
High Throughput Peptide Mass Fingerprinting and Protein Macroarray Analysis Using Chemical Printing...
10
High Throughput Peptide Mass Fingerprinting and Protein Macroarray Analysis Using Chemical Printing Strategies* Andrew J. Sloane‡, Janice L. Duff‡, Nicole L. Wilson‡, Parag S. Gandhi‡, Cameron J. Hill‡, Femia G. Hopwood‡, Paul E. Smith‡, Melissa L. Thomas‡, Robert A. Cole‡, Nicolle H. Packer‡, Edmond J. Breen‡, Patrick W. Cooley§, David B. Wallace§, Keith L. Williams‡, and Andrew A. Gooley‡¶ We describe a chemical printer that uses piezoelectric pulsing for rapid, accurate, and non-contact microdis- pensing of fluid for proteomic analysis of immobilized protein macroarrays. We demonstrate protein digestion and peptide mass fingerprinting analysis of human plasma and platelet proteins direct from a membrane surface subsequent to defined microdispensing of trypsin and matrix solutions, hence bypassing multiple liquid- handling steps. Detection of low abundance, alkaline pro- teins from whole human platelet extracts has been high- lighted. Membrane immobilization of protein permits archiving of samples pre-/post-analysis and provides a means for subanalysis using multiple chemistries. This study highlights the ability to increase sequence coverage for protein identification using multiple enzymes and to characterize N-glycosylation modifications using a com- bination of PNGase F and trypsin. We also demonstrate microdispensing of multiple serum samples in a quantita- tive microenzyme-linked immunosorbent assay format to rapidly screen protein macroarrays for pathogen-derived antigens. We anticipate the chemical printer will be a major component of proteomic platforms for high throughput protein identification and characterization with widespread applications in biomedical and diagnos- tic discovery. Molecular & Cellular Proteomics 1: 490 – 499, 2002. The ability to accurately define protein expression in rela- tionship to physiological changes associated with healthy or diseased states and the potential to discover novel drug targets are emerging themes of proteomic programs (1, 2). Understanding these dynamics is rendered complex given there is often no correlation between mRNA expression levels and protein expression (3), and the paradigm of one gene-one protein is known not to hold (4). The rapid growth in proteomics has resulted in a technology- driven science oriented toward development of automated high throughput platforms (5). Sample prefractionation, advances in solubilization strategies, and improvements in two-dimensional gel electrophoresis (2-DE) 1 are further refining this art (6 – 8). Identification and characterization of proteins is also becom- ing more rapid with the increasing development and applica- tion of mass spectrometry (MS) for peptide mass fingerprint- ing (pmf) and tandem MS sequence analysis (9 –11). Protein arrays are emerging state of the art technologies for high throughput proteomics (12–16). Developments in protein array technology now encompass protein deposition on mem- branes, glass plates, microwells, polystyrene film (12, 17–22), microfluidic chips, and biochips (23–25) for screening complex protein mixtures for binding affinities, protein associations, and disease markers. With a move toward automation, deposition techniques used to produce these arrays now include pin- based or microdispensing liquid-handling robots (14), photo- lithography (26, 27), and ink-jet printing technology (28 –30). Despite advantages of protein array technology such as speed, sensitivity, and multi-screening capabilities (15), chip- based proteomics has the major caveat that the protein arrays are either constructed from known proteins, such as antibod- ies, or an array of proteins derived from a recombinant ex- pression system (15). Hence, the current chip-based ap- proaches ignore the many protein isoforms produced by cells such as different co- and post-translationally modified forms of the same translated gene product, members of gene fam- ilies, and variable spliced variants of mRNA and protein. Recent advances in sample preparation have enabled many of the known protein isoforms to be displayed in 2-DE arrays From ‡Proteome Systems Limited, 1/35– 41 Waterloo Rd., North Ryde, Sydney, New South Wales 2113, Australia and §Microfab Tech- nologies, Inc., Plano, Texas 75074 Received, March 18, 2002, and in revised form, June 26, 2002 Published, MCP Papers in Press, July 3, 2002, DOI 10.1074/ mcp.M200020-MCP200 1 The abbreviations used are: 2-DE, two-dimensional gel electro- phoresis; BSA, bovine serum albumin; CFR, curved field reflectron; ESI, electrospray ionization; FITC, fluorescein isothiocyanate; LC, liquid chromatography; MALDI-TOF, matrix-assisted laser desorp- tion/ionization time of flight; MS, mass spectrometry; OGP, n-octyl -D-glucopyranoside; PBS-TAC, phosphate-buffered saline contain- ing 0.1% (v/v) Tween 20, 0.05% (w/v) NaN 3 , and 0.5% (w/v) casein, pH 7.4; pmf, peptide mass fingerprinting; PSL, Proteome Systems Limited; PVDF, polyvinylidene fluoride; TB, tuberculosis. Research © 2002 by The American Society for Biochemistry and Molecular Biology, Inc. 490 Molecular & Cellular Proteomics 1.7 This paper is available on line at http://www.mcponline.org
Transcript of High Throughput Peptide Mass Fingerprinting and Protein Macroarray Analysis Using Chemical Printing...

High Throughput Peptide Mass Fingerprintingand Protein Macroarray Analysis UsingChemical Printing Strategies*Andrew J. Sloane‡, Janice L. Duff‡, Nicole L. Wilson‡, Parag S. Gandhi‡,Cameron J. Hill‡, Femia G. Hopwood‡, Paul E. Smith‡, Melissa L. Thomas‡,Robert A. Cole‡, Nicolle H. Packer‡, Edmond J. Breen‡, Patrick W. Cooley§,David B. Wallace§, Keith L. Williams‡, and Andrew A. Gooley‡¶
We describe a chemical printer that uses piezoelectricpulsing for rapid, accurate, and non-contact microdis-pensing of fluid for proteomic analysis of immobilizedprotein macroarrays. We demonstrate protein digestionand peptide mass fingerprinting analysis of humanplasma and platelet proteins direct from a membranesurface subsequent to defined microdispensing of trypsinand matrix solutions, hence bypassing multiple liquid-handling steps. Detection of low abundance, alkaline pro-teins from whole human platelet extracts has been high-lighted. Membrane immobilization of protein permitsarchiving of samples pre-/post-analysis and provides ameans for subanalysis using multiple chemistries. Thisstudy highlights the ability to increase sequence coveragefor protein identification using multiple enzymes and tocharacterize N-glycosylation modifications using a com-bination of PNGase F and trypsin. We also demonstratemicrodispensing of multiple serum samples in a quantita-tive microenzyme-linked immunosorbent assay format torapidly screen protein macroarrays for pathogen-derivedantigens. We anticipate the chemical printer will be amajor component of proteomic platforms for highthroughput protein identification and characterizationwith widespread applications in biomedical and diagnos-tic discovery. Molecular & Cellular Proteomics 1:490–499, 2002.
The ability to accurately define protein expression in rela-tionship to physiological changes associated with healthy ordiseased states and the potential to discover novel drugtargets are emerging themes of proteomic programs (1, 2).Understanding these dynamics is rendered complex giventhere is often no correlation between mRNA expression levelsand protein expression (3), and the paradigm of one gene-oneprotein is known not to hold (4).
The rapid growth in proteomics has resulted in a technology-driven science oriented toward development of automated highthroughput platforms (5). Sample prefractionation, advances insolubilization strategies, and improvements in two-dimensionalgel electrophoresis (2-DE)1 are further refining this art (6–8).Identification and characterization of proteins is also becom-ing more rapid with the increasing development and applica-tion of mass spectrometry (MS) for peptide mass fingerprint-ing (pmf) and tandem MS sequence analysis (9–11).
Protein arrays are emerging state of the art technologies forhigh throughput proteomics (12–16). Developments in proteinarray technology now encompass protein deposition on mem-branes, glass plates, microwells, polystyrene film (12, 17–22),microfluidic chips, and biochips (23–25) for screening complexprotein mixtures for binding affinities, protein associations, anddisease markers. With a move toward automation, depositiontechniques used to produce these arrays now include pin-based or microdispensing liquid-handling robots (14), photo-lithography (26, 27), and ink-jet printing technology (28–30).
Despite advantages of protein array technology such asspeed, sensitivity, and multi-screening capabilities (15), chip-based proteomics has the major caveat that the protein arraysare either constructed from known proteins, such as antibod-ies, or an array of proteins derived from a recombinant ex-pression system (15). Hence, the current chip-based ap-proaches ignore the many protein isoforms produced by cellssuch as different co- and post-translationally modified formsof the same translated gene product, members of gene fam-ilies, and variable spliced variants of mRNA and protein.
