
Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice
6
Hepatitis B Virus Promotes Hepatocarcinogenesis in Transgenic Mice Yanyan Zheng, 1 Wen-ling Chen, 1 Stan G. Louie, 2 T. S. Benedict Yen, 3,4 and Jing-hsiung James Ou 1 HBV is a major risk factor for hepatocellular carcinoma (HCC). However, whether HBV can directly cause HCC or only indirectly via the induction of chronic liver inflammation has been controversial. By using transgenic mice carrying the entire HBV genome as a model, we now demonstrate that HBV by itself is an inefficient carcinogen. However, it can efficiently promote hepatocarcinogenesis initiated by the carcinogen diethylnitrosamine (DEN). This effect of HBV does not involve chronic liver inflammation, is apparently due to enhanced hepatocellular apoptosis and compensatory regeneration following DEN treatment, and does not require the HBV X protein. Conclusion: Our results demonstrate a direct role of HBV in a hepatocarcinogenesis pathway that involves the interaction between this virus and a dietary carcinogen. (HEPATOLOGY 2007;45:16-21.) H epatitis B virus (HBV) is a small DNA virus that infects approximately 350 million people worldwide. This virus can cause acute and chronic hepatitis as well as liver cirrhosis. Epidemiological studies have also established that this virus is a major risk factor for HCC. 1,2 A large body of evidence indicates that HBV can indirectly induce HCC by eliciting host im- mune responses against infected hepatocytes. This results in chronic liver inflammation and the eventual develop- ment of HCC. 3,4 In contrast, whether HBV can directly induce hepatocarcinogenesis without invoking chronic liver inflammation has been a subject of much debate. HBV DNA can integrate into host chromosomes during chronic infection and may directly cause HCC via inser- tional mutagenesis. HBV DNA has been shown to inte- grate near erb-A and cyclin A genes. 5,6 However, this integration of HBV DNA near genes that may regulate cell proliferation is a rare event, and no host genes that are consistently activated by HBV DNA integrants have been identified. Another possible way for HBV to directly induce HCC is via a trans-acting viral protein factor. The HBV genome is 3.2 kilobases (kb) in size and contains 4 genes named S, C, P, and X. The S gene codes for 3 forms of the surface (envelope) proteins. The C gene codes for the viral core protein that forms the viral core particle. The P gene codes for the viral DNA polymerase, which is also a reverse transcriptase. The HBV X gene codes for a 16.5 kDa regulatory protein that can enhance HBV replication. 7-10 This protein does not bind to DNA directly, but it can bind to transcription factors to enhance their DNA bind- ing activities. It can also regulate cellular signaling path- ways and bind to a number of cellular factors to affect their activities (for a review, see Ganem and Schneider 11 ). Due to its multiple regulatory activities, it was suggested that HBV X protein (HBx) plays an important role in hepatocarcinogenesis induced by HBV. 12 HBV has a narrow host range, which has hampered its research. For this reason, transgenic mice that expressed HBV gene products had been used to study HBV patho- genesis. For example, mice that expressed the HBV X gene or the X gene and the S gene had been reported to be more likely to develop hepatocellular neoplasia than the control mice after treatment with the carcinogen dieth- ylnitrosamine (DEN). 13,14 However, the biological signif- icance of these previous studies is unclear, because these mice carried only partial HBV genome. More recently, transgenic mice carrying the entire HBV genome have been produced. These mice contain replicating HBV DNA in the liver and carry circulating HBV particles in the blood. This mouse model has been used extensively to Abbreviations: ADV, adefovir dipivoxil; ALT, alanine aminotransferase; BrdU, bromodeoxyuridine; DEN, diethylnitrosamine; HBx, HBV X protein; HCC, hep- atocellular carcinoma. From the 1 Department of Molecular Microbiology and Immunology, 2 Depart- ment of Pharmacy, University of Southern California, Los Angeles, CA; the 3 De- partment of Pathology, University of California, San Francisco, CA; and 4 Pathology Services, Veterans Administration Medical Center, San Francisco, CA. Received August 1, 2006; accepted October 2, 2006. Supported by grants from the National Institutes of Health to J.-H.J.O. (R21RR027812) and T.S.B.Y. (R01CA055578 and P30DK026743). Address reprint requests to: J.-H. James Ou, Ph.D., Department of Molecular Microbiology and Immunology, University of Southern California, Keck School of Medicine, 2011 Zonal Avenue, HMR-401, Los Angeles, CA 90033. E-mail: [email protected]; fax: 323-442-1721. Copyright © 2006 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.21445 Potential conflict of interest: Nothing to report. 16
Transcript of Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice

Hepatitis B Virus Promotes Hepatocarcinogenesis inTransgenic Mice
Yanyan Zheng,1 Wen-ling Chen,1 Stan G. Louie,2 T. S. Benedict Yen,3,4 and Jing-hsiung James Ou1
HBV is a major risk factor for hepatocellular carcinoma (HCC). However, whether HBV candirectly cause HCC or only indirectly via the induction of chronic liver inflammation hasbeen controversial. By using transgenic mice carrying the entire HBV genome as a model, wenow demonstrate that HBV by itself is an inefficient carcinogen. However, it can efficientlypromote hepatocarcinogenesis initiated by the carcinogen diethylnitrosamine (DEN). Thiseffect of HBV does not involve chronic liver inflammation, is apparently due to enhancedhepatocellular apoptosis and compensatory regeneration following DEN treatment, anddoes not require the HBV X protein. Conclusion: Our results demonstrate a direct role ofHBV in a hepatocarcinogenesis pathway that involves the interaction between this virus anda dietary carcinogen. (HEPATOLOGY 2007;45:16-21.)
