
Formation of H2 on graphene using Eley-Rideal and ...
12
Formation of H 2 on graphene using Eley-Rideal and Langmuir-Hinshelwood processes J. Petucci, S. Semone, C. LeBlond, M. Karimi, and G. Vidali Citation: The Journal of Chemical Physics 149, 014702 (2018); doi: 10.1063/1.5026691 View online: https://doi.org/10.1063/1.5026691 View Table of Contents: http://aip.scitation.org/toc/jcp/149/1 Published by the American Institute of Physics
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Formation of H2 on graphene using Eley-Rideal and ...

Formation of H2 on graphene using Eley-Rideal and Langmuir-HinshelwoodprocessesJ. Petucci, S. Semone, C. LeBlond, M. Karimi, and G. Vidali
Citation: The Journal of Chemical Physics 149, 014702 (2018); doi: 10.1063/1.5026691View online: https://doi.org/10.1063/1.5026691View Table of Contents: http://aip.scitation.org/toc/jcp/149/1Published by the American Institute of Physics

THE JOURNAL OF CHEMICAL PHYSICS 149, 014702 (2018)
Formation of H2 on graphene using Eley-Ridealand Langmuir-Hinshelwood processes
J. Petucci,1 S. Semone,2 C. LeBlond,3 M. Karimi,2,a) and G. Vidali41Department of Physics and Astronomy, University of Denver, Denver, Colorado 80208, USA2Department of Physics, Indiana University of Pennsylvania, Indiana, Pennsylvania 15705, USA3Department of Chemistry, Indiana University of Pennsylvania, Indiana, Pennsylvania 15705, USA4Department of Physics, Syracuse University, Syracuse, New York 13244, USA
(Received 22 February 2018; accepted 12 June 2018; published online 3 July 2018)
A hydrogen atom can either physisorb or chemisorb onto a graphene surface. To describe the inter-action of H with graphene, we trained the C−−C, H−−H, and C−−H interactions of the ReaxFF CHObond order potential to reproduce Density Functional Theory (DFT) generated values of graphenecohesive energy and lattice constant, H2 dissociation energy, H on graphene adsorption potentials,and H2 formation on graphene using the Eley-Rideal (ER) and Langmuir-Hinshelwood (LH) pro-cesses. The results, generated from the trained H-graphene potentials, are in close agreement with thecorresponding results from DFT. The advantage of using optimized CH potentials is, for example,the inclusion of physisorption interactions and quantum mechanical features of chemical bonding inthe functional forms of the potentials. The trained CH potentials are utilized to study the energeticsof formation of an H2 molecule on graphene using the Eley-Rideal and Langmuir-Hinshelwood pro-cesses. Potential energy surfaces for the formation of H2 through ER are generated for the collinearand oblique approach of the second hydrogen atom. Energetics of the formation of H2 through LH isstudied for a variety of cases such as when hydrogen atoms are chemisorbed or physisorbed and whenhydrogen occupies ortho, meta, or para chemisorption sites. The likelihood of H2 formation throughLH for various configurations is discussed. Furthermore, the tunneling probability of an atom througha continuous symmetric/asymmetric barrier is calculated and applied to an adsorbed hydrogen atomon graphene. Published by AIP Publishing. https://doi.org/10.1063/1.5026691
I. INTRODUCTION
The interaction of hydrogen with graphene, graphite,PAHs (Polycyclic Aromatic Hydrocarbons), and related mate-rials has been the object of a very large number of theoret-ical studies, for a number of reasons. First, hydrogen inter-action is of importance in hydrogen storage.1 Second, theunusual electronic properties of graphene are amenable tobe tailored by chemical modification.2 Third, hydrogen inter-action with graphitic materials continues to be investigatedfor applications to erosion damage of graphitic tiles in toka-mak fusion reactors.3 Finally, graphitic carbon and PAHs4
are important dust materials in interstellar space;5 their inter-action with hydrogen, by far the most abundant element inspace, is of obvious importance for molecular hydrogen for-mation (which occurs principally on dust grains and possiblyon PAHs)6–8 and for the formation of other molecules aswell.9
Experiments of scattering of low energy atomic andmolecular hydrogen beams from a single crystal graphite ledto the construction of empirical physisorption potentials.10,11
Later, wavefunction-based12 and density functional theory +van der Waals (DFT + vdW)13 calculations confirmed the
a)Author to whom correspondence should be addressed: [email protected]
presence of a shallow (∼40 meV) physisorption well. DFT(Density Functional Theory) calculations of H interacting withgraphite14,15 found that there is a barrier to enter the chemisorp-tion state. This is due to the re-hybridization of C atoms fromsp2 to sp3 due to the chemisorption of H. Most theories showthat the chemisorption state has an ∼0.7–0.8 eV well and thatthe carbon atom lying below the chemisorbed hydrogen atompuckers out of the graphite plane by ∼0.35 Å. Other neighbor-ing carbon atoms are affected as well (extended puckering),but to a lesser extent.16 Although the presence of an entrancebarrier to chemisorption has been verified experimentally,17
there are still uncertainties on how to handle dispersion forcesaccurately and how to model the passage of a hydrogen atomfrom physisorption to chemisorption.18 Besides DFT-basedcalculations of the adsorption process,19–22 other approachesinclude accurate wavefunction calculations on cluster mod-els23,24 in order to improve the handling of the long-rangeforces.
The formation of molecular hydrogen on surfaces of dustgrains is the leading route to H2 formation in the interstel-lar medium.25 Because H2 is continuously destroyed by UVphotons, there is a stringent bound on how much H2 needsto be made to match observations.26 Since H2 formationrequires a third body and three-body gas-phase reactions areexceedingly rare in space, H2 formation must occur on dustgrains.27 Experiments of H2 formation in simulated interstellar
0021-9606/2018/149(1)/014702/11/$30.00 149, 014702-1 Published by AIP Publishing.

014702-2 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
conditions were first done on amorphous silicates28,29 andthen on amorphous carbon,30 PAHs,31 and HOPG.32 In thelatter case, the ro-vibrational distribution of H2 emergingfrom graphite was measured. These studies showed that H2
is formed by the Langmuir-Hinshelwood (LH) mechanismwhere atoms become accommodated on a surface and thendiffuse until they encounter a partner and form a molecule.However, Zecho et al.,33 Hornekaer et al.,34,35 and later Areouet al.17 showed that using energetic hydrogen atoms, hydrogencould chemisorb on the basal plane of graphite.
Several studies using a variety of theoretical methods havetried to capture the processes that were observed experimen-tally. DFT-based calculations, molecular dynamics, and quan-tum dynamic calculations have been employed to study theformation of H2 via the Eley-Rideal (ER) mechanism.15,36–41
In this case, a hydrogen atom coming from the gas phase bindswith a hydrogen atom already on the surface to form H2 with-out becoming accommodated first with the surface; the energygained in the reaction may be used by the molecule to leavethe surface in a ro-vibrational excited state and with superther-mal speed. The formation of H2 from two weakly adsorbedhydrogen atoms was studied by Morisset et al.42–44 using asemiclassical theory and by Martinazzo and Tantardini45 usingDFT.
Because DFT and other first-principles calculationsrequire a heavy use of computational resources, they are mostlyused to explore the formation of H2 on graphene/graphite ina limited number of cases. This has been recognized before,and a number of studies used semi-classical or quasi-classicalapproaches46–48 to obtain a wider understanding of the for-mation process. However, experiments (see Refs. 6 and 49 forrecent reviews) and actual processes in the interstellar medium(see, for example, Ref. 50) demand a holistic approach. Thegoal is to have a theoretical framework that would reliablyhandle, in a seamless fashion, chemisorption and physisorp-tion and processes, such as diffusion, that are needed inaccurately describing H2 formation both qualitatively andquantitatively.
