
assessing antimalarial cidality in plasmodium falciparum ...
Upload
independentCategory
view
0download
0
Heptyl prodigiosin, a bacterial metabolite, is antimalarial in vivo andnon-mutagenic in vitroJ. Enrico H. Lazaro1, Josiane Nitcheu1, Rey Z. Predicala2, Gina C. Mangalindan2, Fabrice Nesslany3, Daniel Marzin3, Gisela P. Concepcion2, and Bertrand Diquet1, 4
1 INSERM U511 Immuno-Biologie Cellulaire et Moléculaire desInfections Parasitaires, Centre Hospitalo-Universitaire Pitié-Salpêtrière, Université Pierre et Marie Curie, 75013 Paris, France.
2 Marine Science Institute, University of the Philippines, Diliman, Quezon City 1101, Philippines3 Institut Pasteur de Lille, 1 Rue du Prof. Calmette, 59019 Lille Cedex, France4 Départemet de Pharmacologie, Hôpital Pitié Salpêtrière, 47 Bd de l'Hôpital, 75013 Paris, France
Running title: Antimalarial heptyl prodigiosin
1

AbstractHeptyl prodigiosin was purified from a culture of -
proteobacteria isolated from a marine tunicate collected inZamboanga, Philippines as part of a program to screen naturalproducts for antiparasitic activity. An in vitro antimalarial activitysimilar to that of quinine was found against the chloroquine-sensitive strain Plasmodium falciparum 3D7. The in vitro antimalarialactivity was about 20 times the in vitro cytotoxic activity againstL5178Y mouse lymphocytes. A single subcutaneous administration of 5and 20 mg/kg significantly extended survival of P. berghei ANKA strain-infected mice but also caused sclerotic lesions at the site ofinjection. A single administration by gavage of 50 mg/kg did notincrease survival time. The compound was not found to be mutagenicusing in vitro micromethods for the Ames Salmonella typhimurium assay andthe micronucleus assay using L5178Y mouse lymphoma cells.
IntroductionResistance to chloroquine and other antimalarial drugs and the
lack of interest by most pharmaceutical companies to develop newcompounds is helping to increase the public health and economicburden of malaria in endemic countries (Breman 2001). Multilateralinitiatives by agencies from both the First and Third Worlds andmostly under the aegis of the World Health Organization have issuedcalls to participate in the discovery of new molecules and in thedevelopment of more effective formulations and combinations ofexisting products.
Prodigiosin, 2-methyl-3-pentyl-6-methoxyprodigiosene (MW 323),is the most well-known representative of a class of tripyrrolebacterial pigments with antibiotic and antiparasitic properties(Williams and Hearn 1967). It was once produced in quantity,clinically tested for treatment of coccidiomycosis, described ascausing dermal sclerosis, and subsequently abandoned (Gerber 1975).Recent evidence showing that some members of the class might induceapoptosis in cancer cell lines (Montaner et al. 2000) and inhibit T-cell proliferation (Fürstner et al. 2001; D'Alessio et al. 2000;Kawauchi et al. 1997) have revived interest in these compounds.
The antimalarial properties of the more common members of theclass have been studied even until recently. Prodigiosin from Serratiamarcescens was described as conferring protection at a singlesubcutaneous dose of 40 mg/kg in an in vivo Plasmodium berghei model, buttoxic effects on the animals were not described (Castro 1967). Gerber(1975) showed that a subcutaneous administration of as low as 10mg/kg of some compounds, including prodigiosin, increased meansurvival time in mice infected with P. berghei. In the most recentstudy, Kim et al. (1999) described the well-known cycloprodigiosinhydrochloride (cf. Gerber 1983; Laatsch and Thomson 1983; Wassermanand Fukuyama 1984; Kawauchi et al. 1997) as delaying increase of
2

parasitemia for up to 10 days in P. berghei-infected mice at a singleintraperitoneal dose of 25 mg/kg.
Heptyl prodigiosin (2-methyl-3-heptyl-6-methoxyprodigiosene, MW351, Figure 2) is a relatively unknown member of the class. Naturalheptyl prodigiosin has been isolated from a culture of Methylosinustrichosporium but no details of its biological activity were given(Strauss & Berger 1983). It was a minor component (1% of the mass,99% being prodigiosin) of a Pseudomonas magnesiorubra extract as wellas that of an unidentified sewage bacteria, but its biologicalactivity was not described (Gerber 1975). No data has been publishedon the antimalarial activities of heptyl prodigiosin in vitro or in vivo.Aside from one study suggesting that prodigiosin intercalates to AT-rich sites in DNA and effects oxidative DNA strand scission, no workhas been published on the mutagenic potential of any prodigiosin andprodigiosin-like compound in living systems (Melvin et al. 1999). Wetherefore decided to study the antimalarial and potential mutagenicproperties of the compound.
Materials and MethodsExtraction and purification
A bright red bacterial culture (Accession no. CNCM I-2907,Pasteur Institute, Paris, France) that was isolated by dilution froma still-unidentified marine tunicate collected in Zamboanga,Philippines, and stored at the Marine Natural Products Laboratory ofthe Marine Science Institute, University of the Philippines, wasmaintained in marine broth 2216 (Difco, Becton Dickinson, MD, USA)supplemented with 2% (w/w) glucose, pH 7.6. Analysis of the DNAsequence of the 16S rRNA gene and fatty acid methyl esters (FAME)suggested an -proteobacteria (unpublished results).
An aliquot of broth was plated on marine agar (marine brothwith 1.5% (w/w) granulated agar (BBL, Becton Dickinson & Co., MD,USA)) and incubated at 27C for two days. A single red colony wasused to inoculate 10 ml of marine broth (glucose 2%, pH 7.6) andincubated in a TEQ orbital shaker (130 rpm, 27C, 10 hours). Then,7.5 ml of this culture was added to 150 ml of sterile marine brothand incubated similarly. Then, 37.5 ml of the latter was added to 750ml of sterile marine broth in a 2-liter Fehrnbach flask and incubated(130 rpm, 27C, 36 hours). The dark red cultures were centrifuged toseparate cells.
Extraction of the pigments was adapted from a procedure topurify prodigiosin 25-C (or undecylprodigiosin, MW 393) andmetacycloprodigiosin (MW 391) as shown in Figure 1 (cf. Nakamura et al.1986). Both cells and broth were extracted twice with an equal volumeof acetone (analytical grade, APS, NSW Australia). The acetoneextract was partitioned with hexane (analytical grade, APS, NSWAustralia, pH adjusted to 4.0 using 20% trifluoroacetic acid, Sigma,MO, USA). The hexane fraction was dried in vacuo using a rotaryevaporator (Buchi RE 111) and gave a dark purple residue.
3

