
The use of trivalent chromium bath to obtain a solar selective black chromium coating
Upload
independentCategory
view
3download
0
This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Toxicology 268 (2010) 31–39
Contents lists available at ScienceDirect
Toxicology
journa l homepage: www.e lsev ier .com/ locate / tox ico l
Genome-wide analysis of BEAS-2B cells exposed to trivalent arsenicals anddimethylthioarsinic acid
Jaya Chilakapati a, Kathleen Wallaceb, Hongzu Renb, Michael Frickec, Kathryn Baileyb,William Wardb, Jack Creedc, Kirk Kitchinb,∗
a Curriculum in Toxicology, University of North Carolina at Chapel Hill, United Statesb Environmental Carcinogenesis Division, National Health and Environmental Effects Research Laboratory, US Environmental Protection Agency, NC, United Statesc Microbiological and Chemical Exposure Assessment Research Division, NERL, USEPA, Cincinnati, OH, United States
a r t i c l e i n f o
Article history:Received 1 September 2009Received in revised form 30 October 2009Accepted 20 November 2009Available online 27 November 2009
Keywords:ArsenicOxidative stressBEAS-2BLungsGenomicsDEGs (differentially expressed genes)
a b s t r a c t
Lung is a major target for arsenic carcinogenesis in humans by both oral and inhalation routes. However,the carcinogenic mode of action of arsenicals is unknown. We investigated the effects of inorganic arsenic(iAsIII), monomethylarsonous acid (MMAIII), dimethylarsinous acid (DMAIII) and dimethylthioarsinicacid (DMTA), a sulfur containing dimethyl arsenic metabolite, in human bronchial epithelial (BEAS-2B)cells. Cells were exposed to 3, 15 �M-iAsIII; 0.3, 1 �M-MMAIII; 0.2, 1 �M-DMAIII; 0.2, 0.9 �M-DMTAas non-cytotoxic and minimally cytotoxic (∼20%) concentrations based on Neutral Red uptake assaysafter 24 h of culture. Total RNA was isolated and gene expression analysis conducted using Affymetrix®
Human Genome 133 Plus 2.0 arrays. Differentially expressed genes (DEGs) were determined using a one-way ANOVA (p ≤ 0.05) by Rosetta Resolver®, a Benjamini–Hochberg FDR (false discovery rate) multipletesting correction (<0.05) followed by a Scheffe’s post hoc test. For all compounds except DMTA, >90%of DEG altered in the low concentration were also changed at the high concentration. There was a cleardose–response seen in the number of DEGs for all four compounds. iAsIII showed the highest numberof DEG at both concentrations (2708 and 123, high and low, respectively). 1749, 420 and 120 DEGswere unique to the high concentrations of iAsIII, MMAIII and DMAIII, respectively. Transferrin receptoris a common DEG in low concentration arsenical treated cells. Ingenuity Pathway AnalysisTM revealedp53 signaling (E2F1 and 2, SERPIN), and cell cycle related genes (cyclin D1) were altered by the highconcentrations of DMTA, MMAIII and iAsIII. Oxidative stress (DUSP1, GPX2, NQO1, GCLC) and NF-�Bsignaling (TLR4, NF-�B) pathways were changed by the high concentrations of MMAIII and iAsIII. Thegenes identified in this study can be a valuable tool to determine the mechanism of arsenic toxicity andcancer formation. A number of similarities were observed in the gene expression profiles of DMAIII andDMTA and also iAsIII and MMAIII. These findings reveal some biological effects of arsenicals that will aidin creating a better risk assessment model for arsenical-induced lung cancer.
Published by Elsevier Ireland Ltd.
1. Introduction
Arsenic is a naturally occurring environmental contaminant, andits inorganic forms, arsenate and arsenite, often occur in drinkingwater particularly in areas with geologic sources of arsenic (IARC,2004; NRC, 1999, 2001a). The current EPA maximum contaminantlevel for iAs in drinking water is 10 ppb. Exposure to arsenic hasbeen associated with lung, skin, urinary bladder, liver, and kid-ney cancer in addition to circulatory, nervous system disorders
∗ Corresponding author at: US Environmental Protection Agency, EnvironmentalCarcinogenesis Division, 109 TW Alexander Drive (B143-06), Durham, NC 27711,United States. Tel.: +1 919 541 7502; fax: +1 919 541 0694.
E-mail address: [email protected] (K. Kitchin).
and diabetes (Bates et al., 1992; Chen et al., 1992; Kitchin, 2001;Navas-Acien et al., 2006; Smith et al., 1992).
Although arsenic can cause lung cancer via inhalation like allother lung carcinogens, it can also cause lung cancer via ingestionof drinking water. Arsenic in drinking water may also cause non-malignant lung disease (Smith et al., 2006). In fact, the main causeof death after long-term exposure to arsenic in the drinking water islung cancer (NRC, 2001b). Most lung cancers arise in the epitheliumof the bronchial tree (Mace et al., 1994). One way to model theeffects of arsenic in the lungs is to use BEAS-2B cell lines which arederived from human lung epithelium. BEAS-2B cells are an SV40immortalized cell line originating from normal human bronchialepithelial cells, which are progenitors of human lung cancer. Thiscell line has been used as a model for studying human lung cancerbut has not been used with arsenic. They are non-tumorigenic and
0300-483X/$ – see front matter. Published by Elsevier Ireland Ltd.doi:10.1016/j.tox.2009.11.018

Author's personal copy
32 J. Chilakapati et al. / Toxicology 268 (2010) 31–39
Fig. 1. The metabolic pathway of inorganic arsenic involves sequential two-electron reduction of pentavalent species [arsenate, (iAsV); monomethylarsonic acid, (MMAV);dimethylarsinic acid, (DMAV)] followed by oxidative methylation of trivalent species [arsenite, (iAsIII); monomethylarsonous acid, (MMAIII); dimethylarsinous acid, (DMAIII)].DMAV can convert to DMTA. In DMTA, arsenic is complexed to sulfur and not oxygen.
remain so up to a very high number of passages (Mace et al., 1994;Reddel et al., 1988). Human bronchial epithelial cells have very lowarsenic methylation capacity when compared to rat and humanhepatocytes and human keratinocytes (Styblo et al., 2000).
The metabolic pathway of inorganic arsenic involves sequen-tial two-electron reduction of pentavalent species [arsenate, (iAsV);monomethylarsonic acid, (MMAV); dimethylarsinic acid, (DMAV)]followed by oxidative methylation of trivalent species [arsenite,(iAsIII); monomethylarsonous acid, (MMAIII); dimethylarsinousacid, (DMAIII)] (Fig. 1) (Thomas et al., 2001; Vahter, 1999). InorganicAsIII has been shown to cause chromosomal damage in a varietyof human tissues. The methylated trivalent species monomethy-larsonous acid (MMAIII) and dimethylarsinous acid (DMAIII) havebeen shown to be more cytotoxic and genotoxic than inorganicarsenic and the pentavalent methylated forms (Ahmad et al., 2002;Andrewes et al., 2003; Kligerman et al., 2003; Mass et al., 2001;Petrick et al., 2000; Styblo et al., 2000). The role of each trivalentarsenical in arsenic-derived lung cancer is unknown.
