.6, NS
77 163
6.2 3.4
- 8 3
0.27, NS
(see text)
Abbreviations: DHEAS, dehydroepiandrosterone sulfate; T, testosterone; E2, estradiol; RA, rheumatoid arthritis; NS, not significant. Summation t test was derived from the difference in grand means of patient groups divided by the pooled SE of all studies according to the formula:
XCN- XRA
~-~cN) 2 + (~RA) 2
The weighted mean SE (SE) of CN and RA was calculated from the formula:
/ ~ x SE) 2
N 2
where n is the number of CN or RA subjects in each study and N 2 is the summed number of subjects in each patient group. *Percentile standardized difference. l"t = 1.84, P = .07, excluding the largest data set from Feher et al. 5° St = 0.46, NS, excluding the largest data set from Feh~r et al.S0
(Table 3). Four studies also reported DHEAS levels. In each study, either serum total T or derived free T was lower in the RA than one of the control groups (Table 3; Fig 3). Differences were relatively greater in the Genoa, Italy, 59,61,65 than Glasgow, U K , 60 o r London, U K , 62"64 studies.
Among RA patients in a London study, 64 serum T was found to be significantly (P < .05) lower in those 15 who were HLA-B15+ (10.6 nmol/L) than in those 56 who were H L A - B 1 5 - (13.1 nmol/L). Among normals segregated by the HLA-B15 marker, the difference in mean serum T levels (13.8 v 16.2 nmol/L, respec- tively) was in the same direction but not quite
significant (P < .07), as shown in Table 3. 64 The RA patients were older (approximately 60 years of age) than the normals (approximately 35 years of age), and statistics were not reported on the serum T differences between these case and control groups.
Serum DHEAS was significantly (P < .05) lower in seven RA than six OA patients 59 and in seven RA than eight OA patients 65 from Genoa, Italy, but not in a third study from that unit 61 or in the largest study from London, U K . 62
Luteinizing hormone (LH) levels were re- ported for male RA versus control subjects in four studies. 6°-62,65 Two reports 6°,65 indicated
SEX H O R M O N E S A N D P R E G N A N C Y IN RA 9
O E
4.5
4
3 .5
3
2 .5
2
1.5
1 0.5
0 Subjects: 19 20
9 Refs: [ 5 3 ]
26 20 14 12 6 15 8 8 18 22 27 49 10 11 13
9 9 9 9 9 9 9 [531 [511 [561 [491 [521 [551 [481
47 15 12 8 8 14 25 27 29 68 7 6 7 7 7 14 8
9 9 9 ~ 9 d' 9 G [50] [521 [491 [621 [50] [59] [511 [65,61]
FP<O.O5 I p < 0.01
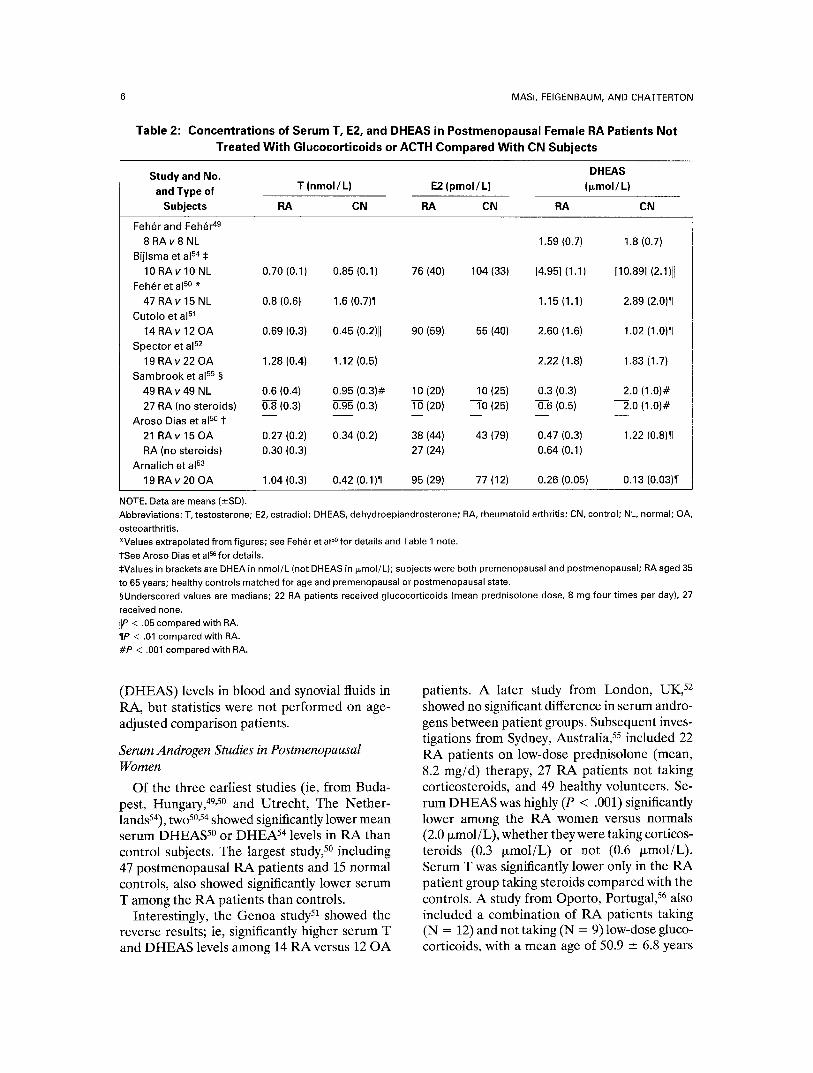
Fig 2: Mean serum DHEAS levels (l~mol/L) of RA patients not treated previously with glucocorticoids versus control (CN} subjects reported in various studies [referenced] of premenopausal and postmeno- pausai women and men. Studies are ordered from left to right according to increasing mean levels of the controls. Significance levels and numbers of subjects are specified in relation to each bar. Note that studies of postmenopausal women concentrate in the left end of the figure, except for one study of premenopausal women. 53 *P < .05; * *P < .01.
higher (P < .05) levels among the RA than AS, normal, or OA controls. The other two stud- ies 61,62 showed similar LH levels among the RA versus CN subjects. Results are markedly influ- enced by values in the largest study. 6° Excluding those results, the mean levels are 8.3 m I U / L versus 7.9 m l U / L [t = .27; not significant (NS)]. Serum LH and FSH levels in eight RA patients and eight age-matched healthy male volunteers showed similarly normal responses after LH- releasing hormone (LH-RH) stimulation. 61
Summary of Case-Control Sex Hormone Studies inRA
Grand mean total sex hormone levels, de- rived by summarizing respective studies in each female and male group, are specified in Table 4. Standardized differences are also indicated; ie, mean levels of the controls minus cases divided by the control mean levels [(CN-RA)/CN]. Results of derived serum free T levels in men, 6°,62,63 as indicated in Table 3 (in brackets), are not included in either the summary Table 4 or Fig 3.
Serum DHEAS levels. The RA women had 39% (premenopausal) and 19% (postmeno- pansal) lower mean serum levels of DHEAS than controls (Table 4; Fig 2). Significant differ- ences (P < .001 and P < .02, respectively)were found among the RA versus CN women. If the majority of the data from premenopausal women 5° is excluded, then the remaining differ- ence in mean DHEAS levels decreased to 20% lower among RA versus CN subjects (P = .07). In men, only borderline (P = .07) difference is found among the limited studies.
Serum total T levels. Among premenopausal women, the significant difference in T could be attributed entirely to one large study 5° (Table 4; Fig 3). Exclusion of this study yielded nearly the same mean serum T values among the premeno- pausal RAversus CN subjects (0.86 nmol /L and 0.89 nmol/L, respectively). In postmenopausal women, no evidence was found of serum T difference among RA versus CN subjects (mean, 0.86 v 0.86 nmol/L, respectively).
In men, decreased serum total T in RA versus CN subjects is strongly (P < .001) supported by

10
2.5
MASI, FEIGENBAUM, AND CHATTERTON
25
20
.-. 1.5
E
"6 E e. ,
0.5
15 _s" E
10 m
o E e-
5
0 Subject=. u , ~ , = = u , ~ , = = u ¢ v , , , v , u = , ~ = , u , , , ~ , = = = , = o , , ~ u , , ~ , ~ u
9 9 9 9 9 9 9 9 9 9~9 a' Refs: [56] [53] [51] [53] [51] [54] [55] [46] [52] [52] [50] [63] [591 [621 [61,651 [611
19 Premenopausal ~ Postmenopausal (~ ialeJ Fig 3: Mean serum total testosterone levels (nmol/L) of RA patients not treated previously with glucocorticoids versus control (CN) subjects reported in various studies [referenced] of premenopausal and postmenopausal women and men. Separate ordinate scales are specified for women (left) and men (right), which differ by a factor of 10. Studies are ordered from left to right according to increasing mean levels of the controls, on their respective scales. Significance levels and numbers of subjects are specified in relation to each bar. Note that studies of males are concentrated at the right side of the figure. *P < .05; * *P < .01; * * *P < .001.
the summary data derived from individual case- control studies (Tables 3 and 4). A similar magnitude of difference is reported in derived free serum T, 6°,62 strengthening the observations.
Serum estradiol levels. No evidence of serum E2 deficiency or excess was found in either the premenopausal or postmenopausal women or the one study in men 61 (Table 4).
Serum L H levels. In men, a higher mean serum LH level was found in RA versus control subjects, but the summary difference was due mainly to one large study. 6° Exclusion of those results yielded a nonsignificant difference (t = .27; Table 4).
Qualifications of case-control serum sex hor- mone studies in RA. Improved case-control studies of serum sex hormones in RA are needed. Better-defined patient clinical subsets 5° and greater comparability of controls 53 might reduce the considerable variability (intra- and interstudy) found in the reported results (Tables 1 through 3; Figs 2 and 3). Studies of early-onset
RA, including CD controls, 48 are needed to address the critical dilemma of primary versus secondary decreases in serum androgens. 3,24,39 Also, other potentially confounding relation- ships with immunologic mediators or HLA types (see below) need further investigation. 3,39
Sex Hormones in Postmenopausal, HLA-Identical, RA-Discordant Sibling Pairs
Sex hormones were studied in a potentially highly informative sample of 50 postmeno- pausal, HLA-identical, RA-discordant sibling pairs. 66 Siblings were matched on HLA-A, -B, and -CW types (with some HLA-DR typing for antigens 1 through 10). The mean age of RA siblings was 62.7 years (range, 45 to 81 years) when studied. They had developed RA at an overall mean age of 47.7 years, ie, 15 years before study. Onsets were between 34 years before to 24 years after menopause. Women who were receiving hormone replacement therapy and RA siblings who had taken oral

