
Nanomechanical properties of multi-block copolymer microspheres for drug delivery applications
Upload
lovely-professional-universityCategory
view
0download
0
Fluorescence Spectroscopic Investigation To Identify the Micelle to Gel Transition ofAqueous Triblock Copolymer Solutions
Sony George,† Manoj Kumbhakar,*,‡ Prabhat Kr. Singh,‡ Rajib Ganguly,§ Sukhendu Nath,‡
and Haridas Pal*,‡
Chemistry Department, UniVersity of Kerala, KariaVattom Campus, ThiruVananthapuram, Kerala 695581, andRadiation & Photochemistry and Chemistry DiVisions, Bhabha Atomic Research Centre, Mumbai 400085, India
ReceiVed: NoVember 07, 2008; ReVised Manuscript ReceiVed: February 06, 2009
Steady-state and time-resolved fluorescence anisotropy measurements using probes coumarin 153 (C153)and 4-heptadecylumbelliferon (HUF) have been carried out to understand the micelle to gel transition of anaqueous triblock copolymer P123 ((EO)20-(PO)70-(EO)20) (EO ) ethylene oxide; PO ) propylene oxide)solution. Anisotropy results with a normal fluorescent probe, C153, do not show a characteristic change dueto the micelle to gel transition. However, the probe HUF having a long hydrocarbon chain that helps itsstrong association with the micelle shows an increase in anisotropy above the sol-gel transition point. Thisdifference has been explained as invoking a substantial contribution from the micellar structural fluctuationsto the depolarization of HUF as its hydrocarbon chain is embedded in the micellar structure, which is notsensed significantly by the normal probe C153. That the extent of change in anisotropy for HUF upon gelationis not that large is possibly caused by the collective motion of the physically interconnected nodes, as observedfrom the dynamic light scattering studies, which acts in favor of a relatively faster depolarization in the gelphase. Similar studies in other copolymers, such as P85 ((EO)26-(PO)40-(EO)26) and F127((EO)100-(PO)65-(EO)100), further demonstrate the potential of probes latched with hydrocarbon chains indisplaying a characteristic change for the micelle to gel transition which otherwise remains obscured fornormal fluorescent probes.
IntroductionIn addition to their widespread industrial applications, the
ability to display a rich structural polymorphism has resultedin numerous investigations involving triblock copolymersystems.1-11 Triblock copolymers comprised of poly(ethyleneoxide) ((EO)x) and poly(propylene oxide) ((PO)y) blocks havethe general structural formula (EO)x-(PO)y-(EO)x, where x andy represent the number of EO and PO units, respectively. Abovea certain temperature, the (PO)y block turns hydrophobic, andconsequently, the triblock copolymers exhibit amphiphiliccharacter. Apart from forming micelles, these triblock copolymersolutions also form a number of lyotropic liquid-crystallinephases with cubic, hexagonal, or lamellar structures at highcopolymer concentrations. In general, an isotropic gel phase isformed at 53% micellar volume fraction, either due to hardsphere crystallization11-16 or due to micellar packing andentanglement.17,18 However, due to the substantial degree ofhydration in the corona region consisting of the EO units, anaqueous solution of these copolymers can also form such anisotropic gel phase at much lower volume fraction than53%.11-16 The stability of this gel phase is determined by twomelting temperatures, namely, the lower and the upper meltingtemperatures.
Even though considerable efforts have been made in elucidat-ing the structures of different phases of block copolymersolutions, attempts to understand their microenvironments or
the local friction offered by these microdomains are only modest.Understanding the local friction experienced by the solutemolecules in confined geometries is essential to get a betterappreciation of many physicochemical processes that transpirein these systems. Rotational diffusion of organic probe moleculessolubilized in these confined systems is one of the convenientmethods to investigate the characteristics of the microenviron-ments. A few such studies have been reported in the literaturewhere different phases of various aqueous triblock copolymershave been examined with the aid of fluorescence anisotropy,19-27
Stokes shift,20 and solvation dynamics and lifetime28-32 mea-surements. According to the literature, the reorientation timeof a neutral probe molecule (e.g., 2,5-dimethyl-1,4-dioxo-3,6-diphenylpyrrolo[3,4-c]pyrrole, DMDPP, coumarin 153, C153,and coumarin 102, C102) increases drastically when there is aunimer to micelle transition for different copolymers (e.g., P123,(EO)20-(PO)70-(EO)20, and F88, (EO)109-(PO)41-(EO)109),because the solute molecule experiences more friction in themicellar environment compared to that in a homogeneousunimer solution.19-21 However, no significant increase has beenobserved in the reorientation time of these neutral probes andeven for ionic probes such as coumarin 343 (C343) andrhodamine 110 (Rh110) in concentrated copolymer (P123 andF88) solutions at the micelle to gel transition temperature.20,21,26
These results also indicate that when probe molecules aresolubilized in the corona region (i.e., for neutral probes) or atthe surface region of the micelle (i.e., for ionic probes), theyexperience almost identical friction in both the micelle and gelphases, and hence, no drastic change is observed in thereorientation time of the solute on transition from the micelleto the gel phase. Recent excitation wavelength dependent
* To whom correspondence should be addressed. E-mail: [email protected], [email protected] (M.K.); [email protected] (H.P.). Fax:91-22-25505151 /25519613.
† University of Kerala.‡ Radiation & Photochemistry Division, Bhabha Atomic Research Centre.§ Chemistry Division, Bhabha Atomic Research Centre.
J. Phys. Chem. B 2009, 113, 5117–5127 5117
10.1021/jp809826c CCC: $40.75 2009 American Chemical SocietyPublished on Web 03/24/2009

rotational dynamic studies of coumarin 480 (C480) in a micelleas well as in the gel phase of P123 also corroborate the aboveliterature findings.28,32 However, an increase in one of thecomponents of the reorientation time for the cationic proberhodamine 123 (Rh123) in 20% aqueous F127((EO)100-(PO)65-(EO)100) solution has been observed by Jeonet al.,22 who ascribed it to the micelle-micelle entanglementabove the sol-gel transition temperature. Mali et al.26 showedlater that the above observation is actually due to the unimer tomicelle transition rather than the sol to gel transition, as initiallysuggested.
