
GENETIC STUDIES OF PRE-ECLAMPSIA - KI Open Archive
74
From DEPARTMENT OF BIOSCIENCES AND NUTRITION Karolinska Institutet, Stockholm, Sweden GENETIC STUDIES OF PRE-ECLAMPSIA Hanna Peterson Stockholm 2010
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of GENETIC STUDIES OF PRE-ECLAMPSIA - KI Open Archive

From DEPARTMENT OF BIOSCIENCES AND NUTRITION
Karolinska Institutet, Stockholm, Sweden
GENETIC STUDIES OF PRE-ECLAMPSIA
Hanna Peterson
Stockholm 2010

2010
Gårdsvägen 4, 169 70 Solna
Printed by
All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by [name of printer] © Hanna Peterson, 2010 ISBN 978-91-7409-795-5

ABSTRACT Pre-eclampsia is a multifactorial, pregnancy-specific vascular disorder characterized by hypertension and proteinuria. It affects around 3-5% of pregnancies worldwide. There is a wide range of phenotypes from mild forms developing in the end of pregnancy, to severe forms with extremely high blood pressure that in worst cases could lead to eclampsia, the occurrence of seizures. Pre-eclampsia and eclampsia account for more than 50 000 maternal deaths per year. The etiology and pathophysiology of pre-eclampsia remain poorly understood, but it is generally accepted that defect placentation during the early stage of pregnancy, most likely in combination with maternal and environmental factors could lead to systemic inflammation, endothelial dysfunction and the manifestation of the clinical symptoms. Both large epidemiological and family studies have demonstrated genetic contribution to susceptibility. Although several loci have been mapped by linkage analysis only a few promising positional candidate genes have been identified so far. A number of functional candidate genes encoding for coagulation factors, oxidative stress and vasoactive substances, have been suggested to mediate susceptibility, but attempts to replicate these findings have yielded inconsistent results. The overall aim of this thesis was to search for genes predisposing for pre-eclampsia using several different approaches. In Paper I we evaluated the role of the first positionally cloned pre-eclampsia candidate gene STOX1 at 10q22 in the Finnish population. We were unable to validate STOX1 as a common pre-eclampsia gene, and our result is in agreement with two other European studies investigating the same gene. An intriguing association in Paper II suggests that pre-eclampsia share a predisposing genetic factor on chromosome 9p21 with coronary artery disease. To the best of our knowledge we were the first to investigate the role of the 9p21 region in pre-eclampsia. The association of this locus has not been confirmed in other populations and further investigations of the genes in this region are warranted. We have previously mapped a candidate susceptibility locus to chromosome 2p25. In Papers III and IV we present our systematic efforts to narrow down the linkage region by fine-mapping followed by association analysis. In conclusion, our investigations provides an insight into a potential role of a new susceptibility locus for pre-eclampsia at 9p21 in the Finnish population. We were able to narrow down the linkage region at 2p25, but found our sample sets underpowered to evaluate the genes residing within it. Finally, there is no conclusive evidence either for or against STOX1 as a susceptibility gene for pre-eclampsia. To further explore the role of STOX1, much larger sample sets are needed.

LIST OF PUBLICATIONS
I. Katja Kivinen, Hanna Peterson, Leena Hiltunen, Hannele Laivuori, Sanna Heino, Inkeri Majuri, Sakari Knuutila, Vesa Rasi and Juha Kere. Evaluation of STOX1 as a preeclampsia candidate gene in a population-wide sample. European Journal of Human Gentics, 2007, 15, 494-497.
II. Hanna Peterson, Katja Kivinen, Leena Hiltunen, Elina Salmela, Tuuli Lappalainen, Vesa Rasi, Ayat Sayed, Lucilla Poston, Matthew P Johnson,
Linda Morgan, Eric K Moses, Juha Kere and Hannele Laivuori. Common variants on chromosome 9p21 are associated with pre-eclampsia in the Finnish population. In manuscript
III. Hanna Peterson, Hannele Laivuori, Erja Kerkelä, Hong Jiao, Leena Hiltunen, Sanna Heino, Inkeri Tiala, Sanna Knuutila, Vesa Rasi, Juha Kere and Katja Kivinen. ROCK2 allelic variants are not associated with pre-eclampsia susceptibility in the Finnish population Molecular Human Reproduction, 2009, 15, 443-449.
IV. Hanna Peterson, Katja Kivinen, Erja Kerkelä, Hannele Laivuori, Leena Hiltunen, Hong Jiao, Ville-Veiko Mäkelä, Risto Kaaja, Olavi Ylikorkala, Vesa Rasi och Juha Kere. Fine-mapping and characterization of pre-eclampsia susceptibility locus on chromosome 2p25. In manuscript

CONTENTS 1 Background......................................................................................................................... 1
1.1 Pre-eclampsia........................................................................................................... 1 1.1.1 Phenotype and symptoms .......................................................................... 1 1.1.2 Definition.................................................................................................... 1 1.1.3 Incidence, prevalence, mortality and morbidity........................................ 2 1.1.4 Long term health effects for mother and child .......................................... 3 1.1.5 Prevention and treatment ........................................................................... 4 1.1.6 Risk factors................................................................................................. 5 1.1.7 Etiology and pathophysiology ................................................................... 6
1.2 The human genome ............................................................................................... 12 1.2.1 The human genome project...................................................................... 12 1.2.2 Sequence variations.................................................................................. 12 1.2.3 The HapMap project ................................................................................ 14 1.2.4 The 1000 Genomes Project...................................................................... 14
1.3 Genetic studies of complex diseases..................................................................... 15 1.3.1 Genome-wide linkage analysis ................................................................ 15 1.3.2 Association analysis and candidate gene studies .................................... 16
1.4 Genetics of pre-eclampsia ..................................................................................... 19 1.4.1 Genetic susceptibility ............................................................................... 20 1.4.2 Susceptibility loci for pre-eclampsia identified by linkage..................... 21 1.4.3 Association-based mapping approaches.................................................. 24
2 Aims of the thesis ............................................................................................................. 28 3 Material and methods....................................................................................................... 29
3.1 Study subjects ........................................................................................................ 29 3.1.1 Finnish families (I-IV) ............................................................................. 29 3.1.2 Finnish case-controls (I-IV)..................................................................... 30 3.1.3 UK case-controls (II) ............................................................................... 30 3.1.4 Australian/New Zealand families (II)...................................................... 31 3.1.5 Finnish placental samples (I-IV).............................................................. 31 3.1.6 Finnish control samples (II) ..................................................................... 31
3.2 Genetic analysis ..................................................................................................... 32 3.2.1 Marker selection (I-IV) ............................................................................ 32 3.2.2 Genotyping: SNPs and microsatellites (I-IV) ......................................... 32 3.2.3 Data analysis (I-IV).................................................................................. 34 3.2.4 DNA re-sequencing (I, III, IV) ................................................................ 35
3.3 Expression analysis ............................................................................................... 36 3.3.1 Microarray expression analysis (I-V) ...................................................... 36 3.3.2 Allele specific expression analysis (III) .................................................. 36
4 Results and discussion...................................................................................................... 38 4.1 Paper I - Evaluation of STOX1 on 10q22 ............................................................. 38 4.2 Paper II - Association in 9p21 CAD risk region................................................... 39 4.3 Paper III, IV - Mapping and characterization of 2p25 linkage region ................. 42 4.4 General aspects ...................................................................................................... 44
5 Concluding remarks and future perspectives................................................................... 47 6 Acknowledgements .......................................................................................................... 49 7 References ........................................................................................................................ 51

LIST OF ABBREVIATIONS ACVR2A Activin receptor type 2A ASSHP Australasian Society for the Study of Hypertension in Pregnancy Aus/NZ Australian/New Zealand BMI Body mass index bp Base pair CAD Coronary artery disease CDCV Common disease, common variant CDKN Cyclin dependent kinase inhibitor cDNA Complementary deoxyribonucleic acid CEPH Centre d'Etude du Polymorphisme Humain CI Confidence interval cM Centimorgan CNV Copy number variant DNA Deoxyribonucleic acid ddNTP Dideoxynucleotide dNTP Deoxynucleotide FINNPEC Finnish Genetics of Pre-eclampsia Consortium GH Gestational hypertension GOPEC The genetics of pre-eclampsia UK Consortium GWAS Genome-wide association study HGP Human genome project HLA Human leukocyte antigen HPM Haplotype pattern mining H/R Hypoxia-reoxygenation HWE Hardy-Weinberg equilibrium ICD International classification of diseases IBD Identical by descent IGF Insulin growth factor IL Interleukin IVF In vitro fertilization ISSHP International Society for the Study of Hypertension in Pregnancy kb Kilo base, 1000 base pairs KIR Killer immunoglobulin-like receptor LD Linkage disequilibrium LDL Low-density lipoprotein LOD Logarithm of odds LPL Lipoprotein lipase MAF Minor allele frequency MALDI-TOF Matrix-assisted laser desorption/ionisation time-of-flight Mb Mega base, 1 000 000 base pairs MHC Major histocompatibility complex MMP Matrix metalloproteinase mRNA Messenger ribonucleic acid NHBPEP National High Blood Pressure Education Program

NK Natural killer NPL Non-parametric linkage OR Odds ratio PCR Polymerase chain reaction PDT Pedigree disequilibrium test PIGF Placental growth factor RAAS Renin-angiotensin-aldosterone system RNA Ribonucleic acid ROCK Rho-associated, coiled-coil containing protein kinase ROS Reactive oxygen species RT-PCR Reverse transcription polymerase chain reaction SNP Single nucleotide polymorphism SSR Simple sequence repeat STMP Syncytiotrophoblast microparticle STOX Storkhead box T2D Type 2 diabetes TDT Transmission disequelibrium test Th T helper cell TNF Tumor necrosis factor TSC The SNP consortium UTR Untranslated region VEGF Vascular endothelial growth factor


1
1 BACKGROUND 1.1 PRE-ECLAMPSIA
1.1.1 Phenotype and symptoms
Pre-eclampsia is a pregnancy-specific vascular disorder manifesting in the latter part of the pregnancy (after 20 weeks of gestation), although the pathophysiological process is thought to take place during placental development in early stages of pregnancy. Pre-eclampsia is characterized by elevated blood pressure and protein in the urine (proteinuria). There is a wide range of phenotypes from mild forms of pre-eclampsia developing in the end of pregnancy, generally with few or no symptoms, to severe forms with extremely high blood pressure that may lead to upper abdominal pain, visual disturbances or headache, problems in the liver, kidneys, brain and clotting abnormalities. The fetus is at risk and the most common abnormalities are intrauterine growth restriction as a result of reduced blood supply through placenta and problems of prematurity. A final and severe phase of pre-eclampsia is called eclampsia. It is a rare complication, characterized by the occurrence of seizures, often leading to changes in the circulatory system and kidney failure, and is associated with an increased risk of maternal death. Although the outcome of pre-eclampsia is often good, it can be in some cases life threatening for both mother and child.
1.1.2 Definition
There is no universal classification system of hypertensive disorders of pregnancy and definition of pre-eclampsia, and there has been a wide diversity of terminology and diagnostic criteria over the years for published studies (Harlow and Brown 2001). Currently there are several internationally recognized definitions available (Brown et al. 2001). Reports widely used for classification and definitions are from the Australasian Society for the Study of Hypertension in Pregnancy (ASSHP) and the National High Blood Pressure Education Program (NHBPEP) (ASSHP 1993, NHBPEP 2000). The definitions of hypertension in pregnancy are identical between the two reports; Systolic blood pressure ≥ 140 mmHg and/or a diastolic blood pressure ≥ 90 mmHg. The classifications of hypertensive disorders during pregnancy are similar with four defined categories; pre-eclampsia or eclampsia, gestational hypertension (GH), chronic hypertension and superimposed pre-eclampsia. The categories are summarized in table 1, including generalized and most commonly used definitions based on diagnostic criteria suggested by different reports. Pre-eclampsia is most often defined as a combination of hypertension and proteinuria, and the NHBPEP definition consists of new onset of hypertension (≥ 140 mmHg and/or a diastolic blood pressure ≥ 90 mmHg) after 20 weeks of gestation in combination with proteinuria which is defined as the appearance of ≥ 0.3g/24h of urinary protein or ≥ 1+ reading on a dipstick that correlates to ≥ 0.3g/L in a random urine determination. Attempt to subdivide pre-eclampsia has been done and severe pre-eclampsia is often defined as systolic blood pressure ≥ 160 mmHg and/or a diastolic blood pressure ≥ 110 mmHg in combination with severe proteinuria of ≥3 g/24h.

2
Table 1. Commonly used diagnostic criteria and classification of hypertensive pregnancy disorders.
Classification Diagnostic criteria Pre-eclampsia
Hypertension: Blood pressure of ≥140 mm Hg systolic or ≥90 mm Hg diastolic that occurs after 20 weeks of gestation in a woman with previously normal blood pressure.
Proteinuria: Defined as urinary excretion of ≥0.3 g protein in a 24-hour urine specimen.
Eclampsia
Occurrence, in a woman with pre-eclampsia, of seizures not attributed to other causes.
Gestational hypertension
Hypertension: Blood pressure of ≥140 mm Hg systolic or ≥90 mm Hg diastolic that occurs after 20 weeks of gestation in a woman with previously normal blood pressure.
Superimposed pre-eclampsia
Chronic hypertension associated with new-onset of proteinuria during pregnancy.
Chronic hypertension
Hypertension before 20 weeks of gestation and/or persistent for more than 6 weeks after delivery.
1.1.3 Incidence, prevalence, mortality and morbidity
Approximately 10 % of women will have high blood pressure at some point before delivery, and pre-eclampsia complicates around 3-5% of pregnancies worldwide (Hogberg 2005). The incidence is higher in the developing world and specific ethnical groups (Duley 2003, Zhang et al. 2001). Eclampsia, the severe end phase of pre-eclampsia, is associated with mortality and accounts for more than 50 000 maternal deaths per year (Duley 2003). It is rare in Europe with 2-3 cases per 10 000 births, but more common in developing countries with an estimated incidence of 16-69 cases per 10 000 births (Frias and Belfort 2003, Knight 2007, Kullberg et al. 2002). Limited access to maternity services and emergency obstetric care in developing regions is a possible explanation and is reflected by the fact that 99 % of all maternal deaths occur in low and middle-income countries (Hogberg 2005). In the countries with low maternal mortality, only a third is associated to eclampsia, compared to countries with high maternal mortality rate where almost all deaths are associated to eclampsia and not pre-eclampsia (Duley 2009). It is noteworthy that the incidence of pre-eclampsia has increased by 40% in the last 15 years (Duley 2009). Plausible causes include worldwide obesity epidemic, a rise in the numbers of older mothers and increase in the frequency of multiple pregnancies. Renal failure, cardiac arrest, stroke, adult respiratory distress syndrome, coagulopathy and liver failure are all severe morbidities associated with eclampsia and pre-eclampsia, and affected women are in need of intensive care (reviewed in (Duley 2009)). There are some studies about the psychological effect of pre-eclampsia on women, since it can be a difficult and

3
unexpected experience with illness, early deliveries and in worse cases fetal deaths. It may increase the risk of post-traumatic stress disorder (van Pampus et al. 2004). More than 10% of infants born small for gestational age are from pre-eclamptic pregnancies (Kramer et al. 2000). Perinatal mortality is high after pre-eclampsia and eclampsia, and 25% of all neonatal deaths and stillbirths are associated to the disorders (Ngoc et al. 2006, Roberts et al. 2005). Mortality rates for infants are several times higher in developing world compared to developed countries for both pre-eclampsia (3 times) and eclampsia (4.5 times) (reviewed in (Duley 2009)). The severity of the disorder affects the outcome for both mother and child, with the highest risk for severe pre-eclampsia or eclampsia (Gaugler-Senden et al. 2006). There are also many complications associated with pre-term birth and the infants require intensive care and neonatal facilities, which again are less common in developing countries and will affect the outcome for the infant.
1.1.4 Long term health effects for mother and child
Epidemiological data on long term effects of pre-eclampsia indicate that affected women may also be at increased risk of cardiovascular or cerebrovascular diseases in later life. One of the first studies investigating the long-term effects of eclampsia was published in 1976 (Chesley et al. 1976). Their findings did not show an association to cardiovascular disease or an increase of morbidity and mortality in women who had pre-eclampsia in their first pregnancy. However, women who had experienced eclampsia more than once had a greater incidence of cardiovascular disease as well as higher death rates. More recent data are in conflict with the early findings, and it is now widely accepted that women who have previously experienced pre-eclampsia or eclampsia, even in the first pregnancy, have an increased long-term risk for remote cardiovascular and cerebrovascular disease, such as hypertension, ischemic heart disease, myocardial infarction, and cerebrovacular accidents (reviewed in (Harskamp and Zeeman 2007)). The individual risk is different when looking at specific subgroups of pre-eclampsia and it has been shown that women with recurrent pre-eclampsia, early/severe forms, pre-eclampsia as multiparas, pre-term delivery and women pregnant at older age seem to be at even greater risk (Arnadottir et al. 2005, Irgens et al. 2001, Jonsdottir et al. 1995, Sibai et al. 1986). Moreover, death resulting from cardiovascular causes among pre-eclamptic women has been reported to be 8-12 fold higher than in normotensive women (Irgens et al. 2001). Many risk factors are shared between pre-eclampsia and coronary artery disease (CAD), including endothelial dysfunction, hypertension, obesity, insulin resistance and dyslipidemia (reviewed in (Garovic and Hayman 2007)). Therefore, it has been suggested that the metabolic syndrome, which refers to a group of conditions including hypertension, dyslipidemia, abdominal fat, and fasting hyperglycemia, may be a possible underlying mechanism common to CAD and pre-eclampsia, and may lead to the different disorders at different time points in a woman’s life (Newstead et al. 2007). Both pre-eclampsia and gestational diabetes (glucose intolerance with onset or first recognition during pregnancy) share features of the metabolic syndrome, and patients with a history of these pregnancy disorders have an increase in the lifelong risk of CAD (Carpenter 2007, Rodie et al. 2004). Supporting the connection between pre-eclampsia

4
and glucose intolerance, the risk of subsequent type 2 diabetes (T2D) is increased more than 3-fold after severe pre-eclampsia (Lykke et al. 2009). An alternative explanation for the exaggerated risk of CAD and T2D after pre-eclampsia is that pre-eclampsia itself may induce irreversible vascular and metabolic changes that may increase the later risk. The majority of children from pre-eclamptic pregnancies survive in countries with good health care, but they may have an increased susceptibility for diseases later in life beyond that mediated by their preterm birth. Higher risk of childhood hypertension have been reported (Seidman et al. 1991, Tenhola et al. 2003, 2006) and it has been found that pre-eclampsia is associated with an increased risk of diabetes in the offspring, although, contradicting results have been published as well (Bache et al. 1999, Dahlquist et al. 1999, Jones et al. 1998, McKinney et al. 1999). In addition, a decreased risk of breast cancer has been reported among female offspring from pre-eclamptic pregnancies, which may be explained by low intrauterine estrogen levels that characterize pre-eclamptic pregnancies (Ekbom et al. 1992, Innes et al. 2000, Sanderson et al. 1998, 2006, Xue and Michels 2007). However, the potential biological mechanisms underlying the association between pre-eclampsia and long-term offspring health remain unknown. One possibility is that genetic factors that predispose the mother for pre-eclampsia and disorders later in life are inherited by the offspring or adaptive responses to the intrauterine environment in pre-eclamptic pregnancy may result in epigenetic changes that affect disease susceptibility later in life (Gluckman et al. 2008, Smith et al. 2001).
1.1.5 Prevention and treatment
Since the etiology and pathogenesis of pre-eclampsia are unclear, the development of strategies for prevention and treatment is difficult. Promising results have been published concerning the use of antiplatelet drugs, primarily low-dose acetylsalicylic acid for prevention and a reduced risk of pre-eclampsia after treatment has been reported (Askie et al. 2007). Treatment with acetylsalicylic acid should be considered for women with a history of severe pre-eclampsia in previous pregnancy. Calcium supplementation is associated with a lower risk of pre-eclampsia as well, however, there is no clear impact on outcome for the infant (Hofmeyr et al. 2007). Treatment is largely directed towards the symptoms, with little evidence that any intervention alters the underlying pathophysiology. Mild pre-eclampsia is often managed with careful observation at maternal care units, at hospital or at home, in combination with activity restriction. Since high blood pressure may lead to direct vascular damage, which in turn could lead to other complications such as renal failure, fetal distress and stroke, antihypertensive drugs are mandatory for women with very high blood pressure (Duley and Henderson-Smart 2000). Magnesium sulfate is known to reduce the risk of eclampsia in pre-eclamptic women and can be injected to prevent eclampsia-related seizures (Duley et al. 2003). To date, the only effective “treatment” is delivery and removal of placenta. It can be challenging to balance between protecting the mother by ending the pregnancy, and maximizing the maturity of the fetus when timing the delivery of women with severe pre-eclampsia before 32 weeks of gestation. Therefore, there is a great need for a safe treatment that would eliminate the need for premature

5
delivery in the severe cases of pre-eclampsia as well as ensure the well-being of the pregnant women.
1.1.6 Risk factors
Risk factors that predispose women for pre-eclampsia are summarized in table 2. Nulliparity is considered to be a powerful predictor of increased risk and a meta-analysis from 2005 reported that women pregnant for the first time are almost three times more likely to get pre-eclampsia than with a second or later pregnancy (Duckitt and Harrington 2005). Increased risk is also observed for multiparous women when there is a change in paternity (Trupin et al. 1996), prior use of barrier contraceptives and with shorter length of sexual relationship (Klonoff-Cohen et al. 1989, Robillard et al. 1994). A plausible explanation for this is that pre-eclampsia may represent a maternal immunological reaction towards paternal antigens (see 1.1.7.4). However, contradicting results have been reported, suggesting that the effect of these factors may be explained by other confounding factors (Ness et al. 2004). Risk associated with change in paternity could also be explained by the fact that longer intervals between pregnancies seems to predispose regardless of paternity (Skjaerven et al. 2002). Age has also been considered as a risk factor, with women ≥40 years old twice as likely to get pre-eclampsia as woman under 40 (Duckitt and Harrington 2005). This may be explained by other age-related risk factors, such as obesity and chronic hypertension. Increasing body mass index (BMI), even in absence of real obesity, is associated with increased risk of pre-eclampsia (Duckitt and Harrington 2005, Eskenazi et al. 1991). Diastolic blood pressure at ≥110 mm Hg increases the risk of developing superimposed pre-eclampsia five-fold and an increased blood pressure within the normal range is also recognized as a risk factor (reviewed in (Duckitt and Harrington 2005). Other conditions linked to pre-eclampsia are insulin dependent diabetes, renal disease, antiphospholipid syndrome (a blood clotting disorder) and autoimmune diseases (Davies et al. 1970, Duckitt and Harrington 2005, Pattison et al. 1993, Stamilio et al. 2000, Yasuda et al. 1995). Prevalence of pre-eclampsia is higher in African-Americans than in other ethnic groups in United States and ethnicity may be one predisposing factor (Sibai et al. 1997). However, the prevalence of chronic hypertension is also higher in this population (Kurian and Cardarelli 2007). Both personal and familial history of pre-eclampsia are considered as true risk factors, with data showing that women with pre-eclampsia in their first pregnancy have seven times the risk of getting pre-eclampsia in a second pregnancy compared to women with healthy first pregnancies (Duckitt and Harrington 2005). Additionally, a family history of pre-eclampsia triples the risk (Duckitt and Harrington 2005). It has been reported that twin pregnancy triples the risk for pre-eclampsia and triplet pregnancy triples the risk of pre-eclampsia compared with twin pregnancy (Duckitt and Harrington 2005, Skupski et al. 1996). This could have an explanation by the increased placental size associated with multiple pregnancies and the fact that decreased placental perfusion is considered as a central feature of pre-eclampsia (Sibai et al. 2000). Furthermore, in vitro fertilization (IVF) is associated to pre-eclampsia and could be explained by immune maladaptation (Kallen et al. 2005, Shevell et al. 2005). An alternative explanation could be that multiple pregnancies are clearly more common after IVF (reviewed in (El-Toukhy et al. 2006)). However, a meta-analysis from 2004 shows that the risk for pre-eclampsia after IVF is higher even in singleton pregnancies (Jackson et al. 2004).