The hexane fraction from the broth (349 mg) was run through asilica gel (70-230 Mesh ASTM, American Scientific Products, IL, USA)in an open glass column (3.0 x 36.0 cm, hexane/acetone 7:3, 3.8ml/min) and produced 9 fractions. These fractions were analyzed usingaluminum-backed silica thin layer chromatography (TLC) plates (Merck60 F254, Darmstadt, Germany) with chloroform/methanol 9:1 as themobile phase. Three fractions contained a bright red spot which onTLC gave an Rf = 0.63. These fractions were pooled and dried to give221.6 mg of material.
The hexane fraction from the cells (740 mg) was run through thesame column as above, except for an elution rate of 4.2 ml/min, andproduced 7 fractions. Three fractions contained a bright red spot atRf = 0.63. The fractions were pooled and dried to give 84.3 mg ofmaterial.
The dried material was pooled (305.9 mg) then run through asecond silica gel column, with the mobile phase chloroform/acetone95:5, pH adjusted to 10 using 23% ammonia solution (APS, NSWAustralia), and with a flow rate of 7 ml/min.
Mass spectrometry (ESI-MS, Finnigan MAT95STrap massspectrometer), 13C and 1H nuclear magnetic resonance spectroscopy(Jeol LA400) were performed at the Ateneo de Manila University(Philippines) and the University of the Philippines. The spectra wereanalyzed and the compound was identified as heptyl prodigiosin(Figure 2) by D. Tasdemir of the University of Utah, Salt Lake City,USA.
Antimalaria in vitro assay
Parasite culture. A chloroquine-sensitive strain of P. falciparum,3D7, was maintained in culture as described by Druilhe and Gentilini(1982). The strain was cultured with erythrocytes (type O) in asolution of RPMI 1640-serum : RPMI (Gibco) supplemented with 8.3 g/lof Hepes (Sigma, St. Louis, MO, USA), 2.1 g/l of NaHCO (Merck) and 10mg/ml of gentamycin. To this solution was added 10% type AB humanserum.
Hypoxanthine assay. The tritiated hypoxanthine assay was doneas described by Mirovsky, et al. (1989). Parasite culture was preparedas a suspension containing: 3H-8-hypoxanthine (Sigma) and 1.8%erythrocytes (v/v), about 0.5% parasitized. Then, 200 l of thesesuspensions (parasitized erythrocytes and healthy erythrocytes) weredispensed in a 96-well flat-bottom plate.
Crude extracts were screened at a concentration of 10,000 ng/mlper compound. Very active pure and semi-pure extracts were tested atnine two-fold concentrations between 10 and 5000 ng/ml to estimatethe median inhibitory concentration, IC50. Controls, consisting of a2-fold dilution series from 2.34 to 600 ng/ml for chloroquine, andfrom 4.78 to 1200 ng/ml for quinine, were included in each plate.Tests were done in duplicate.
4

Incubation was carried out for 48 hours at 37°C in a CO2
incubator. Parasite DNA was collected on filter disks using aSkatron cell harvester. Tritium incorporation was measured using anautomated scintillation counter (Beckman LS 1701, Les Ulis, France).The counts-per-minute (cpm) were adjusted relative to controls toobtain a %inhibition, p, which was then transformed using thefunction
p' = arcsin
as described by various authors (Zar 1999; Finney 1978; Ashton 1972),and expressed in radians. The IC50 was estimated by linear regressionof p' versus log nM of compound for data points between plateaus,including only the highest concentration in the lower plateau and thelowest concentration in the higher plateau, and provided 0.000 < p'<1.571 radians.1 The significance of the differences between thecurves defined by these data points was tested by two-factor ANOVAusing Statview (SAS Institute, USA). The F statistic was used, with = 0.05.
Antimalaria in vivo assay
C57BL/6N CRL FEM mice (18g, age 6 weeks, Charles River, France)were inoculated intraperitoneally with 100 µl of 1x107 P. bergheiparasites/ml (Clone 1.49L of ANKA strain, a gift of Dr. D. Walliker(Institute of genetics, Edinburgh, UK) (day 0). Heptyl prodigiosinwas dissolved in peanut oil (Sigma) at a concentration of 10 mg/mland a dose of 40 mg/kg (n = 8) was injected subcutaneously on days 3and 5 (n = 8 mice). Other mice were injected subcutaneously once atday 3 with doses of 20 mg/kg, 5 mg/kg, and 1 mg/kg (n per group = 8).Administration by gavage of 50 mg/kg was performed on another batch.Parasitemia was counted everyday starting day 3 using thin smear andGiemsa staining. Survival of the no-drug and drug treated groups werecompared.
Survival curves were determined using the method of Kaplan-Meier using Statview. The Mantel-Cox log rank test was used toevaluate the chi-square and the corresponding p values of the curveswith = 0.05.
1 Proportions can be validly used neither for statistics like the F test because they follow a binomial rather than a normal distribution, nor for the calculation of IC50's by linear regression because of heteroscedasticity. The arcsin transformation serves to approximately solve both problems except at values close to 0 and 100%. Only data between 0 and 100 %inhibition (0 < p' <1.571 radians)were considered for analysis. See Finney (1978) and Ashton (1972) formore details on bioassay statistics.
5

In vitro micronucleus assay
A microscale version of the in vitro rodent micronucleus assay wasused as described (Nesslany & Marzin 1999). Briefly, cells weretreated with drug in the presence or absence of microsomal extractS9. Heptyl prodigiosin was dissolved in dimethylsulfoxide (DMSO) to aconcentration of 100 mg/ml (or 2 mg/ml)2, then diluted to 1000 µg/ml(or 20 g/ml), and for 10 successive 2-fold dilutions thereafter,with Fischer’s medium (containing 10% decomplemented horse serum, or"FM10"; Gibco BRL, Paisley, UK) that had or did not have 10% S9 mix,and dispensed (100 µl) into conical-bottom wells of a 96-well plate(Costar, Brumath, France). Mouse lymphoma L5178Y (TK +/- clone3.7.2C, Porton Down, Salisbury, UK) in 100 l of culture (600,000cells/ml for S9 treatment; 650,000 cells/ml for no S9 and 24hincubation; 400,000 cells/ml for no S9 and 24h incubation + 20hrecovery period) were added to all wells. Cells treated in theabsence of S9 were cultured with drug for 24h (37C, 5% CO2, 100%humidity). They were then either washed and incubated for a further20h (recovery period) without drug, or harvested immediately. Cellstreated in the presence of S9 were cultured for 4h with drug, thenwashed to remove S9 and drug, incubated for a further 20h, thenharvested. Positive controls were cyclophosphamide (10 g/ml finalconcentration in wells) for treatment with S9 and mitomycin C (0.05g/ml without recovery period or 0.025µg/ml with a 20-h recoveryperiod) for treatment without S9. Negative control was 1% DMSO inculture medium. Treatments were performed in duplicate and coupled toan assessment of cytotoxicity using MTT (see below). Harvested cellswere fixed and stained. The number of micronuclei-containing cellsper 2000 cells was counted in wells that showed 70% growth inhibitionat most as determined by the MTT assay.3
Cytotoxicity evaluation using MTT
For every 96-well plate prepared above, a parallel 96-wellplate was prepared under the same conditions, except that all volumeswere halved. Survival estimation based on MTT reduction to formazanwas modified from Mosman (1983) and carried out at the time ofharvesting as follows.
Cells were rinsed once then resuspended with 100 l FM10. Toeach well was then added 100 l of 0.5 mg/ml MTT (5 mg/ml MTT in PBS,diluted 10x in DMEM without phenol red), and the cells incubated for2 Two ranges -- a high one going down from 1000 µg/ml, and a low one from 20 µg/ml -- were used because strong toxicity was observed in a preliminary test for most of the concentrations in the high range against cells not exposed to S9. Hence, these cells were treated using the lower range.3 At higher growth inhibitions micronuclei are not only difficult to score but might sometimes be confused with chromosome fragments resulting from apoptosis.
6