Recent advances in sample preparation have enabled manyof the known protein isoforms to be displayed in 2-DE arrays
From ‡Proteome Systems Limited, 1/35–41 Waterloo Rd., NorthRyde, Sydney, New South Wales 2113, Australia and §Microfab Tech-nologies, Inc., Plano, Texas 75074
Received, March 18, 2002, and in revised form, June 26, 2002Published, MCP Papers in Press, July 3, 2002, DOI 10.1074/
mcp.M200020-MCP200
1 The abbreviations used are: 2-DE, two-dimensional gel electro-phoresis; BSA, bovine serum albumin; CFR, curved field reflectron;ESI, electrospray ionization; FITC, fluorescein isothiocyanate; LC,liquid chromatography; MALDI-TOF, matrix-assisted laser desorp-tion/ionization time of flight; MS, mass spectrometry; OGP, n-octyl�-D-glucopyranoside; PBS-TAC, phosphate-buffered saline contain-ing 0.1% (v/v) Tween 20, 0.05% (w/v) NaN3, and 0.5% (w/v) casein,pH 7.4; pmf, peptide mass fingerprinting; PSL, Proteome SystemsLimited; PVDF, polyvinylidene fluoride; TB, tuberculosis.
Research
© 2002 by The American Society for Biochemistry and Molecular Biology, Inc.490 Molecular & Cellular Proteomics 1.7This paper is available on line at http://www.mcponline.org

(8). A Western blot of 2-DE separated proteins onto a mem-brane such as polyvinylidene fluoride (PVDF) or nitrocelluloserepresents a protein chip, albeit a macroarray. The proteinmacroarray differs from a protein chip microarray, becausethe coordinates of each protein are determined by the phys-ical attributes of the isoelectric point and apparent molecularweight of the protein. Once the coordinates of each proteinwithin the protein macroarray are identified by an image-capture device, each protein spot then has a defined X and Yposition and can be manipulated by robotic platforms.
Here we present technology that combines the advantagesof both protein chips and 2-DE, which we have described asa chemical printer (Fig. 1). The chemical printer uses a micro-jet device utilizing piezoelectric drop-on-demand-type ink-jettechnology for rapid liquid microdispensing (31–33). The abil-ity of ink-jet printing to dispense minimal amounts of rarefluids and permit parallel processing of large numbers of testsmeans assay sizes can be decreased whereas assay densityis increased (34, 35). These qualities have lead to increased
applications for dispensing of bioactive materials. Immunodi-agnostics and antibody/antigen dispensing (36), synthesisand deposition of oligonucleotides in microarray formats (29,38, 39), protein and peptide analysis (40), and drug discovery(41) are a number of applications utilizing ink-jet printing tech-nologies. We demonstrate in situ proteinase digests of mem-brane-immobilized protein macroarrays and subsequentMALDI-TOF MS analysis directly from the membrane surface.This approach bypasses the multiple liquid-handling stepsassociated with in-gel digestion procedures. Importantly, theability to analyze immobilized proteins allows for both archiv-ing of samples pre-/post-analysis and for multiple chemicalreactions to be performed at different locations on an individ-ual protein spot. We demonstrate identification of N-linkedglycosylation sites by MALDI-TOF MS using sequential PN-Gase F and trypsin digestion, as well as preparation andextraction of oligosaccharides from PVDF membrane forstructural analysis by LC-ESI MS.
An exciting new high throughput assay also uses the chem-
FIG. 1. A, schematic representation ofthe chemical printer. Piezoelectric mi-crojets are connected to individual rea-gent reservoirs and an air pressure/vac-uum control unit that enables ease ofsample loading and fluid stabilizationwithin the microjet devices. Fluid dis-pensing and motion of the target plat-form in both X and Y directions are con-trolled by integrated software andcontroller boxes. Droplet formation (pan-el B) and a top view of the target can bevisualized using respective cameras andmonitors. Image-capture software isused to define X and Y coordinate listsfor automated fluid delivery. The heightof the microjets (Z-axis) can be adjustedmanually. B, a piezoelectric-generateddroplet of 2-propanol. Sample was mi-crodispensed from a glass capillary pi-ezoelectric device with an orifice diame-ter of 55 �m. The droplet was generatedby pulsing the device at a frequency of240 Hz, with rise and fall times of 3 �s,respectively, a dwell time of 42 �s, and adwell voltage of 30 V. The droplet has avolume of �100 pl. The image was cap-tured by illuminating the droplet streamwith a light-emitting diode (LED) that waspulsed concurrently at a frequency of240 Hz.
Chemical Printing for Analysis of Protein Macroarrays
Molecular & Cellular Proteomics 1.7 491
ical printer to microdispense antibodies onto membrane-im-mobilized proteins to rapidly define immunoreactivity andquantitate signals in a solid phase enzyme-linked immunosor-bent assay-like format. The chemical printer thus represents apowerful tool for identification of novel protein targets forbiomedical and diagnostic purposes.
EXPERIMENTAL PROCEDURES
Materials
Standard laboratory chemicals were obtained from Sigma unlessspecified otherwise. Human serum and plasma samples and 38 kDatuberculosis (TB) protein were gifts from AP Clinical (Sydney, Austra-lia). Where stipulated in the text, human serum albumin was depletedfrom plasma using methods described previously (42, 43). Humanplatelets were purchased from the Red Cross Blood Bank (Sydney,Australia). Contaminating red blood cells were removed from theplatelets by centrifugation at 200 � g for 10 min at 4 °C. The platelet-rich plasma was then centrifuged at 1500 � g for 20 min at 4 °C. Theplatelet component of the pellet was removed gently and then resus-
pended in 50 mM Tris-HCl, 5 mM EDTA, pH 7.4. The platelets werewashed similarly two more times. Whole platelets were finally solubi-lized using a ProteoPrepTM sample extraction kit (Sigma) using thesupplied cellular and organelle membrane solubilizing reagent, to afinal protein concentration of 4 mg/ml. Purified immunoglobulin wasobtained from CSL (Parkville, Australia).
Chemical Printing
Solutions were dispensed using an �-version chemical printer be-ing developed by Proteome Systems Ltd. (PSL) (Sydney, Australia) incollaboration with Shimadzu Biotech (Kyoto, Japan). Solutions werepre-filtered through either 0.22- or 0.45-�m membrane filters (Milli-pore). Glass capillary piezoelectric microjet devices (Microfab Tech-nologies, Inc., Plano, TX) were used to dispense all solutions. X and Ycoordinates of target protein spots were determined using Im-agepIQTM, an in-house image-capture product (PSL).