In most of the literature reports, investigation of the micelleto gel transition has been attempted by temperature-dependentfluorescence anisotropy experiments where no clear transitioncould be observed in the anisotropy results.19-22,26 If the increasein the rigidity of the probe microenvironment is not sufficientto be reflected in the temperature-dependent anisotropy results,studying the concentration-dependent micelle to gel formationseems to be a viable alternative. This approach has also beenattempted but with very limited success by Mali et al.,26 wherethe interfacial probe Rh110 was employed at different concen-trations of P123. However, the uncertainty of the probe locationin these heterogeneous media often impedes the conclusion fromthe fluorescence experiments to indirectly evaluate the sur-rounding microenvironment of the probe.33 Preferential solu-bilization of ionic probes, such as C343, Rh110, or Rh123, onthe micellar surface is not dubious, but small partitioning ofthese probes in the bulk water phase cannot be ruled out.Similarly, neutral probes, such as coumarin 153 (C153),coumarin 102 (C102), etc., may reside either in the core or inthe corona region of the micelle or may be distributed in boththe locations. In fact, in the literature there have been argumentsboth in favor and against the solubilization of C153 dye in thecore of the copolymer micelles.20,21,30,34 Thus, it is important toexplore the micelle to gel transition using a fluorescence probethat has a conspicuous location in the micellar phase, and withthis aim the present study has been undertaken.
In this work we present a systematic investigation on themicelle to gel transition of triblock copolymer P123 solutionusing two coumarin probes, one a functionalized dye (with along alkyl chain), 4-heptadecylumbelliferon (HUF), and theother a normal dye, C153. The structures of these dyes areshown in Chart 1. Structurally, HUF can be envisaged asequivalent to a surfactant molecule with a fluorescent moietyacting as its headgroup. There are several reports in the literaturewhere such a probe has been extensively used to investigatethe surface potential and interface polarity of surfactant ag-gregates from the variations in proton equilibrium.35-39 Fol-lowing recent literature,30,40-53 it can be assumed that thisfunctionalized probe (HUF) will be an integral part of thesupramolecular assemblies when dissolved in low concentrationin copolymer micellar solution, where the -C17H35 chain ofHUF will dissolve in the core of the copolymer micelle and thepolar chromophoric head of HUF will be projected out fromthe micellar core to the hydrated corona region (cf. Chart 2a).
Following the well-known two-step model,54-59 the fluores-cence anisotropy decay of a probe in a micelle is mainlydetermined by the interplay of three different motions. Theyare (i) the wobbling motion of the micellized probe in a smallcone, (ii) the lateral diffusion of the probe along the curvedmicellar surface, and (iii) the rotation of the whole micelle (cf.Chart 2b). In general, with normal fluorescence probes, the slowoverall rotation of the micelle does not give any significantcontribution to the observed anisotropy decay, because the majorpart of the anisotropy decay is caused by the other two motions(wobbling and lateral diffusion), which are much faster thanthe whole micelle rotation.19,26,34,45,46 In spite of this generaltrend, some estimation of reverse micellar sizes from a longerrotational time constant (employing probes with few nanosecondfluorescence lifetimes, e.g., ammonium salt of 1-anilino-8-naphthalenesulfonic acid, ANS, and coumarin 500, C500) hasbeen reported in the literature.60,61 It is expected that, due tothe surfactant-like structure, HUF will experience an exceedinglylarge hindrance to its lateral diffusion owing to its greateranchoring to the micelle compared to a normal probe.58,59
Accordingly, the surfactant-like probe with a hydrophobic tailis often known to exhibit a nonzero residual anisotropy value.58,59
We expect that a small contribution from the micellar rotationto fluorescence depolarization with a long time constant maypossibly be observed with HUF, though the same may not bepossible with normal probe C153. The microenvironments fora probe residing in the corona or interfacial region are reportedto be nearly alike in the gel and micellar phases.20,21,26 Therotational motion of a micelle in the gel phase will however belargely hindered in comparison to that in a simple micellarsolution. This in effect is predicted to cause a significant changein the fluorescence anisotropy of HUF in the gel phase comparedto that in the micellar phase, though such a change may not beexpected with the normal C153 probe for which whole micellerotation does not contribute much in the anisotropy decay.19,34
It is this proposition that motivated us to undertake the presentstudy. Additionally, comparison of anisotropy results of HUFwith those of C153 along with extension of this study to otherblock copolymers, such as P85 ((EO)26-(PO)40-(EO)26) andF127, of different hydrophilic ((EO)x) to hydrophobic ((PO)y)ratios is expected to provide more insight into the micelle togel transition and give a kind of generalized picture in relationto the applicability of the present propositions and inferences.Motivation of the present work is also to characterize theinfluence of the surfactant probe on the aggregation behaviorof the aqueous copolymer solutions. A qualitative structure ofthe micelle and gel phases of a copolymer solution is depictedin Chart 3.
CHART 1: Chemical Structures of HUF and C153 CHART 2: (a) Qualitative Picture for the Probe (HUF)Location in the Micellar Phase and (b) DifferentRotational Motions of a Probe for the Anisotropy Decayin a Micellea
a Wobbling in a cone (1), translational diffusion on the micellarsurface (2), and overall micelle rotation (3).
5118 J. Phys. Chem. B, Vol. 113, No. 15, 2009 George et al.

Methods
The copolymers P123, F127, and P85 were obtained fromAldrich, Sigma, and BASF, respectively, and used withoutfurther purification. Laser grade C153 and HUF dyes wereobtained from Exciton and Sigma, respectively, and used asreceived. The nanopure water, having a conductivity of <0.1µS cm-1, was obtained by passing distilled water through aMillipore gradiant A10 water system and used for the prepara-tion of the aqueous micellar solutions.
In the experimental solutions, the required amounts of theblock copolymers were taken by weighing and were keptovernight under refrigeration after addition of the requisiteamount of water in sealed containers. Samples for measurementswere prepared by adding C153 and HUF dyes in the aqueoussolutions of the block copolymers at low temperatures (∼285K) under stirring for 24 h. An estimate of the micelleconcentrations ([M]) in the solutions was made from theconcentration of the block copolymers ([S]) used, the cmc ofthe copolymer micelles, and the average aggregation number(Nagg) of these micelles, using a simple relation such as [M] )([S] - cmc)/Nagg.62 The probe concentrations (∼1-60 µM) usedwere significantly low, typically on the order of 1/10 to 1/100of the micelle concentrations. With this experimental condition,the possibility of multiple probes occupying a single micellewas negligibly small. Such an experimental condition wasimportant to avoid the effect of any interdye interaction on theobserved anisotropy results.
Steady-state absorption spectra were recorded using a Shi-madzu UV-vis spectrophotometer, model UV-160A. Fluores-cence spectra were recorded using a Hitachi (Tokyo, Japan)model F-4010 spectrofluorimeter. In spectrofluorimetric mea-surements, the desired sample temperatures were achieved ((1°C) with the help of a thermocouple-based temperature control-ler, which is regulated by a microprocessor (Eurotherm).
