
Multi-criteria evaluation of wastewater treatment plant control strategies under uncertainty
Upload
independentCategory
view
2download
0
1. Introduction
2. General considerations
regarding gene therapy for
MPS
3. Viral vectors
4. Non-viral gene therapy
approaches
5. Combined therapies
6. Immune response
7. Conclusion
8. Expert opinion
Review
Gene delivery strategies forthe treatment ofmucopolysaccharidosesGuilherme Baldo, Roberto Giugliani† & Ursula Matte†Programa de Pos-Graduacao em genetica e Biologia Molecular, UFRGS, Porto Alegre, Brazil
Introduction: Mucopolysaccharidosis (MPS) disorders are genetic diseases
caused by deficiencies in the lysosomal enzymes responsible for the degrada-
tion of glycosaminoglycans. Current treatments are not able to correct all
disease symptoms and are not available for all MPS types, which makes gene
therapy especially relevant. Multiple gene therapy approaches have been
tested for different types of MPS, and our aim in this study is to critically
analyze each of them.
Areas covered: In this review, we have included the major studies that
describe the use of adeno-associated retroviral and lentiviral vectors, as well
as relevant non-viral approaches for MPS disorders.
Expert opinion: Some protocols such as the use of adeno-associated vectors
and lentiviral vectors are approaching the clinic for these disorders and, along
with combined approaches, seem to be the future of gene therapy for MPS.
Keywords: lysosomal storage disorders, mucopolysaccharidosis, non-viral vectors, viral vectors
Expert Opin. Drug Deliv. [Early Online]
1. Introduction
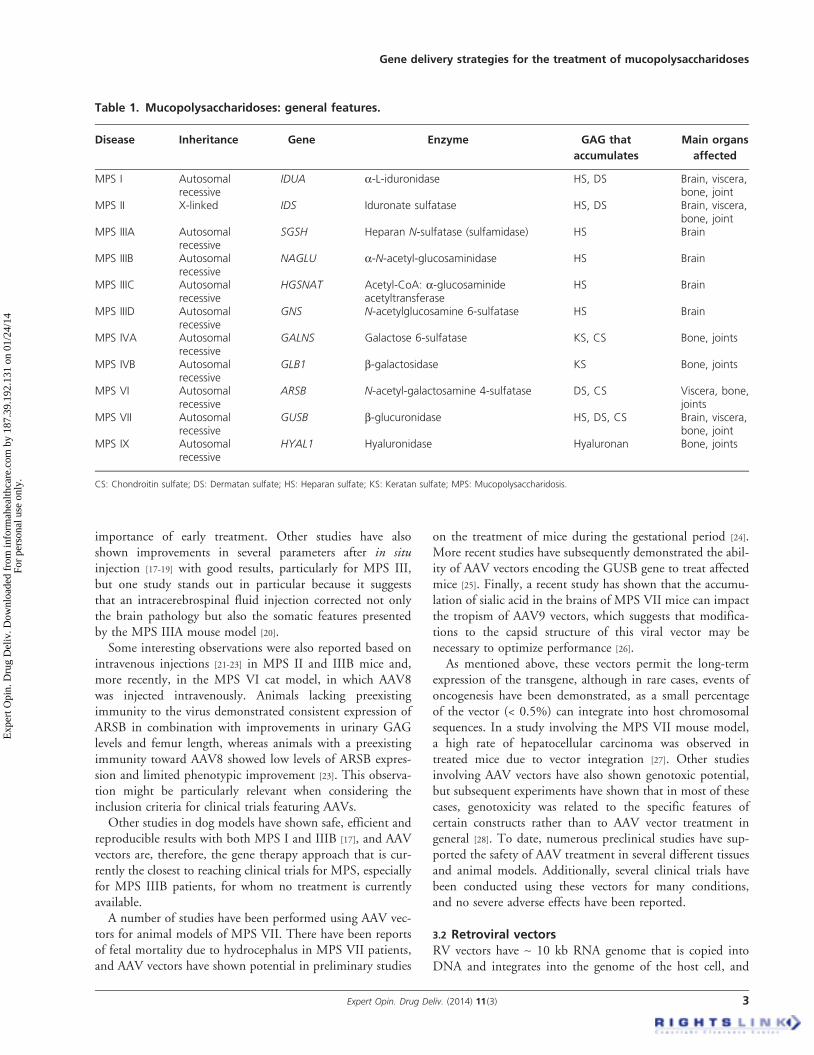
The mucopolysaccharidoses (MPS) are a group of 11 genetic diseases caused by adeficiency in one of the lysosomal enzymes that participate in the multistep degra-dation of a diverse group of sugar polymers called glycosaminoglycans (GAGs).Depending on the enzyme affected, the degradation of different types of GAGs,either alone or in combination, is blocked [1]. Accordingly, different clinical signscan be observed depending on which GAG degradation pathway is involved. Themain characteristics of each MPS disease, including the gene involved, the deficientenzyme, the partially metabolized GAG and the organs affected, are summarizedin Table 1. All MPS disorders are inherited as autosomal recessive traits except forthe X-linked recessive MPS II, and most of these syndromes display a phenotypethat varies from attenuated to severe, which in most of the cases correlates withthe mutations found and the level of enzyme activity produced in each case [2,3].
Therapies designed to treat these disorders must take into consideration thecharacteristics of the enzymes that are deficient in the MPS syndromes: i) theseenzymes are soluble; ii) they are active at lower pH; iii) they can be secreted intothe bloodstream and taken up by adjacent cells and tissues, traditionally viamannose-6-phosphate receptors (M6PR); and iv) they can produce clear benefitseven at expression levels that are 10 -- 20% of normal [4]. These characteristics haveimportant implications in the sense that there is no need to transfect/transduce everysingle cell to obtain an effect, in contrast to other diseases such as cystic fibrosis. Themodified cells can secrete the enzyme into the blood and therefore cross-correct otherdeficient cells and tissues. Furthermore, overexpression does not seem to lead to sideeffects, because these enzymes are active only inside the lysosome [5].