In this paper, we present a computational approach thatcan be used to calculate the diffusion of atoms both by thermalhopping and tunneling and the formation of H2 via the Eley-Rideal (ER) and Langmuir-Hinshelwood (LH) mechanisms.The essence of this new approach is to combine first-principlescalculations with the results from experiments by training a CHbond-order potential so as to reproduce all the input theoreticaland experimental data. With the potential so obtained, calcu-lations of H adsorption, diffusion, and H2 formation becomefeasible over a variety of conditions.
The paper is organized as follows. In Sec. II, we describethe computational methods, i.e., DFT and ReaxFF. In Sec. III,we present the DFT results of the adsorption on graphene ofa single and then a second H atom. Then we introduce theReaxFF potential that has been optimized to reproduce theDFT results and available experimental data. Using the opti-mized ReaxFF, we compute the potential energy surface andmigration of a hydrogen atom on graphene. We then presentthe results for the molecular hydrogen formation using theER and LH mechanisms and a calculation of the tunnelingprobability of hydrogen from a physisorption site to another
physisorption site and from a physisorption site to a chemisorp-tion site. Finally, in Sec. IV, we summarize the most importantfindings.
II. COMPUTATIONAL METHODS
First-principles approaches are, in general, very accuratebut have the limitation that they are computationally intensiveand, as a result, a large system cannot be simulated. Classi-cal approaches are computationally less intensive at the costof sacrificing accuracy. A semi-empirical potential model fit-ted to first principles as well as to experimental data is a goodcompromise choice because it can capture the essential physicswith reasonable accuracy and computing time. Calculations inthis paper are achieved using the classical molecular dynam-ics code LAMMPS (Large-scale Atomic/Molecular MassivelyParallel Simulator).51 The interaction between hydrogen andcarbon atoms is modeled using the ReaxFF potential52 whichis implemented in LAMMPS. Training of the ReaxFF poten-tial is accomplished by the GARFfield program53 against theDFT generated data,54 and the energy barriers are obtainedusing the nudged elastic band (NEB) method55–57 availablewithin LAMMPS. In the physisorption regime, the data thatwere employed to train the adsorption potential were takenfrom the work of Rougeau et al.19 The PBE-D3 functionalwas employed in the work of Rougeau et al.
A. System geometry
The computational box that models graphene has anorthorhombic shape with periodic boundary conditions in allthree directions. The graphene layer consists of a rectangle ofsides 39.4 Å × 34.0 Å, in the x-y plane, with a total numberof 512 carbon atoms. It is located at z = 0 with its x and ysides oriented along the computational box. A periodic lengthof z = 20 Å is considered along the z-direction perpendic-ular to the graphene plane. The large periodic length alongthe z direction is required to avoid the self-interactions dueto the long-range van der Waals forces. A segment of ourgraphene layer is shown in Fig. 1. C0 represents the on-topadsorption site for hydrogen, and C1, C2, and C3 representthe first, second, and third near-neighbor carbon atoms to C0,respectively.
ReaxFF force field parameters were optimized against adiverse set of training data obtained by DFT calculations. TheDFT calculations were performed using the Siesta suite of
FIG. 1. A segment of our graphene layer with the first hydrogen adsorptionsite and first through third near-neighbor carbon atoms.

014702-3 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
programs54 which applies Troullier-Martins norm-conservingpseudopotentials.58 A double-ζ plus polarization (DZP) basisset was chosen since it provides reasonable accuracy withoutan overbearing computational cost. The basis set superpositionerror (BSSE) was determined to be −0.105 eV for the adsorp-tion of a single H atom on a 32-atom periodic graphene sheet.This is within the typical error obtained by DFT (<0.2 eV);therefore, no BSSE corrections were applied to the data. Therevised Perdew−Burke−Ernzerhof (PBE)59 form of the gen-eralized gradient approximation (GGA) functional was imple-mented with a mesh cutoff of 200 Ry. For the bulk Brillouinzone integration, a Monkhorst-Pack (6 × 6 × 1) set of spe-cial k-points was used. The density matrix was minimized toa tolerance of 10−5. All calculations, unless otherwise noted,were done on a 4 × 4 graphene sheet (32 atoms) supercellwith periodic boundary conditions in the x, y, and z direc-tions. A vacuum layer of 15 Å was employed to ensure nointeraction between parallel images. The NEB scheme55,56
as implemented in the atomic simulation environment (ASE)suite of programs was employed to locate minimum energypaths (MEPs) and transition states. To ensure that the highestenergy image would be biased toward the transition state, theclimbing image modification to the NEB was used. Minimumenergy paths were converged until all forces on atoms wereless than a tolerance of 0.04 eV/Å.
B. The ReaxFF CHO potential
A wide variety of bond-order potentials including theBrenner,60 AIREBO,61 and ReaxFF52 are available in the lit-erature. The REAXFF potential attracted our attention due toits parametrization of a diverse array of elements and systems.ReaxFF potential was originally developed by the WilliamA. Goddard group at Caltech62 and is based on bond ordertechnique appropriate for reactive force fields. Even thoughReaxFF was initially developed to model the reactions ofhydrocarbons, its applicability has been extended to metals,63
semiconductors,64 and oxides.52 It has been implemented inLAMMPS65 and is one of the potential models that can betrained by the GarFField program.53 In the present work,applicability of the ReaxFF potential is further extended toadsorption of hydrogen on a graphene surface. In a bond orderpotential, the energy of a bond depends on its environment and,therefore, a bond on the surface could be stronger than the cor-responding one in the bulk. The ReaxFF potential is capable ofmodeling continuous bond forming and bond breaking, a capa-bility that many potentials lack. Furthermore, ReaxFF includesthe van der Waals interaction which is proven to be importantin the study of the H-graphene interaction. The total energy ina ReaxFF potential is the sum of many terms including σ, π,and ππ chemical bonds and long distance van der Waals andCoulomb interactions, to name a few. The functional formsalong with the parameters values of the CHO-ReaxFF potentialare reported in the work of Chenoweth et al.52 The parame-ters of the ReaxFF potential are obtained by fitting to the DFTdata from first-principles calculations. In order to improve thereliability of the CHO-ReaxFF potential for the current surfaceapplications, the parameters of the H−−H, C−−C, and C−−H sec-tions of the potential are retrained against the DFT generateddata for the present system.
C. Training of the ReaxFF CHO potentials
The ReaxFF force field parameters were optimized usingGarFField,53 a genetic algorithm (GA)-based force field opti-mization framework. Genetic algorithms employ stochasticglobal search strategies reminiscent of evolutionary mech-anisms. Unlike Newton-type optimizers, GA’s are particu-larly suited for problems with multiple local minima, non-smooth objective functions, and large parameter sets. UsingGarFField, a 2D graphene crystal of 128 carbon atoms isoptimized against its lattice constant and cohesive energy inconstant pressure mode. In doing so, only C−−C parameters ofSections 3 and 5 of the CHO-ReaxFF potential are allowed tovary (Ref. 52). Finally, the adsorption properties of hydrogenon graphene are optimized by varying the C−−H parameters ofSections 3, 4, 5, and 6 of the ReaxFF potential. The follow-ing DFT calculated adsorption properties of hydrogen (seeSec. III A) are employed in the fitting: physisorption welldepth and its location, chemisorption well depth and its loca-tion, and Langmuir-Hinshelwood (LH) ortho, meta, and parastate energies as well as Eley-Rideal (ER) formation potentialenergy.
