
Histological and proteomic analysis of reversible H-RasV12G expression in transgenic mouse skin
33
Oh et al 6/4/07 7:29 PM 1 Histological and proteomic analysis of reversible H-Ras V12G expression in transgenic mouse skin Won-Jun Oh # , Vikas Rishi # , Steven Pelech $ , and Charles Vinson # * # Laboratory of Metabolism, NCI, CCR, NIH, MD 20892, USA, $ Kinexus Bioinformatics Corporation and the Department of Medicine, University of British Columbia, Vancouver, British Columbia, Canada Running Title: Proteomic targets of H-Ras V12G de-induction *Address correspondence to: Charles Vinson, Bldg. 37, Rm. 3128, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, Tel: 301-496-8753; Fax: 301-496-8419; E- mail: [email protected] Published by Oxford University Press 2007. Carcinogenesis Advance Access published June 5, 2007 by guest on June 7, 2013 http://carcin.oxfordjournals.org/ Downloaded from
Transcript of Histological and proteomic analysis of reversible H-RasV12G expression in transgenic mouse skin

Oh et al 6/4/07 7:29 PM 1
Histological and proteomic analysis of reversible H-RasV12G expression in transgenic mouse
skin
Won-Jun Oh#, Vikas Rishi#, Steven Pelech$, and Charles Vinson#*
# Laboratory of Metabolism, NCI, CCR, NIH, MD 20892, USA, $Kinexus Bioinformatics
Corporation and the Department of Medicine, University of British Columbia, Vancouver,
British Columbia, Canada
Running Title: Proteomic targets of H-RasV12G de-induction
*Address correspondence to: Charles Vinson, Bldg. 37, Rm. 3128, National Cancer Institute,
National Institutes of Health, Bethesda, MD 20892, Tel: 301-496-8753; Fax: 301-496-8419; E-
mail: [email protected]
Published by Oxford University Press 2007. Carcinogenesis Advance Access published June 5, 2007 by guest on June 7, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

Oh et al 6/4/07 7:29 PM 2
Abstract
We have used a two-transgene tetracycline system to reversibly express oncogenic H-RasV12G in
mouse skin and primary keratinocytes culture using the bovine keratin 5 promoter. Induction of
H-RasV12G expression in skin at 30 days after birth causes epidermal basal cell hyperplasia, an
eruption of keratinous cysts, and loss of hair follicles by 3 weeks. Subsequent H-RasV12G de-
induction for 3 days results in massive apoptosis in the non H-RasV12G expressing stroma as well
as in the suprabasal cells of the epidermis. Several procaspases such as CASP-3, -1? , -5, -12
disappeared while the pro-apoptotic proteins AIF, Bax, and Fas ligand were induced in H-
RasV12G de-induction skin. This process is followed by a wave of cell division at 14 days as hair
follicles regrew, returning to near normal histology and skin appearance by 30 days. Using
Kinetworks™ multi- immunoblotting screens, the phosphorylation status of 37 proteins and
expression levels of 75 protein kinases in the skin were determined in three samples: 1) wild-
type skin, 2) hyperplastic H-RasV12G expressing skin, 3) skin where H-RasV12G expression was
suppressed for 7 days. Following H-RasV12G induction, 16 kinases were increased over 2-fold,
and 2 kinases were reduced over 50%. This included increased phosphorylation of both known
downstream H-RasV12G targets and unknown H-RasV12G targets. After H-RasV12G suppression,
many but not all protein changes were reversed. These results from skin and primary
keratinocytes are organized to reflect the molecular events that cause the histological changes
observed. These proteomic changes identify markers that may mediate the oncogenic addiction
paradigm.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 3
Introduction
Expression in transgenic mice of either oncogenic H-Ras or K-Ras results in both cellular
proliferation and tumorigenesis [1] producing a variety of mouse models for human disease
including follicular adenoma [2], melanoma [3] and Costello syndrome [4]. Several
investigators have constitutively expressed Ras or its oncogenic forms in the skin and observed
both hyperproliferation of the epidermis and subsequent formation of squamous cell carcinomas
[5] demonstrating that activated Ras is sufficient to produce a malignant phenotype. Expression
of oncogenic Ras in different compartments of the skin produces different results. Suprabasal
epithelial cell expression using the keratin 10 promoter causes hyperplasia and papillomas which
slowly progress into carcinomas [6] while expression in the hair follicle compartment that
contains skin stem cells using the keratin 5 promoter caused papillomas which readily convert
into carcinomas [5].
Mutations in each Ras gene have been detected at high frequency in many human cancers,
with frequencies ranging from 10 to 90% [7], with different cancers types having reproducible
mutations in specific Ras genes. Mouse models of skin chemical carcinogenesis indicate that
oncogenic mutations in Ras plays a central role in skin tumor development [8] suggesting that
the oncogenic properties of Ras are similar in mice and humans.
There are three Ras proto-oncogenes in mammals [1] which have GTPase activity and
function to activate kinases [9] whose downstream targets are transcription factors culminating in
changes in gene expression [10]. Oncogenic Ras is able to transform cells in culture by both
promoting cell growth and inhibiting anoikis [11], a term for cell death induced in tissue culture
when cells fail to make proper cell-cell and cell-stromal interactions [12]. The inhibition of
anoikis is critical for the ability of Ras transformed cells to grow in soft agar [13].
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 4
Ras-signaling pathways underlie cellular transformation as demonstrated by transfection
of either Ras or its downstream targets [14]. However, in epidermal keratinocytes, conflicting
data have been presented indicating that Ras promotes proliferation and suppress differentiation
[15] or does the opposite, such as cell cycle arrest and induction of apoptosis [16].
More recently, oncogenic H-RasV12G has been reversibly expressed in melanocytes to
show that it is continually needed to maintain the malignant phenotype [17]. Similar
observations were made for the reversible expression of Myc [18] and a mutant EGF receptor
[19]. These observations have been conceptua lized to suggest that cancer cells develop a
dependence or “addiction” to the continued activity of over expressed oncogenes for
maintenance of their malignant phenotype [20,21]. However, the molecular correlates of this
poorly understood phenomenon have not been reported.
To identify such molecular correlates, we have reversibly expressed H-RasV12G in the
basal layer of the mouse epidermis using the K5 promoter. Here we show that the expression of
H-RasV12G induces hyperplasia in the skin by 3 weeks and subsequent de- induction result in a
return to normal skin and histology by 30 days. To help determine the mechanism of tumor
regression, we have examined changes in the concentration of over 100 phosphoproteins and
proteins that occur in the skin when H-RasV12G expression is activated and subsequently
suppressed. The reversibility by H-RasV12G induction and de- induction also was also observed in
primary keratinocytes. These proteomic changes that occur as H-RasV12G is de-induced may be
potential therapeutic targets.
