Formation of crystalline PuO2+x.nH2O nanoparticles upon sorption of Pu(V,VI) onto hematite
12
Formation of crystalline PuO 2+x nH 2 O nanoparticles upon sorption of Pu(V,VI) onto hematite Anna Yu. Romanchuk a , Stepan N. Kalmykov a,b,c,⇑ , Alexander V. Egorov a , Yan V. Zubavichus d , Andrey A. Shiryaev b , Olga N. Batuk e , Steven D. Conradson e , Denis A. Pankratov a , Igor A. Presnyakov a a Lomonosov Moscow State University, Department of Chemistry, 119991 Moscow, Russia b Frumkin Institute of Physical Chemistry and Electrochemistry of RAS, 119071 Moscow, Russia c Vernadsky Institute of Geochemistry and Analytical Chemistry of RAS, 119991 Moscow, Russia d National Research Centre “Kurchatov Institute”, 123182 Moscow, Russia e Los Alamos National Laboratory, Los Alamos, NM 87544, USA Received 5 July 2012; accepted in revised form 14 July 2013; available online 25 July 2013 Abstract It has been recognized that natural aquatic colloids can readily sorb actinide elements, including plutonium, whose behav- ior is complicated by its multiple valence states and the possibility of redox reactions under environmental conditions. In this paper, the sorption and surface-mediated redox transformations of hexavalent plutonium on synthetic well-characterized hematite colloids are studied in a series of batch sorption experiments. The variation in the kinetics of the Pu–hematite inter- actions, Pu-L 3 -XAFS, and HRTEM over a broad range of total concentrations of Pu have been studied in an attempt to define the molecular-level speciation of Pu. The surface-mediated slow reduction of Pu(V/VI) results in the formation of crystalline nanoparticles of PuO 2+x nH 2 O approximately 1.5 nm in size at [Pu] tot P 10 9 M. This result is confirmed indepen- dently by HRTEM images of Pu-containing particles and through the identification in the EXAFS of a Pu neighbor shell at 3.8 A ˚ . The formation of such nanoparticles potentially influences the colloid-mediated transport of Pu in the subsurface environment because of the very slow leaching of Pu from the hematite colloids. Ó 2013 Elsevier Ltd. All rights reserved. 1. INTRODUCTION Plutonium is the chemical element of most concern at the nuclear legacy sites in the USA and the former Soviet Union. Its speciation and migration in subsurface environ- ments is attributed to the presence of colloids of different origins and compositions (McCarthy and Zachara, 1989; Penrose et al., 1990; Mysoedov and Novikov, 1998; Novikov et al., 1998, 2006; Kersting et al., 1999; Za ¨nker et al., 2000; Santschi et al., 2002; Kalmykov et al., 2007; Conradson et al., 2011). Various mineral inorganic colloids play an important role in plutonium migration at several sites, including the Nevada Test Site (Kersting et al., 1999), “Mayak” PA (Novikov et al., 2006; Kalmykov et al., 2007), and Rocky Flats (Santschi et al., 2002; Con- radson et al., 2011). Organic colloids, presumably humic and fulvic acids, were found to contribute to enhanced plu- tonium migration under near-surface conditions (Penrose et al., 1990; Mysoedov and Novikov, 1998; Novikov et al., 1998). The performance assessments at the existing and candidate nuclear repository sites are based on the long-term (time scale of thousands of years) predictions of the migration of various radionuclides; these predictions 0016-7037/$ - see front matter Ó 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.gca.2013.07.016 ⇑ Corresponding author at: Lomonosov Moscow State Univer- sity, Department of Chemistry, 119991 Moscow, Russia. Tel.: +7 495 9393220. E-mail address: [email protected] (S.N. Kalmykov). www.elsevier.com/locate/gca Available online at www.sciencedirect.com ScienceDirect Geochimica et Cosmochimica Acta 121 (2013) 29–40
-
Upload
moscowstate -
Category
Documents
-
view
1 -
download
0
Transcript of Formation of crystalline PuO2+x.nH2O nanoparticles upon sorption of Pu(V,VI) onto hematite

Available online at www.sciencedirect.com
www.elsevier.com/locate/gca
ScienceDirect
Geochimica et Cosmochimica Acta 121 (2013) 29–40
Formation of crystalline PuO2+x�nH2O nanoparticlesupon sorption of Pu(V,VI) onto hematite
Anna Yu. Romanchuk a, Stepan N. Kalmykov a,b,c,⇑, Alexander V. Egorov a,Yan V. Zubavichus d, Andrey A. Shiryaev b, Olga N. Batuk e, Steven D. Conradson e,
Denis A. Pankratov a, Igor A. Presnyakov a
a Lomonosov Moscow State University, Department of Chemistry, 119991 Moscow, Russiab Frumkin Institute of Physical Chemistry and Electrochemistry of RAS, 119071 Moscow, Russiac Vernadsky Institute of Geochemistry and Analytical Chemistry of RAS, 119991 Moscow, Russia
d National Research Centre “Kurchatov Institute”, 123182 Moscow, Russiae Los Alamos National Laboratory, Los Alamos, NM 87544, USA
Received 5 July 2012; accepted in revised form 14 July 2013; available online 25 July 2013
Abstract
It has been recognized that natural aquatic colloids can readily sorb actinide elements, including plutonium, whose behav-ior is complicated by its multiple valence states and the possibility of redox reactions under environmental conditions. In thispaper, the sorption and surface-mediated redox transformations of hexavalent plutonium on synthetic well-characterizedhematite colloids are studied in a series of batch sorption experiments. The variation in the kinetics of the Pu–hematite inter-actions, Pu-L3-XAFS, and HRTEM over a broad range of total concentrations of Pu have been studied in an attempt todefine the molecular-level speciation of Pu. The surface-mediated slow reduction of Pu(V/VI) results in the formation ofcrystalline nanoparticles of PuO2+x�nH2O approximately 1.5 nm in size at [Pu]tot P 10�9 M. This result is confirmed indepen-dently by HRTEM images of Pu-containing particles and through the identification in the EXAFS of a Pu neighbor shell at3.8 A. The formation of such nanoparticles potentially influences the colloid-mediated transport of Pu in the subsurfaceenvironment because of the very slow leaching of Pu from the hematite colloids.� 2013 Elsevier Ltd. All rights reserved.
1. INTRODUCTION
Plutonium is the chemical element of most concern atthe nuclear legacy sites in the USA and the former SovietUnion. Its speciation and migration in subsurface environ-ments is attributed to the presence of colloids of differentorigins and compositions (McCarthy and Zachara, 1989;Penrose et al., 1990; Mysoedov and Novikov, 1998;Novikov et al., 1998, 2006; Kersting et al., 1999; Zanker
0016-7037/$ - see front matter � 2013 Elsevier Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.gca.2013.07.016
⇑ Corresponding author at: Lomonosov Moscow State Univer-sity, Department of Chemistry, 119991 Moscow, Russia. Tel.: +7495 9393220.