D. Energy barrier calculations
Molecular Static (MS) and Molecular Dynamics (MD)techniques are employed to probe the equilibrium and dynam-ical properties of H on graphene. Using a graphene lattice of512 carbon atoms, energy barriers for adsorption, migration,and formation of molecular hydrogen on graphene throughER and LH processes are obtained using the nudged elasticband (NEB) method.52,53,60,61 Within NEB, a series of sys-tem replicas are considered which connect the initial and finalconfigurations on either side of the energy barrier being inves-tigated. These replicas are defined as 3N dimensional pointson the system’s configuration space and are initially linearlyinterpolated between the initial and final states. Introducinginter-replica spring forces that connect each atom to its ownimage in the two most adjacent replicas and minimizing aspecific force quantity will result in replica configurationsfor the minimum energy transition path over the barrier. Theforce quantity minimized for each replica is the sum of theforce due to the interaction potential that is perpendicularto the transition path and the spring force that is parallel tothe transition path. NEB calculations are performed with 16to 64 system replicas (including the initial and final states),with a force tolerance convergence criterion ranging from0.1 to 0.001 (eV/Å). LAMMPS also implements a climb-ing image algorithm in which the replica highest in energyis identified and driven to the top of the energy barrier. Eventhough NEB was originally applied to the calculation of migra-tion barriers,55,56,66 its applicability has been extended to thecalculation of desorption, adsorption, and chemical reactionbarriers.
III. RESULTS
All the calculations in this section are carried out usingthe trained potential (TP) and only some are done using first-principles methods. The CHO-ReaxFF potential is trained
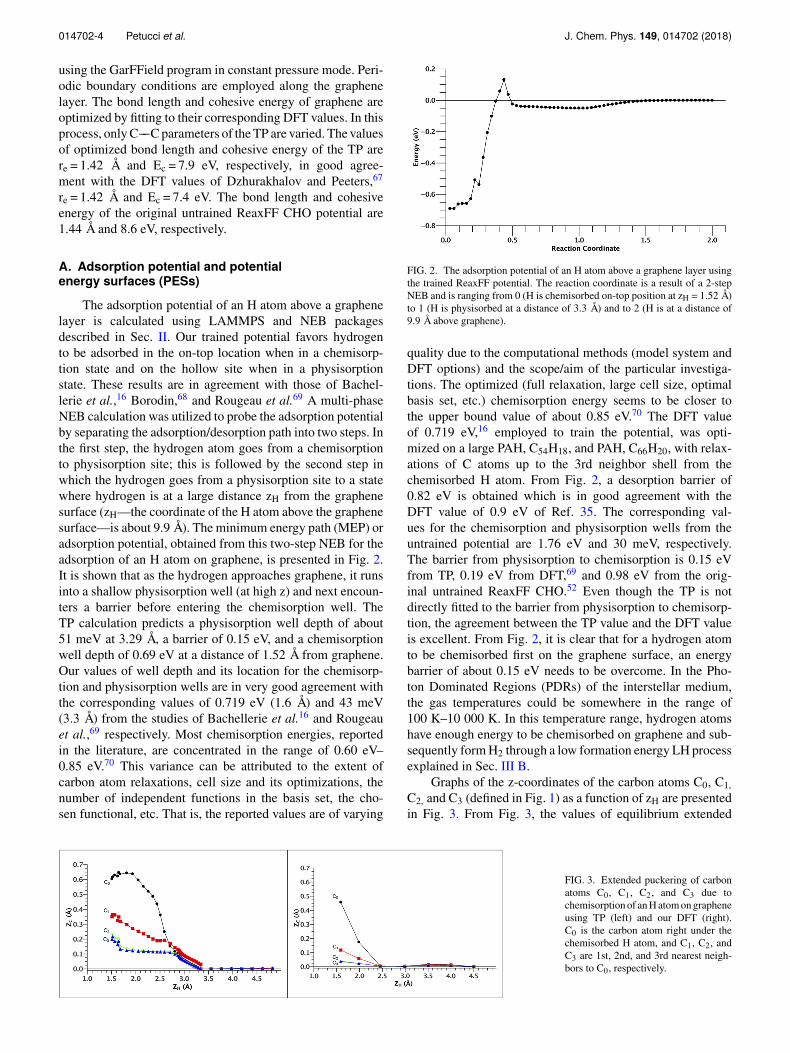
014702-4 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
using the GarFField program in constant pressure mode. Peri-odic boundary conditions are employed along the graphenelayer. The bond length and cohesive energy of graphene areoptimized by fitting to their corresponding DFT values. In thisprocess, only C−−C parameters of the TP are varied. The valuesof optimized bond length and cohesive energy of the TP arere = 1.42 Å and Ec = 7.9 eV, respectively, in good agree-ment with the DFT values of Dzhurakhalov and Peeters,67
re = 1.42 Å and Ec = 7.4 eV. The bond length and cohesiveenergy of the original untrained ReaxFF CHO potential are1.44 Å and 8.6 eV, respectively.
A. Adsorption potential and potentialenergy surfaces (PESs)
The adsorption potential of an H atom above a graphenelayer is calculated using LAMMPS and NEB packagesdescribed in Sec. II. Our trained potential favors hydrogento be adsorbed in the on-top location when in a chemisorp-tion state and on the hollow site when in a physisorptionstate. These results are in agreement with those of Bachel-lerie et al.,16 Borodin,68 and Rougeau et al.69 A multi-phaseNEB calculation was utilized to probe the adsorption potentialby separating the adsorption/desorption path into two steps. Inthe first step, the hydrogen atom goes from a chemisorptionto physisorption site; this is followed by the second step inwhich the hydrogen goes from a physisorption site to a statewhere hydrogen is at a large distance zH from the graphenesurface (zH—the coordinate of the H atom above the graphenesurface—is about 9.9 Å). The minimum energy path (MEP) oradsorption potential, obtained from this two-step NEB for theadsorption of an H atom on graphene, is presented in Fig. 2.It is shown that as the hydrogen approaches graphene, it runsinto a shallow physisorption well (at high z) and next encoun-ters a barrier before entering the chemisorption well. TheTP calculation predicts a physisorption well depth of about51 meV at 3.29 Å, a barrier of 0.15 eV, and a chemisorptionwell depth of 0.69 eV at a distance of 1.52 Å from graphene.Our values of well depth and its location for the chemisorp-tion and physisorption wells are in very good agreement withthe corresponding values of 0.719 eV (1.6 Å) and 43 meV(3.3 Å) from the studies of Bachellerie et al.16 and Rougeauet al.,69 respectively. Most chemisorption energies, reportedin the literature, are concentrated in the range of 0.60 eV–0.85 eV.70 This variance can be attributed to the extent ofcarbon atom relaxations, cell size and its optimizations, thenumber of independent functions in the basis set, the cho-sen functional, etc. That is, the reported values are of varying
FIG. 2. The adsorption potential of an H atom above a graphene layer usingthe trained ReaxFF potential. The reaction coordinate is a result of a 2-stepNEB and is ranging from 0 (H is chemisorbed on-top position at zH = 1.52 Å)to 1 (H is physisorbed at a distance of 3.3 Å) and to 2 (H is at a distance of9.9 Å above graphene).
quality due to the computational methods (model system andDFT options) and the scope/aim of the particular investiga-tions. The optimized (full relaxation, large cell size, optimalbasis set, etc.) chemisorption energy seems to be closer tothe upper bound value of about 0.85 eV.70 The DFT valueof 0.719 eV,16 employed to train the potential, was opti-mized on a large PAH, C54H18, and PAH, C66H20, with relax-ations of C atoms up to the 3rd neighbor shell from thechemisorbed H atom. From Fig. 2, a desorption barrier of0.82 eV is obtained which is in good agreement with theDFT value of 0.9 eV of Ref. 35. The corresponding val-ues for the chemisorption and physisorption wells from theuntrained potential are 1.76 eV and 30 meV, respectively.The barrier from physisorption to chemisorption is 0.15 eVfrom TP, 0.19 eV from DFT,69 and 0.98 eV from the orig-inal untrained ReaxFF CHO.52 Even though the TP is notdirectly fitted to the barrier from physisorption to chemisorp-tion, the agreement between the TP value and the DFT valueis excellent. From Fig. 2, it is clear that for a hydrogen atomto be chemisorbed first on the graphene surface, an energybarrier of about 0.15 eV needs to be overcome. In the Pho-ton Dominated Regions (PDRs) of the interstellar medium,the gas temperatures could be somewhere in the range of100 K–10 000 K. In this temperature range, hydrogen atomshave enough energy to be chemisorbed on graphene and sub-sequently form H2 through a low formation energy LH processexplained in Sec. III B.