Based on the aforementioned mechanism of cross-correction, two main treat-ments have been developed for MPS: i) enzyme replacement therapy (ERT),
10.1517/17425247.2014.880689 © 2014 Informa UK, Ltd. ISSN 1742-5247, e-ISSN 1744-7593 1All rights reserved: reproduction in whole or in part not permitted
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

which is currently available for MPS I, II and VI, and whichis also in development for some other MPS syndromes; andii) hematopoietic stem cell transplantation (HSCT). ERTinvolves the intravenous injection of a recombinant enzyme,whereas HSCT is effective due to the migration of cells intoorgans where they secrete the enzyme both locally and intothe blood [4]. The main limitations of ERT concern theinability of the intravenously administered enzyme to crossthe blood--brain barrier (BBB) and reach the brain, theneed for weekly infusions, the high cost of the drug andthe formation of neutralizing antibodies (ABs) against therecombinant enzyme [6]. On the other hand, HSCT onlyseems advantageous over ERT before the onset of cognitivedecline, and HSCT is not an effective treatment for all formsof MPS [7]. Intrathecal ERT has been proposed as an alterna-tive strategy to bypass the BBB and is presently in develop-ment for the treatment of the severe forms of MPS I, II, aswell as IIIA [8].Considering the limitations of the current treatments, gene
therapy has arisen as a promising alternative approach. How-ever, despite the similarities among the different types ofMPS, there are also prominent differences if one considersthe characteristics of the enzymes and the modificationsneeded for their activity, as well as the tissues and organsaffected in each case. For example, all sulfatases are modifiedin the endoplasmic reticulum by sulfatase-modifying factor1 (SUMF1) or SUMF2, which catalyzes the conversion of acysteine into the c-a-formylglycine residue required forcatalytic activity, and this information is important whenconsidering gene therapy for MPS disorders caused bysulfatase deficiencies (MPS II, IIIA, IIID, IVA and VI) [9].In addition, almost all lysosomal enzymes need to have amannose-6-phosphate marker to be taken up by other cells,and in some cases, it has been shown that overexpression ofa lysosomal enzyme above normal levels may result in theincomplete modification of the enzyme [5]. As another exam-ple, MPS III has a primarily neurocognitive component, yetintravenously administered lysosomal enzymes do not crossthe BBB; therefore, different approaches such as the in situinjection of vectors should also be considered [10].
2. General considerations regarding genetherapy for MPS
There are several possible protocols for gene therapy of MPS.The traditional approach involves the direct injection of avector that carries the transgene and allows for its long-termexpression (in vivo gene therapy). Another possible approachis to harvest deficient cells, correct them in vitro and injectthem back into the patient (ex vivo gene therapy). Differenttypes of vectors can be used, and although viruses usuallyfacilitate the long-term expression of the transgene, severalnon-viral techniques have also recently been discovered andused for the same purpose. Furthermore, different routes ofinjection have been tested, including intravenous [11], intra-muscular [12] and intracranial [10], just to mention a few.Regarding the ideal timing for treatment, neonatal adminis-tration seems to be more efficient and cost-effective [13], butwhen translating this to patients, neonatal treatment wouldrequire a suspected case of MPS based either on a previousMPS case in the family or on the results of neonatalscreening [14].
Below, we will discuss the outcomes obtained using thedifferent gene therapy strategies that have already beenpublished.
3. Viral vectors
The use of viral vectors for gene therapy involves the replace-ment or removal of some viral genes that control replicationand the addition of the therapeutic gene. Viral vectors areusually produced using packaging systems that express theviral proteins in trans (i.e., not on the same plasmid as thetherapeutic gene) to avoid recombination events. Differentvector serotypes express different capsid/envelope proteinsand determine the affinity for different cell types.
The viral vectors used in the gene therapy protocols forMPS are largely derived from adeno-associated viruses(AAVs), retroviruses (RVs) or lentiviruses (LVs). However,some other viral vectors have also been studied.
3.1 Adeno-associated vectorsIn recent years, special emphasis has been placed on the devel-opment of preclinical gene therapy protocols involving AAVvectors. These vectors have ~ 5 kb single-stranded DNAgenome, which is converted into double-stranded DNA thatforms concatamers in the cell nucleus. Although they are main-tained for long periods of time as episomes in non-replicatingcells, they may also integrate into the chromosome [15].
Different serotypes of AAVs have been widely used inpreclinical studies of several types of MPS (Table 2).Wolf et al. [16] achieved high IDUA activity (40-fold normal)in the brains of MPS I mice after a single neonatal intracere-broventricular injection of an AAV8 vector and observed bio-chemical and behavioral improvements that highlighted the
Article highlights.
. Mucopolysaccharidoses (MPS) are lysosomal disordersthat lead to glycosaminoglycan storage.
. Among the gene therapy protocols using viralapproaches for MPS, those using retrovirus, lentivirusand adeno-associated virus have shown the best results.
. Non-viral approaches such as transposons andencapsulated cells have also shown improvements.
. Major concerns include immune responses to thetherapeutic protein or to the vector, as well asinsertional oncogenesis.
This box summarizes key points contained in the article.
G. Baldo et al.
2 Expert Opin. Drug Deliv. (2014) 11(3)
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.
importance of early treatment. Other studies have alsoshown improvements in several parameters after in situinjection [17-19] with good results, particularly for MPS III,but one study stands out in particular because it suggeststhat an intracerebrospinal fluid injection corrected not onlythe brain pathology but also the somatic features presentedby the MPS IIIA mouse model [20].
Some interesting observations were also reported based onintravenous injections [21-23] in MPS II and IIIB mice and,more recently, in the MPS VI cat model, in which AAV8was injected intravenously. Animals lacking preexistingimmunity to the virus demonstrated consistent expression ofARSB in combination with improvements in urinary GAGlevels and femur length, whereas animals with a preexistingimmunity toward AAV8 showed low levels of ARSB expres-sion and limited phenotypic improvement [23]. This observa-tion might be particularly relevant when considering theinclusion criteria for clinical trials featuring AAVs.
Other studies in dog models have shown safe, efficient andreproducible results with both MPS I and IIIB [17], and AAVvectors are, therefore, the gene therapy approach that is cur-rently the closest to reaching clinical trials for MPS, especiallyfor MPS IIIB patients, for whom no treatment is currentlyavailable.
A number of studies have been performed using AAV vec-tors for animal models of MPS VII. There have been reportsof fetal mortality due to hydrocephalus in MPS VII patients,and AAV vectors have shown potential in preliminary studies
on the treatment of mice during the gestational period [24].More recent studies have subsequently demonstrated the abil-ity of AAV vectors encoding the GUSB gene to treat affectedmice [25]. Finally, a recent study has shown that the accumu-lation of sialic acid in the brains of MPS VII mice can impactthe tropism of AAV9 vectors, which suggests that modifica-tions to the capsid structure of this viral vector may benecessary to optimize performance [26].
As mentioned above, these vectors permit the long-termexpression of the transgene, although in rare cases, events ofoncogenesis have been demonstrated, as a small percentageof the vector (< 0.5%) can integrate into host chromosomalsequences. In a study involving the MPS VII mouse model,a high rate of hepatocellular carcinoma was observed intreated mice due to vector integration [27]. Other studiesinvolving AAV vectors have also shown genotoxic potential,but subsequent experiments have shown that in most of thesecases, genotoxicity was related to the specific features ofcertain constructs rather than to AAV vector treatment ingeneral [28]. To date, numerous preclinical studies have sup-ported the safety of AAV treatment in several different tissuesand animal models. Additionally, several clinical trials havebeen conducted using these vectors for many conditions,and no severe adverse effects have been reported.