6
Table 2. Summary of risk factors for pre-eclampsia
Risk factors for pre-eclampsia Maternity age Multiple pregnancy Nulliparity Previous pre-eclampsia Family history of pre-eclampsia Body mass index (BMI) Time between pregnancies Change of partner African-American race In vitro fertilization Pre-existing medical conditions Insulin dependent diabetes Insulin resistance Chronic hypertension Renal disease Antiphospholipid syndrome Autoimmune disease
1.1.7 Etiology and pathophysiology
Pre-eclampsia has been termed the “disease of theories”, reflecting the confusion surrounding the causes and pathophysiology of the disease. Despite the large number of studies, the pathophysiology of this syndrome is not fully understood. However, placental vasculopathy and endothelial dysfunction appear central to the pathogenesis. Pre-eclampsia is considered as a two-stage disorder; Stage I - Defective placentation due to failed remodeling of maternal vessels that results in a poorly perfused placenta. Stage II - Clinical manifestation of pre-eclampsia (Roberts and Hubel 2009). However, the link between the pathophysiology of abnormal placentation and the physiology of the maternal syndrome remains unclear, but it is widely hypothesized that oxidative stress may be one of the important factors (Gupta et al. 2005). The failed remodeling of maternal vessels observed in pre-eclamptic pregnancies in the first stage of the disorder, resulting in a defective placenta, is probably not sufficient to cause pre-eclampsia. Reduced placental perfusion and pathological evidence of failed placental vascular remodeling is also evident in women who had growth-restricted babies and preterm birth with no maternal signs of pre-eclampsia (Roberts and Post 2008). The occurrence of stage II requires interactions with maternal constitutional factors that may be genetic influences, immune maladaptation, and/or environmental factors (reviewed in (Roberts and Post 2008)). Many of those factors leading to the maternal syndrome are also risk factors for cardiovascular disease in later life (Rodie et al. 2004). Pre-eclampsia is a heterogeneous condition, which is consistent with varying degrees of contribution from mother and infant (reviewed in (Ness and Roberts 1996)). Thus, with profoundly reduced placental perfusion the generation of maternal signs may require very little contribution from the mother. Conversely, the woman with extensive predisposing constitutional sensitivity could develop pre-eclampsia with very little reduced perfusion (Roberts and Hubel 2009).

7
Figure 1. The two stage model of pre-eclampsia pathophysiology. (Stage I) Abnormal placentation in the first trimester, followed by reduced placental perfusion, which results in release of factors from the placental unit that will influence maternal physiology, lead to endothelial damage, and the maternal syndrome (Stage II). The placental dysfunction is triggered by poorly understood mechanisms, which may include genetic, environmental and immunological factors. The same type of factors could also have a role in later pathophysiological events and initiating the maternal syndrome. Pre-eclampsia is associated to both growth restriction pregnancies and later maternal disorders, such as cardiovascular disease and type 2 diabetes. (Modified from (Parikh and Karumanchi 2008)) 1.1.7.1 Stage I – Abnormal placentation
Placenta is a temporally vascular organ essential for the exchange of gases, nutrients and waste between fetal and maternal circulatory systems. A variety of metabolic, hormonal and immunological molecules important for the fetal development are synthesized by the placenta. In normal pregnancy, after implantation, cytotrophoblastic cells of fetal origin attach to the uterine wall via so called anchoring villi. The cytotrophoblasts form a shell, lining the uterine cavity and a small proportion invade the uterine wall and its blood vessels called spiral arteries. Around 12 weeks’ gestation, the process where cytotrophoblastic cells invade the spiral arteries of the myometrium peaks and by 18 to 20 weeks, cytotrophoblast cells have replaced endothelial cells lining the vessels and dismantled the muscle and connective tissue. The previous small, high-resistance vessels are converted into large, low resistance vessels that allow for an increase in placental blood flow needed to sustain the fetus throughout the pregnancy (reiewed in (Pijnenborg et al. 2006)) (Figure 2).

8
In pre-eclampsia pathophysiology, inadequate invasion of trophoblasts has been implicated and different kinds of pathophysiological hallmarks could be found in the placenta (reviewed in (Cheng and Wang 2009)). One observation in pre-eclamptic placentas is poor cytotrophoblast differentiation, which leads to reduced trophoblast invasion into the myometrial segments of the spiral arteries that remain narrow and undilated. This has been supported by Doppler ultrasound studies of the maternal uterine blood flow (Papageorghiou et al. 2004). The invasive cytotrophoblast cells is known to adopt a cell surface adhesion phenotype characteristic for endothelial cells, but in pre-eclampsia the cells fails to undergo the switching from epithelial to endothelial characteristics (Zhou et al. 1997). It means that the cells fail to express some of the intergrin, cadherin, selectin or immunoglobulin superfamily members important for vascular adhesion phenotype. The end result after defective vascular transformation of the arteries is insufficient placental circulation leading to hypoxia, or at least intermittent perfusion, oxidative stress and release of soluble “toxic” factors from the ischemic placenta that damage the vasculature of the mother, leading to widespread vascular injury and increased permeability. Figure 2. A summary of the suggested pathophysiological events in pre-eclampsia leading to endothelial dysfunction. The upper part of the figure shows the spiral arteries, the normal adaptation at pregnancy and the incomplete remodeling process in pre-eclampsia, followed by the possible pathophysiological events leading to endothelial dysfunction (Modified from (Moffett-King 2002)).

9
1.1.7.2 The link between stages I and II
The link between abnormal trophoblast invasion, and later generalized endothelial activation and dysfunction, leading to the maternal syndrome is not clear, but it may be via release of placental factors. Microparticles are cellular derived vesicles that are shed from cell membranes, produced in placenta during the continuous process of growth and apoptosis, and mediate cell-to-cell communication with potential role in processes such as thrombosis, homeostasis, angiogenesis and inflammation (reviewed in (Redman and Sargent 2008)). The levels of these kinds of particles are elevated in conditions associated with enhanced systemic inflammation, such as normal pregnancy and at even higher levels in pre-eclampsia. In pre-eclamptic placentas, an increased apoptosis of particular syncytiotrophoblastic cells lining the outer layer of placenta has been observed (Allaire et al. 2000, Leung et al. 2001) and the release of syncytiotrophoblast microparticles (STMP) into the maternal circulation is elevated in pre-eclampsia (Knight et al. 1998). The STMPs are suggested to stimulate inflammatory responses and directly damage endothelial cells (reviewed in (Germain et al. 2007)), since in vitro studies have shown that STMP could simultaneously disrupt endothelial layers and stimulate production of factors that activate leukocytes (Smarason et al. 1993, von Dadelszen et al. 1999). Another interesting factor released from the syncytiotrophoblasts is the soluble receptor for vascular endothelial growth factor 1 (sVEGFR-1), also called sFlt-1. It functions as an antagonist of two angiogenic factors called vascular endothelial growth factor (VEGF) and placental growth factor (PIGF) and has been found to be upregulated in pre-eclamptic placentas. When an excess of sFlt-1 is present, it binds and inactivates VEGF and PIGF, which are needed for endothelial survival, and therefore induces endothelial dysfunction (reviewed in (Myatt and Webster 2009)) . Interestingly, it has been demonstrated that administration of sFlt-1 to pregnant rats induces hypertension, proteinuria and glomerular endotheliosis, which are all classical features of pre-eclampsia (Maynard et al. 2003). Altogether, this strongly supports sFlt1 as a disease predisposing factor. However, what is upregulating sFlt-1 in pre-eclampsia is not clear. Moreover, upregulation of sFlt-1 only explains part of the cases as some pre-eclamptic women have normal levels of gene product (Powers et al. 2005). It has been suggested that reduced blood flow through the spiral arteries leads to chronic hypoxia in placenta. There is also evidence for an alternative mechanism, characterized by hypoxia-reoxygenation (H/R) injury as a result of intermittent placental perfusion, secondary to the abnormal artery remodeling (Hung and Burton 2006). H/R injury could be a possible mechanism causing oxidative stress in placenta. Oxidative stress is excessive in pre-eclampsia, may cause endothelial dysfunction through the action of reactive oxygen species (ROS) and therefore considered to be a key step in the pathogenesis (reviewed in (Poston and Raijmakers 2004)). Oxidative stress has a generally accepted role in the pathogenesis of atherosclerosis. The same dyslipidemia is present in both pre-eclampsia and atherosclerosis, in addition to pathological lesions observed in placenta called atherosis, which have high similarity to atherosclerotic lesions (reviewed in (Belo et al. 2008)). Taken together, this leads to the assumption that oxidative stress could play a significant roll in pre-eclampsia as well. One characteristic of dyslipidemia is increased levels of small dense low-density

10
lipoproteins (LDL) and the H/R in placenta could lead to peroxidation of the LDL particles, which is known to be increased in pre-eclampsia (Atamer et al. 2005, Wang et al. 1992). Subsequently, the toxic products after lipid peroxidation are transported to distant sites in the body and can cause systemic oxidative stress and cellular damage. Many pro-inflammatory cytokines and modulators are found at increased levels in both circulation and placenta during pre-eclamptic pregnancy. Two of them, tumor necrosis factor α (TNFα) and Intreleukin-1 (IL-1) have both been implicated in pre-eclampsia pathophysiology, since they have the ability to stimulate structural and functional alterations in endothelial cells (reviewed in (Conrad and Benyo 1997)). However, the source of these molecules has not yet been identified, but a placental contribution is suggested. Interestingly, infusion of TNFα into rats in their late pregnancy resulted in increased arterial pressure and renal resistance (Alexander et al. 2002, Giardina et al. 2002). 1.1.7.3 Stage II – The maternal syndrome
The end stage of pre-eclampsia is the maternal syndrome defined by cardiovascular and renal features; hypertension and proteinuria. A specific renal endothelial lesion called glomerular endotheliosis is associated to proteinuria and endothelial dysfunction has been implicated in the process leading to hypertension. Injury of the endothelium could lead to a cascade of vasoconstriction, coagulation and redistribution of intravascular fluid and this is the center of the systemic dysfunctions in pre-eclampsia (reviewed in (Hayman et al. 1999)). In normal pregnancy, blood pressure and peripheral vascular resistance are decreased, but in pre-eclampsia the changes are reversed. Vascular constriction is universally present in pre-eclampsia and endothelial dysfunction is believed to be responsible for that. Several markers for endothelial dysfunction and activation are altered and women previously affected by pre-eclampsia are much more responsive to vasopressors (Agatisa et al. 2004, Chambers et al. 2001). Additionally, studies in vitro of pre-eclamptic vessels have shown alterations in endothelial function (reviewed in (Roberts 1998)). Prostaglandin I2 (PGI2) is a vasodilator produced by the endothelial and smooth muscle layers of blood vessels and its expression is lower in pre-eclampsia compared to normal pregnancy (Mills et al. 1999). Vasoconstrictors with a suggested role in pre-eclampsia pathophysiology are Thromboxane A2 (TXA2) and Endothelin-1 (Clark et al. 1992, Slowinski et al. 2002). Due to vasoconstriction and endothelial leakage observed in pre-eclampsia, fluids are lost from the vascular compartment and perfusion of organs is reduced. A systemic inflammatory response is observed in normal pregnancy, but exaggerated in pre-eclampsia. The inflammatory response is generated by different networks, mainly involving endothelial cell activation, maternal leukocytes and complement systems (Redman and Sargent 2003). Activation of the coagulation cascade is likely to further reduce organ perfusion by formation of microthrombi (Roberts and Lain 2002). 1.1.7.4 The immune maladaptation theory
The so called “immune maladaptation hypothesis” is one suggested etiology for pre-eclampsia. It is subject to controversy, but several epidemiological studies are supporting its validity by showing that pre-eclampsia risk is much higher in first pregnancy and that multiparity functions as a protective effect that is lost with a change of partner (reviewed in (Saito et al. 2007)). The hypothesis is based on the fact that the

11
fetoplacental unit contains paternal antigens that are foreign for the mother because of the differences between mother and father in respect to human leukocyte antigens. During early pregnancy, natural killer (NK) cells accumulate around the invading trophoblasts and produce cytokines that are involved in angiogenesis and vascular stability (Parham 2004). The tissues located in the maternal-fetal interface are protected against T-lymphocyte destruction by not expressing major histocompatibility complex (MHC) class Ia and II molecules, except for weak expression of human leukocyte antigen (HLA) C. Instead, invading trophoblasts express an unusual combination of HLA-F, HLA-E and HLA-G (Ishitani et al. 2003, Saftlas et al. 2005). HLA-G is only expressed by the extravillous trophoblasts and may, in part, explain the immune tolerance of the mother to the fetoplacental unit by protecting the cells from lysis by NK cells (O'Brien et al. 2000). A reduction of HLA-G expression on trophoblasts has been observed in pre-eclampsia and that may lead to the impaired trophoblast invasion (Colbern et al. 1994). Additionally, HLA-C is a dominant ligand for NK cells by interaction with killer-cell immunoglobulin-like receptors (KIRs) on the surface of the NK cells. It has been shown that a certain combination of the fetal HLA-C and the maternal KIR genotypes is associated to pre-eclampsia (Hiby et al. 2004). This combination is sending an inhibitory signal from the trophoblast to the NK cell and this inhibition has been suggested to play a role in the defective invasion and transformation of the arteries. However, not all pre-eclamptic pregnancies have this genotype combination and this only indicates that additional factors are most certainly involved in the process leading to defective invasion of cytotrophoblasts and the systemic responses. Several immune cell types, such as NK cells, monocytes, neutrophils, T and B cells, become hyperactivated in pre-eclampsia as a reaction to trophoblastic debris from damaging processes due to hypoxic conditions, oxidative stress or excessive inflammation (reviewed in (Borzychowski et al. 2006)). This activation enhances the production of cytokines, which may play a role in the maternal-fetal interface or in the whole body. T-helper (Th) cells can be classified into two subgroups known as Th1 and Th2, expressing different kinds of cytokines. Th1 cells enhance the cell-mediated immunity, while Th2 are involved in antibody production and repression of cell-mediated immunity. Th2 type immunity is known to play an important role in successful pregnancy by regulating immune response to the fetus. In pre-eclampsia on the other hand, greater amounts of Th1-type cytokines, such as TNFα, are observed in the circulation and the balance has shifted towards Th1 type immunity, which may be harmful for the invading trophoblasts (Saito and Sakai 2003). In summary, immune maladaptation may cause abnormal trophoblast invasion or, alternatively, cause the release of toxic cytokines, free radicals and enzymes from the deciduas, which may cause damage or disturb normal function of the maternal endothelium and the syncytiotrophoblasts.

12
1.2 THE HUMAN GENOME
1.2.1 The human genome project
The sequence of the human genome is rich in information about human evolution and encodes for genetic instructions of human physiology. The DNA double helix was discovered in 1953 by Watson and Crick and made basis for new research in the field of genetics (Watson and Crick 1953). The human genome project (HGP) was launched in 1990 with the goal of determining the sequence of approximately 3 billion base pairs (bp) that make up the human genome, identifying all human genes, store this information in public databases and develop tools for data analysis. The project was completed in 2003, however, drafts of the human genome sequence, both from the publicly funded project and from the private company Celera, were published already in 2001, comprising roughly 90 % of the total sequence (Lander et al. 2001, Venter et al. 2001). The drafts suggested that human genome contained 30 000-40 000 genes (Lander et al. 2001), much less than previous predictions of 60 000-100 000 (Strachan and Read, 2004). The numbers have been revised further after project completion and are now estimated to 20 000-25 000 (IHGSC 2004). Availability of human genome sequence has made it possible to investigate what types of DNA make up our genome and how we are related to other organisms that have been sequenced already. 1.2.2 Sequence variations
Humans are 99.9 % identical with respect to their DNA sequence, but many genetic variations in the human genome have been observed. Genetic variation explains some of the phenotypic differences among people, such as physical traits and whether a person has a higher or lower risk for certain diseases. However, the vast majority of the variants are believed to be neutral with no effect on phenotypic variation. Variations in the genome can be common (polymorphism), defined as genetic variant with minor allele frequencies (MAF) of at least 1 %, or rare (mutation) with MAF less than 1 % (reviewed in (Frazer et al. 2009)). Depending on the nucleotide composition of the variant, they can be subdivided into different classes and two of the most common ones are; Single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs). Both of them have been widely used as genetic markers and have been very important for genetic research. It was not until recently that copy number variants (CNVs) (i.e. DNA segments that present at variable copy number in comparison to a reference genome), which are structural variations, were found to represent a major source for human genetic variation and genome diversity (reviewed in (Conrad et al. 2009)). 1.2.2.1 Simple sequence repeats
SSRs are blocks of tandem repeats consisting of single repetitive nucleotide or di-, tri-, tetra-, or pentanucleotide repeat (dinucleotides are most frequent) (Figure 3B), and constitute around 3 % of the human genome, one every 2 kb (Lander et al. 2001). According to their size, they can be subdivided into two classes; minisatellites (14-500 bp) and microsatellites (1-13 bp). Microsatellite lengths are known to be highly variable between individuals, probably resulting from slippage during the replication process. Therefore, they have been widely used in disease-gene mapping in pedigrees,

13
where they can distinguish between maternally and paternally inherited alleles (see 1.3.1). 1.2.2.2 Single nucleotide polymorphisms
The most common genetic variation in the human genome are SNPs, where a single nucleotide (Adenine (A), cytosine (C), Guanine (G), thymine (T)) is changed (Figure 3A), inserted or deleted. The number of SNPs in a human genome is estimated to be approximately 3.3 million, one every 1000 bp (Levy et al. 2007, Wheeler et al. 2008). SNPs are found in both non-coding and coding regions of the genome, and if a single substitution leads to an amino-acid change (missense), frame shift or termination of translation (nonsense), the variation is called non-synonymous. In reverse, when the variant does not affect the amino-acid sequence, it is referred to as synonymous SNP, however, those could have an effect on mRNA stability or splicing instead. Other regulatory elements such as promoters, enhancers and silencers can be affected by base substitutions located outside coding regions. Information on close to 18 million SNPs have been made publicly via dbSNP database (http://www.ncbi.nlm.nih.gov/snp/). Thus, several of those are spurious findings. SNPs are popular in genome-wide studies because of their high frequency in the genome, providing a dense marker map and availability of powerful methods for large-scale analysis (see 1.3.2.1).
Figure 3. (A) Single nucleotide polymorphism. (B) Simple sequence repeat
1.2.2.3 Copy number variants
CNV results from genomic rearrangements and represent gain or loss of genomic segment of between a few hundred to several million bp and could be generated by normal mechanisms such as replication, recombination and DNA break repair. Generally, genomic segments with variable copy number could encompass parts of genes, reside entirely outside genes or, in the case of larger variants, include several known genes. The most obvious effect of CNVs is gene-dosage effects that could be observed if regulatory elements or genes are located in duplicated/deleted segments. In the past few years, a large number of CNVs have been identified by various genome-wide technologies (reviewed in (Zhang et al. 2009). According to the latest statistics, >29 000 entries of CNVs have been included in the Database of Genomic Variants (http://projects.tcag.ca/variation/).

14
1.2.3 The HapMap project
The International HapMap Project (www.hapmap.org) was initiated in 2002, with the aim to determine common patterns of SNP variation in human genome and to make the information available to the public domain. The goal was to genotype at least one common SNP every 5 kilo base (kb) across the euchromatic regions of the genome in individuals from four geographically diverse populations: mother-father-child trios from Nigeria; trios of northern and western European ancestry in Utah; and unrelated Chinese and Japanese individuals (IHMC 2003, 2005). In the first phase of the study, around 1.3 million SNPs were genotyped, the project continued and today over 4 million SNPs have been analyzed resulting in a SNP density of at least one every 1 kb (Frazer et al. 2007, www.hapmap.org). The extensive genotyping led to an insight into allele frequency differences among populations, along with characterization of the distribution and extent of linkage disequilibrium (LD) across the entire human genome. Genetic variants that are near each other tend to be inherited together, are in LD with each other, and the combination of those linked variants are known as haplotypes (Daly et al. 2001). SNPs in high LD are located in regions of chromosomes that have not been broken up by recombination, and they are separated by places where recombination has occurred. The HapMap project has been able to build a haplotype map of the genome and identified haplotype tagging SNPs that uniquely identify corresponding haplotypes without genotyping the whole combination of SNPs. Therefore, one of the major applications of the HapMap data has been to guide the design and prioritization of SNP genotyping for disease association studies, and making them much more cost effective, since the likelihood of genotyping a monomorphic SNP decreased dramatically, together with the number of SNPs needed for an association studies. Subsequently, the known knowledge about LD structures and tagging SNPs throughout the genome enabled for whole genome-wide association studies (GWASs) (see 1.3.2.1).
1.2.4 The 1000 Genomes Project
The 1000 Genomes Project (http://www.1000genomes.org/) is an international research effort that aims to provide the most comprehensive map of human genetic variation using next-generation sequencing platforms. Advances in sequencing technology have made it possible to process millions of sequence reads in parallel, allowing for whole genomes to be sequenced in an effective way. The 1000 genomes project involves sequencing the genomes of at least 1000 people from a number of different ethnic groups within the next three years The experience gained from this pilot project will hopefully be helpful in the guidance of future large-scale sequencing projects. A new map of the human genome will be developed, including SNPs, CNVs, and short insertion/deletion polymorphisms that appear in at least 1 %, at a resolution unmatched by current resources. Newly identified low-frequency SNPs and CNVs are essential for development of new generation of genotyping arrays, which will enable integrated analysis of SNPs and CNVs, and have better resolution and coverage of the true sequence variation in the human genome (Hurles et al. 2008, McCarroll 2008). As with other major human genome reference projects, data from the 1000 Genomes Project will be made available to the worldwide scientific community through freely accessible public databases and that would hopefully improve the sensitivity of disease discovery efforts.