Experimental procedures
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 5
Transgenic mice and cell culture. The transgenic mouse containing the tetracycline
transactivator under the control of the bovine K5 promoter (K5-tTA) was a kind gift from Dr.
Adam Glick [22]. The tetO-H-RasV12G transgenic mouse [17] were obtained from NCI Mouse
Repository (Frederick, MD). Offspring from the cross between K5-tTA and tetO-H-RasV12G
mice were genotyped using the PCR primers for H-RasV12G transgene: 5'-
GGTCCACTTCGCATATTAAGG and 5'-GCCGGCGGTATCCAGGATGTCCAAC, and for
tTA transgene: 5'- AACAACCCGTAAA-CTCGCCC and 5’-GCAACCTAAAGTAAAAT-
GCCCCAC.
Primary keratinocytes were isolated from wild-type or K5-tTA:tetO-H-RasV12G (K5:H-RasV12G)
transgenic newborn littermates (1day old) as described [23].
Regulated H-RasV12G expression in mouse skin. To generate K5:H-RasV12G double transgenic
mice, heterozygous K5-tTA mice were crossed to homozygous tetO-H-RasV12G mice in the
presence of 200 mg/kg doxycycline feed (Bioserv, Baltimore, MD). To induce H-RasV12G
expression, 30 day old mice were removed from doxycycline food. After 3 weeks when K5:H-
RasV12G mice showed a phenotype, they were feed food containing 200 mg/kg doxycycline to
suppress expression of the H-RasV12G transgene.
Skin histology, measurement of cell proliferation, and apoptosis. Skin tissues were fixed in
10% NBF overnight, transferred to 95% Et-OH and embedded in paraffin. Six-micrometer
sections were cut and stained with hematoxylin and eosin. To label dividing cells, mice were
sacrificed 60 min after a single intraperitoneal injection of a sterile solution of BrdU (Sigma; 10
mg/ml in PBS, 50 mg/kg of body weight). Skin sections were treated for 30 min at 37°C with
2N HCl in PBS containing 0.5% Triton X-100, rinsed in sodium tetraborate buffer (0.1 M, pH
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 6
8.5) and stained using an anti-BrdU antibody (DAKO diagnostics). Apoptosis was determined in
tissue sections using an ApopTag kit (Chemicon, Temecula, CA).
Western blot analysis. Protein extracts were made from whole skin or primary keratinocytes
using RIPA lysis buffer containing 1% SDS, 1% NP-40, and 0.5% Na-deoxycolate and cocktail
of protease inhibitors. Western blots of lysates were performed on 15% SDS-PAGE and probed
with ? -146 anti H-Ras monoclonal antibodies (Quality Biotech, New Jersey), anti-Bax
polyclonal antibodies and anti-VEGF polyclonal antibodies (Santa Cruz Biotechnology),
differentiation-related antibodies (K1, K10, involucrin, and loricrin; Covance, Richmond, CA),
anti-PKC? , and anti-phospho PKC? (Santa Cruz Biotechnology). Protein bands were visualized
using the ECL chemiluminescence detection kit (GE Healthcare, Piscataway, NJ).
Immunohistochemistry. Serial 6 ? m-thick sections were prepared and immunohisto-chemistry
was done using the ABC and DAB kits from Vector Laboratories. Skin was immunostained with
goat anti-CD31 (1:200; Santa Cruz Biotechnology), and rabbit anti-cytokeratin antibodies
(1:2000; DAKO diagnostics). Histological sections were photographed using an Olympus AX70
microscope (Olympus Optical) with analysis software (Soft Imaging System).
Apoptosis, protein kinase, and phospho-protein screening. Whole skin lysate from mice (0.5
mg) was prepared as described previously [24]. The Kinetworks™ apoptosis (KAPS-1.0),
phospho-site (KPSS-1.3), and protein kinase (KPKS-1.2) screens were performed by Kinexus
Bioinformatics (Vancouver, BC) [25].
Results
Regulated expression of activated H-RasV12G in the skin basal epidermis was achieved by
producing a mouse containing two transgenes. One transgene expresses the tetracyc line
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 7
transactivator under the control of the bovine keratin 5 gene promoter (K5-tTA) that is active in
the basal epidermis of the skin both in utero and in the adult. The second transgene contains the
oncogenic H-RasV12G gene driven by a minimal promoter containing seven DNA binding sites
for the tetracycline transactivator (tetO-H-RasV12G). Crosses between K5-tTA and tetO-H-
RasV12G mice did not produce any double transgenic pups indicating that K5 driven expression of
activated H-RasV12G in utero is lethal. However, if the mothers were fed doxycycline (Dox) in
the food to suppress H-RasV12G expression during development, double transgenic K5:H-RasV12G
mice were born at the expected Mendelian frequencies. These double transgenic mice develop
like their wild-type littermates when continually fed Dox in the food.
H-RasV12G expression was induced at 30 days by feeding K5:H-RasV12G double
transgenic mice food without Dox. All the double transgenic but none of the single transgenic
mice (either K5 or tetO-H-RasV12G mice) developed a wide variety of epithelial alterations,
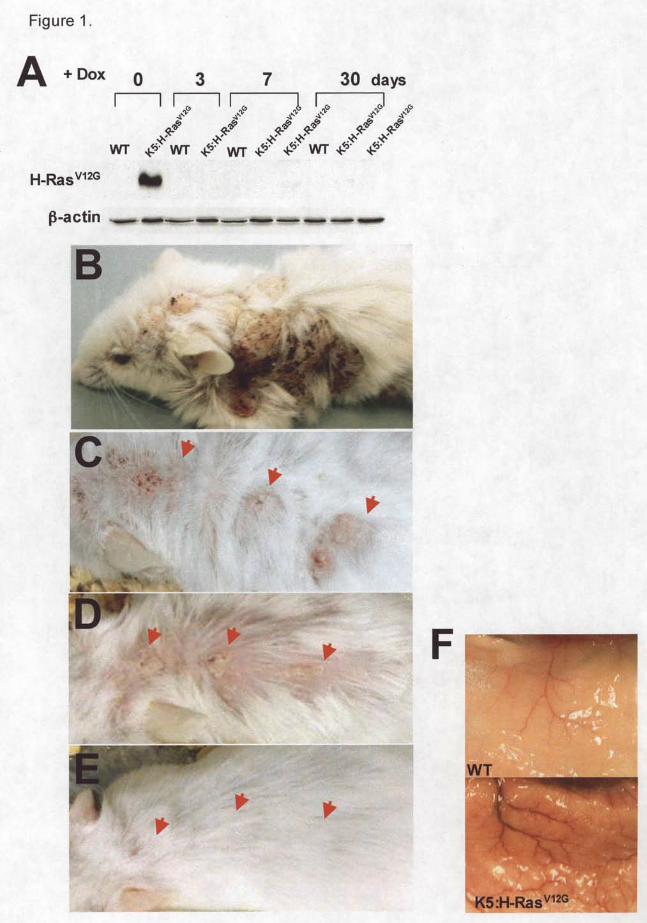
ranging from benign skin hyperplasia to dysplasia, alopecia, keratinous cysts (Figure 1B, 2A)
and swelling of the salivary glands (data not shown) which are consistent with other groups
findings [5] [26]. However, we did not observe any macro-abnormality of other organs (i.e., oral
mucosa, tongue, esophagus, uterine cervix, and fore stomach) as reported by Vitale-Cross L. et al
[5]. Massive angiogenesis was also observed in the skin (Figure 1F). 21 days after H-RasV12G
expression was induced, expression was suppressed by using food containing 200 mg/kg Dox.
