
NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes

International Journal of Cardiology 152 (2011) 218–224
Contents lists available at ScienceDirect
International Journal of Cardiology
j ourna l homepage: www.e lsev ie r.com/ locate / i j ca rd
Free fatty acids induce endothelial dysfunction and activate protein kinase C andnuclear factor-κB pathway in rat aorta
Hua Li a, Hongliang Li a, Yige Bao b, Xiangxun Zhang c, Yerong Yu a,⁎a Department of Endocrinology and Metabolism, West China Hospital, Sichuan University, Chengdu City, Sichuan Province, PR Chinab West China Medical School, Sichuan University, Chengdu City, Sichuan Province, PR Chinac Lab. of Endocrinology and Metabolism, West China Hospital, Sichuan University, Chengdu City, Sichuan Province, PR China
⁎ Corresponding author. #37 Guo Xue Road, ChengduEndocrinology andmetabolism, West China Hospital. Tel28 85423459.
E-mail address: [email protected] (Y. Yu).
0167-5273/$ – see front matter © 2010 Elsevier Irelanddoi:10.1016/j.ijcard.2010.07.019
a b s t r a c t
a r t i c l e i n f oArticle history:
Received 23 February 2010Received in revised form 6 June 2010Accepted 4 July 2010Available online 6 August 2010Keywords:Free fatty acidsEndothelial dysfunctionProtein kinase CNuclear factor-κB
Background: Insulin resistance is associated with an inappropriate elevation of plasma free fatty acids (FFAs)and endothelial dysfunction. In this study, we asked if elevated circulating FFA levels led to impaired insulinsignaling and endothelial dysfunction in-vivo via activation of PKC-mediated inflammatory pathways.Methods: Sprague–Dawley (S–D) rats were infused with 1) 20% intralipid+heparin (FFA group) or 2) salinealone (Control group) for 6 h. The intact aorta thoracica and aorta abdominalis were then removed. Aorticrings were isolated and evaluated for endothelial-dependent and non-dependent relaxation in an organbath. The activities of eNOS and PKC were measured in endothelial homogenates prepared from endothelialcells harvested from the aorta. The expression levels of insulin signaling molecules IRS-1, Akt, eNOS, ERK1/ERK2, PKC-α, NFκB-p65 subunit and IκB-α in rat aortic endothelium were determined by immunohis-tochemistry and Western blot.
Results: Elevation of FFAs resulted in a 35.9% reduction in the response to acetylcholine (pb0.01), a 26%decline in plasma NOx levels (pb0.05), a 53% decrease in eNOS activity and a 34±9% inhibition in IRS-1tyrosine phosphorylation (pb0.05). We also found a 46% decrease in Akt phosphorylation and a 36%decrease in eNOS phosphorylation. FFA-induced endothelial insulin resistance was associated with 82%increase in total membrane-associated PKC activity, a 1.7-fold increase in total PKC-α protein, 1.29-folddecrease in IκB-α expression levels and 1.47-fold increase in NF-κB p65 subunit expression in rat aorticendothelium.Conclusion: The molecular mechanisms underlying FFA-induced endothelial insulin resistance and eNOSinhibition may provide an important link implicating the PKC and IκB-α/NF-κB pathways in FFA-mediatedinhibition of vascular insulin signaling.© 2010 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
Cardiovascular diseases are the leading cause of morbidity andmortality in patients with insulin resistance syndrome. A number ofdisorders associated with endothelial dysfunction, including diabetes,obesity, hypertension, coronary heart disease, and atherosclerosis, arecharacterized by insulin resistance [1]. Conversely, endothelial dysfunc-tion is often present in patients with insulin resistance. Understandingthe mechanisms of endothelial dysfunction in insulin-resistant patientsis important for the development of novel therapeutic strategies toreduce the morbidity and mortality of cardiovascular disease.
Endothelial dysfunction has been described as an impairment inthe generation of nitric oxide (NO) and its function as a vasodilatorand vascular homeostatic agent. Insulin enhances vasculature via the
, China. 610041, Department of.: +86 28 85422357; fax: +86
Ltd. All rights reserved.
activation and expression of endothelial nitric oxide synthase (eNOS),and thus NO bioavailability [2,3]. Insulin resistance has been reportedto induce alterations in insulin signaling within the endothelium,which could account for reduced NO production. Reduced NO actioncould, in turn, be instrumental in accelerating the process ofatherosclerotic disease. Insulin signaling in endothelial cells hasbeen shown to be similar to that seen in classic insulin-responsivecells such as skeletal muscle cells, hepatocytes, and adipocytes [4].Mechanisms of impaired insulin signaling in these better-character-ized insulin-responsive cells are therefore likely to help dissect insulinresistance associated endothelial dysfunction, which is not wellunderstood.
