
Electroosmotic Flow in a Capillary Annulus with High Zeta Potentials

Geochemical characterization of Malm Zeta laminated carbonates
from the Franconian Alb, SW-Germany (II)
L. SCHWARK1*, M. VLIEX1 and P. SCHAEFFER2
1Geological Institute, University of Cologne, ZuÈ lpicher Str. 49a, 50674 Cologne, Germany and 2Institutde Chimie, Universite Louis Pasteur, 1 rue Blaise Pascal, 67000 Strasbourg, France
AbstractÐDuring Jurassic times, especially within the Malm z stage, local depressions formed on theEastern Bavarian Carbonate Platform that were surrounded by wall reefs. This created a unique deposi-tional environment, where in an open-marine setting stagnant and anoxic bottom waters developed inan intra-reef depression. Anoxic conditions were stabilized below wave base by enhanced salinity in thebottom waters and establishment of a density strati®cation. Biomarker analysis was applied to charac-terize palaeosalinity and redox conditions, utilizing organic sulfur compound, methylated chroman,hopanoid and saturated isoprenoid distributions. Prevention of terrigenous in¯ux by protecting reefwalls lead to iron de®ciency in intra-reefal sediments causing early diagenetic sulfurization of functiona-lized lipids. Absence of clay-mineral catalysts and early sulfurization favored an unusual steroid distri-bution lacking rearranged analogues but providing coexistence of saturated steranes with baa-, aaa-and abb-con®guration, D13(17)-spirosterenes, unknown sterenes and steradienes as well as mono-, di-and triaromatic steroids. Desulfurization of the polar fraction was carried out for two samples to verifythat palaeoenvironment reconstruction based on free hydrocarbon and heterocompound distributionwas not invalidated by sulfur quenching of selected compounds. Gammacerane was found to be theonly component to occur exclusively in the sulfur-bound fraction. Only minor amounts of hydrocar-bons were released upon desulfurization implying that free bitumen analysis was applicable for organo-facies characterization. Organic petrological investigation of organic mats revealed the presence of twodi�erent types of such structures. Comparison of organoclast ¯uorescence spectra with those ofextracted porphyrin fractions indicate an origin of porphyrins exclusively from cyanobacterial matswhereas algal dominated mats yield no porphyrins. This aims towards a better localization of speci®cbiomarker origin in geological samples. # 1998 Elsevier Science Ltd. All rights reserved
Key wordsÐJurassic, Malm, Solnhofen Lithographic Limestone, marls, cherts, limestones, palaeoenvir-onment reconstruction, palaeosalinity, organic sulfur compounds, aromatic steroids, sterenes, spiroster-enes, steradienes, hopenes, porphyrins, ¯uorescence properties
INTRODUCTION
The Jurassic age lithographic limestone of the
Solnhofen area, Franconian Alb in SW-Germany
(Fig. 1), is well known for its excellent preservation
of fossils. Vertebrates like Archeopterix, the earliest
bird and Compsognathus, known to be the smallest
dinosaur, were found in the well laminated micritic
limestones of the Solnhofen and Jachenhausen area.
The exquisite preservation of fossils and the extre-
mely ®ne lamination of the matrix rocks indicate
exceptional conditions of sedimentation for the car-
bonates, pointing towards a low energy and poss-
ibly anoxic environment. Several palecological
models have been established, based on micro- and
macrofossil faunal assemblages to de®ne, for
example, water circulation and strati®cation pat-
terns, autochthonous productivity and terrigenous
in¯ux, redox and salinity conditions (von Freyberg,
1966, 1968; Barthel, 1970, 1978; Keupp, 1977, 1994;
Keupp et al., 1993). Reconstruction of palaeoenvir-
onmental conditions by means of organic geochem-
istry, however, is usually hindered because the
entire area was subjected to uplift and erosion
during the Early Cretaeous and late Tertiary, allow-
ing groundwater to penetrate into the limestones
and thus causing oxidation of the organic matter,
especially in the Solnhofen area (Meyer and
Schmidt-Kaler, 1993).
In order to overcome the problem of weathering,
we investigated a core from the Rennertshofen
Trough, south of Solnhofen, where the organic mat-
ter embedded in the Malm limestone was preserved,
allowing for reliable organofacies characterization.
Molecular indicators for palaeoenvironmental
reconstruction in carbonatic/evaporitic depositional
settings, previously developed during phases I and
II of the ENOG programme (ENOG: European
Network of Organic Geochemists; see Huc and
Sinninghe Damste , 1993; Leythaeuser and
Sinninghe Damste , 1995) were applied to character-
ize organic matter incorporated in the Malm z sedi-
ments, in order to verify or re®ne and improve
existing palecological models. This is also regarded
Org. Geochem. Vol. 29, No. 8, pp. 1921±1952, 1998# 1998 Elsevier Science Ltd. All rights reserved
Printed in Great Britain0146-6380/98 $ - see front matterPII: S0146-6380(98)00192-2
*To whom correspondence should be addressed. E-mail:[email protected]
1921

as a test, whether molecular palaeontology couldcontribute to improve environment characterizationin a case where micro- and nannofossil evidence is
limited due to dissolution and in part replacementcrystallization of fossil shells and test (Keupp et al.,
1993; Meyer and Schmidt-Kaler, 1994).In part I of this publication we described in more
detail the geological setting, the basic organic pet-rology and the inorganic and bulk organic geo-
chemistry. In the present contribution we focus onthe biomarker geochemistry to obtain informationabout palaeoenvironmental conditions during sedi-
mentation of the Malm carbonates. We examinedthe composition of the ``free'' solvent extractable
bitumen, including aliphatic and aromatic hydrocar-bons as well as organic sulfur compounds (OSC).In order to account for a potential strong bias in
lipid distribution due to early sulfur incorporation,the polar bitumen fractions of two samples were
desulfurized and the distribution of released hydro-carbons compared with those of the ``free'' bio-
marker composition.
Aiming towards a better coupling of microscopi-cal observations with biomarker geochemistry andan improved understanding of the exact localization
of speci®c extractable compounds within a sedimentsample, we investigated the relationship betweenspectral ¯uorescence characteristics of solventextractable porphyrins and the ¯uoresence proper-
ties of organoclasts and bitumens.
EXPERIMENTAL
Organic petrology
Whole block samples were cut perpendicular tobedding, embedded in epoxy resin and polished.
Organic petrographical investigations were deter-mined by re¯ected light in oil immersion using aZeiss Axioplan microscope using magni®cations of
200� and 500� .The mean random vitrinite (Rr%) or solid bitu-
men re¯ectance (Rb%) was measured to determine
the maturity of organic matter. The measurements
Fig. 1. Location map of study area in southern Germany.
L. Schwark et al.1922

were carried out in comparison to a Saphir stan-dard of Rr=0.500% (546 nm, oil immersion) apply-
ing the methods of DIN 22020 (1986). Bitumenre¯ectance (Rb) was converted into vitrinite re¯ec-tance using the equation of Jacob (1989):
Rv=0.618Rb+0.40 (%, oil). This equation wasestablished for Palaeozoic and Mesozoic rocksamples from the Saxonian Basin of Northwest-
Germany.Spectral ¯uorescence measurements were carried
out with the same equipment but under illumination
applying a HBO 100 W lamp (Zeiss). A spectrum ofa stabilized tungsten lamp served as ¯uorescencestandard and intensity was normalized to an uranylglass (GG-17, 1 mm) at 535 nm. ICI-colourmetric
values were determined after DIN 5033 (1979) andplotted into a two dimensional chromatographicdiagram after DIN 6164 (1978) (for details of the
procedure, see Hagemann and Hollerbach, 1986).
Carbon and sulfur determination and Rock-Eval py-
rolysis
The contents of total carbon (TC) and total sul-fur (Stot) were determined with a LECO CS225 ana-
lyser. The same instrument was used for totalorganic carbon (TOC) measurements after removalof carbonate minerals with hydrochloric acid (32%
v/v) and repetitive washing with distilled water.Carbonate contents were calculated by di�erenceand expressed as percent CaCO3. Rock-Eval analy-
sis was performed according to the methoddescribed by Espitalie et al. (1977) and Bordenave(1993) with a VINCI Rock-Eval-II Analyser. S1, S2
and Tmax-values were measured and corresponding
HI, OI and PI values calculated.
Extraction and chromatography
Finely ground (<0.2 mm) samples were extractedusing a DIONEX ASE 200 extractor with dichloro-
methane for 20 min under a pressure of 50 bar at atemperature of 758C. Total extracts were separatedinto maltenes and asphaltenes by precipitation witha 40 fold excess of n-hexane. Maltenes were separ-
ated into fractions of aliphatic hydrocarbons, aro-matic hydrocarbons and heterocomponents byMPLC (Radke et al., 1982).
Free hydrocarbon analysis
GC±MS analysis of the saturated and aromatichydrocarbons was carried out using a HP 5890Series II gas chromatograph coupled to a HP 5989single quadrupole mass spectrometer. A
50 m*0.20 mm (ID) fused silica column (HP 5),coated with 5% chemically bonded phenyl±methyl±silicon (0.33 mm) was used with helium carrier gas.
The GC oven temperature was programmed from70 to 1408C at a rate of 108C/min followed by asecond gradient from 140 to 3208C at 38C/min. The
mass spectrometer operated in EI mode at 70 eV
with a scan range from m/z= 30 to 600 for identi®-cation of individual compounds of the aliphatic and
aromatic hydrocarbon fractions. Data acquisitionand processing was performed using a HP MS-ChemStation data system. Peak identi®cation was
carried out by comparison of mass spectra withthose of the system library and by comparison withpublished spectra.
Desulfurization experiments
Fractionation. The Raney nickel desulfurization
experiments were carried out on the polar fractionfrom the organic extract of two samples (sampleM110 and sample ML69). Brie¯y, the precipitated
asphaltenes (ca. 100 mg) were adsorbed on silica geland loaded on a silica gel column. A ®rst elutionwith a hexane/dichloromethane mixture (7:3 v/v)yielded some occluded free hydrocarbons and small
amounts of nickel porphyrins (ca. 10±15 mg/g or-ganic extract). A second fraction was then recov-ered by elution with a dichloromethane/methanol
mixture (3:1 v/v), yielding ca. 700±800 mg/g organicextract of polar material.Raney nickel desulfurization of a polar chromato-
graphic fraction. The polar chromatographic frac-tion (see above) of the organic extract was dissolvedin a 1:1 mixture of toluene/ethanol and an excess ofRaney nickel, preextracted with distilled water and
ethanol, was added. The mixture was re¯uxedunder argon for 3 h, after which the supernatantwas recovered. The Raney nickel was extracted with
dichloromethane (�2), the organic extracts com-bined and the solvent removed under reduced press-ure. Puri®cation of the extract by liquid
chromatography (silica gel, hexane as eluant)yielded an (alkane + alkene) fraction (Rf>0.8; ca.15 mg/g polar fraction). The aromatic hydrocarbons
(ca. 25 mg/g polar fraction) were recovered byelution with a hexane/dichoromethane mixture (8:2v/v).Gas chromatography. The hydrocarbons recovered
by Raney nickel desulfurization were analysed on aHewlett Packard gas chromatograph (HP 6890series) equipped with an on-column injector, a FID
detector and a HP5 column (30 m�0.32 mm i.d.;0.25 mm ®lm thickness). H2 was used as carrier gasand the oven was programmed from 70 to 2008C(108C/min), 200 to 3008C (48C/min), then held iso-thermally for 30 min.Gas chromatography±mass spectrometry. Analyses
of the hydrocarbons released by desulfurization
were performed on a Varian 3400 gas chromato-graph equipped with a programmable on-columninjector and a DB5 fused silica column
(30 m�0.25 mm; 0.2 mm ®lm thickness) connectedto a Finnigan MAT INCOS 50 mass spectrometeroperating at 70 eV. The oven temperature was pro-
grammed from 70 to 2008C at 108C/min, 200±
Geochemistry of Malm carbonates 1923

3008C at 58C/min, then held isothermal at 3008Cfor 30 min. He was used as carrier gas.
Carbon isotopic analysis
After organic solvent extraction, determination ofbulk kerogen carbon isotope composition was per-
formed on a Finnigan Delta S mass spectrometer.Standard notation in d13C (-) values relative to thePDB-standard was used. Prior to measurement ofthe organic carbon isotopes all carbonate minerals
were quantitatively removed by treatment with hy-drochloric acid.
RESULTS
Organic petrology and spectral ¯uorimetry ofextracts and porphyrin fractions
The basal interval of the studied core section(Fig. 2) below 135 m belongs to the Malm z1(Meyer and Schmidt-Kaler, 1994) and contains thetypical ``lithographic limestones'', i.e. white to bu�coloured micritic limestone, with excellent beddingand no internal texture (in the following abbre-
viated KW). The micrites are exclusively composedof individual calcite crystals, 1±10 mm in size.Isolated dino¯agellates and coccolithes are enclosed
within the matrix. Pyrite is only present in minoramounts and of diagenetic origin. In all samples ofthe Malm z 1 studied no particulated organic mat-
ter could be detected.
The interval comprising the Malm z 2 (135±67 m)
and z 3 (67±25 m) stage consists of a sequence of
grey and bu� coloured micritic limestones (abbre-
viated as K), which are interbedded with brownish
bituminous, very ®ne laminated marls (ML). Within
the Malm z 2 chert layers (KK) of 1±10 mm thick-
ness occur between 80 and 110 m depth (Fig. 2).
Slumping structures and disturbance of sediment is
observed for the Malm z 3 sediments.
The bituminous marls (ML) of the Malm z 2 and
z 3 show an intensive mm-scaled lamination.
Laminae of ®ne grained carbonate minerals (mainly
calcite) alternate with dark brown laminae com-
posed of a mixture of clay minerals and organic
matter. These shaly layers are rich in framboidal
pyrite formed by bacterial sulphate reduction
(Berner, 1983). The relative amount of pyrite
increases from bottom to top. In the iron-limited
depositional environment of the Rennertshofen
Trough this more likely indicates an increase in the
supply of terrigenous derived dissolved iron than an
increase of anoxicity during sedimentation.
The organic matter of the laminated limestones
shows microscopically identi®able marine and some
very minor terrestrial organic constituents.
Terrestrial organic matter consists of some isolated
tissues with preserved cell structures but preferen-
tially of reworked vitrinitic organoclasts. Main con-
stituents of the marine organic matter within
laminated limestones are unicellular thick-walled
Fig. 2. Stratigraphic cross section of the Upper Malm in South-Germany with schematic representationof reef and trough facies, adopted from Meyer and Schmidt-Kaler (1993).
L. Schwark et al.1924