This allowed us to examine the dependence of the skin phenotype on continual H-RasV12G
expression. The expression of H-RasV12G protein was suppressed in the skin after 3 days (Figure
1A). After 10 days, mouse skin showed intermediate phenotype. Finally, the skin appearance
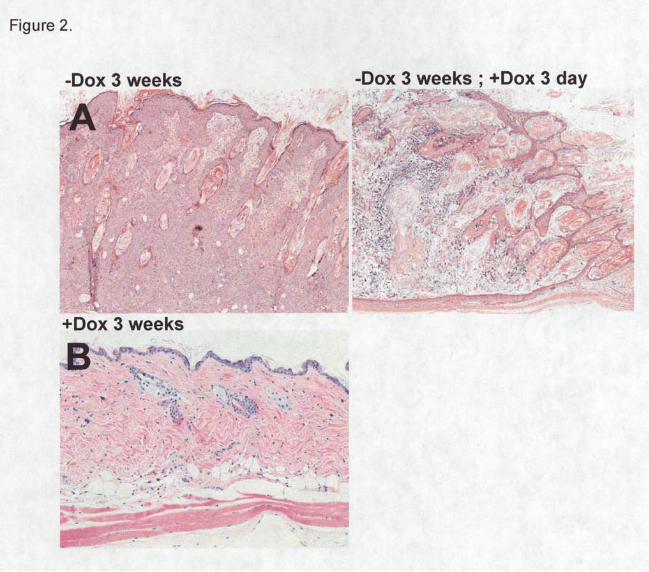
returned to near normal after 30 days (Figure 1C-E). Compare to H-RasV12G expressing skin
(Figure 2A; left panel), dramatic histological changes in the skin were observed at 3 day after
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 8
suppression of H-RasV12G protein (Figure 2A; right panel) and the skin histology became normal
after 30 days (Figure 2C). However, in the presence of Dox when H-RasV12G is not expressed,
WT (Supplementary figure) and K5:H-RasV12G (Figure 2B) skins were histologically
indistinguishable, indicating the tight control of oncogenic H-Ras protein expression on
doxycycline.
Histological analysis of skin during reversible H-RasV12G expression: growth and death
Three types of histological analyses of the skin revealed cellular events involved in both
the H-RasV12G dependent skin hyperplasia and the subsequent regression following H-RasV12G
de-induction. These analyses included; 1) Hematoxylin and eosin-stained sections to examine
general tissue histology, 2) BrdU labeling and immunohistological staining to identify cells
undergoing cell proliferation, and 3) TUNEL analysis to identify cells undergoing apoptosis.
The dorsal skin of H-RasV12G expressing (Figure 2C-E) and wild-type (Supplementary figure)
mice was examined at 8 time points. Four time points were 0, 3, 7, and 21 days of H-RasV12G
expression. Four additional time points examined the consequences of H-RasV12 suppression for
3, 7, 14, and 30 days after being expressed for 21 days. The quantification of cell growth and
apoptosis during this time course is presented for both the K5:H-RasV12G and wild-type mice
(Figure 2F-G).
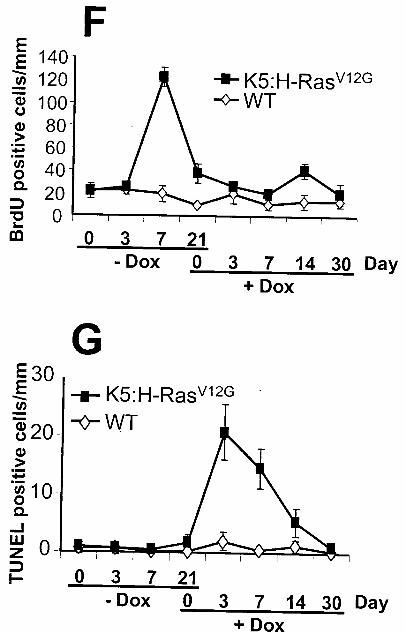
The induction of H-RasV12G expression resulted in hyperproliferation of the epidermis
that was evident as early as 3 days. At 7 day, the hyperplasia is more pronounced in both the
basal layer of the epidermis and in the hair follicles (Figure 2C), something not observed in wild-
type skin (Supplementary figure). At 21 day, the epidermis was hyperplastic and developed
keratinous cysts. BrdU positive cells were abundant in basal cells and in cells of the cyst walls
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 9
(Figure 2D). There were no apoptotic cells observed at either 3 or 7 day although there was a
slight increase in TUNEL positive cells at 21 day (Figure 2E).
The de- induction of H-RasV12G after 21 days of expression resulted in a dramatic shift
from cell growth to apoptosis (Figure 2F-G). Three days after H-RasV12G de- induction, BrdU
incorporation was not detected while apoptosis, as evident by TUNEL staining, occurred
primarily in the stromal cells that do not express H-RasV12G. In some sections, the entire region
of stroma underwent apoptosis. In addition, apoptotic cells also appeared in the suprabasal cells
of the epidermis. On 14 day of H-RasV12G de-induction, apoptosis was lower while proliferating
cells became detectable in the hair follicles. At this time, many of hair follicles were in the
anagen phase as a part of skin recovery.