Elevation in circulating free fatty acids (FFAs) is a characteristicmetabolic abnormality of the insulin-resistant state. The skeletalmuscle, liver and endothelium of insulin-resistant patients areconstantly exposed to high concentrations of FFAs, which areknown to interfere with insulin signaling in the phosphatidylinositol3-kinase (PI3-kinase) pathway via mechanisms that involve en-hanced oxidative stress, activation of the proinflammatory NF-κB

Fig. 1. Concentration–response curves to (A) Ach (10−9–10−4 mol/l) and (B) SNP(10−9–10−4 mol/l) on NE-contracted aortic rings from control and FFA group rats.Relaxation expressed as percentage of maximal NE induced vasoconstriction. Ach-induced vasorelaxation significantly reduced in the FFA group in comparison tocontrol group (Pb0.01), whereas endothelia-independent vasodilation remainedunaffected. Data are expressed as mean±SD for each group (n=10 aortas).
219H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
pathway and activation of protein kinase C (PKC) resulting in skeletaland hepatic insulin resistance [5–8]. However, mechanisms underly-ing FFA-induced endothelial dysfunction are not completely under-stood. Exposure of endothelial cells to high levels of FFAs in-vitroinitiates multiple cellular processes including impaired insulinsignaling [9], activated oxidative stress [10,11] and inflammationpathways [12], leading to reduced NO production. However, theassociation between insulin signaling, oxidative stress and inflam-mation and FFA-induced endothelial dysfunction has not been in-vestigated in-vivo.
Our study was designed to investigate the role of elevated con-centrations of circulating FFAs in-vivo on impaired insulin signalingvia activation of PKC-mediated inflammatory pathways which maycontribute to endothelial dysfunction. We used chronically catheter-ized Sprague–Dawley (S–D) rats in which plasma FFA concentrationswere acutely raised by intralipid plus heparin infusion. Infusedanimals were evaluated for endothelial-dependent relaxation of aor-tic rings. Rat aortic endothelium was also evaluated for activity of theinsulin signaling pathway, and expression of IRS-1, Akt, eNOS, ERK1/2,PKC-α, NFκB-p65 subunit and IκB-α. All experimental procedureswere approved by the Animal Experimentation Ethics Committee(West China Hospital, Sichuan University) and were in accordancewith the Guide for Care and Use of Laboratory Animals of the NationalInstitutes of Health.
2. Methods
2.1. Animals
Male S–D rats (weighing between 250 and 300 g) were obtained from the AnimalResources Centre (Sichuan University, China) and housed individually in environmen-tally controlled conditions at 22 °C with 12-h light–dark cycles. Rats were fed standardrat chow for 1 week and had access to food and water ad libitum. Rats werecatheterized under pentobarbital anesthesia three days before the study. PE-50 tubingwas inserted into the left carotid artery and silastic tubing into the right jugular vein.Both catheters were tunneled under the skin and exteriorized in the nuchal area.Animals were allowed to recover over the following 3 days until their weights werestable. Rats were studied while awake and unstressed after 14 h of food deprivation.Animals were assigned to two groups: the control group (NC; n=30) was infused withnormal saline for 6 h (18 μl/min); the FFA group (n=30) was infused with 20%intralipid (Sino-Swed Pharmaceutical Corp. Ltd.) 18 μl/min plus heparin (NanjingxinbaiPharmaceutical Co., Ltd.) 0.72 IU/min. Blood was obtained at baseline and at the end ofinfusion for estimation of serum FFAs and nitrite/nitrate (NOx) concentrations. Animalswere sacrificed by an overdose of pentobarbital at the end of infusion and the intactaorta thoracica and aorta abdominalis were rapidly removed.
2.2. Assessment of endothelial function
The isolated thoracic aortas (n=10 in each group) were placed in oxygenatedKrebs–Henseleit solution (composition in mM: 115 NaCl, 25 NaHCO3, 1.38 NaH2PO4,2.51 KCl, 2.46 MgSO4, 1.91 CaCl2, and 5.56 dextrose). The aorta was cut into 3-mm ringsegments and placed in an organ bath chamber containing 10 ml Krebs–Henseleitsolution that was bubbled with 5% CO2/95% O2 gas mixture and maintained at 37 °C.The aortic rings were stretched to 1.0-g tension and allowed to equilibrate for 60 min.After equilibration, the rings were precontracted with 1 μmol/l norepinephrine (NE).Endothelium-dependent vessel relaxation was determined as the response toacetylcholine (ACh, 10−9 to 10−4 mol/l). Endothelium-independent vessel relaxationwas measured as the response to sodium nitroprusside (SNP, 10−9 to 10−4 mol/l). NE,Ach and SNP were obtained from Sigma-Aldrich (St. Louis, MO, USA).
2.3. Blood assay
Serum FFAs were measured enzymatically using a commercially available kit(Randox Laboratories Ltd., Co Antrim, UK). Plasma NOx levels were measured usinghigh performance liquid chromatography (HPLC) to analyze blood nitrite/nitrate levels.Briefly, post-column NaNO2 oxidation with a reversed-phase ion-pair LC system wasused to detect NOx fluorometrically at 214 nm (LC-10AT series, Shimadzu, Japan).