Time-resolved fluorescence anisotropy measurements were car-ried out using a diode-laser-based time-correlated single-photon-counting (TCSPC) spectrometer from IBH, United Kingdom. Inthe present work, 373 and 408 nm diode lasers (1 MHz) were usedas the excitation sources for the HUF and C153 dyes, respectively,and an MCP-PMT detector (IBH) was used for the fluorescencedecay measurements. For the present setup, the instrument responsefunction was ∼90 ps at the full width at half-maximum (fwhm).In the present work, the temperature of the solution was variedwith the help of a coldfinger arrangement and the temperature wascontrolled within (1 °C using a microprocessor-based temperaturecontroller (model DS from IBH).
For lifetime measurements, fluorescence decays were recordedat the magic angle (54.7°) with respect to the vertically polarizedexcitation light. Fluorescence anisotropy studies were carried
out by measuring the fluorescence decays for parallel (I|(t)) andperpendicular (I⊥ (t)) polarizations with respect to the verticallypolarized excitation light. The anisotropy decay function, r(t),was constructed using the following relation:63
where G is a correction factor for the polarization bias of thedetection setup. The G factor was independently determined forboth steady-state and time-resolved anisotropy measurements byusing the horizontally polarized excitation light and measuring thetwo perpendicularly polarized fluorescence decays with respect tothe excitation polarization.63 All the measurements were repeatedat least three times, both to check the reproducibility and to obtainthe average values of the relaxation times. As mentioned in theResults and Discussion, due to the involvement of several depo-larizing motions in the overall anisotropy decay, it is erroneous toindividually assign a time constant to each of them. Rather weadopted the concept of comparing the average anisotropy decaytimes to correlate changes during the micelle to gel transition ofcopolymer solutions. It should be noted that the quality of the fits(fluorescence and anisotropy decays) was judged by the reduced�2 values and the distribution of the weighted residuals among thedata channels. For the accepted fits the �2 values were close tounity and the weighted residuals were distributed randomly amongthe data channels.
Dynamic light scattering (DLS) measurements of the solutionswere performed using a Malvern 4800 autosizer employing a7132 digital correlator. The light source was an argon ion laseroperated at 514.5 nm with a maximum output power of 2 W,and the scattered light was detected by an avalanche photodiode(APD). The average decay rates (Γ) of the electric fieldautocorrelation functions, g1(τ), were obtained either by usingthe method of cumulants or from a biexponential analysis.64,65
Diffusion coefficients (D) of the scattering particles werecalculated using the following equation:
q being the magnitude of the scattering vector given by q )[4πn sin(θ/2)]/λ, where n is the refractive index of the solvent,λ is the wavelength of laser light, and θ is the scattering angle).Γ vs q2 plots were generated by carrying out measurements atfive different angles ranging from 50° to 130°. For the diffusivescattering species, these Γ vs q2 plots are linear, and the diffusioncoefficients were calculated from their slopes. In the absenceof translational diffusion, on the other hand, they are of nonlinearnature, and the diffusion coefficients were calculated from thedecay rate obtained at a 90° scattering angle. The hydrodynamicradius, rM, of the scattering particles was calculated using theStokes-Einstein equation
where k is the Boltzmann constant, T is the absolute temperature,and η is the viscosity of the medium.
Results and Discussion
Photophysics and Location of Fluorescence Probes. Thesolubilities of HUF and C153 in water are quite low. In the
CHART 3: Schematic Representation of the Micelle toGel Transition in Triblock Copolymers
r(t) )I|(t) - GI⊥ (t)
I|(t) + 2GI⊥ (t)
Γ ) Dq2
rM ) kT6πηD
Micelle to Gel Transition in Copolymer Solutions J. Phys. Chem. B, Vol. 113, No. 15, 2009 5119
presence of P123 micelles, however, their solubility increasessubstantially. According to the literature reports, these probesreside preferentially in the micellar phase.35-39 It is shown bya detailed spectroscopic investigation of C153 in copolymermicelles that the probe resides preferentially in the micellarcorona region.30,34,40 The spectroscopic investigations of HUFin micelles of common surfactants are also reported in theliterature.35-39 To the best of our knowledge, such studies forthe HUF probe have not been reported in copolymer micelles.Hence, we made an attempt to understand the photophysicalproperties of HUF in P123 micelles before carrying out theanisotropy measurements, which are discussed below.
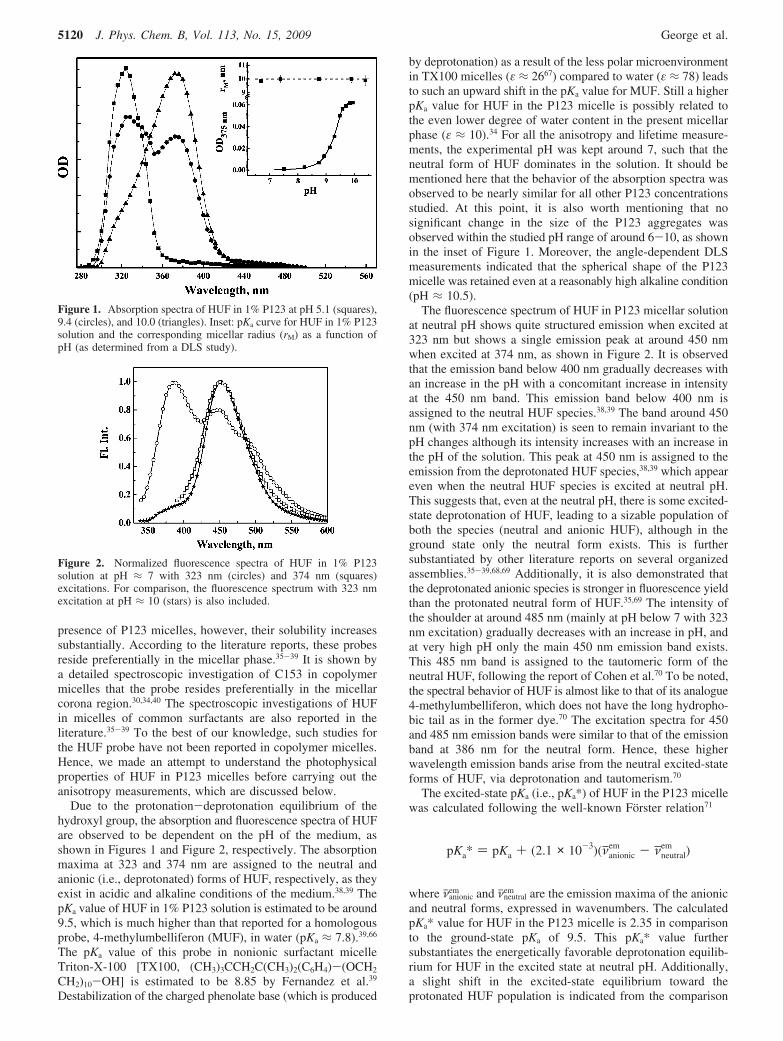
Due to the protonation-deprotonation equilibrium of thehydroxyl group, the absorption and fluorescence spectra of HUFare observed to be dependent on the pH of the medium, asshown in Figures 1 and Figure 2, respectively. The absorptionmaxima at 323 and 374 nm are assigned to the neutral andanionic (i.e., deprotonated) forms of HUF, respectively, as theyexist in acidic and alkaline conditions of the medium.38,39 ThepKa value of HUF in 1% P123 solution is estimated to be around9.5, which is much higher than that reported for a homologousprobe, 4-methylumbelliferon (MUF), in water (pKa ≈ 7.8).39,66
The pKa value of this probe in nonionic surfactant micelleTriton-X-100 [TX100, (CH3)3CCH2C(CH3)2(C6H4)-(OCH2
CH2)10-OH] is estimated to be 8.85 by Fernandez et al.39
Destabilization of the charged phenolate base (which is produced
by deprotonation) as a result of the less polar microenvironmentin TX100 micelles (ε ≈ 2667) compared to water (ε ≈ 78) leadsto such an upward shift in the pKa value for MUF. Still a higherpKa value for HUF in the P123 micelle is possibly related tothe even lower degree of water content in the present micellarphase (ε ≈ 10).34 For all the anisotropy and lifetime measure-ments, the experimental pH was kept around 7, such that theneutral form of HUF dominates in the solution. It should bementioned here that the behavior of the absorption spectra wasobserved to be nearly similar for all other P123 concentrationsstudied. At this point, it is also worth mentioning that nosignificant change in the size of the P123 aggregates wasobserved within the studied pH range of around 6-10, as shownin the inset of Figure 1. Moreover, the angle-dependent DLSmeasurements indicated that the spherical shape of the P123micelle was retained even at a reasonably high alkaline condition(pH ≈ 10.5).
