
ГОДИШНИК ANNUAL - CiteSeerX
236
ГОДИШНИК НА ШУМЕНСКИЯ УНИВЕРСИТЕТ „ЕПИСКОП КОНСТАНТИН ПРЕСЛАВСКИ” Т. ХIX В 2 ПРИРОДНИ НАУКИ ________________________ ANNUAL OF KONSTANTIN PRESLAVSKY UNIVERSITY OF SHUMEN Vol. XIX B 2 FACULTY OF NATURAL SCIENCES ШУМЕН 2009
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of ГОДИШНИК ANNUAL - CiteSeerX

ГОДИШНИК
НА ШУМЕНСКИЯ УНИВЕРСИТЕТ
„ЕПИСКОП КОНСТАНТИН ПРЕСЛАВСКИ”
Т. ХIX В 2
ПРИРОДНИ НАУКИ
________________________
ANNUAL
OF KONSTANTIN PRESLAVSKY UNIVERSITY OF SHUMEN
Vol. XIX B 2
FACULTY OF NATURAL SCIENCES
ШУМЕН 2009

Отговорен редактор: проф. дхн Валерий Христов
ISSN 1311-834X © Шуменски университет, Факултет по природни науки, 2009 © Университетско издателство “Епископ Константин Пре сла вски”

Съдържание
Ivan Bangov, Marina Moskovkina, Charge-Related Molecular Index and its Usage in Computer Chemistry………………………………...6 Gueorgui Vassilev, Vania Gandova, Parvoletka Docheva, Comparison between Cu-Ni-Zn Liquid Phase Thermodynamic Assessments Performed by the CALPHAD Method and by the General Solution Model……………………………………………..15 Milena Spassova, Benoît Champagne, Valentin Monev, Bernard Kirtman, Structure and Nonlinear Optical Properties of Doped Polyacetylene: A Theoretical Study…………………………………28 Viara Ivanova, Immobilization of Trypsin on Silica Carriers………39 Н. Атанасова, В. Иванова, В. Петкова, А. Тонкова, Основни характеристики на циклодекстрин глюканотрансферази от два алкалофилни щама Bacillus pseudoalkalophilus 20RF и Bacillus pseudoalkalophilus 8SB……………………………………………..49 Диляна Иванова, Жана Ангарска, Емил Манев, Пенни филми от разтвори на n-додецил-β-D-малтозид с додецил триметиамониев бромид……………………………………………............................60 Пантелей Денев, Мина Тодорова, Кремена Никовска, Стамен Стамов, Относно получаването на химически модифициран инулин и използването му в O/W емулсии……………………….71 Георги Д. Нейков, Анастасия А. Ляпова, Галин П. Петров, Tеоретично изследване на влиянието на молекулната геометрия на възбудените състояния на някои азини върху флуоресцентните и фосфоресцентните им свойства…………………………………81 Rossen Pl. Hristov, Electro-optic Investigation of Carboxymethyl Cellulose Adsorption on Alumina Nanoscale Particles……………..90 Жана Ангарска, Красимир Тачев, Влияние на Дауфакс върху изтъняването и критичната дебелина на пенни филми, стабилизирани от смес на Динакол и Солвитоза………………...99 Радослав Иванов, Изследване влиянието на вида и количеството на легиращата добавка в изходната алумотермична смес върху някои показатели на получените боридни слоеве………………110

Марина Московкина, Иван Бангов, Сравнителен анализ за възможностите и особеностите на моделиране на хроматографско задържане на пиразини с помощта на молекулни дескриптори………………….........................................................119 S. Ivanova, G. Vasileva, D. Aleksiev, Synthesis and Characterization of some Novel Unsaturated Ketones……………………………….129 Галин П. Петров, Анастасия А. Ляпова, Деница Ц. Борисова, Получаване на нови тетраметилпиперидиноксилови производни на някои природни оксо- и карбоксисъединения……………….134 И. Иванов, Ст. Николова, Е. Кочовска, Ст. Статкова-Абегхе, Синтез на 1,1-дизаместени 1,2,3,4-тетрахидроизохинолини…..143 М. Кашчиева, М. Маринов, Н. Стоянов, В. Матева, Синтез на комплексни съединения на непротеинови аминокарбоксилни киселини с Сu(II).............................................................................151 Маргарита Кашчиева, Нейко Стоянов, Синтез и комплексообразувателна способност на 5-карбоксиметил-2,3-дихидро-1,3-диоксо-2-(2-пиридил)-1Н-инден..............................158 Yolina Hubenova, Mario Mitov, Comparison of Candida melibiosica and Saccharomyces cerevisiae Performance in Experimental Biofuel Cell…………………………………………………………………164 Yolina Hubenova, Marin Marinov, Ognyan Argirov, Investigation of the Biological Activity of Glycation Analogous Product 3-Hydroxy-1-methylpyridinium Chloride on Microorganisms…….......................171 V. B. Konsulov, A. A. Lyapova, G. P. Petrov, P. Saha, Synthesis of Amphiphilic Alternating Copolymers of the Maleic Acid, Containing Nitroxide Radicals………………………………………………….176 V. B. Konsulov, A. A. Lyapova, Z. S. Grozeva, J. I. Tacheva, Synthesis and Characterizations of N-substituted Maleimides….....186 В. Грозев, М. Иванова, З. Грозева, П. Миндова, А. Патлеева, Й. Тачева, Изследване влиянието на антипенители и противоутаители върху някои характеристики на акрилатна вододисперсионна боя……………………………………………195 М. Стоянова, В. Грозев, Проблеми на контрола и управлението на химични вещества в училищата……………............................205 Светлана Христова, Борис Атанасов, Тестване на програми за белтъчна електростатика върху база данни за НМФК................210

Stefan Slavov, Surface Modification of γ-Alumina with Chlorotrimethylsilane. 1. Solid and Gas Products of Silation...........216 Stefan Slavov, Surface Modification of γ-Alumina with Chlorotrimethylsilane. 2. Mechanism of Silation.............................226

Charge-Related Molecular Indices and their Usage in Computer Chemistry
Ivan Bangov,* Marina Moskovkina Konstantin Preslavsky University of Shumen
115 Universitetska Str., Shumen 9712, Bulgaria E-Mail: [email protected]
Abstract: The charge-related molecular indices developed by one of the authors (IB) and their use in chemoinformatics has been discussed. It was shown that they can be successfully employed to isomorphic structure perception, correlations with both structure branching and octane numbers, enthalpies of formation, etc., in 13C NMR chemical shift predictions, and structural fingerprint formation. It was shown that they appear very good descriptors of the polar interactions in the cases of generation of quantitative structure chromatographic retention relationship (QSRR) models. Keywords: CTI, molecular index, isomorphic structures, structure branching, octane number, enthalpy of formation, fingerprint formation, QSSR
Introduction Molecular indices are widely used in modern computer
chemistry and chemoinformatics. They represent numbers characterizing molecular structure. The transformation of the molecular structure into a molecular index is shown in Figure 1. However, the opposite procedure – extraction of the structure from any molecular index is impossible as these numbers do not possess the connectivity information in an explicit manner.
The charge-related topological index and its usage
A charge-related molecular index (CRI) was developed by one of us (I.B.) in the 80s.[1,2] It has the following form:
(1) i j
i j ij
L LCRI
D= ∑∑

Figure 1. Molecular index (CTI in our case) formation of a chemical structure. The structure to index transformation is straightforward, the
opposite transformation from index to structure is not possible.
Here Dij are the inter-atomic distances and Li are local indices featuring each one heavy (non-hydrogen) atom i given as follows: Li=Lo–NH + qi (2)
Lo are constant values for each atom and for each hybridization state (it can be the atom valence in some cases), NH is the number of hydrogen atoms attached to a given heavy atom, and qi are the corresponding charge densities.
Initially, CTI (Charge-related Topological Index) was developed for perception of isomorphic (equivalent) complete molecular structures and substructures (fragments) in the process of 2D structure generation. It manifested extremely good discriminating power and appeared practically an index of no degeneracy. Thus, equivalent (isomorphic) structures produce the same CTI values, and different structures – different values.
The discriminating potentials of this index have been depicted in Figure 2.
Additionally the Li values have been used to the perception of the constitutional molecular structure symmetry (automorphism). Thus, symmetric atoms have the same Li values.
In Table 1 the calculated CTI values for a series of lower alkanes are presented [2]. One can see that the value of the CTI strongly depends on the size n of the molecule on the one hand. On the other hand by inspecting the different isomers of n= 4, 5, 6, and 7
CTI=26.354890123

having the same size we can figure out that the most branched isomers produce higher CTI values. This is better illustrated on the example of the octane isomers the results are compared in Table 2 with those of Bertz [3].
Figure 2. a) Two isomers of different connectivity providing the same Wiener index values W but different CTI and ASIIg (an older form of CTI) values. b) two isomers of different connectivity providing the
same Randic index values χ but different CTI and ASIIg values.
Table 1. Calculated CTI values for a series of lower alkanes. ------------------------------------------------------------------------------------- n Alkane CTI ------------------------------------------------------------------------------------- 2 ethane 0.8682 3 propane 4.0641 4 n-butane 9.5193 4 2-methylpropane 9.6103 5 n-pentane 16.3392 5 2-methylbutane 17.3298 5 2,2-dimethylpropane 17.5292 6 n-hexane 24.1502

6 2-methylpentane 25.5174 6 3-methylpentane 26.4147 6 2,3-dimethylbutane 27.4097 6 2,2-dimethylbutane 27.5181 7 n-heptane 32.7421 7 2-methylhexane 34.3215 7 3-methylhexane 35.5936 7 2,4-dimethylpentane 36.0662 7 3-ethylpentane 36.8652 7 2,2-dimethylpentane 37.0763 7 2,3-dimethylpentane 37.8630 7 3,3-dimethylpentane 38.8731 7 2,2,3-trimethylbutane 39.8726 -------------------------------------------------------------------------------------
The results of Table 2 prove that CTI correlates very well with the structure branching [2]. Although the notion of branching is not well defined, one can see that it provides much more reasonable results than the ranking of Bertz.[3]
Table 2. Charge-related topological index (CTI) calculated for the series of octanes the order of different isomers, according to the
branching index of Bertz is given in brackets. ------------------------------------------------------------------------------------- compound CTI ________________________________________________________
1. octane (1) 41.9796 2. 2-methylheptane (2) 43.6962 3. 3-methylheptane (4) 45.1788 4. 2,5-dimethylhexane (3) 45.4882 5. 4-methylhepthane (5) 45.5535 6. 2,2-dimethylhexane (12) 46.8757 7. 3-ethylhexane (7) 47.0352 8. 2,4-dimethylhexane (6) 47.1357 9. 2,3-dimethylhexane (8) 48.0352 10. 2,2,4-thrimethylpentane (13) 48.9985 11. 3,4-dimethylhexane (9) 49.3077
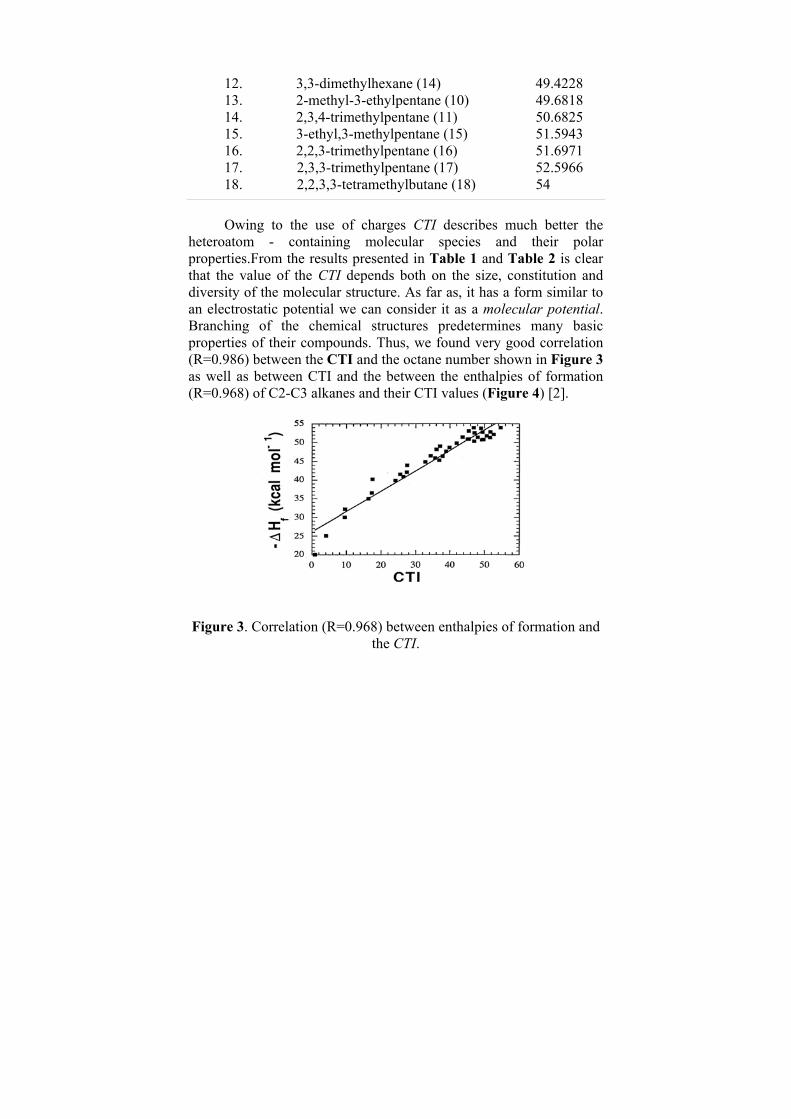
12. 3,3-dimethylhexane (14) 49.4228 13. 2-methyl-3-ethylpentane (10) 49.6818 14. 2,3,4-trimethylpentane (11) 50.6825 15. 3-ethyl,3-methylpentane (15) 51.5943 16. 2,2,3-trimethylpentane (16) 51.6971 17. 2,3,3-trimethylpentane (17) 52.5966 18. 2,2,3,3-tetramethylbutane (18) 54
Owing to the use of charges CTI describes much better the
heteroatom - containing molecular species and their polar properties.From the results presented in Table 1 and Table 2 is clear that the value of the CTI depends both on the size, constitution and diversity of the molecular structure. As far as, it has a form similar to an electrostatic potential we can consider it as a molecular potential. Branching of the chemical structures predetermines many basic properties of their compounds. Thus, we found very good correlation (R=0.986) between the CTI and the octane number shown in Figure 3 as well as between CTI and the between the enthalpies of formation (R=0.968) of C2-C3 alkanes and their CTI values (Figure 4) [2].
Figure 3. Correlation (R=0.968) between enthalpies of formation and
the CTI.
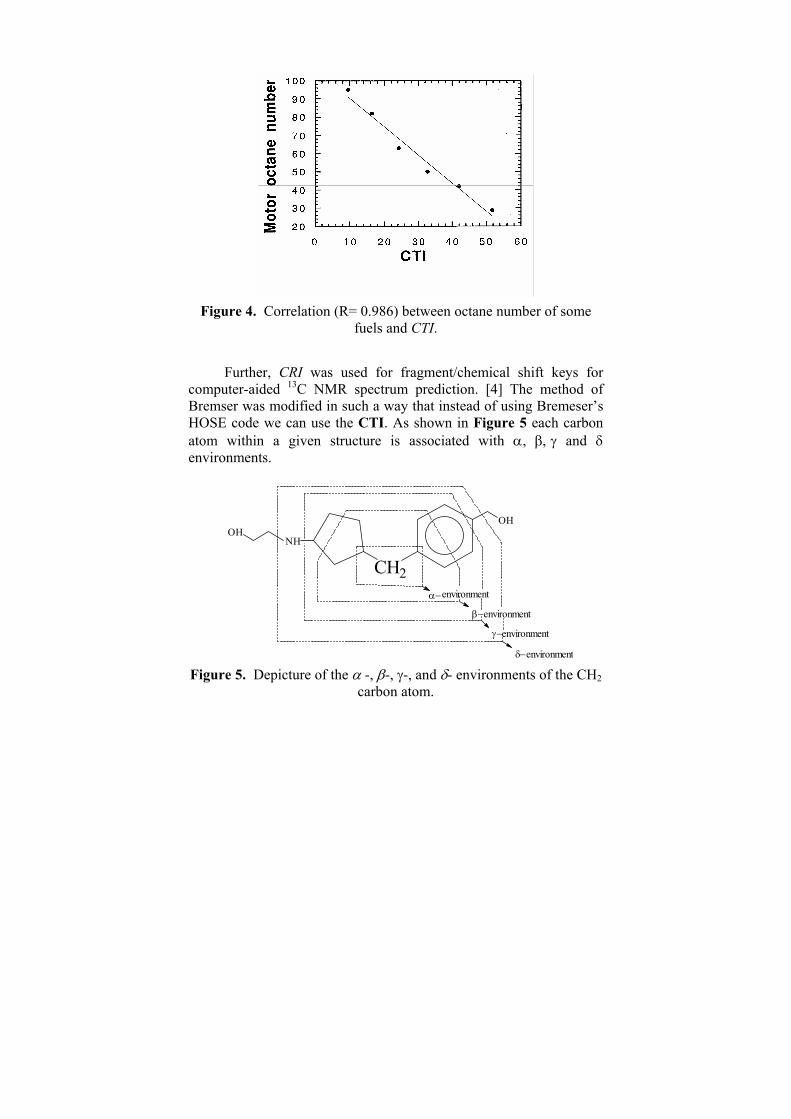
Figure 4. Correlation (R= 0.986) between octane number of some
fuels and CTI.
Further, CRI was used for fragment/chemical shift keys for computer-aided 13C NMR spectrum prediction. [4] The method of Bremser was modified in such a way that instead of using Bremeser’s HOSE code we can use the CTI. As shown in Figure 5 each carbon atom within a given structure is associated with α, β, γ and δ environments.
Figure 5. Depicture of the α -, β-, γ-, and δ- environments of the CH2
carbon atom.
environmentα−
CH2
OH
NH
β−environment
γ−environment
OH
δ−environment

These environments are practically fragments and we can assign
a CTI value to each one of them - α -(CTIα), β- (CTIβ), γ- (CTI γ), и δ- (CTIδ) and related to them 13C chemical shifts and coupling constants. We can use them either in a relational base, or we can form fingerprints similar to these of Daylight and assign to each 1 in the fingerprint both fragment and spectral characteristics.
A charge related agreement factor (CAF) was developed by one of the authors (IB) [5,6]. It is based on the assumption that there exist a linear relationship between 13C NMR chemical shifts and charge densities of the corresponding carbon atoms:
13C CAq Bδ = = (3)
By using the Hamilton Agreement Factor we form the Charge-related Hamilton Agreement Factor, which has the form: (4)
This factor can be used also as a charge-related index. In the same way the CTI can be employed as fragment keys in
structural fingerprints. The α −, β−, γ−, and δ− environments given in Figure 5, can be such fragments. Accordingly, the CTI is calculated for each α −, β−, γ−, and δ− environment and a hashing procedure similar to that of Daylight [7] can be applied.
In a series of papers of the authors the potentials of the CRI as a descriptor in the Quantitative Structure/Retention Relationships (QSRR) in modeling different types of chromatographic behavior have been investigated. The modern state of art in the QSRR approach gives the possibility to generate multivariable regression equations, able to reflect the chromatographic retention behavior for different solute series. The models derived usually include a set of numeric molecular indices to quantitate nonspecific (dispersive) and specific (polar) interactions between the solutes and chromatographic phases.
exp . 2 exp 213 13 13
exp2 exp213 13
( ) ( )calcC C C C
C C
Aq BHAF
δ δ δδ δ− − −
= =∑ ∑∑ ∑

Finely the chromatographic retention data is presented in terms of chemo informatics in the form ofmultivariable linear regression: RI = (5)
Where RI – experimental retention data; Pi and Di are the molecular descriptors of polar and dissuasive interactions; a , b and c are constants reflecting the chromatographic phase properties. Whereas, the quantitation of the non-specific dispersive molecular interactions can be successfully carried out by using some global topological indices, the various specific polar interactions are quantitated less precisely by numeric local molecular indices.
However in cases of polar interactions the CTI prove to give much superior results as shown in Figure 6 for the case of substituted phenols (a generalization based on 90 models, generated for retention of phenols in the SE-30 phase, 138 models on the OV-255 phase and 139 models on the NGA phase) [8] and in Figure 7 for the case of 1,4 benzodiazepines (BDZ).[9]
Figure 6. Statistical features (R) of the regression equations of
substituted phenols in Gas Chromatography.
Заместени фенолиГХ
0.930.940.950.960.970.980.99
1
M MR a V S CTI D
R
SE-30
OV-225NGA
i i i ia D b P c+ +∑ ∑

Figure 7. Statistical features (R) of the regression equations of 1,4
benzodiazepines (BDZ).
One can see from both Figure 6 and Figure 7 that the use of CTI provides the best correlation results.
References 1. I. Bangov, J. Chem. Inf. Coput. Sci., 1990, 30, 277-289. 2. P. .A. Demirev, A. S. Dyulgerov, I. P. Bangov, J. Math. Chem.,
1991, 8, 367-382. 3. St. Bertz, Discr. Appl. Math. 1988, 19, 65-72. 4. I. Bangov, Annuaire Uni. Sofia, 2001, 91, 103-113. 5. I. Bangov, Org. Magn. Reson., 1981, 16(4), 296-303. 6. I. Bangov, Anal. Chim. Acta, 1988, 209, 29-43. 7. http://www.daylight.com/dayhtml/doc/theory/theory.finger. html 8. M. Moskovkiona, I. Bangov, Annual of Konstantin Preslavsky
University, Shumen, 2007, XVII B 1, 189-197. 9. M. Moskovkina, Thesis, 2004, 43-54.
ГХБДЗ
0.85
0.86
0.87
0.88
0.89
0.9
0.91
0.92
0.93
0.94
0.95
K'H I'H ZZinf
o
Randich
Bal D2 W
Winfo
GWGWinfo
Lmax MR M
Vpol
CTI
CTI+GW
CTI+Lmax D
R

Comparison between Cu–Ni–Zn Liquid Phase Thermodynamic Assessments Performed by the CALPHAD Method and by the General Solution
Model Gueorgui Vassilev,a* Vania Gandova,a Parvoletka Dochevab
aUniversity of Plovdiv, Faculty of Chemistry, 24 Tsar Asen str., 4000
Plovdiv, Bulgaria bUniversity of Rousse, Dept. Materials Science, Rousse, Bulgaria
E-mail: [email protected], [email protected]
Abstract: Estimation of the thermodynamic properties of the Cu–Ni–Zn systems has been done by means of the general solution model of Chou. The calculations have been performed in a wide temperature range (1000–2000 K). Ternary interaction parameters for the liquid phase Gibbs excess energy calculation have been determined using thermodynamic data of the binary end–systems (Ni–Zn, Cu–Zn, Cu–Ni). Comparison between calculated results and available literature data was done. Good agreement of the present assessment with thermodynamically optimized values of the system Cu–Ni–Zn (obtained by the CALPHAD approach) was observed. The validity of the general solution model was confirmed, thus the hypothesis that it could be used in cases where no experimental data are avalable was verified. Keywords: thermodynamics, general solution model, ternary interaction parameters, Cu–Ni–Zn, ternary system, CALPHAD method.
Introduction
The Cu–Ni–Zn system is important for the non-ferrous metallurgy [1], while recently is interesting as potential lead-free solder material [2] as well. Few thermodynamic data have been reported for the Cu–Ni–Zn system [1–5]. For example, Chadwick and Argent [3] have done some vapor pressure zinc

activity measurement in the α-field of the Cu-rich region at 1000 K. Using the same technique, Sugino and Hagiwara [4] determined the zinc activities in dilute Cu–Ni–Zn melt, at 1373 and 1423 K. Zinc isoactivity curves at 1048K were evaluated and calculated by Sisson and Dayananda [5]. Recently, Vassilev [6] investigated the phase boundaries in the zinc-rich corners of the Cu–Ni–Zn at 868 K. Intermetallic compounds non-related to the binary systems have not been found but these authors have revealed that the ternary extension of the Cu–Zn ε-phase (around 80–85 at. % Zn) contains up to 4 at. % Ni. The phase boundaries of the zinc-rich Cu-Ni-Zn liquid and γ-phases have been plotted [6].
Miettinen [7] optimized the thermodynamic parameters of the Cu–Ni–Zn system, at zinc contents up to 70 wt. % and at temperatures above 873 K, using experimental thermodynamic and phase equilibrium data. The most recent thermodynamic calculation of the phase equilibria in the Cu–Ni–Zn system is the work of Jiang et al. [1]. In both works, the thermodynamic description of the Ni–Zn system done by Vassilev [8,9] was retained.
The task of the present study was to verify different ways to achieve thermodynamic description of a ternary system (in this case, the system Cu–Ni–Zn) by assessing the thermodynamic properties of the liquid phase using the general solution model (GSM) and comparing to values, obtained by the CALPHAD approach.
Theoretical fundamentals
In order to remind some general notions about the construction of the binary end-systems (Cu–Ni, Cu–Zn, Ni–Zn) they are reproduced in the following three figures (Figs. 1–3, respectively). As one can see the Cu–Ni system [10] is relatively simple while Cu–Zn [11,12] and Ni–Zn [8,9] phase diagrams have a large number of intermediate phases.
Chou [13,14] has developed a method called general solution model (GSM) for calculation of a ternary phase thermodynamic properties. The application of this model requires some

thermochemical information about the three binary end-systems. Such information is essentially known if the composition and temperature dependence of the Gibbs molar excess energy of a phase is available. In the equations below, the following excess energies are represented: ΔGE – Gibbs molar excess energy of the ternary liquid phase; ΔGE
ij, ΔGE
jk, ΔGEki or ΔGE
ij – the respective Gibbs molar excess energy values of the binary liquid phases, where the down indexes indicate the considered binary system. The basic equations of the GSM [13,14] are given as follows:
ΔGE = xixjΔGEij + xjk ΔGE
jk + xkxi ΔGEki + xixjxk fijk (1)
Here, the symbols xi, xj, xk correspond to the mole fractions of the relevant ternary system constituents (i.e Cu, Ni, Zn) and fijk is the ternary interaction coefficient. Further, the composition dependence of the binary Gibbs molar excess energies is given: ΔGE
ij = XiXj (Aoij +A1
ij(Xi – Xj) +A2ij(Xi –Xj)2 + ... +An
ij (Xi –Xj)2) (2) Here, Ao
ij, A1ij, A2
ij are parameters (independent of composition, only relying on temperature, being analogue of the Redlich-Kister parameters) belonging to the binary system "ij"; Xi and Xj indicate the mole fractions of the corresponding constituents.
For the sake ternary Gibbs molar excess energy estimation fijk ought to be assessed. According to CSM [13.14] this coefficient is expressed [13] as: fijk = (2ξij –1){A2
ij ((2ξij –1)xk + 2(xi–xj)) + A1ij} + (2ξjk–1){A2
jk ((2ξjk–1)xi +2(xj–xk)) +A1
jk} + (2ξki–1){A2ki((2ξki–1)xj + 2(xk–xi)) + A1
ki} (3) Here ξij are similarity coefficients, defined by ηi - called
“deviation sum of squares”: ξij = ηi / (ηi + ηj) (4)
where:
iEik
EijI dXGG 2
1
0
)( Δ−Δ= ∫η ; jEjk
EjiII dXGG 2
1
0
)( Δ−Δ=η ∫ ;
kEkj
EkiIII dXGG 2
1
0
)( Δ−Δ= ∫η (5)
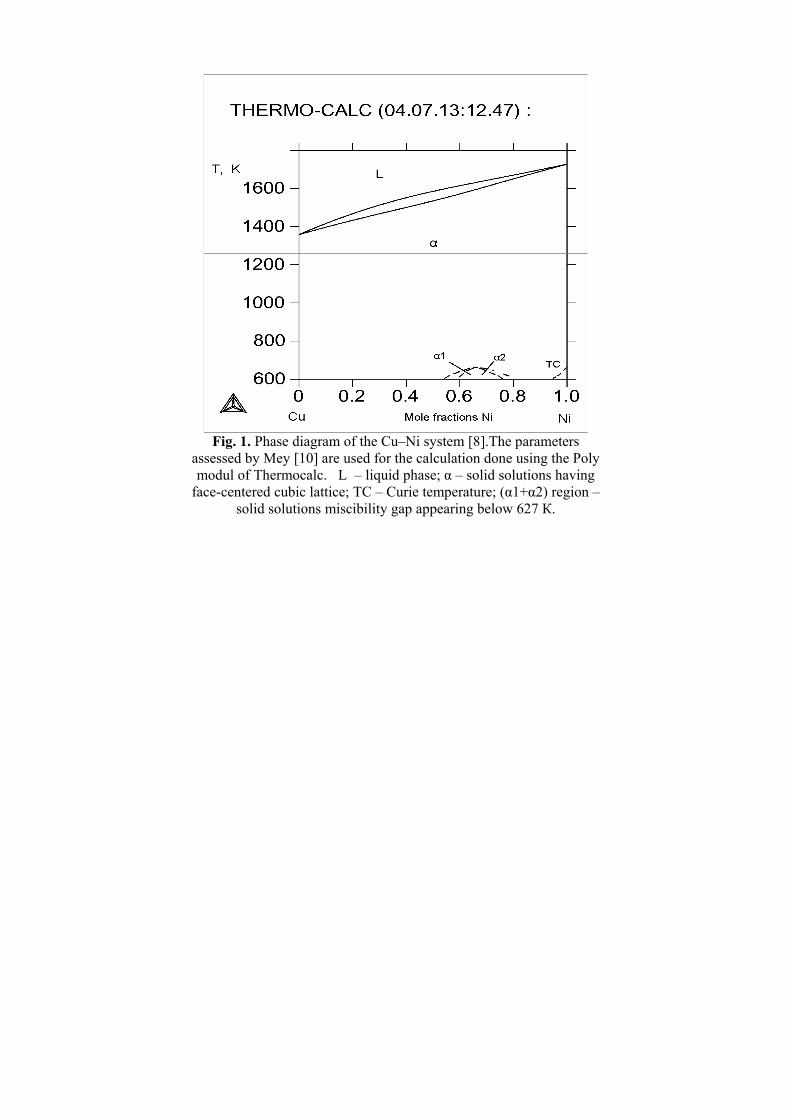
Fig. 1. Phase diagram of the Cu–Ni system [8].The parameters
assessed by Mey [10] are used for the calculation done using the Poly modul of Thermocalc. L – liquid phase; α – solid solutions having
face-centered cubic lattice; TC – Curie temperature; (α1+α2) region – solid solutions miscibility gap appearing below 627 К.
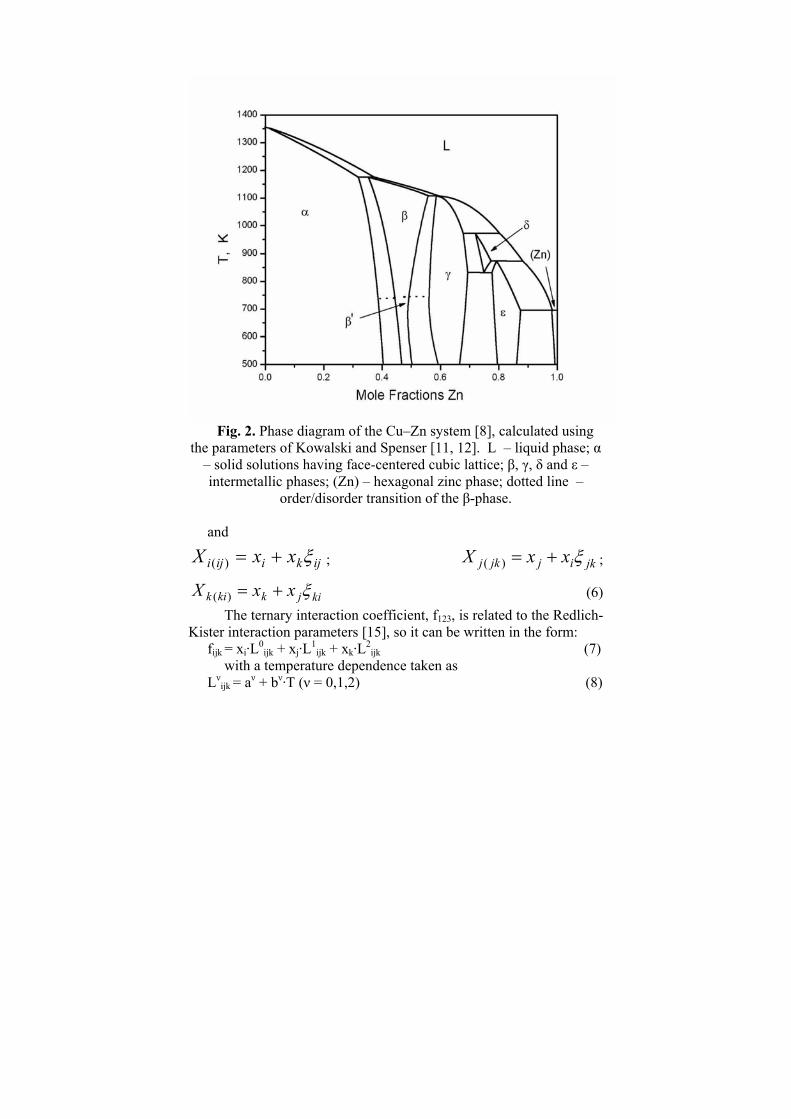
Fig. 2. Phase diagram of the Cu–Zn system [8], calculated using
the parameters of Kowalski and Spenser [11, 12]. L – liquid phase; α – solid solutions having face-centered cubic lattice; β, γ, δ and ε – intermetallic phases; (Zn) – hexagonal zinc phase; dotted line –
order/disorder transition of the β-phase. and
ijkiiji xxX ξ+=)( ; jkijjkj xxX ξ+=)( ;
kijkkik xxX ξ+=)( (6) The ternary interaction coefficient, f123, is related to the Redlich-
Kister interaction parameters [15], so it can be written in the form: fijk = xi·L0
ijk + xj·L1ijk + xk·L2
ijk (7) with a temperature dependence taken as
Lνijk = aν + bν·T (ν = 0,1,2) (8)
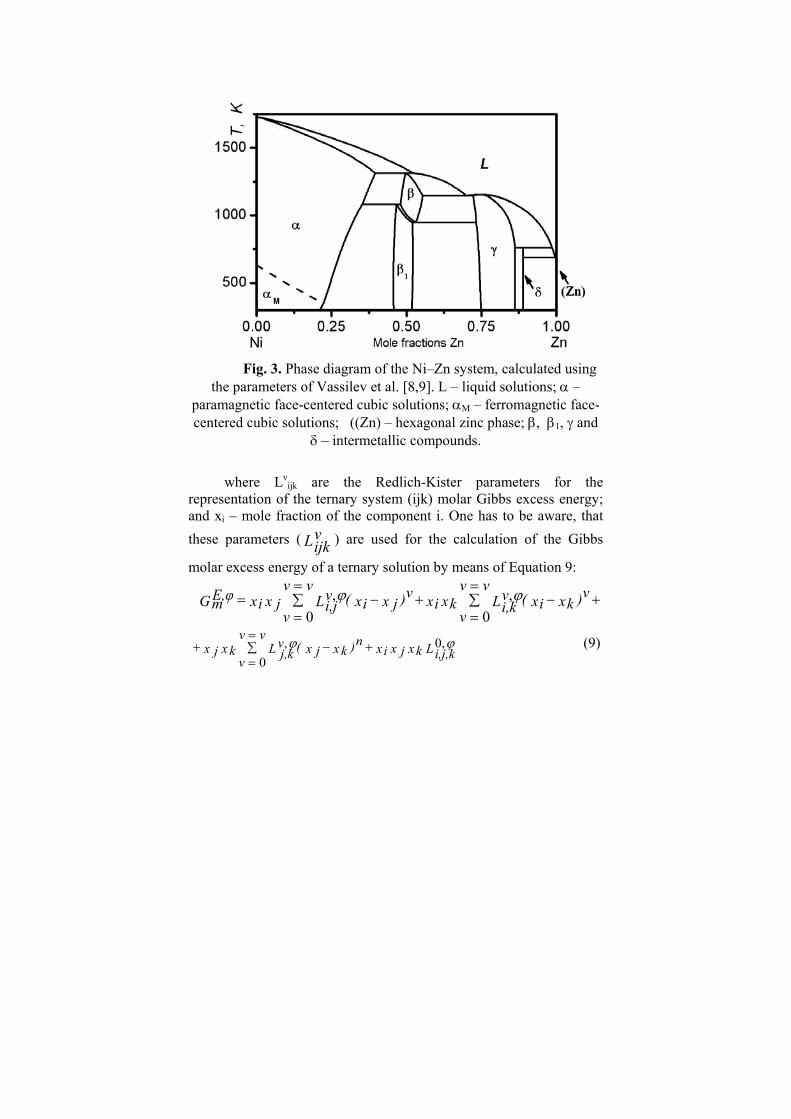
Fig. 3. Phase diagram of the Ni–Zn system, calculated using
the parameters of Vassilev et al. [8,9]. L – liquid solutions; α – paramagnetic face-centered cubic solutions; αM – ferromagnetic face-centered cubic solutions; ((Zn) – hexagonal zinc phase; β, β1, γ and
δ – intermetallic compounds.
where Lν
ijk are the Redlich-Kister parameters for the representation of the ternary system (ijk) molar Gibbs excess energy; and xi – mole fraction of the component i. One has to be aware, that these parameters ( Lνijk ) are used for the calculation of the Gibbs
molar excess energy of a ternary solution by means of Equation 9:
+−∑=
=+−∑
=
== ν)xkxi(
νν
νLνi,kxkxi
ν)x jxi(νν
νLνi,jx jxiG E,φm
0,
0, ϕϕ
Li,j,kx kx jxin)x kx j(
νν
νLνj,kx kx j ϕϕ ,0
0, +−∑
=
=+ (9)

The first three terms represent the contributions of the binary systems and the third – the ternary interactions contributions (in the case when the ternary solution is regular).
Results and Discussion Basic thermodynamic information on the constitutive
subsystems, needed for the assessment, was taken from Refs. [1,7–12,16]. Actually, the optimized Redlich-Kister parameters of each system (Table 1) are essentially needed. They are used for the calculation (see Equation 2) of the molar excess Gibbs energies of the binary end-systems liquid phases.
Table 1. Optimized literature data (Redlich-Kister parameters (Lνij) of
the Cu–Ni [7], Ni–Zn [9] and Cu–Zn [7] systems, ν=0, 1 or 2) of the liquid binary phases, used for the GSM calculation of the molar excess
Gibbs energies, J/mol; T – temperature, K.
System ij
Loij (T) L1
ij (T) L2ij (T)
Cu–Ni +11760+1.084T –1672 0 Ni–Zn –50721.64
+7.34178T +8436.3+1.972T –25136.08
+11.79211T Cu–Zn –40696+12.653T +4403–6.554T +7818–3.254T
For the sake of the assessment, ternary Cu–Ni–Zn alloys, with
copper molar fractions equal to 0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1, were considered along sections with Ni/Zn molar ratio equal to 1/3, 1/1 and 3/1. The calculations were done in the temperature interval from 1000 to 2000 K. The values of the ternary integral Gibbs excess energies, ΔGE, and the ternary interaction coefficients, fijk, have been calculated according to the GSM (Equations 1, 9 and 3) using the parameters given in Table 1.
In Fig. 4a–c, are presented the integral ternary molar Gibbs excess energies (DGxs, J/mol) as calculated along the selected sections and at the retained temperatures.
In order to verify the reliability of the GSM a comparison between the molar Gibbs excess energies, calculated with ternary interaction
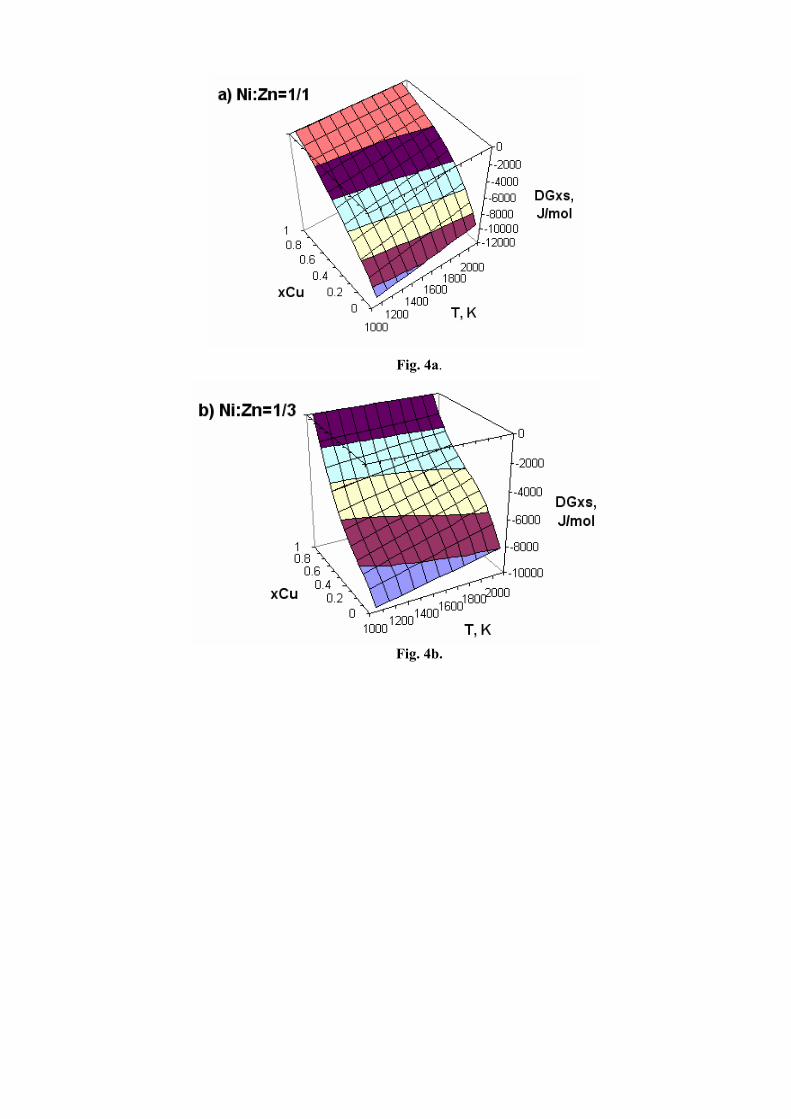
Fig. 4a.
Fig. 4b.
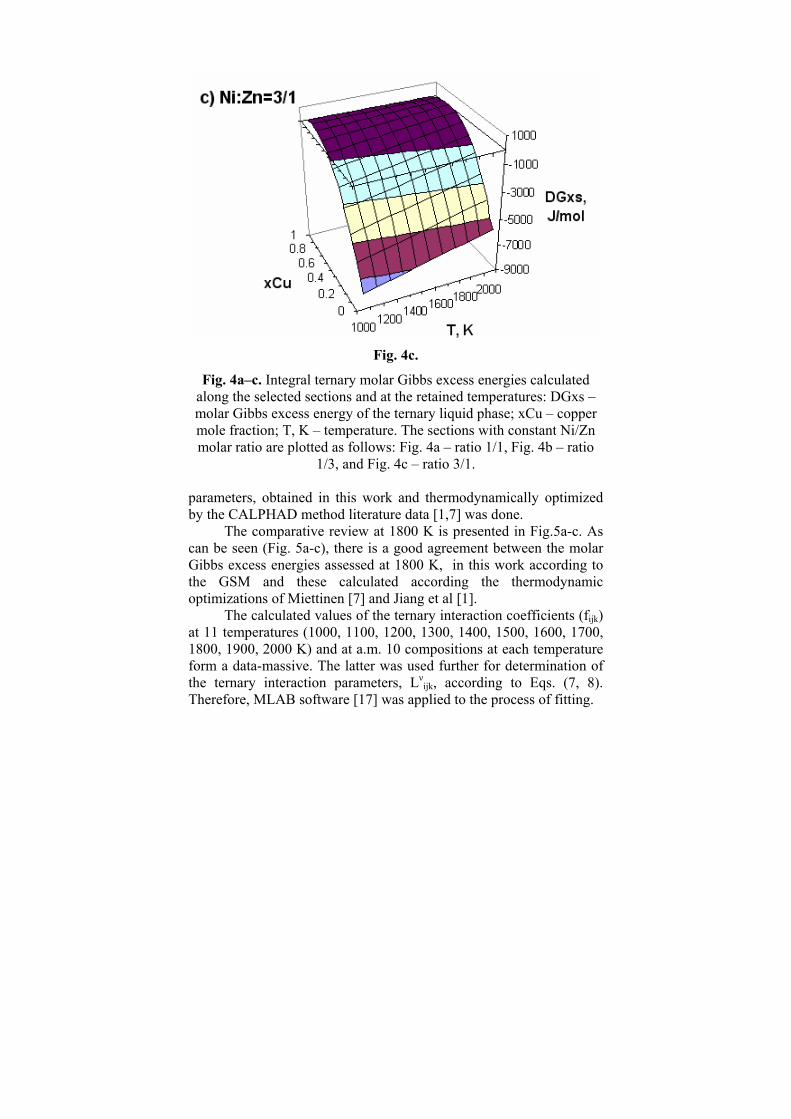
Fig. 4c.
Fig. 4a–c. Integral ternary molar Gibbs excess energies calculated along the selected sections and at the retained temperatures: DGxs – molar Gibbs excess energy of the ternary liquid phase; xCu – copper mole fraction; T, K – temperature. The sections with constant Ni/Zn molar ratio are plotted as follows: Fig. 4a – ratio 1/1, Fig. 4b – ratio
1/3, and Fig. 4c – ratio 3/1.
parameters, obtained in this work and thermodynamically optimized by the CALPHAD method literature data [1,7] was done.
The comparative review at 1800 K is presented in Fig.5a-c. As can be seen (Fig. 5a-c), there is a good agreement between the molar Gibbs excess energies assessed at 1800 K, in this work according to the GSM and these calculated according the thermodynamic optimizations of Miettinen [7] and Jiang et al [1].
The calculated values of the ternary interaction coefficients (fijk) at 11 temperatures (1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000 K) and at a.m. 10 compositions at each temperature form a data-massive. The latter was used further for determination of the ternary interaction parameters, Lν
ijk, according to Eqs. (7, 8). Therefore, MLAB software [17] was applied to the process of fitting.
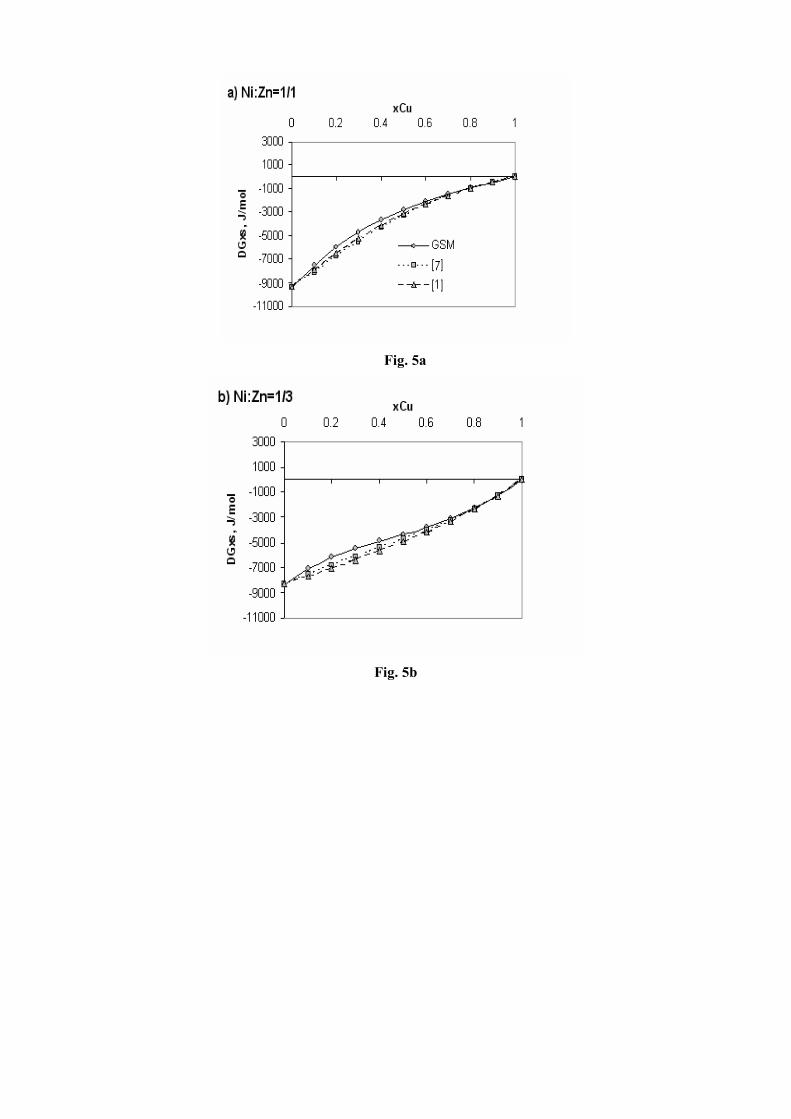
Fig. 5a
Fig. 5b
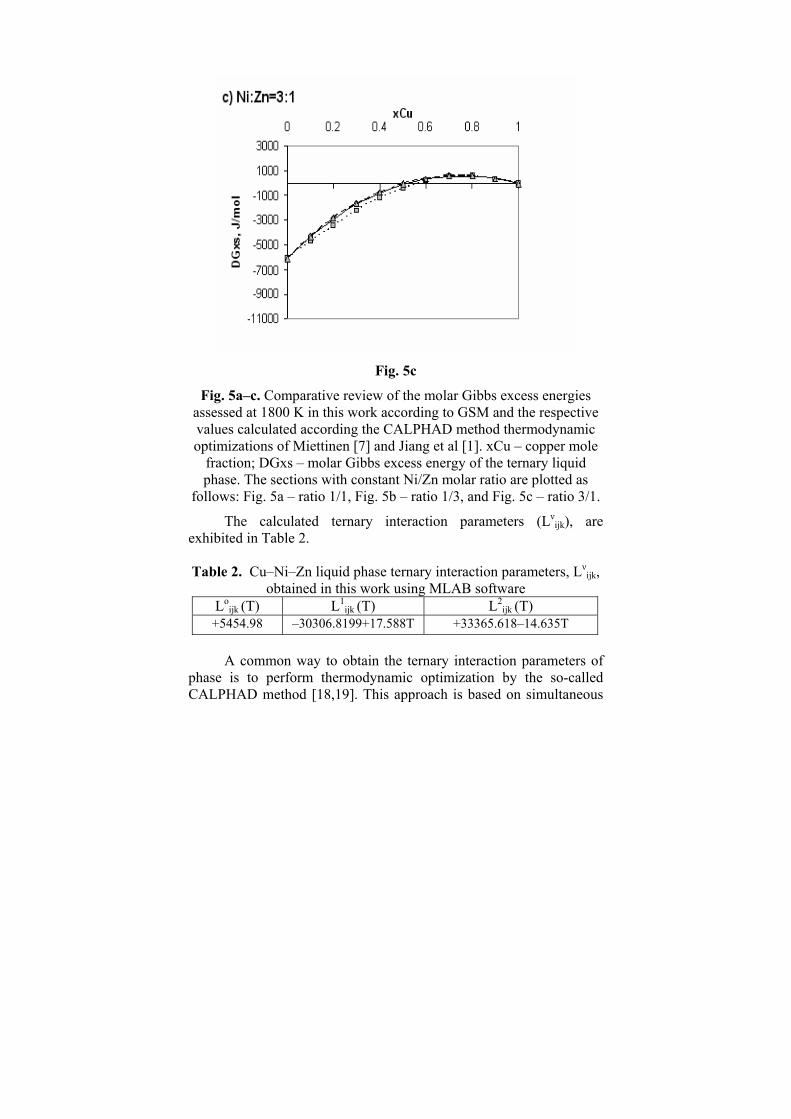
Fig. 5c
Fig. 5a–c. Comparative review of the molar Gibbs excess energies assessed at 1800 K in this work according to GSM and the respective values calculated according the CALPHAD method thermodynamic optimizations of Miettinen [7] and Jiang et al [1]. xCu – copper mole
fraction; DGxs – molar Gibbs excess energy of the ternary liquid phase. The sections with constant Ni/Zn molar ratio are plotted as
follows: Fig. 5a – ratio 1/1, Fig. 5b – ratio 1/3, and Fig. 5c – ratio 3/1.
The calculated ternary interaction parameters (Lνijk), are
exhibited in Table 2. Table 2. Cu–Ni–Zn liquid phase ternary interaction parameters, Lν
ijk, obtained in this work using MLAB software
Loijk (T) L1
ijk (T) L2ijk (T)
+5454.98 –30306.8199+17.588T +33365.618–14.635T
A common way to obtain the ternary interaction parameters of phase is to perform thermodynamic optimization by the so-called CALPHAD method [18,19]. This approach is based on simultaneous

use of experimental thermochemical and phase equilibria data. Anyhow, the calculation procedure, given here, showed that the GSM could also be helpful. Its advantage is that there is no need of experimental data about the ternary system. We have to acknowledge that procedures similar to that used in this work were presented recently in the literature [21,22].
Conclusions Successful thermodynamic properties calculations of the Cu–
Ni–Zn liquid phase were done using the general solution model developed by Chou. For this purpose, the ternary interaction coefficients were obtained at eleven temperatures and at three constant Ni/Zn ratios. Thereafter, Redlich-Kister ternary interaction parameters were assessed in the temperature interval from 1000 to 2000 K.
The values of the ternary liquid phase molar Gibbs excess energies calculated by GSM have been compared to thermodynamically optimized values. Good mutual agreement was found indicating that such an approach is possible in systems where no experimental data are available. The results may serve for further thermodynamic optimization of the Cu-Ni-Zn system by CALPHAD method. Acknowledgments: Thanks are due to Prof. D. Živković (University of Belgrade, Technical faculty of Bor) for very helpful discussions.
References 1. M. Jiang, C.P. Wang, X. J. Liu, I. Ohnuma, R. Kainuma, G. P.
Vassilev, K. Ishida, J. Physics and Chemistry of Solids (2005) 66, 246–250.
1. A. Kroupa, A. Dinsdale, A. Watson, J. Vrestal, J. Vizdal, A. Zemanova, JOM, Journal of the Minerals, Metals and Materials Society, (2007) 59(7), 20–25; see also http://mtdata.software.googlepages.com/soldersdatabase.htm, or A. Dinsdale, A. Watson, A. Kroupa, J. Vrestal, A. Zemanova and J. Vizdal, COST Action 531 - Atlas of Lead free soldering, COST office 2008, Brussels, ISBN 978-80-86292-28-1.

2. G. A. Chadwick, B. B. Argent, Trans. Faraday Soc. (1961) 57, 619–624.
3. S. Sugino, H. Hagiwara, J. Japan Inst. Metals (1986) 50, 1068–1074.
4. R. D. Sisson, M. A. Dayananda, Met. Trans. A (1977) 8A, 1849–1855.
5. G. P. Vassilev, Arch. Metall. and Materials (2004) 49, 903–915. 6. J. Miettinen, CALPHAD, (2003) 27, 263–269. 7. G. P. Vassilev, D. Sc. Thesis, Фазови диаграми на системи на
основа на елементи от четвърти (Ti, Co, Ni, Cu, Zn, Se) и пети (Ag, In, Sn) периоди, СНС по неорганична и аналитична химия, Sofia, 2006.
8. G. Vassilev, T.G. Acebo, J. -C. Tedenac, J. Phase Equibria, (2000) 21, 287–301.
9. S. Mey, CALPHAD, (1992) 16, 255–260. 10. M. Kowalski, P. Spencer, J. Phase Equil., (1993) 14, 432–438. 11. COST 507, Editors I. Ansara, A. T. Dinsdale, M. H. Rand,
Thermochemical database for light metal alloys, Vol. 2, European Communities, Brussels, 1998.
12. K. C. Chou, CALPHAD, (1995) 19, 315–25. 13. K. C. Chou, W. C. Li, F. Li, M. He, CALPHAD, (1996) 20, 395–
406 14. O. Redlich, A. T. Kister, Ind. Eng. Chem., (1948) 24, 345–353. 15. G. P. Vassilev, J. Phase Equil. and Diffusion, (2005) 26, 309–310. 16. MLAB Mathematical Modelling System (www.civilized.com). 17. L. Kaufman, H. Bernstein, Computer Calculation of Phase
Diagrams, Acad. Press, N. Y., 1970. 18. B. Sundman, B. Jansson and J. O. Andersson, CALPHAD, (1985)
9, 153–190. 19. D. Živković, I. Mihajlović, Ž. Živković, Therm. Acta, (2004) 417,
119–125. 20. D. Živković, D. Manasijević, I. Mihajlović, Ž. Živković, J. Serb.
Chem. Soc. (2006) 71, 203–211.

Structure and Nonlinear Optical Properties of Doped Polyacetylene: A Theoretical Study
Milena Spassova,a,* Benoît Champagne,b Valentin Monev,a Bernard Kirtmanc
aInstitute of Organic Chemistry with Centre of Phytochemisty, Bulgarian Academy of Sciences, Sofia 1113, BULGARIA
E-mail: [email protected] bLaboratoire de Chimie Théorique Appliquée, Facultés Universitaires
Notre-Dame de la Paix, rue de Bruxelles, 61, B-5000 Namur, BELGIUM
cDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, CA. 93106, USA
Abstract: The present report summarizes our ab initio investigations that have recently addressed the effects of charging and alkali-doping on the geometrical structure, charge distribution and nonlinear optical properties of increasingly large polyacetyene (PA) chains. The calculations have been performed at the coupled-perturbed Hartree-Fock (CPHF) level of approximation with 6-31G basis set. Additionally, for a selection of small systems the second-order Møller-Plesset (MP2) level of approximation has been employed to assess the impact of the electron correlation effects. It has been found out that charging dramatically enhances the static electronic and vibrational second hyperpolarizabilities whereas the presence of an alkali atom (Li, Na, K) substantially reduces this effect. As the size of the alkali atom increases, most properties, including hyperpolarizabilities, approach those of the isolated chain.
Keywords: nonlinear optics, doped polyacetylene, quantum chemistry.
Introduction The design of new materials for nonlinear optical (NLO)
applications requires molecules possessing large hyperpolarizabilities leading, at the macroscopic scale, to large nonlinear susceptibilities. In addition, these materials have to be transparent to the ingoing and

outgoing waves and be stable with respect to processing conditions and laser irradiation. Quantum chemistry is closely involved in the understanding of the physical phenomena of NLO responses of molecules [1,2] due to its ability to predict the relationships between the structures and their properties.
One of the strategies for optimizing the NLO response consists in charging the chromophores by appropriate chemical or electrochemical dopings [3]. The transfer of a charge to or from the polymer backbone induces structural relaxation due to the strong electron-phonon coupling and results in the creation of localized defects (soliton, polaron) to which the excess charge is confined. These defects perturb the regular periodic character of the undoped polymer electronic structure and are responsible, at least at low doping levels, for the enhancement of the electrical conductivity by 10–15 orders of magnitude [4].
The formation of a soliton in a degenerate ground state polyene like trans-polyacetylene is accompanied by the separation of the chain into two phases where the single and double CC bonds are differently oriented. The determination of the extent of the charge defect and its associated geometrical relaxation in trans-PA could be assessed by bond length alternation (BLA) defined as
1( 1) ( )
−= − −n
n n nBLA R R . The
value of BLA is zero in the region where phases switch.
Structure of alkali-doped polyacetylene chains To model the soliton-bearing chains, a polyene chain containing
an odd number of carbon atoms is employed [5]. This usual procedure is adopted with and without counterion.
Figure 1. Schematic representation of the C23H25M chain doped by one alkali atom.
The HF/6-31G BLA pattern along the chain sketched in Fig. 1
(left) follows the hyperbolic tangent relation of ref. 6
tanh( )n
n CBLA BLA
L∞
−= where n is the position along the chain,
∞BLA
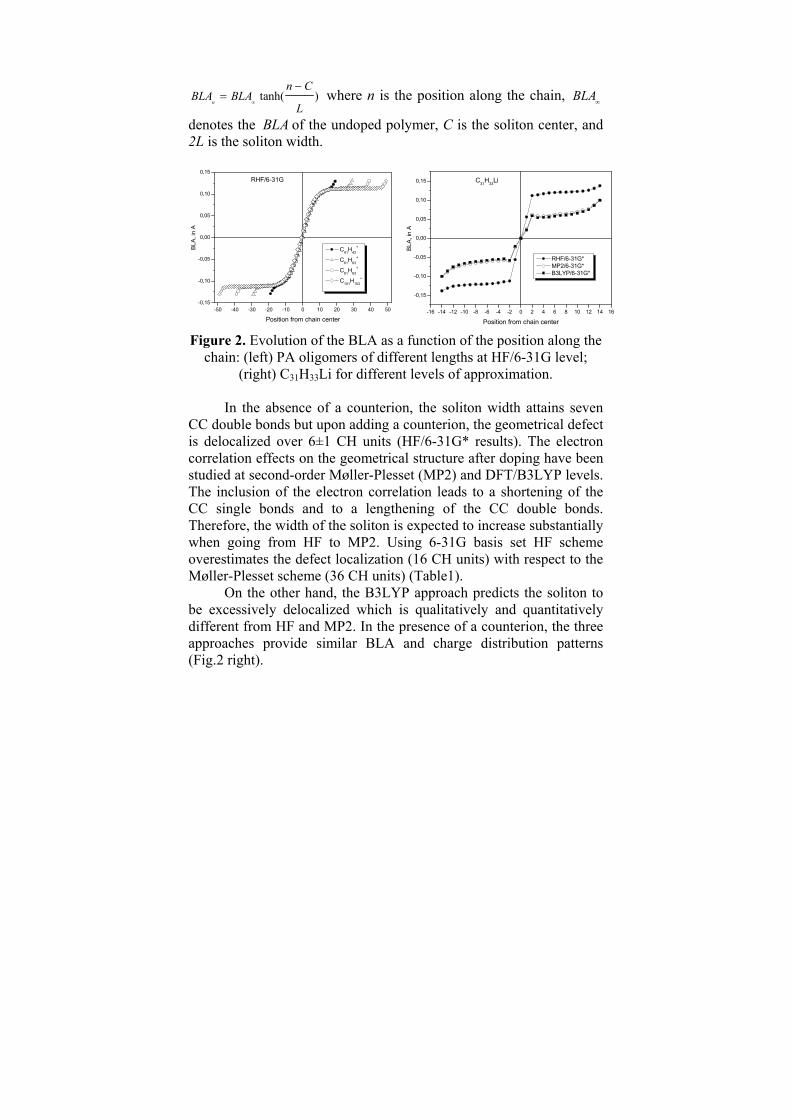
denotes the BLA of the undoped polymer, C is the soliton center, and 2L is the soliton width. Figure 2. Evolution of the BLA as a function of the position along the
chain: (left) PA oligomers of different lengths at HF/6-31G level; (right) C31H33Li for different levels of approximation.
In the absence of a counterion, the soliton width attains seven
CC double bonds but upon adding a counterion, the geometrical defect is delocalized over 6±1 CH units (HF/6-31G* results). The electron correlation effects on the geometrical structure after doping have been studied at second-order Møller-Plesset (MP2) and DFT/B3LYP levels. The inclusion of the electron correlation leads to a shortening of the CC single bonds and to a lengthening of the CC double bonds. Therefore, the width of the soliton is expected to increase substantially when going from HF to MP2. Using 6-31G basis set HF scheme overestimates the defect localization (16 CH units) with respect to the Møller-Plesset scheme (36 CH units) (Table1).
On the other hand, the B3LYP approach predicts the soliton to be excessively delocalized which is qualitatively and quantitatively different from HF and MP2. In the presence of a counterion, the three approaches provide similar BLA and charge distribution patterns (Fig.2 right).
-50 -40 -30 -20 -10 0 10 20 30 40 50-0,15
-0,10
-0,05
0,00
0,05
0,10
0,15
C41H43+
C61H63+
C81H83+
C101H103+
RHF/6-31G
BLA,
in A
Position from chain center-16 -14 -12 -10 -8 -6 -4 -2 0 2 4 6 8 10 12 14 16
-0,15
-0,10
-0,05
0,00
0,05
0,10
0,15
RHF/6-31G* MP2/6-31G* B3LYP/6-31G*
C31
H33
Li
BLA
, in
A
Position from chain center

Table 1. Soliton width (2L) and BLA∞ (Å) (in parenthesis) as a function of the method of calculation
2L (BLA∞) STO-3G 6-31G 6-31G*
HF MP2
B3LYP
10.8 (0.151) 18.1 (0.118) 44 (0.085)
15.6 (0.112) 35.7 (0.086)
14.5 (0.123)
The soliton defect can also be characterized by the charge
distribution along the chain. At HF/6-31G level, the excess positive charge is localized in the central part of the chain and decreases towards the chain ends (Fig. 3 left). In fact, this excess charge is delocalized on every other CH unit, commensurate with the simple organic chemistry picture of moving two π-electrons or the entire π-bond at a time. By least-squares fit of the expression
2
0 sec ( )n
n Cq q h
L
−= (for even values of n) it is obtained that the
charge defect appears to be more extended than the geometrical distortion (2L=21.6 vs. 15.6 obtained from the BLA). Again, the situation is different at the B3LYP/6-31G level (Fig. 3 right) where charge is mostly delocalized over the whole chain. This incorrect behavior is attributed to the ultra-locality of the exchange–correlation potential although it incorporates 20% of Hartree–Fock exchange. Upon considering the counterion, the soliton defect is much more localized and the three approaches provide a similar picture (at least qualitatively) of the defect. In the case of alkali-doping, the interaction between the dopant and the chain presents an important covalent character and the defect spreads over 5-7 CH units.
Another study focuses on the charge density associated to a positively charged soliton in large polyacetylene chains [7]. The investigation is carried out by considering charge distributions obtained using different charge definitions at different levels of approximation: HF, MP2, and DFT/B3LYP. The charge storage in the soliton-bearing systems is explored in detail, including charge magnitude, charge separation, charge alternation, and chain length effects.