Fig. 3. Spectral colourimetry diagram according to DIN 6164 showing two clusters of data points.Cluster one comprises the alginites and porphyrin-free maltene fractions, cluster two contains cyanobac-terial mat derived red bituminites and the free extractable porphyrins. Porphyrins therefore predomi-
nantly originate from cyanobacterial mats.
Geochemistry of Malm carbonates 1925


tasmanites type algae >50 mm in size. They are ran-
domly distributed in the carbonatic sediment or
enriched within the shaly laminae as mats elongated
parallel to bedding. Under blue light excitation the
algal mats and the associated exudated bitumen
reveal an intensive green to yellowish ¯uorescence
indicating low thermal maturity.
In some cases the associated bitumen is of a red
¯uorescence colour and has a negative alteration, a
characteristical feature for the presence of chlorins/
porphyrins within the mats. Keupp (1977), Barthel
(1978), Keupp et al. (1993) and Keupp and
Neumann (1996) proposed a cyanobacterial origin
for the organic mats in case of the laminated lime-
stones from the Solnhofen area, which is located
some tens of kilometers north of the Rennertshofen
Trough. An additional input of cyanobacterial mat-
ter to the algae-bearing mats thus seems likely, but
is impossible to determine by microscopical obser-
vation. In contrast to the shaly laminae, the carbo-
natic laminae of the laminated limestones include
only a few isolated tasmanites type algae and dino-
¯agellates, which include some bitumens with yel-
low ¯uorescence colour.
Especially within the Malm z 2 interval the lami-
nated limestones are interbedded with black cherts
of 1±10 mm in thickness (in the following labelled
MK). The carbonate minerals and organic matter
in those cherts show the same appearence as in the
laminated marls. The cherts are, however, enriched
with spherical radiolaria, 1±10 mm in size and the
matrix and intergranular cavities show a secondary
cementation with amorphous silica. RFA analyses
of chert samples indicate high amounts of SiO2 and
extremely low Al2O3 contents (Vliex and Schwark,
1998). The resulting SiO2/Al2O3-ratios by far exceed
average shale values also indicating a biological ori-
gin of the amorphous silica (Brumsack, 1988), i.e.
from dissolved radiolaria, now often replaced by
carbonate, and from siliceous sponges. Under blue
light excitation the radiolaria o�er a green to yellow
¯uorescence colour that argues for a good preser-
vation of bitumen within the organoclasts. No
microscopical observations of diatomaceous algae
were made and no indications of their presence in
Malmian samples exist in the literature.
Microscopical ¯uorescence measurements were
made on solitaire alginite and red coloured streaky
bituminite within polished block samples. The same
technique was applied to chromatographically sep-
arated Ni- and V.O-porphyrin and porphyrin-free
maltene fractions. Fluorescence spectra of the uni-
cellular tasmanites type algae (type A) were
measured for wavelength intervals from 450 to
700 nm. Main intensities are recognised for the
green range (500±510 nm) of the spectrum, whilst
intensities in the yellow and red range of the spec-
trum (600±650 nm) are low. The visual impression
of a greenish to yellow ¯uorescence colour is also
re¯ected by a red/green ratio varying from 0.23±
0.09. In some cases the same algae illuminate with
bright intensities in the range of 470 nm and exhibit
a light bluish colour (type B). After Hagemann and
Hollerbach (1986) this colour corresponds to high
contents of aliphatic hydrocarbons.
In the case of the organic mats characteristic of
the laminated marls, the visual impression is that of
a reddish colour. The associated ¯uorescence spec-
trum contains two maxima of intensity. The ®rst
one occurs in the range of green to yellow and the
second one between 620 and 720 nm. The spectrum
is the result of a mixture of the ¯uorescence beha-
viour of tasmanites type algae and exsudated red
coloured liquid bitumen. After extended illumina-
tion (>2 min) the red colour shows a strong nega-
tive alteration and only the yellow colour of algae
remains (Vliex and Schwark, 1998). This intensive
red ¯uorescence colour and the strong negative
alteration are characteristical criteria for chloro-
phylls or fossil chlorins/porphyrins.
To verify this observation porphyrin and por-
phyrin-free maltene fractions were isolated by liquid
chromatography and analysed by spectral ¯uor-
escence measurements. The spectra of isolated V.O-
porphyrin fractions comprise a single maximum in
the red range (725 nm) while Ni-porphyrins with
some admixture of V.O-porphyrins reveal an ad-
ditional maximum in the range of 650 nm (red). In
both cases no intensities are recognised within the
green to yellow range of the spectrum.
For a quantitative comparison of the ¯uorescence
properties of algae, bituminites and isolated extract
fractions, the colorimetric values x and y were cal-
culated following DIN 6164 and plotted in Fig. 3.
The ¯uorescence colours of the measured objects
are separated into two clusters. The ®rst cluster
occurs between colour shade lines 21 and 23, repre-
senting the tasmanites type algae of type A and B.
The same ¯uorescence properties were determined
for the porphyrin-free maltene fractions. After
Hagemann and Hollerbach (1986) the position of
this cluster in the discrimination diagram corre-
sponds to that of oils and extracts of low thermal
maturity.
The second cluster is composed of data points
from isolated Ni- and V.O-porphyrin fractions and
is located between the colour shade lines 1 and 5 in-
dicating orange to red colours. This cluster also
contains the data points for red coloured liquid
bitumens, exclusively found in organic mats occur-
ring in laminated marls (Fig. 3).
These results indicate that tasmanites type algae
do not account as a source for the porphyrins
extracted from the laminated limestones. According
to the ¯uorescence behaviour, a close spatial re-
lationship between the occurrence of porphyrin bio-
marker compounds in the extracts and the red-
coloured organoclasts exclusively occurring with the
Geochemistry of Malm carbonates 1927

ML organic mats is attributed to a cyanobacterialcontribution. The ¯uorescence spectrum of these
red streaky bitumens from the bacterial mats showsidentical intensity maxima in the long wave rangeof the spectrum and the same strong negative
alteration as compared to the porphyrin fractions.After Keupp (1977, 1994) and Keupp et al.
(1993) the organic mats of laminated limestones
from the nearby Solnhofen area are composed ofalgae and an additional cyanobacterial input. Theauthors proposed episodic bacterial blooms as a
likely process for the formation of the organicmats. Meyer and Schmidt-Kaler (1994) relate thesedimentation of laminated marls and limestones to``rhythmic algal blooms''. Our results con®rm that
two types of organic mats are present, one com-posed almost exclusively of algae and the othermat-like structures preferentially originate from cya-
nobacteria with algal input in those intervalsstrongly reduced. Algal mats that are mainly pro-duced by Tasmanales-like algae do occur frequently
in the Malm carbonates and may represent regularmarine conditions whereas cyanobacterial matswere build under stressed conditions.
Random re¯ectance measurements have been car-ried out on solid bitumen in order to determinethermal maturity of organic matter in Malmsamples. Re¯ectance values of laminated limestones,
cherts and lithographic limestones are plotted inre¯ectance frequency histograms (re¯ectograms) inFig. 4. In case of the laminated marls (ML) re¯ec-
tance values extend from 0.1 to 0.9% Rb. The max-ima of the unimodal distributions range between0.35 to 0.40% Rb indicating a very low degree of
thermal maturity. In contrast to laminated marls,the nonlaminated carbonates (K) show polymodaldistributions of the re¯ectance classes. The valuesrange up to 1.5% corresponding to vitrinitic and
inertinitic organoclasts of a reworked origin. Inthese samples the terrestrial input is also dominatedby solid bitumen, which shows the same maxima as
the laminated limestones.
Bulk parameters
Bulk parameters determined on an extended
sample set are described by Vliex and Schwark(1998) and therefore, only a short summary is givenhere. The TOC-content is lowest in the pure white
carbonate samples with 0.1±0.5% and in all othersamples ranges from 0.3 to 15% TOC with no sig-ni®cant control by lithofacies. Total sulfur valuesshow excellent positive correlation with TOC and
vary between 0.05 and 3.0% TS. The C/S-ratio onaverage calculates to 3.2 and indicates a high pro-portion of organically bound sulfur as given by the
low Fe/S-ratios of on average 0.05 documentingthat only a minor fraction of the sulfur is bound toiron-sul®des. Excellent quality of the organic matter
is revealed by HI-values ranging from 100 to 1050
(mg HC/g TOC), most samples clustering aroundHI-values of 600 to 800 (mg HC/g TOC). In agree-
ment with microscopical analysis a type II marinealgal kerogen can be attributed to most samples.Extremely lipid-rich samples of type I-kerogen are
characterized by TOC-values below 2.0%. Rock-Eval maturity estimation of organic matter withTmax values of 400 to 4108C and PI-values of <0.1
con®rms the microscopically deduced low matu-ration level. Isotopic characterization of the kero-gens gives d13C ratios ranging from ÿ25.8 to
ÿ29.5- with most values between ÿ28.0 andÿ29.0-, typical of Jurassic marine organic matter.Extract yields show a dependency on lithologies
with limestones and cherts yielding less than
1000 ppm, whereas marls and siliceous carbonateson average provide between 400 to 4000 ppm withseveral samples in the range of 5000 to 16500 ppm.
Due to the low thermal maturity only between 10and 25% of the extractable bitumen is composed ofhydrocarbons.
Biomarker composition
Discussion of biomarker composition follows theclassi®cation into three main lithofacies types micri-
tic limestones, marls and cherts as previouslyreported (Vliex and Schwark, 1998). Where appli-cable, a more detailed grouping into sub-lithofacies
types of white (KW) and grey limestones (K), lami-nated (LM) and non-laminated marls (M) and alter-nations of chert layers (KK) with marls (MK) was
used. Distribution of free lipids is applicable forpalaeoenvironment characterization, even at the lowmaturity of 0.3% Rr because desulfurization exper-iments with Raney Nickel gave very low yields and
hence no strong bias in biomarker distribution dueto sulfur-bonding occurs. The free lipid distributionis therefore discussed ®rst and in detail, followed by
supplementary information derived from desulfur-ized polar fractions.
Free aliphatic hydrocarbons
The fraction of the free aliphatic hydrocarbonspredominantly consists of n-alkanes and acyclic iso-prenoids for most samples. Only for a few samples
of the LM lithofacies type, cyclic terpenoids domi-nate the aliphatics. Reduced amounts, sometimesonly traces, of unsaturated and saturated steroids
and hopanoids occur within the K and KW facieswhereas the M and LM marls are generallyenriched in cyclics that often dominate over the n-alkanes. The KK and MK chert are almost devoid
of hopanoid and steroid biomarkers. Bi-, tri-, ortetracyclic sequi- or diterpenoids are lacking in allsamples. Comparably, tricyclic and tetracyclic triter-
penoids could not be detected but in trace amounts.Distribution of n-alkanes. Solvent extractable ali-
phatic hydrocarbons of the three lithofacies-types
limestones, marls and cherts contain a series of
L. Schwark et al.1928

homologous n-alkanes. Pure cherts are character-
ized by a very smooth n-alkane distribution ranging
from n-C12 to n-C35, maximising at n-C15 and show-
ing a very minor predominance of n-C21 and n-C23
over the neighboring even numbered n-alkanes. Sili-
ci®ed limestones give a comparable distribution in
Fig. 4. Solid bitumen re¯ectograms for three di�erent lithofacies types. MK 122 represents a silici®edcarbonate or chert layer, K11 originates from a lithographic limestones and ML45 is representative ofthe laminated marl lithofacies type. Laminated marls always show a narrow unimodal distribution of
low re¯ectance values around 0.35% Rb indicative of an thermally immature sediment.
Geochemistry of Malm carbonates 1929

the C20+ range with additional high contributions
of even numbered short chain n-alkanes (Fig. 5).
Limestones show n-alkanes ranging from n-C12 to
n-C35 with a pronounced predominance of short
chain even-numbered n-alkanes C14, C16, C18 and to
a lesser extent n-C20. In all KK and KW samples n-
C16 is the most abundant n-alkane. Comparable to
the chert samples, a minor preference of n-C23 and
n-C25 in relation to n-C22, n-C24 and n-C26 is noted
(Fig. 5). Marls showing a much higher lithological
variability are consequently also characterized by
more diverse n-alkane distributions. Non-laminated
marls and silici®ed marls more closely resemble the
distribution noted for limestones, i.e. predominance
of even-numbered n-alkanes in the C14 to C18
range. This feature was not observed for laminated
marls that instead reveal a shift in n-alkane distri-
bution to compounds with longer chain length max-
imising at n-C23 and a preference of odd- over
even-numbered n-alkanes in the C21 to C35 range
(Fig. 5). The silici®ed marls di�er from the non-
laminated marls by showing a predominance of n-
C14 instead of n-C16 as for the latter, by higher rela-
tive contribution of compounds in the C21 to C35
domain and a more pronounced odd over even pre-
dominance in that range.
The n-alkane distribution for all samples thus in-
dicates a fully marine origin for all samples with
virtually no terrigenic input of plant wax derived n-
alkanes. Although deposition in the Rennertshofen
Trough occurred in a shallow water environment
and close to the continent, this is in agreement with
the palaeogeographical situation. Terrigenous input
very e�ectively was restricted by the coral, algal
and spongal reefs providing a perfect barrier sur-
rounding the intra-reef depocenter (Meyer and
Schmidt-Kaler, 1989, 1994; Vliex and Schwark,
1998).
The strong dominance of short chain even num-
bered alkanes (odd over even predominance = 0.2±
0.5 in the C12 to C20 range) argues for a carbonatic
anoxic environment (Hite et al., 1984; Palacas et
al., 1984; ten Haven et al., 1985, 1988; Connan et
al., 1986; Peters and Moldowan, 1993; de las Heras
Fig. 5. Fragmentograms of m/z = 85 for 4 di�erent lithotypes illustrating low end biased distributionof isoprenoid and n-alkanes for cherts (MK122) and limestones (K116) as opposed to long-chain domi-nated non-laminated (M125) and laminated (ML26) marls. Numbers indicate chain length of n-alkanes
or isoprenoids, e.g. pristane, phytane, PME, squalane and lycopane.
L. Schwark et al.1930

et al., 1997). Short chain even-numbered n-alkanes
have been reported from various marine and lacus-
trine systems spanning a wide range of salinities,
where they were related to microbial sources (e.g.
Grimalt and Albaiges, 1990). Even carbon number
preference of n-alkanes is also associated with
enhanced salinity environments (Palacas et al.,
1984; Moldowan et al., 1985; ten Haven et al.,
1985, 1988). Sheng et al. (1980) suggested, that the
reason for the dominance of even numbered alkanes
is a complete reduction of fatty acids. Alternatively,
a selective preservation of naturally occurring even
numbered n-alcohols and alkyl-esters by incorpor-
ation in the high molecular polar fraction by sulfur-
bounding can occur (de Leeuw and Sinninghe
Damste , 1990). The short chain n-alkanes may orig-
inate from marine algae and from cyanobacteria.
Cyanobacteria are reported to produce no car-
boxylic acids and n-alkanes extending beyond n-C18
(Volkman and Maxwell, 1986). Hence, the long-
chain n-alkanes of the Malm z samples must be de-
rived from marine algae with some very minor con-
tribution from land plant waxes. This is based on
microscopical investigations and the low amounts
of plant wax derived n-C27, n-C29, n-C31 as com-
pared to n-C21, n-C23 and n-C25 alkanes. No prefer-
ence of medium chain length even-numbered n-
alkanes n-C22 and n-C24, characteristic of several
carbonatic/evaporitic systems studied by ten Haven
et al. (1985, 1988) was noted.
The predominance of n-C23 and adjacent odd-
numbered n-alkanes is similar to the one observed
for other Jurassic rocks, especially those of the
Posidonia Shale or Kimmerdige Clay Fm.
(Farrimond et al., 1984; van Kaam-Peters et al.,
1997). An excursion to heavier d13C-values for this
particular compound as compared to the adjacent
n-alkanes in the Kimmeridge Clay Fm. suggests an
origin from highly specialized algae (van Kaam-
Peters and Sinninghe Damste , 1997). In analogy, a
signi®cant change in the algal association for the
marl and limestone lithotypes of the Malm z car-
bonates has to be invoked. As a precursor com-
pound for the n-C23-alkanes is still unknown, the
type of algae preferentially contributing to the free
lipids of the marl lithofacies samples could not be
determined.
Free isoprenoid alkanes. Regular isoprenoid
alkanes in the Malm z samples include farnesane
(low amounts), i-C16, norpristane, pristane, phy-
tane, i-C21, PME (2,6,10,15,19-pentamethyleico-
sane), squalane and lycopane. No highly branched
C20 to C25-isoprenoids, often found in carbonatic
evaporitic systems were detected and thus an input
from diatoms is excluded. The occurrence of PME
and squalane indicates a contribution of archaebac-
teria to the organic matter (Holzer et al., 1979;
Risatti et al., 1984), although both compounds were
also suggested to be derived from algal sources
(Kenig et al., 1995). Lycopane, abundant in most
samples, is generally attributed to algal sources
(Kohnen et al., 1993; Salmon et al., 1997). Diage-
netic degradation products of chlorophyll derived
phytol including phytane, pristane, norpristane and
i-C16 occur in all samples and except for the lime-
stones (K, KW) in high abundances. A dominance
of phytane over pristane and shorter chain com-
pounds is always found, with pristane/phytane-
ratios ranging between 0.2 to 0.9. Although the
pristane/phytane-ratios do not strictly follow litho-
facies types, lower values of 0.2 to 0.5 were
observed for the laminated marls and the chert
layers. Limestones generally reveal higher pristane/
phytane-ratios of 0.5 to 0.9 whereas marls and sili-
ci®ed marls show a wide distribution of pristane/
phytane-ratios. No depth trend was recognized for
this ratio and thus the high variability for this par-
ameter re¯ects rapid ¯uctuations in the depositional
conditions of the Malm z. Because terrigenous in¯u-
ences can be ruled out, changes are due to vari-
ations of sealevel and associated biological input,
salinity and redox potential in the intra-reefal set-
ting. The extremely low pristane/phytane ratios of
the laminated marls argue for strictly anoxic con-
ditions during sedimentation (Brooks et al., 1969;
Didyk et al., 1978) possibly coupled to higher sali-
nities. Low pristane/phytane-values were recognized
in a variety of enhanced salinity environments (Hite
et al., 1984; ten Haven et al., 1985, 1988; Schwark
and PuÈ ttmann, 1989; de Leeuw and Sinninghe
Damste , 1990; Kenig et al., 1995). In hypersaline
environments halotolerant bacteria containing high
contents of phytanyl lipids (Albrecht et al., 1976)
can be very abundant and thus a low pristane/phy-
tane ratio in special environments may be re¯ecting
the salinity depended growth rate of bacteria rather
than the degree of anoxicity (de Leeuw and Sin-
ninghe Damste , 1990).
The ratios of pristane/n-C17 and phytane/n-C18
also do not show a distinctive facies control but
reveal a general trend towards higher values around
110 m depth, i.e. in the center of the Malm z 2
interval. Plotted in a pr/n-C17 vs. ph/n-C18-diagram
(Shanmugam, 1985; Talukdar et al., 1993) all except
the white limestone values fall in the range of mar-
ine algal derived organic matter deposited under
strongly reducing conditions. This diagram groups
laminated marl and chert samples at more reducing
and non-laminated marls and limestones at less
reducing conditions.
Hopanes and hopenes. A C27 to C35 series of
hopanes and hop(17,21)enes dominates in all but
the KW samples (Fig. 6), indicating an eubacterial
and/or cyanobacterial input (Ourisson and
Albrecht, 1992; Rohmer et al., 1992; Ourisson et
al., 1994). Both series posses the 17a(H),21b(H)
con®guration and extended C31 to C35 members
occur as doublets of 22S and 22R isomers (Fig. 6).
Geochemistry of Malm carbonates 1931