Dimethylthioarsinic acid (DMTA) is a thioarsenical which canbe formed from DMAV (Fricke et al., 2007; Raml et al., 2007). Itwas identified as a microbial metabolite of DMAV in rat urine anddetermined to be both genotoxic and cytotoxic (Kuroda et al., 2004;Naranmandura et al., 2007; Yoshida et al., 2003). DMTA is a metabo-lite which is found in the urine of rats, hamsters, sheep and humans(Fricke et al., 2007). Recently, it has been identified in the urine ofseveral Bangladeshi women living in an area of arsenic contami-nation in drinking water (Raml et al., 2007). DMTA has also beenfound to be a potent cytotoxicant in HepG2 cells (Raml et al., 2007).Studying the effect of this sulfur containing arsenical on BEAS-2Bcells will shed light on its implications for lung cancer.
The mode of action of arsenicals in causing cancer is not yetknown (Hughes et al., 2007; Kitchin, 2001). There are a num-ber of proposed mechanisms for arsenic-induced cancer. Theseinclude sulfhydryl group occupancy, oxidative stress, altered DNAmethylation, genotoxicity, cell proliferation and inhibition of DNArepair (Kitchin, 2001). However, there are three major ways inwhich arsenic species can interact with biological molecules ofimportance (Kitchin et al., 2002; Kitchin and Wallace, 2008). First,trivalent arsenicals can bind to macromolecule sites, principally thesulfhydryls of peptides and proteins. Second, arsenical exposuresmay generate free radicals and other reactive species in biologi-cal systems (Hughes and Kitchin, 2006; Kitchin and Ahmad, 2003).
Third, arsenic exposures can result in changes in the cellular DNAmethylation state (Waalkes et al., 2004).
Arsenic is registered as one of the most hazardous substancesin ATSDR’s priority list, but relatively few studies have dealt withthe effects of arsenic on lung cells. The major goal of this studywas to investigate the modulation of signaling molecules by arsen-ite, MMAIII, DMAIII and DMTA in human bronchial epithelial cells(BEAS-2B). The second major goal of this study was to determinethe dose–response relationship of arsenic and its metabolites in thiscellular experimental system. For this particular study we wanteda concentration which would not be cytotoxic and a concentrationwhich would elicit 20% cytotoxicity to BEAS-2B cells. The reasonfor this is that genomic studies are highly sensitive. Even a verylow increase in concentration could elicit a significant increase inthe number of genes. We wanted to focus on the genomic changesthat occurred with a slight change in arsenical concentrations.We examined global effects of arsenic on gene expression whichis useful for elucidating toxicity pathways, patterns of biologicalresponse and discovering underlying mechanisms of toxicity. Thegenes identified in this study can be a valuable tool to determinethe mechanism of arsenic toxicity and cancer formation.
2. Materials and methods
2.1. Chemicals
Monomethylarsine iodide and dimethylarsine iodide were synthesized usingpreviously described methods (Le et al., 1994) (all compounds >99% purity). DMTAwas synthesized as previously described (Fricke et al., 2007). Sodium arsenite waspurchased from Sigma–Aldrich (St. Louis, MO). All other chemicals used were thehighest grade commercially available.
2.2. Cell culture
BEAS-2B human bronchial epithelial cells (American Type Culture Collection,Rockville, MD) (Reddel et al., 1989) were used at passage numbers 40–46. The cellswere cultured in Lechner and LaVeck media (LHC-9) containing retinoic acid (33 nM)and epinephrine (2.75 mM). The cells were maintained in 75 cm2 flasks at 37 ◦Cand 6% CO2. Media was replaced every second day, and cells were passaged when>85% confluent by washing with Versene and dislodging with TrypLe. Cell treatmentexperiments used 96-well polystyrene plates (Costar, Fisher Scientific) containing0.3 ml of media per well, with cells seeded at an initial density of 20,000/cm2. After1 day, new media containing the treatments was applied. Experiments used 6 wellsfor each treatment level and 12 wells per culture plate were allocated as controls.Positive controls were included to monitor changes in the BEAS-2B cell response.All experiments were replicated with at least two independent cell passages.

Author's personal copy
J. Chilakapati et al. / Toxicology 268 (2010) 31–39 33
Table 1Concentrations of the four arsenicals used to treat BEAS-2B cells and the respectivenumber of microarray chips used for the two concentrations.
Arsenical Low concentration(�M)
Minimally cytotoxicconcentration (�M)
N
Control 0 0 4iAsIII 3 15 3, 4 respectivelyMMAIII 0.3 1 3DMAIII 0.2 1 3DMTA 0.2 0.9 3
2.3. Cytotoxicity assay
Cytotoxicity toward BEAS-2B cells was determined by the Neutral Red uptakeassay (Fautz et al., 1991). For this assay, 96-well plates were seeded at a density of2.5 × 104 cells/well (in 0.25 ml medium/well). Each plate had a blank and negativecontrol column. After 48 h of growth, cells were dosed with the arsenical, whichwas serially diluted from the high-dose concentration in subsequent microplatecolumns, for 24 h. After the incubation period, the wells were washed with DPBS.Neutral Red media [LHC-9 with 0.003% Neutral Red] was added, and cells were thenincubated for 3 h. Neutral Red media were removed, and wells were again washedwith DPBS before 100 �l of extraction buffer (50% ethanol and 1% acetic acid) wereadded to each well. Plates were shaken for 20 min and read at 540 nm to measure dyeuptake. The measured Neutral Red dye uptake was used to calculate the percentageof survival where percentage of survival = (A540 treatment/A540 control) × 100. Allexperiments were replicated with at least two independent cell passages.