Figure 3. Distribution of the Mulliken C+H charges as a function of the position along the chain: (left) - HF/6-31G level, (right) -
B3LYP/6-31G level.
Since most of the related investigations have so far been limited to short chains and/or, to the Mulliken population analysis scheme, we tackle the charge distributions of rather large chains (up to 41 carbon atoms) using correlated schemes. Definitions of atomic charges that are derived from four different types of schemes are employed. The first type of definition is based on real-space partitioning such as in the quantum theory of atoms in molecules (AIM) due to Bader [8]. The second is derived from Hilbert space partitioning where the charge is assigned to basis functions such as Mulliken [9] and natural population analysis (NPA) [10]. Fitting properties and, in particular, the electrostatic potential is another means for defining charges: according to the Merz–Kollman (MK) scheme [11a], the restrained electrostatic potential (RESP) method [11b], and the charges from electrostatic potential using a grid (CHELPG) method [11c]. The last definition employed originates from the generalized atomic polar tensors (GAPT) [12]. Similarity analysis [13] is used quantitatively to compare the obtained charge distributions.
Generally, HF charges are twice as large as the corresponding B3LYP and MP2 and, as mentioned above, are localized at the center of the chain. They also strongly alternate for all charge definitions. GAPT charges are one order of magnitude larger than the rest and increase with the size of the system which is consistent with the very
-50 -40 -30 -20 -10 0 10 20 30 40 50
-0,05
0,00
0,05
0,10
0,15 RHF/6-31G
char
ge
Position from chain center
C+ H atoms
-30 -25 -20 -15 -10 -5 0 5 10 15 20 25 30-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
B3LYP/ 6-31G
char
ge
Position from chain center
C+ H atoms

slow saturation of the vibrational polarizability of the studied charged systems [14].
Nonlinear optical properties (Hyper)polarizabilities are defined as the coefficients in the
Taylor series expansion of the induced dipole moment or the energy in the presence of static and/or dynamic (oscillating) electric fields:
( ) ( ) ( ) ( ) ( ) ( )( ) ( ) ( ) ( )
01 1 1 2 1 2
1 2 3 1 2 3
; ; ,
; , ,ξ σ ξ σ η σ η ζ
σ η ζ
ξη ξηζ
ξηζ
μ ω μ α ω ω ω ω ω ω ω ω
ω ω ω ω ω ω ω
β
γ +
= + − + − +
+ − …vv
E E E
E E E
where the
ξηα ,
ξηζβ and ξηζγ v components define the linear
polarizability, first and second hyperpolarizability, respectively, describing nonlinear responses to the external field with σω ω= ∑ ii
. The Coupled Perturbed Hartree-Fock method with 6-31G basis set was used for evaluating of the longitudinal second hyperpolarizability
(0; 0, 0, 0) (0)e e
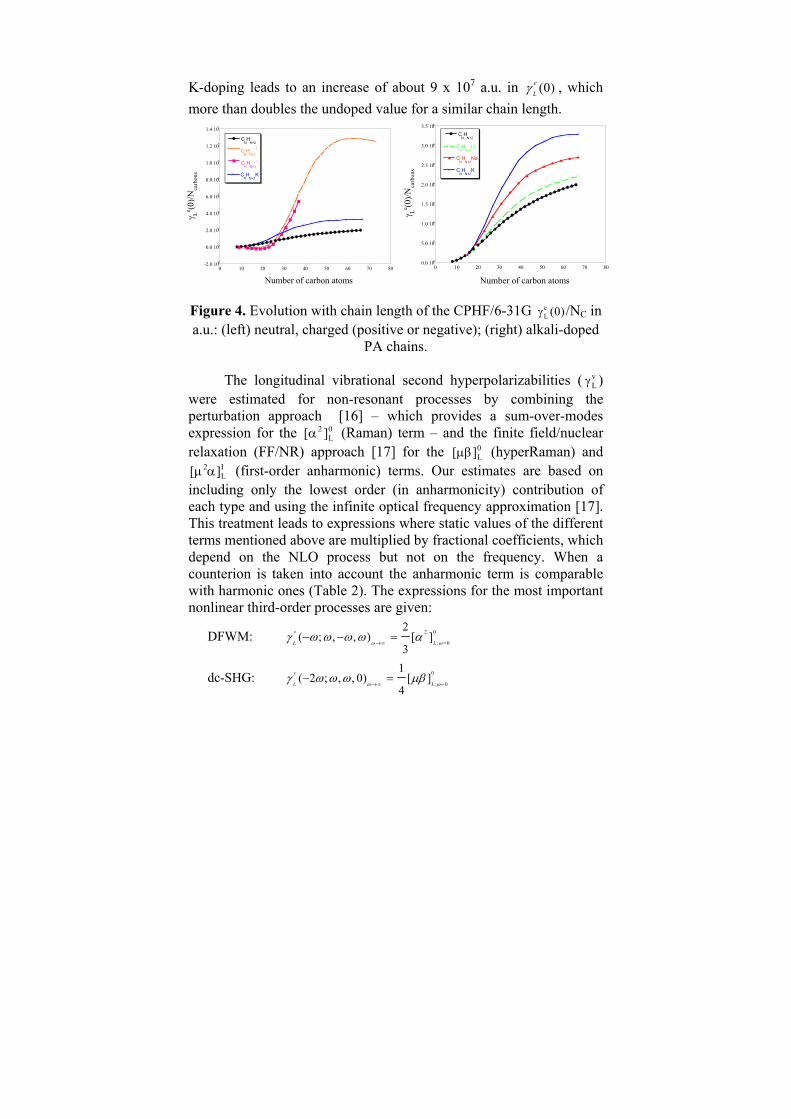
L Lγ γ= of polyacetylene chains containing up to 70 carbon atoms with and without explicit alkali atoms (Li, Na, K) as dopant [15]. Figure 4 (left) displays the evolution of the normalized quantity (0) /e
L CNγ as a function of the PA chain length in comparison with the undoped case and with positively and negatively charged chains without a counterion. (0) /e
L CNγ can exhibit a maximum at about NC = 61 for the isolated charged soliton, and the value of the hyperpolarizability per carbon atom at that point is over four times that of an infinite undoped PA chain. Whereas charging dramatically enhances (0)e
Lγ of an isolated chain at intermediate chain lengths, the presence of an alkali atom counterion substantially reduces this effect (Fig. 4 right). As the size of the alkali atom increases the hyperpolarizabilities approach those of the isolated charged chain. The behavior of (0)e
Lγ is most simply explained in terms of a reduced pinning potential at longer counterion-chain distances. For NC = 50,
K-doping leads to an increase of about 9 x 107 a.u. in (0)e
Lγ , which more than doubles the undoped value for a similar chain length. Figure 4. Evolution with chain length of the CPHF/6-31G γL
e (0)/NC in a.u.: (left) neutral, charged (positive or negative); (right) alkali-doped
PA chains.
The longitudinal vibrational second hyperpolarizabilities ( γLv )
were estimated for non-resonant processes by combining the perturbation approach [16] – which provides a sum-over-modes expression for the [α2 ]L
0 (Raman) term – and the finite field/nuclear relaxation (FF/NR) approach [17] for the [μβ]L
0 (hyperRaman) and [μ2α]L
I (first-order anharmonic) terms. Our estimates are based on including only the lowest order (in anharmonicity) contribution of each type and using the infinite optical frequency approximation [17]. This treatment leads to expressions where static values of the different terms mentioned above are multiplied by fractional coefficients, which depend on the NLO process but not on the frequency. When a counterion is taken into account the anharmonic term is comparable with harmonic ones (Table 2). The expressions for the most important nonlinear third-order processes are given:
DFWM: 2 0
; 0
2( ; , , ) [ ]
3v
L Lω ωγ ω ω ω ω α
→∞ =− − =
dc-SHG: 0
; 0
1( 2 ; , , 0) [ ]
4v
L Lω ωγ ω ω ω μβ
→∞ =− =
-2.0 106
0.0 100
2.0 106
4.0 106
6.0 106
8.0 106
1.0 107
1.2 107
1.4 107
0 10 20 30 40 50 60 70 80
CNH
N+2
CNH
N+2
+
CNH
N+2
-
CNH
N+2K
γ Le (0)/N
carb
ons
Number of carbon atoms
0.0 100
5.0 105
1.0 106
1.5 106
2.0 106
2.5 106
3.0 106
3.5 106
0 10 20 30 40 50 60 70 80
CNH
N+2
CNH
N+2Li
CNH
N+2Na
CNH
N+2K
γ Le (0)/N
carb
ons
Number of carbon atoms
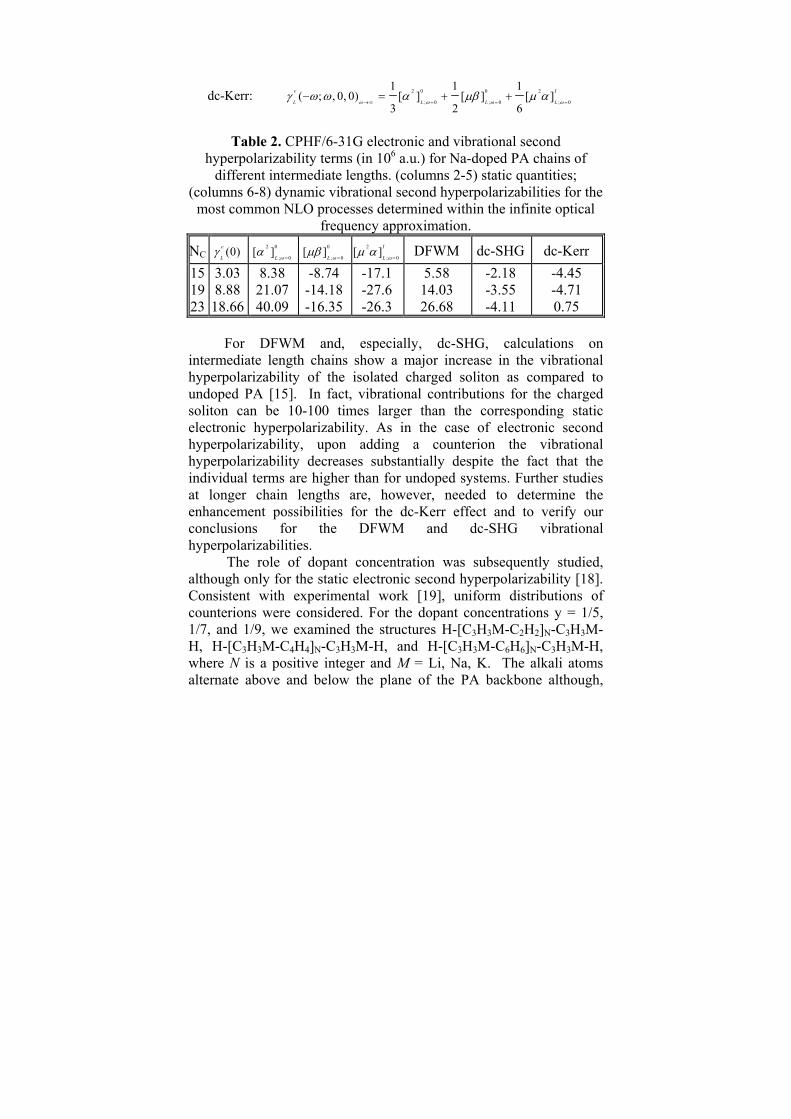
dc-Kerr: 2 0 0 2
; 0 ; 0 ; 0
1 1 1( ; , 0, 0) [ ] [ ] [ ]
3 2 6v I
L L L Lω ω ω ωγ ω ω α μβ μ α
→∞ = = =− = + +
Table 2. CPHF/6-31G electronic and vibrational second
hyperpolarizability terms (in 106 a.u.) for Na-doped PA chains of different intermediate lengths. (columns 2-5) static quantities;
(columns 6-8) dynamic vibrational second hyperpolarizabilities for the most common NLO processes determined within the infinite optical
frequency approximation.
NC (0)e
Lγ 2 0
; 0[ ]
L ωα
= 0
; 0[ ]
L ωμβ
=
2
; 0[ ]I
L ωμ α
=DFWM dc-SHG dc-Kerr
15 19 23
3.03 8.88 18.66
8.38 21.07 40.09
-8.74 -14.18 -16.35
-17.1 -27.6 -26.3
5.58 14.03 26.68
-2.18 -3.55 -4.11
-4.45 -4.71 0.75
For DFWM and, especially, dc-SHG, calculations on
intermediate length chains show a major increase in the vibrational hyperpolarizability of the isolated charged soliton as compared to undoped PA [15]. In fact, vibrational contributions for the charged soliton can be 10-100 times larger than the corresponding static electronic hyperpolarizability. As in the case of electronic second hyperpolarizability, upon adding a counterion the vibrational hyperpolarizability decreases substantially despite the fact that the individual terms are higher than for undoped systems. Further studies at longer chain lengths are, however, needed to determine the enhancement possibilities for the dc-Kerr effect and to verify our conclusions for the DFWM and dc-SHG vibrational hyperpolarizabilities.
The role of dopant concentration was subsequently studied, although only for the static electronic second hyperpolarizability [18]. Consistent with experimental work [19], uniform distributions of counterions were considered. For the dopant concentrations y = 1/5, 1/7, and 1/9, we examined the structures H-[C3H3M-C2H2]N-C3H3M-H, H-[C3H3M-C4H4]N-C3H3M-H, and H-[C3H3M-C6H6]N-C3H3M-H, where N is a positive integer and M = Li, Na, K. The alkali atoms alternate above and below the plane of the PA backbone although,
except for chain bending when all alkali atoms are on the same side, the conclusions change little.
For Li-doping there is a small increase in γLe (0) /NC with respect
to undoped PA (~ 10%) regardless of doping level and chain length. In the long chain limit the largest enhancement is attained for the lowest doping level (1/5). Figure 5 (left) shows a comparison between Li, Na, and K for y = 1/9. There is evidently a substantial increase in γL
e (0) due to replacing Li by Na and, particularly, by K. For the longest chains (93 carbon atoms) the values of γL
e (0) are in the ratio 1.00 : 1.99 : 4.83. A rather large maximum theoretical enhancement factor of ~ 60 occurs for K-doped chains at a length of 24 carbon atoms and doping level y = 1/7 (Fig. 5 right). In general, this factor diminishes with chain length until reaching its asymptotic value. Figure 5. (left) CPHF/6-31G γL
e (0)/NC for different dopants at y = 1/9; (right) enhancement factor for CPHF/6-31G γ of Na- and K-doped vs.
undoped PA chains as a function of chain length.
Conclusions The presented ab initio study points out that a substantial
enhancement of the second hyperpolarizability with respect to the undoped polymer might be achieved for a suitable system and doping strategy. Charging dramatically enhances the static electronic and vibrational second hyperpolarizabilities whereas the presence of alkali atom substantially reduces this effect due to pinning of the charge. Therefore, best enhancement strategy of NLO properties, besides optimizing the chain length, is to maximize the charge transferred to the chain and minimize the electrostatic interaction between dopant
0.00
2.00
4.00
6.00
8.00
10.0
12.0
14.0
0 20 40 60 80 100 120
M = Li
M = Na
M = K
γ Le (0)/N
C
(in 1
06 a.u
.)
NC
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
0 20 40 60 80 100
Na, y = 1/5Na, y = 1/7Na, y = 1/9K, y = 1/7K, y = 1/9
log
[γLe (0
)-PA
-dop
ed /
γ Le (0)-P
A-u
ndop
ed]
NC

and chain - by using a dopant which has a very delocalized charge distribution. This is of crucial importance in the current widespread search for materials having high nonlinear optical coefficients that could be used in optoelectronic and photonic devices.
References 1. Int. J. Quantum. Chem., special issue on Molecular Nonlinear
Optics, 1992 43 (1), edited by M. A. Ratner and P. O. Lowdin. 2. Chemical Review, thematic issue on Optical Nonlinearities in
Chemistry 1994, 94 (1), edited by D. A. Burland and J. Michl. 3. See for instance A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8475. 4. (a) H. Shirikawa, W. J. Louis, A. G. MacDiarmid, C. K. Chiang
and A. J. Heeger, J. Chem. Soc.,Chem. Commun., 1977, 16 578; (b) M. J. Rice, Phys. Lett., 1979, 71, 152.
5. B. Champagne, M. Spassova, PhysChemChemPhys, 2004, 6 3167. 6. a) W. P. Su, J. R. Schrieffer, A. J. Heeger, Phys. Rev. Lett., 1979,
42, 1698; (b) W. P. Su, J. R. Schrieffer and A. J. Heeger, Phys. Rev. B., 1980, 22, 2099.
7. V. Monev, M. Spassova, B. Champagne, Int. J. Quant. Chem., 2005, 104, 354.
8. R. F. W. Bader, Atoms in Molecules - A Quantum Theory; Oxford University Press: Oxford, 1990.
9. R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833. 10. A. E. Reed, R. B. Weinstock, F. Weinhold, J. Chem.Phys., 1985,
83, 735. 11. a) B. H. Besler, K. M. Merz, P. A. Kollman, J. Comp. Chem.
1990, 11, 431; b) C. I. Bayly, P. Cieplak, W. D. Cornell, P. A. Kollman, J. Phys. Chem., 1993, 97, 10269; computer program RESP freely available at http://amber.scripps.edu/ftp/plep.tar.gz, see also http://amber.scripps.edu/doc6/html/AMBER-sh- 19.4.html; c) C. M. Breneman, K. B. Wiberg, J. Comp. Chem., 1990, 11, 361.
12. J. Cioslowski, J. Am. Chem. Soc., 1989, 111, 8333. 13. For an overview of the similarity concept in chemistry, see V.
Monev, Match-Commun Math Comput. Chem., 2004, 51, 7. 14. B. Champagne, E. Deumens, Y. O. hrn, J. Chem. Phys., 1997,
107, 5433.

15. B. Champagne, M. Spassova, J. -B. Jadin, B. Kirtman, J. Chem. Phys., 2002, 116, 3935-3946.
16. D. M. Bishop, B. Kirtman, J. Chem. Phys., 1991, 95, 2646. 17. D. M. Bishop, M. Hasan, B. Kirtman, J. Chem. Phys., 1995, 103,
4157. 18. M. Spassova, B. Champagne, B. Kirtman, Chem. Phys. Lett.,
2005, 412, 217-222. 19. N. S. Murthy, L. W. Shacklette, R. H. Baughman, Phys. Rev. B,
1989, 40, 12550.

Immobilization of Trypsin on Silica Carriers Viara Ivanova
University of Food Technology, Technological Faculty Department of Organic Chemistry and Microbiology
26, Maritza Blvd., 4002 Plovdiv E mail: [email protected]
Abstract: Trypsin (EC 3.4.21.4) was immobilized on silica carriers. The effects of various immobilization parameters on the catalytic properties, the kinetic parameters and the stability of the immobilized enzyme were studied. It was found, that the amount of bound protein and the measured activity (BAPNA) are considerably higher when protein is immobilized on controlled pore glass (CPG) and Spherosil. Taking into account storage stability and activity, the Spherosil 15 was the most effective support for the enzyme immobilization. For this preparation, BAPNA conversion, pH- and T-optima, pH- and thermal stability at 50°C, Km and Vmax were compared with those obtained for the native enzyme.
Keywords: Immobilization; trypsin; silica carriers.
Introduction Immobilized enzymes are defined as “enzymes which are
physically confined or localized in a certain region of space with retention of their catalytic activities, and which can be used repeatedly and continuously” [1]. The main advantages of enzyme immobilization are: low downstream processing cost; better stability; possibility of enzyme recycling; easy realization of continuous process. The main disadvantages are: loss of absolute enzyme activity due to the immobilization process, additional cost of carrier or other reagents, and potential for mass transfer limitations.
Stabilization of proteolytic enzymes by immobilization is of considerable interest because of their potential applications in medicine and in the chemical and pharmaceutical industries. Immobilized trypsin can be used in bioaffinity chromatography and

enzymatic reactors for specific separation and for isolation, purification and analytic characterization of trypsin inhibitors [2,3], in immobilized proteolytic enzyme reactors designed for studies in proteomics, protein digestion and peptide mapping [4-6], for cleavage, synthesis or chemical modification [7], for obtaining phosphopeptides from casein [8]. The most commonly used carrier binding methods are adsorption or covalent linkage to a carrier [9-19]. The choice of the immobilization method depends on the enzyme stability [20,21] and the process of application. Physical adsorption induces only slight modifications on the conformation of the enzyme as the binding mainly results from hydrogen bonds, salt linkages or van der Waal's forces [22]. Too strong interaction between the support material and protein after formation of chemical bounds could lead to variations on tertiary structure and finally to deactivation of enzyme [23].
Materials and Methods
The following porous silica carriers were pre-treated: acid washed CPG (Sigma, USA, average pore diameter 350 nm, specific area 6 m2/g) and porous Spherosil particles (IBF Biotechnics, France) with specific areas 5, 15 and 400 m2/g (Spherosil 5, 15 and 400, respectively) and respective average pore diameters 460, 220 and 8 nm. The first method for pre-treatment was a simple washing with distilled water, followed by drying at 105°C for 24 h. In the second method, the porous silica carriers were treated with 65% (w/v) nitric acid. The resulting mixture was boiled for 5 min. Then the beads were washed with distilled water and dried at 105°C for 24 h. The resulting beads were called acid-treated beads. Hydrophilic silica beads with epoxy groups were prepared by incubating acid-treated silicates with 2% 3-glycidoxypropyltrimethoxysilane (Fluka) in toluene for two weeks at room temperature and then washed with distilled water. Subsequently, they were treated with 10% acetic acid in order to open the epoxy group formed on the surface of the beads. The mixture was heated in a water bath at 50°C for 5 h. After removing the supernatant, the beads were washed with distilled water and dried at room temperature. Hydrophobic silica beads were prepared by incubating acid-treated beads with 5% dichlorodimethylsilane in toluene at room temperature. Then the mixture was shaken for 5 min and the

supernatant was removed. The resulting beads were washed with distilled water and dried at room temperature.
Merckogel SI 1000 – 120-200 mesh (Merck), quartz powder – 0.16-0.20 mm (Riedel-de Haen), pumice stone – 0.125-0.250 mm (Bulgarian product), and Silica gel S 4004 GF-10-40 μ (Sigma) were pre-wetted with 50 ml ethanol for 30 min and then washed with 50 % (v/v) ethanol-water solution. Finally, the mixture was washed several times with distilled water and the wet beads were used for the immobilization process.
Immobilization procedure. Unless otherwise stated, 5 ml of 0.2 M phosphate buffer (pH 7.8) containing 4 mg protein as dissolved trypsin was added to 500 mg of support at 4°C. The resulting mixture was stirred for 2 h with a magnetic stirrer at 100 rpm. After the coupling step, the supports were washed with an abundant quantity of water until no protein was detectable in the filtrates, and then were washed again with 0.2 M phosphate buffer. Immobilized protein quantity was measured after the end of all washing steps. The enzyme-carrier complex was stored at 4 °C in 0.2 M phosphate buffer, pH 7.8.
Effect of immobilization conditions. Three grams of dry support were added to 40 ml buffered trypsin solution (0.05-0.3 M phosphate buffers with pH 6.3 and pH 7.8 and 0.05-0.3 M borate buffers, pH 9.0 and 10.0). The mixture was stirred at 4°C, 15°C or 22°C for 2-18h. The stirrer speed was from 50 to 400 rpm and the trypsin quantity in the solution was from 10 to 40 mg. After the coupling step, the supports were washed with an abundant quantity of water until no protein was detectable in the filtrates, determined by the method of Bradford [24] at 595 nm. Bovine serum albumin (BSA) was used as a standard protein.
Determination of the amount of bound enzyme. The amount of protein loaded onto the dried supports was determined using a modified Bradford assay. This method measures the decrease in the absorbance of the solution at 465 nm due to adsorption of the dye by the bound protein. For this assay, 200 μl of distilled water was added to 4.5 mg of enzyme loaded support and 4.5 mg of blank support, respectively. Then 1800 μl of Bradford reagent, diluted (1:5) with distilled water, was subsequently added to each of these mixtures.

After short agitation of the mixture (3 min) to allow binding of the dye with the protein, the mixtures were centrifuged at 3000 g. The absorbance of the two supernatants was measured at 465 nm. The reading for the enzyme loaded support was then subtracted from the similar reading obtained with the blank support. The calibration curve for quantification of the protein was obtained with BSA as a reference protein at 465 nm. The binding (BY) yield was determined as follows: BY, [%] = (P/Po) x100, where Po and P are the protein added and the measured after immobilization, as [mg protein]. The specific enzyme activity was defined as: Specific enzyme activity = Enzyme activity/mg of protein. The residual activity (RA) of trypsin was calculated as a ratio of the specific enzyme activity measured after immobilization [U/mg protein] to the specific enzyme activity before the immobilization [U/mg protein] and was expressed as percentage.
Assay of enzyme activity. Trypsin activity was determined spectrophotometrically using BAPNA (Nα-bensoyl-DL-arginine-4-nitroanilide hydrochloride, Mw 434.88) as a substrate at 25 °C. Hydrolysis of DL-BAPNA at the bond between the arginine and the p-nitroaniline moieties releases the chromophore p-nitroaniline, which can be detected by colorimetric analysis of reaction products from native (free) [25] and immobilized trypsin [15]. The reaction mixture contained 1 ml 2.2 mM BAPNA, 1.8 ml 0.2 M triethanolamine buffer pH 7.8; 20 mM CaCl2 and 0.2 ml trypsin (50 μg/ml, dissolved in 0.001 M HCl) or 0.01 g beads (w/w). One enzyme unit (U) corresponds to an absorbance change ΔA405 of 3.32 per minute in a 3 ml assay mixture. The activity of the immobilized enzyme was measured under constant mixing. The specific enzyme activity was calculated by the equation: Activity = A405/3.32 x t x a [U/mg], where: A405 is the absorbance at 405 nm; t is reaction time; a is the quantity of immobilized trypsin in the reaction mixture, and 3.32 is a pre-factor.
The thermo-stability in aqueous phase was obtained by subjecting immobilized enzyme to 50°C in a thermo-constant bath. Samples were taken periodically and the residual activity of the enzyme was measured spectrophotometrically as described. Km was assayed for substrate BAPNA at 25°C using the Lineweaver-Burk plot [27] for calculation. The substrate concentrations were from 0.202 to 2.0 mM, dissolved in 0.2M triethanolamine buffer pH 7.8. The quantity of

native protein was 50 μg/ml and the amount of biocatalyst was 10 mg (w/w) per assay. The pH-optima and pH-stability of immobilized and native (free) trypsin were measured using 50 μg/ml protein or 10 mg (w/w) of biocatalysts per assay. 0.2 M phosphate buffer with pH 6.0 and 7.0; 0.2 M triethanolamine buffer pH 7.8 and 0.2 M borate buffer with pH 9.0 and 10.0 were used. The half-life was determined using the immobilized trypsin in consecutive assays.
Results and Discussion The obtained results are summarized on Figures 1-10 and Table
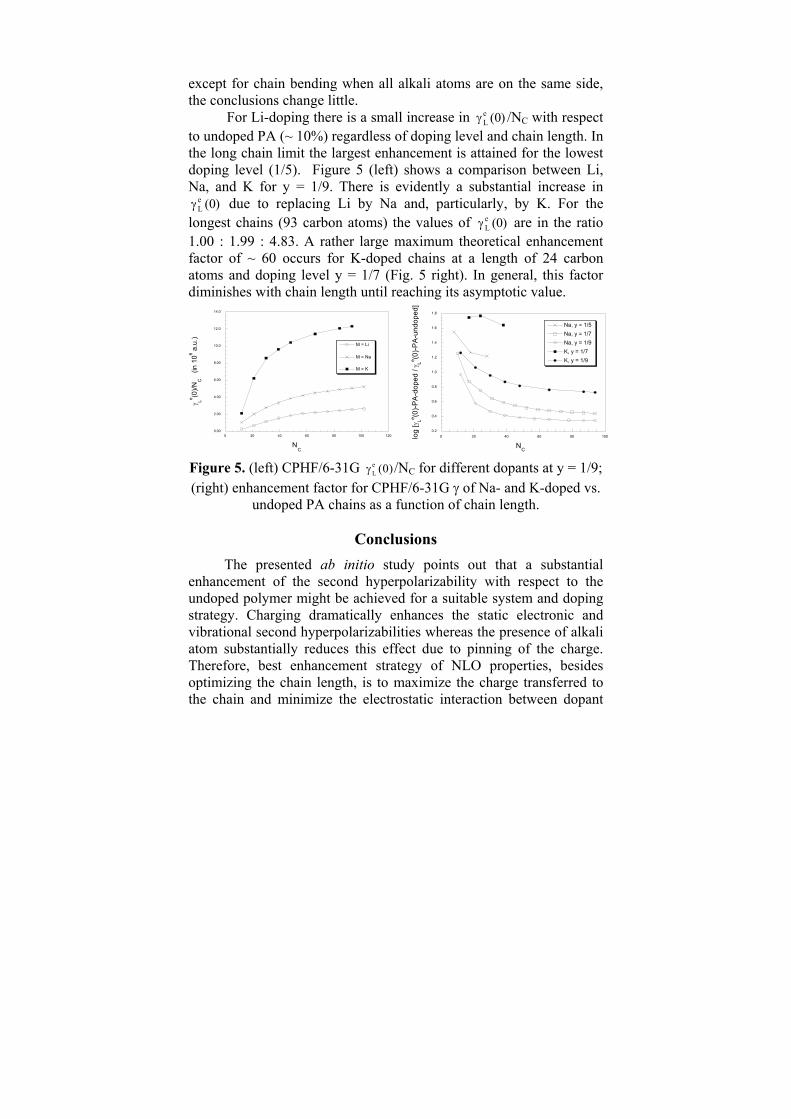
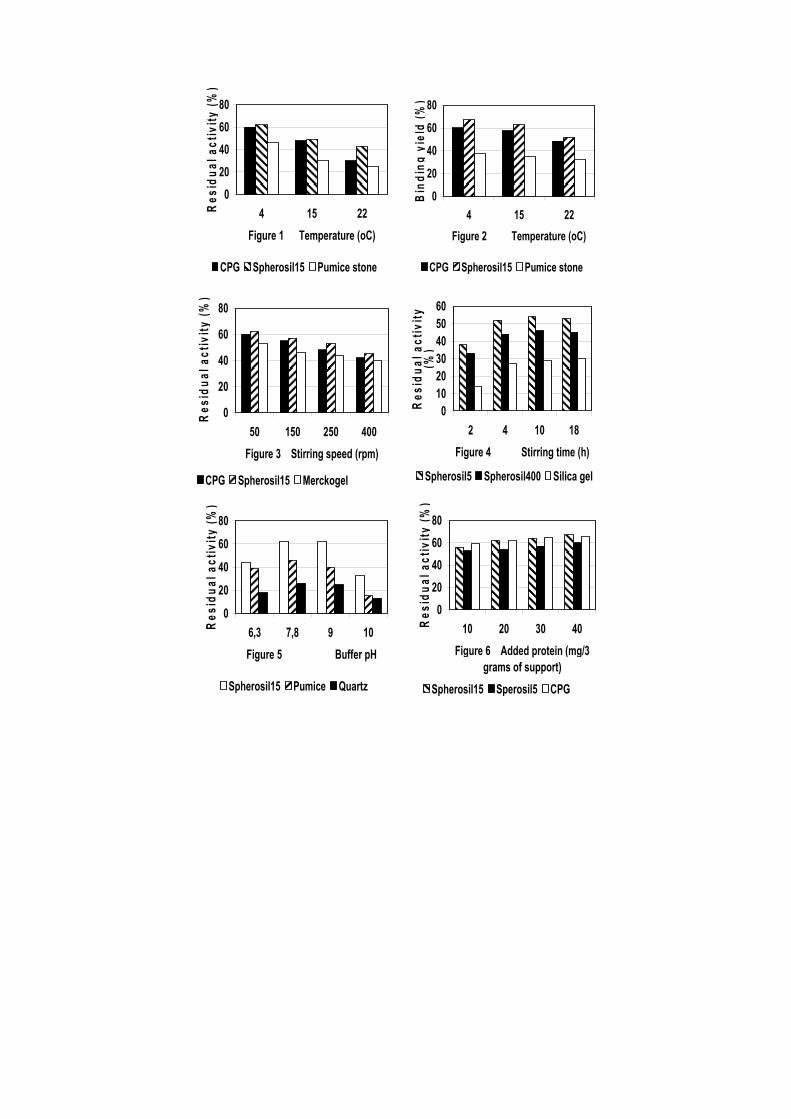
1. The following immobilization parameters were tested: immobilization temperature (Figs 1 and 2); stirring speed (Fig. 3); stirring time (Fig. 4); buffer pH (Fig. 5), concentration (Fig. 7) and type (Fig. 8); amount of added protein (Fig. 6); supports and their treatment (Table 1).
Low temperature was found to be less detrimental to the enzyme (Fig.1) and could be due to reduced thermoinactivation of the enzyme at lower temperature. Up to 67.3% of the initially added protein was adsorbed on the Spherosil 15 beads (Fig.2). The residual activity decreased with increasing stirring speed (Figure3). This is attributed to detrimental shearing effects on the enzyme caused by the magnetic stirrer. Stirring time had slight influence on the protein loading (Fig.4).
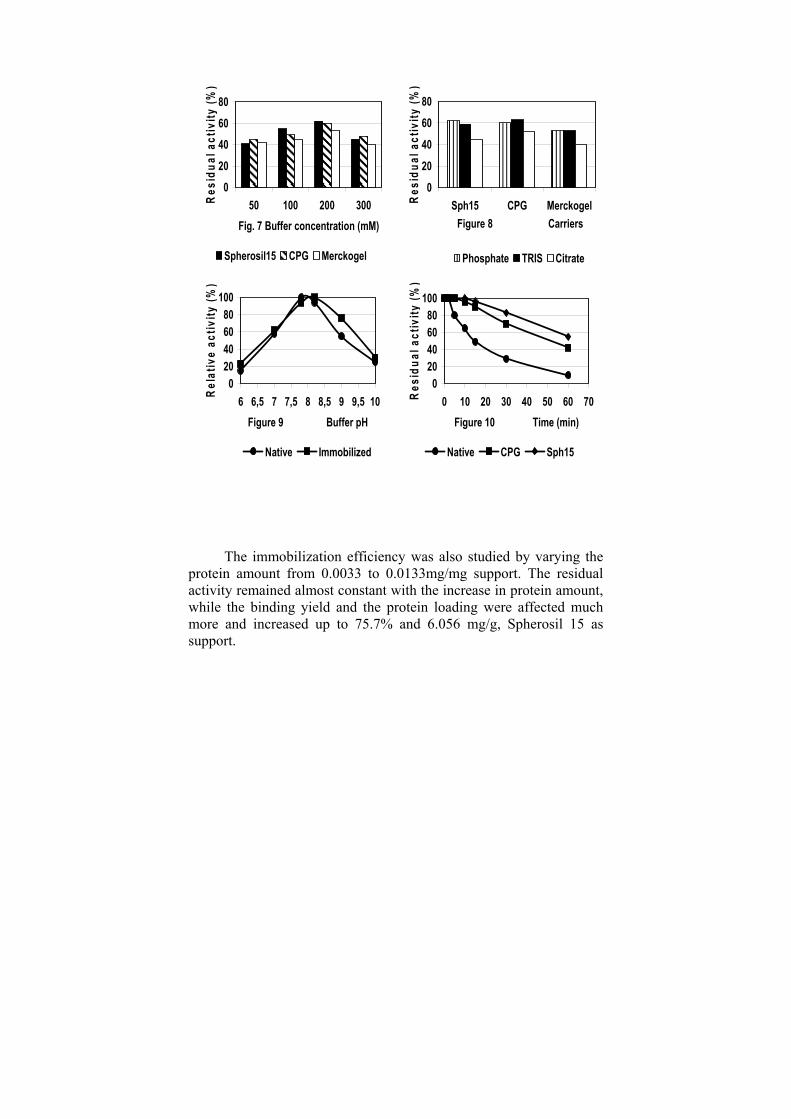
The optimum pH for trypsin immobilization was found to be at pH 7.8-9.0 (Fig.5). The effect of buffer concentration on immobilization efficiency was studied by varying the buffer concentration in the range of 50 mM to 300 mM. The residual activity increased with the increase in buffer concentration from 50 mM to 200 mM and then decreased (Fig.7). This increase might be attributed to structural changes of the enzyme with increase in ionic strength. At concentrations much higher, the decrease might be due to conformational changes leading to loss of enzyme activity. The effect of buffer type on immobilization efficiency of trypsin was also evaluated (Fig. 8). The enzyme was immobilized using 0.2M phosphate, TRIS and citrate buffers, pH 7.8. High residual activity and protein loading were obtained when using phosphate or TRIS buffers.
0
20
40
60
80
4 15 22Figure 1 Temperature (oC)
Res
idua
l act
ivity
(%)
CPG Spherosil15 Pumice stone
0
20
40
60
80
4 15 22Figure 2 Temperature (oC)
Bin
ding
yie
ld (%
)
CPG Spherosil15 Pumice stone
0
20
40
60
80
50 150 250 400Figure 3 Stirring speed (rpm)
Res
idua
l act
ivity
(%)
CPG Spherosil15 Merckogel
0102030405060
2 4 10 18Figure 4 Stirring time (h)
Res
idua
l act
ivity
(%
)
Spherosil5 Spherosil400 Silica gel
0
20
40
60
80
6,3 7,8 9 10Figure 5 Buffer pH
Res
idua
l act
ivity
(%)
Spherosil15 Pumice Quartz
020406080
10 20 30 40Figure 6 Added protein (mg/3
grams of support)
Res
idua
l act
ivity
(%)
Spherosil15 Sperosil5 CPG
0
20
40
60
80
50 100 200 300Fig. 7 Buffer concentration (mM)
Res
idua
l act
ivity
(%)
Spherosil15 CPG Merckogel
0
20
40
60
80
Sph15 CPG MerckogelFigure 8 Carriers
Res
idua
l act
ivity
(%)
Phosphate TRIS Citrate
020406080
100
6 6,5 7 7,5 8 8,5 9 9,5 10Figure 9 Buffer pH
Rel
ativ
e ac
tivity
(%)
Native Immobilized
020406080
100
0 10 20 30 40 50 60 70Figure 10 Time (min)
Res
idua
l act
ivity
(%)
Native CPG Sph15
The immobilization efficiency was also studied by varying the
protein amount from 0.0033 to 0.0133mg/mg support. The residual activity remained almost constant with the increase in protein amount, while the binding yield and the protein loading were affected much more and increased up to 75.7% and 6.056 mg/g, Spherosil 15 as support.
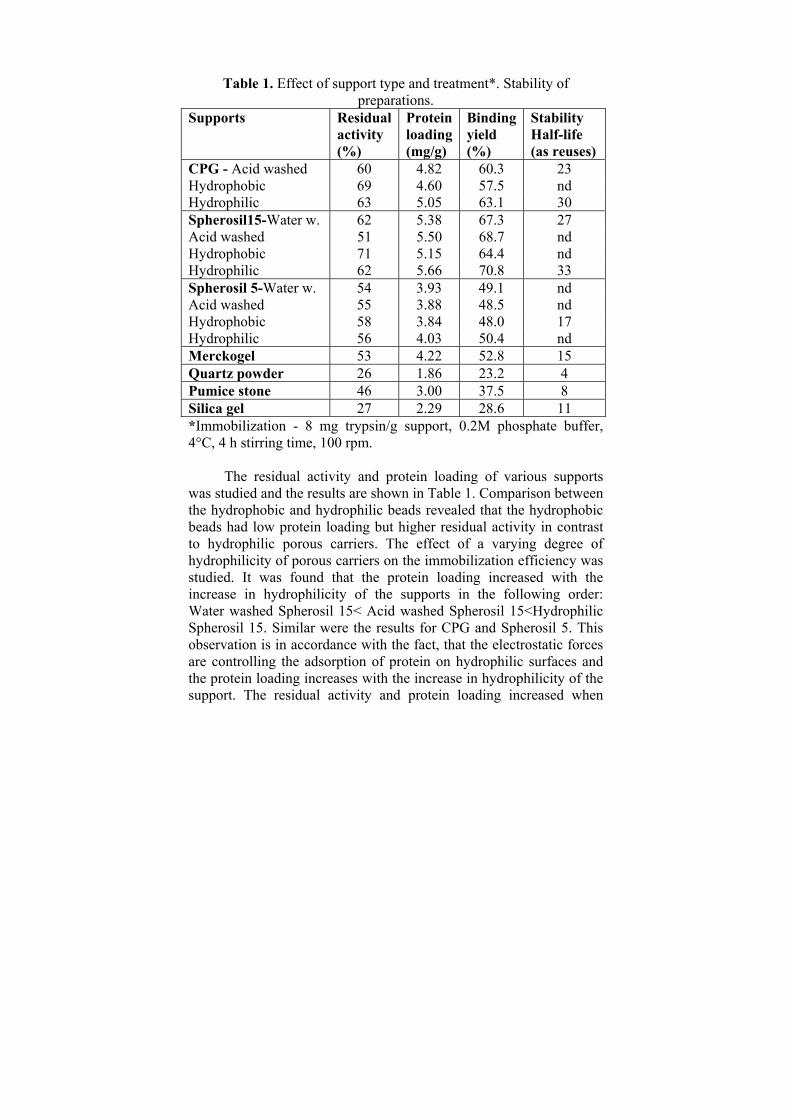
Table 1. Effect of support type and treatment*. Stability of preparations.
Supports Residual activity (%)
Protein loading(mg/g)
Bindingyield (%)
Stability Half-life (as reuses)
CPG - Acid washed 60 4.82 60.3 23 Hydrophobic 69 4.60 57.5 nd Hydrophilic 63 5.05 63.1 30 Spherosil15-Water w. 62 5.38 67.3 27 Acid washed 51 5.50 68.7 nd Hydrophobic 71 5.15 64.4 nd Hydrophilic 62 5.66 70.8 33 Spherosil 5-Water w. 54 3.93 49.1 nd Acid washed 55 3.88 48.5 nd Hydrophobic 58 3.84 48.0 17 Hydrophilic 56 4.03 50.4 nd Merckogel 53 4.22 52.8 15 Quartz powder 26 1.86 23.2 4 Pumice stone 46 3.00 37.5 8 Silica gel 27 2.29 28.6 11 *Immobilization - 8 mg trypsin/g support, 0.2M phosphate buffer, 4°C, 4 h stirring time, 100 rpm.
The residual activity and protein loading of various supports was studied and the results are shown in Table 1. Comparison between the hydrophobic and hydrophilic beads revealed that the hydrophobic beads had low protein loading but higher residual activity in contrast to hydrophilic porous carriers. The effect of a varying degree of hydrophilicity of porous carriers on the immobilization efficiency was studied. It was found that the protein loading increased with the increase in hydrophilicity of the supports in the following order: Water washed Spherosil 15< Acid washed Spherosil 15<Hydrophilic Spherosil 15. Similar were the results for CPG and Spherosil 5. This observation is in accordance with the fact, that the electrostatic forces are controlling the adsorption of protein on hydrophilic surfaces and the protein loading increases with the increase in hydrophilicity of the support. The residual activity and protein loading increased when

porous carriers with pore diameter 220-350 nm were used (Spherosil 15 and CPG). Satisfactory residual activity of 46% was obtained with pumice stone as carrier but the binding yield was only 37.5%. Comparing the other ethanol washed silicates – Merckogel, Silica gel and quartz powder, it was found that the residual activity and protein loading increases with decrease in particle size of the support. Results for Merckogel as carrier were comparable to the obtained with Spherosil 5.
The catalytic properties and kinetic parameters, as well as the stability of the immobilized trypsin were compared to those of the free enzyme. Results showed that the optimum temperature for the immobilized on Spherosil 15 trypsin was 50°C, optimum pH for hydrolysis of BAPNA was 8.2, both of which were higher than that for the free form (Fig.9). The immobilized trypsin exhibited much better thermal stability, its half-life at 50°C reached 50-55 minutes (Fig.10), and maintained over 50% of its initial activity after reusing up to 33 times (Table 1).
It was found that the apparent Km (Michaelis constant) of the immobilized trypsin on Spherosil 15 porous silica beads was 2.1.10-3 M (BAPNA) compared to the value of 1.5.10-3 M for the native trypsin, Vmax being similar, unaffected by the immobilization. Km values can significantly vary from enzyme to enzyme and even for different substrates of the same enzyme. Hence, the immobilization of trypsin to silica carriers affected the enzyme affinity to its substrate BAPHA. Thus, the results proved that immobilization by adsorption on porous silica beads caused low accessibility of the substrate to the enzyme active sites, as it was reported also for bacterial proteases and trypsin [14,24].
References 1. Katchalski-Katzir, E., et al. J Mol Catalysis B: Enzymatic, 2000,
10, 157-176. 2. Kasai, K. J Chromatography, 1992, 597, 3-18. 3. Thelohan, S., et al. Chromatographia, 1989, 28, 551-555. 4. Svec, F. Electrophoresis, 2006, 27, 947-961. 5. Migneault, I., et al. Electrophoresis, 2004, 25, 1367-1378. 6. Ye, M., et al. Electrophoresis, 2004, 25, 1319-1326.

7. Kenkova, J., Electrophoresis, 2004, 25, 3550-3563. 8. Lorenzen, P.C., et al. Z Ernahrungswiss, 1995, 34, 118-30. 9. Hartmeier, W. Immobilized Biocatalysts. Berlin: Springer-Verlag,
1988. 10. Weetall, H.H. In: Methods Enzymol. (Mosbach K., ed.), New
York, Academic Press, 1976, 44, 134-140. 11. Xue, B., et al. Sep Sci Technol., 2001, 36, 2449-2461. 12. Kang, K., et al. Macromol Biosci. 2005, 5, 344-451. 13. Liu, C.G., et al. J Agric Food Chem., 2005, 53, 1728-33. 14. Hu, J., et al. Biotechnol J., 2006, 1, 75-79. 15. Jiang, H., et al. J. Chromatogr A, 2000, 903, 77-84. 16. Bryjak, J., et al. Proc. Biochem., 1998, 33, 409-417. 17. Hong, J., et al. J Mol Catalysis B: Enzymatic, 2007, 45, 84-90. 18. Nonaimi, M., et al. Enzyme Microb Technol., 2001, 29, 567-574. 19. Keinan, E., et al. J Am Chem Soc., 1986, 108, 162-169. 20. Kelly, N., et al. Biotechnol Bioeng., 1977, 19, 1211-1213. 21. Kierstan, M.P.J., et al. In: Protein immobilization: Fundamentals
and applications, Taylor, RF., ed., Marcel Dekker, New York, 1991, 13-71
22. Buchloz K., et al. Biokataysatoren und enzymtechnologie. VCH, Weinheim, 1997.
23. Mansfeld J., et al. Biotechnol Appl Biochem, 2000, 32,189-195. 24. Bradford M.M. Analytical Biochem , 1976, 72,248-254. 25. Geiger R., Fritz H. In: Methods of Enzymatic Analysis, 3rd edn.,
Verlag Chemie, Germany,1984, 5, 119-128. 26. Dixon M., Webb E. (eds) Enzymes. 3rd edn. Longman, London,
UK, 1979.

Основни характеристики на циклодекстрин глюканотрансферази от два алкалофилни щама
Bacillus pseudoalkalophilus 20RF и Bacillus pseudoalkalophilus 8SB
Н. Атанасова, В. Иванова,* В. Петкова, А. Тонкова Институт по микробиология, БАН, секция “Екстремофилни
бактерии”, ул. Акад. Г. Бончев бл. 26, 1113 София * Университет по хранителни технологии
Технологичен факултет Катедра “Органична химия и микробиология”
бул Марица 26, 4002 Пловдив, E mail: [email protected]
Abstract: Partially purified by ultrafiltration CGT-ase preparations were obtained. Some of their characteristics were determined – the temperature optimum was at 65°C, thermostability, their two pH-optima were at pH 6.0 and at pH 9.0 and the pH-stability.
Keywords: CGT-ase, Bacillus, characterization.
Увод
Ензимът циклодекстрин глюканотрансфераза (CGT-за, EC 2.4.1.19) превръща нишестето и други сродни α-глюкани в циклодекстрини (CDs), притежаващи хидрофилна външна страна на пръстена и хидрофобна централна кухина. CD-те са хомогенни циклични нередуциращи олигозахариди, в които глюкозните единици са свързани чрез α-1,4-глюкозидна връзка. Повечето от CGT-зите продуцират голямо количество циклодекстрини със степен на полимеризация (DP) 6, 7 и 8 глюкозни единици, които се наричат съответно α-, β- и γ–циклодекстрини. CGT-те също продуцират и циклодекстрини със степен на полимеризация от 9 до 60 глюкозни единици [1]. Способноста на CD-те да капсулират различни химични съединения и така да променят техните физични и химични свойства, определя тяхното широко приложение и значение за опазването на околната среда,

медицината, фармацевтичната и химическата промишлености. Освен циклизация - превръщане на нишестето и сродни α-1,4 глюкани в CD-ни чрез вътрешномолекулна трансглюкозилираща реакция, ензимите също катализират и купелуваща реакция - отваряне на пръстените на CD-ните, образуване на линейни малтоолигозахариди и прехвърлянето им към акцептори, и диспропорционална реакция - прехвърляне на линейните малтоолигозахариди към акцептори чрез междумолекулна трансглюкозилираща реакция. Освен това CGT-те притежават слаба хидролитична активност и превръщат CD-ните един в друг [2,3]. Установено е, че тези четири реакции са взаимно свързани [4] (Фиг. 1).
Фиг. 1. Реакции, катализирани от ензима CGT-а. Gn, Gm, Gx, Gy - - линейни олигозахариди с n, m, x, y глюкозни единици; cGx и cGy - циклодекстрини, съставени от x и y глюкозни единици.
В зависимост от ензимната и субстратната концентрации и
присъствието на малтоолигозахариди с различна степен на полимеризация в реакционната смес, всяка една от тези реакции може да бъде доминантна, независимо, че диспропорционалната реакция е основната реакция, катализирана от CGT-зите [5]. Експерименталните данни за различни бактериални CGT-зи показват, че когато в реакционната смес се натрупат малтоолигозахариди със степен на полимеризация повече от 7
cGy + G(n-y)
cyclization
G(n-x) +Gm+x
Gn G(n-x) + cGx

глюкозни единици, тези малтоолигозахариди са директни субстрати за циклизиращата реакция. Нискомолекулните олигозахариди не могат да бъдат директни субстрати за образуването на циклодекстрини. Когато тяхната концентрация в реакционната смес се повиши, тогава се активира диспропорционалната реакция [6-10].
Ензимът се продуцира от голям брои бактерии: (а) аеробни мезофилни бактерии като напр.: Bacillus macerans [11,12], Bacillus megaterium [13,14], Bacillus cereus [15], Bacillus firmus NCIM 5119 [16], Bacillus circulans DF 9R [17], Bacillus amyloliquefaciens [18], Klebsiella pneumonie [19], Klebsiella oxytoca [14], Micrococcus luteus [20]; (b) аеробни термофили Bacillus stearothermophilus [21,22]; (c) анаеробни термофили: Thermoanaerobacterium thermosulfurigenes [23]; (d) аеробни алкалофили Bacillus sp. AL-6 [24], Bacillus firmus 324 [25]; Bacillus firmus var. alkalophilus [26], Bacillus agaradhaerens [27], Bacillus оhbensis [28], Bacillus sp. No 38-2 [29], Bacillus sp. 1011 [30], Bacillus sp. A2-5a [31]; (e) анаеробни термоалкалофили Anaerobranca gottschalkii [32]; (f) хипертермофилни архебактерии Thermococcus sp. B-1001 [9].
CGT-те показват максимална активност в рН граници от 4.0 до 9.0 и в температурни граници от 40ºC - 100ºС. Първата изолирана, пречистена и кристализирана CGT-за е тази от Bacillus macerans [12], която е стабилна при рН между 8.0-10.0 [11] и запазва 100% от първоначалната си активност при температура под 60ºС [33].
CGT-зите от някои алкалофили притежават два или три рН оптимума в киселата и алкална област на рН. Например, суров ензим от B. circulans ATCC 21783 показва три pH оптимума, при pH 5.5, 6.0 и 8.5 [34]. Ензимът с рН оптимум в алкалната област е по-лабилен при топлинно третиране, за разлика от ензимът с рН оптимум в киселата област [35]. CGT-за от алкалофила Bacillus sp. AL-6 е стабилна в рН граници от 5.0 до 8.0 при 40ºС за 3 часа [24]. Пречистена CGT-за от Bacillus firmus 324 е стабилна между рН 6.5 и 9.0 [25]. Ензимът е стабилен при 55ºС, но неговата термостабилност се повишава до 70ºС чрез добавяне на 1mM CaCl2 към реакционната смес. Пречистена CGT-а от Bacillus

firmus NCIM 5119 в отсъствие на субстрат бързо губи своята активност над 30ºС [16] т.е. ензимът е термолабилен. CGT-та от Bacillus agaradhaerens е стабилна в много широки рН граници - от 5.0 до 11.4 при 25ºС [27] като термалната стабилност се повишава в присъствието на субстрат и CaCl2.
CGT-та от алкалофила Bacillus ohbensis sp. nov C-1400 продуцира основно γ-CD-ни. Ензимът продуцира и малко количество β-CD-ни, но не и α-CD-ни за разлика от други CGT-зи [36]. Клонирането на CGT-ият ген от B. ohbensis в E. coli не само повишава продукцията на ензима, но също подобрява и свойствата му за по ефективна γ-CDs продукция. Пречистени ензимни препарати от културалната супернатанта на B. оhbensis и от периплазматичната фракция от трансформанта E. coli показват еднакви ензимни свойства: оптимално рН за циклизираща активност 5.0 (суб оптимална 10.0), pH-стабилност между рН 6.5 и 10.0, оптимален температурен оптимум 55оС и температурна стабилност под 45 оС.
За разлика от много бактериални CGT-зи, ензимът, продуциран от термофилния анаероб Thermoanaerobacterim thermosulfurigenes EM1 се характеризира с висока термостабилност и температурен оптимум [23]. След 2 часа инкубиране при 90оС, ензимът запазва 100% от цикличната си активност в присъствие на 10 mM CaCl2 и 1% нишесте. При 100ºС ензимът запазва 50% от своята първоначална активност след третиране в продължение на 30 минути. Хипертермофилния анаеробен археон Thermococcus sp. B-1001, изолиран от горещи извори в Япония, продуцира екстремно термостабилна CGT-за [9]. Eнзимът образува главно α-CD и малко количество от β- и γ-CD. Този ензим може да действа при кисело рН в границите от рН 4.0 до 5.0 и затова е приложим както за втечняване на нишестето, така и за образуване на CD-ни, без да се изисква корекция на рН.
Материали и методи Микроорганизми и хранителна среда. Използвани са два облигатни алкалофилни щама Bacillus, изолирани от български региони, от колекцията на секция Екстремофилни бактерии,

Институт по микробиология, БАН. Направени са 16S rRNA анализи и пълните секвенции са публикувани в Ген-банката на NCBI (Национален център за биотехнологична информация), САЩ. Двата щама са представители на Bacillus pseudoalkalophilus - Bacillus pseudoalkalophilus 8SB (No на секвенцията EF589780) и Bacillus pseudoalkalophilus 20RF (No на секвенцията EF589779). В някои от графичните резултати са представени съответно като Bacillus sp. 8SB и Bacillus sp. 20 RF. Хранителната среда за поддържане и култивиране е следния състав: 0.5% пептон, 0.5% дрождев екстракт, 0.02% MgSO4 безводен, 0.1% K2HPO4 безводен, 0.2% нишесте. След автоклавиране pH на средата се довежда до 9.8-10.0 със стерилен Na2CO3. Определяне β-CGT-зна активност. Ензимната активност е определена по метода на Kaneko et al. [34]. Една единица ензимна активност е дефинирана като количеството ензим, което формира 1 μg β-циклодекстрин за 1 мин при условията на ензимната реакция (60ºC, фосфатен буфер pH 6.0, 20 мин реакционно време). Получаване на частично пречистен ензим. След центрофугиране на културалната течност при 4000 rpm за 20 мин., получената бистра супернатанта е концентрирана и частично пречистена чрез пропускане през мембрани PM30 (апарат за ултрафилтрация Millipore, Massachsetts, USA). Определяне на температурен оптимум (профил). Ензимната реакция е проведена при pH 6.0, за време 20 мин при различни температури: 40, 50, 60, 65, 70, 75 и 80ºC. Определяне на pH-оптимум. Ензимната реакция е проведена при 60ºC за време 20 мин с буфери с различни pH-стойности: 0.1M Na-ацетат-CH3COOH-буфер (pH 5.0-6.0), 1/15M калиево-натриев фосфатен буфер (pH 6.0-8.0) и 0.1M глицин-NaOH-NaCl-буфер (pH 8.5-10.0). Определяне на pH-стабилност. pH-Стабилността на ензимите е определена чрез предварително инкубиране в различни буфери с pH от 4 до 11 за 30 мин при 25ºC. Остатъчната ензимна активност е определена при стандартните условия на ензимната реакция. Активността на нетретирана ензимна проба се приема за 100% и служи за контрола.
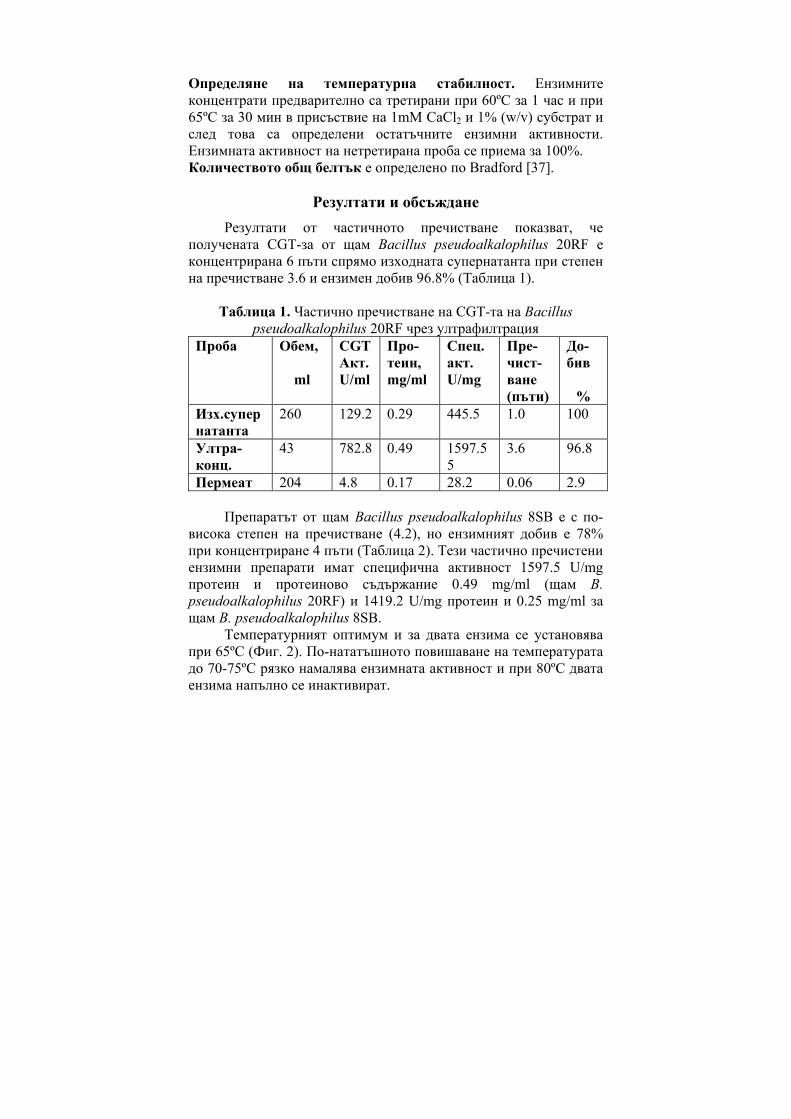
Определяне на температурна стабилност. Ензимните концентрати предварително са третирани при 60ºC за 1 час и при 65ºC за 30 мин в присъствие на 1mM CaCl2 и 1% (w/v) субстрат и след това са определени остатъчните ензимни активности. Ензимната активност на нетретирана проба се приема за 100%. Количеството общ белтък е определено по Bradford [37].
Резултати и обсъждане Резултати от частичното пречистване показват, че
получената CGT-за от щам Bacillus pseudoalkalophilus 20RF е концентрирана 6 пъти спрямо изходната супернатанта при степен на пречистване 3.6 и ензимен добив 96.8% (Таблица 1).
Таблица 1. Частично пречистване на CGT-та на Bacillus
pseudoalkalophilus 20RF чрез ултрафилтрация Проба
Обем,
ml
CGT Акт. U/ml
Про-теин, mg/ml
Спец. акт. U/mg
Пре-чист-ване (пъти)
До-бив
% Изх.супер натанта
260 129.2 0.29 445.5 1.0 100
Ултра-конц.
43 782.8 0.49 1597.55
3.6 96.8
Пермеат 204 4.8 0.17 28.2 0.06 2.9
Препаратът от щам Bacillus pseudoalkalophilus 8SB e с по-висока степен на пречистване (4.2), но ензимният добив е 78% при концентриране 4 пъти (Таблица 2). Тези частично пречистени ензимни препарати имат специфична активност 1597.5 U/mg протеин и протеиново съдържание 0.49 mg/ml (щам B. pseudoalkalophilus 20RF) и 1419.2 U/mg протеин и 0.25 mg/ml за щам B. pseudoalkalophilus 8SB.
Температурният оптимум и за двата ензима се установява при 65ºC (Фиг. 2). По-нататъшното повишаване на температурата до 70-75ºC рязко намалява ензимната активност и при 80ºC двата ензима напълно се инактивират.
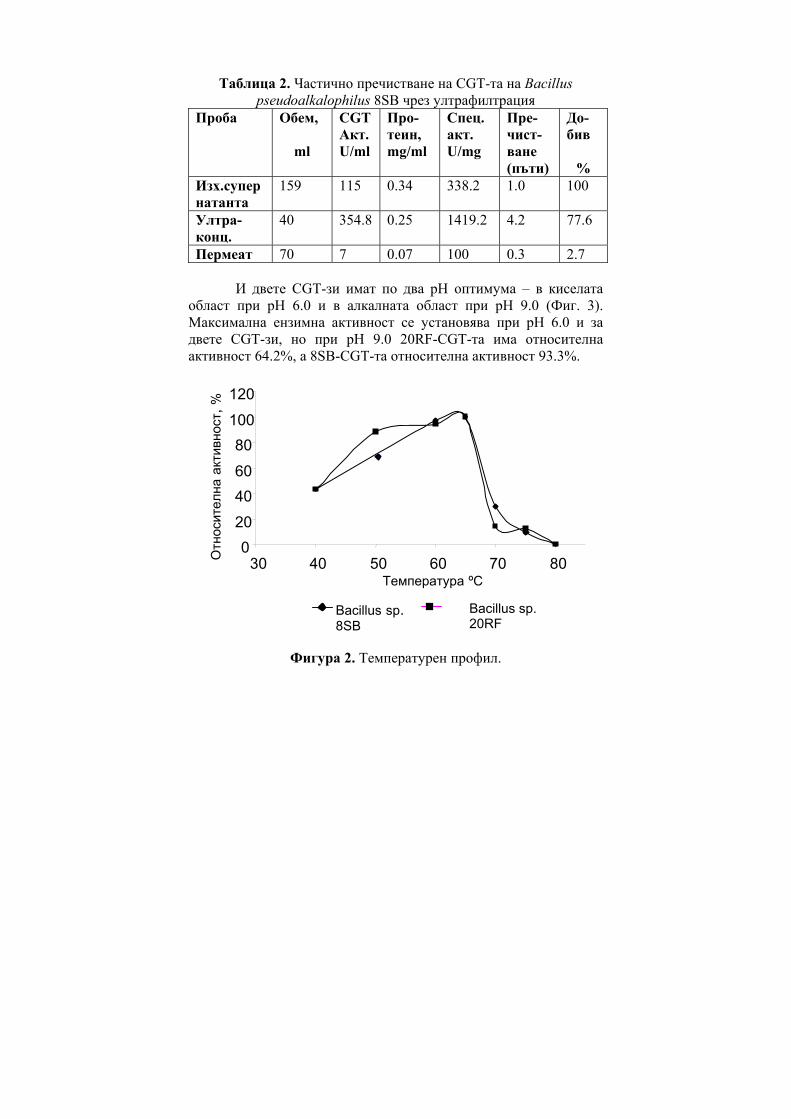
Таблица 2. Частично пречистване на CGT-та на Bacillus pseudoalkalophilus 8SB чрез ултрафилтрация
Проба Обем,
ml
CGT Акт. U/ml
Про-теин, mg/ml
Спец. акт. U/mg
Пре-чист-ване (пъти)
До-бив
% Изх.супер натанта
159 115 0.34 338.2 1.0 100
Ултра-конц.
40 354.8 0.25 1419.2 4.2 77.6
Пермеат 70 7 0.07 100 0.3 2.7
И двете CGT-зи имат по два pH оптимума – в киселата област при pH 6.0 и в алкалната област при pH 9.0 (Фиг. 3). Максимална ензимна активност се установява при pH 6.0 и за двете CGT-зи, но при pH 9.0 20RF-CGT-та има относителна активност 64.2%, а 8SB-CGT-та относителна активност 93.3%.
Фигура 2. Температурен профил.
0
20
4060
80100
120
30 40 50 60 70 80Температура ºC
Относителна
активност
, %
Bacillus sp. 8SB
Bacillus sp. 20RF
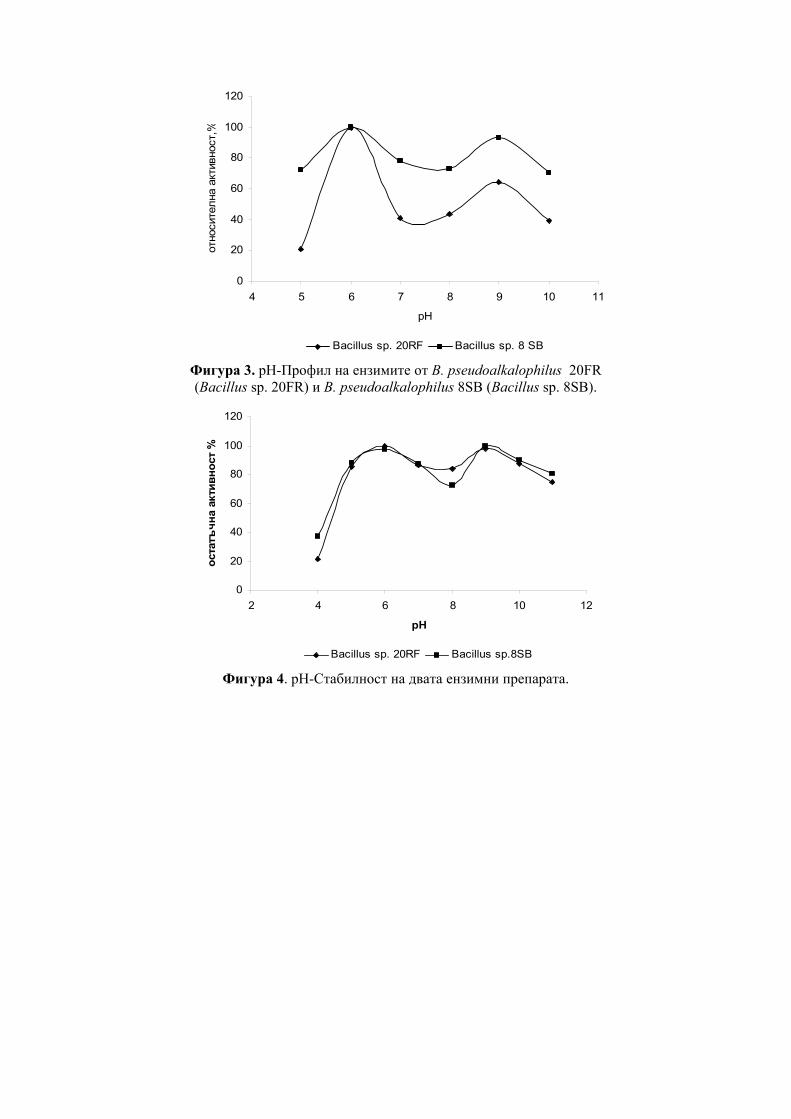
0
20
40
60
80
100
120
4 5 6 7 8 9 10 11
pH
относителна активност,
%
Bacillus sp. 20RF Bacillus sp. 8 SB
Фигура 3. pH-Профил на ензимите от B. pseudoalkalophilus 20FR (Bacillus sp. 20FR) и B. pseudoalkalophilus 8SB (Bacillus sp. 8SB).
0
20
40
60
80
100
120
2 4 6 8 10 12
pH
остатъ
чна активн
ост
%
Bacillus sp. 20RF Bacillus sp.8SB
Фигура 4. pH-Стабилност на двата ензимни препарата.