Besides the two dominating hopanoid types, more-
tanes occur in the C27 to C31 range and neohop-
(13,18)ene accompanied by 30nor-neohop(13,18)eneis present in most samples.
The relative amounts of hopenes to hopanes is
fairly constant for the laminated marls (2.0 to 2.5)and more variable for the non-laminated marls (0.8
to 4.5) with a tendency to higher values above
110m depth. Data frequency for limestones and sili-
ci®ed marls (K, KM) is insu�cient for interpret-ation and only trace amounts of hopanoids occur in
KK and KW samples.
The high abundance of unsaturated hopenes con-
®rms a low level of maturity and has been noted
for a variety of hypersaline environments (Boonet al., 1983; ten Haven et al., 1985, 1988; Kohnen
et al., 1991; de las Heras et al., 1997). The low
maturity is in accordance with petrological results(Vliex and Schwark, 1998). In contrast, the degree
of isomerisation at the C22 position of the
hop(17,21)enes has already reached equilibrium
values of (22S/(22S+22R))=0.50±0.53. Accordingto Sinninghe Damste et al. (1995) the isomerisation
for homohop(17,21)enes reaches an end point at
0.52±0.53 for the 22S/(22S+22R) ratio. This anda C27 a/(a+b) ratio of 0.80±0.82 would either
correspond to higher level of maturity or point
towards a speci®c depositional environment and
diagenetic history.
Comparable distributions of C31 to C35-
hop(17,21)enes and hopanes showing similar con-
versions rates at C22 were identi®ed by Boon et al.(1983), ten Haven et al. (1985, 1988) and Kohnen et
al. (1991) in hypersaline environments. Ten Haven
et al. (1985, 1988) concluded that in hypersaline en-vironments hopanes were formed from hopenes by
direct reduction rather than by isomerisation of the
unstable 17b(H),21b(H)hopanes and regard this
speci®c pattern as characteristic for an enhancedsalinity environment.
The distribution pattern of the C31 to C35
analogues provides further arguments for high
reduction potential and salinity in the palaeo-
depositional environment during sedimentation ofthe marls due to high abundance of the extended
hop(17,21)enes. Sample ML2 is exceptional for con-
taining no hopenes and samples ML5 and ML125show only trace amounts. Despite of this, a clear
di�erentiation between laminated and non-lami-
nated marls can be achieved by extended hopene
distribution (Fig. 6). Laminated marls show a moresymmetrical distribution with a maximum at the
C34-hopenes whereas non-laminated marls systema-
tically show a decrease from C31 to C32 membersfollowed by maximum at C33 and then a constant
decline of the C34 and C35 members. Silici®ed marls
reveal an extended hopene distribution maximising
at C31 and then smoothly declining to the C35
Fig. 6. Mass fragmentograms of m/z= 191 and 367 showing distribution of hopanes and hopenes forthree lithofacies types marls, silici®ed carbonates and laminated marls. Numbers indicate numbers ofcarbon atoms. Filled triangles represent hop(17,21)enes, stars denote neohop(13,18)enes, open circles in-
dicate moretanes and ®lled circles ab-hopanes.
L. Schwark et al.1932

member (Fig. 6). Pure limestones and cherts do not
contain hopenes.
Steranes and sterenes. The saturated and unsatu-
rated steroids consist of C27 to C29 compounds with
only traces of C30 and no C26 components (Fig. 7).
Diasterenes are missing in the samples as character-
istic for many clay-depleted carbonatic environ-
ments. However, D13(17)-spirosterenes (Fig. 8) occur
in signi®cant amounts and monoaromatic des-
methyl- and methylsteranes are major and often the
dominating members of aromatic fractions. Due to
the low maturity and possibly due to the low
amount of clay mineral catalysts, the steroid distri-
bution is dominated by instable intermediate pro-
ducts of steroid diagenesis. Unusual distributions of
steroids were encountered that are related to vari-
ations in (i) biological input into an environment
stressed by high salinity and anoxia or (ii) the early
diagenetic conditions. These will be in¯uenced by
the special mineralogy of the sediments and possibly
by exceptional microbial mediation pathways of
early diagenetic reactions.
The distribution pattern of the saturated steranes
is strongly a�ected by these in¯uences and reveals
an unusual pattern of aaa- and abb-steranes (Fig. 7),with diagenetically intermediate baa-steranes miss-
ing, except for traces of the C27baa-sterane. The
aaa-steranes are dominated by C27- and C29-com-
pounds (40±50% and 35±45%, respectively) with
C28-steranes comprising only 10±15%. The abb-steranes are composed of lower amounts of C27-
and C29 steranes (25±30%) but contain up to 50%
C28-steranes. Consistent with the low maturity of the
bitumens, the ratio of aaa/abb-steranes is very high
for cholestane and ethylcholestane. Accordingly, the
ratio of 20S to 20R isomers of those compounds is
particularly low, with 20S-members often missing.
The methylcholestanes, on the contrary, show a
pronounced dominance of the abb- over aaa-ster-anes (Fig. 7). This deviation can not be related to
an increased thermal maturity exclusively for the
methylcholestanes (e.g. due to impregnation with
more mature migrated bitumens) but rather re¯ects
a speci®c input, possibly from D7-sterenes (de
Leeuw and Sinninghe Damste , 1990) via intermedi-
ate formation of D13(17)-spirosterenes (Peakman et
al., 1989).
These labile compounds occur with large vari-
ations of the 4 most prominent isomers 5b20R-,5b20S-, 5a20R and 5a20S-D13(17)-spirosterenes
(Fig. 8). For C27-members, a ratio of 0.8 was deter-
mined for the relative abundance of 5b/5a-isomers
(based on intensities in m/z = 206 mass fragmento-
grams). The C28-compounds give a 5b/5a-isomer
ratio of 5.0, whereas the C29-spirosterenes again
reveal a 5b/5a-isomer ratio of only 0.4 (Fig. 8). The
diagenesis of spirosteroids must have proceeded
di�erently for the C28-member as compared to the
C27- and C29-compounds and most likely was
triggered by variable contributions from a speci®c
precursor sterol.
The sterene distribution con®rms the observation,
that C27- and C29-steroids show similarities but dif-
fer strongly from C28-steroids. No regular sterenes
and only trace amounts of diasterenes were detected
in the samples but a series of compounds eluting
prior to the regular steranes occurs in signi®cant
abundance (Fig. 8). These were tentatively identi®ed
as C27 to C30-sterenes by mass spectra [Fig. 9(a)].
The mechanism of formation for these sterenes is
not clear and an origin from other precursors, e.g.
norlanostenes might not be excluded but is not in
agreement with the C27 to C29 distribution pattern.
Four isomers occur for each sterene, all compounds
characterized by a strong M+-43 ion, indicating
loss of an isopropyl-group and a strong signal at
M+-56 giving ions of m/z = 314, 328 and 342,
respectively [Fig. 9(a)].
The compound distribution for this steroid
type is C29>C28>C27>>C30 and thus di�ers
markedly from those of saturated steranes
Fig. 7. Distribution of steranes and sterenes exemplarilyshown for sample M110 as given by mass fragmentogramsof m/z = 217, 218 and 215. Numbers indicate total num-ber of carbon atoms per molecule. Triangles representsterenes, star indicates baa-cholestane, ®lled and opensquares denote abb-steranes with 20R and 20S con®gur-ation and ®lled and open circles indicate aaa-steranes with
20S and 20R con®guration, respectively.
Geochemistry of Malm carbonates 1933

Fig.8.(a)Mass
fragmentogramsofm/z
=206,220and234indicate
distributionofC27to
C29spiro(13,17)enes.Open
circlesandsquaresindicate
5b-isomerswith20S
and20R
con®guration
and
®lled
circlesandsquaresindicate
5a-isomerswith
20S
and20R
con®guration,respectively.Shaded
peaksin
them/z
=234-fragmentogram
depictdialkylthiophenes.(b)Mass
fragmentogramsofm/z
=370,384,398indicatingaseries
ofunknownC27to
C29sterenes,each
comprising4isomersthatelute
shortly
after
thespirosterenes.TheC27andC29mem
bersshow
similarisomer
distributionpatterns,whereastheC28analogues
displayadi�erentisomer
distribution.Mass
spectra
ofcompoundsare
shownin
Fig.9(a).(c)Mass
fragmentogramsofm/z
=368,382,396.Thesterenes
shownin
(b)are
accompaniedbyaseries
ofunknownsteradienes
elutingin
thearomaticfraction.Theoccurrence
ofthesecompoundswaspreviouslyreported
fortheOrbanouxsite
byvanKaam-PetersandSinningheDamsteÂ(1997).
Correspondingmass
spectraare
shownin
Fig.9(b).
L. Schwark et al.1934

(C27rC29>>C28>>C30). As already noted for thespirosterenes and regular steranes, the C27- and C29-compound show a similar distribution of isomers
that is quite distinguishable from those of the C28-members [Fig. 8(b)]. Therefore, although the precisestructure of the modi®ed sterenes has still to be
fully elucidated, the biological precursors of theC28-steroids must be di�erent from those of theC27- and C29-members and consequently undergo a
di�erent diagenesis route.Because of low thermal maturity of Malm z bitu-
mens, deviations in early diagenesis pathways mustaccount for the irregular isomer pattern of satu-
rated steranes. Enhancement in the abundance ofabb-steranes has been previously observed in hyper-saline environments (ten Haven et al., 1985, 1988;
de Leeuw and Sinninghe Damste , 1990) and a poss-ible input of D7-sterols was proposed by Peakmanet al. (1989) and de Leeuw and Sinninghe DamsteÂ
(1990). In the case of the Malm carbonates sucha sterol input seems likely and this diageneticroute is supported by the concomitant occurrence
of spirosterenes. Furthermore, the similar isomerdistribution pattern of the unknown sterenes andthe spirosterenes might indicate a microbial me-
diation that preferentially a�ects the C28-memberdue to its biological inheritance of the double bondposition.
A marine origin has to be encountered forall steroids because due to the absence of otherterrestrially derived biomarkers and microscopicalobservations the terrigenous input for this deposi-
tional setting was extremely low. A large varietyof C28-sterols is produced by autotrophic plank-tonic organisms (Scheuer, 1978; Volkman, 1986,
1988) that will allow for di�erent precursors anddiagenetic routes as compared to C27- and C29-steranes.
Fig. 9. (a) Mass spectra of unknown sterenes shown in Fig. 8(b). Mass spectra were recorded from thehighest peak, i.e. the third eluting isomer. (b) Mass spectra of unknown steradienes shown in Fig. 8(c).
Mass spectra are shown for the ®rst eluting isomer.
Geochemistry of Malm carbonates 1935

Aromatic fractions
Aromatic fractions of Malm z carbonates o�er a
spectrum of components that is dictated by lowmaturity and high availability of reduced sulfur
species. Due to the low thermal maturity and thelack of terrigenic input, naphthalenes, phenan-
threnes, dibenzothiophenes, ¯uorenes, biphenyles
and their alkylated homologues are found only intrace amounts.
As a result the aromatic fraction is mainly com-
posed of aromatic steroids, benzohopanes, alkyl-chromans, perylene, an aromatized triterpenoid and
minor amounts of alkylbenzenes. In the intra-reefal
iron-limited depositional environment sulfur incor-poration into functionalized lipids (Sinninghe
Damste et al., 1990) occurred frequently and severalseries of organic sulfur compounds are present
including various acyclic thiophenes and thienylho-panes. No sulfur bearing steroids or carotenoids
were detected.
Benzohopanes are typical constituents of carbona-
tic depositional environments and are suggested tobe formed by cyclisation of bacterial C35-hopanoids
during very early stages of diagenesis (Hussler etal., 1984; Wei and Songnian, 1990; Schae�er, 1993).
These compounds occur ubiquitously and do not
o�er much potential for environmental characteriz-ation. In signi®cant abundances a B-ring monoaro-
matic triterpenoid of the fernane/arborane type(Hauke et al., 1992) occurs in all samples. A mi-
crobial as well as an angiospermal origin for the
arborene/fernene precursor is possible (Hauke et al.,1992, 1995) but due to the widespread occurrence in
pre-Cretaceous sediments a bacterial or even algalsource has been postulated for these compounds.
This bacterial input also has to be encountered forthe Malm z samples which predate the evolution of
angiosperms. None of the A-ring degraded or dia-
romatic analogues often co-occurring with themonoaromatic pentacyclic triterpenoid were
detected and no further paleoenvironmental infor-mation can be derived from the occurrence of the
ferna/arboratriene.
Steroids. Monoaromatic desmethyl and 4-methyl-steroids occur in large concentrations in the Malm
samples and often comprise the most abundantcompounds in the aromatic fraction. The C27 to C29
desmethyl monoaromatic steroid distribution shows
no contribution of rearranged steroids comparableto the lack of diasterenes in the aliphatic fraction.
Due to co-elution of the C28abR- with the C29bbR-isomer a precise calculation of the relative contri-
butions is hindered but the ethylcholestanes always
dominate the monoaromatic steroid distributionpattern. On average a distribution of 30:45:25 for
the C27:C28:C29 monoaromatic steroids occurs,which is directly opposite to the one observed for
the saturated steroids. Biological input estimation
based on steroid distribution following the ternary
discrimination plot of Huang and Meinschein
(1979), as often applied in paleoenvironment assess-
ment (Peters and Moldowan, 1993), will thus give
highly unreliable results.
The ratio of desmethyl- vs. 4-methyl-monoaro-
matic steroids varies between 1.3 and 2.8. The high
amount of 4-methyl-monoaromatic steroids indi-
cates a major production of organic matter by mar-
ine algae, including those from microscopically
identi®ed dino¯agellates, in the Rennertshofen
Trough.
A series of C26 to C29-steradienes was detected in
the aromatic fraction [Fig. 8(c)], which was pre-
viously reported by van Kaam-Peters and Sinninghe
Damste (1997) for the Jurassic Carbonates at
Orbanoux. The structures of these compounds are
still unknown and the mass spectra exhibit a strong
similarity with those of the unknown sterenes found
in the aliphatic fraction [Fig. 9(c)]. The distribution
pattern shows no clear relationship to any of the
other steroid classes.
Three C27 to C29-compounds occuring in the aro-
matic fraction were tentatively identi®ed as posses-
sing a diaromatic steroid skeleton based on the
occurrence of a m/z= 249 mass fragment and mol-
ecular ions reduced by 4 Da as compared to mono-
aromatic steroids. A/B diaromatic steroids were
tentatively identi®ed in Monterey Formation by de
Lemos Sco®eld (1990) and di�erent MS±MS exper-
iments as well as chemical ionisation was carried
out on these components. However, the mass spec-
tra (EI, 70 eV) of those diaromatic steroids do not
fully match with those from the Malm samples and
therefore a related, e.g. anthrasteroidal or regular
vs. rearranged, structure has to be assumed. The
distribution of the diaromatic steroids shows a clear
dominance of the C27 and C29 compounds, whereas
the diaromatic methyl cholestane occurs in modest
amounts only. This pattern is comparable to the
one of the saturated steranes and opposite to that
of the monoaromatic steroids. This indicates a
further complication of the diagenetic history of
steroids in the Malm samples from the
Rennertshofen Trough. Palaeoenvironmental assess-
ment based on one of the standard saturated or
monoaromatic steroid composition parameters can
not provide a representative picture of biological
input and early diagenetic conditions (Figs 7, 8 and
10).
Perylene. The most prominent fully aromatized
constituent of the aromatic fractions is perylene, a
compound that frequently occurs in marine and
lacustrine environments, even if other PAH-
compounds are missing and therefore a combustion
origin is excluded. Aizenshtat (1973), La¯amme
and Hites (1978), Gschwend et al. (1983) and
Louda and Baker (1984) suggested, that perylene
may be directly derived from biogenic precursors.
Aizenshtat (1973) described that the reduction of
L. Schwark et al.1936