2.4. Gene expression analysis
BEAS-2B cells were grown in 25 cm2 flasks and then subjected to arsenical treat-ment (see Table 1 for detailed treatment plan) for 24 h. Total RNA was isolated fromBEAS-2B cells according to the TRIzol RNA extraction protocol (Invitrogen) followedby RNeasy Mini Kit (RNeasy® , Qiagen, Valencia, CA). RNA quality was determinedspectrophotometrically (ratio of A260 to A280) using Nanodrop ND-1000 spec-trophotometer (Nano-Drop Technologies, Wilmington, DE) and also by checkingthe ratio of 18S to 28S RNA using the RNA Nano Chip on a 2100 Bioanalyzer (AgilentTechnologies, Palo Alto, CA). Total RNA (3 �g) was labeled using the Affymetrix®
One-Cycle cDNA Synthesis protocol and hybridized to Affymetrix® Gene ChipHuman 133 Plus 2.0 arrays (containing 54,675 probesets) as described by the manu-facturer. Biotin-labeled complementary RNA (cRNA) was produced from 15 �g totalRNA using an Affymetrix “one-way” labeling kit (cat# 900493). Total cRNA wasthen quantified using a Nano-Drop and evaluated for quality after fragmentation ona 2100 Bioanalyzer. Following overnight hybridization at 45 ◦C to Affymetrix® GeneChip Human 133 Plus 2.0 arrays in an Affymetrix Model 640 GeneChip hybridizationoven, the arrays were washed and stained using an Affymetrix 450 fluidics station asrecommended by the manufacturer and scanned on an Affymetrix Model 3000 scan-ner. After scanning, raw data (Affymetrix .cel files) were obtained using AffymetrixGeneChip Operating Software (version 1.4). Data (.cel files) was analyzed and sta-tistically filtered using Rosetta Resolver version 6.0 software (Rosetta Informatics,Kirkland, WA). Principal component analyses (PCA) reduce the dimensionality ofthe data wherein the multidimensional gene expression data is reduced to a three-dimensional space to allow for visualization of the variability of the data. PCA wasapplied to statistically significant genes of all treated and control groups of the data.Differentially expressed genes (DEGs) were determined using a one-way ANOVA(p ≤ 0.05) by Rosetta Resolver® , a Benjamini–Hochberg FDR (false discovery rate)multiple testing correction (<0.05) followed by a Scheffe’s post hoc test. DEGs wereevaluated for canonical pathways and biological function using Ingenuity PathwayAnalysis (IPA) (Ingenuity, Mountain View, CA, USA). A data set containing gene iden-tifiers and their corresponding expression values such as fold-changes and p-valueswas uploaded as a tab-delimited text file. Each gene identifier was mapped to itscorresponding gene object in the Ingenuity Pathways Knowledge Base. A fold changecutoff of 1.5-fold and p ≤ 0.05 was set to identify genes whose expression was differ-entially regulated. These genes were then used as the starting point for generatingbiological networks. Venn analysis was done from Affymetrix online data analysisresource NetAffx (http://www.affymetrix.com/analysis/index.affx).
3. Results
3.1. Cytotoxicity of arsenicals in human BEAS-2B cell line
Cytotoxicity assays were carried out for all four arsenicals after a24 h exposure at 37 ◦C. The low concentrations of arsenicals shownin Table 1 were non-cytotoxic as measured by neutral red as wellas by visual observations. The high concentrations showed around20% cytotoxicity. Arsenite showed the lowest cytotoxicity of thefour as expected. DMTA, a pentavalent sulfur containing arsenical,shows similar cytotoxicity potency to DMA III.
3.2. Principal component analyses
A good separation was observed between different compoundsand control groups as well as good separation between the dif-ferent concentration groups (Fig. 2). The high concentrations ofiAsIII and MMAIII clustered very well when compared to DMAIIIand DMTA.
3.3. Dose response in number of genes
There is a clear dose–response relationship in gene expressionfor all four compounds (Fig. 3). Exposure to 15 �M sodium arsen-ite resulted in the greatest number (5997) of DEGs compared tothe control. Exposure to 1.0, 1.0 and 0.9 �m MMAIII, DMAIII andDMTA resulted in 4001, 2349 and 2155 genes, respectively. The lowconcentrations of DMAIII and DMTA yielded only 18 and 4 DEGsrespectively.
3.4. Venn analysis
A ±1.5-fold change cut-off was applied to the DEGs to conductVenn analysis. Over 90% DEGs found in low concentrations werealso found in the respective high concentrations for the trivalentarsenicals (Fig. 4A). 344 and 2 DEGs were found common amongtrivalent arsenicals for high and low concentrations respectively(Fig. 4B). Transferrin receptor (p90, CD71) (TFRC) is the commonDEG among all the trivalent arsenicals after exposure to low con-centrations. It was upregulated in all three arsenicals (3.2-fold byiAsIII, 4.7-fold by MMAIII and 2.5-fold by DMAIII). The high con-centrations of DMAIII and DMTA had 302 common DEGs betweenthem (Fig. 4C).
3.5. Pathway analysis
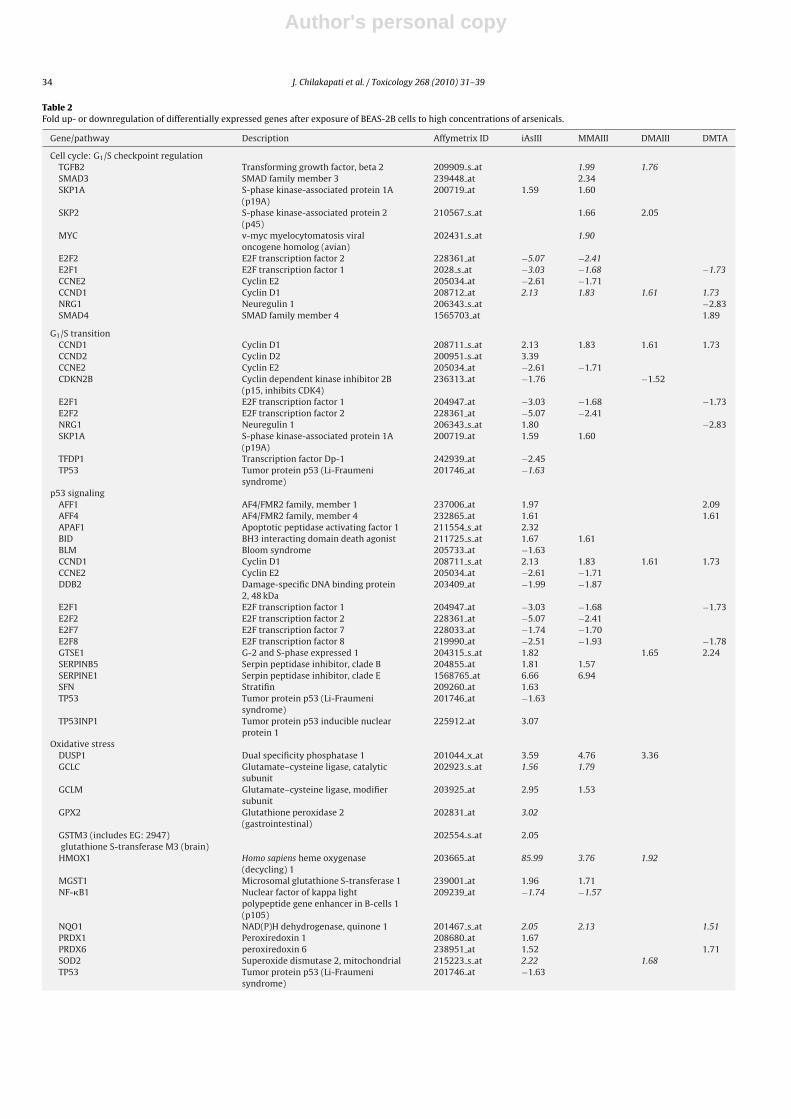
Differentially expressed genes were mapped to known com-pendia of physiological processes (Ingenuity) and are presented inTable 2.