И двата ензима са стабилни в широки граници на pH – от 5.0 до 11.0 (Фиг.4). Различията между двата ензима се състоят в това, че този, получен от B. pseudoalkalophilus 20RF показва 100% остатъчна активност при pH 6.0, докато ензима, получен от B. pseudoalkalophilus 8SB e с 95.2% остатъчна активност при тази стойност на pH. Обратно, в алкалната област pH 9.0, 8SB-CGT-та е 100% стабилна и при pH 11.0 запазва 81% от активността си, докато 20RF-CGT-та има 98% остатъчна активност при pH 9.0 и 75 % - при pH 11.0.
Термалната стабилност беше определена в присъствието на Ca2+ и субстрат, които имат протективен ефект при топлинна денатурация, без да са извършени подробни изследвания относно влиянието на различни концентрации Ca2+, други реагенти, субстрат. Установените остатъчни активности след 1 час третиране при 60ºC в присъствие на 1m Ca2+ и 1% субстрат са съответно 73.3% за CGT-за от B. pseudoalkalophilus 29RF и 100% за CGT- за от щам 8SB, докато например CGT-зата от Bacillus firmus NCIМ е термолабилна и бързо губи активността си при температури над 30ºC [16]; CGT-зата от Bacillus agaradhaerens LS-3C след 1 час третиране в присъствие на 5% малтодекстрин или 5 mM Ca2+ е стабилна при температури до 45-50ºC [27]; CGT-зата на Bacillus ohbensis е стабилна само при температури под 45ºC [36].
В заключение може да се каже, че предварителната оценка на двата частично пречистени ензимни препарати показва възможността за успешното им приложение в конверсията на нишестето до циклодекстрини.
Това изследване е спонсорирано от МОН чрез ФНИ, грант ВУ-Х 301/07.
Литература
1. Terada, Y., et al. Appl. Environ. Microbiol. 2001, 67:1453-1460. 2. Palohemio, M. et al. Appl. Microbiol. Biotechnol. 1992, 36:584-
591. 3. Nakamura, A., Haga, K., Yamane, K. FEBS Letters. 1994,
337:66-70. 4. Tonkova A. Enzyme Microb. Technol., 1998, 22:678-686.

5. Nakamura, A., Haga, K., Yamane, K. Biochemistry. 1994, 33:9929-9936.
6. Abelian, V. A., Adamian , et al. Biokhimiya 1995, 60:891-897. 7. Abelian, V. A. et al. Biokhimiya 1994, 59:1122-1129. 8. Okada, T., Ito, M., Hibino, K. J .Ferment. Bioeng. 1994, 77:264-
267. 9. Tachibana, Y. et al. Appl. Environ. Microbiol. 1999, 65:1991-
1997. 10. Yoon, S-H., and Robyt, J. F. Carbohydrate Res. 2002, 337:2245-
2254 11. Kitahata, S. et al. Agric. Biol. Chem. 1974, 38:387-393. 12. Takano, T., Fukuda et al. J. Bacteriol . 1986, 166:1118-1122. 13. Kitahata, S., and Okada, S. Agric. Biol. Chem. 1974, 38:2413-
2417. 14. Lee, J., et al. Enzyme Microbi. Technol. 1992, 14:1017-1021. 15. Jamuna, R. et al. Appl. Biochem. Biotechnol. 1993, 43:163-176. 16. Gawande, B. N. et al. Appl. Microbiol. Biotechnol. 1999, 51:504-
509. 17. Rosso, A. M. et al. Microbial Cell Factories 2002, 1:1-11. 18. Abdel-Naby, M. A. et al. Biochem. Engineering J. 2000, 5:1-9. 19. Fogarty, W.M. In: Microbial Enzymes and Biotechnology (W.M.
Fogarty, Ed.) 1983, Applied Science Publishers Ltd, Essex, UK,pp.1-92
20. Lee, Y. and Kim, H. Biotechnol. Bieng. 1992, 39:977-983. 21. Fujiwara, S., Kakihara, H. et al. J. Bacteriol. 1992, 174:7478-7481 22. Chung, H-J. et al. J. Agric. Food Chem. 1998, 61:1502-1506. 23. Wind, R. D. et al. Appl. Environ. Microbiol. 1995, 61:1257-1265. 24. Fujita, Y., Tsubouchi, H. et al. J. Ferment Bioeng. 1990, 70:150-
154. 25. Yim, D.G. et al. J. Ind. Microbiol. Biotechnol. 1997, 18:402-405. 26. Park, T., et al. J. Microbiol . Biotechnol. 1999, 9:811-819. 27. Martins, R. F., et al. Enzyme Microb Technol.2002, 30:116-124. 28. Nishida, T. et al. FEMS Microbiol. Lett. 1997, 149:221-226. 29. Horikoshi, K. FEMS Microbiology review. 1996, 18:259-270. 30. Haga, K. et al. J. Mol .Biol. 1994, 237:163-164. 31. Ohdan, K. et al. Appl. Microbiol. Biotechnol. 2000, 53:430-434. 32. Prowe, S. G., et al. Int. Sys. Evol. Microbiol. 2001, 51:457-465.

33. Yu, E. K. S. et al. Appl. Microbiol. Biotechnol. 1988, 28:377-379. 34. Kaneko, T., Kato, T. et al. J.Jpn.Soc.Starch Sci. 1987, 34:45-48. 35. Nakamura, N., Horikoshi, K. Agric. Biol. Chem. 1976, 40:935-
941. 36. Sin, K-A.et al. Appl. Microbiol. Biotechnol. 1991, 35:600-605. 37. Bradford, M.M. Annal. Biochem. 1976, 72:248-254.

Пенни филми от разтвори на n-додецил-β-D-малтозид с додецил триметиламониев бромид Диляна Иванова,* Жана Ангарска,* Емил Манев**
*Факултет по природни науки ШУ „Епископ Константин. Преславски”, 9712 Шумен
E-mail: [email protected] **Химически факултет, СУ „Св. К. Охридски”, 1164 София
E-mail: [email protected]
Резюме: Изследвано е изтъняването на пенни филми, образувани от смесени разтвори на n-додецил-β-D-малтозид (C12G2) и додецил триметиламониев бромид (C12TAB) с молни съотношения 1:0, 50:1, 1:1, 1:50, 0:1 в отсъствието и в присъствието на NaBr. Интерферометрично са определени критичната и равновесна дебелина на филмите. Тоталната концентрация на ПАВ за всяка от смесите е равна на критичната ú концентрация на мицелообразуване (ККМ). Чрез молното съотношение на сместа са контролирани повърхностния товар, типа и електростатиката във филма.
Филмите от разтвори с молни съотношения 1:0, 50:1, 1:1 (без NaBr) не изтичат с времето и са с дебелина около 100 nm, докато при 1:50 е намерен скок в дебелината им.
За филмите при йонна сила (0.1 mol/l) е намерена разлика в типа на филма (NBF или CBF) в зависимост от молното съотношение на компонентите в сместа, но е установено, че природата на ПАВ не влияе върху стойността на коефициента на изтъняване. Влиянието на радиуса на филма върху скоростта на изтъняване и критичната дебелина е в съответствие с теорията на Манев и съавт.[5], което е доказателство, че тя е валидна и за филми от смесени разтвори.
Keywords: n-dodecyl-β-D-maltoside; foam films; drainage constant; equilibrium thickness; surface charge.

Обща част
Пяната е колоидно-дисперсна система (КДС), използвана в технологичната схема на процеса флотация, при получаване на биопродукти, храни, лекарства, козметични препарати, хартия, пласмаси и др. Съществена е и ролята ú за процеса миене, пречистване на водите, извличане на петрол и в пожарогасенето. Във всички тези области на науката и технологията, често е необходим начин за контролиране на стабилността ú Стабилността на пяната се определя от свойствата на тънките течни филми, образувани между мехурчетата в нея, наречени пенни филми (ПФ). Физикохимичната наука, изследвайки кинетичната и термодинамична стабилност на ПФ, предлага подходи за управляване на стабилността на пяната чрез подбор на вида и концентрацията на ПАВ и електролита, pH на средата и налягането [1]. Стабилността на ТТФ се обуславя от възникването на повърхностни сили в тях, водещи до появата на положителен компонент на разклинящото им налягане. Според класическата DLVO-теорията, такъв е електростатичният компонент (Пel), породен от електростатични сили на отблъскване при припокриване на опашките на двойните електрични слоеве, образувани около филмовите повърхности.
hel e
kTze
nkT κϕ −⎟⎠
⎞⎜⎝
⎛=Π
4tanh64 002 (1),
където: n е частичковата йонна концентрация; k е константа на Болцман; Т е абсолютната температура; ϕ0 е потенциала на фазовата граница; h е равновесната дебелина на филма; κ-1 е дебелината на дифузния слой. Връзката между потенциала на фазовата граница и повърхностния товар q0 се дава чрез уравн.. 2.
⎥⎦⎤
⎢⎣⎡=
RTFRTcq el 2
sinh8 000
ϕεε (2),
където: ε0 и ε са диелектричната проницаемост във вакуум (8.85×10-12 C2/J×m) и диелектричната константа на водата (ε = 80.0); R e газовата константа, равна на 8.3 Jmol-1K-1 и F е фарадеевото число = 95000С. Пel зависи от повърхностния товар, определен от природата на фазовите граници и от адсорбцията и

природата на ПАВ. Експериментално е показано, че филмите от разтвори на йонни ПАВ са стабилни при повърхностен товар от порядъка на 10-40 mC/m2 , докато при нейонни ПАВ той е от порядъка на 0.5-2.5 mC/m2.
В зависимост от природата и концентрацията си ПАВ по различен начин създават или променят товара на филмовите повърхности. За йонните ПАВ, отрицателният товар (1.6 mC/m2) на фазова граница вода/въздух е пренебрежимо нисък и поради това товарът на фазовите ата граници във филма (отрицателен или положителен) преимуществено се определя от тяхната адсорбция. Следователно увеличаването на концентрацията на йонните ПАВ води до нарастване на електростатичната стабилизация.
Поради редица предимства нейонните ПАВ са по-предпочитаните в практиката [2]. Тяхната адсорбцията към отрицателно заредената фазова граница вода/въздух води до намаляване на повърхностния товар и филмовата дебелина т.е създават се условия за електростатична дестабилизация. Това налага използването им в комбинация с йонни ПАВ, което позволява едно прецизно контролиране на повърхностния товар, а от там и на филмовата стабилност. Следва, че за практиката е актуален подборът на ПАВ в използваните смеси като стабилизатори на КДС. От екологична гледна точна ПАВ трябва да са биоразграждащи се, нетоксични, нечувствителни към температура, pH и твърдостта на водата. Захаридното нейонно ПАВ n-додецил-β-D-малтозид отговаря на тези условия [3], поради което както индивидуалната му повърхностна активност, така и тази на сместа му с катионното ПАВ додецил триметил амониев бромид е широко изследвана [4]. Подборът на това катионно ПАВ, не е случаен. Чрез промяна на молната му част в сместа може да бъде контролирана плътността на повърхностния товар, т.е дебелината и вида на пенния филм, с други думи контролира се енергетичния бариер, определящ стабилността на филма. Освен това, изследването на тази смес като стабилизатор е актуално, поради антибактериалното, антифунгативно и антисептично действие на C12TAB. Очаква се с успех тя да бъде използвана в детергентната химия, селското стопанство и

козметиката. Изследванията на пенни филми от смес на C12G2 с C12TAB в [4] са насочени към равновесното им състояние. Известно е, че филмите не винаги успяват да достигнат до равновесно състояние, дори и да са на лице факторите, обуславящи термодинамичната им стабилност. Възможно е да се скъсат още в процеса на изтичането си или при прехода към ново равновесно състояние (критична дебелина на скъсване), поради хидродинамични смущения, пораждащи се в тях. Факторите, предпазващи филмите от скъсване по време на тяхното из-тъняване определят кинетичната им стабилност. Тя зависи от свойствата на адсорбционните слоеве, образуващи се на филмовите фазови граници и в много случаи е определящата. Малко са изследванията, свързани с кинетичната стабилност на тези филми, което определи и целта на настоящото изследване.
Целта на настоящата работа е да се изследва кинетичната стабилност на пенни филми от смес на n-додецил-β-D-малтозид (C12G2) с додецил триметиламониев бромид (C12TAB) и да се покаже как чрез вариране на количеството на C12TAB в сместа се променя повърхностния товар, типа на филма, т.е. се контролира електростатичния бариер, определящ стабилността на пяната.
Еволюцията и скоростта на филмовото изтъняване могат да бъдат описани и определени чрез регистриране на кинетиката на изтичане, т.е. чрез експериментално измерване на дебелината на филма с времето t. Според Манев и съавт. [5] за скоростта на изтъняване на хоризонтален пенен филм с радиус r, образуван от разтвор на ПАВ с вискозитет μ и повърхностно напрежение σ под действието на смучещата сила ΔP =Pσ-Π е в сила уравн. 3.
( )543
812
MTR46
1r
hV
σμσ Π−Ρ
= (3)
Скоростта на изтъняване може да бъде зададена и чрез коефициента на изтъняване α, определящ се от наклона на линейната зависимост dthd /ln , следваща от уравн. (4) (процедурата е подробно описана в [6]):
thh lnln α−= (4)

където: ho е началната реална дебелина или условно избраната (1 s след максимума в кинетичната крива); h е реална дебелина в даден момент t; t е време за изтъняване от ho до h;.
В настоящото изследване константата α e използвана за анализиране на кинетичното поведение на изследваните пенни филми. Чрез нея се изчисляват и критичните дебелини на филмите. Това ú приложение е важно, защото изчислените дебелини с реалната скорост на изтъняване не са зависими от специален модел, използван в уравненията за изтъняването.
Експериментални резултати и обсъждане В настоящата работа е изследвано кинетичното поведение
на пенни филми, образувани от смесени разтвори на C12G2 с C12TAB (50:1; 1:1; 1:50) с тотална концентрация равна на ККМ на сместа. Молните съотношения са заимствани от [4] с цел получе-ните от нас данни да бъдат корелирани с данните за равновесните филми. При тези условия са проведени две серии експерименти с филми: от разтвори в отсъствие на електролит, в които йонната сила се определя от концентрацията на C12TAB и от разтвори при постоянна йонна сила 1×10-1 mol/l, създадена от концентрацията на C12TAB и добавено количество NaBr.
Пенните филми са получени при 25°C±1°C в капилярна клетка [1] и наблюдавани в отразена монохроматична светлина (551 nm). Кинетиката на изтъняване на получените пенни филми е регистрирана до критичното или равновесното им състояние чрез интерферометричен метод. От регистрираните кинетичните криви на изтъняване на филми са определени:
коефициента на изтъняване, α; критичната (hcr ) и равновесна (heq) дебелина. Експериментално е проверено влиянието на: тоталната
концентрация на ПАВ в сместта; съотношението между ПАВ в сместа; йонната сила и радиуса на филма (0.05, 0.1, 0.15 mm) върху коефициента на изтъняване на филма, критичната му и равновесна дебелина. Проверена е и специфичната роля на C12G2 или C12TAB върху кинетичните свойства на филмите.
Получените резултати са систематизирани в Таблици 1 и 2.
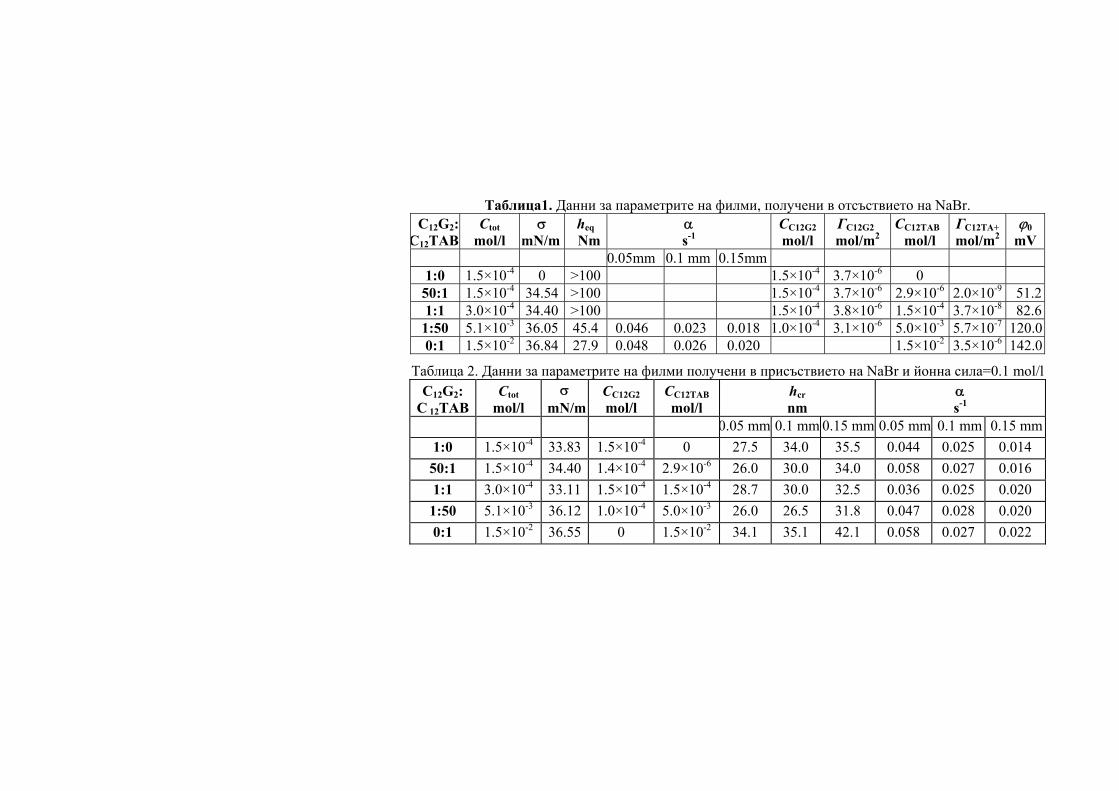
Таблица1. Данни за параметрите на филми, получени в отсъствието на NaBr. C12G2:
C12TABCtot
mol/l σ
mN/mheq Nm
α s-1
CC12G2mol/l
ГC12G2 mol/m2
CC12TAB mol/l
ГC12TA+ mol/m2
ϕ0 mV
0.05mm 0.1 mm 0.15mm 1:0 1.5×10-4 0 >100 1.5×10-4 3.7×10-6 0
50:1 1.5×10-4 34.54 >100 1.5×10-4 3.7×10-6 2.9×10-6 2.0×10-9 51.2 1:1 3.0×10-4 34.40 >100 1.5×10-4 3.8×10-6 1.5×10-4 3.7×10-8 82.6
1:50 5.1×10-3 36.05 45.4 0.046 0.023 0.018 1.0×10-4 3.1×10-6 5.0×10-3 5.7×10-7 120.0 0:1 1.5×10-2 36.84 27.9 0.048 0.026 0.020 1.5×10-2 3.5×10-6 142.0
Таблица 2. Данни за параметрите на филми получени в присъствието на NaBr и йонна сила=0.1 mol/l C12G2:
C 12TABCtot
mol/l σ
mN/mCC12G2 mol/l
CC12TAB mol/l
hcr nm
α s-1
0.05 mm 0.1 mm 0.15 mm 0.05 mm 0.1 mm 0.15 mm 1:0 1.5×10-4 33.83 1.5×10-4 0 27.5 34.0 35.5 0.044 0.025 0.014
50:1 1.5×10-4 34.40 1.4×10-4 2.9×10-6 26.0 30.0 34.0 0.058 0.027 0.016 1:1 3.0×10-4 33.11 1.5×10-4 1.5×10-4 28.7 30.0 32.5 0.036 0.025 0.020
1:50 5.1×10-3 36.12 1.0×10-4 5.0×10-3 26.0 26.5 31.8 0.047 0.028 0.020 0:1 1.5×10-2 36.55 0 1.5×10-2 34.1 35.1 42.1 0.058 0.027 0.022

В колона 1 на тези таблици е даден съставът на използваните смеси, представен чрез молното съотношение между двете ПАВ. В началото и в края на колоната са включени и индивидуални ПАВ (1:0 е за разтвор само от C12G2, а 0:1 е за разтвор само от C12TAB). Тоталната сърфактантна концентрация е ККМ на всяка смес. Оценените адсорбции , Г, на фазовата граница течност/газ и повърхностният товар, ϕ0, са получени от изотермите за повърхностно напрежение [4], анализирани в приближение на ван дер Ваалс. В колона 4 на таблица 1 сме представили равновесните дебелини на филмите. Вижда се, че филмите от разтвори с молно съотношение 1:0, 50:1 и 1:1 в отсъствие на NaBr са с дебелина около 100 nm и за тях липсва стойност на α, тъй като след тази дебелина те не изтичат с времето. При тези съотношения концентрацията на C12G2 съответства на неговата ККМ, а концентрацията на C12TAB, който играе ролята на електролит е изключително ниска. Освен това от адсорбциите следва, че поради ниската си повърхностна активност, сравнена с тази на C12G2 той остава в разтвора. Следва, получените дебели филми са електростатично стабилизирани, т.е. C12G2 не е успяло съществено да неутрализира отрицателния товар на филмовите повърхности. Скок във дебелината на филмите е намерен при съотношение 1:50. Оценената за това съотношение адсорбция е с порядък по-висока 5.7×10-7 mol/m2 от тази при 1:1. Филмите са с дебелина около 45 nm, което означава, че адсорбиралият се C12TAB не само е неутрализира отрицателния товар на филмовите повърхности, но и ги презарежда. Основанието за този извод следва и от направената от нас оценка, според която адсорбция от 1.7×10-7 mol/m2 е достатъчна за неутрализиране на отрицателния товар на границата вода/газ (1.6 mC/m2). Дебелината на филмите от разтвор само на C12TAB намалява два пъти, поради действието на C12TAB като електролит в разтвора. От приведените и обсъдени резултати следва, че доста голямо количество C12TAB е нужно, за да се промени кинетиката на филмите, при това обаче, видът им се запазва (CF). Това се потвърждава и от еднаквата скорост на изтичане при постоянен филмов радиус (виж данни за α в Табл.
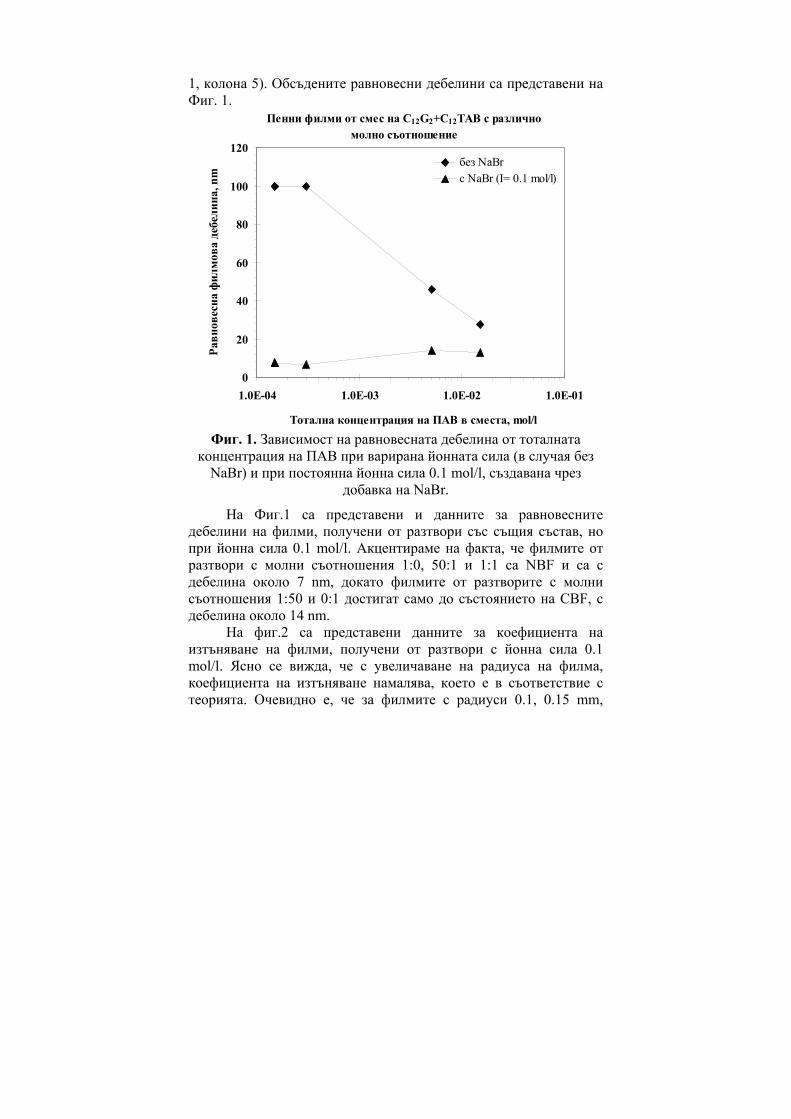
1, колона 5). Обсъдените равновесни дебелини са представени на Фиг. 1.
Пенни филми от смес на C12G2+C12TAB с различно молно съотношение
0
20
40
60
80
100
120
1.0E-04 1.0E-03 1.0E-02 1.0E-01
Тотална концентрация на ПАВ в сместа, mol/l
Равновесна
филмова дебелина
, nm
без NaBrс NaBr (I= 0.1 mol/l)
Фиг. 1. Зависимост на равновесната дебелина от тоталната
концентрация на ПАB при варирана йонната сила (в случая без NaBr) и при постоянна йонна сила 0.1 mol/l, създавана чрез
добавка на NaBr.
На Фиг.1 са представени и данните за равновесните дебелини на филми, получени от разтвори със същия състав, но при йонна сила 0.1 mol/l. Акцентираме на факта, че филмите от разтвори с молни съотношения 1:0, 50:1 и 1:1 са NBF и са с дебелина около 7 nm, докато филмите от разтворите с молни съотношения 1:50 и 0:1 достигат само до състоянието на CBF, с дебелина около 14 nm.
На фиг.2 са представени данните за коефициента на изтъняване на филми, получени от разтвори с йонна сила 0.1 mol/l. Ясно се вижда, че с увеличаване на радиуса на филма, коефициента на изтъняване намалява, което е в съответствие с теорията. Очевидно е, че за филмите с радиуси 0.1, 0.15 mm,
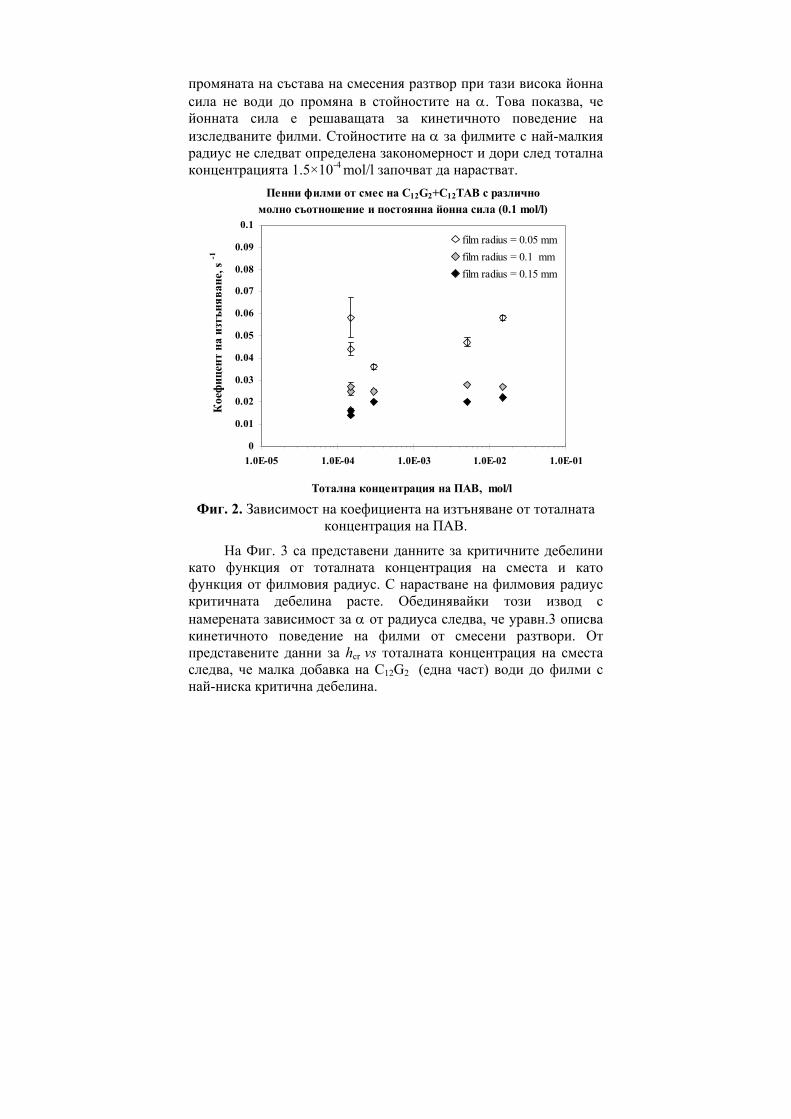
промяната на състава на смесения разтвор при тази висока йонна сила не води до промяна в стойностите на α. Това показва, че йонната сила е решаващата за кинетичното поведение на изследваните филми. Стойностите на α за филмите с най-малкия радиус не следват определена закономерност и дори след тотална концентрацията 1.5×10-4 mol/l започват да нарастват.
Пенни филми от смес на C12G2+C12TAB с различно молно съотношение и постоянна йонна сила (0.1 mol/l)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.1
1.0E-05 1.0E-04 1.0E-03 1.0E-02 1.0E-01
Тотална концентрация на ПАВ, mol/l
Коефицент на
изтъняване,
s-1
film radius = 0.05 mmfilm radius = 0.1 mmfilm radius = 0.15 mm
Фиг. 2. Зависимост на коефициента на изтъняване от тоталната
концентрация на ПАВ.
На Фиг. 3 са представени данните за критичните дебелини като функция от тоталната концентрация на сместа и като функция от филмовия радиус. С нарастване на филмовия радиус критичната дебелина расте. Обединявайки този извод с намерената зависимост за α от радиуса следва, че уравн.3 описва кинетичното поведение на филми от смесени разтвори. От представените данни за hcr vs тоталната концентрация на сместа следва, че малка добавка на C12G2 (една част) води до филми с най-ниска критична дебелина.
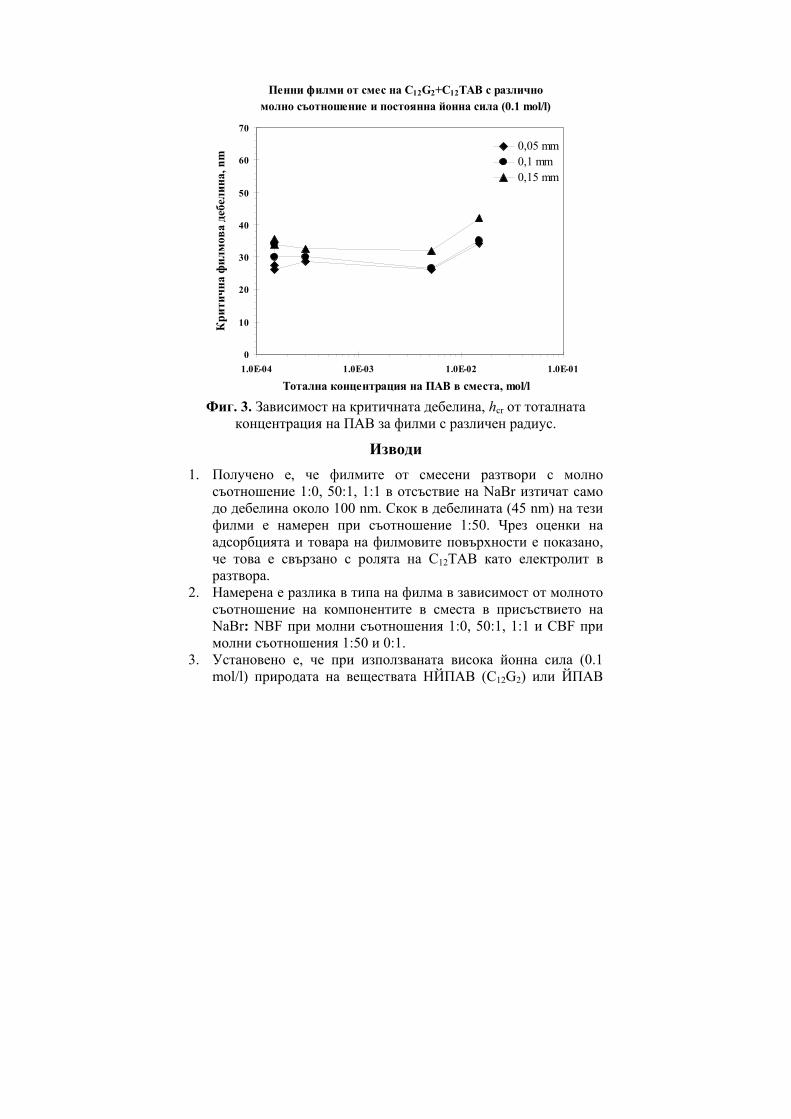
Пенни филми от смес на C12G2+C12TAB с различно молно съотношение и постоянна йонна сила (0.1 mol/l)
0
10
20
30
40
50
60
70
1.0E-04 1.0E-03 1.0E-02 1.0E-01
Тотална концентрация на ПАВ в сместа, mol/l
Критична филмова дебелина
, nm
0,05 mm0,1 mm0,15 mm
Фиг. 3. Зависимост на критичната дебелина, hcr от тоталната
концентрация на ПАB за филми с различен радиус.
Изводи 1. Получено е, че филмите от смесени разтвори с молно
съотношение 1:0, 50:1, 1:1 в отсъствие на NaBr изтичат само до дебелина около 100 nm. Скок в дебелината (45 nm) на тези филми е намерен при съотношение 1:50. Чрез оценки на адсорбцията и товара на филмовите повърхности е показано, че това е свързано с ролята на C12TAB като електролит в разтвора.
2. Намерена е разлика в типа на филма в зависимост от молното съотношение на компонентите в сместа в присъствието на NaBr: NBF при молни съотношения 1:0, 50:1, 1:1 и CBF при молни съотношения 1:50 и 0:1.
3. Установено е, че при използваната висока йонна сила (0.1 mol/l) природата на веществата НЙПАВ (C12G2) или ЙПАВ

(C12TAB) не влияе върху стойността на коефициента на изтъняване.
4. Данните за влиянието на радиуса на филма върху стойностите на скоростта на изтъняване и критичната дебелина показаха, че теоретичните зависимости са валидни и за описание на кинетичното поведение на филми от смесени разтвори.
Литература 1. Кругляков, П. М., Ексерова, Д. Р., “Пена и пенные пленки”,
Химия, 1991, Москва. 2. Manev, E. D., Pugh, R. J., Langmuir, vol. 7, (1992) 2253-2260. 3. Stubenrauch, C., Schlarmann, J., Strey R., Phis. Chem. Chem.
Phis., vol. 4, (2002) 4504-4513. 4. Buchavzov, N., and C. Stubenrauch, Langmuir vol. 23 (2007)
5315-5323 5. Manev, E., Tsekov, R., Radoev, B., J. Colloid and Interface Sci.,
vol. 18, (1997) 769-788. 6. Angarska, J., Stubenrauch, C., and E. Manev, Colloids&Surfaces
A; vol. 309, 1-3 (2007) 189-197.

Относно получаването на химически модифициран инулин и използването му в O/W
емулсии Пантелей Денев, Мина Тодорова, Кремена Никовска,
Стамен Стамов Университет по хранителни технологии
4002 Пловдив, бул. Марица 26, Е-Мail: [email protected]
Abstract: The possibilities of obtaining chemical modified inulin with improved hydrophobic properties were demonstrated. Esters of inulin with different fatty acids (acetic, lauric, palmitic, stearic) were obtained. HLB of the inulin products were determined. The emulsifying and stabilizing properties of part of them were determined and analyzed. The rheological properties, dispersion and emulsion stability of O/W emulsions were studied. The effect of the inulin products concentration and of the oil phase volume upon them was determined.
Key words: modified inulin, emulsions, rheological properties.
Въведение
Инулинът е линеен растителен полизахарид. Веригата му е изградена от β-фруктофуранозилни звена (F), свързани (2→1), а в редуциращия й край се намира α-глюкопиранозилен остатък (G) – (1→2).
Степента на полимеризация на инулина варира между n=2÷70. Тя зависи от вида на растението, от климатичните условия по време на растежа му и от физиологичната възраст на растението. Повишеният интерес към инулина се дължи на неговите пребиотични и функционални свойства.[4-6] В хранителната промишленост инулинът е текстуриращ компонент, има добра разтворимост, подобрява вкуса, замества захарта и мазнините.[6] Тези свойства дават възможност за включването му в редица технологични решения за производство на разнообразни

храни. [4,7] Синтезирането на модифицирани хидроколоиди с изразени повърхностно-активни свойства представлява особен интерес в хранителната технология за получаване на стабилни структурирани впенени и емулсионни продукти.[11]
Разширяват се и се задълбочават изследванията върху ма-леноводните емулсии, приготвяни с инулинови препарати с повишени хидрофобни свойства. Хидрофобно модифицираният инулин представлява амфофилна макромолекула с хидрофилна главна верига и хидрофобни странични ацилни групи. Във водна среда такива полимери участват във вътрешно- и междумолекулни хидрофобни взаимодействия, в резултат на които се проявяват необичайни свойства, определящи многообразни практически приложения. В зависимост от строежа на молекулите – степен на заместване и природа на хидрофобните групи, разпределението по дължината на главната верига, молекулната маса на главната верига – различните деривати притежават специфичен хидрофилен-липофилен баланс (HLB). Приложението им във фармацията, козметиката, хранителната промишленост като емулгатори, сгъстители, структурообразуватели зависи от HLB, концентрацията, температурата, разтворителя и т.н.
Познати са различни методи за получаването на такива амфофилни деривати, като хидроксилните групи от хидрофилния скелет на инулина взаимодействат с производните на карбоксилните киселини (киселинни халогениди, анхидриди, естери, фосфонати и изоцианати).[8-11]
Нейоногенни повърхностно-активни вещества от инулин с желани свойства се получават като се варира с време, температура, молни съотношения както между реагиращите вещества, така и между реагиращите вещества и съответния катализатор.
Целта на настоящата работа е да се получат модифицирани инулинови препарати с различен хидрофилен-липофилен баланс и изследване на възможността за приложението им в моделни М/В емулсии.

Материал и методи Използван е инулин от цикория Frutafit® TEX! на Foods
Consulting: сухо в-во – 95-99 %; съдържание на фруктоза, глюкоза, захароза ≤ 0.5 %; пепел ≤ 0.2 %; средна дължина на веригата ≥ 23.
HLB е определен по метода на Грифин прилаган за нейоногенни повърхностно-активни вещества [12].
Дериватите на инулина са получени чрез преестерификация с метилови естери на висшите масти киселини (ВМК) или естерификация с оцетен анхидрид. Реакцията се извършва при кипене в среда от метанол за 180 min. Като катализатори са използвани натриев метилат за естерите на ВМК и натриев ацетат за оцетния анхидрид.
Степента на естерификация (СЕ) и степента на заместване (СЗ) на естерифицираните инулинови препарати са определяни титриметрично.
Моделните М/В емулсии с емулгатор естерифициран инулинов препарат – инулин стеарат (ИС) (СЕ=18 и HLB 16,5) са приготвяни с концентрация на препарата (в % спрямо водата) – 5, 10 и 15% и маслена фаза (МФ) слънчогледово масло, в количество – 30, 50 и 70%. Емулгирането е извършвано на миксер тип Taurus, при 523 rad.s-1.
Реологичните характеристики на емулсиите са определяни на ротационен вискозиметър тип “RV 2”.[1] Дисперзитетът е оце-няван по светлопропускливост (Т), определена спектро-фотометрично, при λ = 540 nm.[3] Емулсионната стабилност е определяна чрез центрофугиране при 314 rad.s-1 за 15 min и отчитане на отделените фази.[2]
Резултати и обсъждане
Получаването на инулиновите препарати се извършваше по схемата на реакцията (фиг. 1):

RCOX
X = OCH3, OOCCH3R = C-C17n H
H
O
HO
O
CH2OH
CH2
O
RCOO
O
CH2OH
CH2
O
O
O
HO
O
CH2OH
CH2OH
O
HO
O
CH2OH
CH2
O
O
OH
HO
HOOH
O
H
H
Фиг.1. Получаване на естери на инулин
Условията на получаване и характеристиката на химично модифицираните инулини са показани в табл. 1. Вижда се, че степента на естерификация и на заместване на препаратите зависи от вида на киселината и молното съотношение на реагентите. Таблица 1. Характеристика на химично модифициран инулин
Реакционна смес Молноотношен
ие
СЕ, %
СЗ, % HLB
Инулин/(CH3CO)2O/CH3COONa 1:2:2 8,89 0,25 19,2 Инулин/C11H23COOCH3/CH3COONa 1:1:0,4 18 0,17 16.5 Инулин/C13H27COOCH3/CH3COONa 1:1:0,4 16 0,11 17.3 Инулин/C17H35COOCH3/CH3COONa 1:2:0,3 14 0,11 17.1
Структурата на получените производни обуславя и по-осо-
бените им отнасяния и възможността за приложението им като пеногасители, емулгатори (за емулсии от типа В/М или М/В), омокрящи вещества, детергенти и солюбилизатори. Идея за това към коя група се причислява веществото дава емпиричният метод за оценка на бифилния характер на ПАВ, определен чрез HLB. Синтезираните от нас деривати са насочени към ползване като емулгатори в М/В емулсии, поради което се търсеха продукти с

HLB 8-18 (табл. 1). В този интервал на дифилност според W. Griffin [15] се стимулира образуването на маслено-водни дисперсни системи
Представляваше интерес да се изследват емулгиращите и структурно-стабилизиращите свойства на получените инулинови препарати. За целта бе определен един от тях – инулин стеарат (ИС), имащ много подходящи характеристики (HLB=16,5) за ползването му като емулгатор в М/В емулсии.
На емулсиите с 50% маслена фаза бе оценен дисперзитета спектрометрично – чрез стойностите за светлопропускливост (Т, в %). При емулсиите с 5, 10 и 15% инулин стеарат, стойностите на Т са съответно 63,3; 49,6 и 42,4%. Увеличаване концентрацията на ИС води до получаване на по-фино дисперсни системи, което илюстрира добрите емулгиращи свойства на изследвания инулинов дериват.
На табл. 2 са данните за структурния вискозитет на моделните емулсии с ИС.
При всички изследвани емулсии, структурният вискозитет (η) зависи от скоростния градиент (D), което ги характеризира като ненютонови течности. Реологичният им тип (фиг. 2 до 5) мо-же да се характеризира като псевдопластично тяло, с тенденция към неидеално пластично тяло – при емулсиите с по-високите маслени фази и концентрации на ИС.
Увеличаване концентрацията на инулин стеарат (ИС) от 5 на 15% повишава вискозитетите на емулсиите. Така при емулсиите с маслени фази 30, 50 и 70%, η (D=40,5 s-1) нараства съответно 8,7; 7,0 и 3,5 пъти. Аналогична е тенденцията в нарастването и при другите скоростни градиенти. Данните показват, че с увеличаване количеството на ИС се постигат по-вискозни емулсионни системи, което се дължи на по-финодисперсната маслена фаза и вероятно структуриране на дисперсната среда.

Таблица 2. Структурен вискозитет (η) на моделни емулсии с 5, 10 и 15% ИС и 30 до 70% маслена фаза, в Ра.s (х102), при 20оС
Скоростен градиент (D), s-1 ИС, % 2,7 4,5 8,1 13,5 24,3 40,5 72,9 121,5 218,7
30 % маслена фаза 5 32,9 26,3 14,6 8,8 4,9 3,7 2,4 1,9 1,9
10 32,9 32,9 29,2 26,3 25,6 24,8 21,1 18,5 15,7 15 65,8 52,6 51,2 35,1 34,1 32,2 26,0 21,4 17,3
50 % маслена фаза 5 21,9 13,2 11,0 8,8 7,3 7,3 6,5 5,8 5,7
10 65,8 52,6 51,2 48,2 39,0 27,8 31,7 24,4 17,3 15 65,8 65,8 58,5 57,0 53,6 51,2 41,4 28,3 19,5
70 % маслена фаза 5 32,9 26,3 21,9 17,5 17,1 16,1 14,6 10,7 7,9
10 65,8 65,8 58,5 48,2 46,3 42,4 34,1 27,3 21,4 15 109,6 92,1 73,1 83,3 68,2 57,0 45,5 39,5 24,6
Резултатите корелират с тези, получени при оценяване на
дисперзитета. Данните от табл. 2 и показаните на фиг. 2 до 5 реологични криви доказват добрите структурно-механични свойства на изследвания инулинов препарат. При това се получават по-вискозни системи при по-високите концентрации инулин стеарат и по-големите обеми на маслена фаза (фиг. 5). Така при емулсии с 15% ИС, вискозитетът (при D=40,5 s-1) нараства с увеличаване на МФ от 30 на 70% – 1,8 пъти (от 30 на 50% и от 50 на 70%, съответно 1,6 и 1,1 пъти).
Сравнително по-нестабилни са емулсиите с 5% ИС – при тях се отделя 6 до 16% “масло”, а запазената “емулсия” е 19 до 67%. Повишаване количеството на инулиновия препарат подобрява значително дисперсната устойчивост. При 10% ИС се получават емулсии с добра стабилност, особено при висомаслените. Не се отделя “масло”, а количеството запазена “емулсия” е 37 до 87%, в зависимост от маслената фаза. Следващото увеличаване на ИС – на 15%, почти не изменя стабилитетните характеристики – не се отделя “масло”, а количеството на запазената “емулсия” е 33 до 92%; стойностите са дори по-ниски, от тези при емулсиите с 30 и 50% МФ.
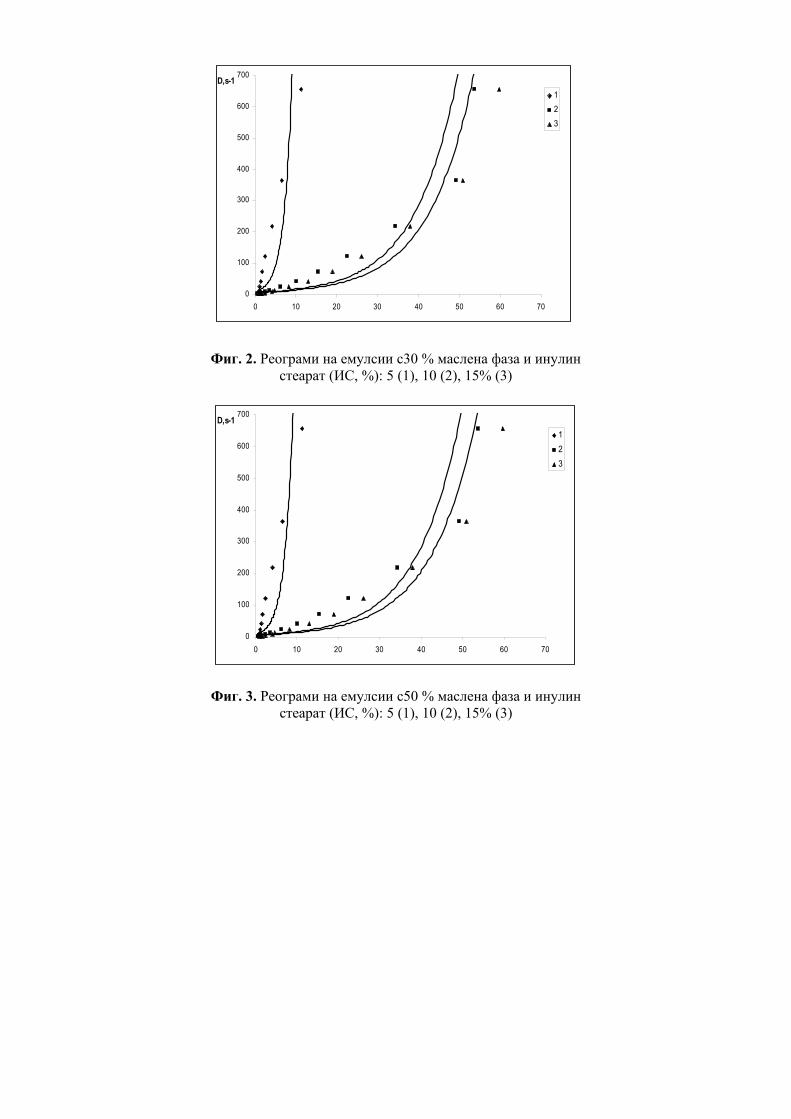
0
100
200
300
400
500
600
700
0 10 20 30 40 50 60 70
D,s-1123
0
100
200
300
400
500
600
700
0 10 20 30 40 50 60 70
D,s-1 123
Фиг. 2. Реограми на емулсии с30 % маслена фаза и инулин стеарат (ИС, %): 5 (1), 10 (2), 15% (3)
0
100
200
300
400
500
600
700
0 10 20 30 40 50 60 70
D,s-1123
Фиг. 3. Реограми на емулсии с50 % маслена фаза и инулин стеарат (ИС, %): 5 (1), 10 (2), 15% (3)
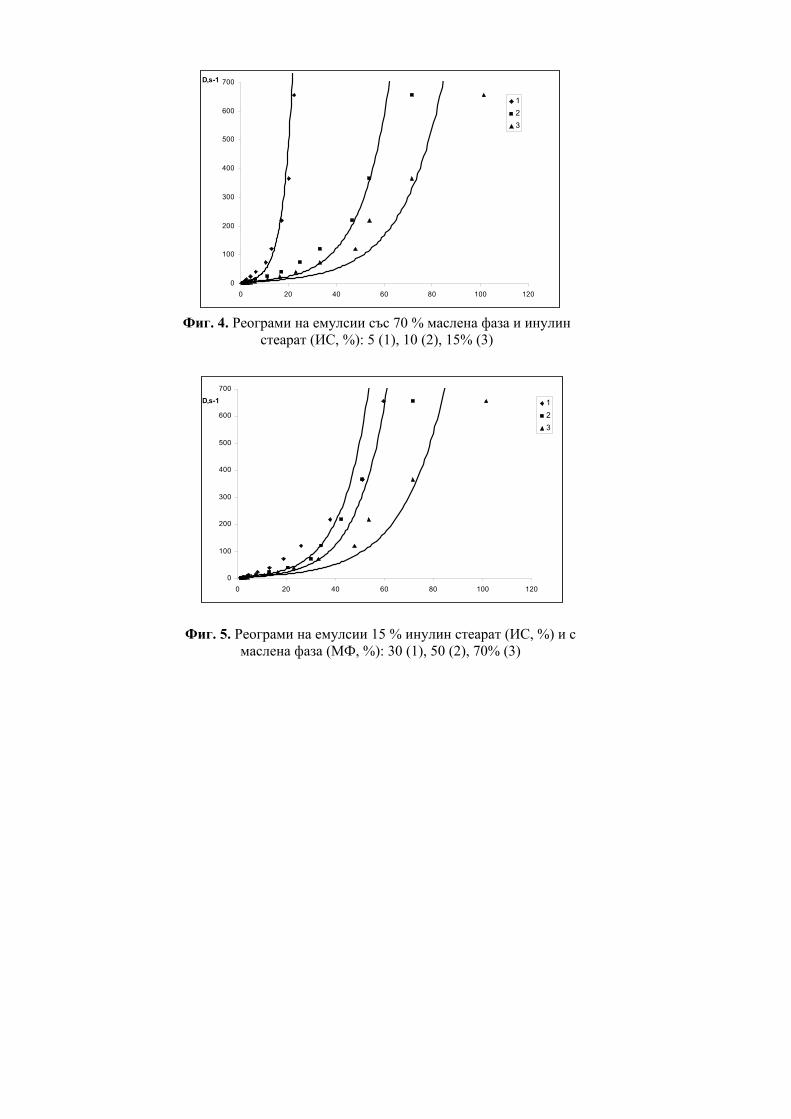
0
100
200
300
400
500
600
700
0 20 40 60 80 100 120
D,s-1
123
0
100
200
300
400
500
600
700
0 20 40 60 80 100 120
D,s-1 123
Фиг. 4. Реограми на емулсии със 70 % маслена фаза и инулин стеарат (ИС, %): 5 (1), 10 (2), 15% (3)
Фиг. 5. Реограми на емулсии 15 % инулин стеарат (ИС, %) и с маслена фаза (МФ, %): 30 (1), 50 (2), 70% (3)
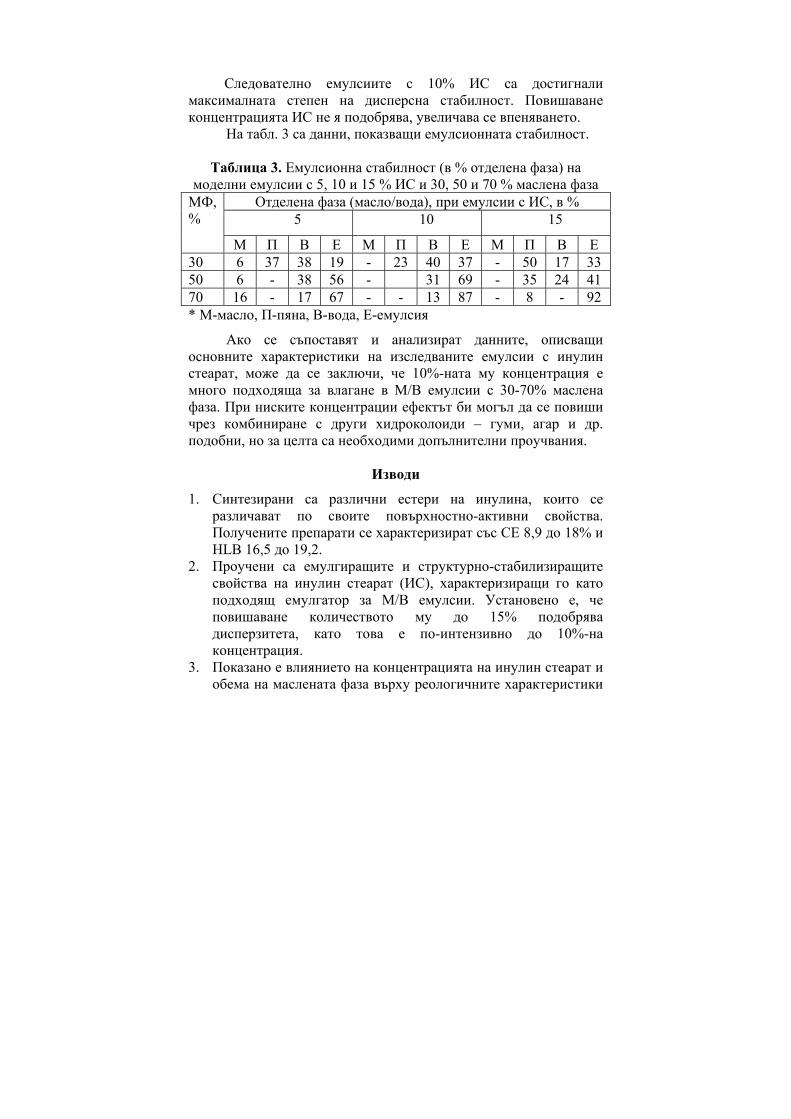
Следователно емулсиите с 10% ИС са достигнали максималната степен на дисперсна стабилност. Повишаване концентрацията ИС не я подобрява, увеличава се впеняването.
На табл. 3 са данни, показващи емулсионната стабилност.
Таблица 3. Емулсионна стабилност (в % отделена фаза) на моделни емулсии с 5, 10 и 15 % ИС и 30, 50 и 70 % маслена фаза
Отделена фаза (масло/вода), при емулсии с ИС, в % 5 10 15
МФ, %
М П В Е М П В Е М П В Е 30 6 37 38 19 - 23 40 37 - 50 17 33 50 6 - 38 56 - 31 69 - 35 24 41 70 16 - 17 67 - - 13 87 - 8 - 92 * М-масло, П-пяна, В-вода, Е-емулсия
Ако се съпоставят и анализират данните, описващи основните характеристики на изследваните емулсии с инулин стеарат, може да се заключи, че 10%-ната му концентрация е много подходяща за влагане в М/В емулсии с 30-70% маслена фаза. При ниските концентрации ефектът би могъл да се повиши чрез комбиниране с други хидроколоиди – гуми, агар и др. подобни, но за целта са необходими допълнителни проучвания.
Изводи
1. Синтезирани са различни естери на инулина, които се различават по своите повърхностно-активни свойства. Получените препарати се характеризират със СЕ 8,9 до 18% и HLB 16,5 до 19,2.
2. Проучени са емулгиращите и структурно-стабилизиращите свойства на инулин стеарат (ИС), характеризиращи го като подходящ емулгатор за М/В емулсии. Установено е, че повишаване количеството му до 15% подобрява дисперзитета, като това е по-интензивно до 10%-на концентрация.
3. Показано е влиянието на концентрацията на инулин стеарат и обема на маслената фаза върху реологичните характеристики

на изследваните емулсии. Увеличаването на концентрацията на ИС от 5 до 15% повишава стойностите на структурния вискозитет 1,9 до 9,1 пъти, в зависимост от МФ и D. Подобно, но по-слабо изразено е и влиянието на маслената фаза – повишаването й увеличава вискозитета до 1,8 пъти.
4. Установено е, че повишаване количеството на инулин стеарат до 10% подобрява стабилитетните характеристики изслед-ваните емулсии.
5. Обобщавайки основните характеристики на изследваните емулсии може да се заключи, че 10%-ната концентрация на получения инулин стеарат е много подходяща за влагане в М/В емулсии с 50-70% маслена фаза.
Литература 1. Белкин, И. и кол. Ротационные приборы измерения вязкости и
физико-механических материалов, Москва, 1968. 2. Козин, Н. Пищевые эмульсии, Пищепромиздат, М., 1950. 3. Govin R., J. Leeder, J. Food Sci., 5, 718, 1971. 4. Roberfroid, M. In “Inulin-Type Fructans. Functional Food
Ingredients”. CRC PRESS, 2005. 5. Van Loo, J., P. Coussement, L. De Leenheer, H. Hoebregs, G. Smits..
Rev. Food Sci. Nutr., 35, 525–552, 1995. 6. Prosky, L. JAOAC International 82, 223–226, 1999. 7. Roberfroid M.B., J.A. E. Van Loo, and G.R. Gibson, J. Nutrition
128, 11-19, 1998. 8. Stevens, C., A. Meriggi, K. Booten, Biomacromol., 2, 1-16, 2001. 9. Rogge, T.; C.V Stevens. Biomacromol., 5, 1799-1803, 2004. 10. Rogge, T., C.V. Stevens, A. Colpaert, B. Levecke and K. Booten,
Biomacromol., 8, 485-489, 2007. 11. Tadros Th.F., A. Vandamme, B. Levecke, K. Booten, C.V. Stevens,
Adv. Colloid & Interface Sci., 108-109, 207-226, 2004. 12. Griffin W.C., J. Soc. Cosmetic Chem., 5, 259, 1954.