15
1.3 GENETIC STUDIES OF COMPLEX DISEASES
Many common complex diseases are known to cluster in families and are believed to be influenced by several genetic and environmental factors as well as interactions among those. The common disease, common variant (CDCV) hypothesis suggests that variants with relatively high frequency, but low penetrance, are the major contributors to common complex diseases (Altshuler et al. 2008). A late onset is common among complex diseases, with modest or no impact on reproductive fitness (pregnancy specific disorder such as pre-eclampsia and dystocia are exceptions). Therefore, mildly deleterious alleles can rise to moderate frequencies in the population compared to mutations that cause strongly deleterious phenotypes and are lost by natural selection. Moreover, some alleles that were beneficial or neutral during human evolution may now confer susceptibility to disease because of changes in our environment. However, it has been argued that multiple rare variants contributing to the disease are more consistent with pathobiology than common variants (Schork et al. 2009). The genetic etiology is most likely based on a combination of multiple rare and common susceptibility loci. Efforts to investigate complex diseases initially adopted strategies similar to those employed for the successful mapping of Mendelian disorders. The two most commonly used methods are linkage and association studies. Positional cloning, which has been extremely successful for mapping Mendelian diseases (Jimenez-Sanchez et al. 2001), has been the most common approach for identifying genes in complex disorders during many years and has been successful in some cases. However, even if susceptibility regions are detected by linkage, extensive candidate gene studies are often needed to narrow down causal genes in the broad linkage regions. Another limitation of the linkage studies is that they lack power to identify common genetic variants with modest genetic risk on disease (Hirschhorn and Daly 2005). Association studies are one strategy for refining the linkage regions and to search for genetic variants of modest effects associated to disease. However, during the time of positional cloning, the association studies were restricted to specific genes or loci, and whole genome analyses were not an option. When GWAS become possible in 2006, it opened new frontiers in the understanding of many complex diseases and it is the most widely used approach for genetic mapping today (McCarthy and Hirschhorn 2008). However, the execution and analysis of those studies require big efforts and so far GWAS have only identified a small fraction of the genetic variance underlying the heritable component of complex diseases (Manolio et al. 2009).
1.3.1 Genome-wide linkage analysis
Two loci are linked because of their physical proximity along a chromosome, which means that they are so close together that their alleles tend to cosegregate within families. Cosegregating loci can be broken up by recombination during meiosis, and the probability of recombination increases the further apart two loci are from each other. Subsequently, the probability, referred to as recombination fraction, is a function of the genetic distance between loci, which is expressed in centimorgans (cM). One cM is defined by 1% recombination chance between two loci and represents approximately 1 mega base (Mb). In linkage analysis, the recombination fraction between individual

16
markers and the disease locus are estimated. Logarithm of odds (LOD) score compares the likelihood that a locus is linked to the likelihood that the observation is purely by chance and not due to linkage, and is an often used test in linkage analysis. The ratio between the two likelihoods gives the odds of linkage and the linkage is reported as LOD score (Morton 1955). If a positive LOD score is observed the presence of linkage is suggested, whereas negative LOD scores indicate that linkage is less likely. Standard LOD score analyses, also called parametric, require a precise genetic model detailing the mode of inheritance, gene frequencies and penetrance of each genotype and are therefore suitable for Mendelian traits. For complex diseases with no clear inheritance pattern, model-free (non-parametric) methods are preferred. Non-parametric linkage (NPL) analysis ignores unaffected people, and looks for chromosomal segments or alleles that are shared by affected individuals. In families, shared segment analysis can be conducted using identical by descent (IBD) data and NPL LOD score could be calculated with a method based on calculating the extent to which affected relatives share alleles IBD (Kruglyak et al. 1996). Families with increased occurrence of a certain disease are utilized in linkage studies, since affected family individuals are most likely to carry the same genetic predisposition. Genome-wide linkage studies, also called genome-wide scans, have been used over the last two decades to map disease predisposing loci. Markers, preferably microsatellites (see 1.2.2.1) are genotyped across de genome, regularly spaced, and the segregation through families is studied (reviewed in (Borecki and Province 2008)). Linkage analysis identifies large genomic regions that are often tens of Mb and may contain hundreds of genes (Boehnke 1994). Further investigations are needed to be able to map predisposing gene(s) and causal allele(s) and for those purposes, association analysis approaches could be useful (see 1.3.2).
1.3.2 Association analysis and candidate gene studies
Association studies seek a correlation between a specific genetic variation and a trait in a sample of individuals. There are three types of association analysis including the two hypothesis driven approaches focusing on candidate genes (i) or candidate susceptibility regions (ii), often as a result from genome-wide linkage analysis in a complex disease, with linked regions of several Mb in need of several steps of fine-mapping, and the non-hypothesis driven approach GWAS (iii) (see 1.3.2.1). Candidate genes are often chosen for their relevance to pathophysiology of the disease of interest, or they may be picked from previously determined linkage regions. It is generally accepted that susceptibility of complex diseases involves, to different extent, multiple genes with genetic effect size most likely being small to modest. For those variants, association are much more powerful than linkage analysis (Cardon and Bell 2001) and the importance of association analysis has increased during the past year as a consequence of the large number of SNPs mapped and development of the technology, which enable genotyping of millions of SNPs simultaneously. Both association and linkage analysis rely on the coinheritance of adjacent variants, which are separated primarily by recombination, but other factors such as population growth and admixture, natural selection, genetic drift and mutation, could affect LD patterns (Ardlie et al. 2002). Since linkage is focusing on families and rather small recombination events have taken place, the disease loci will often be large. In contrast, association analysis utilizes the recombination history over many hundreds or thousands of generations in

17
the population and disease loci would be comparatively small (reviewed in (Ardlie et al. 2002, Boehnke 1994)). Association between a marker and a phenotype could appear under two different circumstances, either by typing the causal allele itself (Figure 4A) or, more likely in complex diseases, by typing neighboring variants in LD (Figure 4B). However, in the latter case the power to detect association depends on the strength of the LD (Borecki and Province 2008). The statistical power in association studies is not only depending on the proximity of the marker and causal allele, but also on the contribution of a specific allele to the phenotype (effect size), allele frequencies and sample size. At weak effect sizes, low allele frequencies or modest LD, there is a need for large sample sets. Closely located genetic markers, showing strong intermarker LD, are often transmitted as haplotype blocks and different blocks are suggested to be separated by recombination hot-spots (Daly et al. 2001). LD patterns across the human genome have been characterized in multiple populations by the HapMap project (see 1.2.3), which has made it possible to detect association with only a few tagging SNPs genotyped when LD is high. However, in regions with low LD, dense SNP maps and higher numbers of genotyped markers are needed to find potential effects. The evolutionary history of LD patterns and haplotypes can vary in different ethnical populations (Gu et al. 2007, Service et al. 2006). It has been suggested that the choice of particular ethnic groups might facilitate association studies as these might arise from limited numbers of founders and would provide less disease heterogeneity and larger regions of LD. Sardinia, Iceland and certain areas of Finland, have all been considered as suitable (Peltonen 1996). Figure 4. (A) Direct association analysis by genotyping the causal SNP. (B) The causal SNP is tested indirectly by genotyping neighboring SNPs in linkage disequilibrium (LD). Case-control study design has been the most widely applied strategy of association analysis. Briefly, a number of unrelated affected and unaffected from a population has been collected, a set of markers at a locus or loci are genotyped, and subsequently, the genotypes are tested to evaluate their frequencies between cases and controls (McCarthy and Hirschhorn 2008). The selection of controls is crucial because any systematic allele frequency differences between cases and controls can appear as disease association, even though they may reflect differences in for example evolutionary or migratory history. Therefore, controls must be carefully matched to reflect the ethnic and genetic composition of cases. In order to reduce the effect of

18
population stratification, various family-based association approaches have been developed that use controls selected from the families of affected probands (Cardon and Palmer 2003). At present, one of the most popular approaches is the transmission disequilibrium test (TDT), providing a joint test of linkage and association, focusing only on heterozygous parental genotypes and is applied to single probands and parents (Spielman et al. 1993). TDT compares the frequencies of transmitted vs untransmitted alleles in affected offspring and uses untransmitted parental alleles as controls. Limitations of TDT include not utilizing all genotype data since transmissions from homozygote parents are not used and not being applicable for extended pedigrees. Pedigree disequilibrium test (PDT) is an extension of TDT that evaluates evidence of linkage disequilibrium (LD) in general pedigrees by using data from both related nuclear families and discordant sib pairs (Martin et al. 2000). To evaluate association results and get conclusive proof of an effect, associated variants need to be replicated in several independent populations. The SNP showing association in a study is often not the causal one and instead only reflects existing LD between variants. It is a difficult task to identify a causal allele in a susceptibility gene or region, thus the ultimate proof that a gene is connected to a disease phenotype is the identification of a variant clustering in affected subjects and with a plausible effect on normal gene function, affecting for example protein function, gene expression or mRNA splicing etc. Variants in coding regions affecting amino acid sequences or at splice sites can generally easily be assessed and mutational screening is often exon-centric. However, in complex diseases it is highly plausible to find causal variants in regulatory regions, such as the promoter, in introns or at more distal regulatory element that could be find several kb away from the gene (Frazer et al. 2009). Other variants with an likely effect on disease could affect localization, stability and translation of mRNA. Functional studies are required to determine the consequences of a causal allele. 1.3.2.1 Genome-wide association - Past and future
GWASs allow a hypothesis free approach for finding novel loci and genes associated with diseases. This has been facilitated by the development of tools and methods required, such as mapping of millions of common variants in the human genome and LD information, techniques to capture all of those variants in thousands of individuals, analytical approaches to handle huge amount of data and to distinguish true associations. Although, several new candidate genes and loci have been suggested, some replicated and accepted, these only explain a small proportion of the observed phenotypic variation (reviewed in (Manolio, 2009)). Additionally, GWAS are still missing for many complex diseases such as pre-eclampsia. For the published GWAS, it is unlikely that common variants of large effect have been missed, however, effect sizes for common variants are typically modest, often between 1.1-1.3 (Wray et al. 2008), and achieving enough power for a common SNP with 20% frequency requires over 8000 samples (Altshuler et al. 2008). Thus, most of the original GWAS were clearly underpowered. The importance of less common SNPs (MAF of 0.5-1%) with modest effects, which are not well covered by current GWAS chips that are restricted to high frequency variants, and variants to rare to prove statistical evidence of association, must be considered. Fine-mapping around GWAS hits, denser SNP panels and gene-centric

19
approaches could be an alternative that will capture variants not always represented on GWAS platforms and resulting in higher locus risk than estimated for the original GWAS. Data from the 1000 genomes project (see 1.2.4) will aid further identification of less common variants (both SNPs and structural variants) and a more comprehensive catalog of genomic variation. In order to identify disease specific rare variants influencing risk, the ultimate approach would be deep sequencing in thousands of cases and controls, but not the most cost-effective. There are many aspects to consider such as how to select the most appropriate cases, how many to sequence per group and is it possible to lower the coverage and still getting useful information. Hopefully, some knowledge will be gained from the first pilot projects that will be helpful for guidance of future studies. It is most likely that common diseases are affected by gene-gene and gene-environment interactions. However, these are difficult to identify with current methods and data set sizes and further investigations are needed (Altshuler et al. 2008). Large scale deletions and insertions, known as CNVs (see 1.2.2.3), are known to account for a substantional fraction of human genetic variation (Conrad et al. 2009, Hurles et al. 2008), and have been shown to play a role in human evolution and variation in gene expression (Perry et al. 2007, Stranger et al. 2007). Therefore, the involvement of CNVs in common diseases is highly plausible. The SNP microarrays that have been used in GWAS can be used to detect a small portion of CNVs indirectly, however, the mast majority remain invisible. Instead, high-resolution tiling arrays have been used for exploring CNVs in some areas, but they break down for the large fraction containing repetitive elements (Emanuel and Saitta 2007). The new generation of genotyping arrays already contains some CNVs, and the 1000 genomes project will be essential for developing arrays with even better coverage of the structural variants (McCarroll 2008). Epigenetics represent inherited information not carried in the DNA, usually in the form of chemical modification of DNA without changing the sequence and may influence human disease risk (reviewed in (Soejima 2009)), but are not detectable by GWAS, since the technology is based on DNA sequence and epigenetics needs to be tackled with a new type of high-throughput technology. Despite the failure to uncover the majority of genetic risk for common diseases, GWAS has contributed substantially to our understanding of disease mechanisms and we are now approaching the “post-GWAS era”, collectively with new challenges. An explosion of targeted deep sequencing will be seen in near future and finally, complete sequencing of hundreds to thousands of individuals will hopefully help us to pinpoint new disease variants and give us further insight into pathophysiological pathways of complex diseases. 1.4 GENETICS OF PRE-ECLAMPSIA
The identification of genes predisposing for pre-eclampsia has not been very successful over the years, with no universally accepted susceptibility gene. However, both large epidemiological and family studies demonstrate genetic contribution to the pre-eclampsia susceptibility. Genome-wide linkage analysis is one strategy for identifying candidate loci. So far, three loci showing significant linkage to pre-eclampsia have been identified on chromosomes 2p13, 2p25 and 9p13 (Arngrimsson et al. 1999, Laivuori et al. 2003) and evidence of suggestive linkage has been reported for eight additional chromosomal loci (Arngrimsson et al. 1999, Harrison et al. 1997, Hayward et al. 1992,

20
Lachmeijer et al. 2001, Laivuori et al. 2003, Moses et al. 2000). A summary of all loci can be found in table 3. Although several loci have been mapped, only a few promising positional candidate genes have been identified so far. Parent-of-origin effect to the storkhead box 1 gene (STOX1) on chromosome 10 was suggested in the Dutch population (see 1.4.2.3) and evidence for association has been reported in two separate populations for activin receptor type II (ACVR2A) at 2q22 and endoplasmic reticulum aminopeptidase 2 (ERAP2) at 5q (see 1.2.2.2). (Fitzpatrick et al. 2009, Roten et al. 2009). Several genes have been suggested to mediate susceptibility based on functional properties and a number of studies have reported association to genes encoding for coagulation factors, oxidative stress and vasoactive substances, but attempts to replicate these findings have yielded inconsistent results. The lack of consistent reproducibility and convincing results has been observed for many complex disorders and pre-eclampsia is no exception. Possible explanations include genetic heterogeneity, phenotyping, possible involvement of fetal genes and study power (reviewed in (Chappell and Morgan 2006)). Efforts are required to carefully select the study population and to minimize phenotypic differences. Several classifications of pre-eclampsia are used still used and the definition does not always have to reflect the different pathogenic mechanisms. For example, it has been suggested that familial and sporadic representation of pre-eclampsia have different genetic background (Oudejans et al. 2007). Furthermore, the severity of the disorder as well as involvement of various organs is highly variable between women. Most of the genetic studies in pre-eclampsia until today have focused on maternal genes, but it is possible that both maternal and fetal genes play a role and it is not possible to distinguish between these using a case-control design. Although there are few genes and gene variants that look promising, allelic and locus genetic heterogeneity can be a plausible explanation for the lack of replication. Finally, larger studies of statistical power to detect small genetic effects are needed in combination with comprehensive SNP typing in order to pinpoint the causal allele to reliably identify or exclude susceptibility genes in pre-eclampsia.
1.4.1 Genetic susceptibility
The incidence of pre-eclampsia is three times higher among women with a family history of pre-eclampsia than among women without, suggesting a strong familial predisposition (Esplin et al. 2001). The past research has focused primarily on the maternal factors that predispose to pre-eclampsia. However, the observation that a high number of monozygotic twin sets are discordant for the development of pre-eclampsia during their own pregnancies suggests that environmental factors and the fetal genotype are also important in determining susceptibility (Thornton and Macdonald 1999, Thornton and Onwude 1991, Treloar et al. 2001). A study including both monozygotic and dizygotic twin pairs estimated that genetic and environmental effects are of equal importance (Salonen Ros et al. 2000). Additionally, Cnattingius et al analyzed over 200 000 sibling pairs and their pregnancy outcomes and estimated that genetic factors contribute to the incidence of pre-eclampsia for more than 50% of the total phenotypic variance, with 35% of this variance originating from maternal effects and 20% from fetal effects combining genetic factors originating from both parents (Cnattingius et al. 2004). The contribution of the paternal genotype to pre-

21
eclampsia is supported by large database studies. One of them shows that mothers who become pregnant by a man who has fathered a pre-eclamptic pregnancy in another woman have nearly a double risk in their own pregnancies (Lie et al. 1998). Additionally, children of a man born from a pre-eclamptic pregnancy have twice the risk to be born from a pre-eclamptic pregnancy themselves than children of men born from an uncomplicated pregnancy (Esplin et al. 2001). Pre-eclampsia in the second-generation has also been studied and men born from a pre-eclamptic pregnancy have 1.5-fold increased risk of fathering a pre-eclamptic pregnancy (Skjaerven et al. 2005). No clear inheritance pattern of pre-eclampsia is known. Family studies designed to test different models of inheritance have reported inconclusive findings, including a single maternal recessive gene (Sutherland et al. 1981), homozygosity for a single recessive gene shared by mother and fetus (Liston and Kilpatrick 1991), and either recessive or dominant gene inheritance (Arngrimsson et al. 1990). Adding to the genetic complexity of pre-eclampsia, possible involvement of mitochondrial inheritance has been reported (Folgero et al. 1996). However, a further study has excluded direct contribution of mitochondrial genes to pre-eclampsia (Lie et al. 1998). Finally, it has also been proposed that the unclear inheritance pattern might be related to maternally or paternally imprinted genes (Graves 1998).
1.4.2 Susceptibility loci for pre-eclampsia identified by linkage 1.4.2.1 Chromosome 2p25 and 9p13
The most recent genome-wide scan in 15 Finnish pre-eclamptic multiplex families found two loci exceeding the threshold for significant linkage on chromosome 2p25 (NPL score 3.77; P=0.000761) and 9p13 (NPL score 3.74; P=0.000821 (Laivuori et al. 2003). Two other linkage studies from Iceland and Australia/New Zealand have reported significant linkage to this chromosome, but the 2p25 loci is clearly different from 2p12 and 2q22-23 (Arngrimsson et al. 1999, Moses et al. 2000). The families in the study were recruited predominantly from the Kainuu province in Finland, a known founder population, to reduce genetic heterogeneity. Significant linkage was only observed when a general diagnostic criteria (pre-eclampsia, eclampsia or GH without proteinuria) were used. However, it is likely that a woman in a pre-eclampsia family with GH shares genetic risk factors with her affected relatives with diagnosed pre-eclampsia. The chromosome 2p25 region directly overlaps a previously reported risk locus for general hypertension (Angius et al. 2002, Zhu et al. 2001). The gene for Rho-associated coiled-coil protein kinase 2 (ROCK2), previously implicated in general hypertension, is located in the region of overlap. ROCK2 is widely expressed, but appears to be most abundant in smooth muscle cells, suggesting a role in vascular contraction (Riento and Ridley 2003). Interestingly, expression levels of RhoA mRNA have been shown to be significantly higher in pre-eclamptic placentas compared to placentas from normotensive women (Friel et al. 2008). However, the gene did not show any evidence for association to pre-eclampsia in a Finnish population (Paper III) (Peterson et al. 2009). Another plausible candidate gene found in the 2p25 region is lipin 1 (LPIN1). LPIN1 gene encodes for phosphatidate phosphatase (PAP) enzyme required for glycerolipid biosynthesis (Carman and Han 2006). Several recent studies have

22
confirmed the relationship between adipose tissue lipin-1 levels and insulin sensitivity in humans (Reue 2009). Metabolic changes, such as reduced glucose utilization, hyperinsulinemia and hyperlipidemia are characteristic for pre-eclampsia, making LPIN1 an interesting candidate gene (Kaaja et al. 1999, Kaaja et al. 1995). The second significant finding at 9p13 has been shown to be a candidate region for T2D in two genome-wide studies from Finland and China (Lindgren et al. 2002, Luo et al. 2001). Additionally, chromosome 9p21, near the pre-eclampsia linkage borders, has shown association for both T2D and CAD in several GWAS during the past 3 years (Helgadottir et al. 2007, McPherson et al. 2007, Samani et al. 2007, Saxena et al. 2007, Scott et al. 2007, WTCCC 2007, Zeggini et al. 2007). There is a clear relationship between pre-eclampsia, CAD, and T2D. For example, pre-eclampsia predisposes women for elevated lifelong risk of CAD (Carpenter 2007, Rodie et al. 2004) and the risk of subsequent T2D is increased more than 3-fold after severe pre-eclampsia (Lykke et al. 2009). Therefore, it is possible that the three different disorders share genetic risk factors. 1.4.2.2 Chromosome 2q22 and 5q - ACVR2A and ERAP2
Moses et al identified a suggestive linkage region at 2q23 in Australian/New Zealand families and they concluded that their findings were consistent with the Arngrimssom et al study and therefore proposed that the linked locus should be designated the “PREG1” (pre-eclampsia, eclampsia gene 1) locus (Fitzpatrick et al. 2004, Moses et al. 2000). However, none of the other linkage studies have pointed to this region. The 2q locus was then strengthened and resolved further to 2q22 by fine-mapping and the application of a genetic linkage method assuming that the underlying liability of pre-eclampsia susceptibility is inherently quantitative (Moses et al. 2006). The same approach was used by Johnson et al for re-analysis of the original genome-wide scan data and led to the identification of two additional susceptibility regions at 5q and 13q (Johnson et al. 2007). Subsequently, a gene priorization strategy assigned the highest priority to the ACVR2A gene at 2q22, which is a receptor for the cell signaling protein activin A (Moses et al. 2006). Activin A is known to play a role in cell differentiation, implantation and decidualization (Caniggia et al. 1997, Jones et al. 2006). Evidence for association was found for ACVR2A in both an extended cohort of the Aus/NZ families and an independent Norwegian case-control cohort (Fitzpatrick et al. 2009, Roten et al. 2009). At the 5q locus, several genes were investigated and significant association for the endoplasmatic reticulum aminopeptidase 2 (ERAP2) was found in both families and Norwegian case-control data set (Johnson et al. 2009). The ERAP2 gene encode for an enzyme that play a role in blood pressure regulation, in addition to a suggested role in innate immune and inflammatory responses, which are known pathways involved in pre-eclampsia pathophysiology (Johnson et al. 2009, Tanioka et al. 2003). 1.4.2.3 Chromosome 10q22 - STOX1
Lachmeijer et al studied affected sib-pairs families and found suggestive linkage with pre-eclampsia to chromosomes 10q22 and 22q12 (Lachmeijer et al. 2001). The 10q22 locus was confirmed and maximal allele sharing was shown between pre-eclamptic sisters for the maternal, but not paternal alleles (Oudejans et al. 2004). Van Dijk et al identified five different missense mutations in the gene STOX1 at 10q22, co-