Two-dimensional Gel Electrophoresis
Sample Preparation—36 �l of whole human plasma was made upto a final volume of 490 �l in 7 M urea, 2 M thiourea, 2% (w/v) CHAPS,
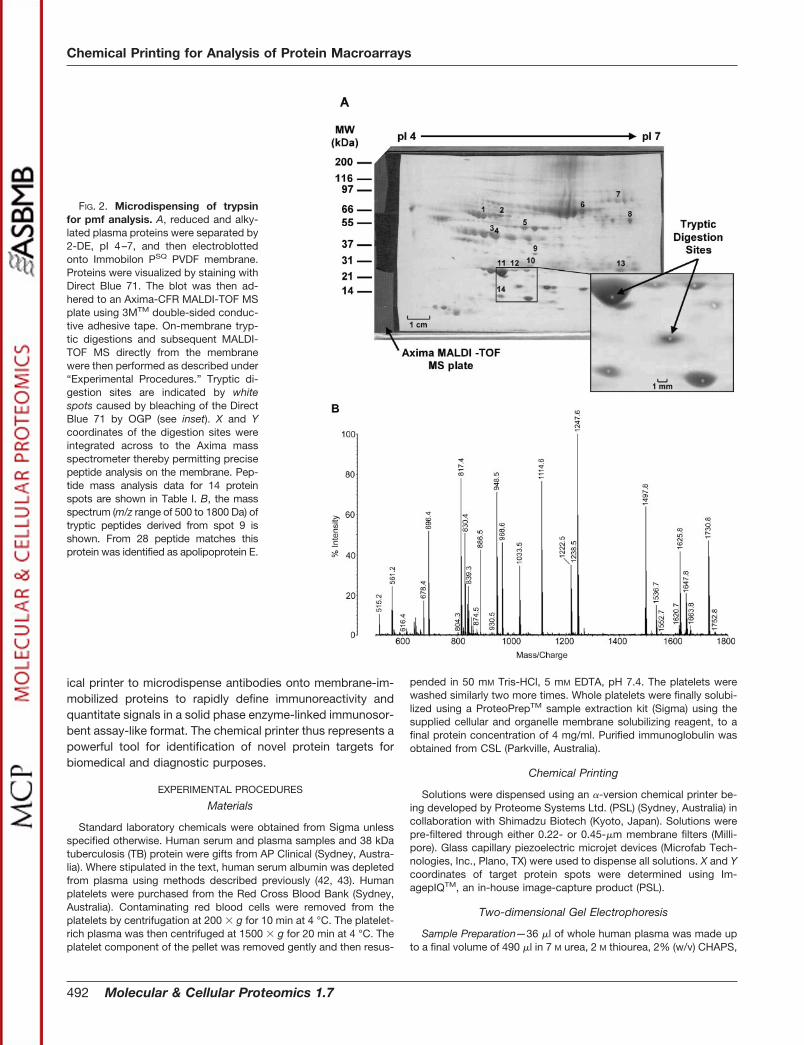
FIG. 2. Microdispensing of trypsinfor pmf analysis. A, reduced and alky-lated plasma proteins were separated by2-DE, pI 4–7, and then electroblottedonto Immobilon PSQ PVDF membrane.Proteins were visualized by staining withDirect Blue 71. The blot was then ad-hered to an Axima-CFR MALDI-TOF MSplate using 3MTM double-sided conduc-tive adhesive tape. On-membrane tryp-tic digestions and subsequent MALDI-TOF MS directly from the membranewere then performed as described under“Experimental Procedures.” Tryptic di-gestion sites are indicated by whitespots caused by bleaching of the DirectBlue 71 by OGP (see inset). X and Ycoordinates of the digestion sites wereintegrated across to the Axima massspectrometer thereby permitting precisepeptide analysis on the membrane. Pep-tide mass analysis data for 14 proteinspots are shown in Table I. B, the massspectrum (m/z range of 500 to 1800 Da) oftryptic peptides derived from spot 9 isshown. From 28 peptide matches thisprotein was identified as apolipoprotein E.
Chemical Printing for Analysis of Protein Macroarrays
492 Molecular & Cellular Proteomics 1.7

and 5 mM Tris, pH 10.2. The sample was then ultrasonicated for 30 s,reduced with 3 mM tributylphosphine for 2 h, and then alkylated with15 mM iodoacetamide for 1 h. Before rehydration of immobilized pHgradient strips, sample was ultrasonicated for 2 min and then centri-fuged at 21000 � g for 5 min. The supernatant was collected, and 10�l of Orange G was finally added as an indicator dye. Sample pre-fractionation into narrow range pI fractions was performed with anElectrophoretIQTM multicompartment electrolyzer (8) (PSL).
First Dimension—Dry 11-cm immobilized pH gradient strips (Am-ersham Biosciences) were rehydrated for 6 h with 200 �l of proteinsample. Rehydrated strips were focused on a Protean IEF cell (Bio-Rad) or PSL prototype IsoElectrIQTM electrophoresis equipment for120 kV-h at a maximum of 10 kV. Focused immobilized pH gradientstrips were equilibrated for 20 min in 6 M urea, 2% (w/v) SDS, 50 mM
Tris-HCl, pH 7.0.Second Dimension—Equilibrated strips were inserted into loading
wells of 6–15% (w/v) Tris acetate SDS-PAGE pre-cast prototype 10 �15-cm GelChipsTM (PSL). Electrophoresis was performed at 50 mAfor 1.5 h. Proteins were electroblotted onto 0.45-�m nitrocellulose(Bio-Rad) or Immobilon PSQ PVDF membranes (Millipore) using aprototype ElectrophoretIQTM electroblotting apparatus (PSL) at 400mA for 1.3 h and methods described by Kyhse-Andersen (44). Pro-teins were finally stained using Direct Blue 71.
One-dimensional SDS-PAGE Analysis of BSA—Titrated amountsof BSA were prepared in SDS-PAGE sample buffer containing 2%(w/v) SDS, 20% (v/v) glycerol, 0.025% (w/v) bromphenol blue, 50 mM
dithiothreitol, 10 mM acrylamide, 0.375 M Tris, pH 8.8. Samples wereallowed to reduce and alkylate for 1 h at room temperature prior toelectrophoresis using a 6–15% (w/v) polyacrylamide ProteoGelTM
(Sigma) and the conditions described above. Protein was finally elec-trotransferred onto an Immobilon PSQ PVDF membrane as describedabove.
On-membrane Protein Digestions
Immobilon PSQ PVDF membranes were first adhered to an Axima-CFR MALDI-TOF target plate (Kratos, Manchester, United Kingdom)using 3MTM electrically conductive tape 9703 (St. Paul, MN). Porcine
FIG. 3. On-membrane tryptic digestion of low abundance, alka-line whole-platelet proteins. Reduced and alkylated whole-plateletproteins were separated by 2-DE, pI 3–10, and then electroblottedonto an Immobilon PSQ PVDF membrane. Proteins were visualized bystaining with Direct Blue 71. The blot was then adhered to an Axima-CFR MALDI-TOF MS plate as described previously. On-membranetryptic digestion using the chemical printer and subsequent MALDI-TOF MS analysis directly from the membrane surface were thenperformed as described under “Experimental Procedures.” Peptidemass analysis for the six proteins (spots 15–20) is shown in Table I.
TABLE IComparison of pmf analysis of proteins identified from on-membrane versus in-gel tryptic digestions
Protein spots 1–14, as shown in Fig. 2, and spots 15–20, as shown in Fig. 3, represent a range of proteins present in variable amounts asindicated by Direct Blue 71 staining intensity in human plasma and platelets, respectively. A comparison of pmf results for these proteins,digested either in-gel or on a PVDF membrane surface, is shown. Peptide hits include peptides containing carboxyamidomethylated cysteineand oxidized methione modifications.
Spotnumber Protein name Accession
number
In-gel digest On-membrane digest
Peptidehits
%Coverage
Peptidehits
%Coverage
1 �-1-Antitrypsin P01009 27 62.4 11 31.72 Antithrombin-III P01008 16 43.8 16 31.03 Apolipoprotein A-IV P06727 26 64.1 22 53.74 Haptoglobin-1 �-chain P00737 22 60.0 12 39.2
Haptoglobin-2 �-chain P00738 22 60.0 12 39.25 Fibrinogen �-chain P04469 24 64.6 18 34.46 Serum albumin P02768 25 44.8 32 45.07 Serotransferrin chain 1 P02787 33 55.4 32 48.28 Fibrinogen � chain P02675 15 38.8 22 53.59 Apolipoprotein E P02649 27 81.3 28 74.9
10 Serum amyloid P-component P02743 8 35.3 10 34.811 Apolipoprotein A-I P02647 20 79.4 9 29.612 Apolipoprotein A-I P02647 26 84.4 11 39.513 Ig � chain C region P01834 5 67.0 8 76.414 Transthyretin P02766 11 88.2 8 65.415 Pyruvate kinase P14618 20 44.1 15 20.516 Fructose bisphosphate aldolase P04075 14 53.2 6 20.117 Fructose bisphosphate aldolase P04075 12 46.3 8 25.618 Glyceraldehyde 3-phosphate dehydrogenase P04406 13 48.2 17 46.119 Peptidyl prolyl cis-trans isomerase chain 1
(cyclophilin A)P23284 6 28.4 4 18.6
20 Nucleotide diphosphate kinase B P22392 9 62.5 5 36.2
Chemical Printing for Analysis of Protein Macroarrays
Molecular & Cellular Proteomics 1.7 493
trypsin (Promega, Madison, WI) at 200 �g/ml in 25 mM NH4HCO3, pH8.5, or Staphylococcus aureus V8 endoproteinase Glu-C (Roche Mo-lecular Biochemicals) at 200 �g/ml in 5 mM NaHPO4, pH 7.8, weredispensed as 50 or 25 iterations, respectively, onto protein spots at 1nl per iteration. Prior to dispensing trypsin or Glu-C, either 3 � 1.5-nliterations of 1% (v/v) n-octyl �-D-glucopyranoside (OGP) or 5 nl of 1%(w/v) polyvinylpyrrolidone (PVP40) in 50% (v/v) methanol, respec-tively, were printed to pre-wet the PVDF membrane. Excess PVP40was removed by washing with water using a transfer pipette. Diges-tion was performed for 3 h at 37 °C in a humidified environment. Afterdigestion, 50 � 2-nl iterations of 10 mg/ml matrix solution, �-cyano-4-hydroxycinnamic acid in a methanol/2-propanol/2-butanol/0.5%(v/v) formic acid solution was then dispensed on top of the digestionzone of each spot. Digests were analyzed using an Axima-CFRMALDI-TOF mass spectrometer (Kratos). All spectra underwent aninternal two-point calibration using autodigested trypsin peakmasses, m/z 842.51 and 2211.10 Da. Software designed by PSL wasused to resolve isotopic peaks from MS spectra (45). In-house data-bases and tools (PSL) and PeptIdent from the ExPASy molecularbiology server (www.expasy.ch/tools/peptident.html) were used forpmf analysis using a mass tolerance of 100 ppm.