Tеоретично изследване на влиянието на молекулната геометрия на възбудените
състояния на някои азини върху флуоресцентните и фосфоресцентните им
свойства Г. Д. Нейков, А. А. Ляпова, Г. П. Петров
Факултет по природни науки Шуменски университет “Епископ Константин Преславски”
9712 Шумен, ул. “Университетска”115 Abstract: The cycle ground state-absorption-fluorescence (resp. phosphorescence)-ground state, can be represented as follow:
1 S eqfcheqfcn
heq SSSS 01
01'
111
0 ∼∼→⎯→⎯∼∼→⎯→⎯ νν
eqfcheq SST 01'
01'
13 ∼∼→⎯→⎯ ν
еq-equilibrium geometry and fc-non- equilibrium. Taking into account the change in molecular geometry upon
exitation the magnitute of the stokes shift and position of the luminescent Franck-Condon trangtition were calculated.
It is shown that the molecular equilibrium geometry of nπ* and ππ*- states exerts a great influence on the luminescent properties of the azines.
Keywords: absorption, fluorescence, phosphorescence, azines.
Увод
Структурата на електронно възбудените състояния на молекулите както и някои други фактори, като вида на състоянията, относителното разположениe на синглетните и триплетните състояния по енергия, разпределението на електронната и вибрационната енергия при възбуждане на молекулите [1-3] и др. определят както луминисцентните

свойства на молекулите, така и техните физични и химични свойства в основно и възбудено състояние.
Цикълът: основно състояние - абсорбция на електромагнитни кванти - флуoресценция (съответно фосфоресценция) - основно състояние, схематично се представя по следния начин:
)()( 1
11j
fchi
eqo vSvS ⎯→⎯ ν ~ )()( 1
11 '
efc
oh
keq vSvS ⎯→⎯→ ν ~ )(1
ieqo vS→
(1)
)()( 111 ''
jfch
ieqo vSvS ⎯→⎯ ν ~ )()( 1'
13 '''
efc
oh
k vSvT ⎯⎯→⎯→ ν ~ )(1i
eqo vS→
(2) В горната схема с vi ….ve са означени съответните
вибрационни състояния, а с еq и fc –равновесните или Франк-Кондоновите синглетни (S) или триплетни (T) състояния. Вибрационната компонента на енергията не се разглежда, тъй като изчисленията са проведени в приближението на Born-Oppenheimer.
В циклите (1) и (2) еднозначно са дефинирани само 0-0 – преходите, извършващи се от или до най-ниското вибрационно ниво на съответното електронно възбудено състояние, т.е vi =0… vj =0.
Ако равновесните възбудени състояния 1S eq1 и 3T eq
1 се реализират, то енергията на излъчените кванти в по-голяма или по-малка степен се различава от енергията на абсорбираното лъчение.
Обекти и цели на изследването Азините с шестатомен пръстен (пиридин, пиридазин,
пиримидин, пиридин, s-триазин, s-тетразин) са обект на изследване в настоящата работа (фиг.1).

N
HH
H
N
N
NNN
NN
N
N
N
N
NN
HH H
H
H
H
H
1 1 1 1
11
2 2 2 2
2
2
3 3 3 3
33
4 4 4 4
4
4
5
5
5
566 67
Фиг. 1. Изследвани молекули и номерация на атомите.
Симетрично разположените атоми не са означени. При
някои съединения за удобство, не са спазени правилата на UPAC за номериране на атомите. Тези азини бяха избрани за обект на изследване от една страна затова,че някои от тях са структурни компоненти на голям брой природни продукти, и от друга страна, като изключим някои публикации върху техните спектрални свойства, данните за геометричната и електронна структура на електронно-възбудените им състояния в литературата са оскъдни.
Изчислителен метод
Всички изчисления са проведени с MNDO+CI варианта на полуемперичния метод.[5] Като стартовa равновеснa геометриq на основното състояние на изследваните съединения (фиг.1) е използвана опитната геометрия от литературни източници.[7]
Тази геометрия на основното състояние (1So) се явява всъщност неравновесната Франк-Кондонова геометрия на възбудените състояния - 1S fc
1 и 3Т fc1 .
Равновесната геометрия на възбудените състояния 1S eq1 и
3Т eq1 бе изчислена чрез CI-вълнови функции. Базисните функции,
използвани в конфигурационното взаимодействие са получени в MNDO.[5] В конфигурационното взаимодействие са включени всички едновъзбудени конфигурации и някои избрани

двувъзбудени, определени въз основа на критерии, формулирани в работа [7].
Ф ∑ ∑++=
m n
qpjin
pkmCI VCVCVC ,
,00 ,
където V0 e конфигурацията на основното състояние, а V pk и
V pqji,, са съотвтно едно- и двувъзбудените конфигурации.
Енергиите на емисионните (флуоресцентни и фосфоресцентни) преходи са изчислявани с равновесната геометрия на възбудените 1S eq
1 и 3Т eq1 състояния.
Резултати и обсъждане
Геометрична структура. Равновесната геометрия на основното състояние (1S eq
0 ) съгласно принципа на Франк-Кондон се явява като неравновесна Франк-Кондонова геометрия на възбудените състояния fcS1
1 и fcТ13 състояния, а равновесната
геометрия на eqS11 и eqT1
3 е неравновесната Франк-Кондонова
геометрия на основното състояние fcS01 и
'
01 fcS (вж. Фиг.2).
Както се вижда от табл.1 между изчислените с MNDO+CI метода и опитните стойности [6] има добро съответствие. Разликата между изчислените и опитните стойности за Rа,b не надвишава 0.01Å, а за валентните ъгли разликата не повече от 1-2 о. Опитната геометрия на възбудените (1S1) и (3T1) състояния на тези съединения не е известна, но добрите резултати за енергията на луминисцентните преходи, които са изчислени с тази геометрия, е една гаранция за доброто съвпадение на изчислената геометрия на възбудените състояния с реалната.
Изчисленията показват, че изследваните молекули както в основно (1So), така и в електронно-възбудените състояния (1S1) и (3T1) са планарни. Възбудените състояния запазват съответната симетрична група, към която принадлежат тези молекули в основно състояние (вж. табл.1), независимо от измененията в дължината на валентните връзки и ъгли.
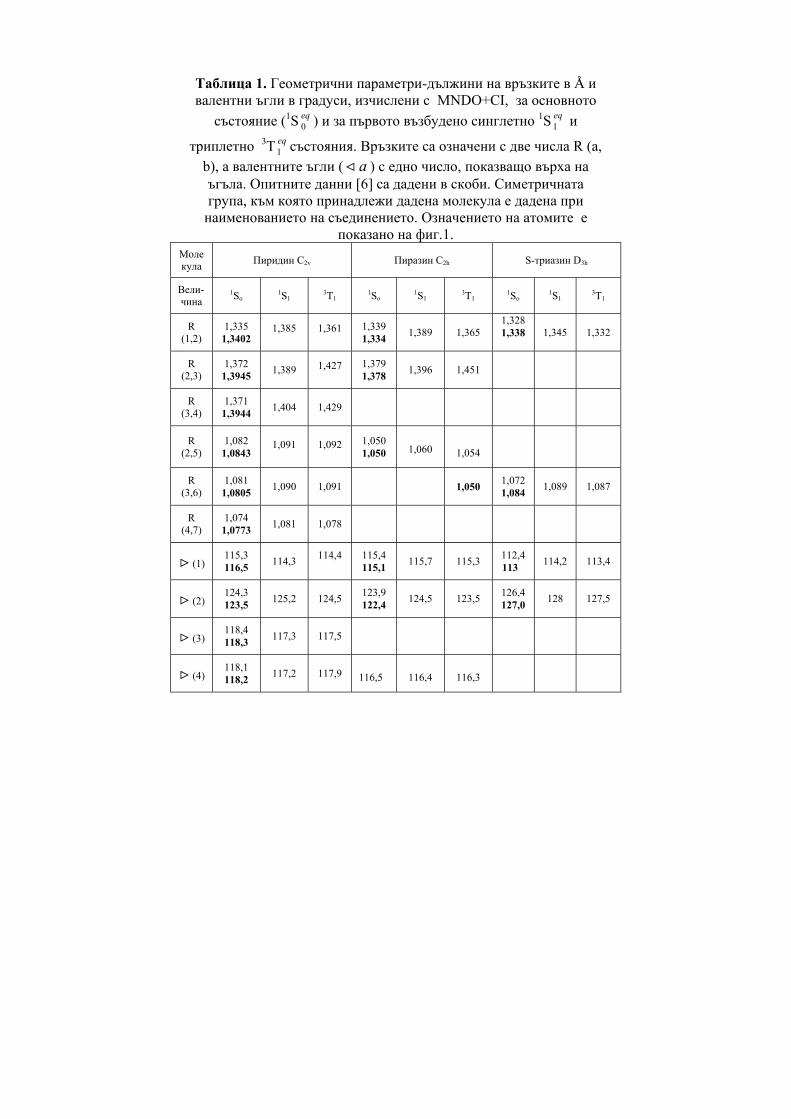
Таблица 1. Геометрични параметри-дължини на връзките в Å и валентни ъгли в градуси, изчислени с MNDO+CI, за основното
състояние (1S eq0 ) и за първото възбудено синглетно 1S eq
1 и
триплетно 3T eq1 състояния. Връзките са означени с две числа R (a,
b), а валентните ъгли ( а ) с едно число, показващо върха на ъгъла. Опитните данни [6] са дадени в скоби. Симетричната група, към която принадлежи дадена молекула е дадена при наименованието на съединението. Означението на атомите е
показано на фиг.1. Молекула Пиридин C2v Пиразин C2h S-триазин D3h
Вели- чина
1So 1S1 3T1 1So 1S1 3T1 1So 1S1 3T1
R (1,2)
1,335 1,3402
1,385 1,361 1,339 1,334 1,389 1,365
1,328 1,338
1,345 1,332
R (2,3)
1,372 1,3945
1,389
1,427 1,379
1,378 1,396 1,451
R (3,4)
1,371 1,3944
1,404
1,429
R (2,5)
1,082 1,0843
1,091
1,092 1,050
1,050 1,060
1,054
R (3,6)
1,081 1,0805
1,090
1,091 1,050 1,072
1,084 1,089 1,087
R (4,7)
1,074 1,0773
1,081
1,078
(1)
115,3 116,5
114,3
114,4
115,4 115,1 115,7 115,3 112,4
113 114,2 113,4
(2)
124,3 123,5
125,2
124,5 123,9
122,4 124,5 123,5 126,4 127,0 128 127,5
(3)
118,4 118,3
117,3
117,5
(4)
118,1 118,2
117,2
117,9 116,5 116,4 116,3

*Не всички данни за изследваните съединения са показани в таблицата. Те се намират при авторите и могат да бъдат получени при поискване.
Йонизационни потенциали. За спектралните свойства на веществата с n-електрони от съществено значение е типа на най-ниско енергетичния преход.
В табл. 2 са сравнени изчислените по абсолютна стойност енергии на най-високозаетата π-орбитала и на n-орбиталата с опитните стойности за фотойонизационните потенциали от работата.[8]
Нашите резултати показват, че най-високозаетата орбитала при всички изследвани съединения е π-орбитала. Този резултат е в съответствие с резултатите от други изследователи, получени с CNDO-метода.[9] Те обаче са в противоречие с резултатите получени чрез аb initio квантовохимични изследвания, съгласно които най-високозаетата е n-орбиталата.
Таблица 2. Изчислени орбитални енергии по абсолютна стойност на най-високо заетата π-орбитала и на n-орбиталата и опитни
стойности на йонизационния потенциал в eV. Молекула /En / /Eπ / Iexp. [d] Пиридин 10.36 9.21 9.23 Пиридазин 10.34 8.95 9.91 Пиримидин 10.37 8.87 9.47 Пиразин 10.60 8.54 9.27
S-триазин 10.53 9.98 10.00 S-тетразин 10.93 9.68 -
При пиридина, пиримидина и пиразина опитно е показано,
че първият йонизационен потенциал се дължи на избиване на електрон от π-МО [11,12]. Oт табл. 2 се вижда, че нашите резултати съвпадат с опитно установените факти. Изказаното предположение в работата [12], че първият йонизационен потенциал при пиридазина се дължи на избиване на електрон от n(-)-орбиталата не се потвърждава от други изследователи и се

подлага на сериозни съмнения [8]. Нашите изследвания показват, че най-високо заетата MO е от π-тип и при пиридазина.
Енергия на абсорбционните и емисионните електронни преходи. На фиг.2 е показана модифицираната диаграма на Яблонски на пиридина за циклите (1) и (2).
3T1fc
1S0eq
1S0eq
1S0fc
1S1eq
1S1fc
1S2fc
3T1eq
1S0fc'
1S0eq
1S0eq
νh νhhν'hν'4,59
(4,59)4,88
(4,59)3,51
(3,58)3,68
Модифицираната диаграма на Яблонски е използвана от
Тютюлков и сътр. за обяснение на някои особености в спектралните свойства на молекули с делокализирани електрони [13]. От фиг. 2 се вижда, че съвпадението между изчислените и опитните стойности за енергиите на абсорбционните преходи е в добро съответствие. Използването на молекулната геометрия на възбудените равновесни състояния позволява не само да се предскаже енергията на флуоресцентния, респ. на фосфоресцентния 0-0-преход, но и да се предвиди Стоксовото отместване. В табл. 3 са дадени изчислените енергии на няколко абсорбционни прехода fc
mS1 eqS01← и силата на осцилатора (f) и
енергията на флуоресцентния fceq SS 01
11 → и фосфоресцентен
fceq ST 013 → преход.
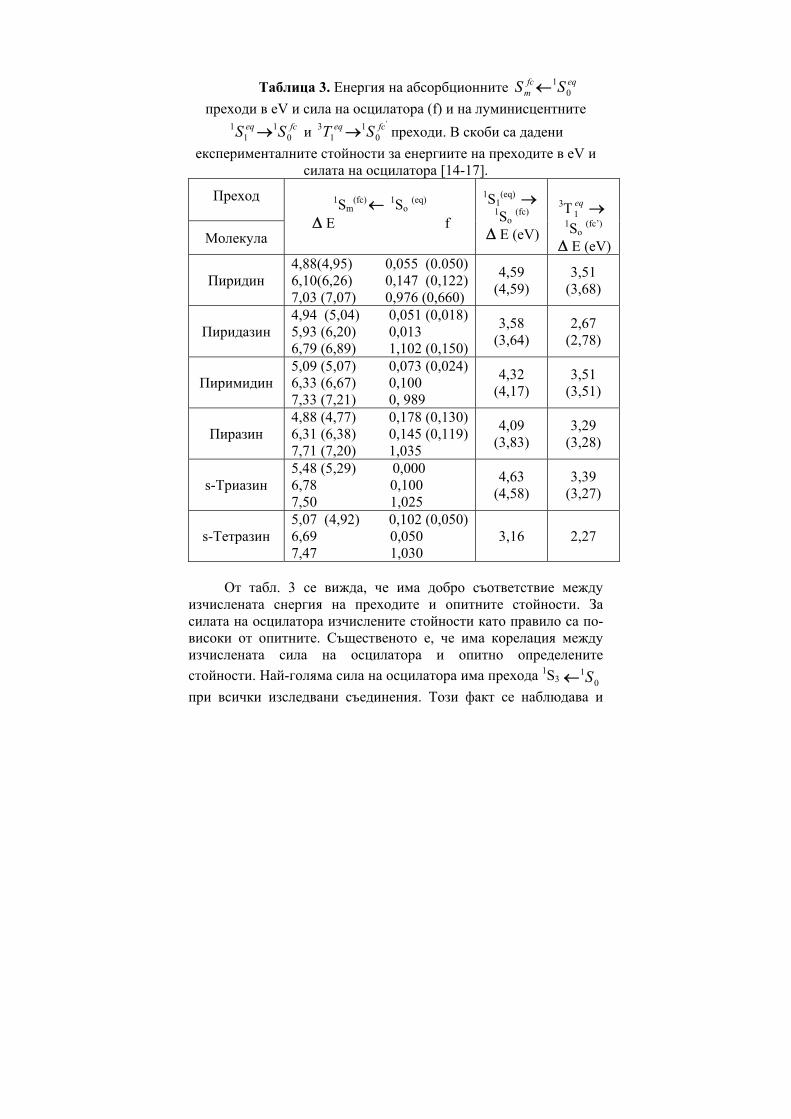
Таблица 3. Енергия на абсорбционните eqfcm SS 0
1← преходи в eV и сила на осцилатора (f) и на луминисцентните
fceq SS 01
11 → и
'
01
13 fceq ST → преходи. В скоби са дадени
експерименталните стойности за енергиите на преходите в eV и силата на осцилатора [14-17].
Преход
Молекула
1Sm(fc) ← 1So (eq)
Δ E f
1S1(eq) →
1So (fc)
Δ E (eV)
3T eq1 →
1So (fc’) Δ E (eV)
Пиридин 4,88(4,95) 0,055 (0.050) 6,10(6,26) 0,147 (0,122) 7,03 (7,07) 0,976 (0,660)
4,59 (4,59)
3,51 (3,68)
Пиридазин 4,94 (5,04) 0,051 (0,018) 5,93 (6,20) 0,013 6,79 (6,89) 1,102 (0,150)
3,58 (3,64)
2,67 (2,78)
Пиримидин 5,09 (5,07) 0,073 (0,024) 6,33 (6,67) 0,100 7,33 (7,21) 0, 989
4,32 (4,17)
3,51 (3,51)
Пиразин 4,88 (4,77) 0,178 (0,130) 6,31 (6,38) 0,145 (0,119) 7,71 (7,20) 1,035
4,09 (3,83)
3,29 (3,28)
s-Триазин 5,48 (5,29) 0,000 6,78 0,100 7,50 1,025
4,63 (4,58)
3,39 (3,27)
s-Тетразин 5,07 (4,92) 0,102 (0,050) 6,69 0,050 7,47 1,030
3,16 2,27
От табл. 3 се вижда, че има добро съответствие между
изчислената снергия на преходите и опитните стойности. За силата на осцилатора изчислените стойности като правило са по-високи от опитните. Същественото е, че има корелация между изчисленaта сила на осцилатора и опитно определените стойности. Най-голяма сила на осцилатора има прехода 1S3 0
1S←
при всички изследвани съединения. Този факт се наблюдава и

опитно. Най-голяма екстинкция има третата абсорбционна ивица при изследваните съединения.
Стоксовото отместване се предвижда с голяма точност. Това се дължи на използването на равновесната геометрия на възбудените eqS1
1 и eqT13 състояния при изчисляване на
енергиите на флуоресцентните и фосфоресцентните преходи.
Литература 1. Stevens B., J. Chem. Phys., 1956, 24, 488. 2. Hепорент Б. С., Ж. Физ. Химии, 1956, 30, 1048. 3. Пикок Т., Электронные свойства ароматических и
гетероциклических молекул, “Мир”, Москва, 1969. 4. Kuehulenz G.W., C. A. Masmaidi, H. Jaffe, J. Mol. Structure,
1973, 15, 445. 5. Dewar M. J. S., W. Thiel, J. Am. Chem. Soc., 1977, 99, 4899,
4907. 6. Tables of Interatomic Distances and Configuration in Molecules
and Iones, Burlington Hause, London, 1958, 1965. 7. Тютюлков Н., Г. Д. Нейков, И. Н. Кънев, Теорет. и эксперим.
химия, 1977, 13, 386. 8. Turner D. W., Adv. Phys. Org. Chem., 1966, 4, 31. 9. Del-Bene J., H. Jaffe, J. Chem. Phys., 1968, 48, 1807, 4050; 1969,
49, 1221; 1967, 65, 463. 10. Lencha A. J., M. A. El-Sayed, J. Chem. Phys., 1968, 48, 3469. 11. El-Sayed, M. A., M. Kasha, L. Tanaka, J.Chem. Phys., 1961, 34,
334. 12. Lencha A. J., M. A. El-Sayed, J. Chem. Phys., 1968, 48, 34. 13. Тютюлков Н., Р. Воденичаров, Изв. на отделението за хим.
науки-БАН, 1971, 4, 653. 14. UV-Atlas of Organic Compounds, Butterworths Verlag Chemie,
London and Weinheim, 1966, 1999. 15. Innes K. K., J. P. Byrne, I.G. Ross, J.Mol. Spectr., 1967, 22, 125. 16. Goodman L., J. Mol. Spectrosc., 1961, 6, 109. 17. El-Sayed, M. A., J. Chem. Phys., 1962, 36, 573.

Electro-optic investigation of carboxymethyl cellulose adsorption on alumina nanoscale particles
Rossen Pl. Hristov Institute of Physical Chemistry, Bulgarian Academy of Sciences
Acad. G. Bonchev St., Bl. 11, 1113 Sofia, Bulgaria E-Mail: [email protected]
Abstract: Adsorption of polyelectrolyte (PE) molecules on colloid nanoparticles in water solutions is in theoretical and practical interest. In this study adsorption of sodium carboxymethyl cellulose (NaCMC) on gamma-aluminium oxide (γ-Al2O3) particles with nanoscale dimensions at different PE concentrations by means of electro-optics (electric light scattering) was investigated. Dispersion dependencies were obtained to identify a frequency at which the electro-optical effect (EOE) of all solutions is on a plateau so they can be compared properly.
Electrophoresis was also applied as a method for analyzing the changes of particles’ charge caused by the PE adsorption. Charge overcompensation was observed above concentration of 3⋅10−3 g/L NaCMC and anti-flocculation stability was reached at concentration 4⋅10−2 g/L NaCMC.
I was interested in the kinetics of the adsorption process as well. Increasing of relaxation time values and decreasing in EOE were found out when increasing the NaCMC concentration at constant electric field strengths. Measurements for analysis of particles’ optical properties changes caused by already completed PE adsorption were also made at maximal degree of orientation.
Introduction
Alumina nanoscale colloid particles are amphoteric with point of zero charge (PZC) at pH 8.5.[1] At lower pH values they get positively charged and at higher ones – negatively which makes them ideal for adsorption of variously charged PE at suitable pH values, and investigation of their properties.

NaCMC is a sodium salt of the carboxymethyl derivative of cellulose. Dissolved in water, this salt – product of the neutralization of the strong NaOH and acidic carboxymethyl groups – dissociates entirely and releases all sodium ions in the water medium. As a result the carboxymethyl substitutes get negatively charged and so does the macromolecule as a whole. This process does not depend on pH, i.e. NaCMC is a strong PE.
The maximal positive charge which a γ-Al2O3 suspension can possess is at pH 4.5. At lower pH values this compound do not exists as a suspension but as a molecular solution instead. The latter is not proper to be investigated by our method – electric light scattering, suitable to be applied only for studies of colloidal systems. Because of that lower pH values were not taken into account in this research.
The research was made at constant ionic strength as well. PEs are polyions so their charge is taken into account for the calculation of that parameter. Its value was chosen to be the value of the most concentrated PE solution possessing the biggest charge. The ionic strengths of the other solutions were equalized with that value by adding proper quantity NaCl, as indifferent electrolyte. Working in that way I varied only the NaCMC concentration to find out how does its increasing changes the properties of the colloidal particles.
The dispersion degree (and the specific surface of the oxide, respectively) was enlarged by ultrasound applied to the suspension before its mixing with a NaCMC solution.[2] I made a special experiment to check if and to what extend this manipulation is really necessary and found that it betters the results unneglectably.
Measurements of the electro-optic behaviour of the suspensions after the completion of the adsorption proccess are provided as well as ones of the kinetics of the latter itself. I observed the changes of both steady-state EOE value and relaxation time of the particles during adsorption.
All measurements of colloidal oxide particles covered with PE layer formed by various NaCMC concentrations were compared with ones of control suspensions of particles uncoated with PE adsorption layer.

Materials and Methods The γ-Al2O3 used in the present study is product of “Degussa”
with catalogue number P110C1. It consists of single spherical particles, 20 nm in diameter, stuck together in anisodiametric aggregates of submicron size. Тhe mean specific surface is 200 m2/g. The aggregates are rigid structures formed by the spheres as a result of the industrial preparation itself – Al2Cl3 flame hydrolysis. They do not vary with time, do not depend on initial light concentration and presence of EOE is observed even after additional dispersing by application of ultrasound. The fact that the aggregates are anisodiametric is proved by both electron micrographs [3] and by the presence of EOE unobservable in solutions with single spherical particles.
The NaCMC (commercial name “Blanose 12M31FD”) is also a product of “Degussa” with molecular mass 250,000. The number of its monomers is 1,000 and its degree of substitution is 1.2. This data is of an importance for the calculations needed to determine the proper volume of NaCl, added for equalizing the ionic strength.
The pH value (4.5) was fixed by titration of both solutions (particles’ and PE’s) with 0.2 M HCl before mixing. Such was the concentration of the NaCl applied for ionic strength adjusting.
By electrophoresis was found that suitable final NaCMC concentrations are the ones in the range between 5⋅10-4 – 5⋅10-2 g/L. The natural ionic strength of the highest PE concentration, 5⋅10-2 g/L, was calculated to be 2.5⋅10-4 mol/L so it was chosen as a constant for all the solutions.
Adsorption was achieved by mixing equal volumes of NaCMC and γ-Al2O3. Treatment of colloidal particles with ultrasound was applied for 30 seconds (Ultrasonic Disintegrator UD-11 automatic, working frequency 22 ± 1.65 kHz) before pH and ionic strength adjusting, followed by mixing and stirring for 30 minutes with magnetic bar.
The electro-optic method applied for that type of investigation is based on the orientation of charged colloid particles in electric field. The orientation changes are detected by a white unpolarized light beam, propagating through the suspension. The observation angle is

900. That is why the method is called electric light scattering. The particles present in the system, scatter the light differently according to their orientation. The bigger the degree of particle orientation, the bigger the difference between the intensities of the light scattered by orientated (IE) and nonorientated (I0) particles – absolute EOE (ΔI). The ratio of that difference and I0 is called relative electro-optic effect α=ΔI/I0. At constant optical coefficient (stable system) the variation of the EOE reflects directly the changes in particles’ electric polarizability. At full orientation (α∞) – the saturation of EOE at high electric field strength – the electro-optic effect reflects only the geometrical and optical properties of the particles.
When the electric field is turned off, the particles get disorientated because of their Brownian motion. The characteristic time a particle needs to disorientate is called relaxation or decay time (τ). The bigger is the particle, the bigger is the relaxation time. Thus, decay time changes give information about particles size by certain formula.[4,5]
Results and Discussion At the beginning the EOE dependence on uncoated γ-Al2O3
particles’ concentration was measured and nonlinearity appearing at 0.13 g/L was observed. As a result 0.1 g/L was chosen as a suitable concentration for PE adsorption in the linear region. This concentration is high enough to register a reliable EOE at big signal/noise ratio but far enough from nonlinearity (Fig.1).
0,0 0,2 0,40
75
150
225
300
ΔI, m
V
[γ-Al2O3], g/L
0,1 g/L
Fig. 1. Concentration dependence of the absolute EOE forγ-Al2O3 in
particles in water medium (concentration range 0.05-0.4 g/L)

Ultrasound treatment of γ-Al2O3 suspensions showed up to 21% increase of ΔI/I according to the electric field intensity (Fig.2). That is why sonication was applied at the preparation of all the solutions in that research.
0 2 4 10 20 300
15
30
45
60
E2.10‐10, V2.m‐2
α, %
Fig. 2. Comparison between relative EOE of γ-Al2O3 suspensions
sonicated (solid circles) and not sonicated (open circles)
Electrophoretic measurements proved that the adsorption process really happens because electrophoretic mobility (respectively surface electric charge of the oxide particles) decreases with NaCMC concentration increase. Low PE concentrations change slightly particles’ surface charge and electrophoretic mobility in contrast to the high ones. At 3⋅10-3 g/L NaCMC, particles are uncharged as a whole because the electric charges of the latter and the PE are equal. In this point the particles do not move electrophoretically and flocculate because of lacking electrostatic repulsion. PE amounts adsorbed above PZC overcompensate the positive surface charge of the oxide. Complexes get negatively charged just as the macromolecules themselves and their absolute charge values begin to increase. At 4⋅10-2 g/L NaCMC coated particles reach a plateau of their electrophoretic mobility (Fig. 3).
Frequency dependence (Fig. 4) of EOE of oxide–PE complexes manifested appearance of a plateau within the low kilohertz range (1–10 kHz). Except that EOE increases proportionally to high PE concentration and in inverse proportionality with low ones as has been observed in previous works with other oxide particles .[6] The plateau of the γ-Al2O3 water solution, however, is much wider, 1- 40 kHz than for the ones of suspensions with ionic strength adjusted, including zero NaCMC concentration. The latter has wider plateau than the
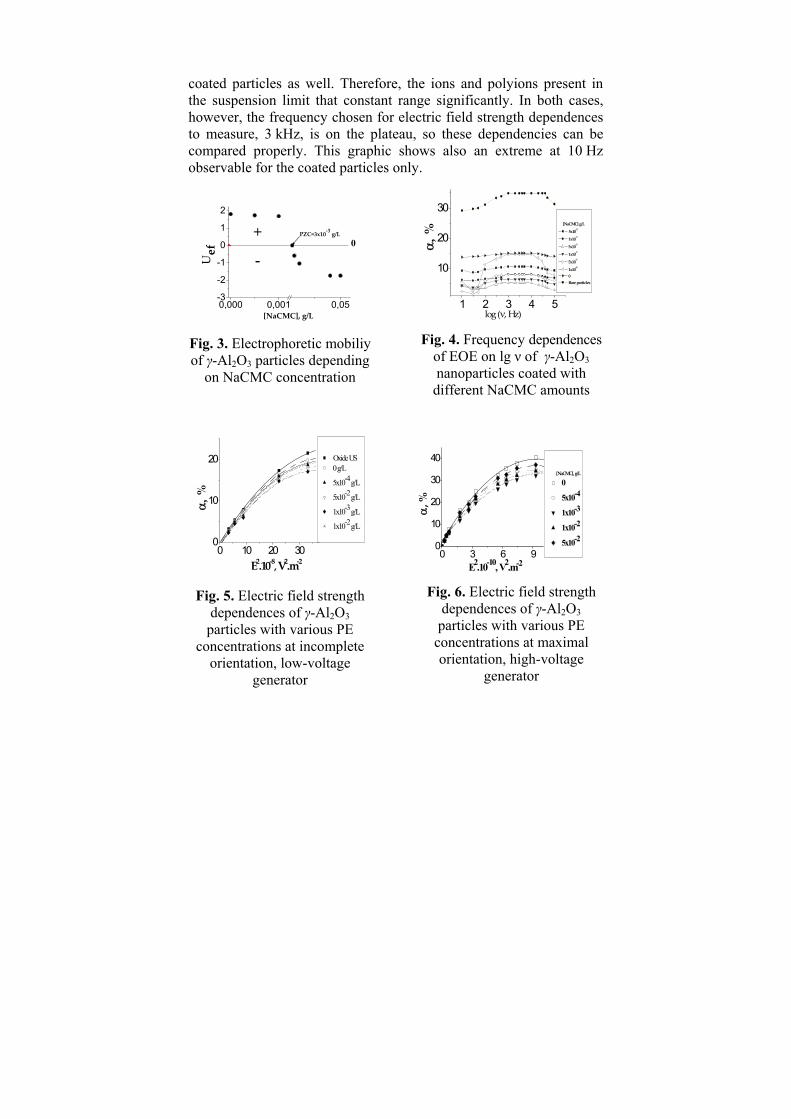
coated particles as well. Therefore, the ions and polyions present in the suspension limit that constant range significantly. In both cases, however, the frequency chosen for electric field strength dependences to measure, 3 kHz, is on the plateau, so these dependencies can be compared properly. This graphic shows also an extreme at 10 Hz observable for the coated particles only.
0,000 0,001 0,05-3
-2
-1
0
1
2
-
+0
Uef
[NaCMC], g/L
PZC=3x10‐3 g/L
1 2 3 4 5
10
20
30
α, %
log (ν, Hz)
[NaCMC] g/L 5x10‐2
1x10‐2
5x10‐3
1x10‐3
5x10‐4
1x10‐4
0 Bare particles
Fig. 3. Electrophoretic mobiliy of γ-Al2O3 particles depending
on NaCMC concentration
Fig. 4. Frequency dependences of EOE on lg ν of γ-Al2O3 nanoparticles coated with
different NaCMC amounts
0 10 20 300
10
20 Oxide US 0 g/L
5x10-4 g/L
5x10-2 g/L
1x10-3 g/L
1x10-2 g/L
α, %
E2.10‐8, V2.m‐2
0 3 6 90
10
20
30
40
α, %
E2.10-10, V2.m-2
[NaCMC], g/L
0
5x10-4
1x10-3
1x10-2
5x10-2
Fig. 5. Electric field strength
dependences of γ-Al2O3 particles with various PE
concentrations at incomplete orientation, low-voltage
generator
Fig. 6. Electric field strength dependences of γ-Al2O3
particles with various PE concentrations at maximal orientation, high-voltage
generator
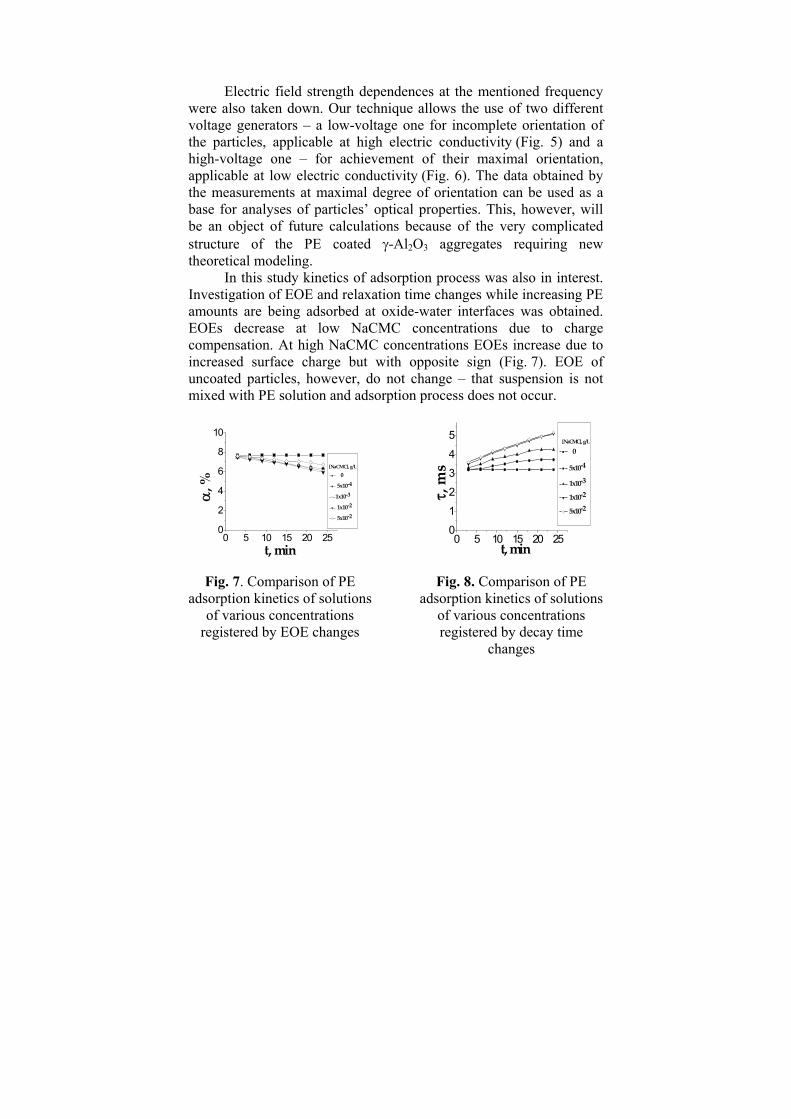
Electric field strength dependences at the mentioned frequency were also taken down. Our technique allows the use of two different voltage generators – a low-voltage one for incomplete orientation of the particles, applicable at high electric conductivity (Fig. 5) and a high-voltage one – for achievement of their maximal orientation, applicable at low electric conductivity (Fig. 6). The data obtained by the measurements at maximal degree of orientation can be used as a base for analyses of particles’ optical properties. This, however, will be an object of future calculations because of the very complicated structure of the PE coated γ-Al2O3 aggregates requiring new theoretical modeling.
In this study kinetics of adsorption process was also in interest. Investigation of EOE and relaxation time changes while increasing PE amounts are being adsorbed at oxide-water interfaces was obtained. EOEs decrease at low NaCMC concentrations due to charge compensation. At high NaCMC concentrations EOEs increase due to increased surface charge but with opposite sign (Fig. 7). EOE of uncoated particles, however, do not change – that suspension is not mixed with PE solution and adsorption process does not occur.
0 5 10 15 20 250
2
4
6
8
10
[NaCMC], g/L
0 5x10‐4
1x10‐3
1x10‐2
5x10‐2
α, %
t, min
0 5 10 15 20 250
1
2
3
4
5 [NaCMC], g/L
0
5x10‐4
1x10‐3
1x10‐2
5x10‐2
τ, ms
t, min
Fig. 7. Comparison of PE adsorption kinetics of solutions
of various concentrations registered by EOE changes
Fig. 8. Comparison of PE adsorption kinetics of solutions
of various concentrations registered by decay time
changes

Concentration 5⋅10-3 g/L is not represented on that graphic – it does not give a measurable values because it is too close to the PZC and electric polarizability is too small.
Unlike EOE that decreases with time because of PE adsorption caused decreased polarizability of the particles, relaxation time increases within the whole concentration range due to increase of particles dimensions caused by the NaCMC adsorption (Fig. 8).
The two maximal NaCMC concentrations give insignificant difference between relaxation time values unlike the significant difference between their EOEs. That discrepancy can be interpreted as observation of PE layer densification.
Conclusions
The experimental results provided in this paper make us capable to conclude that: 1. Adsorption of NaCMC causes an appearance of an extreme at low
frequency region (10 Hz) that lacks in the frequency dependence of bare oxide particles suspension. PE adsorption strengthens the plateau narrowing effect of the indifferent electrolyte.
2. Adsorption process and electric field strength changes do not shift the chosen for the study frequency of 3 kHz out of the plateau, so the graphics are assumed to be correctly interpreted.
3. The usage of ultrasound gives an unneglectable increase of the electro-optic effect.
4. Electrophoretic analysis represents that high concentrations of NaCMC change strongly the electrophoretic mobility of the colloid particles and overcompensate their positive charge in contrast to the low ones. The zero charge point is at 3·10-3 g/L NaCMC.
5. The EOE of the particles decreases at low adsorbed NaCMC amounts, insufficient for entire particles charge compensation in contrast to overcompensation obtaining quantities causing increasing EOE.
6. Relaxation times increase with the increase of NaCMC concentration due to the bigger adsorbed amounts. Comparison of relaxation times at 1⋅10-2 g/L and 5⋅10-2 g/L NaCMC shows

ignorable difference despite the big concentration difference. Apparently this effect is due to adsorption layer densification instead to increased adsorption layer thickness and particles size respectively.
Acknowledgements: The author is grateful to Assoc. Prof. Alexandar M. Zhivkov and Dr. M. Buleva from the Rostislaw Kaischew Institute of Physical Chemistry, Bulgarian Academy of Sciences for the help and stimulating discussions. I also gratefully acknowledge the financial support received from FP6 European project NANOPHEN (INCO-CT-2005-016696).
References 1. Buleva, M., Thesis: Електрични моменти на колоидни частици
и строеж на двойния електричен слой, 1982, Sofia. 2. Starchev, K. N., Thesis: Структура и кинетика на образуване на
фрактални агрегати, изследвани чрез електрично светоразсейване, 1993, Sofia.
3. Buleva, M., Stoimenova, M., Electro-optic Investigation of Oxide Suspensions – Mechanism of Formation of Induced Dipole Moment, Journal of Colloid and Interface Science, , 1991,vol. 141, pp. 426-432.
4. Stoylov, Stoyl P., Colloid Electro-Optics – Theory, Techniques, Applications, Academic Press Ltd, 1991, Colloid Science - Series of Monographs 129, pp.
5. Stoylov, Stoyl P., Stoimenova, Maria V., Molecular and Colloid Electro-optics, Taylor & Francis, 2005, surfactant science series vol.134, pp. 17-38.
6. Kamburova, K., Radeva, Ts., Electro-optics of Colloid-Polyelectrolyte Complexes: Counterion Condensation on Free and Adsorbed Sodium Carboxymethyl Cellulose, Journal of Colloid and Interface Science, 2007, vol. 313 (2), pp. 398-404.

Влияние на Дауфакс върху изтъняването и критичната дебелина на пенни филми,
стабилизирани от смес на Динакол и Солвитоза Жана Ангарска*, Красимир Тачев**
ШУ “Епископ Константин Преславски” Факултет по природни науки
Катедри *Обща химия, **Биология, 9712 Шумен E-Мail: [email protected], [email protected]
Резюме: Изследвана е кинетичната стабилност на пенни филми (с радиуси 0.005, 0.01, 0.015 cm), получени от смесени разтвори на на Динакол и Солвитоза (1:10) и Динакол, Солвитоза и Дауфакс при 40°C. Тя е съпоставена с тази на филмите от разтвори на Дауфакс и смесените разтвори на Динакол, Солвитоза и Афранил.
Установено е, че вида и стабилността на пенния филм зависи от количеството на Дауфакса (0.005, 0.05%) в смесения разтвор. С повишаване на концентрацията на Дауфакс, филмите от късащи се в момента на получаване на черно петно, стават стабилни нютонови.
Съпоставката между данните за скоростта на изтъняване на филмите от смеси показа, че най-бързо изтъняват филмите от Динакол и Солвитоза, дори по-бързо от филмите от разтвор на чист Динакол; скоростта на изтъняване на филмите от Дина-кол:Солвитоза:Афранил = 1:10:10 е близка до тази на филмите от 0.005% Дауфакс, което подкрепя издигната хипотеза, че Дауфаксът, излъчен от восъците, съдържащи се в Афранила, определя поведението и стабилността на филмите. Корелацията между критичните дебелини на филмите от Динакол:Солви-тоза:Дауфакс = 1:10:1 и Динакол:Солвитоза:Афранил = 1:10:10 показва, че излъченият Дауфакс е в количество, близко до наличния в сместа Динакол:Солвитоза: Дауфакс = 1: 10:1.
Keywords: paper surfactants; foam films; drainage; critical thickness.

Увод Експериментално е наблюдавано, че в пулпата при
производството на целулоза се образуват различни по размер газови мехурчета, присъствието на които остава до последния етап от технологичния процес. Наличието на тези мехурчета води до намаляване на качеството на продукта. За да се потисне процеса на образуването им, към пулпата се добавят антипенители. За оптимизирането на тяхното действие е важно да се знае от какво се определя стабилността на образуващата се пяна, т.е. стабилността на пенните филми, образувани между мехурчетата.
В пулпата се съдържат веществата Динакол, Лигнинсулфат и Солвитоза, които имат свойства на ПАВ и често са наричани “paper surfactants”. Тези ПАВ оказват нееднопосочно влияние върху свойствата на пенния филм по следните причини; известно е, че Динаколът се адсорбира на фазовата граница относително бързо и понижава повърхностното напрежение, но няма пенообразуваща способност; Солвитозата няма нито висока адсорбционна способност на фазовата граница вода/въздух, нито пенообразуваща способност, но според [1] тя съществено влияе върху адсорбционното поведение на Динакола. Показано е, че от смесен разтвор на Динакол и Солвитоза се образуват стабилни пени, което е доказателство за ролята на Солвитозата върху пенообразуването. В [2] е установено, че стабилността и критичната дебелина на скъсване на филмите от Динакол и Солвитоза зависи както от съотношението между Динакола и Солвитозата, така и от концентрация на електролит в разтвора. Изказано е предположение, че това е свързано с възможните взаимодействия между Динакола и Солвитозата (с образуването на комплекси) и влиянието на електролита върху тях.
В пулпата се съдържат и парченца восъци, чиито частици могат да променят филмовата стабилност по два начина: като забавят изтъняването поради натрупването им извън филма или поради излъчването от тях на допълнителни вещества като мастни алкохоли, бустери или Дауфакс в разтвора. В условията на технологичния процес (при висока температура) е много вероятно от тези восъци да се излъчва определено количество

Дауфакс - йоногенно ПАВ, чието наличие в разтвора може да благоприятства пенообразуването. В [2] са изследвани пенни филми от филтрувани (за да се изключи стабилизиращата роля на частиците от Афранил) смесени разтвори на Динакол, Солвитоза и Афранил (с молни съотношения 1:10:1; 1:10:10) при температура 25° и 40°C и е показано, че филмовата стабилност действително нараства с повишаване на температурата и количеството на Афранила. Издигната е хипитеза за ролята на излъчения в разтвора Дауфакс от Афранила. За да бъде доказана тази хипотеза, е нужно да се изследват пенни филми от Динакол, Солвитоза и Дауфакс и да се съпоставят с данните за филми от чист Дауфакс и от смесени разтвори на Динакол, Солвитоза и Афранил. Това определи целта на настоящото изследване: да се изследва влиянието на Дауфакс върху кинетичната стабилност на пенни филми от смесен разтвор на Динакол и Солвитоза при 40ºС и получените данни да се съпоставят с параметрите за филми от смес на Динакол, Солвитоза и Афранил.
Експериментална част
А. Вещества. В настоящата работа са използвани следните вещества [3]:
Дауфакс 2A1 от BASF, който представлява смес от 12% Дауфакс 1 [динатриева сол на оксо - бис додецил бензен сулфонова киселина, С36Н56S2O7Na2 (710.2)] и 32% Дауфакс 2 [динатриева сол на окси додецил бензен сулфоновата киселина С24Н32S2O7Na2 (542.2)].
Динакол VS 50 FS от Akzo Nobel Chemicals GmbH. Той има отнасяния на анионно ПАВ, защото е смес от соли на така наречените „смолисти киселини”, които са под формата на смес от изомерни форми с обща формула C19H29COOH. Преоб-ладаващите киселини в тази смес са от абеитинов тип - абеитинова киселина и левопиринова киселина.
Солвитоза BRN от “AVEBE” Dеutschland GmbH, която представлява нишесте, върху веригата на което са закачени молекули от типа на алкил амониевите халогениди, поради което тя се разглежда като катионно ПАВ.

Натриев хлорид [NaCl, М = 58.45] с квалификация р.а., от Sigma, накален при 600°C за 4 часа, с концентрация 0.5 mol/l във всички изследвани разтвори.
Б. Експериментални условия. Използваните вещества са с концентрации: Динакол - 0.005%, Солвитоза - 0.05% и Дауфакс - 0.005 и 0.05%, подбрани така, че смесените разтворите да са със съотношения Динакол:Солвитоза = 1:10 и Динакол:Солвитоза:Дауфакс = 1:10:1 или 1:10:10. Тези концентрации и съотношенията между веществата в смесените разтвори са избрани в съответствие с условията, при които са изследвани адсорбционните свойства на използваните вещества и стабилизираните с тях пенни филми, налични в литературата [1,2].
Динаколът с концентрация 0.005% не може да бъде отговорен за стабилността на филмите, защото при молекулна маса на Динакола (307 g/mol), използваната концентрация ще съответства на 1.62×10-4 mol/l. Ако между Динакола и Солвито-зата (0.05%) се образува комплекс, следва че концентрацията на свободния в разтвора Динакол е 4×10-5 mol/l. Ясно е, че при тази ниска концентрация даже и да има електростатична стабилиза-ция, в присъствието на 0.5 mol/l NaCl, тя ще е напълно потисната.
Солвитозата с концентрация 0,05% (при молекулна маса за мономер равна на 160 g/mol) е 3.12×10-3 mol/l. Вземайки предвид, че молното съотношение между мономерите и закачените молекули на алкиламониевите халогениди в нея са 0.04 – 0.045 следва, че 4% от мономерите са заредени, т.е. концентрацията им е 12.5×10-5 mol/l. Според (Фиг. 1) в изотермата на повърхностното напрежение на смес от Динакол и Солвитоза, в отсъствие на електролит, се образува плато точно при съотношение 1:10, което е индикация за образуването на комплекс. В изотермата, получена в присъствие на 0.5 mol/l електролит, това плато изчезва, което означава, че се потиска образуването на комплекса Динакол/Солвитоза. Ако това става и във филма, то стеричната му стабилизация, определена от страна на Динакола и Солвитозата, намалява.
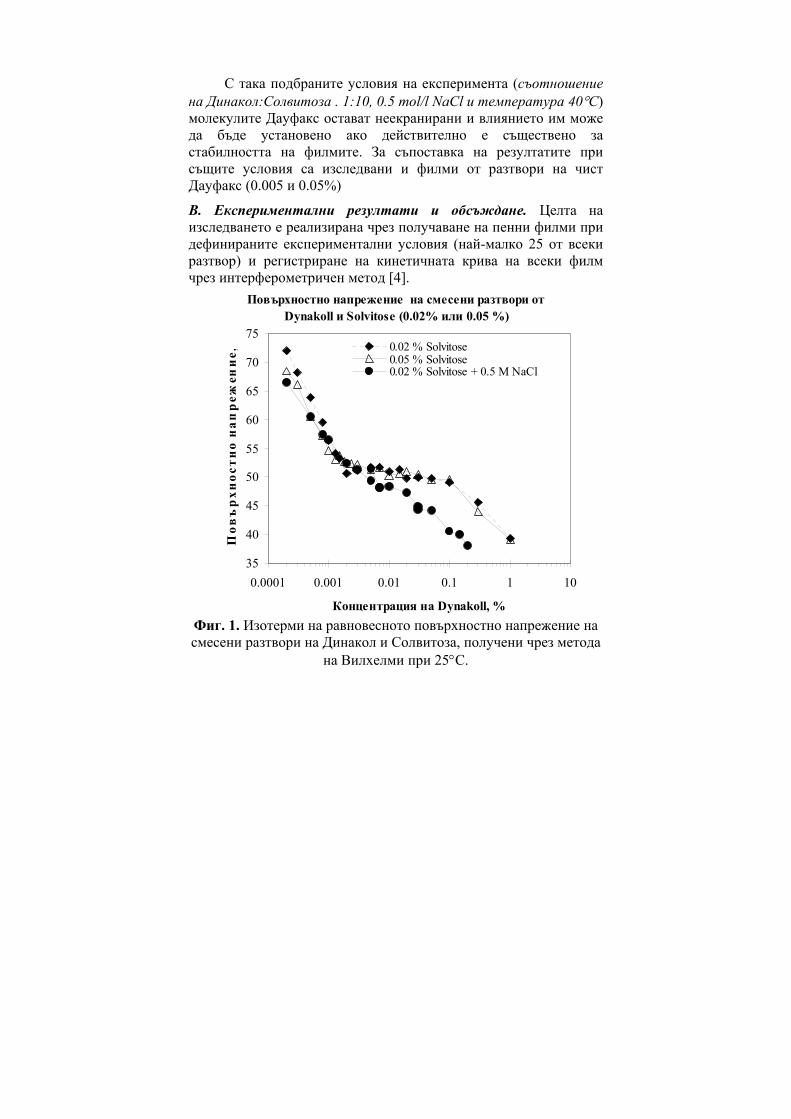
С така подбраните условия на експеримента (съотношение на Динакол:Солвитоза . 1:10, 0.5 mol/l NaCl и температура 40°C) молекулите Дауфакс остават неекранирани и влиянието им може да бъде установено ако действително е съществено за стабилността на филмите. За съпоставка на резултатите при същите условия са изследвани и филми от разтвори на чист Дауфакс (0.005 и 0.05%)
В. Експериментални резултати и обсъждане. Целта на изследването е реализирана чрез получаване на пенни филми при дефинираните експериментални условия (най-малко 25 от всеки разтвор) и регистриране на кинетичната крива на всеки филм чрез интерферометричен метод [4].
Повърхностно напрежение на смесени разтвори от Dynakoll и Solvitose (0.02% или 0.05 %)
35
40
45
50
55
60
65
70
75
0.0001 0.001 0.01 0.1 1 10
Концентрация на Dynakoll, %
Пов
ърхн
остн
о нап
реж
ение, 0.02 % Solvitose
0.05 % Solvitose0.02 % Solvitose + 0.5 M NaCl
Фиг. 1. Изотерми на равновесното повърхностно напрежение на смесени разтвори на Динакол и Солвитоза, получени чрез метода
на Вилхелми при 25°C.
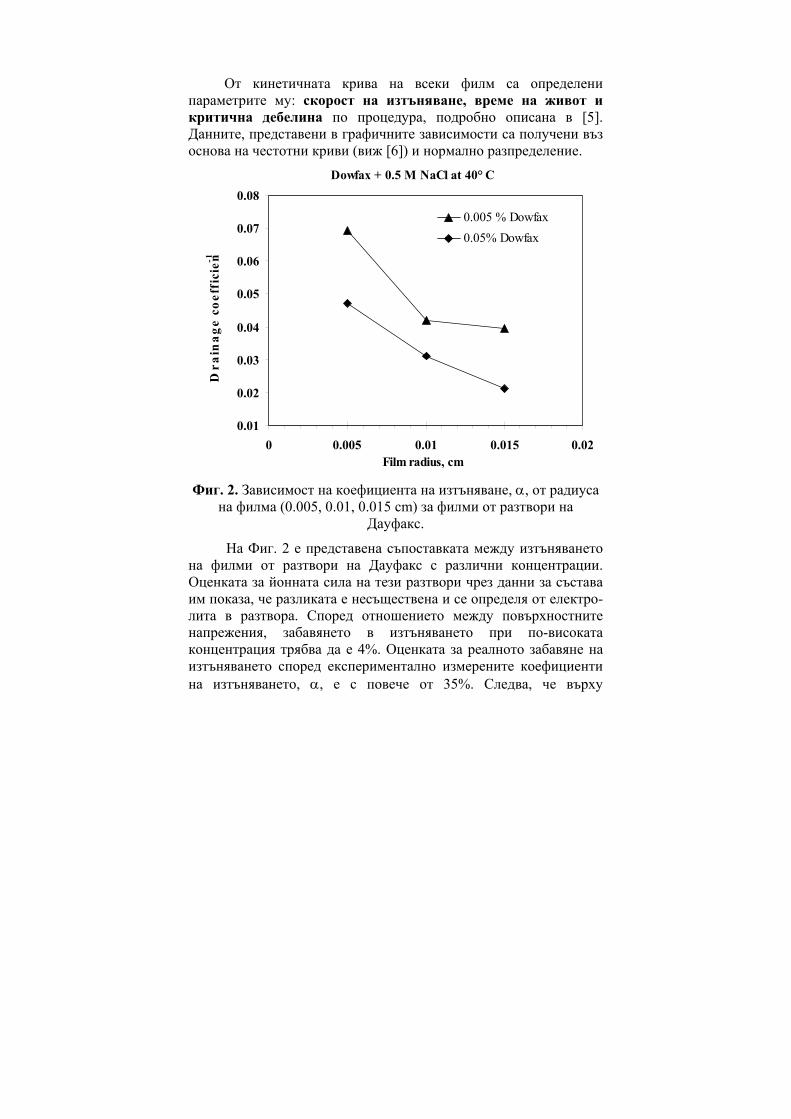
От кинетичната крива на всеки филм са определени параметрите му: скорост на изтъняване, време на живот и критична дебелина по процедура, подробно описана в [5]. Данните, представени в графичните зависимости са получени въз основа на честотни криви (виж [6]) и нормално разпределение.
Dowfax + 0.5 M NaCl at 40° C
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 0.005 0.01 0.015 0.02Film radius, cm
Dra
inag
e co
effi
cien-1
0.005 % Dowfax0.05% Dowfax
Фиг. 2. Зависимост на коефициента на изтъняване, α, от радиуса
на филма (0.005, 0.01, 0.015 сm) за филми от разтвори на Дауфакс.
На Фиг. 2 е представена съпоставката между изтъняването на филми от разтвори на Дауфакс с различни концентрации. Оценката за йонната сила на тези разтвори чрез данни за състава им показа, че разликата е несъществена и се определя от електро-лита в разтвора. Според отношението между повърхностните напрежения, забавянето в изтъняването при по-високата концентрация трябва да е 4%. Оценката за реалното забавяне на изтъняването според експериментално измерените коефициенти на изтъняването, α, е с повече от 35%. Следва, че върху
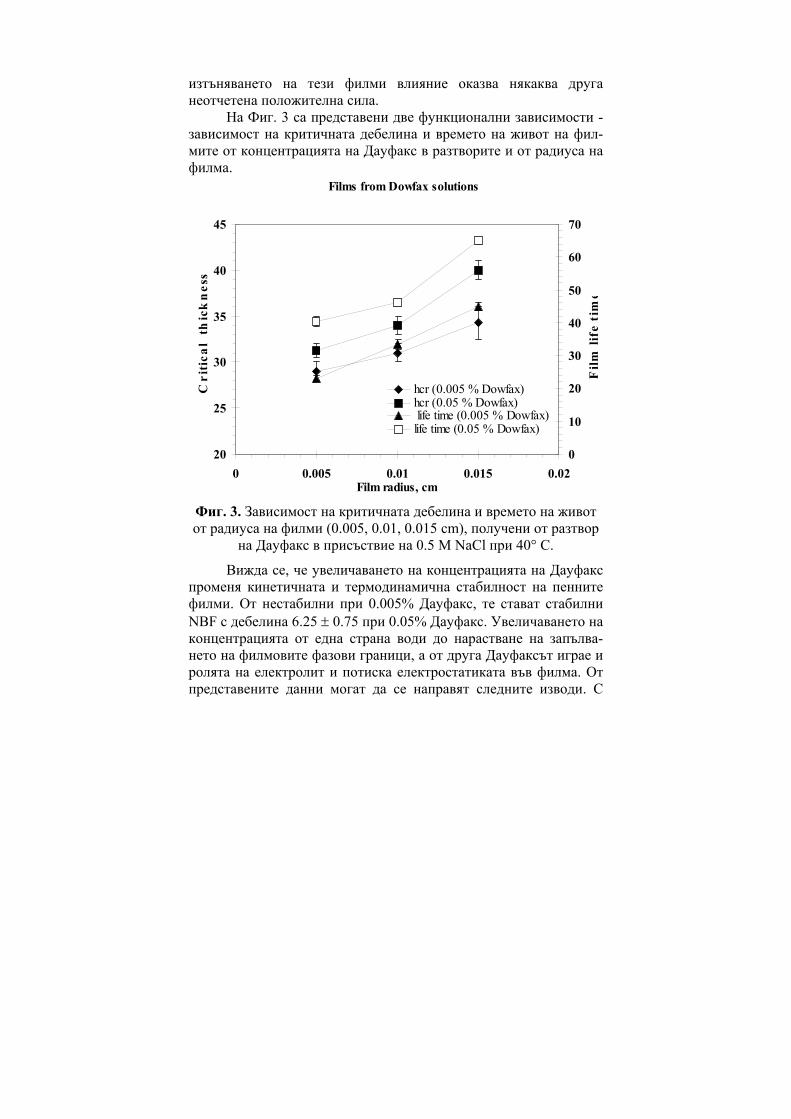
изтъняването на тези филми влияние оказва някаква друга неотчетена положителна сила.
На Фиг. 3 са представени две функционални зависимости - зависимост на критичната дебелина и времето на живот на фил-мите от концентрацията на Дауфакс в разтворите и от радиуса на филма.
Films from Dowfax solutions
20
25
30
35
40
45
0 0.005 0.01 0.015 0.02Film radius, cm
Cri
tica
l th
ick
nes
s
0
10
20
30
40
50
60
70
Film
life
tim
e
hcr (0.005 % Dowfax)hcr (0.05 % Dowfax) life time (0.005 % Dowfax)life time (0.05 % Dowfax)
Фиг. 3. Зависимост на критичната дебелина и времето на живот от радиуса на филми (0.005, 0.01, 0.015 cm), получени от разтвор
на Дауфакс в присъствие на 0.5 M NaCl при 40° С.
Вижда се, че увеличаването на концентрацията на Дауфакс променя кинетичната и термодинамична стабилност на пенните филми. От нестабилни при 0.005% Дауфакс, те стават стабилни NBF с дебелина 6.25 ± 0.75 при 0.05% Дауфакс. Увеличаването на концентрацията от една страна води до нарастване на запълва-нето на филмовите фазови граници, а от друга Дауфаксът играе и ролята на електролит и потиска електростатиката във филма. От представените данни могат да се направят следните изводи. С

увеличаване на концентрацията на Дауфакс расте критичната дебелина на филмите. За филмите от разтвори на 0.005% Дауфакс тя е дебелина на скъсване, докато за филмите при 0.05 % това е дебелината, при която се появява вторично черно петно и филма със скок преминава в NB. Ясно е, че електростатиката във филма е потисната и изтъняването на филмите ще се управлява от смучещата сила vwPP Π+=Δ σ , еластичността и повърхностния вискозитет на филмовите повърхности. При големи дебелини основния принос идва от капилярното налягане, което зависи от повърхностното напрежение, а то с нарастване на концентрацията на Дауфакс намалява и това води до намаляване на скоростта на изтъняване и до повишаване на hcr. Приближена оценка на базата на σ, съгласно формулата на Радоев и съавт. показа, че повишението на hcr трябва да бъде с около 1%. От експерименталните стойности за hcr увеличението е от 8 до 17% в зависимост от размера на филма. Това означава, че има неотчетен фактор, който забавя изтъняването.
Тенденцията за нарастване на критичната дебелина с увеличаване на радиуса на филма е в съответствие с теорията, но намерената тенденция за увеличаване на времето на живот на филмите с нарастване на радиуса е противоположна на очаквана-та. Такава тенденция е намерена и за филмите от смес на Динакол и Солвитоза - критичните дебелини и времената на живот на филмите нарастват с нарастване на радиуса им. В този случай нарастването на времето на живот се дължи на частиците от Солвитоза, които изпълняват ролята на стеричен стабилизатор.
Обобщените данни за изтъняването и критичната дебелина на пенните филми от разтворите на индивидуалните вещества Динакол и Дауфакс, смесите от Динакол и Солвитоза със съотношение 1:10 и от Динакол, Солвитоза и Дауфакс със съотношение 1:10:1 и 1:10:10 са представени на Фигури 4 и 5.
От кривите, представени Фиг. 4 се вижда, че филмите от бинарната смес Динакол и Солвитоза изтъняват най-бързо и се късат още докато са дебели (тъмно сиви), а филмите от тройната смес Динакол, Солвитоза и Дауфакс със съотношение 1:10:10 имат най-малък коефициент на изтъняване и са равновесни NF при всички изследвани радиуси (най-ниско разположената зави-
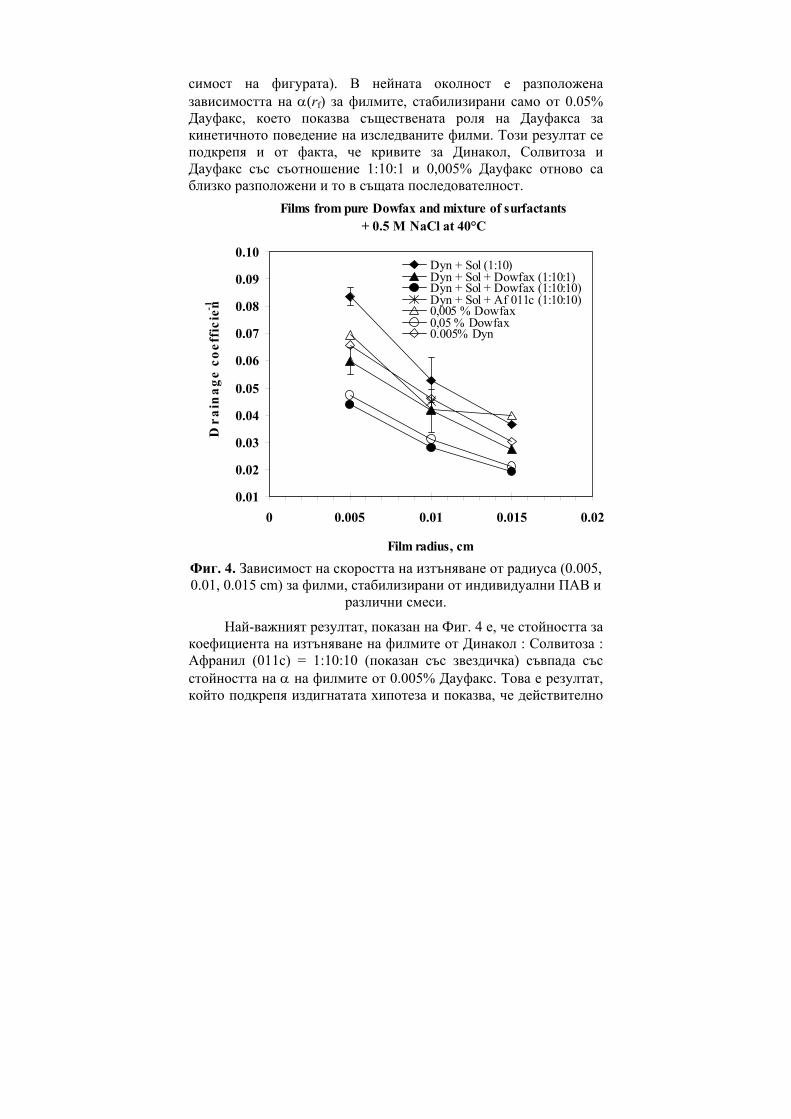
симост на фигурата). В нейната околност е разположена зависимостта на α(rf) за филмите, стабилизирани само от 0.05% Дауфакс, което показва съществената роля на Дауфакса за кинетичното поведение на изследваните филми. Този резултат се подкрепя и от факта, че кривите за Динакол, Солвитоза и Дауфакс със съотношение 1:10:1 и 0,005% Дауфакс отново са близко разположени и то в същата последователност.
Films from pure Dowfax and mixture of surfactants + 0.5 M NaCl at 40°C
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0 0.005 0.01 0.015 0.02
Film radius, cm
Dra
inag
e co
effi
cien-1
Dyn + Sol (1:10)Dyn + Sol + Dowfax (1:10:1)Dyn + Sol + Dowfax (1:10:10)Dyn + Sol + Af 011c (1:10:10)0,005 % Dowfax0,05 % Dowfax0.005% Dyn
Фиг. 4. Зависимост на скоростта на изтъняване от радиуса (0.005, 0.01, 0.015 cm) за филми, стабилизирани от индивидуални ПАВ и
различни смеси.
Най-важният резултат, показан на Фиг. 4 е, че стойността за коефициента на изтъняване на филмите от Динакол : Солвитоза : Афранил (011c) = 1:10:10 (показан със звездичка) съвпада със стойността на α на филмите от 0.005% Дауфакс. Това е резултат, който подкрепя издигнатата хипотеза и показва, че действително
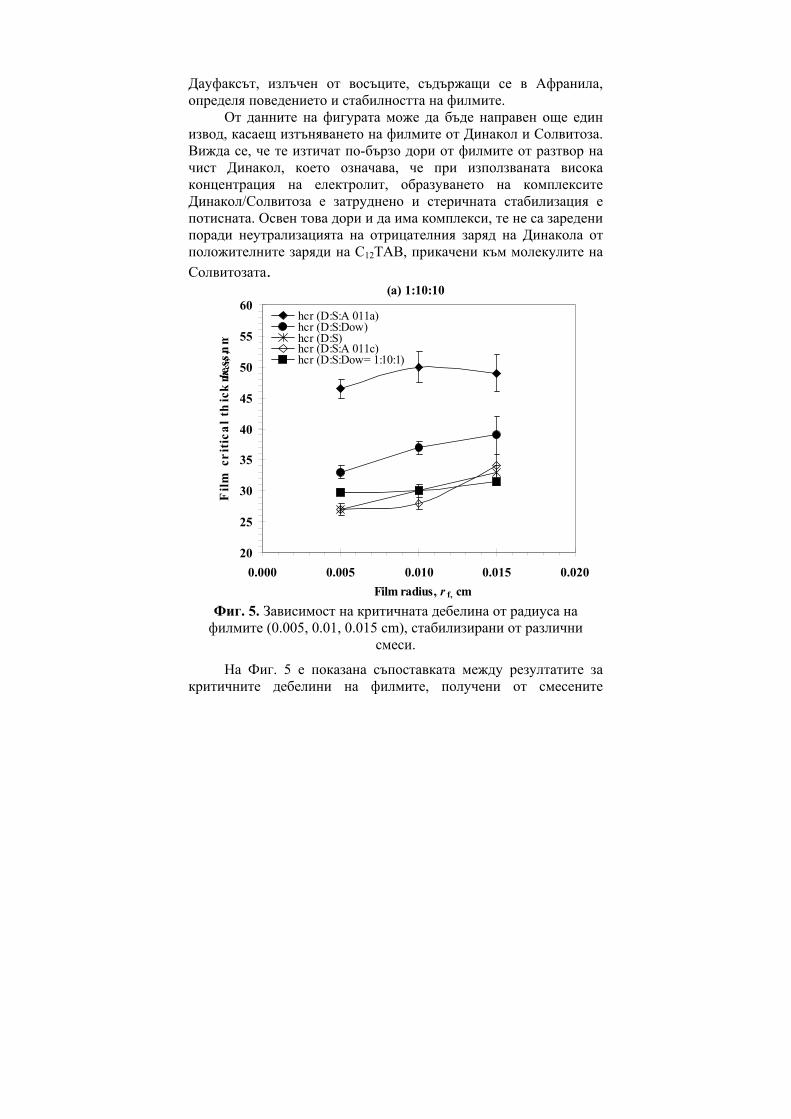
Дауфаксът, излъчен от восъците, съдържащи се в Афранила, определя поведението и стабилността на филмите.
От данните на фигурата може да бъде направен още един извод, касаещ изтъняването на филмите от Динакол и Солвитоза. Вижда се, че те изтичат по-бързо дори от филмите от разтвор на чист Динакол, което означава, че при използваната висока концентрация на електролит, образуването на комплексите Динакол/Солвитоза e затруднено и стеричната стабилизация е потисната. Освен това дори и да има комплекси, те не са заредени поради неутрализацията на отрицателния заряд на Динакола от положителните заряди на C12TAB, прикачени към молекулите на Солвитозата.
(a) 1:10:10
20
25
30
35
40
45
50
55
60
0.000 0.005 0.010 0.015 0.020Film radius, r f, cm
Film
cri
tica
l th
ick
nes
s,
hcr
, nm
hcr (D:S:A 011a)hcr (D:S:Dow)hcr (D:S)hcr (D:S:A 011c)hcr (D:S:Dow= 1:10:1)
Фиг. 5. Зависимост на критичната дебелина от радиуса на филмите (0.005, 0.01, 0.015 cm), стабилизирани от различни
смеси.
На Фиг. 5 е показана съпоставката между резултатите за критичните дебелини на филмите, получени от смесените

разтвори с различен състав и молно съотношение. Вижда се, че при всички смеси критичната дебелина нараства с увеличаване на филмовия радиус. По стойност критичните дебелини на филмите от смесите Динакол:Солвитоза = 1:10; Динакол:Солвитоза: Дауфакс = 1:10:1; Динакол:Солвитоза:Афранил (011c)= 1:10:10 са близки в рамките на грешката. Това означава, че при най-ниската концентрация на Дауфакс, влиянието му върху критичната дебелина на филмите от Динакол:Солвитоза = 1:10 не е съществено. Добрата корелация между стойностите за критичната дебелина на филмите от разтвори на Динакол:Солвитоза:Дауфакс = 1:10:1 с тези на филмите от Динакол:Солвитоза:Афранил (011c без бустер) = 1:10:10 показва, че при 40°C отделеният Дауфакс от Афранила е в количество, съответстващо на наличния в сместа Динакол:Солвитоза:Дауфакс = 1:10:1.
Високите стойности на критичната дебелина на филмите от Динакол:Солвитоза:Афранил (011a) = 1:10:10 се дължат на наличието на бустери (нейонни ПАВ) в този Афранил.
Литература
1. Report of Laboratory of Chemical Physics & Engineering, University of Sofia presented to BASF AG, Deutschland, Mechanistic studies and Optimization of Deaerators and Defoamers, Nov. (2005).
2. Angarska, J., Tachev, K. and B. Dimitrova, Annual of Shumen University, vol. XVIII B2 (2008) 132-145.
3. Гюлхан Расим, Дипломна работа, ФПН, ШУ, 2008. 4. Кругляков, П. М., Ексерова, Д. Р., “Пена и пенные пленки”,
Химия, (1991), Москва. 5. Angarska, J., Stubenrauch, C., and E. Manev, Colloids&Surfaces
A; vol. 309, 1-3 (2007) 189-197. 6. Angarska, J., B. Dimitrova and K. Tachev, Annual of Shumen
University, vol. XVII B1 (2007)147-159.