3.2 Retroviral vectorsRV vectors have ~ 10 kb RNA genome that is copied intoDNA and integrates into the genome of the host cell, and
Table 1. Mucopolysaccharidoses: general features.
Disease Inheritance Gene Enzyme GAG that
accumulates
Main organs
affected
MPS I Autosomalrecessive
IDUA a-L-iduronidase HS, DS Brain, viscera,bone, joint
MPS II X-linked IDS Iduronate sulfatase HS, DS Brain, viscera,bone, joint
MPS IIIA Autosomalrecessive
SGSH Heparan N-sulfatase (sulfamidase) HS Brain
MPS IIIB Autosomalrecessive
NAGLU a-N-acetyl-glucosaminidase HS Brain
MPS IIIC Autosomalrecessive
HGSNAT Acetyl-CoA: a-glucosaminideacetyltransferase
HS Brain
MPS IIID Autosomalrecessive
GNS N-acetylglucosamine 6-sulfatase HS Brain
MPS IVA Autosomalrecessive
GALNS Galactose 6-sulfatase KS, CS Bone, joints
MPS IVB Autosomalrecessive
GLB1 b-galactosidase KS Bone, joints
MPS VI Autosomalrecessive
ARSB N-acetyl-galactosamine 4-sulfatase DS, CS Viscera, bone,joints
MPS VII Autosomalrecessive
GUSB b-glucuronidase HS, DS, CS Brain, viscera,bone, joint
MPS IX Autosomalrecessive
HYAL1 Hyaluronidase Hyaluronan Bone, joints
CS: Chondroitin sulfate; DS: Dermatan sulfate; HS: Heparan sulfate; KS: Keratan sulfate; MPS: Mucopolysaccharidosis.
Gene delivery strategies for the treatment of mucopolysaccharidoses
Expert Opin. Drug Deliv. (2014) 11(3) 3
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

therefore the modification is maintained even after cell repli-cation occurs. These vectors only transduce dividing cells,and depending on the tissue to be transduced, growth factorstimulation (e.g., hepatocyte growth factor in liver cell trans-duction) may increase the transduction efficiency [29]. Themain disadvantage of RVs is the possibility of insertionalmutagenesis, which has occurred in patients in some previoustrials due to activation of proto-oncogenes [29-31]. Also becausethese vectors are only able to transduce dividing cells, their useis limited for certain MPS that are mainly characterized bybrain complications (such as MPS III). A summary of themain studies involving g-RV and LV vectors can be foundin Table 3.Initial studies using a g-RV in MPS I and VII mice and
dogs have shown promising results, achieving high serumenzyme levels and major improvements in somatictissues [32-34]. Interestingly, this vector had no expression inbrain tissue, although some enzyme activity, as well as areduction in GAG levels, was detected [35]. This could beexplained by two main mechanisms: first, cells from theimmune system could be carrying the enzyme through theBBB and secreting it into the brain. Alternatively, whenhigh levels of the enzyme are present in the serum, a smallfraction of the enzyme can cross the BBB using a somewhatinefficient and as-yet-unknown mechanism. This latter
hypothesis has recently been strengthened by studies of MPSI mice with a recombinant enzyme used in ERT [36,37].
Despite these promising results, the initial design of thevector contained a complete long-terminal repeat (LTR)region, which could potentially activate the expression ofother genes after integration. Therefore, a second vector wasdesigned containing a deletion in the enhancer region of the3¢ LTR that results in self-inactivation on transduction bytransferring the deletion at the 3¢ end of the LTR to the 5¢end of the LTR [13]. This second vector was tested in MPS Imice, and neonatal treatment resulted in enzyme levels thatwere 50-fold higher than normal but still smaller than theresults obtained with the LTR-intact vector [13]. Furthermore,a second study using this vector demonstrated stable serumIDUA levels after 8 months, with reductions in brain GAGcontent and improvements in behavioral function [38]. Studiesin the dog model will address the question of long-termefficacy and safety of this vector.
Even though the results from the LTR-intact vector did notreach the clinic due to safety concerns, this approach could beused as a surrogate for the long-term results of ERT. The vec-tor had a liver-specific promoter, which caused hepatocytes toproduce and secrete a large amount of enzyme into the circu-lation, which is similar to the scenario obtained after ERT.With that in mind, dogs were kept alive until they reached
Table 2. Main studies using adeno-associated vectors for mucopolysaccharidoses.
Disease Animal
model
Vector Route and age
of treatment
Outcomes Observation Ref.
MPS I Dog AAV2/5 Intracerebral, 4 -- 10 months Reduction in GAG, increasedenzyme activity
None [17]
MPS I Mouse AAV8 Intracerebroventricular,neonatal
Improvement in behavior,GAG level, enzyme activity
None [16]
MPS II Mouse AAV2/8 Intravenous, 20 weeks Improvement in tissueenzyme and GAG levels
None [21]
MPS IIIA Mouse AAV2/5 Intracerebral, neonatal Improvement in GAG,enzyme activity,inflammation
Delivery of sulfamidaseand SUMF1 genes
[18]
MPS IIIA Mouse AAV9 Intracerebrospinal fluid,2 months
Increase in serum and brainenzyme activity, survival,behavior, reduced GAG
Intracerebrospinal fluidinjection correctedsomatic pathology
[20]
MPS IIIB Dog AAV2/5 Intracerebral, 7 -- 10 months Reduction in GAG, increasedenzyme activity
None [17]
MPS IIIB Mouse AAV9 Intravenous, 4 -- 6 weeks Improvement in GAG,enzyme activity,inflammation, behavior,survival
AAV9 crosses the BBB [22]
MPS IIIB Mouse AAV2/5 Intracerebral, neonatal Improvement in lifespan,motor function and hearing
AAV combined withHSCT had benefits onfew parameters.
[19]
MPS VI Cat AAV2/8 Intravenous, 50 days Improvement in bone lengthand urinary GAG
Better results in catswithout preexistingimmunity to AAV8
[23]
MPS VII Mice AAV9 Intravenous, 6 weeks Improvement in GAGstorage, behavior
Sialic acid storage impairsAAV9 from work properly
[26]
AAV: Adeno-associated virus; BBB: Blood--brain barrier; GAG: Glycosaminoglycans; HSCT: Hematopoietic stem cell transplantation; MPS: Mucopolysaccharidosis;
SUMF1: Sulfatase-modifying factor 1.
G. Baldo et al.
4 Expert Opin. Drug Deliv. (2014) 11(3)
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

up to 10 years of age, yet recent results suggest that someorgans, specially the spine and the heart valves, might behard to correct even with high serum enzyme levels. Research-ers should thus give special consideration to these results,because it is evident that other strategies, including the appli-cation of gene therapy in situ or any other therapy, should beconsidered for the MPS cases involving these hard-to-treatorgans [11,39,40].