2.4. Preparation of endothelial cell homogenate samples
In order to harvest endothelial cells, the entire aorta was opened longitudinally andcarefully rinsed free of blood with cold 0.01 PBS. The inner surface was gently scrapedwith the tip of a scalpel in a single movement to selectively isolate endothelial cells. Theendothelial scrapings from each rat aorta were sonicated in 200 μl of ice-coldhomogenizing buffer. The aortic endothelial homogenate was clarified by centrifuga-
tion (13,800 g for 10 mins, at 4 °C), snap-frozen and stored at−80 °C for eNOS activityassays, PKC activity assays and Western blot analysis. Protein concentrations weredetermined by the Coomassie brilliant blue G-250 dye-binding method.
2.5. eNOS activity measurements
Endothelial cell extracts from rats in the FFA group (n=8) and the control group(n=8) were assayed for eNOS enzymatic activity. Conversion of L-arginine to L-citrulline was determined in triplicate for each sample. Endothelial cell homogenate(100 μl) was incubated with 200 μl buffer A (50 mM Tris/HCl, pH 7.5, containing 1 mMEDTA, 2 mMCaCl2, 1 mMDTT, 0.1 mM β-NADPH and 0.1 mM L-Arg) or buffer B (50 mMTris/HCl, pH 7.5, containing 1 mM EDTA, 2 mM CaCl2, 1 mM DTT, and 0.1 mMβ-NADPH). The reaction was incubated for 30 min at 37 °C, terminated by boiling for5 min, and then clarified by centrifugation (8000 g for 5 min). The supernatant (50 μl)was assayed for citrulline using HPLC as described previously [13]. Nitrite wasquantitated as previously described [14] and eNOS enzymatic activity was calculatedand expressed in picomoles per minute per milligram of protein. NaNO2 was used as astandard.
2.6. PKC activity measurements
Aortic endothelial homogenate samples were centrifuged in a Beckman airfuge at100,000 g for 1 h to separate the cytosolic fraction. The pellet was re-suspended withthe homogenization buffer described above, with 1% Triton added. Pellets were lysed bysonication (Sonics VibraCell) and incubated on ice for 1 h. The homogenates were thenre-centrifuged to obtain the membrane (particulate) fraction in the supernatant. PKCactivity was determined in the membrane and cytosolic fractions of the rats in the FFAand control groups by liquid scintillation counting using a commercially available PKCactivity assay kit (Promega Corporation, Madison, USA).
220 H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
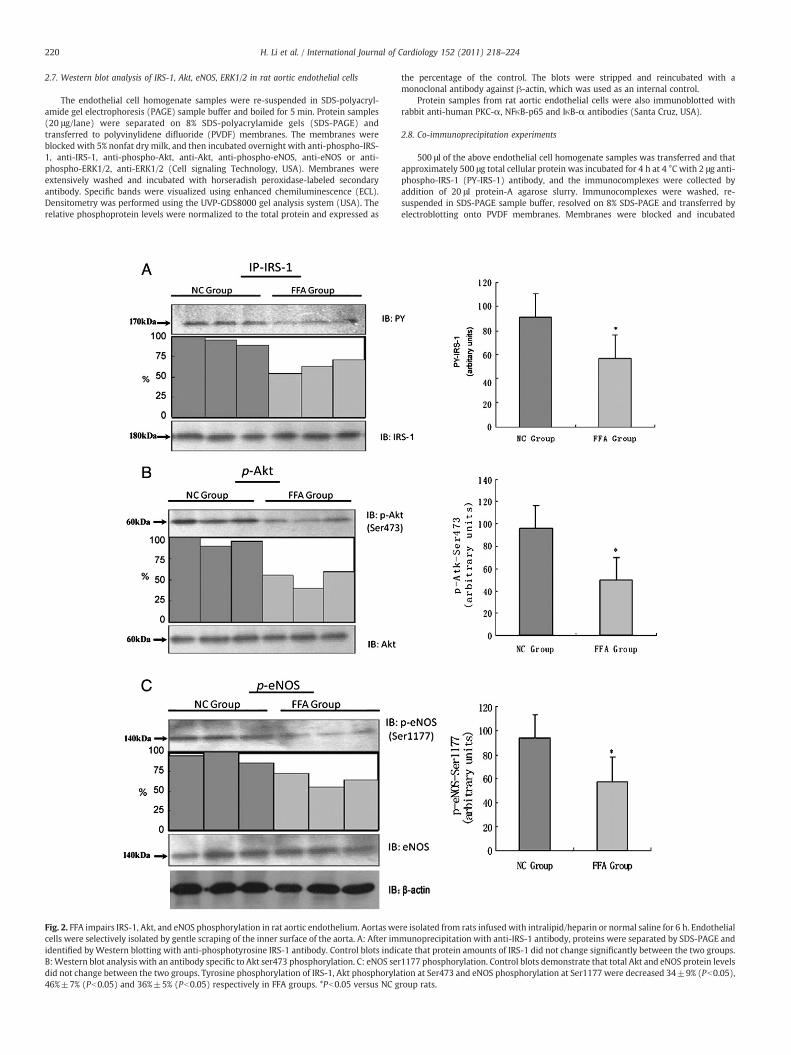
2.7. Western blot analysis of IRS-1, Akt, eNOS, ERK1/2 in rat aortic endothelial cells
The endothelial cell homogenate samples were re-suspended in SDS-polyacryl-amide gel electrophoresis (PAGE) sample buffer and boiled for 5 min. Protein samples(20 μg/lane) were separated on 8% SDS-polyacrylamide gels (SDS-PAGE) andtransferred to polyvinylidene difluoride (PVDF) membranes. The membranes wereblocked with 5% nonfat dry milk, and then incubated overnight with anti-phospho-IRS-1, anti-IRS-1, anti-phospho-Akt, anti-Akt, anti-phospho-eNOS, anti-eNOS or anti-phospho-ERK1/2, anti-ERK1/2 (Cell signaling Technology, USA). Membranes wereextensively washed and incubated with horseradish peroxidase-labeled secondaryantibody. Specific bands were visualized using enhanced chemiluminescence (ECL).Densitometry was performed using the UVP-GDS8000 gel analysis system (USA). Therelative phosphoprotein levels were normalized to the total protein and expressed as