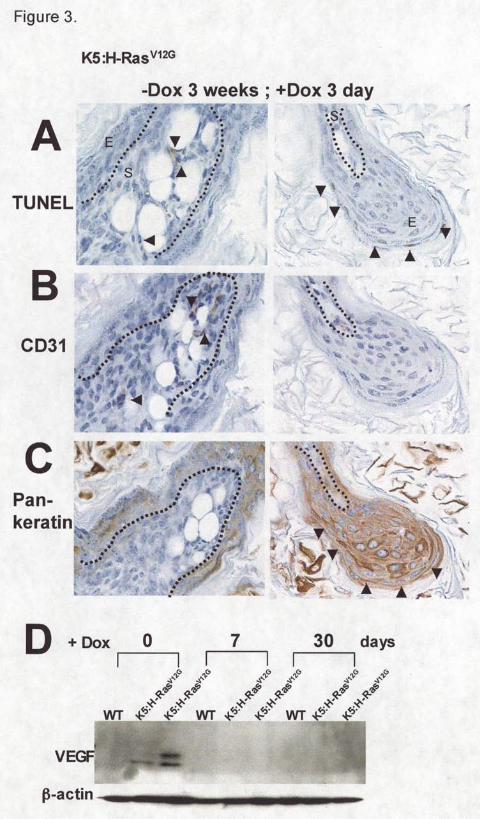
To identify if the apoptotic cells are epidermal in origin, serial sections of skin were
stained using a pan-keratin antibody to detect epidermis cells and a CD31 antibody to detect
endothelial cells. Within three days of H-RasV12G de-induction, apoptotic cells were observed
that are pan-keratin negative (Figure 3A, C; left panel). They are near CD31 positive cells
adjacent to blood vessels (Figure 3A, B; left panel). In addition, vascular endothelial growth
factor (VEGF) protein was strongly expressed in H-RasV12G expressing skin, but completely
disappeared within one week of H-RasV12G de-induction (Figure 3D). The significant apoptotic
rate of cells lining tumor vessels suggest that continued H-RasV12G expression is required for the
critical host-tumor symbiotic interaction that sustains stable tumor vasculature and is consistent
with the reports that K- and H-RasV12G can stimulate VEGF expression [27]. Apoptotic cells
were also detected in epidermal cells expressing keratin protein (Figure 3A, C; right panel).
Increase in proapoptotic proteins without effecting the expression of differentiation
markers in H-RasV12G suppressing skin
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 10
It has been reported previously that tumor regression upon oncogene de- induction is due
to apoptosis and differentiation [28]. To identify molecular pathways involved in the induction
of apoptosis in skin after H-RasV12G de-induction, the Kinetworks™ Apoptosis Protein Screen
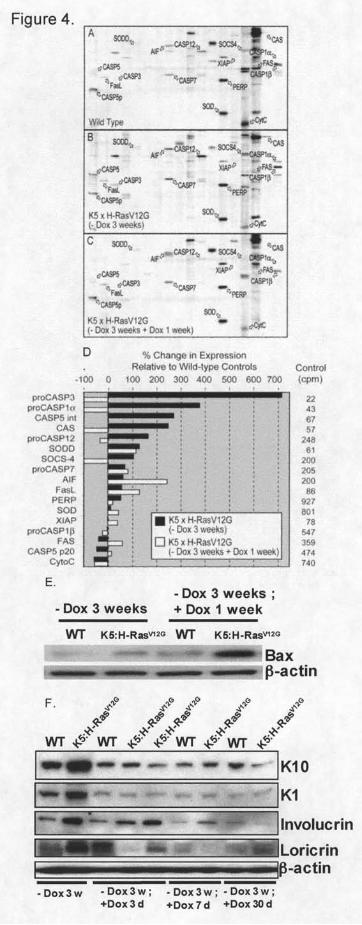
(KAPS-1.0) that determines protein levels of 25 candidate apoptosis proteins (Figure 4) was used.
The induction of H-RasV12G for 3 weeks increased concentrations of several proteins
including the procaspases (proCASP1? , proCASP3, CASP5 int, and proCASP12) and decreased
concentrations of others including CytoC, Fas and CASP5 p20, a matured form of procaspase 5
(Figure 4D). The increase in procaspase level in H-RasV12G expressing human ovarian cancer
model has been described earlier [29].
When H-RasV12G is de- induced, the four procaspases that were most strongly induced by
H-RasV12G expression decreased to below wild-type levels. In contrast, several proteins that are
proapoptotic were induced including AIF, Fas, and FasL (Figure 4D). Bax protein, that induces
the releases of mitochondrial AIF and is a member of Bcl-2 protein family [30], increased over
three fold in mice after H-RasV12G de-induction (Figure 4E).
To evaluate if the observed apoptosis coincided with cell differentiation as occurs in
normal skin [31], we examined the levels of differentiation markers such as keratin 1, keratin 10,
involucrin, and loricrin. These markers increased with H-RasV12G expression but decreased after
H-RasV12G suppression indicating that the observed apoptosis was not part of the normal
differentiation program (Figure 4F).
Phosphoprotein changes in skin after reversible H-RasV12G expression
Ras act via downstream effectors cascades to alter cellular growth and apoptosis [32]. To
identify kinases and protein phosphorylation changes that accompany H-RasV12G activation and
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 11
subsequent suppression, we used four Kinetworks™ multi- immunoblotting screens to examine
the phosphorylation of 37 proteins and expression levels of 75 protein kinases.
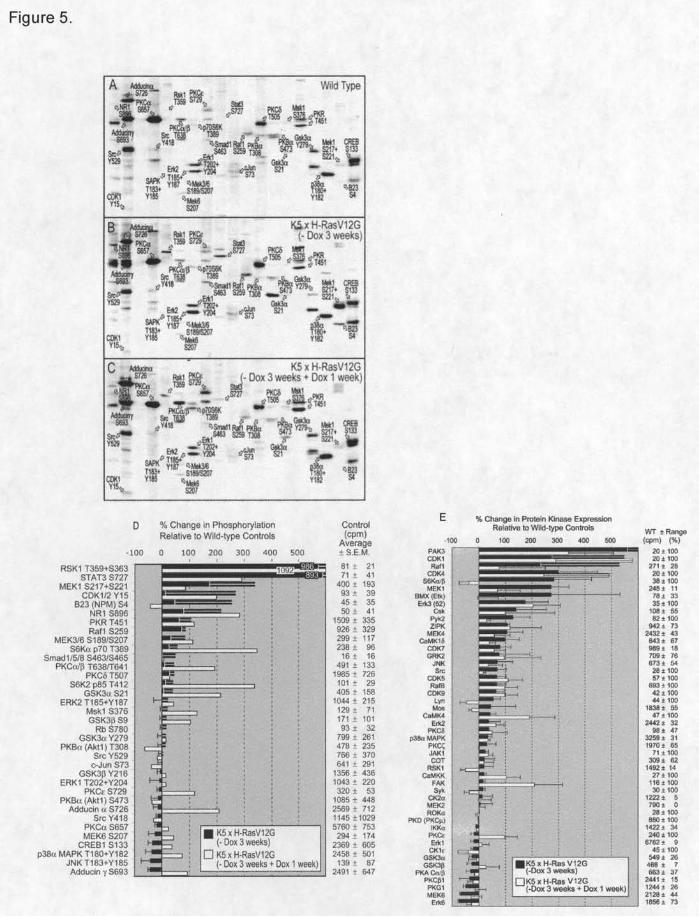
We first utilized a KinetworksTM Phospho-Site Screen (KPSS-1.3) to identify changes in
the phosphorylation status of 37 protein phosphorylation sites after H-RasV12G expression in the
three skin samples described above (Figure 5A-C). These results are summarized in Figure 5D
organized to show the most striking increases in protein phosphorylation at the top and the
largest decreases at the bottom of the bar graph. It should be appreciated that the most reliable
results are provided when the intensity of the ECL signals from the immunoblots are higher.