Изследване влиянието на вида и количеството на легиращата добавка в изходната
алумотермична смес върху някои показатели на получените боридни слоеве
Радослав Иванов Катедра “Органична химия и технология”
Факултет по природни науки Шуменски университет “Епископ Константин Преславски”
Университетска, № 115, Е- Мail: [email protected]
Abstract: The present work is devoted to the research on diffusive saturation of carbon steel with boron, boron and alluminium, boron and copper, boron and titanium, and boron and vanadium. The boriding is done using a powder alluminothermic mixture with major composition (В2О3 + 7%Al + 72,5%Al2O3 + 0,5%Na2O.4BF3), alloyed separately with Cu2O, ТiO2, and V2O5, treating at 1100 K for 6 hours. Analysis is performed on the thickness and microhardness of the obtained diffusive layers, their structure and composition, corrosive resistance and wearing quality, as well as the quantity of the alloying elements in them.
Keywords: carbon steel, diffusive saturation, boriding, boride layers, thickness, microhardness.
Въведение Получените при дифузионно насищане на стоманените
изделия с бор дифузионни слоеве се характеризират с висока микротвърдост, износоустойчивост, корозионна устойчивост, но и с висока крехкост.[1,2] С цел понижаване на крехкостта на слоя се прибягва до неговото легиране, но в някои случаи се стига до чувствително намаляване на дебелината му и понижаване на някои технологични показатели.[1-4] Насищането на стоманените изделия с бор и легиращи елементи може да се извърши по различни методи, като един от най-често използваните е алумотермията.[5,6] При алумотермичното бориране на стомани
в подходящи алумотермични смеси биха могли да се получат легирани дифузионни слоеве с желана дебелина и добри технологични показатели.[7]
Изложение Целта на настоящата работа е изследване влиянието на
дифузионното насищане на стомана 45 с бор, бор и алуминий, бор и мед, бор и титан, и бор и ванадий върху състава, структурата и някои показатели на получените дифузионни слоеве. Съставът на използваните алумотермични смеси е посочен в таблица 1.
Таблица 1. Състав на изходните смеси*
№
B2O3, мас.%
Cu2O, мас.%
TiO2, мас.%
V2O5, мас.%
B2O3/ Al; (Σ MeO/Al)
1 20 - - - 2,0:0,7 (2,0:0,7) 2 10 - - - 2,0:1,4 (2,0:0,7) 3 5 - - - 2,0:2,8 (2,0:0,7) 4 17 3,0 - - 1,7:0,7 (2,0:0,7) 5 15 5,0 - - 1,5:0,7 (2,0:0,7) 6 17 - 3,0 - 1,7:0,7 (2,0:0,7) 7 15 - 5,0 - 1,5:0,7 (2,0:0,7) 8 17 - - 3,0 1,7:0,7 (2,0:0,7) 9 15 - - 5,0 1,5:0,7 (2,0:0,7)
*Постоянни съдържания на: Al – 7,0%; Al2O3 – 72,5%; Na2O.4BF3 – 0,5%.
Химико-термичната обработка (ХТО) на образците от стомана 45 е проведена в херметизиран стоманен контейнер [8] от неръждаема стомана при температура 1100 К и продължителност 6 h. Схемата на лабораторната апаратура е показана на фиг.1.

Фиг. 1. Схема на лабораторната апаратура.
1-контейнер; 2-капак; 3-уплътнение; 4-подвижна скоба; 5-винт; 6-прахообразна алумотермична смес; 7-образци; 8-междинен
капак; 9 пълнител от Al2O3 ; 10-електросъпротивителна пещ; 11-кожух на термометъра; 12-термодвойка; 13-кожух на
термодвойката; 14-миливолтметър.
Протичат следните реакции: 2/3 B2O3 + 4/3 Al → 4/3 B + 2/3 Al2O3, ΔG = -350 kJ/mol; (1) 3Cu2O + 2Al → 6Cu + Al2O3, ΔG = - 138 kJ/mol; (2) TiO2 + 4/3 Al → Ti + 2/3 Al2O3 , ΔG = -155 kJ/mol; (3) 2/5V2O5 + 4/3 Al → 4/5 V + 2/3 Al2O3, ΔG = -425 kJ/mol (4)
Обработените образци са подложени на следните анализи: Микроструктурен анализ на черно-бяло и цветно проявени микрошлифове, измерване дебелината на формирания

дифузионен слой на микроскоп “Neophot-2”, измерване на микротвърдостта на слоевете на микротвърдомер ПМТ-3, рентгенофлуоресцентен анализ (РФА) за съдържание на легиращите елементи в слоевете, изпитване за корозионна устойчивост в 10% HCl в продължение на 240 h; изпитване на устойчивост на абразивен износ на машина за полиране “Мinosupan” с използване на шлифовъчна хартия № 280 в продължение на 10 min (дължина на пътя – 400m) при натиск 10 kg/mm² [9]. Обработените резултати са представени в таблица 2.
От данните в таблици 1 и 2 се вижда, че при 20%-но съдържание на В2О3 (съотношение В2О3 : Al = 2,0 : 0,7), което според [2] гарантира получаването на дифузионен слой, състоящ се само от железни бориди [10]. Показателни за това са резултатите от цветното проявяване на микрошлифовете (FeB се оцветява в синьо-виолетово, а Fe2B в жълто-кафяво) и най-вече тези от измерването на микротвърдостта (H0,1). Действително, полученият слой е с дебелина 167±35μm, иглеста структура с микротвърдост в отделните зони на слоя: 1980±140MPa, 1297±147MPa и 482±162MPa, стойности, които съответстват на боридните фази FeB, Fe2B и α-твърд разтвор на бор в желязото.
При 10%-но съдържание на B2O3 в изходната смес съотношението В2О3 : Al вече става 2 : 1,4, при което според [2] вече се получава бороалитиран дифузионен слой с преобладаване на боридните фази. Полученият при насищането дифузионен слой е с по-голяма дебелина - 236±10 μm. Тук обаче съдържанието на Al в изходната смес съставлява 70% от това на B2O3, което според [3] предполага получаването на бориден слой без съдържание на Al в него. Измерените по сечението на формирания дифузионен слой стойности за микротвърдостта са: 2236±165 MPa, 1539±56 MPa и 794±38MPa, които съответстват на боридните фази – FeB и Fe2B и на α-твърд разтвор на бор в желязото.

Фиг. 2. Микроструктура (х100) на дифузионен слой, получен в
алумотермична смес с 10% B2O3.
Действително, полученият слой (фиг.2) е с характерната за боридите иглеста структура, равномерен фронт на растеж и най-голяма дебелина. Според класификацията, дадена в [3], този слой може да се отнесе към първия тип бороалитирани слоеве, малко отличаващ се от боридния слой. Вероятно има известно количество Al, разтворен в боридите, което е довело и до повишаване на микротвърдостта.
При 5%-но съдържание на B2O3 в алумотермичната смес съотношението B2O3 : Al става 2,0 : 2,8, което според [2] предполага формирането на бороалитиран слой с преобладаване на алитирането. Полученият при това насищане дифузионен слой е със значително по-малка дебелина - 138±21 μm, плътен, хомогенен, без иглеста структура, с бяла ивица от страна на металната основа. Според класификацията, дадена в [3] основната част на този слой се състои от алуминидни фази - Fe2Al5 + свръхструктури (FeAl+Fe3Al) + α-фаза. Бялата ивица, разположена между алуминидните фази и основния метал, вероятно е боридна фаза, представена от Fe2B, чиято микротвърдост достига 1598±188 MPa, благодарение на разтварянето на Al в него. Микротвърдостта на алуминидните фази е 827±83MPa, а на α-фазата - 254±45MPa.
От данните в таблица 2 се вижда, че с увеличаване на съдържанието на Cu2O в изходната алумотермична смес дебелината на боридния слой (δ) намалява до 146 μm при 3% и

отново нараства и то значително – достигайки стойности около и над 220 μm. Получаваните дифузионни слоеве са двуфазни, включващи и двата железни борида (FeB и Fe2B). Микротвърдостта на FeB е около 2000-2100 МРа и съвсем слабо се понижава с увеличаване на съдържанието на легиращата добавка в насищащата смес. Микротвърдостта на Fe2B е значително по-ниска (1200-1480 МРа) и слабо нараства при увеличаване на съдържанието на Cu2O в изходната алумотермична смес. Преходната зона, представляваща вероятно α-тв. р-р, се характеризира с ниска микротвърдост (500-600 МРа), която все пак е по-висока от тази на металната основа (около 120 МРа).
Получаваните дифузионни слоеве са с характерната за боридните слоеве иглеста структура, която се запазва в целия интервал на изменение на съдържанието на Cu2O в изходната насищаща смес.
Оскъдните данни от изследвания по титаниране и още повече по боротитаниране на стомани правят много труден анализа на получените резултати. Така, на пръв поглед трудно може да се обясни първоначалното намаляване на дебелината (δ) на получавания дифузионен слой при въвеждане на TiO2 в изходната борираща смес и следващото и нарастване при увеличаване съдържанието на TiO2 в сместа. Това изменение в дебелината може да се обясни по следния начин: Титанът, отделен при металотермичната редукция дифундира в образците заедно с бора. При ниски съдържания на TiO2 в сместа мощността и по отношение на титана е ниска и насищането с него е слабо. Насищането на образците е предимно с бор. Едновременно с това титанът силно намалява скоростта на дифузия на бора в стоманата, а оттам намалява и дебелината на дифузионния слой. При по-високо съдържание на TiO2 в изходната смес нараства мощността и по отношение на титана, което води и до нарастване дебелината на дифузионния слой. От микроструктурния анализ е установено, че дифузионният слой е разнороден. На повърхността се разполага сравнително тънък подслой от железни бориди и вътрешен подслой със значително по-голяма дебелина и структура, различна от тази на

сърцевината. В боридния подслой вероятно преобладава FeB, тъй като титанът повишава съдържанието му и се концентрира в него. Доказателство за това е значителното съдържание на титан в повърхностния подслой (1,65%), установено чрез РФА. Този подслой е и с високи стойности на микротвърдостта (2943 МРа). Освен, че се концентрира в FeB титанът повишава и неговата микротвърдост. Вътрешният подслой вероятно представлява твърд разтвор на бор и титан в α-Fe, характеризиращ се с ниски стойности на микротвърдостта (613МРа). Междинните стойности за микротвърдостта (1100-1800 МРа) съответстват на Fe2B, който се разполага в началото на вътрешния подслой. Малко вероятно е образуването на съединения между желязото и титана като FeTi и FeTi2, тъй като съдържанието на титан в тях е високо, а от друга страна в диаграмата на състоянието на системата Fe-Ti съществува широка област на твърд разтвор на титан в α-Fe [11].
От данните в таблица 2 се вижда, че с повишаване съдържанието на V2O5 в изходната алумотермична смес от 0 до 5% дебелината (δ) на дифузионния слой намалява от 167 до 104 μm. Това може да се обясни със затормозяващото действие, което оказва въглерода върху дифузията на двата насищащи елемента – B и V. Резултатите от РФА на обработените образци показват сравнително ниско съдържание на V в дифузионния слой. При такива ниски съдържания (0,05-0,1%) е възможно образуване само на твърд разтвор на V в α-Fe. Вероятно слоят е бориден, легиран с V. Това се потвърждава и от стойностите за микротвърдостта (H0,1), които съответстват по скоро на тези за микротвърдостта на железните бориди, отколкото на съединения на бора с ванадия (микротвърдостта на V3B2 е над 2300 MPa, а на VB2 – до 2800 MPa [10]). Много ниските стойности за микротвърдостта на дифузионния слой за образци, обработени в смес с 5% V2O5 вероятно отговарят на твърд разтвор на В и V в α-Fe. Ванадият дифундира на значително по-голяма дълбочина в образците, а борът образува тънък бориден слой на повърхността, чиято микротвърдост трудно може да се измери. От микроструктурния анализ е установено, че дифузионният слой е разнороден. Непосредствено до повърхността се разполага светъл подслой от железни бориди (най-вероятно FeB) и вътрешен

хомогенен, различаващ се по структура от ферито-перлитната сърцевина. Потвърждение за значителната дълбочина на дифундиране на ванадия са резултатите от послойния анализ на образци, подложен само на ванадиране. Дифузионният слой достига дебелина над 200μm, а съдържанието на ванадий на дълбочина 150μm от повърхността е около 0,1%.
Изводи
1. С понижаване съдържанието на В2О3 в изходната алумотермична смес от 20% до 10% (увеличаване на съотношението В2О3 : Al от 2,0:0,7 до 2,0:1,4) включително, формираният дифузионен слой е с нарастваща дебелина , иглеста структура и микротвърдост, характерни за боридните слоеве, състоящи се от двата борида (FeB + Fe2B).
2. При 5% -но съдържание на В2О3 (съотношение В2О3 : Al = 2,0 : 2,8) полученият дифузионен слой е бороалитиран, включващ алуминидни фази (Fe2Al5+ FeAl+Fe3Al+α-фаза). Бялата ивица, разположена между алуминидните фази и основния метал, вероятно е боридна фаза, представена от Fe2B.
3. При съвместното насищане на стоманените образци с бор и мед получаваните дифузионни слоеве са двуфазни (FeB + Fe2B), със значителна дебелина, с характерната за тях иглеста структура и микротвърдост, съответстваща на двата борида.
4. С повишаване съдържанието на TiO2 в изходната смес дебелината на дифузионния слой нараства значително. Формираните дифузионни слоеве са с разнородна структура. На повърхността се намира подслой с микротвърдост 2650 - 2940 МРа, характерна за FeB. Под него се намира друг, представляващ вероятно твърд разтвор на B и Ti в α-Fe с ниска микротвърдост – 730МРа.
5. При повишаване съдържанието на V2O5 дебелината на слоя намалява. Формираните дифузионни слоеве са с разнородна структура. На повърхността се разполага тънък бориден подслой (FeB) с висока микротвърдост (до 2160 МРа), а под него друг, представляващ вероятно твърд разтвор на B и V в α-Fe с ниска микротвърдост – до 320 МРа.

Литература 1. Ворошнин Л. Г., Борирование промышленных сталей и
чугунов, Минск, Наука и техника, 1985. 2. Ляхович Л. и др., Многокомпонентные диффузионные
покрытия, Минск, Науковая думка, 1974, с. 99-118. 3. Борисенок Г. и др., Химико-термическая обработка
металлов и сплавов Справочник, М, Металлургия, 1981, с. 296-312.
4. Полевой С. Н., В. Д. Евдокимов, Упрочнение металлов, М. Машиностроение, 1986.
5. Chchłowski M., K. Przybyłowicz, M. Chochlowska, Metaloznawstwo i obrobka cieplna, 1986, 79, s.12-17.
6. Гузанов Б. и др., Защитные покрытия на металлах. 1991. в. 25. с.34-36.
7. Uzunov N., R. Ivanov, Applied Surface Science 225 (2004), p.72-77.
8. Иванов Р. И., Авт. св-во РБ № 51308. 9. Ivanov R., T. Ignatova, UNITECH`04, Gabrowo, 2004, vol.II,
190-193. 10. Самсонов Г. В., и др., Бориды, М., Атомиздат, 1975. 11. Kubaschewski O., Iron-Binary Phase Diagrams, M., 1982.

Сравнителен анализ за възможностите и особеностите на моделиране на хроматографско
задържане на пиразини с помощта на молекулни дескриптори
Марина Московкина, Иван Бангов
Катедра „Обща химия”, Факултет по природни науки Шуменски университет “Епископ Константин Преславски”
ул. Университетска, 115, 9712 Шумен Е-Мail: [email protected]
Аbstract: The Quantitative Structure Retention Relationship (QSRR) approach has been applied to model the gas chromatographic retention of 111 substituted pyrazines, separated on the columns with different polarities (unpolar phase OV-101 and polar phase CW-20). The experimental retention data used for QSRRs deriving were taken from literature [1].
Comparison of parametric values of regression coefficients in the similar QSRR models for chromatographic phases with different polarities exhibits the specific features of molecular interactions between the solutes and stationary phases in both cases. The statistical precision of the various QSRRs derived with different global descriptors for the both phases was used to compare their ability to reflect the intermolecular interactions between the solutes and the stationary phases.
Keywords: pyrazines, molecular indices, QSRR.
Увод Пиразините като хетероциклени ароматни вещества имат
широко приложение в промишлеността, а именно в хранително-вкусовата, фармацевтичната и медицинската, поради което интереса към тях е голям. При анализиране на пиразин-съдържащи вещества особено често се използват газхроматографски методи. Като основният подход за газ-хроматографско идентефициране на пиразините се предпочита

методът на сравнение на ретентното им свойство със това на стандартните (еталонни) смеси, специфични за всеки един анализиран материал. Трудностите при подобен подход са в това, че не винаги е възможно да се създаде точно необходимата стандартна смес, състояща се от чисти еталонни компоненти. По тази причина е много наложително и актуално да се създават теоретични методи за прогнозиране на ретентни свойства, в основата на които да присъстват структурни характеристики на анализиращите се вещества. Изследванията от вида “ретентно свойство – структура” се отнасят към хемометрични проблеми и представляват отделна част от общия хемометричен подход, известен като “QSPR” - подход (Quantitative Structure - Property Relationships). Целта на настоящата работа е чрез използване на компютърен метод да се създадат регресионни модели свързващи ретентните характеристики на заместените пиразини и да научим колко различия в модела може да опишат полярните различия между две стационарни фази.
Теоретична и експериментална част
В анализираната група влизат 111 вещества, които са разделени в две газхроматографски колони с различни полярности: OV-101 и CW-20M. Експерименталните данни са взети от литература [1]; ретентното свойство е представено като индекси на Kovats, получени при изотермично газхроматографско разделяне.
Съединенията включени в изследването имат структури показани схематично на фиг.1. Неподвижните фази, върху които са разделени пиразините са с различна полярност: неполярна OV-101 и полярна CW-20M. Метилсиликоновите фази са слабополярни фази; взаимодействията с тях се определят от дисперсионни сили. Полярната фаза CW-20M е от групата на полиетиленгликолевите фази. В структурите на тези фази присъстват етерни кислородни атоми (в централния фрагмент), които са протоноакцепторни, и хидроксилните групи в крайните фрагменти – протонни донори.

R2 - H, R, O-R, SR, O-Ph, S-Ph, Racetyl, Cl R3 - H, R, Cl
R5 - H, R R6 - H, R
Фиг.1. Обща структура на заместени пиразини
В зависимост от съотношението между двата вида кислородни атоми (зависи от дължината на централния полиетиленов фрагмент) се създава различна селективност, в основата на която е концентрация на хидроксилните групи, достъпни за междумолекулни взаимодействия (ММВ). Доминиращ вид ММВ е чрез създаване на водородна връзка.
За създаване на корелации между ретентното свойство и молекулни характеристики на разделящите се вещества бе необходимо да се изчисляват различни молекулни индекси, които да се използват за дескриптори в линейни регресии. За всяка една молекулна структура са изчислени по 25 индекса, които пренадлежат към различни групи молекулни дескриптори. Приоритетно са изчислявани физикохимичните (конституционалните) дескриптори като молекулната маса М, моларната рефракция MR, поляризуемост, молекулната хидрофобност, Ван-дер-Ваалсовите обем V и повърхност S и няколко топологични индекса- индексът на Винер W и тополого-електронен индекс CTI; всички квантови индекси – глобалните и локалните и индикаторните дескриптори, които отразяват присъствие на различни структурни фрагменти в анализираните молекули. Квантовохимичните молекулни индекси са изчислявани с помощта на програмния пакет HyperChem по полуемпиричния метод АМ1 след геометрична оптимизация. Изчисляваните индекси са: обща молекулна енергия, Etot ; енергиите на висшата заета молекулна орбитала, EHOMO и на нисшата незаета орбитала, ELUMO; тяхната полусума - молекулната електроотрицателност, EN и молекулната реакционоспособност, HARD, която се изчислява като полуразлика между EHOMO и ELUMO ; молекулен диполен момент, μ ; атомни заряди на всички атоми в молекулата, qi.
N
NR2
R3R5
R6

Със същата програма са получени и някои физико-химични молекулни индекси, а именно: молекулната рефракция, MR, молекулна маса, М, хидрофобност, logP , поляризуемост, α, Ван-дер-Ваалсова повърхност, S и Ван-дер-Ваалсов обем, V.
Тополого-електронния индекс CTI и топологичния индекс на Винер W са изчислявани с помощта на програмата STRMAN, разработена от И. Бангов.
В настоящото изследване регресионния анализ е извършен с помощта на софтуерни продукти EXCEL и STATISTIKA.
Резултати и обсъждане
За всички глобални молекулни дескриптори стойностите на корелационните коефициенти за неполярната фаза са по-високи, отколкото за полярната фаза. Изключение прави индекс CTI, за който те са еднакви (R=0.91). Най-големи стойности на R за неполярната фаза имат глобалните дескриптори, които описват екстензивни молекулни свойства – молекулната маса M, моларната рефракция MR и поляризуемост α. Тези свойства определят силата на дисперсионните взаимодействия. При хроматографско разделяне в неполярната фаза задържането се определя предимно от дисперсионни взаимодействия. Информация за разпределение на електронната плътност значимо се използва за описание на полярни взаимодействия, участието на които е доминиращо за полярната фаза; в неполярната фаза то е нюансиращо. Именно присъствие на информация за разпределение на електронната плътност в индекса CTI повишава неговата корелация със задържане в полярната фаза и го прави най-предпочитан глобален молекулен дескриптор.
Общо при моделиране за двете фази е характерно, че с всеки от проверените глобални дескриптори се създават регресионни модели. Типове модели, създадени за един глобален екстензивен дескриптор се възпроизвеждат и при комбинации с други дескриптори.
При моделиране за OV-101 фаза се наблюдават следните особености: от всички проверени глобални дескриптори не може да се избере един, който да участва във всички видове модели и формира адекватни уравнения.

Въвеждане само на един нюансиращ дескриптор повишава статистическите показатели и води до получаване на адекватни регресии. Модели с два параметъра в които се постига точност R>0.9 се създават, когато към един глобален дескриптор се добавя един от квантовите индекси (Elumo, или на висшата заета АО Ehomo, ΔHform, Hard, q2 или μ). От индикаторните дескриптори значими модели се формират само при включване на тиофенилен структурен фрагмент S-Ph и оксоалкилен структурен фрагмент O-R.
При оценяване на статистическите показатели (R) се вижда, че най-слабо нюансиращо действие се оказва от въвеждане на диполен момент μ като параметър. В модели от този тип се постига точност 0.91<R<0.95. Добавяне на други полярни дескриптори влияе по-значимо и сформираните с тях модели имат по-висока точност (0.94< R<0.99). Най-значими уравнения с два параметъра са регресии с общ вид:
RI = D + ΔHform, и RI = D + q2 . Въвеждане на трети параметър доведе до повишаване на
статистическите показатели, когато в регресиите са включени комбинации от RI = (D+ΔHform+q2) или изцяло с изчислителни дескриптори RI = (D + q2+ q3). Показателите за R стигат до R=0.989.
Уравненията от всички типове имат най високи стойности за R, ако за глобални дескриптори са включени индексите M и MR.
При създаване на регресии с по-голям брой параметри се установи, че адекватни модели се получават при комбиниране с гореописаните полярни дескриптори. Статистическите показатели в многовариационни регресии показват, че се постига висока точност (0.988< R<0.995).
Интересно е да се знае, дали има необходимост да се използват многобройни изчислителни дескриптори или аналогична (адекватна) статистическа точност може да бъде получена като в регресиите се включват индикаторни структурни локални дескриптори. С тази цел бе направено групиране на уравнения в три групи – в първа група попадат модели, в които към глобален дескриптор са добавени само изчисляеми индекси;
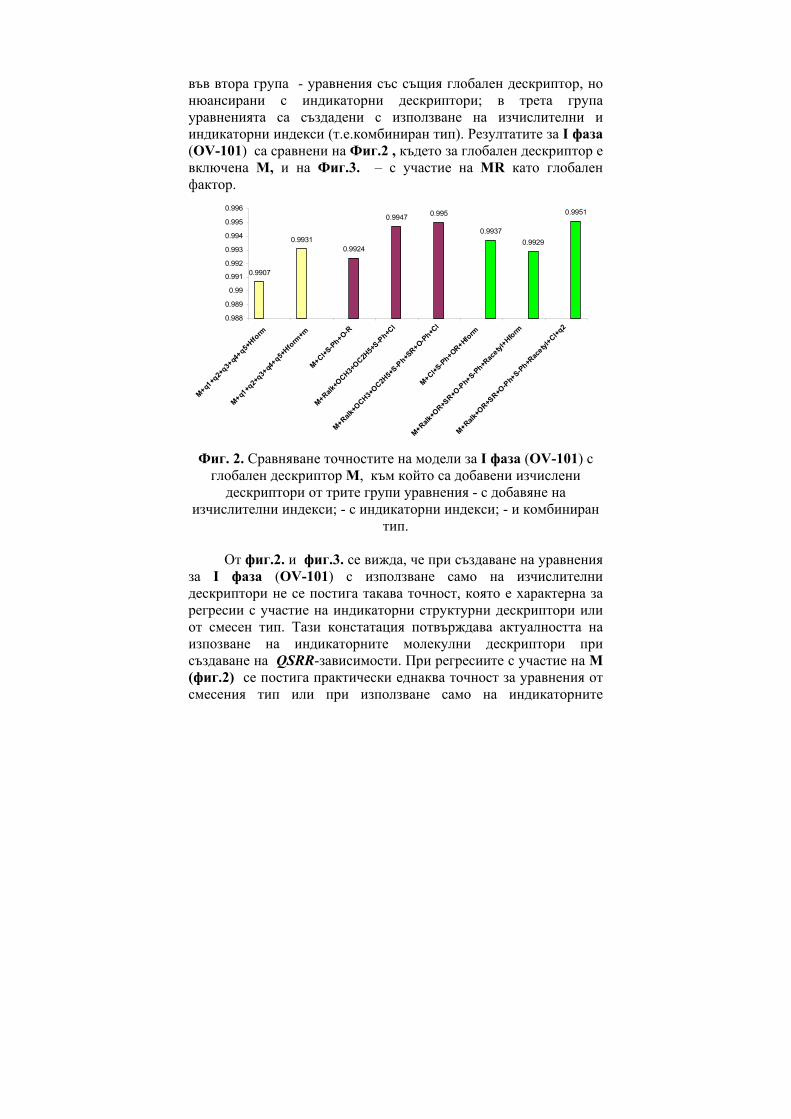
във втора група - уравнения със същия глобален дескриптор, но нюансирани с индикаторни дескриптори; в трета група уравненията са създадени с използване на изчислителни и индикаторни индекси (т.е.комбиниран тип). Резултатите за I фаза (OV-101) са сравнени на Фиг.2 , където за глобален дескриптор е включена М, и на Фиг.3. – с участие на МR като глобален фактор.
0.9907
0.99310.9924
0.9947 0.995
0.99370.9929
0.9951
0.988
0.989
0.99
0.991
0.992
0.993
0.994
0.995
0.996
M+q1+q
2+q3+
q4+q5+
Hform
M+q1+q
2+q3+
q4+q5+
Hform
+m
M+Cl+S-P
h+O-R
M+Ralk+O
CH3+OC2H
5+S-P
h+Cl
M+Ralk+O
CH3+OC2H
5+S-P
h+SR+O-P
h+Cl
M+Cl+S-P
h+OR+Hform
M+Ralk+O
R+SR+O
-Ph+
S-Ph+R
acetyl
+Hform
M+Ralk+O
R+SR+O
-Ph+
S-Ph+R
acetyl
+Cl+q2
Фиг. 2. Сравняване точностите на модели за I фаза (OV-101) с глобален дескриптор М, към който са добавени изчислени дескриптори от трите групи уравнения - с добавяне на
изчислителни индекси; - с индикаторни индекси; - и комбиниран тип.
От фиг.2. и фиг.3. се вижда, че при създаване на уравнения
за I фаза (OV-101) с използване само на изчислителни дескриптори не се постига такава точност, която е характерна за регресии с участие на индикаторни структурни дескриптори или от смесен тип. Тази констатация потвърждава актуалността на изпозване на индикаторните молекулни дескриптори при създаване на QSRR-зависимости. При регресиите с участие на М (фиг.2) се постига практически еднаква точност за уравнения от смесения тип или при използване само на индикаторните
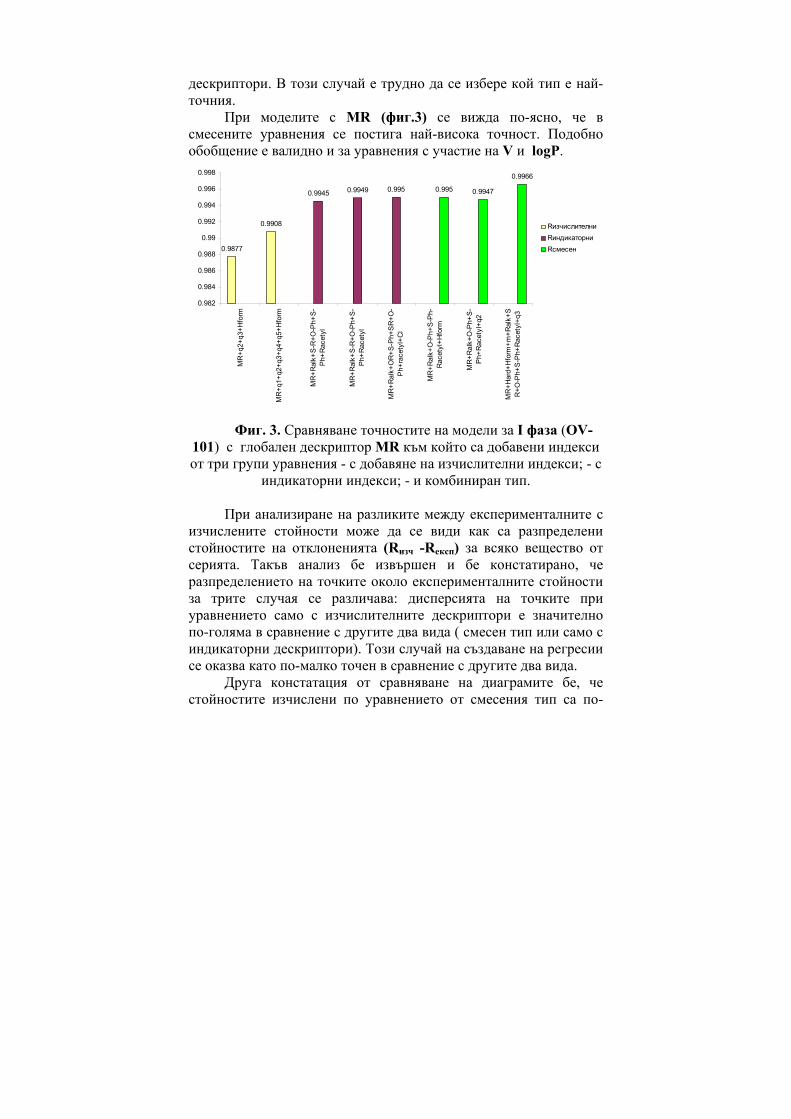
дескриптори. В този случай е трудно да се избере кой тип е най-точния.
При моделите с МR (фиг.3) се вижда по-ясно, че в смесените уравнения се постига най-висока точност. Подобно обобщение е валидно и за уравнения с участие на V и logP.
0.9877
0.9908
0.9945 0.9949 0.995 0.995 0.9947
0.9966
0.982
0.984
0.986
0.988
0.99
0.992
0.994
0.996
0.998
MR
+q2+
q3+H
form
MR
+q1+
q2+q
3+q4
+q5+
Hfo
rm
MR
+Ral
k+S
-R+O
-Ph+
S-
Ph+
Rac
etyl
MR
+Ral
k+S
-R+O
-Ph+
S-
Ph+
Rac
etyl
MR
+Ral
k+O
R+S
-Ph+
SR
+O-
Ph+
race
tyl+
Cl
MR
+Ral
k+O
-Ph+
S-P
h-R
acet
yl+H
form
MR
+Ral
k+O
-Ph+
S-
Ph+
Rac
etyl
+q2
MR
+Har
d+H
form
+m+R
alk+
SR
+O-P
h+S
-Ph+
Rac
etyl
+q3
RизчислителниRиндикаторниRсмесен
Фиг. 3. Сравняване точностите на модели за I фаза (OV-
101) с глобален дескриптор МR към който са добавени индекси от три групи уравнения - с добавяне на изчислителни индекси; - с
индикаторни индекси; - и комбиниран тип.
При анализиране на разликите между експерименталните с изчислените стойности може да се види как са разпределени стойностите на отклоненията (Rизч -Rексп) за всяко вещество от серията. Такъв анализ бе извършен и бе констатирано, че разпределението на точките около експерименталните стойности за трите случая се различава: дисперсията на точките при уравнението само с изчислителните дескриптори е значително по-голяма в сравнение с другите два вида ( смесен тип или само с индикаторни дескриптори). Този случай на създаване на регресии се оказва като по-малко точен в сравнение с другите два вида.
Друга констатация от сравняване на диаграмите бе, че стойностите изчислени по уравнението от смесения тип са по-

близки до експерименталните, отколкото точките получени по регресията с индикаторните дескриптори (близката област Rексп±30 е по-плътно населена).
Аналогичен анализ бе проведен за сравняване на уравнения, получени за II фаза (CW-20M) е показана на фиг.4. (с М като глобален фактор) и фиг.5. (с участие на МR )
0.9627
0.970.9737
0.9947
0.9833 0.9846 0.9857 0.9833
0.9948
0.9864
0.94
0.95
0.96
0.97
0.98
0.99
1
M+q1+
q2+q3
+Hform
M+Ralk
+O-Ph+
S-Ph+
Racety
l
M+Ralk
+OCH3+O-C
2H5+Cl+S-P
h
M+Ralk
+OCH3+OC2H
5+S-P
h+Cl
M+Ralk
+OCH3+OC2H
5+S-P
h+SR+O-P
h+Cl
MR+Ralk+O
R+S-R
+OPh+
Sph+Rac
etyl
M+Ralk
+OR+SR+O-Ph+
S-Ph+R
acetyl
+Cl
M+Ralk
+O-Ph+
S-Ph+
Racety
l+q2
M+Ralk
+OR+SR+O-Ph+
S-Ph+R
acetyl
+Hform
M+Ralk
+OR+SR+O-Ph+
S-Ph+R
acetyl
+Cl+q2
RизчислителниRиндикаторниRсмесен
Фиг. 4. Сравняване точностите на модели за II фаза (CW-20M) ) с глобален дескриптор М към който са добавени молекулни
дескриптори от три групи - с добавяне на изчислителни индекси; - с индикаторни индекси; - и комбиниран тип.
За полярната фаза още по-ясно се вижда, че най-точните регресии се създават при използване на комбиниране на изчислителните и индикаторни дескриптори.

0.95
66
0.9861 0.98470.9914
0.9860.98160.9745
0.930.940.950.960.970.980.99
1
MR+
q1+q
2+q3
+q4+
q5.. .
MR+
Ralk
+O-P
h,+S
-Ph.
..M
R+Ra
lk+O
-Ph+
S-R+
S...
MR+
Ralk
+O-R
+O-P
h+...
MR+
Hfor
m+R
alk+
O-P
...M
R+Ra
lk+O
-Ph,
+S-P
h...
MR+
Hard
+Hfo
rm+m
+...
Rсмесен
Rиндикаторни
Rсмесен
Фиг. 5. Сравняване точностите на модели за II фаза (CW-20M) с
глобален дескриптор МR към който са добавени изчислени дескриптори от три групи уравнения: - с добавяне на
изчислителни индекси; - с индикаторни индекси; - и комбиниран тип.
Бе показано, че подобно на уравненията за неполярната
фаза, при моделиране за II фаза (CW-20M) изчислените стойности за индекса на Kovats попадат близко до експерименталните, но в този случай присъстват няколко явни отклонения. При модела съставен само от изчислителни индекси те са по-многобройни, докато в уравненията с индикаторните дескриптори и при регресията от смесен тип се забелязват по едно отклонение.
Оценяване на регресиите по стойности на отклонения между експерименталните и изчислените ретентни свойства за II фаза (CW-20M) бе извършено и показани предимствата на уравненията с използване на комбинации от изчислителни и индикаторни дескриптори.
Изводи
1. Извършен е химометричен анализ за установяване на количествени връзки между структурата на група от 111

заместени пиразини и тяхното задържане в газова хроматография, на базата на създадени QSRR модели.
2. Изчислени са молекулни индекси от топологичен, физикохимичен, квантово химичен и стеричен произход за 111 заместени пиразина.
3. Получени са адекватни математически модели за изчисляване индексите на Kovats за ГХ на заместени пиразини върху две фази, неполярна фаза OV-101 и полярна фаза CW-20M.
4. Направен е сравнителен анализ за възможностите и особеностите на моделиране на хроматографското задържане с използваните дескриптори.
5. Установена е връзка между задържането и структурата на пиразините.
6. Параметричните оценки за дескрипторите в регресии могат да служат като количествени изрази за различни видове ММВ, описвани в тях.
Литература
1. M. David, P. Jure. Anal. Chem., 61, (1989), 1328.

Synthesis and Characterization of some Novel Unsaturated Ketones
S. Ivanova, G. Vasileva*, D. Aleksiev
Dept. of Organic Chemistry, University of Bourgas BG-8000, Bourgas, Bulgaria
*Martin Lutter Universitat, Halle-Wittenberg, Germany E-Mail: [email protected]
Abstract: A number of unsaturated ketones, some of which have not been described in the literature so far, have been synthesized. Studies on both the chemical structure and molecular geometry were conducted by employing sophisticated instrumental and quantum-chemical methods.
Keywords: 4-aryl-3-nitro-3-butene-2-on, 3-phenyl-2-nitro-3-propenone, synthesis, structure.
Introduction Studies on the methods of obtaining and the chemical behavior
of chalcones date back to the second half of the last century when these compounds became known as chemotherapenticaly agents. A number of 2’-hydroxychalcones occur in Nature.[1] They constitute the primary precursors of all important naturale flavonoides and can be converted in the laboratory into other flavanoides and dihydrochalcones.[2] Chalcones have different physicological activites and act as antibiotic. They find application in industries [3] and as artificial sweetners.[4] The objective of the present work was to synthesize novel unsaturated ketones, which could be used as additives for preparation of some pharmaceutical.
Discussion
The studies on the interaction of aromatic aldehydes with nitroacetone conducted so far have shown that various products can be obtained.

ArCHO + NO2CH2COCH3
ArCH = CNO2COMe(Ph)
I. 1 - 11
ArCH = CHCOCH2NO2
II 11 examples, yield 70-75% [1-5,9,10]; 60-62% [6-8,11] Ar = Ph (1); Ar = 4-MeC6H4 (2); Ar = 4-MeOC6H4 (3);
Ar = 4-NO2C6H4 (4); Ar = 2-C10H7 (5); 1-thienyl (6); N-methyl-2-pirolyl (7); 4-Me2NC6H4 (8); Ar = Ph , COPh (9); Ar = 4-MeOC6H4,
COPh (10); Ar = 4-Me2NC6H4, COPh (11)
Compounds 1-3 and 9-11 were only described in the literature until now. As a result from the conjugation of the nitro group with the carbon-carbon double bond, the frequency of both the asymmetric and symmetric vibrations in the IR spectra is, generally, decreased. On the other hand, however, the presence of electron-withdrawing substituents such as carbonyl group at α-position with respect to the nitro group acts in a more complicated manner. More specifically, as a result from these electronic effects, the frequency of the asymmetric vibrations of the nitro group is increased, whereas the corresponding symmetric vibrations are characterized by reduced frequency. The principal factor, affecting the position of the bands, which correspond to the symmetric vibration appears to be stereochemistry. Compounds such as chalcons are characterized by mutual trans-location of the nitro group and arene nucleus, which was confirmed by both the UV- and NMR spectroscopy methods. Two characteristic single bands were observed for compound 4, which corresponded to asymmetric and symmetric stretching vibrations of nitro group. This proved the existence of two different types of this functionality. The characteristic band for the carbon-carbon double bond had low intensity within the 1605 - 1600 cm-1 interval. For the carbonyl group, the observed absorption bands were at 170-1690 cm-1.

UV-spectra of the compounds studied showed strong K-type bands, affected essentially by the type of substituents. The principal trans-form of existence of these compounds resulted in an increased planarity of molecule and, therefore, in more efficient conjugation of the double bonds. These effects led to decreased energy of the K-type transition and batochromic shift of the corresponding band as well as to reducted molar absorptivity, associated again with stereochemical factors. On the basis of the UV-spectra obtained, it could be concluded that the molecules of this type of compounds are more polarized in the direction of the nitro vinyl fragment. It could be assumed that the sterical “strain” is decreased at the expense of the changes in C1-C2-aryl and C1-C2-N valence angles, with preservation of the molecular planarity. In the corresponding NMR spectra, chemical shifts, characteristic for aromatic multiplet at 7.26-7.95 ppm were observed. The value for vinyl proton was shifted towards 8.10 ppm. The observed constants for spin – spin interaction confirmed the stereochemical E-configuration of the molecules of compounds studied.
Geometrical configuration of compound 6 was made by using semiempirical methods.
HC - CH
HC C - C = C S
NOO
C
O
CH
HH
(6)H
1 2
Calculations by these methods were applied for a molecule
model which was optimized beforehand by molecular mechanics. It was found that the molecule is flat. The geometric parameters of the carbonyl- and nitro group are close to those reported in the literature. The geometry of the molecule 6 shows that the values calculated by the three methods (AM1, PM3, ZIND01) of molecule configuration are very close, and that the main skeletion of the molecule lies on one plane.

The results obtained for the calculated relative charges at the C{sp
2} carbon atoms by using quantum chemistry methods were
experimentally confirmed by the behavior of these compounds with respect to nucleophilic and electrophilic addition reactions.[4]
In order to study the electron transitions by semiempirical methods it is best to use ZIND0/S. At first, the molecule was configurated and then the electron transitions were analyzed by means of the geometry having minimum energy. A comparison of the calculated electron transitions and the corresponding spectral bands shows that the best results are obtained when four bonding and four antibonding molecular orbitals take part in the configuration reaction. The torsion angle O=C–C=C is 115.06o.
Experimental
IR- and UV-spectra were obtained using a Bruker and Specord UV-VIS. 1H-NMR chemical shifts measured in deuterated solvent are given in ppm from TMS) spectra were recorded with Bruker 250MHz spectrometer using CDCl3 solution.
Selected data
Compounds I.4: m.p. 118 oC; Found (%): C 48.20, H 2.70, N 19.50; for C10H8N2O5 (245.99) Calc. (%): C 48.79, H 3.25, N 15.45. IR (KBr, ν, cm-1): 1530-1350 (NO2-Ar); 1540-1310 (NO2); 1690 (CO); 1605 (C=C); 810 (γ=C). NMR (δ, ppm, CDCl3/TMS): 7.30-7.80 (m, 4H, C6H4); 8.07 (s, 1H, =CH); 2.40 (s, 3H, CH3). UV (λmax, nm (lg ε), EtOH): 210 (2.35); 252 (2.47); 362 (2.15). Compounds I.5: m.p. 126 oC; Found (%): C 68.95, H 4.10, N 19.50; for C14H11NO3 (241.00) Calc. (%): C 69.72, H 4.55, N 19.98. IR (KBr, ν, cm-1): 1550-1360 (NO2); 1695 (CO); 1605 (C=C); 815 (γ=C). NMR (δ, ppm, CDCl3/TMS): 7.20-7.95 (m, 7H, C10H7); 8.10 (s, 1H, =CH); 2.42 (s, 3H, CH3). UV (λmax, nm (lg ε), EtOH): 212 (2.30); 256 (2.50); 370 (2.21). Compounds I.6: m.p. 136 oC; Found (%): C 47.90, H 3.30, N 6.75; for C8H7NO3S (197.06) Calc. (%): C 48.73, H 3.55, N 7.18. IR (KBr, ν, cm-1): 1555-1365 (NO2); 1700 (CO); 1602 (C=C); 805 (γ=C); 3100 (C-H). UV (λmax, nm (lg ε), EtOH): 220 (2.15); 263 (2.30); 373 (2.08).

Compounds I.7: m.p. 138 oC; Found (%): C 48.20, H 2.75, N 7.15; for C9H10NO3 (179.91) Calc. (%): C 48.79, H 3.25, N 7.80. IR (KBr, ν, cm-1): 1560-1350 (NO2); 1702 (CO); 1605 (C=C); 805 (γ=C); 3485 (NH). UV (λmax, nm (lg ε), EtOH): 216 (2.10); 260 (2.36); 375 (2.18). Compounds I.8: m.p. 154 oC; Found (%): C 60.65, H 5.45, N 11.26; for C12H14N2O3 (234.00) Calc. (%): C 61.54, H 6.00, N 11.97. IR (KBr, ν, cm-1): 1555-1350 (NO2); 1695 (CO); 1605 (C=C); 805 (γ=C). UV (λmax, nm (lg ε), EtOH): 212 (2.18); 254 (2.30); 386 (2.10).
References 1. J. H. Harbone, The flavonoids. Advances in research (Chapman
and Hall, London and New York), 348, 1998. 2. C. Jain, A. Sharma, Tetrahedron, 41, 5933, 1985. 3. D. N. Dhar, The Chemistry of Chalcones and Related Compounds
(John Wiley & Sons, New York, Chichester, Brisbane, Toronto), 241, 1981.
4. D. I. Aleksiev, S. Ivanova, G. Vasileva, Phosphorus, Sulfur and Silicon, 181, 7, 1601, 2006.

Получаване на нови тетраметилпиперидиноксилови производни на някои природни оксо- и карбоксисъединения
Галин П. Петров, Анастасия А. Ляпова, Деница Ц. Борисова
Факултет по природни науки, ШУ “Епископ Константин Преславски”, 9712 Шумен
Университетска 115, Е-Mail: [email protected]
Abstract: New compound containing stable nitroxyle radicals by reaction of nucleofilic addition and elimination have been synthesized. Their structure has been established through IR, 1H-NMR and EPR spectroscopy.
Keywords: Penicillin, salicylaldehyde, 4-amino-2,2,6,6-tetramethyl-piperidine, 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyle.
Въведение Изследвана е биологичната активност на значителен брой
нитроксили и е установено, че 2,2,6,6-тетраметилпиперидин-1-оксила (TЕMPO) има хипотензивно действие, а неговият аналог 4-хидрокси-TЕMPO, освен хипотензивен и антипиретичен ефект, има и брадикардично действие.
Няколко обстоятелства са дали основание за използването на нитроксилните радикали като противотуморни средства. На първо място свойството на нитроксилите да улавят образуваните предимно в раковите клетки свободни нестабилни радикали, както и свойството им да инхибират окислително редукционните ензими, участващи в гликолизата, доставяща значителна част от енергията, необходима за злокачествените клетки.[1] Пеницилините са производни на тетрахидротиазола или тиазолидина. Пеницилините се произвеждат от доста щамове на Penicillium и действат унищожаващо на много болестотворни микроорганизми. Установяването на тяхната структура се дължи главно на Р. Робинсън и последователите от неговата школа, а Du

Vigneaux успява да синтезира, макар и с нисък добив бензилпеницилин.[2] Известни са доста производни на пеницилина - както природни, така и полусинтетични (синтезирани около 15000). Основната част от тяхната структура е кондензиран тиазолидинов с β-лактамен пръстен. Пеницилинът представлява киселина, която е разтворима в органични разтворители, а като сол на алкални метали (K, Na) се разтваря във вода и под тази форма се използва в терапията. Пеницилините имат малка токсичност и се използват много широко за лечение на пневмонии, менингити и много инфекциозни заболявания, например-инфекциозен ендокардит, инфекциозна мононуклеоза, Лаймска болест (борелиоза), сифилис, перикардити и други.[2]
Карбонилните съединения, които се използват в настоящата работа са: 2-хидроксибензалдехид (Салицилалдехид) и 4-изопропилбензалдехид (Куменал). Салициловият алдехид (салицилалдехид) се намира в етеричното масло Spiraea ulmaria и представлява течност с миризма на бензалдехид. Използва се за производството на кумарин. Куминалдехидът (р-изопропил-бензалдехид) се съдържа в етеричното масло на обикновения анасон и действа върху храносмилателната система, белите дробове и млечните жлези, като има и слабо противомикробно действие. Етеричното масло се използва още и като сироп против кашлица.
Целта на настоящата работа е: Да се изследват реакциите на 4-амино-2,2,6,6-тетраметилпиперидин (А-ТЕМР) и 4-амино-2,2,6,6-тетраметилпиперидин-1-оксил (А-ТЕМРО) със салицилз-алдехид, куменал и Пеницилин G с цел получаване на техни производни с вероятна биологична противотуморна активност.
Резултати и дискусия Настоящите изследвания са продължение на осъществените
досега синтези на утвърдения реагент 4-амино-2,2,6,6-тетраметилпиперидин (А-ТEМР) и неговия азоксилов радикал (А-ТEМРО) с различни полифункционални алдехиди с цел получаване на кондензационни азоксилови продукти (наричани спин белязани органични съединения). Тяхната биологична

активност най-вероятно би се насочила към противотуморно действие. Такова действие притежава самият реагент А-ТEМРО. Неговата синтеза се извършва чрез няколко известни методи на окисление на А-ТEМР с различни окислители - главно водороден пероксид.[3]
С двата реагента А-ТEМР и А-ТEМРО извършихме
синтези със следните моделни съединения: 4-изопропил-бензалдехид (куменал), 2-хидроксибензалдехид (салицилалдехид) и Пеницилин-G.
Синтезите с куменал (К) се провеждат по следната реакционна схемa. Реакциите се извършват в среда от диетилов етер при стайна температура за 60 мин. време. Продуктът К-ТЕМP се изолира във вид на бели кристали из етилацетат, а продуктът К-ТЕМРО като светло оранжево вещество. Структурата на получените съединения е доказана с елементен анализ, ИЧ-, 1Н-ЯМР или ЕПР-спектроскопия.
В ИЧ-спектъра на К-ТЕМР снет в нуйол не се наблюдава характеристична ивица при 1690 сm-1 отнасяща се за валентни трептения на карбонила от алдехидната група, която се открива в спектъра на изходния куменал. Появява се ивица за С=N връзка при 1620 сm-1. За ароматния пръстен се отнасят ивиците при 1600, 1510 сm-1, характеризиращи скелетните трептения за СН=СН двойните връзки от ядрото, γAr=820, 770 сm-1 характерни за р-дизаместено ароматно ядро.
В 1Н-ЯМР-спектъра на продукта в ДМСО се откриват дублетни сигнали на изопропиловите метилови групи δ=1,33 м.ч. с J=3,0 Hz, мултиплетен сигнал на четирите метилови групи към пиперидиловия пръстен при δ=1,24 м.ч., мултиплет за двете метиленови групи в този пръстен при δ=3,10 м.ч. с J=3,0-5,0 Hz, дублети за Н-атомите на бензеновия пръстен при δ=7,0 и 7,7 м.ч. с J ~5,0 Hz и синглет при δ=8,0 м.ч. за азометиновия протон.
NH
H3CH3C
CH3
CH3
NH2
N
NH2
H3CH3C
CH3CH3
O

CO
H
CHCH3H3C
N
CH3H3C
H3C CH3
HH2N
CHCH3H3C
CH N
CH3H3C
H3C CH3
HN
CHCH3H3C
COH
HN
CH3H3C
H3C CH3
HHN
H2O
K TEMPN
CH3H3C
H3C CH3
OH2N
H2O
CHCH3H3C
CH N
CH3H3C
H3C CH3
ON
K TEMPO
CHCH3H3C
COH
HN
CH3H3C
H3C CH3
OHN
В ИЧ-спектъра на К-ТЕМРО в нуйол се наблюдават същите характеристични ивици както за предходния продукт, само че се появява и нова ивица за оксиловия радикал N-О. при 1230 сm-1 .
ЕПР-спектърът на продукта в разтвор на етанол е комбиниран триплетен сигнал, характерен за иминоксилов радикал /фиг.1/. Не се появяват три отделни сигнала, както е при други нитроксилни радикали, тъй като те се припокриват в общ триплетен сигнал, поради ограничената свобода на електронния спин в пространството.

Фиг. 1. ЕПР-спектър на N-(4-изопропилбензилиден)-4-амино-
2,2,6,6-тетраметилпиперидин-1-оксил (К-ТЕМPО).
Взаимодействието на двата изследвани реагента със салицилов алдехид (С) протича по схемата.
Продуктът С-ТЕМР се получава във вид на жълти, прозрачни кристали след прекристализация из диетилов етер, а С-ТЕМРО се получава във вид на светлокафяви кристали.
В ИЧ-спектъра на С-ТЕМР снет в нуйол не се наблюдава характеристична ивица при 1680 сm-1 отнасяща се за валентни трептения на карбонила от алдехидната група, която се открива в спектъра на изходния салицилалдехид. Появява се ивица за С=N връзка при 1620 сm-1. За ароматния пръстен се отнасят ивиците при 1590, 1500 сm-1, характеризиращи скелетните трептения за СН=СН двойните връзки от ароматното ядро, γAr=750сm-1 за о-дизаместено ароматно ядро, широка ивица за валентни трептения за хидроксилна група при νОН = 2700-3300 сm-1 .
В 1Н-ЯМР-спектъра на продукта в ДМСО се наблюдават сигнали за: мултиплет от синглети за 4-те метилови групи при δ = 1,20 м.ч., мултиплет за двете метиленови групи при δ =2,9 м.ч. с J= 3,0-5,0 Hz, мултиплет на бензеновите протони в интервала δ = 6,6-7,2 м.ч. с J= 3,0-7,0 Hz и синглет за азометиновия протон при δ =7,5 м.ч.
В ИЧ-спектъра на С-ТЕМРО снет в нуйол вместо ивица за карбонила от алдехидната група, се появава ивица за С=N връзка при 1630 сm-1. Валентните трептения за N-О. връзката се наблюдават при 1230 сm-1.
В ЕПР-спектъра на продукта в твърдо състояние има два комбинирани триплетни сигнала /фиг.2/.
CO
HOH
N
CH3H3C
H3C CH3
H2N H
CH N
CH3H3C
H3C CH3
NOH
O
H2O
COH
HN
CH3H3C
H3C CH3
HN H
C TEMP
CH N
CH3H3C
H3C CH3
NOH
H
OH
OH
COH
HN
CH3H3C
H3C CH3
HN O
N
CH3H3C
H3C CH3
H2N O
H2O
C TEMPO
Фиг. 2. ЕПР-спектър на N-(2-хидроксибензилиден)-4-амино-
2,2,6,6-тетраметилпиперидин-1-оксил (С-ТЕМPО).