SEX HORMONES AND PREGNANCY IN RA 11
glucocorticoids within the previous year or intra- articular steroids in the previous 6 months were excluded. This study was not included among the previously described controlled surveys (Tables 1, 2, and 4), as serum sampling was not performed during the premenopausal years among the sizable proportion (46%) of women having such onsets.
The RA probands had a significantly (P < .01) lower mean serum DHEAS level Qxmol/L) than the non-RA siblings; ie, 1.27 versus 1.97, respec- tively. Although not exactly equivalent, these values may be compared with the previously summarized mean serum DHEAS levels Qxmol/L) reported on women with postmeno- pausal onset of RA versus CN (Table 4); ie, 1.32 versus 1.63, respectively. The percentile stan- dardized differences between RA and CN val- ues for the sibling 66 and summarized studies (Tables 1, 2, and 4) are 36% and 19%, respec- tively.
These data 66 support a concept of lower DHEAS levels in postmenopausal women with RA versus CN (or sibling) subjects. They fur- ther suggest that factors other than the shared HLA-A, -B, and -CW types account for the significant differences found. Further HLA stud- ies are needed, with careful matching of class II alloantigens (ie, HLA-DRB1, -DQA, and -DQB). These markers were shown to associate with disease severity 13,14 and pregnancy-induced remissions 41 in RA.
The primary question persists as to whether the difference found in DHEAS (or other AA steroids) may be a primary or secondary phe- nomenon. The serum level of DHEAS corre- lated inversely with disease duration, a radio- graphic grading score as well as a clinical score of disease activity and severity. 66 Data provided on the RA siblings allowed estimation (A.T.M.) of the mean DHEAS levels (~zmol/L) for women with premenopausal (1.07) versus postmeno- pausat (1.37) onsets. However, respective ages at the time of serum sampling were not pro- vided.
The investigators 66 suggested that the low levels of DHEAS found in the RA women may have been a consequence of RA rather than a predisposing factor to disease. No significant difference in serum levels of total T, derived
free T, A, E2, or sex hormone-binding globulin (SHBG) was found between the RA and nonaf- fected siblings. The conclusion of this study was that abundant evidence exists to suggest that sex hormones are important in the etiology and pathogenesis of RA. However, clarification of their role must be approached in a manner different from that adopted in the report. 66
Cross-sectional case-control studies cannot clearly distinguish determinants of risk or predic- tive markers from associated or secondary dis- ease effects, especially when sampling occurs some 15 years after onset. 66 Early-onset, longitu- dinal case-control studies would be more infor- mative. However, discrimination of primary ver- sus secondary effects are best accomplished in prospective analyses (see below).
Preliminary, Prospective, Controlled Studies of Hormone Levels in RA
To test the hypothesis that decreased AA steroids may precede and possibly predispose to onset of RA in menstruating women, 24 available sera from a community-wide prospective study were analyzed. 67,68 The prospective study (Op- eration CLUE) was initiated in 1974, when the sera were obtained from participants. For pur- poses of this prospective study, an RA case is defined as a women who developed RA, accord- ing to ACR criteria, after entry to study. For each case, four control women were selected who did not develop any known systemic rheu- matic disease and who participated in this study in the same manner as did the cases. Both cases and controls donated serum at the start of the 1974 community survey. Controls were matched to the cases at the time of entry to study (ie, the pre-RA women on entry to study) for race (all White), age (+6 years) and pre- versus post- menopausal status. Menopausal status was based on histories of the last menstrual period (LMP) on study entry plus assays of plasma follMe stimulating hormone and LH on sera donated at that time. Levels of AA hormones (ie, DHEA, DHEAS, A, T) and cortisol and 12 other adre- nal-gonadal-pituitary hormones are being blindly assayed on case and control sera. The mean (_SE) serum AA steroid and cortisol levels (all in nmol/L) plus P values (two-tailed) are as follows for the initial 14 RA and 56 CN subjects

12 MASI, FEIGENBAUM, AND CHATTERTON
having sera fully assayed: RA: DHEA, 6.5 _ 0.9; DHEAS, 2,235 _+ 608; A, 4.9 _+ 0.6; T, 0.75 + 0.08; cortisol, 242.0 _+ 36; CN: DHEA, 12.7 + 1.1 (P < .05); DHEAS, 2,966 + 269 (P < .39); A, 8.2 _ 0.5 (P < .01); T, 0.95 _+ 0.06 (P = .17); cortisol, 269.7 _+ 12 (P -- .40).
The preliminary mean serum DHEA and A levels were significantly (P < .05 and P < .01, respectively) lower in the women who subse- quently (median, 9 years; range, 4 to 15 years) developed RA than the matched control sub- jects. The five women who developed RA at a younger premenopausal age (mean, 35.5 years) showed greater decreases in AA levels than either the four women who had onset at an older premenopausal age (43.3 years) or the five women with postmenopausal onsets (53.4 years). No other hormone studied differed significantly between RA versus CN groups.
Serum levels of DHEAS and cortisol have now been analyzed on an additional 21 RA cases and 84 controls (CN), making a total of 35 RA and 140 CN subjects having these steroids assayed. 6s The Wilcoxon signed-rank test was used to derive two-tail probability levels (Table 5). The DHEAS and cortisol data were ana- lyzed in total and within three subgroups, accord- ing to entry menopausal status (EMS) plus onset age of RA, ie, before age 50 versus 50+ years. The three groups analyzed were (1) cases who were premenopausal (pre-MS) at entry and had onset of RA before age 50 years; (2) cases
who were pre-MS at entry and had onset of RA at age 50+ years; and (3) cases who were postmenopausal (post-MS) at entry (all of whom had onset at age 50+ years).
The 11 younger premenopausal RA cases had a greater (P < .05) decrease in DHEAS versus their 44 CN (41%) than the 11 older pre-MS versus their 44 CN (14%) or the 13 oldest post-MS RA versus their 52 CN (15%) women (Table 5). Mean serum cortisol levels were similar between the RA and CN subjects in total and in each of the three subgroups.
Low serum DHEAS levels may be a long- term risk factor or marker for RA in younger, premenopausal, white women. Assays of the remaining 15 hormones are being performed in the additional 21 RA and 84 CN subjects. These preliminary prospective data 67,6s support a hy- pothesis that decreased AA hormones may be either a determinant or a predictive marker for subsequent onset of RA in women. Further prospective studies are needed with larger num- bers of subjects to better define age, race, gender, menopausal status, HLA types, and other demographic factors that may influence the relationships of pre-existing AA hormone levels to subsequent onset of RA.
Limitations of Studies of Total Serum Steroid Levels in RA
Measurement of random serum total steroid concentrations do not, of themselves, indicate
Table 5: Serum DHEAS and Cortisol Levels Among 35 RA Cases Before (mean, 12 years) Onset of Disease Compared With 140 CN Women Without Rheumatic Disease and Matched on Entry Age,
Race, and MS
Pre-MS and Pre-MS and Post-MS and RA Onset at RA Onset at RA Onset at Total Study Age < 50 yr Age 50+ yr Age 50+ yr Subjects (RA = 11) (RA = 11) (RA = 13) (RA = 35)
Mean age at entry of RA (yr) 29.3 46.2 54.1 43.8
Mean age of RA onset (yr) 41.2 58.1 65.6 55.5
Entry DHEAS levels (nmol /L)
RA 2,145 _+ 475* 2,435 + 603 1,388 _+ 238 1,955 _+ 261"
CN 3,623 _+ 363* 2,819 _+ 613 1,639 _+ 185 2,633 _+ 243*
Entry cort isol levels (nmol /L)
RA 312 _+ 62 224 -4- 36 253 _+ 31 262 +_ 25
CN 245 _+ 19 239 -4- 20 255 + 17 246 _+ 10
NOTE. Data are means -+ SE. Four CN women were matched to each pre-RA case. Abbreviations: DHEAS, dehydroepiandrosterone sulfate; RA, rheumatoid arthritis; CN, control; MS, menopausal status. *Wilcoxon signed-rank test, P < .05.

SEX HORMONES AND PREGNANCY IN RA 13
the net effects of these hormones or their milieu upon target cells. 3,39 Most serum total steroid levels have diurnal fluctuations. Also, they can vary with changes in the concentration of their respective serum binding proteins. Further- more, even serum free hormone levels can have varying effects and concentrations in tissues. 69 Complex intracellular steroid receptor binding dynamics 7° or antagonistic actions at the ge- nomic or transcriptional levels (eg, cortisol and DHEA interactions on the thymus 71) can affect target tissue responses, acutely or chronically. Other mediators, eg, dopamine, may activate steroid receptors (eg, the progesterone recep- tor) in the absence of ligand binding. 72 Other hormones may have synergistic actions with steroids, eg, those of glucagon and cortisol on gluconeogenesis. Prostaglandins, growth fac- tors, and cytokines may also modify the effects of individual hormones on target tissues, 7° be- sides various medications which active RA pa- tients may be taking.
For these various reasons, one must cau- tiously interpret results of serum hormone stud- ies in RA, even if found in controlled, prospec- tively designed investigations .67,68 Serum differences found in specific hormones (eg, AA) may reflect unrecognized dysfunctions in other biologic systems and, therefore, may be markers of disease rather than causal determinants.
Interpretation of Available Controlled Serum Sex Hormone Studies in RA
Summary interpretation of these available case-control sex hormone data are difficult be- cause of the many above-specified qualifica- tions. Previous review of earlier-vintage, con- trolled studies of serum glucocorticoid levels of RA versus control subjects revealed relatively small differences. 24 However, adrenocortico- tropic hormone controls glucocorticoid produc- tion as a primary function and is not influenced by adrenal cortical androgen levels. 3,39 Also, various inflammatory mediators in RA [espe- cially interleukin-1 (IL-1)] can stimulate gluco- corticoid secretion. 3,25-3° Thus, important, under- lying glucocorticoid deficiencies may exist in RA but may be masked and not detected by random serum cortisol assays (see below).
Given the total available reported data, one may suspect that preexisting androgenic hor-
mone deficiencies do occur in selected RA subgroups. 3,38,39,67,68 Deficiencies seem to exist in serum DHEA and DHEAS, particularly in pre- menopausal-onset RA, 3,38,39,68 as well as in se- rum T in males. 38
Findings are inconclusive, however, in terms of relative degrees of adrenal versus gonadal deficiencies of AA hormone synthesis in women versus men, or their mechanisms. 3,39 Women depend more on adrenal than gonadal contribu- tions to androgen synthesis than men. 3,39 One might suspect relatively greater adrenal insuffi- ciency in women and greater gonadal deficiency in men with RA. Such deficiencies might be due to primary, enzymatic steroidogenesis factors in the endocrine organs or secondary effects from H-P dysfunctions or other control mediators/ mechanisms. Androgenic deficiencies may occur independently or in association with possible dysfunctions of adrenocortical glucocorticoid physiology or HPA responsiveness (see below).
Biologic Plausibility of Sex Hormones as Potential Risk Factors in RA
HLA and Sex Hormone Risk Factor Associafions
Class I HLA-B15 antigen was associated with lower mean serum testosterone levels than any other HLA type in men, 64 both in RA and normal control subjects (Table 3). This antigen may be more frequent in men than women with RA. 64 Class I HLA-B8 antigen was associated with lower than expected (P < .05) mean serum testosterone levels in women, v3
Male gender predominated among RA pa- tients at a tertiary medical referral center who were homozygous for HLA-DRB1 alleles of the -04 type (old terminology: -DR4, -Dw4, or -Dw14). These men had more aggressive nodu- lar, joint, and systemic disease than other pa- tient groups. 13 HLA-DR4 status was not found to be associated with low serum testosterone in one study of male RA cases. 62 However, testos- terone synthesis is genetically influenced in men, as blood levels were correlated in haplo- type identical brothers. 74 HLA-DR4 was found to be associated with low free and total testoster- one in both healthy women and postmeno- pausal women with R A Y ,75
HLA-DR4 associations with RA are com- plex; variations were found between the sexes and by age in women, v6 No association was