Fig. 2. FFA impairs IRS-1, Akt, and eNOS phosphorylation in rat aortic endothelium. Aortas wecells were selectively isolated by gentle scraping of the inner surface of the aorta. A: After imidentified byWestern blotting with anti-phosphotyrosine IRS-1 antibody. Control blots indicB:Western blot analysis with an antibody specific to Akt ser473 phosphorylation. C: eNOS serdid not change between the two groups. Tyrosine phosphorylation of IRS-1, Akt phosphoryla46%±7% (Pb0.05) and 36%±5% (Pb0.05) respectively in FFA groups. *Pb0.05 versus NC g
the percentage of the control. The blots were stripped and reincubated with amonoclonal antibody against β-actin, which was used as an internal control.
Protein samples from rat aortic endothelial cells were also immunoblotted withrabbit anti-human PKC-α, NFκB-p65 and IκB-α antibodies (Santa Cruz, USA).
2.8. Co-immunoprecipitation experiments
500 μl of the above endothelial cell homogenate samples was transferred and thatapproximately 500 μg total cellular protein was incubated for 4 h at 4 °C with 2 μg anti-phospho-IRS-1 (PY-IRS-1) antibody, and the immunocomplexes were collected byaddition of 20 μl protein-A agarose slurry. Immunocomplexes were washed, re-suspended in SDS-PAGE sample buffer, resolved on 8% SDS-PAGE and transferred byelectroblotting onto PVDF membranes. Membranes were blocked and incubated
re isolated from rats infused with intralipid/heparin or normal saline for 6 h. Endothelialmunoprecipitation with anti-IRS-1 antibody, proteins were separated by SDS-PAGE andate that protein amounts of IRS-1 did not change significantly between the two groups.1177 phosphorylation. Control blots demonstrate that total Akt and eNOS protein levelstion at Ser473 and eNOS phosphorylation at Ser1177 were decreased 34±9% (Pb0.05),roup rats.

221H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
overnight with anti-phospho-IRS-1. Membranes were washed and incubated with thehorseradish peroxidase-labeled secondary antibody. Protein bands were visualizedusing the ECL method.
2.9. Immunohistochemical staining of PKCα
PKC-α expression was evaluated immunohistochemically in rat aortic endothelialcells using the streptavidin/peroxidase kit (Zhongshan Biotek Co., China) according tothe manufacturer's instructions. Briefly, sections were deparaffinized, rehydrated andthen soaked in antigen retrieval buffer (0.01 mmol/l Tris-base, 1 mmol/l EDTA, 0.05%Tween 20, pH 6.0) for 3 min at 95 °C. Endogenous peroxidase activity was blocked byincubating the sections in 3% H2O2 for 20 min at room temperature. The sections wererinsed thrice with PBS, and then incubated with normal rabbit serum or 100 μl rabbitanti rat PKC-α antibody (Santa Cruz, USA). The sections were rinsed with PBS andincubated with 100 μl biotinylated goat anti-rabbit IgG (1:100 dilution in PBS). Proteinwas visualized with DAB (diaminobenzidine) substrate solution. Primary antibody wassubstituted by PBS in the negative controls.
2.10. Statistical analysis
Data were analyzed with SPSS for Windows Version 10.0 (SPSS Inc., Chicago, IL,USA). All results were presented as means±SD. Statistical evaluations of the data wereperformedwith one-way analysis of variance (ANOVA) and t test. Significancewas set apriori at Pb0.05.
3. Results
3.1. In-vivo studies
Basal serum FFA levels were similar in the two groups (controlgroup: 337.9±97.7 μmol/l, FFA group: 345.4±86.6 μmol/l, pN0.05).Serum FFA levels increased dramatically after intralipid plus heparininfusion (1146.4±336.6 μmol/l; pb0.01). Animals in the FFA groupexhibited 3.4 times higher levels of serum FFA when compared withsaline-treated rats.