3.5.1. Cell cycle related genes
Cyclin D1 was upregulated by all four arsenicals after adminis-tration of the high concentration (Table 2). The low concentrationtreatment did not differentially express this gene. E2F1 was down-regulated after minimally cytotoxic concentrations of iAsIII, MMAIIIand DMAIII.
3.5.2. P53 signaling
SERPINE1 was upregulated in iAsIII (low and minimally cyto-toxic concentrations) and MMAIII (high concentration). TP53 wasdownregulated only in iAsIII group (minimally cytotoxic concen-tration).
3.5.3. Oxidative stress pathways
It is known from the existing literature that HMOX1 is induceddose dependently after exposure to arsenic. As expected, HMOX1showed a 6.5-fold upregulation after a low concentration and an86-fold upregulation after the minimally cytotoxic concentrationof iAsIII. NQO1 was upregulated in iAsIII, MMAIII and DMTA whileDUSP1 was up in iAsIII, MMAIII and DMAIII after minimally cyto-toxic concentration. GCLC was upregulated in iAsIII (1.56-fold) andMMAIII (1.79) while GPX2 was upregulated in iAsIII (3-fold) afterthe minimally cytotoxic concentration.
Author's personal copy
34 J. Chilakapati et al. / Toxicology 268 (2010) 31–39
Table 2Fold up- or downregulation of differentially expressed genes after exposure of BEAS-2B cells to high concentrations of arsenicals.
Gene/pathway Description Affymetrix ID iAsIII MMAIII DMAIII DMTA
Cell cycle: G1/S checkpoint regulationTGFB2 Transforming growth factor, beta 2 209909 s at 1.99 1.76SMAD3 SMAD family member 3 239448 at 2.34SKP1A S-phase kinase-associated protein 1A
(p19A)200719 at 1.59 1.60
SKP2 S-phase kinase-associated protein 2(p45)
210567 s at 1.66 2.05
MYC v-myc myelocytomatosis viraloncogene homolog (avian)
202431 s at 1.90
E2F2 E2F transcription factor 2 228361 at −5.07 −2.41E2F1 E2F transcription factor 1 2028 s at −3.03 −1.68 −1.73CCNE2 Cyclin E2 205034 at −2.61 −1.71CCND1 Cyclin D1 208712 at 2.13 1.83 1.61 1.73NRG1 Neuregulin 1 206343 s at −2.83SMAD4 SMAD family member 4 1565703 at 1.89
G1/S transitionCCND1 Cyclin D1 208711 s at 2.13 1.83 1.61 1.73CCND2 Cyclin D2 200951 s at 3.39CCNE2 Cyclin E2 205034 at −2.61 −1.71CDKN2B Cyclin dependent kinase inhibitor 2B
(p15, inhibits CDK4)236313 at −1.76 −1.52
E2F1 E2F transcription factor 1 204947 at −3.03 −1.68 −1.73E2F2 E2F transcription factor 2 228361 at −5.07 −2.41NRG1 Neuregulin 1 206343 s at 1.80 −2.83SKP1A S-phase kinase-associated protein 1A
(p19A)200719 at 1.59 1.60
TFDP1 Transcription factor Dp-1 242939 at −2.45TP53 Tumor protein p53 (Li-Fraumeni
syndrome)201746 at −1.63
p53 signalingAFF1 AF4/FMR2 family, member 1 237006 at 1.97 2.09AFF4 AF4/FMR2 family, member 4 232865 at 1.61 1.61APAF1 Apoptotic peptidase activating factor 1 211554 s at 2.32BID BH3 interacting domain death agonist 211725 s at 1.67 1.61BLM Bloom syndrome 205733 at −1.63CCND1 Cyclin D1 208711 s at 2.13 1.83 1.61 1.73CCNE2 Cyclin E2 205034 at −2.61 −1.71DDB2 Damage-specific DNA binding protein
2, 48 kDa203409 at −1.99 −1.87
E2F1 E2F transcription factor 1 204947 at −3.03 −1.68 −1.73E2F2 E2F transcription factor 2 228361 at −5.07 −2.41E2F7 E2F transcription factor 7 228033 at −1.74 −1.70E2F8 E2F transcription factor 8 219990 at −2.51 −1.93 −1.78GTSE1 G-2 and S-phase expressed 1 204315 s at 1.82 1.65 2.24SERPINB5 Serpin peptidase inhibitor, clade B 204855 at 1.81 1.57SERPINE1 Serpin peptidase inhibitor, clade E 1568765 at 6.66 6.94SFN Stratifin 209260 at 1.63TP53 Tumor protein p53 (Li-Fraumeni
syndrome)201746 at −1.63
TP53INP1 Tumor protein p53 inducible nuclearprotein 1
225912 at 3.07
Oxidative stressDUSP1 Dual specificity phosphatase 1 201044 x at 3.59 4.76 3.36GCLC Glutamate–cysteine ligase, catalytic
subunit202923 s at 1.56 1.79
GCLM Glutamate–cysteine ligase, modifiersubunit
203925 at 2.95 1.53
GPX2 Glutathione peroxidase 2(gastrointestinal)
202831 at 3.02
GSTM3 (includes EG: 2947)glutathione S-transferase M3 (brain)
202554 s at 2.05
HMOX1 Homo sapiens heme oxygenase(decycling) 1
203665 at 85.99 3.76 1.92
MGST1 Microsomal glutathione S-transferase 1 239001 at 1.96 1.71NF-�B1 Nuclear factor of kappa light
polypeptide gene enhancer in B-cells 1(p105)
209239 at −1.74 −1.57
NQO1 NAD(P)H dehydrogenase, quinone 1 201467 s at 2.05 2.13 1.51PRDX1 Peroxiredoxin 1 208680 at 1.67PRDX6 peroxiredoxin 6 238951 at 1.52 1.71SOD2 Superoxide dismutase 2, mitochondrial 215223 s at 2.22 1.68TP53 Tumor protein p53 (Li-Fraumeni
syndrome)201746 at −1.63

Author's personal copy
J. Chilakapati et al. / Toxicology 268 (2010) 31–39 35
Table 2 (Continued )
Gene/pathway Description Affymetrix ID iAsIII MMAIII DMAIII DMTA
NF-�B signalingBCL10 B-cell CLL/lymphoma 10 1557257 at 1.61CARD10 Caspase recruitment domain family,
member 10210026 s at 1.57
CSNK2A1 Casein kinase 2, alpha 1 polypeptide 239228 at 1.92 2.47EGFR Epidermal growth factor receptor 232541 at 1.58 1.66IKBKB inhibitor of kappa light polypeptide
gene enhancer in B-cells, kinase beta209341 s at −1.58
IL1A Interleukin 1, alpha 210118 s at 1.60 −2.20 −2.40IL1B Interleukin 1, beta 205067 at 3.92 2.49IL1R1 Interleukin 1 receptor, type I 202948 at 1.64MALT1 Mucosa associated lymphoid tissue
lymphoma translocation gene 1210018 x at 1.51
NF-�B1 Nuclear factor of kappa lightpolypeptide gene enhancer in B-cells 1(p105)
209239 at −1.74 −1.57
NRAS Neuroblastoma RAS viral (v-ras)oncogene homolog
202647 s at 1.53
PIK3CD Phosphoinositide-3-kinase, catalytic,delta polypeptide
203879 at 1.75 1.58
PIK3R1 Phosphoinositide-3-kinase, regulatorysubunit 1 (p85 alpha)
212239 at −1.61 −1.70
TGFA Transforming growth factor, alpha 205016 at 1.55TLR4 Toll-like receptor 4 1552798 a at 4.66 7.21 3.51 2.44ZCCHC2 Zinc finger, CCHC domain containing 2 222816 s at 1.85 1.88 1.60
Genes italicised are those already known to be of biological significance to arsenical toxicity.