23
segregating with pre-eclampsia in the families, identical between affected sisters and following matrilineal inheritance (van Dijk et al. 2005). One of the mutations, Y153H, was suggested to be highly mutagenic according to conservation analysis of the protein. They hypothesize that STOX1 could be involved in the transition process of noninvasive to invasive extravillous trophoblasts, known to be central in normal placental development. However, Iglesias-Platas et al showed that expression of STOX1 was very low in placentas and invading trophoblastic cells, and detected biallelic expression in both normal and pre-eclamptic placentas (Iglesias-Platas et al. 2007). Additionally, two other studies investigating STOX1 variants in Dutch and Finnish (Paper I) populations failed to detect any association to pre-eclampsia (Berends et al. 2007, Kivinen et al. 2007). However, Rigourd et al suggested that STOX1 should still be considered as a key factor in pre-eclampsia, since their study strongly suggests that STOX1 plays a major part in normal placental development (Rigourd et al. 2008). They hypothesize that STOX1 is able to act directly on specific gene targets and induce a transcriptional profile approximating that observed in pre-eclampsia. A recent study comparing gene expression of early placentas (10-12 weeks of gestation) of women later developing pre-eclampsia or leading to a normal gestation, showed that STOX1 was induced 2-fold in pre-eclampsia, suggesting that a dysregulation of STOX1 occurs early in pre-eclampsia (Founds et al. 2009). 1.4.2.4 Other reported linkage based studies
The first genome-wide study for pre-eclampsia was designed as an exclusion map, and suggested a role for a gene or genes on chromosomes 1, 3, 9 and 18 (Hayward et al. 1992). The second scan suggested the presence of a candidate region on chromosome 4q34 in an Australian population (Harrison et al. 1997). An Icelandic study, comprising a large number of affected women, revealed a maternal susceptibility locus for pre-eclampsia on chromosome 2p13 and was the first one to meet the criteria for genome-wide significance (Arngrimsson et al. 1999). However, findings were mostly supported by two large pedigrees and exclusion of those lead to identification of a suggestive locus on 2q23, the same region later found linked to pre-eclampsia in Australian/New Zealand families, where ACTRV2 was identified as a susceptibility gene. Additionally, Angrimsson el al has reported linkage to both the Angiotensinogen gene (AGT) at 1q42-43 and Endothelial nitric oxide synthase (eNOS3) at 7q36 in two separate studies before their whole-genome screen was published (Arngrimsson et al. 1993, 1997). Table 3. Summary of the genome-wide linkage studies in pre-eclampsia.
Country Criteria Markers Families Chromosome with nominal Chromosome with S=Strict G=General or suggestive linkage significant linkage Scotland S 43 35 3, 9 Australia S, G 90 15 4q34 (G) Iceland S, G 440 124 2q23 (G) 2p13 (G) Aus,NZ S, G 400 34 2q23 (S), 11q23 (G) Netherlands S, HELLP 293 67 10q, 22q (S), 12q (HELLP) Finland S, G 435 15 4q32 (G) 2p25 (G), 9p13 (G) (Aus, NZ) Australia, New Zealand (HELLP) Hemolysis, elevated liver enzymes, and low platelets

24
1.4.3 Association-based mapping approaches
Candidate gene studies have failed to identify universally accepted susceptibility genes for pre-eclampsia, but studies of more than 50 candidates have been reported. However, around 70% of the published studies have focused on only eight genes (Factor V Leiden (F5), Methylenetetrahydrofolate (MTHFR), Prothrombin (F2), Angiotensin converting enzyme (ACE), Angiotensin II type 1 and type 2 receptor (AGTR1, AGTR2), Angiotensinogen (AGT), Endothelial nitric oxide synthetase 3 (eNOS3) and Tumor necrosis factor α (TNFα) (reviewed in (Chappell and Morgan 2006, Mutze et al. 2008)). The candidate genes studied belongs to different groups according to their functional properties and plausible role in the pathophysiology. The different subgroups and most important genes are reviewed below and are summarized in table 4.
1.4.3.1 Trombophilia
Since several studies have shown that pre-eclampsia is associated with underlying thrombophilic disorders (Dekker 2005, Kupferminc et al. 1999, van Pampus et al. 1999), placental interactions and villous thrombosis are characteristic for pre-eclampsia, genes in this category have been suggested as candidates and have been investigated in a large number of association studies. F5, MTHFR and F2 are three of the most widely studied genes in pre-eclampsia and thrombophilia mutations have been identified in those genes in pre-eclampsia, but several studies have also provided contradictory results (reviewed in (Mutze et al. 2008). However, a meta analysis reported by Lin et al, including all studies for F5 up to November 2002, showed a 2-fold increase in pre-eclampsia risk for a 1691G>A mutation, but no effect for mutations in MTHFR or F2 (Lin and August 2005). To date, the number of studies not confirming the mutations as pre-eclampsia risk factors for all three genes is much higher than the ones confirming. A 4G/5G deletion/insertion polymorphism of the plasminogen activator factor-1 gene (PAI-1), an inhibitor of fibrinolysis, has been associated to pre-eclampsia, but several replication attempts have failed (Dalmaz et al. 2006, Fabbro et al. 2003, Gerhardt et al. 2005, Morrison et al. 2002). The β integrin protein has a suggested role in the process of cytotrophoblast failing to adopt a vascular phenotype in pre-eclampsia (Zhou et al. 1997). Therefore, the Integrin glycoprotein IIIa gene (GPIIIa), encoding for a subunit of the β integrin, has been investigated, but with conflicting results (O'Shaughnessy et al. 2001, Pegoraro et al. 2003).
1.4.3.2 Hemodynamics, endothelial function and angiogenesis
The renin-angiotensin-aldosterone system (RAAS) is known to play an important role in regulation of renal function and arterial pressure during pregnancy and several studies have implicated the RAAS in pre-eclampsia pathophysiology (reviewed in (Shah et al. 2006). Therefore, genes in this system have been considered as plausible candidates for pre-eclampsia (Gilbert et al. 2008). ACE, AGTR1, AGTR2, and AGT have all been studied extensively in pre-eclampsia with inconsistent results (reviewed in (Mutze et al. 2008)). A gene associated to RAAS is the eNOS3, widely expressed in placental tissue (Myatt et al. 1991), but with reduced activity in pre-eclamptic placentas (Brennecke et al. 1997), which could lead to reduced vasodilation, and vascular remodeling. Association studies in populations with different ethnical background have yielded both positive and negative findings. A meta-analysis including studies up to

25
2006 investigating a E298D polymorphism, initially associated to pre-eclampsia in Colombian women, did not report an increased risk for that particular polymorphism (Medica et al. 2007, Serrano et al. 2004). Vascular endothelial growth factor (VEGF) is an angiogenic factor and plays an important role in endothelial cell proliferation, migration, survival and regulation of vascular permeability. Soluble fms-like tyrosine kinase-1 (sFlt-1) is the soluble form of the VEGFR-1, an antagonist for VEGF, and up-regulated in pre-eclampsia (Levine et al. 2004, 2006, Maynard et al. 2003, Staff et al. 2005, Thadhani et al. 2004). Interestingly, during placental development, the expression of VEGFR-1 is not only detected in vascular endothelial cells, but also in the developing trophoblasts (Clark et al. 1996) and it has been shown that increased sFlt-1 levels can be detected approximately five to six weeks before the onset of clinical symptoms of pre-eclampsia. The number of studies that has investigated SNPs in the genes involved in the VEGF system is very small. Two polymorphisms of the VEGF, 405G>C and 936C>T were found associated to the severe form of the disease in two small studies (Banyasz et al. 2006, Papazoglou et al. 2004) and are not considered as major risk factors.
1.4.3.3 Oxidative stress and lipid metabolism
Placental oxidative stress, which is the result of abnormal placentation, reduced blood flow and subsequently a hypoxic placenta, is reported to play a central role in the pathogenesis of pre-eclampsia (Gupta et al. 2005). Several genes involved in the generation of ROS could, if they are defective in some way, increase endothelial dysfunction through lipid peroxidation. Despite the strong correlation between oxidative stress and pre-eclampsia, only a few genes have been investigated. Polymorphisms in the genes microsomal epoxide hydrolase (EPHX) that catalyzes the hydrolysis of certain oxides and may produce toxic intermediates that could be involves in pre-eclampsia, and glutathione S-transferase (GST), a protein protecting cells against ROS, have shown association. However, conflicting results have been reported (Canto et al. 2008, Gebhardt et al. 2004, Laasanen et al. 2002, Ohta et al. 2003, Zusterzeel et al. 2000, 2001). Lipid metabolism is associated with oxidative stress and abnormal lipid profiles have been observed in pre-eclampsia (Sattar et al. 1997). Lipoprotein lipase (LPL) and Apolipiprotein E (ApoE) are the two major regulators of lipid metabolism, abundantly expressed in placenta and have therefore been proposed as possible candidate genes (Descamps et al. 2005, Kim et al. 2001). A missense mutation Asn291Ser in LPL seems to be the most promising variant, since it is associated with lowered LPL activity and increased dyslipidemia in two separate studies. However, others have failed to replicate these findings (Hubel et al. 1999, Kim et al. 2001, Zhang et al. 2006). It has also been reported that the fetal genotype of the two genes could contribute to the metabolism of maternal lipoproteins (Descamps et al. 2005).
1.4.3.4 Immunoregulation
It has been suggested that a maternal-fetal (paternal) immune maladaptation is central in pre-eclampsia pathogenesis and genes encoding for various factors of the immune system, therefore, appear to be good candidates. Several HLA molecules have been

26
suggested, but the data remains inconclusive (reviewed in (Saftlas et al. 2005)). TNFα, a pro-inflammatory cytokine, has been implicated in the etiology. It has been shown that an excessive release of TNFα by activated macrophages and monocytes may contribute to endothelial activation and damage that later result in the maternal syndrome of pre-eclampsia (LaMarca et al. 2007). Interestingly, TNFα induces hypertension in pregnant, but not in non-pregnant rats (Alexander et al. 2002) and plasma levels of TNFα in pre-eclamptic women are significantly higher compared to matched controls (Sharma et al. 2007). TNF is also involved in the generation of ROS and subsequently oxidant-mediated endothelial cell injury (Chen et al. 1996). One of the most commonly studied variants in pre-eclampsia is -308G>A transition in the promoter region that is associated with higher TNF levels and an increased risk for pre-eclampsia linked disorders T2D, CAD and dyslipidemia (Elahi et al. 2009, Saarela et al. 2005). A meta-analysis from 2008 combined sixteen studies investigating the promoter SNP, but did not detect significant association to pre-eclampsia (Bombell Mcguire 2008). Several cytokines are produced at the maternal-fetal interface and have an impact on trophoblast invasion. It has been suggested that deficiency of IL-10 may contribute to an enhanced inflammatory response towards trophoblast cells, resulting in defective transformation of spiral arteries, hypoxia, thrombosis and infarction of the placenta (Renaud et al. 2007). The gene expression of IL-10 is reduced in pre-eclamptic placentas (Makris et al. 2006) and studies have investigated variants in the gene, but with conflicting results (Daher et al. 2006, Goddard et al. 2007, Haggerty et al. 2005, Kamali-Sarvestani et al. 2006). Association has been detected to two additional pro-inflammatory genes, Interleukin 1α (IL-1α ) and the interleukin 1 receptor antagonist (IL1Ra), but few studies have addressed the role of polymorphisms in the genes so far (Faisel et al. 2003, Haggerty et al. 2005).
1.4.3.5 Placentation
Defective placentation is known to be central in pre-eclampsia pathophysiology, as a result of defective transformation of spiral arteries, reduced blood flow through the placenta and subsequently ischemia. Therefore, placental genes appear to be good candidates, however, not many genes have been studied and the results are not convincing (reviewed in (Roberts and Cooper 2001)). Expression studies of pre-eclamptic placentas show evidence for matrix metalloproteinases (MMPs) and apoptosis related gene, both with a possible implication in trophoblast invasion (Pang and Xing 2003, 2004, Sitras et al. 2009). However, association studies of those genes are very limited and effects of variants in the genes need to be further investigated. The best candidate that can be implicated in placentation to date is STOX1 that was positionally cloned following a genome-wide linkage scan (see 1.4.2.3). Interestingly, a study by Rigourd et al suggests that STOX1 plays a major part in normal placental development and is able to act directly on specific gene targets, and induce a transcriptional profile approximating that observed in pre-eclampsia (Rigourd et al. 2008). It is plausible that genes working in the same pathway as STOX1 could influence pre-eclampsia risk as well. One interesting candidate is glial cells missing homolog 1 (GCM1), which is a crucial transcription factor for placental development, down-regulated in pre-eclamptic placentas (Chen et al. 2004) and there is evidence suggesting that GCM1 could be under regulation of STOX1 at both a transcriptional level and by proteasome-mediated post-translational degradation (Rigourd et al. 2009).

27
Table 4. Summary of investigated functional candidate genes in pre-eclampsia confirmed at least in one study Gene name (symbol) Trombophilia Immunoregulation Factor V Leiden (F5) Tumor necrosis factor α (TNFα) Methylenetetrahydrofolate (MTHFR) Interleukin 1 receptor antagonist (IL1Ra) Prothrombin (F2) Interleukin 1α (IL-1α) Plasminogen activator factor-1 (PAI-1) Interleukin 10 (IL-10) Integrin glycoprotein IIIa (GPIIIa) T-lymphocyte-associated protein 4 (CTLA4) Oxidative stress and lipid metabolism Hemodynamics Aplolipoprotein E (ApoE) Angiotensinogen (AGT) Lipoprotein lipase (LPL) Angiotensin converting enzyme (ACE) Microsomal epoxide hydrolase (EPHX) Angiotensin II type 1 receptor (AGTR1) Glutathione S-transferase (GST) Angiotensin II type 2 receptor (AGTR1) Endothelial function and angiogenesis Endothelial nitric oxide synthetase 3 (eNOS3) Vascular endothelial growth factor receptor 1 (VEGFR-1) Vascular endothelial growth factor (VEGF)

28
2 AIMS OF THE THESIS The overall aim of this study was to search for genes that may predispose to pre-eclampsia. The specific aims were: I. To asses the previously published pre-eclampsia candidate STOX1 for
associations in Finnish population. (Paper I) II. To evaluate previously published type 2 diabetes (T2D) and coronary artery
disease (CAD) susceptibility loci on chromosome 9p21 for increased pre-eclampsia risk. (Paper II)
III. Fine-map the previously implicated pre-eclampsia susceptibility locus on
chromosome 2p25 and identify the most likely susceptibility gene within the region. (Papers III and IV)

29
3 MATERIAL AND METHODS
3.1 STUDY SUBJECTS
Individuals were recruited mainly from Finland, but samples from United Kingdom and Australia/New Zealand were utilized in Paper II. The number of families and independent cases and controls from each population used in each study are presented in Table 5 and the study materials are described in detail below. Table 5. Sample collections and numbers of individuals used in each of the studies
Finland United Kingdom Australia/New Zealand
Genomic DNA Placental samples Genomic DNA Genomic DNA
Families Case-controls Case-controls Case-controls Families 15 340/357 8/6 251/198 74
Study I-IV Study I-IV Study I-IV Study II Study II
3.1.1 Finnish families (I-IV)
In total, the family cohort consisted of 15 multiplex families recruited predominantly from the Kainuu province in central eastern Finland and in the Helsinki region for the original genome-wide scan that formed the basis of this thesis. The families (174 individuals) were recruited through pre-eclampsia probands by sending letters to patients who had been hospitalized with the diagnosis of severe pre-eclampsia in their first pregnancies in Kainuu Central Hospital (Kajaani, Finland) or the Department of Obstetrics and Gynecology, Helsinki University Central Hospital (Helsinki, Finland) and by advertisement for the study, between 1988 and 1997. Ten multiplex families were recruited from Kainuu and five from Helsinki. To verify the origin of families, the birthplaces of parents and grandparents were traced by using church records. The reproductive medical records of all women were reviewed to confirm or exclude the diagnosis. Pre-eclampsia was defined as systolic blood pressure >140 mm Hg or diastolic blood pressure >90 mm Hg in a woman who was normotensive before 20 weeks of gestation, and the urinary excretion of ≥0.3 g protein in a 24-h specimen or ≥1+ reading on dipstick in a random urine determination at least twice with no evidence of urinary tract infection. For more detailed description of definitions, see (Laivuori et al. 2003). In families that included at least one patient with pre-eclampsia, women with gestational hypertension (GH) without significant proteinuria were considered to have mild pre-eclampsia under the general diagnostic criteria. In total, fifty women met the criteria of pre-eclampsia or eclampsia and severe pre-eclampsia was diagnosed in 29 of these 50 women (58%). Thirteen women met the criteria of GH in their first pregnancies. Each family was ascertained on the basis of at least one proteinuric pre-eclampsia case. Clinical characteristics for patients are summarized in Table 6.

30
3.1.2 Finnish case-controls (I-IV)
A nation-wide sample set consisting of 340 cases and 357 controls were recruited by combining the National Hospital Discharge register with diagnosis classified according to International Classification of Diseases (ICD-10 1996) with The National Register of Blood Groups and Blood Group Antibodies of Pregnant, from which data were obtained for 100.000 consecutive pregnancies during 1997–1998. In the cohort of 100.000 pregnancies, 686 were picked as case-candidates, medical records were reviewed by two clinicians and pre-eclampsia was defined according the definitions of the NHBPEP (NHNPEP, 2000). Phenotypes were ascertained by two clinicians and further subdivided into pre-eclampsia (N=245), eclampsia (N=5), superimposed pre-eclampsia (N=46) and GH without proteinuria (N=44). Controls were matched with cases for the province of residence, number of pregnancies, age of mother and number of fetuses. The cases and controls were invited by letters and reminders. All participants gave their written informed consent and the study was approved by the ethics committee of Finnish Red Cross Blood Service, and Ministry of Social Affairs and Health. For more detailed description of the recruitment process and diagnostic criteria see (Hiltunen et al. 2009). Clinical characteristics for cases and controls are summarized in Table 6. Table 6. Clinical characteristics for the two Finnish sample sets studied in this thesis
Finnish families Finnish case-controls Strict General Strict General
Cases (N=50) Cases (N=63) Cases (N=250) Cases (N=294) Controls (N=357)
Mean ±SD Mean ±SD Mean ±SD Mean ±SD Mean ±SD
Age (years) 27.2 ± 4.4 26.7 ± 4.4 28.9 ± 5.59 29.1 ± 5.57 28.8 ± 4.99
Body mass index (kg/m2)* 21.2 ± 2.3 21.4 ± 2.4 24.2 ± 4.6 24.3 ± 4.5 22.8 ± 3.6
Systolic BP (mmHg) 156.5 ± 27.1 155.8 ± 24.8 169.8 ± 16.4 168.3 ± 16.4 NA
Diastolic BP (mmHg) 103.6 ± 16.3 103.5 ± 14.8 105.6 ± 9.5 105.3 ± 9.2 NA
Proteinuria (g/24h) ** NA 4.3 ± 4.2 NA NA
Birth weight 2482.3 ± 929.2 2615.5 ± 895.8 2622.3 ± 835.6 2709.3 ± 847.9 3569.2 ± 410.4
(Strict criteria) Women with pre-eclampsia or eclampsia (General criteria) Women with pre-eclampsia, eclampsia or gestational hypertension without proteinuria ** ≥ 0.3 g protein in 24-h specimen or ≥ plus 1 or more reading on dipstick in a random urine determination at least twice with no evidence of urinary tract infection. * Pre-pregnancy body mass index
3.1.3 UK case-controls (II)
The sample set consisting of 102 women with pre-eclampsia and 198 women with healthy normotensive pregnancies that were recruited during pregnancy in maternity units in Nottingham, UK. All patients were of white Western European origin and were recruited according to a definition from the International Society for the Study of Hypertension in Pregnancy (ISSHP) (Brown et al. 2001). A further 149 cases were white Western European women recruited from 25 UK hospitals, using the same diagnostic criteria, to a study investigating prophylactic oral vitamins C and E in women at high risk of pre-eclampsia. In these women, a history of eclampsia or pre-eclampsia in a previous pregnancy leading to delivery before the 37th week of gestation was used as the sole risk factor for pre-eclampsia. Exclusion criteria applied to both

31
cases and controls were: previous chronic hypertension, renal or cardiovascular disease, hydatidiform mole in the index or previous pregnancy and multiple pregnancy. Detailed information for the two sample collections can be found here (Morgan et al. 1999, Poston et al. 2006).
3.1.4 Australian/New Zealand families (II)
The cohort consists of 480 individuals from 74 pre-eclampsia families recruited over a period of 15 years through the Royal Women's Hospital and the Monash Medical Centre in Melbourne, Australia, through print and electronic advertisements in Sydney and through the National Women's Hospital in Auckland, New Zealand. Thirty-four of the families have been used to identify a susceptibility locus for pre-eclampsia on chromosome 2q22 locus (Moses et al. 2000). In summary, the cohort included 20 women with eclampsia, 120 women with pre-eclampsia, 56 women who had GH without proteinuria, and 90 women who had normal pregnancies. Diagnosis was based on clinical assessment, using criteria of the ASSHP (ASSHP 1993). Women who met these criteria and experienced convulsions or unconsciousness in the prenatal period were classified as having eclampsia. Women with the pattern of hypertension outlined above but with no proteinuria were classified as mild pre-eclampsia. Women with pre-existing hypertension or other medical conditions known to predispose them to pre-eclampsia (e.g. renal disease, diabetes, twin pregnancies or fetal chromosomal abnormalities) were excluded. Ethical approval was obtained from the research and ethics committees of the Royal Women’s Hospital, Melbourne, and written informed consent was obtained from family members.
3.1.5 Finnish placental samples (I-IV)
Placental samples were obtained immediately after delivery from eight primiparous pre-eclamptic women and six primiparous controls at the Department of Obstetrics and Gynecology, Helsinki University Central Hospital, Finland. Samples were included only if there was no indication of diabetes, renal disease, twin pregnancy, smoking, or any other condition known to affect pre-eclampsia risk or placental function. Pre-eclampsia was defined according to the NHBPEP (NHBPEP 2000). The biopsies were taken in a separate room dedicated for handling of tissue samples, snap frozen in liquid nitrogen and stored at -80°C until further analysis.
3.1.6 Finnish control samples (II)
A Finnish population-wide sample set consisting of 260 male blood donors who had given informed consent and were collected through the Finnish Red Cross Blood Service. This sample set was collected for the study of population genetic variation in Finland (Salmela et al. 2008).