In-gel Tryptic Digestion
Protein gel pieces were excised using a prototype XciseTM system(PSL and Shimadzu Biotech) and then washed with 25 mM NH4HCO3,pH 8.5. Gel pieces were then dehydrated under vacuum for 15 minand digested with 10 �l of 20 �g/ml porcine trypsin in 25 mM
NH4HCO3, pH 8.5, for 3 h at 37°C. Peptides were extracted from gelpieces with 10 �l of 50% (v/v) acetonitrile, 0.5% (v/v) formic acid andsonication for 10 min. Prior to MALDI-TOF MS analysis, peptideswere concentrated and purified using a C18 ZipTip® (Millipore) andeluted onto a target plate in 2 �l of matrix solution and allowed to dry.
N-Linked Oligosaccharide Release
N-linked oligosaccharides were cleaved on the PVDF membrane,which was adhered to a MALDI-TOF plate by printing 50 � 10-nl
iterations of 5 units/�l PNGase F (Roche Molecular Biochemicals) perprotein spot and incubation for 3 h at 37°C in a humidified environ-ment. Released oligosaccharides were extracted into 1 �l of water bypipette for LC-ESI MS analysis. The membrane was then washed withwater using a transfer pipette. PNGase F-treated protein was subse-quently digested on the membrane with trypsin using chemical print-ing followed by MALDI-TOF MS analysis.
LC-ESI MS
Sample was loaded onto a ThermoHypersil 5-�m Hypercarb col-umn (Keystone Scientific Operations, Bellefonte, PA) using a Surveyorautosampler connected to an LCQ Deca mass spectrometer (Ther-moFinnigan, San Jose, CA). Oligosaccharides were separated andeluted using a 30-min 0–25% (v/v) acetonitrile gradient in 10 mM
NH4HCO3. Tandem MS analysis was performed in negative ion modeover a m/z range of 320 to 2000 Da. Oligosaccharide structures werepredicted using the GlycoSuiteTM database (PSL).
Antigen Screening
Human serum was diluted 1:3 with phosphate-buffered saline con-taining 0.1% (v/v) Tween 20, 0.05% (w/v) NaN3, and 0.5% (w/v)casein, pH 7.4 (PBS-TAC), and then filtered through a 0.22-�m mem-brane (Millipore). Five applications of 10 nl of serum were printed ontoproteins immobilized on a nitrocellulose membrane after blockingnonspecific binding sites with PBS-TAC for 15 min at room temper-ature. The membrane assay area was clamped onto a layer of ab-sorbent tissue paper to prevent dispersal of fluid over the membranesurface during microdispensing. Excess antibody was removed bywashing twice with 2 � 50-�l drops of PBS-TAC using a transferpipette. Bound antibody was detected by pipetting 20 �l of goatanti-human IgG conjugated to fluorescein isothiocyanate (FITC)(Zymed Laboratories Inc., San Francisco, CA) diluted 1/10 with PBS-TAC. Fluorescence was detected using a Bio-Rad FluorSTM system.
RESULTS
Microdispensing Trypsin for High Throughput On-mem-brane pmf Analysis—Conventional pmf analysis of purifiedproteins involves isolation of individual protein spots from agel (or membrane) followed by in-gel tryptic digestion, peptideextraction, peptide clean up, and finally loading the extractsonto a MALDI-TOF target (46). As an alternative rapid highthroughput approach, we demonstrate dispensing of trypsinonto a macroarray of human plasma proteins on a PVDFmembrane that had been adhered to a MALDI-TOF targetplate (Fig. 2A). The most efficient digestion conditions wereachieved by jetting 200 �g/ml trypsin in 50 � 1-nl iterationsonto each protein spot. The small amount of drying timebetween each iteration increased the digestion efficiency bypreventing excessive diffusion of trypsin solution across themembrane surface. Digestion sites on the hydrophobic PVDFmembrane surface were pre-wet by printing either 4.5 nl of thenon-ionic detergent n-octyl �-D-glucopyranoside or 5 nl ofPVP40. This also prevented nonspecific binding of proteinaseto the membrane. After digestion, matrix solution was dis-pensed directly onto the tryptic peptides prior to MALDI-TOFMS analysis directly from the membrane surface. Fig. 2Aillustrates the relative size of the digestion zones (�200–
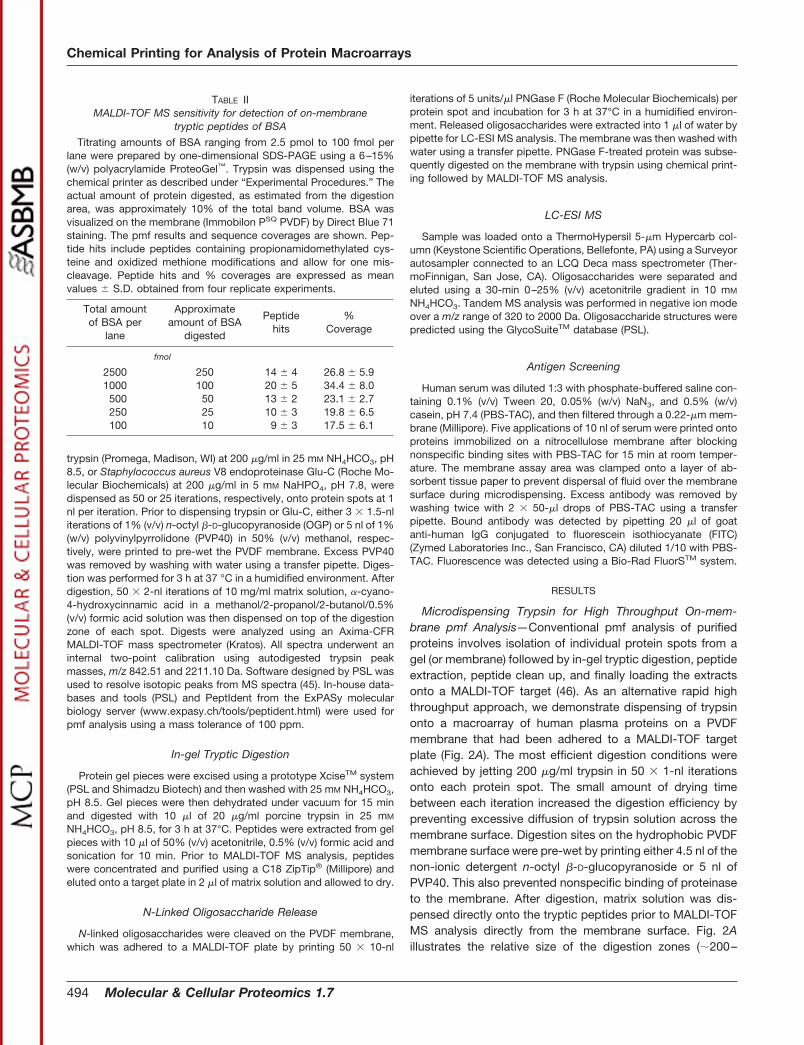
TABLE IIMALDI-TOF MS sensitivity for detection of on-membrane
tryptic peptides of BSA
Titrating amounts of BSA ranging from 2.5 pmol to 100 fmol perlane were prepared by one-dimensional SDS-PAGE using a 6–15%(w/v) polyacrylamide ProteoGel™. Trypsin was dispensed using thechemical printer as described under “Experimental Procedures.” Theactual amount of protein digested, as estimated from the digestionarea, was approximately 10% of the total band volume. BSA wasvisualized on the membrane (Immobilon PSQ PVDF) by Direct Blue 71staining. The pmf results and sequence coverages are shown. Pep-tide hits include peptides containing propionamidomethylated cys-teine and oxidized methione modifications and allow for one mis-cleavage. Peptide hits and % coverages are expressed as meanvalues � S.D. obtained from four replicate experiments.
