
Genistein protects pancreatic β cells against cytokine-mediated toxicity
11
Molecular and Cellular Endocrinology 278 (2007) 18–28 Genistein protects pancreatic cells against cytokine-mediated toxicity Eun-Kyung Kim a,1 , Kang-Beom Kwon b,1 , Mi-Young Song a , Sang-Wan Seo c , Sung-Joo Park c , Sun-O Ka a , Lv Na a , Kyung-Ah Kim a , Do-Gon Ryu b , Hong-Seob So d , Raekil Park d , Jin-Woo Park a , Byung-Hyun Park a,∗ a Department of Biochemistry, Medical School and Institute for Medical Sciences, Chonbuk National University, Jeonju, Jeonbuk, 561-756, Republic of Korea b Department of Physiology, School of Oriental Medicine, Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Korea c Department of Herbology, School of Oriental Medicine, Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Korea d Vestibulocochlear System Research Center, Department of Microbiology, College of Medicine, Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Korea Received 26 May 2007; received in revised form 3 August 2007; accepted 10 August 2007 Abstract In the past few decades, the use of genistein as an anti-inflammatory agent has gained much attention. Our current study focuses on the preventive effects of genistein on cytokine-induced pancreatic -cell damage. Treatment of RINm5F (RIN) rat insulinoma cells with interleukin (IL)-1 and interferon (IFN)- induced cell damage, which was correlated with nitric oxide (NO) production. Genistein completely prevented cytokine-mediated cytotoxicity and NO production, a finding that correlated well with reduced levels of the inducible form of NO synthase (iNOS) mRNA and protein. The molecular mechanism of genistein inhibition of iNOS gene expression appeared to involve the inhibition of NFB activation. The cytokine induced increases in NFB binding activity, nuclear p50 and p65 subunit levels, and IB degradation in cytosol compared to unstimulated cells; genistein abolished all of these parameters. The cytoprotective effects of genistein are also mediated through the suppression of ERK-1/2 and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways. In a second set of experiments, rat islets were used. The findings on -cell protective effects of genistein were essentially the same as for the RIN cell data, namely genistein prevented cytokine-induced NO production, iNOS expression, ERK-1/2 activation, JAK/STAT activation, and impairment of glucose-stimulated insulin secretion. Collectively, these results suggest that genistein might be used to preserve functional -cell mass. © 2007 Elsevier Ireland Ltd. All rights reserved. Keywords: Genistein; Cytokine; NFB; ERK-1/2; JAK-STAT; Pancreatic -cell 1. Introduction Type 1 diabetes mellitus is an autoimmune disease causing selective destruction of insulin producing cells of the Langer- Abbreviations: IL-1, interleukin-1; IFN-, interferon-; NO, nitric oxide; iNOS, inducible form of nitric oxide synthase; NFB, nuclear factor B; IB, inhibitory factor of NFB; RIN, RINm5F; JAK, Janus kinase; STAT, signal transducer and activator of transcription; ERK, extracellular signal- regulated kinase; JNK, c-Jun N-terminal kinase; l-NAME, N w -nitro-l-arginine methylester; GAS, -activated sequence; MTT, 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium; BrdU, 5-bromo-2-deoxyuridine; PCNA, proliferating cell nuclear antigen; MAPK, mitogen-activated protein kinase; PMSF, phenyl- methylsulfonyl fluoride; DMSO, dimethyl sulfoxide ∗ Corresponding author. Tel.: +82 63 270 3139; fax: +82 63 274 9833. E-mail address: [email protected] (B.-H. Park). 1 These authors contributed equally to this work. hans islets (Nossal et al., 1992). Much evidence supports a crucial role for infiltrated immune cells in and around pancre- atic islets early in pathogenesis (Jorns et al., 2005; Kanazawa et al., 1984). Cytokines such as interleukin (IL)-1 and inter- feron (IFN)- have been implicated as effector molecules that participate in the initial destruction of cells leading to the development of disease (Eizirik et al., 1996; Mandrup-Poulsen, 1996; Southern et al., 1990). IL-1 in combination with IFN- produces excess nitric oxide (NO) by the inducible form of nitric oxide synthase (iNOS) and causes apoptosis of rat and human pancreatic cells (Cetkovic-Cvrlje and Eizirik, 1994; Corbett and McDaniel, 1995; Eizirik et al., 1996; Heitmeier et al., 1997). Nitric oxide is a short-lived and highly reactive rad- ical, which inhibits the Krebs-cycle enzyme aconitase and the electron transport chain complexes I and II leading to decreased glucose oxidation rates, ATP generation and insulin production 0303-7207/$ – see front matter © 2007 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.mce.2007.08.003
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Genistein protects pancreatic β cells against cytokine-mediated toxicity

A
eicTigJfiNt©
K
1
s
iIsrm2cm
0d
Molecular and Cellular Endocrinology 278 (2007) 18–28
Genistein protects pancreatic � cells against cytokine-mediated toxicity
Eun-Kyung Kim a,1, Kang-Beom Kwon b,1, Mi-Young Song a, Sang-Wan Seo c, Sung-Joo Park c,Sun-O Ka a, Lv Na a, Kyung-Ah Kim a, Do-Gon Ryu b, Hong-Seob So d,
Raekil Park d, Jin-Woo Park a, Byung-Hyun Park a,∗a Department of Biochemistry, Medical School and Institute for Medical Sciences, Chonbuk National University,
Jeonju, Jeonbuk, 561-756, Republic of Koreab Department of Physiology, School of Oriental Medicine, Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Koreac Department of Herbology, School of Oriental Medicine, Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Korea
d Vestibulocochlear System Research Center, Department of Microbiology, College of Medicine,Wonkwang University, Iksan, Jeonbuk, 570-749, Republic of Korea
Received 26 May 2007; received in revised form 3 August 2007; accepted 10 August 2007
bstract
In the past few decades, the use of genistein as an anti-inflammatory agent has gained much attention. Our current study focuses on the preventiveffects of genistein on cytokine-induced pancreatic �-cell damage. Treatment of RINm5F (RIN) rat insulinoma cells with interleukin (IL)-1� andnterferon (IFN)-� induced cell damage, which was correlated with nitric oxide (NO) production. Genistein completely prevented cytokine-mediatedytotoxicity and NO production, a finding that correlated well with reduced levels of the inducible form of NO synthase (iNOS) mRNA and protein.he molecular mechanism of genistein inhibition of iNOS gene expression appeared to involve the inhibition of NF�B activation. The cytokine
nduced increases in NF�B binding activity, nuclear p50 and p65 subunit levels, and I�B� degradation in cytosol compared to unstimulated cells;enistein abolished all of these parameters. The cytoprotective effects of genistein are also mediated through the suppression of ERK-1/2 andanus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways. In a second set of experiments, rat islets were used. The
ndings on �-cell protective effects of genistein were essentially the same as for the RIN cell data, namely genistein prevented cytokine-inducedO production, iNOS expression, ERK-1/2 activation, JAK/STAT activation, and impairment of glucose-stimulated insulin secretion. Collectively,hese results suggest that genistein might be used to preserve functional �-cell mass.2007 Elsevier Ireland Ltd. All rights reserved.
ll
h
eywords: Genistein; Cytokine; NF�B; ERK-1/2; JAK-STAT; Pancreatic �-ce
. Introduction
Type 1 diabetes mellitus is an autoimmune disease causingelective destruction of insulin producing � cells of the Langer-
Abbreviations: IL-1�, interleukin-1�; IFN-�, interferon-�; NO, nitric oxide;NOS, inducible form of nitric oxide synthase; NF�B, nuclear factor �B;�B, inhibitory factor of NF�B; RIN, RINm5F; JAK, Janus kinase; STAT,ignal transducer and activator of transcription; ERK, extracellular signal-egulated kinase; JNK, c-Jun N-terminal kinase; l-NAME, Nw-nitro-l-arginineethylester; GAS, �-activated sequence; MTT, 3-(4,5-dimethylthiazol-2-yl)-
,5-diphenyltetrazolium; BrdU, 5-bromo-2-deoxyuridine; PCNA, proliferatingell nuclear antigen; MAPK, mitogen-activated protein kinase; PMSF, phenyl-ethylsulfonyl fluoride; DMSO, dimethyl sulfoxide∗ Corresponding author. Tel.: +82 63 270 3139; fax: +82 63 274 9833.
E-mail address: [email protected] (B.-H. Park).1 These authors contributed equally to this work.
caefpd1�nhCaieg
303-7207/$ – see front matter © 2007 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.mce.2007.08.003
ans islets (Nossal et al., 1992). Much evidence supports arucial role for infiltrated immune cells in and around pancre-tic islets early in pathogenesis (Jorns et al., 2005; Kanazawat al., 1984). Cytokines such as interleukin (IL)-1� and inter-eron (IFN)-� have been implicated as effector molecules thatarticipate in the initial destruction of � cells leading to theevelopment of disease (Eizirik et al., 1996; Mandrup-Poulsen,996; Southern et al., 1990). IL-1� in combination with IFN-produces excess nitric oxide (NO) by the inducible form of
itric oxide synthase (iNOS) and causes apoptosis of rat anduman pancreatic � cells (Cetkovic-Cvrlje and Eizirik, 1994;orbett and McDaniel, 1995; Eizirik et al., 1996; Heitmeier et
l., 1997). Nitric oxide is a short-lived and highly reactive rad-cal, which inhibits the Krebs-cycle enzyme aconitase and thelectron transport chain complexes I and II leading to decreasedlucose oxidation rates, ATP generation and insulin production
llular
(WNdde
(kLa(tate�aopttsBs1aatototdbma
itClsetia
2
2
l1bsS
PC(d
2
omi(3ts
2a
spcbwo((waafTss
2
sss1maaSft
2
rDDaC
Bs
E.-K. Kim et al. / Molecular and Ce
Corbett and McDaniel, 1994; Cunningham and Green, 1994;elsh et al., 1991). It has been reported that the iNOS inhibitors,
w-nitro-l-arginine methylester (l-NAME) and aminoguani-ine, attenuate cytokine-induced �-cell dysfunction and isletegeneration (Eizirik et al., 1996; Southern et al., 1990; Welsht al., 1991).