E-mail address: [email protected] (S.N. Kalmykov).
et al., 2000; Santschi et al., 2002; Kalmykov et al., 2007;Conradson et al., 2011). Various mineral inorganic colloidsplay an important role in plutonium migration at severalsites, including the Nevada Test Site (Kersting et al.,1999), “Mayak” PA (Novikov et al., 2006; Kalmykovet al., 2007), and Rocky Flats (Santschi et al., 2002; Con-radson et al., 2011). Organic colloids, presumably humicand fulvic acids, were found to contribute to enhanced plu-tonium migration under near-surface conditions (Penroseet al., 1990; Mysoedov and Novikov, 1998; Novikovet al., 1998). The performance assessments at the existingand candidate nuclear repository sites are based on thelong-term (time scale of thousands of years) predictionsof the migration of various radionuclides; these predictions

30 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40
require a detailed understanding of the behavior of theseradionuclides at the molecular scale. The interaction ofradionuclides with colloids can be described by surfacecomplexation constants (Davis and Kent, 1990; Alonsoand Degueldre, 2003; Bradbury and Baeyens, 2006), whichare the basis for calculating the distribution coefficients, Kd,for given environmental conditions. Correct surface com-plexation models should be based on the chemical specia-tion data from spectroscopy measurements. However, forplutonium in neutral pH solutions, this description isimpossible because of the extremely low solubility of tetra-valent plutonium (610�10 M), which is below the detectionlimit for XAFS, XPS, and most other methods that are sen-sitive to valence and other chemical speciation parameters.
The behavior of Pu is further complicated by the forma-tion of polynuclear species associated with the hydrolysis ofPu(IV); these species further agglomerate through the for-mation of intrinsic nanocolloids. Recently the speciationof such polynuclear complexes and primary nanoparticleswas examined by nano-ESI-TOF and EXAFS (Rotheet al., 2004, 2009). However, large uncertainties remain con-cerning the structure of intrinsic colloids of Pu(IV), i.e.,whether the structure is polymeric or crystalline.
The behavior of Pu is even more complicated in mineralcolloidal suspensions, in which the formation of intrinsiccolloids could be accompanied by redox reactions, surfacesorption, bulk diffusion and other processes that effect thePu speciation and migration. Previously, the interactionof Pu with Fe(III) oxide colloids, including the speciationand surface complexation of Pu, was extensively examined(Keeney-Kennicutt and Morse, 1985; Sanchez et al., 1985;McCubbin et al., 2002; Powell et al., 2005, 2011; Khasano-va et al., 2007; Hixon et al., 2010; Hu et al., 2010; Kirschet al., 2011; Nitsche et al., 2011; Romanchuk et al., 2011)and showed the stabilization of Pu(IV) on the surface.
Previously proposed reasons for Pu(V/VI) reduction in-clude the effects of radiolysis and Pu(V) disproportionationin the electrical double layer (Sanchez et al., 1985; Silver,2000). However, measurements at [Pu]tot of approximately10�14 M with the short-lived 237Pu tracer, which decaysthrough electron capture, contradict the radiolytic reduc-tion of Pu(V/VI) Romanchuk et al., 2011. The dispropor-tionation of Pu(V) in an electrical double layer is also notpossible at such extremely low concentrations. Nitscheet al. (2011) attributed the reduction of Pu(V/VI) to thepresence of admixtures, including Fe(II), and demonstratedthat Pu(VI) sorption on goethite results in reduction toPu(V) rather than to Pu(IV). No reductant in a hematitesuspension was identified in any of the previous studies.However, one could not exclude the presence of very lowconcentrations of Fe(II).
Several papers (McCubbin et al., 2002; Powell et al.,2005) have described a possible role for light in the kineticsof Pu(V/VI) reduction in Fe(III) oxide suspensions. Theseexperiments demonstrated that the surface-mediated reduc-tion of Pu(V/VI) is because of the semiconductor propertiesof hematite, whose band gap of 2.2 eV lies within the rangeof visible light. Evidence in hematite for electron transfercatalyzed by dissolved Fe(II) was also reported earlier(Yanina and Rosso, 2008; Handler et al., 2009). In
oxygen-free solutions, Fe(III) oxides undergo reductive dis-solution according to the reaction:
a� Fe2O3 þ 6Hþ þ 2e� ¡ 2Fesol2þ þ 3H2O
with a standard redox potential of approximately 0.7 V.The dissolved Fe(II) could be sorbed onto the hematite sur-face, preferentially onto the (001) crystal edge, with furtherelectron transfer in the bulk and reductive dissolution of the(hk0) crystal edge, as shown by Yanina and Rosso (2008).A similar mechanism was also proposed by Handler et al.(2009). Such electron transfer processes would create anopportunity for Pu(V/VI) surface-mediated reduction.
Thermodynamically, the formation of Pu(IV) surfacespecies, i.e., �Fe–O–PuIV(OH)n, is much more energeticallyfavorable than the formation of Pu(V) surface species, i.e.,�Fe–O–PuVO2(OH)m, and could possibly result in the shiftof the formal redox potential of the Pu(IV)/Pu(V) pairbased on the Nernst formalism. The values of the equilib-rium constants for the surface complexation reactions oftetravalent and pentavalent actinides reported for analogsof the Pu valence state, i.e., Th(IV) and Np(V), arelogbTh(IV) = 10.5 for the reaction �FeOH + Th4+
M �FeOTh3+ + H+ from Cromieres et al. (1998) andlogbNp(V) = �2.09 from Kohler et al. (1999) for the reaction�FeOH + NpO2
+M �FeONpO2 + H+ and differ by
approximately 12 orders of magnitude.An important observation was made previously (Kee-
ney-Kennicutt and Morse, 1985; Powell et al., 2004;Romanchuk et al., 2011) demonstrating that Pu(IV) be-comes more stable on the surface over time. Although thereason for such behavior remains unknown, several possi-bilities were proposed (Powell et al., 2004), such asintraparticle diffusion, surface precipitation, chemisorption,or surface complexation. In our previous paper (Roman-chuk et al., 2011), which describes batch sorption data forPu at various concentrations, the precipitation of intrinsicPu(IV) colloids, which have high kinetic stabilities, is pro-posed to be a reason for the very slow leaching of Pu fromthe hematite surface. In a recent paper by Powell et al.(2011), the formation of Pu4O7 crystalline nanoparticlesin goethite suspensions was established to be a result of epi-taxial crystal growth upon the addition of Pu(IV) solutionand colloids to a goethite suspension at pH 7.
In this paper, we describe the interaction of oxidizedplutonium, Pu(V/VI), with hematite colloids through batchsorption experiments accompanied by XAFS and HRTEMas spectroscopic nanoscale characterization techniques todetermine the Pu speciation and local distribution. Bydoing these experiments in combination with Pu leachingstudies and comparing the data with those reported forthe solubility of PuO2(am, hyd), we were able to suggestthe factors that control Pu concentration in solution. An-other important question addressed in the paper is thestructure of the Pu(IV) nanoparticles formed in the suspen-sion, i.e. whether the structure is polymeric or crystalline.
Oxidized Pu species were added to solutions and suspen-sions for the following reasons. First, Pu(V) and Pu(VI) arerelatively soluble and could be added as truly soluble spe-cies compared with Pu(IV), which easily forms intrinsic col-loids. Through this addition, we can trace the kinetics of the

A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40 31
sorption/reduction/precipitation processes. Second, thepresence of oxidized Pu species has been reported earlierat very low concentrations in sea water (Nelson and Lovett,1978) and for 240Pu, which is a decay product of 244Cmfrom the Szilard-Chalmers process (Dai et al., 2002).
2. MATERIALS AND METHODS
2.1. Hematite preparation and characterization
Hematite was synthesized with the forced hydrolysisreaction as described by Burukhin et al. (2000). The samplewas characterized by XRD, TEM, Mossbauer spectroscopyand the BET-technique. No Fe2+ phases were observed inthe synthesized sample; however, the presence of traceFe2+ (<4 at.%) cannot be ruled out (see on-line supporting
information for more details on sample characterization).
2.2. Sorption experiments
Radiochemically pure 239Pu was used in all the experi-ments; its purity was ascertained by alpha- and gamma-spectrometry. The sorption experiments were performedwith an the initial oxidation state of Pu(VI) because bothPu(VI) and Pu(V) are orders of magnitude more solublethan Pu(IV). To prepare the Pu(VI) stock solution, aPu(IV) nitrate solution was fumed with concentrated per-chloric acid. The Pu valence was monitored by UV–visspectrophotometry. The absorption band at 831 nm wasused to monitor Pu(VI).