Hepatitis B virus (HBV) is a small DNA virus thatinfects approximately 350 million peopleworldwide. This virus can cause acute and
chronic hepatitis as well as liver cirrhosis. Epidemiologicalstudies have also established that this virus is a major riskfactor for HCC.1,2 A large body of evidence indicates thatHBV can indirectly induce HCC by eliciting host im-mune responses against infected hepatocytes. This resultsin chronic liver inflammation and the eventual develop-ment of HCC.3,4 In contrast, whether HBV can directlyinduce hepatocarcinogenesis without invoking chronicliver inflammation has been a subject of much debate.HBV DNA can integrate into host chromosomes duringchronic infection and may directly cause HCC via inser-tional mutagenesis. HBV DNA has been shown to inte-grate near erb-A and cyclin A genes.5,6 However, thisintegration of HBV DNA near genes that may regulatecell proliferation is a rare event, and no host genes that are
consistently activated by HBV DNA integrants have beenidentified.
Another possible way for HBV to directly induce HCCis via a trans-acting viral protein factor. The HBV genomeis 3.2 kilobases (kb) in size and contains 4 genes named S,C, P, and X. The S gene codes for 3 forms of the surface(envelope) proteins. The C gene codes for the viral coreprotein that forms the viral core particle. The P gene codesfor the viral DNA polymerase, which is also a reversetranscriptase. The HBV X gene codes for a 16.5 kDaregulatory protein that can enhance HBV replication.7-10
This protein does not bind to DNA directly, but it canbind to transcription factors to enhance their DNA bind-ing activities. It can also regulate cellular signaling path-ways and bind to a number of cellular factors to affecttheir activities (for a review, see Ganem and Schneider11).Due to its multiple regulatory activities, it was suggestedthat HBV X protein (HBx) plays an important role inhepatocarcinogenesis induced by HBV.12
HBV has a narrow host range, which has hampered itsresearch. For this reason, transgenic mice that expressedHBV gene products had been used to study HBV patho-genesis. For example, mice that expressed the HBV Xgene or the X gene and the S gene had been reported to bemore likely to develop hepatocellular neoplasia than thecontrol mice after treatment with the carcinogen dieth-ylnitrosamine (DEN).13,14 However, the biological signif-icance of these previous studies is unclear, because thesemice carried only partial HBV genome. More recently,transgenic mice carrying the entire HBV genome havebeen produced. These mice contain replicating HBVDNA in the liver and carry circulating HBV particles inthe blood. This mouse model has been used extensively to
Abbreviations: ADV, adefovir dipivoxil; ALT, alanine aminotransferase; BrdU,bromodeoxyuridine; DEN, diethylnitrosamine; HBx, HBV X protein; HCC, hep-atocellular carcinoma.
From the 1Department of Molecular Microbiology and Immunology, 2Depart-ment of Pharmacy, University of Southern California, Los Angeles, CA; the 3De-partment of Pathology, University of California, San Francisco, CA; and 4PathologyServices, Veterans Administration Medical Center, San Francisco, CA.
Received August 1, 2006; accepted October 2, 2006.Supported by grants from the National Institutes of Health to J.-H.J.O.