Graphs of the z-coordinates of the carbon atoms C0, C1,
C2, and C3 (defined in Fig. 1) as a function of zH are presentedin Fig. 3. From Fig. 3, the values of equilibrium extended
FIG. 3. Extended puckering of carbonatoms C0, C1, C2, and C3 due tochemisorption of an H atom on grapheneusing TP (left) and our DFT (right).C0 is the carbon atom right under thechemisorbed H atom, and C1, C2, andC3 are 1st, 2nd, and 3rd nearest neigh-bors to C0, respectively.

014702-5 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
TABLE I. Values of the equilibrium extended puckering (EEP) of carbonatoms C0, C1, C2, and C3 from our and other trained potentials and DFTresults.
EEP values C0 (Å) C1 (Å) C2 (Å) C3 (Å)
Our TP 0.58 0.30 0.21 0.19Our DFT 0.46 0.12 0.03 0.04Other DFTa 0.48 0.13 0.06 0.06Other TPa 0.68 0.21 0.04 0.005
aBachellerie et al. (2009)16
puckering (EEP) of carbon atoms in graphene are extracted forour TP and DFT results and are reported in Table I. For furthercomparison, other TP and DFT results16,68 are also reported inTable I. From this table, a close agreement between our TP andDFTs is observed for C0 puckering. The favorable quantitativeagreement does not carry over to include higher order neigh-bors (C1, C2, and C3). This is expected since these quantitieswere not included in the training set data. Furthermore, onecan only expect limited transferability of classical potentialsoutside of the fitting criteria, especially for quantities relatedto chemical bonding. Our TP and DFT extended puckeringvalues (C0, C1, C2, and C3) as a function of zH (hydrogendistance from graphene) both approach zero in the limit of alarge value of zH. The barrier between the physisorption andchemisorption well is attributed to these puckerings. The goodagreement between our values of extended puckering and ofa barrier with the corresponding values from DFTs supportsthis correlation.
The physisorption potential energy surface (PES) of anH atom on graphene is obtained by placing the H atom at acoordinate (x, y, z = 3-4 Å) above the surface and then min-imizing the potential energy V(x, y, z) with respect to z.This process is continued for various values of x and y. Theminimized V(x, y, z) is then plotted as a function of x and y ofthe H atom using a density plot where the darkest color repre-sents the lowest energy. By calculating the minimum energyat each (x, y) position in this regime, the density plot depictsthe energy landscape that a hydrogen atom encounters as itmoves laterally along the surface. The calculated PES of thephysisorption states, using our trained CHO ReaxFF poten-tial (TP), is presented in Fig. 4. The calculated PES induces
FIG. 4. PES for an H atom in the physisorption states on a graphene layer.
insignificant relaxation in the carbon atoms of the graphenelayer. One important aspect of this PES is that the H atomwould like to be adsorbed at the hollow site. Furthermore, thebarrier for an H atom to diffuse from a hollow site to its nearestneighbor hollow site is about 5-6 meV. This small barrier todiffusion of an H atom confirms that an H atom physisorbed ongraphene has a high mobility. These results are corroboratedby those in the work of Bonfanti et al.12
Figure 5 presents the TP results of the NEB MEP of achemisorbed hydrogen atom migrating from a top site to anearest neighbor top site. From Fig. 5, a value of about 1.2eV is obtained for the migration barrier, which is in closeagreement with the values of 0.99 eV and 1.14 eV of Borodinet al.’s Climbing NEB-DFT.68 The value of the migrationbarrier, obtained from the untrained potential, is about 2.09eV. It is important to emphasize that the migration barrier dataare not employed in the training of the potential. Therefore,close agreement between our TP value of the migration barrierand other DFT values of Bachellerie et al.16 is a result of thereliability of the potential model. From the adsorption potentialgraph, a value of 0.82 eV is obtained for the desorption barrierof hydrogen from graphene. This value is much smaller thanthe value of 1.2 eV for the diffusion barrier obtained fromFig. 5. This implies that from an energetic viewpoint, an Hatom chemisorbed on graphene will desorb before it has anychance of diffusing. This result is in excellent agreement withthe work of Hornekaer et al.35
B. H2 formation1. Eley-Rideal process
The formation of H2 on graphene through the ER pro-cess is studied using the trained ReaxFF potential. A NEB runwith appropriate initial and final states is carried out to obtainthe reaction barrier to the formation of H2 through collinearER. The initial state includes the graphene lattice, described inSec. II A, plus two H atoms, one of which (Ha) is chemisorbed(on the top site) and the second one (Hb) is at a distance 4.8 Åfrom graphene and above the first one (Ha−−Hb is perpendicu-lar to graphene, and Hb is frozen). The final state is similar tothe initial state, except that the second H atom is close enoughto the first H atom to form an H2 molecule. C atoms of grapheneas well as Ha and Hb hydrogen atoms are allowed to relax along
FIG. 5. Migration energy barrier of a hydrogen atom to its nearest neighborsite.

014702-6 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
FIG. 6. TP potential results for theMEP and distances of Ha, Hb, and C0atoms from graphene during the for-mation of H2 on graphene through theER collinear mechanism. Left and rightcartoons correspond to the initial andfinal configurations of the NEB MEPcalculations.
the z-direction. Our graphs of the collinear ER potential energyas a function of the reaction coordinate calculated using ourTP are presented in Fig. 6. Reaction barriers for the forma-tion of H2, using the ER collinear approach, are about 100meV and 0 meV from our TP and DFT results, respectively.Other DFT results similarly predict a barrier-less ER formationenergy.71,72
Distances of the three atoms C0, Ha, and Hb with respectto the graphene plane are also presented in Fig. 6. Once H2 isabout to be formed, the bond length between the two hydrogensHa and Hb remains roughly the same and C0 moves into theplane of graphene (zero puckering) and hybridization changesfrom sp3 to sp2.
The Potential Energy Surface (PES) for the formation ofH2 as a function of zHa and zHb–zHa is presented in Fig. 7.In constructing this PES, constraints are imposed to allowthe relaxation/motion of both hydrogen atoms only along thez-direction. C0, C1, C2, and C3 are frozen at their equilib-rium values. The two H atoms (Ha and Hb) can move on aline perpendicular to graphene and on top of C0. In Fig. 7,two regions of low energy are noticeable, parallel to the
FIG. 7. PES for the collinear approach of a hydrogen (Hb) toward achemisorbed hydrogen (Ha) (see Fig. 6 for configuration of collinearapproach). zHa and zHb are vertical distances of Ha and Hb from the graphenesurface. The energy reference (0 eV) corresponds to the system configurationof large hydrogen-hydrogen and hydrogen-graphene separation (i.e., largevalues of zHa and zHb–zHa).
vertical and horizontal axes. The region parallel to the verticalis around zHb–zHa = 1 Å, while the one parallel to the hori-zontal is around zHa = 1.5 Å. Vertical and horizontal regionscorrespond to the ejected H2 molecule and chemisorbed Ha
on graphene, respectively. The vertical band is when H2 isformed.