IL-1� activates the extracellular signal-regulated kinaseERK)-1/2 and p38 kinase, as well as the c-Jun N-terminalinase (JNK) in pancreatic � cells (Ammendrup et al., 2000;arsen et al., 1998). Combined inhibition of ERK-1/2 and p38lso blocks IL-1�-induced NO production and iNOS inductionLarsen et al., 1998). Inhibition of JNK activation also pro-ected � cells against IL-1�-induced apoptosis (Ammendrup etl., 2000; Bonny et al., 2001) and human islets against destruc-ion mediated by IL-1� and IFN-� (Aikin et al., 2004). IL-1�xerts its main effects through the transcriptional nuclear factorB (NF�B) pathway. NF�B is initially located in the cytosols an inactive form complexed with I�B, an inhibitory factorf NF�B. Various inducers cause dissociation of this com-lex, presumably by phosphorylation of I�B, allowing NF�Bo be released from the complex. NF�B then translocates tohe nucleus, where it interacts with specific DNA recognitionites to mediate gene transcription (Baeuerle and Henkel, 1994;aldwin, 1996; May and Ghosh, 1998). IFN-� alone does not
timulate iNOS expression in rat islets, but does prime for IL-�-induced iNOS expression (Heitmeier et al., 1999). IFN-�cts mostly via the Janus kinase (JAK)/signal transducer andctivator of transcription (STAT-1) pathway. Binding of IFN-�o its receptor induces receptor oligomerization and activationf the receptor-associated JAK1 and JAK2 by transphosphoryla-ion. The activated JAKs phosphorylate the intracellular domainf the receptor, which serves as the binding sites for STAT pro-eins. Phosphorylated STAT-1 proteins at tyrosine residue formimers, translocate to the nucleus, and regulate gene expressiony binding to �-activated sequence (GAS) elements in the pro-oters of IFN-�-regulated genes, including iNOS (Aaronson
nd Horvath, 2002; Ramana et al., 2002).Genistein, a soybean isoflavone with diverse biological activ-
ties, is a potent antioxidant, a nonselective inhibitor of proteinyrosine kinase, and a phytoestrogen (Akiyama et al., 1987;heng et al., 1953; Sarkar and Li, 2003). Evidence has accumu-
ated that this natural ingredient protects � cells from cytokine byuppressing iNOS expression in human and rat islets (Corbettt al., 1994, 1996). In the present study, we have investigatedhe preventive mechanism of genistein on IL-1� and IFN-�-nduced �-cell damage models using a rat insulinoma cell linend isolated rat islets.
. Materials and methods
.1. Cell culture and reagents
RINm5F (RIN) cells were purchased from the American Type Culture Col-
ection and grown at 37 ◦C under a humidified, 5% CO2 atmosphere in RPMI640 medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetalovine serum and 2 mM glutamine, 100 units/ml of penicillin, 100 �g/ml oftreptomycin, and 2.5 �g/ml of amphotericin B. Genistein was purchased fromigma (St. Louis, MO) and dissolved at 40 mM in dimethyl sulfoxide (DMSO).5caur
Endocrinology 278 (2007) 18–28 19
D98059, SP600125, and SB203580, purchased from Calbiochem (San Diego,A), were resuspended at 10 mM in DMSO. IL-1� and IFN-� were from R&D
Minneapolis, MN). All reagents were purchased from Sigma unless specificallyescribed.
.2. MTT assay for cell viability
The viability of cultured cells was determined by assaying for the reductionf 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to for-azan as described previously (E.K. Kim et al., 2007a). In brief, after 48 h of
ncubation, cells (105/well) in 96-well plates were washed twice with PBS. MTT100 �g/0.1 ml of PBS) was added to each well. Cells were then incubated at7 ◦C for 1 h and DMSO (100 �l) was added to dissolve the formazan crys-als. Absorbance was measured at 570 nm with a model Spectra MAX PLUSpectrophotometer (Molecular Devices, Sunnyvale, CA).
.3. 5-Bromo-2-deoxyuridine (BrdU)-labeling cell proliferationssay
A cell proliferation enzyme-linked immunosorbant assay (BrdU kit; Amer-ham Biosciences, Piscataway, NJ) was used, according to the manufacturer’srotocol, to measure incorporation of BrdU during DNA synthesis. Briefly,ells were seeded overnight in 96-well tissue culture plates having clear, flatottoms (Becton Dickinson, Franklin Lakes, NJ) at a density of 105 cells perell in 100 �l of medium. Cells were pretreated with varying concentrationsf genistein (5–40 �M), PD98059 (10 �M), SB2035580 (10 �M), SP60012510 �M), l-NAME (1 mM) or DMSO (0.1% (v/v)) for 3 h, and then IL-1�
2 ng/ml) and IFN-� (100 U/ml) were added for 48 h. Thereafter, BrdU (10 �M)as added to the culture medium for 2 h, the BrdU-labeled cells were fixed,
nd the DNA was denatured in fixative solution for 30 min at room temper-ture. Cells were incubated with peroxidase-conjugated anti-BrdU antibodyor 2 h at room temperature and washed three times with washing solution.he immune complex was detected by the 3,3′,5,5′-tetramethylbenzidine sub-trate reaction and absorbance measured at 405 nm with a Spectra MAX PLUSpectrophotometer.
.4. Nitrite measurement
Biologically produced NO is rapidly oxidized to nitrite and nitrate in aqueousolutions (Moncada et al., 1991). Nitrite concentrations in the cell-free cultureupernatant, therefore, served as a reflection of NO production and were mea-ured using a colorimetric assay (Green et al., 1982). Following 48 h incubation,00 �l aliquots of the culture supernatants were incubated with 100 �l of aodified Griess reagent (1:1 mixture of 1% sulfanilamide in 30% acetic acid
nd 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 60% acetic acid)t room temperature. After 5 min, absorbance was measured at 540 nm using apectra MAX PLUS spectrophotometer. Concentrations of NO were determinedrom a linear standard curve obtained from serial dilutions of sodium nitrite inhe working medium.
.5. RNA isolation and real-time PCR
Total RNA was isolated from RIN cells or islets using Trizol reagent (Invit-ogen, Carlsbad, CA). RNA was precipitated with isopropanol and dissolved inEPC-treated distilled water. Total RNA (2 �g) was treated with RNase-freeNase (Invitrogen) and first-strand cDNA was generated with the random hex-
mer primer in the first-strand cDNA synthesis kit (Applied Biosystems, Fosterity, CA).
Specific primers were designed using primer express software (Appliediosynthesis) and their primer sequences were followings; iNOS (Acces-
ion No. NM 012611), 5′-TGTGCTAATGCGGAAGGTCAT-3′ (forward), and
′-CGACTTTCCTGTCTCAGTAGCAAA-3′ (reverse). The sequence for theontrol 18S ribosomal RNA was purchased from Applied Biosystems and useds the invariant control. The real-time PCR reaction contained in a final vol-me of 10 �l, 10 ng of reverse transcribed total RNA, 167 nM of forward andeverse primers and 2 × PCR master mix. PCR reaction was carried out in 384-
2 llular
wB
2
p(t1aspwBSHuli
2
2(dcta1iTaT(Eoea4nb
2
nd5cotgalTc
2
ntg1
ta
2
g(2goE
2
t
3
3g
nffaabITcRptItSma(dn
3p
taaIrp
0 E.-K. Kim et al. / Molecular and Ce
ell plates using the ABI Prism 7900HT Sequence Detection System (Appliediosystems). All reactions were done in triplicate.
.6. Western blot analysis
RIN cells or islets were homogenized in cold lysis buffer (20 mM HEPES,H 7.2, 1% Triton X-100, 10% glycerol, 1 mM phenylmethylsulfonyl fluoridePMSF), 10 �g/ml leupeptin, and 10 �g/ml aprotinin). The homogenates con-aining 20 �g of protein were separated by SDS-PAGE with 7% (for iNOS),0% (for STATs), or 12% (for I�B�, p50 p65, JNK, ERK, p38, and PCNA)crylamide resolving and 3% stacking gels, and transferred to nitrocelluloseheets in a Western blot apparatus (Bio-Rad, Hercules, CA). The nitrocelluloseaper was blocked with 2% bovine serum albumin and then incubated for 4 hith 1 �g/ml of primary antibodies for iNOS, p50, p65, PCNA (Santa Cruziochemicals, Santa Cruz, CA), p38, p-p38, JNK, p-JNK, STAT-1, -3, and -5, p-TAT-1, -3, and -5, ERK, or p-ERK, (Cell Signaling Technology, Danvers, MA).orseradish peroxidase-conjugated IgG (Zymed, South San Francisco, CA) wassed as secondary antibody. Protein expression levels were determined by ana-yzing the signals captured on the nitrocellulose membranes using a Chemi-docmage analyzer (Bio-Rad).
.7. Preparation of nuclear extracts
Nuclear extracts were prepared as described previously (E.K. Kim et al.,007b). RIN cells (5 × 106) were treated with IL-1� (2 ng/ml) and IFN-�100 U/ml) in the presence or absence of genistein for 30 min. Cells were imme-iately washed twice, scraped into 1.5 ml of cold PBS (pH 7.9), and pelleted byentrifugation (12,000 × g, 30 s). The cell pellet was suspended in cold hypo-onic lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 0.2 mM KCl, 0.2 mM PMSF,nd 0.5 mM dithiothreitol), vortexed for 10 s, and then incubated on ice for5 min. The packed cells were resuspended in ice-cold hypotonic lysis buffern the presence of 50 �l of 10% Nonidet P-40 and incubated on ice for 25 min.he nuclear fraction was precipitated by centrifugation at 13,000 × g for 1 mint 4 ◦C. The supernatants (cytosol extracts) were collected and stored at −80 ◦C.he pelleted nuclei were resuspended in 50–100 �l of low salt extraction buffer
20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 25% glycerol, 20 mM KCl, 0.2 mMDTA, 0.2 mM PMSF, and 0.5 mM dithiothreitol) and added to an equal volumef high salt extraction buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 25% glyc-rol, 80 mM KCl, 0.2 mM EDTA, 0.2 mM PMSF, and 0.5 mM dithiothreitol) indrop wise fashion, and then incubated under continuous shaking at 4 ◦C for
5 min. The sample was centrifuged for 20 min at 12,000 × g. Aliquots of theuclear extracts were stored at −80 ◦C. Protein concentration was determinedy the Bradford method (Bradford, 1976).