(R21RR027812) and T.S.B.Y. (R01CA055578 and P30DK026743).Address reprint requests to: J.-H. James Ou, Ph.D., Department of Molecular
Microbiology and Immunology, University of Southern California, Keck School ofMedicine, 2011 Zonal Avenue, HMR-401, Los Angeles, CA 90033. E-mail:[email protected]; fax: 323-442-1721.
Copyright © 2006 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.21445Potential conflict of interest: Nothing to report.
16

study HBV pathogenesis (for an example, see Isogawa etal.15). In this report, we used HBV transgenic mice toinvestigate whether HBV can directly cause HCC and thepossible role of HBx in hepatocarcinogenesis. Our resultsindicate that HBV is an inefficient carcinogen by itself.However, it can greatly increase the risk for HCC if theanimals are exposed to the dietary carcinogen DEN. Thisincrease of HCC risk is independent of HBx and does notinvolve chronic liver inflammation.
Materials and Methods
Transgenic Mice. Five different HBV transgenicmouse lines were used in our studies: wtTg05 andwtTg08 were 2 independent mouse lines that carried thewild-type HBV genome; mtTg04 and mtTg31 were 2independent mouse lines that carried the X-negativeHBV genome; and T22 was the mouse line that carriedonly the HBV X gene, which was under the expressioncontrol of its own promoter. The production of thesetransgenic mouse lines has been described.9 All of ourmouse lines except T22 have the B6 genetic background.The T22 mouse line has the CD1 background. For thisreason, we ensured that all of the mice used in our studieshave the same CD1/B6 hybrid genetic background. Thehomozygous wtTg05, wtTg08, mtTg04, and mtTg31mice, which had the B6 genetic background, were crossedto the control CD1 mice to generate the hemizygousHBV transgenic mice with the CD1/B6 genetic back-ground. The X-negative mtTg04 and mtTg31 mouselines were also crossed to the T22 mouse line to generatethe double transgenic mice with the CD1/B6 back-ground. The control mice were produced by crossing thenontransgenic B6 mouse line to the CD1 mouse line togenerate mice with the same CD1/B6 genetic back-ground. For simplicity, only male mice were used in ourstudies. For hepatocarcinogenesis studies, HBV trans-genic mice or their control nontransgenic mice wereinjected with phosphate-buffered saline (PBS) or dieth-ylnitrosamine (DEN) (2 mg/kg body weight) at 16 daysof age. The mice were killed at 7-8 months of age foranalysis of hepatocellular neoplasia. All of the studies in-volving animals were conducted in compliance with theGuide for the Care and Use of Laboratory Animals pub-lished by the National Institutes of Health.
TUNEL Assay and the BrdU-Staining Experiment.Hemizygous wtTg05 mice were crossed to CD1 mice togenerate wtTg05 hemizygous mice and the control non-transgenic littermates. These mice were injected withDEN (100 mg/kg body weight) at 16 days of age. Ahigher dose of DEN was needed for injection in this study
to increase the number of TUNEL-positive cells for sta-tistical analysis.
Liver tissues were isolated from wtTg05 and controlmice 24 hours after DEN injection and fixed in 4% para-formaldehyde for 2 hours before paraffin-embedding.TUNEL assay was conducted on liver tissue sections us-ing the TACS TDT kit (R&D Systems, Minneapolis,MN) following the manufacturer’s protocol. For bro-modeoxyuridine (BrdU) staining, mice were injectedwith DEN followed by the i.p. injection of BrdU (100mg/kg body weight; Sigma, St. Louis, MO) on the secondday. Liver tissues were isolated 2 hours after the BrdUinjection, fixed in 4% paraformaldehyde and stored in80% ethanol. Liver tissue sections were stained with themouse monoclonal anti-BrdU antibody (1:100 dilution,DAKO, Carpinteria, CA). TUNEL-positive and BrdU-positive cells were counted blindly by a research memberwho did not know the codes of the samples. The data werethen analyzed by Student’s t test.
Results and DiscussionThe wtTg05 and wtTg08 mouse lines are 2 indepen-
dent transgenic mouse lines that carry the wild-type HBVgenome.9 The HBV DNA fragment used to producethese mouse lines is 1.3 times the size of the viral genomeand contains the entire genomic information of HBV.Both of these mouse lines contains replicating HBV DNAin the liver and produce circulating virions in the blood.9
As shown in Table 1, only 1 of these mice developed asingle liver tumor nodule when they were killed and ex-amined at 8 months of age. This result indicates that HBVis an inefficient carcinogen by itself and is consistent witha previous report,16 which indicated that no pathologicalchanges were observed in HBV transgenic mice during a1-year follow-up study (also unpublished observation).