The aforementioned approach was also used to treat MPSVI cats; however, arylsulfatase B and the other sulfatasesrequire post-translational modification by SUMF1 and 2 tobe active. Transduction of hepatocytes with the ARSB geneled to an increase in enzyme activity similar to that previouslyobserved in MPS I and VII animal models, suggesting that theSUMF1 activity in the liver was sufficient to result in correctARSB modification and activity. RV-treated MPS VI catsshowed improvements in bone, cartilage and aortic abnormal-ities and reduced aortic valve thickening, whereas spine abnor-malities were not improved [41]. These results are in agreementwith those from MPS I and VII animal models, suggestingthat some organs, especially the spine, receive little to nobenefit from this treatment.
3.3 Lentiviral vectorsLV vectors are integrative vectors that belong to theRetroviridae family; however, unlike the g-RV, they are ableto transduce both dividing and non-dividing cells. Thischaracteristic allows these vectors to exhibit tropism towarda series of tissues including, for example, brain cells. It isalso worth mentioning that, although they are integrative,LV vectors offer safer integration site selection profiles and a
lower degree of genotoxicity compared with g-RV vectors,as discussed in a recent review [42]. Two recent clinical trialshave successfully used LVs for the treatment of two otherconditions --metachromatic leukodystrophy [43] and Wiskott--Aldrich syndrome [44] -- with no signs of aberrant clonalbehavior after 18 months. This ex vivo approach seems partic-ularly promising for the treatment of MPS diseases as well.
These vectors have been extensively used in situ to delivertherapeutic proteins in mouse models of MPS, especially inMPS III. One initial study [45] performed a single intracranialinjection of the LV-NAGLU vector in the fimbria, which ledto sustained and widespread enzyme delivery in the mousebrain over the long term. The following year, a group study-ing MPS IIIB mice showed that whereas the intravenousinjection of an LV vector produced improvements in somaticparameters and conflicting results in the brain [46], betterresults were achieved in the brain after intracranial injectionof the vector [47], which indicates that multisite injectionmight be necessary to completely correct the phenotype.
More recently, a study performed in MPS VII mice frombirth or from week 7 evaluated the use of an LV vector,with specific emphasis on the prevention of bone disease.Two weeks after intravenous injection, expression wasobserved in the liver, spleen and bone marrow as well as inthe kidney, lung and heart, along with GAG clearance in theseorgans. However, only mice treated from birth had any clear-ance of neuronal storage. Additionally, significant decreases inbone mineral volume and bone surface density and thicknesswere observed in both treatment groups [48]. However, lumbarand femoral bone lengths did not change upon treatment,suggesting, as shown by the studies using RV vectors, that
Table 3. Main studies using retroviral vectors for gene therapy of mucopolysaccharidoses.
Condition Animal
model
Vector Route and age
of treatment
Outcomes Observation Ref.
MPS I Mouse RV i.v., neonatal or4 weeks
Improvement in brain, ear,eye and visceral pathology
Self-inactivating RV vectorused
[13,38]
MPS I Dog RV i.v., neonatal Improvement in bone, joint,aorta, viscera
No brain study performed [34]
MPS IIIA Mouse LV i.v., 5 weeks Improvement in somatictissues, partially in the brain
No benefits on the joint [46]
MPS IIIA Mouse LV i.v. or i.c.v.,6 weeks
Improvement in somatictissues (i.v.) and in the brain(i.c.v.)
Different outcomes based onroute of injection
[47]
MPS IIIB Mouse LV Intracranial,10 weeks
Improvement in enzymeactivity, GAG, behavior
Identifies Bdnf as a possiblebiomarker
[45]
MPS VI Cat RV i.v., neonatal Improvement in bone, joint,aorta, viscera
No benefits on spine [41]
MPS VII Mouse RV i.v., neonatal Partial improvement in aortadiameter, increased serumGUSB
Other organs not analyzed [11]
MPS VII Mouse LV i.v., neonatal orat 7 weeks
Partial improvement inskeletal abnormalities
Neuronal benefits only inneonatal-treated mice
[48]
Bdnf: Brain derived neurotrophic factor; GAGs: Glycosaminoglycans; i.v.: Intravenous; i.c.v.: Intracerebroventricular; LV: Lentiviral vector;
MPS: Mucopolysaccharidosis; RV: g-retroviral vector.
Gene delivery strategies for the treatment of mucopolysaccharidoses
Expert Opin. Drug Deliv. (2014) 11(3) 5
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

some bone abnormalities cannot be ameliorated using theseapproaches.
4. Non-viral gene therapy approaches
Classical plasmid-based vectors have some characteristics thatmake them especially attractive for gene therapy: their ease ofproduction, their capacity for carrying large DNA moleculesand the fact that they are maintained episomally, whichreduces the chances of a deleterious insertional event.Although these vectors are safer, they usually do not havehigh transfection efficiency nor do they allow long-term trans-gene expression due to gene silencing, which hampers the useof these types of vectors for MPS and most other inheriteddiseases as well.A few studies using hydrodynamic injection of a naked
DNA plasmid have shown some improvements in mousemodels of MPS I and VII (Table 4) [49,50]. However, theexpression of the transgene is lost after a few weeks, andhydrodynamic injection cannot be easily performed inhumans, although a recent study described the use of thistype of injection in humans [51]. Therefore, modifications tothese techniques, as well as newly developed gene therapyalternatives using non-viral vectors, have been tested forMPS treatment over the past decade.One interesting approach is the use of minicircle-based vec-
tors, which are resistant to epigenetic gene silencing in vivo.This approach is based on the hypothesis that the loss ofplasmid expression occurs due to the deposition of repressiveheterochromatin on the noncoding bacterial backbonesequences, which are required for plasmid production, butnot for transfection. Therefore, in a study involving MPS Imice, the authors used a system relying on phiC31integrase-mediated recombination to remove the bacterialbackbone elements of the plasmid. This technology, coupledwith immunomodulation, was able to achieve stable IDUAlevels and correct GAG storage in several organs in IDUA
knockout mice [52]. However, one must again note thathydrodynamic injection was performed to deliver the plasmidand that the results were not as promising in the absenceof immunomodulation.
Another way of achieving long-term transgene expressionusing a non-viral approach is by using transposable elements.These plasmid-based vectors carry a transposase, an enzymecapable of integrating DNA into host cells. This technologywas tested in NOD-SCID MPS I mice and was able to pro-duce stable IDUA levels for 18 weeks after hydrodynamicdelivery. The transposon system proved efficacious in correct-ing several clinical manifestations, including hepatomegaly,thickening of the zygomatic arch and accumulation of foamymacrophages in the synovium [53]. However, as it involvesintegration into the host genome, it is still uncertain if thisapproach is safer than viral vectors or if it confers any otheradvantages regarding transfection efficiency.