14 MASI, FEIGENBAUM, AND CHATTERTON
found in men, unlike women. The strength of association decreased significantly with aging in women. 76 These 76 and other studies 13,64,73,75 indi- cate interactions between HLA antigens and age-related and sex-related factors in RA that may affect susceptibility or outcome. Low serum androgens in younger female RA patients is consistent with the findings of HLA-DR4 asso- ciation with RA in this subgroup 76 and low serum T observed in women with this allele. 75 In the mouse, the H-2 complex [analogous to the major histocompatibility complex (MHC) in humans] encodes gene(s) responsible for regu- lating testosterone and testicular size. 9 The biologic plausibility of genetic linkages between the immunogenetic and endocrine systems are supported by available data, as has been sepa- rately reviewed. 3,33,36,38,39 Such complex interac- tions may be conjointly contributing to suscepti- bility and outcome in RA.
Experimental Data
Estrogen administration at physiologic levels. The development and severity of type II colla- gen-induced arthritis in mice and rats can be suppressed'by physiologic levels of estrogens, as recently reviewed. 77 This experimental arthritis model is T cell-dependent and is believed to simulate human RA. Also, estrogen has been demonstrated to enhance B-cell activities. 3,33,34,78 These findings suggested 77 a dualistic action of estrogens; ie, reduction of T cell-dependent disorders (eg, RA) but enhancement of B cell- mediated processes leg, circulating immune complex formation in systemic lupus erythmato- sus (SLE)].
Female Lewis rat. The female Lewis rat (LEW/N) rat is vulnerable to streptococcal cell wall (SCW)-mediated arthritis, a mainly B cell- mediated model. 79,s° Blunted glucocorticoid pro- duction has been found in this strain, secondary to dysregulation of its corticotropin-releasing hormone (CRH) response. 79,s° In contrast, con- trol histocompatible Fisher (F 344/N) rats show robust CRH responsiveness and active hypothal- mic-pituitary-adrenal (HPA) axis function. Their greater corticosterone responsiveness effec- tively suppresses the development of chronic inflammation after the same early, acute reac- tion to SCW immunization. 8° Male sex hor- mones promote resistance to SCW-mediated
arthritis in LEW/N rats. Either castration or estrogen treatment of males was found to in- crease severity of this disorder. 77
Prolactin
Prolactin, pregnancy, and autoimmune dis- ease was reviewed recently. 81 A concept was developed that the pituitary gland secretes both immunostimulatory hormones [eg, prolactin (PRL) and growth hormone (GH)] and immuno- suppressive hormones (eg, ACTH). PRL recep- tors have been found on human T and B lymphocytesY Decreased bioactivity of circulat- ing prolactin 83 and prolactin deficiency 84 have been reported in RA. Multiple endocrine altera- tions in RA patients were cited 81 and reviewed. 85
Cortisol Circadian Rhythms
Although serum cortisol levels were found to be within the normal range in RA, they were considered to be inappropriately low for the degree of ongoing inflammationY Circadian variation in disease activity in RA 87 is believed to be influenced by diurnal fluctuations in circu- lating plasma cortisol levels and their antiinflam- matory effects. 88
Hypothalamic and Neuroendocrine Axis Deficiency
Deficiencies in central nervous system (CNS) stress response to immune and inflammatory stimuli were proposed in patients with RA, 89 analogous to abnormalities found in the SCW- induced arthritis of LEW/N. 79,s° The deficient HPA response to inflammation is believed to permit an excessive immune/inflammatory reac- tion, which may then switch from acute to chronic inflammation.
Patients with RA showed an insignificant increase in plasma cortisol levels 1 and 2 days after hip or knee arthroplasty, in marked con- trast to patients with OA. 89 Changes in ACTH and serum cortisol were measured in 10 RA and 10 healthy control subjects after ovine CRH infusions over 180 minutes. Similar temporal profile and quantitative changes were noted in both groups. However, the RA patients showed slightly lower serum cortisol levels than the control subjects in the later response period (ie, 90 to 180 minutes), in the presence of slightly higher or equal ACTH concentrations (see Fig

SEX HORMONES AND PREGNANCY IN RA 15
5 in Chikanza et al89). Further controlled stud- ies discriminating the degree of hypothalamic- pituitary (H-P) vis-a-vis adrenal cortical gluco- corticoid responsiveness to various stimuli are indicated in RA. Such data can determine the level(s) of possible deficiency in the HPA stress system in RA. 27,3°,9°
Adrenal Androgenic Deficiency Versus Gonadal Abnormality
In menstruating RA women, significantly low urinary excretion of adrenal AA hormone me- tabolites suggested a preexisting deficiency. 24 Alternatively, a gonadal hypothesis for the devel- opment of RA was raised. 33 Women were pro- posed to be "more female" and men "less male" than their unaffected counterparts. 33 The above-reviewed data do not support a concept of greater femaleness in RA women, but rather decreased androgenicity. 3,38-4° However, less maleness is consistent with the findings re- viewed in RA men (Table 4). Whether the sex hormone findings result from adrenal, gonadal, neuroendocrine, or other combined endocrine and immunologic system interactions remains to be determined. 3
Oral Contraceptives and the Risk of Developing RA
The use of oral contraceptives (OCs) and other female sex hormones has been studied in relation to either the risk of developing RA or treating active disease. 91-97 Such observational studies of cause-effect relationships have signifi- cant, inherent self-selection and ascertainment biases.38.97-100
Metaanalyses were performed on some 13 studies pertaining to the question of whether or not use of OCs protects against the develop- ment of RA. 96,1°° A lower use of OCs in RA patients than in normal comparison women would infer a protective effect against RA among the users. However, self-selection fac- tors influencing the use of OCs must be consid- ered before drawing conclusionsY ,39 Only two studies reported a 95% confidence interval (CI) of their odds ratio (OR) below 1.0 among RA patients. Metaanalysis of available data in the two separate reports 96,100 resulted in differing conclusions with respect to a protective effect of OCs on the development of RA.
Hospital-based studies showed a protective effect of OCs (OR, 0.49; 95% CI 0.39 to 0.63), but population-based samples did not (OR, 0.95; 95% CI, 0.78 to 1.16). l°° Use of OCs was inferred to protect against progression of RA to severe disease in the hospital sample. 1°° How- ever, unknown selection and comorbidity fac- tors may be operating that could bias those results. 38,39,1°° Overall, available data indicate both the complexity of the issues and lack of definitive conclusions.
Any associations observed with use (or non- use) of OCs and the development of RA seem more likely to be due to self-selection or index- ing by the type of women using these prod- ucts, 3s,39,1°1 rather than a true protective effect. This assessment also seems to apply to women who develop more severe RA. Similar conclu- sions have been drawn about peri- or postmeno- pausal women using noncontraceptive replace- ment hormones. 97
Sex Hormones in the Treatment of RA
Effects of Androgens on RA
An open 6-month clinical trial 65 of testoster- one undecanoate (TU; 40 mg orally, three times per day with meals) was performed in seven male RA patients (mean age, 57.9 years + 9.1 SD). Baseline serum sex hormone concentra- tions in these RA patients were compared pretherapy with an age-matched control group of eight men with secondary OA 65 (Table 3). Results in the latter controls are the same as previously reported by this group. 61 At baseline, percentages of CD8+ T cells were significantly (P < .05) low in the RA patients. The ratio of CD4+ to CD8+ T cells was significantly (P < .01) high versus controls (2.2 _+_ 0.2 v 1.5 - 0.2, respectively).
The TU therapy fully normalized the low baseline mean serum T concentrations among the RA patients within 3 months (P < .05). The originally lower serum DHEAS level (1.71 p~moi/L) was also increased (P < .05) some- what (to 2.8 ~xmol/L) by TU therapy. No signifi- cant effects were found on other hormones reported. Surprisingly, free T and SHBG levels were not significantly affected by the treatment.
At the end of 6 months, the mean number of tender joints decreased (P < .01) to five from seven at baseline. The IgM rheumatoid factor

16 MASI, FEIGENBAUM, AND CHATTERTON
(RF) decreased significantly (P < .05) by 6 months from baseline levels. Both CD4+ and CD8+ T-cell measures normalized at the 3- and 6-month evaluations while on TU therapy. In this uncontrolled trial, the overall clinical status was considered to be improved (by 60%) with no relevant side effects from TU therapy.
Miscellaneous Other Therapeutic Observations
Estrogen, progestogen, or androgen ~°2J°3 therapy of RA patients in the preglucocorticoid era produced variable results, as previously reviewed, 24 and was not further promoted. How- ever, intraarticular progesterone has more re- cently been demonstrated to be effective in R A . 104
PREGNANCY AND REPRODUCTIVE HISTORY IN RA
A high proportion (approximately 75%) of RA patients substantially improve or remit during pregnancy, starting after the first month o r tWO. 32,41,42,105"108 Also, the onset of RA is delayed or less frequent during pregnancy ~°9 and increased in the year postpartum. H° For these reasons and because of the important effects of pregnancy on the immunologic and microvascular systems (see below), studies of reproductive events related to RA in women deserve priority attention.
Fecundity, Fertility, and Pregnancy Outcomes Before Onset of RA
Reproductive history before onset of RA, ie, fecundity (probability of conception), fertility (ability to conceive a child), and pregnancy outcomes, may offer clues to factors that affect host susceptibility to or mechanisms of RA. m However, as with observational studies of OC use and RA, undefined self-selection and demo- graphic factors complicate analysis and interpre- tation of such results. 3s,39
Reproductive and contraceptive history was analyzed in a cohort of women with recent- onset RA, recruited from 1986 to 1991 from the Seattle, WA, area, and compared with commu- nity or clinic controls. 211 Only cases and controls who had an opportunity for pregnancy were analyzed. Decreased fecundity was defined as "unprotected intercourse on a regular basis for
> 12 months without pregnancy." Among 259 RA cases, 42% indicated having had an episode of decreased fecundity compared with 34% of 1,258 controls (OR, 1.44; 95% CI, 1.10 to 1.91). These results were interpreted to indicate dimin- ished fecundity (ie, probability for conception). However, 88% of cases and 89% of controls did become pregnant during their reproductive life span. Thus, fertility (ie, ability to conceive a child) was equivalent among the RA cases and controls. No difference was found either in the frequency distribution of numbers of pregnan- cies or in pregnancy outcomes among these subjects.
The interval from the episode of suspected decreased fecundity until the subsequent onset of RA was substantial, being 20+ years in 71%. Numerous hormonal, 3,24,38,39 immunologic, 3,34-39 and behavioraP s factors may have interacted to influence both the frequency of "unprotected intercourse on a regular basis" and the risk of developing RA. Issues of libido and frequency of intercourse were not addressed in the article. Both low androgen concentrations and de- creased libido (as well as lower use of OCs) were suggested to precede the onset of RA in women. 3a This hypothesis 3a is consistent with low serum DHEAS levels in younger menstruat- ing women before onset of RA. 6s Should de- creased fecundity (rather than frequency of intercourse) be confirmed in women antecedent to RA, the cause(s) may offer clues to disease susceptibility.
Fertility and Pregnancy in RA Patients
After disease onset, women with RA have reported lessened sexual desire as well as sub- stantial reduction of coital frequency, u2 Subfer- tility has been reported in RA, both before and after development of RA. 113
Increased nulliparity has been reported in women with RA versus comparison women in a majority of epidemiologic studies. 43 The mecha- nisms that may be operating to explain this reproductive finding in women who develop RA are unknown. Decreased serum AA hormone levels preceding onset of RA 38,67,68 and de- creased libido 38,m could affect sexual behaviors and the probability of nulliparity among women with RA.