Сложната структура на ЕПР-сигнала е указание за наличието на различни видове радикали. Най-вероятното обяснение в случая е възможността за наличие на иминоксилов и на феноксилов радикал.[4] Последният се получава чрез миграция на Н-атом от хидроксилната група, свързана с бензеновото ядро при О-атом на нитроксиловата група:
CH N
CH3H3C
H3C CH3
ON
OH
CH N
CH3H3C
H3C CH3
OHN
O
нитроксилов радикал феноксилов радикал
Като интересен биосубстрат за взаимодействие с използваните реагенти бе избран Пеницилин G като алкална сол.
CH2C
OS
NC
O
O
H3C
H3CC
C
H
O
NH
Na
Съществува проблем за избор на ефективна реакция за
такова взаимодействие. Една възможност е получаване на N-бензоил-пенициланова киселина и синтези на съответните амиди с всеки от двата реагента. Това трябва да се осъществи при подходящи условия, тъй като е известно, че производните на тази киселина се разпадат термично и с алкални основи с разкъсване на азетидиноновия пръстен и други връзки до пенициламин и пеналдинови киселини [2].

CO
HCH
COOH
NH C
O
CH2
CH3C
CHH3CSH NH2
COOH
пеналдинова киселина пенициламин
За да получим свободна карбоксилна група неутрализи-
рахме водния разтвор на натриевата сол, екстрахирахме с етилацетат и изолирахме Пеницилановата киселина във вид на жълти кристали.
1
2
3 45
67
a b
CO
CH2
N
S
CO
HOH3C
H3C
CC
O
H
NH
Взаимодействието на Пеницилановата киселина с А-ТЕМРО проведохме при стайна температура в среда от метилов алкохол и разбъркване. След престояване една нощ получената амониева сол, нагрявахме на обратен хладник в продължение на 3 часа. Изолирахме амида на Пеницилановата киселина във вид на бледожълти кристали, след прекристализация из хлороформ / петролев етер.

NO
CH3H3C
H3CCH3
NH2 NO
CH3H3C
H3CCH3
NH3
H2O
S
NC
O
HO
H3C
H3C
C
C
O
NHCH2
H
S
NCO
H3CH3C
C
C
O
HO
NHCH2
NO
CH3H3C
H3CCH3
NH
S
NC
O
H3C
H3C
C
C
O
HNH
CH2
Полученото съединение е охарактеризирано с елементен
анализ за азот и ИЧ-спекроскопия. В ИЧ-спектъра на продукта изчезва ивицата за валентни
трептения за карбонилна група νС=О при 1720 сm-1, която се наблюдава в спектъра на изходната киселина. Поради наличие на няколко амидни карбонилни групи новата амидна група не може точно да се идентифицира, тъй като се наслагва в широката амидна ивица в интервала от 1620-1690 сm-1. Появява се нова ивица за нитроксилов радикал N-О. при 1190 сm-1 .
Литература
1. Райков, З. Д., В. А. Спасов, Е. Т. Райкова, Спин белязани прозводни на хидразинобарбитуравата киселина, метод за тяхното получаване и приложението им, Авт. свид. № 27813, 1978.
2. Янков Л., Б. Месроб, Ч. Иванов, Л. Младенова-Орлинова, Органична химия, 1982, 2.
3. Розанцев Э Г., В. Д. Шолле, Нитроксильнье радикалы. Синтез, химия, приложения, М., 1987.
4. Нонхибел Д., Д.Уолтон, Химия свободных радикалов, прев. с англ., изд. Мир, Москва, 1977.

Синтез на 1,1-дизаместени 1,2,3,4-тетрахидроизохинолини
И. Иванов, Ст. Николова, Е. Кочовска, Ст. Статкова-Абегхе
Катедра Органична химия, Химически Факултет ПУ "П. Хилендарски", Пловдив 4000, ул. "Цар Асен" 24
Е-Mail: [email protected]
Abstract: The synthesis of 1,1-disubstituted 1,2,3,4-tetrahydro-isoquinolines from ortho-acylated 2-phenethylamides was investigated. The reaction of ortho-acylated 2-phenyethylamides with Grignard reagent and following cyclization of the newly synthesized hydroxyamides was studied. The newly obtained 1,1-disubstituted isoquinolines are of considerable interest of their biological activity.
Keywords: ortho-acylated 2-phenethylamides, Grignard reagent, cyclization, 1,1-disubstituted 1,2,3,4-tetrahydroisoquinolines, p-toluenesulfonic acid (PTSA).
Изохинолиновият скелет участва в изграждането на една от
най-големите и разнообразни групи от алкалоиди, като до сега са открити и изследвани над шест хиляди изохинолинови съединения. Изохинолиновите алкалоиди са обект на изследване още от края на XIX век, като интересът към тях не отслабва и до днешни дни.[1-4] Той се дължи преди всичко на разнообразната биологична активност на различните групи изохинолинови съединения и приложението на редица от тях в медицинската практика.[5-13]
Тези насоки на изследвания и приложения, както и в получаването на нови изохинолинови производни с биологична активност са причина за търсенето на нови подходи за конструиране на изохинолиновия пръстен, наред с широко използваните за тези цел методи на Bishler-Napieralski, Pictet-Spengler и Pomeranz-Fritsch.
Синтетичните резултати получени при взаимодействието на 2-фенилетиламини, триптамин, фенилацетамиди, фенилацет-

нитрили и карбамиди с карбоксилни киселини в РРА и получаването на разнообразни изохинолинови съединения, β-карболини, хиназолини и изохромани,[14-20] чрез едноетапна синтеза при сравнително меки реакционни условия, ни подтикна към разширяване възможността за приложение на реакцията, с оглед използването им за синтез на нови 1,1-дизаместени изохинолинови съединения. Решаването на тази задача е мотивирано от литературни данни за силно антиконвулсантно действие при редица 1,1-дизаместени тетрахидроизохинолини. Още повече, 1,1-дизаместените 1,2,3,4-тетрахидроизохинолини са синтетични интермедиати за синтеза на пиримидо[6,1-a]изохинолинов скелет, който притежава широк набор от биологични активности като антихипертензивно, бронхо-дилаторно и противоалергично действие.[21] В същото време, предходни изследвания в тази насока [22] добре демонстрират осъществимостта на предлагания подход. В този смисъл усилията ни са насочени към разширяване областта на приложимост на методиката и синтез на разширен набор от нови вещества с потенциална биологична активност.
Решението на задачата включва получаване на орто-ацилирани 2-фенилетиламини и взаимодействието им с Гринярови реагенти, последвано от циклизация на получените съединения до съответните 1,1-дизаместени 1,2,3,4-тетрахидроизохинолини.
За целта първоначално синтезирахме изходни кетоамиди на 3,4-диметокси-2-фенилетиламиди с оцетен анхидрид на орто-място в бензеновото ядро. Реакцията бе проведена при 80°С в полифосфорна киселина (PPA).
Следваща стъпка в синтеза беше проучване условията на взаимодействие на кетоамидите 5 с Гриняров реагент. Установихме, че взаимодействието на кетоамидите с метилмагнезиев йодид в безводен етер води до получаване на съответните хидроксиамиди 6, които след пречистване от нереагиралия изходен кетоамид чрез колонна хроматография се получават с ~50 % добив.

CH3O
CH3O
NH2 CH3O
CH3O
NH R
R= COCH3, COPh, COBn SO2CH3, COOC2H5
CH3O
CH3O
NH CORO
CH3MgI
CH3O
CH3O
NH COROH
CH3O
CH3O
N CORH+
CH3COOHor
(CH3CO)2O
PPA
Установихме условията на взаимодействие на кетоамидите с Гриняров реагент като варирахме вида на амидната група при еднаква ацилираща компонента (оцетен анхидрид) и един и същи алкилхалогенид (метилйодид). При посочените условия реакцията протича със запазване на амидната група, т.е. засяга само орто-ацетилния остатък, докато амидната група не встъпва във взаимодействие с органомагнезиевото съединение.
6 a-д5 a-д
CH3O
CH3O
NH COR1O
CH3MgI
CH3O
CH3O
NH COR1OH
Изследванията бяха свързани с вариране вида на амидната
група - ацетилна, бензоилна, бензилова, уретанова, сулфамидна и формилна, при еднаква ацилираща компонента (оцетен анхидрид) и един и същи алкилхалогенид (метилйодид). Установихме, че при посочените условия реакцията протича със запазване на амидната група, т.е. засяга само орто-ацетилния остатък, докато амидната група не встъпва във взаимодействие с органомагнезиевото съединение.
Последен етап в синтеза беше циклизацията на получените алкохоли до 1,1-дизаместени 1,2,3,4-тетрахидроизохинолини. От гледна точка на удобство при работа като циклизиращ агент бе предпочетена р-толуенсулфонова киселина.

CH3O
CH3O
N COR1CH3O
CH3O
NH COR1OH
PTSA
6 а-д 7 а-д Установихме, че оптималните условия за циклизация е
разбъркване на хидроксиамид 6 при стайна температура в разтвор на дихлорометан в присъствие на каталитично количество р-толуенсулфонова киселина за 20-30 минути. След филтруване на реакционната смес на къса хроматографска колонка с неутрален Al2O3, се отстранява катализатора р-толуенсулфонова киселина и се получава съответният чист продукт 7. При така подбраните реакционни условия 1,1-дизаместените 1,2,3,4-тетрахидро-изохинолинови производни 7 се получават с добиви от ~95 %.
Експериментална част
Температурите на топене са определени на апарат Boetius РНМКО5 без корекция. 1Н-ЯМР- и 13С-ЯМР-спектрите са снети с Bruker Avance DRX250 като за разтворител е използван CDCl3. Химичните отмествания са изразени с ppm като за стандарт е използван тетраметилсилан (TMS), а J-константа е измерена в Hz. Всички NMR са снети при RT.
Получаване на N-{2-[2-(1-хидрокси-1-метил-етил-4,5-диметоксифенил]-етил}-амиди (6а-д): 15 mmol (0.36 g) магнезиеви стружки се поставят в тригърлена колба, снабдена с делителна фуния, бъркалка и обратен воден хладник с хлоркалциева тръбичка, и се заливат с 50 ml безводен етер. Към сместа се добавят ~1/20 от общото количество (15 mmol) метилйодид. Реакцията се провежда в атмосфера от инертен газ. След започването на реакцията, към реакционната смес се добавя на капки останалото количество метилйодид, разтворено в 30 ml диетилов етер. След пълното разтваряне на магнезия (около 1 час), започва прибавянето на 3 mmol от съответния кетоамид 5, разтворен в равен обем C2H4Cl2 или CH2Cl2. След края на реакцията (получаването на продукта се следи тънкослойно-хроматографски) пробата се излива във вода и се екстрахира четирикратно с хлороформ, като получената утайка се разтваря с наситен разтвор на амониев хлорид. Обединените органични

екстракти се сушат с безв. Na2SO4 и се концентрират, а продуктът се пречиства от нереагиралия изходен кетоамид чрез колонна хроматография (силикагел; n-хексан, диетилов етер).
N-{2-[2-(1-хидрокси-1-метил-етил)-4,5-диметокси-фенил]-етил}-ацетамид (6а): Добив 52 %, масло, 1H-NMR(CDCl3): 1.67 (s, 6H, 2xCH3), 1.89 (s, 3H, CH3), 2.61 (broad s, 1H, OH), 3.14 (t, J=7.12 Hz, 2H, CH2), 3.49 (dd, J= 12.37, 6.75 Hz, 2H, CH2), 3.87 (s, 6H, 2xOCH3), 5.31 (s, 1H, NH), 6.73 (s, 1H, Ar), 6.89 (s, 1H, Ar); 13C-NMR (CDCl3): 170.4, 147.7, 146.5, 138.0, 129.8, 114.7, 109.8, 56.0, 55.85, 42.1, 32.6, 29.4, 23.1; елементен анализ: изч. за C15H23NO4: C 64.03, H 8.24, N 4.98. намерено: C 64.18, H 8.38, N 4.89.
N-{2-[2-(1-хидрокси-1-метил-етил)-4,5-диметокси-фенил]-етил}-бензамид (6в): Добив 50 %, Тт=160-162ºС, 1H-NMR(CDCl3): 1.71 (s, 6H, 2xCH3), 2.74 (s, 1H, OH), 3.24 (t, J=6.79 Hz, 2H, CH2), 3.69 (dd, J= 11.69, 6.82 Hz, 2H, CH2), 3.81 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.74 (s, 1H, NH), 6.86 (s, 1H, Ar), 7.26 (s, 1H, Ar), 7.33-7.42 (m, 3H, Ar), 7.69 (d, J=1.58 Hz, 1H, Ar), 7.72 (t, J=1.42 Hz, 1H, Ar); 13C-NMR (CDCl3): 167.6, 147.8, 146.6, 137.7, 134.6, 131.1, 129.98, 128.3, 126.9, 114.7, 109.8, 56.1, 55.8, 42.6, 32.7, 32.2; елементен анализ: изч. за C20H25NO4: C 69.95, H 7.34, N 4.08. намерено: C 70.08, H 7.48, N 4.19.
N-{2-[2-(1-хидрокси-1-метил-етил)-4,5-диметокси-фенил]-етил}-2-фенил-ацетамид (6г): Добив 50 %, масло, 1H-NMR(CDCl3): 1.58 (s, 6H, 2xCH3), 1.88 (s, 1H, OH), 3.08 (t, J=6.85 Hz, 2H, CH2), 3.49-3.53 (m, 4H, 2xCH2), 3.85 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 6.68 (s, 1H, NH), 6.81 (s, 1H, Ar), 7.14 (s, 1H, Ar), 7.15-7.18 (m, 2H, Ar), 7.29-7.33 (m, 3H, Ar); 13C-NMR (CDCl3): 171.3, 147.7, 146.4, 137.9, 135.1, 129.7, 128.8, 127.1, 114.5, 109.6, 56.1, 55.8, 43.7, 42.1, 32.3, 31.9; елементен анализ: изч. за C21H27NO4: C 70.56, H 7.61, N 3.92. намерено: C 70.68, H 7.72, N 4.11.
Циклизация на хидроксиамиди 6 до получаване на 1,1-дизаместени 6,7-диметокси-1,2,3,4-тетрахидроизохинолини (7а-д): 1 mmol от съответния хидроксиамид 6 се разтваря в дихлорометан. Към разтвора се добавя каталитично количество р-толуенсулфонова киселина при стайна температура и разбъркване. След 20-30 минути реакционната смес се филтрува

на къса хроматографска колонка с неутрален Al2O3 за отстраняване на катализатора р-толуенсулфонова киселина, в резултат на което се получава съответният чист продукт 7.
1-(6,7-диметокси-1,1-диметил-3,4-дихидро-1H-изохинолин-2-ил)-етанон (7а): Добив 96 %, бели кристали, Тт=123-125ºС, 1H-NMR(CDCl3): 1.82 (s, 6H, 2xCH3), 2.21 (s, 3H, CH3), 2.79 (t, J=5.5 Hz, 2H, CH2), 3.56 (dt, J=5.5, 3.75 Hz, 2H, CH2), 3.87 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 6.57 (s, 1H, Ar), 6.77 (s, 1H, Ar); 13C-NMR (CDCl3): 170.3, 147.9, 147.1, 137.0, 126.3, 110.5, 109.6, 59.85, 56.1, 55.8, 44.1, 30.4, 27.99, 25.6; елементен анализ: изч. за C15H21NO3: C 68.42, H 8.04, N 5.32. намерено: C 68.54, H 8.18, N 5.44
(6,7-диметокси-1,1-диметил-3,4-дихидро-1H-изохинолин-2-ил)-фенил-метанон (7в): Добив 97 %, бели кристали, Тт=143-146ºС, 1H-NMR(CDCl3): 1.96 (s, 6H, 2xCH3), 2.80 (t, J=5.5 Hz, 2H, CH2), 3.52 (dt, J= 5.5, 3.5 Hz, 2H, CH2), 3.89 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 6.59 (s, 1H, Ar), 6.84 (s, 1H, Ar), 7.42-7.46 (m, 3H, Ar), 7.48-7.52 (m, 2H, Ar); 13C-NMR (CDCl3): 172.6, 147.9, 147.2, 138.9, 136.8, 129.6, 128.5, 126.7, 126.1, 110.8, 109.7, 59.9, 56.1, 55.9, 45.3, 30.4, 27.7; елементен анализ: изч. за C20H23NO3: C 73.82, H 7.12, N 4.30. намерено: C 73.94, H 7.24, N 4.42.
1-(6,7-диметокси-1,1-диметил-3,4-дихидро-1H-изохинолин-2-ил)-2-фенил-етанон (7г): Добив 97 %, масло, 1H-NMR(CDCl3): 1.84 (s, 6H, 2xCH3), 2.49 (t, J=5.5 Hz, 2H, CH2), 3.47 (dt, J=5.5, 3.75, 2H, 2xCH2), 3.82 (s, 3H, OCH3 припокрит с 2Н, СН2), 3.86 (s, 3H, OCH3), 6.47 (s, 1H, Ar), 6.75 (s, 1H, Ar), 7.23-7.27 (m, 2H, Ar), 7.295-7.33 (m, 3H, Ar); 13C-NMR (CDCl3): 170.9, 147.8, 147.1, 136.8, 135.6, 128.6, 128.4, 126.6, 126.3, 110.5, 109.6, 60.1, 56.1, 55.8, 44.8, 43.8, 30.1, 27.9; елементен анализ: изч. за C21H25NO3: C 74.31, H 7.42, N 4.13. намерено: C 74.44, H 7.52, N 4.24. Благодарност: Настоящата работа е част от научно-изследователски проект, финансиран от фонд “Научни изследвания” при ПУ “Паисий Хилендарски”, както и на ИС-Х-2/2008.

Литература 1. Bentley, K. The Isoquinoline Alkaloids, Vol.1. Ed.: Ravindranath,
B.; United Kingdom, Harwood Academic, Bangalore, India, 1998. 2. Herbert, R. The Chemistry and Biology of Isoquinoline Alkaloids.
Ed.: Philipson, J., Roberts, M., Zenk, M.; Springer-Verlag, Berlin, Heidelberg, New York, Tokyo, 1985, p. 213.
3. Shamma, M. The Isoquinoline Alkaloids, Chemistry and Pharmacology, Academic Press, New York, 1972.
4. Lundström, J. In The Alkaloids. Simple Isoquinoline Alkaloids. Ed.: Brossi, A.; Academic Press, London, 1983, p. 21, 255.
5. Shamma, M. and Moniot, J. L. In Isoquinoline Alkaloids Research, 1972–1977; Plenum, New York, 1978.
6. Kametani, T. The Total Syntheses of Natural Products, Vol. 3. Ed.: ApSimon, J.; Wiley, New York, 1977, pp 1-272.
7. Dulenco, V.; Comissarov, I.; Doljenko, A. and Nikolukin, U. β-Carbolines, Chemistry and Neurobiology. Ed.: Andronati, C. A.; Naukova Dumka, Kiev, 1992, pp. 88-188.
8. Love, B. and Raje, P. J. Org. Chem. 1994, 59, 3219. 9. Collins, M. Parkinsonism Relat. Disord. 2002, 8, 417. 10. Yousaf, M.; Hammond, N.; Peng, J.; Wahyuono, S.; McIntosh,
K.; Charman, W.; Mayer, A.M. and Hamann, M. J. Med. Chem. 2004, 47, 3512.
11. Shen, Y. C.; Tai, H. R. and Duh, C. Y. Chin. Pharm. J. 1996, 48, 1.
12. Shen, Y. C.; Chen, C. Y.; Hsieh, P. W.; Duh, C. Y.; Lin, Y. M. and Ko, C. L. Chem. Pharm. Bull. 2005, 53, 32.
13. Cui, W.; Iwasa, K.; Tokuda, H.; Kashihara, A.;Mitani, Y.; Hasegawa, T.; Nishiyama, Y.; Moriyasu, M.; Nishino, H.; Hanaoka, M.; Mukai, C. and Takeda, K. Phytochemistry 2006, 67, 70.
14. Venkov, A., Ivanov, I. Synth. Comm. 1994, 24, 1145. 15. Venkov, A., Ivanov, I. Tetrahedron 1996, 52, 12299. 16. Ivanov, I.; Nikolova, St.; Statkova-Abeghe, St., Angelov P.
Heterocycles 2006, 68, 387. 17. Ivanov, I.; Nikolova, St., Statkova-Abeghe, St. Heterocycles 2006,
68, 369.

18. Ivanov, I.; Nikolova, St., Statkova-Abeghe, St. Heterocycles 2005, 65, 2483.
19. Ivanov, I.; Nikolova, St.; Kochovska, E., Statkova-Abeghe, St. Heterocycles 2006, 68, 1443.
20. [20] Ivanov, I., Nikolova, S., Kochovska, E., Statkova-Abeghe S., ARKIVOC, 2007, (xv) 31.
21. (a) B. Lal, A. N. Dohadwalla, N. K. Dadkar, A. D'Sa, N. J. de Souza, J. Med. Chem., 1984, 27, 1470; (b) E. H. F. Wong, J. A. Kemp, T. Priestley, A. R. Knight, G. N. Woodruff, L. L. Iversen, Proc. Natl. Acad. Sci. USA, 1986, 83, 7104; (c) M. Ohkubo, A. Kuno, K. Katsuta, Y. Ueda, K. Shirakawa, H. Nakanishi, I. Nakanishi, T. Kinoshita, H. Takasugi, Chem. Pharm. Bull., 1996, 44, 95.
22. (a) A. Venkov, P. Angelov, Synth. Comm., 2003, 17, 3025; (b) A. Venkov, P. Angelov, Synthesis, 2003, 14, 2221; (c) P. Angelov, I. Ivanov, A. Venkov, Molecules, 2004, 9, 694.

Синтез на комплексни съединения на непротеинови аминокарбоксилни киселини с
Сu(II) М. Кашчиева1, М. Маринов2, Н. Стоянов3, В. Матева3
1Катедра по обща химия, Факултет по природни науки ШУ “Епископ Константин Преславски”, Шумен
ул. “Университетска” № 115, 9712, Е-Mail:[email protected] 2Катедра по органична химия, Химически факултет,
ПУ “Паисий Хилендарски”, Пловдив, ул. “Цар Асен” № 24 E-Mail: [email protected]
3Катедра по химия и химични технологии, Филиал-Разград към РУ “Ангел Кънчев”
Abstract: The synthesis of non-protein Cα,α-asymmetrical aminocarboxylic acids is realized through hydrolyze of 5-aryl-5-methyl hydantoins with Ba(OH)2.2H2O in autoclave under pressure and 160°C temperature. Complexes of 2-amino-2-arylpropane acids with Cu (II) are obtained. The newly obtained compounds are characterized through m.p., elemental analysis, IR- and UV-Vis spectral data.
Key words: Cu (II) complexes; hydantoin; aminocarboxylic acids.
Сред многото синтетични α-аминокарбоксилни киселини особено място заемат Сα,α-симетрично дизаместените амино-карбоксилни киселини, които са подробно изучени и сред тях са открити съединения с разнообразно биологично действие, включително и такива с антиепилептично и антиконвулсивно действие [1,2], докато несиметрично заместените са значително по-слабо проучени.
В научната литература липсват данни за получаването на комплексни съединения на несиметрично Сα,α-дизаместените аминокарбоксилни киселини с йони на преходни метали. Оскъдни данни има само за получаването на Cu(II) комплексни съединения с 1-амино-1-циклоалкан карбоксилни киселини.[3]

Тази предпоставка до голяма степен определи и целта на настоящата работа а именно получаване и охарактеризиране на Сu(II) комплексни съединения на 2-амино-2-(2;4-Rфенил)-пропанови киселини (дизаместени глицини), където R=Н; CH3; OCH3; F.
Прототипът на тези аминокарбоксилни киселини е аминоизомаслената киселина, характеризираща важна група антибиотици, които променят йонната пропускливост на биологичните мембрани чрез образуване на канали. Освен това тази аминокиселина е съставна част на природния циклотетрапептиден метаболит хламидоцин [3], като латентен инхибитор на протеолитични ензими [3,4], остатъци от нея са включени и в структурата на много биоактивни пептиди.[5]
За синтез на непротеиновите Сα,α-несиметрични амино-карбоксилови киселини използвахме схема, основаваща се на образуването на хидантоини [6] и следваща хидролиза до съответната α-аминокарбоксилова киселина:
NaCN(NH4)2CO3 N C
NCC
H
H
O
O
R1
R2
CR1
R2
O
I
Ba(OH)2.2H2O
H2OC
R1
R2
COO
NH3
II
a) R1=CH3; R2=C6H5b) R1=CH3; R2=4-CH3C6H4
c) R1=CH3; R2=4-CH3OC6H4d) R1=CH3; R2=2F-C6H4
Изолираните кристални аминокарбоксилови киселини
охарактеризирахме спектрално. В UV-Vis спектрите на посочените киселини се наблюдават
характерни ивици на поглъщане за фениловото ядро при 205 нм и характерните по-слабо интензивни ивици в интервалаа от 230-270 нм.
В IR-спектрите на лигандите II се наблюдават ивици на поглъщане за NH-частица – характерна широка абсорбционна ивица в интервала 3100-2500 см-1. В областта 1800-1500 см-1 са

налице ивици на поглъщане за карбоксилатния йон и за двойните връзки от ароматната система. Тъй като част от спектрите са снети в суспензия от нуйол, не може да се отчете ивица за поглъщане на метиловата група, но ясно се вижда ивица за р-заместено ароматно ядро при 850 см-1. Слабоинтензивната ивица в IR-спектъра в областта 1760-1700 см-1 би могла да се отнесе за карбонилната група от карбоксилната група. Явно е, че в случая на неприродните аминокиселини II в твърдо състояние преобладава цвитерйонната структура, но присъства и известно количество от нейонизираната форма.
Елементният анализ потвърждава състава на изследваните съединения.
Синтезираните и охарактеризирани лиганди подложихме на взаимодействие с CuCl2.2H2O в среда от метанол. Лигандите се разтварят в метанол, разтворът се алкализира с натриев хидроксид и бавно на капки се прибавя разтвора на комплексообразувателя, разтворен в метанол в съотношение метал към лиганд 1:2. При тези условия се получават синьо оцветени разтвори, из които след концентриране се отделят добре кристализиращи комплексни съединения ІІІ на разглежданите аминокоселини с Cu (II).
Елементният анализ на новополучените съединения сочи, че настъпва комплексообразуване в съотношение метал към лиганд 1:2 и вероятния начин на координиране е следния:
CH3C
NH2
CO
O
R
Cu CCH3C
OO
RNH2
ІІІ
В случая на лиганд IIa допълнително се координира молекула метанол, а при останалите лиганди не се наблюдава такава координация.

В UV-Vis спектрите на комплексните съединения III се наблюдава съществена разлика в сравнение със спектрите на самите лиганди. Абсорбционните максимуми за комплексното съединение IIIа са при 256 нм и 620 нм, за IIIб 260 нм и 625 нм, IIIв 262 нм и 628 нм и IIIг 263 нм и 625 нм.В тези случаи новопоявилата се абсорбция при 620, 625 и 628 нм е твърде далече от абсорбционната ивица на поглъщане при 860 нм за медния дихлорид в метанол.
IR-спектрите на комплексните съединения III показват три ивици на поглъщане в интервала 3600-3100 см-1 (NH2 група). От друга страна в областта 1700-1500 см-1 се наблюдава съществена разлика в ивиците на поглъщане на лигандите II и техните комплексни съединения III.
Eкспериментална част За синтеза на лигандите бяха използвани вещества с
класификация “р.а”. Елементният анализ е определен на автоматичен анализатор Carlo Erba-1106. Чистотата на лигандите беше контролирана, чрез тънкослойна хроматография в система: С4Н9ОН:СН3СООН:Н2О = 4:1:5.
Спектрите в ултравиолетовата и видимата област са снети на спектрофотометър “SPEKORD UV-Vis”. IR-спектрите са снети на спектрофотометър “SPEKORD-75R” в суспензия от нуйол или KBr.
1.Синтезът на изходните лиганди е извършен по методика [6]. 1.1.Получаване на 5-метил-5-фенилхидантоин (Ia). Добив:
63%; т.т. 194-50С; Rf = 0,22; IR-спектрални данни см-1 (табл. KBr): 3282.3 (N3-H); 3207.4 (N1-H); 1769.3 (C2=O); 1715 (C4=O), 2990.9; 2937.7; 1400.5; 1360.9 (CH3); за аренова част: 3066.1; 1598.4; 1495.3; 1444.6; моносубституирано бензеново ядро: 751.7; 733.9; 695.2.
1.2.Получаване на 5-метил-5-толилхидантоин (Iб). Добив: 65%; т.т. 195-60С; Rf = 0,31; IR-спектрални данни см-1 (табл. KBr): 3291.3 (N3-H); 3206.1(N1-H); 1781.2 (C2=O); 1731.3 (C4=O), 2994.5; 2927.4; 1397.6; 1367.7 (CH3); за аренова част: 3067.2; р-дисубсти-туирано бензеново ядро: 1509.3; 1448.6; 819.4.

1.3. Получаване на 5-метил-5-(4-метоксифенил)-хидантоин (Iв). Добив: 72%; т.т. 200-10С; Rf = 0,33; IR-спектрални данни см-1
(табл. KBr): 3295.1(N3-H); 3207.1 (N1-H); 1780.3 (C2=O); 1725.4 (C4=O); 2996.5; 2925.3; 1411.1; 1367.7 (CH3); 1250 (OCH3); за аренова част: 3067.3; р-дисубституирано бензеново ядро: 1510.2; 1442.4; 820.1.
1.4. Получаване на 5-метил-5-(2-флуорофенил)- хидантоин (Iг). Добив: 84%; т.т. 187-80С; Rf = 0,28; IR-спектрални данни см-1
(табл. KBr): 3293.2 (N3-H); 3206.2 (N1-H); 1775.6 (C2=O); 1712.5 (C4=O); 2995.2; 2918.6; 1398.1; 1368.3 (CH3); за аренова част: 3065.2; 1596.5; 1495.3; 1442.8; о-дисубституирано бензеново ядро: 1509.3; 1448.6; 849.4.
2.Синтез на непротеинови аминокарбоксилни киселини по методика [6].
2.1.Получаване на 2-амино-2-фенилпропанова киселина (IIа). Добив: 57%; т.т. 317-80С; Rf = 0,43; IR-спектрални данни см-1 (нуйол): 3230 (NH2); 3030 (CH3); 1680 (C=O); аренова част: 1598; Елементен анализ за N%: изчислен: 9,32%; намерен: 9,16% за С8Н8О2N.
2.2. Получаване на 2-амино-2-толилпропанова киселина (IIб). Добив: 54%; т.т. 286-70С; Rf = 0,39. IR-спектрални данни см-1 (нуйол): 3180 (NH2); 3029 (CH3); 1680 (C=O); аренова част: 1600; Елементен анализ за N%: изчислен: 8,52%; намерен: 8,48% за С9Н10О2N.
2.3. Получаване на 2-амино-2-(4-метоксифенил)-пропанова киселина (IIв). Добив: 65%; т.т. 293-40С; Rf = 0,45; IR-спектрални данни см-1 (нуйол): 3216 (NH2); 3025 (CH3); 1680 (C=O); 1250 (OCH3); аренова част: 1601; р-дисубституирано бензеново ядро: 820; Елементен анализ за N%: изчислен: 7,77%; намерен: 7,58% за С9Н10О3N.
2.4. Получаване на 2-амино-2-(2-флуорофенил)-пропанова киселина (IIг). Добив: 50%; т.т. 266-70С; Rf = 0,48; IR-спектрални данни см-1 (нуйол): 3220 (NH2); 3020 (CH3); 1681 (C=O); аренова част: 1600; о-дисубституирано бензеново ядро: 848; Елементен анализ за N%: изчислен: 8,33%; намерен: 8,16% за С8Н7О2NF.
3. Обща методика за получаване на комплексни съединения (III). 2 mmоl 2-амино-2-арил-пропанова киселина се разтваря при

разбъркване в 240 см3. Към получения разтвор се прибавят 2 mmоl натриев хидроксид. 1 mmоl CuCl2.2H2O се разтваря в 300 см3 метанол и разтвора се прикапва бавно при постоянно разбъркване към разтвора на лиганда. Получава се син разтвор, който се концентрира на водна баня с помоща на вакуум-ротационен изпарител. Получените сини кристали се филтруват, промиват се с метанол и се сушат в ексикатор над Р4О10.
3.1. Комплексно съединение на 2-амино-2-фенил-пропанова киселина с Cu(II). Добив: 85%; т.т. >3000С; IR-спектрални данни см-1 (нуйол): 3290, 3220, 3140(NH2); 1650 (C=O); бензеново ядро: 1605; елементен анализ: за Cu(C9H10NO2)2.CH3OH: N% изч. 6,60; нам. 6,37. Cu% изч.14,67; нам. 15,03.
3.2. Комплексно съединение на 2-амино-2-толил-пропанова киселина с Cu(II). Добив: 62%; т.т. >3000С; IR-спектрални данни см-1 (нуйол): 3510, 3415, 3340 (NH2); 1650 (C=O); р-дизам. бензеново ядро: 820; елементен анализ: за Cu(C10H12NO2)2:
С% Н% N% Cu% изч. 56,89 5,60 6,06 15,35 нам. 57,12 5,73 6,46 15,13 3.3. Комплексно съединение на 2-амино-2-(4-метокси-
фенил)-пропанова киселина с Cu(II). Добив: 77%; т.т. >3000С; IR-спектрални данни см-1 (нуйол): 3480, 3390, 3220 (NH2); 1650 (C=O); 1250 (OCH3); р-дизам.бензеново ядро: 820; елементен анализ: за Cu(C10H12NO3)2: С% Н% N% Cu%
изч. 53,15 5,35 6,20 14,06 нам. 53,33 5,41 6,18 13,95 3.3. Комплексно съединение на 2-амино-2-(2-флуоро-
фенил)-пропанова киселина с Cu(II). Добив: 90%; т.т. >3000С; IR-спектрални данни см-1 (нуйол): 3465, 3385, 3240 (NH2); 1648 (C=O); о-дизам.бензеново ядро: 850; елементен анализ: за Cu(C9H9NO2F)2:
С% Н% N% Cu% изч. 50,52 4,24 6,55 14,85 нам. 50,78 4,27 6,63 14,75

Литература 1. J. Tsang, B. Schmied, R. Nyfeler, M. Goodman, J. Med. Chem.,
1984, 27 (12), 1663. 2. C. Snckling, Angew. Chem. Int. Ed. Engl., 1988, 27, 537. 3. R.Cremlin, R. Ellam, T. Mitra, Ind. J. Chem., 1970, 8, 218. 4. K. Chacko, R. Zand, Act. Cryst., 1973, 29, 2684. 5. C. Toniolo, M. Crisma, Pept. Res., 1989, 2, 275. 6. H. Bucherer, V. Lieb, J. Pract. Chem., 1934, 5, 141.

Синтез и комплексообразувателна способност на 5-карбоксиметил-2,3-дихидро-1,3-диоксо-2-(2-
пиридил)-1Н-инден М. Кашчиева1, Н. Стоянов2
1Катедра обща химия, Факултет по природни науки ШУ “Епископ Константин Преславски”
ул. “Университетска” № 115, 9712 Шумен Е-Mail: [email protected]
2Катедра химия и химични технологии, Филиал-Разград към РУ “Ангел Кънчев”
Abstract: Synthesis of 5-carboxymethyl-2,3-dihydro-1,3-dioxo-2-(2-pyridyl)-1H-inden from 5-carboxy-2,3-dihydro-1,3-dioxo-2-(2-pyridyl)-1H-inden is realized in two ways – through thyonil chloride in methanol and through esterification with catalyst Н2SО4. Сu(II) сomplex of 5-carboxymethyl-2,3-dihydro-1,3-dioxo-2-(2-pyridyl)-1H-inden is obtained. The newly obtained compounds are characterized through m.p., elemental analysis, IR- and UV-Vis spectral data.
Key words: Сu(II) complex; indandione; pyrophtalon.
Въведение В предишно изследване [1] изолирахме стабилно
комплексно съединение на 5-карбокси-2,3-дихидро-1,3-диоксо-2-(2-пиридил)-1Н-инден в съотношение М:L=1:2. Чрез карбоксилното производно търсехме по-добра разтворимост на лиганда както в обичайните органични разтворители, така и в слабо алкална и водно-алкохолна среда, още повече че при 2-(2-пиридил)-1,3-индена [2,3,4] това не е възможно.
В нашия случай поради наличието на спрегната цвитерйонна структура честотата на трептене на тази ивица пада до 1577 см-1 и ние отдаваме това на съществуването на цвитерйонни структури, от които по-вероятна е II.

Тази силно полярна структура е нелетлива в условията на газ-хроматографията.
В търсене на ново доказателство за съществуването на структура I е получаването на неговия метилов естер, който би трябвало да е летлив в условията на газ-хроматографията, за разлика от I. Освен това II ще се различава по разтворимост от I.
C
COH
O
CO
O
NH
C
CO
O
CO
HO
NH
I II Експериментална част
Използвани са материали с чистота ”р.а.”. Температурите на топене са определени на Кофлеров микроскоп. Електронните и ИЧ спектри на съединенията са регистрирани със спектро-фотометри “Specord UV-Vis” и ”Perkin Elmer 580” (в табл. KBr). ЕПР спектъра е направен с ЕПР-спектрометър “Bruker B-ER” 420 със стандарт DPPH. Елементния анализ за азот е направен по Келдал след 15 часово горене, а елементния анализ за мед е направен комплексометрично.
1. Получаване на метилов естер на 5-карбокси-2,3-дихидро-1,3-диоксо-2-(2-пиридил)-1 Н-инден.
а) посредством тионилхлорид в метанол. Към 15 см3 сух метанол, охладен до -20 °С, при непрекъснато разбъркване се прикапва 0,72 см3 (0.01mol) прясно дестилиран тионилхлорид. След това се прибавят 1,335 g (0.005 mol) 5-карбокси-2,3-дихидро-1,3-диоксо-2-(2-пиридил)-1Н-инден. Суспензията се бърка 2 часа при -5 °С, като температурата на реакционната смес бавно се повишава до 40 °С и тази температура се поддържа още 2 часа. Падат кристали, които са главно от естера. След филтруване твърдият продукт се разтваря в етилацетат и разтворът се промива с наситен разтвор на NаНСОз. След

отстраняване на разтворителя, твърдият остатък се прекристализира из етилацетат или етилацетат - петролев етер.
б) чрез естерификация с катализатор Н2SО4. Смесват се 2,67 g (0.01 mol) 5-карбокси-2,3-дихидро-1,3-диоксо-2-(2-пиридил)-1Н-инден, 10 см3 метанол, 1,07 см3 (0.02 mol) к. Н2SО4 и реакционната смес се кипи на обратен хладник без достъп на влага 5 часа. След това излишният алкохол се отдестилира, а остатъкът се излива в петкратен обем вода. Органичният слой се отделя, а водният се екстрахира три пъти с етилацетат. Обединените органични фракции се промиват с наситен разтвор на NаНСО3, за да се отдели нереагиралия изходен продукт и се промива с вода. След сушене разтворителят се изпарява, а твърдият остатък се прекристализира из етилацетат-петролев етер.
Полученият естер III показва Тт=330°С. Добивът по методика а) е 35%, а по методика б) e 71,1%. Rf стойностите му в система А=0.001 и В=0.85. Анализът за N% е 5.10 (изчислен - 4.98%). В ИЧ спектъра се наблюдават ивици при: 1710, 1577, 2310 и 1200 см-1. В газ-спектъра на съединението се наблюдава пик т/z 281, отговарящ на молекулния йон [С14Н9О2N-СООСН3]+.
2. Синтез на Сu(II) комплекс на 5-карбоксиметил-2,3-дихидро-1,3 -диоксо-2-(2-пиридил)-1 Н-инден. Комплексното съединение се получава при постепенно смесване на наситен етанолов разтвор на лиганда 1.10-3 М с 5. 10-3 М воден разтвор на Сu(NО3)2.ЗН2О в съотношение М:L = 1:2 при непрекъснато разбъркване. Температурата на разтвора по време на комплексообразуването се поддържа около 40°С. След филтруване, комплексното съединение се промива неколкократно с етанол и се суши в ексикатор над Р4О10.
Комплексното съединение не се стапя до 350 °С. Елементният анализ за N% е 4.55% (изчислен 4.50%) и за
Сu% - 9.80% (изчислен -10.18%); UV-Vis спектрални данни: 365, 350, 295(280) нм. ИЧ-спектрални данни: 1705, 1550, 1210 см-1. ЕПР спектърът на комплекса е асиметричен сигнал с gII=2,063± 0,001.

Резултати и дискусия Естерът III синтезирахме по два начина: посредством SОСl2
и чрез естерификация в присъствието на катализатор к. Н2SО4. И в двата случая се получава един и същ продукт, но във втория случай се получава по-чист и с по-висок добив продукт (71,1%).
В ИЧ-спектъра на съединението III се появява ивица за валентно трептене на естерна карбонилна група при 1710 см-1. Няма промяна за спрегнатата система
HO C C C O O C C C O
в сравнение с тази при I. Отново се наблюдава ивицата за -NН+ при 2310 см-1 (в нуйол), както и ивици в областта 1630-1610 см-1, които се дължат на трептенията, характерни за спрегнатата ароматна система. Резултатите показват, че съединението е със следната структура:
C
CO
O
CO
CH3O
NH
III Данните се подтвърждават от газ-мас спектъра на
съединението III. Наблюдава се пик m/z 281, отговарящ на молекулния йон, който фрагментира до получаване на пик m/z 222, отговарящ на молекулния йон на 2-(2-пиридил)-1,3-инден по следната схема на фрагментиране:

C
COH
O
CO
NH3CO
281
C16H11O4N Mm=281
C
COH
O
CO
N
250
CO
OCH3
C
COH
O
N
222
C
COH
O
N
222
C
COH
O
CO
NO
266
CH3COOCH3
В електронния спектър на Сu(II) комплекса с III се
наблюдават две нови ивици при 355 и 370 нм, докато в спектъра на лиганда абсорбционния максимум е при 390 нм. Същият не се наблюдава в спектъра на комплекса.
В ИЧ-спектъра на комплекса с Сu(II) няма промяна в честотата на ивицата на естерната карбонилна група и за С=O групата от спрегнатата система, но ивиците са по-малко интензивни. Вероятната структура на съединението е следната:
C
CO
O
CO
CH3O
N
Cu
C
CO
O
CO
CH3ON
Потвърждава се фактът, че координирането на металния
йон става с кислородния атом на спрегнатата система III и пиридиновия пръстен.

Литература 1. Kashchieva М., Nikolova St., Minchev St., Сотрt. Rend. Асаd.
Виlg. Sci. 2003, 56(1), 45-48. 2. Воnchev Р., Мitewa М., Мinchev St., Каshchieva М.,
Мehandgiev D., J. Pract. Chemie, 1983, 325, 803-810. 3. Мitewa М., Воnchev Р., Маlinovski А., Каshchieva М., Мinchev
St., Сотрt. Rend. Асаd. Виlg. Sci., 1983, 36, 1311- 1313. 4. Бончев П., Кашчиева М., Митева М., Минчев Ст., Сборник
доклади на ВПИ-Шумен, Юбилейна научна сесия, 1982, 20-32.

Comparison of Candida melibiosica and Saccharomyces cerevisiae Performance in
Experimental Biofuel Cell Yolina Hubenova1, Mario Mitov2,3
1Department of Biochemistry and Microbiology Plovdiv University “Paisii Hilendarski”, 4000 Plovdiv
24 Tzar Asen str., Bulgaria, E-Mail: [email protected] 2Department of Chemistry, South-West University “Neofit Rilski”
Blagoevgrad, Bulgaria 3Institute of Electrochemistry and Energy Systems Bulgarian Academy of Sciences, Sofia, Bulgaria
Abstract: The aim of this study is to compare the performance of both Candida melibiosica and Saccharomyces cerevisiae yeast as a microreactor in experimental biofuel cell. For this purpose different modified yeast media were developed so that the microbiological requirements of these strains could be coupled with the electrochemical ones. Various carbohydrate sources, cultivation temperatures and pH of media were examined. Polarization measurements with suspension of yeast cells, carbohydrate, phosphate buffer and mediator methylene blue at variable as well as at constant load resistance were carried out in a two-chamber electrochemical cell. Higher electric characteristics (generated current, voltage and power) values were achieved with Saccharomyces. cerevisiae. However, much more stable characteristics within time were obtained with Candida melibiosica yeast during long-term polarization tests. Keywords: yeast, electricity generation, biofuel cells
Introduction Biofuel cells represent an innovative technology for overcoming
both the dramatically increasing energy demands and the irreversible environmental pollution caused by the use of carbon-based fuel resources (Bennetto, 1990; Bullen et al., 2006). A biofuel cell employs

the catalytic action of living organisms to convert the chemical energy of a fuel into electrical energy. Most of the up-to-now developed biofuel cells are designed on a base of prokaryotic cells utilization (Rabaey et al., 2005; Kim et al., 2007).
The aim of this investigation is to compare the performance of eukaryotic Candida melibiosica and Saccharomyces cerevisiae yeast as a microreactor into experimental biofuel cell. For this purpose different modified yeast media were developed so that the microbiological requirements of these strains could be coupled with the electrochemical ones. Various carbohydrate sources, cultivation temperatures, aeration degree and pH of media were examined. The electric output characteristics of the biofuel cell obtained at optimized conditions for both yeast species were compared and discussed.
Experimental Microbiological and Biochemical Methods
The yeast Candida melibiosica and Saccharomyces cerevisiae were originally cultivated on YP (yeast peptone) media with a carbohydrate as an additive. 1,2 g/L yeast cells were transferred into electrochemical media containing 250 mM solutions of glucose, fructose or sucrose and 67 mM phosphate buffer. The yeast strains were cultivated at different temperatures (22 - 28°C) and pH of media in separate experiments. The rate of carbohydrate assimilation within time was determined by DNS method. The conversion of inorganic phosphate into organic one during yeast cell growth was quantitatively measured by means of Molybdenum blue phosphorous method. The amount of yeast cells was monitored by using the functional dependence between cell suspension OD600 (optical density) and dry biomass as well as by cell counting in hemocytometer. Tests about non toxicity of the mediator were also performed.
Electrochemical Methods
The electrochemical experiments were carried out in a two-chamber experimental cell with a salt bridge and graphite electrodes. Suspension of yeast cells, carbohydrate, phosphate buffer and

mediator methylene blue was used as an anolyte. 0,1 M K3[Fe(CN)6] served as an electron acceptor. Polarization measurements at variable as well as at constant load resistance were carried out.
Results and Discussion
Recently, we have examined the possibility for use Saccharomyces cerevisiae yeast as a microrector into biofuel cell. The lag/log-phase of growth of Saccharomyces cerevisiae was determined under both aerobic and anaerobic conditions (Hubenova and Mitov, 2008a, 2008b). The three stages of cellular respiration - glycolysis, citric acid cycle and oxidative phosphorylation in eukaryotic cells, were traced out, aspiring to shuttle the electrons between the biological system and the fuel cell anode by using appropriate mediator. The biochemical results with regard to iso-osmotic conditions and carbohydrate source showed that the glucose was almost completely assimilated at the 40th minute of incubation. The comparison with the sucrose showed that for the same time its concentration decreased up to 40 mM, probably due to the lower expression of invertase of this strain.
Verification of possibility for biofuel cell usage of another yeast Candida melibiosica included the implementation of parallel microbiological and electrochemical experiments. Summary of the biochemical results about yeast suspension media, modified for biofuel cell application, is given in Table 1.
In previous investigation (Georgiev and Gargova, 2007), the fructose was proven to be the most appropriate carbohydrate for Candida melibiosica development. Almost the same levels of carbohydrate and phosphate assimilation were found at two examined pH 5,5 and 7, which corresponds to the wide pH range of living of this yeast strain. For further electrochemical experiments, the neutral pH was chosen. The biochemical comparison of both yeast species performance showed that the carbohydrate assimilation by Candida melibiosica was quite lower within time.
Table 1. Quantitative determination of fructose and inorganic phosphate by DNS and Molybdenum blue phosphorous methods
during Candida melibiosica yeast cultivation.
Candida melibiosica
0.1 g/ml yeast + 125 mM fructose/ 67 mM
phosphate buffer, pH 5,5
0.1 g/ml yeast + 125 mM fructose/ 67 mM
phosphate buffer, pH 7 Conditions Fermentation without
aeration Fermentation without
aeration Incubation time, hours
Conc. fructose
mM
Conc. PO4
3- mM
Conc. fructose
mM
Conc. PO4
3- mM
1 50 65 49 60 2 49 60 49 56 3 47 57 47 51 4 44 53 44 47 5 44 50 44 43 6 37 45 36 38
24 36 37 36 32
Optimization of anolyte composition for biofuel cell application was done on the base of results from polarization measurements with variation of yeast cell content, carbohydrate type and concentration.
An optimal cell number corresponding to 1,2 g/l Candida melibiosica yeast cells was found – Figure 1:
0 20 40 60 80 1000
50
100
150
200
250
300
350 0.6 g/l 1.2 g/l 2.4 g/l 6.0 g/l 12.0 g/l
I, mA x 10-3
U, m
V
0
2
4
6
8
10
0 20 40 60 80 100
I, mA x 10-3
P, m
W x
10-3
0.6 g/l 1.2 g/l 2.4 g/l 6.0 g/l 12.0 g/l
Fig. 1. Polarization and power curves obtained with different content
of Candida melibiosica yeast cells.
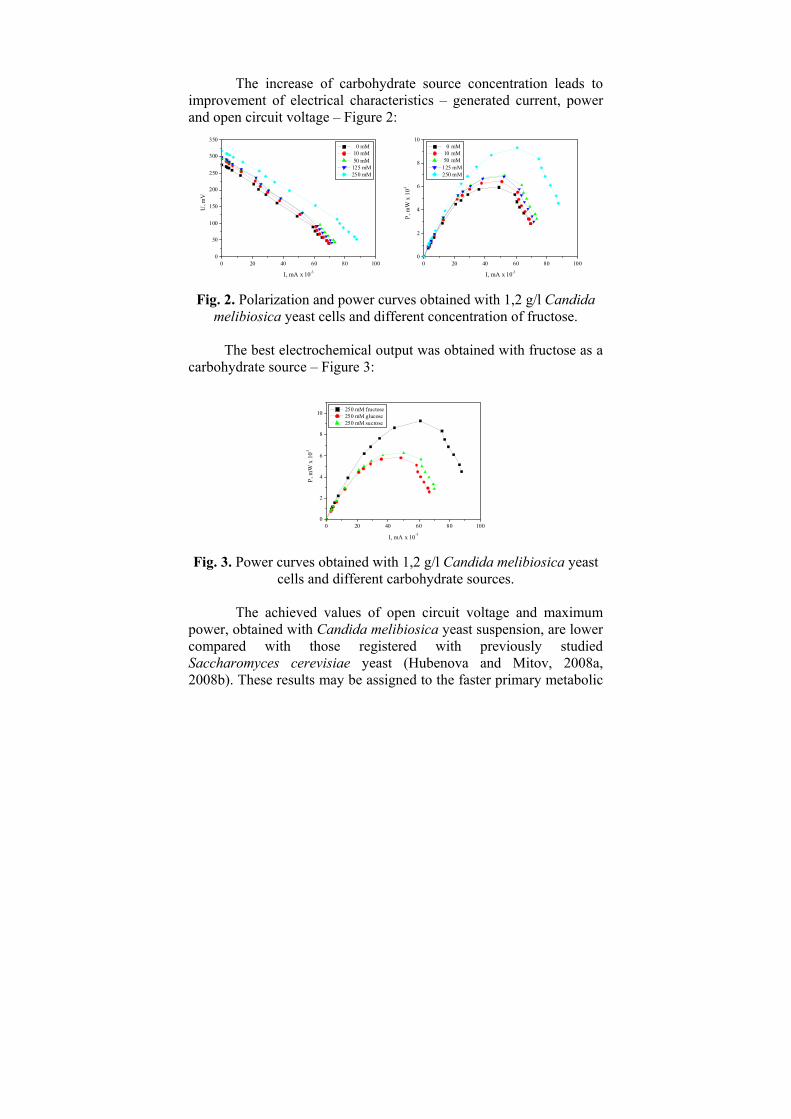
The increase of carbohydrate source concentration leads to improvement of electrical characteristics – generated current, power and open circuit voltage – Figure 2:
0 20 40 60 80 1000
50
100
150
200
250
300
350
U, m
V
I, mA x 10-3
0 mM 10 mM 50 mM 125 mM 250 mM
0 20 40 60 80 1000
2
4
6
8
10
P, m
W x
10-3
I, mA x 10-3
0 mM 10 mM 50 mM 125 mM 250 mM
Fig. 2. Polarization and power curves obtained with 1,2 g/l Candida
melibiosica yeast cells and different concentration of fructose.
The best electrochemical output was obtained with fructose as a carbohydrate source – Figure 3:
0 20 40 60 80 1000
2
4
6
8
10
P, m
W x
10-3
I, mA x 10-3
250 mM fructose 250 mM glucose 250 mM sucrose
Fig. 3. Power curves obtained with 1,2 g/l Candida melibiosica yeast
cells and different carbohydrate sources.
The achieved values of open circuit voltage and maximum power, obtained with Candida melibiosica yeast suspension, are lower compared with those registered with previously studied Saccharomyces cerevisiae yeast (Hubenova and Mitov, 2008a, 2008b). These results may be assigned to the faster primary metabolic
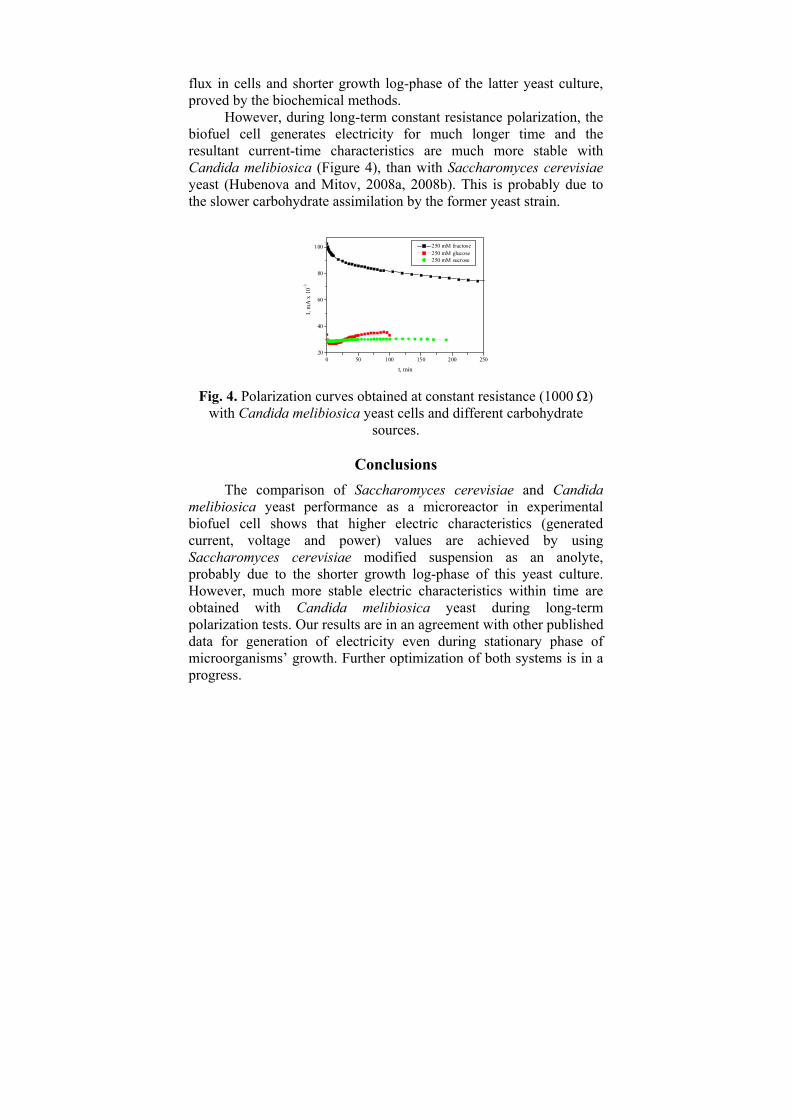
flux in cells and shorter growth log-phase of the latter yeast culture, proved by the biochemical methods.
However, during long-term constant resistance polarization, the biofuel cell generates electricity for much longer time and the resultant current-time characteristics are much more stable with Candida melibiosica (Figure 4), than with Saccharomyces cerevisiae yeast (Hubenova and Mitov, 2008a, 2008b). This is probably due to the slower carbohydrate assimilation by the former yeast strain.
0 50 100 150 200 25020
40
60
80
100
I, m
A x
10-3
t, min
250 mM fructose 250 mM glucose 250 mM sucrose
Fig. 4. Polarization curves obtained at constant resistance (1000 Ω)
with Candida melibiosica yeast cells and different carbohydrate sources.
Conclusions
The comparison of Saccharomyces cerevisiae and Candida melibiosica yeast performance as a microreactor in experimental biofuel cell shows that higher electric characteristics (generated current, voltage and power) values are achieved by using Saccharomyces cerevisiae modified suspension as an anolyte, probably due to the shorter growth log-phase of this yeast culture. However, much more stable electric characteristics within time are obtained with Candida melibiosica yeast during long-term polarization tests. Our results are in an agreement with other published data for generation of electricity even during stationary phase of microorganisms’ growth. Further optimization of both systems is in a progress.

References 1. Bennetto HP (1990) Electricity generation by microorganisms.
Biotechnology Education 1 (4): 163-168. 2. Bullen RA, Arnot TC, Lakeman JB, Walsh FC (2006) Biofuel
cells and their development. Biosens Bioelectron. 21 (11): 2015-2045.
3. Georgiev D, Gargova S (2007) Influence of carbon and phosphorous sources on the biosynthesis of phytase by Candida melibiosica. Report in Plovdiv University of Nutritious Technologies.
4. Hubenova Y, Mitov M (2008a) The new era has begun: renewable bioenergy production as a progressing interdisciplinary research approach. Proceedings of COST conference „Global change and sustainable development in mountain regions“, Innsbruck University Press - in press.
5. Hubenova Y, Mitov M (2008b) Biochemical study of Saccharomyces cerevisiae yeast as a microreactor for biofuel cells. Proceedings of South-Eastern European Regional Symposium of Electrochemistry, 260 – 262.
6. Kim J, Cheng S, Gadd G (2007) Challenges in microbial fuel cell development and operation. Appl Microbiol Biotechnol 76: 485–494.
7. Rabaey K, Verstraete W (2005) Microbial fuel cell: novel biotechnology for energy generation. Trends in Biotechnology 23: 292–298.

Investigation of the Biological Activity of Glycation Analogous Product 3-Hydroxy-1-methylpyridinium
Chloride on Microorganisms Yolina Hubenova1, Marin Marinov2, Ognyan Argirov2
1Department of Biochemistry and Microbiology Plovdiv University “Paisii Hilendarski”, 4000 Plovdiv
24 Tzar Asen str., Bulgaria, E-Mail: [email protected] 2Department of Organic Chemistry
Plovdiv University “Paisii Hilendarski”, 4000 Plovdiv 24 Tzar Asen str., Bulgaria
Abstract: The Maillard reaction is an important factor in the post-translational modifications of proteins in the living organisms. It is proved that the modified amino acid residues have biological activity towards various microorganisms and yeasts. The aim of this study is to determine the biological activity of a glycation product analogue on some microorganisms. The kinetic dependence of its influence on the base of different concentration proportions as well as growth stage and vitality of microorganisms is found out. The toxicity of the glycation analogous product 3-hydroxy-1-methylpyridinium chloride was proven for eukaryotes. The toxicity level of this analogue is a function of its concentration.
Keywords: glycation, microorganisms, toxicity, Maillard reaction.
Introduction The nonenzymatic reaction of the amino groups of amino acids,
peptides, and proteins with reducing sugars is known as a Maillard reaction or glycation. This process also occurs slowly in vivo. The Maillard reaction is an important factor in the post-translational modifications of proteins in the living organisms (Bhattacharyya et al., 2007). The glycation process, however, may cause metabolic changes and even pathological diversion in some diseases. The reasons why glycation products are formed in organisms are not fully clarified yet.

It is proved, however, that the modified amino acid residues have biological activity (Argirov et al., 2003, 2004, 2005).
The aim of this study is to synthesize 3-hydroxy-1-methylpyridinium chloride (Fig. 1) as an analogue of the glycation product hydroxy-pyridinium-lysine as well as to determine its biological activity towards various microorganisms: Gram-positive (Bacillus cereus), Gram-negative (Escherichia coli) microorganisms and yeast species Candida melibiosica and Saccharomyces cerevisiae.
Fig.1. Scheme of synthesis of 3-hydroxy-1-methylpyridinium chloride.
Materials and methods Synthesis and analysis
The starting compounds 3-hydroxy pyridine (5.7 g, 60 mmols) and methyl iodide (3.72 ml, 60 mmols) (Fig. 1) were refluxed for one hour in 250 ml 1-propanol. The reaction product crystallized overnight at -32oC. The product was dissolved in 30 ml water. Silver oxide (3.6 g, 15.4 mmols) was added to the solution in a centrifuge tube and stirred under sonication. The solid was separated by centrifugation and washed with 15 ml water. Silver oxide (3.3g, 14 mmols) was added to the combined water supernatants and stirred under sonication again. The solid was separated by centrifugation. The supernatant was filtered and extracted with 50 ml ethylacetate twice. The water phase was acidified with 30% HCl to pH 2 (white precipitate appeared immediately) and the precipitate was eliminated by filtration. The filtrate was concentrated under reduced pressure. After precipitation with acetone, the target compound 3-hydroxy-1-methylpyridinium chloride was obtained. Its structure was analyzed by means of elemental analysis, 1H-NMR and 13C-NMR spectroscopy.
CH3 I +N
OH
N+
OH
CH3
I-
N+
OH
CH31. Ag2O
2. HCl Cl-

Biological activity
The microorganisms Escherichia coli and Bacillus cereus were cultivated on Mueller-Hinton-medium at 37°C. The yeast Candida melibiosic and Saccharomyces cerevisiae were cultivated on YPfru – medium (yeast peptone medium with fructose as additive) at 28°C. From each starter culture 1x106cells/ml were placed on agar media in petri dish. The test for determination of their biological activity was made by means of the agar-diffusion-method on the 18-th hour from the beginning of cultivation. 10; 1; 0,1; 0,01mg absolute amount of 3-hydroxy-1-methylpyridinium chloride (100 μl water solution) was filled into agar`s axillae. The diameter of sterile zone was measured.
Results The yield of 3-hydroxy-1-methylpyridinium chloride in the
above described synthesis procedure was 0.91 g (10 %). The newly synthesized substance was confirmed by elemental analysis (Tab. 1).
Tab.1. Elemental analysis of 3-hydroxy-1-methylpyridinium chloride.
The definitive structure determination of 3-hydroxy-1-methylpyridinium chloride was acchived by the spectral data received as follows: 1H-NMR (D2O, ppm): δ 4.28 (s, 3H), 7.81-8.29 (m, 4H). 13C-NMR (D2O, ppm): δ 156.5, 136.7, 133.5, 131.5, 128.3, 48.1.
The chemical shifts and the intensities of the peaks of NMR data confirmed the structure suggested.
Testing the biological activity of this glycation analogue, it was ascertained that it does not influence the bacterial growth in the examined concentrations. One of the both yeast strains Candida melibiosica showed also negative results. However, the budding yeast` Saccharomyces cerevisiae development was inhibited. A sterile zone of 23 mm diameter was observed (Fig. 2).
C % H % N % Cl % Calc. Exp. Calc. Exp. Calc. Exp. Calc. Exp. 49.65 48.83 5.56 5.44 49.65 48.83 5.56 5.44

Fig. 2. Biological activity of 3-hydroxy-1-methylpyridinium chloride
towards Saccharomyces cerevisiae.
Discussion It was recently proven, that the hydroxy-pyridinium-lysine is
located in the cataractous lenses (Argirov et al., 2003, 2004). A new glycation product derivative 3-hydroxy-1-methylpyridinium chloride was synthesized and proved by means of EA, 1H-NMR and 13C-NMR. This compound is a model of the structure, which is formed on protein molecules due to glycation. It was interesting to study this heterocycle providing that it is a substructure in living systems. Its biological activity towards microorganisms was investigated. Both prokaryotes Escherichia coli and Bacillus cereus were resistant to this substance in the range of concentrations applied. The glycation analogous product did not influence the growth and development of Candida melibiosica yeast, the strain previously shown the highest phytase activity. However, a sterile zone with diameter 23 mm was observed when 10 mg 3-hydroxy-1-methylpyridinium chloride absolute was supplied to Saccharomyces cerevisiae culture. This result argues that there is a receptor or receptors (Argirov et al., 2004), which are sensitive toward the compound of interest. Lower concentration did not inhibit the yeast growth. This preliminary study shows the necessity of further investigations about the precise determination of the biological activity of 3-hydroxy-1-methylpyridinium chloride towards different eukaryotic tissues (Teruyuki et al., 2004). Further investigations for the induced cytotoxicitiy of this product are in progress.