3 hours (37C, 5% CO2, 100% humidity). The plates were centrifuged,the liquid decanted, and the residue agitated in 100 µl of acidisopropanol (1 volume 1N HCl to 23 volumes isopropanol) for 10minutes. The plates were read in an ELISA plate reader (550 nmabsorbance wavelength, 620 nm reference wavelength). Absorbance wasdenoted as "A550". The %inhibition was determined relative to DMSOcontrols, and the IC50 determined by linear regression of p' versuslog concentration as described for the in vitro malaria test. ANOVA wasdone as described above.
Ames assay
A standard protocol described by Maron and Ames (1983) wasadapted in microscale. Salmonella typhimurium mutant strains used wereTA 1537, TA 98, TA 100, and TA 102. The strains had been controlledfor dependence on histidine, the presence of R-factor plasmids codingfor ampicillin resistance (TA 98, TA 100 and TA 102) and resistanceto ampicillin + tetracycline (TA 102), and the presence of the uvrBmutation. The compounds were tested with and without activation byS9.
Preparation of cell cultures. Cells (1 ml) stored in liquidnitrogen were thawed and preincubated to the late exponential orearly stationary phase under the following conditions: incubation 12hat 37C, in 40 ml of nutrient broth (2.5% w/v of nutrient broth“Oxoid” No. 2, and containing 12.5 g/ml ampicillin (except for TA1537), and, in addition, 0.5 g/ml of tetracycline (in the case of TA102), 35 rpm in a water bath shaker. Then cells were centrifuged 3000rpm for 20 minutes except for TA 102, resuspended in either 40 mlnutrient broth (TA 1537, TA 98, TA 100) or 20 ml nutrient broth (TA102), and kept at 4C until use.
Drug preparations. Heptyl prodigiosin was dissolved in DMSO(100 mg/ml). A three-fold dilution series of 10 concentrations foreach drug was prepared by dilution in DMSO. Positive controlsdepended on the strain and on exposure to S9. These controls were
Control (µg/plate),with S9
Control (µg/plate),without S9
TA 1537 2-aminoanthramine, 0.5 9-aminoacridine, 1.56TA 98 2-aminoanthramine, 0.5 2-nitrofluorene, 0.5TA 100 2-aminoanthramine, 0.5 sodium azide, 2.0TA 102 benzo[a]pyrene, 0.5 g/ml mitomycin C,
0.0625
Negative control was DMSO, not known to react with the testcompound or to be toxic to bacteria at the concentrations used here.
Then, 5 l of each drug and control was dispensed into 96-wellplates, three trials per dose per strain. Then, 100 l of suspension
7

of bacterial strain was added to the wells, followed by 95 l of S9mix (or 95 l of PBS in the case of no activation). The plates wereagitated for 90 min at 37C. Then the contents of each well wereadded to 2.2 ml of molten top agar (supplemented with 10% of asolution of histidine-biotin 0.5mM and maintained at 45C in a waterbath), vortexed, and dispensed into 55 mm plastic petri dishescontaining 10 ml of bottom agar. The dishes were incubated at 37Cfor 48h.
Sterility tests. The following reagents were tested on sterilepetri dishes containing the same agar medium as above: compounds (atthe highest concentration), solvent (duplicate), and PBS or S9 mix(triplicate). The reagents proved sterile.
Induced revertants. The number of colonies in the presence ofpositive controls was compared with historical data extending overtwo years.
Scoring. The number of colonies per dish was scored.Cytotoxicity of the test material was indicated by the absence ofbackground growth.4 The results were expressed as the mean number ofmutant colonies per dose of product, divided by the number ofspontaneous revertants in the DMSO negative control. A product wasconsidered mutagenic if there was a dose response over at least 3concentrations and if the largest increase in revertants for any ofthese concentrations was at least 3 times that of spontaneousrevertants in the case of TA 1537, and twice in the case of the otherstrains.
4 Background growth refers to the 2 or 3 generations of bacteria thatcan be supported by the trace amount of histidine in the medium.
8