SEX HORMONES AND PREGNANCY IN RA 17
Age at menopause is normal in women with RA and approximated the mean or modal onset of disease in several studies. 66,114 Such data suggest that menopause may be related to the onset risk and age-specific incidence of RA in women. 4° One may suspect that different combi- nations of determinants may exist in women having onset of RA during their premeno- pausal, perimenopausal, or postmenopausal years . 3,3s,39,66-68,114 HLA-DR4-related mecha- nisms and low androgens may be relatively more important risk factors before than after meno- pause. 3'38-40'68'76 Physiologic aging mechanisms, eg, decreased microvascularity and cartilage degenerative processes, may assume a relatively greater role in onset risk of RA at older ages. 3
Theories of How Pregnancy Affects RA
Pregnancy induces profound changes in the hormonal, immunologic, and microcirculatory systems of the mother. 42 Multiple hypotheses have been proposed to explain gestational im- provement in R A . 41-43,108,115"117
Immunomodulatory Effects
Autoimmunity is a state of abrogation of normal tolerance barriers with autologous anti- body formation and failure in discrimination of self from nonself antigens. Autoimmunity does not necessarily imply immunopathogenesis. 3 Im- munosuppressive mechanisms in pregnancy may modulate immunopathogenetic processes in RA. 4t-43'108'115"117 However, generalized theories of pregnancy-induced immunosuppression do not explain this phenomenon. 10s Gestation may worsen SLE 42,1°8 and does not usually affect AS.11s
Humoral immunity is not usually altered during pregnancy. 10s Cell-mediated immunity (CMI), however, is profoundly affected in preg- nancy, both in the fetoplaeental unit and, more generally, in the mother, l°s Pregnancy may in- duce increased susceptability, H9,120 reactiva- tion, 121 and virulence 119-121 of certain viral and other ~19 infections. Depression of CMI may be due partly to decreased levels of T-helper cells, 122 on the order of 25% reduction during early pregnancy. 1:3 However, maternal T-helper lym- phocytes have normal sensitivity to pokeweed mitogen during pregnancy.10s Additionally, selec-
tive suppression of polymorphonuclear 124 and macrophage 125 function as well as ovarian- derived and placentally derived immunosuppres- sive mediators l°s and other complex mecha- nisms, may contribute to depressed CMI during pregnancy. HLA class II surface antigen expres- sion is suppressed on villous trophoblast cells by an IgG fraction from maternal serum. 126 Preg- nancy-associated alpha 2-glycoprotein (PAG) is synthesized by mononuclear leukocytes in the maternal decidua. This protein was reported to be inversely correlated with an index of RA activity during pregnancy in one study, 127 but this was not confirmed in another study. 128 PAG is one example of a rapidly growing list of early pregnancy factors (EPFs) that are heteroge- neous molecules, many of which have immuno- suppress ive activity. 129
Pregnancy sera contain other factors (eg, cortisol) that are believed to suppress T- lymphocyte response to mitogens.130 Mixed lym- phocyte reactions (MLR) are also suppressed by pregnancy plasma 131 and can be reversed with addition of recombinant IL-2.132 Interest- ingly, IL-2 secreted by large granular lympho- cytes in the pregnant uterus may help promote neovascularization at the maternal-placental in- terface. ~33 Evidence of direct estrogenic regula- tion of human CRH gene expression ~34 may indicate another pathway of immunomodula- tion during pregnancy, when estrogen levels increase considerably.
Angiogenesis Mechanisms in RA and Angiostatic Steroids
Angiogenesis is believed to be an integrat part of the pathogenesis of RA, as recently reviewed. 21 It is also an essential process in normal placental function. 133 One provocative question in RA is its primary event: do the vascular changes 15-21 initiate or follow the syno- vial inftammationU -s The answer is not pres- ently known. 3,39
Regulation of blood vessel reactivity 46 and growth 45,135 in health and disease is complex. Angiogenesis in RA is believed to be primarily under the control of peptide mediators, includ- ing cytokines. 21 Importantly, angiostatic ste- roids, eg, tetrahydrocortisol, have been de- scribed in the chorioallantoic membrane.45,136,137

18 MASI, FEIGENBAUM, AND CHATTERTON
Potential effects of such steroids and other hormones generated in pregnancy on induced remissions of RA deserve further attention.
Hormonal Influences of Pregnancy That May Affect RA
Integrated fetoplacental-maternal unit. Avari- ety of steroid and polypeptide hormones and other mediators are produced by the placenta in abundance during pregnancy and may affect RA. These include CRH, proopiomelanocortin (POMC), RNA, and their biologic and immuno- logically active products, eg, ACTH. 138-141 Mater- nal ACTH levels are usually increased in preg- nancy, 14° which may be related to estrogen effects. 134 Immunomodulation, intravascular pro- tein production, increased microcirculation, tis- sue/vessel permeability changes, and other path- way alterations occur in pregnancy and may also affect RA. 42 The placenta does not produce mineralocorticoids or glucocorticoids directly.
Cortisol levels. In pregnancy, both free and total cortisol levels are elevated, us,116,14° Al- though circadian secretion of cortisol (F) per- sists, evening levels do not decline as much as in nonpregnant women, n6 As may occur in the nongravid female, 8s inflammatory cytokine pro- duction is downregulated by glucocorticoids in the pregnant woman. 13°
Reproductive steroid hormones. Estrogens, pri- marily estrone (El) and progesterone, 117,13s-14° are synthesized in great amounts during preg- nancy and have also been suggested as factors ameliorating RA. Estrogens decrease metabolic clearance of F by direct action on liver enzymes responsible for inactivation of cortisol. By mid- pregnancy, over 90% of maternal estriol (E3) production is derived from fetal adrenal precur- sors. 142
PRL and human placental lactogen. During pregnancy, PRL and hPL reach highest levels in the third trimester. The role of human placental lactogen (hPL), which has both GH- and PRL- like activities, on the immune system has not been determined. However, PRL has profound effects on lymphocytes, sl,s2,85 A specific receptor for PRL has been identified on human T- and B-lymphocyte cell membranes. 82 PRL was lower than normal in patients with RA. s4 Prolactin appears to have a bell-shaped, dose-response action; optimum concentrations are required
for maximal lymphocyte stimulation, but lower or higher levels cause decreased or negative responses. 143 Pathologic hyperprolactinemia re- duces the numbers of natural killer cells. 144 Similarly, elevated PRL levels in pregnancy and early stages of lactation may contribute to suppression of immune responses.
Pregnancy is profoundly complex biologically. Factors are produced in abundance in preg- nancy that are either different or more potent than OCs and that may suppress RA. 145,146 Although OCs are examples of synthetic estro- genic-progestogenic hormones, they do not seem to affect the risk of developing RA or its c o u r s e . 38,39,42,96,100,145 Female sex hormone levels are higher in pregnancy than obtained with use of OCs. Progesterone production rate may reach 250 mg daily by the end of pregnancy and reach plasma levels of 130 ng/mL (413 nmol/L). 147 Progesterone appears to be immunomodula- tory. 148 In humans, progesterone binds almost as well as cortisol to transcortin or corticosteroid- binding globulin (CBG). 146,148 One may suspect that high serum free progesterone levels in pregnancy compete with cortisol for CBG sites. Such receptor-binding dynamics might contrib- ute to the elevated free cortisol levels in preg- nancy, a15,116 if not the elevated ACTH levels34°
Androgenic steroid serum levels in pregnancy. Maternal serum DHEA levels do not vary significantly during pregnancy, whereas serum DHEAS levels were reported to decrease 30% to 50% in one study. 139 Maternal serum DHEAS levels were found to be unchanged during pregnancy in another study. 149 However, mater- nal serum levels are difficult to interpret due to increased placental clearance and hepatic me- tabolism53s-14° Serum levels do not adequately reflect production rates of these hormones, which increase during pregnancy. 139 A maternal production rate (PR) of DHEAS was estimated at 20 to 40 mg/24 h, whereas the normal menstruating female PR was 10 to 15 mg/24 h. 139
Fetal DHEAS. Quantitatively, DHEAS is the major steroid produced by the fetal adre- nal. 149 It is the major androgenic precursor in placental estrogen formation538 Almost all DHEAS is produced in the inner (fetal) zone of the fetal adrenal cortex, whereas cortisol is the primary product of the outer, definitive (adult) z o n e . 138