3.2. Elevated FFA concentration impaired endothelial function of theaorta
There was no significant difference in the contraction amplitude ofaortic rings preconstricted with 1 μmol/L NE. We showed (Fig. 1A),that acetylcholine (10−9–10−4M) caused a concentration-dependentrelaxation in preconstricted aortic rings. The maximal endothelium-dependent relaxation was impaired in the FFA group when comparedwith the control group (54.5±2.5% vs. 85%±3.2%; pb0.01). Westudied endothelium-independent relaxation and showed that there
Fig. 3. ERK 1/2 phosphorylation is not involved in FFA-induced endothelial dysfunction. AoEndothelial cells were selectively isolated by gentle scraping of the inner surface of the aorta.(A, B) and ERK (C, D) were not changed between the two groups.
was no statistically significant difference between the two groups insodium nitroprusside (SNP, 10−9–10−4 M) induced potent relaxationof rat aortic rings (Fig. 1B).
3.3. Elevated FFA levels decreased NO production and inhibited eNOSactivation
We analyzed blood nitrite/nitrate (NOx) concentrations to deter-mine NO levels in the two groups. There was no significant differencein total NOx level between the two groups at baseline (FFA group:20.1±3.9 μmol/l vs. NC group: 19.6±3.5 μmol/l, pN0.05). However,the FFA group showed a slight (26%) decrease post-infusionwhen compared with the control group (14.3±1.9 μmol/l vs. 19.3±2.9 μmol/l; pb0.05). Endothelial homogenates from aortas of bothgroups were analyzed by HPLC in order to determine the effect ofelevated FFA levels on eNOS activity. We showed a significantdecrease in the eNOS activity in the FFA group when compared withthat in NC group (1.15±0.42 pmol/min/mg vs. 2.43±0.56 pmol/min/mg; pb0.01).
3.4. FFAs inhibited IRS-1-Akt-eNOS signaling pathway activities
We evaluated tyrosine phosphorylation of IRS-1(PY-IRS-1) andserine phosphorylation of Akt (Ser473) and eNOS (Ser1177) in orderto determine the role of elevated FFAs levels on impaired insulinsignal transduction in vascular endothelium. We showed a 34±9%(pb0.05) inhibition in tyrosine phosphorylation of IRS-1 in the FFAgroup when compared with the NC group. We also showed that Aktphosphorylation at Ser473 and eNOS phosphorylation at Ser1177were decreased by 46%±7% (pb0.05) and 36%±5% (pb0.05)respectively, while protein expressions of IRS-1, Akt and eNOS werenot changed in the FFA group when compared with the control group(Fig. 2).
3.5. The ERK1/2 pathway is not involved in FFA-induced endothelialdysfunction
We showed that 20% intralipid/heparin infusion did not stimulatephosphorylation of ERK-1/2 in aortic endothelium (Fig. 3). The FFAand NC groups also showed no significant differences in the proteinlevels of ERK1/2 in the aortic endothelium.
rtas were isolated from rats infused with intralipid+heparin or normal saline for 6 h.Western blot analysis with an antibody specific to anti-ERK1/2 antibody, levels of p-ERK

Fig. 4. FFA treatment induces membrane-associated PKC activity significantly increasedand cystolic-associated PKC activity slightly reduced (Pb0.05). Aortas were isolatedfrom rats infused with intralipid/heparin or normal saline for 6 h. Endothelial cells wereselectively isolated by gentle scraping of the inner surface of the aorta.
222 H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
3.6. Increased PKC activity and PKC-α expression are involved in FFA-induced impaired insulin signaling pathway activity in aorticendothelium
We assessed PKC activity and the distribution of specific PKCisoforms in aortic endothelium of both groups in order to determine
Fig. 5. FFA increases PKC-α expression in rat aortic endothelium. Aortas were isolated fro(original magnification ×400) of a representative PKC-α immunohistochemistry assay demlocalization of PKA-α in aortic endothelium. B. Western blot analysis of PKC-α in rat aortic eshowing increases in total PKC-α expression in rats infused with intralipid/heparin.
the effect of FFA-induced endothelial dysfunction on PKC. We showed(Fig. 4) a slight reduction in cytosolic PKC activity in the FFA groupwhen compared with the NC group (8.4±1.9 pmol/min/mg proteinvs. 12.3±2.7 pmol/min/mg protein; pb0.05). However, we foundsignificantly higher PKC activity in cell membrane fractions of the FFAgroup when compared with the control group (20.4±4.3 pmol/min/mg protein vs. 11.2±2.1 pmol/min/mg protein; pb0.05). We alsoshowed an increase in total PKC activity in the FFA group (28.8±3.3 pmol/min/mg protein vs. 23.5±2.4 pmol/min/mg protein;pb0.05). We used immunohistochemistry to localize PKC-α inendothelium from the FFA and NC groups. Although this methodwas not quantitative, we demonstrated higher levels of PKC-α inendothelium of the FFA group rats when compared with the NC grouprats (Fig. 5A). These data were confirmed by Western blotting anddensitometry, which indicated a 1.7-fold increase in total PKC-αprotein in the FFA group when compared with the NC group (Fig. 5B).