The fluorescence spectrum of HUF in P123 micellar solutionat neutral pH shows quite structured emission when excited at323 nm but shows a single emission peak at around 450 nmwhen excited at 374 nm, as shown in Figure 2. It is observedthat the emission band below 400 nm gradually decreases withan increase in the pH with a concomitant increase in intensityat the 450 nm band. This emission band below 400 nm isassigned to the neutral HUF species.38,39 The band around 450nm (with 374 nm excitation) is seen to remain invariant to thepH changes although its intensity increases with an increase inthe pH of the solution. This peak at 450 nm is assigned to theemission from the deprotonated HUF species,38,39 which appeareven when the neutral HUF species is excited at neutral pH.This suggests that, even at the neutral pH, there is some excited-state deprotonation of HUF, leading to a sizable population ofboth the species (neutral and anionic HUF), although in theground state only the neutral form exists. This is furthersubstantiated by other literature reports on several organizedassemblies.35-39,68,69 Additionally, it is also demonstrated thatthe deprotonated anionic species is stronger in fluorescence yieldthan the protonated neutral form of HUF.35,69 The intensity ofthe shoulder at around 485 nm (mainly at pH below 7 with 323nm excitation) gradually decreases with an increase in pH, andat very high pH only the main 450 nm emission band exists.This 485 nm band is assigned to the tautomeric form of theneutral HUF, following the report of Cohen et al.70 To be noted,the spectral behavior of HUF is almost like to that of its analogue4-methylumbelliferon, which does not have the long hydropho-bic tail as in the former dye.70 The excitation spectra for 450and 485 nm emission bands were similar to that of the emissionband at 386 nm for the neutral form. Hence, these higherwavelength emission bands arise from the neutral excited-stateforms of HUF, via deprotonation and tautomerism.70
The excited-state pKa (i.e., pKa*) of HUF in the P123 micellewas calculated following the well-known Forster relation71
where νjanionicem and νjneutral
em are the emission maxima of the anionicand neutral forms, expressed in wavenumbers. The calculatedpKa* value for HUF in the P123 micelle is 2.35 in comparisonto the ground-state pKa of 9.5. This pKa* value furthersubstantiates the energetically favorable deprotonation equilib-rium for HUF in the excited state at neutral pH. Additionally,a slight shift in the excited-state equilibrium toward theprotonated HUF population is indicated from the comparison
Figure 1. Absorption spectra of HUF in 1% P123 at pH 5.1 (squares),9.4 (circles), and 10.0 (triangles). Inset: pKa curve for HUF in 1% P123solution and the corresponding micellar radius (rM) as a function ofpH (as determined from a DLS study).
Figure 2. Normalized fluorescence spectra of HUF in 1% P123solution at pH ≈ 7 with 323 nm (circles) and 374 nm (squares)excitations. For comparison, the fluorescence spectrum with 323 nmexcitation at pH ≈ 10 (stars) is also included.
pKa* ) pKa + (2.1 × 10-3)(νanionicem - νneutral
em )
5120 J. Phys. Chem. B, Vol. 113, No. 15, 2009 George et al.
of intensity ratios of the protonated to deprotonated HUF speciesin P123 micelles and water under the neutral pH condition. Thisis in accordance with the fact that the pKa* value of HUF inwater (∼1.86) is reasonably less than that in P123 micelles.This is certainly due to the higher polarity of water than P123micelles, which further substantiates the location of HUF insidethe micelles rather than in the bulk water. Another observationto note is that the fluorescence spectra of HUF for both theexcitation wavelengths show a marginal blue shift in P123micelles compared to water. These observations correlate nicelywith the fact that the extent of hydration in P123 micelles issubstantially less, as inferred in our earlier studies.34
In essence, from the preliminary spectral studies we can inferthat, under the working neutral pH condition, the ground-statepopulation is predominantly due to the neutral HUF species,and upon excitation a deprotonated anionic species also evolveswhich mainly emits around 450 nm. In the following experi-ments we have recorded the fluorescence signal of HUF above440 nm only. Due to limited excitation laser diodes, we haveused 373 nm for HUF excitation in TR studies. It should benoted that, due to a poor excitation efficiency, the fluorescencesignal was comparatively weak at neutral pH compared to higherpH in TR studies, although in the case of steady-state anisotropyexperiments 323 nm excitation was used to enhance the signalstrength from the deprotonated HUF species around 450 nm.The trends in the steady-state anisotropy values were observedto be identical irrespective of the excitation wavelength. Thus,small variations that might arise due to the differences in theprimary excitation species are neglected in the present study.Further, to discard the possibility of any discrepancy in the time-resolved anisotropy measurements arising due to the excited-state deprotonation process, we have also carried out somecontrol experiments at higher pH (∼10.5), where only thedeprotonated HUF species prevails in the ground state. Theoverall results were observed to be quite similar both at pH 7and at pH 10.5, as discussed below. Therefore, we can safelyassume that at higher wavelengths (>440 nm) emission is mostlydue to the anionic HUF form and the relatively weak emissionfrom the keto tautomeric form of HUF does not complicate ourinferences.