32
3.2 GENETIC ANALYSIS
3.2.1 Marker selection (I-IV)
In Paper IV, microsatellite markers in the 2p25 linkage region were selected primarily from the deCODE genetic map (Kong et al. 2002). The Marshfield (http://research. arshfieldclinic.org/genetics/) genetic map was used to fill the gaps when no deCODE markers were available. The order and physical distances between markers were established using the NCBI (http://www.ncbi.nlm.nih.gov/unists/) and Ensembl (http://www.ensembl.org/) public databases. SNPs in Paper IV were chosen from the SNP consortium (TSC) database and dbSNP based on availability and information content. Information regarding variant tagging properties and population frequencies were limited at the time the study was designed, but re-sequencing a genomic region allowed us to identify novel SNPs that were chosen for genotyping. Coding SNPs in STOX1 (I) were selected based on the work by van Dijk and others (van Dijk et al. 2005). An additional SNP was selected for the 3' end of the gene based on its tagging properties to cover the whole gene region. Tagging SNPs were chosen for the plausible candidate genes ROCK2 (III), TRIB2, GREB1, LPIN1 and E2F6 (IV) at the 2p25 locus. The SNP selection was performed in the CEU population genotype data downloaded from HapMap (http://www.hapmap.org) with the Tagger program implemented in Haploview (Barrett et al. 2005, de Bakker et al. 2005). Two SNPs in GREB1 were chosen based on previously reported association to hypertension (Kamide et al. 2005). Additionally, ROCK2 (III), LPIN1 and TRIB2 (IV) were re-sequenced in affected individuals from a subset of families contributing to the 2p25 linkage signal to increase our chances of identifying the most relevant functional variants and other novel variants. SNPs were selected for genotyping based on the sequencing results (non-synonymous coding, splice site, other functional SNPs, insertion/deletion poly-morphisms or observed allele frequency differences between cases and controls). Markers at the 9p21 locus (II) were selected based on previous GWA studies showing association for T2D and CAD, and tagging properties to cover majority of common variation within the region (Helgadottir et al. 2007, McPherson et al. 2007, Samani et al. 2007, Saxena et al. 2007, Scott et al. 2007, WTCCC 2007, Zeggini et al. 2007).
3.2.2 Genotyping: SNPs and microsatellites (I-IV)
SNPs and microsatellites were genotyped using Sequenom (I-IV) (Sequenom Inc. San Diego, CA, USA), TaqMan (II) (Applied Biosystems, Foster City, CA, USA), Illumina (II), Affymetrix (II) and MegaBACE (I, III, IV) (Amersham Biosciences, Uppsala, Sweden) platforms. The Sequenom methodology is based on matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry and it was our method of choice for SNP genotyping in all studies (I-IV). The typing procedure is based on an allele specific primer extension reaction of an oligonucleotide probe over a SNP site in a PCR product, with a mixture of deoxynucleotides (dNTPs) and dideoxynucleotides (ddNTPs), to produce different size products for each allele of a SNP. The extension products are analyzed by MALDI-TOF mass spectrometry, and the time-of-flight is proportional to mass, permitting precise determination of the size of products

33
generated, which can be converted into genotype information. This genotyping assay allows multiplexing of up to 40 SNPs simultaneously, where differentiation of genotypes for each of the SNPs in one assay is a result of unique mass ranges for the extension primers. An overview of the assay technology is shown in Figure 5. PCR assays and extension primers were designed using the SPECTROdesigner software (Sequenom). Extension products were identified by a MassARRAY mass spectrometer (Bruker, Daltonik GmbH, Bremen, Germany) and resulting mass spectra were analyzed for peak identification using the SpectroTYPER RT software (Sequenom Inc). Genotypes were verified independently by two investigators, and Hardy-Weinberg equilibrium (HWE) was assessed for each marker as a quality control procedure. To identify possible genotyping errors in the families, data were checked for Mendelian consistency using Pedcheck (O'Connell and Weeks 1998). Figure 5. Schematic overview of the Sequenom methodology based on allele specific primer extension over a SNP site in a PCR product and MALDI-TOF mass spectrometry. In Paper II, one SNP was genotyped using the TaqMan chemistry, which is based on commercially available on-demand SNP genotyping assays with fluorescent-labelled allele-specific probes provided by Applied Biosystems (Applied Biosystems, Foster City, CA, USA). The PCR reactions and allelic discriminations were performed using ABI PRISM 7900 Sequence Detection System instrument (Applied Biosystems, Foster City, CA, USA) at the Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, USA. In the same study (II) one SNP could not be genotyped with TaqMan, and Illumina’s GoldenGate assay was used instead. It is based on a combination of oligo ligation assay (OLA), allele specific extension reactions and universal PCR.

34
Automated sequencing was utilized to genotype microsatellites (IV) and SNPs (I, III, IV). The technique this method is based on is described in more detailed under the paragraph “DNA re-sequencing” (see 3.2.4). Genotyping of microsatellites was performed using PCR with fluorescently labeled primers for amplifying the microsatellite region, followed by capillary electrophoresis on a MegaBACE 500 sequencer (Amersham Biosciences). A labeled size standard was co-injected and allowed accurate size estimation of the microsatellites. Allele calling was carried out using the Genetic Profiler v 2.0 software. SNP genotyping was performed by direct sequencing of PCR products including the SNP site utilizing the DYEnamic ET Dye Terminator Cycle Sequencing Kit for MegaBACE DNA Analysis Systems (Amersham Biosciences) and a MegaBACE 1000 instrument. The sequences were analyzed using the MegaBACE Sequence Analyser software (3.0) (Amersham Biosciences) and the Staden Package computer programs Pregap4 and Gap4 (http://staden.sourceforge.net/). 3.2.3 Data analysis 3.2.3.1 Linkage analysis (IV)
In Paper IV, linkage was assessed by the affecteds-only non-parametric multipoint linkage (NPL) analysis method using Genehunter 2.1 software, which performs reconstruction of haplotypes and complete multipoint analysis of allele sharing IBD among all affected family members at each location in the genome (Kruglyak et al. 1996). 3.2.3.2 Association analysis
Both family (II, IV) and case-control (I-IV) based association analysis have been performed using several different statistical approaches depending on the study design. Power calculations (1-IV) We used the Genetic Power Calculator program (http://pngu.mgh.harvard.edu/~pur ell/gpc/) and the case-control for discrete traits option to evaluate our ability to detect association to certain markers in the case-control cohorts (Purcell et al. 2003). We used pre-eclampsia prevalence of 8% as reported previously in the Finnish population (Kaaja et al. 2005). Haplotype pattern mining (IV) Haplotype pattern mining (HPM) was used in Paper IV for the initial association analysis of SNPs genotyped in the Finnish families at the 2p25 locus, when the markers were sparsely distributed over the studied region. HPM is a data mining-based algorithm that searches for shared patterns among haplotypes, scores each marker location for the number of shared patterns, and evaluates empirically the significance of the scores by a permutation test (Toivonen et al. 2000). The same genotype data was also analyzed using both TDT and PDT. Transmission and pedigree disequilibrium test (II, IV) TDT is the simplest and one of the best known family-based association tests using genotype data from trios, which consist of an affected offspring and the two parents. It

35
was first developed to test for linkage between alleles and phenotypes in qualitative traits (Spielman et al. 1993). TDT compares the frequencies of transmitted vs untransmitted alleles in affected offspring and uses untransmitted parental alleles as controls. In paper IV, we used TDT as implemented in the Haploview software 4.1 to analyze SNP genotype data from our Finnish families. One limitation of TDT is that, although it remains a valid test of linkage, it is not a valid test of association if related nuclear families and/or sibships from extended pedigrees, such as our Finnish families, are used. The PDT on the other hand can use data from extended pedigrees and is valid, just like TDT, even when there is population substructure (Martin et al. 2003). This test uses data from related nuclear families in combination with discordant sibships. Therefore, PDT and PDTPHASE, which is an extension of PDT that can deal with missing data and haplotype analysis, were used to analyze the family data in paper II and IV (Dudbridge 2003). Additionally, LD between markers in paper IV was estimated using PDTPHASE. Haploview (I-IV) The Haploview software was used to assess linkage disequilibrium (LD) between markers, calculate allele and haplotype frequencies, and to run single-marker and haplotype association tests in the Finnish (I-IV) and British (II) case-control materials (Barrett et al. 2005). The Finnish material has been subdivided into pre-eclampsia, eclampsia, superimposed pre-eclampsia and GH without proteinuria. Therefore, that sample set was analyzed by using different inclusion/exclusion criteria. We used chi-square as the test statistic and corrected the overall significance for multiple testing using 10,000 permutations. Haplotype blocks were defined according to the confidence interval (CI) of D’ as implicated in Haploview (Gabriel et al. 2002). Meta-analysis (II) In Paper II, combined Odds Ratios (OR) for marker rs7865618 were calculated by using data from all four pre-eclampsia materials. Mantel-Haenszel meta-analysis Forest plots were constructed using rmeta v2.14 on the R platform (http://www.r-project.org/) and significance was determined by a two-sided t-test.
3.2.4 DNA re-sequencing: Genomic and gene-centric mutational screening (I, III, IV)
The Sanger sequencing method is based on a technique utilizing 2',3'-dideoxy-nucleotide triphosphate (ddNTP) termination of in vitro replication of single stranded DNA (Sanger et al. 1977). A mixture of ddNTP and dNTP is utilized in sequence reactions and by including a DNA polymerase, elongation of a DNA strand will take place and terminates whenever a ddNTP is incorporated into the growing strand. ddNTPs terminate the elongation since they cannot form a phosphodiester bond with the next dNTP, resulting in DNA fragments of varying length that could be separated by size with gel electrophoresis. By fluorescently labeling the four different ddNTPs, automated sequencing was made possible with parallel termination for all ddNTPs followed by capillary electrophoresis. The automated sequencing in this study was performed utilizing the DYEnamic ET Dye Terminator Cycle Sequencing Kit for MegaBACE DNA Analysis Systems (Amersham Biosciences). All fragments were

36
PCR-amplified and sequenced in both directions. Purified sequencing products were resolved using a MegaBace 1000 instrument. The sequences were analyzed using the MegaBACE Sequence Analyser software 3.0 (Amersham Biosciences) and the Staden Package computer Programs; Pregap4 for preparing the sequence trace data before entry into the assembly program and Gap4 for analysing, since it allows careful and efficient alignment of all corresponding sequences (http://staden.sourceforge.net/). To explore the genetic variation in a 75 kb region at the 2p25 locus (IV), the non-repetitive DNA segments were PCR-amplified and re-sequenced in one pre-eclamptic woman, homozygous for the associated risk haplotype observed after the first round of SNP fine-mapping in Paper IV. The genes ROCK2 (III), LPIN1 and TRIB2 (IV) at the 2p25 locus were screened for polymorphisms by direct sequencing exons, including 100 bp flanking sequence on both sides, untranslated regions (UTRs) and 1 kb upstream of the first exon in genomic DNA from pre-eclampsia cases and controls. 3.3 EXPRESSION ANALYSIS
3.3.1 Microarray expression analysis (I-V)
Microarrays were used for genome-wide gene expression profiling in pre-eclamptic cases and controls. The expression data for the genes studied in paper I-IV are collected from the same study using placental samples from eight pre-eclamptic and six normotensive placentas analyzed on an Affymetrix GeneChip® Human Genome U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA, USA) for the analysis of over 47 000 transcripts simultaneously. Affymetric GeneChip® technology is based on oligonucleotide probes and complementary hybridization. Placental RNA was isolated using TRIzol kit (Invitrogen, Carlsbad, CA, USA) and purified with RNAeasy total RNA isolation kit (Qiagen, Hilden, Germany), 6 µg of total RNA was reverse transcribed, amplified, labeled according to the alternative protocol for one-cycle cDNA synthesis and hybridized on the chip. Arrays were stained and scanned according to Affymetrix protocols. Transcript levels were determined from data images with Affymetrix GeneChip® DNA analysis software (GDAS). Arrays were background corrected and normalised using GC Robust Multi-array Average (GCRMA) adjustment, and differential expression was assessed using linear models from Limma. Both software packages are available from the open-source Bioconductor project, and were used under the R environment for statistical computing (Gentleman et al. 2004).
3.3.2 Allele specific expression analysis (III)
Allele specific mRNA expression of ROCK2 in Paper III was assessed in three pre-eclamptic women heterozygous for marker rs17366517. The assay was based on the comparison of allelic signal intensities in cDNA and genomic sequence from each individual. RNA isolated from our placental samples was reverse-transcribed and both cDNA and placental genomic DNA from each individual were sequenced in six independent reactions, originating from three separate PCR amplifications. Signal

37
intensities in each channel were compared and an allelic ratio was calculated for each sequence. To estimate the degree of attenuation of one allele, the average cDNA allelic ratio was divided by the average genomic DNA allelic ratio. Standard deviation of the measurements was calculated on replicated experiments.

38
4 RESULTS AND DISCUSSION
The aims of this thesis were to further investigate a previously identified linkage region for pre-eclampsia, evaluate the pre-eclampsia candidate STOX1, and a locus identified by GWAS associated to CAD and T2D. We found association to the CAD locus at 9p21 (II) and narrowed down the linkage region at 2p25 (IV). We did not find any evidence for STOX1 as a susceptibility gene in the Finnish population (I) and failed to confirm or reject any of the genes within 2p25 linkage region as risk factors for pre-eclampsia (III-IV). Our studies highlight the challenges of gene mapping in complex diseases and more specifically in pre-eclampsia. Implications of our results, limitations of the separate studies and challenges in genetic studies of pre-eclampsia will be discussed on the following pages.
4.1 PAPER I - EVALUATION OF STOX1 ON 10q22
In Paper I, we used three methods to assess STOX1 as a candidate gene for pre-eclampsia. STOX1 was one of the first positionally cloned susceptibility genes for pre-eclampsia, with seven coding variations co-segregating with the pre-eclamptic phenotype in the Dutch population (van Dijk et al. 2005). The suggestion of STOX1 as a susceptibility gene for pre-eclampsia was followed by attempts to replicate results in different populations. At the time our study begun, no other studies of this gene in pre-eclampsia had been reported. Today, additional reports have rejected STOX1 as a common European pre-eclampsia susceptibility factor, based on both association studies and by investigating the suggested maternal inheritance (Berends et al. 2007, Iglesias-Platas et al. 2007). No evidence for STOX1 in the Finnish population We aimed to genotype all seven markers co-segregating with disease in the Dutch study and one additional SNP for covering the 3’ end of the gene. Only five markers were successfully genotyped, but covered all common haplotype variants even in the absence of the three failed markers, and were therefore likely to represent a comprehensive set to be tested for association. The five markers had similar allele frequencies in Finland and in the CEPH reference material obtained from the HapMap project, and formed a single LD block with identical haplotypes in both data sets. One marker was excluded from the haplotype analysis due to its low minor allele frequency (4%). In summary, none of the single markers or haplotypes tested showed significant association in our case-control data set. It is evident that additional population samples are needed to increase the power further for detecting genetic effects that may be weaker than those that we could assess with good power. It is also possible that the specific mutations observed in the Dutch families are causal in only a subset of families, and therefore not easily detected by large population-based analyses. In that case, linkage approaches should be more powerful to implicate this locus. Therefore, we re-examined our linkage analysis results in the extended Finnish families, that had been published previously (Laivuori et al. 2003), but there was no evidence for linkage (linkage scores were negative or just above zero throughout chromosome 10).

39
The expression pattern of STOX1 were investigated in placental biopsies taken from pre-eclamptic and uncomplicated deliveries using Affymetrix Human Genome arrays, but neither of the two STOX1 specific probe sets showed statistically significant difference in expression levels between cases and controls. We searched for genes that had sequence similarity to STOX1 within the linkage peaks in our genome-wide scan, since it has been suggested that paralogous genes located on different chromosomes may be responsible for the same disease, although they are likely to segregate in different populations due to different founders (Leegwater et al. 2001). We did not find obvious paralogs to STOX1 within our linkage peak regions; the closest candidate was STOX2 (28% identical to STOX1) on chromosome 4q35, but it was located >20 Mb downstream from the 3’ end of our candidate region. If other paralogs exist, their sequences may have diverged too far from STOX1 to be recognized by standard similarity search` methods. In summary, none of our three tests showed any evidence in support of STOX1 as a pre-eclampsia susceptibility gene, and we were therefore unable to confirm the association of STOX1 with pre-eclampsia in the Finnish population. This is in agreement with two other studies reporting bi-allelic expression of STOX1 in the placenta and no association to pre-eclampsia (Berends et al. 2007, Iglesias-Platas et al. 2007). However, a study by Rigourd et al suggested that STOX1 should still be considered as a key factor in pre-eclampsia, since their study strongly suggest that STOX1 plays a major part in normal placental development (Rigourd et al. 2008) Furthermore, evidence for an increased STOX1 expression in early pre-eclamptic placentas has been shown (Founds et al. 2009). Altogether, there is no conclusive evidence either for or against STOX1 as a susceptibility gene, but much larger sample sets than previously used in any study are needed to understand its role in pre-eclampsia.
4.2 PAPER II - ASSOCIATION IN 9p21 CAD RISK REGION
The number of published GWAS has been growing rapidly since 2007 leading to the discovery and replication of many new disease loci. Interestingly, several GWAS have identified a region on chromosome 9p21 that is associated with both CAD and T2D. Since there is a clear link between pre-eclampsia, CAD and T2D, and our linkage peak borders the GWA signals, we decided to genotype previously reported associated and tagging SNPs in our 15 families and the nation-wide case-control cohort. To our knowledge, we are the first to examine an association between this chromosomal region and pre-eclampsia. Association in Finnish, but not in UK and Australian/New Zealand populations Twenty-three markers were successfully genotyped and single marker association test revealed four markers with nominal association in the case-controls when GH women were excluded from the analysis (Table 7). We observed a trend towards association for the same four markers in the extended families. However, this association was very weak due to the small number of families available for the analysis. We included
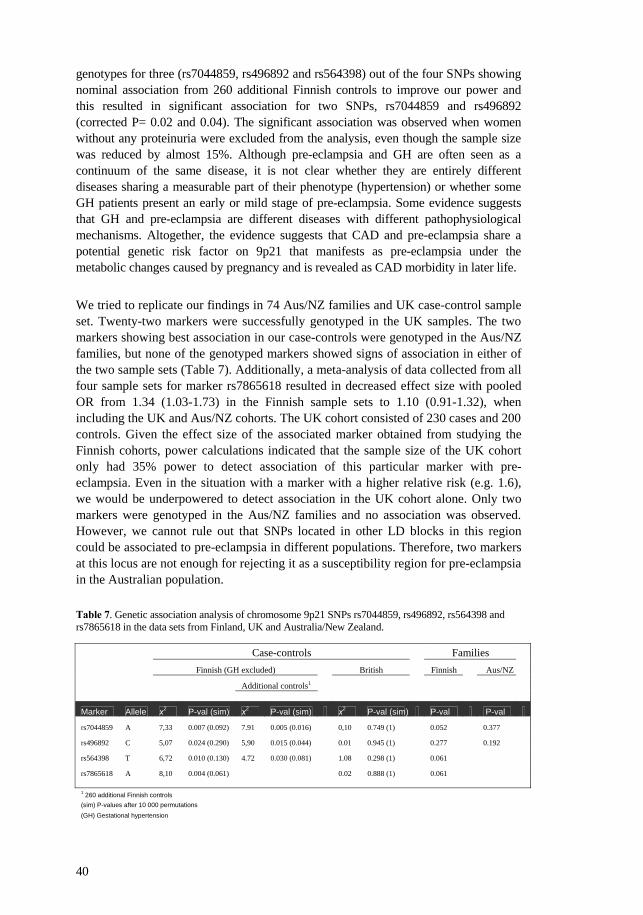
40
genotypes for three (rs7044859, rs496892 and rs564398) out of the four SNPs showing nominal association from 260 additional Finnish controls to improve our power and this resulted in significant association for two SNPs, rs7044859 and rs496892 (corrected P= 0.02 and 0.04). The significant association was observed when women without any proteinuria were excluded from the analysis, even though the sample size was reduced by almost 15%. Although pre-eclampsia and GH are often seen as a continuum of the same disease, it is not clear whether they are entirely different diseases sharing a measurable part of their phenotype (hypertension) or whether some GH patients present an early or mild stage of pre-eclampsia. Some evidence suggests that GH and pre-eclampsia are different diseases with different pathophysiological mechanisms. Altogether, the evidence suggests that CAD and pre-eclampsia share a potential genetic risk factor on 9p21 that manifests as pre-eclampsia under the metabolic changes caused by pregnancy and is revealed as CAD morbidity in later life.
We tried to replicate our findings in 74 Aus/NZ families and UK case-control sample set. Twenty-two markers were successfully genotyped in the UK samples. The two markers showing best association in our case-controls were genotyped in the Aus/NZ families, but none of the genotyped markers showed signs of association in either of the two sample sets (Table 7). Additionally, a meta-analysis of data collected from all four sample sets for marker rs7865618 resulted in decreased effect size with pooled OR from 1.34 (1.03-1.73) in the Finnish sample sets to 1.10 (0.91-1.32), when including the UK and Aus/NZ cohorts. The UK cohort consisted of 230 cases and 200 controls. Given the effect size of the associated marker obtained from studying the Finnish cohorts, power calculations indicated that the sample size of the UK cohort only had 35% power to detect association of this particular marker with pre-eclampsia. Even in the situation with a marker with a higher relative risk (e.g. 1.6), we would be underpowered to detect association in the UK cohort alone. Only two markers were genotyped in the Aus/NZ families and no association was observed. However, we cannot rule out that SNPs located in other LD blocks in this region could be associated to pre-eclampsia in different populations. Therefore, two markers at this locus are not enough for rejecting it as a susceptibility region for pre-eclampsia in the Australian population.
Table 7. Genetic association analysis of chromosome 9p21 SNPs rs7044859, rs496892, rs564398 and rs7865618 in the data sets from Finland, UK and Australia/New Zealand.
Case-controls Families
Finnish (GH excluded) British Finnish Aus/NZ
Additional controls1
Marker Allele x2 P-val (sim) x2 P-val (sim) x2 P-val (sim) P-val P-val
rs7044859 A 7,33 0.007 (0.092) 7.91 0.005 (0.016) 0,10 0.749 (1) 0.052 0.377
rs496892 C 5,07 0.024 (0.290) 5,90 0.015 (0.044) 0.01 0.945 (1) 0.277 0.192
rs564398 T 6,72 0.010 (0.130) 4.72 0.030 (0.081) 1.08 0.298 (1) 0.061
rs7865618 A 8,10 0.004 (0.061) 0.02 0.888 (1) 0.061 1 260 additional Finnish controls (sim) P-values after 10 000 permutations (GH) Gestational hypertension

41
Genes in the associated region The two associated SNPs in the Finnish cohort are both located in the same LD region previously known to harbour CAD associated SNPs (Samani et al. 2007, Schunkert et al. 2008). The region contains the cyclin-dependent kinase inhibitors 2A/2B genes (CDKN2A/2B) and a antisense noncoding RNA in the INK4 locus gene (ANRIL), but none of the genes has previously been implicated in the pathogenesis of pre-eclampsia. Interestingly, the protein p16INK4a encoded by CDKN2A has been shown to inhibit smooth muscle cell proliferation and is therefore an interesting functional candidate since inadequate transformation and replacement of the smooth muscle cells of spiral arteries in the placental bed has been considered to be a major pathophysiological event in pre-eclampsia. It has recently been shown that ANRIL is expressed in vascular endothelial cells, monocyte derived macrophages, and coronary smooth muscle cells, all of which could play a role in the pathophysiology (Broadbent et al. 2008). Additionally, an investigation of the functional significance of conserved sequences within the 9p21 risk locus for CAD demonstrated an effect of genetic variation on ANRIL expression (Jarinova et al. 2009). It is plausible that ANRIL could regulate the two genes CDKN2A/2B through RNA interference, gene co-suppression, gene silencing, chromatin structure maintenance, imprinting, or DNA demethylation. Our two associated SNPs are not in strong LD with markers showing an effect on ANRIL expression and the corresponding conserved sequence with enhancer activity. However, given the structural similarity to other long ncRNA genes, ANRIL is likely to rely on multiple transcriptional and splicing regulators (Ng et al. 2007, Storz et al. 2005) and other sequences within the 9p21 locus may modulate activity of other ANRIL splicing enhancer(s). Another plausible explanation for the association within this region is that a functional variant could have a potential role in transcriptional control of CDKN2B, without any involvement of ANRIL. It has been reported that our best associated SNP, located in the putative promoter region of CDNK2B, could alter the binding of the transcription factor hepatic nuclear factor (HNF) 1α, known to regulate the expression of a number of liver genes and may play a key role in linking metabolic and inflammatory pathways underlying the pathogenesis of CAD (Armendariz and Krauss 2009, Ruiz-Llorente et al. 2007). This could be the variant responsible for the observed association, but without further analysis we cannot say that a putative causal variants at this locus has been identified. This will require follow-up studies involving deep re-sequencing in preferably extreme phenotypes (e.g. severe cases) in the Finnish population to define the full allele spectrum and uncover functional variants. Finally, our gene expression analysis did not detect any differences between cases and controls at the 9p21 locus using the microarray data.