In the past few years, nucleases have been widely used forgenome editing and gene therapy. These synthetic enzymesare designed to cut the host cell DNA at specific points, afterwhich a second plasmid with the correct version of the genecan integrate into the chromosome via homologous recombi-nation. Zinc finger nucleases have been designed to target theIDUA gene [54], but despite its potential, this technology hasyet to be used for therapeutic purposes in any of the MPS syn-dromes or in any other lysosomal storage disorder for thatmatter.
Cell microencapsulation is an approach in which cells aretrapped within a semipermeable membrane, thereby allowingfor the exchange of metabolites and nutrients with the exter-nal environment, while simultaneously isolating the cellsfrom the immune system [55]. Alginate microcapsules contain-ing genetically modified cells that overexpress the deficientenzyme have been used both in vitro and in vivo to correctthe metabolic defect in different types of MPS.
Barsoum et al. [56] used Madin-Darby canine kidney cellsgenetically modified to overexpress canine Idua enclosed in
Table 4. Non-viral approaches tested in gene therapy studies for mucopolysaccharidoses.
Approach Description Disease Animal
model
Outcomes Observation Ref.
Naked DNA Direct injection of a plasmidinto the circulation
MPS I,MPS VII
Mouse Partial non-lastingimprovements
Hydrodynamic injection [49,50]
Minicirclevector
DNA vector designed to beresistant to epigenetic genesilencing
MPS I Mouse Long-term improvementwhen coupled withimmunomodulation
Hydrodynamic injection [52]
Transposons Use of transposable elementsto integrate exogenous DNAinto host cell
MPS I Mouse Long-term improvementwhen coupled withimmunomodulation
Hydrodynamic injection [53]
Microcapsules Implant in situ ofsemipermeable capsulescontaining cells that secreteenzyme inside
MPS I,MPS II
Mouse Partial improvement insomatic disease
Intraperitoneal implant [58,61]
MPS: Mucopolysaccharidosis.
G. Baldo et al.
6 Expert Opin. Drug Deliv. (2014) 11(3)
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

alginate-poly-l-lysine-alginate microcapsules, which were thenimplanted into the brains of MPS I dogs. Enzyme activity inthe plasma and cerebrospinal fluid through 21 days was lowbut detectable. However, lysosomal inclusions persisted,even in the immediate vicinity of the capsule-implantationsite. Accordingly, no improvements in clinical signs weredetected. On the other hand, Piller Puicher et al. [57], usinga murine myoblast cell line implanted intraperitoneally,showed significant induction of enzyme activity and reductionof GAG accumulation in the MPS I mouse model. Indeed,complete normalization of deposits was achieved in 2 out of12 animals. Intermediate results have been reported byBaldo et al. [58], who implanted microencapsulated recombi-nant baby hamster kidney cells in the peritoneum of thesame mouse model. A transient increase in serum enzymeactivity was observed with decreased, but not normal, GAGlevels in tissues 5 months after treatment. The same groupreported the successful correction of MPS I fibroblastsin vitro for up to 45 days [59]. Additionally, studies in MPSII and MPS VII in vitro and in vivo showed improvementssuch as reductions in the amount of GAG accumulated invisceral organs on peritoneal injection [60-62], as well as inthe brain on injection into the CNS [63,64]. The results fromthis study make this approach particularly interesting for thetreatment of organs that neither ERT nor HSCT are able toreach, such as the brain and the joints.
Despite promising results in vitro, the host responseinduced by the polymers used for encapsulation is still a majorchallenge facing the microencapsulation technique. Microcap-sule graft failure is usually associated with a foreign bodyreaction, with fibrotic overgrowth around the capsules [55].Polymer modifications can be made to reduce the inflamma-tory reaction [65]. In addition, the administration of anti-inflammatory drugs together with the capsules has alsoresulted in improvements in biocompatibility and enzymerelease [66,67].
5. Combined therapies
As denoted by the animal model studies, it is clear that genetherapy alone will not be able to correct all disease symptoms,especially in cases of multisystemic MPS. In particular, theskeletal system, the joints, the heart valves and the brainhave often showed minor or no improvements after treat-ment. With this in mind, scientists have tried some alterna-tives to boost gene therapy protocols with relative success.
As mentioned above, one simple alternative is to performmultiple-site injections of the vector. A recent study comparedintravenous, intracranial or combined injection of an LVvector in MPS IIIB. The combined treatment showed thegreatest increase in lifespan, as well as other improvementssuch as in the circadian rhythm [19].
A second protocol tested the combination of HSCT withgene therapy. A study conducted in MPS IIIA mice comparedcombining HSCT using wild-type donor cells transduced
ex vivo with a LV vector-expressing SGSH versus eitherwild-type donor cell transplant or LV-SGSH-transducedMPS IIIA cells alone [68]. The combined therapy was moreeffective at reducing GAG levels and ameliorating abnormalGAG sulfation. An important point to be considered here isthat HSCT transplantation alone is not efficient in patientswith MPS IIIA, but according to this study, it might be effec-tive if the transplanted cells overexpress the enzyme. A similarapproach was tested in MPS I mice. In this study, HSCs fromnormal mice were transduced with a LV vector to overexpressIDUA, and the combined HSCT + gene therapy treatmentwas able to correct all disease symptoms, whereas HSCT orgene therapy alone presented limitations [69]. This study isespecially important because it is one of the studies that servedas the basis for the two recent successful clinical trials,using LV vectors for metachromatic leukodystrophy andWiskott--Aldrich syndrome, mentioned above.
6. Immune response
The immune system, either through cytotoxic T lympho-cytes (CTL) or AB responses, may impose an additionaldifficulty on successful gene therapy. The immune responsecan be against the therapeutic protein itself or against thevector, and both scenarios have been described in preclinicalstudies of gene therapy for MPS. A CTL response wasdetected when performing gene therapy in adult MPS Imice and cats [70,71], and neonatal treatment seems to reducethis adverse effect, most likely due to the immature immunesystem of the newborn. AB responses were detected afterboth viral and non-viral gene therapy approaches for differ-ent types of MPS [58,72]. In both cases, immunosuppressiveagents can be used to reduce or prevent these adverse effects[70]. A few studies have coupled gene therapy with immuno-modulation to prevent most manifestations of MPS [67,70].These results hold out the promise that gene therapy maybe efficacious in older patients if transient immunosuppres-sion is given.
7. Conclusion
Several viral and non-viral gene therapy approaches have beentested in animal models of MPS in recent years, with relativesuccess and the best results have been obtained after viraltransduction. Systemic administration, ex vivo gene therapycoupled with HSC and in situ injection have all been shownto reduce GAG storage in multiple organs, including the liver,heart, joints and, in some cases, even the brain. A few sites,such as the spine, remain hard to correct, and otherapproaches might be used to achieve better results. At thismoment, at least two protocols seem to hold great promisefor these disorders, and efforts should be made by researchersto bring these to the clinic.