Results Antimalarial activity
Figure 1 shows the steps for purifying heptyl prodigiosin. Thefinal step using a silica column gave a bright red compound (95:5CHCl3/acetone, flow rate = 7.0 ml), an Rf = 0.68 (TLC, 9:1 CHCl3/MeOH)with a dry weight of 162 mg, about 15% of the crude hexane extractsfrom both cells and broth. The distribution of the red compound,however, was not similar in both sources. Preliminary experiments onseparate extraction of broth and cell material showed thedistribution to be between 60-70% red compound in broth extract, andless than 10% in the cell extract, suggesting that the compound ismostly extruded in the medium. NMR showed a heptyl prodigiosin (cf.Strauss and Berger 1983; MS m/z 351; Figure 2).5 The extractcontained about 50% fats by weight, and these were removed byrepeated solvent partitioning between CHCl3 and hexane.
Heptyl prodigiosin had an in vitro antimalarial IC50 of about 24ng/ml (0.07 µM). Its curve was significantly different (p = 0.0022)from that of chloroquine (IC50 = 0.015 µM) and not significantly (p =0.5016) different from that of quinine (IC50 = 0.3 µM) against thechloroquine-sensitive strain 3D7 (the chloroquine curve issignificantly different from that of quinine, p = 0.0046) (Figure 3).In contrast, Kim et al.'s (1999) IC50 of cycloprodigiosin against theresistant strain FCR3 was 0.011 µM. However, this result can not becompared with ours since their chloroquine control had an IC50 of0.018 µM, similar to our IC50 against 3D7, and at least an order ofmagnitude less than what is typical for what we find with FCR3.Gerber (1975), on the other hand, did not perform in vitro assays on P.falciparum as these were not yet available before 1979 (cf. Desjardinset al. 1979).
The activity was confirmed in an in vivo mouse model using thevirulent P. berghei ANKA strain that causes fatal cerebral malariaabout a week after inoculation in the strain of mouse used (Amani etal. 1998). Two subcutaneous injections of 40 mg/kg (days 3 and 5),gave a median survival time of 29 days as compared to 7 days forcontrols treated with solvent. Figure 4a shows the survival curvesof both groups and the log-rank test suggested that the curves weresignificantly different (p < 0.001). In contrast, the best meansurvival time obtained by Gerber (1975) was about 20 days formethylcyclodecylprodiginine (versus 6 days for untreated mice) at adose of 160 mg/kg. Unspecified toxic effects were noted at 160 mg/kg,and 320 mg/kg was lethal for cyclononylprodiginine andmethylcyclodecylprodiginine. In another batch of mice,cyclononylprodiginine effected cure in some and killed others at 160mg/kg.
5 Strauss and Berger's NMR data is incomplete. We are preparing a manuscript on the complete spectra.
9

Since dermal sclerosis was observed at the site of injection inthe treated mice, the experiment was repeated by administering 20mg/kg, 5 mg/kg and 1 mg/kg subcutaneously once (on day 3). Figure 4bshows the survival curves. The log rank test of all groups showedthem to be significantly different (p = 0.0012). Median survivaltimes were 26 days (20 mg/kg group), 24 days (5 mg/kg group), 7 days(1 mg/kg group), and 6 days (0 mg/kg and untreated groups). Log rankanalysis of the 1 mg/kg, 0 mg/kg and the untreated groups gave a 2
of 0.453 and a p = 0.7972 suggested no significant differences amongthe survival curves of the said groups: administration of 1 mg/kgdid not increase survival time compared to no-drug controls. Log rankanalysis of the 20 mg/kg and 5 mg/kg groups gave a 2 of 0.421 (p =0.5164) suggesting no significant difference between their survivalcurves. A single subcutaneous dose of 5 mg/kg was as effective inextending survival time as 20 mg/kg. However, a single dose bygavage of 50 mg/kg did not increase survival time compared to solventcontrols, suggesting poor bioavailability by this route.
No treatment was curative.Increased survival appears to have resulted from a delay in the
rapid rise in parasitemia during the first week followinginoculation, as shown by the series of graphs in Figure 5. Mice whichhad a parasitemia below 6% by the end of the first week afterinoculation survived longer and with much higher parasitemias thanmice in which parasitemia reached 6% within the first week.Administration of the compound at 20 mg/kg and 5 mg/kg could havekept parasitemia low during the first week of infection, probablythrough a cytotoxic activity. A dose of 1 mg/kg did not protect themice, all of which had died by day 10.
Three mice treated with solvent (two subcutaneously and one bygavage) and one untreated mouse survived beyond the critical period.They appeared to have been able to keep parasitemia low, probablythrough a better defense response. These mice later died with severeanemia, as did all remaining mice from the treated groups, and aroundthe same time.
In all cases of surviving mice, parasitemia started to increasedramatically around the second week after infection. Increasinglyanemic blood (revealed by the low density of thin smears) wasobserved qualitatively until the time of death. In contrast, healthy(non-parasitized control) mice, whether they were administered with20 mg/kg or with solvent, did not develop anemia.
A low parasitemia during the first week was also observed withmice treated with 40 mg/kg (x2) followed by a dramatic rise beginningon the second week. All the solvent-treated mice in this group diedtowards the end of the first first week with parasitemias between 6and 7% on the day before they were found dead.
Kim et al. (1999) also described the low level of parasitemia intreated mice until day 10 and the rapid rise of parasitemia inuntreated mice (reaching up to 50% in day 7), but they did not
10

discuss mean survival time. All untreated mice seem to have died byday 8.
Heptyl prodigiosin at low doses still caused sclerosis at thesite of injection in a roughly dose-dependent manner. Dermalirritation followed by hardening of the skin at the site of injectionwas evident within 3 days of injection with 40 or 20 mg/kg but not atlower concentrations. These lesions began appearing early in somemice, but by day 15 lesions appeared in all treated mice includingnon-parasitized controls. The mice treated with 1 mg/kg had developedneither hardening of the skin nor sclerotic lesions at the time ofdeath. Mice treated by gavage did not show any behavioral or physicalchanges that suggested toxicity.
Mutagenic potential
The mutagenic potential of heptyl prodigiosin was studied sinceprodigiosin might intercalate at AT sites or cause DNA strandscission in vitro as suggested by Melvin et al. (1999). Two tests werecarried out with and without exogenous metabolic activation. Originalmicromethods were used that allowed small quantities of compound tobe tested: a gene mutation assay, i.e. the Ames's test performed onfour Salmonella typhimurium strains, and the in vitro micronucleus test, achromosomal mutation assay, performed on L5178Y mouse lymphoma cells.
No concentration of heptyl prodigiosin gave a mean number ofrevertant colonies that was more than twice that of the negativecontrol dimethylsulfoxide (background mutation) for any of the S.typhimurium strains, with or without S9 activation (Table 1). Themicronucleus assay did not reveal any significant increase in meannumber of micronuclei per 2000 cells resulting from treatment withheptyl prodigiosin (Table 2). The compound was, therefore, notgenotoxic in these two tests. To our knowledge, there have been noprevious published works on the in vitro or in vivo mutagenic potential ofthis compound.
Heptyl prodigiosin had an in vitro IC50 against L5178Y cells of1.4 µM (without S9 treatment), which is 20 times the IC50 against P.falciparum in vitro. In contrast to earlier studies, this activity wasalmost similar to that of cycloprodigiosin which had an IC50 of about6 to 7 µM against a variety of non-mouse cell lines (Kim et al. 1999).If not for the difficulties described earlier as regards the latterstudy, these IC50's would be about 500 times the antimalarial IC50's.The activity we determined for heptyl prodigiosin was less than thatof prodigiosin (0.06 µM) and more than that of prodigiosene (80 µM)and 2-methyl-3-pentylprodigiosene (40 µM, Figure 2) against L1210mouse lymphocyte leukemia cells (Boger and Patel 1988). The averageactivity of 2-heptyl-6-methoxyprodigiosene against murine restinglymphocytes and two other cells lines, murine melanoma and humanerythroleukemia, was 0.5 µM ( = 0.03 µM) (D'Alessio et al. 2000).There were no corresponding malaria IC50's for these figures.
11