.8. Electrophoretic mobility shift assay (EMSA)
The activation of NF�B was assayed by a gel mobility shift assay usinguclear extracts from control and treated cells. As a probe for the gel retar-ation assay, an oligonucleotide containing the �-chain binding site (�B,′-CCGGTTAACAGAGGGGGCTTTCCGAG-3′) was synthesized. The twoomplementary strands were annealed and labeled with [�-32P]dCTP. Labeledligonucleotides (approximately 10,000 cpm), 10 �g of nuclear extracts, andhe binding buffer (10 mM Tris–HCl, pH 7.6, 500 mM KCl, 10 mM EDTA, 50%lycerol, 100 ng poly(dI·dC), and 1 mM dithiothreitol) were incubated for 30 mint room temperature in a final volume of 20 �l. The reaction mixtures were ana-yzed by electrophoresis on 4% polyacrylamide gels in 0.5 × Tris-borate buffer.he gels were dried and examined by autoradiography. Specific binding wasonfirmed by competition with a 50-fold excess of unlabeled �B oligonucleotide.
.9. Isolation of islets
Pancreatic islets were isolated from male Sprague–Dawley rats by collage-ase digestion as described previously (Kim et al., 1994). Following isolation,he islets were cultured overnight in RPMI-1640 supplemented with 2 mM l-lutamine, 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and00 �g/ml streptomycin in humidified air containing 5% CO2 at 37 ◦C. Prior
iNai
Endocrinology 278 (2007) 18–28
o each experiment, the islets were washed three times in RPMI-1640, counted,nd then cultured overnight.
.10. Insulin secretion assay
The islets were cultured for 24 h with cytokine in the presence or absence ofenistein. The islets were washed three times in Krebs-Ringer bicarbonate buffer25 mM HEPES, 115 mM NaCl, 24 mM NaHCO3, 5mM KCl, 1 mM MgCl2,.5 mM CaCl2, and 0.1% bovine serum albumin, pH 7.4) containing 3 mM d-lucose; insulin secretion assays were performed in the presence of either 5.5r 20.0 mM d-glucose. The insulin content of the medium was determined byLISA (Park et al., 1995).
.11. Statistical analysis
Statistical analysis of the data was performed with ANOVA and Duncan’sest. Differences with P < 0.05 were considered statistically significant.
. Results
.1. Prevention of IL-1β and IFN-γ-induced cell death byenistein
RIN cells from a rat pancreatic �-cell line were cultured toear confluence. The cells pretreated with or without genisteinor 3 h were exposed to IL-1� (2 ng/ml) and IFN-� (100 U/ml)or 48 h, at which time they were harvested and their viability wasssessed using the MTT assay. Compared to control, the IL-1�nd IFN-� combination caused a significant reduction in cell via-ility to 26 ± 1.1% (Fig. 1A). Genistein restored the viability ofL-1� and IFN-�-treated RIN cells in a dose-dependent manner.he protective effect of genistein on IL-1� and IFN-�-inducedytotoxicity was further confirmed by BrdU incorporation inIN cells. BrdU is a thymidine analog that is incorporated intoroliferating cells during DNA synthesis; thus BrdU incorpora-ion reflects the proliferation potential of the cells. IL-1� andFN-� reduced the level of BrdU incorporation, hence prolifera-ion, at 48 h incubation to 47.9 ± 4.5% of control level (Fig. 1B).imilar to the MTT assay data, genistein prevented the cytokine-ediated decrease in cell proliferation potential. DMSO, used
s diluents for genistein and mitogen-activated protein kinaseMAPK) inhibitors did not affect the viability. Genistein aloneid not affect the viability at all concentrations used, either (dataot shown).
.2. Effect of genistein on IL-1β and IFN-γ-induced NOroduction by RIN cells
It has been reported that IL-1� and IFN-�-mediated destruc-ion of � cells is caused by an increase of NO (Cetkovic-Cvrljend Eizirik, 1994; Corbett and McDaniel, 1995; Eizirik etl., 1996; Heitmeier et al., 1997; E.K. Kim et al., 2007c).ncubation of RIN cells with IL-1� and IFN-� for 48 hesulted in significant production of nitrite (a stable oxidizedroduct of NO) by the � cells. Addition of l-NAME, an
nhibitor of iNOS, completely prevented the production ofO (Fig. 1C) and cytokine-mediated cytotoxicity (Fig. 1A)s expected. Pretreatment with genistein inhibited cytokine-nduced NO production in a dose-dependent manner, with
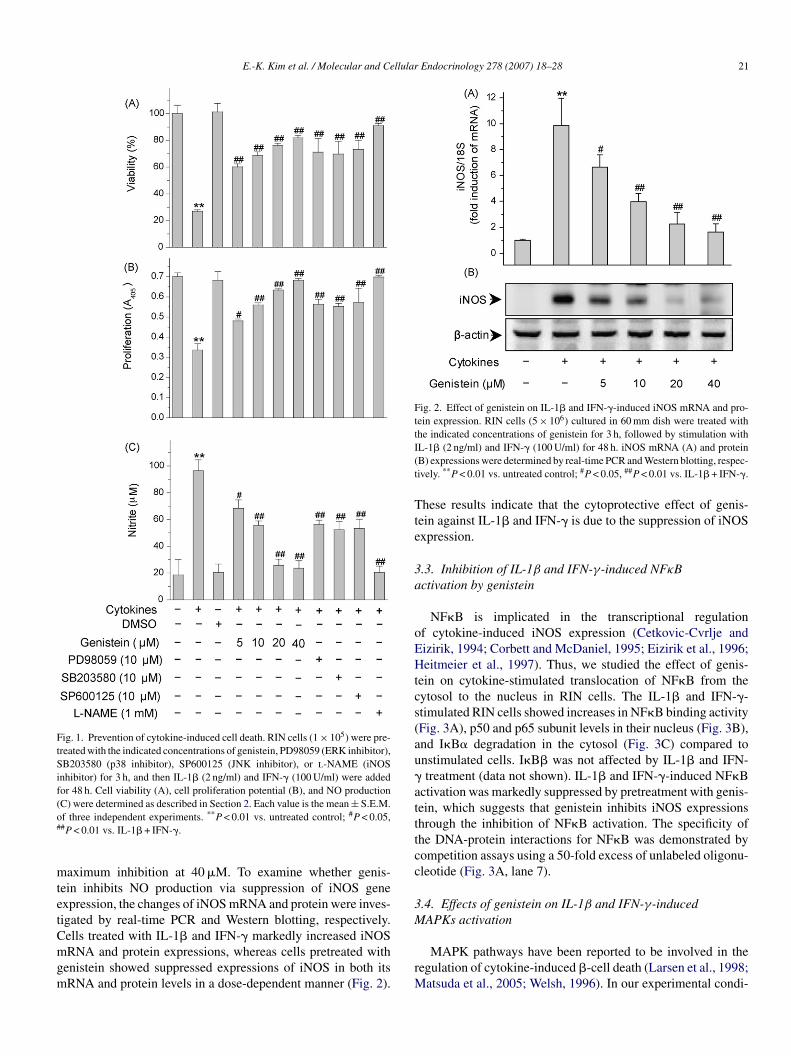
E.-K. Kim et al. / Molecular and Cellular Endocrinology 278 (2007) 18–28 21
Fig. 1. Prevention of cytokine-induced cell death. RIN cells (1 × 105) were pre-treated with the indicated concentrations of genistein, PD98059 (ERK inhibitor),SB203580 (p38 inhibitor), SP600125 (JNK inhibitor), or l-NAME (iNOSinhibitor) for 3 h, and then IL-1� (2 ng/ml) and IFN-� (100 U/ml) were addedfor 48 h. Cell viability (A), cell proliferation potential (B), and NO production(C) were determined as described in Section 2. Each value is the mean ± S.E.M.o#
mtetCmgm
Fig. 2. Effect of genistein on IL-1� and IFN-�-induced iNOS mRNA and pro-tein expression. RIN cells (5 × 106) cultured in 60 mm dish were treated withthe indicated concentrations of genistein for 3 h, followed by stimulation withI(t
Tte
3a
oEHtcs(au�atttcc
3M
f three independent experiments. **P < 0.01 vs. untreated control; #P < 0.05,#P < 0.01 vs. IL-1� + IFN-�.
aximum inhibition at 40 �M. To examine whether genis-ein inhibits NO production via suppression of iNOS genexpression, the changes of iNOS mRNA and protein were inves-igated by real-time PCR and Western blotting, respectively.ells treated with IL-1� and IFN-� markedly increased iNOS
RNA and protein expressions, whereas cells pretreated withenistein showed suppressed expressions of iNOS in both itsRNA and protein levels in a dose-dependent manner (Fig. 2).
rM
L-1� (2 ng/ml) and IFN-� (100 U/ml) for 48 h. iNOS mRNA (A) and proteinB) expressions were determined by real-time PCR and Western blotting, respec-ively. **P < 0.01 vs. untreated control; #P < 0.05, ##P < 0.01 vs. IL-1� + IFN-�.
hese results indicate that the cytoprotective effect of genis-ein against IL-1� and IFN-� is due to the suppression of iNOSxpression.