SEX HORMONES AND PREGNANCY IN RA 19
Serum free and total testosterone. Serum to- tal T levels increase throughout pregnancy, reaching four to five times higher levels than in menstruating women. 139 However, serum free T levels increase only near term, x39 as may also occur with serum A levels. 139,14° Detailed and controlled hormone studies are needed of preg- nant RA women who do and do not remit in pregnancy. Also, results should be correlated with maternal-fetal class II alloantigens (see below).
Maternal-Fetal Disparity in HIM Class H Alloantigens and Remission in RA
Interestingly, in one study, maternal-fetal disparities for class II alloantigens occurred significantly (P < .01) more frequently in preg- nancies during which RA remitted or improved than did not improve. 41 In this study, disparities in alleles of HLA-DRB1, -DQA, and -DQB occurred in 26 (76%) of 34 pregnancies in which RA improved versus only 3 (25%) of 12 pregnan- cies in which RA remained active (OR, 9.7; P = .003). The basis for such HLA-linked find- ings is inferred to relate to maternal immune responses to paternal HLA antigens.
Effects of Delivery or Termination of Pregnancy on RA
Marked reductions in serum progesterone and E2 occur within hours after delivery. 138- 140,149 PRL also decreases, with the degree of reduction depending on whether or not the mother is breastfeeding. 15° The inhibition of HLA class II expression by maternal serum was no longer evident in specimens examined at 2 weeks postpartum. 126 After natural or elective termination of pregnancy, RA exacerbates to the p repar tum status within 1 to several months. 32,1°6 About a quarter of RA patients do not improve 32,41 or may worsen during preg- nancy, 1°6 especially in the early stages. Similar course patterns tend to recur with subsequent pregnancies.106,108
An increased incidence of onset of RA has been reported within 6151 or 1211°,152 months postpartum. It is not known if this phenomenon is due to a postponement of RA onsets during the pregnancy period 1°9 or a truely increased predisposi t ion during the pos tpar tum months. 11°,15~,~52 Interestingly, maternal serum
DHEAS decreases significantly after a first pregnancy, and the duration of the effect is at least 150 months after delivery. ~53
Fetal Outcome and Pregnancy Risks to Developing RA
Fetal Outcome in RA Patients
Fetal morbidity or mortality is not generally increased in RA, 1°6,1°8 unlike other CTDs. 42 However, few data are available on women with severe disease. Fetal complications may occur in such pregnancies, especially in women with systemic manifestations, due to either the moth- er's status or aggressive pharmacologic therapy for RA. 105,106,108
Pregnancy as a Risk Factor to Developing RA
The question has been raised as to whether pregnancy is a risk factor to developing RA. 154 Multiparity has been associated with RA, par- ticularly among women having more than four children. 155 Multiparity may also be associated with more severe R A ) °6 Long-term decreases in serum DHEAS resulting from pregnancy 153 may affect risk of subsequently developing RA. 11°,151,~52 Properly controlled data are not available on this relevant issue.
DISCUSSION
This review attempts to integrate recent data and knowledge on physiopathogenic aspects of RA from the perspective of dynamic factors operating on the affected persons. 4° This ap- proach complements investigations of the experi- mental or in vitro pathophysiology and immuno- pathogenesis of the disease. 1 Without knowledge of the etiology of RA or its related CTDs, investigations in various promising research pathways wilt likely be most informative and clinically rewarding. Critical analysis of physio- pathogenic concepts 3 is needed, as they may corroborate or conflict with current knowledge of the pathogenesis of RA. Essential facts of RA need to be differentiated from widely held beliefs. 14 Also, major determinants of disease must be differentiated from associated or sec- ondary phenomena in the pathogenic process. 39
The major determinants of increased risk in RA are advancing age, female gender, and a positive family history of erosive, seropositive dis- ease in a first-degree relative (FDR). 12,4°,156-16°

20 MASl, FEIGENBAUM, AND CHATTERTON
O C
C i
100
90
80
70
60
50
40
30
20
10
0 ml-- --
_.mm .~
/
/
[] I
I mr"
/ ~ Females .~ ~ Males
i I I I I i I
0 10 20 30 40 50 60 70+
Age At Onset
Fig 4: Composite, age-specific incidence of RA per 100,000 person-years in females and males. See Masi 4° for referenced data and explanations. Figure reproduced with permission of Oxford University Press.
These factors are illustrated in Fig 4 and summa- rized in Table 6. Several epidemiologic charac- teristics may be considered facts of RA, ie, observations or relationships that are consistent with all available data. 4° These include the important patterns of increasing onset risk (or incidence) during aging and greater risk in the
female sex, as well as their significant interac- tions during the female reproductive years (Fig 4). Collectively, aging and female sex, both nongenomic traits, exert greater influence on the risk of developing RA than genetics, and these processes need to be determined.
Familial aggregation of seropositive, erosive
Table 6: Estimated Relative Risks of the Aging (70+ years), Female Gender, and Heritability Accentuating Factors Compared With Baseline Frequencies of RA
Accentuating Risk Factors Accentuated Frequencies Baseline Frequencies Accentuating RRs
Age (70+ yr) Incidence (70+ yr)* Incidence (20 yr)* F 80 10 8
M 60 =3 =20
Female gender Incidence (female)t Incidence (male)t Adult ages 45 15 3
Reproductive ages 20 4 5
Heritability RR¢ Prevalence (total)§ DZ twin pairs 3.4 1% 3.4
MZ twin pairs 8.6 1% 8.6
Abbreviations: RR, relative risks; RA, rheumatoid arthritis; DZ, dizygotic; MZ, monozygotic. *Female and male age-specific incidence rates per 100,000 person-years. 4° 1"Female and male incidence rates estimated from Fig 4 composite data. 4° $Observed/expected RR of RA among those DZ and monozygotic MZ cotwin pairs having an affected member with RA, adjusted by age and gender. 16° §Prevalence of RA in the total twin sample studied. 16°

SEX HORMONES AND PREGNANCY IN RA 21
RA has been long-established. 156 However, its mechanisms are complex and heterogeneous. 1°44 Familial aggregation of RA varies by age and sex of the probands. Heritability of RA is more likely when the proband has disease onset under age 45 years 157,~5s or is male 158 than the converse. Twin studies 12 document heritability of RA, but the maximum genomic contribution is estimated to be 15%. The ratio of MZ to DZ twins, an indicator of heritability, may vary from approximately 6 for persons affected with RA requiring hospital attendance 156,a59 to approxi- mately 2 within a nation-wide twin registry s tudy. 16° Heritability has not been documented for seronegative, milder RA. 12-14,156 Available data suggest that for erosive, seropositive RA, the relative risk of genomic determinants may be approximately threefold for FDRs (as DZ twins) and a maximum of eightfold for MZ co-twins, independent of age- and gender- related factors. 12
The heritability 12,39,157,158 and immunogenet- ics 10-14'38'39'76 of RA cannot be explained on the basis of a single, autosomal dominant HLA- DRB1 gene locus controlling antigen binding and presentation of arthritogenic peptides.14 To better explain the considerable heterogeneity in clinical expression of RA, a two-haplotype ge- netic model was proposed. 14 Homozygosity of RA-linked alleles at the HLA-DRB1 locus correlates strongly with aggressive, erosive forms of RA, but this occurs in a minority subset of patients, 14 particularly among men. HLA-linked factors were deemed to have a limited pathoge- netic role in the overall spectrum of RA and to not explain the onset risks of disease] 4 as was also indicated in twin studies. 12
A decade ago, 24 AA steroid deficiency was proposed as a possible predisposing factor to RA in menstruating women. That suspicion was based on results of clinical research center (CRC)-protocol studies of early-diagnosed RA in women treated with only relatively modest doses of aspirin or other nonsteroidal antiinflam- matory drugs (NSAIDs) compared with matched, normal controls. Subtle delays in HPA glucocorticoid responsiveness were observed af- ter induced hypocortisolemia. 24 However, far more significant deficiencies of adrenal AA urinary steroid metabolites (ie, the ll-deoxy-17- ketosteroids) were found in the RA patients than controls. Over the past decade, numerous
controlled investigations 4s47,s9-65 have been pub- lished on serum sex hormone levels in RA patients who were not treated with glucocorti- coids. Those results are analyzed in this review. An independent assessment of this literature was also published recently, 3s in which James also proposed that preexisting androgen deft- ciency exists in RA.
The aggregate data indicate that low andro- gen concentrations as well as HLA-DR4- related factors correlate with the onset of RA in premenopausal w o m e n . 24,38,39,67,68,76 Deficiency of AA steroids in younger women with RA seems to result mainly from adrenal 3,24,3s,39 rather than gonadaP 3 sources, in view of normal ovar- ian function. Whether or not low adrenal AA steroid concentrations predispose to RA in premenopausal women is not yet proved. How- ever, a lower mean serum concentration of DHEAS was found in such women considerably before onset of disease, as compared with con- trols (Table 5). Serum DHEAS levels were also recently reported to be lower in a large series of postmenopausal RA cases versus comparison women 161 not included among the studies re- viewed.
Available data indicate normal ovarian func- tion in women with RA, as indicated by their serum E2 levels (Table 4), reproductive histo- ries, 42,111 and normal mean age of meno- pause. 66,114 The somewhat lower serum total T levels in premenopausal RA cases versus con- trol women (22%), but not among postmeno- pausal women (Table 4), could be explained by deficiency of adrenal androgen precursors con- verted peripherally to T among the premeno- pausal RA patients. 3
Anaong males, the available studies 59-65 show consistently lower serum concentrations of se- rum free and total T in RA patients versus control subjects (Tables 3 and 4). On the contrary, results for serum DHEAS were nei- ther consistent (Table 3) nor statistically signifi- cant when summated (Table 4). In men, unlike women, the great majority of serum T is se- creted by the gonads, ie, the Leydig cells of the testis. 3 Only a minor contribution is derived from peripheral conversion of adrenal andro- gens. If significantly low serum androgens do precede onset of RA in males, the hypophyseal- pituitary-gonadal (HPG) axis could be suspect. However, no deficiency of serum LH was found

22 MASI, FEIGENBAUM, AND CHATTERTON
in RA versus control subjects in the studies reviewed. 6°-62,65 Low concentrations of serum T in male RA patients could result from gonadal rather than H-P mechanisms, be they primary or secondary to inflammatory processes. 3°,162 Low serum T correlates with HLA-B15 in male RA patients and, to some extent, in normal men.
Vascular alterations in RA may be regarded as characteristic features of the disease, both in joints 5,15-21,39 and systemically. 22 Available data indicate that the vascular system assumes an important role in RA, but its relationship can- not yet be considered a physiopathogenic risk factor. Nevertheless, important questions need to be addressed with regard to microvascular interactions in RA. Do the early noninflamma- tory vascular changes, eg, microvasculopathy or insufficiency of tissue perfusion, 16,2° precede and contribute to the physiopathogenesis of synovitis and other tissue alterations in RA? Alternatively, are the diffuse microvascular changes observed in this disease 3,22 a result of independent pathogenetic processes?
The dramatic ameliorating effects of preg- nancy on RA 23,41-43 may be considered a funda- mental fact in this disease. Pregnancy is a physiologic state that promotes tissue perfusion generally. 42 On the contrary, the onset risk of RA increases markedly with aging, 4° a state during which microvascularity and tissue perfu- sion generally decrease. A central role for the vascular system in the physiopathogenesis of RA is suspected. 3
Interactions likely exist among the aging, female sex and genomic-accentuating factors in RA. How and to what extent do sex hormone changes and, particularly, low androgens relate to each of these significant risk factors? Plau- sible biologic mechanisms exist for multiple protective actions of androgens that might apply to RA. They are immunomodulatory, 3,26,31,34,35,77 have chondroprotective effects against inflam- mation-induced cartilage degeneration in a ro- dent model, 163 and are believed to be important factors in many physiologic processes during aging. 164,165 The potential vasculoprotective ef- fects of DHEA and DHEAS were recently reviewed. 3 However, no quantitative estimate can yet be given as to the degree that androgens might protect against RA. Nevertheless, com- pounding of risk factors greatly magnifies the
baseline frequencies of RA, ie, from the ex- tremely low incidence in young men with a negative family history 4°,159,16° to the highest risk among elderly women with a positive family history (Fig 4, Table 6). Critical research on interactions between the hormonal, immuno- logic, and vascular systems promises to reveal mechanisms that may explain these vast discrep- ancies in risk of RA. 3,166
CONCLUSION
Evidence is mounting that sex hormones and glucocorticoids act in concert to modulate immu- noreactivity and influence the vascular system. Available data suggest that the triad of hor- monal, immunologic, and microvascular interac- tions operates in RA in the setting of complex genetic control.
Serum AA hormones are decreased in RA women and men, but in differing patterns. Controlled studies indicate that serum DHEAS levels are significantly lower than normal in RA women, particularly premenopausal RA women, but not in men. On the contrary, serum T levels are highly significantly decreased in RA men, but not impressively in women. No evidence was found of an association between serum E2 levels and RA in women (or men). Ovarian function appears normal in women, both before and after the onset of disease.
Long-term, antecedent decreases in AA ste- roids are suspected among younger menstruat- ing women who later develop RA. Data do not answer whether the observed sex hormone defi- ciencies in RA might be primary or secondary hormonal abnormalities. Deficiencies of AA steroids may possibly be combined with rela- tively deficient glucocorticoid responsiveness in the development or progression of RA, as has been demonstrated in murine experimental models.
Unlike pregnancy, use of OCs (ie, estrogens and progestrogens) does not seem to influence susceptibility or course of RA. Whether or not reproductive experience affects the predisposi- tion to RA is reviewed and is presently unre- solved. The pregnancy-ameliorating effect in RA is dramatic and characteristic of this diffuse CTD. Multiple immunosuppressive, hormonal, and microvascular circulatory alterations occur in pregnancy and may contribute to the improve-