Discussion
The in vitro antimalarial activity of heptyl prodigiosinappeared to be strong at concentrations below what might be toxic tononmalarial cells. This result encourages us to think that even morespecific analogs might eventually be designed. However, it is alsonot known whether the compound undergoes metabolic activation orinactivation. Furthermore, an analytical method for determiningheptyl prodigiosin metabolites in body fluids has not yet beendeveloped that will permit us to correlate the in vivo concentration ofthe active metabolite with its in vitro activity and to monitor thebioavailability of the compound with each route of administration.
What are the current hypotheses regarding the activity of theprodigiosins that might be relevant to malaria? It was suggested thatprodigiosin and metacycloprodigiosin might inhibit lysosomalacidification driven by vacuolar-type ATPase. The compounds mightpromote H+/Cl- symport which has the effect of decoupling vacuolartype ATPase (Ohkuma et al. 1998). Kim et al. (1999) have shown that thefluorescent cycloprodigiosin was accumulated in infected mouseerythrocytes in vitro. This accumulation was inhibited by pretreatmentof infected erythrocytes with concanamycin A, a compound known toinhibit vacuolar H+-ATPase, and carbonylcyanide m-chlorphenylhydrazone, a proton conductor. Vacuolar-type ATPases arepresent in plasmodial membranes and serve as the major route for theefflux of H+ ions and the regulation of parasite cytoplasmic pH(Saliba and Kirk 1999). Aminoquinolines like chloroquine, quinine,and mefloquine might also induce alkalinization of the plasmodialfood vacuole (Zarchin et al. 1986). Alkalinization, however, does notappear to be the major mode of action of these drugs because itoccurs at concentrations one to two orders of magnitude larger thanthe growth IC50 (Ginsburg et al. 1989). It is, therefore, not likelythat pH change is the major mode of action for prodigiosin compounds,although such a change might be one step in a toxic process.
The possible inhibition of tyrosine kinase by heptylprodigiosin might be relevant. Recent in vitro studies on theimmunosuppresive properties of synthetic prodigiosins suggested thatthe compounds inhibited a protein tyrosine kinase, JAK-3. This kinaseis associated with a cell surface receptor component (common chain)that is exclusive of all IL-2 cytokine family receptors (D'Alessio etal 2000). The malaria toxin glycosylphosphotidylinositol (GPI) andderived products happen to activate protein kinases that regulate thedownstream NF-B/rel-dependent gene expression of interleukin 1 ,tumor necrosis factor (TNF) , and inducible NO synthase, all ofwhich play a role in the pathology of severe malaria (Gowda andDavidson, 1999; Schofield et al., 2002).
The protection afforded by heptyl prodigiosin in vivo may,therefore, be due to two modes: the first, a direct cytotoxic effectagainst the parasite, and the second, protection against the effects
12

of induced cytokines. Distinguishing the second mode of protectionfrom the first will involve injecting mice with malaria toxin (notthe parasite), assaying the levels of cytokines, and correlating theresults with blood levels of heptyl prodigiosin and its metabolites.
The sclerotic activity at the lowest concentration capable ofincreasing survival in P. berghei-infected mice, the uncertainusefulness of performing repeated subcutaneous doses to effect cure,and the likely inefficacy of an oral route probably preclude the useof heptyl prodigiosin as an antimalarial. Nevertheless, the moleculemight be used as a lead compound. Its structure might be modified toimprove its solubility (a significant property in the context ofpublic health) and its specificity of action against malaria or otherdisease targets. Data from these synthetic products have shown that,among other structural features, the length and cyclization of theside chains significantly affect specific immunosuppresive action,for example. Similar strategies can be foreseen for improvingantimalarial effects.
13

AcknowledgementsWe thank D. Tasdemir (Department of Medicinal Chemistry,
University of Utah) for identification of the compound; A. Sertan(Marine Science Institute, Philippines), E. Bernardo, and B. Neilan(University of New South Wales, Australia) for identification of thebacterial strain; and M. Tefit of INSERM Unité 511 for animal care.This study was made possible through grants from the PhilippineCouncil for Aquatic and Marine Research and Development (GPC),International Foundation for Science (GPC), the US National CancerInstitute NCDDG (GPC), the French Embassy in Manila (JEHL), and theParasitology Laboratory of the Pitié-Salpêtrière Hospital, Paris,France (M. Danis, chief of service).
14

ReferencesAmani, V., Boubou, M.I., Pied, S., Marussig, M., Walliker, D.,Mazier, D., and Renia, L. (1998) Cloned lines of Plasmodium bergheiANKA differ in their abilities to induce experimental cerebralmalaria. Infect Immun 66 (9):4093-9.Ashton, W. (1972) The Logit Transformation, With Special Reference toIts Uses in Bioassay, 88 p. Charles Griffin & Co., Ltd., London.Bennett, J.W. and Bentley, R. (2000) Seeing red: the story ofprodigiosin. Adv Appl Microbiol 47:1-32.Boger, D.L. and Patel, M. (1988) Total synthesis of prodigiosin,prodigiosene, and desmethoxyprodigiosin: Deis-Alder reactions ofheterocyclic azadienes and development of an effective palladium(II)-promoted 2,2'-bipyrrole coupling procedure. J Org Chem 53: 1405-1410.Breman, J.G. (2001) The ears of the hippopotamus: manifestations,determinants, and estimates of the malaria burden. Am J Trop Med Hyg64 (1,2)S: 1-11.Castro, A.J. (1967) Antimalarial activity of prodigiosin, Nature213(79): 903-4.D’Alessio, R., Bargiotti, A., Carlini, O., Colotta, F., Ferrari, M.,Gnocchi, P., Isetta, A., Mongelli, N., Motta, P., Rossi, A., Tibolla,M., and Vanotti, E. (2000) Synthesis and immunosuppressive activityof novel prodigiosin derivatives. J Med Chem 43: 2557-2565.Desjardins, R.E., Canfield, C.J., Haynes, J.D. and Chulay, J.D.(1979) Quantitative assessment of antimalarial activity in vitro by asemiautomated microdilution technique. Antimicrob Agents Chemotherapy16:710-718.Druilhe, P. and Gentilini, M. (1982) Culture in vitro de Plasmodiumfalciparum: interêt et limites, méthodologie. Medicine Tropicale 42:437-458.Finney, D.J. (1978) Statistical Method in Bioassay 3rd ed., 508 p.Charles Griffin & Co., Ltd., London.Fürstner, A., Grabowski, J., Lehmann, C.W., Kataoka, T., and Nagai,K. (2001) Synthesis and biological evaluation of nonylprodigiosin andmacrocyclic prodigiosin analogues. Chembiochem 2: 60-68.Gerber, N.N. (1975) A new prodiginne (prodigiosin-like) pigment fromStreptomyces. Antimalarial activity of several prodiginnes. J Antibiot(Tokyo) 28(3): 194-99.Gerber, N.N. (1983) Cycloprodigiosin from Beneckea gazogenes.Tetrahedron Lett 24(27): 2797-98.Ginsburg, H., Nissani, E., and Krugliak, M. (1989) Alkalinization ofthe food vacuole of malaria parasites by quinoline drugs andalkylamines is not correlated with their antimalarial activity.Biochem Pharmacol 38(16): 2645-54.
15