Table 1. Liver Tumor Incidence in HBV TransgenicMice and Control Mice
Mouse Groups*
Numberof MiceStudied
Number ofMice with
Liver Tumors Tumor Incidence
DEN� wtTg05 9 1 11%wtTg08 8 0 0%
DEN� CD1/B6 32 1 3%wtTg05 11 10 91% (P � 0.0001)†wtTg08 15 12 80% (P � 0.0001)†
*Unless otherwise stated, all the mice used in our studies had the sameCD1/B6 genetic background. The control mice were produced by crossing thenontransgenic B6 mouse line to the CD1 mouse line to generate mice with thesame CD1/B6 genetic background. Only male mice were used in the studies. Micewere i.p. injected with phosphate-buffered saline (PBS) (�) or DEN (2 mg/kgbody weight) (�) at 16 days of age.
†The P value was determined by comparing with the control CD1/B6 mousegroup.
HEPATOLOGY, Vol. 45, No. 1, 2007 ZHENG ET AL. 17

To further investigate the possible role of HBV in thedevelopment of HCC induced by carcinogens, wtTg05,wtTg08 and the control nontransgenic mice were injectedonce with DEN, a carcinogen that is widespread in waterand food,17 at 16 days of age. These mice were killed at7-8 months of age for examination of liver tumors. Underthis experimental condition, only 1 of 32 control mice(3%) developed visible liver tumor nodules. Histologicalanalysis of several livers removed from the other 31 micefailed to reveal any microscopic tumors (data not shown).In contrast, wtTg05 and wtTg08 HBV transgenic micedeveloped liver tumors at a significantly higher rate (P �0.0001, Fisher’s exact test against the control group) of91% and 80%, respectively (Table 1). This represents anapproximately 30-fold increase of tumor risk. The tumornodules observed in these HBV transgenic mice were usu-ally multifocal and ranged from 2 mm to more than 10mm in diameter (Fig. 1A). Histological analysis revealedthat these tumor nodules represented hepatocellular ade-nomas or carcinomas (Fig. 1B, panels b and d). Our re-sults thus indicate that HBV can promotehepatocarcinogenesis initiated by DEN.
The histological analysis of the non-neoplastic livertissues of our transgenic mice that developed liver tumorsdid not reveal the infiltration of lymphocytes (Fig. 1B,panels b and c), which is a hallmark of liver inflammation.The same negative result was obtained when the micewere killed at 4 months of age after the DEN or PBSinjection (Fig. 2A). These results were not surprising, asHBV antigens were expressed from the HBV DNA inte-grated in the host chromosome (the “transgene”) andwere not expected to stimulate any significant immuneresponses due to tolerance. To further rule out the possi-bility that there was chronic liver injury in our transgenicmice after the DEN injection, blood samples were ran-domly collected from DEN-injected or saline-injectedwtTg05 mice on a periodical basis and used for the anal-ysis of the levels of alanine aminotransferase (ALT), whichis a marker for liver injury. The ALT levels of most of theblood samples from either DEN-treated or saline-treatedtransgenic mice were below 50 IU/L, while a positivecontrol blood sample collected from a control nontrans-genic mouse 2 days after the injection of CCl4 was over3000 IU/L (data not shown) (Fig. 2B). There were 2blood samples isolated from the DEN-treated HBV micethat had an ALT level slightly higher than the others (Fig.2B, denoted by circles). These 2 blood samples were de-rived from the same mouse, which had a large tumornodule (�10 mm in diameter) when the mouse waskilled. There was also 1 blood sample in the saline-in-jected transgenic mouse group that had a slightly higherALT level (Fig. 2B, denoted by a square). This blood
sample was derived from the sole saline-injected trans-genic mouse that developed a liver tumor nodule (Table1). These slight increases of ALT levels were likely due tothe death of tumor cells and/or the liver injury caused bythe growth of the tumor nodules.