3.7. FFA-induced impaired insulin signaling pathway activity is mediatedthrough NF-κB activation in aortic endothelium
We showed that the relative expression of NF-κB-p65 subunit inrat aortic endothelial cell homogenates from the FFA group waselevated by 1.47-fold (pb0.05, Fig. 6A), while the expression of IκB-αwas decreased by 1.29-fold (pb0.05, Fig. 6B) when comparedwith theNC group.
m rats infused with intralipid+heparin or normal saline for 6 h. A. Photomicrographonstrating the increases of PKC-α production by FFA. The brown color indicates the
ndothelial cell homogenates. Values are means±SD, *Different from NC rats, Pb0.05. It

Fig. 6. A. FFA treatment increases NF-κB p65 subunit expression in rat aortic endothelial cells. Aortas were isolated from rats infused with intralipid/heparin or normal saline for 6 h.This plot is representative of three different experiments with three different S–D rats (*Pb0.05). B. Western blot showing the relative expression of IκB-α in rat aortic endothelialcell homogenates were decreased after intralipid/heparin infusion (*Pb0.05).
223H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
4. Discussion
In this study, we acutely elevated circulating FFA concentrationsin S–D rats to levels seen in postabsorptive, obese individuals(1146.4±336.6 μmol/l). Our results agreed with previous reports[15–18] and showed an impairment in the vasodilator response toacetylcholine, but not to sodium nitroprusside, in aortic rings afteradministration of the lipid emulsion. These data suggested that theresponse to acetylcholine, representing endothelium-dependentvasodilatation, was impaired, but the response to sodium nitroprus-side, representing endothelium-independent vasodilatation, waspreserved after exposure to high concentrations of circulating FFAs.Excessive and persistent supply of FFAs to the bloodstream fromvisceral fat tissues in subjects with insulin resistance/visceral fatobesity suggests that an elevation of circulating FFA may be causallyrelated to the onset and progression of endothelial dysfunction.
Although FFAs have been shown to have a direct inhibitory effecton endothelial function in human and animal studies, themechanismsunderlying this inhibition are not clear. A number of studies onperipheral insulin resistance have suggested a role for elevated serumFFAs in decreased insulin-induced phosphorylation of the insulinreceptor, and insulin receptor substrate (IRS)-1 [19] and/or of proteinkinase B/Akt [20,21]. A 5-h lipid infusion reduced both glucoseoxidation and glycogen synthesis in associationwith impaired glucosetransport in rats [22]. This was accompanied by decreased insulin-stimulated tyrosine phosphorylation of IRS-1 and reduced IRS-1-associated PI3-kinase activity. Elevated plasma FFAs were also shownto result in impaired IRS-1-associated PI3-kinase activity in skeletalmuscle and inhibited insulin-stimulated glucose transport and phos-phorylation in humans [8]. Progressive increase in plasma FFAs wasrecently shown to cause a dose-dependent inhibition of insulin-stimulated glucose disposal and insulin signaling [23].
Since the PI3-kinase-dependent insulin signaling pathways inendothelium which are related to NO production share strikingsimilarities with metabolic pathways in skeletal muscle that promote
glucose uptake, we hypothesized that high FFA levels could causeendothelial dysfunction by impairing vascular insulin signaling. Ourdata showed a decrease in 1) tyrosine phosphorylation of IRS-1, 2) Aktphosphorylation at Ser473 and 3) eNOS phosphorylation at Ser1177after administration of the lipid emulsion. We also found a correlationbetween impaired insulin signaling and decreased endothelial eNOSactivity and NO production. We did not find a significant inhibition ofERK-1/2 in the lipid infusion studies, suggesting that theMAPK branchof the insulin signaling pathway was not involved in FFA-inducedendothelial insulin resistance. Our results are consistentwith previousin-vitro studies showing that treatment of vascular endothelial cellswith FFAs reduced basal and insulin-stimulated eNOS activity and NOproduction, and inhibited the insulin-stimulated activation of PI3K,PDK1, Akt, and eNOS [9]. Based on these data, we suggest that FFA-induced endothelial dysfunction is related with pathway-specificinsulin resistance.