SEX HORMONES AND PREGNANCY IN RA 23
ments in RA. Their interactions and primary influences on RA deserve priority research attention.
The common threads that connect these mul- tifactorial and polygenic factors contributing to RA are not clearly defined at present, although their interactions are being increasingly de- fined. Further research along these broad fronts is needed to explain the complex mechanisms and diverse manifestations in RA. Elucidation of mechanisms underlying the major accentuat- ing determinants of RA risk--aging, female gender, and heritability--promises avenues of
improved management and ultimate measures of prevention.
ACKNOWLEDGMENT
We express sincere gratitude to Debbie Harper for her outstanding contributions to preparing this manuscript and appreciation to Margaret Walsh for her generous assis- tance, to Jo Dorsch and Walter Wilkins for their valuable and diligent library research support, to the University of Illinois College of Medicine at Peoria (UICOM-P), and to Jon Spacht, for his persistent, expert graphics and photogra- phy services, at the Methodist Medical Center of Illinois (MMCI). Dr Thomas Dorsch, endocrinologist associate, is also thanked for his astute comments on the manuscript.
REFERENCES 1. Harris ED Jr: Etiology and pathogenesis of rheuma-
toid arthritis, in Kelly WN, Harris ED Jr, Ruddy S, et al (eds): Textbook of Rheumatology, ed 4. Philadelphia, PA, Saunders, 1993, pp 833-873
2. Harris ED Jr: Clinical features of rheumatoid arthritis, in Kelly WN, Harris ED, Jr, Ruddy S, et al (eds): Textbook of Rheumatology, ed 4. Philadelphia, PA, Saunders, 1993, pp 874-911
3. Masi AT, Feigenbaum SL, Chatterton RT, et al: Integrated hormonal-immunological-vascular ("H-I-V" triad) systems interactions in rheumatic diseases. Clin Exper Rheumatol 13:203-216, 1995
4. Decker JL, and the Glossary Subcommittee of the ARA Committee on Rheumatologic Practice: American Rheumatism Association nonmenclature and classification of arthritis and rheumatism (1983). Arthritis Rheum 26:1029- 1032, 1983
5. Ziff M: Role of the endothelium in chronic inflamma- tory synovitis. Arthritis Rheum 34:1345-1352, 1991
6. Crofford LJ, Sano H, Karalis K, et al: Local secretion of corticotropin-releasing hormone in the joints of Lewis rats with inflammatory arthritis. J Clin Invest 90:2555-2564, 1992
7. Crofford LJ, Wilder RL: Arthritis and autoimmunity in animals, in McCarty DJ, Koopman WJ (eds): Arthritis and Allied Conditions: A Textbook of Rheumatology, ed 12. Philadelphia, PA, Lea & Febiger, 1993, pp 525-539
8. Panayi GS: The immunopathogenesis of rheumatoid arthritis. Clin Exper Rheum 10:305-307, 1992
9. Ivanyi P, Hampl R, Starka L, et al: Genetic association between H-2 gene and testosterone metabolism in mice. Nature New Bio1238:280-281, 1972
10. de Vries RRP, Nijenhuis LE, Khan MA, et al: Paradoxical inheritance of HLA-linked susceptibility to rheumatoid arthritis. Tissue Antigens 26:286-292, 1985
11. Khan MA, Khan MK: HLA studies in familial and sporadic rheumatoid arthritis. Dis Markers 4:67-77, 1986
12. Jfirvinen P, Aho K: Twin studies in rheumatic dis- eases. Semin Arthritis Rheum 24:19-28, 1994
13. Weyand CM, Hicok KC, Conn DL, et al: The influence of HLA-DRB1 genes on disease severity. Ann Intern Med 117:801-806, 1992
14. Weyand CM, Goronzy JJ: Disease mechanisms in
rheumatoid arthritis: Gene dosage effect of HLA-DR haplo- types. J Lab Clin Med 124:335-338, 1994
15. Kulka JP: Vascular derangement in rheumatoid arthri- tis, in Hill A (ed): Modern Trends in Rheumatology. London, UK, Butterworths, 1966, pp 49-69
16. Rothschild B, Masi AT: Pathogenesis of rheumatoid arthritis: A vascular hypothesis. Semin Arthritis Rheum 12:11-31, 1982
17. James ML, CMand LG, Rofe AM, et al: Intraarticu- lar pressure and the relationship between synovial perfu- sion and metabolic demand. J Rheumatol 17:521-527, 1990
18. Levick JR: Hypoxia and acidosis in chronic inflamma- tory arthritis; Relation to vascular supply and dynamic effusion pressure. J Rheumatol 17:579-582, 1990
19. Stevens CR, Blake DR, Merry P, et al: A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum 34:1508-1513, 1991
20. Stevens CR, Williams RB, Farrell AJ, et al: Hypoxia and inflammatory synovitis: Observations and speculation. Ann Rheum Dis 50:124-132, 1991
21. Cotvitle-Nash PR, Scott DL: Angiogenesis and rheu- matoid arthritis: Pathogenic and therapeutic implications. Ann Rheum Dis 51:919-925, 1992
22. Bacon PA, Kitas GD: The significance of vascular inflammation in rheumatoid arthritis. Ann Rheum Dis 53:621-623, 1994
23. Hench PS, Kendall EC, Slocumb CH, et al: The effect of a hormone of the adrenal cortex (17-hydroxy-11- dehydrocorticosterone: compound E) and of pituitary adre- nocorticotropic hormone on rheumatoid arthritis: Prelimi- nary report. Mayo Clin Proc 24:181-197, 1949
24. Masi AT, Jos~povic DB, Jefferson WE: Low adrenal androgenic-anabolic steroids in women with rheumatoid arthritis (RA): Gas-liquid chromatographic studies of RA patients and matched normal control women indicating decreased 11-deoxy-17-ketosteroid excretion. Semin Arthri- tis Rheum 14:1-23, 1984
25. Bateman A, Singh A, Kral T, et al: The immune- hypothalamic-pituitary-adrenal axis. Endocrine Rev !0:92- 112, 1989
26. Homo-Delarche F, Fitzpatrick F, Christeff N, et al:

24 MASI, FEIGENBAUM, AND CHATTERTON
Sex steroids, glucocorticoids, stress and autoimmunity. J Steroid Biochem Molec Bio140:619-637, 1991
27. Chrousos GP, Gold PW: The concepts of stress and stress system disorders: Overview of physical and behavioral homeostasis. JAMA 267:1244-1252, 1992
28. Sternberg EM, Chrousos GP, Wilder RL, et al: The stress response and the regulation of inflammatory disease. Ann Intern Med 110:854-866, 1992
29. Sternberg EM, Wilder RL: Corticosteroids, in Mc- Carty D J, Koopman WJ (eds): Arthritis and Allied Condi- tions: A Textbook of Rheumatology, 12th ed. Philadelphia, PA, Lea & Febiger, 1993, pp 665-682
30. Wilder RL: Neuroendocrine-immune interactions in autoimmunity. Annu Rev Immunol 13:307-338, 1995
31. Da Silva JAP: Sex hormones, glucocorticoids and autoimmunity: Facts and hypotheses. Ann Rheum Dis 54:6-16, 1995
32. Hench PS: The ameliorating effect of pregnancy on chronic atrophic (infectious rheumatoid) arthritis, fibrositis, and intermittent hydrathrosis. Mayo Clin Proc 13:161-167, 1938
33. Spector TD: Sex hormone measurements in rheuma- toid arthritis. Br J Rheumatol 28:62-71, 1989 (suppl 1)
34. Schuurs AH, Verheul HA: Effects of gender and sex steroids on the immune response. J Steroid Biochem 35:157-172, 1990
35. Grossman CJ, Roselle GA, Mendenhall CL: Sex steroid regulation of autoimmunity. J Steroid Biochem Mol Biol 40:649-659, 1991
36. Cutolo M, Accardo S: Sex hormones, HLA and rheumatoid arthritis. Clin Exper Rheumatol 9:641-646, 1991
37. Da Silva JAP, Hall GM: The effects of gender and sex hormones on outcome in rheumatoid arthritis. Baillidre's Clin Rheumatol 6:193-219, 1992
38. James WH: Rheumatoid arthritis, the contraceptive pill, and androgens. Ann Rheum Dis 52:470-474, 1993
39. Masi AT: Sex hormones and rheumatoid arthritis: Cause or effect relationships in a complex pathophysiology? Clin Exper Rheumatol 13:227-240, 1995
40. Masi AT: Incidence of rheumatoid arthritis: Do the observed age-sex interaction patterns support a role of androgenic-anabolic (AA) steroid deficiency in its pathogen- esis? Br J Rheumato133:697-699, 1994
41. Nelson JL, Hughes KA, Smith AG, et al: Maternal- fetal disparity in HLA class II alloantigens and the preg- nancy-induced amelorization of rheumatoid arthritis. N Engl J Med 329:466-471, 1993
42. Masi AT, Feigenbaum SL: Rheumatic diseases, in Winn HN, Petrie RH (eds): Clinical Maternal-Fetal Medi- cine. Oradell, NJ, Medical Economics Books (in press)
43. da Silva JAP, Spector TD: The role of pregnancy in the course and aetiology of rheumatoid arthritis. Clin Rheum 11:189-194, 1992
44. Irey NS, Norris H J: Intimal vascular lesions associ- ated with female reproductive steroids. Arch Patho196:227- 234, 1973
45. Folkman J, Ingber DE: Angiostatic steroids: Method of discovery and mechanism of action. Ann Surg 206:374- 382, 1987
46. Kennedy RL, Haynes WG, Webb DJ: Endothelins as
regulators of growth and function in endocrine tissues. Clin Endocrino139:259-265, 1993
47. Cervera R, Khamashta MA, Hughes GRV: Antibod- ies to Endothelial Cells and Vascular Damage. Boca Raton, FL, CRC Press, 1994
48. Dougados M, Nahoul K, Benhamou L, et al: Andro- gen plasma levels in female rheumatoid arthritis patients. Arthritis Rheum 26:935-936, 1983 (letter)
49. Feh6r KG, Feh6r T: Plasma dehydroepiandros- terone, dehydroepiandrosterone sulphate and androsterone sulphate levels and their interaction with plasma proteins in rheumatoid arthritis. Exp Clin Endocrinol 84:197-202, 1984
50. Fehdr KG, Feh~r T, Mer6tey K: Interrelationship between the immunological and steroid hormone param- eters in rheumatoid arthritis. Exp Clin Endocrino187:38-42, 1986
51. Cutolo M, Balleari E, Giusti M, et al: Sex hormone status in women suffering from rheumatoid arthritis. J Rheumatol 13:1019-1023, 1986
52. Spector TD, Perry LA, Tubb G, et al: Androgen status of females with RA. Br J Rheumato126:316-318, 1987 (letter)
53. Arnalich F, Benito-Urbina S, Gonzalez Gancedo P, et al: E16vations des androg~nes plasmatiques chez les femmes menopausdes atteintes de polyarthrite rhumatoide. Rev Rhum Mal Osteoartic 57:509-512, 1990
54. Bijlsma JWJ, Huber-Bruning O, Thijssen JHH, et al: Sex hormones in active rheumatoid arthritis. Proceedings of the XVI International Congress of Rheumatology, Sydney, Australia, 1985 (abstr 683)
55. Sambrook PN, Eisman JA, Champion GD, et al: Sex hormone status and osteoporosis in postmenopausal women with rheumatoid arthritis. Arthritis Rheum 31:973-978, 1988
56. Aroso Dias A, Lopes Vaz A, Hargreaves M, et al: Biomarks in secondary osteoporosis. Clin Rheumatol 8:89- 94, 1989 (suppl 2)
57. Benito Urbina S, Arnalich Fernfindez F, Gonzfilez- Gancedo P, et at: Alteraciones hormonales en mujeres post-menopfiusicas con artritis reumatoide. Rev Clin Esp 190:181-183, 1992
58. Hedman M, Nilsson E, de la Torre B: Low blood and synovial fluid levels of sulpho-conjugated steroids in rheuma- toid arthritis. Clin Exper Rheumatol 10:25-30, 1992
59. Cutolo M, Balleari E, Accardo S, et al: Preliminary results of serum androgen level testing in men with rheuma- toid arthritis. Arthritis Rheum 27:958-959, 1984 (letter)
60. Gordon D, Beastall GH, Thomson JA, et al: Andro- genic status and sexual function in males with rheumatoid arthritis and ankylosing spondylitis. Q J Med 60:671-679, 1986
61. Cutolo M, Balleari E, Giusti M, et al: Sex hormone status of male patients with rheumatoid arthritis: Evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheum 31:1314-1317, 1988
62. Spector TD, Perry LA, Tubb G, et al: Low free testosterone levels in rheumatoid arthritis. Ann Rheum Dis 47:65-68, 1988
63. Spector TD, Oilier W, Perry LA, et al: Free and serum testosterone levels in 276 males: A comparative study