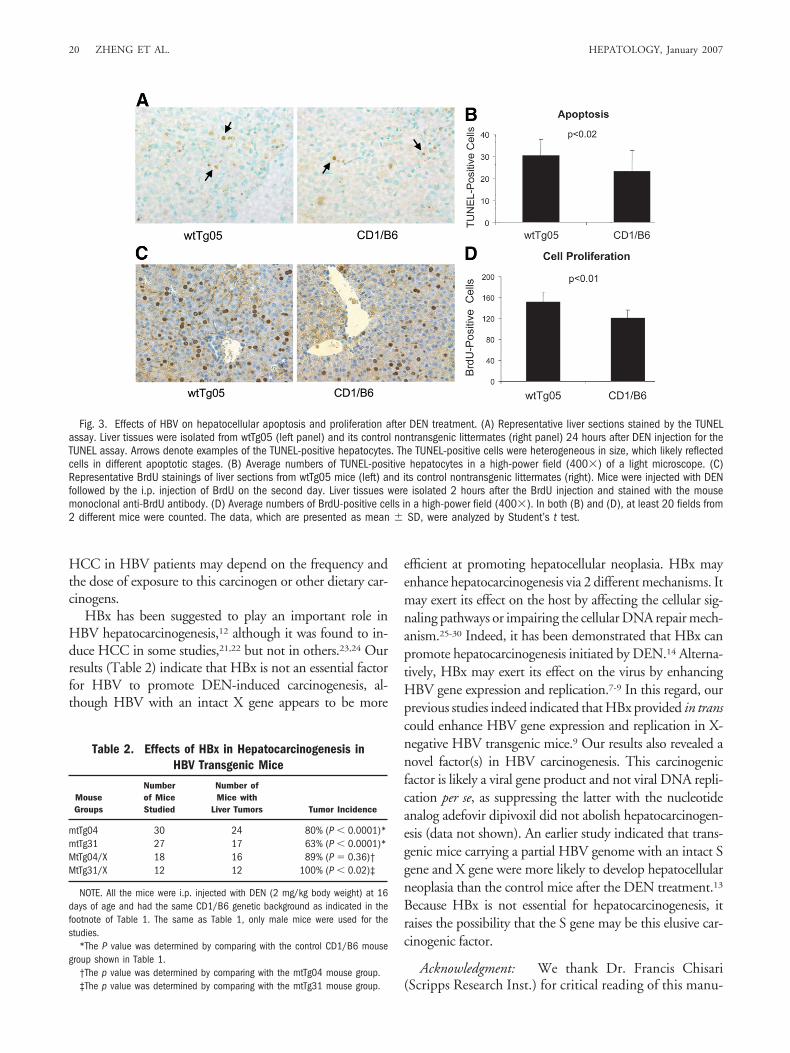
DEN is metabolically converted to electrophilic formsin hepatocytes by CYP2E1 and other enzymes. This con-version causes DNA damage,18,19 which can in turn in-duce cell death and the subsequent hepatocellularregeneration.20 To investigate whether HBV affects hep-atocellular turnover rates caused by DEN, we isolatedliver tissues from mice 24 hours after the DEN injectionand performed the TUNEL assay and the BrdU-labelingassay. The former is an assay for apoptosis whereas thelatter measures cellular proliferation. Typical liver sec-tions stained by the TUNEL assay are shown in Fig. 3Aand stained by BrdU shown in Fig. 3C. Their respectivelyquantitative results are shown in Fig. 3B,D. As shown inFig. 3B, more apoptotic hepatocytes were detected inwtTg05 HBV transgenic mice than in its control non-transgenic CD1/B6 mice (P � 0.02). This result indicatesthat HBV sensitized hepatocytes to apoptosis induced byDEN. Similarly, there were more BrdU-positive hepato-cytes in wtTg05 mice than in control nontransgenic mice(P � 0.01) (Fig. 3C,D), indicating a higher hepatocellu-lar proliferation rate in HBV mice after the DEN treat-ment. Taken together, these results indicated that HBVincreased the hepatocellular turnover rate caused byDEN.
To investigate whether the HBx protein was responsi-ble for the induction of hepatocarcinogenesis, we in-cluded 2 additional transgenic mouse lines, mtTg04 andmtTg31, in our studies. These 2 mouse lines carried thesame HBV genome as that in wtTg05 and wtTg08, withthe exception that the expression of HBx was abolished bya missense mutation in its translation initiation codonand a nonsense mutation in its coding region.9 As shownin Table 2, mtTg04 and mtTg31 developed hepatocellu-lar neoplasia with a frequency of 80% and 63%, respec-tively. These tumor frequencies were still significantlyhigher than that of the control CD1/B6 mice shown inTable 1 (P � 0.0001), indicating that HBx was not re-quired for hepatocarcinogenesis after the DEN treatment.To further examine whether HBx could enhance hepato-carcinogenesis, we crossed mtTg04 and mtTg31 mouselines to the HBV X gene transgenic mouse line T22.9 Thetumor frequency for the double transgenic micemtTg04/X and mtTg31/X were 89% and 100%, respec-tively (Table 2). These tumor frequencies were bothhigher than their respective single transgenic parentalmice. The difference between 89% for mtTg04/X and80% for mtTg04 was statistically insignificant (P �
18 ZHENG ET AL. HEPATOLOGY, January 2007

0.36). However, we cannot rule out the possibility thatthis insignificance was due to the relatively small samplesize, which did not have enough power to analyze thelower than 10% increase. In contrast, the difference be-tween 100% for mtTg31/X and 63% for mtTg31 wasstatistically significant (P � 0.02), suggesting that HBxmight play an accessory role in hepatocarcinogenesis, atleast for this mouse lineage.