For all the sorption experiments, the hematite suspen-sion was prepared with 0.1 M NaClO4 as a backgroundnon-complexing electrolyte. The experiments were per-formed under ambient atmospheric conditions at25 ± 2 �C with continuous agitation of the suspension.The hematite suspension was sustained for several days inthe background electrolyte solution prior to the sorptionexperiments. Immediately after the addition of a known ali-quot of Pu(VI) stock solution, the pH value was measuredand was adjusted by small amounts with diluted HClO4
(ultrapure grade) or NaOH (ultrapure grade). For variousreaction times, the solid phase was separated by centrifuga-tion at 40,000 g for 15 min (Allegra 64R, Beckman Coul-ter). Liquid scintillation spectroscopy (TriCarb 2700TR,Canberra Packard Ind.) was used for the 239Pu radioactiv-ity counting. The sorption was calculated from the differ-ence between the radioactivity of the 239Pu initially addedto the suspension and the radioactivity remaining afterthe equilibration.
The redox speciation of Pu in solution was analyzedwith parallel solvent extractions: 0.5 M thenoyltrifluoroace-tone (TTA) (Lancaster) in toluene at pH 0 (Nitsche et al.,1988) (the TTA was purified by vacuum sublimation at45 �C prior to use and only freshly prepared TTA solutionswere used in all the experiments) and 0.05 M bis(2-ethyl-hexyl) phosphate (HDEHP) (Merck) in toluene at pH 0.The required pH was adjusted by adding the requiredamount of HNO3 or HCl to the TTA and HDEHP, respec-tively. At this pH, TTA extracts Pu(IV) leaving Pu(V) andPu(VI) in the aqueous phase whereas HDEHP extracts
Pu(IV) and Pu(VI), leaving Pu(V). A blank experiment withno hematite added to the solution was also performed un-der exactly the same experimental conditions to accountfor the sorption of Pu onto the vial walls and to studythe redox behavior of Pu in the absence of hematite.
Leaching tests were performed by the addition of con-centrated HClO4 to make a pH 1.3 suspension. After vari-ous time intervals, aliquots of the suspension were collectedand centrifuged for 15 min at 40,000 g. Afterwards, the plu-tonium in solution was measured to obtain the fraction ofPu that leached from the hematite.
The redox speciation of Pu in the leachate was analyzedby the same procedure as described above. The mass bal-ance for the different Pu redox states was calculated withthe following equation (Morgenstern and Choppin, 2002):
½Pu�total ¼ ½PuðIVÞ�unleached þ ½PuðVÞ�solution þ ½PuðVIÞ�solution
þ ½PuðIVÞ�solution
The redox potentials, E(mV) were measured with a Pt-electrode relative to a Ag/AgCl reference electrode andwere converted to Eh through Eh(mV) = E(mV) + 208 mV.ZoBell’s standard solution was used for the control. The Ehwas directly measured in the suspension without filtrationand separation of the Pu. In each case, the measurementswere performed until constant values were obtained. Thedrift in the Eh values during the experiment was 20–30 mV, which is most likely within the measurement uncer-tainty and the difficulty in quantifying the Eh for redoxpairs at low concentrations.
2.3. HRTEM examination
The nanoscale distribution of the Pu on the hematitesurface and the possible formation of Pu(IV) intrinsic col-loids was examined with HRTEM and with the resultingelectron diffraction patterns and EDX spectrometry. Tothis end, after the equilibration with Pu, a single drop ofthe hematite suspension was deposited onto a carbon-coated TEM grid for the HRTEM. The HRTEM imageswere obtained with an aberration-corrected JEOL 2100Foperated at 200 kV, yielding an information limit of0.8 A. The DF images and EDX analysis were performedin STEM mode; the spot size was 1 nm with the HAADFand JED 2300 (JEOL) detectors.
2.4. XAFS data collection and analysis
The hematite suspensions examined by XAFS wereequilibrated with Pu for 6 months ([Pu]tot = 2.6 � 10�6 M,[a-Fe2O3] = 2 m2/L, V = 1 L). The pH and Eh were mea-sured periodically, and small adjustments in the pH wereobtained through the addition of a few drops of diluteNaOH or HClO4. The changes in the pH and Eh valuescorrespond to the line presented in Fig. 1B, which will bediscussed further. XAFS measurements were performedfor two samples with different pH values: the suspensionsSample 1 and Sample 2 had final pH values of 6.6 and6.0, respectively. After 24 h and 3 weeks of equilibration,an aliquot of the Sample 1 suspension was collected for
2360
20
40
60
80
100
236236 103103103
pH = 6.0 pH = 6.0Eh = 0.38 VEh = 0.38 V
pH = 3.5 pH = 3.5Eh = 0.48 VEh = 0.48 V
4545 3131 33
Frac
tion,
%
Pu(VI) Pu(V) Pu(IV)
331 45
Time, d.
pH = 2.0 pH = 2.0Eh = 0.57 VEh = 0.57 V
A B
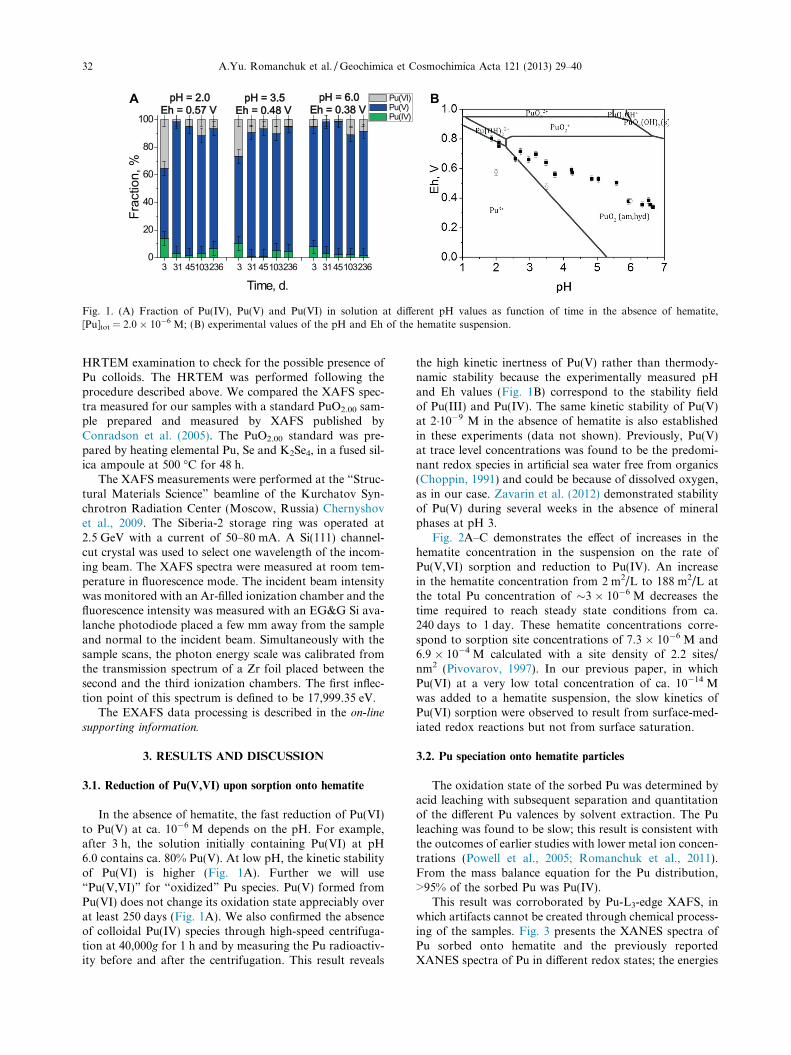
Fig. 1. (A) Fraction of Pu(IV), Pu(V) and Pu(VI) in solution at different pH values as function of time in the absence of hematite,[Pu]tot = 2.0 � 10�6 M; (B) experimental values of the pH and Eh of the hematite suspension.