SEX HORMONES AND PREGNANCY IN RA 25
of rheumatoid arthritis, ankylosing spondylitis and healthy controls. Clin Rheumatol 8:37-41, 1989
64. Oilier W, Spector T, Silman A, et ah Are certain HLA haplotypes responsible for low testosterone levels in males? Dis Markers 7:139-143, 1989
65. Cutolo M, Balleari E, Giusti M, et al: Androgen replacement therapy in male patients with rheumatoid arthritis. Arthritis Rheum 34:1-5, 1991
66. Deighton CM, Watson M J, Walker D J: Sex hor- mones in postmenopausal HLA-identical rheumatoid arthri- tis discordant sibling pairs. J Rheumatol 19:1663-1667, 1992
67. Masi AT, Chatterton RT, Comstock GW, et al: Decreased androgenic-anabolic (AA) hormone levels in sera of white women before onset of RA: A controlled, prospective study. Arthritis Rheum 36:$176, 1993 (abstr)
68. Masi AT, Chatterton RT, Comstock GW, et al: Decreased serum dehydroepiandrosterone sulfate (DHEAS) levels before onset of RA in younger, pre-menopausal women: A controlled, prospective study. Arthritis Rheum 37:$315, 1994 (abstr)
69. Boumpas DT, Paliogianni F, Anastassiou ED, et ah Glucocorticosteroid action on the immune system: Molecu- lar and cellular aspects. Clin Exper Rheumatol 9:413-423, 1991
70. Distelhorst CW: Steroid hormone receptors. J Lab Clin Med 122:241-244, 1993
71. May M, Holmes E, Rogers W, et al: Protection from glucocorticoid induced thymic involution by dehydroepi- androsterone. Life Sci 46:1627-1631, 1990
72. Power RF, Mani SK, Codina J, et al: Dopaminergic and ligand-independent activation of steroid hormone recep- tors. Science 254:1636-1639, 1991
73. Geren~er M, Tajic M, Kerhin-Brklja~ic V, et al: An association between serum testosterone level and HLA phenotype. Immunol Lett 4:155-158, 1982
74. Spector TD, Ollier WER, Perry LA, et al: Evidence for similarity in testosterone levels in haplotype identical brothers. Dis Markers 6:119-125, 1988
75. Deighton CM, Watson M, Walker D J: Rheumatoid arthritis, sex ratios, HLA-DR, and testosterone. Ann Rheum Dis 52:244, 1993
76. Jaraquemada D, Ollier W, Awad J, et al: HLA and rheumatoid arthritis: A combined analysis of 440 patients. Ann Rheum Dis 45:627-636, 1986
77. Holmdahl R, Carlsten H, Jansson L, et al: Oestrogen is a potent immunomodulator of murine experimental rheumatoid disease. Br J Rheumatol 28:54-58, 69-71, 1989 (suppl 1)
78. Cutolo M, Sulli A, Seriolo B, et al: Estrogens, immune response and autoimmunity. Clin Exper Rheuma- tol 13:217-226, 1995
79. Aksentijevich S, Whitfield HJ, Jr, Young WS III, et al: Arthritis-suspectible Lewis rats fail to emerge from the stress hyporesponsive period. Dev Brain Res 65:115-118, 1992
80. Sternberg EM, Glowa JR, Smith MA, et al: Cortico- tropin releasing hormone related behavioral and neuroendo- crine responses to stress in Lewis and Fisher rats. Brain Res 570:54-60, 1992
81. Berczi I: Prolactin, pregnancy and antoimmune dis- ease. J Rheumato120:1095-1100, 1993
82. Russell DH, Kibler R, Matrisian L, et al: Prolactin receptors on human T- and B-lymphocytes. Antagonism of prolactin binding by cyclosporine. J Immunol 134:3027- 3031, 1985
83. Berczi I, Cosby H, Hunter T, et al: Decreased bioactivity of circulating prolactin in patients with rheuma- toid arthritis. Br J Rheumato126:433-436, 1987
84. Nagy E, Chalmers IM, Baragar FD, et al: Prolactin deficiency in rheumatoid arthritis. J Rheumatol 18:1662- 1668, 1991
85. Berczi I, Baragar FD, Chalmers IM, et al: Hormones in self tolerance and autoimmunity: A role in the pathogen- esis of rheumatoid arthritis? Autoimmunity 16:45-56, 1993
86. Neeck G, Federlin K, Graef V, et al: Adrenal secretion of cortisol in patients with rheumatoid arthritis. J Rheumatol 17:24-29, 1990
87. Harkness JAL, Richter MP, Panayi GS, et al: Circa- dian variation in disease activity in rheumatoid arthritis. Br Med J 284:551-553, 1982
88. Saldanha C, Touzas G, Grace E: Evidence for the anti-inflammatory effects of normal circulating plasma corti- sol. Clin Exp Rheumatol 4:365-366, 1986
89. Chikanza IC, Petrou P, Kingsley GH, et al: Defective hypothalamic response to immune and inflammatory stimuli in patients with rheumatoid arthritis. Arthritis Rheum 35:128141288, 1992
90. Wilder RL, Sternberg EM: Neuroendocrine hor- monal factors in rheumatoid arthritis and related condi- tions. Curr Opin Rheumatol 2:436-440, i990
91. Linos A, Worthington JW, O'Fallon MW, et al: The epidemiology of rheumatoid arthritis in Rochester, Minne- sota: A study of incidence, prevalence and mortality, Am J Epidemiol 1:87-98, 1980
92. Dugowson CE, Koepsell TD, Voigt LF, et al: Rheu- matoid arthritis in women. Incidence rates in Group Health Cooperative, Seattle, Washington, 1987-1989. Arthritis Rheum 34:1502-1507, 1991
93. Wingrave SJ, Kay CR: Reduction in incidence of rheumatoid arthritis associated with oral contraceptives. Lancet 1:569-571, 1978
94. Silman AJ: Has the incidence of RA declined in the United Kingdom? Br J Rheumatol 27:77-78, 1988
95. Silman AJ, Vandenbroucke JP (eds): Female sex hormones and rheumatoid arthritis. Proceedings of an International Workshop. Leiden, The Netherlands, 20-21, March 1989. Br J Rheumatol 28:1-73, 1989 (suppl 1)
96. Hernandez-Avila M, Liang MH, Willett WC, et al: Exogenous sex hormones and the risk of rheumatoid arthri- tis. Arthritis Rheum 33:947-953, 1990
97. Spector TD, Brennan P, Harris P, et al: Does estrogen replacement therapy protect against rheumatoid arthritis? J Rheumatol 18:1473-1476, 1991
98. Silman AJ: The role for epidemiology in rheumatol- ogy. Clin Exper Rheumatol 10:105-108, 1992
99. Esdaile JM, Horwitz RI: Observational studies of cause-effect relationships: An analysis of methodological problems as illustrated by the conflicting data for the role of oral contraceptives in the etiology of rheumatoid arthritis. J Chron Dis 39:841-852, 1986
100. Spector TD, Hochberg MC: The protective effect of the oral contraceptive pill on rheumatoid arthritis: An