Gowda, D.C., Davidson, E.A. (1999) Protein glycosylation in themalaria parasite. Parasitol Today 15(4):147-52.Kawauchi, K., Shibutani, K., Yagisawa, H., Kamata, H., Nakatsuji, S.,Anzai, H., Yokoyama, Y., Ikegami, Y., Moriyama, Y., Hirata, H. (1997)A possible immunosuppresant cycloprodigiosin hydrochloride, obtainedfrom Pseudoalteromonas denitrificans. Biochem Biophys Res Commun 237: 543-47.Kim, H.S., Hayashi, M., Shibata, Y., Wataya, Y., Mitamura, T., Horii,T., Kawauchi, K., Hirata, H., Tsuboi, S., and Moriyama, Y. (1999)Cycloprodigiosin hydrochloride obtained from Pseudoalteromonasdenitrificans is a potent antimalarial agent. Biol Pharm Bull 22(5): 532-4.Laatsch, H. and Thomson, R.H. (1983) A revised structure forcycloprodigiosin. Tetrahedron Lett 24(26): 2701-04.Maron, D.M. and Ames, B.N. (1983) Revised methods for the Salmonellamutagenicity test. Mutation Research 113: 173-215.Melvin, M.S., Ferguson, D.L., Lindquist, N., and Manderville, R.A.(1999) DNA-binding by 4-methoxypyrrolic natural products: preferencefor intercalation at AT sites by tambjamine E and prodigiosin. J OrgChem 64(18): 6861-6.Mirovsky, P., Gay, F., Bustos, F., Mazier, D., and Gentilini, M.(1989) Cloning of a fresh isolate of Plasmodium falciparum and drugsensitivity of the clones. Trans R Soc Trop Med Hyg 84: 511-515Montaner, B., Navarro, S., Piqué, M., Vilaseca, M., Martinell, M.,Giralt, E., Gil, J., and Pérez-Tomas, R. (2000) Prodigiosin from thesupernatant of Serratia marcescens induces apoptosis in haematopoieticcancer cell lines. Br J Pharmacol 131: 585-593.Mosmann, T. (1983). Rapid colorimetric assay for cellular growth andsurvival : application to proliferation and cytotoxicity assays. JImmunol Methods 65(1-2): 55-63.Nakamura, A., Nagai, K, Kunio, A., and Tamura, G. (1986) Selectivesuppression by prodigiosin of the mitogenic response of murinesplenocytes. J Antibiot (Tokyo) 39(8): 1155-9.Nesslany, F. and Marzin, D. (1999) A micromethod for the in vitromicronucleus assay. Mutagenesis 14(4): 403-410.Ohkuma, S., Sato, T., Okamoto, M., Matsuya, H., Arai, K., Kataoka,T., Nagai, K., Wasserman, H.H. (1998) Prodigiosins uncouple lysosomalvacuolar-type ATPase through promotion of H+/Cl- symport. Biochem J334(Pt 3): 731-41.Ritchie, G.Y., Mungthin, M., Green, J.E., Bray, P.G., Hawley, S.R.,Ward, S.A. (1996) In vitro selection of halofantrine resistance inPlasmodium falciparum is not associated with increased expression ofPgh1. Mol Biochem Parasitol 83(1): 35-46.
16

Saliba, K.J. and Kirk, K. (1999) pH regulation in the intracellular malaria parasite, Plasmodium falciparum. H(+) extrusion via a V-type H(+)-ATPase. J Biol Chem 274(47): 33213-9.Schofield, L., Hewitt, M.C., Evans, K., Siomos, M.A., Seeberger, P.H.(2002) Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature 418(6899), 785-9.Strauss, D.G. und Berger, U. (1983) Methylosin A und B, pigmente ausMethylosinus trichosporium. Zeitschrift für Allgemeine Mikrobiologie23(10): 661-668.Wasserman, H.H. and Fukuyama, J.M. (1984) The synthesis of () -cycloprodigiosin. Tetrahedron Lett 25(13): 1387-88.Williams, R.P. and Hearn, W.R. (1967) Prodigiosin. Antibiotics 2:410-32.Xu, K.J., Winter, R., Riscoe, M., Peyton, D.H. (2001) A spectroscopicinvestigation of the binding interactions between 4,5-dihydroxyxanthone and heme. J Inorg Biochem 86(2-3): 617-25.Zar, J.H. (1999) Biostatistical Analysis 4th ed., 700 p. PrenticeHall, New Jersey.Zarchin, S., Krugliak, M., and Ginsburg, H. (1986) Digestion of thehost erythrocyte by malaria parasites is the primary target forquinoline-containing antimalarials. Biochemical Pharmacology 35(14):2435-2442.
17