Two-step NEB-MEP calculations are carried out, usingour TP, on a 2D ER system. The 2D system is defined as a planezHb–yHb, where the zHb axis is perpendicular to graphene at C0
and the yHb axis is in the plane of graphene along the C0-C3
direction (see Fig. 1). The initial configuration of the first stepNEB includes physisorbed hydrogen Hb somewhere alongthe C0−−C3 line and chemisorbed hydrogen Ha on top ofC0. The final configuration of this NEB step has the hydro-gen Hb somewhere directly above Ha on the zHb axis andis exactly the same as the initial state of the collinear ER.The second step NEB is the carbon copy of the collinearER calculation given in Fig. 6. The results of these two-stepNEB MEP calculations are connected together seamlesslyin Fig. 8. The formation energy of this two-step NEB MEPcalculation is about 0.11 eV. This is only 0.01 eV higher than
FIG. 8. MEP calculation of migration of the incident H atom (xHb = 0, yHb,zHb) in the yHb–zHb plane along the y-axis in Fig. 1 (from C3 to C1) wherezHb is the distance of hydrogen Hb from the graphene surface. Coordinates ofthe chemisorbed hydrogen (Ha) as well as carbon atoms in graphene are setat their equilibrium values. The graph from 0 to 1 represents the barrier forthe physisorption state, and the barrier from 1 to 2 is for the formation of H2through a collinear approach.

014702-7 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
FIG. 9. Chemisorbed states of H dimers on graphene [ortho (O), meta(M), and para (P) from lower-left to lower-right] along with their final H2orientations (upper).
the corresponding value from the collinear ER calculation inFig. 6. Based on this 2D ER model, hydrogen physisorbednear a chemisorbed hydrogen could overcome a small bar-rier of about 0.01 eV to go right above the chemisorbedhydrogen and subsequently form H2 through a collinear ERprocess.
2. Langmuir-Hinshelwood process
The formation of molecular hydrogen through theLangmuir-Hinshelwood mechanism is calculated for the caseswhere H atoms are chemisorbed on the graphene surface.The initial and final configurations of the three NEB runs areshown in Fig. 9. The initial configurations include H atomschemisorbed in ortho, meta, or para states, and the final stateincludes H2 molecules on the hollow site with its axis paral-lel to the graphene surface as well as to the H−−H bonds ofthe chemisorbed H atoms. Minimum energy path NEB resultsfor the formation of H2 through LH are presented in Fig. 10.Forward and reverse barriers of H2 formation on graphene,using TP and DFT, are summarized in Table II under “LHformation barriers.” E→ and E← are the forward and reversebarriers for the formation of H2 and dissociative chemisorp-tion of H dimers, respectively. E→ is the barrier to form H2
from a chemisorbed hydrogen dimer in ortho, meta, or parastates. E←, on the other hand, is the reverse of that process, i.e.,dissociative chemisorption of H2 into chemisorbed hydrogendimer ortho, meta, or para states. Our DFT results, for LH E→,are in reasonable agreement with the other DFTs reported inTable II. The agreement is better for the ortho and para cases.
TABLE II. Values for the forward and reverse H2 formation (left column) andsingle H atom desorption (right column) from ortho, meta, and para dimerson graphene. Data without any superscripts are our TP or DFT results.
LH formation Dimer barriersbarriers (eV) (eV)
Method (dimer type) E→ E← E→ E←
Trained potential (ortho) 1.63 4.60 1.38 0.16Trained potential (meta) 1.49 4.85 0.75 0.17Trained potential (para) 1.02 4.34 1.14 0.08DFT (ortho) 2.57, 2.50a 4.14, 4.53a 1.9b 0.08b
DFT (meta) 1.13, 1.32a 3.94 0.95b 0.15b
DFT (para) 1.39, 1.40a, 0.8c 3.18, 3.44a,c 2.1b 0.0b, 0.0d
aSljivancanin et al.73
bHornekaer et al.34
cCostanzo et al.74
dRougeau et al.19
Our TP results, for LH E→, although is not that close to theDFT findings, represents the correct trend in predicting thelargest forward barrier for the ortho dimer among the threechemisorbed dimers.
Based on our DFT and TP calculations, formation ofH2 through the LH process, when H atoms are chemisorbedon the graphene surface, is very unlikely, if not impossible.Furthermore, dissociative chemisorption of H2 on grapheneinto ortho, meta, and para dimers is even more unlikely tooccur.
An alternative low energy route for the formation of H2,from the meta state hydrogen dimer, is a combination of twotransitions: the diffusion of a H dimer from the meta to parastate followed by a subsequent transition from the para state toform H2. Based on our TP calculations, EM-P = 1.07 eV (fromtwo-step NEB, see Fig. 13) and EP = 1.02 eV (see Table II).This in turn provides, from a two-step NEB, a LH formationbarrier from the meta state to be about 1.07 eV. This indirectroute to form H2 only requires overcoming a barrier of 1.07 eVas compared to 1.49 eV for the direct transition.
Other H2 formation routes via LH are when one H ischemisorbed and the other H is physisorbed or when bothatoms are physisorbed. Barriers for the formation of H2 ongraphene for these two cases are much smaller than the corre-sponding values when both hydrogen atoms are chemisorbed.This in turn provides a higher probability for the formation ofmolecular hydrogen.
Initial and final states of H dimers on graphene [O (ortho),M (meta), and P (para) from lower-left to lower-right] along
FIG. 10. MEP NEB results for the for-mation of H2 on graphene through theLH mechanism using trained potential(left) and DFT (right).

014702-8 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
FIG. 11. Chemisorbed states of H dimers on graphene (O, M, and P fromlower-left to lower-right) along with the desorbed H atom from O, M, and Pdimers.
with the desorbed H atom from O, M, and P dimers are pre-sented in Fig. 11. The results of desorption of an H atom froman H dimer chemisorbed on graphene in ortho, meta, or parastates are given in Fig. 12 and Table II under “dimer barriers.”E→ is a forward barrier for desorption of a hydrogen from anH ortho, meta, or para dimer. E← is the reverse of that, i.e.,the barrier for the adsorption of an H atom in the vicinity ofan already chemisorbed hydrogen to form an H ortho, meta,or paradimer. From Fig. 12 and Table II, several conclusionsare drawn for the dimer barriers using the TP. (a) Energy bar-riers to desorption of an H atom from ortho and para dimersare much higher than the corresponding value for a single Hatom chemisorbed on graphene. (b) Binding energies of orthoand para dimers are significantly larger than the correspondingvalues for a single H atom on graphene. (c) Binding energiesof ortho and para dimers are lower than the correspondingmeta dimer and, therefore, more stable than the meta dimer.(d) Barriers for adsorption of a second H atom to the ortho andpara sites are about the same as for chemisorption of a singleH atom on clean graphene. (e) The barrier for adsorption of asecond H atom to the para site is small but not zero comparedto the barrierless results from DFT.19 Points (a)–(d) are con-sistent with the DFT results in the work of Hornekaer et al.34
Our conclusion (e) indicating a small barrier for adsorption to
FIG. 12. MEP NEB results for the desorption of one H atom from ortho,meta, and para states. The curves in the graph are the results generated fromthe TP.
the para site differs from the barrierless result predicted by theDFT.19,34
Figure 13 presents the TP results of a two-step NEB fordiffusion of an H dimer from an ortho to a meta to a para state.The energy barriers for the conversions of meta to ortho or paradimers are about 0.97 eV and 1.07 eV, respectively. Likewise,the barriers for diffusion from ortho to meta and para to metaare 1.60 eV and 1.37 eV, respectively. These values are inexcellent agreement with the corresponding DFT values of1.63 eV and 1.40 eV from the work of Sljivancanin et al.73
From Fig. 13, it is clear that the O dimer is more stable thanM and P dimers. The M dimer is the least stable one of thethree. These trends are in agreement with the TPD analysisof Hornekaer et al.34 Our M → O and M → P barriers areabout 1 eV which is larger than 0.5 eV obtained in the workof Hornekaer et al.34
C. Tunneling of hydrogen
The main contribution to the migration of a hydrogen atomon a graphene surface at low temperatures is due to the quantumtunneling through a potential barrier. At higher temperatures,thermal hopping of the hydrogen is the main contribution tothe migration. In this section, we extend a simple quantumtunneling approach73 to model the tunneling of a hydrogenatom on graphene at low temperatures. Using the potentialbarrier V (x), a transmission coefficient Tij is obtained,
FIG. 13. MEP for the diffusion ofdimers chemisorbed on graphene froman ortho (O) to a meta (M) state and thento a para (P) state.