32 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40
HRTEM examination to check for the possible presence ofPu colloids. The HRTEM was performed following theprocedure described above. We compared the XAFS spec-tra measured for our samples with a standard PuO2.00 sam-ple prepared and measured by XAFS published byConradson et al. (2005). The PuO2.00 standard was pre-pared by heating elemental Pu, Se and K2Se4, in a fused sil-ica ampoule at 500 �C for 48 h.
The XAFS measurements were performed at the “Struc-tural Materials Science” beamline of the Kurchatov Syn-chrotron Radiation Center (Moscow, Russia) Chernyshovet al., 2009. The Siberia-2 storage ring was operated at2.5 GeV with a current of 50–80 mA. A Si(111) channel-cut crystal was used to select one wavelength of the incom-ing beam. The XAFS spectra were measured at room tem-perature in fluorescence mode. The incident beam intensitywas monitored with an Ar-filled ionization chamber and thefluorescence intensity was measured with an EG&G Si ava-lanche photodiode placed a few mm away from the sampleand normal to the incident beam. Simultaneously with thesample scans, the photon energy scale was calibrated fromthe transmission spectrum of a Zr foil placed between thesecond and the third ionization chambers. The first inflec-tion point of this spectrum is defined to be 17,999.35 eV.
The EXAFS data processing is described in the on-line
supporting information.
3. RESULTS AND DISCUSSION
3.1. Reduction of Pu(V,VI) upon sorption onto hematite
In the absence of hematite, the fast reduction of Pu(VI)to Pu(V) at ca. 10�6 M depends on the pH. For example,after 3 h, the solution initially containing Pu(VI) at pH6.0 contains ca. 80% Pu(V). At low pH, the kinetic stabilityof Pu(VI) is higher (Fig. 1A). Further we will use“Pu(V,VI)” for “oxidized” Pu species. Pu(V) formed fromPu(VI) does not change its oxidation state appreciably overat least 250 days (Fig. 1A). We also confirmed the absenceof colloidal Pu(IV) species through high-speed centrifuga-tion at 40,000g for 1 h and by measuring the Pu radioactiv-ity before and after the centrifugation. This result reveals
the high kinetic inertness of Pu(V) rather than thermody-namic stability because the experimentally measured pHand Eh values (Fig. 1B) correspond to the stability fieldof Pu(III) and Pu(IV). The same kinetic stability of Pu(V)at 2�10�9 M in the absence of hematite is also establishedin these experiments (data not shown). Previously, Pu(V)at trace level concentrations was found to be the predomi-nant redox species in artificial sea water free from organics(Choppin, 1991) and could be because of dissolved oxygen,as in our case. Zavarin et al. (2012) demonstrated stabilityof Pu(V) during several weeks in the absence of mineralphases at pH 3.
Fig. 2A–C demonstrates the effect of increases in thehematite concentration in the suspension on the rate ofPu(V,VI) sorption and reduction to Pu(IV). An increasein the hematite concentration from 2 m2/L to 188 m2/L atthe total Pu concentration of �3 � 10�6 M decreases thetime required to reach steady state conditions from ca.240 days to 1 day. These hematite concentrations corre-spond to sorption site concentrations of 7.3 � 10�6 M and6.9 � 10�4 M calculated with a site density of 2.2 sites/nm2 (Pivovarov, 1997). In our previous paper, in whichPu(VI) at a very low total concentration of ca. 10�14 Mwas added to a hematite suspension, the slow kinetics ofPu(VI) sorption were observed to result from surface-med-iated redox reactions but not from surface saturation.
3.2. Pu speciation onto hematite particles
The oxidation state of the sorbed Pu was determined byacid leaching with subsequent separation and quantitationof the different Pu valences by solvent extraction. The Puleaching was found to be slow; this result is consistent withthe outcomes of earlier studies with lower metal ion concen-trations (Powell et al., 2005; Romanchuk et al., 2011).From the mass balance equation for the Pu distribution,>95% of the sorbed Pu was Pu(IV).
This result was corroborated by Pu-L3-edge XAFS, inwhich artifacts cannot be created through chemical process-ing of the samples. Fig. 3 presents the XANES spectra ofPu sorbed onto hematite and the previously reportedXANES spectra of Pu in different redox states; the energies

0 50 100 150 2000
20
40
60
80
100
pH = 6.6 ± 0.1 pH = 6.2 ± 0.1
Sorp
tion,
%
Time, d.
A
0 2 4 6 8 100
20
40
60
80
100
pH = 6.7 ± 0.1 pH = 6.4 ± 0.1
Sorp
tion,
%
Time, d.
B
0 1 2 3 4 5 6 7 8 90
20
40
60
80
100
pH = 6.1 ± 0.1
Sorp
tion,
%
Time, d.
C
0.0 0.5 1.0 1.5 2.0 2.50
20
40
60
80
100
pH = 6.1 ± 0.1
Sorp
tion,
%Time, d.
D
Fig. 2. Sorption of Pu(VI) (initial oxidation state) onto hematite as a function of time at various hematite concentrations and at constant[Pu]tot concentration (2.3 � 10�6 – 4 � 10�6 M): A – [a-Fe2O3] = 2 m2/L; B – [a-Fe2O3] = 11 m2/L; C – [a-Fe2O3] = 21 m2/L, and D –[a-Fe2O3] = 188 m2/L.
18060 18080 18100
0.5
1.0
1.5
18040 18060 18080 18100
dμ/d
eV Pu(IV)aquo sample1 sample2
eV
Pu L
III N
orm
aliz
ed A
bsor
banc
e Pu(IV)aquo Pu(V)aquo Pu(VI)aquo sample1 sample2
Fig. 3. Normalized XANES Pu-L3-edge spectra compared tostandard solutions of Pu(IV), Pu(V) and Pu(VI) as prepared andmeasured by Conradson et al. (2004).
A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40 33
and shapes of the previously reported spectra can be used toidentify the valence of the Pu species, albeit with someambiguity (Conradson et al., 2004). Both the white lineshape and the features (first inflection point at18,063 ± 0.5 eV and peak energy at 18,068 ± 0.5 eV) ofthe spectra of our samples fall within the range found forPu(IV) compounds. As is common of all AnO2
+/AnO22+
actinyl complexes, the XANES of the Pu(V) and Pu(VI)trans dioxo species exhibit diminished amplitude for thewhite line and peak absorbance and exhibit a feature justbeyond the peak absorbance, referred to as the “actinyl”shoulder, which originates in the single and multiple scat-
tering from the two distinct apical and equatorial neighborshells with differences of 0.5–0.6 A in the Pu–O distance.The absence of the shoulder in these spectra indicates thatthe Pu valence in these samples is, at the highest, of thePuO2+x type and only a fraction exists as a Pu(V)-oxo moi-ety, which has a relatively long bond length when averagedover the entire sample.