Gene delivery strategies for the treatment of mucopolysaccharidoses
Expert Opin. Drug Deliv. (2014) 11(3) 7
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

8. Expert opinion
Gene therapy for the MPS disorders has shown goodpre-clinical results using different approaches. Viral vectorshave shown the best transduction results, and it is reasonableto think that a relatively simple procedure such as systemicgene therapy could be used to treat these disorders. Withthat in mind, g-RV vectors are unlikely to be used in thenear future, due to their oncogenic potential. Several clinicalstudies with AAV vectors have demonstrated the safety ofthis approach for other conditions and therefore seem to bethe best option for the in vivo gene therapy of MPS disordersat this moment. Importantly, the two recently published clin-ical trials using ex vivo gene therapy with LV-transducedHSCs have demonstrated the safety and efficacy of this proce-dure, and the same approach could easily be translated to thetreatment of MPS disorders. Based on this, it is our opinionthat clinical trials using AAVs or LVs should be consideredif there are reasonable safety data in animal models of MPS.Based on preclinical data, it seems that for the MPS diseases
involving multiple organs, no single gene therapy protocolwill be able to correct all disease symptoms. It is possiblethat a combination of different gene therapy approaches,or the combination of a gene therapy approach with anestablished treatment, will lead to a better outcome.
As an example, the results obtained using intrathecal ERTare promising, and a combined intrathecal ERT + systemicgene therapy approach could be tested for MPS disordersthat involve both visceral organs and the brain.
Even though some studies in animal models have shownthat some enzymes can reach the brain under gene therapyconditions that achieve very high serum enzyme levels, thisscenario is unlikely to happen in patients, because gene ther-apy is not expected to achieve supernormal levels when scalingto humans. Therefore, it is our opinion that in situ vectorinjections can serve as another alternative for reaching thesedifficult-to-correct sites.
Finally, given that these are such rare diseases, effortsshould be made to have researchers in the field join forcesand collaborate in multicenter clinical trials once the mostsuitable protocols are chosen. Based on the results from ani-mal models, early (neonatal) treatment would most likelylead to the best results. The challenge would be to diagnosepatients early enough, which is difficult unless there is aprevious case in the family or the disorder is detected byneonatal screening.
Declaration of interest
Authors have received sponsorship from CNPq.
BibliographyPapers of special note have been highlighted as
either of interest (�) or of considerable interest(��) to readers.
1. Giugliani R. Mucopolysacccharidoses:
from understanding to treatment, a
century of discoveries. Genet Mol Biol
2012;35:924-31
2. de Ru MH, Teunissen QG,
van der Lee JH, et al. Capturing
phenotypic heterogeneity in MPS I:
results of an international consensus
procedure. Orphanet J Rare Dis
2012;7-22
3. Matte U, Yogalingam G, Brooks D,
et al. Identification and characterization
of 13 new mutations in
mucopolysaccharidosis type I patients.
Mol Genet Metab 2003;78:37-43
4. Beck M. Therapy for lysosomal storage
disorders. IUBMB Life 2010;62:33-40
5. Xu L, Mango RL, Sands MS, et al.
Evaluation of pathological manifestations
of disease in mucopolysaccharidosis VII
mice after neonatal hepatic gene therapy.
Mol Ther 2002;6:745-58
6. Giugliani R, Federhen A, Rojas MV,
et al. Mucopolysaccharidosis I, II, and
VI: brief review and guidelines for
treatment. Genet Mol Biol
2010;33:589-604
7. Lau AA, Hannouche H, Rozaklis T,
et al. Allogeneic stem cell transplantation
does not improve neurological deficits
inmucopolysaccharidosis type IIIA mice.
Exp Neurol 2010;225:445-54
8. Munoz-Rojas MV, Vieira T, Costa R,
et al. Intrathecal enzyme replacement
therapy in a patient with
mucopolysaccharidosis type I and
symptomatic spinal cord compression.
Am J Med Genet A 2008;146A:2538-44
9. Ponder KP, Haskins ME. Gene therapy
for mucopolysaccharidosis. Expert Opin
Biol Ther 2007;7(9):1333-45
. An excellent review of early attempts
of gene therapy for MPS.
10. Lau AA, Rozaklis T, Ibanes S, et al.
Helper-dependent canine adenovirus
vector-mediated transgene expression in a
neurodegenerative lysosomal storage
disorder. Gene 2012;491:53-7
11. Baldo G, Wu S, Howe RA, et al.
Pathogenesis of aortic dilatation in
mucopolysaccharidosis VII mice may
involve complement activation.
Mol Genet Metab 2011;104:608-19
12. Tessitore A, Faella A, O’Malley T.
Biochemical, pathological, and skeletal
improvement of mucopolysaccharidosis
VI after gene transfer to liver but not to
muscle. Mol Ther 2008;16:30-7
13. Metcalf JA, Ma X, Linders B, et al.
A self-inactivating gamma-retroviral
vector reduces manifestations of
mucopolysaccharidosis I in mice.
Mol Ther 2010;18:334-42
. This paper reports the results from
a self-inactivating RV vector in
MPS I mice, indicating some
hard-to-treat organs.
14. Giugliani R. Newborn screening for
lysosomal diseases: current status and
potential interface with population
medical genetics in Latin America.
J Inherit Metab Dis 2012;35:871-7
15. Logan GJ, Alexander IE.
Adeno-associated virus vectors:
immunobiology and potential use for
immune modulation. Curr Gene Ther
2012;12:333-43
16. Wolf DA, Lenander AW, Nan Z, et al.
Direct gene transfer to the CNS prevents
emergence of neurologic disease in a
murine model of mucopolysaccharidosis
type I. Neurobiol Dis 2011;43:123-33
G. Baldo et al.
8 Expert Opin. Drug Deliv. (2014) 11(3)
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

17. Ellinwood NM, Ausseil J, Desmaris N,
et al. Safe, efficient, and reproducible
gene therapy of the brain in the dog
models of Sanfilippo and Hurler
syndromes. Mol Ther 2011;19(2):251-9
.. A very complete report of safety and
efficacy of gene therapy in the dog
models of MPS I and III using
AAV vectors.
18. Fraldi A, Hemsley K, Crawley A, et al.
Functional correction of CNS lesions in
an MPS-IIIA mouse model by
intracerebral AAV-mediated delivery of
sulfamidase and SUMF1 genes.
Hum Mol Genet 2007;16:2693-702
19. Heldermon CD, Ohlemiller KK,
Herzog ED, et al. Therapeutic efficacy of
bone marrow transplant, intracranial
AAV-mediated gene therapy, or both in
the mouse model of MPS IIIB.
Mol Ther 2010;18(5):873-80
20. Haurigot V, Marco S, Ribera A, et al.
Whole body correction of
mucopolysaccharidosis IIIA by
intracerebrospinal fluid gene therapy.
J Clin Invest 2013. [Epub ahead of print]
. This paper suggests that injection of
an AAV vector into the cerebrospinal
fluid can achieve not only the brain
but also whole body correction in
MPS IIIA mice.