014702-9 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
TABLE III. Parameters for tunneling; see Equations (1)–(6). ν is the attemptfrequency.
Constant Value
V0 0.3577 (eV)x0 0.00d 1.0 (Å)µ 0.402V1 �0.16 (eV)V3 �0.80 (eV)ν 1012 (Hz)
V (x) = −V0
sinh2(
x−x0d
)cosh2
(x−x0
d − µ) , (1)
Tij =2 sinh(πdk1)sinh(πdk3)
cosh(πd(k1 + k3)) + cosh(πγ)) , (2)
where
γ =
√(8md2
~2
)V0 cosh2(µ) − 1, (3)
k1 =
√2m(E − V1)~
, k3 =
√2m(E − V3)~
, (4)
V1 = limx→−∞
V (x) = −Voe−2µ, V3 = limx→∞
V (x) = −Voe2µ. (5)
The probability, Pij, and mobility, αij, of migration of ahydrogen atom from site i to j are obtained by Cazaux andTielens,76
Pij =1
kBT
∫ ∞0
Tije−E/kBT dE, αij = vPij. (6)
Parameters Vo, xo, d, µ, V1, and V3 are determined byfitting V (x) to the barrier between the two nearest neighborphysisorption sites or the barrier between a physisorption siteto a nearest neighbor chemisorption site. The values of thebarrier parameters are listed in Table III. Here, the parame-ters reflect the case of tunneling from a physisorption site to achemisorption site. The potential barrier function V (x), usingthese parameters, is presented in Fig. 14. While the barrier
FIG. 14. Potential energy barrier that hydrogen has to overcome to bechemisorbed into a nearby adsorption site. The origin of energy is the maxi-mum of the transition barrier connecting the physisorption and chemisorptionstates.
FIG. 15. Mobility αij (see the text for definition) of a hydrogen atom goingfrom a physisorbed site on graphene to a nearby chemisorption site bytunneling (blue) and by thermal hopping (black).
between two physisorption sites is symmetric, the chemisorp-tion site introduces asymmetry. In Fig. 15, mobility and ther-mal diffusion are plotted as a function of temperature. Theadvantage of these simple models, as compared to others76
(e.g., Cazaux et al.), is that the potential barriers are continu-ous (symmetric or asymmetric) and have analytical solutionsfor the transition probability. From Fig. 15, it is observed thatthe tunneling contribution to the migration of hydrogen ongraphene becomes important at temperatures below 20 K. Thedashed line (thermal hopping) is included only as a compara-tive reference to the classical migration process, as above 20 Kthermal desorption processes from physisorption dominate dueto the lower energy barrier.
IV. SUMMARY AND CONCLUSIONS
We obtained a ReaxFF potential for H interaction with agraphene sheet by retraining the ReaxFF bond-order potentialof hydrocarbons against the DFT generated data of hydro-gen adsorption on graphene. The retrained ReaxFF poten-tial has the advantage over similar bond order potentials toinclude both chemisorption and physisorption interactions.The adsorption potential obtained using the TP is in excellentagreement with the DFT and experimental data. In agreementwith DFT, the potential favors on-top and hollow sites for thechemisorption and physisorption of hydrogen on graphene,respectively. The equilibrium extended puckering (up to the3rd nearest neighbors of a carbon atom C0) and extendedpuckering as a function of hydrogen-graphene distance areobtained. The equilibrium extended puckering results are inclose agreement with our and other DFT results. The extendedpuckering as a function of hydrogen distance from grapheneis in reasonable agreement with the corresponding resultsfrom our DFT. The calculated migration barrier is in reason-able agreement with other DFT values. The inclusion of thephysisorption well has resulted in a low barrier for the for-mation of H2 through a 2D ER mechanism. This result is inexcellent agreement with those of other DFTs.70 The potentialalso predicts, in close agreement with the DFT, that the for-mation of H2, through the collinear ER mechanism, requiresthe first H atom to overcome a barrier of about 0.16 eV tochemisorb and the second H atom to overcome a barrier of

014702-10 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
about 0.10 eV to form H2. The potential, furthermore, pre-dicts a weak preferential adsorption of H on graphene; i.e.,the first H atom requires overcoming a barrier to chemisorbon graphene and the subsequent chemisorption of H atomsrequires equal or slightly smaller barriers. Physisorption inter-actions are included in the ReaxFF potential model and arenecessary to explain the formation of H2 through the LH pro-cess. The potential predicts likely routes for the formation ofH2 through LH, when one or two of the H atoms are adsorbedin the physisorption states.
The present model potential has the flexibility not only toreproduce some of the DFT results that were employed in itsoptimization but also to make predictions for cases where thereare no DFT results or for cases where it would be computation-ally very cumbersome to obtain DFT results. This flexibilitycould be exploited to study the dynamics of the formation ofH2 on graphene. We have extended two quantum mechanicalmodels of tunneling of a particle through a simple barrier tothe cases of a hydrogen atom going through barriers betweenphysisorption-physisorption and physisorption-chemisorptionstates. The results indicate that contribution of tunneling tothe migration of hydrogen on graphene is quite importantfor hydrogen at low temperatures. This static model could beextended to the dynamical diffusion of hydrogen on graphene.Retraining of the ReaxFF bond-order potential for the ener-getics of adsorption of hydrogen on graphene has proven tobe a desirable platform for narrowing the gap between thepotential and the DFT results. Furthermore, with the retrain-ing approach, full relaxations, dynamical calculations, andreasonable system sizes could be studied.
SUPPLEMENTARY MATERIAL
See supplementary material for tabulation of the final fullset of trained ReaxFF CHO potential parameters that are avail-able free of charge. The format of the original ReaxFF potentialalong with the definition of each parameter is given in thesupplementary material section of Ref. 52.
ACKNOWLEDGMENTS
G.V. would like to acknowledge financial support fromNSF Astronomy and Astrophysics Research Grant No.1615897. M.K. would like to acknowledge partial financialsupport through a grant from the University Senate.
1T. Heine, L. Zhechkov, and G. Seifert, “Hydrogen storage by physisorptionon nanostructured graphite platelets,” Phys. Chem. Chem. Phys. 6, 980–984(2004).
2D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake,M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim,and K. S. Novoselov, “Control of graphene’s properties by reversiblehydrogenation: Evidence for graphane,” Science 323, 610–613 (2009).
3J. Roth, “Chemical erosion of carbon based materials in fusion devices,”J. Nucl. Mater. 266, 51–57 (1999).
4J. C. Weingartner and B. T. Draine, “Dust grain-size distributions and extinc-tion in the Milky Way, large magellanic cloud, and small magellanic cloud,”Astrophys. J. 548, 296–309 (2001).
5B. Draine, “Interstellar dust grains,” Annu. Rev. Astron. Astrophys. 41,241–289 (2003).
6G. Vidali, “H2 formation on interstellar grains,” Chem. Rev. 113, 8762–8782(2013).