Additional information on the speciation of Pu can beobtained from analysis of the EXAFS extracted from thehigh-energy region of the spectrum beyond the edge(Fig. 4). The Fourier transforms of the EXAFS, v(R),which bear some resemblance to the partial pair distribu-tion function of Pu, can be compared to the EXAFS ofthe PuO2.00 standard (Conradson et al., 2005) to give someinformation about whether and how these samples may dif-fer from fully ordered PuO2.00 and may exhibit the struc-tural features that occur in the various forms ofPuO2+x�y(OH)2y type of species. All three spectra showtwo large peaks in v(R) at � R = 1.9 A and 3.6 A and asmaller peak at approximately R = 3.0 A (Fig. 4A). ForPuO2.00, these peaks originate from the phase-shifted pri-mary contributions of the O neighbor at 2.34 A and thePu neighbor at 3.82 A and from the Ramsauer-Townsendresonance in the Pu–Pu EXAFS amplitude that causes thecontribution from this shell to have two peaks in v(R).The principal peak is near its actual distance, and the smal-ler peak is below the principal peak at approximatelyR = 3.0 A. However, this region also contains contributionsfrom the additional neighbor shells that occur when thePuO2 is disordered. Such disorder in these samples is indi-cated by the relative amplitude of this feature, which wouldscale with the amplitude of the Pu-Pu peak if the localstructure was identical to PuO2.00 but instead is much

0 2 4 6
2.2 2.4 2.6 2.8 3.0 3.2 3.4
Sample 2 Sample 1 PuO2.00
Pu
L 3�(
R, k
3 �(k
))
Sample 2
data fit
R- , Å
A
PuO2.00
Sample 1
2 4 8 10
k3
k(Å-1)
data fit
Sample 1
Sample 2
PuO2.00
B
5 6 8
Sample 2
data fit O fit Fek3
Sample 1
C
k(7Å
-1)
χχ
Δ
Pu L
3χ(R
,k3 χ
(k))
R-Δ, Å
Fig. 4. Pu-L3-edge EXAFS for our experimental samples and a crystalline PuO2.00 sample: (A and B) Fourier transform and v(R). The blackline is the modulus and the imaginary component of the data, and the red line is the fit. The inset in the upper right corner of figure A showsthe imaginary parts for all the samples from 2.2 to 3.5 A. (C) The black Fourier-transformed shell at a distance of approximately 3.3–3.4 A fitwith both the Fe and O phases and amplitudes. (For interpretation of the references to colour in this figure legend, the reader is referred to theweb version of this article.)
34 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40
larger. It is more directly apparent in comparisons of thereal component of v(R) (Fig. 4A insert); these comparisonsshow that the spectra in this region all differ from eachother despite the superficial resemblance of their moduli.Close inspection of these data shows that the spectra ofSample 1 and Sample 2 differ near R = 2.2 A at the localminimum in the modulus; at this point, the imaginary partfor Sample 1 displays a kink whereas that for Sample 2 isasymmetric but smooth. There must therefore be at leastone additional neighbor shell around the Pu in these sam-ples that has different characteristics in our samples.Although this neighbor shell is most likely an O with aPu–O distance of approximately 3.3 A, the recent reportson Th, U, and Pu sorbed onto Fe-containing minerals withAn–Fe distances from 3.4–3.7 A (Bargar et al., 2000; Secoet al., 2009; Hu et al., 2010) or even the Fe incorporationdiscovered in environmental samples of PuO2 (Batuk
et al., in preparation) indicate that this possibility shouldbe checked as well.
The possibility of Fe incorporation was checked withcurves fit to the spectra with various combinations of O,Pu, and Fe neighbor shells. The evidence that this featureoriginates with a neighbor shell in the Pu environment iscorroborated by the large residual in this region when fit-ting with only PuO2.00 shells. The fit is greatly improved,and this residual is eliminated through the inclusion of anadditional O or Fe shell that gives chemically reasonablemetric parameters (Table 1); these results lead to the princi-pal conclusion that the Pu speciation in these samples is thePuO2+x�y(OH)2y type we have observed previously. Testingfor O or Fe was performed by Fourier filtering the EXAFSfrom this region after the principal structural componentswere subtracted so the quality of the fit with both oxygenand iron amplitudes and phases could be compared

Table 1EXAFS structural parameters of the measured Sample 1 and Sample 2 and comparison with the crystallographic data and the crystallinePuO2.00 sample.
Sample O shells, Pu–O < 3.6 A Fe/O Pu
Crystal structure Belin et al. (2004) 2.3368(1) 3.81612(1)PuO2.00 Conradson et al. (2005) R 2.34 ± 0.02 3.39 ± 0.02 3.83 ± 0.01
N 7.4 ± 2.0 1.3 ± 0.5 10 ± 2.4r 0.076 ± 0.01 0.08 ± 0.02 0.067 ± 0.006
Sample 1 2.22 ± 0.02 2.37 ± 0.02 3.42 ± 0.03 3.79 ± 0.0283.3 ± 1.1 3.8 ± 1.1 0.7 ± 0.04 4.4 ± 1.30.07* 0.07* 0.048 ± 0.02 0.087 ± 0.016
Sample 2 2.24 ± 0.02 2.40 ± 002 3.33 ± 0.02 3.81 ± 0.025.0 ± 1.6 4.3 ± 1.6 1.4 ± 0.5 4.7 ± 1.00.07* 0.07* 0.035 ± 0.016 0.076 ± 0.015
* Set to 0.07.
A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40 35
(Fig. 4C). However, over this limited range, these values arequite similar, and differences in the amplitude functions areeasily compensated by changes in the Debye Waller factor.The Pu speciation in these two samples is therefore consis-tent with the structures of the PuO2+x-type compoundsfound previously in a large number of laboratory preparedmaterials. The 0.09 A difference in the Pu–O distance in theshell at 3.3–3.4 A is also consistent with these previous re-sults although it is at the high end of the previous results.
The sample analyzed by XAFS was further character-ized by HRTEM and EDX. The corresponding HRTEMimages and EDX data for Sample 1 are presented inFig. 5. The aggregates of the Pu-containing crystallinenanoparticles are clearly seen on the hematite surface. Ascalculated from several TEM images, the average size ofthese nanoparticles is 1.5 nm (Fig. 5). The cubic crystalstructure typical of Pu(IV) dioxide is confirmed by theFFT and HRTEM images presented in Fig. 5B and C.The d-spacing determined from the ca. 30 independentHRTEM images are presented in Table 2 and indicate val-ues close to those of the crystal structure of PuO2. Theuncertainty in the d-spacing determined from the HRTEMmake it impossible to draw conclusions about the possibledifferences with the ideal PuO2 crystal structure. The differ-ence in these values also could arise from variations of x inPuO2+x and from the nanoparticle effect. Several papershave reported the difference between the lattice parametersof metal or metal oxide nanoparticles and those of micro-crystalline phases (Woltersdorf et al., 1981; Ayyub et al.,1995; Lamber et al., 1995; Tsunekawa et al., 1999; Zhanget al., 2001, 2002; Tonejc et al., 2002; Utsunomiya et al.,2002; Rajh et al., 2003; Wu et al., 2004; Qi and Wang,2005; Djerdj and Tonejc, 2006). The decrease in the latticeparameter compared to the bulk values (Woltersdorf et al.,1981; Lamber et al., 1995; Qi and Wang, 2005) is generallyexplained by the internal pressure produced by surfacestress in small particles. The inverse phenomenon, an in-crease in the lattice parameters for metal oxide nanoparti-cles, has been reported (Ayyub et al., 1995; Tonejc et al.,2002; Zhang et al., 2002; Wu et al., 2004; Djerdj and Tone-jc, 2006) and is explained in terms of compensation for thesurface charge of the oxide nanoparticles through increasesin the distances between atoms. The increased solubility
that has been reported for ThO2 and other oxides is animportant macro-scale behavior of nanoparticles comparedto the microcrystalline solids of the same compositions (Ba-tuk et al., in press).