Total amountof BSA per
lane
Approximateamount of BSA
digested
Peptidehits
%Coverage
fmol
2500 250 14 � 4 26.8 � 5.91000 100 20 � 5 34.4 � 8.0500 50 13 � 2 23.1 � 2.7250 25 10 � 3 19.8 � 6.5100 10 9 � 3 17.5 � 6.1
Chemical Printing for Analysis of Protein Macroarrays
494 Molecular & Cellular Proteomics 1.7
300-�m diameter). Of 14 protein spots representing a range ofproteins present in various amounts (Fig. 2A) all were identi-fied successfully by pmf analysis after on-membrane diges-tion (Table I). Fig. 2B shows a representative spectrum ofapolipoprotein E generated after on-membrane digestion.
Detection of low abundance proteins is often a limitation inproteomic studies. Furthermore, alkaline proteins are evenmore difficult to identify given the difficulty in focusing suchhighly positive charged proteins. For this reason we haveanalyzed several low abundance alkaline platelet proteins todemonstrate that identification of such proteins is achievableusing on-membrane digestion methods with the chemicalprinter (see Fig. 3 and Table I). Positive identification of nu-cleoside diphosphate kinase B (Table I) was confirmed follow-
ing post-source decay analysis of the 1175.76 ion (data notshown).
The sequence coverages from peptides generated by on-membrane digestion were usually less than those from in-geldigests (Table I). This was not unexpected given the in-geldigests were each purified using a C18 ZipTip®, and theexcised area of the in-gel digest was 1.2 mm in diameter,more than five times the area digested on-membrane. In somecases increased numbers of peptide hits were observed offthe membrane, but lower sequence coverages were obtainedrelative to in-gel digests. This was a result of the higherabundance of smaller peptides extracted from the membranesurface (Table I). Nevertheless, with respect to sensitivity ofpeptide detection, analysis of on-membrane tryptic digests of
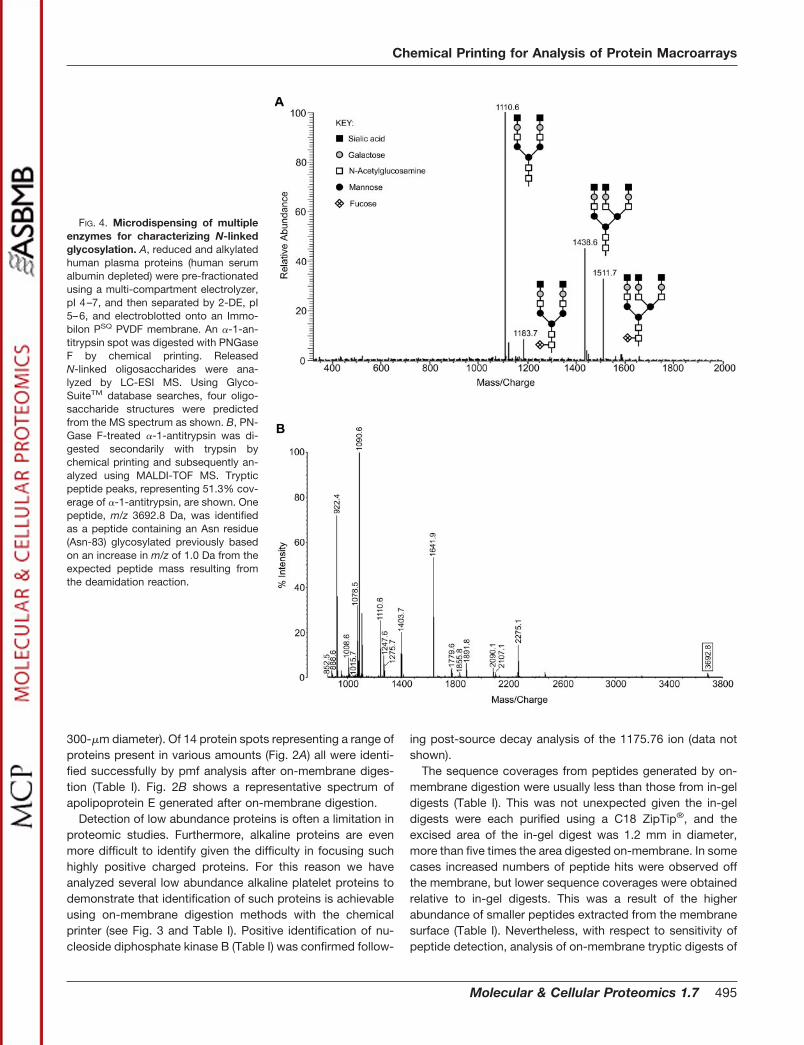
FIG. 4. Microdispensing of multipleenzymes for characterizing N-linkedglycosylation. A, reduced and alkylatedhuman plasma proteins (human serumalbumin depleted) were pre-fractionatedusing a multi-compartment electrolyzer,pI 4–7, and then separated by 2-DE, pI5–6, and electroblotted onto an Immo-bilon PSQ PVDF membrane. An �-1-an-titrypsin spot was digested with PNGaseF by chemical printing. ReleasedN-linked oligosaccharides were ana-lyzed by LC-ESI MS. Using Glyco-SuiteTM database searches, four oligo-saccharide structures were predictedfrom the MS spectrum as shown. B, PN-Gase F-treated �-1-antitrypsin was di-gested secondarily with trypsin bychemical printing and subsequently an-alyzed using MALDI-TOF MS. Trypticpeptide peaks, representing 51.3% cov-erage of �-1-antitrypsin, are shown. Onepeptide, m/z 3692.8 Da, was identifiedas a peptide containing an Asn residue(Asn-83) glycosylated previously basedon an increase in m/z of 1.0 Da from theexpected peptide mass resulting fromthe deamidation reaction.
Chemical Printing for Analysis of Protein Macroarrays
Molecular & Cellular Proteomics 1.7 495

BSA demonstrated that a targeted protein amount of 10 fmolof BSA was still sufficient for generating pmf data that enabledreliable protein identification (Table II).
Multiple Chemistry Subanalysis of PVDF Membrane-immo-bilized Proteins—The ability to dispense small volumes ofreagents to specific locations permitted multiple chemicalanalyses of a single protein spot. Here we demonstrate re-lease of N-linked oligosaccharides from human �-1-antitryp-sin after chemical printing and on-membrane digestion withthe endoglycosidase PNGase F. Released oligosaccharideswere subsequently extracted from the membrane surface.LC-ESI MS analysis of these extracted oligosaccharides iden-tified four different carbohydrate moieties on �-1-antitrypsin(Fig. 4A). Deglycosylation of Asn results in deamidation con-verting the Asn to an Asp and consequently increasing them/z of the parent protein by 1.0 Da. This enables identificationof sites of N-linked glycosylation following tryptic digestionand pmf analysis. This study revealed one of three predictedpeptides, peptide 70–101, with an observed m/z 3692.8 Da,implicating Asn-83 as an N-linked glycosylation site (Fig. 4B).This is the largest of the expected tryptic peptides of �-1-antitrypsin, which contained N-linked glycosylation. The othertwo glycosylation sites occur on peptide 40–69 (predicted
m/z 3181.6 Da) and peptide 244–259 (predicted m/z 1755.9Da). It is possible that sites Asn-46 and Asn-247 were notdeglycosylated and hence not desorbed in reflectron mode.The peptide containing Asn-46 was definitely cleaved by tryp-sin as both peptides N- and C-terminal to peptide 40–69 wereidentified in the MALDI-TOF spectrum. No peptides betweenresidues 218 and 274 were identified, so it is possible that thetryptic peptide containing Asn-247 was never cleaved.
The ability to dispense multiple enzymes for increasingprotein sequence coverage, and thus increase confidence inprotein identification, has also been demonstrated (Table III).On-membrane digestion of a single protein spot, apolipopro-tein A1 (a different sample from that described in Table I), withboth Glu-C and trypsin, resulted in sequence coverages of41.6 and 46.1%, respectively. The combined sequence cov-erages represented 66.7% of the total sequence of apoli-poprotein A1.