chinone pigments would produce perylenes. These
pigments have been found in insects (Cameron et
al., 1964), fungi (Thompson, 1979) but also in leafs
of terrestrial plants (Orr and Grady, 1967). Because
chinone pigments are sensitive to oxidation, a for-
mation of perylene under anoxic condition was pro-
posed (Orr and Grady, 1967; Wakeham et al., 1979;
Louda and Baker, 1984). However, chinone pig-
ments with a perylene skeleton are not abundant in
nature and there is a contrast between pigment con-
centration and abundance of perylene of sediments
(Watts and Maxwell, 1977). Wakeham et al. (1979)
proposed that precursors of perylene are not necess-
arily of terrestrial origin, because the occurrence of
high amounts of perylene in marine sediments (Orr
and Grady, 1967; La¯amme and Hites, 1978; Wake-
ham et al., 1979; Louda and Baker, 1984; Venkate-
san, 1988).
In case of the Malm z carbonates a terrestrial ori-
gin of perylene is not likely, because of a lack of
other terrestrial biomarkers. A marine precursor, as
suggested by Wakeham et al. (1979) could be an
Fig. 10. In addition to the steradienes shown in Figs 8 and 9 and the dominating monoaromatic ster-oids (accompanied by traces of triaromatic steroids), the aromatic fractions of the Malm z samplesreveal the presence of tentatively identi®ed diaromatic steroids as shown by a) the mass fragmentogramof base peaks m/z = 210. Mass spectra recorded for the C27 and C29 diaromatic steroids are shown in
(b) and (c), respectively.
Geochemistry of Malm carbonates 1937
alternative explanation for the high concentration.
Louda and Baker (1984) proposed sul®de sensitive
marine microbes or algae (diatoms, which are unli-
kely for Malm z samples) as possible precursors.
This explanation would be in accordance with the
facies conditions of Malmian samples, where main
producers of organic matter are algae or bacteria. It
is generally accepted that high abundances of pery-
lene are associated with strictly anoxic conditions
(Venkatesan, 1988; Peters and Moldowan, 1993)
although the potential biogenic origin and the diag-
entic route for perylene formation are still not unra-
velled (Silliman et al., 1997).
Chromans. Four alkylated 2-methyltrimethyltride-
cylchromans (MTTCs) were detected in Malm zsamples by ion chromatograms of m/z =
121 + 135 + 149. These compounds were ®rst dis-
covered in sediments and crude oils by Sinninghe
Damste et al. (1987a) and it was proposed that
MTTCs are directly biosynthesised because of their
limited numbers of potential isomers. Widespread
tocopheroles, although structurally closely related
to chromans, were excluded to be precursors of
MTTCs. Although biological precursors for
MTTCs could not be identi®ed until today, based
on compound speci®c carbon isotope data, an ori-
gin from eu- or archaebacteria has been proposed
(de Leeuw and Sinninghe Damste , 1990; Sinninghe-
Damste et al., 1993; Kenig et al., 1995). An alterna-
tive formation of chromans via condensation of al-
kylated phenols with phytol was postulated by Li et
al. (1995) and the generation mechanism of MTTCs
is still under debate (Li and Larter, 1995; Sinninghe
Damste and de Leeuw, 1995).
Based on empirical observations it was suggested
that in sediments from non-hypersaline environ-
ments 5,7,8-trimethyl-MTTC dominates and 8-
methyl-MTTC is completely missing. For the inves-
tigated samples 5,7,8-trimethyl-MTTC is the most
prominent compound, but 8-methyl-MTTC also
occurs, which infers enhancement of salinity but no
severe hypersalinity (i.e. >120-). Similar obser-
vations were made for the Permian Kupferschiefer
(Schwark and PuÈ ttmann, 1989; Grice et al., 1997) in
agreement with slightly enhanced ``mesosaline'' con-
ditions. Mesosaline refers to a salinity between 40
and approximately 120-, i.e. before onset of gyp-
sum precipitation at 140- (Kirkland and Evans,
1981). For a more elaborated characterisation of
palaeosalinities the MTTC ratio was de®ned by
Sinninghe Damste et al. (1989); Sinninghe-DamsteÂ
et al. (1993) as: MTTC-ratio = 5,7,8-trimethyl-
MTTC/total MTTCs. For the Malm z sediments
investigated here, the ratio was calculated to vary
from 0.55±0.61, indicating no signi®cant changes in
palaeosalinity.
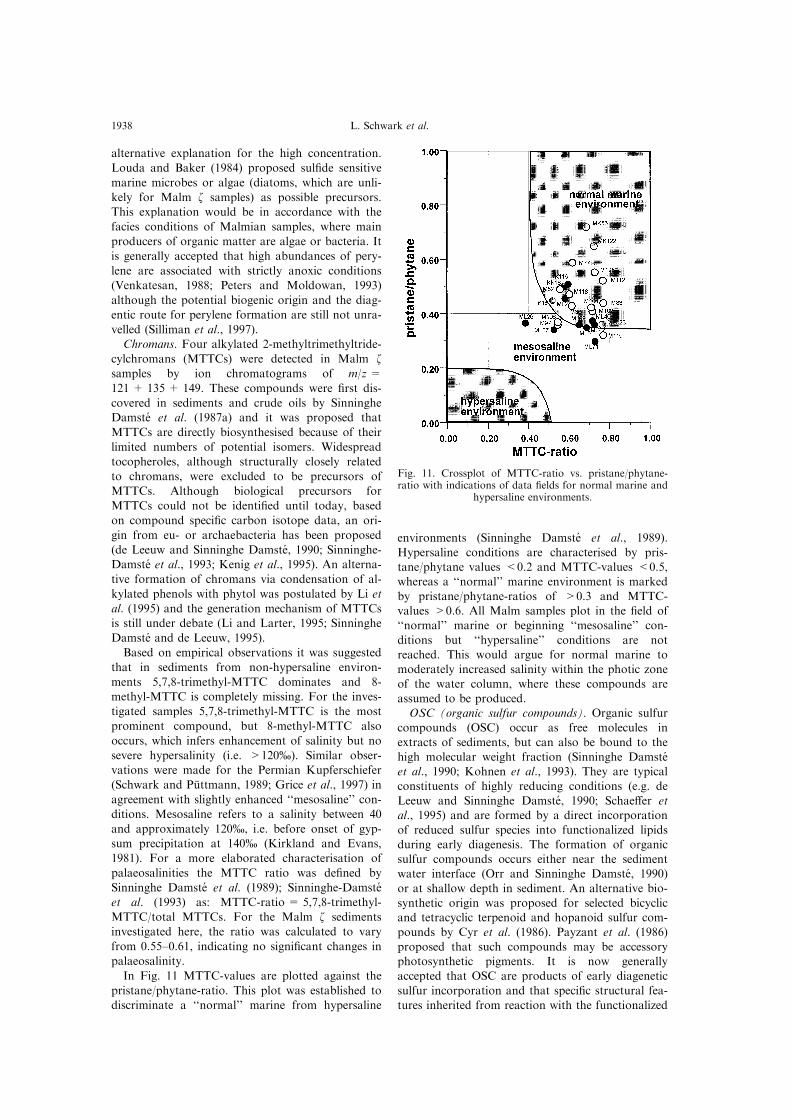
In Fig. 11 MTTC-values are plotted against the
pristane/phytane-ratio. This plot was established to
discriminate a ``normal'' marine from hypersaline
environments (Sinninghe Damste et al., 1989).
Hypersaline conditions are characterised by pris-tane/phytane values <0.2 and MTTC-values <0.5,
whereas a ``normal'' marine environment is marked
by pristane/phytane-ratios of >0.3 and MTTC-values >0.6. All Malm samples plot in the ®eld of
``normal'' marine or beginning ``mesosaline'' con-
ditions but ``hypersaline'' conditions are not
reached. This would argue for normal marine tomoderately increased salinity within the photic zone
of the water column, where these compounds are
assumed to be produced.OSC (organic sulfur compounds). Organic sulfur
compounds (OSC) occur as free molecules in
extracts of sediments, but can also be bound to the
high molecular weight fraction (Sinninghe DamsteÂ
et al., 1990; Kohnen et al., 1993). They are typical
constituents of highly reducing conditions (e.g. de
Leeuw and Sinninghe Damste , 1990; Schae�er etal., 1995) and are formed by a direct incorporation
of reduced sulfur species into functionalized lipids
during early diagenesis. The formation of organicsulfur compounds occurs either near the sediment
water interface (Orr and Sinninghe Damste , 1990)
or at shallow depth in sediment. An alternative bio-
synthetic origin was proposed for selected bicyclicand tetracyclic terpenoid and hopanoid sulfur com-
pounds by Cyr et al. (1986). Payzant et al. (1986)
proposed that such compounds may be accessoryphotosynthetic pigments. It is now generally
accepted that OSC are products of early diagenetic
sulfur incorporation and that speci®c structural fea-
tures inherited from reaction with the functionalized
Fig. 11. Crossplot of MTTC-ratio vs. pristane/phytane-ratio with indications of data ®elds for normal marine and
hypersaline environments.
L. Schwark et al.1938

lipids are preserved in the OSC structures thus
allowing to reconstruct original biological input
(Kohnen et al., 1993). The free OSC occuring in the
``aromatic'' fractions were identi®ed based on com-
parison of mass spectra and ion chromatograms
with reference spectra in the literature.
Thienyl hopanes. The C35-thienyl hopane isomers
17a(H),21b(H), 17b(H),21a(H) and 17b(H),21b(H)
commonly found in sulfur-rich environments (Vali-
solalao et al., 1984) were identi®ed in all samples
investigated. It was suggested that these hopanes
derive directly from sulfur incorporation into bac-
teriohopanetetrol, the reason for high C35-concen-
tration being a spontaneous reaction of the alcohol
with reduced sulfur. This reaction occurs at early
stages of diagenesis in strongly anoxic environments
preventing biotransformation or mineralisation (de
Leeuw and Sinninghe Damste , 1990). High concen-
trations of thienyl- and benzohopanes indicate an
intensive reworking of organic matter or a deri-
vation of hopanoids from cyanobacteria (Schae�er
et al., 1995).
The occurence of C35-thienyl-hopanes is in agree-
ment with the dominance of extended hop(17,21)-
enes in the aliphatic fraction. Ten Haven et al.
(1985, 1988) reported, that in many samples re¯ect-
ing high saline environments extended C35-members
preferentially occur. High amounts of benzo- and
thienylhopanes together with the increased abun-
dance of C35-hopanes indicate strongly reducing
conditions and may also point towards a higher
saline environment.
Alkylthiophenes. Incorporation of sulfur in unsa-
turated aliphatic precursors produces substituted
thiophenes including 2-alkylthiophenes, 2-alkyl-5-
methylthiophenes, 2-alkyl-5-ethylthiophenes, various
mid chain 2,5- and 3,4-dialkylthiophenes and mono-
methyl-thiophenes (Sinninghe Damste et al., 1986,
1987a, 1989, 1990; Peakman and Kock-van Dalen,
1990; Kohnen et al., 1991, 1993; Russell et al.,
1997, de las Heras et al., 1997). Except for the long-
chain 3,4-dialkylthiophenes all compound classes
are present in the Malm samples investigated.
Inspection of the m/z = 111 mass fragmentogram
shows 2-alkyl-5-methylthiophenes (referred to as
MATP) with the n-alkyl side chain ranging from C9
to C27 dominating in all samples (Fig. 12). A trimo-
dal distribution is observed in most samples, the
®rst maximum occuring at the C18-, the second at
the C21- and the third at C24- or C26-MATP.
Di�erences in chain length distribution can be used
to group samples into two classes of long chain
dominated (LC-MATP) and short chain (SC-
MATP) methyl-alkylthiopenes. Samples ML26 and
ML 80 represent SC-MATP, whereas samples
ML69 and ML71 belong to the LC-MATP group
with sample ML67 taking an intermediate position.
The nonlaminated marls can be subgrouped accord-
ingly with samples M102, M104 and M106 repre-
senting SC-MATP, samples M52, M110, M112,
M118 representing LC-MATP and samples M30
and M77 showing equally distributed MATP.
Intervals of similar chain length distribution over
the studied core section seem to be indicated but
the current database does not allow for further in-
terpretations due to the high variability of sedi-
ments and organic fractions. Presently, it can only
be speculated that the controls on chain length dis-
tribution of the MATP may be related to the degree
of sulfurization and thus the proximity of the che-
mocline to the bioproductive photic zone (e.g.
Kenig et al., 1995). The distribution pattern of
MATP can however be shown to be largely inde-
pendent from lithofacies type. Although lower
amounts of MATP were found in the non-marly
lithologies, the K samples show comparable distri-
butions and MK also show identical patterns. A
possible explanation might be that after sulfuriza-
tion of lipids had already proceeded, a sili®cation
of porous carbonate layers occurred, which thus did
not a�ect the MATP distribution. The MATP-pat-
tern shows a slight even predominace in the C18 to
C27 range which is in contrast to n-alkanes charac-
terized by a moderate odd over even predominance
(Fig. 5). Various ``shift'' phenomena between n-
alkanes distribution and patterns of methyl-, ethyl-
and propyl-substituted n-alkyl-thiophenes were
recognised by Sinninghe Damste et al. (1986) but a
satisfying explanation for the di�erent behaviour of
alkylthiophenes is still missing.
The characteristic low CPI-values of the free n-
alkanes with special preference of the C22 and C24
members observed in other enhanced salinity en-
vironments (ten Haven et al., 1985, 1988; Barbe et
al., 1990), might be obscured by the preferential sul-
furization of these compounds and thus their occur-
ence as MATP. Upon release of these compounds
from the thiophene fraction the expected hypersa-
line n-alkane distribution will be generated. A selec-
tive incorporation of the C22-precursor into the
kerogen matrix is unlikely due to the free occurence
of these compound in other settings (Barbe et al.,
1990).
Non-substituted and ethyl- or propyl-substituted
alkylthiophenes occur in minor concentrations as
revealed by inspection of m/z = 97, 125 and 139
mass chromatograms. Due to comparably low
abundance and and multiple isomers for the substi-
tuted series those compounds are not further dis-
cussed here.
A second series of 2,5-di-n-alkylthiophenes or
midchain dialkylthiophenes (referred to as DATP)
is very prominent in the higher molecular weight
region (Fig. 12). The peaks labelled w in Fig. 12
comprise a series of three DATP-isomers per peak.
The C27-compound mixture for example consists of
three isomers possessing a n-C9/n-C14-, n-C10/n-C13-
and n-C11/n-C12-di-n-alkylsubstitution pattern. A
Geochemistry of Malm carbonates 1939