Figures and TablesFigure 1. Scheme for the purification of heptyl prodigiosin, the mainproduct of an unidentified -proteobacteria (accession no. CNCM I-2907, Pasteur Institute, Paris, France). The scheme follows the method of Nakamura et al. 1986 for extracting undecylprodigiosin and metacycloprodigiosin as major products from Streptomyces hiroshimensis.
18
CNCM I-2907, 9 L
Broth,about 9 L
cells, 50g+
acetone
31-3a104mg
31-3b136 mg
31-3c109 mg
31-3d740 mg
hexane fractions
hexane fraction
silica gel, 3x36 cmhexane/acetone 7:3,
pH 431-3, 31-4 &
31-7221.6 mg
32-4 to 32-684.3 mg
silica gel 3x36 cm
CHCl3/acetone 95:5,pH 10
34-8, 34-9162 mg(heptyl
34-1414.2mg
CHCl3/acetone 9:1,pH 10
repeated solvent partitioning between CHCl3
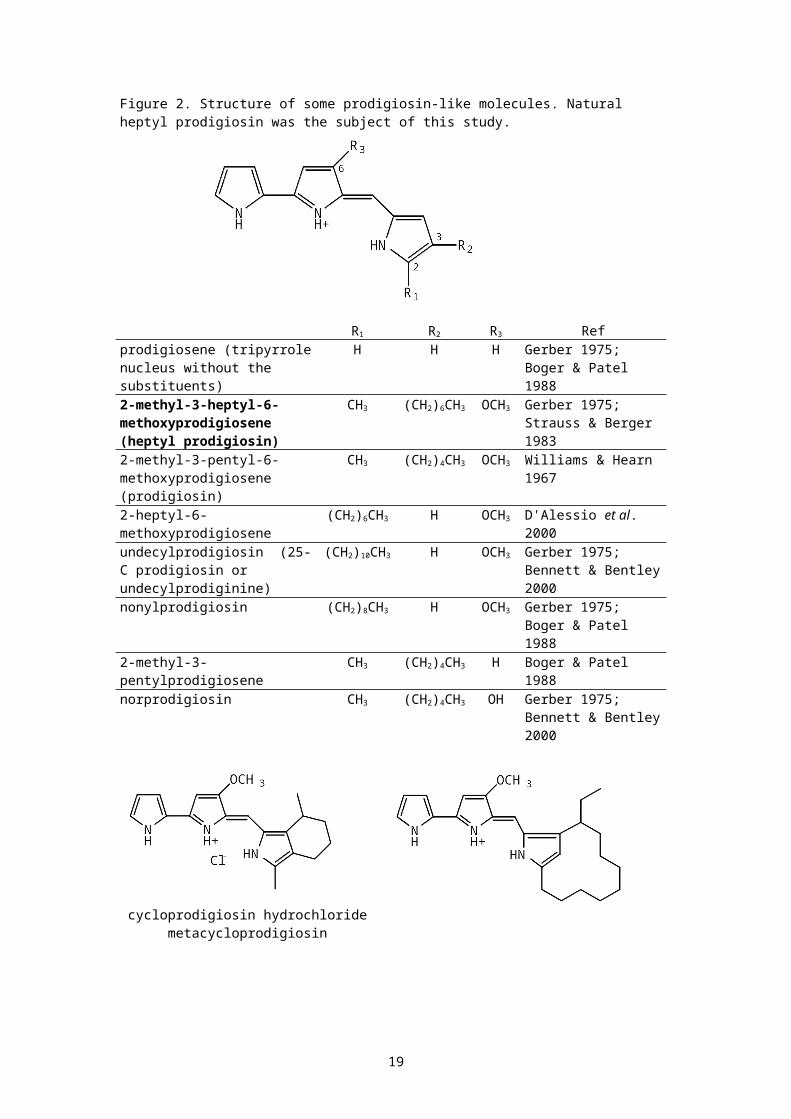
Figure 2. Structure of some prodigiosin-like molecules. Natural heptyl prodigiosin was the subject of this study.
R1 R2 R3 Refprodigiosene (tripyrrolenucleus without the substituents)
H H H Gerber 1975; Boger & Patel 1988
2-methyl-3-heptyl-6-methoxyprodigiosene (heptyl prodigiosin)
CH3 (CH2)6CH3 OCH3 Gerber 1975; Strauss & Berger 1983
2-methyl-3-pentyl-6-methoxyprodigiosene (prodigiosin)
CH3 (CH2)4CH3 OCH3 Williams & Hearn 1967
2-heptyl-6-methoxyprodigiosene
(CH2)6CH3 H OCH3 D'Alessio et al. 2000
undecylprodigiosin (25-C prodigiosin or undecylprodiginine)
(CH2)10CH3 H OCH3 Gerber 1975; Bennett & Bentley2000
nonylprodigiosin (CH2)8CH3 H OCH3 Gerber 1975; Boger & Patel 1988
2-methyl-3-pentylprodigiosene
CH3 (CH2)4CH3 H Boger & Patel 1988
norprodigiosin CH3 (CH2)4CH3 OH Gerber 1975; Bennett & Bentley2000
cycloprodigiosin hydrochloridemetacycloprodigiosin
19

(Gerber 1983; Kim et al. 1999; Kawauchi et al. 1997) (Gerber
1975; Bennett & Bentley 2000)
cyclomethyldecylprodiginine cyclononylprodiginine(Gerber 1975) (Gerber 1975)
Figure 3. In vitro inhibition of malaria reproduction by heptyl prodigiosin versus quinine and chloroquine sulfate evaluated using the tritiated hypoxanthine assay. Regression lines from which estimated IC50's were calculated are boldened and span only the data points that were used. Certain pharmacological properties might explain why the slopes are different. For example, chloroquine and quinine have almost the same affinity for the proposed drug target heme (cf. Xu et al. 2001) but they probably do not share the same transporter (cf. Ritchie et al. 1996). In contrast, little is known about the target or the transporter of heptyl prodigiosin.
20
-0,2
0
0,2
0,4
0,6
0,8
1
1,2
%inh
ibition
0 0,5 1 1,5 2 2,5 3 3,5 4 4,5log nM
heptyl prodigiosinquininechloroquine
CourbeEclaté par : Treatm ent

Figure 4. Kaplan-Meier survival curves of a) treated (40mg/kg injected twice: day 3 and day 5) and solvent (0mg/kg) groups. Mantel-Cox log rank 2 = 15.319, p < 0.001; and b) 4 treated groups (20 mg/kg; 5 mg/kg; 1 mg/kg; 0 mg/kg or solvent) and untreated control. Mantel-Cox log rank 2 = 18.071, p = 0.0012.
A.
B.
21
0
,2
,4
,6
,8
1Survie Cu
m.
0 5 10 15 20 25 30 35Temps
Délais de Survie (Traité)Survie Cum. (Traité)
Délais de Survie (Témoin)Survie Cum. (Témoin)
Kaplan-M eier Graphe de Survie Cum . pour Durée de surveillance (jours)Variable censure : <Sans>Facteur : Traitem ent
0
,2
,4
,6
,8
1
Survie Cum.
0 5 10 15 20 25 30Temps
Délais de Survie (Zeromg/kg)Survie Cum. (Zeromg/kg)
Délais de Survie (Unmg/kg)Survie Cum. (Unmg/kg)
Délais de Survie (Cinqmg/kg)Survie Cum. (Cinqmg/kg)
Délais de Survie (Vingtmg/kg)Survie Cum. (Vingtmg/kg)
Délais de Survie (Témoin)Survie Cum. (Témoin)
Kaplan-M eier Graphe de Survie Cum . pour Durée de surveillance (jours)Variable censure : CensureFacteur : Traitem ent
Days
Days
40 mg/kg x 2
solvent
20 mg/kg
5 mg/kg
solvent
untreated
1 mg/kg
Fraction
surviving
Fraction
surviving