26 MASI, FEIGENBAUM, AND CHATTERTON
overview of the analytical epidemiological studies using meta-analysis. J Clin Epidemio143:1221-1230, 1990
101. Silman A J: The genetic epidemiology of rheumatoid arthritis. Clin Exper Rheumatol 10:309-312, 1992
102. Kuzell WC, Glover RP, Bruns DL, et al: Methandro- stenolone in rheumatic diseases and osteoporosis. Geriat- rics 17:428-44l, 1962
103. West HF, Lewis-Farting E: Controlled trial of methe- nolone acetate in prednisolone-treated rheumatoid arthritis patients. Ann Rheum Dis 24:576-580, 1965
104. Cuchacovich M, Tchernitchin A, Gatica H, et al: Intraarticular progesterone: Effects of a local treatment for rheumatoid arthritis. J Rheumatol 15:561-565, 1988
105. Bulmash JM: Rheumatoid arthritis and pregnancy. Obstet Gynecol Annu 8:223-276, 1979
106. Cecere FA, Persellin RH: The interaction of preg- nancy and the rheumatic diseases. Clin Rheum Dis 7:747- 768, 1981
107. Ostensen M, Aune B, Husby G: Effect of pregnancy and hormonal changes on the activity of rheumatoid arthri- tis. Scand J Rheumatol 12:69-72, 1983
108. Klipple GL, Cecere FA: Rheumatoid arthritis and pregnancy. Rheum Dis Clin North Am 15:213-239, 1989
109. Silman A, Kay A, Brennan P: Timing of pregnancy in relation to the onset of rheumatoid arthritis. Arthritis Rheum 35:152-154, 1992
110. Suarez-Almazor ME, A1-Arfag AS, Kendall C, et al: Risk of developing post-partum rheumatoid arthritis (RA). Arthritis Rheum 37:$370, 1994
111. Nelson JL, Koepsell TD, Dugowson CE, et al: Fecundity before disease onset in women with rheumatoid arthritis. Arthritis Rheum 36:7-14, 1993
112. Yoshino S, Uchida S: Sexual problems of women with rheumatoid arthritis. Arch Phys Med Rehab 62:122- 123, 1981
113. Kay A, Bach F: Subfertility before and after the development of rheumatoid arthritis in women. Ann Rheum Dis 24:169-173, 1965
114. Goemaere S, Ackerman C, Goethals K, et al: Onset of symptoms of rheumatoid arthritis in relation to age, sex and menopausal transition. J Rheumatol 17:1620-1622, 1990
115. Nolten WE, Rueckert PA: Elevated free cortisol index in pregnancy: Possible regulatory mechanisms. Am J Obstet Gynecol 139:492-498, 1981
116. Abou-Samra AB, Pugeat M, Dechaud H, et al: Increased plasma concentration of N-terminal lipotrophin and unbound eortisol during pregnancy. Clin Endocrinol 20:221-228, 1984
117. Ansar Ahmed S, Penhale WJ, Talal N: Sex hor- mones, immune responses, and autoimmune diseases: Mechanisms of sex hormone action. Am J Pathol 121:531- 551, 1985
118. Ostensen M, Husby G: A prospective clinical study of the effect of pregnancy on rheumatoid arthritis and ankylosing spondylitis. Arthritis Rheum 26:1155-1159, 1983
119. Brabin BJ: Epidemiology of infection in pregnancy. Rev Infect Dis 7:579-603, 1985
120. Waterson AP: Virus infections (other than rubella) during pregnancy. Br Med J 2:564-566, 1979
121. Sakamoto K, Greally J, Gilfillan RF, et al: Epstein- Barr virus in normal pregnant women. Am J Reprod Immunol 2:217-221, 1982
122. Sridama V, Pacini F, Yang SL, et al: Decreased levels of helper T cells: A possible cause of immunodefi- ciency in pregnancy. N Engl J Med 307:352-356, 1982
123. Degenne D, Canepa S, Lecomte C, et al: Serial study of T-lymphocyte subsets in women during very early pregnancy. Clin Immunol Immunopatho148:187-191, 1988
124. Krause PJ, Ingardia CJ, Pontius LT, et al: Host defense during pregnancy: Neutrophil chemotaxis and ad- herence. Am J Obstet Gynecol 157:274-280, 1987
125. Ostensen M, Revhaug A, Volden G, et al: The effect on pregnancy of functions of inflammatory cells in healthy women and in patients with rheumatic diseases. Acta Obstet Gynecol Scand 66:247-253, 1987
126. Vanderbeeken YE, Duchateau J, Calletl H, et al: Modulation of the HLA class II antigen at a molecular level by maternal serum. Immunopharmacol Immunotoxicol 12: 679-695, 1990
127. Unger A, Kay A, Griffin A J, et al: Disease activity and pregnancy associated c~ 2-glycoprotein in rheumatoid arthritis during pregnancy. Br Med J 286:750-752, 1983
128. Ostensen M, von Schoultz B, Husby G: Comparison between serum alpha 2-pregnancy-associated globulin and activity of rheumatoid arthritis and ankylosing spondylitis during pregnancy. Scand J Rheumatol 12:315-318, 1983
129. Bose R: Properties of human pre- and post- implantation embryo-associated immunosuppressor fac- tor(s). Immunol Lett 30:325-332, 1991
130. Wajner M, Papiha S, Arruda N: Inhibition of phytohaemagglutinin-induced lymphocyte blastogenesis by serum from pregnant women: Correlation between cortisol level and in vitro immunosuppression. Acta Endocrinol 109:411-417, 1985
131. Kasakura S: A factor in maternal plasma during pregnancy that suppresses the reactivity of mixed leukocyte cultures. J Immunol 107:1296-1301, 1971
132. Nicholas N, Panayi G: Inhibition of interleukin-2 production by retroplacental sera: A possible mechanism for human fetal allograft survival. Am J Reprod Immunol 9:6-11, 1985
133. Loke YW, King A: Recent developments in the human maternal-fetal immune interaction. Curt Opin Immu- nol 3:762-766, 1991
134. Vamvakopoulos NC, Chrousos GP: Evidence of di- rect estrogenic regulation of human corticotropin-releasing hormone gene expression. Potential implications for the sexual dimorphism of the stress response and immune/ inflammatory reaction. J Clin Invest 92:1896-1902, 1993
135. Folkman J: How is blood vessel growth regulated in normal and neoplastic tissue? Cancer Res 467-473, 1986
136. Crum R, Szabo S, Folkman J: A new class of steroids inhibits angiogenesis in the presence of heparin or a heparin fragment. Science 230:1375-1378, 1985
137. Folkman J, Ingber DE: Angiostatic steroids: Method of discovery and mechanism of action. Ann Surg 206:374- 382, 1987
138. Jaffe RB: Endocrine physiology of the fetus and fetoplacenial unit, in Yen SSC, Jaffe RB (eds): Reproduc- tive Endocrinology: Physiology, Pathophysiology, and Clini-

SEX HORMONES AND PREGNANCY IN RA 27
cal Management, ed 3. Philadelphia, PA, Saunders, 1991, pp 888-919
139. Pasquatini JR, Kincl FA: Biosynthesis and Metabo- lism of Different Hormones in the Fetal and Placental Compartments. Hormones and the Fetus; vol I. Oxford, UK, Pergamon Press 1:73-172, 217-329, 1985
140. Lobo RA: Endocrinology of pregnancy, in Mishell DR Jr, Davajan V, Lobo RA (eds): Infertility, Contracep- tion & Reproductive Endocrinology. Boston, MA, Black- well Scientific, 1991, pp 150-179
141. Fujieda K, Faiman C, Feyes FI, et al: The control of steroidogenesis by human fetal adrenal cells in tissue culture. IV. The effects of exposure to placental steroids. J Clin Endocrinol Metab 54:89-94, 1982
142. Siiteri PK, MacDonald PC: Placental estrogen bio- synthesis during human pregnancy. J Clin Endocrinol Metab 26:751-761, 1966
143. Spangelo BL, Hall RS, Ross PC, et al: Stimulation of in vivo antibody production and concanavalin-A-induced mouse spleen cell mitogenesis by prolactin. Immunopharma- col 14:11-20, 1987
144. Gerli R, Rambotti P, Nicoletti I, et al: Reduced number of natural killer cells in patients with pathological hyperprolactinemia. Clin Exp Immunol 64:399-406, 1986
145. Romieu I, Hernandez-Avila M, Liang MH: Oral contraceptives and the risk of rheumatoid arthritis: A meta-analysis of a conflicting literature. Br J Rheumatol 28:13-23, 1989 (suppl 1)
146. Stites D, Siiteri P: Steroids as immunosuppressants in pregnancy. Immunol Rev 75:117-138, 1983
147. Johansson ED: Plasma levels of progesterone in pregnancy measured by a rapid competitive protein binding technique. Acta Endocrinot (Copenh) 61:607-617, 1969
148. Szekeres-Bartho J, Varga P, Kinsky R, et at: Proges- terone-mediated immunosuppression and the maintenance of pregnancy. Res Immunol 141:175-181, 1990
149. Donaldson A, Nicolini U, Symes EK, et al: Changes in concentrations of cortisol, dehydroepiandrosterone sul- fate and progesterone in fetal and maternal serum during pregnancy. Clin Endocrino135:447-451, 1991
150. Mishell DR Jr: Endocrinology of lactation and the puerperium, in Mishell DR Jr, Davajan V, Lobo RA (eds): Infertility, Contraception & Reproductive Endocrinology, ed 3. Boston, MA Blackwell Scientific Publications, 1991, pp 180-203
151. Oka M: Effect of pregnancy on the onset and course of rheumatoid arthritis. Ann Rheum Dis 12:227-229, 1953
152. Felbo M, Snorrason E: Pregnancy and the place of therapeutic abortion in rheumatoid arthritis. Acta Obstet Gynecot Scand 40:116-126, 1961
153. Musey VC, Collins DC, Brogan DR, et al: Long term effects of a first pregnancy on the hormonal environ- ment: Estrogens and androgens. J Clin Endocrinol Metab 64:111-118, !987
154. Silman AJ: Is pregnancy a risk factor in the causa- tion of rheumatoid arthritis? Ann Rheum Dis 45:103 !- 1034, 1986
155. Engel A: Rheumatoid arthritis in U.S. adults 1960- 1962, in Bennet PH, Wood PHN (eds): PopuIation Studies in the Rheumatic Diseases. Amsterdam, The Netherlands, Excerpta Medica Foundation 1968, pp 83-89
156. Lawrence JS: Rheumatoid arthritis--Nature or nur- ture? Ann Rheum Dis 29:357-359, 1970
157. Del Junco DJ, Luthra HS, Annegers JF, et al: The familial aggregation of rheumatoid arthritis and its relation- ship to the HLA-DR4 association. Am J Epidemiol 119:813- 829, 1984
158. Kwoh CK, Venglish CM, Whitley DM, et at: Famil- ial risk of rheumatoid arthritis. Arthritis Rheum 36:$98, 1993
159. Lawrence JS: Rheumatism in Populations. London, UK, William Heineman Medical Books, pp. 16-20, 231-263, 1977
160. Aho K, Koskenvuo M, Tuominen J, et al: Occur- rence of rheumatoid arthritis in a nationwide series of twins. J Rheumatot 13:899-902, 1986
161. Hall GM, Perry LA, Spector TD: Depressed levels of dehydroepiandrosterone sulfate in postmenopausal women with rheumatoid arthritis but no relation to axial bone density. Ann Rheum Dis 52:211-214, 1993
162. Gordon D, Beastall GH, Thomson JA, et al: Pro- longed hypogonadism in male patients with rheumatoid arthritis during flares in disease activity. Br J Rheumatol 27:440-444, 1988
163. Da Silva JAP, Larbre J-P, Spector TD, et al: Protective effects of androgens against inflammation in- duced cartilage degradation in male rodents. Ann Rheum Dis 52:285-291, 1993
164. Morales AJ, Nolan JJ, Nelson JC, et al: Effects of replacement dose of dehydroepiandrosterone in men and women of advancing age. J Clin Endocrinol 78:1360-1367, 1994
165. B61anger A, Candas B, Dupont A, et al: Changes in serum concentrations of conjugated and unconjugated ste- roids in 40- to 80-year-old men. J CIin Endocrinol Metab 79:1086-1090, 1994
166. Chrousos GP: V-hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332: 1351-1362, 1995
Copyright © 2022 FDOKUMEN