A growing body of evidence indicates that PKC may play animportant role in FFA-induced insulin resistance in the muscle andliver [22,24,25]. PKC activity was upregulated and insulin-stimulatedincrease in Akt phosphorylation was blunted in the aorta of Zuckerfatty rats. These effects were partly normalized after 2 weeks oftreatmentwith a PKC-β inhibitor [26]. In viewof these data implicatingan important role for PKC inmediating FFA-induced insulin resistance,we examined the associationbetweenendothelial FFA-induced insulinresistance and altered PKC activity. We demonstrated that impairedendothelial-dependent vasodilation in this settingwas associatedwith1) an 82% increase in total membrane-associated PKC activity and PKCtranslocation from the cytosol to the cell membrane, and 2) a 1.7-foldincrease in total PKC-α protein levels. Our results showing that FFAsstimulated the activity and translocation of PKC, agreewith thewidelyheld concept that PKC activation may directly phosphorylate serineresidues of the insulin receptor and its substrates. This phosphoryla-tion event is known to inhibit the activity of IRS-associated PI3K,whichis mainly responsible for the FFA-induced endothelial insulin resis-tance. Inhibition of insulin-stimulated PI3-kinase activation and

224 H. Li et al. / International Journal of Cardiology 152 (2011) 218–224
decreased IRS tyrosyl phosphorylation has previously been shown toaccompany the increase inmembrane-associated PKC-θ in rats infusedwith lipid during euglycemic-hyperinsulinemic clamping [22].
Activation of PKC has also been suggested to activate the IKK-β/IκB-α/NF-κB pathway. IκB is known to bind NF-κB in the cytoplasmand inhibits its nuclear translocation. IκB-α is phosphorylated by IKK-β or other serine kinases such as PKC, is released from the NF-κBcomplex, is ubiquitinated and degraded [27]. NF-κB now enters thenucleus where it transactivates its target genes that are responsiblefor synthesis of a number of inflammatory proteins [28]. PKC is knownto activate NF-κB [29] which has been linked to fatty acid-inducedimpairment of insulin action in muscle in rodents [30,31]. FFA-induced insulin resistance in skeletal muscle and liver was also shownto be associated with increases in DAG mass and PKC activity and adecrease in IκB-α levels [5,6]. FFAs increased oxidative stress andactivated NF-κB in endothelial cells in-vitro and these effects weremediated by PKC [10,32,33]. Our in-vivo study showed a 1.29 folddecrease in IκB-α expression and a 1.47 fold increase in NF-κB p65subunit expression after administration of the lipid emulsion.Inflammatory processes are now recognized to play a pivotal role inthe pathogenesis of endothelial dysfunction and the IKK-β/IκB-α/NF-κB pathway is a major proinflammatory pathway [28]. Our datashowing that acute elevation of plasma FFA concentrations reducedthe activity of the insulin signaling pathway via the IKK-β/IκB-α/NF-κB, suggested that PKC-mediated activation of the IKK-β/IκB-α/NF-κBpathway may be related with FFA-induced endothelial dysfunction.
In summary, we found that physiological elevations of plasma FFAlevels for 6 h caused impaired endothelial-dependent vasodilation,inhibited IRS-1, Akt and eNOS phosphorylation, resulted in activationand translocation of PKC, and activation of the classic proinflammatoryIκB-α/NF-κB pathway in the endothelium. These results suggested thatFFA-induced insulin resistance and eNOS inhibition in endotheliummay play an important role in impaired endothelial function inmetabolic syndrome. The characterization of molecular mechanismsinvolved in FFA-induced insulin resistance in endothelium provides animportant mechanistic link implicating PKC and the IκB-α/NF-κBpathway in the inhibitory effects of FFAs on vascular insulin signaling.
Acknowledgments
This work was supported by the National Nature ScienceFoundation of China (Grant number 30370679).
The authors of this manuscript have certified that they complywith the Principles of Ethical Publishing in the International Journal ofCardiology [34].
References
[1] Cersosimo E, DeFronzo R. Insulin resistance and endothelial dysfunction: the roadmap to cardiovascular diseases. Diabetes Metab Res Rev 2006;22:423–36.
[2] Aljada A, Dandona P. Effect of insulin on human aortic endothelial nitric oxidesynthase. Metabolism 2000;49:147–50.
[3] Zeng G, Nystrom F, Ravichandran L, et al. Roles for insulin receptor, PI3-kinase, andAkt in insulin-signaling pathways related to production of nitric oxide in humanvascular endothelial cells. Circulation 2000;101:1539–45.
[4] Muniyappa R, Montagnani M, Koh K, Quon M. Cardiovascular actions of insulin.Endocr Rev 2007;28:463–91.
[5] Itani S, Ruderman N, Schmieder F, Boden G. Lipid-induced insulin resistance inhuman muscle is associated with changes in diacylglycerol, protein kinase C, andIkappaB-alpha. Diabetes 2002;51:2005–11.
[6] Boden G, She P, Mozzoli M, et al. Free fatty acids produce insulin resistance andactivate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes2005;54:3458–65.
[7] Bachmann O, Dahl D, Brechtel K, et al. Effects of intravenous and dietary lipidchallenge on intramyocellular lipid content and the relation with insulinsensitivity in humans. Diabetes 2001;50:2579–84.
[8] Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucosetransport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest1999;103:253–9.
[9] Wang X, Zhang L, Youker K, et al. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTENor inhibiting Akt kinase. Diabetes 2006;55:2301–10.
[10] Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulatereactive oxygen species production through protein kinase C-dependentactivation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000;49:1939–45.