distribution starting with three coeluting C20-iso-
mers: n-C6/n-C10-, n-C7/n-C9- and n-C8/n-C8-di-n-
alkylthiopene continues to C29 by addition of one
acetyl-group alternatingly to both side chains.
The DATP distribution extends from C22 to C30
in most samples and is characterized by a strong
preference of the C26- followed by the C24 com-
pound. The pattern reveals a strong predominance
of the even-numbered homologues with OEP-values
of 0.55 to 0.95 in the C23- to C28-DATP range. No
dependence of OEP-values calculated from DATP-
distributions with lithology was observed. This
again demonstrates that organic matter input and
early diagenesis was not primarily controlled by
Fig. 12. Mass fragmentograms of m/z= 111 indicative for alkylthiophenes plotted for three samples ofthe marl lithofacies type. Numbers indicate total numbers of carbon atoms per molecule. Filled circlesdenote 2-methyl-5-alkylthiophenes, open circles indicate dialkylthiophenes occurring as mixtures of
three di�erent isomers (see text).
L. Schwark et al.1940
lithofacies and that organic matter distribution will
provide additional palaeoenvironmental infor-
mation.
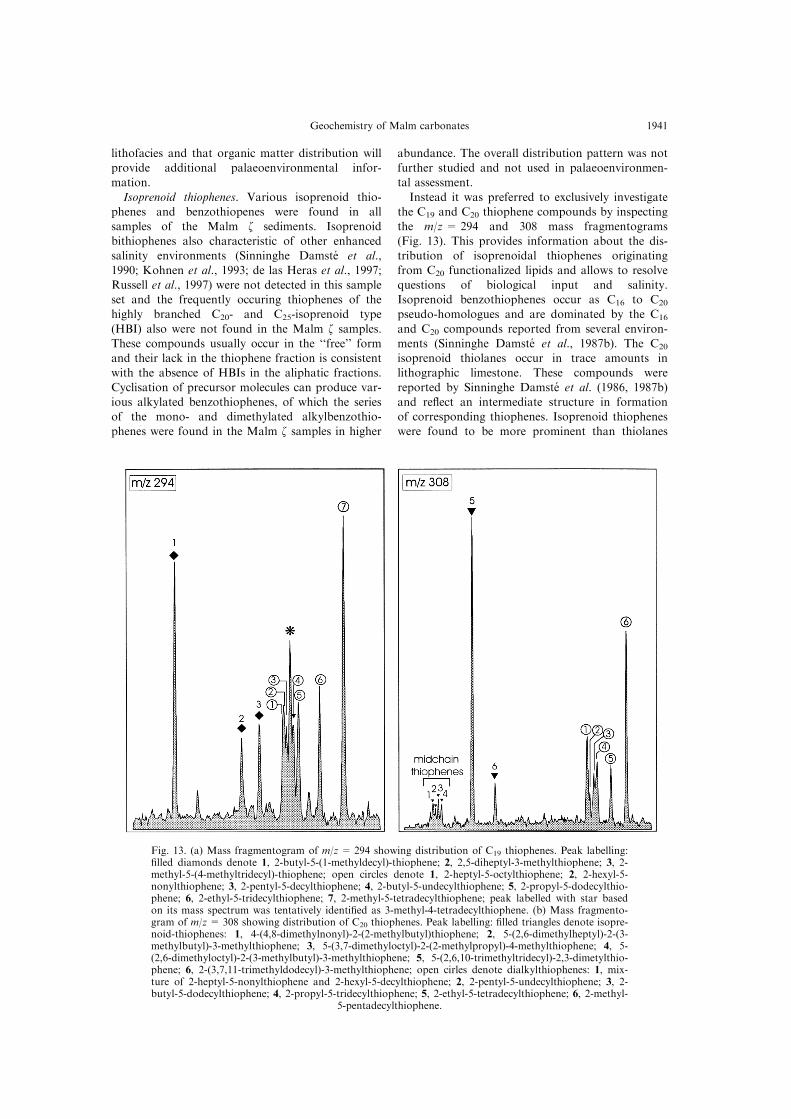
Isoprenoid thiophenes. Various isoprenoid thio-
phenes and benzothiopenes were found in all
samples of the Malm z sediments. Isoprenoid
bithiophenes also characteristic of other enhanced
salinity environments (Sinninghe Damste et al.,
1990; Kohnen et al., 1993; de las Heras et al., 1997;
Russell et al., 1997) were not detected in this sample
set and the frequently occuring thiophenes of the
highly branched C20- and C25-isoprenoid type
(HBI) also were not found in the Malm z samples.
These compounds usually occur in the ``free'' form
and their lack in the thiophene fraction is consistent
with the absence of HBIs in the aliphatic fractions.
Cyclisation of precursor molecules can produce var-
ious alkylated benzothiophenes, of which the series
of the mono- and dimethylated alkylbenzothio-
phenes were found in the Malm z samples in higher
abundance. The overall distribution pattern was not
further studied and not used in palaeoenvironmen-
tal assessment.
Instead it was preferred to exclusively investigate
the C19 and C20 thiophene compounds by inspecting
the m/z = 294 and 308 mass fragmentograms
(Fig. 13). This provides information about the dis-
tribution of isoprenoidal thiophenes originating
from C20 functionalized lipids and allows to resolve
questions of biological input and salinity.
Isoprenoid benzothiophenes occur as C16 to C20
pseudo-homologues and are dominated by the C16
and C20 compounds reported from several environ-
ments (Sinninghe Damste et al., 1987b). The C20
isoprenoid thiolanes occur in trace amounts in
lithographic limestone. These compounds were
reported by Sinninghe Damste et al. (1986, 1987b)
and re¯ect an intermediate structure in formation
of corresponding thiophenes. Isoprenoid thiophenes
were found to be more prominent than thiolanes
Fig. 13. (a) Mass fragmentogram of m/z = 294 showing distribution of C19 thiophenes. Peak labelling:®lled diamonds denote 1, 2-butyl-5-(1-methyldecyl)-thiophene; 2, 2,5-diheptyl-3-methylthiophene; 3, 2-methyl-5-(4-methyltridecyl)-thiophene; open circles denote 1, 2-heptyl-5-octylthiophene; 2, 2-hexyl-5-nonylthiophene; 3, 2-pentyl-5-decylthiophene; 4, 2-butyl-5-undecylthiophene; 5, 2-propyl-5-dodecylthio-phene; 6, 2-ethyl-5-tridecylthiophene; 7, 2-methyl-5-tetradecylthiophene; peak labelled with star basedon its mass spectrum was tentatively identi®ed as 3-methyl-4-tetradecylthiophene. (b) Mass fragmento-gram of m/z= 308 showing distribution of C20 thiophenes. Peak labelling: ®lled triangles denote isopre-noid-thiophenes: 1, 4-(4,8-dimethylnonyl)-2-(2-methylbutyl)thiophene; 2, 5-(2,6-dimethylheptyl)-2-(3-methylbutyl)-3-methylthiophene; 3, 5-(3,7-dimethyloctyl)-2-(2-methylpropyl)-4-methylthiophene; 4, 5-(2,6-dimethyloctyl)-2-(3-methylbutyl)-3-methylthiophene; 5, 5-(2,6,10-trimethyltridecyl)-2,3-dimetylthio-phene; 6, 2-(3,7,11-trimethyldodecyl)-3-methylthiophene; open cirles denote dialkylthiophenes: 1, mix-ture of 2-heptyl-5-nonylthiophene and 2-hexyl-5-decylthiophene; 2, 2-pentyl-5-undecylthiophene; 3, 2-butyl-5-dodecylthiophene; 4, 2-propyl-5-tridecylthiophene; 5, 2-ethyl-5-tetradecylthiophene; 6, 2-methyl-
5-pentadecylthiophene.
Geochemistry of Malm carbonates 1941
and isoprenoidal benzothiophenes in the Malm zsamples. Isoprenoid thiophenes have been reported
from oils and sediments from a variety of environ-
ments by Brassell et al. (1986), Sinninghe Damste et
al. (1986, 1987b, 1989, 1990); Orr and Sinninghe
Damste (1990); Peakman and Kock-van Dalen
(1990); Kohnen et al. (1993) and Russell et al.
(1997). Most prominent isoprenoid thiophenes are
the C20 homologues, which show a characteristic
pattern in the m/z = 308 ion chromatogram. After
Orr and Sinninghe Damste (1990) the structure of
C20 isoprenoids strongly suggests an origin from in-
corporation of reduced inorganic sulfur into phytol
and/or archeobacterial phytenes or their diagenetic
products. Sulfurisations reactions of phytenic acid
and phytenol take place in sediments (Kohnen et
al., 1993). C20 isoprenoid thiophenes occur in sedi-
ments of ``normal'' marine to ``hypersaline'' en-
vironments in varying concentrations and were
therefore introduced as indicators for palaeosalinity
(Sinninghe Damste et al., 1989; Kohnen et al.,
1990; de Leeuw and Sinninghe Damste , 1990).
In Fig. 13 the ion chromatograms of m/z = 294
and 308 illustrate the distribution of C19 and C20
thiophenes. Terminal isoprenoid thiophenes V, VI
and mid chain thiophene isoprenoids I±IV accord-
ing to the notation used by de Leeuw and
Sinninghe Damste (1990) and Kohnen et al. (1990)
for calculations of the isoprenoid thiophene ratio
ITR, are identi®ed in the m/z= 308 fragmentogram
by comparison with published mass spectra and
retention times (Sinninghe Damste et al., 1986,
1987a, 1989). In addition to the commonly found 2-
methyl-5-pentadecyl- and 2-ethyl-5-tetradecylthio-
phenes two chromatographically resolved di-n-
alkylthiophenes occur (2-propyl-5-tridecyl- and 2-
butyl-5-dodecylthiophene) and the m/z = 308 mass
fragmentogram shows a further peak containing a
mixture of 2-octyl-5-nonyl-, 2-heptyl-5-decyl- and 2-
hexyl-5-undecyl-thiophenes. HBI-thiophenes as
mentioned above are missing. The C19 compound
distribution as illustrated by the m/z = 294 mass
chromatogram is devoid of any isoprenoid thio-
phenes (Fig. 13) but in addition to a series of 2,5-
dialkylthiophenes three methyl-branched thiophenes
are present (Fig. 13). These branched thiophenes
most probably originate from 9-methyl-nonadeca-
diene precursors (Sinninghe Damste et al., 1989)
and thus provide evidence for a speci®c microbial
input, probably from sulfur bacteria (Sinninghe
Damste et al., 1989).
Isoprenoid thiophene V is the most prominent
constituent of the C20 thiophenes and together with
the C20 isoprenoids possessing a midchain thio-
phene moiety, occurs especially in environments of
enhanced salinity. Isoprenoid thiophene VI occurs
in minor amounts in the Malm samples. In normal
marine environments it usually is accompanied by
the isoprenoid VII carrying the thiophene moiety in
a terminal position. For assessment of palaeosali-
nity the isoprenoid thiophene ratio is calculated as
ITR = (VI + VII)/(I + II + III + IV + V)
according to Sinninghe Damste et al. (1987b) and
de Leeuw and Sinninghe Damste (1990).
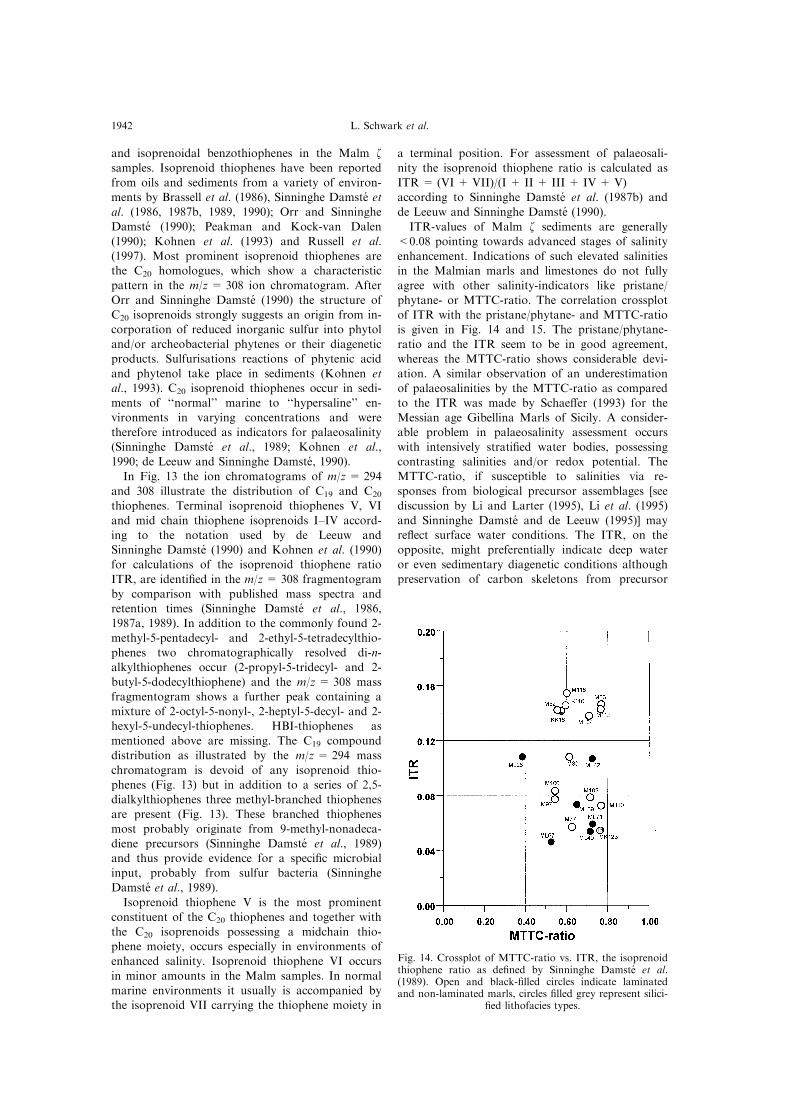
ITR-values of Malm z sediments are generally
<0.08 pointing towards advanced stages of salinity
enhancement. Indications of such elevated salinities
in the Malmian marls and limestones do not fully
agree with other salinity-indicators like pristane/
phytane- or MTTC-ratio. The correlation crossplot
of ITR with the pristane/phytane- and MTTC-ratio
is given in Fig. 14 and 15. The pristane/phytane-
ratio and the ITR seem to be in good agreement,
whereas the MTTC-ratio shows considerable devi-
ation. A similar observation of an underestimation
of palaeosalinities by the MTTC-ratio as compared
to the ITR was made by Schae�er (1993) for the
Messian age Gibellina Marls of Sicily. A consider-
able problem in palaeosalinity assessment occurs
with intensively strati®ed water bodies, possessing
contrasting salinities and/or redox potential. The
MTTC-ratio, if susceptible to salinities via re-
sponses from biological precursor assemblages [see
discussion by Li and Larter (1995), Li et al. (1995)
and Sinninghe Damste and de Leeuw (1995)] may
re¯ect surface water conditions. The ITR, on the
opposite, might preferentially indicate deep water
or even sedimentary diagenetic conditions although
preservation of carbon skeletons from precursor
Fig. 14. Crossplot of MTTC-ratio vs. ITR, the isoprenoidthiophene ratio as de®ned by Sinninghe Damste et al.(1989). Open and black-®lled circles indicate laminatedand non-laminated marls, circles ®lled grey represent silici-
®ed lithofacies types.
L. Schwark et al.1942