.3. Inhibition of IL-1β and IFN-γ-induced NFκBctivation by genistein
NF�B is implicated in the transcriptional regulationf cytokine-induced iNOS expression (Cetkovic-Cvrlje andizirik, 1994; Corbett and McDaniel, 1995; Eizirik et al., 1996;eitmeier et al., 1997). Thus, we studied the effect of genis-
ein on cytokine-stimulated translocation of NF�B from theytosol to the nucleus in RIN cells. The IL-1� and IFN-�-timulated RIN cells showed increases in NF�B binding activityFig. 3A), p50 and p65 subunit levels in their nucleus (Fig. 3B),nd I�B� degradation in the cytosol (Fig. 3C) compared tonstimulated cells. I�B� was not affected by IL-1� and IFN-treatment (data not shown). IL-1� and IFN-�-induced NF�B
ctivation was markedly suppressed by pretreatment with genis-ein, which suggests that genistein inhibits iNOS expressionshrough the inhibition of NF�B activation. The specificity ofhe DNA-protein interactions for NF�B was demonstrated byompetition assays using a 50-fold excess of unlabeled oligonu-leotide (Fig. 3A, lane 7).
.4. Effects of genistein on IL-1β and IFN-γ-inducedAPKs activation
MAPK pathways have been reported to be involved in theegulation of cytokine-induced �-cell death (Larsen et al., 1998;
atsuda et al., 2005; Welsh, 1996). In our experimental condi-
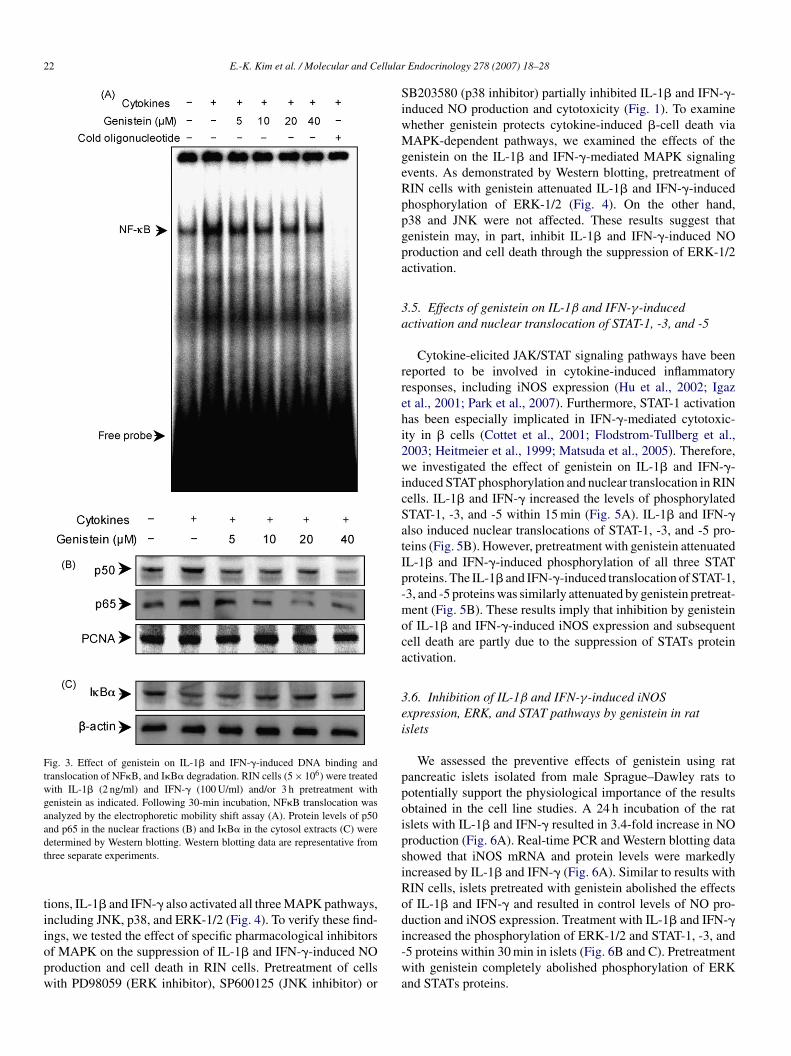
22 E.-K. Kim et al. / Molecular and Cellular
Fig. 3. Effect of genistein on IL-1� and IFN-�-induced DNA binding andtranslocation of NF�B, and I�B� degradation. RIN cells (5 × 106) were treatedwith IL-1� (2 ng/ml) and IFN-� (100 U/ml) and/or 3 h pretreatment withgenistein as indicated. Following 30-min incubation, NF�B translocation wasanalyzed by the electrophoretic mobility shift assay (A). Protein levels of p50and p65 in the nuclear fractions (B) and I�B� in the cytosol extracts (C) weredt
tiiopw
SiwMgeRppgpa
3a
rrehi2wicSatIp-moca
3ei
ppoipsiRoduction and iNOS expression. Treatment with IL-1� and IFN-�
etermined by Western blotting. Western blotting data are representative fromhree separate experiments.
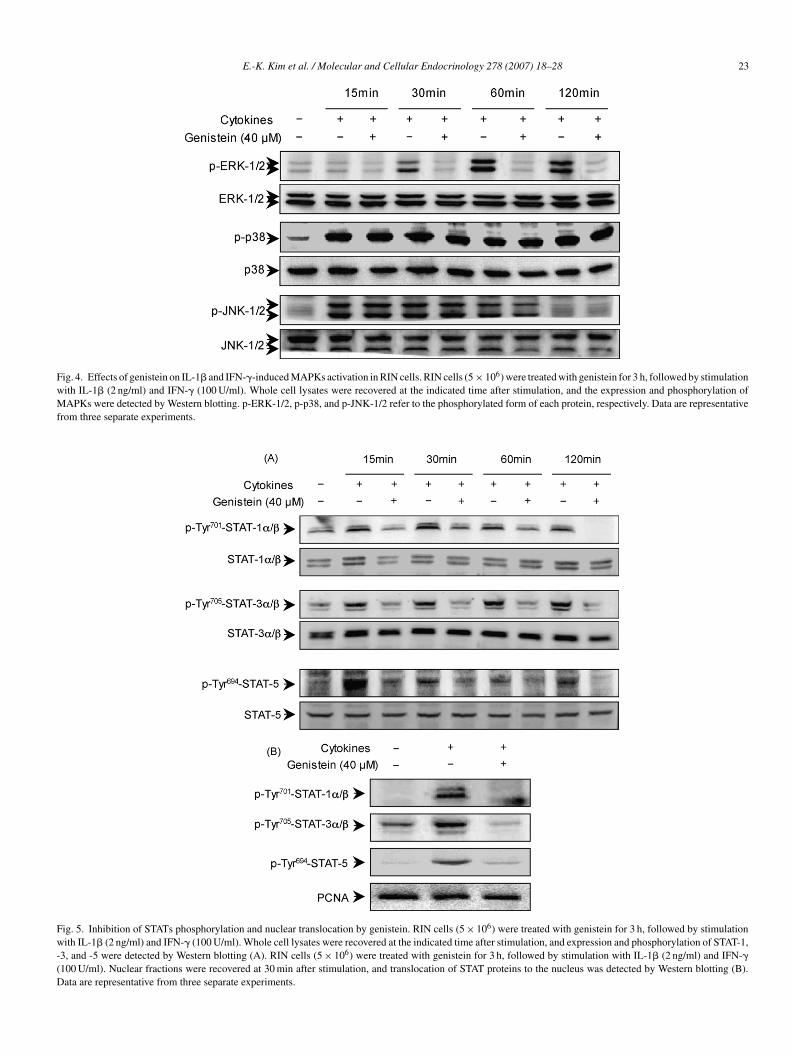
ions, IL-1� and IFN-� also activated all three MAPK pathways,ncluding JNK, p38, and ERK-1/2 (Fig. 4). To verify these find-
ngs, we tested the effect of specific pharmacological inhibitorsf MAPK on the suppression of IL-1� and IFN-�-induced NOroduction and cell death in RIN cells. Pretreatment of cellsith PD98059 (ERK inhibitor), SP600125 (JNK inhibitor) ori-wa
Endocrinology 278 (2007) 18–28
B203580 (p38 inhibitor) partially inhibited IL-1� and IFN-�-nduced NO production and cytotoxicity (Fig. 1). To examinehether genistein protects cytokine-induced �-cell death viaAPK-dependent pathways, we examined the effects of the
enistein on the IL-1� and IFN-�-mediated MAPK signalingvents. As demonstrated by Western blotting, pretreatment ofIN cells with genistein attenuated IL-1� and IFN-�-inducedhosphorylation of ERK-1/2 (Fig. 4). On the other hand,38 and JNK were not affected. These results suggest thatenistein may, in part, inhibit IL-1� and IFN-�-induced NOroduction and cell death through the suppression of ERK-1/2ctivation.
.5. Effects of genistein on IL-1β and IFN-γ-inducedctivation and nuclear translocation of STAT-1, -3, and -5
Cytokine-elicited JAK/STAT signaling pathways have beeneported to be involved in cytokine-induced inflammatoryesponses, including iNOS expression (Hu et al., 2002; Igazt al., 2001; Park et al., 2007). Furthermore, STAT-1 activationas been especially implicated in IFN-�-mediated cytotoxic-ty in � cells (Cottet et al., 2001; Flodstrom-Tullberg et al.,003; Heitmeier et al., 1999; Matsuda et al., 2005). Therefore,e investigated the effect of genistein on IL-1� and IFN-�-
nduced STAT phosphorylation and nuclear translocation in RINells. IL-1� and IFN-� increased the levels of phosphorylatedTAT-1, -3, and -5 within 15 min (Fig. 5A). IL-1� and IFN-�lso induced nuclear translocations of STAT-1, -3, and -5 pro-eins (Fig. 5B). However, pretreatment with genistein attenuatedL-1� and IFN-�-induced phosphorylation of all three STATroteins. The IL-1� and IFN-�-induced translocation of STAT-1,3, and -5 proteins was similarly attenuated by genistein pretreat-ent (Fig. 5B). These results imply that inhibition by genistein
f IL-1� and IFN-�-induced iNOS expression and subsequentell death are partly due to the suppression of STATs proteinctivation.