Figure 5D reveals tha t several known downstream effectors of H-RasV12G including RSK1 (10-
fold), STAT3 (9-fold), and MEK1 (3.5-fold) have increased phosphorylation levels following H-
RasV12G expression. Other proteins, not previously associated with Ras activation, such as NR1
and nucleophosmin (B23), had over a 2-fold increase in their phosphorylation levels. Decreased
phosphorylation is observed with adducin ? and ?, CREB1, p38MAPK and JNK. CREB does
not show increased phosphorylation even though it is a direct target of RSK1, which is induced
10-fold [33].
Following H-RasV12G de- induction, the phosphorylation states of many proteins returned
to normal (Raf1, MEK1, and B23). However, several proteins had increased phosphorylation
(adducin? /?, S6K? ??, PKC? /? /?, GSK3? /? ) and may mediate the increased apoptosis observed
as the hyperplasic state reverts to a normal.
Protein kinase changes in skin after reversible H-RasV12G expression
To evaluate the consequences of H-RasV12G induction and de-induction on protein kinase
expression levels, we employed a Kinetworks™ protein kinase screen, which detected 48 out of
a possible 75 protein kinases in the mouse skin samples. Following H-RasV12G induction, 16
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 12
kinases increased over 2-fold while only two decreased more than 2-fold. Seven kinases
increased 3-fold or more following H-RasV12G expression (PAK3, CDK1/4, BMX, S6K? ?? , Csk,
Raf1 and MEK1) (Figure 5E).
When H-RasV12G is de- induced, only three (Pak3, BMX, and S6K? ?? ) of the seven
kinases that increase 3-fold revert to near normal while CDK4 concentration actually increases.
Several additional kinases also increase with H-RasV12G de-induction (ZIPK, CaMK4, CaMKK,
FAK, and PKC??? ) (Figure 5E).
The protein kinase screen (Figure 5E) and the phosphoprotein screen (Figure 5D) contain
signals for 16 common proteins allowing one to determine if the fraction of protein that is
phosphorylated changes in the different samples. The stoichiometry of phosphorylation (the
ratio of total phospho-site signal divided by the total protein signal) was substantially reduced
with H-RasV12G de-induction for Raf1 (by 54%), MEK1 (by 76%), ERK2 (by 55%), GSK3? ?(by
54%), and PKC? (by 55%). In contrast, H-RasV12G de- induction increases the stoichiometry of
phosphorylation for S6K? /? ?T389 (by greater than 1000%) and Rsk 1/3 T359+S363/T356+S360
(by 330%), but there was a compensatory reduction in the total protein levels of these kinases at
the same time.
Reversible morphological changes following H-RasV12G suppression in primary
keratinocytes.
To determine if the reversibility observed in adult mice skin after H-RasV12G suppression
was also observed in cell culture, we prepared primary keratinocytes cultures from the K5:H-
RasV12G new-born pups. 2 days after induction of H-RasV12G expression in primary keratinocytes,
distinct morphological changes were observed including swollen cytoplasm with vacuoles. 9
days of H-RasV12G expression causes dramatic morphological changes (Figure 6A; upper panel).
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 13
These morphological changes were totally reversed within 7 days after suppression of H-RasV12G
expression (Figure 6A; lower panel). We also found that expression of H-RasV12G protein was
dramatically decreased within 5 days after addition of doxycycline (Figure 6B). To confirm the
skin screening results, we examined the levels of PKC? in the keratinocytes. Compare to H-
RasV12G expressing keratinocytes, the phosphorylation of PKC? protein dramatically increased
>5-fold on 5 day after H-RasV12G suppression and then decreased on 7 day (Figure 6B).
However, compare to H-Ras expressing keratinocytes, total PKC? protein level was not
significantly different in the H-RasV12G suppressed keratinocytes (Figure 6B).
Discussion
We present for the first time a histological and proteomic examination of the reversible
expression of oncogenic H-RasV12G in the mouse epidermis under keratin 5 promoter control.
This study shows that the H-RasV12G induced epidermal hyperplasia is reversible after H-RasV12G
de-induction, a process that involves apoptosis in both the stroma and the suprabasal cells of the
epidermis. The massive induction of apoptosis in the stroma within 3 day of H-RasV12G
suppression occurred near blood vessels in an “all or none” type pattern. These results indicate
that H-RasV12G expression is needed in a non-cell autonomous (paracrine) manner to maintain the
non-H-RasV12G expressing cells. Apoptosis also occurred in the suprabasal cells with individual
cells being TUNEL positive. To initiate a molecular characterization of this poorly understood
phenomenon of cell death induced by oncogene withdrawal, we have identified changes in
protein expression and phosphorylation status in the skin following H-RasV12G de-induction.
These proteins are potential biomarkers and molecular mediators of oncogenic addiction.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 14
Oncogenic dependence or oncogenic addiction is a term used to describe the observation
that tumors initiated by oncogene expression are dependent on their continual expression for
tumor stability [20] [21]. When the oncogene is de-induced, the tumors swiftly regress mainly
by apoptosis or differentiation. For example, H-RasV12G induced mouse melanomas regressed
via apoptosis when H-RasV12G expression was suppressed [17] or partially regress when K-Ras is
suppressed in lung cancers [34]. A similar reversal of the tumor phenotype via apoptosis is
observed when the expression of the c-Myc oncogene [18] or mutant EGF receptor [19] is de-
induced. However, this phenomenon has never been described in the epidermis.
The induction of apoptosis in the stroma where H-RasV12G is not expressed suggests a
paracrine function for cells that express H-RasV12G. One of these proteins could be the secreted
VEGF that is upregulated by H-RasV12G induction in skin and substantially downregulated when
H-RasV12G is suppressed (Figure 3). Down regulation of VEGF following H-RasV12G
suppression is also observed in melanocytes [17]. In that study, forced expression of VEGF was
not sufficient to reverse the induction of apoptosis when H-RasV12G was de- induced suggesting
that additional H-RasV12G targets are critical for the induction of apoptosis in melanomas.
Using the Kinetworks apoptosis protein screen (KAPS-1.0) (Figure 4), expression
profiles of number of apoptotic proteins were studied. Several procaspases (1? , 3, 5, and 12)
that were induced following H-RasV12G induction decreased or disappeared in the skin after H-
RasV12G de-induction as the skin switches from cell growth to apoptosis (Figure 4). Several
proapoptotic proteins increased including Bax and AIF involved in the intrinsic apoptosis
pathway and FAS and FAS ligand involved in the extrinsic apoptosis pathway [35]. An increase
in AIF level suggests that a caspase- independent mechanism may also play a role to induce
apoptosis. However, unlike the previous reports showed that marked differentiation in oncogene
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 15
inactivation [18] [36], H-RasV12G suppressing skin did not show any increase in differentiation
(Figure 4), suggesting that differentiation may not be a contributing factor in reversible
phenotype.