42
4.3 PAPER III, IV - MAPPING AND CHARACTERIZATION OF 2p25 LINKAGE REGION
Our previously identified susceptibility locus on chromosome 2p25 directly overlaps with a risk locus for general hypertension (Angius et al. 2002, Zhu et al. 2001, Laivuori et al. 2003). The linkage region contains several plausible susceptibility genes based on their functional properties. In Paper III, we first focused on ROCK2, which has been implicated in general hypertension and is located in the region of overlap between pre-eclampsia and hypertension linkage peak (see Figure 6B). In Paper IV, we present our efforts to narrow down the linkage region at the 2p25 locus and evaluate genes within it by association test, re-sequencing and gene expression analysis. Significant linkage, but no association in families Genotyping of eight new microsatellites between markers D2S398 and D2S305 (28cM-42cM) was done at approximately 1 cM intervals (Paper IV) (Figure 6A), and the region was narrowed down to 1.4 Mb with the highest peak showing significant linkage (NPL score 4.09, P= 0,00036) using general diagnostic criteria (women with GH but without proteinuria were considered as cases). We attempted to refine the 1.4 Mb region further by genotyping more SNPs in our 15 families, but ended up with several clearly separated potential hits showing weak association signals. However, we may have missed associated variants by chance since our first stages of fine-mapping were inefficient due to absence of knowledge of tagging SNP and allele frequency information. Even after it became possible to select reasonably common tagging SNPs to cover variation within the region of interest there were gaps in our genotyping. Preferably, the whole region of 1.4 Mb and corresponding LD blocks should have been covered more densely with tagging SNP to increase the power of the study. No significant evidence for the genes investigated We decided to focus on the genes in the 1.4 Mb region; E2F6, GREB1, LPIN1 and TRIB2 (Figure 6B). The four genes, and ROCK2 investigated in Paper III, were tested for association by genotyping tagging SNPs in our case-control sample set. Single marker and haplotype analysis did not reach statistical significance. However, markers in TRIB2 and GREB1 were nominally associated (P<0.04) and a common haplotype in TRIB2 (P=0.01). SNPs with MAF <5% were not considered due to lack of power to detect association. However, rare SNPs are more likely to be functionally deleterious than common SNPs and it would be important to cover them (Gorlov et al. 2008). Our association studies were gene-centric, however, intronic or intergenic variants could modulate disease risk by affecting gene expression pattern (tissue specificity, timing, expression level) and these are missed by our approach that focuses on the genes only. The weak association could be a reflection of a causal allele that is located somewhere else in the region and is not in complete LD with the associated SNPs. The nominally associated genes need to be further investigated in well powered sample sets and with comprehensive SNP typing over the gene and plausible regulatory regions. Re-sequencing the genes could have helped us to identify rare variants that confer susceptibility to pre-eclampsia. Based on both functional properties and association results, the genes TRIB2, LPIN1 and ROCK2 were re-sequenced in pre-eclamptic

43
women from families contributing to the 2p25 linkage signal to improve our chances of identifying the most relevant functional variants and healthy controls. ROCK2 is a widely expressed gene, but appears to be most abundant in smooth muscle cells, suggesting a role in vascular contraction (Riento and Ridley 2003). Interestingly, expression levels of RhoA mRNA have been shown to be significantly higher in pre-eclamptic placentas compared to placentas from normotensive women (Friel et al. 2008). However, we were not able to detect any expression differences for any of the specific probes included in the microarray expression analysis between the case-control groups. This is in agreement with a previous study that evaluated ROCK2 expression levels in placental tissues using RT-PCR and did not detect alterations between pre-eclamptic women and controls (Friel et al. 2008). However, the lack of alteration in mRNA expression levels does not rule out the possibility of alterations in protein levels that have been reported using immunohistochemistry and Western blot for ROCK2 (Ark et al. 2005). LPIN1 encodes for a phosphatidate phosphatase (PAP) enzyme required for glycerolipid biosynthesis (Carman and Han 2006). Several recent studies have confirmed the relationship between adipose tissue lipin-1 levels and insulin sensitivity in humans (Reue 2009). Interestingly, LPIN1 variants have previously shown association to pre-eclampsia risk factors such as BMI (Fawcett et al. 2008, Wiedmann et al. 2008), high insulin levels at resting metabolic rate (Loos et al. 2007) and the metabolic syndrome (Wiedmann et al. 2008). LPIN1 and ROCK2 were obvious candidates based on their involvement in lipid metabolism and general hypertension. TRIB2 on the other hand encodes a signaling protein that may regulate a number of physiological and developmental processes. However, the cellular and molecular basis for its action is still poorly characterized and it is therefore impossible to speculate about any plausible involvement in pre-eclampsia pathophysiology. We identified several novel variants in ROCK2 by re-sequencing and selected five for genotyping in our case-control data set based on their potential functional role (non-synonymous coding, splice site, other functional SNPs and insertion/deletion polymorphisms), but none showed association. Re-sequencing TRIB2 and LPIN1 resulted in identification of several variants, but none co-segregated with pre-eclampsia in the families when they were further investigated. Figure 6. Mapping stages, re-sequenced genes and peak-markers within the 2p25 linkage region (A) Non-parametric linkage (NPL) analysis in 15 pre-eclampsia families. The NPL results were obtained by increasing the marker density in the previously mapped candidate susceptibility loci for pre-eclampsia on chromosomes 2p25 to approximately 1 cM intervals by genotyping 8 new microsatellites between markers D2S398 and D2S305. (28cM-42cM) (gray horizontal box). Vertical dotted lines highlights the emplacement of peak observed for marker D2S262 (NPL=4.09) and the borders of the region subjected for SNP fine mapping (black horizontal box). (B) Enlarged ~ 1.8 Mb interval between D2S2278 and D2S2199. ROCK2, E2F6, GREB1, NTSR2, LPIN1 and TRIB2 are the protein coding genes located in the region. Rectangles highlights the peak marker D2S262 and the borders of the region subjected for SNP fine mapping (~ 1.4 Mb ). 11 genotyped SNPs are marked on the x-axis. A thickened portion of the x-axis denotes the associated 3-marker haplotype region of ~290 kb. The arrow points out the peak marker rs1453489. Genes that were re-sequenced are colored in gray and genes tested for association in cases and controls are marked with asterisk. Connected arrows point out a susceptibility locus for hypertension (Angius et al. 2002). Marker D2S168, peak marker in genome-wide linkage studies of pre-eclampsia and hypertension (Laivuori et al. 2003, Zhu et al. 2001).

44
4.4 GENERAL ASPECTS
Replication analyses in pre-eclampsia have often yielded inconsistent results, however, in order for association to be valid, the results need to be replicated in independent sample sets to distinguish between false and true association. There is substantial phenotypic and most likely genetic heterogeneity within and across populations for pre-eclampsia. Thus, it is not surprising that different research groups have identified unique loci and associations, especially since there must exist a strong natural selection against predisposing variants for pre-eclampsia until very recently due to the documented increased mortality for both mother and infant in the absent of effective obstetric care. Utilizing a founder population, such as our Finnish families, is likely to reduce genetic heterogeneity and aid genetic studies. In paper II, we demonstrated some evidence for association for markers at 9p21, but were not able to replicate in the two other populations, which could be due to locus or allelic heterogeneity, especially in the Aus/NZ families where only two markers were genotyped. Although multiple linkage regions have been identified in pre-eclampsia, the mapping of genes has been unsuccessful, with only a few promising candidates showing association in separate populations. Linkage is known to be more robust against heterogeneity than association and we see a convincing linkage signal at 2p25 in our extended families. However, when we attempt to fine-map the region and test single markers for association in paper III and IV, the signal becomes diluted and disappears. When investigating linkage region or more refined areas, gene-centric approaches are

45
very commonly used, but intergenic variants that could have an effect on the phenotype will be missed. To complicate things further, there is evidence that even within a causal locus, not all markers show association (Kotowski et al. 2006). Therefore, it is important to perform as exhaustive and comprehensive mapping of variants as possible when investigating candidate genes or loci. Weak associations may be a result of rare variants or causal alleles not in complete LD with the associated variant. Rare SNPs have generally been ignored in association studies because sample size needed to detect association to a rare variant has been too expensive to study. Also, common SNPs are more likely to be shared between populations enabling design of commercial genotyping arrays. However, rare SNPs are more likely to be functionally deleterious than common SNPs and it would be important to cover them (Gorlov et al. 2008). Definition of the phenotype is crucial in genetic studies, and an accurate phenotype definition is necessary to increase the chance of finding risk genes and to minimize the genetic heterogeneity in the collection of samples. Misclassification can reduce the power to detect association and increase the probability of spurious findings as a result of biased classification or confounding of the phenotype (Ellsworth and Manolio 1999). In general, defining phenotypes for complex diseases is difficult due to issues of heterogeneity, gene-gene and gene-environment interactions. As a likely example of multiple biological pathways leading to disease in pre-eclampsia, defective vascular remodeling is not observed in all affected women (Egbor et al. 2006). It is also possible that familial and sporadic representation of pre-eclampsia have different genetic background. Oudejans et al suggest that pre-eclampsia comes in an early form with a clear familial component, associated with growth restricted babies and abnormal placentation, and a much more common, late form, that arises from interaction between a normal placenta and maternal predisposing factors (Oudejans et al. 2007). The complexity of pre-eclampsia with a heterogeneous genetic background and different manifestations may explain the failures to detect association in linkage regions. Unfortunately, there is no universal definition of pre-eclampsia and inclusion criteria are in some cases inconsistent. For our Finnish sample sets, we have used an internationally accepted definition and all medical records were reviewed by experienced clinicians. Additionally, the risk for population stratification should be small as the controls have been carefully matched according living area and pregnancy number. Sample size is a clear determinant of a successful association study and it is apparent that majority of the published candidate gene studies and replication attempts in pre-eclampsia have been underpowered, at least for rare variants and variants with low genetic effects. Our case-control data set consists of around 350 cases and controls and gives us 80% power to detect association to common variants (MAF>0.3) with genetic effects of 1.6. If we subdivided the cases into pre-eclampsia/eclampsia, superimposed pre-eclampsia, GH and twin pregnancy to reduce the possibility of heterogeneity, we will end up with approximately 250 pre-eclampsia/eclampsia cases, which reduces the power even more. There is no evidence that pre-eclampsia would be driven by common markers with such strong effects. In fact, genome-wide association studies of many multifactorial diseases have highlighted that most associations are to markers with high MAF and low-medium OR (1.1-1.3) (Wray et al. 2008).

46
An alternative to association studies for investigating complex diseases is to examine gene expression profiles in diseased and healthy tissues. Pre-eclampsia only occurs in the presence of placenta and abnormal placentation is a known characteristic for the disease. Therefore, placental gene expression profiles may help to identify genes and pathways likely to be involved in the development of the disease. Previous expression study findings on genes and pathways are not consistent, possibly due to small sample size, different maternal ethnicity, different types of arrays and matching variability (Founds et al. 2008). We did not detect significant expression differences when examined our term placentas. However, we found that delivery method (vaginal, acute section, elective section) had a larger impact to differential expression than sample case-control status. True variation in expression levels may also have been missed due to small numbers of samples in each group studied. Moreover, the factors that trigger pre-eclampsia are likely to be expressed in early pregnancy and by examined gene expression later in gestation, as we have done, may only trace the mechanisms that have already occurred. Optimally, we would follow gene expression levels throughout gestation by taking placental biopsies longitudinally during different timepoints, but this is of course ethically unacceptable.

47
5 CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Despite the tremendous technological development in the field of genetics during the past few years, the numbers of identified genetic predisposing factors for pre-eclampsia have not increased as dramatically as for many other complex diseases, with only a few promising susceptibility genes identified so far. This is most likely explained by the fact that there is no GWAS performed in pre-eclampsia so far compared to many other complex diseases. Therefore, the genetics of pre-eclampsia is still an extremely important field to explore and a GWAS in a well characterized population is eagerly awaited, especially since the condition is both life threatening and resource demanding responsible for most of the maternal deaths worldwide. Right now the geneticists in the field are still investigating linkage regions identified almost a decade ago and most of the susceptibility loci are likely to be population-specific and will not allow identification of common predisposing factors with an effect in several populations. In this thesis, one of our efforts was to investigate the first positionally cloned pre-eclampsia candidate gene STOX1. We were unable to find any evidence for STOX1 association to pre-eclampsia in the Finnish population. Two independent studies have also rejected the impact of STOX1 in pre-eclampsia and its suggested maternal inheritance. Since the link between pre-eclampsia and cardiovascular diseases is well established, it was intriguing to observe the association to the previously reported CAD locus at 9p21, which further strengthen the theory that the different disorders share risk factors that could manifest differently during a woman’s life. However, genetic evidence for this locus in other populations is still lacking and further investigations of genes CDNK2B, ANRIL and plausible regulatory elements are needed. We were able to refine the susceptibility region at 2p25 identified by a genome-wide linkage analysis, but found ourselves underpowered to implicate individual genes residing within this region. Taken together, all our studies highlight the challenges of gene mapping in complex diseases and pre-eclampsia is characterized by a substantial phenotypic and, most likely, genetic heterogeneity within and across populations. To further elaborate the genetic contribution to pre-eclampsia in the future, carefully chosen definitions and focus on different pre-eclampsia phenotypes are most likely needed. Most important, the suggestion that pre-eclampsia could come in two forms, probably with different genetic background and underlying pathophysiology must be considered. Sample size clearly needs to be increased since we are currently underpowered to detect association to rare variants and variants with low genetic effects that could be expected for a complex disease such as pre-eclampsia. An ongoing project by the Finnish Genetics of Pre-eclampsia Consortium (FINNPEC) is recruiting pre-eclamptic women and healthy pregnant controls, as both trios and separate case-controls. The trio design will make it possible to study the paternal vs maternal contribution in the fetus and plausible effects, which have been implicated in pre-eclampsia. This project is aiming for over 3000 cases and will have sufficient power to detect association to common (MAF >2%) variants with medium effect size (OR > 1.3). A large nationwide clinical and genetic-epidemiological database will be

48
established, which will be used for studying molecular mechanisms and later consequences of pre-eclampsia and related traits. FINNPEC collaborates with UK Genetics of Pre-eclampsia Consortium (GOPEC), which includes families of over 1000 women whose pregnancies were affected by pre-eclampsia and collaborations as this will enable well-powered future GWAS and replication attempts in pre-eclampsia. GWAS represent an alternative approach to the previous hypothesis free genome-wide linkage studies, with the ability to detect common markers with moderate effect associated to disease and could lead to an increased understanding of diverse molecular pathways underlying pre-eclampsia. Thus, the causal variant are rarely revealed and the statistical power to detect small gene-gene and gene-environment interactions are limited. Furthermore, variants identified in one population are not always transferable to other and up until today, they have not been able to capture information about rare variants or a large fraction of the existing CNVs. The new sequencing technologies with a dramatic increase in throughput will hopefully overcome many of the challenges of traditional mapping and cloning. Massive cost reduction in sequencing has the potential to accelerate the identification of predisposing genetic variants and genes in pre-eclampsia. One potential approach would be to sequence complete linkage peaks identified in pre-eclampsia, including our susceptibility loci 2p25 and the less studied 9p13, in extended families to discover causal variants in affected individuals. Susceptibility genes found this way may help discover signaling and metabolic pathways involved in pre-eclampsia risk and guide further studies to prevent or treat this disorder. Rare markers, that have been hard to identify with past technologies and limited sample sets, could be discovered by sequencing extreme phenotypes (e.g. individuals suffering from severe pre-eclampsia), are likely to have a stronger effect to susceptibility than common markers tested by genotyping, and may prove more successful in diagnostic screening. Hopefully, international collaborations in the future, in combination with new technologies will shed further light on genetic susceptibility factors and increase the understanding of the underlying pathophysiology in pre-eclampsia.

49
6 ACKNOWLEDGEMENTS The work presented in this thesis has been carried out at the Department of Biosciences and Nutrition, Karolinska Institutet. I wish to express my sincere gratitude to all colleagues, collaborators, friends and family who in one way or another have contributed to this thesis. Först och främst ett stort tack till min handledare Juha för att du gav mig möjlighet att fortsätta som doktorand efter avslutat examensarbete. Ditt stora intresse, samt kunnande inom området har bidragit till den fina vägledning du givit mig genom åren som jag är mycket tacksam för. Trots både lite upp- och nedgångar i projektet så har du alltid visat upp en entusiasm och optimism som varit beundransvärd och väldigt inspirerande. Katja for being a fantastic co-supervisor! Thank you for all your help and advise through these years, and finding the time to work with this project although you were working fulltime elsewhere during the last years. I have very much appreciated your encouragement and support in everything. I also want to thank my second co-supervisor Hannele, for introducing me into this field of genetics. You have been a great support to me, and I really appreciated your company at the ISSHP meetings. I am grateful to everyone that have contributed in the PE-project. Thanks to Erja for guidance through my exam-work, introducing me to this project and for helping me out in the lab during my first time in Novum. Always with a smile on your face ☺. Many thanks to Hong for the initial statistical help in the project and Marco for introducing med to statistical analysis. I also want to acknowledge Hanna N for genotyping assistance. I would like to express my thanks to all co-authors for excellent collaborations and contributions to the papers in this thesis. Vesa and Leena at the Finnish Red Cross for making our research possible by providing us with samples. I also want to thank our collaborators Eric and Matt in the US and Linda M in UK for letting us investigate our findings in your samples and for always being very helpful. Tack till Michael och Magnus för ett lärorikt och intressant Dystoci-samarbete. Jag vill passa på att tacka alla på MAF för genotypning och goda råd genom åren: Kristina, Linda, Astrid, Ville, Päivi, Aida, Malin, Lotta, Tiina, Cissi och Eva. Many thanks to the past and present members of the JKE-group: Juha, Katja, Hannele, Myriam, Isabel, Ulf, Kristiina, Sara, Virpi, Cilla, Per, Marie, Ingegerd, Heidi, Gustaf, Hans, Christina, Liselotte, Hong, Marco, Tiina S, Mauro, Lovisa, Linda, Astrid, Rezin, Ville, Malin, Aida, Eva, Kristina, Päivi, Erik, Tiina, Lotta, Fransesca A, Cissi, Rasko, Fransesca B, Erja, Thomas, Jingwen, Jun and Pu. TMK-tjejerna: Heidi, Sara, Christina, Kristiina, Linda, Astrid och Anna. Utsökt mat, vin och ”lite” skvaller... kan inte bli så mycket bättre! ☺ Anna, tack för allt stöd under dessa år vi följts åt som doktorander och inte minst alla trevliga fikastunder.

50
Biomed-vännerna, ett stort tack till er alla för att ni funnits där genom åren. Mycket roligt har vi upplevt tillsammans. Allt tog sin början i Scheele-labb... Johanna, Susanne, Ying, Maria L och Andreas för alla härliga resor, fester, trevliga fikastunder och mycket mer ☺ Så tacksam att jag har er som vänner! Ett hjärtligt tack till övriga vänner. Ingrid, fint att du tar dig an TM-uppdraget. Monika, du förgyllde både helg och vardagen i Novum. Tack Angelica för att du följde med till DC/NY, det blev så mkt roligare. Eva-Lena, Karin, Maria W, Helena, Torbjörn, Micke och Nisse. Tack till alla mina släktingar i Södertälje, Huddinge och närliggande trakter för trevliga sammankomster genom åren. Slutligen vill jag tacka mina fantastiska föräldrar för all er omtanke, samt ert stöd och uppmuntran under alla år. Ni ska veta hur mycket det har betytt för mig!!!

51
7 REFERENCES Agatisa PK, Ness RB, Roberts JM, Costantino JP et al. (2004) Impairment of endothelial
function in women with a history of preeclampsia: an indicator of cardiovascular risk. Am J Physiol Heart Circ Physiol 286, H1389-1393.
Alexander BT, Cockrell KL, Massey MB, Bennett WA et al. (2002) Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens 15, 170-175.
Allaire AD, Ballenger KA, Wells SR, McMahon MJ et al. (2000) Placental apoptosis in preeclampsia. Obstet Gynecol 96, 271-276.
Altshuler D, Daly MJ and Lander ES (2008) Genetic mapping in human disease. Science 322, 881-888.
Ananth CV, Savitz DA and Bowes WA, Jr. (1995) Hypertensive disorders of pregnancy and stillbirth in North Carolina, 1988 to 1991. Acta Obstet Gynecol Scand 74, 788-793.
Angius A, Petretto E, Maestrale GB, Forabosco P et al. (2002) A new essential hypertension susceptibility locus on chromosome 2p24-p25, detected by genomewide search. Am J Hum Genet 71, 893-905.
Ardlie KG, Kruglyak L and Seielstad M (2002) Patterns of linkage disequilibrium in the human genome. Nat Rev Genet 3, 299-309.
Ark M, Yilmaz N, Yazici G, Kubat H et al. (2005) Rho-associated protein kinase II (rock II) expression in normal and preeclamptic human placentas. Placenta 26, 81-84.
Armendariz AD and Krauss RM (2009) Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol 20, 106-111.
Arnadottir GA, Geirsson RT, Arngrimsson R, Jonsdottir LS et al. (2005) Cardiovascular death in women who had hypertension in pregnancy: a case-control study. Bjog 112, 286-292.
Arngrimsson R, Bjornsson S, Geirsson RT, Bjornsson H et al. (1990) Genetic and familial predisposition to eclampsia and pre-eclampsia in a defined population. Br J Obstet Gynaecol 97, 762-769.
Arngrimsson R, Hayward C, Nadaud S, Baldursdottir A et al. (1997) Evidence for a familial pregnancy-induced hypertension locus in the eNOS-gene region. Am J Hum Genet 61, 354-362.
Arngrimsson R, Purandare S, Connor M, Walker JJ et al. (1993) Angiotensinogen: a candidate gene involved in preeclampsia? Nat Genet 4, 114-115.
Arngrimsson R, Sigurard ttir S, Frigge ML, Bjarnad ttir RI et al. (1999) A genome-wide scan reveals a maternal susceptibility locus for pre-eclampsia on chromosome 2p13. Hum Mol Genet 8, 1799-1805.
Askie LM, Duley L, Henderson-Smart DJ and Stewart LA (2007) Antiplatelet agents for prevention of pre-eclampsia: a meta-analysis of individual patient data. Lancet 369, 1791-1798.
Atamer Y, Kocyigit Y, Yokus B, Atamer A et al. (2005) Lipid peroxidation, antioxidant defense, status of trace metals and leptin levels in preeclampsia. Eur J Obstet Gynecol Reprod Biol 119, 60-66.
Australasian Society for the Study of Hypertension in Pregnancy (ASSHP) (1993) Management of hypertension in pregnancy: executive summary. Med J Aust 158, 700-702.
Bache I, Bock T, Volund A and Buschard K (1999) Previous maternal abortion, longer gestation, and younger maternal age decrease the risk of type 1 diabetes among male offspring. Diabetes Care 22, 1063-1065.