compounds should still re¯ect responses of biologi-cal communities to salinity variations.
Bound hydrocarbons released by desulfurization ofpolar fractions
One laminated and one nonlaminated marlsample were desulphurized to check whether a sig-
ni®cant bias in the biomarker distribution due toselective sulfur bonding might occur. A drastic biasin the compound distribution could invalidate the
environment characterization based on free bio-marker analysis or at least a�ord a combined in-terpretation of both compound pools.Desulfurization of polar fractions of both samples
a�orded less than 2% of hydrocarbon products andthus the amount of sulfur-bound lipids in the Malmz samples is insigni®cant. No experiments have yet
been carried out to desulfurize the kerogens andthus it is still possible that a considerable pro-portion of labile lipids is still attached to the kero-
gen matrix.Saturated hydrocarbons. The saturated hydrocar-
bons released by Raney nickel desulfurization ofthe polar chromatographic fraction from sample
M110 consist mainly of n-alkanes, steranes andhopanes (Fig. 16). In the case of sample ML69, n-alkanes largely predominate the hydrocarbon distri-
bution, and polycyclic components occur only intrace amounts. The alkane fraction of the latersample is also characterized by the occurrence of an
unresolved complex mixture.
n-Alkanes from sample M110 show a monomodal
distribution and range from C14 to C40 maximising
at C19. A slight odd over even carbon number pre-
dominance can be noted for the components in the
C21±C32 range. For sample ML69, the n-alkane dis-
tribution is monomodal centered around the C17-
and C18-components. A very slight odd over even
carbon number predominance is present is the C27±
C31 range.
In both samples, phytane and pristane are present
in trace amounts. In sample M110, the pristane/
phytane ratio is ca. 0.4, whereas this ratio is close
to 1.2 in sample ML69. This could re¯ect di�er-
ences in the degree of anoxicity between the two
samples with sample M110 more anoxic or just
re¯ect di�erent degrees of sulfurization.
Hopanes are by far more abundant in sample
M110 (Fig. 16). In the later case, the m/z = 191
chromatogram is dominated by gammacerane and
C35 ab-hopane with the 22R con®guration (Fig. 17).
A complete suite of C30- to C35 ab-hopanes is pre-
sent. For this series the 22R components clearly
predominate over the 22S isomers, with 22R/
(22R+ 22S) ratios of ca. 0.64 (C32 and C34 homol-
ogues), 0.56 (C33 component) and 0.84 (C35 homol-
ogues). The high abundance of gammacerane would
be in agreement with the low pristane/phytane-ratio
of the desulfurized polar fraction and indicate depo-
sition under strictly anoxic conditions. Gamma-
cerane has been widely used as an empirical
indicator of palaeosalinity (ten Haven et al., 1985,
1988). Recently it has been postulated that gamma-
cerane originates from bacterivorous ciliates and
might more likely be an indicator of intense water
column strati®cation and possibly a marker for
photic zone anoxia (Sinninghe Damste et al., 1996).
Salinity strati®cation implies circulation restriction
and possibly temperature strati®cation which in
turn cause an enhancement of redox strati®cation in
the water body. A combination of slightly enhanced
salinity corresponding to the ``mesosaline stage'' and
strictly anoxic conditions is almost inevitable and
gammacerane might be an indicator of coinciding
mesosaline and strictly anoxic conditions. Occur-
rence of gammacerane in non-hypersaline settings
has been reported as well as a lack of gammacerane
in hypersaline settings (ten Haven et al., 1985, 1988;
Sinninghe Damste et al., 1995b) favoring the view
of a combined salinity/anoxicity e�ect. Gamma-
cerane is the major compound that occurs exclu-
sively in a sulfur-bound mode in the extracts and its
detection in the polar fraction clearly supports the
view derived from the free lipid distribution. In
summary, the m/z = 191 trace of the desulfurized
sample M110 clearly resemble those from organic-
rich marls deposited in a hypersaline context (e.g.
Kenig et al., 1995; Schae�er et al., 1995).
In sample ML69, hopanes occur in trace amounts
only (Fig. 16). The distribution is again predomi-
Fig. 15. Crossplot of ITR, the isoprenoid thiophene ratioas de®ned by Sinninghe Damste et al. (1989) against thepristane/phytane-ratio. Black-®lled and open circles indi-cate laminated and non-laminated marls, circles ®lled greyrepresent silici®ed lithofacies types. Laminated marls pre-
ferentially plot in the region of enhanced salinity.
Geochemistry of Malm carbonates 1943

nated by the 22R C35 ab-component (Fig. 17). The
main di�erence in sample ML69 consists in the low
relative abundance of gammacerane which is almost
absent. If gammacerane is considered as a marker
for water column strati®cation and possibly photic
zone anoxia (cf. Sinninghe Damste et al., 1995), this
could re¯ect di�erences between the depositional
environment of the laminated and non-laminated
marls. This would however indicate high anoxicity
and intense strati®cation of the water column for
the non-laminated marl (M110) and more oxic con-
ditions or less marked strati®cation for the lami-
nated marls (ML69). This seems to be in
contradiction with the assumption that laminated
sediments were deposited in a low energy environ-
ment in contrast to sediments where lamination has
been destroyed by water turbulence or benthic
activity. The gammacerane distribution is however
in agreement with the ITR and pristane/phytane-
ratio of the free lipid hydrocarbon fraction that
also indicate higher salinity and anoxicity for the
M110 vs. the M69 sample (Fig. 15).
An interesting feature in sample ML69 is pre-
sence of C30-neohop-12-ene being the predominant
polycyclic component in the alkane fraction. This
compound is also present in sample M110, but in
lower relative abundance. Further investigations
will be aimed towards a closer inspection of the dis-
tribution of that compound in free and bound frac-
tions because currently its palaeoenvironmental
signi®cance remains unclear.
Steranes exclusively occur as baa- and aaa-iso-mers (Fig. 17). In the sterane distribution from
sample M110, the distribution of homologues is
C29>C27>>C28. It is also possible that a C30
sterane occurs in this distribution, but it coelutes
with a C30 methyl sterane. In sample ML69, the
sterane distribution resembles that of sample M110
Fig. 16. GC/MS total ion trace for aliphatic hydrocarbons obtained from desulphurized polar fractionsof a non-laminated (M110) and a laminated marl (ML69). Filled circles represent n-alkanes with num-bers indicate total number of carbon atoms. Open circles and squares denote hopanes and steranes, re-
spectively.
L. Schwark et al.1944

Fig.17.Mass
fragmentogramsofm/z
=191and217shownforthealiphaticfractionofdesulfurizedpolarfractionsofsamplesML69andM110.
Geochemistry of Malm carbonates 1945

(Fig. 17) with a slightly higher relative abundanceof baa- vs. aaa-isomers.
Methylsteranes (presumably 4-methylsteranes) arepresent in both samples (C30>C29>C28), but inmuch lower relative abundance in sample ML69.
Two dinosterane isomers only occur in sampleM110 where they represent the predominant 4-methylsteranes.
The presence of additional ``di''- and ``tri''-meth-ylated steranes indicated by m/z= 245 and 259 asmain fragment in the mass spectra was noted. It
was not determined whether these components rep-resent 3-alkylated steranes (i.e. 3-ethyl- and 3-pro-pyl steranes) or polymethylated components (e.g.2,3-dimethylsteranes or 4,4-dimethylsteranes)
because of low quantities and unknown origin andsigni®cance of theses compounds. Their presence,however, con®rms the highly diverse input of ster-
oidal compounds and the complexity of diageneticproducts observed in the free steroid distribution.Dimethylsteroids could be possible precursors for
the modi®ed sterenes found in the free hydrocar-bons.With both samples, the aromatic fractions of
desulfurized polars consist predominantly of a hugeunresolved complex mixture. With the exception ofalkylbenzenes recorded at m/z = 91, 105 and 133occuring in trace amounts in the C13±C30 range, no
aromatic components could be identi®ed. Due tomultiple coelutions, it was di�cult to obtain massspectra of the alkylbenzenes and therefore proble-
matic to determine if the m/z = 133 componentshave an arylisoprenoid structure. This possibilityseems rather unlikely, as the distributions observed
on the m/z = 133 + 134 mass chromatograms donot show the pattern usually expected for isoprenoi-dal components (i.e. absence or lower abundance ofC12-, C17- and C23-homologues). In addition, no
C40 diarylisoprenoids were observed in the twosamples investigated.
DISCUSSION
In thermally immature, sulfur-rich depositionalsettings, the molecular characterization of free
hydrocarbons and OSC for palaeoenvironmentreconstruction requires the complementary study ofsulfur-bound lipids, if those contribute signi®cantly
to the total extract, in order to avoid any bias fromselective removal of biomarkers from the free hy-drocarbon fractions. Bitumens extracted from theMalm z carbonates in the Rennertshofen Trough,
located in the Franconian Alb, SW-Germany, gaveonly minor quantities of hydrocarbons releasedupon desulfurization with Raney nickel. The com-
position of free hydrocarbons and OSC is thus suit-able for palaeoenvironment characterization.Variations in biomarker distribution between free
and bound lipids provide some additional infor-
mation on biological input, palaeomilieu and dia-
genesis.
The Malmian samples were previously categor-
ized in the three main lithofacies types limestones,
marls and cherts and subdivisions into laminated
and non-laminated marls were made. Biomarker
distribution of aliphatic, aromatic and OSC frac-
tions does not follow the lithofacies development.
Therefore, geochemical results provide signi®cant
additional information on palaeodepositional con-
ditions. The main point of debate in palaeoenviron-
mental assessment of Malm z sediments has been
the characterization of palaeosalinities and in part
of hydrodynamic energy levels, which in turn are
correlated to the redox potential (e.g. Keupp, 1977,
1994; Barthel, 1978; Meyer and Schmidt-Kaler,
1989, 1993; Keupp et al., 1993). One reason being
that hypersaline conditions during deposition would
terminate the growth of sponge and algal reefs on
the carbonate platform and reduce biological pro-
duction of normal marine macrofauna found in
outcrop studies. The palaeogeographical setting in
the Malm of SW-Germany, however, may lead to a
bias in palaeoenvironmental assessment using exclu-
sively macrofaunal assemblages because those are
preferentially preserved in reefal build-ups accessible
in outcrops. Marls and limestones from intra-reef
depressions are more subjective to intensive weath-
ering, not as well exposed and thus less well stu-
died. The reefs, although characteristic present-day
morphological features of the area are not fully
representative of the palaeoenvironments in local
depressions on the Eastern Bavarian Malm
Carbonate Platform, the so-called lagoons, troughs
or ``Wannen''.
Our samples are derived from such a location
where reef girdles surrounding an intra-reef de-
pression restricted water circulation in the lower
water body of the reef trough (Fig. 18). In the
upper water body above the reef top and above fair
weather wave base, free water circulation under
normal marine conditions prevailed and thus high
bioproductivity occurred. On the long term, only
minor salinity enhancement could develop on the
extended carbonate platform because episodic
storm events mixed the surface waters with normal
marine waters from the Tethys Ocean. In contrast,
within the sheltered intra-reef depression the waters
below storm wave based were continuously enriched
in salinity and density strati®cation developed. The
wall-reef surrounding the trough also e�ectively
prevented the input of terrigenous derived minero-
genic and organogenic debris which di�erentiates
this open marine from most other hypersaline
depositional settings.
In many other studies of hypersaline settings [see
thematic issues of Organic Geochemistry (1993) Vol.
20/8 and Organic Geochemistry (1995) Vol. 26/3] a
strong terrigenous derived organic component was
L. Schwark et al.1946

noted because of a restricted, land-locked lagoonal
environment. Such (sub)basins are often (i) almost
completely surrounded by land, (ii) connected tothe open sea by only a narrow and shallow passage
and (iii) separated from deeper marine settings by a
sill and thus susceptive to sealevel changes by eva-
poritic drawdowns and tectonic movements. Those
factors will inevitably lead to the accumulation ofterrigenous derived plant material, even if arid con-
ditions in the surrounding are not favorable for
extensive plant cover (Meyer and Schmidt-Kaler,
1989). Due to the special intra-reef setting of our
case study the terrigenous component is minimaland long chain n-alkane and alkylthiophene distri-
butions consequently re¯ect algal sources. A mi-
crobial rather than an algal contribution might be
indicated by the high abundance of C14-, C16- and
C18-n-alkanes. These compounds are likely to havebeen directly biosynthesized as saturated hydrocar-
bons because no functionalized lipids that incorpor-
ated reduced sulfur are present in the OSC.
Isolation of the intra-reef basin from terrigenous
minerogenic input consequently lead to an iron-
depleted environment that favoured incorporation
of sulfur in organic matter to produce OSC. In ad-dition, the protection of intra-reef sediments from
wave action and water column mixing by the sur-
rounding sponge reef girdle, stimulated oxygen de-
pletion and the establishment euxinic conditions via
activity of sulfate reducing bacteria. Surprisingly, aclear relationship between degree of sediment lami-
nation and degree of sulfurization could not be
detected. Comparable to a restricted lagoonal set-
ting the environmental changes must have occured
very frequently and de®nitely above the temporal
resolution covered by our sample set. This will be
one possible explanation why lithofacies does not
follow organic matter variations. Another pointmay be seen in the very rapid and irregular depo-
sition of micritic carbonates which seems to be
decoupled from main phases of organic matter sedi-
mentation. For the non-laminated marls and the
limestones it can be assumed that a major part ofsedimentation took place when storm events stirred-
up carbonate oozes produced on surrounding open
marine platform areas, which were then washed
over the reef-walls into the intra-reef depression.
The hopene distribution, the composition of iso-
prenoid thiophenes, the pristane/phytane-ratio and
also the partially the MTTC-ratio do indicate``higher than normal marine salinities''. Although
this is the exact de®nition of the term ``hypersaline''
(Kirkland and Evans, 1981), ``hypersaline'' has been
used to describe various other degrees of salinity.
We therefore de®ne the salinity range during depo-sition of the Malm z sediments as mesosaline sensu
Kirkland and Evans (1981) (for clarity note that the
term ``mesohaline'' in a Remane diagram de®nes a
hyposaline regime of 5 to 18-). We further restrict
the range of possible salinity variations to 40±80-,episodically reaching maxima of up to approxi-
mately 100-. On average, salinities >40- in the
uppermost water body of the intra-reef lagoon are
unlikely because they would have terminated
growth of the surrounding algal/spongal reefs. Apeak salinity of approximately 100- in the bottom
waters is assumed because higher values would ulti-
mately lead to gypsum deposition even during
short-term salinity oscillations and no indication of
such sulfate deposition is known. Some redissolu-
Fig. 18. Schematic diagram of the depositional setting during Malm z on the Southern BavarianCarbonate Platform, illustrating control of reef morphology on restriction. Surface waters on the plat-form are episodically mixed with fresh marine waters from the southernly Tethys Ocean during stormevents, which also supply large amounts of carbonatic oozes into the reef trough. During periods ofreduced mixing, slightly enhanced salinity develops on the platform and higher-density waters sinkdown and accumulate in the intra-reef depression. Morphology induced stagnant bottom water con-ditions are thus density-stabilized and anoxic conditions are easily established. Reef tops reach well intothe highly productive zone of surface water above fair weather wave base, where algae and cyanobac-teria may thrive. Microbial mats may ¯oat at the pycnocline and/or develop on the sediment surface.
This scenario ®ts an open water marine setting with local development of hypersaline conditions.
Geochemistry of Malm carbonates 1947