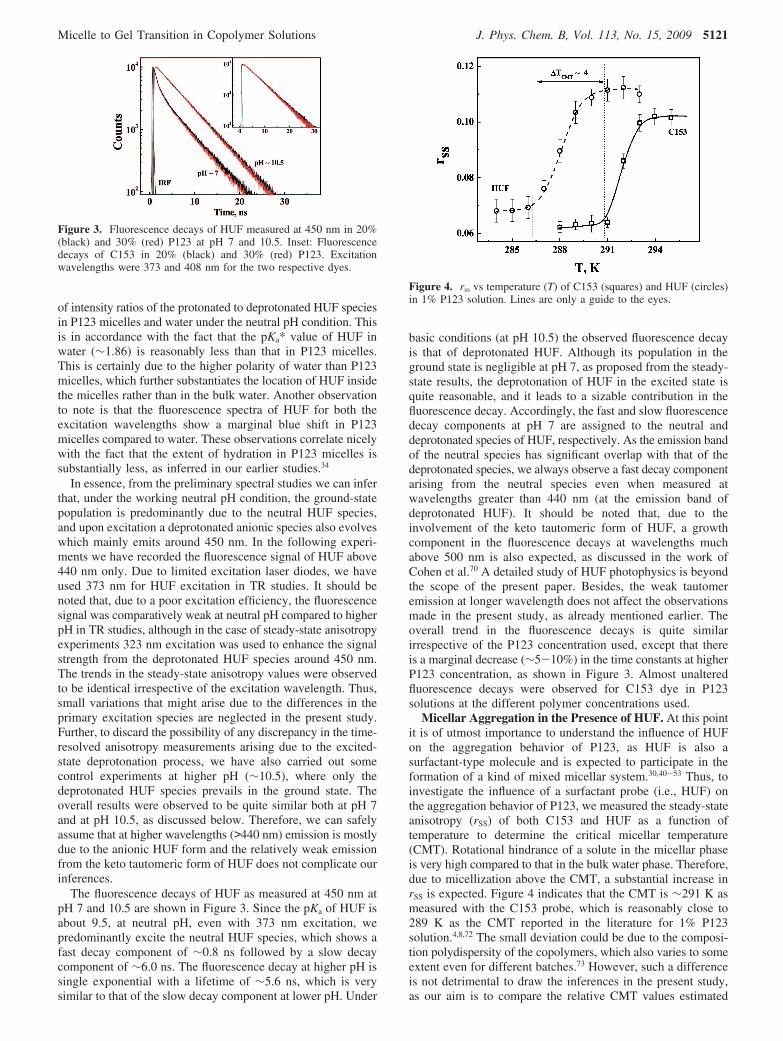
The fluorescence decays of HUF as measured at 450 nm atpH 7 and 10.5 are shown in Figure 3. Since the pKa of HUF isabout 9.5, at neutral pH, even with 373 nm excitation, wepredominantly excite the neutral HUF species, which shows afast decay component of ∼0.8 ns followed by a slow decaycomponent of ∼6.0 ns. The fluorescence decay at higher pH issingle exponential with a lifetime of ∼5.6 ns, which is verysimilar to that of the slow decay component at lower pH. Under
basic conditions (at pH 10.5) the observed fluorescence decayis that of deprotonated HUF. Although its population in theground state is negligible at pH 7, as proposed from the steady-state results, the deprotonation of HUF in the excited state isquite reasonable, and it leads to a sizable contribution in thefluorescence decay. Accordingly, the fast and slow fluorescencedecay components at pH 7 are assigned to the neutral anddeprotonated species of HUF, respectively. As the emission bandof the neutral species has significant overlap with that of thedeprotonated species, we always observe a fast decay componentarising from the neutral species even when measured atwavelengths greater than 440 nm (at the emission band ofdeprotonated HUF). It should be noted that, due to theinvolvement of the keto tautomeric form of HUF, a growthcomponent in the fluorescence decays at wavelengths muchabove 500 nm is also expected, as discussed in the work ofCohen et al.70 A detailed study of HUF photophysics is beyondthe scope of the present paper. Besides, the weak tautomeremission at longer wavelength does not affect the observationsmade in the present study, as already mentioned earlier. Theoverall trend in the fluorescence decays is quite similarirrespective of the P123 concentration used, except that thereis a marginal decrease (∼5-10%) in the time constants at higherP123 concentration, as shown in Figure 3. Almost unalteredfluorescence decays were observed for C153 dye in P123solutions at the different polymer concentrations used.
Micellar Aggregation in the Presence of HUF. At this pointit is of utmost importance to understand the influence of HUFon the aggregation behavior of P123, as HUF is also asurfactant-type molecule and is expected to participate in theformation of a kind of mixed micellar system.30,40-53 Thus, toinvestigate the influence of a surfactant probe (i.e., HUF) onthe aggregation behavior of P123, we measured the steady-stateanisotropy (rSS) of both C153 and HUF as a function oftemperature to determine the critical micellar temperature(CMT). Rotational hindrance of a solute in the micellar phaseis very high compared to that in the bulk water phase. Therefore,due to micellization above the CMT, a substantial increase inrSS is expected. Figure 4 indicates that the CMT is ∼291 K asmeasured with the C153 probe, which is reasonably close to289 K as the CMT reported in the literature for 1% P123solution.4,8,72 The small deviation could be due to the composi-tion polydispersity of the copolymers, which also varies to someextent even for different batches.73 However, such a differenceis not detrimental to draw the inferences in the present study,as our aim is to compare the relative CMT values estimated
Figure 3. Fluorescence decays of HUF measured at 450 nm in 20%(black) and 30% (red) P123 at pH 7 and 10.5. Inset: Fluorescencedecays of C153 in 20% (black) and 30% (red) P123. Excitationwavelengths were 373 and 408 nm for the two respective dyes.
Figure 4. rss vs temperature (T) of C153 (squares) and HUF (circles)in 1% P123 solution. Lines are only a guide to the eyes.