The molecular changes that accompany tumor regression when the activating oncogene is
de-induced have not previously been described. To begin to address this issue, we investigated
protein changes in the skin after H-RasV12G induction and subsequent de- induction. We
examined the phosphorylation status of 37 phosphoproteins and the expression levels of 75
protein kinases. The proteomic analysis examined the entire skin and describes a global change
in protein concentrations while our histological analysis identified apoptosis in two distinct cell
populations following H-RasV12G suppression, the stroma and the suprabasal epidermis. Future
studies will address where in the skin these proteomic changes occur.
Targets of H-RasV12G activity that induce cell proliferation are readily identifiable in the
hyperplastic skin. These include phosphorylation of several well-known downstream effectors
of Ras signaling via Raf1, including Raf1 itself, MEK1, RSK1, and STAT3. There was also a
decrease in the phosphorylation of MEK6 and its target p38 MAP kinase as well as JNK MAP
kinase following H-RasV12G induction (Figure 5) consistent with repression of the inhibitory roles
that these protein kinases normally play in cell proliferation and their proapoptotic functions. In
contrast, ERK phosphorylation was not changed much in H-RasV12G induced skin (Figure 5D).
These results have precedence. Mao and co-workers [37] have shown that phophorylated ERK
proteins are barely detectable in skin papilloma with the oncogenic H-Ras protein. We speculate
that our results may indicate an imbalance of phophotases and oncogene signaling. We also
measured the expression levels of 25 different phosphatases using a Kinetworks™ protein
phosphatase screen (KPPS-1.2; unpublished work), H-RasV12G induced greater than a 2-fold
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 16
elevation of the catalytic and regulatory A’2 subunit levels of protein-serine phosphatase 4 (PP4)
and the dual specificity MAP kinase phosphatase 2 (MKP2). With H-RasV12G suppression, there
was a complete loss of PP4-A’2 expression and a reduction of PP4-C levels to even below the
wild-type values. By contrast, H-RasV12G suppression also caused even more marked increases in
the expressions of MKP2. The finding that the concentrations of diverse protein phosphatases
were also markedly altered with the gain of Ras function as well as its subsequent repression
adds an extra dimension to the complexity in interpreting the observed changes in protein
phosphorylation. The induction of the dual specificity MAP kinase phosphatase (MKP2) may
partly account for why the phosphorylation levels of ERK1 and ERK2 were not significantly
affected in the skin of H-RasV12G expressing and repressed animals, despite the increases in
expression and activating phosphorylation of the upstream kinase MEK1.
When H-RasV12G is de- induced, signaling pathways upstream of the apoptotic machinery
(discussed above) also changes. These changes have previously been shown to have either anti-
apoptotic or proapoptotic consequences. Anti-apoptotic signals are increased phosphorylation of
the?? ? and??? ??subunits of S6K which increase ribosomal translation. An additional anti-apoptotic
signal is the increase in FAK protein, a kinase involved in organizing stress fibers in cell culture
[38].
Several proapoptotic changes are evident. These include decreased phosphorylation of
Akt1, a survival factor, that has also been shown to respond to VEGF to promote endothelial cell
survival [39] thus contributing to apoptosis in the stroma. In addition, an increase in p38 MAPK
phosphorylation level [40] and ZIP kinase protein after H-RasV12G de-induction is consistent with
previously reported work [41].
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 17
Proapoptotic signals that increase include phosphorylation of PKC? ?? ?and PKC?. PKC’s
promote apoptosis in human gastric cancer cells [42] and mouse keratinocytes [43]. Our studies
indicate PKC? activation after H-RasV12G suppression could also contribute to cell killing that is
often required before the recovery process. The result showing that the phosphorylation of
PKC? decreased in H-RasV12G expressing skin (Figure 5D) was consistent with the previous
reports that downregulation of PKC protein level and activity are seen in cells expressing
activated Ras genes [44-48]. The PKC phosphorylation targets adducin? ???also increases which
weaken cell-cell interactions resulting in apoptosis [49]. Furthermore, the phenotypic
reversibility and an increase in PKC? phosphorylation also occurred in the primary keratinocytes
system after H-RasV12G suppression (Figure 6) indicating that it is a cell autonomous property.
More work will be needed to determine if these pathways are active in apoptotic cells.
Interestingly, in primary keratinocytes cell culture (Figure 6), the significant apoptotic event did
not occur in the reversible manner upon H-Ras suppression (data not shown), suggesting the
possibility that the reversibility following doxycycline addition may reflect a continued
requirement for H-Ras in promoting and sustaining cell-autonomous hyperplasia–host
interactions that are essential for the skin phenotype and maintenance that are not faithfully
mimicked in tissue culture.
In summary, we have identified protein and phosphoprotein changes that accompany H-
RasV12G induction and de-induction in mouse skin. These molecular changes may mediate the
poorly understood phenomenon of oncogenic addiction.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 18
Acknowledgements : We thank Stuart Yuspa, Ronald Wolf, Christophe Cataisson, and Lyuba
Varticovski for insights into Ras signaling.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 19
Figure legends
Figure 1. A) Change in H-RasV12G protein in K5:H-RasV12G mice when de-induced for 0, 3, 7, or
30 days following expression for 3 weeks (0 days). B) Dorsal view of mouse following 3 weeks
of H-RasV12G expression. Reversal of skin hyperplasia (arrows) following suppression of H-
RasV12G expression. C) H-RasV12G expression for 3 weeks. H-RasV12G expression for 3 weeks
and subsequently suppression for D) 10 days and E) 30 days. F) Dissected skin expressing H-
RasV12G for 3 weeks showing extensive angiogenesis.
Figure 2. Histological differences in K5:H-RasV12G mice skin during H-RasV12G induction and
de-induction. Hematoxylin and eosin-stained sections from A) H-RasV12G expressing skin for 3
weeks (left), H-RasV12G suppression for 3 days after 3 week induction (right panel), and B) H-
RasV12G suppression for 3 weeks. Time-dependent changes in skin histology of K5:H-RasV12G
mice. Eight time points are presented. H-RasV12G expression was induced and samples were
examined at 0, 3, 7, and 21 days. H-RasV12G expression was suppressed and samples were
examined at 3, 7, 14, and 30 days. C) H&E. D) BrdU immunohistochemistry to monitor cell
proliferation. E) TUNEL assay to monitor apoptosis. Number of positive cells/mm at the eight
time points presented in Figure 2D-E and supplementary figure for F) BrdU positive cells. G)
TUNEL positive cells.