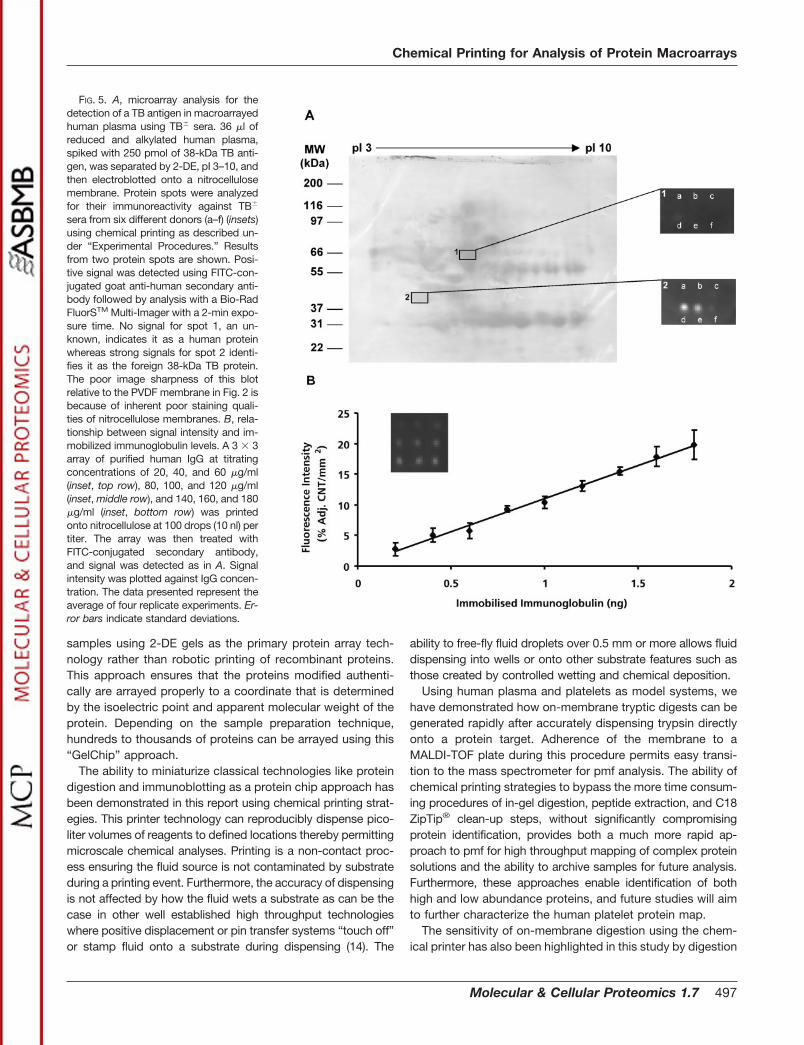
Mapping Immunodiagnostic Markers—From a biomedicalperspective we have demonstrated the ability to identify im-munoreactive pathogen-derived antigens in human plasmaand simultaneously compare immunoreactivity of multiple pa-tient sera. Sera from six different patients (a–f) were sequen-tially printed onto a macroarray of plasma proteins that hadbeen spiked with a 38-kDa mycobacterium TB protein. Theresults from two protein spots (1, a human (control) proteinand 2, the TB protein) are shown (Fig. 5A). Antigen recognitionwas determined after subsequent application of anti-humanIgG conjugated to FITC in reaction times of less than 3 min.Patients d and e demonstrated strong recognition of the38-kDa TB antigen whereas patients c and f reacted weakly.Patients a and b were TB-negative controls. These findingsdemonstrate how potentially new pathogen-specific antigenscan be identified using patient serum antibodies. In additionalstudies, titrated amounts of human immunoglobulin antigenwere printed onto nitrocellulose. Subsequent to detectionusing excess anti-human IgG-FITC-conjugated antibody,comparison of relative signal intensities versus antigen con-centration demonstrated a linear relationship, thus highlight-ing the potential for quantitating signal responses derivedfrom chemical printing strategies (Fig. 5B).
DISCUSSION
Although DNA microarrays have been heralded as a revo-lution in genomic studies, the role of protein microarrays inproteomic studies is not as simple, principally because of thechallenges of developing high throughput strategies for pre-paring proteins prior to their deposition onto chip surfaces.Generally, protein arrays use recombinant proteins that arearrayed robotically onto surfaces. However, a fundamentalproblem with this approach is that the recombinant proteinsare not unique, lacking the myriad co- and post-translationalmodifications that lead to protein isoforms, many of which arecell type-specific.
The strategy we have presented here is to array authentic
TABLE IIIIncreased sequence coverage using on-membrane
multiple enzyme digestion strategies
Reduced and alkylated human plasma proteins were separated by2-DE, pI 4–7, and then electroblotted onto Immobilon PSQ PVDFmembrane. Proteins were visualized by staining with Direct Blue 71.An apolipoprotein A-1 spot (SwissProt accession number P02647)was then digested by using the chemical printer to microdispense 5 nlof PVP40 followed by either 25 nl of 200 �g/ml Glu-C (inset, G) or 50nl of 200 �g/ml trypsin (inset, T) in 1-nl iterations. Peptides generatedafter digestion with Glu-C (light grey boxes) and trypsin (dark greyboxes) resulted in sequence coverages of 41.6 and 46.1%, respec-tively. Italics and black boxes indicate the overlapping peptide se-quences. The total sequence coverage after combining both sets ofpmf data was 66.7%.
Chemical Printing for Analysis of Protein Macroarrays
496 Molecular & Cellular Proteomics 1.7
samples using 2-DE gels as the primary protein array tech-nology rather than robotic printing of recombinant proteins.This approach ensures that the proteins modified authenti-cally are arrayed properly to a coordinate that is determinedby the isoelectric point and apparent molecular weight of theprotein. Depending on the sample preparation technique,hundreds to thousands of proteins can be arrayed using this“GelChip” approach.
The ability to miniaturize classical technologies like proteindigestion and immunoblotting as a protein chip approach hasbeen demonstrated in this report using chemical printing strat-egies. This printer technology can reproducibly dispense pico-liter volumes of reagents to defined locations thereby permittingmicroscale chemical analyses. Printing is a non-contact proc-ess ensuring the fluid source is not contaminated by substrateduring a printing event. Furthermore, the accuracy of dispensingis not affected by how the fluid wets a substrate as can be thecase in other well established high throughput technologieswhere positive displacement or pin transfer systems “touch off”or stamp fluid onto a substrate during dispensing (14). The
ability to free-fly fluid droplets over 0.5 mm or more allows fluiddispensing into wells or onto other substrate features such asthose created by controlled wetting and chemical deposition.
Using human plasma and platelets as model systems, wehave demonstrated how on-membrane tryptic digests can begenerated rapidly after accurately dispensing trypsin directlyonto a protein target. Adherence of the membrane to aMALDI-TOF plate during this procedure permits easy transi-tion to the mass spectrometer for pmf analysis. The ability ofchemical printing strategies to bypass the more time consum-ing procedures of in-gel digestion, peptide extraction, and C18ZipTip® clean-up steps, without significantly compromisingprotein identification, provides both a much more rapid ap-proach to pmf for high throughput mapping of complex proteinsolutions and the ability to archive samples for future analysis.Furthermore, these approaches enable identification of bothhigh and low abundance proteins, and future studies will aimto further characterize the human platelet protein map.
The sensitivity of on-membrane digestion using the chem-ical printer has also been highlighted in this study by digestion
FIG. 5. A, microarray analysis for thedetection of a TB antigen in macroarrayedhuman plasma using TB� sera. 36 �l ofreduced and alkylated human plasma,spiked with 250 pmol of 38-kDa TB anti-gen, was separated by 2-DE, pI 3–10, andthen electroblotted onto a nitrocellulosemembrane. Protein spots were analyzedfor their immunoreactivity against TB�
sera from six different donors (a–f) (insets)using chemical printing as described un-der “Experimental Procedures.” Resultsfrom two protein spots are shown. Posi-tive signal was detected using FITC-con-jugated goat anti-human secondary anti-body followed by analysis with a Bio-RadFluorSTM Multi-Imager with a 2-min expo-sure time. No signal for spot 1, an un-known, indicates it as a human proteinwhereas strong signals for spot 2 identi-fies it as the foreign 38-kDa TB protein.The poor image sharpness of this blotrelative to the PVDF membrane in Fig. 2 isbecause of inherent poor staining quali-ties of nitrocellulose membranes. B, rela-tionship between signal intensity and im-mobilized immunoglobulin levels. A 3 � 3array of purified human IgG at titratingconcentrations of 20, 40, and 60 �g/ml(inset, top row), 80, 100, and 120 �g/ml(inset, middle row), and 140, 160, and 180�g/ml (inset, bottom row) was printedonto nitrocellulose at 100 drops (10 nl) pertiter. The array was then treated withFITC-conjugated secondary antibody,and signal was detected as in A. Signalintensity was plotted against IgG concen-tration. The data presented represent theaverage of four replicate experiments. Er-ror bars indicate standard deviations.