Micelle to Gel Transition in Copolymer Solutions J. Phys. Chem. B, Vol. 113, No. 15, 2009 5121
using normal probe C153 and surfactant probe HUF. As shownin Figure 4, the micellization starts (CMT) at around 286 K inthe presence of surfactant probe HUF, which is ∼5 K lowerthan that estimated with C153. Since HUF is expected to formmixed micellar assemblies when dissolved in a low concentra-tion in copolymer micellar solution, the present results indicatethat the interaction between the surfactant probe HUF and P123favors the micellar aggregation in the solution. Due to synergisticinteraction, many cosurfactants are in fact reported to cause adecrease in the critical micellar concentration of copolymers,which results in the formation of mixed copolymer-surfactantmicelles. Similar lowering of CMT by ∼3 K for F127 in thepresence of SDS or TTAB (tetradecyltrimethylammoniumbromide) as cosurfactant is also reported in the literature.74,75
In the present context, however, it is also important to knowthe extent of changes in the fluorescence lifetimes (τ0) of theprobes for the temperature range studied, because a change inthe τ0 values can also modulate the rSS values. This is becausethe rSS value is inversely related to the fluorescence lifetimes(τ0) of the probe, and the latter for many organic dyes is oftenseen to change quite significantly by changing the experimentaltemperature.63,71 For the present probes, C153 and HUF,however, it is observed that the changes in the τ0 values arequite insignificant for the studied temperature range. Conse-quently, our inferences based on the observed ∼65% increasein the rSS values do not seem to have any influence from thenegligible changes in the τ0 values.
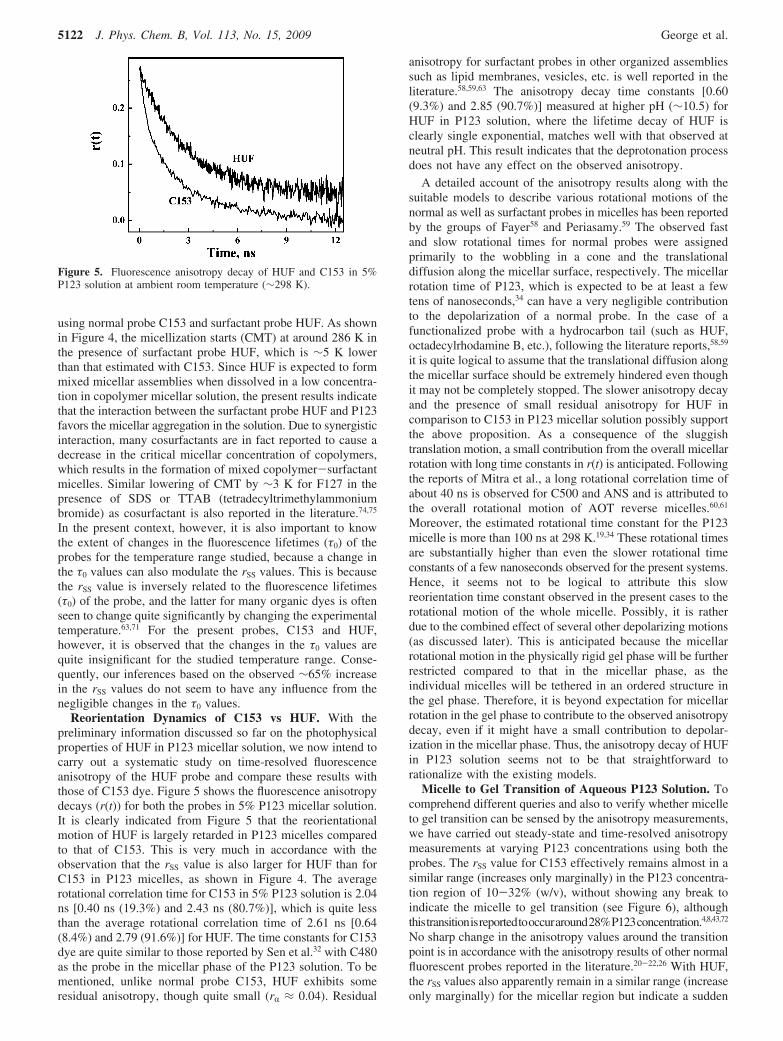
Reorientation Dynamics of C153 vs HUF. With thepreliminary information discussed so far on the photophysicalproperties of HUF in P123 micellar solution, we now intend tocarry out a systematic study on time-resolved fluorescenceanisotropy of the HUF probe and compare these results withthose of C153 dye. Figure 5 shows the fluorescence anisotropydecays (r(t)) for both the probes in 5% P123 micellar solution.It is clearly indicated from Figure 5 that the reorientationalmotion of HUF is largely retarded in P123 micelles comparedto that of C153. This is very much in accordance with theobservation that the rSS value is also larger for HUF than forC153 in P123 micelles, as shown in Figure 4. The averagerotational correlation time for C153 in 5% P123 solution is 2.04ns [0.40 ns (19.3%) and 2.43 ns (80.7%)], which is quite lessthan the average rotational correlation time of 2.61 ns [0.64(8.4%) and 2.79 (91.6%)] for HUF. The time constants for C153dye are quite similar to those reported by Sen et al.32 with C480as the probe in the micellar phase of the P123 solution. To bementioned, unlike normal probe C153, HUF exhibits someresidual anisotropy, though quite small (rR ≈ 0.04). Residual
anisotropy for surfactant probes in other organized assembliessuch as lipid membranes, vesicles, etc. is well reported in theliterature.58,59,63 The anisotropy decay time constants [0.60(9.3%) and 2.85 (90.7%)] measured at higher pH (∼10.5) forHUF in P123 solution, where the lifetime decay of HUF isclearly single exponential, matches well with that observed atneutral pH. This result indicates that the deprotonation processdoes not have any effect on the observed anisotropy.
A detailed account of the anisotropy results along with thesuitable models to describe various rotational motions of thenormal as well as surfactant probes in micelles has been reportedby the groups of Fayer58 and Periasamy.59 The observed fastand slow rotational times for normal probes were assignedprimarily to the wobbling in a cone and the translationaldiffusion along the micellar surface, respectively. The micellarrotation time of P123, which is expected to be at least a fewtens of nanoseconds,34 can have a very negligible contributionto the depolarization of a normal probe. In the case of afunctionalized probe with a hydrocarbon tail (such as HUF,octadecylrhodamine B, etc.), following the literature reports,58,59
it is quite logical to assume that the translational diffusion alongthe micellar surface should be extremely hindered even thoughit may not be completely stopped. The slower anisotropy decayand the presence of small residual anisotropy for HUF incomparison to C153 in P123 micellar solution possibly supportthe above proposition. As a consequence of the sluggishtranslation motion, a small contribution from the overall micellarrotation with long time constants in r(t) is anticipated. Followingthe reports of Mitra et al., a long rotational correlation time ofabout 40 ns is observed for C500 and ANS and is attributed tothe overall rotational motion of AOT reverse micelles.60,61
Moreover, the estimated rotational time constant for the P123micelle is more than 100 ns at 298 K.19,34 These rotational timesare substantially higher than even the slower rotational timeconstants of a few nanoseconds observed for the present systems.Hence, it seems not to be logical to attribute this slowreorientation time constant observed in the present cases to therotational motion of the whole micelle. Possibly, it is ratherdue to the combined effect of several other depolarizing motions(as discussed later). This is anticipated because the micellarrotational motion in the physically rigid gel phase will be furtherrestricted compared to that in the micellar phase, as theindividual micelles will be tethered in an ordered structure inthe gel phase. Therefore, it is beyond expectation for micellarrotation in the gel phase to contribute to the observed anisotropydecay, even if it might have a small contribution to depolar-ization in the micellar phase. Thus, the anisotropy decay of HUFin P123 solution seems not to be that straightforward torationalize with the existing models.