Fig. 2. Principle components analysis (PCA) demonstrating the separation of control (black), low (green) and high (red) concentrations of the arsenicals. PCA plots werecreated in Rosetta Resolver. All human probesets on the arrays were used to generate these plots. (For interpretation of the references to color in the citation of this figure,the reader is referred to the web version of the article.)
3.5.4. NF-�B pathwayToll-like receptor 4 (TLR4) was upregulated in all four arsenicals
after administration of the high concentration (Table 2) while it wasupregulated only in iAsIII and MMAIII after the low concentration.NF-�B was downregulated in iAsIII and MMAIII.
4. Discussion
This is the first gene expression study that involves a compre-hensive microarray that utilizes arsenic related changes in humanbronchial epithelial cells. Gene expression changes in bronchialepithelial cells after arsenical exposure are consistent with sev-
eral of the proposed mechanisms of arsenic carcinogenesis (Fig. 5)(Kitchin, 2001).
We are aware of the fact that the best models for in vitro assayswould be primary human cultures of untransformed cells. SinceBEAS-2B cells are SV40 immortalized cells, it would be interestingto study the effects of arsenic and its metabolites in primary normalhuman bronchial epithelial cells.
Inorganic arsenic is the most predominant form of arsenic towhich humans are exposed to via drinking water (NRC, 1999).Although the methylated trivalent arsenicals can be direct act-ing genotoxins (Kitchin and Ahmad, 2003; Mass et al., 2001), theyare not present in high concentrations in drinking water. Cultured

Author's personal copy
36 J. Chilakapati et al. / Toxicology 268 (2010) 31–39
Fig. 3. Total number of differentially expressed genes after arsenical exposure toBEAS-2B cells for 24 h.
human cells derived from liver, skin, urinary bladder and lung wereexposed to iAs and trivalent and pentavalent methylated arsenicals(Styblo et al., 2000). Trivalent methylated arsenicals were signif-icantly more toxic than pentavalent arsenicals for normal humanhepatocytes, epidermal keratinocytes, bronchial epithelial cells andurinary bladder cells (Styblo et al., 2000). Trivalent arsenicals havebeen shown to be more potent than pentavalent arsenicals (Dopp etal., 2004). Cohen et al. (2002) reported that the LC50 of DMAIII in ratand human urothelial cell lines to be less than 1 �M. The LC50 forDMAV was in the millimolar range. Our study showed that DMAIII,MMAIII and DMTA were more toxic to BEAS-2B cells than iAsIII.
The low concentrations that we used in our study (225 ppbfor iAs, 22.5 for MMAIII and 15 ppb for DMAIII) are higher thanthe 10 ppb arsenic drinking water standard (MCL of the USEPA).However, the low concentration of arsenicals utilized in this studyrarely produced a significantly altered pathway. The minimallycytotoxic concentration of trivalent arsenicals resulted in signifi-cantly altered pathways including p53 signaling, oxidative stress,G1/S transition of the cell cycle, NF-�B signaling, and mechanismof gene regulation by peroxisome proliferators via PPAR� (Fig. 5).
Transferrin receptor (p90, CD71) (TFRC) is involved in cellulariron ion homeostasis and endocytosis. TfR1 (transferrin receptor 1)mediates the uptake of transferrin-bound iron and thereby plays acritical role in cellular iron metabolism. Its expression is coupled tocell proliferation/differentiation and controlled in response to ironlevels and other signals by transcriptional and post-transcriptionalmechanisms (Wang et al., 2005). We found TFRC to be a commongene differentially expressed in low concentration arsenical treatedcells or cultures. This might be a protective response to arsenic-induced stress.
TLR4 is a member of the Toll-like receptor (TLR) family whichplays a fundamental role in pathogen recognition and activation ofinnate immunity (Cartwright et al., 2007). TLR4 was upregulatedby the minimally cytotoxic concentration of all four arsenicals.
DUSP1 is upregulated in response to a variety of cellular stressconditions including oxidative stress and DNA-damaging agents(Arkell et al., 2008). In our study, the minimally cytotoxic concen-trations of iAsIII, MMAIII and DMAIII upregulated DUSP1.
E2F transcription factors control the expression of numerousgenes involved in the G1/S cell cycle transition, initiation of DNAsynthesis, and mitosis and also link DNA-damage recognition path-ways to key cell cycle checkpoints (Cam and Dynlacht, 2003). E2F1is a positive regulator of S phase entry (Wilson, 2007). E2F1 wasdownregulated by iAsIII, MMAIII and DMTA.
Cyclin D1 is a member of the cyclin family of proteins whichfunction as the regulatory subunits of cyclin/cyclin dependent
kinase holoenzymes that regulate entry into and progressionthrough the cell cycle (Sherr, 1996). Overexpression of cyclin D1has been found in many cancers including breast, prostate, colonand lymphoma (Rodriguez-Puebla et al., 1999). Induction of cyclinD1, which is a cell cycle promoter, can cause lung tumorigenesis.Cyclin D1 has been shown to be an important protein implicatedin cell transformation in JB6 Cl41 cells (mouse epidermal cells)exposed to low concentration arsenic (Zhang et al., 2009). Overex-pression of cyclin D1 has been reported in arsenic transformed cells(Chen et al., 2001). Our study showed upregulation of cyclin D1 inBEAS-2B cells after treatment with minimally toxic concentrationof arsenite. Thus, cell cycle dysregulation as manifested by cyclin D1overexpression appears to be a consistent event in arsenic-inducedcarcinogenesis.