Figure 3. Serial sections of skin showing TUNEL assay and immunohistochemistry using CD31
or pan-keratin antibody in samples expressing H-RasV12G for 3 weeks and subsequent de-
induction for 3 days. A) TUNEL assay to identify apoptotic cells (arrows). B) CD31
immunohistochemistry to identify endothelial cells (arrows). E, epidermis; S, stroma. Dotted line,
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 20
epidermis-stroma borderline. C) Pan-Keratin immunohistochemistry to identify epidermis shown
by arrows. D) Western analysis of VEGF following H-RasV12G suppression.
Figure 4. KinetworksTM KAPS1.0 analysis of 25 apoptosis related proteins for three skin
samples. A) wild-type skin. B) Skin expressing H-RasV12G for 3 weeks, and C) Skin following
H-RasV12G expression for 3 weeks and then suppression for 1 week. D) The percent change in
concentration for 17 detected proteins relative to wild-type skin with H-RasV12G expression
(black bars) and H-RasV12G de-induction (white bars) are shown. The average intensity of the
ECL signal (counts per minute) from wild-type skin is in the right column. E) Bax protein
concentration at the same time points used in B and C. F) Protein concentrations of four
epidermal differentiation markers at 0, 3, 7, and 30 days following H-RasV12G suppression.
Figure 5. KinetworksTM KPSS1.3 analysis of 37 phospho-sites in cell signaling proteins for
three skin samples (A-C) described in Figure 4. The positions of detected target
phosphoproteins are indicated with arrows. D) The percent changes in expression for 17
detected target proteins with H-RasV12G expression (black bars) and H-RasV12G de-induction
(white bars) are shown. Error bars indicate the standard error of the means from four separate
experiments performed on different samples. E) The percent change in concentration for 48
detected proteins in KinetworksTM KPKS 1.2 analysis. Error bars indicate the standard error of
the means from two separate experiments performed on different samples.
Figure 6. Effect of H-RasV12G expression and suppression on primary keratinocytes morphology
and expression profile of PKC? . A) Primary keratinocytes in which H-RasV12G protein was
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 21
expressed for 2 days. After 2 days, H-RasV12G was suppressed or expressed for another 7 day
and cell morphologies were monitored by phase-contrast microscopy. B) Time-dependent
expression levels of H-RasV12G , phospho PKC? , and total PKC? protein.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 22
References
1. Malumbres, M. and Barbacid, M. (2003) RAS oncogenes: the first 30 years. Nat Rev
Cancer, 3, 459-65. 2. Nikiforova, M.N., Lynch, R.A., Biddinger, P.W., Alexander, E.K., Dorn, G.W., 2nd,
Tallini, G., Kroll, T.G. and Nikiforov, Y.E. (2003) RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab, 88, 2318-26.
3. Sekiya, T., Fushimi, M., Hori, H., Hirohashi, S., Nishimura, S. and Sugimura, T. (1984) Molecular cloning and the total nucleotide sequence of the human c-Ha-ras-1 gene activated in a melanoma from a Japanese patient. Proc Natl Acad Sci U S A, 81, 4771-5.
4. Aoki, Y., Niihori, T., Kawame, H., Kurosawa, K., Ohashi, H., Tanaka, Y., Filocamo, M., Kato, K., Suzuki, Y., Kure, S. and Matsubara, Y. (2005) Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet, 37, 1038-40.
5. Vitale-Cross, L., Amornphimoltham, P., Fisher, G., Molinolo, A.A. and Gutkind, J.S. (2004) Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res, 64, 8804-7.
6. Bailleul, B., Surani, M.A., White, S., Barton, S.C., Brown, K., Blessing, M., Jorcano, J. and Balmain, A. (1990) Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell, 62, 697-708.
7. Bos, J.L. (1989) ras oncogenes in human cancer: a review. Cancer Res, 49, 4682-9. 8. Yuspa, S.H. (1994) The pathogenesis of squamous cell cancer: lessons learned from
studies of skin carcinogenesis--thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res, 54, 1178-89.
9. Marshall, M.S. (1995) Ras target proteins in eukaryotic cells. Faseb J, 9, 1311-8. 10. Shuman, J.D., Sebastian, T., Kaldis, P., Copeland, T.D., Zhu, S., Smart, R.C. and
Johnson, P.F. (2004) Cell cycle-dependent phosphorylation of C/EBPbeta mediates oncogenic cooperativity between C/EBPbeta and H-RasV12. Mol Cell Biol, 24, 7380-91.
11. McFall, A., Ulku, A., Lambert, Q.T., Kusa, A., Rogers-Graham, K. and Der, C.J. (2001) Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Mol Cell Biol, 21, 5488-99.
12. Aplin, A.E., Short, S.M. and Juliano, R.L. (1999) Anchorage-dependent regulation of the mitogen-activated protein kinase cascade by growth factors is supported by a variety of integrin alpha chains. J Biol Chem, 274, 31223-8.
13. Eckert, L.B., Repasky, G.A., Ulku, A.S., McFall, A., Zhou, H., Sartor, C.I. and Der, C.J. (2004) Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res, 64, 4585-92.
14. Shields, J.M., Pruitt, K., McFall, A., Shaub, A. and Der, C.J. (2000) Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol, 10, 147-54.
15. Yuspa, S.H., Dlugosz, A.A., Cheng, C.K., Denning, M.F., Tennenbaum, T., Glick, A.B. and Weinberg, W.C. (1994) Role of oncogenes and tumor suppressor genes in multistage carcinogenesis. J Invest Dermatol, 103, 90S-95S.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 23
16. Roper, E., Weinberg, W., Watt, F.M. and Land, H. (2001) p19ARF-independent induction of p53 and cell cycle arrest by Raf in murine keratinocytes. EMBO Rep, 2, 145-50.
17. Chin, L., Tam, A., Pomerantz, J., Wong, M., Holash, J., Bardeesy, N., Shen, Q., O'Hagan, R., Pantginis, J., Zhou, H., Horner, J.W., 2nd, Cordon-Cardo, C., Yancopoulos, G.D. and DePinho, R.A. (1999) Essential role for oncogenic Ras in tumour maintenance. Nature, 400, 468-72.
18. Felsher, D.W. and Bishop, J.M. (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell, 4, 199-207.
19. Politi, K., Zakowski, M.F., Fan, P.D., Schonfeld, E.A., Pao, W. and Varmus, H.E. (2006) Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev, 20, 1496-510.
20. Weinstein, I.B. (2002) Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science, 297, 63-4.
21. Varmus, H. (2006) The new era in cancer research. Science, 312, 1162-5. 22. Diamond, I., Owolabi, T., Marco, M., Lam, C. and Glick, A. (2000) Conditional gene
expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol, 115, 788-94.