Micelle to Gel Transition of Aqueous P123 Solution. Tocomprehend different queries and also to verify whether micelleto gel transition can be sensed by the anisotropy measurements,we have carried out steady-state and time-resolved anisotropymeasurements at varying P123 concentrations using both theprobes. The rSS value for C153 effectively remains almost in asimilar range (increases only marginally) in the P123 concentra-tion region of 10-32% (w/v), without showing any break toindicate the micelle to gel transition (see Figure 6), althoughthistransitionisreportedtooccuraround28%P123concentration.4,8,43,72
No sharp change in the anisotropy values around the transitionpoint is in accordance with the anisotropy results of other normalfluorescent probes reported in the literature.20-22,26 With HUF,the rSS values also apparently remain in a similar range (increaseonly marginally) for the micellar region but indicate a sudden
Figure 5. Fluorescence anisotropy decay of HUF and C153 in 5%P123 solution at ambient room temperature (∼298 K).
5122 J. Phys. Chem. B, Vol. 113, No. 15, 2009 George et al.
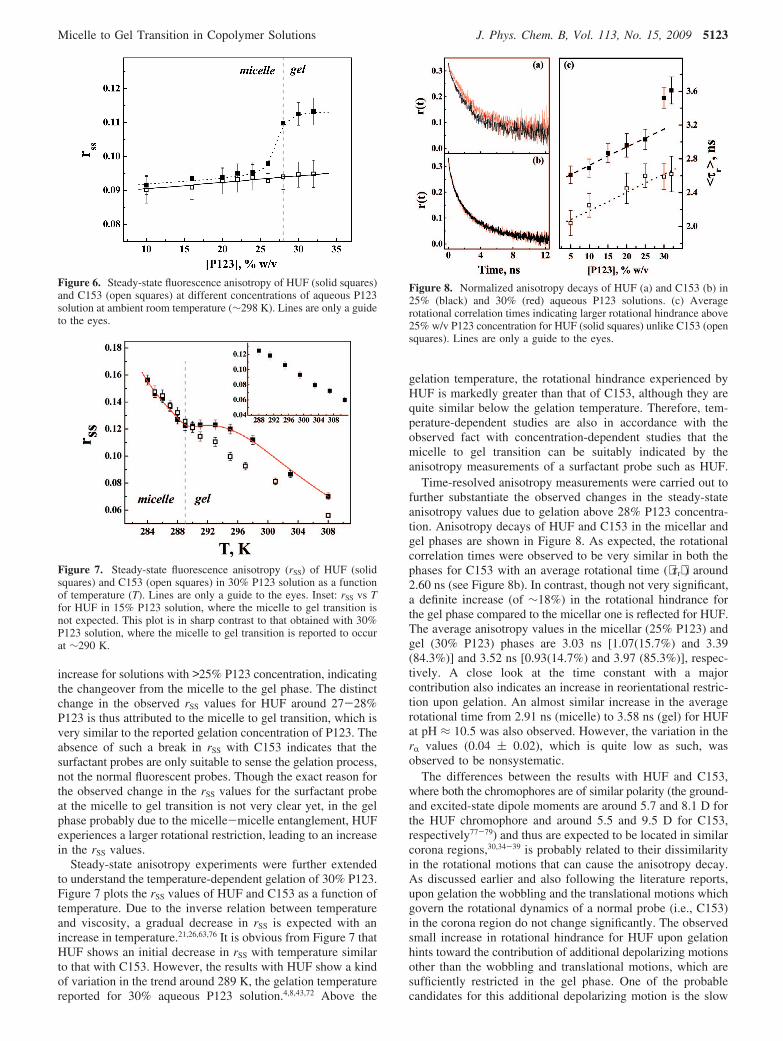
increase for solutions with >25% P123 concentration, indicatingthe changeover from the micelle to the gel phase. The distinctchange in the observed rSS values for HUF around 27-28%P123 is thus attributed to the micelle to gel transition, which isvery similar to the reported gelation concentration of P123. Theabsence of such a break in rSS with C153 indicates that thesurfactant probes are only suitable to sense the gelation process,not the normal fluorescent probes. Though the exact reason forthe observed change in the rSS values for the surfactant probeat the micelle to gel transition is not very clear yet, in the gelphase probably due to the micelle-micelle entanglement, HUFexperiences a larger rotational restriction, leading to an increasein the rSS values.
Steady-state anisotropy experiments were further extendedto understand the temperature-dependent gelation of 30% P123.Figure 7 plots the rSS values of HUF and C153 as a function oftemperature. Due to the inverse relation between temperatureand viscosity, a gradual decrease in rSS is expected with anincrease in temperature.21,26,63,76 It is obvious from Figure 7 thatHUF shows an initial decrease in rSS with temperature similarto that with C153. However, the results with HUF show a kindof variation in the trend around 289 K, the gelation temperaturereported for 30% aqueous P123 solution.4,8,43,72 Above the
gelation temperature, the rotational hindrance experienced byHUF is markedly greater than that of C153, although they arequite similar below the gelation temperature. Therefore, tem-perature-dependent studies are also in accordance with theobserved fact with concentration-dependent studies that themicelle to gel transition can be suitably indicated by theanisotropy measurements of a surfactant probe such as HUF.
Time-resolved anisotropy measurements were carried out tofurther substantiate the observed changes in the steady-stateanisotropy values due to gelation above 28% P123 concentra-tion. Anisotropy decays of HUF and C153 in the micellar andgel phases are shown in Figure 8. As expected, the rotationalcorrelation times were observed to be very similar in both thephases for C153 with an average rotational time (⟨τr⟩) around2.60 ns (see Figure 8b). In contrast, though not very significant,a definite increase (of ∼18%) in the rotational hindrance forthe gel phase compared to the micellar one is reflected for HUF.The average anisotropy values in the micellar (25% P123) andgel (30% P123) phases are 3.03 ns [1.07(15.7%) and 3.39(84.3%)] and 3.52 ns [0.93(14.7%) and 3.97 (85.3%)], respec-tively. A close look at the time constant with a majorcontribution also indicates an increase in reorientational restric-tion upon gelation. An almost similar increase in the averagerotational time from 2.91 ns (micelle) to 3.58 ns (gel) for HUFat pH ≈ 10.5 was also observed. However, the variation in therR values (0.04 ( 0.02), which is quite low as such, wasobserved to be nonsystematic.