Chemical Printing for Analysis of Protein Macroarrays
Molecular & Cellular Proteomics 1.7 497

and subsequent successful pmf identification of �10 fmol ofBSA immobilized on an Immobilon PSQ PVDF membrane. Thislevel of sensitivity is equal to, if not higher than, other reportedin-gel digestion procedures (47, 48).
Another emerging technology for automated pmf analysis isthe molecular scanner, which simultaneously digests andelectrotransfers peptides onto a PVDF membrane (49). Thestrategy of the molecular scanner is not to visualize thepolypeptides on the electroblot but to rapidly scan the mem-brane to detect the presence of peptides. The process isinefficient, because digested proteins only occupy a low per-centage of the entire area that is scanned. Unlike the chemicalprinter, the molecular scanner cannot control delivery of dif-ferent amounts of trypsin to different protein spots. Further-more, the digestion of all of the sample on the membraneabolishes the ability to archive untreated samples. Subse-quent analyses using alternate chemistries are therefore lim-ited. The ability to archive and also subject a protein to mul-tiple analyses are advantages of the chemical printerapproach. Furthermore, the option to specifically select indi-vidual proteins for subanalysis using multiple chemistries in-creases both time efficiency and conservation of valuablereagents and samples.
Protein transfer onto a membrane is an inherent componentof the experimental strategies presented here. Importantly, wehave found routinely that protein transfer of a specific sampleonto a particular membrane type is a highly reproducibleprocess. However, it must be appreciated that buffer condi-tions should be optimized for analysis of a particular pro-teome (50). The studies presented here have used the transferconditions described by Kyhse-Andersen (44) that were opti-mized for proteins with molecular mass less than 200 kDa andpI 4–7. Thus, other buffer systems, as well as membranetypes, should be considered if particular proteins with specificcharacteristics, such as size, antigenicity, pI, or hydrophobic-ity are required for analysis (51).
Although other studies have demonstrated remarkable ex-amples of large scale protein identification (52), the ability formultiple analyses on a specific protein(s) within the samesample, such as mapping post-translational modifications, islimited in these approaches. We have demonstrated successfulon-membrane digestion by sequentially delivering PNGase Fand trypsin for mapping sites of N-glycosylation on human�-1-antitrypsin. With the increasing interest in post-translationalmodifications, particularly glycosylation (37), this technologyalso represents a powerful tool, particularly when coupled withLC-ESI MS, for structural analyses of oligosaccharides releasedfrom a solid-phase membrane. Furthermore, the ability to ana-lyze a single protein spot using multiple endoproteinases hasdemonstrated the ability to increase markedly the sequencecoverage of a protein, thus increasing confidence of a success-ful identification. In principle it should be possible to achievecomplete characterization of proteins with such an approach.This is a significant advantage, particularly when considering
characterization of low abundance proteins or proteins thatcontain minimal cleavage sites for a single enzyme.
One of the major outcomes of the proteomic era will be theidentification of novel protein targets useful for biomedicalapplications, particularly in the diagnostic arena. In this studywe have printed nanoliter volumes of serum from differentpatients onto individual plasma protein spots of a macroarrayin a quantitative miniaturized enzyme-linked immunosorbentassay format. Unlike Western blotting protocols practicedcurrently that routinely require at least 3 h per analysis, rapiddispensing of multiple antibodies can be used to screen forantigens in several minutes. Using chemical printing, Westernblotting becomes a rapid and quantitative user-independenttechnology. We have thus demonstrated one strategy thatpermits mapping immunoreactive antigens using human serafor subsequent identification by pmf analysis with implicationsfor identifying vaccine and diagnostic targets.
Understanding the dynamic and complex nature of theproteome of an organism will require the development of highthroughput multi-tasking proteomic platforms to cope withthis enormous task. The chemical printer represents a tech-nology that can function as a component of a proteomicplatform working in conjunction with the powerful resolvingtools of 2-DE and MS or as a standalone work station.
Acknowledgments—We thank Dr. Tracey Edgell for assistance inplasma sample preparation and Dr. Alice Zhou (AP Clinical, Sydney,Australia) for the generous supply of serum and plasma samples andTB protein.
* This work was supported in part by an AusIndustry R and DSTART program and by Shimadzu Corporation, Kyoto, Japan. Thecosts of publication of this article were defrayed in part by the pay-ment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
¶ To whom correspondence should be addressed. Tel.: 61-2-9889-1830; Fax: 61-2-9889-1805; E-mail: [email protected].
REFERENCES
1. Abbott, A. (1999) A post-genomic challenge: learning to read patterns ofprotein synthesis. Nature 402, 715–720
2. Banks, R. E., Dunn, M. J., Hochstrasser, D. F., Sanchez, J. C., Blackstock,W., Pappin, D. J., and Selby, P. J. (2000) Proteomics: new perspectives,new biomedical opportunities. Lancet 356, 1749–1756
3. Anderson, L., and Seilhamer, J. (1997) A comparison of selected mRNA andprotein abundances in human liver. Electrophoresis 18, 533–537
4. Wilkins, M. R., Sanchez, J. C., Williams, K. L., and Hochstrasser, D. F.(1996) Current challenges and future applications for protein maps andpost-translational vector maps in proteome projects. Electrophoresis 17,830–838
5. Lee, K. H. (2001) Proteomics: a technology-driven and technology-limiteddiscovery science. Trends Biotechnol. 19, 217–222
6. Gygi, S. P., Corthals, G. L., Zhang, Y., Rochon, Y., and Aebersold, R. (2000)Evaluation of two-dimensional gel electrophoresis-based proteome anal-ysis technology. Proc. Natl. Acad. Sci. U. S. A. 97, 9390–9395
7. Herbert, B. (1999) Advances in protein solubilization for two-dimensionalelectrophoresis. Electrophoresis 20, 660–663
8. Herbert, B., and Righetti, P. G. (2000) A turning point in proteome analysis:sample prefractionation via multicompartment electrolyzers with isoelec-tric membranes. Electrophoresis 21, 3639–3648
Chemical Printing for Analysis of Protein Macroarrays
498 Molecular & Cellular Proteomics 1.7

9. Gevaert, K., and Vandekerckhove, J. (2000) Protein identification methodsin proteomics. Electrophoresis 21, 1145–1154
10. Goodlett, D. R., Keller, A., Watts, J. D., Newitt, R., Yi, E. C., Purvine, S., Eng,J. K., von Haller, P., Aebersold, R., and Kolker, E. (2001) Differentialstable isotope labeling of peptides for quantitation and de novo se-quence derivation. Rapid Commun. Mass Spectrom. 15, 1214–1221
11. Gygi, S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H., and Aebersold,R. (1999) Quantitative analysis of complex protein mixtures using iso-tope-coded affinity tags. Nat. Biotechnol. 17, 994–999
12. Ge, H. (2000) UPA, a universal protein array system for quantitative detec-tion of protein-protein, protein-DNA, protein-RNA and protein-ligandinteractions. Nucleic Acids Res. 28, e3
13. Bussow, K., Cahill, D., Nietfeld, W., Bancroft, D., Scherzinger, E., Lehrach,H., and Walter, G. (1998) A method for global protein expression andantibody screening on high density filters of an arrayed cDNA library.Nucleic Acids Res. 26, 5007–5008