.6. Inhibition of IL-1β and IFN-γ-induced iNOSxpression, ERK, and STAT pathways by genistein in ratslets
We assessed the preventive effects of genistein using ratancreatic islets isolated from male Sprague–Dawley rats tootentially support the physiological importance of the resultsbtained in the cell line studies. A 24 h incubation of the ratslets with IL-1� and IFN-� resulted in 3.4-fold increase in NOroduction (Fig. 6A). Real-time PCR and Western blotting datahowed that iNOS mRNA and protein levels were markedlyncreased by IL-1� and IFN-� (Fig. 6A). Similar to results withIN cells, islets pretreated with genistein abolished the effectsf IL-1� and IFN-� and resulted in control levels of NO pro-
ncreased the phosphorylation of ERK-1/2 and STAT-1, -3, and5 proteins within 30 min in islets (Fig. 6B and C). Pretreatmentith genistein completely abolished phosphorylation of ERK
nd STATs proteins.
E.-K. Kim et al. / Molecular and Cellular Endocrinology 278 (2007) 18–28 23
Fig. 4. Effects of genistein on IL-1� and IFN-�-induced MAPKs activation in RIN cells. RIN cells (5 × 106) were treated with genistein for 3 h, followed by stimulationwith IL-1� (2 ng/ml) and IFN-� (100 U/ml). Whole cell lysates were recovered at the indicated time after stimulation, and the expression and phosphorylation ofMAPKs were detected by Western blotting. p-ERK-1/2, p-p38, and p-JNK-1/2 refer to the phosphorylated form of each protein, respectively. Data are representativefrom three separate experiments.
Fig. 5. Inhibition of STATs phosphorylation and nuclear translocation by genistein. RIN cells (5 × 106) were treated with genistein for 3 h, followed by stimulationwith IL-1� (2 ng/ml) and IFN-� (100 U/ml). Whole cell lysates were recovered at the indicated time after stimulation, and expression and phosphorylation of STAT-1,-3, and -5 were detected by Western blotting (A). RIN cells (5 × 106) were treated with genistein for 3 h, followed by stimulation with IL-1� (2 ng/ml) and IFN-�(100 U/ml). Nuclear fractions were recovered at 30 min after stimulation, and translocation of STAT proteins to the nucleus was detected by Western blotting (B).Data are representative from three separate experiments.
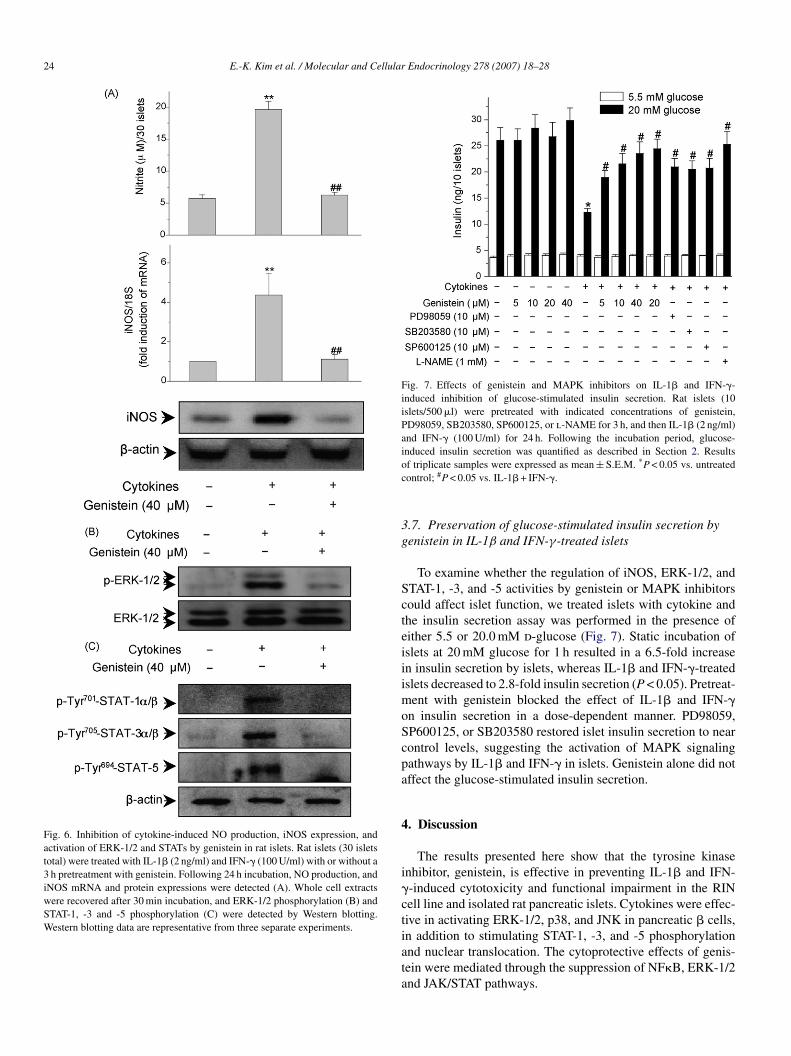
24 E.-K. Kim et al. / Molecular and Cellular Endocrinology 278 (2007) 18–28
Fig. 6. Inhibition of cytokine-induced NO production, iNOS expression, andactivation of ERK-1/2 and STATs by genistein in rat islets. Rat islets (30 isletstotal) were treated with IL-1� (2 ng/ml) and IFN-� (100 U/ml) with or without a3 h pretreatment with genistein. Following 24 h incubation, NO production, andiNOS mRNA and protein expressions were detected (A). Whole cell extractswere recovered after 30 min incubation, and ERK-1/2 phosphorylation (B) andSTAT-1, -3 and -5 phosphorylation (C) were detected by Western blotting.Western blotting data are representative from three separate experiments.
Fig. 7. Effects of genistein and MAPK inhibitors on IL-1� and IFN-�-induced inhibition of glucose-stimulated insulin secretion. Rat islets (10islets/500 �l) were pretreated with indicated concentrations of genistein,PD98059, SB203580, SP600125, or l-NAME for 3 h, and then IL-1� (2 ng/ml)and IFN-� (100 U/ml) for 24 h. Following the incubation period, glucose-induced insulin secretion was quantified as described in Section 2. Resultsoc
3g
ScteiiimoScpa
4
i�ctiata
f triplicate samples were expressed as mean ± S.E.M. *P < 0.05 vs. untreatedontrol; #P < 0.05 vs. IL-1� + IFN-�.
.7. Preservation of glucose-stimulated insulin secretion byenistein in IL-1β and IFN-γ-treated islets
To examine whether the regulation of iNOS, ERK-1/2, andTAT-1, -3, and -5 activities by genistein or MAPK inhibitorsould affect islet function, we treated islets with cytokine andhe insulin secretion assay was performed in the presence ofither 5.5 or 20.0 mM d-glucose (Fig. 7). Static incubation ofslets at 20 mM glucose for 1 h resulted in a 6.5-fold increasen insulin secretion by islets, whereas IL-1� and IFN-�-treatedslets decreased to 2.8-fold insulin secretion (P < 0.05). Pretreat-
ent with genistein blocked the effect of IL-1� and IFN-�n insulin secretion in a dose-dependent manner. PD98059,P600125, or SB203580 restored islet insulin secretion to nearontrol levels, suggesting the activation of MAPK signalingathways by IL-1� and IFN-� in islets. Genistein alone did notffect the glucose-stimulated insulin secretion.
. Discussion
The results presented here show that the tyrosine kinasenhibitor, genistein, is effective in preventing IL-1� and IFN--induced cytotoxicity and functional impairment in the RINell line and isolated rat pancreatic islets. Cytokines were effec-ive in activating ERK-1/2, p38, and JNK in pancreatic � cells,n addition to stimulating STAT-1, -3, and -5 phosphorylation
nd nuclear translocation. The cytoprotective effects of genis-ein were mediated through the suppression of NF�B, ERK-1/2nd JAK/STAT pathways.
llular
ocd(oiticap
onashitsitgeIdIvibIdn
iet(aocmNoimeavTagA(ef
Iw
ppedid(�t�r1ethfososieTnbd
1aT1cPtllMcaAtJpWtJSti
E.-K. Kim et al. / Molecular and Ce
Previous reports provided evidence that chemical inhibitorsf NO production protected insulin-secreting cells againstytokine-mediated toxicity but with variable efficacies depen-ent on species and the combination of the cytokinesCetkovic-Cvrlje and Eizirik, 1994; Corbett et al., 1996). Forur experimental conditions, genistein as well as l-NAME, anNOS inhibitor prevented IL-1� and IFN-�-induced NO produc-ion and �-cell destruction. This finding indicates that NO is anndispensable component of cytokine-induced toxicity of RINells, and that the protective effect of genistein against IL-1�nd IFN-�-mediated cell death is due to the inhibition of NOroduction.
Exposure of the � cells to IL-1� has been shown to induce notnly iNOS but also heat shock protein 70 (hsp70), heme oxyge-ase, manganese-superoxide dismutase (Mn-SOD) (Strandell etl., 1995), and COX-2 (Sorli et al., 1998; Tran et al., 1999). Thesetress response proteins are known to be induced by cytokines,eat shock and oxidative stresses, and it is conceivable thatnduction of these genes also requires NF�B, either directly byrans-activation or indirectly by interacting with some other tran-cription factor (Renard and Raes, 1999). As a result, NF�B orts downstream iNOS expression has been targeted for studies ofhe prevention of islet destruction against a variety of diabeto-enic agents (Eldor et al., 2006; E.K. Kim et al., 2007c; Mableyt al., 2002). Genistein inhibited NO production in IL-1� andFN-�-stimulated RIN cells through a suppression of NF�B-ependent iNOS expression, thereby protecting RIN cells fromL-1� and IFN-� cytotoxicity. In addition to increased RIN celliability, we also observed the preservation of insulin secretionn rat islets pretreated with genistein. The molecular mechanismy which genistein inhibits NF�B activation against IL-1� andFN-� appears to involve the inhibition of both I�B� degra-ation and the parallel translocation of p65 and p50 into theucleus.