7L. Boschman, S. Cazaux, M. Spaans, R. Hoekstra, and T. Schlatholter,Astron. Astrophys. 579, A72 (2015).
8V. Wakelam, E. Bron, S. Cazaux, F. Dulieu, C. Gry, P. Guillard, E. Habart,L. Hornekær, S. Morisset, G. Nyman, V. Pirronello, S. D. Price, V. Val-divia, G. Vidali, and N. Watanabe, “H2 formation on interstellar dust grains:The viewpoints of theory, experiments, models and observations,” Mol.Astrophys. 9, 1–36 (2017).
9G. Vidali, “Cosmic low temperature physics: Making molecules on star-dust,” J. Low Temp. Phys. 170, 1–30 (2013).
10E. Ghio, L. Mattera, V. SaIvo, F. Tommasini, and U. Valbusa, “Vibra-tional spectrum of H and D on the (0001) graphite surface from scatteringexperiments,” J. Chem. Phys. 73, 556–561 (1980).
11L. Mattera, R. Rosatelli, C. Salvo, F. Tommasini, U. Valbusa, and G. Vidali,“Selective adsorption of 1H2 and 2H2 on the (0001) graphite surface,” Surf.Sci. 93, 515–525 (1980).
12M. Bonfanti, R. Martinazzo, G. Tantardini, and A. Ponti, “Physisorptionand diffusion of hydrogen atoms on graphite from correlated calcula-tions on the H-coronene model system,” J. Phys. Chem. C 111, 5825(2007).
13R. M. Ferullo, N. F. Domancich, and N. J. Castellani, “On the perfor-mance of van der Waals corrected-density functional theory in describingthe atomic hydrogen physisorption on graphite,” Chem. Phys. Lett. 500,283–286 (2010).
14L. Jeloaica and V. Sidis, “DFT investigation of the adsorption of atomichydrogen on a cluster-model graphite surface,” Chem. Phys. Lett. 300,157–162 (1999).
15X. Sha and B. Jackson, “First-principles study of the structural and energeticproperties of H atoms on a graphite (0001) surface,” Surf. Sci. 496, 318–330(2002).
16D. Bachellerie, M. Sizun, F. Aguillon, D. Teillet-Billy, N. Rougeau,and V. Sidis, “Unstricted study of the Eley–Rideal formation of H2 ongraphene using a new multidimensional graphene–H–H potential: Role ofthe substrate,” Phys. Chem. Chem. Phys. 11, 2715–2729 (2009).
17E. Areou, G. Cartry, J. M. Layet, and T. Angot, “Hydrogen-graphite inter-action: Experimental evidences of an adsorption barrier,” J. Chem. Phys.134, 014701 (2011).
18M. Bonfanti, B. Jackson, K. H. Hughes, I. Burghardt, and R. Martinazzo,J. Chem. Phys. 143, 124703 (2015).
19N. Rougeau, D. Teillet-Billy, and V. Sidis, “Double H atom adsorptionon a cluster model of a graphite surface,” Chem. Phys. Lett. 431, 135(2006).
20S. Casolo, O. M. Lovvik, R. Martinazzo, and G. F. Tantardini, “Under-standing adsorption of hydrogen atoms on graphene,” J. Chem. Phys. 130,054704 (2009).
21Y. Ferro, S. Morisset, and A. Allouche, “Evidence of hydrogenatedhexamers on graphite,” Chem. Phys. Lett. 478, 42–44 (2009).
22M. Bonfanti, S. Casolo, G. F. Tantardini, and R. Martinazzo, “Surfacemodels and reaction barrier in Eley-Rideal formation of H2 on graphiticsurfaces,” Phys. Chem. Chem. Phys. 13, 16680 (2011).
23M. Bonfanti, R. Martinazzo, R. F. Tantardini, and A. Ponti, “Physisorptionand diffusion of hydrogen atoms on graphite from correlated calculationson the H-coronene model system,” J. Phys. Chem. C 111, 5825–5829(2007).
24Y. Wang, H.-J. Qian, K. Morokuma, and S. J. Irle, “Coupled cluster anddensity functional theory calculations of atomic hydrogen chemisorptionon pyrene and coronene as model systems for graphene hydrogenation,”Phys. Chem. A 116, 7154–7160 (2012).
25G. Vidali, “H2 formation on interstellar grains,” Chem. Rev. 113, 8762–8782(2013).
26W. W. Duley and D. A. Williams, Interstellar Chemistry (Academic Press,Orlando, FL, 1984).
27D. Hollenbach and E. Salpeter, “Surface recombination of hydrogenmolecules,” Astrophys. J. 163, 155–164 (1971).
28V. Pirronello, C. Liu, L. Y. Shen, and G. Vidali, “Laboratory synthesis ofmolecular hydrogen on surfaces of astrophysical interest,” Astrophys. J.475, L69–L72 (1997).
29V. Pirronello, O. Biham, C. Liu, L. Y. Shen, and G. Vidali, “Efficiency ofmolecular hydrogen formation on silicates,” Astrophys. J. 483, L131–L134(1997).
30V. Pirronello, C. Liu, J. E. Roser, and G. Vidali, “Measurements of molec-ular hydrogen formation on carbonaceous grains,” Astron. Astrophys. 344,681–686 (1999).
31V. Mennella, L. Hornekær, J. Thrower, and M. Accolla, “The catalytic role ofcoronene for molecular hydrogen formation,” Astrophys. J. 745, L2 (2012).

014702-11 Petucci et al. J. Chem. Phys. 149, 014702 (2018)
32E. R. Latimer, F. Islam, and S. D. Price, “Studies of HD formed in excitedvibrational states from atomic recombination on cold graphite surfaces,”Chem. Phys. Lett. 455, 174–177 (2008).
33T. Zecho, A. S. Guattler, X. Sha, B. Jackson, and J. Kuappers, “Adsorptionof hydrogen and deuterium atoms on the (0001) graphite surface,” J. Chem.Phys. 117, 8486–8492 (2002).
34V. Hornekaer, Z. Sljivancanin, W. Xu, R. Otero, E. Rauls, I. Stensgaard,E. Laegsgaard, B. Hammer, and F. Besenbacher, “Metastable structuresand recombination pathways for atomic hydrogen on the graphite (0001)Surface,” Phys. Rev. Lett. 96, 156104 (2006).
35V. Hornekaer, E. Rauls, W. Xu, Z. Sljivancanin, R. Otero, I. Stensgaard,E. Laegsgaard, B. Hammer, and F. Besenbacher, “Clustering of chemisorbedH(D) atoms on the graphite (0001) surface due to preferential sticking,”Phys. Rev. Lett. 97, 186102 (2006).
36S. Morisset, F. Aguillon, M. Sizun, and V. Sidis, “Quantum wavepacketinvestigation of Eley-Rideal formation of H2 on a relaxing graphite surface,”Chem. Phys. Lett. 378, 615–621 (2003).
37R. Martinazzo and G. F. Tantardini, “Quantum effects in an exoergic, barri-erless reaction at high collision energies,” J. Phys. Chem. A 109, 9379–9383(2005).
38R. Martinazzo and G. F. Tantardini, “Quantum study of Eley-Rideal reactionand collision induced desorption of hydrogen atoms on a graphite surface.II. H-physisorbed case,” J. Chem. Phys. 124, 124702 (2006).
39Y. Ferro, D. Teillet-Billy, N. Rougeau, S. Morriset, and A. Allouche, “Sta-bility and magnetism of hydrogen dimers on graphene,” Phys. Rev. B 78,085417 (2008).
40R. Martinazzo, S. Casolo, and L. H. Hornekær, “Hydrogen recombinationon graphitic surfaces,” Springer Ser. Surf. Sci. 50, 157–177 (2013).