tion and carbonate replacement of gypsum crystals
may take place under extremely reducing con-
ditions, but a portion of originally precipitated gyp-
sum would persevere and no ghost crystals are
reported. Due to density strati®cation, salinities in
the lower water column of the intra-reef depressions
were much higher than in the surrounding plat-
form.
The organisms contributing to the organic matter
deposited were strictly marine and included photo-
synthetic algae and cyanobacteria as well a hetero-
trophic microbial community. Algal and microalgal
input is indicated by n-alkanes and isoprenoid thio-
phenes in the C21 to C27 range, steranes, sterenes
and especially the abundant monoaromatic steroids.
A dino¯agellate input is indicated by some minor
occurence of dinosterane in the desulphurized polar
fraction and by the high concentration of 4-methyl-
monoaromatic steroids in the free bitumen and is in
agreement with microscopically identi®able dino¯a-
gellate-derived calci®ed cysts (Keupp and
Neumann, 1996; Vliex and Schwark, 1998).
Algal and zooplanktonic contribution to the or-
ganic matter as revealed by steroid distribution is
extremly di�cult to assess for the Malm z due to
complex biological input and early diagenesis. The
steroids extracted from the immature organic mat-
ter (approximately 0.3% Rr) include a coexisting
suite of steranes, sterenes, spirosterenes, steradienes,
monoaromatic desmethyl- and 4-methylsteranes,
diaromatic steranes and some minor triaromatic
steranes (sterols and steroidal ketones are the sub-
ject of ongoing studies and will add even more com-
plexity to this picture). Several of the above
mentioned steroid classes were not previously
reported in the literature or their structures could
not be fully elucidated. Thus very special conditions
must have prevailed during deposition of the Malm
z sediments.
All observations point to an enhanced salinity
and strong reducing conditions. The paucity of
minerogenic detritus, in accordance with the lack of
rearranged steroids, leads to intensive sulfur incor-
poration into organic matter. Sulfur incorporation
into steroids however, as evidenced by the complete
lack of thiophenic steroids, either did not take place
or intermediately formed sulfurized steroids already
have had their sulfur released. The high amount of
available sulfur is furthermore assumed to have
promoted aromatization of steroids at this low
stage of maturity. Depending on double bond pos-
itions of biological precursor sterols, early diagen-
esis proceeded di�erently for C28-steroids as
compared to C27 and C29-counterparts. We noticed
signi®cant di�erences in the relative abundance of
the C27, C28 and C29 members for various steroid
classes as determined from the integration of
characteristic fragment ions. Most similar patterns
occur for the aaa-steranes, 5a-spirosterenes and dia-
romatic steroids with a clear predominance of C27
and C29 over the C28 analogues. The opposite pat-
tern with a predominance of C28 over C27 and C29
steroids is noted for the abb-steranes, the 5b-spiros-terenes and the regular monoaromatic steroids. The
sterenes and steradienes reveal a more evenly distri-
bution with a slight reduction in the amount of the
C27 compounds. Ongoing studies aimed to elucidate
the structure of the unknown sterenes and stera-
dienes will probably allow a better understanding
and re-interpretation of the early diagentic path-
ways prevailing during deposition of the Malm zsediments.
Although frequently silici®ed carbonates, alterna-
tions of cherts and carbonates and pure chert layers
occur, a diatomaceous input can be excluded for
the Malm z sediments, which is in accordance with
the total lack of saturated C20 and C25 HBI and
their respective thiophenic diagenesis products.
These compounds are very abundant in other
hypersaline settings but diatoms did not become
major contributors until the late Cretaceous and
thus were not present in the Jurassic samples stu-
died. Radiolaria and siliceous sponge rhaxes often
well preserved and identi®ed by REM as well as by
¯uorescence microscopy on polished blocks (Keupp
and Neumann, 1996; Vliex and Schwark, 1998) are
considered to be the source of biogenic silica.
Biomarkers speci®c for sponges and radiolaria are
not known and their contribution to extractable or-
ganic matter therefore can not be determined.
Cyanobacterial input is represented by short
chain n-alkanes and extended hopenes. For marl
lithologies, microscopical observations established a
close relationship between ¯uorescence behaviour of
cyanobacterial mats and extractable porphyrin ¯u-
orescence properties. Algal mats present in the car-
bonates contrastingly show a ¯uorescence
behaviour that is compatible with that of por-
phyrin-free maltene fractions. Hence, we consider
cyanobacteria as the main source for porphyrins in
the Malm M and ML lithofacies types.
The lack of aryl isoprenoids and di-aromatic
carotenoids or their condensation products in the
free aromatic as well as in the desulphurized polar
fractions indicates that during deposition of Malm
z marls no chlorobiaceae would thrive in the water
column of the intra-reef depression. This is surpris-
ing in that the relatively low water depth (<60 m)
and the high degree of sulfurization point towards
an extension of the euxinic realm well into the pho-
tic zone. Preliminary investigations of porphyrin
distributions (Turner, 1998, personal communi-
cation) revealed the presence of extended porphyr-
ins. Although precise structural elucidation is not
completed and coelution of chlorobiaceae derived
porphyrins with other isomers can not be fully
excluded, it thus seems very likely that photic zone
anoxia at least periodically prevailed in the Malm z
L. Schwark et al.1948

environment. The presence of gammacerane in thesulfur bound fraction would ®t into a scheme of
well-strati®ed water column and the existence of abacterial plate ¯oating at the chemocline.
CONCLUSIONS
Biomarker distribution of free and sulfur boundlipids indicates deposition of marine derived organic
matter for the Malm z sediments in theRennertshofen Trough in SW-Germany. Lack ofterrigenous input as revealed by n-alkane and terpe-
noid distribution lead to iron-limitation and prefer-ential incorporation of reduced sulphur intofunctionalized lipids. Local palaeogeography of the
intra-reef depression provided a restricted environ-ment on the open marine platform, thus leading tothe establishment of oxygen-depleted conditionsand via bacterial sulfate reduction to highly euxinic
conditions in the lower water body. Sulfurization oflipids was thus favoured by close spatial relation-ship between the euxinic and the photic zone.
An enhanced salinity of approximately 40±80-was recognized based on saturated isoprenoid, al-kylated thiophene and alkylated chroman distri-
bution. Correlation of pristane/phytane-ratios andITR is better than with the MTTC-ratio indicatingthat the ITR and the MTTC-ratio might re¯ect sal-inity conditions in di�erent parts of the water body.
High abundance of abb-steranes in the immaturesediments, as previously noted in hypersaline set-tings is restricted to the methylcholestanes, whereas
cholestanes and ethylcholestanes show maturityconcordant isomer distributions. Diagenetically in-termediate and labile sterene distributions are domi-
nated by spirosterens and unknown sterenes thatshow similar isomer distribution patterns for C27-and C29-members but a di�erent distribution for
the C28-compounds. A comparable compound pat-tern is noticed for structurally equivalent stera-dienes, occuring in the aromatic fraction. Di�erentearly diagenetic and/or dietary pathways further-
more lead to very low abundances of the C28-com-pound in a series of tentatively identi®ed A/B-ringdiaromatic steroids. Input of an speci®c C28 steroid
from a possibly more halotolerant precursor organ-ism seems likely, although particularly high cumu-lative abundances of C28 steroids due to improved
salinity adaption of the precursor were not encoun-tered. Alternatively a speci®c dietary pathway lead-ing to unusual C28-steroid distribution can not beexcluded. D7 sterenes quoted as possible precursor
for abb-steranes in hypersaline settings as well as D5
sterenes were not detected in the immature samples.Biological input from photosynthetic algae and
cyanobacteria was recognized by distribution ofsteroids and n-alkanes as well as alkyl-thiophenes.Organic matter derived from cyanobacteria is also
represented by abundant porphyrins. Diatoms often
signi®cant contributers to organic matter found inhypersaline settings were not fully evolved in the
Jurassic and thus C20 and C25 HBI compounds werenot to be detected. Terrigenous input at the studysite was extremely low but microbially derived or-
ganic matter is evident by hopanoids and especiallyby branched C19-thiophenes that originate fromstraight chain precursors with a 9-methyl-skeleton.
Biomarker distribution of desulphurized polarfractions show patterns compatible with free hydro-carbon fractions except for the exclusive occurence
of gammacerane in the bound fractions. Presence ofgammacerane might be taken as indication ofenhanced salinity coupled with highly reducing con-ditions. Complete quenching of the functionalized
gammacerane precursor by sulfurization in thepolar fraction points towards a close spatial re-lationship between habitat of precursor organism
and the euxinic zone, making an origin from ciliatesthriving from a bacterial plate ¯oating at the che-mocline most likely.
Organic petrology using UV-excitation revealedtwo types of organic mats present in the laminatedmarls. One type consists exclusively of macroalgae
the second contains algae and cyanobacteria. The¯uorescence properties of the cyanobacterial matorganoclasts are identical to the ¯uorescence prop-erties of isolated Ni- and V.O-porphyrins, whereas
the alginite ¯uorescence is identical to those of por-phyrin-free maltene fractions. Thus we concludethat porphyrins in the Malm z extracts originate
predominantly from cyanobacteria. We assume thatthis observation is a ®rst step into an improvedmicroscale localization of speci®c biomarkers in
complex built geological samples.
AcknowledgementsÐProvision of samples and discussionwith Dr R. K. F. Meyer and Dr. H. Schmidt-Kaler during®eld-work and core studies is most gratefully acknowl-edged. We thank R. Loesing and B. Spittho� for analyti-cal assistance. This work was carried out under auspicesof the ENOG (European Network of OrganicGeochemists) and bene®tted from discussion within thisgroup. ENOG consists of Laboratoire de Geochimie, IFP,Rueil Malmaison, France; Institute of Petroleum andOrganic Geochemistry, FZ Juelich, Germany; UniversiteÂLouis Pasteur, Strasbourg, France; NIOZ, Texel, TheNetherlands; Department of Environmental Chemistry,CID-CISC, Barcelona, Spain; Organic Geochemistry Unit,University of Bristol, U.K. and Geological Department,University of Cologne, Germany and receives fundingfrom the European Community.
REFERENCES
Aizenshtat, Z. (1973) Perylene and its geochemical signi®-cance. Geochimica et Cosmochimica Acta 37, 559±567.
Albrecht, P., Vandenbroucke, M. and Mandenque,M. (1976) Geochemical studies on the organic matterfrom the Douala Basin (Cameroon). I. Evolution of theextractable organic matter and the formation of pet-roleum. Geochimimica et Cosmochimimica Acta 40, 791±799.
Geochemistry of Malm carbonates 1949

Barbe, A., Grimalt, J. O., Pueyo, J. J. and Albaiges,J. (1990) Characterization of model evaporitic environ-ments through study of lipid components. In Advancesin Organic Geochemistry 1989, eds. B. Durand and F.Behar. Organic Geochemistry 16, 815±828.
Barthel, K. W. (1970) On the deposition of the Solnhofenlithographic limestone (Lower Thithonian, Bavaria,Germany). Neues Jahrbuch fuÈr Geologie undPalaÈontologie 135, 1±18.
Barthel, K. W. (1978) Solnhofen: Ein Blick in dieErdgeschichte. Ott Verlag, Thun, p. 392.
Berner, R. A. (1983) Sedimentary pyrite formation: Anupdate. Geochimica et Cosmochimimica Acta 48, 605±615.
Boon, J. J., Hines, H., Burlingame, A. L., Klok, J.,Rijpstra, W. I. C., de Leeuw, J. W., Emunds, K. E. andEglinton, G. (1983) Organic geochemical studies ofSolar Lake laminated cyanobacterial mats. In Advancesin Organic Geochemistry 1981, eds. M. Bjorùy et al. pp.207±227.
Bordenave, M. L. (1993) Applied Petroleum Geochemistry.Editions Technip, Paris, p. 528.
Brassell, S. C., Lewis, C. A., de Leeuw, J. W., de Lange,F. and Damste , J. S. (1986) Isoprenoid thiophenes:novel products of sediment diagenesis? Nature 320, 160±162.
Brooks, J. D., Could, K. and Smith, J. W. (1969)Isoprenoid hydrocarbons in coal and petroleum. Nature222, 257±259.
Brumsack, H.-J. (1988) Rezente, Corg.: reiche Sedimenteals SchluÈssel zum VerstaÈndnis fossiler Schwarzschiefer.Habilitationsschrift UniversitaÈ t GoÈ ttingen, 126 S.GoÈ ttingen.
Cameron, D. D., Cromate, R. I. T and Todd, L. (1964)Colouring matters of aphididae. Part XVI. Journal ofthe Chemical Society D, 48±50.
Connan, J., Bouroullec, J., Dessort, D. and Albrecht, P.(1986) The microbial input in carbonate-anhydrite faciesof a sabkha palaeoenvironment from Guatemala: Amolecular approach. In Advances in OrganicGeochemistry 1985, eds. D. Leythaeuser andJ. RullkoÈ tter. Pergamon, Oxford. Organic Geochemistry10, 29±50.
Cyr, T. D., Payzant, J. D., Montgomery, D. S. andStrauzs, O. P. (1986) A homologous serie of novelhopane sul®des in petroleum. Organic Geochemistry 9,139±143.
Didyk, B. M., Simoneit, B. R., Brassell, S. C. andEglinton, G. (1978) Organic geochemical indicators ofpalaeoenvironmental conditions of sedimentation.Nature 272, 216±222.
DIN 22020 (1986) Mikroskopische Untersuchungen anSteinkohle, Koks und Briketts, Teil 1±5. Berlin, p. 31S.
DIN 5033 (1979) Farbmessung: Grundbegri�e derFarbmetrik, Teil 1±9. Berlin/Cologne.
DIN 6164 (1978) DIN-FarbKarte: System derDIN_Farbkarte fuÈr den 28-Normalbetrachter, Teil 1.Berlin/Cologne.
Espitalie , J., Laporte, J. L., Madec, M., Marquis, F.,Leplat, P., Paulet, J. and Boutefeu, A. (1977) Me thoderapide de caracte risation des roches meÁ tres de leurpotentiel pe trolier et de leur degre d'e volution. Reviewd'Institute Francaise de PeÂtroleum 32, 23±42.
Farrimond, P., Comet, P., Eglinton, G., Evershed, R. P.,Hall, M. A., Park, D. W. and Wardroper, A. M.K. (1984) Organic geochemical study of the UpperKimmeridge Clay of the Dorset type area. MarinePetroleum Geology 1, 340±354.
von Freyberg, B. (1966) Der Faziesverband im UnterenMalm Frankens. Ergebnisse der Stromatometrie.Erlanger geologische Abhandlungen 62, 1±92.
von Freyberg, B. (1968) UÈ bersicht uÈ ber den Malm derAltmuÈ hl-Alb. Erlanger geolologische Abhandlungen 70,1±40.
Grice, K., Schae�er, P., Schwark, L. and Maxwell,J. R. (1997) Molecular indicators of palaeoenvironmen-tal conditions in an immature Permian shale(Kupferschiefer, Lower Rhine Basin, North-WestGermany) from free and S-bound lipids. OrganicGeochemistry 25, 131±147.
Grimalt, J. O. and Albaiges, J. (1990) Charcterization ofthe depositional environments of the Ebro Delta (wes-tern Mediterranean) by the study of sedimentary lipidmarkers. Marine Geology 95, 207±224.
Gschwend, P. M., Chen, P. H. and Hites, R. A. (1983) Onthe formation of perylene in recent sediments: kineticmodels. Geochimica et Cosmochimica Acta 47, 2115±2119.
Hagemann, H. W. and Hollerbach, A. (1986) The ¯uor-escence behavior of cfude oils with respect to their ther-mal maturation and degradation. In Advances inOrganic Geochemistry 1985, eds. D. Leythaeuser andJ. RullkoÈ tter. Pergamon, Oxford. Organic Geochemistry10, 473±480.
Hauke, V., Gra�, R., Wehrung, P., Trendel, J., Albrecht,P., Riva, A., Hopfgartner, G., GuÈ lacar, F., Buchs,A. and Eakin, P. (1992) Novel triterpene-derived hydro-carbons of the arborane/fernane series in sediments.Part II. Geochimica et Cosmochimica Acta 56, 3595±3602.
Hauke, V., Adam, P., Trendel, J.-M., Albrecht, P.,Schwark, L., Vliex, M., Hagemann, H. and PuÈ ttmann,W. (1995) Isoraborinol trough geological times:Evidence for its presence in the Permian and Triassic.Organic Geochemistry 23, 91±93.
ten Haven, H. L., de Leeuw, J. W. and Schenk, P.A. (1985) Organic geochemical studies of a Messinianevaporitic basin, northern Apennines (Italy). I:Hydrocarbon biomarkers for a hypersaline environment.Geochimica et Cosmochimica Acta 49, 2181±2191.
ten Haven, H. L., de Leeuw, J. W., RullkoÈ tter, J. andSinninghe Damste , J. S. (1988) Restricted utility of thepristane/phytane ratio as a palaeoenvironmental indi-cator. Nature 330, 641±643.
de las Heras, F. X. C., Grimalt, J. O., Lopez, J. F.,Albaiges, J., Sinnighe Damste , J. S., Schouten, S. andde Leeuw, J. W. (1997) Free and sulphurized hopanoidsand highly branhced isoprenoids in immature lacustrineoil shales. Organic Geochemistry 27, 41±64.
Hite R. J., Anders, D. E. and King, T. G. (1984) Organic-rich source rocks of Pennsylvanian age in the ParadoxBasin of Utah and Colorado. In Hydrocarbon SourceRocks of the Greater Rocky Mountain Region, eds. F. F.Woodward et al. Rocky Mountains Association ofGeology, Denver.
Holzer, G., Oro, J. and Tornabene, J. (1979) Gas chroma-tographic±mass spectrometric analysis of neutral lipidsfrom methanogenic and thermoacidophilic bacteria.Journal of Chromatography 186, 795±809.
Huc, A. Y. and Sinninghe Damste , J. S. (eds.) (1993)Origin and signi®cance of the organic matter inn eva-porites: Application to the Mulhouse Basin. OrganicGeochemistry 20/8.
Hussler, G., Conan, J. and Albrecht, P. (1984) Novelfamilies of tetra- and hexacyclic aromatic hopanoidspredominant in carbonate rock and crude oils. InAdvances in Organic Geochemistry 1983, eds. P. A.Schenk, J. W. de Leew and G. W. M. Lijmbach.Pergamon, Oxford. Organic Geochemistry 6, 39±49.
Huang, W. J. and Meinschein, W. G. (1979) Sterols asecological indicators. Geochimica et Cosmochimica Acta43, 739±745.
L. Schwark et al.1950