HRTEM has been performed for hematite suspensionsequilibrated with Pu(V/VI) for one day and three weeks,at which times steady state conditions are not reached.These measurements were performed to demonstrate thatno Pu-containing precipitates are formed upon additionof Pu and to prove that the formation of PuO2+x�nH2Onanoparticles is a slow surface-mediated reaction.
The redox speciation of plutonium in a solution equili-brated with hematite was studied by solvent extraction.Fig. 6A presents the pH sorption edge after 720 days ofequilibration. Fig. 6B presents the data on the redox speci-ation of plutonium in solution equilibrated with hematite;and these data are compared with the calculated solubilitydiagrams for Pu(IV) and Pu(V). The experimental pointswith arrows correspond to plutonium concentrations belowthe detection limit. At pH > 1.5, the concentration of pluto-nium is dominated by Pu(V) (corresponds to >95%) andonly at pH 1.1 fraction of Pu(IV) reached approximately60%. The distribution of plutonium according to the redoxstates follows the same trend as the oxidative solubility ofPuO2; however, in our case, the Pu(V) concentration insolution is systematically lower. This result could be ex-plained by the decrease of plutonium concentration becauseof sorption onto the hematite.
The kinetics of the leaching of plutonium from hematiteat micro- and nanomolar concentrations depends on theequilibration time in the sorption experiments and have verysimilar dependencies at 22 and 35 days of equilibration andat 33 and 7 months of equilibration, respectively (Fig. 7).This trend also follows the slow kinetics of leaching of thePu(IV) intrinsic colloids as reported earlier (Romanchuket al., 2011). This is indirect evidence of the same behaviorat total Pu concentrations of 10�9 M and 10�6 M whichare indicative of the formation of PuO2+x�nH2O crystallinenanoparticle in both cases. The aging time of the Pu/hema-tite suspension has a clear effect on the Pu-leaching rate andthe dissolved Pu-fraction. A possible explanation for the ef-fect of the aging time on the Pu-leaching rate is the increase

Fig. 5. HRTEM examination of Sample 1: (A) STEM image and EDX spectra from the regions marked by the numbers. Nanoparticles ofPuO2+x�nH2O are clearly seen in the STEM image. (B and C) HRTEM images demonstrating the PuO2+x�nH2O nanoparticles. The FFT ispresented for the boxes indicated on the HRTEM image.
36 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40

Table 2Experimentally determined from HRTEM, d-spacing for the PuO2+x�nH2O crystalline nanoparticles formed in the hematite suspension([Pu]tot = 1.8 � 10�6 M, [a-Fe2O3] = 2 m2/L and pH 6.0), and comparison with crystallographic data.
d1, A d2, A d3, A d4, A d5, A
This work 2.60 ± 0.05 1.80 ± 0.05 1.25 ± 0.02 1.06 ± 0.02 0.88 ± 0.02PuO2 crystal structure Belin et al. (2004) 2.6991 1.9085 1.3495 1.1019 0.8997
1 2 3 4 5 60
20
40
60
80
100
Sorp
tion,
%
pH
A
0 1 2 3 4 5 6 7 8 9 10-12
-10
-8
-6
-4
calculation for 0.1 M NaClO4
log[
Pu]
pH
Pu(IV) Pu(V)
Pu(IV)
Pu(V)
B
Fig. 6. (A) Sorption of Pu onto hematite as a function of pH after an equilibration period of 720 days and (B) redox speciation of plutoniumin solution studied by solvent extraction and the calculated solubility values for Pu(IV) and Pu(V) ([Pu]tot = 1.8 � 10�6 M, [a-Fe2O3] = 10 m2/L). The dash-dot line corresponds to the total concentration of Pu in solution. For the solubility calculations, the thermodynamic data fromNeck et al. (2007) have been used.
0 1 2 25 50 75 100 125 1500
20
40
60
80
100 [Pu] = 10-9 М, 22 d. [Pu] = 10-9 М, 33 m. [Pu] = 10-6 М, 35 d. [Pu] = 10-6 М, 7 m.
Frac
tion
of le
acha
ble
Pu, %
Time, h.
Fig. 7. Kinetics of Pu leaching from the hematite suspension aftersorption. The time in the legend corresponds to the sorptionequilibration time before leaching.
Fig. 8. HRTEM image of the PuO2+x�nH2O nanoparticles, whichare formed from the interaction of Pu(VI) (initial oxidation state)with hematite and a typical FFT of these particles. [Pu]tot =8 � 10�9 M, [a-Fe2O3] = 11 m2/L, pH = 6.1.
A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40 37
the degree of crystallinity of the PuO2+x�nH2O nanoparticlesor the slow diffusion into the micropores.
HRTEM was also applied to the hematite suspensionwith a lower total Pu concentration to provide direct proofof the formation of PuO2+x�nH2O nanoparticles. Theexperimental setup was the same as that for the micromolarconcentrations. Fig. 8 shows the HRTEM image of a hema-tite surface equilibrated with 8 � 10�9 M of Pu(VI) for13 months at pH 6.1. As for the micromolar concentrationsdescribed above, the formation of crystalline nanoparticleswith an average size of 1–2 nm is established; however, theconcentration of the nanoparticles is much lower. Theseparticles also exhibit the cubic crystal structure typical ofPuO2+x�nH2O and have similar d-spacing.
The formation of Pu(IV) nanocolloids is favored forthermodynamic reasons as demonstrated by the Pourbaixdiagram (Fig. 1B); however, it is important that in hema-tite-free solutions under close pH–Eh conditions, Pu(V)
demonstrates high kinetic stability. The effect of the forma-tion of Pu(IV) nanocolloids is clearly demonstrated in thisstudy by HRTEM and EXAFS at a total Pu concentrationof ca. 10�6 M. This mechanism remains feasible even atmuch lower concentrations, e.g., 10�9 M. Although thedetermination of Pu speciation by EXAFS is limited to

Fig. 9. The general scheme for the interaction of Pu with hematite colloids at different total concentrations of Pu, and the distribution ofdifferent Pu redox states.
38 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40
plutonium concentrations higher than ca. 10�6 M,HRTEM could be successfully applied because of the highlocal concentration.
Based on the experimental data presented here and inour previous paper (Romanchuk et al., 2011), the reductionof Pu(V,VI) is always mediated by the hematite surface.The interaction mechanism of Pu with hematite strongly de-pends on the Pu/hematite ratio and the total Pu concentra-tion. At very low [Pu]tot, i.e., approximately 10�14 M, whichis close to the concentration of the global fallout level, therelatively fast sorption kinetics and leaching behavior indi-cate the formation of monomeric �FeO–Pu(OH)x surfacespecies (Romanchuk et al., 2011). At concentrations>10�9 M, the surface-mediated reduction of Pu(V/VI) re-sults in the formation of crystalline PuO2+x�nH2O nanopar-ticles as the main Pu species with a possible small fractionof chemisorbed species. The generalized scheme for theinteraction of Pu with hematite is presented in Fig. 9.
In conclusion, the observed surface-mediated redoxreactions and the formation of crystalline PuO2+x�nH2Onanoparticles can affect the potential colloid-facilitatedtransport of actinides in subsurface environments. The pri-mary macroscale observation is that even at relatively lowtotal Pu concentrations, the formation of the intrinsic crys-talline nanoparticles inhibits or decelerates Pu leaching. Asa result, the modeling for colloid-facilitated transport of Puis more complicated than for other cations, for which sur-face complexation modeling could be applied.
ACKNOWLEDGEMENTS
We acknowledge a RF President stipend to AYuR (SP-173.2012.2), the Russian Basic Research Foundation (12-03-
31370 and 11-04-92116) and the Ministry of Education and Scienceof Russian Federation (contract 11.519.11.5011). Measurements atthe User Facility, Kurchatov Synchrotron Radiation Center, aresupported by the Russian Federal Contract 16.552.11.7003. Wealso acknowledge the “Nanochemistry and nanomaterials” userfacility of the Department of Chemistry of MSU for providingthe HRTEM measurements. Work at Los Alamos National Labo-ratory was supported by the DOE OBES Heavy Element Chemis-try and the Laboratory Directed Research and Developmentprograms.