Acknowledgments: This study was supported by project IS-H-2/08, project TK-H-1612/06 and 07-H-50.
References 1. Argirov O., Lin B., Olesen P., Ortwerth B. (2003) Isolation and
characterization of a new advanced glycation endproduct of dehydroascorbic acid and lysine. Biochimica et Biophysica Acta 1620: 235–244.
2. Argirov O., Lin B, Ortwerth B. (2004) 2-Ammonio-6-(3-oxidopyridinium-1-yl)hexanoate (OP-lysine) is a newly identified advanced glycation end product in cataractous and aged human lenses. JBC, 279 (8): 6487–6495.
3. Argirov O., Lin B., Ortwerth B. (2005) Phototransformations of advanced glycation end products in the human eye lense due to ultraviolet A light irradiation. Ann N Y Acad Sci. 1043:166-73.
4. Bhattacharyya J, Shipova EV, Santhoshkumar P, Sharma KK, Ortwerth BJ. (2007) Effect of a single AGE modification on the structure and chaperone activity of human alphaB-crystallin. Biochemistry 46 (50):14682-92.
5. Teruyuki U., Satomi S., Hirohito W., Fumitaka H. (2004) Cytotoxicity and oxidative stress induced by the glyceraldehyde-related Maillard reaction products for HL-60 cells. Biosci. Biotechnol. Biochem., 68 (2): 333-340.

Synthesis of Amphiphilic Alternating Copolymers of the Maleic Acid, Containing Nitroxide Radicals V. B. Konsulov1, A. A. Lyapova1, G. P. Petrov1, P. Saha2
1Konstantin Preslavsky University of Shumen 9712 Shumen, Universitetska str. 115, Bulgaria
2Tomas Bata University in Zlin, nam. T. G. Masaryka 5555 76001 Zlin, Czech Republik
Abstract: The alternating copolymers poly(α-methylstyrene-alt-maleic anhydride), poly(N-vinylpyrrolidone-alt-maleic anhydride) and poly(acrylic acid-alt-maleic anhydride) were prepared by solution copolymerization. The composition analysis of the copolymers is determined by NMR spectra.The data shows that the molar fraction of MA in the obtained copolymers is almost equal to 0.5 throughout the alternating copolymerization.The monomer reactivity ratios were determined by the Kelen-Tudös method: r1 =0.09, r2=0.05 for MS-MA; r1 =0.08, r2=0.07 for VP-MA and r1 =0.11, r2=0.06 for AA-MA systems. The polymeranalogous reaction of obtained MA-copolymers with 4-amino-2,2,6,6-tetramethylpiperidin-1-oxyl (A-TEMPO) was investigated. New kind of water soluble polyelectrolite copolymers, containing carboxyle and stable piperidine-oxyl radicals, were prepared and investigated for biological activity properties. The structure of the resulting functional copolymers was proved by FT-IR, NMR and EPR spectroscopy. The typical triplet signal proof including of stable nitroxyl radicals in the macromolecules.
Keywords: alternating copolymers; copolymerization; α-methyl styrene; N-vinylpyrrolidone; acrylic acid; maleic anhydride; polymeranalogous reaction; 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl; nitroxyl radicals.
Introduction The established antitumour and radio-sensibilizing effects of the
nitroxylic radicals, and also their low toxicity gibes us the grownds to enlist them in their molecule of the citoxic groups.[1] The avalability of the nitroxylic radical in the molecule of the newly formed

compounds disclose read opportunities for solving many biomolecular and biopharmaceutical problems by using EPR method and also for making biopharmaceutical applications.[1-3]
The mostly applied nitroxyles from 4-amino-2,2,6,6-trtramethylpiperidine-1-oxyl (A-TEMPO). As a stable radical TEMPO was uncovered by Lebedev and Kazanski [4] in 1960. It is prepared by oxydation of 2,2,6,6-trtramethylpiperidine. TEMPO is widely used as a radical trap, and also as a structural probe for biological systems in conjucation, as a reagent in organic synthesis, and in polymer chemistry.
Amino-, hydroxy- and propargylamine derivatives of TEMPO are of great interest. The structure of 4-amino-2,2,6,6-trtramethylpiperidine-1-oxyl (A-TEMPO) is examined in details [5]. In our search for novel, low-toxic, cell-penetrable and neuroprotective antioxidants.[6] The novel class of N- propargylamine nitroxyls may have potential implications for the experimental therapies of Parkinson disease.[7]
The functional copolymers (polyanions) show biologicaly activity properties.[8-13] The kind of copolymer poly(maleimide-co-2-ethylacrylicacid) show low toxicity and significant curative effects on Lewis lung carcinoma and S180.[10]
Amphiphilic alternating copolymers of maleic anhydride and hydrophobic monomers form an important class of polymeric materials with many applications.[11,12] Biologicalactivity amphiphilic maleimide polyanions were synthesised by the copolymerization of N-(4-carboxyphenyl)maleimide with acrylic and methacrylic acid.[11] The hydrolized form of the copolymer of divinyl ether with maleic anhydride (DIVEMA) shows various biological activities such as interferon induction, antiviral and antitumor activity as well as immunostimulating effects.[8, 9] Maeda reported [12, 13] of neocarzinostatin SMANCS/Lipiodol (S/L drugs) on the base of poly(styrene-co-maleic acid anhydride). Formulations based on S/L have shown to be effective both as a diagnostic tool and for therapeutic use in solid tumors.
Free nitroxilic radicals 4-R-2,2,6,6-tetramethyl-piperidine-1-oxyl show antitumor activity.[1] Their toxicity and effectivity depend on the substituent R.

In this work aim to obtain and characterize news functional copolymers, as a macromolecular substituent in TEMPO with functional carboxylic groups, containing stable piperidine oxyl radicals.
Experimental Preparation of 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl
(A-TEMPO): To the solution of 1.5 g 4-amino-TEMP (0.01mol) in 20 ml methanole in flask was addided 10 ml 30 % H2O2 at room temperature:
H3CH3C
NH2
CH3
CH3N
H
H2O2
NCH3
CH3
NH2
H3CH3C
O
On the reaction mixture was operated with ultrawave (40 kHz) for about maximum time 30 min. The reaction of oxidation was controlled through TLC with movable system chloroform-ethanol (4:1) and immovable phase silicagel. After that the solution was concentrate on the rotational vacuumevaporator and after cooling TEMPO was obtaned: orange to red solid crystals or liquid: Yield 82 %; C9H19N2O (Mw = 171.26 g/mol); N=16.24 % (calc. N=16.36 %).
Copolymerization: All reactions of copolymerization of N-vinylpirrolidone (VP) with Maleic anhydride (MA) and vinylbutyl ether (VBE) were carred aut in solution with AIBN as radical initiator at 60 or 70°C under nitrogen atmosphere. The obtained copolymers were purified twice by repricipitation and extraction in Socsle aparatus. The copolymer composition was determined by elemental analysis and 1H NMR.
Results and Discussion Synthesis of poly(α-Methyl Styrene-alt-Maleic Anhydride)
The alternating copolymer poly(α-methylstyrene-alt-maleic anhydride) were prepared by solution copolymerization:
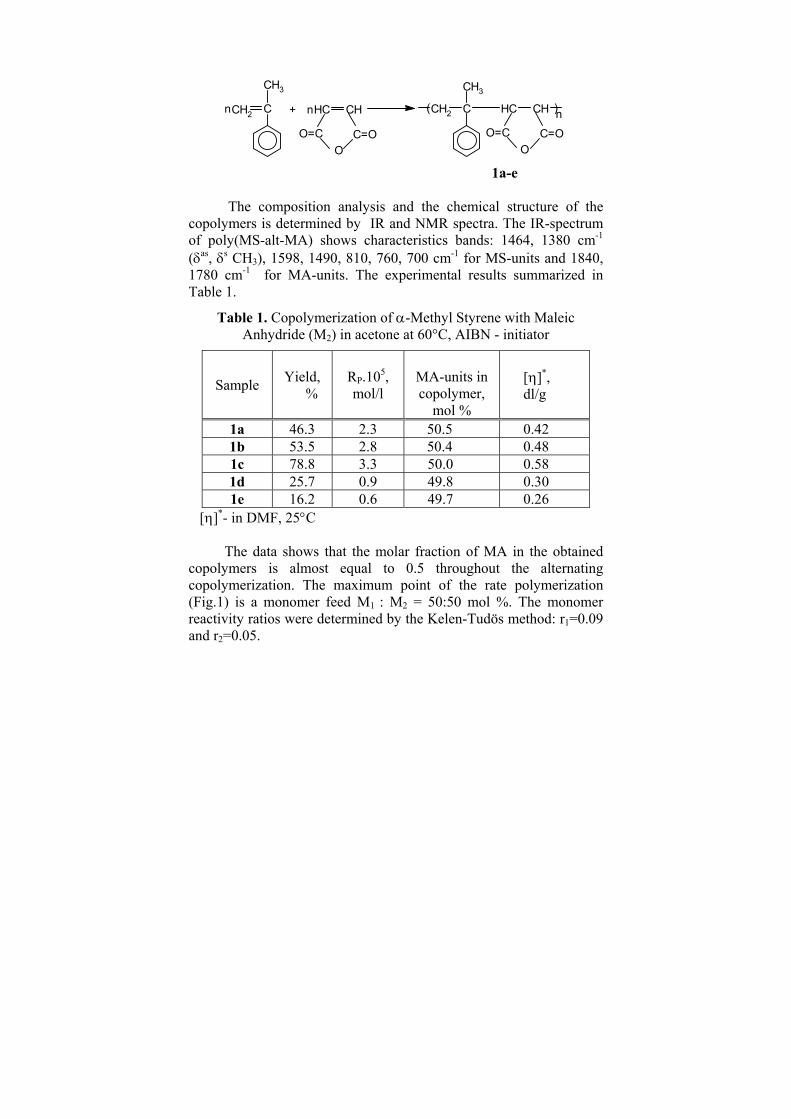
nC C
CH3 CH3
O O
( )
C=OO=C
CHHCCH2HC CH
O=C C=O
nn +CH2
1a-e
The composition analysis and the chemical structure of the copolymers is determined by IR and NMR spectra. The IR-spectrum of poly(MS-alt-MA) shows characteristics bands: 1464, 1380 cm-1 (δas, δs CH3), 1598, 1490, 810, 760, 700 cm-1 for MS-units and 1840, 1780 cm-1 for MA-units. The experimental results summarized in Table 1.
Table 1. Copolymerization of α-Methyl Styrene with Maleic Anhydride (M2) in acetone at 60°C, AIBN - initiator
Sample Yield, %
RP.105, mol/l
MA-units in copolymer,
mol %
[η]*, dl/g
1a 46.3 2.3 50.5 0.42 1b 53.5 2.8 50.4 0.48 1c 78.8 3.3 50.0 0.58 1d 25.7 0.9 49.8 0.30 1e 16.2 0.6 49.7 0.26
[η]*- in DMF, 25°C
The data shows that the molar fraction of MA in the obtained copolymers is almost equal to 0.5 throughout the alternating copolymerization. The maximum point of the rate polymerization (Fig.1) is a monomer feed M1 : M2 = 50:50 mol %. The monomer reactivity ratios were determined by the Kelen-Tudös method: r1=0.09 and r2=0.05.
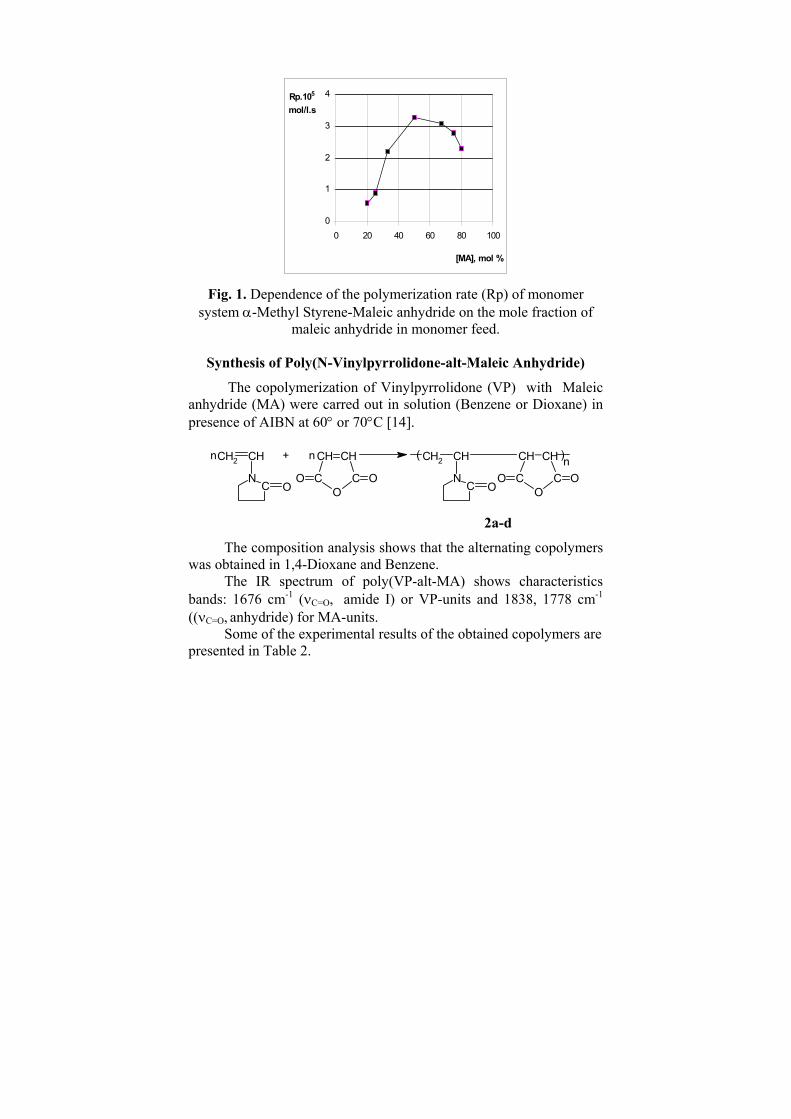
0
1
2
3
4
0 20 40 60 80 100
[MA], mol %
Rp.105
mol/l.s
Fig. 1. Dependence оf the polymerization rate (Rp) of monomer
system α-Methyl Styrene-Maleic anhydride on the mole fraction of maleic anhydride in monomer feed.
Synthesis of Poly(N-Vinylpyrrolidone-alt-Maleic Anhydride)
The copolymerization of Vinylpyrrolidone (VP) with Maleic anhydride (MA) were carred out in solution (Benzene or Dioxane) in presence of AIBN at 60° or 70°C [14].
+ CHCH
CO
C OO
CH2 CH
CN
O
n n CH2 CH
CN
O
CHCH
CO
C OO
_( )n
2a-d
The composition analysis shows that the alternating copolymers was obtained in 1,4-Dioxane and Benzene.
The IR spectrum of poly(VP-alt-MA) shows characteristics bands: 1676 cm-1 (νC=O, amide I) or VP-units and 1838, 1778 cm-1 ((νC=O, anhydride) for MA-units.
Some of the experimental results of the obtained copolymers are presented in Table 2.
Table 2. Copolymerization of N- vinylpyrrolidone (VP) (M1) with Maleic anhydride (M2) in 1,4-Dioxane (DO) or Benzene (Bz); M1 /
M2 =50 : 50 mol ; CM =3.0 mol/l ; 2 h.
Copolymer compozition, mol
% Sam- ple
solvent
Yield, %
N, %
m1 m2
[η]*
dl/g
2a DO 46.0 6.60 49.3 50.7 0.65 2b DO 78.5 6.73 50.5 49.5 0.70 2c Bz 42.5 6.55 49.2 50.8 0.55 2d Bz 72.8 6.64 49.8 50.2 0.60
[η]*- in 1,4-Dioxane, 25°C
Synthesis of Poly(Acrylic Acid-alt-Maleic Anhydride)
Alternating copolymer poly(AA-alt-MA) was synthesised by copolymerization of acrylic acid (AA) with maleic anhydride (MA) in the presence of 2,2’-azo-bisizobutironitrile (AIBN) as an initiator in benzene at 60°C:
n
OCOOC
CHHCCH2 CH
COOHO
COOC
CHHC n+nCH2 CH
COOH
( )
3a-d
The structure of the obtained poly(AA-alt-MA) has been proved by NMR and FT-IR spectroscopy.
IR-spectrum: AA-units: 1718 cm-1(νC=O COOH), MA-units: 1838 and 1780 cm-1(νC=O, anhydride).
1H NMR spectrum (CDCl3) δ,ppm: 1,51-1,66 (2H, -CH2-) and 11,2 (1H, COOH) for AA-units; 2,73-2,88 (2H, -CH-CH-) for MA-units.
13C NMR spectrum (CDCl3): 34,9-37,1 ppm(-CH2) and 175,8 ppm(-COOH) for AA-units; 166,8 ppm (C=O) for MA-units.
Best results were given in equimolar proportions because with high speed have been obtained product. The formation of two types complexes through H-bond and CTC in the maleic anhydride-acrylic

acid monomer system determine alternating copolymetization. Some of the experimental results are presented in Table 3.
Table 3. Copolymerization of Acrylic acid (M1) with Maleic
anhydride (M2) in benzene, CM=2 or 4 mol/l; AIBN=0.75 mass %; 60°C; 3h)
Monomer feed, mol %
Copol. compozition,
mol.%
№ M1 M2
CM, mol/l
m1 m2
[η]*
g/dl
3a 66.7 33.3 2.0 50.4 49.6 0.30 3b 50.0 50.0 2.0 50.0 50.0 0.38 3c 25.0 75.0 2.0 49.7 50.3 0.32 3d 50.0 50.0 4.0 50.5 49.5 0.45
[η]* in 1,4-Dioxane, 25°C, Ubbelode d=0.54mm
Polymeranalogous reactions
By the polymeranalogous reactions of alternating MA-copolymers with 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl (A-TEMPO) in DMF were prepared news polymers containing stable nitroxile radicals. The reactions of N-acylation are presented in scheme:
4a-c
O
N
NH
CO
CH
OH
COR
CHCHCH2
.O
N
NH2+
OCOOC
CHCH
R
CHCH2
Table 4. Synthesis and characteristics of alternating copolymers of maleic acid, containing stable piperidine-1-oxyl radicals (TEMPO).
Elemental analysis Sam-ple Formula
% С % Н % N х,* %
[η] dl/g
4a
O
N
NHOH
CO
CH
CO
CH
CH3
CCH2
66.95 8.16 6.54 90.8 0.70
4b
CH2 CH CH
C=ON CO
OH
CH
CO
NH
N
O
51.48 7.98 9.37 84.7 0.48
4c
O
N
NHOH
CO
CH
CO
CH
COOH
CHCH2
48.77 7.04 5.69 92.8 0.54
х,* % - conversion of anhydride units.
For the modification copolymers 4 the anhydride bands (1778 and 1838 cm-1) are completely displaced to the stretching vibration of the carboxyle groups at 1705 cm-1(νC=O) and of the carboxylate (-COO-) at 1526 cm-1. The intensive band at 1658cm-1 (νC=O, amide I) proves the formation of an amide group –CO-NH- connecting TEMPO with the polymer chain. The typical signals in the EPR- spectra (Fig. 2 and 3) and bande about 1198-1230 cm-1 in FT-IR spectra of the obtained polynitroxyles 4a,b,c containing TEMPO is a proof for stable piperidiloxide radicals in the macromolecules. The obtained nitroxide-containing copolymers of α-Methylstyrene, N-vinylpirrolidone and Acrylic acid are spin labelled compound.
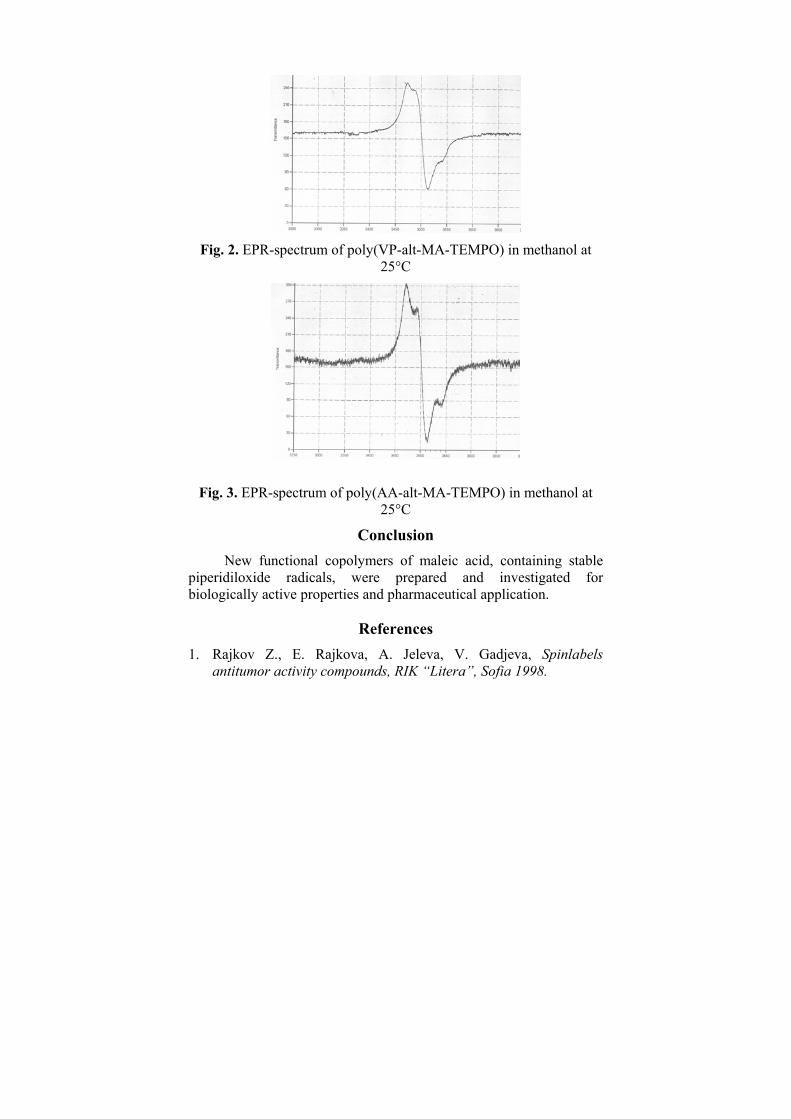
Fig. 2. EPR-spectrum of poly(VP-alt-MA-TEMPO) in methanol at
25°C
Fig. 3. EPR-spectrum of poly(AA-alt-MA-TEMPO) in methanol at 25°C
Conclusion New functional copolymers of maleic acid, containing stable
piperidiloxide radicals, were prepared and investigated for biologically active properties and pharmaceutical application.
References
1. Rajkov Z., E. Rajkova, A. Jeleva, V. Gadjeva, Spinlabels antitumor activity compounds, RIK “Litera”, Sofia 1998.

2. Rajkov Z. Z., K. Vassilev, G. Grigorova, A. Lyapova, A. Alexiev, G. Petrov Pharmazie, 2008, 63, 61.
3. Gőttinger H. A., V. E. Zubarev, O. Brede, J. Chem. Soc. Perkin Trans. 1997, 2 ,2167.
4. Lebedev O. L., S. N. Kazanovski, Zhur. Obshch. Khim. 1960, 30, 1631.
5. Legros J. -P., D. De Caro, H. Casselas, Acta Cryst. 2007, E 63. 6. Udassin R., J. Haskel, A. Samuni, GUT, 1998, 42, 623-627. 7. Cochman A., J. Skolimovski, L. Gebicka, D. Metodieva, Pol. J.
Pharmacol. 2003, 55, 389-400. 8. Ghosh, Polymer News, 1988, 13, 71-77. 9. Hirano T., W. Klesse, H. Ringsdorf, Makromol. Chem., 1979,
180, 1125-1131. 10. Zhu Z., L. Shi, J. Huang, Bioorganic & Medicinal Chem. Lett.,
2002, 12, 2843-2846. 11. Konsulov V. B., in Polymeric Materials Encyclopedia (Ed. J. C.
Salamone): Maleimide Copolymers (N-substituted), CRC Press, Boca Raton, New York, London, Tokyo, Vol. 6, 1996, 3996-3413.
12. Maeda H., Advanced Drug Delivery Reviews, 2001, 46, 165-185. 13. Maeda H., T. Sawa, T. Konno, J. Controlled Release 2001, 74,
47-61. 14. Konsulov V. B., A. Lyapova, G. Petrov, B. Eremieva, P. Saha, J.
Univ. Chem. Technol. Met., 2008, 43, 297-302.

Synthesis and Characterizations of N-substituted Maleimides
V. B. Konsulov, A. A. Lyapova, Z. S. Grozeva, J. I. Tacheva Department of Organic Chemistry and Technology
Faculty of Natural Sciences Konstantin Preslavsky University of Shumen
115 Universitetska Str., 9712 Shumen, Bulgaria E-Mail: [email protected]
Abstract: N-substituted maleimides (RMI) are functional monomers for new polymer synthesis and polymer modification reaction [1]. RMI were synthesized by two step reaction of p-substituted anilines and maleic anhydride (MA) in the presence of acetic anhydride and sodium acetate. The structure of RMI was determined by the 1H, 13C NMR and FT-IR spectroscopy.
Keywords: N-substituted maleimides, monomers, polymer synthesis, polymer modification reaction, p-substituted anilines, maleic anhydride.
Introduction The synthesis of differently structured N-substituted maleimides
(RMI) [1,2] are realized by Searle´s [3] and Ivanov´s [4] methods mainly in interaction with primary aliphatic and aromatic amines with maleic anhydride (MA). Kretov and Kulchitskaya [5] stu-died the influence of substituent in the phenyl ring on the acetyleniza-tion and determined that the reaction speed depends on the amine basis. A dehydrocyclization of the heating of solution in the presence of a dehydrating agents (e.g. NR3, thionylchloride, chloroformiates, acid chlorides, dicyclohexylcarbodiimide, PCl3, P2O5, etc.) have been tried. Using Searle´s sodium acetate – acetic anhydride usually gives a good RMI yield.
Yang and Wang [6] determined during an imidization study that an intermediate product is constituted at the second stage- anhydride of maleamic acid (MAA). Optimal result have been reached with a molecular proportion MAA: CH3COONa=1.0 : 0.1. The acetate ion

acts as catalyst separating a proton from MAA and forming the maleamic ion. It intergoes a nucleophilic attack and contributes to formation the anhydride:
C=OO=C
CHHC
NH
R
O-
CH3COO -(CH3CO)2O
O
O=C CH3
C=OO=C
CHHC
NH
R
CH3COOH CH3COOH
R
HC CH
O=C C=ON
+
R
OHNH
HC CH
O=C C=O
The cyclization of MAA flows increases with increasing in
temperature. The N-substituted maleimides are known to homopolymerize
both free-radically and anionically to give low-molecular weights polymers.[7] The copolymerization of N-phenylmaleimide with vinyl monomers (styrene, vinyl acetate and methyl methacrylate) using free radical initiators have been reported in the past.[8-11] New functional maleimide (polyanions) copolymers was synthesized via the copolymerization N-(4-carboxyplenyl)maleimide with styrene, α-methylstyrene, acrylic acid and methacrylic acid.[11]
The maleimide copolymers are characterized by high thermostability, liquid-cristaline and electro optical properties that used or preparing a resists, selective membranes, polyhelates, in the peptide and organic synthesis.
Results and Discussion
Synthesis and characterization of N-substituted maleimides
The N-substituted maleimides (RMI) were synthesised by two step reaction of aniline derivativies and maleic anhydride (MA) in the presence of acetic anhydride and sodium acetate at 25°C and 95°C [1]:
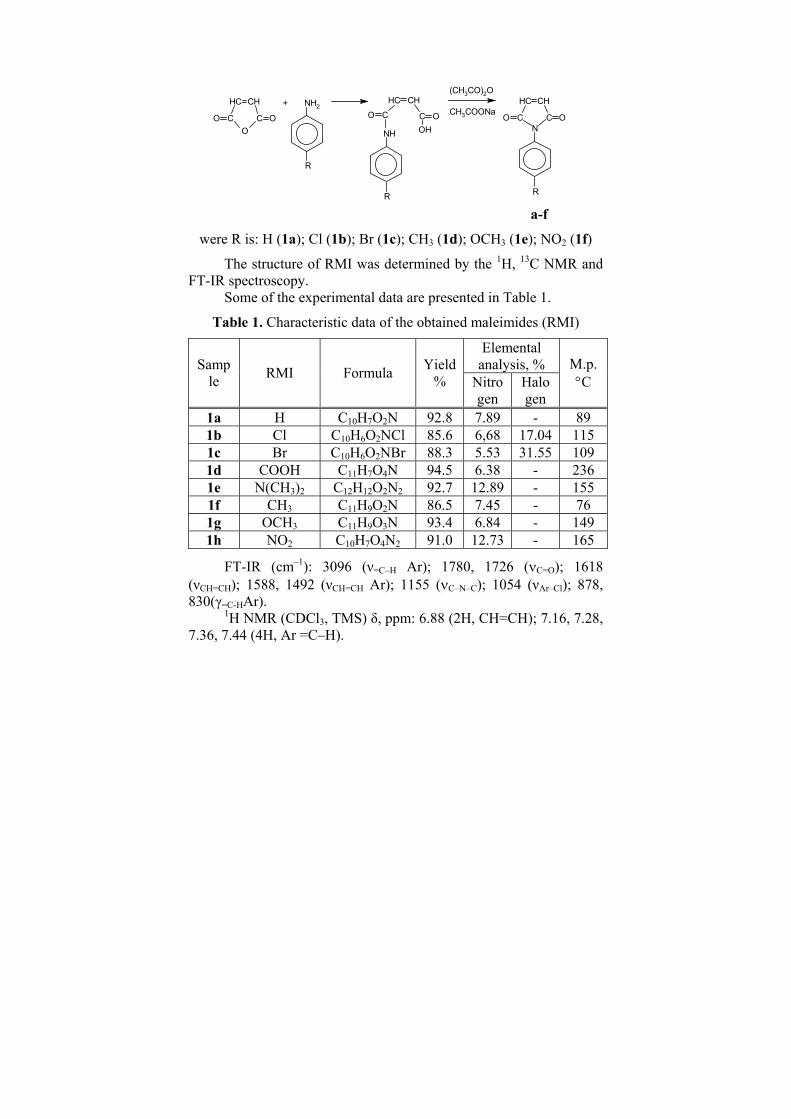
RR
R
NH2CHHC
CN
C OO
(CH3CO)2O
CH3COONa+
CO
HC CH
OHOC
NHO
CHHC
C C OO
a-f
were R is: H (1a); Cl (1b); Br (1c); CH3 (1d); OCH3 (1e); NO2 (1f)
The structure of RMI was determined by the 1H, 13C NMR and FT-IR spectroscopy.
Some of the experimental data are presented in Table 1.
Table 1. Characteristic data of the obtained maleimides (RMI)
Elemental analysis, % Samp
le RMI Formula Yield % Nitro
gen Halogen
M.p. °C
1a H C10H7O2N 92.8 7.89 - 89 1b Cl C10H6O2NCl 85.6 6,68 17.04 115 1c Br C10H6O2NBr 88.3 5.53 31.55 109 1d COOH C11H7O4N 94.5 6.38 - 236 1e N(CH3)2 C12H12O2N2 92.7 12.89 - 155 1f CH3 C11H9O2N 86.5 7.45 - 76 1g OCH3 C11H9O3N 93.4 6.84 - 149 1h NO2 C10H7O4N2 91.0 12.73 - 165
FT-IR (cm–1): 3096 (ν=C–H Ar); 1780, 1726 (νC=O); 1618 (νCH=CH); 1588, 1492 (νCH=CH Ar); 1155 (νC–N–C); 1054 (νAr–Cl); 878, 830(γ=C-HAr).
1H NMR (CDCl3, TMS) δ, ppm: 6.88 (2H, CH=CH); 7.16, 7.28, 7.36, 7.44 (4H, Ar =C–H).
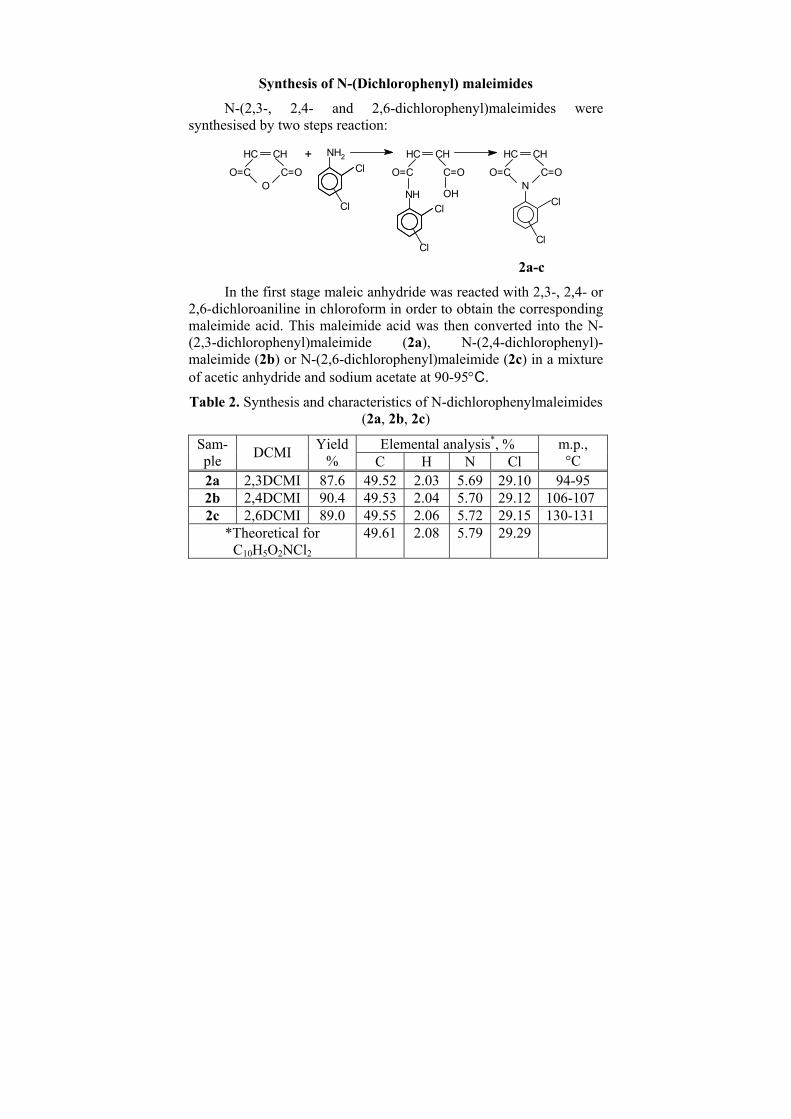
Synthesis of N-(Dichlorophenyl) maleimides
N-(2,3-, 2,4- and 2,6-dichlorophenyl)maleimides were synthesised by two steps reaction:
HC CH
O=C C=OO
+
NH OH
HC CH
O=C C=ON
Cl
Cl
C=OO=C
CHHC
Cl
ClCl
ClNH2
2a-c
In the first stage maleic anhydride was reacted with 2,3-, 2,4- or 2,6-dichloroaniline in chloroform in order to obtain the corresponding maleimide acid. This maleimide acid was then converted into the N-(2,3-dichlorophenyl)maleimide (2a), N-(2,4-dichlorophenyl)-maleimide (2b) or N-(2,6-dichlorophenyl)maleimide (2c) in a mixture of acetic anhydride and sodium acetate at 90-95°C.
Table 2. Synthesis and characteristics of N-dichlorophenylmaleimides (2a, 2b, 2c)
Elemental analysis*, % Sam-ple DCMI Yield
% C H N Cl m.p., °C
2a 2,3DCMI 87.6 49.52 2.03 5.69 29.10 94-95 2b 2,4DCMI 90.4 49.53 2.04 5.70 29.12 106-107 2c 2,6DCMI 89.0 49.55 2.06 5.72 29.15 130-131
*Theoretical for C10H5O2NCl2
49.61 2.08 5.79 29.29
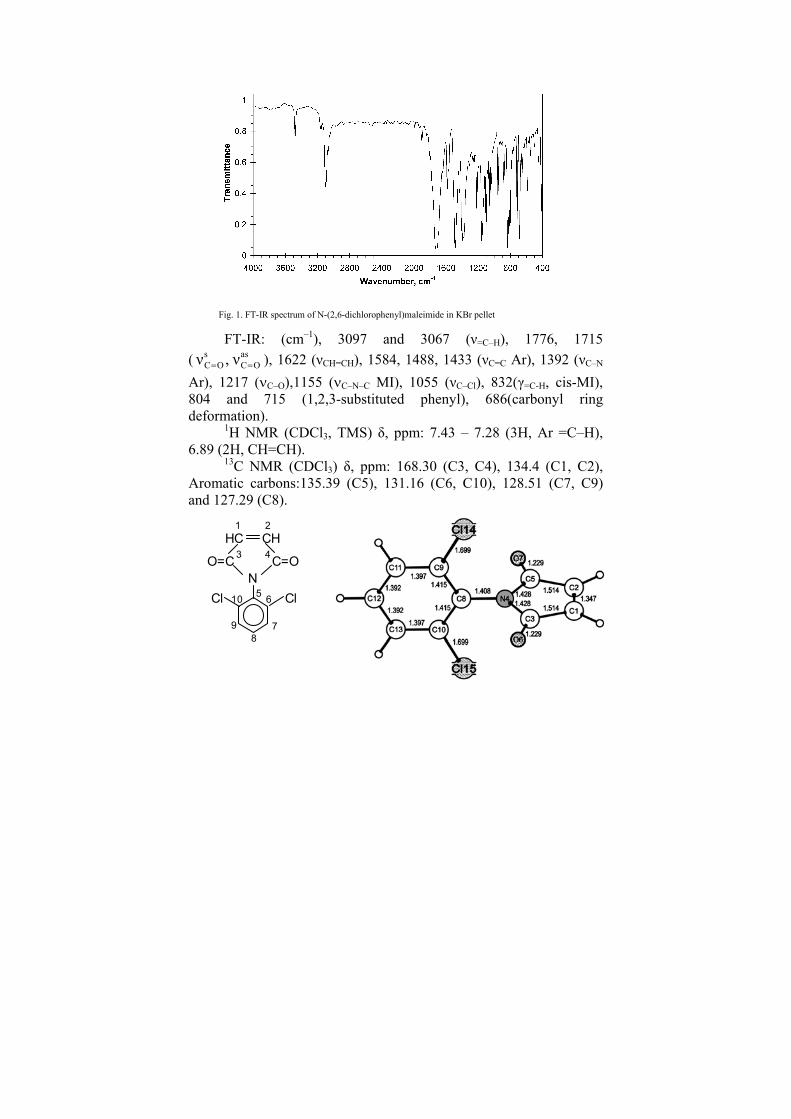
Fig. 1. FT-IR spectrum of N-(2,6-dichlorophenyl)maleimide in KBr pellet FT-IR: (cm–1), 3097 and 3067 (ν=C–H), 1776, 1715
( asOC
sOC ν ,ν == ), 1622 (νCH=CH), 1584, 1488, 1433 (νC=C Ar), 1392 (νC–N
Ar), 1217 (νC–O),1155 (νC–N–C MI), 1055 (νC–Cl), 832(γ=C-H, cis-MI), 804 and 715 (1,2,3-substituted phenyl), 686(carbonyl ring deformation).
1H NMR (CDCl3, TMS) δ, ppm: 7.43 – 7.28 (3H, Ar =C–H), 6.89 (2H, CH=CH).
13C NMR (CDCl3) δ, ppm: 168.30 (C3, C4), 134.4 (C1, C2), Aromatic carbons:135.39 (C5), 131.16 (C6, C10), 128.51 (C7, C9) and 127.29 (C8).
1
3
2
4
5 6
78
9
10
HC CH
O=C C=ON
ClCl
Synthesis of new functional N-substituted maleimides
The new functional (N-substituted maleimides) maleimide monomers N-(2-methoxy-4-nitrophenyl)maleimide, N-(4-methoxy-phenyl)maleimide, N-(4-nitrophenyl)maleimide, N-(2-nitro-4-methyl-phenyl)maleimide, N-(2,4-dinitrophenyl)maleimide, N-(4-carboxy-phenyl)maleimide and N-(4-N´,N´-dimethylaminophenyl)maleimide were synthesized (Table 3 ) by the reaction:
HC CH
O=C C=O
HC CH
O=C C=OO
+ NH2R1
R2
HC CH
O=C C=ON
(CH3CO)2O
CH3COONaHC CH
O=C C=O
HC CH
O=C C=O
R1
R2
NH OHR1
R2
3a-g
Table 3. Characteristic data of the obtained maleimides (3a-g)
N-substituted maleimides Sam-ple R1 R2
Formula % N
found (calc.)
M.w m.p °C
3a -OCH3 -NO2 C11H8O5N2 11.04
(11.28) 248.2 146
3b -H -OCH3 C11H9O3N2 6.78
(6.92) 203.2 136
3c -H -NO2 C10H6O4N2 12.52
(12.84) 218.1 162
3d -NO2 -CH3 C11H8O4N2 11.96
(12.06) 232.2 98
3e -NO2 -NO2 C10H6O6N3 15.82
(15.91) 278.2 193
3f -H -COOH C11H7O4N 6.38 (6.45) 217.2 228
3g -H -N(CH3) C12H12O2N212.78
(12.96) 216.1 155
Table 4. Spectral characteristics of RMI (3a,b,d)
IR, cm-1 1H NMR, ppm Sample cm-1 bond δ, ppm H
3a
1618 1710 1500 1340 1258 1376 1460 800 820 880
-HC=CH- >C=O -NO2
-OCH3
-Ar
6.84
3.45
7.18 7.32
-HC=CH-
-OCH3
Ar =CH-
3b
1608 1700 1240 1378 1456 838 940
-HC=CH- >C=O
-OCH3
-Ar
6.71
3.78
7.14 7.24
-OCH3
Ar =CH-
3d
1608 1700 1340 1510 850 940
-HC=CH- >C=O -NO2
-Ar
6.9
7.54 7.65 8.36 8.48
-HC=CH-
Ar =CH-
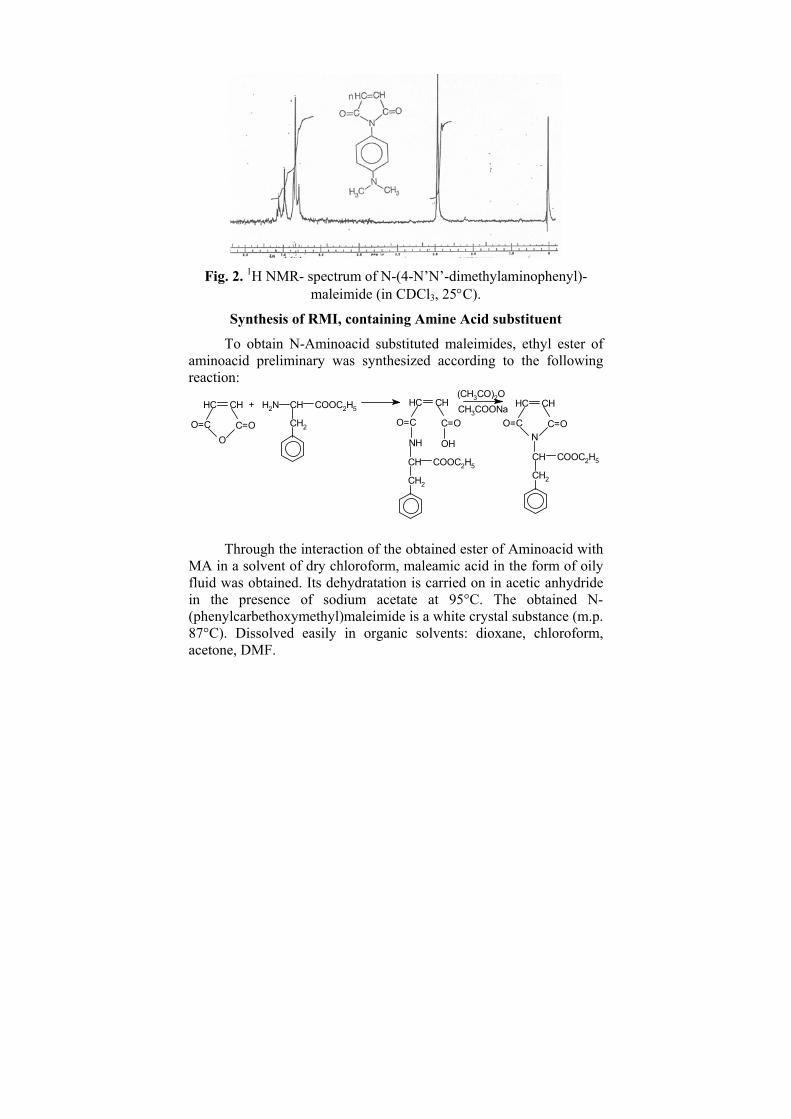
Fig. 2. 1H NMR- spectrum of N-(4-N’N’-dimethylaminophenyl)-
maleimide (in CDCl3, 25°C).
Synthesis of RMI, containing Amine Acid substituent
To obtain N-Aminoacid substituted maleimides, ethyl ester of aminoacid preliminary was synthesized according to the following reaction:
NC=OO=C
CHHC
OHNH
C=OO=C
CHHC(CH3CO)2OCH3COONaCHH2N COOC2H5
CH2
+
C=OO=C
CHHC
O
CH COOC2H5
CH2
CH COOC2H5
CH2
Through the interaction of the obtained ester of Aminoacid with
MA in a solvent of dry chloroform, maleamic acid in the form of oily fluid was obtained. Its dehydratation is carried on in acetic anhydride in the presence of sodium acetate at 95°C. The obtained N-(phenylcarbethoxymethyl)maleimide is a white crystal substance (m.p. 87°C). Dissolved easily in organic solvents: dioxane, chloroform, acetone, DMF.

References 1. V. Konsulov, Z. Grozeva, IUPAC 32-nd Int. Symp. On
Macromolecules MACRO´88, Kyoto, Japan, 1988, p. 680. 2. V. B. Konsulov, in Polymeric Materials Encyclopedia (Ed. J. C.
Salamone): Maleimide copolymers (N-substituted), 1996, v. 6, p 3996-4013. CRC Press, Boca Raton, New York, London, Tokyo.
3. N. E. Searle, US Pat. 2 444 536, 1948. 4. V. S. Ivanov, USSR Pat. 166 832, 1967. 5. D. Kretov, N. E. Kulchiskaya, J. Gen. Chem., 1955, 25, 2363. 6. C. -P. Yang, S. -S. Wang, J. Polym. Sci.: Polym. Chem. Ed., 1989,
A 27, 15. 7. R. C. Cubbon, Polymer, 1965, 6, 419. 8. M. Barales-Rienda, J. I. G. De La Campa, I. Gonzales Ramos, J.
Macromol. Sci. Chem. 1977, A11, 267. 9. V. B. Konsulov, Z. S. Grozeva, T. Kelen, J. Borbely, Makromol.
Chem. 1992, 193, 847. 10. Tz. Godjevargova, R. Nenkova, V. Konsulov, J. Mol. Cat. B:
Enzimatic, 2006, 38, 59. 11. V. Konsulov, D. Diakova, A. Lyapova, Z. Grozeva, Bulg. Chem.
Commun., 2007, 39, 72.

Изследване влиянието на антипенители и противоутаители върху някои характеристики
на акрилатна вододисперсионна боя В. Грозев*, М. Иванова**, З. Грозева*, П. Миндова**, А. Патлеева*, Й. Тачева*
*Факултет по природни науки Шуменски университет “Епископ Константин Преславски” Шумен 9712, Университетска 115,Е-Mail: [email protected]
**Оргахим АД, бул. "Трети Март" № 21, 7000 Русе
Abstract: Paints are complex, multi-component systems. Their properties are determined not only by their building components, but also by the interaction between them. Therefore, for a complete study of the characteristics, multi-factorial experiments are most suitable. The results presented, are inferred, by the use of experimental modelling and analyses of the influence of the concentration of the antifoaming and antisettling agents, on the quality of the coating and layering of the latex paints. The so derived mathematical models could be applied to the optimisation of the paint composition, by using various criteria.
Keywords: latex paint, antifoaming agent, antisettling agent, mathematical model, optimisation.
Въведение Фирмите-производители на адитиви за бои постоянно
разработват и предлагат на пазара нови продукти, включително антипенители и противоутаители. В техническите описания се препоръчват ориентировъчни минимална и максимална стойности на тяхната концентрация в боите. Това налага производителите на бои непрекъснато да актуализират рецептурите си с оглед подобряване на техните експлоатационни характеристики и минимизиране разходите за производство. Липсват количествени зависимости, които описват процесите на филмообразуване и устойчивост на боите, както и техните

реологични характеристики. Поради това изследванията за използване на нови антипенители и противоутаители като компоненти на вододисперсионна боя трябва да се извършат по експериментален път. Най-подходящ метод за намиране на оптимален състав на боята при различна концентрация на антипенители и противоутаители е експерименталното моделиране и оптимизация [1] на получения математичен модел. Негови основни преимущества са:
- получаване на богата информация за характеристиките на боята при различни видове и концентрации на антипенители и противоутаители при минимален брой експерименти;
- възможност за намиране на оптимален състав на боите при различни критерии на оптимизация.
Въпреки безспорните предимства на метода той намира ограничено приложение за разработка на лакобояджийски материали. В [2] чрез дробен факторен експеримент е намерен оптимален състав на боя за маркировка на пътища по предварително зададени характеристики. Чрез моделиране и оптимизация [3] е решен проблемът за недостатъчната устойчивост на автомобилна боя в производствени условия
Целта на разработката е чрез математични модели да се изследва влиянието на антипенители и противоутаители върху качеството на покритието и разслояването на вододисперсионна боя при изкуствено стареене. Експериментална част и планиране на експеримента
Приготвяне на акрилатната боя
Използваните компоненти са основна бяла паста за акрилатна боя, вода, противоутаител, антипенител, акрилатна дисперсия, фин пълнител, коалесценти. Получаването се извършва в дисолвер – метален съд, снабден с бъркалка с регулируеми обороти. Към водата, дозирана в дисолвера, при разбъркване, се добавят последователно пастата и антипенителят. Сместа се хомогенизира 10-15 минути и се добавят акрилатната дисперсия и финият пълнител. Сместа се хомогенизира отново, добавят се коалесцентите, сгъстителят, противоутаителят и се разбърква добре в продължение на 15-30 минути, докато

нанесена върху стъкло капка от сместа не образува повече кратери (петна от мазни капки).
Определяне на качеството на покритието
За определяне качеството на покритието проби боя се нанасят върху стандартни подложки при стандартни условия със стандартни инструменти. Според регламентиращите документи качеството на покритието (външен вид на филма) се оценява визуално. По такъв начин не може да се получи количествена оценка за качеството на покритието, необходима за решаване на оптимизационната задача. Затова е приложен подход, при който качеството на покритието се оценява визуално по десетобална система. Бал 0 получават пробите с най-ниско качество (голям брой кратери, стичания, неравности и др. забележими дефекти), бал 10 получават пробите с идеален, равен и гладък филм. В зависимост от броя и големината на забележимите дефекти на покритието, пробите могат да получат оценка от 0, 1, 2 и т.н. до 10 бала. По такъв начин всички проби от проведените експерименти бяха разделени на 10 групи вследствие оценката на 5 наблюдатели ( I група, оценка за качеството на покритието от 0 до 1 бала, II група – от 1 до 2 бала, III група – от 2 до 3 бала, ... X група – от 9 до 10 бала). Във всяка група оценката за качеството на покритието се прецезира до първия знак след десетичната запетая, като се използва компютърната програма ImageJ. След сканиране на боядисаните подложки програмата дава възможност да се отчетат броя на кратерите и тяхната площ.
Определяне на разслояването на боята при изкуствено стареене
Проби от получената акрилатна боя се поставят в сушилен шкаф при 50оС за един месец. След изваждането им всяка проба се окачествява визуално за разслояване и наличие на утайка. Дебелината на разслояването се измерва в cm.
Планиране на експеримента
Във всички изследвани проби количеството на пигментната паста, акрилатната дисперсия, коалесцента и сгъстителя е
еднакво, променят се само видът и концентрациите на антипенителя и противоутаителя. Между изследваните антипенители и противоутаители са възможни следните комбинации:
- комбинация 1 (К1) – акрилатна боя с антипенител 1 (А1) и противоутаител 1 (П1);
- комбинация 2 (К2) - акрилатна боя с антипенител 2 (А2) и противоутаител 1 (П1);
- комбинация 3 (К3) - акрилатна боя с антипенител 2 (А2) и противоутаител 2 (П2);
- комбинация 4 (К4) - акрилатна боя с антипенител 1 (А1) и противоутаител 2 (П2).
За описание на изследваните зависимости избирамe математични модели от втора степен:
y=b0+b1.x1+b2.x2+b12.x1.x2+b11.x2+b22.x22 (1) където: y – критерии за оптимизация, който може да бъде оценка качеството на покритието в балове или разслояване при изкуствено стареене, cm; x1, – управляващ фактор концентрация на антипенителите, кодирани стойности; x2 – управляващ фактор концентрация на противо-утаителите, кодирани стойности; b0, b1, b2, b12, b11, b22 - регресионни коефициенти.
Таблица 1. Натурални и кодирани стойности на независимите променливи
Фактори
Основ-но
ниво, Xi = 0
Интер-вал
варира-не, λi
Долно ниво,
Xi = -1
Горно ниво,
Xi = 1
Kонцентрация на А1, % 0,8 0,7 0,1 1,5 Kонцентрация на А2, % 0,55 0,45 0,1 1 Kонцентрация на П1 % 1,25 0,75 0,5 2 Kонцентрация на П2, % 0,75 0,25 0,5 1
Резултати от експеримента. Математически модели
При брой на факторите m = 2 оптималният композиционен план [1] и резултатите от експериментите са представени в таблица 2.
Таблица 2. Оптимален композиционен план и резултати от експеримента
Кодирани стойности X1 X2
Качество на покритието, бала
Разслояване, cm
Комбинация 1 -1 -1 9,75 2,0 1 -1 2,5 1,0 -1 1 9,5 0 1 1 8,6 0,2 -1 0 9,0 0,5 0 -1 3,0 1,5 1 0 2,75 0,5 0 1 9,25 0,2 0 0 3,4 0,5
Комбинация 2 -1 -1 4,0 2,0 1 -1 0 2,0 -1 1 3,2 0,2 1 1 3,6 0,2 -1 0 1,5 0,5 0 -1 2,2 2,5 1 0 1,0 0,5 0 1 3,8 0,2 0 0 2,0 0,5
Комбинация 3 -1 -1 0,5 0 1 -1 8,2 0 -1 1 7,0 0 1 1 4,5 0 -1 0 5,0 0 0 -1 6,0 0 1 0 8,8 0 0 1 5,7 0 0 0 5,2 9
Комбинация 4 -1 -1 8,4 0,2 1 -1 8,0 0,1 -1 1 4,3 0 1 1 7,4 0 -1 0 6,5 0 0 -1 7,6 0,1 1 0 7,8 0 0 1 4,8 0 0 0 5,5 0
За определяне на математичните модели (уравнение1) са
изчислени стойностите на регресионните коефициенти b по методика, описана в [1]. Данните са представени в таблица 3.
Оценка за съвпадението между предсказаните по модела и опитно определени стойности на величините може да се извърши чрез корелационния коефициент на Пирсън. Всички неговите стойности са по – високи от 0,90 (таблица 3), което показва много силно изразена положителна линейна корелация. Следователно представените математични модели описват с висока точност изследваните зависимости.
Таблица 3. Регресионни коефициенти и коефициенти на
детерминация R2
Комбинация Коефициенти К1 (A1,
П1) К2 (A2, П1)
К3(A2, П2)
К4 (A1, П2)
Качество на покритието, бала b0 3,8499 1,8111 6,3499 5,8611 b1 -2,3999 -0,6833 1,4999 0,6749 b2 2,0166 0,7249 0,4249 -1,2666 b12 1,5875 1,1000 -2,5500 0,8875 b11 1,8000 -0,4666 0 1,1083 b22 2,0500 1,3083 -1,0249 0,1333 R2 0,94 0,9600 0,9100 0,98
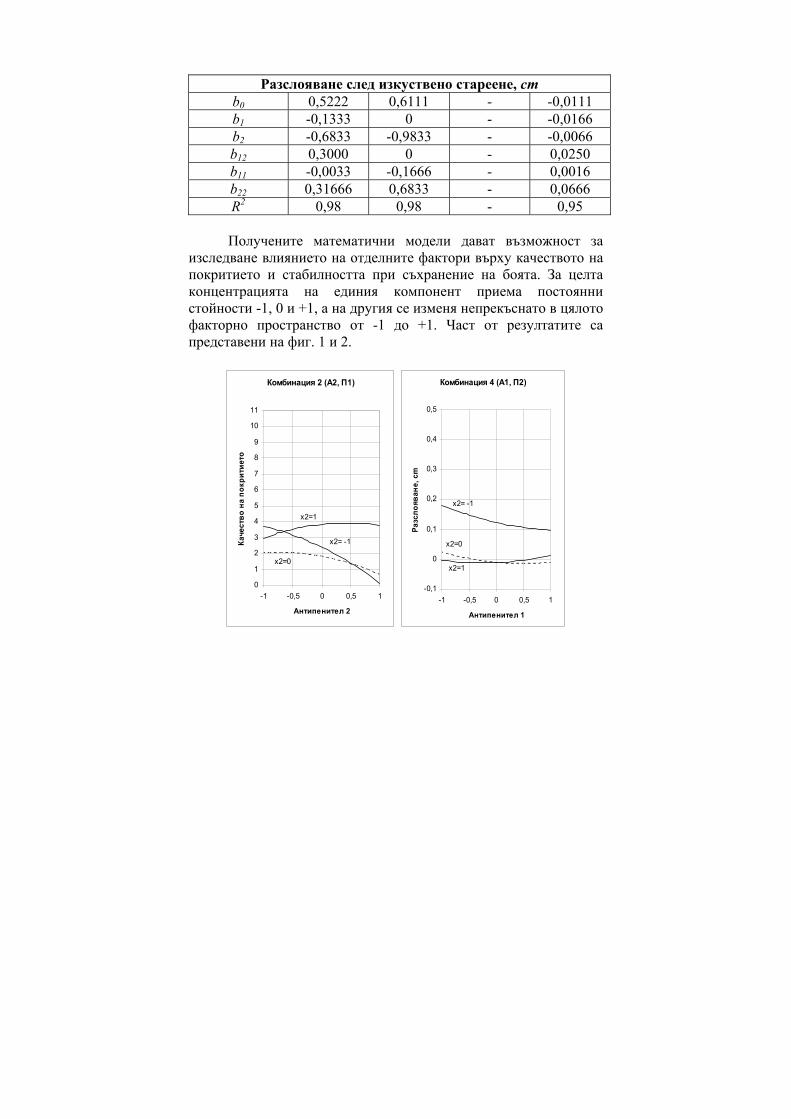
Разслояване след изкуствено стареене, cm b0 0,5222 0,6111 - -0,0111 b1 -0,1333 0 - -0,0166 b2 -0,6833 -0,9833 - -0,0066 b12 0,3000 0 - 0,0250 b11 -0,0033 -0,1666 - 0,0016 b22 0,31666 0,6833 - 0,0666 R2 0,98 0,98 - 0,95
Получените математични модели дават възможност за
изследване влиянието на отделните фактори върху качеството на покритието и стабилността при съхранение на боята. За целта концентрацията на единия компонент приема постоянни стойности -1, 0 и +1, а на другия се изменя непрекъснато в цялото факторно пространство от -1 до +1. Част от резултатите са представени на фиг. 1 и 2.
Комбинация 4 (А1, П2)
-0,1
0
0,1
0,2
0,3
0,4
0,5
-1 -0,5 0 0,5 1
Антипенител 1
Разсло
яван
е, c
m
х2= -1
х2=0
х2=1
Комбинация 2 (А2, П1)
0
1
2
3
4
5
6
7
8
9
10
11
-1 -0,5 0 0,5 1
Антипенител 2
Качество
на по
критие
то
х2=1
х2=0
х2= -1
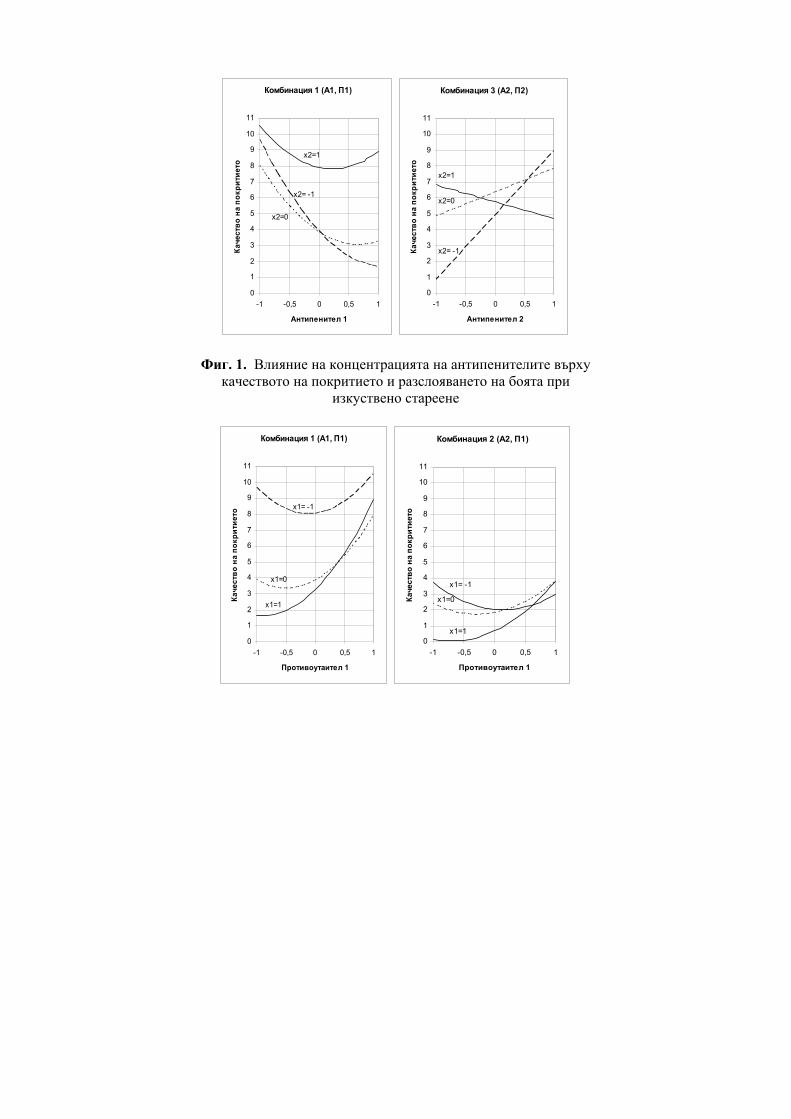
Фиг. 1. Влияние на концентрацията на антипенителите върху качеството на покритието и разслояването на боята при
изкуствено стареене
Комбинация 2 (А2, П1)
0
1
2
3
4
5
6
7
8
9
10
11
-1 -0,5 0 0,5 1
Противоутаител 1
Качество
на по
критие
то
х1= -1
х1=1
х1=0
Комбинация 1 (А1, П1)
0
1
2
3
4
5
6
7
8
9
10
11
-1 -0,5 0 0,5 1
Противоутаител 1
Качество
на по
критие
то
х1= -1
х1=0
х1=1
Комбинация 3 (А2, П2)
0
1
2
3
4
5
6
7
8
9
10
11
-1 -0,5 0 0,5 1
Антипенител 2
Качество
на по
критие
то
х2= -1
х2=0
х2=1
Комбинация 1 (А1, П1)
0
1
2
3
4
5
6
7
8
9
10
11
-1 -0,5 0 0,5 1
Антипенител 1
Качество
на по
критие
то
x2=1
x2= -1
x2=0
Фиг. 2. Влияние на концентрацията на противоутаителите върху качеството на покритието и разслояването на боята при
изкуствено стареене
Изводи От получените математични модели и построените графики
могат да се направят следните изводи: 1. Антипенителите са основен фактор, от който зависи качеството на покритията. Комбинация К1 достига максимално качество 10 бала при минимална концентрация на А1 и максимална концентрация на П1. Стойност на качеството на покритието от 9 бала се постига с използване на висока концентрация А2 при ниски концентрации на П2 (комбинация 3). В цялото факторно пространство на комбинацията А2 - П1 качеството на покритието не отговаря на изискванията на стандарта. 2. При промяна на концентрацията на П1 от минимална до максимална стойност разслояването се променя около 1,8 cm без значение на вида на антипенителя. Липса на разслояване при
Комбинация 4 (А1, П2)
-0,1
0
0,1
0,2
0,3
0,4
0,5
-1 -0,5 0 0,5 1
Противоутаител 2
Разсло
яван
е, c
m
х1= -1
х1=0
х1=1
Комбинация 2 (А2, П1)
0
0,5
1
1,5
2
2,5
3
-1 -0,5 0 0,5 1
Противоутаител 1
Разсло
яван
е, c
m
х1= -1х1=1
х1=0

комбинация 1 се получава при високи концентрации на П1 и ниски концентрации на антипенителите. 3. Присъствието на П2 във всички комбинации определя разслояване под 0,2 mm, което отговаря на вътрешните норми за вододисперсионните бои. Качеството на покритието 8,5 – 8,8 бала се получава само при ниски концентрации на този противоутаител.
4. Получените високи стойности на коефициента b12 показват силно изразено взаимно влияние между някои фактори: А1 и П1 (комбинация 1), А2 и П2 (комбинация 2) върху качеството на покритието, както А1 и П1 върху разслояването на боята при изкуствено стареене.
Литература 1. Вучков И., С. Стоянов, Математическо моделиране и
оптимизация на технологични обекти, Техника, София, 1986. 2. Fatemi S., M. Khakbaz, Z. Ranjbar., S. Bastani, Progress in
Organic Coatings, 2006, 55, 337-344. 3. Gupta A., Journal of Applied Statistics, 2001, 28, 199-213.