Arsenic has been shown to disrupt cellular levels of p53 in theimmortalized keratinocyte cell line HaCaT (Hamadeh et al., 1999).Our study also showed inhibition of p53 with minimally toxic con-centration of arsenite. In contrast, arsenite exposure increased p53protein levels in HeLa, Jurkat and lymphoblast cell lines (Salazar etal., 1997).
NF-�B plays a critical role in modulating several cell survival andcell death mechanisms. NF-�B is known to mediate G1 to S progres-sion by regulating cyclin D1 expression. Exposure to 1 �M arseniteinhibits the NF-�B pathway functionally in lung macrophages withLPS-induced TNF-alpha production (Lantz and Hays, 2006). Consis-tent with previous reports, arsenic treatment inhibited NF-�B.
The differential expression of oxidative stress-related genes isassociated with arsenic toxicity and cell proliferation (Liu et al.,2001; Pi et al., 2003). The oxidation of CuZn superoxide dismutaseprotein is evident in arsenic-exposed cells (Pi et al., 2003). In thepresent work, expression of SOD was increased in minimally toxicconcentration of iAsIII and DMAIII exposed BEAS-2B exposed cells.
GPX2, which is not exclusively expressed in the gastrointestinalsystem, is upregulated in colon and skin cancers and in certain cul-tured cancer cells (Brigelius-Flohe, 2006). GPX2 is upregulated in avariety of cancer cells with thus far unknown consequences. GPX2supports growth of established tumors (Brigelius-Flohe and Kipp,2009). We found a 3-fold induction of GPX2 after a minimally toxicconcentration of iAsIII.
GSH plays a key role in antioxidant defense. The first and rate-limiting step in GSH synthesis is catalyzed by glutamate–cysteineligase (GCL, previously known as gamma-glutamylcysteine syn-thetase). GCL is a heterodimeric protein composed of catalytic(GCLC) and modifier (GCLM) subunits that are expressed from dif-ferent genes (Franklin et al., 2009). GCLC was upregulated afterexposures of minimally toxic concentrations of iAsIII and MMAIII.
The marked dose dependent increase in heme oxygenase(HMOX1) induction after arsenite exposure is supported by theliterature (Menzel et al., 1998; Taketani et al., 1989; Yih et al.,2002). In the present study, HMOX1 was induced 86-fold after theminimally toxic concentration of arsenite. This remarkably highdegree of induction was not found with MMAIII (3.8-fold) or DMAIII(2-fold). HMOX1 was not differentially expressed from control inDMTA group.
iAsIII and MMAIII had a lot of DEGs in common both at low andhigh concentrations when compared to the other two compounds.MMAIII has been indicated as a likely candidate to cause cancer inhumans as humans excrete more MMAIII than any other species(Vahter, 1994).
One highlight of this study is the use of the sulfur containingarsenical DMTA. Recent work shows that an arsenical may havebeen misidentified as DMAIII when it was really DMTA becausethe chromatographic properties are similar (Raml et al., 2007). Thisshows that DMTA might have escaped detection in many samples.Little is known about the contribution of DMTA to the toxic effectsassociated with arsenic exposures and especially towards lungs.

Author's personal copy
J. Chilakapati et al. / Toxicology 268 (2010) 31–39 37
Fig. 4. Venn analysis showing the common and unique number of significant DEGs (with a 1.5-fold cut-off) between high and low concentrations. Venn analysis showing thecommon and unique number of significant DEGs (with a 1.5-fold cut-off) across the trivalent arsenicals. Venn analysis showing the common and unique number of significantDEGs (with a 1.5-fold cut-off) between high concentrations of DMTA and DMAIII.
Studying the gene expression changes due to DMTA may providemuch needed new information as relatively little data is availablefor sulfur containing arsenicals. DMTA showed a similar cytotoxic-ity profile to DMAIII and also shared 302 common DEGs with DMAIIIafter a minimally toxic concentration exposure. We found that theminimally toxic concentration of DMTA differentially expressedgenes related to cell cycle and p53 signaling.
In conclusion, this study shows BEAS-2B cells exposed to arsenicand its metabolites had differentially expressed genes linked toarsenic-induced carcinogenesis. Our analysis links several of theidentified genes with potential biological pathways that includep53 signaling, oxidative stress, cell cycle transition and NF-�B sig-
naling. Briefly, this study reveals a number of important findings.First, iAsIII downregulated p53 and NF-�B and upregulated CCND1,SERPINE 1, HMOX1, GCLC, GPX2, and SOD. iAsIII, MMAIII and DMAIIIall upregulated CCND1, TLR4, DUSP1 and HMOX1. Second, MMAIIIaltered most genes altered by iAsIII. Third, iAsIII and MMAIII sharedthe largest number of common genes. Fourth, all four of the com-pounds chosen exhibited a dose response in the number of DEGsexpressed. Fifth, there was a good separation between high andlow concentrations as observed by PCA analysis. Sixth, we not onlysaw similarities between iAsIII and MMAII but also between DMAIIIand DMTA in terms of the number of genes shared as well as theirgenomic profiles. Finally, the results obtained from DMTA expo-

Author's personal copy
38 J. Chilakapati et al. / Toxicology 268 (2010) 31–39
Fig. 5. Comparison analysis of three trivalent arsenicals at high concentrations using Ingenuity Pathway Analysis. The contributions of each trivalent arsenical to the topnine pathways are shown in this figure. The blue line represents threshold of statistical significance at p < 0.05. (For interpretation of the references to color in the citation ofthis figure, the reader is referred to the web version of the article.)
sures can be used to further study its role in arsenic-induced lungcarcinogenesis. Overall, the findings of this study are in agreementwith the existing literature about arsenic toxicity and mechanismsproposed for carcinogenicity. These data also provide importantinsights into genomic effects related to arsenic-induced humanlung cancer.
Conflict of interest statement
The authors declare that they have no potential conflicts of inter-est.
Acknowledgments
We thank Gail Nelson and Janice S. Lee for reviewing thismanuscript as part of EPA clearance procedures.
References
Ahmad, S., Kitchin, K.T., Cullen, W.R., 2002. Plasmid DNA damage caused by methy-lated arsenicals, ascorbic acid and human liver ferritin. Toxicol. Lett. 133, 47–57.
Andrewes, P., Kitchin, K.T., Wallace, K., 2003. Dimethylarsine and trimethylarsineare potent genotoxins in vitro. Chem. Res. Toxicol. 16, 994–1003.
Arkell, R.S., Dickinson, R.J., Squires, M., Hayat, S., Keyse, S.M., Cook, S.J., 2008.DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. CellSignal 20, 836–843.