NF�B governs both cell death and survival responses accord-ng to the modes of insults in � cells. NF�B regulates thexpressions of multiple proinflammatory genes that contributeo �-cell destruction such as Fas, iNOS and cyclooxygenase-2Cetkovic-Cvrlje and Eizirik, 1994; Eizirik et al., 1996; Darvillend Eizirik, 1998; Sorli et al., 1998). In addition, the promotersf other proinflammatory genes induced in � cells, includinghemokines and adhesion molecules, also possess binding ele-ents for NF�B (May and Ghosh, 1998). The importance ofF�B in �-cell damage is underscored by the fact that inhibitionf NF�B activation or translocation prevents IL-1� and IFN-�-nduced �-cell dysfunction and death both in vitro and in vivoodels (Eldor et al., 2006; Giannoukakis et al., 2000; Heimberg
t al., 2001; E.K. Kim et al., 2007c). In contrast, the defensivend protective role of NF�B is also reported. Blockage of NF�Bia the use of an I�B� super-repressor also sensitized � cells toNF-�-mediated apoptosis (Chang et al., 2003). NF�B regulatespoptosis by controlling the expression of multiple antiapoptoticenes, including the inhibitors of apoptosis protein (IAP) and
20/tumor necrosis factor (TNF)-induced protein 3 (TNFAIP3)Karin and Lin, 2002; Liuwantara et al., 2006). Recently, Kimt al. (Kim et al., 2007) reported that NF�B had an antiapoptoticunction in � cells incubated with a combination of TNF and
ermi
Endocrinology 278 (2007) 18–28 25
FN-�, and that it showed a proapoptotic effect in � cells treatedith IL-1 and IFN-� as our results showed.Genistein also suppressed IL-1� and IFN-�-induced phos-
horylation and nuclear translocation of STAT-1, -3, and -5roteins. By using specific pharmacologic inhibitors, Blanchettet al. (2003) suggested that JAK2/STAT-1 and ERK-1/2-ependent pathways were the main factors in IFN-�-inducibleNOS induction. All mammalian iNOS promoters studied toate contain the binding site for the IFN-�-regulated STAT-1GAS) (Kleinert et al., 2004). Deleterious effects of IFN-� on
cells, even in the absence of increased NO production, arehought to be mediated by STAT-1 activation. Although IFN-
alone does not stimulate iNOS expression in islets from theodent and human, IFN-� does reduce the concentration of IL-� required to induce iNOS expression in rat islets (Heitmeiert al., 1999), and a combination of IL-1� and IFN-� is requiredo induce iNOS expression and �-cell dysfunction in mouse anduman islets (Corbett et al., 1993; Thomas et al., 2002). There-ore, the JAK/STAT-1 pathway plays a key role in this primingr synergistic action of IFN-� for IL-1�-induced iNOS expres-ion. The JAK/STAT-1 pathway also activates the expressionf IFN regulatory factor-1, which, through binding to IFN-�-timulated response elements and interaction with NF�B sitesn the iNOS promoter, would augment IL-1�-induced iNOSxpression (Darville and Eizirik, 1998; Paludan et al., 2001).herefore, the suppressive effect of genistein on activation anduclear translocation of STAT-1 possibly contributes to the inhi-ition of IL-1� and IFN-�-induced iNOS expression, �-celleath, and islet dysfunction.
In addition to NF�B and JAK/STAT, genistein inhibited IL-� and IFN-�-induced ERK-1/2 activation. Actually, ERK, p38,nd JNK were activated in IL-1� and IFN-�-treated RIN cells.o investigate the possible causal relationship between the IL-� and IFN-�-induced MAPKs activation and susceptibility toytokine toxicity, specific MAPK inhibitors SB203580 (p38),D98059 (ERK-1/2), and SP600125 (JNK) were used to pro-
ect against IL-1� and IFN-�-induced cytotoxicity. Compared to-NAME, MAPK inhibitors partially restored cell viability, pro-iferation potential, and NO production. It is clear that when one
APK signaling pathway is inhibited, there is still an appre-iable cytokine induction of iNOS, presumably via continuedctivation of the other signaling pathway such as JAK/STAT.mong the tested MAPK signaling pathways, ERK-1/2 was the
argets for genistein. In addition to tyrosine phosphorylation byAK, maximal STAT-1 activation has been found to require phos-horylation on a single serine residue, Ser727 (David et al., 1995;en and Darnell, 1997; Zhang et al., 1995). ERK-1/2 is reported
o phosphorylate STAT-1 at a serine residue in IFN-�-stimulated774 macrophages cells, thus contributing to full activation ofTAT-1 (Blanchette et al., 2003). Therefore, we cannot exclude
he possibility that genistein might inhibit STAT-1 activation bynhibiting ERK-1/2 dependent signaling pathways.
IL-1� activates MAPK and NF�B pathways (Giannoukakis
t al., 2000; Mandrup-Poulsen, 2001) and both pathways areequired for expression of iNOS expression. Even though theolecular mechanisms are unclear, recent studies demonstrat-ng that ERK can increase NF�B transactivational activity

2 llular
s(a1asrvad
lITIgtadcavsceIotfakiawSe3sealSotSenii
tvpiatm
bmmmo
A
Tt(b&
R
A
A
A
A
B
B
B
B
B
B
C
C
C
C
6 E.-K. Kim et al. / Molecular and Ce
uggest that there is some cross-talk between two pathwaysLarsen et al., 2005; Vanden Berghe et al., 1998). ERK islso known to phosphorylate JunD and FosB leading to AP-
induction (Biggs et al., 1999; Rosenberger et al., 1999),nd the iNOS promoter is known to contain two recognitionites for AP-1 (Xie and Nathan, 1993). Taken together, ouresults demonstrate that IL-1� and IFN-� induce ERK-1/2 acti-ation and require an upstream tyrosine kinase, but the naturend location of this genistein-inhibitable kinase remains to beetermined.
We also have observed that STAT-3 and STAT-5 phosphory-ation and nuclear translocation were increased by IL-1� andFN-�, and that genistein prevented STAT-3 and -5 activations.he molecular basis of the STAT-3 and STAT-5 activations by
L-1� and IFN-� treatment and inhibition of phosphorylation byenistein is not clear. Our results are in agreement, however, withhose from a recent study showing that IFN-� activates STAT-3nd -5 in RIN cells (Matsuda et al., 2005). These investigatorsemonstrated that silymarin protected pancreatic � cells fromytokine toxicity by suppressing JAK-dependent STAT-1, -3,nd -5 activations. Although both genistein and silymarin pre-ented cytokine-induced cytotoxicity in pancreatic � cells byuppressing JAK-dependent STAT activation, they had differentytoprotective mechanisms. Unlike genistein, silymarin had noffect on IL-1� and IFN-�-induced NF�B and ERK activation.t rather induced mild activation of ERK-1/2 and attenuationf JNK. One mechanism by which silymarin exerts its protec-ive effect against cytokine toxicity would be due to the oxygenree radical scavenging ability (Comoglio et al., 1990; de Grootnd Rauen, 1998). Because genistein is a nonselective tyrosineinase inhibitor as well as an antioxidant, it might prevent thenhibitory and destructive effect of cytokines on �-cell functionnd viability by inhibiting tyrosine kinase activity, in accordanceith previous reports (Kwon et al., 1995). Concerning to theTAT-3 phosphorylation and iNOS expression, accumulatingvidence also suggests that tyrosine phosphorylation of STAT-is absolutely required for the binding of STAT-3 to a GAS
equence (Kaptein et al., 1996; Wen and Darnell, 1997; Zhangt al., 1999). Recently, Yoshida et al. (2004) reported that IL-1ctivated crosstalk between p65 and non-tyrosine phosphory-ated STAT-3, and that this interaction resulted in the inability ofTAT-3 to bind and to activate GAS site-dependent genes. Basedn these reports, inhibition of tyrosine kinase activity by genis-ein might suppress STAT-3-dependent iNOS expression. UnlikeTAT-3, the evidence for the involvement of STAT-5 in iNOSxpression has not been reported. Therefore, further studies areecessary to elucidate whether and how the STAT-5 protein isnvolved in cytokine-induced iNOS expression and dysfunctionn � cells.