We thank the anonymous referees for their very useful com-ments and suggestions that helped improve the quality of themanuscript.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data associated with this article can befound, in the online version, at http://dx.doi.org/10.1016/j.gca.2013.07.016.
REFERENCES
Alonso U. and Degueldre C. (2003) Modelling americium sorptiononto colloids: effect of the redox potential. Colloids Surf., A 217,49–54.
Ayyub P., Palkan V. R., Chattopadnyay S. and Multani M. (1995)Effect of crystal size reduction on lattice symmetry andcooperative properties. Phys. Rev. B 51, 6135–6138.
Bargar J., Reitmeyer R., Lenart J. and Davis J. (2000) Character-ization of U(VI)-carbonato ternary complexes on hematite:EXAFS and electrophoretic mobility measurements. Geochim.
Cosmochim. Acta 64(16), 2737–2749.Batuk O. N. and Conradson S. D., et al. (in preparation)
Environmental and forensic chemistry of U and Pu at

A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40 39
Chernobyl, McGuire AFB, Mayak, Rocky Flats, Hanford andLos Alamos.
Batuk O. N., Szabo D. V., Denecke M. A., Vitova T. andKalmykov S. N. (in press) Synthesis and characterization ofthorium, uranium and cerium oxide nanoparticles. Radiochim.
Acta.Belin C., Valenza P. J., Reynaud M. A. and Raison P. E. (2004)
New hermetic sample holder for radioactive materials fitting toSiemens D5000 and Bruker D8 X-ray diffractometers: applica-tion to the Rietveld analysis of plutonium dioxide Renaud. J.
Appl. Crystallogr. 37, 1034–1037.Bradbury M. H. and Baeyens B. A. (2006) Quasi-mechanistic non-
electrostatic modeling approach to metal sorption on clayminerals. In Surface Complexation Modeling (ed. J. Lutzenkir-chen). Academic Press, New York, p. 518.
Burukhin A., Churagulov B., Oleynikov N., Knot’ko V. (2000)Hydrothermal synthesis of mesoporous iron oxide powders.Joint 6th International Symposium on Hydrothermal Reactions
and 4th Conference on Solvo-Thermal Reactions. Extended
Abstracts. #194.Chernyshov A. A., Veligzhanin A. A. and Zubavichus Y. V. (2009)
Structural Materials Science end-station at the KurchatovSynchrotron Radiation Source: recent instrumentationupgrades and experimental results. Nucl. Instrum. Methods
Phys. Res. A 603, 95–98.Choppin G. R. (1991) Redox speciation of plutonium in natural
waters. J. Radioanal. Nucl. Chem. 147(1), 109–116.Conradson S. D., Abney K. D., Begg B. D., Brady E. D., Clark D.
L., Auwer C., Ding M., Dorhout P. K., Espinosa-Faller F. J.,Gordon P. L., Haire R. G., Hess N. J., Hess R. F., Keogh W.,Lander G. H., Lupinetti A. J., Morales L. A., Neu M. P.,Palmer P. D., Paviet-Hartmann P., Reilly S. D., Runde W. H.,Tait D., Veirs K. and Wastin F. (2004) Higher order speciationeffects on plutonium L3 X-ray absorption near edge spectra.Inorg. Chem. 43, 116–131.
Conradson S. D., Begg B. D., Clark D. L., den Auwer C., Ding M.,Dorhout P. K., Espinosa-Faller F. J., Gordon P. L., Haire R.G., Hess N. J., Hess R. F., Keogh D. W., Lander G. H.,Manara D., Morales L. A., Neu M. P., Paviet-Hartmann P.,Rebizant J., Rondinella V. V., Runde W., Tait C. D., Veirs D.K., Villella P. M. and Wastin F. (2005) Charge distribution andlocal structure and speciation in the UO2+x and PuO2+x binaryoxides for x 6 0.25. J. Solid State Chem. 178, 521–535.
Conradson S. D. et al. (2011) X-ray absorption spectroscopy ofplutonium particles at the rocky flats US nuclear weaponsproduction site. In Actinides Nanoparticles Research (eds. S. N.Kalmykov and M. A. Denecke). Springer, Heidelberg, p. 377.
Cromieres L., Moulin V., Fourest B. and Guillaumont R. (1998)Sorption of thorium onto hematite colloids. Radiochim. Acta
88, 249–255.Dai M., Kelley J. M. and Buesseler K. O. (2002) Sources and
migration of plutonium in groundwater at the Savannah RiverSite. Environ. Sci. Technol. 36, 3690–3699.
Davis J. A. and Kent D. B. (1990) Surface complexation modelingin aqueous geochemistry. In Mineral-Water Interface Geochem-
istry, Reviews in Mineralogy, vol. 23 (eds. M. F. Hochella andA. F. White). Mineralogical Society of America, Washington,D.C., p. 177.
Djerdj I. and Tonejc A. M. (2006) Structural investigationsof nanocrystalline TiO2 samples. J. Alloys Compd. 413, 159–174.
Handler R. M., Beard B. L., Jonson C. M. and Scherer M. M.(2009) Atom exchange between aqueous Fe(II) and goethite: anFe isotope tracer study. Environ. Sci. Technol. 43, 1102–1107.
Hixon A. E., Hu Y.-J., Kaplan D. I., Kukkadapu R. K., NitscheH., Qafoku O. and Powell B. A. (2010) Influence of iron redox
transformations on plutonium sorption to sediments. Radio-
chim. Acta 98, 685–692.Hu Y.-J., Schwaiger L. K., Booth C. H., Kukkadapu R. K.,
Cristiano E., Kaplan D. and Nitsche H. (2010) Molecularinteractions of plutonium(VI) with synthetic manganese-substi-tuted goethite. Radiochim. Acta 98, 655–663.
Kalmykov S. N., Kriventsov V. V., Teterin Yu. A. and Novikov A.P. (2007) Plutonium and neptunium speciation bound tohydrous ferric oxide colloids. C. R. Chim. 10, 1060–1066.
Keeney-Kennicutt W. L. and Morse J. W. (1985) The redoxchemistry of Pu(V)O2
+ interaction with common mineralsurfaces in dilute solutions and seawater. Geochim. Cosmochim.
Acta 49, 2577–2588.Kersting A. B., Efurd D. W., Finnegan D. L., Rokop D. J., Smith
D. K. and Thompson J. L. (1999) Migration of plutonium ingroundwater at the Nevada Test Site. Nature 397, 56–59.
Khasanova A. B., Shcherbina N. S., Kalmykov S. N., Teterin Yu.A. and Novikov A. P. (2007) Sorption of Np(V), Pu(V), andPu(IV) on colloids of Fe(III) oxides and hydrous oxide andMnO2. Russ. Radiochem. 49, 419–425.
Kirsch R., Fellhauer D., Altmaier M., Rossberg A., FandhanelTh., Charlet L. and Scheinost A. C. (2011) Plutonium redoxreactions with iron oxides under anoxic conditions. Mineral.
Mag. 75, 1194.Kohler M., Honeyman B. and Leckie J. (1999) Neptunium(V)
sorption on hematite (a-Fe2O3) in aqueous suspension: theeffect of CO2. Radiochim. Acta 85, 33–48.