A large body of evidence indicated that HBV couldinduce HCC indirectly by inducing chronic liver inflam-mation.3,4 However, whether HBV can directly partici-pate in hepatocarcinogenesis has been a subject of debate.In this report, we demonstrate that HBV is an inefficientcarcinogen by itself. However, it can promote DEN-ini-tiated hepatocarcinogenesis in the absence of apparent
chronic liver inflammation. Our further studies indicatedthat HBV could enhance apoptosis of hepatocytes afterDEN treatment. Consequently, there was an increasedproliferation of DEN-damaged hepatocytes and the de-velopment of hepatocellular neoplasia. Recently, it wasdemonstrated that the knockout of the IKK� gene led toincreased hepatocellular death and compensatory hepato-cellular growth after the DEN treatment, which resultedin a marked increase of hepatocarcinogenesis in the IKK�knockout mice.20 It is likely that HBV also promoteshepatocarcinogenesis by enhancing hepatocellular celldeath and proliferation after exposure to DEN.
Our results have important implications, because theyindicate that HBV carriers who are exposed to DEN willhave a significantly increased risk for HCC. DEN is a wide-spread carcinogen.17 It can also be converted from nitrite,which is frequently used in meat curing processes,17 andamines. The time period required for the development of
Fig. 1. Development of hepatocellular neoplasia in HBV transgenicmice. HBV transgenic mice were injected with DEN (2 mg/kg bodyweight) at 16 days of age and killed at 7-8 months of age. (A)Comparison of a normal liver and a typical liver with multiple tumornodules. (B) Hematoxylin and eosin (H&E) staining of liver tissue sec-tions. Panel a, normal liver; panel b, a typical liver tumor nodule(denoted by an arrow) from HBV transgenic mice; panel c, a highermagnification of a nontumorous section of a liver with tumor nodules;and panel d, a higher magnification of a liver tumor section. In panel d,the arrowhead denotes a cell undergoing mitosis.
Fig. 2. Analysis of liver injury markers. (A) Liver tissue sections of a4-month-old HBV transgenic mouse that had been treated with DEN (left)or PBS (right). No lymphocyte infiltration was observed. (B) ALT levels inwtTg05 HBV transgenic mice. The blood samples were collected from thetail vein from randomly selected animals at the age indicated. The ALTlevels were measured using an enzymatic assay kit (Thermo ElectronCorp.). Left panel, mice injected with DEN (mean � SD, 27.2 � 16.0IU/L); right panel, mice injected with PBS (mean � SD, 22.6 � 12.7IU/L). The difference of the ALT levels was statistically insignificantbetween these 2 mouse groups (P � 0.24, Student’s t test) or betweenthese 2 mouse groups and the control nontransgenic mouse group nottreated with DEN (data not shown).
HEPATOLOGY, Vol. 45, No. 1, 2007 ZHENG ET AL. 19
HCC in HBV patients may depend on the frequency andthe dose of exposure to this carcinogen or other dietary car-cinogens.
HBx has been suggested to play an important role inHBV hepatocarcinogenesis,12 although it was found to in-duce HCC in some studies,21,22 but not in others.23,24 Ourresults (Table 2) indicate that HBx is not an essential factorfor HBV to promote DEN-induced carcinogenesis, al-though HBV with an intact X gene appears to be more
efficient at promoting hepatocellular neoplasia. HBx mayenhance hepatocarcinogenesis via 2 different mechanisms. Itmay exert its effect on the host by affecting the cellular sig-naling pathways or impairing the cellular DNA repair mech-anism.25-30 Indeed, it has been demonstrated that HBx canpromote hepatocarcinogenesis initiated by DEN.14 Alterna-tively, HBx may exert its effect on the virus by enhancingHBV gene expression and replication.7-9 In this regard, ourprevious studies indeed indicated that HBx provided in transcould enhance HBV gene expression and replication in X-negative HBV transgenic mice.9 Our results also revealed anovel factor(s) in HBV carcinogenesis. This carcinogenicfactor is likely a viral gene product and not viral DNA repli-cation per se, as suppressing the latter with the nucleotideanalog adefovir dipivoxil did not abolish hepatocarcinogen-esis (data not shown). An earlier study indicated that trans-genic mice carrying a partial HBV genome with an intact Sgene and X gene were more likely to develop hepatocellularneoplasia than the control mice after the DEN treatment.13
Because HBx is not essential for hepatocarcinogenesis, itraises the possibility that the S gene may be this elusive car-cinogenic factor.