Collectively, the findings reported in the present study suggesthat the effect of genistein on cytokine toxicity may be mediatedia inhibition of signaling by NF�B, JAK/STAT, and ERK-1/2athways. Accumulating evidence indicates that the regulatory
ntracellular signaling network engaged by IL-1� and potenti-ting cytokines such as TNF-� and IFN-� represents a potentialarget for the development of novel therapies. Even though theechanism of cytokine-induced �-cell damage was different
C
Endocrinology 278 (2007) 18–28
etween species, an improved knowledge of the transductionechanisms by which IL-1� and IFN-� trigger cell death inurine � cells could lead to the development of more selectiveolecules, with immunomodulatory capacities favoring control
ver cytokine toxicities in pancreatic � cells.
cknowledgements
This work was supported by the Ministry of Science &echnology (MoST)/Korea Science & Engineering Founda-
ion (KOSEF) through the Vestibulocochlear Research CenterVCRC) at Wonkwang University (R13-2002-055-0000-0), andy a grant from the Basic Research Program of the Korea Science
Engineering Foundation (R01-2005-000-11028-0).
eferences
aronson, D.S., Horvath, C.M., 2002. A road map for those who don’t knowJAK-STAT. Science 296, 1653–1655.
ikin, R., Maysinger, D., Rosenberg, L., 2004. Cross-talk between phos-phatidylinositol 3-kinase/AKT and c-jun NH2-terminal kinase mediatessurvival of isolated human islets. Endocrinology 145, 4522–4531.
kiyama, T., Ishida, J., Nakagawa, S., Ogawara, H., Watanabe, S., Itoh, N.,Shibuya, M., Fukami, Y., 1987. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 262, 5592–5595.
mmendrup, A., Maillard, A., Nielsen, K., Aabenhus Andersen, N., Serup, P.,Dragsbaek Madsen, O., Mandrup-Poulsen, T., Bonny, C., 2000. The c-Junamino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic �-cells. Diabetes 49,1468–1476.
aeuerle, P.A., Henkel, T., 1994. Function and activation of NF-�B in theimmune system. Annu. Rev. Immunol. 12, 141–179.
aldwin Jr., A.S., 1996. The NF-�B and I�B proteins: new discoveries andinsights. Annu. Rev. Immunol. 14, 649–683.
iggs, T.E., Cooke, S.J., Barton, C.H., Harris, M.P., Saksela, K., Mann,D.A., 1999. Induction of activator protein 1 (AP-1) in macrophages byhuman immunodeficiency virus type-1 NEF is a cell-type-specific responsethat requires both hck and MAPK signaling events. J. Mol. Biol. 290,21–35.
lanchette, J., Jaramillo, M., Olivier, M., 2003. Signalling events involvedin interferon-�-inducible macrophage nitric oxide generation. Immunology108, 513–522.
onny, C., Oberson, A., Negri, S., Sauser, C., Schorderet, D.F., 2001. Cell-permeable peptide inhibitors of JNK: novel blockers of �-cell death. Diabetes50, 77–82.
radford, M.M., 1976. A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72, 248–254.
etkovic-Cvrlje, M., Eizirik, D.L., 1994. TNF-� and IFN-� potentiate the dele-terious effects of IL-1� on mouse pancreatic islets mainly via generation ofnitric oxide. Cytokine 6, 399–406.
hang, I., Kim, S., Kim, J.Y., Cho, N., Kim, Y.H., Kim, H.S., Lee, M.K., Kim,K.W., Lee, M.S., 2003. Nuclear factor �B protects pancreatic �-cells fromtumor necrosis factor-�-mediated apoptosis. Diabetes 52, 1169–1175.
heng, E., Story, C.D., Yoder, L., Hale, W.H., Burroughs, W., 1953. Estrogenicactivity of isoflavone derivatives extracted and prepared from soybean oilmeal. Science 118, 164–165.
omoglio, A., Leonarduzzi, G., Carini, R., Busolin, D., Basaga, H., Albano,E., Tomasi, A., Poli, G., Morazzoni, P., Magistretti, M.J., 1990. Studies onthe antioxidant and free radical scavenging properties of IdB 1016: a new
flavanolignan complex. Free Rad. Res. Commun. 11, 109–115.orbett, J.A., Kwon, G., Marino, M.H., Rodi, C.P., Sullivan, P.M., Turk, J.,McDaniel, M.L., 1996. Tyrosine kinase inhibitors prevent cytokine-inducedexpression of iNOS and COX-2 by human islets. Am. J. Physiol. 270,C1581–C1587.

llular
C
C
C
C
C
C
D
D
d
E
E
F
G
G
H
H
H
H
I
J
K
K
K
K
K
K
K
K
K
K
L
L
L
M
M
M
M
M
M
N
E.-K. Kim et al. / Molecular and Ce
orbett, J.A., Kwon, G., Misko, T.P., Rodi, C.P., McDaniel, M.L., 1994. Tyrosinekinase involvement in IL-1�-induced expression of iNOS by �-cells purifiedfrom islets of Langerhans. Am. J. Physiol. 267, C48–C54.
orbett, J.A., McDaniel, M.L., 1994. Reversibility of interleukin-1�-inducedislet destruction and dysfunction by the inhibition of nitric oxide synthase.Biochem. J. 299 (Pt 3), 719–724.
orbett, J.A., McDaniel, M.L., 1995. Intraislet release of interleukin 1 inhibits �
cell function by inducing � cell expression of inducible nitric oxide synthase.J. Exp. Med. 181, 559–568.
orbett, J.A., Sweetland, M.A., Wang, J.L., Lancaster Jr., J.R., McDaniel, M.L.,1993. Nitric oxide mediates cytokine-induced inhibition of insulin secre-tion by human islets of Langerhans. Proc. Natl. Acad. Sci. U.S.A. 90,1731–1735.
ottet, S., Dupraz, P., Hamburger, F., Dolci, W., Jaquet, M., Thorens, B., 2001.SOCS-1 protein prevents Janus Kinase/STAT-dependent inhibition of � cellinsulin gene transcription and secretion in response to interferon-�. J. Biol.Chem. 276, 25862–25870.
unningham, J.M., Green, I.C., 1994. Cytokines, nitric oxide and insulin secret-ing cells. Growth Regul. 4, 173–180.
arville, M.I., Eizirik, D.L., 1998. Regulation by cytokines of the induciblenitric oxide synthase promoter in insulin-producing cells. Diabetologia 41,1101–1108.
avid, M., Petricoin III, E., Benjamin, C., Pine, R., Weber, M.J., Larner, A.C.,1995. Requirement for MAP kinase (ERK2) activity in interferon �- andinterferon �-stimulated gene expression through STAT proteins. Science269, 1721–1723.
e Groot, H., Rauen, U., 1998. Tissue injury by reactive oxygen species and theprotective effects of flavonoids. Fundam. Clin. Pharmacol. 12, 249–255.
izirik, D.L., Flodstrom, M., Karlsen, A.E., Welsh, N., 1996. The harmony ofthe spheres: inducible nitric oxide synthase and related genes in pancreatic� cells. Diabetologia 39, 875–890.
ldor, R., Yeffet, A., Baum, K., Doviner, V., Amar, D., Ben-Neriah, Y., Christo-fori, G., Peled, A., Carel, J.C., Boitard, C., Klein, T., Serup, P., Eizirik, D.L.,Melloul, D., 2006. Conditional and specific NF-�B blockade protects pan-creatic � cells from diabetogenic agents. Proc. Natl. Acad. Sci. U.S.A. 103,5072–5077.
lodstrom-Tullberg, M., Yadav, D., Hagerkvist, R., Tsai, D., Secrest, P., Stotland,A., Sarvetnick, N., 2003. Target cell expression of suppressor of cytokinesignaling-1 prevents diabetes in the NOD mouse. Diabetes 52, 2696–2700.
iannoukakis, N., Rudert, W.A., Trucco, M., Robbins, P.D., 2000. Protection ofhuman islets from the effects of interleukin-1� by adenoviral gene transferof an I�B repressor. J. Biol. Chem. 275, 36509–36513.
reen, L.C., Wagner, D.A., Glogowski, J., Skipper, P.L., Wishnok, J.S., Tannen-baum, S.R., 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biologicalfluids. Anal. Biochem. 126, 131–138.
eimberg, H., Heremans, Y., Jobin, C., Leemans, R., Cardozo, A.K., Darville,M., Eizirik, D.L., 2001. Inhibition of cytokine-induced NF-�B activation byadenovirus-mediated expression of a NF-�B super-repressor prevents �-cellapoptosis. Diabetes 50, 2219–2224.
eitmeier, M.R., Scarim, A.L., Corbett, J.A., 1997. Interferon-� increases thesensitivity of islets of Langerhans for inducible nitric-oxide synthase expres-sion induced by interleukin 1. J. Biol. Chem. 272, 13697–13704.
eitmeier, M.R., Scarim, A.L., Corbett, J.A., 1999. Prolonged STAT1 activationis associated with interferon-� priming for interleukin-1-induced induciblenitric-oxide synthase expression by islets of Langerhans. J. Biol. Chem. 274,29266–29273.
u, X., Herrero, C., Li, W.P., Antoniv, T.T., Falck-Pedersen, E., Koch, A.E.,Woods, J.M., Haines, G.K., Ivashkiv, L.B., 2002. Sensitization of IFN-� Jak-STAT signaling during macrophage activation. Nat. Immunol. 3, 859–866.
gaz, P., Toth, S., Falus, A., 2001. Biological and clinical significance of the JAK-STAT pathway; lessons from knockout mice. Inflamm. Res. 50, 435–441.
orns, A., Gunther, A., Hedrich, H.J., Wedekind, D., Tiedge, M., Lenzen, S.,2005. Immune cell infiltration, cytokine expression, and �-cell apoptosis
during the development of type 1 diabetes in the spontaneously diabeticLEW.1AR1/Ztm-iddm rat. Diabetes 54, 2041–2052.anazawa, Y., Komeda, K., Sato, S., Mori, S., Akanuma, K., Takaku, F., 1984.Non-obese-diabetic mice: immune mechanisms of pancreatic �-cell destruc-tion. Diabetologia 27 (Suppl.), 113–115.