21. Jung SC, Park ES, Choi EN, et al.
Characterization of a novel
mucopolysaccharidosis type II mouse
model and recombinant AAV2/8
vector-mediated gene therapy. Mol Cells
2010;30:13-18
22. Fu H, Dirosario J, Killedar S, et al.
Correction of neurological disease of
mucopolysaccharidosis IIIB in adult mice
by rAAV9 trans-blood-brain barrier gene
delivery. Mol Ther 2011;19:1025-33
23. Ferla R, O’Malley T, Calcedo R, et al.
Gene therapy for mucopolysaccharidosis
type VI is effective in cats without
pre-existing immunity to AAV8.
Hum Gene Ther 2013;24:163-9
.. This paper suggests that successful
gene therapy using AAV vectors may
rely on the preexistence of
anti-AAV ABs.
24. Karolewski BA, Wolfe JH. Genetic
correction of the fetal brain increases the
lifespan of mice with the severe
multisystemic disease
mucopolysaccharidosis type VII.
Mol Ther 2006;14:14-24
25. Cearley CN, Wolfe JH. A single
injection of an adeno-associated virus
vector into nuclei with divergent
connections results in widespread vector
distribution in the brain and global
correction of a neurogenetic disease.
J Neurosci 2007;27:9928-40
26. Chen YH, Claflin K, Geoghegan JC,
Davidson BL. Sialic acid deposition
impairs the utility of AAV9, but not
peptide-modified AAVs for brain gene
therapy in a mouse model of lysosomal
storage disease. Mol Ther
2012;20:1393-9
27. Donsante A, Miller DG, Li Y, et al.
AAV vector integration sites in mouse
hepatocellular carcinoma. Science
2007;317(5837):477
28. Rosas LE, Grieves JL, Zaraspe K, et al.
Patterns of scAAV vector insertion
associated with oncogenic events in a
mouse model for genotoxicity. Mol Ther
2012;20:2098-110
29. Cavazza A, Moiani A, Mavilio F.
Mechanisms of retroviral integration and
mutagenesis. Hum Gene Ther
2013;24(2):119-31
30. Cavazzana-Calvo M, Fischer A. Gene
therapy for severe combined
immunodeficiency: are we there yet?
J Clin Invest 2007;117:1456-65
31. Stein S, Ott MG, Schultze-Strasser S.
Genomic instability and myelodysplasia
with monosomy 7 consequent to
EVI1 activation after gene therapy for
chronic granulomatous disease. Nat Med
2010;16:198-204
32. Mango RL, Xu L, Sands MS, et al.
Neonatal retroviral vector-mediated
hepatic gene therapy reduces bone, joint,
and cartilage disease in
mucopolysaccharidosis VII mice and
dogs. Mol Genet Metab 2004;82:4-19
33. Wang B, O’Malley TM, Xu L, et al.
Expression in blood cells may contribute
to biochemical and pathological
improvements after neonatal intravenous
gene therapy for mucopolysaccharidosis
VII in dogs. Mol Genet Metab
2006;87:8-21
34. Traas AM, Wang P, Ma X, et al.
Correction of clinical manifestations of
canine mucopolysaccharidosis I with
neonatal retroviral vector gene therapy.
Mol Ther 2007;15:1423-31
35. Chung S, Ma X, Liu Y, et al. Effect of
neonatal administration of a retroviral
vector expressing alpha-L-iduronidase
upon lysosomal storage in brain and
other organs in mucopolysaccharidosis I
mice. Mol Genet Metab 2007;90:181-92
36. Baldo G, Mayer FQ, Martinelli BZ,
et al. Enzyme replacement therapy
started at birth improves outcome in
difficult-to-treat organs in
mucopolysaccharidosis I mice.
Mol Genet Metab 2013;109:33-40
37. Ou L, Herzog T, Koniar BL, et al.
High-dose enzyme replacement therapy
in murine Hurler syndrome.
Mol Genet Metab
2013. [Epub ahead of print]
38. Baldo G, Wozniak DF, Ohlemiller KK,
et al. Retroviral-vector-mediated gene
therapy to mucopolysaccharidosis I mice
improves sensorimotor impairments and
other behavioral deficits. J Inherit
Metab Dis 2013;36:499-512
. This paper suggests that, if high
IDUA activity is achieved after hepatic
gene transfer, a small portion of the
enzyme reaches the brain and corrects
biochemical and behavioral defects.
39. Bigg PW, Sleeper MM, O’Donnell PA,
et al. The effect of neonatal gene therapy
with a gamma retroviral vector on
cardiac valve disease in
mucopolysaccharidosis VII dogs after a
decade. Mol Genet Metab
2013;110:311-18
40. Smith LJ, Martin JT, O’Donnell P, et al.
Effect of neonatal gene therapy on
lumbar spine disease in
mucopolysaccharidosis VII dogs.
Mol Genet Metab 2012;107:145-52
41. Ponder KP, O’Malley TM, Wang P,
et al. Neonatal gene therapy with a
gamma retroviral vector in
mucopolysaccharidosis VI cats. Mol Ther
2012;20:898-907
. This study describes that
overexpression of ARSB can correct
most abnormalities in MPS VI cats,
without the need of
SUMF1 coexpression.
42. Papayannakos C, Daniel R.
Understanding lentiviral vector
chromatin targeting: working to reduce
insertional mutagenic potential for gene
therapy. Gene Ther 2013;20:581-8
43. Biffi A, Montini E, Lorioli L, et al.
Lentiviral hematopoietic stem cell gene
therapy benefits metachromatic
Gene delivery strategies for the treatment of mucopolysaccharidoses
Expert Opin. Drug Deliv. (2014) 11(3) 9
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

leukodystrophy. Science
2013;341:1233158
.. In this clinical trial, a LV vector was
used to transduce hematopoietic stem
cells from patents with a lysosomal
storage disorder. The therapy was
efficacious and safe.
44. Aiuti A, Biasco L, Scaramuzza S, et al.
Lentiviral hematopoietic stem cell gene
therapy in patients with Wiskott-Aldrich
syndrome. Science 2013;341:1233151
.. In this study the authors included a
comparison of integration sites
observed after both RV and LV
transduction of cells, suggesting that
LV might be safer than g-RV.
45. Di Domenico C, Villani GR,
Di Napoli D, et al. Intracranial gene
delivery of LV-NAGLU vector corrects
neuropathology in murine MPS IIIB.
Am J Med Genet A 2009;149A:1209-18
46. McIntyre C, Derrick Roberts AL,
Ranieri E, et al. Lentiviral-mediated gene
therapy for murine
mucopolysaccharidosis type IIIA.