[11] Du X, Edelstein D, Obici S, Higham N, Zou M, Brownlee M. Insulin resistancereduces arterial prostacyclin synthase and eNOS activities by increasingendothelial fatty acid oxidation. J Clin Invest 2006;116:1071–80.
[12] Kim F, Tysseling K, Rice J, et al. Free fatty acid impairment of nitric oxideproduction in endothelial cells is mediated by IKKbeta. Arterioscler Thromb VascBiol 2005;25:989–94.
[13] Qureshi G, Baig S. Application of high performance liquid chromatography instudy of sulphur amino acid metabolism in uremic patients. Biochem Mol Biol Int1993;29:359–68.
[14] Kaku S, Tanaka M, Muramatsu M, Otomo S. Determination of nitrite by high-performance liquid chromatography system with electrochemical detector:measurement of nitric oxide synthase activity in rat cerebellum cytosol. BiomedChromatogr 1994;8:14–8.
[15] Steinberg H, TarshobyM,Monestel R, et al. Elevated circulating free fatty acid levelsimpair endothelium-dependent vasodilation. J Clin Invest 1997;100:1230–9.
[16] Watanabe S, Tagawa T, Yamakawa K, Shimabukuro M, Ueda S. Inhibition of therenin–angiotensin system prevents free fatty acid-induced acute endothelialdysfunction in humans. Arterioscler Thromb Vasc Biol 2005;25:2376–80.
[17] Pleiner J, Schaller G, Mittermayer F, Bayerle-Eder M, Roden M, Wolzt M. FFA-induced endothelial dysfunction can be corrected by vitamin C. J Clin EndocrinolMetab 2002;87:2913–7.
[18] Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids inducesinflammation and impairs vascular reactivity in healthy subjects. Diabetes2003;52:2882–7.
[19] Storz P, Döppler H, Wernig A, Pfizenmaier K, Müller G. Cross-talk mechanisms inthe development of insulin resistance of skeletal muscle cells palmitate ratherthan tumour necrosis factor inhibits insulin-dependent protein kinase B (PKB)/Akt stimulation and glucose uptake. Eur J Biochem 1999;266:17–25.
[20] Chavez J, Summers S. Characterizing the effects of saturated fatty acids on insulinsignaling and ceramide and diacylglycerol accumulation in 3 T3-L1 adipocytes andC2C12 myotubes. Arch Biochem Biophys 2003;419:101–9.
[21] Powell D, Turban S, Gray A, Hajduch E, Hundal H. Intracellular ceramide synthesisand protein kinase Czeta activation play an essential role in palmitate-inducedinsulin resistance in rat L6 skeletal muscle cells. Biochem J 2004;382:619–29.
[22] Griffin M, Marcucci M, Cline G, et al. Free fatty acid-induced insulin resistance isassociated with activation of protein kinase C theta and alterations in the insulinsignaling cascade. Diabetes 1999;48:1270–4.
[23] Belfort R, Mandarino L, Kashyap S, et al. Dose–response effect of elevated plasmafree fatty acid on insulin signaling. Diabetes 2005;54:1640–8.
[24] Cortright R, Azevedo JJ, Zhou Q, et al. Protein kinase C modulates insulin action inhuman skeletal muscle. Am J Physiol Endocrinol Metab 2000;278:E553–62.
[25] Itani S, Zhou Q, Pories W, MacDonald K, Dohm G. Involvement of protein kinaseC in human skeletal muscle insulin resistance and obesity. Diabetes 2000;49:1353–8.
[26] Naruse K, Rask-Madsen C, Takahara N, et al. Activation of vascular protein kinaseC-beta inhibits Akt-dependent endothelial nitric oxide synthase function inobesity-associated insulin resistance. Diabetes 2006;55:691–8.
[27] Karin M, Yamamoto Y, Wang Q. The IKK NF-kappa B system: a treasure trove fordrug development. Nat Rev Drug Discov 2004;3:17–26.
[28] Barnes P, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronicinflammatory diseases. N Engl J Med 1997;336:1066–71.
[29] Luberto C, Yoo D, Suidan H, Bartoli G, Hannun Y. Differential effects ofsphingomyelin hydrolysis and resynthesis on the activation of NF-kappa B innormal and SV40-transformed human fibroblasts. J Biol Chem 2000;275:14760–6.
[30] Kim J, Kim Y, Fillmore J, et al. Prevention of fat-induced insulin resistance bysalicylate. J Clin Invest 2001;108:437–46.
[31] Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-inducedinsulin resistance with salicylates or targeted disruption of Ikkbeta. Science2001;293:1673–7.
[32] Dichtl W, Nilsson L, Goncalves I, et al. Very low-density lipoprotein activatesnuclear factor-kappaB in endothelial cells. Circ Res 1999;84:1085–94.
[33] Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation inducedby T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. ProcNatl Acad Sci USA 2000;97:3394–9.
[34] Coats A. Ethical authorship and publishing. Int J Cardiol 2009;131:149–50.
Copyright © 2022 FDOKUMEN




