Bates, M.N., Smith, A.H., Hopenhayn-Rich, C., 1992. Arsenic ingestion and internalcancers: a review. Am. J. Epidemiol. 135, 462–476.
Brigelius-Flohe, R., 2006. Glutathione peroxidases and redox-regulated transcrip-tion factors. Biol. Chem. 387, 1329–1335.
Brigelius-Flohe, R., Kipp, A., 2009. Glutathione peroxidases in different stages ofcarcinogenesis. Biochim. Biophys. Acta.
Cam, H., Dynlacht, B.D., 2003. Emerging roles for E2F: beyond the G1/S transitionand DNA replication. Cancer Cell 3, 311–316.
Cartwright, N., McMaster, S.K., Sorrentino, R., Paul-Clark, M., Sriskandan, S., Ryffel, B.,Quesniaux, V.F., Evans, T.W., Mitchell, J.A., 2007. Elucidation of toll-like receptorand adapter protein signaling in vascular dysfunction induced by gram-positiveStaphylococcus aureus or gram-negative Escherichia coli. Shock 27, 40–47.
Chen, C.J., Chen, C.W., Wu, M.M., Kuo, T.L., 1992. Cancer potential in liver, lung,bladder and kidney due to ingested inorganic arsenic in drinking water. Br. J.Cancer 66, 888–892.
Chen, H., Liu, J., Zhao, C.Q., Diwan, B.A., Merrick, B.A., Waalkes, M.P., 2001. Asso-ciation of c-myc overexpression and hyperproliferation with arsenite-inducedmalignant transformation. Toxicol. Appl. Pharmacol. 175, 260–268.
Cohen, S.M., Arnold, L.L., Uzvolgyi, E., Cano, M., St John, M., Yamamoto, S., Lu, X.,Le, X.C., 2002. Possible role of dimethylarsinous acid in dimethylarsinic acid-induced urothelial toxicity and regeneration in the rat. Chem. Res. Toxicol. 15,1150–1157.
Dopp, E., Hartmann, L.M., Florea, A.M., von Recklinghausen, U., Pieper, R., Shokouhi,B., Rettenmeier, A.W., Hirner, A.V., Obe, G., 2004. Uptake of inorganic and organicderivatives of arsenic associated with induced cytotoxic and genotoxic effectsin Chinese hamster ovary (CHO) cells. Toxicol. Appl. Pharmacol. 201, 156–165.
Fautz, R., Husein, B., Hechenberger, C., 1991. Application of the neutral red assay(NR assay) to monolayer cultures of primary hepatocytes: rapid colorimetricviability determination for the unscheduled DNA synthesis test (UDS). Mutat.Res. 253, 173–179.
Franklin, C.C., Backos, D.S., Mohar, I., White, C.C., Forman, H.J., Kavanagh, T.J.,2009. Structure, function, and post-translational regulation of the catalyticand modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 30,86–98.
Fricke, M., Zeller, M., Cullen, W., Witkowski, M., Creed, J., 2007. Dimethylthioarsinicanhydride: a standard for arsenic speciation. Anal. Chim. Acta 583, 78–83.
Hamadeh, H.K., Vargas, M., Lee, E., Menzel, D.B., 1999. Arsenic disrupts cellular levelsof p53 and mdm2: a potential mechanism of carcinogenesis. Biochem. Biophys.Res. Commun. 263, 446–449.
Hughes, M., Kitchin, K.T., 2006. Arsenic, oxidative stress and carcinogenesis. In:Singh, K. (Ed.), Oxidative stress, Disease and Cancer. Imperial College Press,London, pp. 825–850.
Hughes, M.F., Kenyon, E.M., Kitchin, K.T., 2007. Research approaches to addressuncertainties in the risk assessment of arsenic in drinking water. Toxicol. Appl.Pharmacol..
IARC, 2004. Some Drinking Water Disinfectants and Contaminants, includingArsenic, International Agency for Research on Cancer, Lyon, France.
Kitchin, K.T., 2001. Recent advances in arsenic carcinogenesis: modes of action,animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Phar-macol. 172, 249–261.
Kitchin, K.T., Ahmad, S., 2003. Oxidative stress as a possible mode of action for arseniccarcinogenesis. Toxicol. Lett. 137, 3–13.
Kitchin, K.T., Wallace, K., 2008. Evidence against the nuclear in situ binding ofarsenicals-oxidative stress theory of arsenic carcinogenesis. Toxicol. Appl. Phar-macol..
Kitchin, K.T., Wallace, K., Andrewes, P., 2002. Some chemical properties underlyingarsenic’s biological activity. In: Chapell, C.R. (Ed.), Arsenic Exposure and HealthEffects, vol. V. Elsevier, San Diego, pp. 345–354.
Kligerman, A.D., Doerr, C.L., Tennant, A.H., Harrington-Brock, K., Allen, J.W., Wink-field, E., Poorman-Allen, P., Kundu, B., Funasaka, K., Roop, B.C., Mass, M.J.,DeMarini, D.M., 2003. Methylated trivalent arsenicals as candidate ultimategenotoxic forms of arsenic: induction of chromosomal mutations but not genemutations. Environ. Mol. Mutagen. 42, 192–205.
Kuroda, K., Yoshida, K., Yoshimura, M., Endo, Y., Wanibuchi, H., Fukushima, S., Endo,G., 2004. Microbial metabolite of dimethylarsinic acid is highly toxic and geno-toxic. Toxicol. Appl. Pharmacol. 198, 345–353.
Lantz, R.C., Hays, A.M., 2006. Role of oxidative stress in arsenic-induced toxicity.Drug Metab. Rev. 38, 791–804.
Le, X.C., Cullen, W.R., Reimer, K.J., 1994. Speciation of arsenic compounds by HPLCwith hydride generation atomic absorption spectrometry and inductively cou-pled plasma mass spectrometry detection. Talanta 41, 495–502.
Liu, S.X., Athar, M., Lippai, I., Waldren, C., Hei, T.K., 2001. Induction of oxyradicals byarsenic: implication for mechanism of genotoxicity. Proc. Natl. Acad. Sci. U.S.A.98, 1643–1648.
Mace, K., Gonzalez, F.J., McConnell, I.R., Garner, R.C., Avanti, O., Harris, C.C., Pfeifer,A.M., 1994. Activation of promutagens in a human bronchial epithelial cell linestably expressing human cytochrome P450 1A2. Mol. Carcinog. 11, 65–73.

Author's personal copy
J. Chilakapati et al. / Toxicology 268 (2010) 31–39 39
Mass, M.J., Tennant, A., Roop, B.C., Cullen, W.R., Styblo, M., Thomas, D.J., Kliger-man, A.D., 2001. Methylated trivalent arsenic species are genotoxic. Chem. Res.Toxicol. 14, 355–361.