P
Endocrinology 278 (2007) 18–28 27
aptein, A., Paillard, V., Saunders, M., 1996. Dominant negative stat3 mutantinhibits interleukin-6-induced Jak-STAT signal transduction. J. Biol. Chem.271, 5961–5964.
arin, M., Lin, A., 2002. NF-�B at the crossroads of life and death. Nat.Immunol. 3, 221–227.
im, E.K., Kwon, K.B., Han, M.J., Song, M.Y., Lee, J.H., Lv, N., Choi, K.B.,Ryu, D.G., Kim, K.S., Park, J.W., Park, B.H., 2007a. Inhibitory effect ofArtemisia capillaris extract on cytokine-induced nitric oxide formation andcytotoxicity of RINm5F cells. Int. J. Mol. Med. 19, 535–540.
im, E.K., Kwon, K.B., Han, M.J., Song, M.Y., Lee, J.H., Lv, N., Yeom, S.R.,Kown, Y.D., Ryu, D.G., Kim, K.S., Park, R., Park, J.W., Park, B.H., 2007b.Coptidis rhizoma extract protects RINm5F cells from cytokine-inducedcytotoxicity through suppression of NF-�B activation. Exp. Mol. Med. 39,149–159.
im, E.K., Kwon, K.B., Koo, B.S., Han, M.J., Song, M.Y., Song, E.K., Han,M.K., Park, J.W., Ryu, D.G., Park, B.H., 2007c. Activation of peroxisomeproliferator-activated receptor-� protects pancreatic �-cells from cytokine-induced cytotoxicity via NF�B pathway. Int. J. Biochem. Cell. Biol. 39,1260–1275.
im, H.R., Rho, H.W., Park, B.H., Park, J.W., Kim, J.S., Kim, U.H., Chung,M.Y., 1994. Role of Ca2+ in alloxan-induced pancreatic �-cell damage.Biochim. Biophys. Acta 1227, 87–91.
im, S., Millet, I., Kim, H.S., Kim, J.Y., Han, M.S., Lee, M.K., Kim, K.W.,Sherwin, R.S., Karin, M., Lee, M.S., 2007. NF-�B prevents � cell deathand autoimmune diabetes in NOD mice. Proc. Natl. Acad. Sci. U.S.A. 104,1913–1918.
leinert, H., Pautz, A., Linker, K., Schwarz, P.M., 2004. Regulation of theexpression of inducible nitric oxide synthase. Eur. J. Pharmacol. 500,255–266.
won, G., Corbett, J.A., Rodi, C.P., Sullivan, P., McDaniel, M.L., 1995.Interleukin-1�-induced nitric oxide synthase expression by rat pancreatic�-cells: evidence for the involvement of nuclear factor �B in the signalingmechanism. Endocrinology 136, 4790–4795.
arsen, C.M., Wadt, K.A., Juhl, L.F., Andersen, H.U., Karlsen, A.E., Su,M.S., Seedorf, K., Shapiro, L., Dinarello, C.A., Mandrup-Poulsen, T., 1998.Interleukin-1�-induced rat pancreatic islet nitric oxide synthesis requiresboth the p38 and extracellular signal-regulated kinase 1/2 mitogen-activatedprotein kinases. J. Biol. Chem. 273, 15294–15300.
arsen, L., Storling, J., Darville, M., Eizirik, D.L., Bonny, C., Billestrup, N.,Mandrup-Poulsen, T., 2005. Extracellular signal-regulated kinase is essentialfor interleukin-1-induced and nuclear factor �B-mediated gene expressionin insulin-producing INS-1E cells. Diabetologia 48, 2582–2590.
iuwantara, D., Elliot, M., Smith, M.W., Yam, A.O., Walters, S.N., Marino, E.,McShea, A., Grey, S.T., 2006. Nuclear factor-�B regulates �-cell death: acritical role for A20 in �-cell protection. Diabetes 55, 2491–2501.
abley, J.G., Hasko, G., Liaudet, L., Soriano, F., Southan, G.J., Salzman,A.L., Szabo, C., 2002. NF�B1 (p50)-deficient mice are not susceptibleto multiple low-dose streptozotocin-induced diabetes. J. Endocrinol. 173,457–464.
andrup-Poulsen, T., 1996. The role of interleukin-1 in the pathogenesis ofIDDM. Diabetologia 39, 1005–1029.
andrup-Poulsen, T., 2001. �-Cell apoptosis: stimuli and signaling. Diabetes50 (Suppl. 1), S58–S63.
atsuda, T., Ferreri, K., Todorov, I., Kuroda, Y., Smith, C.V., Kandeel, F.,Mullen, Y., 2005. Silymarin protects pancreatic �-cells against cytokine-mediated toxicity: implication of c-Jun NH2-terminal kinase and januskinase/signal transducer and activator of transcription pathways. Endocrinol-ogy 146, 175–185.
ay, M.J., Ghosh, S., 1998. Signal transduction through NF-�B. Immunol.Today 19, 80–88.
oncada, S., Palmer, R.M., Higgs, E.A., 1991. Nitric oxide: physiology, patho-physiology, and pharmacology. Pharmacol. Rev. 43, 109–142.
ossal, G.J., Herold, K.C., Goodnow, C.C., 1992. Autoimmune tolerance and
type 1 (insulin-dependent) diabetes mellitus. Diabetologia 35 (Suppl. 2),S49–S59.aludan, S.R., Malmgaard, L., Ellermann-Eriksen, S., Bosca, L., Mogensen,S.C., 2001. Interferon (IFN)-� and Herpes simplex virus/tumor necrosisfactor-� synergistically induce nitric oxide synthase 2 in macrophages

2 llular
P
P
R
R
R
S
S
S
S
T
T
V
W
W
W
X
Y
Z
8 E.-K. Kim et al. / Molecular and Ce
through cooperative action of nuclear factor-�B and IFN regulatory factor-1.Eur. Cytokine Netw. 12, 297–308.
ark, B.H., Rho, H.W., Park, J.W., Cho, C.G., Kim, J.S., Chung, H.T., Kim, H.R.,1995. Protective mechanism of glucose against alloxan-induced pancreatic�-cell damage. Biochem. Biophys. Res. Commun. 210, 1–6.
ark, J.S., Lee, E.J., Lee, J.C., Kim, W.K., Kim, H.S., 2007. Anti-inflammatoryeffects of short chain fatty acids in IFN-�-stimulated RAW 264.7 murinemacrophage cells: involvement of NF-�B and ERK signaling pathways. Int.Immunopharmacol. 7, 70–77.
amana, C.V., Gil, M.P., Schreiber, R.D., Stark, G.R., 2002. Stat1-dependentand -independent pathways in IFN-�-dependent signaling. Trends Immunol.23, 96–101.
enard, P., Raes, M., 1999. The proinflammatory transcription factor NF�B:a potential target for novel therapeutical strategies. Cell. Biol. Toxicol. 15,341–344.
osenberger, S.F., Finch, J.S., Gupta, A., Bowden, G.T., 1999. Extracellularsignal-regulated kinase 1/2-mediated phosphorylation of JunD and FosBis required for okadaic acid-induced activator protein 1 activation. J. Biol.Chem. 274, 1124–1130.
arkar, F.H., Li, Y., 2003. Soy isoflavones and cancer prevention. Cancer Invest.21, 744–757.
orli, C.H., Zhang, H.J., Armstrong, M.B., Rajotte, R.V., Maclouf, J., Robertson,R.P., 1998. Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 inthe pancreatic islet. Proc. Natl. Acad. Sci. U.S.A. 95, 1788–1793.
outhern, C., Schulster, D., Green, I.C., 1990. Inhibition of insulin secretionby interleukin-1� and tumour necrosis factor-� via an l-arginine-dependentnitric oxide generating mechanism. FEBS Lett. 276, 42–44.
trandell, E., Buschard, K., Saldeen, J., Welsh, N., 1995. Interleukin-1� induces
the expression of hsp70, heme oxygenase and Mn-SOD in FACS-purifiedrat islet �-cells, but not in �-cells. Immunol. Lett. 48, 145–148.homas, H.E., Darwiche, R., Corbett, J.A., Kay, T.W., 2002. Interleukin-1 plus�-interferon-induced pancreatic �-cell dysfunction is mediated by �-cellnitric oxide production. Diabetes 51, 311–316.
Z
Endocrinology 278 (2007) 18–28
ran, P.O., Gleason, C.E., Poitout, V., Robertson, R.P., 1999. Prostaglandin E2
mediates inhibition of insulin secretion by interleukin-1�. J. Biol. Chem.274, 31245–31248.
anden Berghe, W., Plaisance, S., Boone, E., De Bosscher, K., Schmitz, M.L.,Fiers, W., Haegeman, G., 1998. p38 and extracellular signal-regulated kinasemitogen-activated protein kinase pathways are required for nuclear factor-�B p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem.273, 3285–3290.
elsh, N., 1996. Interleukin-1�-induced ceramide and diacylglycerol gener-ation may lead to activation of the c-Jun NH2-terminal kinase and thetranscription factor ATF2 in the insulin-producing cell line RINm5F. J. Biol.Chem. 271, 8307–8312.
elsh, N., Eizirik, D.L., Bendtzen, K., Sandler, S., 1991. Interleukin-1�-inducednitric oxide production in isolated rat pancreatic islets requires gene tran-scription and may lead to inhibition of the Krebs cycle enzyme aconitase.Endocrinology 129, 3167–3173.
en, Z., Darnell Jr., J.E., 1997. Mapping of Stat3 serine phosphorylation to asingle residue (727) and evidence that serine phosphorylation has no influ-ence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 25, 2062–2067.
ie, Q.W., Nathan, C., 1993. Promoter of the mouse gene encodingcalcium-independent nitric oxide synthase confers inducibility by interferon-� and bacterial lipopolysaccharide. Trans. Assoc. Am. Phys. 106,1–12.
oshida, Y., Kumar, A., Koyama, Y., Peng, H., Arman, A., Boch, J.A., Auron,P.E., 2004. Interleukin 1 activates STAT3/nuclear factor-�B cross-talk viaa unique TRAF6- and p65-dependent mechanism. J. Biol. Chem. 279,1768–1776.
hang, X., Blenis, J., Li, H.C., Schindler, C., Chen-Kiang, S., 1995. Require-
ment of serine phosphorylation for formation of STAT-promoter complexes.Science 267, 1990–1994.hang, X., Wrzeszczynska, M.H., Horvath, C.M., Darnell Jr., J.E., 1999.Interacting regions in Stat3 and c-Jun that participate in cooperative tran-scriptional activation. Mol. Cell. Biol. 19, 7138–7146.




