Jacob, H. (1989) Classi®cation, structure, genesis andpractical importance of natural solid oil bitumen(``migrabitumen''). International Journal of Coal Geology11, 65±79.
van Kaam-Peters, H. M. E. and Sinninghe Damste ,J. S. (1997) Characterisation of an extremely organicsulphur-rich, 150 Ma old carbonaceous rock: palaeoen-vironmental implications. Organic Geochemistry 27,371±397.
van Kaam-Peters, H. M. E., Schouten, S., de Leeuw,J. W. and Sinninghe Damste , J. S. (1997) A molecularand carbon isotope biochemical study of biomarkersand kerogen pyrolysates of the Kimmeridge ClayFacies: palaeoenvironmental implications. OrganicGeochemistry 27, 1201±1215.
Kenig, F., Sinninghe Damste , J. S., Frewin, N. L., Hayes,J. M. and de Leeuw, J. W. (1995) Molecular indicatorsfor palaeoenvironmental change in a Messinian evapori-tic sequence (Vena del Gesso, Italy). II. High-resolutionvariations in abundances and 13C contents of free andsulphur-bound carbon skeletons in a single marl bed.Organic Geochemistry 23, 485±526.
Keupp, H. (1977) Ultrafazies und Genese der SolnhofenerPlattenkalke. Abh. Naturhist. Ges. NuÈrnberg 37, 1±128.
Keupp, H. (1994) Aspects of the origin of the SolnhofenLithographic limestone facies based on a new core dril-ling in the Maxberg quarry. Geobios 16, 71±80.
Keupp, H. and Neumann, Ch. (1996) Calcareous dino¯a-gellate cysts from the Albian boreholes Kirchrode I andII (central part of the Lower Saxonian Basin, NWGermany): factors controlling their vertical succession.In DFG-Schwerpunk-tprogramm, Glob. u. Region.Steuerungsfaktoren biogener Sedimentation, II, eds.J. Reitner, F. Neuweiler and F. Gunkel. GoÈ ttingen, pp.17±22.
Keupp, H., Jenisch, A., Herrmann, R., Neuweiler, F. andReitner, J. (1993) Microbial carbonate crusts: a key tothe environmental analysis of fossil spongiolites. Fazies29, 41±54.
Kirkland, D. W. and Evans, R. (1981) Source-rock poten-tial of evaporitic environment. Bulletin of the AmericanAssociation of Petroleum Geologists 65, 181±190.
Kohnen, M. E. L., Sinninghe Damste , J. S., Rijpstra, W.I. C. and de Leeuw, J. W. (1990) Alkylthiophenes assensitive indicators of palaeoenvironmental changes.Geochemistry of Sulfur in Petroleum Systems, ACSSymposium Series 42. pp. 417±443.
Kohnen, M. E. L., Sinninghe Damste , J. S., ten Haven, H.L., Kock-Van Dalen, A. C., Schouten, S. and de Leeuw,J. W. (1991) Identi®cation and geochemical signi®canceof cyclic di- and trisulphides with linear and acyclic iso-prenoid carbon skeletons in immature sediments.Geochimica et Cosmochimica Acta 55, 3685±3695.
Kohnen, M. E. L., Sinninghe Damste , J. S., Baas, M.,Kock-van Dalen, A. C. and de Leeuw, J. W. (1993)Sulfur-bound steroid and phytane carbon skeletons ingeomacromolecules: Implications for the mechanism ofincorporation of sulfur into organic matter. Geochimicaet Cosmochimica Acta 57, 2515±2528.
La¯amme, R. E. and Hites, R. A. (1978) The global distri-bution of polycyclic aromatic hydrocarbons in recentsediments. Geochimica et Cosmochimica Acta 42, 289±303.
de Leeuw, J. W. and Sinninghe Damste , J. S. (1990)Organic sulfur compounds and other biomarkers as in-dicators of palaeosalinity. Geochemistry of Sulfur inPetroleum Systems, ACS Symposium Series 429. pp.417±443.
Li, M., Larter, S. R., Taylor, P., Jones, D. M., Bowler,B. and Bjorùy, M. (1995) Biomarkers or not bio-markers? A new hypothesis for the origin of pristaneinvolving derivation from methyltrimethyltridecylchro-
mans (MTTCs) formed during diagenesis from chloro-phyll and alkylphenols Organic Geochemistry 23, 159±167.
Li, M. and Larter, S. R. (1995) Reply to comments bySinninghe Damste and de Leeuw (1995) on Li et al.(1995) Organic Geochemistry 23, 159±167. OrganicGeochemistry 23, 1089±1093.
Louda, J. W. and Baker, E. W. (1984) Perylene occurence,alkylation and possible sources in deep-ocean sediments.Geochimica et Cosmochimica Acta 48, 1043±1058.
Leythaeuser, D. and Sinninghe Damste , J. S. (1995) Originand signi®cance of the organic matter in evaporites II:From modern to ancient. Organic Geochemistry 23/6.
Meyer, R. K. F. and Schmidt-Kaler, H. (1989)PalaÈ ogeographischer Atlas des suÈ ddeutschen Oberjuras(Malm). Geologisches Jahrbuch A 115, 1±77.
Meyer, R. K. F. and Schmidt-Kaler, H. (1993) SchwarzeKalke im Weiûen Jura (uÈ ber die Bitumenfazies imMalm der SuÈ dlichen Frankenalb). Geologica Bavarica97, 155±166.
Meyer, R. K. F. and Schmidt-Kaler, H. (1994)Fazieswandel und Probleme der Stratigraphie imObermalm (Titho) zwischen Solnhofen und Neuburg/D.(Bayern). Erlangener Geologische Abhandlungen 123, 1±83.
Moldowan, J. M., Seifert, W. K. and Gallegos, E. J. (1985)Relationship between petroleum composition anddepositional environment of petroleum source rocks.Bulletin American Association of Petroleum Geolology69, 1255±1268.
Orr, W. L. and Sinninghe Damste , J. S. (1990)Geochemistry of Sulfur in Petroleum SystemsGeochemistry of Sulfur in Petroleum Systems. pp. 2±29.
Orr, W. L. and Grady, J. R. (1967) Perylene in basin sedi-ments o� southern California. Geochimimica etCosmochimica Acta 31, 1201±1209.
Ourisson, G. and Albrecht, P. (1992) Hopanoids. 1.Geohopanoids: The most abundant natural products ofthe earth. Chemical Research 25, 398±402.
Ourisson, G., Albrecht, P. and Rohmer, M. (1994) Themicrolial origin of fossil fuels. Scienti®c American 251,44±51.
Palacas, J. G., Anders, D. E. and King, J. D. (1984)South Florida Basin: Prime example of carbonate sourcerocks of petroleum. In Petroleum Geochemistry andSource Rock Potential of Carbonate Rocks, Studies inGeology 18, ed. J. G. Palacas. American Association ofPetroleum Geologists, Tulsa.
Payzant, J. D., Montgomery, D. S. and Strausz, O.P. (1986) Sul®ds in petroleum. Organic Geochemistry 9,357±369.
Peakman, T. M. and Kock-van Dalen, A. C. (1990)Identi®cation of alkylthiophenes occurring in the geo-sphere by synthesis of authentic standards. Geochemistryof Sulfur in Petroleum Systems, 397±416.
Peakman, T. M., Ten Haven, H. L., Rechka, J. R., deLeeuw, J. W. and Maxwell, J. R. (1989) The occurenceof (20R)- and (20S-) d8(14) and d8(14) 5a(H)-steranes andthe origin of 5a(H),14b(H),17b(H)-steranes in an imma-ture sediment. Geochimica et Cosmochimica Acta 53,2001±2009.
Peters, K. E. and Moldowan, J. M. (1993) The BiomarkerGuide: Interpreting Molecular Fossils in Petroleum andAncient Sediments. Prentice Hall, New Jersey, p. 363S.
Radke, M., Willsch, H., Leythaeuser, D. and TeichmuÈ ller,M. (1982) Aromatic components of coal: relation of dis-tribution pattern to rank. Geochimica et CosmochimicaActa 46, 1831±1848.
Risatti, J. B., Rowland, S. J., Yan, D. A. and Maxwell,J. R. (1984) Stereochemical studies of acyclic isopre-noids. XII. Lipids of methanogenic bacteria and poss-
Geochemistry of Malm carbonates 1951

ible contributions to sediments. Organic Geochemistry 6,93±104.
Rohmer, M., Bisseret, P. and Neunlist, S. (1992) Thehopanoids, prokaryotic triterpenoids and precursors ofubiquitous molecular fossils. In Biological Markers inSediments and Petroleum, eds. J. M. Moldowan et al.Prentice Hall, Englewood Ci�s, New York.
Russell, M., Grimalt, J., Hartgers, W. A., Taberner, C. andRouchy, J. M. (1997) Bacterial and algal markers insedimentary organic matter deposited unter natural sul-phurization conditions (Lorca Basin, Murca, Spain).Organic Geochemistry 26, 605±625.
Salmon, V., Derenne, S., Largeau, C., Beaudoin, B.,Bardoux, G. and Mariotti, A. (1997) Kerogen chemicalstructure and source organisms in a Cenomanian or-ganic-rich black shale (Central Italy): Indications for animportant role of the ``sorptive protection'' pathway.Organic Geochemistry 27, 423±438.
Schae�er, P., (1993) Marqueres biologiques de milieuxevaporitiques. Ph.D. thesis, Universite Loius Pasteur,Strasbourg.
Schae�er, P., Reiss, C. and Albrecht, P. (1995)Geochemical study of macromolecular organic matterfrom sulfur-rich sediments of evaporitic origin(Messinian of Sicily) by chemical degradations. OrganicGeochemistry 23, 567±581.
Scheuer, P. J. (1978) Chemistry of Marine NaturalProducts. Academic Press, New York, p. 176.
Schwark, L. and PuÈ ttmann, W. (1989) Aromatic hydro-carbon composition of the Permian Kupferschiefer inthe Lower Rhine Basin, NW Germany. In Advances inOrganic Geochemistry 1989, eds. B. Durand and F.Behar. Organic Geochemistry 16, 749±761.
Shanmugam, G. (1985) Signi®cance of coniferous rain for-ests and related organic matter in generating commercialquantities of oil, Gippsland Basin, Australia. AmericanAssociation of Petroleum Geologists Bulletin 69, 1241±1254.
Sheng, G., Fan, S., Lin, D., Su, N. and Zhou, H. (1980)The geochemistry of n-alkanes with an even±odd predo-minance in the Tertiary Shahalejie Formation of north-ern China. In Advances in Organic Geochemistry 1979,eds. A. G. Douglas and J. R. Maxwell. Pergamon,Oxford, pp. 115±121.
Silliman, J. E., Meyers, P. A. and Eadie, B. J. (1997)Perylene: an indicator of alteration processes or precur-sor materials. Abstract in 18th International Meeting onOrganic Geochemistry, Part II, Vol. C76.Forschungszentrum Julich.
Sinninghe Damste , J. S., and de Leeuw, J. W. (1995)Comments on Biomarkers or not biomarkers. A new hy-pothesis for the origin of pristane involving derivationfrom methyltrimethytridecyl-chromans (MTTCs) formedduring diagenesis from chlorophyll and alkylphenols, M.Li, S. R. Larter, P. Taylor, D. M. Jones, B. Bowler andM. Bjorùy. Organic Geochemistry 23, 1085±1087.
Sinninghe Damste , J. S., ten Haven, H. L., de Leeuw,J. W. and Schenck, P. A. (1986) Organic geochemicalstudies of a Messinian evaporitic basin, northernApennines (Italy). II. Isoprenoid and n-alkyl thiopenesand thiolanes. Organic Geochemistry 10, 791±805.
Sinninghe Damste , J. S., Kock-Van Dalen, A. C., deLeeuw, J. W., Schenck, P. A., Guoying, S. and Brassell,S. C. (1987a) The identi®cation of mono-, di-and tri-methyl 2-methyl-2-(4,8,12-trimethyltridecyl)chromansand their occurrence in the geosphere. Geochimica etCosmochimica Acta 51, 2393±2400.
Sinninghe Damste , J. S., Kock-Van Dalen, A. C., deLeeuw, J. W. and Schenck, P. A. (1987b) The identi®-cation of 2,3-dimethyl-5-(2,6,10-trimethylundecyl)-thio-phene, a novel sulfur containing biological marker.Tetrahedron Letters 28, 957±960.
Sinninghe Damste , J. S., Rijpstra, I., de Leeuw, J. W. andSchenck, P. A. (1989) The occurrence and identi®cationof series of organic sulfur compounds in oils and sedi-ment extracts. II. Their presence in samples from hyper-saline and non-hypersaline palaeoenvironmental andmaturity indicators. Geochimica et Cosmochimica Acta53, 1323±1341.
Sinninghe Damste , J. S., Kohnen, M. E. L. and de Leeuw,J. W. (1990) Thiophenic biomarkers for palaeoenviron-mental assessment and molecular stratigraphy. Nature345, 609±611.
Sinninghe-Damste , J. S., Keely, B. J., Betts, S. E., Baas,M., Maxwell, J. R. and de Leeuw, J. W. (1993)Variations in abundances and distributions of isopre-noid chromans and long-chain alkylbenzenes in sedi-ments of the Mulhouse Basin: a molecular sedimentaryrecord of palaeosalinity. Organic Geochemistry 20,1201±1215.
Sinninghe Damste , J. S., van Duin, A. C. T., Hollander,D., Kohnen, M. E. L. and de Leeuw, J. W. (1995) Earlydiagenesis of bacteriohopanepolyol derivatives:Formation of fossil homohopanoids. Geochimica etCosmochimica Acta 59, 5141±5156.
Talukdar, S. C., De Toni, B., Marcano, F., Sweeney,J. and Rangel, A. (1993) Upper Cretaceous source rocksof northern South America. American Association ofPetroleum Geologists Bulletin 71, 967±985.
Thompson, R. H. (1979) Recent advances in the chemistryand biochemistry of quinone pigments. In: RecentAdvances in Phytochemistry, eds. T. Swain, J. B.Harborne and van Sumere, Vol. 12. Plenum Press, pp.287±312.
Valisolalao, J., Perakis, B., Chappe, B. and Albrecht,P. (1984) A novel sulfur containing C35 hopanoid insediments. Tetrahedron Letters 25, 1183±1186.
Vliex, M. and Schwark, L. (1998) Geochemical character-ization of Malm Zeta laminated carbonates from theFranconian Alb, SW-Germany (I). Palaeogeography,Paleoclimatology and Paleoecology, submitted for publi-cation.
Volkman, J. K. (1986) A review of sterol markers for mar-ine and terrigenous organic matter. OrganicGeochemistry 9, 83±99.
Volkman, J. K. (1988) Biological marker compounds asindicators of the depositional environments of pet-roleum source rocks. In Lacustrine Petroleum SourceRocks, eds. A. J. Fleet et al., Geological Society SpecialPublication 40. Blackwell Scienti®c Publications,Oxford.
Volkman, J. K. and Maxwell, J. M. (1986) Acyclic isopre-noids as biological markers. In Biological Markers in theSedimentary Record, ed. R. B. Johns, Methods inGeochemistry and Geophysics, Vol. 24. Elsevier,Amsterdam, p. 364.
Wakeham, S. G., Scha�ner, C., Giger, W., Boon, J. J. andde Leeuw, J. W. (1979) Perylene in sediments from theNamibian Shelf. Geochimica et Cosmochimica Acta 43,1141±1144.
Watts, C. and Maxwell, J. (1977) Carotenoid diagenesis ina marine sediment. Geochimica et Cosmochimica Acta41, 493±497.
Wei, H. and Songnian, L. (1990) A new maturity par-ameter based on monoaromatic hopanoids. In Advancesin Organic Geochemistry 1989, eds. B. Durand and F.Behar. Pergamon, Paris. Organic Geochemistry 16,1007±1013.
de Lemos Sco®eld (1990) Author please supply details.Venkatesan (1988) Author please supply details.Sinninghe Damste et al. (1996) Author please supplydetails.
L. Schwark et al.1952
Copyright © 2022 FDOKUMEN




