Menzel, D.B., Rasmussen, R.E., Lee, E., Meacher, D.M., Said, B., Hamadeh, H., Vargas,M., Greene, H., Roth, R.N., 1998. Human lymphocyte heme oxygenase 1 as aresponse biomarker to inorganic arsenic. Biochem. Biophys. Res. Commun. 250,653–656.
Naranmandura, H., Ibata, K., Suzuki, K.T., 2007. Toxicity of dimethylmonothioarsinicacid toward human epidermoid carcinoma A431 cells. Chem. Res. Toxicol. 20,1120–1125.
Navas-Acien, A., Silbergeld, E.K., Streeter, R.A., Clark, J.M., Burke, T.A., Guallar, E.,2006. Arsenic exposure and type 2 diabetes: a systematic review of the exper-imental and epidemiological evidence. Environ. Health Perspect. 114, 641–648.
NRC, 1999. Mechanisms of toxicity. In: Arsenic in Drinking Water. National AcademyPress, Washington, DC, pp. 194–196.
NRC, 2001a. Arsenic in Drinking Water 2001 Update. National Academy Press, Wash-ington, DC, pp. 1–225.
NRC, 2001b. Arsenic in Drinking Water 2001 Update. National Academy Press, Wash-ington, DC, pp. 194–196.
Petrick, J.S., Ayala-Fierro, F., Cullen, W.R., Carter, D.E., Vasken Aposhian, H., 2000.Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Changhuman hepatocytes. Toxicol. Appl. Pharmacol. 163, 203–207.
Pi, J., Qu, W., Reece, J.M., Kumagai, Y., Waalkes, M.P., 2003. Transcription factorNrf2 activation by inorganic arsenic in cultured keratinocytes: involvement ofhydrogen peroxide. Exp. Cell Res. 290, 234–245.
Raml, R., Rumpler, A., Goessler, W., Vahter, M., Li, L., Ochi, T., Francesconi, K.A., 2007.Thio-dimethylarsinate is a common metabolite in urine samples from arsenic-exposed women in Bangladesh. Toxicol. Appl. Pharmacol. 222, 374–380.
Reddel, R.R., Ke, Y., Gerwin, B.I., McMenamin, M.G., Lechner, J.F., Su, R.T., Brash,D.E., Park, J.B., Rhim, J.S., Harris, C.C., 1988. Transformation of human bronchialepithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, ortransfection via strontium phosphate coprecipitation with a plasmid containingSV40 early region genes. Cancer Res. 48, 1904–1909.
Reddel, R.R., Malan-Shibley, L., Gerwin, B.I., Metcalf, R.A., Harris, C.C., 1989. Tumori-genicity of human mesothelial cell line transfected with EJ-ras oncogene. J. Natl.Cancer Inst. 81, 945–948.
Rodriguez-Puebla, M.L., Robles, A.I., Conti, C.J., 1999. ras activity and cyclin D1expression: an essential mechanism of mouse skin tumor development. Mol.Carcinog. 24, 1–6.
Salazar, A.M., Ostrosky-Wegman, P., Menendez, D., Miranda, E., Garcia-Carranca, A.,Rojas, E., 1997. Induction of p53 protein expression by sodium arsenite. Mutat.Res. 381, 259–265.
Sherr, C.J., 1996. Cancer cell cycles. Science 274, 1672–1677.Smith, A.H., Hopenhayn-Rich, C., Bates, M.N., Goeden, H.M., Hertz-Picciotto, I., Dug-
gan, H.M., Wood, R., Kosnett, M.J., Smith, M.T., 1992. Cancer risks from arsenicin drinking water. Environ. Health Perspect. 97, 259–267.
Smith, A.H., Marshall, G., Yuan, Y., Ferreccio, C., Liaw, J., von Ehrenstein, O., Stein-maus, C., Bates, M.N., Selvin, S., 2006. Increased mortality from lung cancer andbronchiectasis in young adults after exposure to arsenic in utero and in earlychildhood. Environ. Health Perspect. 114, 1293–1296.
Styblo, M., Del Razo, L.M., Vega, L., Germolec, D.R., LeCluyse, E.L., Hamilton, G.A., Reed,W., Wang, C., Cullen, W.R., Thomas, D.J., 2000. Comparative toxicity of trivalentand pentavalent inorganic and methylated arsenicals in rat and human cells.Arch. Toxicol. 74, 289–299.
Taketani, S., Kohno, H., Yoshinaga, T., Tokunaga, R., 1989. The human 32-kDa stressprotein induced by exposure to arsenite and cadmium ions is heme oxygenase.FEBS Lett. 245, 173–176.
Thomas, D.J., Styblo, M., Lin, S., 2001. The cellular metabolism and systemic toxicityof arsenic. Toxicol. Appl. Pharmacol. 176, 127–144.
Vahter, M., 1994. What are the chemical forms of arsenic in urine, and what can theytell us about exposure? Clin. Chem. 40, 679–680.
Vahter, M., 1999. Methylation of inorganic arsenic in different mammalian speciesand population groups. Sci. Prog. 82 (Pt 1), 69–88.
Waalkes, M.P., Liu, J., Chen, H., Xie, Y., Achanzar, W.E., Zhou, Y.S., Cheng, M.L., Diwan,B.A., 2004. Estrogen signaling in livers of male mice with hepatocellular carci-noma induced by exposure to arsenic in utero. J. Natl. Cancer Inst. 96, 466–474.
Wang, J., Chen, G., Pantopoulos, K., 2005. Inhibition of transferrin receptor 1 tran-scription by a cell density response element. Biochem. J. 392, 383–388.
Wilson, A.C., 2007. Setting the stage for S phase. Mol. Cell 27, 176–177.Yih, L.H., Peck, K., Lee, T.C., 2002. Changes in gene expression profiles of human
fibroblasts in response to sodium arsenite treatment. Carcinogenesis 23,867–876.
Yoshida, K., Kuroda, K., Zhou, X., Inoue, Y., Date, Y., Wanibuchi, H., Fukushima, S.,Endo, G., 2003. Urinary sulfur-containing metabolite produced by intestinal bac-teria following oral administration of dimethylarsinic acid to rats. Chem. Res.Toxicol. 16, 1124–1129.
Zhang, D., Li, J., Gao, J., Huang, C., 2009. c-Jun/AP-1 pathway-mediated cyclin D1expression participates in low dose arsenite-induced transformation in mouseepidermal JB6 Cl41 cells. Toxicol. Appl. Pharmacol. 235, 18–24.
Copyright © 2022 FDOKUMEN











![financial management 2b [bsr2b01, fnm02b2] last assessment ...](https://static.fdokumen.com/doc/165x107/631aa5b5c51d6b41aa04e9e3/financial-management-2b-bsr2b01-fnm02b2-last-assessment-.jpg)