52
Banyasz I, Szabo S, Bokodi G, Vannay A et al. (2006) Genetic polymorphisms of vascular endothelial growth factor in severe pre-eclampsia. Mol Hum Reprod 12, 233-236.
Barrett JC, Fry B, Maller J and Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263-265.
Belo L, Santos-Silva A, Quintanilha A and Rebelo I (2008) Similarities between pre-eclampsia and atherosclerosis: a protective effect of physical exercise? Curr Med Chem 15, 2223-2229.
Berends AL, Bertoli-Avella AM, de Groot CJ, van Duijn CM et al. (2007) STOX1 gene in pre-eclampsia and intrauterine growth restriction. Bjog 114, 1163-1167.
Boehnke M (1994) Limits of resolution of genetic linkage studies: implications for the positional cloning of human disease genes. Am J Hum Genet 55, 379-390.
Borecki IB and Province MA (2008) Linkage and association: basic concepts. Adv Genet 60, 51-74.
Borzychowski AM, Sargent IL and Redman CW (2006) Inflammation and pre-eclampsia. Semin Fetal Neonatal Med 11, 309-316.
Brennecke SP, Gude NM, Di Iulio JL and King RG (1997) Reduction of placental nitric oxide synthase activity in pre-eclampsia. Clin Sci (Lond) 93, 51-55.
Broadbent HM, Peden JF, Lorkowski S, Goel A et al. (2008) Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet 17, 806-814.
Brown MA, Lindheimer MD, de Swiet M, Van Assche A et al. (2001) The classification and diagnosis of the hypertensive disorders of pregnancy: statement from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertens Pregnancy 20, IX-XIV.
Caniggia I, Lye SJ and Cross JC (1997) Activin is a local regulator of human cytotrophoblast cell differentiation. Endocrinology 138, 3976-3986.
Canto P, Canto-Cetina T, Juarez-Velazquez R, Rosas-Vargas H et al. (2008) Methylenetetrahydrofolate reductase C677T and glutathione S-transferase P1 A313G are associated with a reduced risk of preeclampsia in Maya-Mestizo women. Hypertens Res 31, 1015-1019.
Cardon LR and Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2, 91-99.
Cardon LR and Palmer LJ (2003) Population stratification and spurious allelic association. Lancet 361, 598-604.
Carman GM and Han GS (2006) Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem Sci 31, 694-699.
Carpenter MW (2007) Gestational diabetes, pregnancy hypertension, and late vascular disease. Diabetes Care 30 Suppl 2, S246-250.
Chambers JC, Fusi L, Malik IS, Haskard DO et al. (2001) Association of maternal endothelial dysfunction with preeclampsia. Jama 285, 1607-1612.
Chappell S and Morgan L (2006) Searching for genetic clues to the causes of pre-eclampsia. Clin Sci (Lond) 110, 443-458.
Chen CP, Chen CY, Yang YC, Su TH et al. (2004) Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta 25, 413-421.
Chen G, Wilson R, Wang SH, Zheng HZ et al. (1996) Tumour necrosis factor-alpha (TNF-alpha) gene polymorphism and expression in pre-eclampsia. Clin Exp Immunol 104, 154-159.
Cheng MH and Wang PH (2009) Placentation abnormalities in the pathophysiology of preeclampsia. Expert Rev Mol Diagn 9, 37-49.
Chesley SC, Annitto JE and Cosgrove RA (1976) The remote prognosis of eclamptic women. Sixth periodic report. Am J Obstet Gynecol 124, 446-459.

53
Clark BA, Halvorson L, Sachs B and Epstein FH (1992) Plasma endothelin levels in preeclampsia: elevation and correlation with uric acid levels and renal impairment. Am J Obstet Gynecol 166, 962-968.
Clark DE, Smith SK, Sharkey AM and Charnock-Jones DS (1996) Localization of VEGF and expression of its receptors flt and KDR in human placenta throughout pregnancy. Hum Reprod 11, 1090-1098.
Cnattingius S, Reilly M, Pawitan Y and Lichtenstein P (2004) Maternal and fetal genetic factors account for most of familial aggregation of preeclampsia: a population-based Swedish cohort study. Am J Med Genet A 130A, 365-371.
Colbern GT, Chiang MH and Main EK (1994) Expression of the nonclassic histocompatibility antigen HLA-G by preeclamptic placenta. Am J Obstet Gynecol 170, 1244-1250.
Conrad DF, Pinto D, Redon R, Feuk L et al. (2009) Origins and functional impact of copy number variation in the human genome. Nature
Conrad KP and Benyo DF (1997) Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol 37, 240-249.
Daher S, Sass N, Oliveira LG and Mattar R (2006) Cytokine genotyping in preeclampsia. Am J Reprod Immunol 55, 130-135.
Dahlquist GG, Patterson C and Soltesz G (1999) Perinatal risk factors for childhood type 1 diabetes in Europe. The EURODIAB Substudy 2 Study Group. Diabetes Care 22, 1698-1702.
Dalmaz CA, Santos KG, Botton MR, Tedoldi CL et al. (2006) Relationship between polymorphisms in thrombophilic genes and preeclampsia in a Brazilian population. Blood Cells Mol Dis 37, 107-110.
Daly MJ, Rioux JD, Schaffner SF, Hudson TJ et al. (2001) High-resolution haplotype structure in the human genome. Nat Genet 29, 229-232.
Davies AM, Czaczkes JW, Sadovsky E, Prywes R et al. (1970) Toxemia of pregnancy in Jerusalem. I. Epidemiological studies of a total community. Isr J Med Sci 6, 253-266.
de Bakker PI, Yelensky R, Pe'er I, Gabriel SB et al. (2005) Efficiency and power in genetic association studies. Nat Genet 37, 1217-1223.
Dekker G (2005) Prothrombotic mechanisms in preeclampsia. Thromb Res 115 Suppl 1, 17-21. Descamps OS, Bruniaux M, Guilmot PF, Tonglet R et al. (2005) Lipoprotein metabolism of
pregnant women is associated with both their genetic polymorphisms and those of their newborn children. J Lipid Res 46, 2405-2414.
Duckitt K and Harrington D (2005) Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. Bmj 330, 565.
Dudbridge F (2003) Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol 25, 115-121.
Duley L (2003) Pre-eclampsia and the hypertensive disorders of pregnancy. Br Med Bull 67, 161-176.
Duley L (2009) The global impact of pre-eclampsia and eclampsia. Semin Perinatol 33, 130-137.
Duley L, Gulmezoglu AM and Henderson-Smart DJ (2003) Magnesium sulphate and other anticonvulsants for women with pre-eclampsia. Cochrane Database Syst Rev CD000025.
Duley L and Henderson-Smart DJ (2000) Drugs for rapid treatment of very high blood pressure during pregnancy. Cochrane Database Syst Rev CD001449.
Egbor M, Ansari T, Morris N, Green CJ et al. (2006) Morphometric placental villous and vascular abnormalities in early- and late-onset pre-eclampsia with and without fetal growth restriction. Bjog 113, 580-589.
Ekbom A, Trichopoulos D, Adami HO, Hsieh CC et al. (1992) Evidence of prenatal influences on breast cancer risk. Lancet 340, 1015-1018.

54
Elahi MM, Asotra K, Matata BM and Mastana SS (2009) Tumor necrosis factor alpha -308 gene locus promoter polymorphism: an analysis of association with health and disease. Biochim Biophys Acta 1792, 163-172.
Ellsworth DL and Manolio TA (1999) The emerging importance of genetics in epidemiologic research II. Issues in study design and gene mapping. Ann Epidemiol 9, 75-90.
El-Toukhy T, Khalaf Y and Braude P (2006) IVF results: optimize not maximize. Am J Obstet Gynecol 194, 322-331.
Emanuel BS and Saitta SC (2007) From microscopes to microarrays: dissecting recurrent chromosomal rearrangements. Nat Rev Genet 8, 869-883.
Eskenazi B, Fenster L and Sidney S (1991) A multivariate analysis of risk factors for preeclampsia. Jama 266, 237-241.
Esplin MS, Fausett MB, Fraser A, Kerber R et al. (2001) Paternal and maternal components of the predisposition to preeclampsia. N Engl J Med 344, 867-872.
Fabbro D, D'Elia AV, Spizzo R, Driul L et al. (2003) Association between plasminogen activator inhibitor 1 gene polymorphisms and preeclampsia. Gynecol Obstet Invest 56, 17-22.
Faisel F, Romppanen EL, Hiltunen M, Helisalmi S et al. (2003) Polymorphism in the interleukin 1 receptor antagonist gene in women with preeclampsia. J Reprod Immunol 60, 61-70.
Fawcett KA, Grimsey N, Loos RJ, Wheeler E et al. (2008) Evaluating the role of LPIN1 variation in insulin resistance, body weight, and human lipodystrophy in U.K. Populations. Diabetes 57, 2527-2533.
Fitzpatrick E, Goring HH, Liu H, Borg A et al. (2004) Fine mapping and SNP analysis of positional candidates at the preeclampsia susceptibility locus (PREG1) on chromosome 2. Hum Biol 76, 849-862.
Fitzpatrick E, Johnson MP, Dyer TD, Forrest S et al. (2009) Genetic association of the activin A receptor gene (ACVR2A) and pre-eclampsia. Mol Hum Reprod 15, 195-204.
Folgero T, Storbakk N, Torbergsen T and Oian P (1996) Mutations in mitochondrial transfer ribonucleic acid genes in preeclampsia. Am J Obstet Gynecol 174, 1626-1630.
Founds SA, Dorman JS and Conley YP (2008) Microarray technology applied to the complex disorder of preeclampsia. J Obstet Gynecol Neonatal Nurs 37, 146-157.
Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A et al. (2009) Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta 30, 15-24.
Frazer KA, Ballinger DG, Cox DR, Hinds DA et al. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861.
Frazer KA, Murray SS, Schork NJ and Topol EJ (2009) Human genetic variation and its contribution to complex traits. Nat Rev Genet 10, 241-251.
Frias AE, Jr. and Belfort MA (2003) Post Magpie: how should we be managing severe preeclampsia? Curr Opin Obstet Gynecol 15, 489-495.
Friel AM, Hynes PG, Sexton DJ, Smith TJ et al. (2008) Expression levels of mRNA for Rho A/Rho kinase and its role in isoprostane-induced vasoconstriction of human placental and maternal vessels. Reprod Sci 15, 179-188.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM et al. (2002) The structure of haplotype blocks in the human genome. Science 296, 2225-2229.
Garovic VD and Hayman SR (2007) Hypertension in pregnancy: an emerging risk factor for cardiovascular disease. Nat Clin Pract Nephrol 3, 613-622.
Gaugler-Senden IP, Huijssoon AG, Visser W, Steegers EA et al. (2006) Maternal and perinatal outcome of preeclampsia with an onset before 24 weeks' gestation. Audit in a tertiary referral center. Eur J Obstet Gynecol Reprod Biol 128, 216-221.

55
Gebhardt GS, Peters WH, Hillermann R, Odendaal HJ et al. (2004) Maternal and fetal single nucleotide polymorphisms in the epoxide hydrolase and gluthatione S-transferase P1 genes are not associated with pre-eclampsia in the Coloured population of the Western Cape, South Africa. J Obstet Gynaecol 24, 866-872.
Gentleman RC, Carey VJ, Bates DM, Bolstad B et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80.
Gerhardt A, Goecke TW, Beckmann MW, Wagner KJ et al. (2005) The G20210A prothrombin-gene mutation and the plasminogen activator inhibitor (PAI-1) 5G/5G genotype are associated with early onset of severe preeclampsia. J Thromb Haemost 3, 686-691.
Germain SJ, Sacks GP, Sooranna SR, Sargent IL et al. (2007) Systemic inflammatory priming in normal pregnancy and preeclampsia: the role of circulating syncytiotrophoblast microparticles. J Immunol 178, 5949-5956.
Giardina JB, Green GM, Cockrell KL, Granger JP et al. (2002) TNF-alpha enhances contraction and inhibits endothelial NO-cGMP relaxation in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol 283, R130-143.
Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M et al. (2008) Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol 294, H541-550.
Gluckman PD, Hanson MA, Cooper C and Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359, 61-73.
Goddard KA, Tromp G, Romero R, Olson JM et al. (2007) Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Hum Hered 63, 1-16.
Gonzalez-Juanatey C, Llorca J, Miranda-Filloy JA, Amigo-Diaz E et al. (2007) Endothelial dysfunction in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum 57, 287-293.
Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR et al. (2008) Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am J Hum Genet 82, 100-112.
Graves JA (1998) Genomic imprinting, development and disease--is pre-eclampsia caused by a maternally imprinted gene? Reprod Fertil Dev 10, 23-29.
Gu S, Pakstis AJ, Li H, Speed WC et al. (2007) Significant variation in haplotype block structure but conservation in tagSNP patterns among global populations. Eur J Hum Genet 15, 302-312.
Gupta S, Agarwal A and Sharma RK (2005) The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet Gynecol Surv 60, 807-816.
Haggerty CL, Ferrell RE, Hubel CA, Markovic N et al. (2005) Association between allelic variants in cytokine genes and preeclampsia. Am J Obstet Gynecol 193, 209-215.
Hanis CL, Boerwinkle E, Chakraborty R, Ellsworth DL et al. (1996) A genome-wide search for human non-insulin-dependent (type 2) diabetes genes reveals a major susceptibility locus on chromosome 2. 13, 161-166.
Harlow FH and Brown MA (2001) The diversity of diagnoses of preeclampsia. Hypertens Pregnancy 20, 57-67.
Harrison GA, Humphrey KE, Jones N, Badenhop R et al. (1997) A genomewide linkage study of preeclampsia/eclampsia reveals evidence for a candidate region on 4q. Am J Hum Genet 60, 1158-1167.
Harskamp RE and Zeeman GG (2007) Preeclampsia: at risk for remote cardiovascular disease. Am J Med Sci 334, 291-295.
Hayman R, Brockelsby J, Kenny L and Baker P (1999) Preeclampsia: the endothelium, circulating factor(s) and vascular endothelial growth factor. J Soc Gynecol Investig 6, 3-10.

56
Hayward C, Livingstone J, Holloway S, Liston WA et al. (1992) An exclusion map for pre-eclampsia: assuming autosomal recessive inheritance. Am J Hum Genet 50, 749-757.
Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S et al. (2007) A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316, 1491-1493.
Hiby SE, Walker JJ, O'Shaughnessy K M, Redman CW et al. (2004) Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med 200, 957-965.
Hiltunen LM, Laivuori H, Rautanen A, Kaaja R et al. (2009) Blood group AB and factor V Leiden as risk factors for pre-eclampsia: a population-based nested case-control study. Thromb Res 124, 167-173.
Hirschhorn JN and Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6, 95-108.
Hofmeyr GJ, Duley L and Atallah A (2007) Dietary calcium supplementation for prevention of pre-eclampsia and related problems: a systematic review and commentary. Bjog 114, 933-943.
Hogberg U (2005) The World Health Report 2005: "make every mother and child count" - including Africans. Scand J Public Health 33, 409-411.
Hubel CA, Roberts JM and Ferrell RE (1999) Association of pre-eclampsia with common coding sequence variations in the lipoprotein lipase gene. Clin Genet 56, 289-296.
Hung TH and Burton GJ (2006) Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol 45, 189-200.
Hurles ME, Dermitzakis ET and Tyler-Smith C (2008) The functional impact of structural variation in humans. Trends Genet 24, 238-245.
Iglesias-Platas I, Monk D, Jebbink J, Buimer M et al. (2007) STOX1 is not imprinted and is not likely to be involved in preeclampsia. Nat Genet 39, 279-280; author reply 280-271.
Innes K, Byers T and Schymura M (2000) Birth characteristics and subsequent risk for breast cancer in very young women. Am J Epidemiol 152, 1121-1128.
International HapMap Consortium (IHMC) (2003) The International HapMap Project. Nature 426, 789-796.
International HapMap Consortium (IHMC) (2005) A haplotype map of the human genome. Nature 437, 1299-1320.
International Human Genome Sequencing Consortium (IHGSC) (2004) Finishing the euchromatic sequence of the human genome. Nature 431, 931-945.
Irgens HU, Reisaeter L, Irgens LM and Lie RT (2001) Long term mortality of mothers and fathers after pre-eclampsia: population based cohort study. Bmj 323, 1213-1217.
Ishitani A, Sageshima N, Lee N, Dorofeeva N et al. (2003) Protein expression and peptide binding suggest unique and interacting functional roles for HLA-E, F, and G in maternal-placental immune recognition. J Immunol 171, 1376-1384.
Jackson RA, Gibson KA, Wu YW and Croughan MS (2004) Perinatal outcomes in singletons following in vitro fertilization: a meta-analysis. Obstet Gynecol 103, 551-563.
Jarinova O, Stewart AF, Roberts R, Wells G et al. (2009) Functional Analysis of the Chromosome 9p21.3 Coronary Artery Disease Risk Locus. Arterioscler Thromb Vasc Biol
Jimenez-Sanchez G, Childs B and Valle D (2001) Human disease genes. Nature 409, 853-855. Johnson MP, Fitzpatrick E, Dyer TD, Jowett JB et al. (2007) Identification of two novel
quantitative trait loci for pre-eclampsia susceptibility on chromosomes 5q and 13q using a variance components-based linkage approach. Mol Hum Reprod 13, 61-67.
Johnson MP, Roten LT, Dyer TD, East CE et al. (2009) The ERAP2 gene is associated with preeclampsia in Australian and Norwegian populations. Hum Genet 126, 655-666.

57
Jones ME, Swerdlow AJ, Gill LE and Goldacre MJ (1998) Pre-natal and early life risk factors for childhood onset diabetes mellitus: a record linkage study. Int J Epidemiol 27, 444-449.
Jones RL, Findlay JK and Salamonsen LA (2006) The role of activins during decidualisation of human endometrium. Aust N Z J Obstet Gynaecol 46, 245-249.
Jonsdottir LS, Arngrimsson R, Geirsson RT, Sigvaldason H et al. (1995) Death rates from ischemic heart disease in women with a history of hypertension in pregnancy. Acta Obstet Gynecol Scand 74, 772-776.
Kaaja R, Kinnunen T and Luoto R (2005) Regional differences in the prevalence of pre-eclampsia in relation to the risk factors for coronary artery disease in women in Finland. Eur Heart J 26, 44-50.
Kaaja R, Laivuori H, Laakso M, Tikkanen MJ et al. (1999) Evidence of a state of increased insulin resistance in preeclampsia. Metabolism 48, 892-896.
Kaaja R, Tikkanen MJ, Viinikka L and Ylikorkala O (1995) Serum lipoproteins, insulin, and urinary prostanoid metabolites in normal and hypertensive pregnant women. Obstet Gynecol 85, 353-356.
Kaibuchi K, Kuroda S, Fukata M and Nakagawa M (1999) Regulation of cadherin-mediated cell-cell adhesion by the Rho family GTPases. Curr Opin Cell Biol 11, 591-596.
Kallen B, Finnstrom O, Nygren KG, Otterblad Olausson P et al. (2005) In vitro fertilisation in Sweden: obstetric characteristics, maternal morbidity and mortality. Bjog 112, 1529-1535.
Kamali-Sarvestani E, Kiany S, Gharesi-Fard B and Robati M (2006) Association study of IL-10 and IFN-gamma gene polymorphisms in Iranian women with preeclampsia. J Reprod Immunol 72, 118-126.
Kamide K, Kokubo Y, Yang J, Tanaka C et al. (2005) Hypertension susceptibility genes on chromosome 2p24-p25 in a general Japanese population. J Hypertens 23, 955-960.
Kim WY and Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell 127, 265-275.
Kim YJ, Williamson RA, Chen K, Smith JL et al. (2001) Lipoprotein lipase gene mutations and the genetic susceptibility of preeclampsia. Hypertension 38, 992-996.
Kivinen K, Peterson H, Hiltunen L, Laivuori H et al. (2007) Evaluation of STOX1 as a preeclampsia candidate gene in a population-wide sample. Eur J Hum Genet 15, 494-497.
Klonoff-Cohen HS, Savitz DA, Cefalo RC and McCann MF (1989) An epidemiologic study of contraception and preeclampsia. Jama 262, 3143-3147.
Knight M (2007) Eclampsia in the United Kingdom 2005. Bjog 114, 1072-1078. Knight M, Redman CW, Linton EA and Sargent IL (1998) Shedding of syncytiotrophoblast
microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol 105, 632-640.
Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM et al. (2002) A high-resolution recombination map of the human genome. Nat Genet 31, 241-247.
Kotowski IK, Pertsemlidis A, Luke A, Cooper RS et al. (2006) A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 78, 410-422.
Kramer MS, Seguin L, Lydon J and Goulet L (2000) Socio-economic disparities in pregnancy outcome: why do the poor fare so poorly? Paediatr Perinat Epidemiol 14, 194-210.
Kruglyak L, Daly MJ, Reeve-Daly MP and Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58, 1347-1363.
Kullberg G, Lindeberg S and Hanson U (2002) Eclampsia in Sweden. Hypertens Pregnancy 21, 13-21.

58
Kupferminc MJ, Eldor A, Steinman N, Many A et al. (1999) Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 340, 9-13.
Kurian AK and Cardarelli KM (2007) Racial and ethnic differences in cardiovascular disease risk factors: a systematic review. Ethn Dis 17, 143-152.
Laasanen J, Romppanen EL, Hiltunen M, Helisalmi S et al. (2002) Two exonic single nucleotide polymorphisms in the microsomal epoxide hydrolase gene are jointly associated with preeclampsia. Eur J Hum Genet 10, 569-573.
Lachmeijer AM, Arngrimsson R, Bastiaans EJ, Frigge ML et al. (2001) A genome-wide scan for preeclampsia in the Netherlands. Eur J Hum Genet 9, 758-764.
Laivuori H, Lahermo P, Ollikainen V, Widen E et al. (2003) Susceptibility loci for preeclampsia on chromosomes 2p25 and 9p13 in Finnish families. Am J Hum Genet 72, 168-177.
LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR et al. (2007) Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep 9, 480-485.
Lander ES, Linton LM, Birren B, Nusbaum C et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860-921.
Leegwater PA, Vermeulen G, Konst AA, Naidu S et al. (2001) Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet 29, 383-388.
Leung DN, Smith SC, To KF, Sahota DS et al. (2001) Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol 184, 1249-1250.
Levine RJ, Lam C, Qian C, Yu KF et al. (2006) Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 355, 992-1005.
Levine RJ, Maynard SE, Qian C, Lim KH et al. (2004) Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350, 672-683.
Levy S, Sutton G, Ng PC, Feuk L et al. (2007) The diploid genome sequence of an individual human. PLoS Biol 5, e254.
Lie RT, Rasmussen S, Brunborg H, Gjessing HK et al. (1998) Fetal and maternal contributions to risk of pre-eclampsia: population based study. Bmj 316, 1343-1347.
Lin J and August P (2005) Genetic thrombophilias and preeclampsia: a meta-analysis. Obstet Gynecol 105, 182-192.
Lindgren CM, Mahtani MM, Widen E, McCarthy MI et al. (2002) Genomewide search for type 2 diabetes mellitus susceptibility loci in Finnish families: the Botnia study. Am J Hum Genet 70, 509-516.
Liston WA and Kilpatrick DC (1991) Is genetic susceptibility to pre-eclampsia conferred by homozygosity for the same single recessive gene in mother and fetus? Br J Obstet Gynaecol 98, 1079-1086.
Loos RJ, Rankinen T, Perusse L, Tremblay A et al. (2007) Association of lipin 1 gene polymorphisms with measures of energy and glucose metabolism. Obesity (Silver Spring) 15, 2723-2732.
Luo TH, Zhao Y, Li G, Yuan WT et al. (2001) A genome-wide search for Type II diabetes susceptibility genes in Chinese Hans. Diabetologia 44, 501-506.
Lykke JA, Langhoff-Roos J, Sibai BM, Funai EF et al. (2009) Hypertensive pregnancy disorders and subsequent cardiovascular morbidity and type 2 diabetes mellitus in the mother. Hypertension 53, 944-951.
Makris A, Xu B, Yu B, Thornton C et al. (2006) Placental deficiency of interleukin-10 (IL-10) in preeclampsia and its relationship to an IL10 promoter polymorphism. Placenta 27, 445-451.
Manolio TA, Collins FS, Cox NJ, Goldstein DB et al. (2009) Finding the missing heritability of complex diseases. Nature 461, 747-753.