23. Dlugosz, A.A., Glick, A.B., Tennenbaum, T., Weinberg, W.C. and Yuspa, S.H. (1995) Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol, 254, 3-20.
24. Nunez, N.P., Oh, W.J., Rozenberg, J., Perella, C., Anver, M., Barrett, J.C., Perkins, S.N., Berrigan, D., Moitra, J., Varticovski, L., Hursting, S.D. and Vinson, C. (2006) Accelerated tumor formation in a fatless mouse with type 2 diabetes and inflammation. Cancer Res, 66, 5469-76.
25. Pelech, S., Sutter, C. and Zhang, H. (2003) Kinetworks protein kinase multiblot analysis. Methods Mol Biol, 218, 99-111.
26. Raimondi, A.R., Vitale-Cross, L., Amornphimoltham, P., Gutkind, J.S. and Molinolo, A. (2006) Rapid development of salivary gland carcinomas upon conditional expression of K-ras driven by the cytokeratin 5 promoter. American Journal of Pathology, 168, 1654-65.
27. Okada, F., Rak, J.W., Croix, B.S., Lieubeau, B., Kaya, M., Roncari, L., Shirasawa, S., Sasazuki, T. and Kerbel, R.S. (1998) Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc Natl Acad Sci U S A, 95, 3609-14.
28. Felsher, D.W. (2004) Reversibility of oncogene-induced cancer. Curr Opin Genet Dev, 14, 37-42.
29. Young, T., Mei, F., Liu, J., Bast, R.C., Jr., Kurosky, A. and Cheng, X. (2005) Proteomics analysis of H-RAS-mediated oncogenic transformation in a genetically defined human ovarian cancer model. Oncogene, 24, 6174-84.
30. Cregan, S.P., Fortin, A., MacLaurin, J.G., Callaghan, S.M., Cecconi, F., Yu, S.W., Dawson, T.M., Dawson, V.L., Park, D.S., Kroemer, G. and Slack, R.S. (2002) Apoptosis-
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 24
inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol, 158, 507-17.
31. Maruoka, Y., Harada, H., Mitsuyasu, T., Seta, Y., Kurokawa, H., Kajiyama, M. and Toyoshima, K. (1997) Keratinocytes become terminally differentiated in a process involving programmed cell death. Biochem Biophys Res Commun, 238, 886-90.
32. Downward, J. (1998) Ras signalling and apoptosis. Curr Opin Genet Dev, 8, 49-54. 33. Song, K.S., Seong, J.K., Chung, K.C., Lee, W.J., Kim, C.H., Cho, K.N., Kang, C.D., Koo,
J.S. and Yoon, J.H. (2003) Induction of MUC8 gene expression by interleukin-1 beta is mediated by a sequential ERK MAPK/RSK1/CREB cascade pathway in human airway epithelial cells. J Biol Chem, 278, 34890-6.
34. Fisher, G.H., Wellen, S.L., Klimstra, D., Lenczowski, J.M., Tichelaar, J.W., Lizak, M.J., Whitsett, J.A., Koretsky, A. and Varmus, H.E. (2001) Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev, 15, 3249-62.
35. Raj, D., Brash, D.E. and Grossman, D. (2006) Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol, 126, 243-57.
36. Shachaf, C.M., Kopelman, A.M., Arvanitis, C., Karlsson, A., Beer, S., Mandl, S., Bachmann, M.H., Borowsky, A.D., Ruebner, B., Cardiff, R.D., Yang, Q., Bishop, J.M., Contag, C.H. and Felsher, D.W. (2004) MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature, 431, 1112-7.
37. Mao, J.H., To, M.D., Perez-Losada, J., Wu, D., Del Rosario, R. and Balmain, A. (2004) Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes & Development, 18, 1800-5.
38. Thannickal, V.J., Lee, D.Y., White, E.S., Cui, Z., Larios, J.M., Chacon, R., Horowitz, J.C., Day, R.M. and Thomas, P.E. (2003) Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem, 278, 12384-9.
39. Fujio, Y. and Walsh, K. (1999) Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem, 274, 16349-54.
40. Shimizu, H., Banno, Y., Sumi, N., Naganawa, T., Kitajima, Y. and Nozawa, Y. (1999) Activation of p38 mitogen-activated protein kinase and caspases in UVB-induced apoptosis of human keratinocyte HaCaT cells. J Invest Dermatol, 112, 769-74.
41. Kawai, T., Akira, S. and Reed, J.C. (2003) ZIP kinase triggers apoptosis from nuclear PML oncogenic domains. Mol Cell Biol, 23, 6174-86.
42. Okuda, H., Adachi, M., Miyazawa, M., Hinoda, Y. and Imai, K. (1999) Protein kinase Calpha promotes apoptotic cell death in gastric cancer cells depending upon loss of anchorage. Oncogene, 18, 5604-9.
43. Cataisson, C., Joseloff, E., Murillas, R., Wang, A., Atwell, C., Torgerson, S., Gerdes, M., Subleski, J., Gao, J.L., Murphy, P.M., Wiltrout, R.H., Vinson, C. and Yuspa, S.H. (2003) Activation of cutaneous protein kinase C alpha induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol, 171, 2703-13.
44. Guillem, J.G., Hsieh, L.L., O'Toole, K.M., Forde, K.A., LoGerfo, P. and Weinstein, I.B. (1988) Changes in expression of oncogenes and endogenous retroviral-like sequences during colon carcinogenesis. Cancer Res, 48, 3964-71.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Oh et al 6/4/07 7:29 PM 25
45. Colapietro, A.M., Goodell, A.L. and Smart, R.C. (1993) Characterization of benzo[a]pyrene-initiated mouse skin papillomas for Ha-ras mutations and protein kinase C levels. Carcinogenesis, 14, 2289-95.
46. Weyman, C.M., Taparowsky, E.J., Wolfson, M. and Ashendel, C.L. (1988) Partial down-regulation of protein kinase C in C3H 10T 1/2 mouse fibroblasts transfected with the human Ha-ras oncogene. Cancer Res, 48, 6535-41.
47. Haliotis, T., Trimble, W., Chow, S., Bull, S., Mills, G., Girard, P., Kuo, J.F. and Hozumi, N. (1990) Expression of ras oncogene leads to down-regulation of protein kinase C. Int J Cancer, 45, 1177-83.
48. Wolfman, A. and Macara, I.G. (1987) Elevated levels of diacylglycerol and decreased phorbol ester sensitivity in ras-transformed fibroblasts. Nature, 325, 359-61.
49. Imamdi, R., de Graauw, M. and van de Water, B. (2004) Protein kinase C mediates cisplatin-induced loss of adherens junctions followed by apoptosis of renal proximal tubular epithelial cells. J Pharmacol Exp Ther, 311, 892-903.
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

by guest on June 7, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from




