Mol Genet Metab 2008;93:411-18
47. McIntyre C, Byers S, Anson DS.
Correction of mucopolysaccharidosis type
IIIA somatic and central nervous system
pathology by lentiviral-mediated gene
transfer. J Gene Med 2010;12:717-28
48. Macsai CE, Derrick-Roberts AL, Ding X,
et al. Skeletal response to lentiviral
mediated gene therapy in a mouse model
of MPS VII. Mol Genet Metab
2012;106:202-13
49. Camassola M, Braga LM,
Delgado-Canedo A, et al. Nonviral
in vivo gene transfer in the
mucopolysaccharidosis I murine model.
J Inherit Metab Dis 2005;28:1035-43
50. Richard M, Arfi A, Seguin J, et al.
Widespread biochemical correction of
murine mucopolysaccharidosis type VII
pathology by liver hydrodynamic plasmid
delivery. Gene Ther 2009;16:746-56
51. Khorsandi SE, Bachellier P, Weber JC,
et al. Minimally invasive and selective
hydrodynamic gene therapy of liver
segments in the pig and human.
Cancer Gene Ther 2008;15:225-30
52. Osborn MJ, McElmurry RT, Lees CJ,
et al. Minicircle DNA-based gene
therapy coupled with immune
modulation permits long-term expression
of alpha-L-iduronidase in mice with
mucopolysaccharidosis type I. Mol Ther
2011;19:450-60
53. Aronovich EL, Bell JB, Khan SA, et al.
Systemic correction of storage disease in
MPS I NOD/SCID mice using the
sleeping beauty transposon system.
Mol Ther 2009;17:1136-44
54. Osborn MJ, DeFeo AP, Blazar BR,
Tolar J. Synthetic zinc finger nuclease
design and rapid assembly.
Hum Gene Ther 2011;22:1155-65
55. Matte U, Lagranha VL, de Carvalho TG,
et al. Cell microencapsulation: a potential
tool for the treatment of neuronopathic
lysosomal storage diseases. J Inherit
Metab Dis 2011;34:983-90
56. Barsoum SC, Milgram W, Mackay W,
et al. Delivery of recombinant gene
product to canine brain with the use of
microencapsulation. J Lab Clin Med
2003;142:399-413
57. Piller Puicher E, Tomanin R,
Salvalaio M, et al. Encapsulated
engineered myoblasts can cure Hurler
syndrome: preclinical experiments in the
mouse model. Gene Ther
2012;19:355-64
58. Baldo G, Mayer FQ, Martinelli B, et al.
Intraperitoneal implant of recombinant
encapsulated cells overexpressing
alpha-L-iduronidase partially corrects
visceral pathology in
mucopolysaccharidosis type I mice.
Cytotherapy 2012;14:860-7
59. Baldo G, Quoos Mayer F, Burin M,
et al. Recombinant encapsulated cells
overexpressing alpha-L-iduronidase
correct enzyme deficiency in human
mucopolysaccharidosis type I cells.
Cells Tissues Organs 2012;195:323-9
60. Tomanin R, Friso A, Alba S, et al.
Non-viral transfer approaches for the
gene therapy of mucopolysaccharidosis
type II (Hunter syndrome).
Acta Paediatr Suppl 2002;91:100-4
61. Friso A, Tomanin R, Alba S, et al.
Reduction of GAG storage in MPS II
mouse model following implantation of
encapsulated recombinant myoblasts.
J Gene Med 2005;7:1482-91
62. Ross CJ, Bastedo L, Maier SA, et al.
Treatment of a lysosomal storage disease,
mucopolysaccharidosis VII, with
microencapsulated recombinant cells.
Hum Gene Ther 2000;11:2117-27
63. Ross CJ, Ralph M, Chang PL, et al.
Somatic gene therapy for a
neurodegenerative disease using
microencapsulated recombinant cells.
Exp Neurol 2000;166:276-86
64. Nakama H, Ohsugi K, Otsuki T, et al.
Encapsulation cell therapy for
mucopolysaccharidosis type VII using
genetically engineered immortalized
human amniotic epithelial cells.
Tohoku J Exp Med 2006;209:23-32
65. Mazumder MA, Burke NA, Shen F,
et al. Core-cross-linked alginate
microcapsules for cell encapsulation.
Biomacromolecules 2009;10:1365-73
66. Baruch L, Benny O, Gilert A, et al.
Alginate-PLL cell encapsulation system
Co-entrapping PLGA-microspheres for
the continuous release of anti-
inflammatory drugs.
Biomed Microdevices 2009;11:1103-13
67. Lagranha VL, de Carvalho TG,
Giugliani R, Matte U. Treatment of
MPS I mice with microencapsulated cells
overexpressing IDUA: effect of the
prednisolone administration.
J Microencapsul 2013;30:383-9
68. Langford-Smith A, Wilkinson FL,
Langford-Smith KJ, et al. Hematopoietic
stem cell and gene therapy corrects
primary neuropathology and behavior in
mucopolysaccharidosis IIIA mice.
Mol Ther 2012;20:1610-21
69. Visigalli I, Delai S, Politi LS, et al. Gene
therapy augments the efficacy of
hematopoietic cell transplantation and
fully corrects mucopolysaccharidosis type
I phenotype in the mouse model. Blood
2010;116:5130-9
.. This study describes the transduction
of normal HSCs with a LV, and
injection of these cells into MPS I
mice, achieving complete correction of
the phenotype.
70. Ma X, Liu Y, Tittiger M, et al.
Improvements in mucopolysaccharidosis
I mice after adult retroviral vector-
mediated gene therapy with
immunomodulation. Mol Ther
2007;15:889-902
71. Di Domenico C, Villani GR,
Di Napoli D, et al. Gene therapy for a
mucopolysaccharidosis type I murine
model with lentiviral-IDUA vector.
Hum Gene Ther 2005;16:81-90
72. Gao C, Sands MS, Haskins ME,
Ponder KP. Delivery of a retroviral vector
expressing human beta-glucuronidase to
the liver and spleen decreases lysosomal
storage in mucopolysaccharidosis VII
mice. Mol Ther 2000;2:233-44
G. Baldo et al.
10 Expert Opin. Drug Deliv. (2014) 11(3)
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.

AffiliationGuilherme Baldo1, Roberto Giugliani†1,2 &
Ursula Matte1
†Author for correspondence1Centro de Terapia Genica- Hospital de Clinicas
de Porto Alegre, Ramiro Barcelos, 2350,
Porto Alegre, RS 90035-903, Brazil2Programa de Pos-Graduacao em genetica e
Biologia Molecular, UFRGS, Ramiro Barcelos,
2350, Porto Alegre, RS 90035-903, Brazil
Tel: +55 513 359 8010;
E-mail: [email protected]
Gene delivery strategies for the treatment of mucopolysaccharidoses
Expert Opin. Drug Deliv. (2014) 11(3) 11
Exp
ert O
pin.
Dru
g D
eliv
. Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
187.
39.1
92.1
31 o
n 01
/24/
14Fo
r pe
rson
al u
se o
nly.
Copyright © 2022 FDOKUMEN




