Проблеми на контрола и управлението на химични вещества в училищата
М. Стоянова*, В. Грозев** *РИОСВ, ул. “Съединение”№ 71, ет. 3, 9700 Шумен
**Шуменски университет “Епископ Константин Преславски”, ул. “Университетска”№ 115, 9712 Шумен
Abstract: The unfit for use chemical substances and compounds from school’s chemical practical lessons, are classified as dangerous waste. The school’s governing bodies could deal with this problem, by temporarily storing them for up to three years; treating them; storage (i.e. in BB-cubes) and sequestration. The issue of managing such chemical substances, should be dealt in a centralized manner for all schools in a municipality, with the help of subsidiary financial resources.
Keywords: chemicals, substances, compounds, schools, management.
Въведение Европейското законодателство обръща сериозно внимание
на контрола и управлението на химичните вещества и препарати. На 01. 06. 2007 година влиза в сила регламент REACH (ЕС 1907/2006) [1], отнасящ се до регистрация, оценка, разрешаване и ограничаване на химическите вещества, произвеждани, използвани или внасяни над един тон годишно. Целта на REACH e по-добро опазване на човешкото здраве и околната среда.
В българските училища съществуват кабинети по химия, в които се съхраняват химични вещества в неголеми киличества. Много от тях са опасни, други са с изтекъл срок на годност или не могат да бъдат идентифицирани поради липса на етикети. Констатирани са случаи, при които провеждането на учебни занятия и съхранението на лабораторните реактиви се извършва в едно и също помещениe. През 2008 година в град Търговище избухва пожар в кабинет по химия, при който възниква реална опасност от обгазяване. Този инцидент още веднъж показва, че натрупаните в училищата проблеми със залежалите химични

вещества по отношение на тяхното съхранение, контрол, превенция на инциденти и опазване здравето на ученици и учители трябва да се реши.
Подобни проблеми съществуват и пред малките неакредитирани лаборатории в предприятия и други институции.
Целта на настоящата разработка е да предложи подход за управление на химичните вещества и препарати в училищните кабинети по химия съобразно законовата рамка, регламентираща задълженията по отношение на тяхното съхранение и употреба.
Законова рамка Правата и задълженията на лицата, които съхраняват и
употребяват химичните вещества и препарати, се уреждат със Закона за защита от вредното въздействие на химичните вещества и препарати (ЗЗВВХВП) [2] с цел защита на човешкото здраве и опазване на околната среда.
В допълнителните разпоредби на ЗЗВВХВП се дефинират понятията:
- химични вещества - химични елементи и техните съединения в естествено състояние или получени чрез производствен процес, който включва и добавки, необходими за стабилизация на продуктите, и примеси, възникнали при използвания производствен процес, но изключва всеки разтворител, който може да бъде отделен, без това да повлияе на стабилността на веществото или да промени неговият състав;
- препарати са смеси или разтвори, съставени от две или повече химични вещества.
Опасните химични вещества и препарати са категоризирани в 15 групи като: експлозивни, запалими, токсични, дразнещи, канцерогенни и др.
Съгласно Наредбата за реда и начина на класифициране, опаковане и етикетиране на химични вещества и препарати [3] опаковката на опасните химични вещества и препарати трябва да отговаря на определени изисквания. Тя трябва да бъде конструирана по начин, който не позволява разпиляване на съдържанието и да не взаимодейства със съхраняваните в нея химични вещества. Голяма част от реактивите, съхранявани в
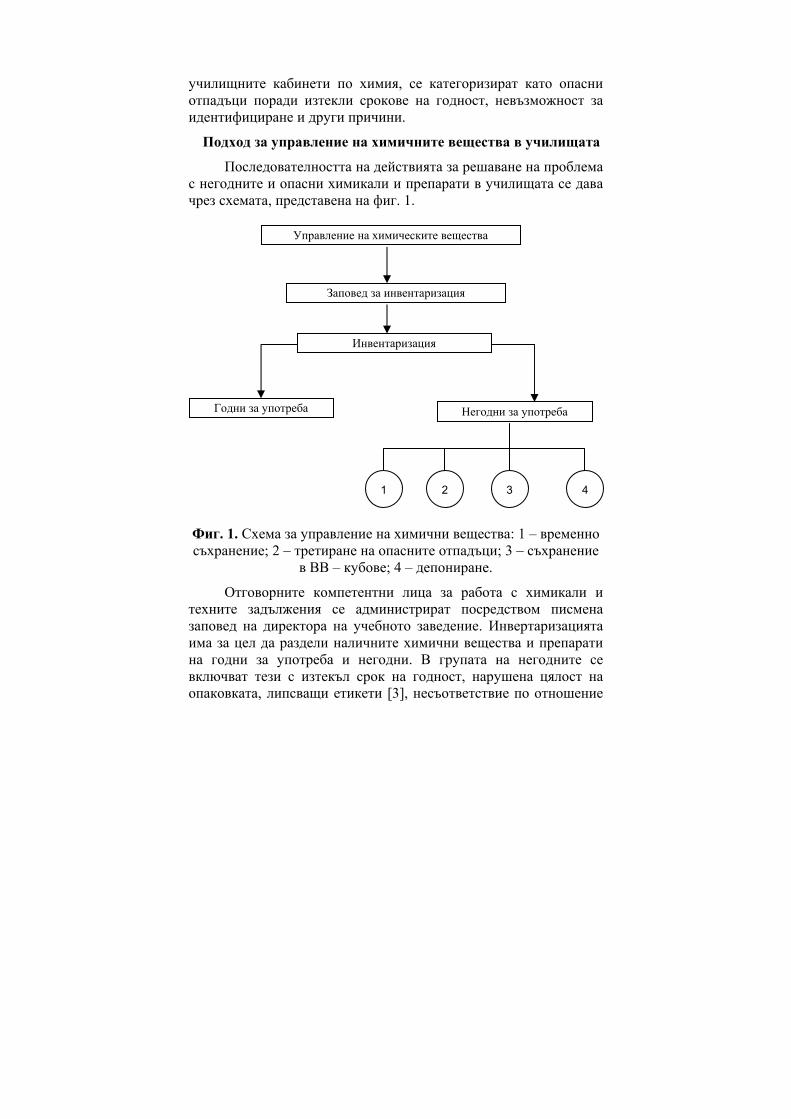
училищните кабинети по химия, се категоризират като опасни отпадъци поради изтекли срокове на годност, невъзможност за идентифициране и други причини.
Подход за управление на химичните вещества в училищата
Последователността на действията за решаване на проблема с негодните и опасни химикали и препарати в училищата се дава чрез схемата, представена на фиг. 1.
Фиг. 1. Схема за управление на химични вещества: 1 – временно съхранение; 2 – третиране на опасните отпадъци; 3 – съхранение
в ВВ – кубове; 4 – депониране.
Отговорните компетентни лица за работа с химикали и техните задължения се администрират посредством писмена заповед на директора на учебното заведение. Инвертаризацията има за цел да раздели наличните химични вещества и препарати на годни за употреба и негодни. В групата на негодните се включват тези с изтекъл срок на годност, нарушена цялост на опаковката, липсващи етикети [3], несъответствие по отношение
Управление на химическите вещества
Заповед за инвентаризация
Инвентаризация
Годни за употреба Негодни за употреба
21 3 4

на физичните и химическите характеристики. При съмнение за годност се препоръчва анализ от акредитирана лаборатория. В общия случай негодните за употреба химични вещества и препарати се класифицират като опасни отпадъци и се третират съгласно Закона за управление на отпадъците (ЗУО) [4]. Според този закон отпадъците се определят като собственост на училището и от това произтичат редица задължения и отговорности. Училищните ръководства имат четири разрешени от закона възможности за решавне на проблема:
1. Временно съхранение на опасните отпадъци до три години. Съгласно ЗУО се извършва разделно в специализирани съдове на територията, върху която притежателят упражнява вещно право.
2. Третиране на опасните отпадъци. Обработката им може да се извършва само от лица, притежаващи разрешение по член 37 от ЗУО или комплексно разрешително съгласно Закона за опазване на околната среда [5].
3. Разполагане на негодните химични вещества и препарати в ВВ-кубове, аналогично на негодните пестициди.
4. Депониране на ненужните химикали в специализирани депа за опасни отпадъци. В последните два случая трябва да се търси съдействието на
общинската управа, тъй като съгласно ЗУО кметът на общината е отговорен за управлението на дейностите по отпадъците, образувани на съответната общинска територия (чл. 16 (1)).
Заключение
1. Според действащото законодателство негодните за употреба химични вещества и препарати са опасни отпадъци и са ангажимент на техния собственик. Възможностите на училищните ръководства за справяне с проблема са временно съхранение до три години, третиране, съхранение в BB – кубове и депониране.
2. Законосъобразното освобождаване от опасните химични вещества е свързано с необходимост от финансов ресурс, с който училищата обикновено не разполагат. Количествата на опасните химикали са незначителни. Поради това проблемът за управление

на химичните вещества следва да се решава централизирано за всички училища в общините с привличане на допълнителни средства.
Литература 1. Регламент (EО) 1907/2006 на Европейския Парламент
относно регистрацията, оценката, разрешаването и ограничаването на химикали (REACH).
2. Закон за защита от вредното въздействие на химичните вещества и препарати / ДВ бр. 10/2000 и изм./.
3. Закон за управлението на отпадъците /ДВ бр.86/2003 и изм./.
4. Наредба за реда и начина на класифициране, опаковане и етикетиране на химични вещества и препарати / ДВ бр. 86/ 2003 и изм./
5. Закон за опазване на околната среда /ДВ бр.91/2002 и изм./.

Тестване на програми за белтъчна електро-статика върху база данни за НМФК
Светлана Христова, Борис Атанасов Институт по физикохимия акад. Ростислав Каишев (ИФХ) БАН
1113 София, ул. Акад. Г. Бончев, бл. 11 Е-Mail: [email protected]
Институт по органична химия с център по фитохимия, БАН 1113 София, ул. Акад. Г. Бончев, бл.9
E-Mail: [email protected]
Резюме: Белтъчната електростатика е мощен способ за изучаване на структурните и функционални свойства на белтъчните молекули. Особен интерес представлява взаимодействието на белтъчни молекули с добре описани електростатични свойства (напр. цитохром с) със струкрурно-дефинирани колоидни частици (напр. от бентонит). Необходимо е да се знае достоверността на предсказаните рН-зависими свойства. Затова цел на това изследване е да се направи количествена оценка на стойностите на зряд-зависими величини генерирани с програмата PHEPS и детайлно сравнени с съшите получени с програмата МССЕ базирана на общоприетата DELPHI. Като обекти на изследването бяха избрани нуклеозид монофосфат кинази (НМФК) от E. coli, поради наличието на база от данни за тях (BANMOKI), където са описани стойностите на тяхните рКа получени с програмата МССЕ. Направеният сравнителен анализ показва, че двете програми генерират съпоставими резултати, но програмата PHEPS претендира за по-галяма точност и достоверност поради отчитането на рН-зависимите свойства на белтъчните молекули.
Ключови думи: рКа, РНЕPS, МССЕ, NMPK.
Увод pKa-стойностите на йоногенните групи са измеряема
количествена мярка на протонния афинитет (ΔGоРА = 2.3RTpKa),

и дават значителна информация за биофизичните свойства на белтъците.[1,2] Промяната на pKa-стойностите на една йонизационна група в зависимост от съответната й стойност в модела, показва приноса на този остатък в белтъчната стабилност [3]. Измененията на йонизационните състояния на титруемите остатъци, индуцирани при дадена реакция, довеждат до свързване/освобождаване на протон и така могат да бъдат наблюдавани чрез промените на съответните pKa стойности.[4] Изчисленията на pKa дават сведения за механизма на ензимните реакции и помагат за охарактеризирането на активния център.[5] Някои йонизационни групи показват нестандартни титрувални криви и това се използва, за да се предскажат функционално-важни остатъци.[6] Може да се заключи, че един йоногенен аминокиселинен остатък, който има необичайно титрувално поведение свързано с друга титруема група от каталитичния център е също част от него.[7,8,9]
Особен интерес представлява взаимодействието на белтъчни молекули с добре описани електростатични свойства (напр. цитохром с) със струкрурно-дефинирани колоидни частици (напр. от бентонит). Необходимо е да се знае достоверността на предсказаните рН-зависими свойства на адсорбирания протеин, за да се предсказват свойствата на протеин-натоварените частици. Затова целта на нашето изследване е да се тестват детайлно два рзлични подхода: PHEPS и.МССЕ. Програмата МССЕ (Multi Conformation Continuum Electrostatics) комбинира континуална електростатика (от популярната DELPHI) с молекулна динамика и физическа статистика (Болтцманова с Мoнте-Карло). Пресметнатите стойности на рКа по МССЕ, са направени при фиксирани рН около 7.0. Известно е, че стойностите на рКа са рН-зависими, защото ΔGо
РА,i са pH-зависими (брой и конфигурация на молекулния мултипол). Именно това предизвика нашият интерес, тези стойности да бъдат пресметната чрез програма, която отчита влиянието на pH (каквато е PHEPS [10] прилагана на повече от 100 протеина), върху електростатичните свойства на МНФK от E.coli, за която разполагаме с широка база от данни (BANMOKI). Обшото при тези програмни пакети е, че те работят в

макроскопско приближение (водата като солвент се описва като континуална диелектрична среда), ползва се реалната молекулна структура с фиксирани атомни детайли и два типа заряди: (i) постоянни (рН-независими) парциални товари (par) и (ii) протон-свързващи места с рН-зависими протонни заряди (tit). Главното различие между PHEPS и МССЕ е, че в пърата границата между белтъка и разтворителя се описва чрез нормирана за всеки i-ти атом статична дoстъпност (SAi), докато при втората се ползва «мрежовия подход» на метода на крайните разлики. В двата метода се ползват експериментално измерените стойности на рКа от от прости модели (напр. стойностите на рКа на N-ацетиламид амидите на съответните йоногенни аминокиселинхи остатъци) се приема за собствено рКmod,i и се отчита работата, която е необходима за пренасянето на заряд от високо полярния воден разтвор (εw=80) къмто белтъчната макромолекула (4 εp,i 40) – т.н.Борнова енергия. Във PHEPS Борновия член, пропорционален на [1-( εp,i/ εw)], е грубо оценен като Wij(1-‹SAij›). И двата пакета приемат парциални заряди от програмите AMBER и PARSE. Отношението между йоногенните аминокиселинни радикали и общия брой аминокиселинни радикали rel=Nion/Ntot e относително високo за НМФК, които са с малък радиус (Rp), така че взаимодействето между всяка i-та и j-та йоногенна група се изчислява като сума от заряд-зарядовите, заряд-диполните и дипол-диполните взаимодействия.
Материали и методи
Изходните файлове са получени от сървера Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do) и от сървер BANMOKI (http://www.ces.clemson.edu/compbio/banmoki). Файловете са обработени частично без да се променя белтъчната структура, като са премахнати всички водни молекули. Използваните изчислителни програми за белтъчна електростатика са PHEPS (лабораторен пакет, разработен в ИОХЦФ-БАН) http://pheps.orgchm.bas.bg/home.html и МССЕ (самостоятелен и сърверен вариант) от лаб. на проф. М. Гуннер (Университет на Ню Йорк, САЩ) при съдействието на проф. Е. Алексов, САЩ) (www.ces.clemson.edu/compbio/databases/kinases).
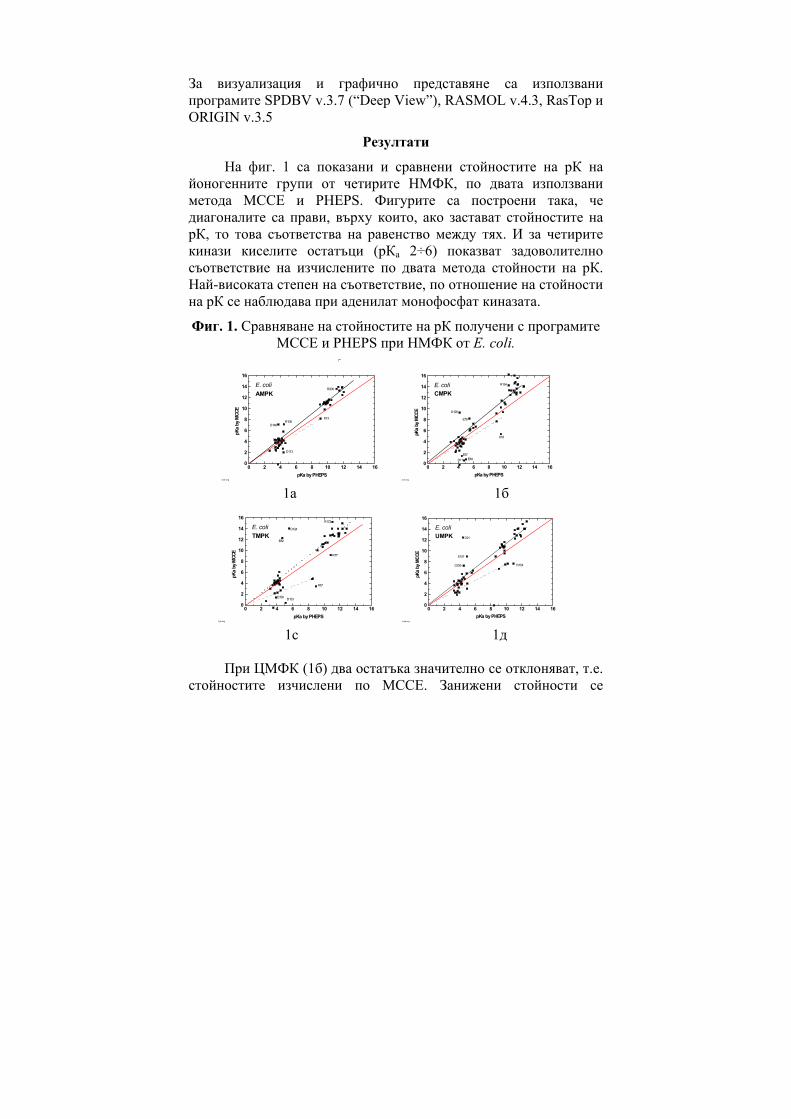
За визуализация и графично представяне са използвани програмите SPDBV v.3.7 (“Deep View”), RASMOL v.4.3, RasTop и ORIGIN v.3.5
Резултати
На фиг. 1 са показани и сравнени стойностите на рК на йоногенните групи от четирите НМФК, по двата използвани метода МССЕ и РНЕРS. Фигурите са построени така, че диагоналите са прави, върху които, ако застават стойностите на рК, то това съответства на равенство между тях. И за четирите кинази киселите остатъци (рКа 2÷6) показват задоволително съответствие на изчислените по двата метода стойности на рК. Най-високата степен на съответствие, по отношение на стойности на рК се наблюдава при аденилат монофосфат киназата.
Фиг. 1. Сравняване на стойностите на рК получени с програмите МССЕ и РНЕРS при НМФК от Е. coli.
0 2 4 6 8 10 12 14 160
2
4
6
8
10
12
14
16
K13
R206
D113
E108D146
a-pk.org
E. coliAMPK
pKa
by M
CCE
pKa by PHEPS0 2 4 6 8 10 12 14 16
0
2
4
6
8
10
12
14
16
K18
E83D11E57
R154
E79
D129
c-pk.org
E. coliCMPK
pKa
by M
CCE
pKa by PHEPS 1а 1б
0 2 4 6 8 10 12 14 160
2
4
6
8
10
12
14
16
E159 D133
K67
R77
R172
D124
E82
t-pk.org
E. coliTMPK
pKa
by M
CCE
pKa by PHEPS 0 2 4 6 8 10 12 14 16
0
2
4
6
8
10
12
14
16
D21
R154D235
E157
U-pk.org
E. coliUMPK
pKa
by M
CCE
pKa by PHEPS 1с 1д
При ЦМФК (1б) два остатъка значително се отклоняват, т.е.
стойностите изчислени по МССЕ. Занижени стойности се

наблюдават при остатъците Glu57, Asp11 и Glu83. Тези отклонения се засилват при ТМФК, където Asp124 и Glu82 имат аномално високи стойности на рК изчислени по МССЕ, в сравнение с PHEPS. Аналогична е картината и при УМФК, където МССЕ дава завишени стойности на рКа на остатъците: Asp21 и Asp235. В областта на максимална населеност на данните, отклонението е с около ±1,5 рКа единици. За базичните групи най-голямо съвпадение на стойностите на рКа изчислени по двата метода има при АМФК (най-добри структури!). Най-силно отклонение се наблюдава при Arg206. При ЦМФК силно нараства различието в рКа стойностите изчислени по МССЕ и РНЕРS. Така например, при остатък Lys18 отклонението спрямо средното е от 3 рК единици, а за точките около Arg154 стойностите са завишени с 2 рКа единици при МССЕ. При ТМФК, остатъкът Lys67 има аномално ниски стойности на рКа в сравнение с очакваното 9. При УMФК, с изключение на няколко АК остатъка, повечето групи показват завишени стойности на рКа.
Представените на Фиг.1 корелационни графики позволяват да се сравнят по прецизност на изчисляване на рКа два много различни метода: МССЕ и PHEPS. Те са реализация на два различни подхода, така че несъвпаденията в оценките на рКа-та е по-разбираемо от доброто съответствие. Сравнението е напълно коректно, защото рКа-та са подбрани за eдин и същи обект (ензим), от един и същи източник (E. coli). Най-добро е съответствието при апо-АМФК и то намалява в реда на влошаване качеството на ренгеновите структури. Това налага да се отделят две различни причини на несъответствия: прецизност на изчислителните методи и прецизност на структурите подложени на анализ. Така налюдаваме понижаване на прецизността в реда:
АМФК > ЦМФК > УМФК > ТМФК Прави впечатление, че данните се групират в по два набора
за всеки тип групи (кисели и базични). Двата набора имат тенденция да лежат върху прави различни от “ъглополвящата” – киселите групи с тенденция върху права с по-малък наклон (“преоценка” по PHEPS), а базичните групи с тенденция върху

права с по-голям наклон (“преоценка” по МССЕ). Това организиране по знак на товарите (зарядите) подсказва различие в оцека на десолватационните енергии по двата метода – тези енергии са леко недооценени във PHEPS-процедурата, а са леко преоценени в МССЕ-процедурата. Изпадането от корелационните прави подсказва силни различия в локални структурни фактори, които се третират по различен начин в двете програми. Може да приемем, че съответствието между тях е задоволително.
Изводи
Показано е, че два електростатични метода (МССЕ и PHEPS) са сравними по оценка на локалните рКа-та, като вторият има предимство в получаване на пълните рН-зависимости на всички функции и претендира за по-висока бързина, достъпност и по-добри оценки. Това дава надежда той да бъде успешно ползван за изследване белтъчната електростатика в по-сложни системи.
Литература 1. Honig B. & Nicholls A., Science, (1995), 268:1144-1149. 2. Warshel, A. & Papazyan, A., Curr Opin Struct Biol., (1998) 8:
211-217. 3. Langsetmo, K., Fuchs, J.A., Woodward, C. & Sharp, K.A.,
Biochemistry, (1991) 30: 7609-7614. 4. Alexov, E. & Gunner, M., Biochemistry, (1999), 38: 8253-8270. 5. Nielsen, J.E. & McCammon, J.A., Protein Science, (2003) 12:
1894-1901. 6. Ondrechen, M.J., Clifton, J.G. & Ringe D., Proc Natl Acad Sci U
S A., (2001) 98: 12473-12478. 7. Shehadi, I.A., Abyzov, A., Uzun, A., Wei, Y., Murga, L.F., Ilyin,
V.& Ondrechen, M.J., Bioinform Comput Biol., (2005) 3: 127-143. 8. Ko, J., Murga, L.F., Andre, P., Yang, H., Ondrechen, M.J.,
Williams, R.J., Agunwamba, A. & Budil, D.E., Proteins, (2005) 59: 183-195.
9. Shehadi, I.A., Yang, H. & Ondrechen, M. J., Mol Biol Rep., (2002) 29: 329-335.
10. Kantardjiev, A & Atanasov, B., Nucleic Acid Research, (2006) 34: 43-47.

Surface Modification of γ-Alumina with Chlorotrimethylsilane.
1. Solid and Gas Products of Silation
Stefan Slavov Faculty of Natural Sciences
Konstantin Preslavsky University of Shumen 115 Universitetska Str., 9712 Shumen
E-mail: [email protected] Abstract: The solid and gaseous products from silation of partially dehydroxylated γ-alumina with chlorotrimethylsilane have been determined for a wide range of pretreatment and reaction conditions. Unexpectedly and contrary to previous assumption, in all cases there was no HCl produced. Methane was the only gaseous product detected for silation reactions up to 500oC. Under very severe conditions a very small amount of higher hydrocarbons were also obtained. A minimum of 1 mol of CH4 is produced per mol of (CH3)3SiCl reacted, and all chlorine remains firmly bound to the silated alumina.
Key words: γ-alumina, (CH3)3SiCl, dehydroxylation, surface modification.
Introduction Alumina and silica are widely used in analytical and separation
techniques and as support for metallic, organometallic, and metal oxide catalysts. Alumina continues to be the focus of several investigations not only as a catalysts support, but also because the nature of its surface is complex and remains incompletely understood.[1] Alumina possesses a defect spinel lattice structure, which is determined by surface hydroxyl groups.[2,3] Numerous investigations have been conducted with the objective of modifying the surface in a controlled manner. The principal approach has been to utilize or modify the surface hydroxyl groups which play an important role in determining the surface chemistry of the resulting alumina based materials. The removal or modification of surface hydroxyls can

result in an increase in the hydrophobic nature of the catalysts, especially its capability to resist adsorption of water vapor.
Organosilanes have been used effectively as modifying agents to achieve the target characteristics of the substrate surface.[4,5] Since the abundance of surface hydroxyl groups and the necessity of their removal or modification is a common feature of the most widely used catalyst supports Al2O3 and SiO2, the application of organosilanes as modifying agents attains wider significance. Effective modification of the surface depends in general in two factors: the structure of an organosilicon compound and the type of surface hydroxyl groups.
The surface hydroxyl groups fall into several mutually connected classifications in accordance with different criteria and properties.[6,7] Some hydroxyls are described as “isolated”, and when two (or more) hydroxyl groups are adjacent, a hydrogen bond is assumed to exist between them, and these are termed “associated” hydroxyl groups. Regarding their ability to be removed - approximately 67% of hydroxyl groups are described as removable without defect formation, 25% - removable with “defects”, and the rest 8% - unremovable (or removable with migration). The coordination number, net electrical charge, and pH character determine whether hydroxyls are effectively negative, neutral, or positive surface sites.
Infrared spectroscopic measurements[4,8] indicated that an irreversible chemical reaction occurred at an adsorption temperature equal to or greater than about 450 K, between silane and the surface hydroxyl groups, and that strongly bound aluminasiloxane surface species (CH3)3Si-O-Al= were formed. Aluminasiloxane compounds were produced as the hydroxyl groups were consumed. At these temperatures the reaction involves the surface hydroxyls and it assumed that HCl was eliminated. The fate of this HCl was not fully understood; it was proposed that it was either liberated as a gas or adsorbed on the alumina surface.
There exist some commonly found features which characterize all results reported to date concerning silation of alumina. Very thorough and detailed studies have been made only for the changes of the solid phase. In all discussion a complete analogy in the behavior of alumina and silica has been assumed, without any recognition of the

significant specific differences in the chemical and surface nature of the two materials. A priory acceptance has been made of the generally of the reaction between chlorosilanes and the surface hydroxyl groups with elimination of HCl which assumption has unfortunately resulted in a lack of interest in the gas phase products of the reaction.
The purpose of the present work is to study the silation of γ-alumina surface with (CH3)3SiCl, paying special attention to carrying out the reaction in dynamic conditions - in a continuous flow fixed bed reactor that allows us to use larger amount of alumina and (CH3)3SiCl, permanent control of the gas phase products of the reaction, and variation of the reaction parameters over a wide range. To our knowledge this is the first examination of γ-alumina modification with (CH3)3SiCl under dynamic conditions, aiming at characterization of both the solid and gaseous reaction products.
Experimental Equipment. The dynamic method was used to study the silation
of γ-alumina with (CH3)3SiCl. Experiments were performed in equipment (Figure 1) consisting of three parts: a gas handling system, a reactor, and an exit control and monitoring system. For the gas handling system the flow rates and exact proportions of the gases were adjustable.

The velocity of the nitrogen stream was regulated by means of two parallel connected mass flow controllers. The concentration of (CH3)3SiCl in the nitrogen-silane gas mixture was controlled through the equilibrium vapor pressure of silane at 0oC by use of an ice bath and checked by weight measurement. The reaction vessel was a glass U-shaped reactor (100 cm3) equipped with temperature programmed heating. A system of four-way and three-way valves provided for the distribution and removal of the gas-phase reaction products. The reactor was placed in a movable electrical furnace for which the temperature was controlled within ±1oC. The temperature profile along the length of the reactor bed was monitored by means of movable thermocouples. Concurrent analyses of gas phase products using gas chromatography and Fourier transform IR were performed.
Materials. Commercially available granulated alumina (La Roche) was used as received. The supplier reported a BET area of 325 m2 g-1 and the size of granules as 1-3 mm. Fumed alumina (Alon) with BET area 100.4 m2g-1 was also used for comparison purposes. The alumina was first dried at 120oC for 3 h and then dehydrated by heating in air at 500oC for 6 h, followed by storage in an oven at 125oC. (CH3)3SiCl (Aldrich, purity >99.0%) was double distilled, followed by subsequent vacuum distillation. It was stored under a N2 atmosphere and transferred to a glass evaporator purged constantly with N2 gas during use. Both N2 and He (Ultra high purity) were obtained in high pressure cylinders.
Results and discussion 1. Gaseous products of the silation reaction. Since earlier
investigations had led to the conclusion (or assumption) that HCl was a product of the silation of alumina, but its fate remained unknown, analyses of the gas phase products were included. However, all of our attempts to establish the formation of HCl were unsuccessful. The major gaseous product of the reaction of (CH3)3SiCl with the surface of partially dehydroxylated γ-alumina is methane, CH4, as determined by infrared and NMR spectroscopy, mass spectrometry, and GC analyses.
Methane is the only detectable gas phase product produced over the course of the reaction.[1] There was no significant difference

between the infrared spectra of the gaseous products taken at 10 min interval throughout the course of the reaction until silane was detected in the exit gases. In each spectrum there were no bands arising from HCl. In the spectral range 2800-3200 cm-1 the bands of CH4 and HCl are partially superimposed.
Comparative analyses were performed using GC. For each sample taken the only gaseous product detected was methane. The silation reaction was terminated when (CH3)3SiCl was identified in the exit gases by detection of a GC peak with the same retention time as that for (CH3)3SiCl taken from the inlet of the reactor. The GC MS spectra of exit gases for a variety of conditions showed only peaks arising from methane in the carrier gas.
The presence of methane was also confirmed by analyses using NMR. The separated gas phase methane from the reactor outlet was collected and dissolved in two different solutions and the results of analyses in each solvent showed the presence of only methane and for high temperatures (300-500oC) reactions (vide infra) trace amounts of higher hydrocarbons.
The absence of detectable amounts of HCl in the gas phase product was further established by means of sensitive qualitative contact reactions with pH paper and a fresh solution of NH4OH. The eliminated methane was captured at liquid nitrogen temperature in a glass trap, quantified and identified as above.
2. Solid phase studies. After dehydroxylation of alumina in the temperature range 400-500oC, the concentration of isolated OH groups left on the surface is between 6.4 x 1014 and 8.2 x 1014 OH groups/cm2, based on DTG analyses. The number of hydroxyls on partially dehydroxylated alumina depends on the source, its chemical purity, BET area, and the experimental conditions for dehydroxylation.
The degree of silation for either granulated or pulverized alumina was the same. Decrease in the BET area (7-13%) and pore volume (16-20%), as well as the amount of supported silane on the surface of granulated or pulverized alumina, were essentially the same for each sample. The peak value of surface concentration of (CH3)3SiCl absorbed or partially dehydroxylated alumina was determined gravimetrically. Depending on the silation temperature, a
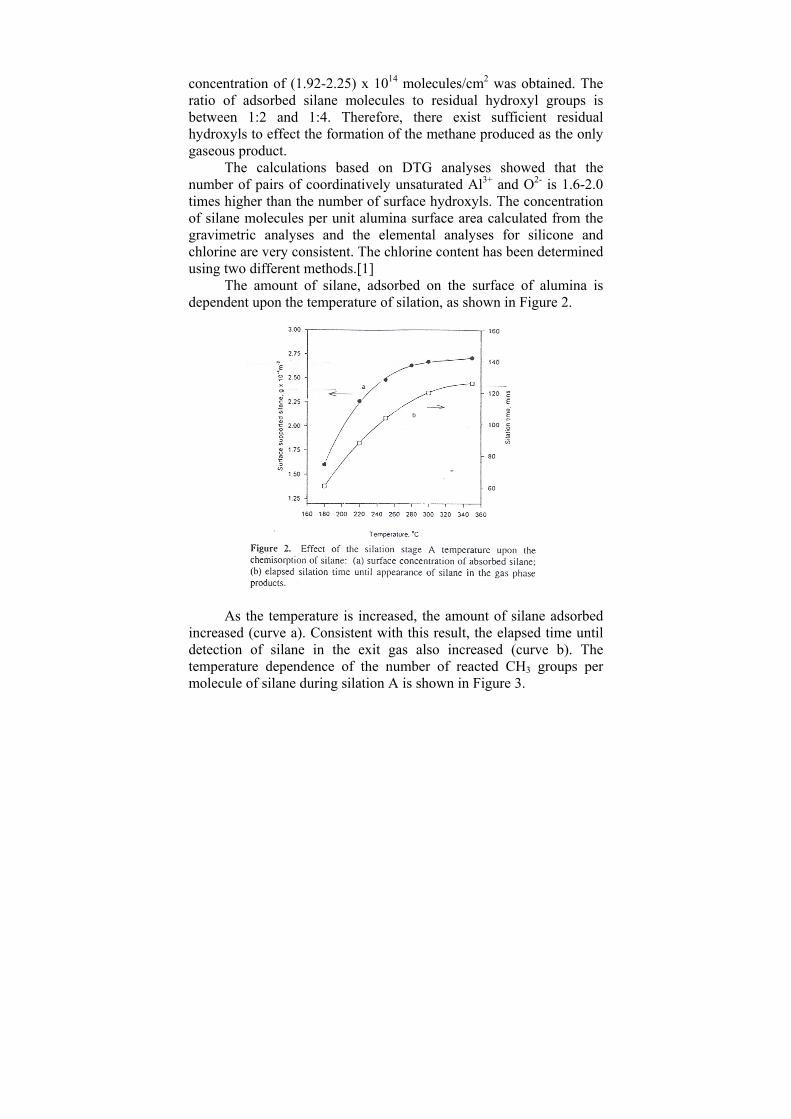
concentration of (1.92-2.25) x 1014 molecules/cm2 was obtained. The ratio of adsorbed silane molecules to residual hydroxyl groups is between 1:2 and 1:4. Therefore, there exist sufficient residual hydroxyls to effect the formation of the methane produced as the only gaseous product.
The calculations based on DTG analyses showed that the number of pairs of coordinatively unsaturated Al3+ and O2- is 1.6-2.0 times higher than the number of surface hydroxyls. The concentration of silane molecules per unit alumina surface area calculated from the gravimetric analyses and the elemental analyses for silicone and chlorine are very consistent. The chlorine content has been determined using two different methods.[1]
The amount of silane, adsorbed on the surface of alumina is dependent upon the temperature of silation, as shown in Figure 2.
As the temperature is increased, the amount of silane adsorbed
increased (curve a). Consistent with this result, the elapsed time until detection of silane in the exit gas also increased (curve b). The temperature dependence of the number of reacted CH3 groups per molecule of silane during silation A is shown in Figure 3.

The number of reacted CH3 groups has been determined by
elemental analyses of carbon. The results of elemental analyses for hydrogen are consistent with the analyses for carbon and show that hydrogen content continuously decreases with increasing temperature. In the temperature range from 140 to 250 oC, the number of reacted CH3 groups per molecule of silane is between 1.0 and 1.8. When the temperature is increased to 300 oC and higher, the ratio of reacted methyl groups per molecule of silane is greater then two. The data show that the number of reacted CH3 groups per molecule of silane is close to 2.5 for samples heated to 500 oC over 3-5 days. In these cases oxidative pyrolysis also probably takes place. The location of silane moiety on the alumina surface could be presented as I for the silation temperature to 160oC and as II for temperatures over 200oC.
The analytical data for the solid products are consistent with the CO coordination reaction. All chloride from the silating agent remains firmly bound to the silated alumina. It is therefore highly probable that the silating agent reacts with available tetrahedral Al3+ sites and the chloride binds irreversibly to these sites.
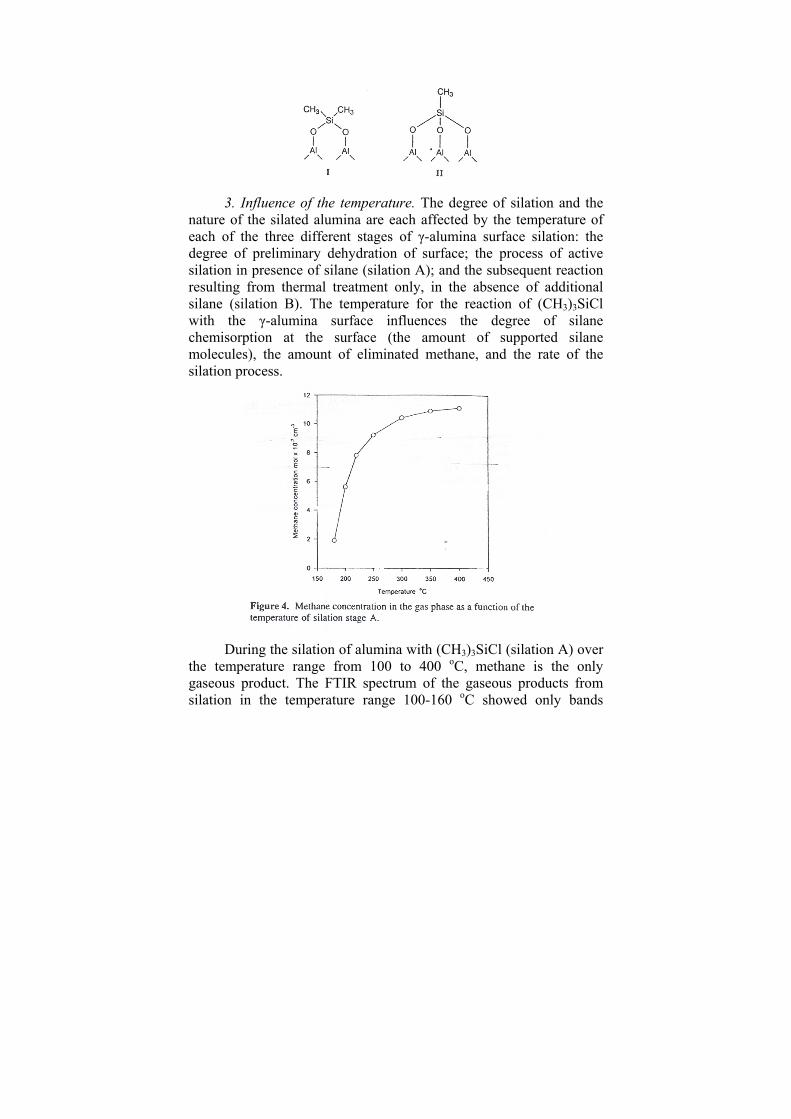
3. Influence of the temperature. The degree of silation and the
nature of the silated alumina are each affected by the temperature of each of the three different stages of γ-alumina surface silation: the degree of preliminary dehydration of surface; the process of active silation in presence of silane (silation A); and the subsequent reaction resulting from thermal treatment only, in the absence of additional silane (silation B). The temperature for the reaction of (CH3)3SiCl with the γ-alumina surface influences the degree of silane chemisorption at the surface (the amount of supported silane molecules), the amount of eliminated methane, and the rate of the silation process.
During the silation of alumina with (CH3)3SiCl (silation A) over
the temperature range from 100 to 400 oC, methane is the only gaseous product. The FTIR spectrum of the gaseous products from silation in the temperature range 100-160 oC showed only bands
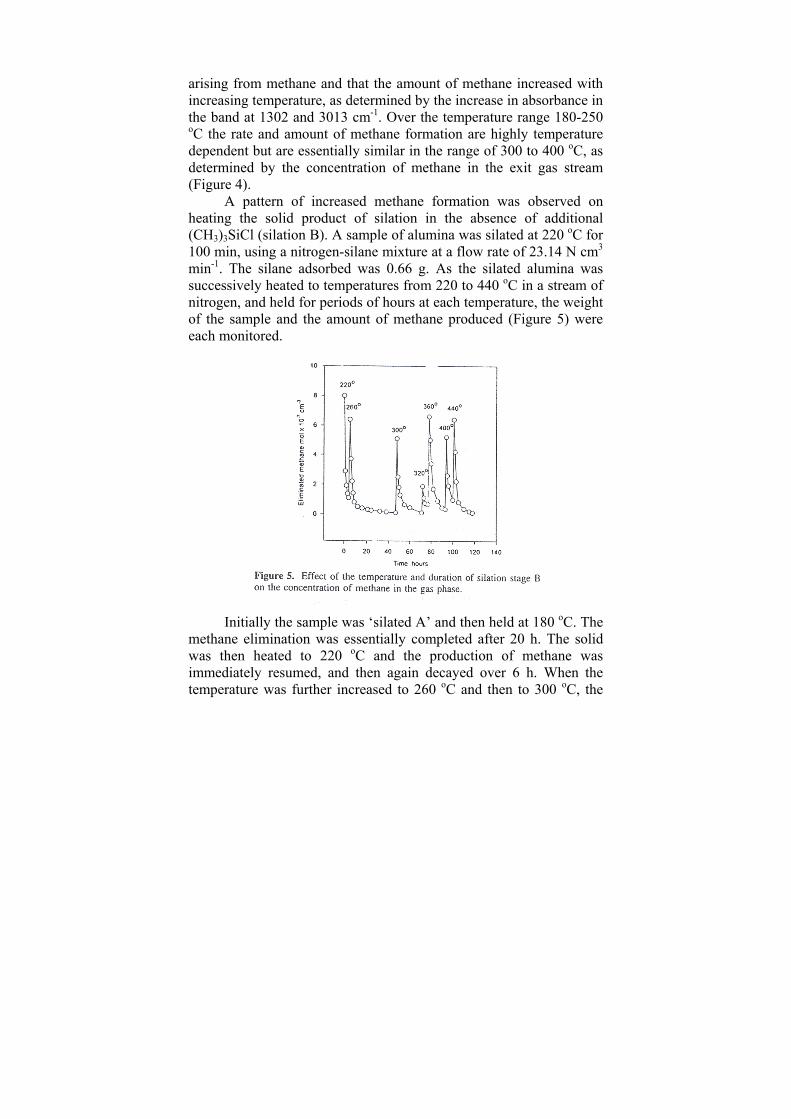
arising from methane and that the amount of methane increased with increasing temperature, as determined by the increase in absorbance in the band at 1302 and 3013 cm-1. Over the temperature range 180-250 oC the rate and amount of methane formation are highly temperature dependent but are essentially similar in the range of 300 to 400 oC, as determined by the concentration of methane in the exit gas stream (Figure 4).
A pattern of increased methane formation was observed on heating the solid product of silation in the absence of additional (CH3)3SiCl (silation B). A sample of alumina was silated at 220 oC for 100 min, using a nitrogen-silane mixture at a flow rate of 23.14 N cm3
min-1. The silane adsorbed was 0.66 g. As the silated alumina was successively heated to temperatures from 220 to 440 oC in a stream of nitrogen, and held for periods of hours at each temperature, the weight of the sample and the amount of methane produced (Figure 5) were each monitored.
Initially the sample was ‘silated A’ and then held at 180 oC. The
methane elimination was essentially completed after 20 h. The solid was then heated to 220 oC and the production of methane was immediately resumed, and then again decayed over 6 h. When the temperature was further increased to 260 oC and then to 300 oC, the

same pattern of behavior was observed. The sample was then successively heated to 400 oC, held for 6 h, and after a further 20 h heated to 440 oC. In each case, an increase in temperature caused further rapid reaction and production of additional methane, but no other gaseous product. The initial increase in methane concentration was similar for each step of 40 oC increase in temperature. The amount of methane produced at each temperature can be determined by integration of the area under the respective peaks. Methane is the only product in the absence of fresh gas phase silane at higher temperatures.
Conclusions The reaction of (CH3)3SiCl with the surface of γ-alumina forms
neither HCl nor pendant -O-Si(CH3)3 groups. Unexpectedly and contrary to previous assumption, in all cases there was no HCl produced. Methane is the only gaseous product from the silation of the surface of γ-alumina using (CH3)3SiCl in a wide temperature range. The silation reaction is strongly affected by the temperature. The degree of silation for either granulated or pulverized alumina was the same. The number of reacted CH3 groups per molecule of silane is between 1.0 and 1.8 for reaction temperatures of 160-240 oC, greater than 2.0 at 300 oC, and close to 2.5 for samples heated to 500 oC. A minimum of 1 mol of CH4 is produced per mol of (CH3)3SiCl reacted, and all chloride remains firmly bound to the silated γ-alumina.
References 1. Slavov, S. V., Chuang K. T., Sanger A. R., Langmuir, 1995, 11,
No 10. 2. Zecchina, A., Otero Arean, C., Catal. Rev. Sc. Eng., 1993, 35,
261. 3. Peri, J. B., J. Phys. Chem. 1966, 70, 3168. 4. Paul D. K., Yates, J. T., J. Phys. Chem. 1991, 95, 1699. 5. Angst, D. L., Simmons, G. W., Langmuir 1991, 7, 2236. 6. Peri, J. B. J. Phys. Chem. 1995, 69, 211. 7. Peri, J. B. J. Phys. Chem. 1995, 69, 220. 8. Paul D. K., Ballinger T. H., Yates, J. T., J. Phys. Chem. 1990, 94,
4617.

Surface Modification of γ-Alumina with Chlorotrimethylsilane
2. Mechanism of Silation Stefan Slavov
Faculty of Natural Sciences Konstantin Preslavsky University of Shumen
115 Universitetska Str., 9712 Shumen E-mail: [email protected]
Abstract: The mechanism of reaction of (CH3)3SiCl with the surface of alumina is a sequential reaction which affords methane as the only gaseous product. The silane is initially chemisorbed at the coordinatively unsaturated aluminum cations and oxygen anions surface sites. This has the effect of modifying the surface by chloride, and the isolated hydroxyl groups are thus made more acidic. The hydroxyl groups the react with CH3-Si groups to eliminate methane and to form bridging silyl moieties.
Key words: mechanism of silation, (CH3)3SiCl, γ-alumina, surface modification.
Introduction The reaction of (CH3)3SiCl with silica is known to occur by
direct reaction with the residual surface hydroxyl groups and consequent elimination of HCl and formation of “umbrella structure” pendant groups: surface-O-Si(CH3)3.[1-4] T he formation of HCl has been determined and quantified by these authors, and our date are in agreement with these results. The formation of HCl by the reaction with silica is both immediate and rapid upon exposure of the silica surface to (CH3)3SiCl.
Paul and co-workers[5,6] have thoroughly studied the silation of Al2O3 and Rh/Al2O3 catalyst for controlling automotive exhaust emissions. They determined[5] that, surprisingly, the silation of the supported catalysts by extensive exposure to (CH3)3SiCl does not result in deactivation of the metallic Rh particles by chlorosilane chemisorption. This finding enabled them to discussed the interaction

of (CH3)3SiCl with surface hydroxyl groups, while disregarding the presence of the small amount of Rh on the alumina surface. Infrared spectroscopic measurements indicated that an irreversible chemical reaction occurred at an adsorption temperature equal to or greater than about 450 K, between the silane and the surface hydroxyls, and that strongly bound aluminasiloxane surface species (CH3)3Si-O-Al= were formed. These authors stated that aluminasiloxane compounds were produced as the hydroxyl groups were consumed. At these temperatures the reaction involved the surface hydroxyl groups, and it was assumed that HCl was eliminated. It was proposed that the reaction being observed involved, for both isolated or associated OH groups, the elimination of gaseous products as the reaction proceeded according to the following general scheme, in which specific stoichiometry is not implied:
This reaction scheme involving surface hydroxyl groups has
been widely published in the area of the organosilicon compounds. Although this reaction seemed to occur for both isolated and associated OH groups on the Al2O3 surface, it is clear from the sequence of changes in the spectra as the reaction progressed, that the isolated OH groups are more reactive since they disappear first.
The above data on the volatile products from the reactions of partially dehydroxylated alumina with the same silating reagent preclude any similar reaction for alumina. No HCl is detected in the gaseous products, even though molar amounts as low as 10-3 times the CH4 generated would have been easily detectable and quantifiable. Even if HCl had been initially generated and then reacted with other surface sites, it is difficult to envisage such a reaction, trapping all HCl with such high efficiency throughout the reactions as to totally remove it from the gas stream in a dynamic system. Further, the chlorine from the silating reagent remains firmly bound to the silated alumina.[7] Thus, it is concluded that HCl is not generated as either a product or an intermediate.
The silation does, however, cause a reduction in intensity of the IR bands associated with the surface hydroxyls. Thus, it is clear that

the mechanism requires the participation of more than one kind of surface site on the alumina, including the hydroxyls.
Mechanism of silation of γ-alumina with (CH3)3SiCl
Of primary importance with respect to the surface chemistry of oxide supports, and in particular of alumina, is the presence on the surface of exposed aluminum cations, oxide anions, and very often hydroxyl groups[8,9]. The creation of these coordinatively unsaturated sites (CUS) is caused most often by dehydration of surface. These ions seem to be directly involved in sites for both chemisorption and catalytic activity. Dehydration of the alumina surface has been postulated to generated “strained oxide linkages”,[10,11] a reaction which may be depicted as follows:
However, it is now agreed[12,11,4] that the dehydroxylation
process leaves coordinatively unsaturated oxygen anions in the outermost surface layer and exposed, incompletely coordinated, aluminum cations in the next lower layer, shown schematically as follows
Exposed or coordinatively unsaturated metal ions act as Lewis
acid sites and can thus provide interaction points for Lewis bases, such as electron donor ligands. Surface anion possess Lewis base properties and can effect nucleophilic attack at appropriate centers of supported molecules.[8,13,14] In this sense the “the strained oxide linkages” could be treated as coordinatively unsaturated sites as well, if we accept that CUS aluminum atom (Ala) is a Lewis acid center and the CUS oxygen bonded to the other one (Alb) is a Lewis base center.

The Si-C bond in organosilicon compounds has significant
polarity due to the great difference between the Pauling electronegativities of silicon (1.8) and chlorine (3.0). This is why the Si-C bond normally undergoes heterolytic rather than homolytic fission. On the other hand, the Si-C bond is easily cleaved by means of nucleophylic ionic agents, which attack the silicon atom15. Thus, the first stage of interaction of (CH3)3SiCl with γ-alumina surface can be understood as the dissociative chemosorption of the silane molecule (Scheme 1). The ionic character of the Si-C bond and the presence of three available unshared electron pairs in sp3 orbitals cause the chlorine to behave as a Lewis base. The silane molecule coordinates via the chlorine center to the coordinatively unsaturated Al3+ cation, which is a Lewis acid center (III). Nucleophilic attack by an adjacent oxygen anion then occurs at the silicon atom (IV), resulting in formation of a surface alumina-siloxane compounds (V).[15]

The dissociative adsorption and aluminasiloxane species formation are consistent with the known chemistry of both alumina surface and organosilicon compounds. The reaction is readily conceptualized as resulting from the action of both the Lewis acid and base centers in adjacent position on the surface, with the corresponding Lewis base and acid sites of the organosilicon compound.
Although the exiting evidence does not permit distinction between possible concerted or sequential interactions between the pertinent pairs of Lewis acids and bases, a concerted mechanism is considered the most probable. In a concerted mechanism there is no need to invoke free silyl cations, and the actions of the surface Lewis acid and base sites will effect cleavage of the Si-C bond in a synergistic manner.
In discussion of the role of polarity and the ionic character of the Si-C bond concerning dissociative chemisorption of silane and formation of surface aluminasiloxane species, a characteristic of this bond should be noted. From the earlier work it has been shown that the polarity of Si-halogen bond is lower than that expected from the theoretical value, due to the partial back (π) bond formation between the available electron pairs of the chlorine atom and the vacant silicon atom (3d) orbitals. This effect, however, is evinced mainly in low dipole moments of organosilyl halides.[15]
The stability of the Si-C bond towards hemolytic fission parallels that of the C-C bonds, and there is only a small difference between the bond energies. In accordance with the difference in the electronegativities, the Si-C bond is generally polarized as Si+ and C- and consequently can react through simultaneous nucleophilic attack on silicon and an electrophilic attack on carbon. Based on the above data, the mechanism we propose for methane formation during the interaction of (CH3)3SiCl with γ-alumina surface includes participation of surface CUS oxide, located proximate to a coordinatively unsaturated aluminum cation Al3+ and an isolated OH group that is capable of supplying proton necessary for methane formation. The elimination of CH4 molecule is accompanied by formation of a surface aluminasiloxane-bridged structure.

The key point of the methane molecule formation in the reaction of (CH3)3SiCl with γ-alumina surface is the incorporation of the proton from surface hydroxyl and the methyl group from the silane. The source of protons is hydroxyl groups left on the surface after partial dehydration. The surface of γ-alumina is dominated by basic acidic hydr acid oxyl groups, and the concentration of both neutral and weakly acidic hydroxyls is significantly smaller. The suggested mechanism needs participation of hydroxyl groups with acidic character, i.e., those in which the OH hydrogens show a stronger proton-donor character and which are adjacent to vacant sites. The isolated OH species react first, followed reaction of some associated OH species.
The adjustment of surface acidity is of significant importance to the silation of γ-alumina and the resulting formation of methane. The overall acidity of the surface is determined by the number of Lewis and Brønsted acid sites. Whereas the Lewis acidity of the Al2O3 surface is considered well established, the existence of intrinsic Brønsted acid sites is controversial. Pure alumina exhibits little or no Brønsted acidity,[16] in a sense, the surface acidity of alumina is “relative”, depending on the relative basicity of probe molecules. The incorporation of fluoride into Al2O3, however, creates some Brønsted acid sites, as has been proved by protonation of pyridine.[16] Thus, it is known that one of the major effects of incorporating fluoride into Al2O3 is the increase of the overall acidity of the Al2O3 surface. The increase in the strength of Brønsted acidity is thought to be due to the electronegativity of fluorine, which makes the OH hydrogens more protonic.[17] This inductive effect might also be expected to increase the strength of both Lewis and Brønsted acid sites. The trend is toward increased acidity and toward increased Brønsted character of the acid sites.
A consequence of silation of γ-alumina with (CH3)3SiCl is the impregnation of the alumina surface with another strong halide – chloride. The elemental analyses of our silated aluminas show that the average content of chloride on the alumina surface is between 2.4 and 3.0 wt %. Thus, our data are completely commensurable with the amount of fluoride determined elsewhere for other treated aluminas.[16] The specific feature of the work described herein is that

the alumina surface has not been pre-modified with the halogen and treated later with a chemisorbing compound, but rather that the halogen modification of the surface has been effected as a consequence of the silation. The advantage of this latter course of action is that the chloride is always located in proximity in the silane moiety, which we propose promotes the inductive effect and accelerates the reaction to form the silicon-oxygen bond. The disadvantage is that chlorine has a lower Pauling electronegativity (3.0), compared with that of fluorine (4.0), and consequently any inductive effect will be more moderate.
It is expected that the impregnation of the alumina surface with chloride will lead to an increase in the overall acidity. Brønsted acidity (as well as Lewis acidity) is associated with formation of an amorphous phase, most probably a chloro-aluminum moiety which is not present in sufficient amount to be detectable using XRD (X-ray diffraction). For fluoride loading of 2 and 5 wt % only the typical diffraction peaks of γ-alumina are detectable.[16]
The fluorination of the alumina surface leads to a change in the surface concentration of hydroxyl groups. The increase in the strength of Brønsted acid sites is accompanied by a decrease in the density of alumina hydroxyl groups, due to replacement of OH groups with fluoride anions.[16,17] In our study the chlorination of the alumina surface is not accompanied by any significant changes in the density of OH groups other than through the direct reaction, because of the different chemical nature of the reactant and the greater number of coordinatively unsaturated sites on the surface, compared with the number of chloride anions. A change of number of surface hydroxyl groups was observed only in the case of silation of alumina, dehydrated at temperature of 200 oC and lower, of which the surface is insufficiently dehydrated.
The location of halogen atoms on the alumina surface leads to an increase in the total acidity due to either the through-lattice inductive effect or the electron-withdrawing effect of a proximate halogen, resulting in a reduction of electron density of the neighboring chemical environment. However, the increase in the alumina surface acidity appears to be moderate, since the relatively higher acidity of

silica surface does not effect methane formation reaction to any comparable degree.
The acquired acidity of isolated hydroxyl groups affects the steric orientation of the chemisorbed aluminasiloxane species and formation of the methane molecules. Acid OH groups (as those found on Al2O3) can interact with weak basic (B) molecules, giving hydrogen-bonded OH…..B species, whereas with strong bases, ionic species are formed by proton transfer, [8] O-…..HB+.
The reaction involving surface hydroxyl groups (eq.1) has been assumed a priory in the area of organosilicon compounds. However, we have not observed production of HCl, and therefore have found no evidence for the occurrence of the above reaction. The previous interpretation appears to be based upon the erroneous assumption that the behavior of Al2O3 and SiO2 surfaces will be similar. It must be born in mind that two significant differences exist between the natures of these catalyst supports. The first difference concerns the unsaturated nature of the alumina surface. As a result of dehydration of alumina surface, a large number of coordinatively unsaturated sites, aluminum cations Al3+ and oxygen anions O2- are created. In contrast, heating the silica surface under vacuum progressively eliminates OH groups, which condense to form H2O and bridged oxides at surface, Si-O-Si. These oxide bridges are relatively unstrained and consequently homopolar in character. They are not as readily susceptible to either nucleophilic or electrophilic attack. The silica surface does not possess significant numbers of coordinatively unsaturated sites, and relatively few isolated hydroxyls remains.
The second distinguishing characteristic concerns the difference in the acid-base character of surface hydroxyl groups. Pure alumina contains mostly basic and some neutral and week acidic groups. All hydroxyl groups at the silica surface possess weak acidic character. The isoelectric point of alumina is 8.3 and that of silica is about 2.0, which determines the different charge of the surface. Al2O3 has a positive charged surface, and SiO2 has a negative charged one. The SiO2 surface contains OH groups which are less reactive than those of AL2O3.[18]
Thus, the initial reaction of alumina is not with surface hydroxyl groups but with coordinatively unsaturated Al3+ and O2- centers. The

irreversibly modification of the surface with chloride increases the effective positive charge of the isolated hydroxyls to an acidity between that of hydroxyl groups on the neutral alumina surface and acidic hydroxyls on the surface of silica. These hydroxyl groups then reacted with the pendant –O-Si(CH3)3 groups, formed as intermediates, to eliminate methane as the only gaseous product.
Conclusions The reaction of (CH3)3SiCl with the surface of alumina forms
neither HCl nor pendant –O-Si(CH3)3 groups. Instead, the mechanism is a sequential reaction which affords methane as the only gaseous product. The silane is initially chemisorbed at the coordinatively unsaturated surface sites. This has the effect of modifying the surface by chloride, and the isolated hydroxyl groups are thus made more acidic. The hydroxyl groups then react with CH3-Si groups to eliminate methane and to form bridging silyl moieties. Methane is the only gaseous product during the silation of alumina surface over the temperature range 140-260 oC as well as during thermal treatment of silated samples in a nitrogen atmosphere up to 500 oC. Methane production increases with each increment in temperature.
References 1. Armistead, C. G., Hockey, J. A., Trans, Faraday Soc., 1967, 63,
2549. 2. Hair M. L., Hertl, W., J. Phys. Chem., 1969, 73, 2372. 3. Sutsumi, K., Takahashi, H., Collod. Polym. Sci., 1985, 263, 506. 4. Knozinger, H., Adv. Catal., 1976, 25, 184. 5. Paul, D. K., Yates, Y. T. Jr., J. Phys. Chem., 1991, 95, 1699. 6. Paul, D. K., Ballinger, T. H., Yates, Y. T. Jr., J. Phys. Chem.,
1990, 94, 4617. 7. Slavov, S. V., Chuang K. T., Sanger A. R., Langmuir, 1995,
Vol.11, No 10. 8. Zecchina, A., Otero Arean, C., Catal. Rev.-Sc. Eng., 1993, 35,
261. 9. Knozinger, H., Ratnasami, P., Catal Rev.- Sc. Eng., 1978, 17, 31. 10. Cornelius, E. B., Milliken, T. H., Mills, G., Oblad, A., J. Phys.
Chem., 1955, 59, 809.

11. Hsu, L. -Y, Shore S. G., D’Ornelas, L., Basset, J. M., J. Catal., 1994, 149, 159.
12. Peri, J. B., J. Phys. Chem. 1965, 69, 220. 13. Davydov A. A., IR Spectroscopy Applied to Surface Chemistry of
Oxides, Nauka, Novosibirsk, 1984. 14. Zecchina, A., Soluccia, S., Morterra, C., Appl. Spectrosc. Rev.,
1985, 21, 259. 15. Bazant, V., Chvalovsky, V., Rathousky, J., Organosilicon
Compounds, Academic Press, New York, 1965. 16. DeCanio, E. C., Bruno J. W., Nero, V. P., Edwards, J. C., J.
Catal., 1993, 140, 84. 17. Ghosh, A. K., Kidd, R. A., Catal Rev.- Sc. Eng., 1985, 27, 539. 18. Rajagopal, S., Marini H. J., Miranda, R., J. Catal., 1994, 147, 417.

ГОДИШНИК
НА ШУМЕНСКИЯ УНИВЕРСИТЕТ
„ЕПИСКОП КОНСТАНТИН ПРЕСЛАВСКИ”
Т. ХIX В 2
ФАКУЛТЕТ ПО ПРИРОДНИ НАУКИ
Формат 16/60/84 Тираж 70
ISSN 1311-834Х Университетско издателство
“Епископ Константин Преславски“




