Acknowledgment: We thank Dr. Francis Chisari(Scripps Research Inst.) for critical reading of this manu-
Fig. 3. Effects of HBV on hepatocellular apoptosis and proliferation after DEN treatment. (A) Representative liver sections stained by the TUNELassay. Liver tissues were isolated from wtTg05 (left panel) and its control nontransgenic littermates (right panel) 24 hours after DEN injection for theTUNEL assay. Arrows denote examples of the TUNEL-positive hepatocytes. The TUNEL-positive cells were heterogeneous in size, which likely reflectedcells in different apoptotic stages. (B) Average numbers of TUNEL-positive hepatocytes in a high-power field (400�) of a light microscope. (C)Representative BrdU stainings of liver sections from wtTg05 mice (left) and its control nontransgenic littermates (right). Mice were injected with DENfollowed by the i.p. injection of BrdU on the second day. Liver tissues were isolated 2 hours after the BrdU injection and stained with the mousemonoclonal anti-BrdU antibody. (D) Average numbers of BrdU-positive cells in a high-power field (400�). In both (B) and (D), at least 20 fields from2 different mice were counted. The data, which are presented as mean � SD, were analyzed by Student’s t test.
Table 2. Effects of HBx in Hepatocarcinogenesis inHBV Transgenic Mice
MouseGroups
Numberof MiceStudied
Number ofMice with
Liver Tumors Tumor Incidence
mtTg04 30 24 80% (P � 0.0001)*mtTg31 27 17 63% (P � 0.0001)*MtTg04/X 18 16 89% (P � 0.36)†MtTg31/X 12 12 100% (P � 0.02)‡
NOTE. All the mice were i.p. injected with DEN (2 mg/kg body weight) at 16days of age and had the same CD1/B6 genetic background as indicated in thefootnote of Table 1. The same as Table 1, only male mice were used for thestudies.
*The P value was determined by comparing with the control CD1/B6 mousegroup shown in Table 1.
†The p value was determined by comparing with the mtTg04 mouse group.‡The p value was determined by comparing with the mtTg31 mouse group.
20 ZHENG ET AL. HEPATOLOGY, January 2007

script prior to its submission, and Drs. John Morrey(Univ. of Utah) and Shelly Xiong (Gilead Sciences) forhelpful discussions on the use of adefovir dipivoxil. Wealso thank the Transgenic Mouse Core Facility of theUSC Norris Cancer Center for the production of trans-genic mice and Dr. Samuel French and the MorphologyCore of the USC Alcoholic Liver and Pancreatic DiseasesResearch Center for the help on TUNEL and BrdU-stain-ing assays.
References1. Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carci-
noma. Cancer 1988;61:1942-1956.2. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and
hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet1981;2:1129-1133.
3. Robinson WS. Molecular events in the pathogenesis of hepadnavirus-as-sociated hepatocellular carcinoma. Annu Rev Med 1994;45:297-323.
4. Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immunepathogenesis of hepatocellular carcinoma. J Exp Med 1998;188:341-350.
5. Dejean A, Bougueleret L, Grzeschik KH, Tiollais P. Hepatitis B virusDNA integration in a sequence homologous to v-erb-A and steroid recep-tor genes in a hepatocellular carcinoma. Nature 1986;322:70-72.
6. Wang J, Chenivesse X, Henglein B, Brechot C. Hepatitis B virus integra-tion in a cyclin A gene in a hepatocellular carcinoma. Nature 1990;343:555-557.
7. Melegari M, Wolf SK, Schneider RJ. Hepatitis B virus DNA replication iscoordinated by core protein serine phosphorylation and HBx expression.J Virol 2005;79:9810-9820.
8. Reifenberg K, Nusser P, Lohler J, Spindler G, Kuhn C, von Weizsacker F,Kock J. Virus replication and virion export in X-deficient hepatitis B virustransgenic mice. J Gen Virol 2002;83:991-996.