59
Martin ER, Bass MP, Gilbert JR, Pericak-Vance MA et al. (2003) Genotype-based association test for general pedigrees: the genotype-PDT. Genet Epidemiol 25, 203-213.
Martin ER, Monks SA, Warren LL and Kaplan NL (2000) A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet 67, 146-154.
Maynard SE, Min JY, Merchan J, Lim KH et al. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111, 649-658.
McCarroll SA (2008) Extending genome-wide association studies to copy-number variation. Hum Mol Genet 17, R135-142.
McCarthy MI and Hirschhorn JN (2008) Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet 17, R156-165.
McKinney PA, Parslow R, Gurney KA, Law GR et al. (1999) Perinatal and neonatal determinants of childhood type 1 diabetes. A case-control study in Yorkshire, U.K. Diabetes Care 22, 928-932.
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A et al. (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316, 1488-1491.
Medica I, Kastrin A and Peterlin B (2007) Genetic polymorphisms in vasoactive genes and preeclampsia: a meta-analysis. Eur J Obstet Gynecol Reprod Biol 131, 115-126.
Mills JL, DerSimonian R, Raymond E, Morrow JD et al. (1999) Prostacyclin and thromboxane changes predating clinical onset of preeclampsia: a multicenter prospective study. Jama 282, 356-362.
Moffett-King A (2002) Natural killer cells and pregnancy. Nat Rev Immunol 2, 656-663. Morgan L, Crawshaw S, Baker PN, Broughton Pipkin F et al. (1999) Maternal and fetal
angiotensinogen gene allele sharing in pre-eclampsia. Br J Obstet Gynaecol 106, 244-251.
Morrison ER, Miedzybrodzka ZH, Campbell DM, Haites NE et al. (2002) Prothrombotic genotypes are not associated with pre-eclampsia and gestational hypertension: results from a large population-based study and systematic review. Thromb Haemost 87, 779-785.
Morton NE (1955) Sequential tests for the detection of linkage. Am J Hum Genet 7, 277-318. Moses EK, Fitzpatrick E, Freed KA, Dyer TD et al. (2006) Objective prioritization of positional
candidate genes at a quantitative trait locus for pre-eclampsia on 2q22. Mol Hum Reprod 12, 505-512.
Moses EK, Lade JA, Guo G, Wilton AN et al. (2000) A genome scan in families from Australia and New Zealand confirms the presence of a maternal susceptibility locus for pre-eclampsia, on chromosome 2. Am J Hum Genet 67, 1581-1585.
Mutze S, Rudnik-Schoneborn S, Zerres K and Rath W (2008) Genes and the preeclampsia syndrome. J Perinat Med 36, 38-58.
Myatt L, Brewer A and Brockman DE (1991) The action of nitric oxide in the perfused human fetal-placental circulation. Am J Obstet Gynecol 164, 687-692.
Myatt L and Webster RP (2009) Vascular biology of preeclampsia. J Thromb Haemost 7, 375-384.
National High Blood Pressure Education Program (NHBPEP) (2000) Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 183, S1-S22.
Ness RB, Markovic N, Harger G and Day R (2004) Barrier methods, length of preconception intercourse, and preeclampsia. Hypertens Pregnancy 23, 227-235.
Ness RB and Roberts JM (1996) Heterogeneous causes constituting the single syndrome of preeclampsia: a hypothesis and its implications. Am J Obstet Gynecol 175, 1365-1370.
Newstead J, von Dadelszen P and Magee LA (2007) Preeclampsia and future cardiovascular risk. Expert Rev Cardiovasc Ther 5, 283-294.

60
Ng K, Pullirsch D, Leeb M and Wutz A (2007) Xist and the order of silencing. EMBO Rep 8, 34-39.
Ngoc NT, Merialdi M, Abdel-Aleem H, Carroli G et al. (2006) Causes of stillbirths and early neonatal deaths: data from 7993 pregnancies in six developing countries. Bull World Health Organ 84, 699-705.
O'Brien M, Dausset J, Carosella ED and Moreau P (2000) Analysis of the role of HLA-G in preeclampsia. Hum Immunol 61, 1126-1131.
O'Connell JR and Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63, 259-266.
Ohta K, Kobashi G, Hata A, Yamada H et al. (2003) Association between a variant of the glutathione S-transferase P1 gene (GSTP1) and hypertension in pregnancy in Japanese: interaction with parity, age, and genetic factors. Semin Thromb Hemost 29, 653-659.
O'Shaughnessy KM, Fu B, Downing S and Morris NH (2001) Thrombophilic polymorphisms in pre-eclampsia: altered frequency of the functional 98C>T polymorphism of glycoprotein IIIa. J Med Genet 38, 775-777.
Oudejans CB, Mulders J, Lachmeijer AM, van Dijk M et al. (2004) The parent-of-origin effect of 10q22 in pre-eclamptic females coincides with two regions clustered for genes with down-regulated expression in androgenetic placentas. Mol Hum Reprod 10, 589-598.
Oudejans CB, van Dijk M, Oosterkamp M, Lachmeijer A et al. (2007) Genetics of preeclampsia: paradigm shifts. Hum Genet 120, 607-612.
Pang ZJ and Xing FQ (2003) Expression profile of trophoblast invasion-associated genes in the pre-eclamptic placenta. Br J Biomed Sci 60, 97-101.
Pang ZJ and Xing FQ (2004) DNA microarrays detect the expression of apoptosis-related genes in preeclamptic placentas. J Perinat Med 32, 25-30.
Papageorghiou AT, Yu CK and Nicolaides KH (2004) The role of uterine artery Doppler in predicting adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol 18, 383-396.
Papazoglou D, Galazios G, Koukourakis MI, Panagopoulos I et al. (2004) Vascular endothelial growth factor gene polymorphisms and pre-eclampsia. Mol Hum Reprod 10, 321-324.
Parham P (2004) NK cells and trophoblasts: partners in pregnancy. J Exp Med 200, 951-955. Parikh SM and Karumanchi SA (2008) Putting pressure on pre-eclampsia. Nat Med 14, 810-
812. Pasmant E, Laurendeau I, Heron D, Vidaud M et al. (2007) Characterization of a germ-line
deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 67, 3963-3969.
Pattison NS, Chamley LW, McKay EJ, Liggins GC et al. (1993) Antiphospholipid antibodies in pregnancy: prevalence and clinical associations. Br J Obstet Gynaecol 100, 909-913.
Pegoraro RJ, Hira B, Rom L and Moodley J (2003) Plasminogen activator inhibitor type 1 (PAI1) and platelet glycoprotein IIIa (PGIIIa) polymorphisms in Black South Africans with pre-eclampsia. Acta Obstet Gynecol Scand 82, 313-317.
Peltonen L (1996) Identification of Disease Genes in Genetic Isolates. Methods 9, 129-135. Perry GH, Dominy NJ, Claw KG, Lee AS et al. (2007) Diet and the evolution of human
amylase gene copy number variation. Nat Genet 39, 1256-1260. Peterson H, Laivuori H, Kerkela E, Jiao H et al. (2009) ROCK2 allelic variants are not
associated with pre-eclampsia susceptibility in the Finnish population. Mol Hum Reprod 15, 443-449.
Pijnenborg R, Vercruysse L and Hanssens M (2006) The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 27, 939-958.

61
Poston L, Briley AL, Seed PT, Kelly FJ et al. (2006) Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomised placebo-controlled trial. Lancet 367, 1145-1154.
Poston L and Raijmakers MT (2004) Trophoblast oxidative stress, antioxidants and pregnancy outcome--a review. Placenta 25 Suppl A, S72-78.
Powers RW, Evans RW, Ness RB, Crombleholme WR et al. (2001) Homocysteine and cellular fibronectin are increased in preeclampsia, not transient hypertension of pregnancy. Hypertens Pregnancy 20, 69-77.
Powers RW, Roberts JM, Cooper KM, Gallaher MJ et al. (2005) Maternal serum soluble fms-like tyrosine kinase 1 concentrations are not increased in early pregnancy and decrease more slowly postpartum in women who develop preeclampsia. Am J Obstet Gynecol 193, 185-191.
Purcell S, Cherny SS and Sham PC (2003) Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149-150.
Redman CW and Sargent IL (2003) Pre-eclampsia, the placenta and the maternal systemic inflammatory response--a review. Placenta 24 Suppl A, S21-27.
Redman CW and Sargent IL (2008) Circulating microparticles in normal pregnancy and pre-eclampsia. Placenta 29 Suppl A, S73-77.
Renaud SJ, Macdonald-Goodfellow SK and Graham CH (2007) Coordinated regulation of human trophoblast invasiveness by macrophages and interleukin 10. Biol Reprod 76, 448-454.
Reue K (2009) The lipin family: mutations and metabolism. Curr Opin Lipidol 20, 165-170. Riento K and Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol
Cell Biol 4, 446-456. Rigourd V, Chauvet C, Chelbi ST, Rebourcet R et al. (2008) STOX1 overexpression in
choriocarcinoma cells mimics transcriptional alterations observed in preeclamptic placentas. PLoS One 3, e3905.
Rigourd V, Chelbi S, Chauvet C, Rebourcet R et al. (2009) Re-evaluation of the role of STOX1 transcription factor in placental development and preeclampsia. J Reprod Immunol 82, 174-181.
Roberts CL, Algert CS, Morris JM, Ford JB et al. (2005) Hypertensive disorders in pregnancy: a population-based study. Med J Aust 182, 332-335.
Roberts DJ and Post MD (2008) The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol 61, 1254-1260.
Roberts JM (1998) Endothelial dysfunction in preeclampsia. Semin Reprod Endocrinol 16, 5-15.
Roberts JM and Cooper DW (2001) Pathogenesis and genetics of pre-eclampsia. Lancet 357, 53-56.
Roberts JM and Hubel CA (2009) The two stage model of preeclampsia: variations on the theme. Placenta 30 Suppl A, S32-37.
Roberts JM and Lain KY (2002) Recent Insights into the pathogenesis of pre-eclampsia. Placenta 23, 359-372.
Robillard PY, Hulsey TC, Perianin J, Janky E et al. (1994) Association of pregnancy-induced hypertension with duration of sexual cohabitation before conception. Lancet 344, 973-975.
Rodie VA, Freeman DJ, Sattar N and Greer IA (2004) Pre-eclampsia and cardiovascular disease: metabolic syndrome of pregnancy? Atherosclerosis 175, 189-202.
Roten LT, Johnson MP, Forsmo S, Fitzpatrick E et al. (2009) Association between the candidate susceptibility gene ACVR2A on chromosome 2q22 and pre-eclampsia in a large Norwegian population-based study (the HUNT study). Eur J Hum Genet 17, 250-257.

62
Ruiz-Llorente S, Montero-Conde C, Milne RL, Moya CM et al. (2007) Association study of 69 genes in the ret pathway identifies low-penetrance loci in sporadic medullary thyroid carcinoma. Cancer Res 67, 9561-9567.
Saarela T, Hiltunen M, Helisalmi S, Heinonen S et al. (2005) Tumour necrosis factor-alpha gene haplotype is associated with pre-eclampsia. Mol Hum Reprod 11, 437-440.
Saftlas AF, Beydoun H and Triche E (2005) Immunogenetic determinants of preeclampsia and related pregnancy disorders: a systematic review. Obstet Gynecol 106, 162-172.
Saito S and Sakai M (2003) Th1/Th2 balance in preeclampsia. J Reprod Immunol 59, 161-173. Saito S, Shiozaki A, Nakashima A, Sakai M et al. (2007) The role of the immune system in
preeclampsia. Mol Aspects Med 28, 192-209. Salmela E, Lappalainen T, Fransson I, Andersen PM et al. (2008) Genome-wide analysis of
single nucleotide polymorphisms uncovers population structure in Northern Europe. PLoS One 3, e3519.
Salonen Ros H, Lichtenstein P, Lipworth L and Cnattingius S (2000) Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am J Med Genet 91, 256-260.
Samani NJ, Erdmann J, Hall AS, Hengstenberg C et al. (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357, 443-453.
Sanderson M, Daling JR, Doody DR and Malone KE (2006) Perinatal factors and mortality from breast cancer. Cancer Epidemiol Biomarkers Prev 15, 1984-1987.
Sanderson M, Williams MA, Daling JR, Holt VL et al. (1998) Maternal factors and breast cancer risk among young women. Paediatr Perinat Epidemiol 12, 397-407.
Sanger F, Nicklen S and Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74, 5463-5467.
Sattar N, Bendomir A, Berry C, Shepherd J et al. (1997) Lipoprotein subfraction concentrations in preeclampsia: pathogenic parallels to atherosclerosis. Obstet Gynecol 89, 403-408.
Saxena R, Voight BF, Lyssenko V, Burtt NP et al. (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316, 1331-1336.
Schork NJ, Murray SS, Frazer KA and Topol EJ (2009) Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev 19, 212-219.
Schunkert H, Gotz A, Braund P, McGinnis R et al. (2008) Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation 117, 1675-1684.
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ et al. (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316, 1341-1345.
Seidman DS, Laor A, Gale R, Stevenson DK et al. (1991) Pre-eclampsia and offspring's blood pressure, cognitive ability and physical development at 17-years-of-age. Br J Obstet Gynaecol 98, 1009-1014.
Serrano NC, Casas JP, Diaz LA, Paez C et al. (2004) Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension 44, 702-707.
Service S, DeYoung J, Karayiorgou M, Roos JL et al. (2006) Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 38, 556-560.
Sevon P, Ollikainen V, Onkamo P, Toivonen HT et al. (2001) Mining associations between genetic markers, phenotypes, and covariates. Genet Epidemiol 21 Suppl 1, S588-593.
Shah NC, Pringle S and Struthers A (2006) Aldosterone blockade over and above ACE-inhibitors in patients with coronary artery disease but without heart failure. J Renin Angiotensin Aldosterone Syst 7, 20-30.

63
Sharma A, Satyam A and Sharma JB (2007) Leptin, IL-10 and inflammatory markers (TNF-alpha, IL-6 and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am J Reprod Immunol 58, 21-30.
Shevell T, Malone FD, Vidaver J, Porter TF et al. (2005) Assisted reproductive technology and pregnancy outcome. Obstet Gynecol 106, 1039-1045.
Sibai BM, el-Nazer A and Gonzalez-Ruiz A (1986) Severe preeclampsia-eclampsia in young primigravid women: subsequent pregnancy outcome and remote prognosis. Am J Obstet Gynecol 155, 1011-1016.
Sibai BM, Ewell M, Levine RJ, Klebanoff MA et al. (1997) Risk factors associated with preeclampsia in healthy nulliparous women. The Calcium for Preeclampsia Prevention (CPEP) Study Group. Am J Obstet Gynecol 177, 1003-1010.
Sibai BM, Hauth J, Caritis S, Lindheimer MD et al. (2000) Hypertensive disorders in twin versus singleton gestations. National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. Am J Obstet Gynecol 182, 938-942.
Sitras V, Paulssen RH, Gronaas H, Leirvik J et al. (2009) Differential placental gene expression in severe preeclampsia. Placenta 30, 424-433.
Skjaerven R, Vatten LJ, Wilcox AJ, Ronning T et al. (2005) Recurrence of pre-eclampsia across generations: exploring fetal and maternal genetic components in a population based cohort. Bmj 331, 877.
Skjaerven R, Wilcox AJ and Lie RT (2002) The interval between pregnancies and the risk of preeclampsia. N Engl J Med 346, 33-38.
Skupski DW, Nelson S, Kowalik A, Polaneczky M et al. (1996) Multiple gestations from in vitro fertilization: successful implantation alone is not associated with subsequent preeclampsia. Am J Obstet Gynecol 175, 1029-1032.
Slowinski T, Neumayer HH, Stolze T, Gossing G et al. (2002) Endothelin system in normal and hypertensive pregnancy. Clin Sci (Lond) 103 Suppl 48, 446S-449S.
Smarason AK, Sargent IL, Starkey PM and Redman CW (1993) The effect of placental syncytiotrophoblast microvillous membranes from normal and pre-eclamptic women on the growth of endothelial cells in vitro. Br J Obstet Gynaecol 100, 943-949.
Smith GC, Pell JP and Walsh D (2001) Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129,290 births. Lancet 357, 2002-2006.
Soejima H (2009) [Epigenetics-related diseases and analytic methods]. Rinsho Byori 57, 769-778.
Somlyo AP and Somlyo AV (2000) Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 522 Pt 2, 177-185.
Spielman RS, McGinnis RE and Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52, 506-516.
Staff AC, Braekke K, Harsem NK, Lyberg T et al. (2005) Circulating concentrations of sFlt1 (soluble fms-like tyrosine kinase 1) in fetal and maternal serum during pre-eclampsia. Eur J Obstet Gynecol Reprod Biol 122, 33-39.
Stamilio DM, Sehdev HM, Morgan MA, Propert K et al. (2000) Can antenatal clinical and biochemical markers predict the development of severe preeclampsia? Am J Obstet Gynecol 182, 589-594.
Storz G, Altuvia S and Wassarman KM (2005) An abundance of RNA regulators. Annu Rev Biochem 74, 199-217.
Strachan T and Read AP (2004) Human momecular genetics. New York (NY): Garland Press Stranger BE, Forrest MS, Dunning M, Ingle CE et al. (2007) Relative impact of nucleotide and
copy number variation on gene expression phenotypes. Science 315, 848-853.

64
Sutherland A, Cooper DW, Howie PW, Liston WA et al. (1981) The indicence of severe pre-eclampsia amongst mothers and mothers-in-law of pre-eclamptics and controls. Br J Obstet Gynaecol 88, 785-791.
Tanioka T, Hattori A, Masuda S, Nomura Y et al. (2003) Human leukocyte-derived arginine aminopeptidase. The third member of the oxytocinase subfamily of aminopeptidases. J Biol Chem 278, 32275-32283.
Tenhola S, Rahiala E, Halonen P, Vanninen E et al. (2006) Maternal preeclampsia predicts elevated blood pressure in 12-year-old children: evaluation by ambulatory blood pressure monitoring. Pediatr Res 59, 320-324.
Tenhola S, Rahiala E, Martikainen A, Halonen P et al. (2003) Blood pressure, serum lipids, fasting insulin, and adrenal hormones in 12-year-old children born with maternal preeclampsia. J Clin Endocrinol Metab 88, 1217-1222.
Thadhani R, Mutter WP, Wolf M, Levine RJ et al. (2004) First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab 89, 770-775.
Thornton JG and Macdonald AM (1999) Twin mothers, pregnancy hypertension and pre-eclampsia. Br J Obstet Gynaecol 106, 570-575.
Thornton JG and Onwude JL (1991) Pre-eclampsia: discordance among identical twins. Bmj 303, 1241-1242.
Toivonen HT, Onkamo P, Vasko K, Ollikainen V et al. (2000) Data mining applied to linkage disequilibrium mapping. Am J Hum Genet 67, 133-145.
Treloar SA, Cooper DW, Brennecke SP, Grehan MM et al. (2001) An Australian twin study of the genetic basis of preeclampsia and eclampsia. Am J Obstet Gynecol 184, 374-381.
Trupin LS, Simon LP and Eskenazi B (1996) Change in paternity: a risk factor for preeclampsia in multiparas. Epidemiology 7, 240-244.
van Dijk M, Mulders J, Poutsma A, Konst AA et al. (2005) Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet 37, 514-519.
van Pampus MG, Dekker GA, Wolf H, Huijgens PC et al. (1999) High prevalence of hemostatic abnormalities in women with a history of severe preeclampsia. Am J Obstet Gynecol 180, 1146-1150.
van Pampus MG, Wolf H, Weijmar Schultz WC, Neeleman J et al. (2004) Posttraumatic stress disorder following preeclampsia and HELLP syndrome. J Psychosom Obstet Gynaecol 25, 183-187.
Venter JC, Adams MD, Myers EW, Li PW et al. (2001) The sequence of the human genome. Science 291, 1304-1351.
von Dadelszen P, Hurst G and Redman CW (1999) Supernatants from co-cultured endothelial cells and syncytiotrophoblast microvillous membranes activate peripheral blood leukocytes in vitro. Hum Reprod 14, 919-924.
Wang Y, Walsh SW and Kay HH (1992) Placental lipid peroxides and thromboxane are increased and prostacyclin is decreased in women with preeclampsia. Am J Obstet Gynecol 167, 946-949.
Watson JD and Crick FH (1953) The structure of DNA. Cold Spring Harb Symp Quant Biol 18, 123-131.
Wellcome Trust Case Control Consortium (WTCCC) (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661-678.
Wheeler DA, Srinivasan M, Egholm M, Shen Y et al. (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872-876.

65
Wiedmann S, Fischer M, Koehler M, Neureuther K et al. (2008) Genetic variants within the LPIN1 gene, encoding lipin, are influencing phenotypes of the metabolic syndrome in humans. Diabetes 57, 209-217.
Wikstrom AK, Haglund B, Olovsson M and Lindeberg SN (2005) The risk of maternal ischaemic heart disease after gestational hypertensive disease. Bjog 112, 1486-1491.
Wray NR, Goddard ME and Visscher PM (2008) Prediction of individual genetic risk of complex disease. Curr Opin Genet Dev 18, 257-263.
Xue F and Michels KB (2007) Intrauterine factors and risk of breast cancer: a systematic review and meta-analysis of current evidence. Lancet Oncol 8, 1088-1100.
Yasuda M, Takakuwa K, Tokunaga A and Tanaka K (1995) Prospective studies of the association between anticardiolipin antibody and outcome of pregnancy. Obstet Gynecol 86, 555-559.
Zeggini E, Weedon MN, Lindgren CM, Frayling TM et al. (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316, 1336-1341.
Zhang C, Austin MA, Edwards KL, Farin FM et al. (2006) Functional variants of the lipoprotein lipase gene and the risk of preeclampsia among non-Hispanic Caucasian women. Clin Genet 69, 33-39.
Zhang F, Carvalho CM and Lupski JR (2009) Complex human chromosomal and genomic rearrangements. Trends Genet 25, 298-307.
Zhang J, Klebanoff MA and Roberts JM (2001) Prediction of adverse outcomes by common definitions of hypertension in pregnancy. Obstet Gynecol 97, 261-267.
Zhou Y, Damsky CH and Fisher SJ (1997) Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest 99, 2152-2164.
Zhu DL, Wang HY, Xiong MM, He X et al. (2001) Linkage of hypertension to chromosome 2q14-q23 in Chinese families. J Hypertens 19, 55-61.
Zusterzeel PL, Peters WH, Visser W, Hermsen KJ et al. (2001) A polymorphism in the gene for microsomal epoxide hydrolase is associated with pre-eclampsia. J Med Genet 38, 234-237.
Zusterzeel PL, Visser W, Peters WH, Merkus HW et al. (2000) Polymorphism in the glutathione S-transferase P1 gene and risk for preeclampsia. Obstet Gynecol 96, 50-54.





