41S. Casolo, G. F. Tantardini, and R. Martinazzo, “Hydrogen recombina-tion and dimer formation on graphite from ab initio molecular dynamicssimulations,” J. Phys. Chem. A 120, 5032–5040 (2016).
42S. Morisset, F. Aguillon, M. Sizun, and V. Sidis, “Quantum dynamics ofH2 formation on a graphite surface through the Langmuir-Hinshelwoodmechanism,” J. Chem. Phys. 121, 6493 (2004).
43S. Morisset, F. Aguillon, M. Sizun, and V. Sidis, “Role of surface relaxationin the Eley−Rideal formation of H2 on a graphite surface,” J. Phys. Chem.A 108, 8571–8579 (2004).
44S. Morisset, F. Aguillon, M. Sizun, and V. Sidis, “Wave-packet study ofH2 formation on a graphite surface through the Langmuir-Hinshelwoodmechanism,” J. Chem. Phys. 122, 194702 (2005).
45R. Martinazzo and G. F. Tantardini, “Quantum study of Eley-Rideal reactionand collision induced desorption of hydrogen atoms on a graphite surface.I. H-chemisorbed case,” J. Chem. Phys. 124, 124703 (2006).
46P. Parneix and B. Brechignac, “Molecular dynamics simulation of the H2recombination on a graphite surface,” Astron. Astrophys. 334, 363–375(1998).
47R. Papoular, “Molecular dynamics simulation of gaseous atomic hydro-gen interaction with hydrocarbon grains,” Mon. Not. R. Astron. Soc. 359,683–687 (2005).
48M. Cacciatore and M. Rutigliano, “Recombination processes involving Hand D atoms interacting with a graphite surface: Collisional data relevant tofusion plasma devices,” Phys. Scr. T124, 80–85 (2006).
49T. Hama and N. Watanabe, “Surface processes on interstellar amorphoussolid water: Adsorption, diffusion, tunneling reactions, and nuclear-spinconversion,” Chem. Rev. 113, 8783–8839 (2013).
50A. G. G. M. Tielens, The Physics and Chemistry of the Interstellar Medium(Cambridge University Press, 2005).
51S. J. Plimpton, “Fast parallel algorithms for short-range molecular dynam-ics,” J. Comput. Phys. 117, 1–19 (1995).
52K. Chenoweth, A. C. T. Van Duin, and W. A. Goddard, “ReaxFF reactiveforce field for molecular dynamic simulations of hydrocarbons oxidation,”J. Phys. Chem. A 112, 1040–1053 (2008).
53A. Jaramillo-Botero, S. Naserifar, and W. A. Goddard III, “A general multi-objective force field optimization framework, with application to reactiveforce fields for silicon carbide,” J. Chem. Theory Comput. 10, 1426–1439(2014).
54J. M. Soler, E. Artacho, J. D. Gale, A. Garcıa, J. Junquera, P. Ordejon, andD. J. Sanchez-Portal, “The SIESTA method for ab initio order-N materialssimulation,” J. Phys.: Condens. Matter 14, 2745–2779 (2002).
55G. Henkelman, B. P. Huberuaga, and H. Jonsson, “A climbing image nudgedelastic band method for finding saddle points and minimum energy path,”J. Chem. Phys. 113, 9901–9905 (2000).
56G. Henkelman and H. Jonsson, “Improved tangent estimate in the nudgedelastic band method for finding minimum energy paths and saddle points,”J. Chem. Phys. 113, 9978–9985 (2000).
57D. Sheppard, R. Terrell, and G. J. Henkelman, “Optimization methods forfinding minimum energy paths,” Chem. Phys. 128, 134106 (2008).
58N. Troullier and J. L. Martins, “Efficient pseudo-potentials for pane-wavecalculations,” Phys. Rev. B 43, 1993–2006 (1991).
59J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approxi-mation made simple,” Phys. Rev. Lett. 77, 3865–3868 (1996).
60D. W. Brenner, “Empirical potential for hydrocarbons for use in simulat-ing the chemical vapor-deposition of diamond films,” Phys. Rev. B 42,9458–9471 (1990).
61J. Stuart, A. B. Tutein, and J. A. Harrison, “A reactive potential forhydrocarbons with intermolecular interactions,” J. Chem. Phys. 112, 6472(2000).
62W. A. Goddard, K. Chenoweth, S. Pudar, A. C. T. van Duin, and M. J. Cheng,“Structures, mechanisms, and kinetics of selective ammoxidation and oxi-dation of propane over multi-metal oxide catalysts,” Top. Catal. 50, 2–18(2008).
63T. T. Jarvi, A. Kuronen, M. Hakala, K. Nordlund, A. C. T. Van Duin, W.A. Goddard, and T. Jacob, “Development of a ReaxFF description for gold,”Eur. Phys. J. B 66, 75 (2008).
64S. Naserifar, L. C. Liu, W. A. Goddard, T. T. Tsotsis, and M. Sahimi, “Towarda process-based molecular model of SiC membranes. 1. Development of areactive force field,” J. Phys. Chem. C 117, 3308 (2013).
65S. J. Plimpton, “Fast parallel algorithms for short-range molecular dynam-ics,” J. Comput. Phys. 117, 1–19 (1995).
66D. Sheppard, R. Terrell, and G. J. Henkelman, “Optimization methods forfinding minimum energy paths,” Chem. Phys. 128, 134106 (2008).
67A. Dzhurakhalov and F. M. Peeters, “Structure and energetics of hydrogenchemisorbed on a single graphene layer to produce graphene,” Carbon 49,3258–3266 (2011).
68V. A. Borodin, T. T. Vehvilainen, M. G. Ganchenkova, and R. M. Nieminen,“Hydrogen transport on graphene: Competition of mobility and desorption,”Phys. Rev. B 84, 075486 (2011).
69N. Rougeau, D. Teillet-Billy, and V. Sidis, “On the PES for the interactionof an H atom with an H chemisorbate on a graphenic platelet,” Phys. Chem.Chem. Phys. 13, 17579–17587 (2011).
70V. Ivanovskaya, A. Zobelli, D. Teillet-Billy, N. Rougeau, V. Sidis, andP. R. Briddon, “Hydrogen adsorption on graphene: A first principles study,”Eur. Phys. J. B 76, 481–486 (2010).
71D. Bachellerie, M. Sizun, D. Teillet-Billy, N. Rougeau, and V. Sidis, “Eley-Rideal formation of H2 involving one of two para-chemisorbed H atoms ona graphite surface,” Chem. Phys. Lett. 448, 223–227 (2007).
72D. Teillet-Billy, N. Rougeau, V. Ivanovskaya, and V. Sidis, “Interactionof atoms with graphenic-type surfaces for the chemistry of the interstellarmedium: New properties of H dimers on the surface,” Int. J. Quantum Chem.110, 2231 (2010); Erratum 111, 4504 (2011).
73Z. Sljivancanin, M. Andersen, L. Hornekaer, and B. Hammer, “Structureand stability of small H clusters on graphene,” Phys. Rev. B 83, 205426(2011).
74F. Costanzo, P. L. Silvestrelli, and F. Ancilotto, “Physisorption, diffusion,and chemisorption pathways of H2 molecule on graphene and on (2, 2)carbon nanotube by first principles calculations,” J. Chem. Theory Comput.8, 1288–1294 (2012).
75H. Rouzo, “Variational R-matrix method for quantum tunneling problems,”Am. J. Phys. 71, 273 (2003).
76S. Cazaux and A. G. G. M. Tielens, “H2 formation on grain surfaces,”Astrophys. J. 604, 222 (2004); “Erratum: H2 formation on grain surfaces,”Astrophys. J. 715, 698 (2010).




