9. Xu Z, Yen TS, Wu L, Madden CR, Tan W, Slagle BL, et al. Enhancementof hepatitis B virus replication by its X protein in transgenic mice. J Virol2002;76:2579-2584.
10. Zhang Z, Torii N, Hu Z, Jacob J, Liang TJ. X-deficient woodchuck hep-atitis virus mutants behave like attenuated viruses and induce protectiveimmunity in vivo. J Clin Invest 2001;108:1523-1531.
11. Ganem D, Schneider R. Hepadnaviridae: the viruses and their replication.In: Knipe DM, Howley PM, eds. Fields Virology, 4th ed. Philadelphia:Lippincott Williams & Wilkins; 2001:2923-2969.
12. Feitelson MA, Reis HM, Liu J, Lian Z, Pan J. Hepatitis B virus X antigen(HBxAg) and cell cycle control in chronic infection and hepatocarcinogen-esis. Front Biosci 2005;10:1558-1572.
13. Dragani TA, Manenti G, Farza H, Della Porta G, Tiollais P, Pourcel C.Transgenic mice containing hepatitis B virus sequences are more suscepti-ble to carcinogen-induced hepatocarcinogenesis. Carcinogenesis 1990;11:953-956.
14. Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as atumor promoter in development of diethylnitrosamine-induced preneo-plastic lesions. J Virol 2001;75:3851-3858.
15. Isogawa M, Furuichi Y, Chisari FV. Oscillating CD8(�) T cell effectorfunctions after antigen recognition in the liver. Immunity 2005;23:53-63.
16. Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis Bvirus replication in transgenic mice. J Virol 1995;69:6158-6169.
17. Xue W, Warshawsky D. Molecular mechanisms of action of selected or-ganic carcinogens. In: Warshawsky D, Landolph JR Jr, eds. MolecularCarcinogenesis and the Molecular Biology of Human Cancer. Boca Raton,FL: CRC Press; 2006:45-77.
18. Yoo JS, Ishizaki H, Yang CS. Roles of cytochrome P450IIE1 in the deal-kylation and denitrosation of N-nitrosodimethylamine and N-nitrosodi-ethylamine in rat liver microsomes. Carcinogenesis 1990;11:2239-2243.
19. Yamazaki H, Inui Y, Yun CH, Guengerich FP, Shimada T. CytochromeP450 2E1 and 2A6 enzymes as major catalysts for metabolic activation ofN-nitrosodialkylamines and tobacco-related nitrosamines in human livermicrosomes. Carcinogenesis 1992;13:1789-1794.
20. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepa-tocyte death to cytokine-driven compensatory proliferation that promoteschemical hepatocarcinogenesis. Cell 2005;121:977-990.
21. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis Bvirus induces liver cancer in transgenic mice. Nature 1991;351:317-320.
22. Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, et al.High-level expression of hepatitis B virus HBx gene and hepatocarcino-genesis in transgenic mice. HEPATOLOGY 1994;19:810-819.
23. Tralhao JG, Roudier J, Morosan S, Giannini C, Tu H, Goulenok C, et al.Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) onliver cell proliferation: an alternative mechanism of HBx-related pathogen-esis. Proc Natl Acad Sci U S A 2002;99:6991-6996.
24. Choo KB, Liew LN, Chong KY, Lu RH, Cheng WT. Transgenome tran-scription and replication in the liver and extrahepatic tissues of a humanhepatitis B virus transgenic mouse. Virology 1991;182:785-792.
25. Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTPcomplex formation and establishes a Ras, Raf, MAP kinase signaling cas-cade. Proc Natl Acad Sci U S A 1994;91:10350-10354.
26. Klein NP, Schneider RJ. Activation of Src family kinases by hepatitis Bvirus HBx protein and coupled signaling to Ras. Mol Cell Biol 1997;17:6427-6436.
27. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus.J Virol 2004;78:12725-12734.
28. Mathonnet G, Lachance S, Alaoui-Jamali M, Drobetsky EA. Expression ofhepatitis B virus X oncoprotein inhibits transcription-coupled nucleotideexcision repair in human cells. Mutat Res 2004;554:305-318.
29. Lee AT, Ren J, Wong ET, Ban KH, Lee LA, Lee CG. The hepatitis B virusX protein sensitizes HepG2 cells to UV light-induced DNA damage. J BiolChem 2005;280:33525-33535.
30. Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx proteinin hepatitis B virus DNA replication. Science 2001;294:2376-2378.
HEPATOLOGY, Vol. 45, No. 1, 2007 ZHENG ET AL. 21




