Figure 5. Parasitemia (%) versus days. One curve represents one animal. Except for one mouse in the 5 mg/kg group (which died on day 7), the final parasitemia was that taken the day before the animals were found dead.
22

23

Table I. Number of revertant colonies of Salmonella typhimurium as a function of µg of heptyl prodigiosin (hep prod) per dish with and without S9 activation. This number is given as the range found in theseries of concentrations shown in the second column. None of the concentrations of heptyl prodigiosin gave a mean number of revertantsthat was at least twice that of the DMSO control.
Strain S9 hep prodµg/plate
Number of colonies pertreatment
hep prod DMSO +control
TA1537 - 0.025-18.52
1-15 9-13 124-167
+ 0.025-18.52
1-8 3-9 497-521
TA98 - 0.025-55.56
8-58 36-42 707-752
+ 0.025-167.7
7-30 21-25 2666-2907
TA100 - 0.025-55.56
38-132 108-144 1128-1542
+ 0.025-167.7
10-82 77-79 2214-2349
TA102 - 0.025-18.52
136-272 315-360 2303-2480
+ 0.025-6.17
286-452 337-379 758-808
Table II. Micronucleus assay of heptyl prodigiosin with and without S9. Cells not exposed to S9 were exposed to drug for 24h, then eitherharvested (24h no S9), or washed then incubated for a recovery time of another 20h (24h + 20h no S9) before harvesting. Only treatments that resulted in at most 70% inhibition (assayed by MTT) were used for scoring. The mean micronucleus count (Mean MN) is per 2000 cells.Significance (S) is indicated on the last column.
Product µg/ml
Treatment %inhibition
Mean MN Chi-square versusDMSO
p Signif
DMSO 0.000
with S9 0.0999 3
CPA 10.000
with S9 0.0 79 71.913 1.000
S
hep prod 3.900
with S9 34.1 2 0.000 . NS
hep prod 1.950
with S9 5.1 5 0.126 0.277
NS
hep prod 0.987
with S9 0.0 7 1.604 0.795
NS
24

DMSO 0.000
24h no S9 0.0 1
mitomycin C
0.050
24h no S9 7.0 31 28.352 1.000
S
hep prod 0.156
24h no S9 51.8 4 0.802 0.630
NS
hep prod 0.078
24h no S9 43.2 3 0.251 0.384
NS
hep prod 0.039
24h no S9 25.1 4 0.802 0.630
NS
DMSO 0.000
24h + 20hno S9
0 5
mitomycin C
0.025
24h + 20hno S9
5.7 47 34.370 1.000
S
hep prod 0.077
24h + 20hno S9
58.0 8 0.695 0.596
NS
hep prod 0.039
24h + 20hno S9
18.0 4 0.000 . NS
hep prod 0.019
24h + 20hno S9
8.0 6 0.091 0.237
NS
hep prod 0.010
24h + 20hno S9
8.5 3 0.125 0.276
NS
25

12 July 2002
Dr. A.T. TuEditorJournal of Natural ToxinsDepartment of Biochemistry and Molecular BiologyColorado State UniversityFort Collins, ColoradoUSA
Dear Sir:
On behalf of all co-authors I am submitting this revisedcopy of the paper Heptyl prodigiosin, a bacterialmetabolite, is antimalarial in vivo and non-mutagenic invitro. This paper has not been submitted to any otherjournal. I hereby agree to abide by the copyrightpolicies of the Journal if the article is accepted forpublication.
Please find encloseda) Referees' comments and our remarks (in upper case);b) NMR profiles;c) An original printout of the article and 2 photocopies;d) An IBM-formatted disk containing 1 copy of the article
in Word 97.
Sincerely,
J. Enrico H. Lazaro
26

Corresponding authors
27

Reviewer I: I have read the manuscript by Lazaro et al. The manuscript describes original and new data on the anti-malarial activity of heptyl prodigiosin. Their observation is interesting and useful. I have the following minor suggestions: 1. Add a reference to the NMR data mentiioned on page 11, para. 1.
REFERENCE IS MADE TO STRAUSS AND BERGER 1983. THEIR NMR WAS INCOMPLETE. WE ARE PREPARING A MANUSCRIPT ON THE NMR OF HEPTYL PRODIGIOSIN. ENCLOSED ARE OUR DATA, ONE FOR THE IMPURE EXTRACT ANDONE AFTER SOLVENT REMOVAL OF THE FATS.
2. The figure legend to Figure 1 should elaborate further to make it a stand alone exhibit. Identify Z143. Is this a typical purification protocol, especially the weights and volumes? THIS HAS BEEN DONE. THE MAIN DIFFERENCE IN OUR PROTOCOL IS THAT OUR COMPOUND WAS FOUND IN THE PLACE OF NAKAMURA'S. OF COURSE, THE ORGANISM IS DIFFERENT.
Reviewer II: The findings presented in the manuscript "Heptyl prodigiosin..." may be so interesting for readers of J. of Natural Toxins. Namely, the manuscript is suitable for the journal. However, some modifications are needed for publication. 1. the purity of the final preparation of heptyl prodigiosin, which
was used in the experiments, was not shown in the text. However, to confirm the biological activities are surely due to heptyl prodigiosin, the homogeneity of the final preparation must be represented. THE PURITY BEFORE FAT REMOVAL IS NOW INDICATED. IT IS50%.
2. The slope of the dose-response curve of chloroquine differs from that of quinine or heptyl prodigiosin (Fig. 3). The authors shoulddiscuss why the slopes were different from each other. IN A GENERAL WAY, THE DIFFERENCES ARE BRIEFLY DISCUSSED IN THE LEGEND TO THE FIGURE.
3. I recommend the section of "Results and Discussion" is divided into "Results" (p. 11, line 1 to p. 14, line 15) and "Discussion" (p. 14, line 16 to p. 15, line 22). "Results" may be further divided into the antimalarial part (p. 11, line 1 to p. 13, line 13) and the mutagenic part (p. 13, line 14 to p. 14, line 15) by subtitles. THIS HAS BEEN DONE.
4. Page 11, line 2: The term "prodigisin 351" is better to be rewritten to "heptyl prodigiosin." THIS HAS BEEN DONE.
5. Page 15, line 5: The sentence, "The malaria toxin glycosylphosphatidylinositol (GPI) and GPI-derived products...." might miss some words. THE WORDING HAS BEEN SHORTENED.
6. The third reference, Bennett, J. W., and Bentley, R. (2000), is not cited in the text. BENNETT AND BENTLEY ARE CITED IN FIGURE 2, NOT IN THE TEXT.
7. Figure 1: "ph 10" is an error of "pH 10." THIS HAS BEEN DONE.
28

29
Copyright © 2022 FDOKUMEN




