14. Lueking, A., Horn, M., Eickhoff, H., Bussow, K., Lehrach, H., and Walter, G.(1999) Protein microarrays for gene expression and antibody screening.Anal. Biochem. 270, 103–111
15. Cahill, D. J. (2001) Protein and antibody arrays and their medical applica-tions. J. Immunol. Methods 250, 81–91
16. Emili, A. Q., and Cagney, G. (2000) Large-scale functional analysis usingpeptide or protein arrays. Nat. Biotechnol. 18, 393–397
17. Mendoza, L. G., McQuary, P., Mongan, A., Gangadharan, R., Brignac, S.,and Eggers, M. (1999) High throughput microarray-based enzyme-linkedimmunosorbent assay (ELISA). Biotechniques 27, 778–780, 782–786,788
18. Ekins, R. P. (1998) Ligand assays: from electrophoresis to miniaturizedmicroarrays. Clin. Chem. 44, 2015–2030
19. Michael, K. L., Taylor, L. C., Schultz, S. L., and Walt, D. R. (1998) Randomlyordered addressable high density optical sensor arrays. Anal. Chem. 70,1242–1248
20. Holt, L. J., Bussow, K., Walter, G., and Tomlinson, I. M. (2000) By-passingselection: direct screening for antibody-antigen interactions using pro-tein arrays. Nucleic Acids Res. 28, e72
21. Sanders, G. H. W., and Manz, A. (2000) Chip-based microsystems forgenomic and proteomic analysis. Trends Anal. Chem. 19, 364–378
22. MacBeath, G., and Schreiber, S. L. (2000) Printing proteins as microarraysfor high throughput function determination. Science 289, 1760–1763
23. Figeys, D., and Pinto, D. (2001) Proteomics on a chip: promising develop-ments. Electrophoresis 22, 208–216
24. Senior, K. (1999) Fingerprinting disease with protein chip arrays. Mol. Med.Today 5, 326–327
25. Xiao, Z., Jiang, X., Beckett, M. L., and Wright, G. L., Jr. (2000) Generationof a baculovirus recombinant prostate-specific membrane antigen andits use in the development of a novel protein biochip quantitative immu-noassay. Protein Expr. Purif. 19, 12–21
26. Mooney, J. F., Hunt, A. J., McIntosh, J. R., Liberko, C. A., Walba, D. M., andRogers, C. T. (1996) Patterning of functional antibodies and other pro-teins by photolithography of silane monolayers. Proc. Natl. Acad. Sci.U. S. A. 93, 12287–12291
27. Jones, V. W., Kenseth, J. R., Porter, M. D., Mosher, C. L., and Henderson,E. (1998) Microminiaturized immunoassays using atomic force micros-copy and compositionally patterned antigen arrays. Anal. Chem. 70,1233–1241
28. Cooley, P., Hinson, D., Trost, H. J., Antohe, B., and Wallace, D. (2001)Ink-jet-deposited microspot arrays of DNA and other bioactive mole-cules. Methods Mol. Biol. 170, 117–129
29. Hughes, T. R., Mao, M., Jones, A. R., Burchard, J., Marton, M. J., Shannon,K. W., Lefkowitz, S. M., Ziman, M., Schelter, J. M., Meyer, M. R.,Kobayashi, S., Davis, C., Dai, H., He, Y. D., Stephaniants, S. B., Cavet,G., Walker, W. L., West, A., Coffey, E., Shoemaker, D. D., Stoughton, R.,Blanchard, A. P., Friend, S. H., and Linsley, P. S. (2001) Expressionprofiling using microarrays fabricated by an ink-jet oligonucleotide syn-thesizer. Nat. Biotechnol. 19, 342–347
30. Roda, A., Guardigli, M., Russo, C., Pasini, P., and Baraldini, M. (2000)Protein microdeposition using a conventional ink-jet printer. Biotech-niques 28, 492–496
31. Adams, R. L., and Roy, J. (1984) A one-dimensional numerical model of adrop-on-demand ink jet. J. Appl. Mech. 53, 193–197
32. Bogy, D. B., and Talke, F. E. (1984) Experimental and theoretical study ofwave propagation phenomena in drop-on-demand ink jet devices. IBM J.Res. Dev. 29, 314–321
33. Dijksman, J. F. (1984) Hydrodynamics of small tubular pumps. J. FluidMech. 139, 173–191
34. Ekins, R. (1994) Immunoassay: recent developments and future directions.Nucl. Med. Biol. 21, 495–521
35. Wallace, D. B., Cox, W. R., and Hayes, D. J. (2001) in Direct-write Tech-nologies for Rapid Prototyping of Sensors, Electronics, and PassivationCoatings (Pique, A. and Chrisey, D. B., eds) 1st Ed., pp. 213–220,Academic Press, San Diego
36. Hayes, D. J., Wallace, D. B., VerLee, D., and Houseman, K. (October 31,1989) U. S. Patent 4,877,745
37. Dell, A., and Morris, H. R. (2001) Glycoprotein structure determination bymass spectrometry. Science 291, 2351–2356
38. Goldmann, T., and Gonzalez, J. S. (2000) DNA printing: utilization of astandard inkjet printer for the transfer of nucleic acids to solid supports.J. Biochem. Biophys. Methods 42, 105–110
39. Okamoto, T., Suzuki, T., and Yamamoto, N. (2000) Microarray fabricationwith covalent attachment of DNA using bubble jet technology. Nat.Biotechnol. 18, 438–441
40. Mueller, U., Nyarsik, L., Horn, M., Rauth, H., Przewieslik, T., Saenger, W.,Lehrach, H., and Eickhoff, H. (2001) Development of a technology forautomation and miniaturization of protein crystallization. J. Biotechnol.85, 7–14
41. Lemmo, A. V., Rose, D. J., and Tisone, T. C. (1998) Inkjet dispensingtechnology: applications in drug discovery. Curr. Opin. Biotechnol. 9,615–617
42. Cohn, E. J., Strong, L. E., Hughes, W. L., Jr., Mulford, D. J., Ashworth, J. N.,Melin, M., and Taylor, H. L. (1946) Preparation and properties of serumand plasma proteins. IV. A system for the separation into fractions of theprotein lipoprotein components of biological tissues and fluids. J. Am.Chem. Soc. 68, 459–475
43. Oncley, J. L., Melin, M., Richert, D. A., Cameron, J. W., and Gross, P. M.,Jr. (1949) The separation of the antibodies, isoagglutinins, prothrombin,plasminogen and �1-lipoprotein into sub-fractions of human plasma.J. Am. Chem. Soc. 71, 541–550
44. Kyhse-Andersen, J. (1984) Electroblotting of multiple gels: a simple appa-ratus without buffer tank for rapid transfer of proteins from polyacryl-amide to nitrocellulose. J. Biochem. Biophys. Methods 10, 203–209
45. Breen, E. J., Hopwood, F. G., Williams, K. L., and Wilkins, M. R. (2000)Automatic poisson peak harvesting for high throughput protein identifi-cation. Electrophoresis 21, 2243–2251
46. Henzel, W. J., Billeci, T. M., Stults, J. T., Wong, S. C., Grimley, C., andWatanabe, C. (1993) Identifying proteins from two-dimensional gels bymolecular mass searching of peptide fragments in protein sequencedatabases. Proc. Natl. Acad. Sci. U. S. A. 90, 5011–5015
47. Katayama, H., Nagasu, T., and Oda, Y. (2001) Improvement of in-geldigestion protocol for peptide mass fingerprinting by matrix-assistedlaser desorption/ionization time-of-flight mass spectrometry. RapidCommun. Mass Spectrom. 15, 1416–1421
48. Lopez, M. F., Berggren, K., Chernokalskaya, E., Lazarev, A., Robinson, M.,and Patton, W. F. (2000) A comparison of silver stain and SYPRO Rubyprotein gel stain with respect to protein detection in two-dimensionalgels and identification by peptide mass profiling. Electrophoresis 21,3673–3683
49. Bienvenut, W. V., Sanchez, J. C., Karmime, A., Rouge, V., Rose, K., Binz,P. A., and Hochstrasser, D. F. (1999) Toward a clinical molecular scannerfor proteome research: parallel protein chemical processing before andduring Western blot. Anal. Chem. 71, 4800–4807
50. Jin, Y., and Cerletti, N. (1992) Western blotting of transforming growthfactor �2, optimization of the electrophoretic transfer. Appl. Theor. Elec-trophor. 3, 85–90
51. Dunn, S. D. (1986) Effects of the modification of transfer buffer compositionand the renaturation of proteins in gels on the recognition of proteins onWestern blots by monoclonal antibodies. Anal. Biochem. 157, 144–153
52. Washburn, M. P., Wolters, D., and Yates, J. R., III (2001) Large-scaleanalysis of the yeast proteome by multidimensional protein identificationtechnology. Nat. Biotechnol. 19, 242–247
Chemical Printing for Analysis of Protein Macroarrays
Molecular & Cellular Proteomics 1.7 499




