Lamber R., Wetjen S. and Jaeder N. I. (1995) Size dependence ofthe lattice parameter of small palladium particles. Phys. Rev. B
51, 10968–10971.McCarthy J. F. and Zachara J. M. (1989) Subsurface transport of
contaminants. Environ. Sci. Technol. 23, 496–502.McCubbin D., Leonard K. and Emerson H. (2002) Influence of
thermal and photochemical reactions upon the redox cycling ofPu between solid and solution phases in seawater. Mar. Chem.
80, 61–77.Morgenstern A. and Choppin G. R. (2002) Kinetics of the
oxidation of Pu(IV) by manganese dioxide. Radiochim. Acta
90, 69–74.Mysoedov B. F. and Novikov A. P. (1998) Main sources of
radioactive contamination in Russia and methods for theirdetermination and speciation. J. Radioanal. Nucl. Chem. 229,33–38.
Neck V., Altmaier M. and Fanghanel T. (2007) Solubility ofplutonium hydroxides/hydrous oxides under reducing condi-tions and in the presence of oxygen. C. R. Chim. 10, 959–977.
Nelson D. M. and Lovett M. B. (1978) Oxidation state ofplutonium in the Irish Sea. Nature 276, 599–600.
Nitsche H., Lee S. C. and Gatti R. C. (1988) Determination ofplutonium oxidation states at trace levels pertinent to nuclearwaste disposal. J. Radioanal. Nucl. Chem. 124, 171–185.
Nitsche H., Hu Y.-J., Wang D. L. and Schwaiger L. K. (2011)Plutonium interaction with pure and substituted iron andmanganese oxyhydroxide minerals. Mineral. Mag. 75, 1545.
Novikov A. P., Pavlotskaya F. I., Goryachenkova T. A., SmaginA. I., Emelyanov V. V., Kyzovkina E. V., Barsykova K. B.,Lavrinovich E. A., Korovaykov P. A., Drozko E. G., Rovniy S.I., Posokhov A. K. and Mysoedov B. F. (1998) The content anddistribution of radionuclides in waters and bottom sedimentsfrom some industrial reservoirs of PA “Mayak”. Russ. Radio-
chem. 40, 453–461.Novikov A. P., Kalmykov S. N., Utsunomiya S., Ewing R. C.,
Horreard F., Merkulov A., Clark S. B., Tkachev V. V. andMyasoedov B. F. (2006) Colloid transport of plutonium in thefar-field of the Mayak Production Association, Russia. Science
314, 638–641.

40 A.Yu. Romanchuk et al. / Geochimica et Cosmochimica Acta 121 (2013) 29–40
Penrose W. R., Polzer W. L., Essington E. H., Nelson D. M. andOrlandini K. A. (1990) Mobility of plutonium and americiumthrough a shallow aquifer in a semiarid region. Environ. Sci.
Technol. 24, 228–234.Pivovarov S. (1997) Surface and site density of the oxide – solution
interface. J. Colloid Interface Sci. 196, 321–323.Powell B., Fjeld R., Kaplan D., Coates J. and Serkiz S. (2004)
Pu(V)O2+ adsorption and reduction by synthetic magnetite
(Fe3O4). Environ. Sci. Technol. 38, 6016–6024.Powell B., Fjeld R., Kaplan D., Coates J. and Serkiz S. (2005)
Pu(V)O2+ adsorption and reduction by synthetic hematite and
goethite. Environ. Sci. Technol. 39, 2107–2114.Powell B. A., Dai Z., Zavarin M., Zhao P. and Kersting A. B.
(2011) Stabilization of plutonium nano-colloids by epitaxialdistortion on mineral surfaces. Environ. Sci. Technol. 45, 2698–2703.
Qi W. H. and Wang M. P. (2005) Size and shape dependent latticeparameters of metallic nanoparticles. J. Nanopart. Res. 7, 51–67.
Rajh T., Poluektov O. I. and Thurnauer M. C. (2003) Chargeseparation in titanium oxide nanocrystalline semiconductorsrevealed by magnetic resonance. In Chemical Physics of
Nanostructured Semiconductors (eds. A. I. Kokorin and D. W.Bahnemann). NOVA Science, New York, pp. 1–34.
Romanchuk A. Yu., Kalmykov S. N. and Aliev R. A. (2011)Plutonium sorption onto hematite colloids onto femto- andnanomolar concentrations. Radiochim. Acta 99, 137–144.
Rothe J., Walther C., Denecke M. A. and Fanghanel Th. (2004)XAFS and LIBD investigation of the formation and structureof colloidal Pu(IV) hydrolysis products. Inorg. Chem. 43, 4708–4718.
Rothe J., Walther C., Brendebach B., Buchner S., Fuss M.,Denecke M. A. and Geckeis H. (2009) A combined XAFS, ESITOF-MS and LIBD study on the formation of polynuclearZr(IV), Th(IV) and Pu(IV) species. J. Phys: Conf. Ser. 190,012188.
Sanchez A. L., Murray J. W. and Sibley T. H. (1985) Theadsorption of plutonium IV and V on goethite. Geochim.
Cosmochim. Acta 49, 2297–2307.Santschi P. H., Roberts K. A. and Guo L. (2002). Environ. Sci.
Technol. 36, 3711–3719.
Seco F., Hennig C., Pablo J., Rovira M., Rojo I., Marti V.,Gimenez J., Duro L., Grive M. and Bruno J. (2009) Sorption ofTh(IV) into iron corrosion products: EXAFS study. Environ.
Sci. Technol. 43, 2825–2830.Silver G. L. (2000) The auto-reduction of hexavalent 239Pu in acid
solution. J. Radioanal. Nucl. Chem. 246, 429–430.Tonejc A. M., Djerdj I. and Tonejc A. (2002) An analysis of
evolution of grain size-lattice parameters dependence in nano-crystalline TiO2 anatase. Mater. Sci. Eng., C 19, 85–89.
Tsunekawa S., Sivamohan R., Ito S., Kasuya A. and Fukuda T.(1999) Structural study on monosize CeO2�x nano-particles.Nanostruct. Mater. 11, 141–147.
Utsunomiya S., Jensen K. A., Keeler G. J. and Ewing R. C. (2002)Uraninite and fullerene in atmospheric particulates. Environ.
Sci. Technol. 36, 4943–4947.Woltersdorf J., Nepijko A. S. and Pippel E. (1981) Dependence of
lattice parameters of small particles on the size of the nuclei.Surf. Sci. 106, 64–69.
Wu L., Wiesmann H. J., Moodenbaugh A. R., Klie R. F., Zhu Y.,Welch D. O. and Suenaga M. (2004) Oxidation state and latticeexpansion of CeO2�x nanoparticles as a function of particlesize. Phys. Rev. B 69, 125415-1–125415-9.
Yanina S. V. and Rosso K. M. (2008) Linked reactivity at mineral-water interfaces through bulk crystal conduction. Science 320,218–222.
Zanker H., Richter W., Brendler V. and Nitsche H. (2000) Colloid-borne uranium and other heavy metals in the water of minedrainage gallery. Radiochim. Acta 88, 619–624.
Zavarin M., Powell B. A., Bourbin M., Zhao P. and Kersting A. B.(2012) Np(V) and Pu(V) ion exchange and surface-mediatedreduction mechanisms on montmorillonite. Environ. Sci. Tech-
nol. 46, 2692–2698.Zhang J., Wu Z., Liu T., Hu T., Wu Z. and Ju X. (2001) XANES
study on the valence transitions in cerium oxide nanoparticles.J. Synchroton Radiat. 8, 531–532.
Zhang F., Chan S. W., Spanier J. E., Apak E., Jin Q., Robinson R.D. and Herman I. P. (2002) Cerium oxide nanoparticles: size-selective formation and structure analysis. Appl. Phys. Lett. 80,127–129.
Associate editor: Jon Chorover