The differences between the results with HUF and C153,where both the chromophores are of similar polarity (the ground-and excited-state dipole moments are around 5.7 and 8.1 D forthe HUF chromophore and around 5.5 and 9.5 D for C153,respectively77-79) and thus are expected to be located in similarcorona regions,30,34-39 is probably related to their dissimilarityin the rotational motions that can cause the anisotropy decay.As discussed earlier and also following the literature reports,upon gelation the wobbling and the translational motions whichgovern the rotational dynamics of a normal probe (i.e., C153)in the corona region do not change significantly. The observedsmall increase in rotational hindrance for HUF upon gelationhints toward the contribution of additional depolarizing motionsother than the wobbling and translational motions, which aresufficiently restricted in the gel phase. One of the probablecandidates for this additional depolarizing motion is the slow
Figure 6. Steady-state fluorescence anisotropy of HUF (solid squares)and C153 (open squares) at different concentrations of aqueous P123solution at ambient room temperature (∼298 K). Lines are only a guideto the eyes.
Figure 7. Steady-state fluorescence anisotropy (rSS) of HUF (solidsquares) and C153 (open squares) in 30% P123 solution as a functionof temperature (T). Lines are only a guide to the eyes. Inset: rSS vs Tfor HUF in 15% P123 solution, where the micelle to gel transition isnot expected. This plot is in sharp contrast to that obtained with 30%P123 solution, where the micelle to gel transition is reported to occurat ∼290 K.
Figure 8. Normalized anisotropy decays of HUF (a) and C153 (b) in25% (black) and 30% (red) aqueous P123 solutions. (c) Averagerotational correlation times indicating larger rotational hindrance above25% w/v P123 concentration for HUF (solid squares) unlike C153 (opensquares). Lines are only a guide to the eyes.
Micelle to Gel Transition in Copolymer Solutions J. Phys. Chem. B, Vol. 113, No. 15, 2009 5123

rotational motion of the micelle. However, in the absence of asubstantial increase in rR upon gelation there is doubt about itslikely role in the observed effect during the micelle to geltransition. If micellar rotation had any participation in theanisotropy decay of HUF, a large increase in rSS and rR upongelation was also expected, as micellar rotation was expectedto be effectively frozen in the gel phase. Further, the decay rateconstant corresponding to the micellar rotation could be muchhigher than the few nanosecond decay times observed. Thus,the observed small increase in the average rotational timeconstant for HUF upon gelation essentially echoes against theinvolvement of micellar rotation in the depicted micelle to geltransition. The other likely candidate for the additional depo-larizing motion is the structural fluctuation of the micellaraggregates themselves, which is often called the tumblingmotion. Such structural fluctuation is expected to be much fasterthan the micellar rotation and can be responsible for significantfluorescence depolarization. Recent NMR studies of biosurfac-tant surfactin in SDS and DPC (dodecylphosphocholine) mi-celles by Tsan et al. indicate a correlation time constant of 10.9ns for the overall tumbling of the micelles at 288 K, whichdecreases to 6.4 ns at 308 K.80,81 These values are similar tothose reported for other biosurfactant systems (such as 22-residue motilin,82 30 N-terminal residues of HIV-I gp41,83 etc.)in SDS micelles. These tumbling time constants are in the rangeof the slow rotational relaxation time constant of a fewnanoseconds observed in the present cases. Drawing an analogyfrom these results, we thus expect that the structural fluctuationof the micellar aggregates actually participates in the overallanisotropy decay of HUF. Following this proposition, anincrease in the average rotational correlation time upon gelationis quite obvious, as in the gel phase the micellar structuralfluctuations will be significantly retarded compared to those ofthe free micelle due to entanglement and packing of micellarunits.17 This hypothesis is in accordance with the similar rR andincreased ⟨τr⟩ values for HUF in the gel phase compared to thosein the micellar phase. The absence of this additional depolarizingmotion for C153 is perhaps related to the substantial contributionof the translational motion of the normal probe to the observedanisotropy decay, which is just negligible in the case ofsurfactant probe HUF. Besides, the reorientational dynamics ofthe EO block itself (which has a time constant of around a fewnanoseconds84,85) can also contribute to the anisotropy decay.However, this motion alone cannot explain the observed increasein restriction upon gelation above the critical gelation concentra-tion (cgc) or the critical gelation temperature (cgt) for HUF incomparison to C153.
The gradual increase in ⟨τr⟩ of HUF from 5% to 25% P123concentration (cf. Figure 8c) could be due to a sphere-to-rodshape transition of either the micelle or the micelle-micelleentanglement. Increased packing in the corona region due tothe sphere-to-rod shape transition in the present case is ruledout following the report of Ganguly et al., which reveals auniform micellar size and shape of P123 micelles in the aboveconcentration range.43 However, micelle-micelle entanglementis reported at high copolymer concentrations.17 As HUF isproposed to experience additional tumbling motion of themicellar aggregates, the increase in the micelle-micelle en-tanglement with copolymer concentration can be responsiblefor the observed increase in the average reorientation time. Itshould be mentioned that a similar increase in reorientation timefor the micellar surface probe Rh110 has also been observedby Mali et al.26 Additionally, an increase in shear viscosity due
to such micellar entanglement with an increase in copolymerconcentration has also been reported by Ganguly et al.43,86,87
Besides micelle-micelle entanglement, an increased reori-entation time could also be due to the crowding of EO blocksin the corona region as a result of an increased micellaraggregation number with P123 concentration, although in thepresent systems the micellar size remains almost similar.43 Suchan increase in the aggregation number with P123 concentrationhas been demonstrated by the double-electron resonance andneutron scattering experiments by Ruthstein et al.88 and Soni etal.,89 respectively. Unlike HUF where micelle-micelle en-tanglement as well as reduced mobility due to increasedaggregation number is responsible for the increase in ⟨τr⟩ evenin the micellar phase from 2.61 to 3.03 ns, it is only the lattereffect that is expected to cause the observed increase in ⟨τr⟩ forC153 (2.04 and 2.63 ns at 5% and 32% P123 concentration)with P123 concentration (cf. Figure 8c). It should be noted thatthe observed small increase in rSS with an increase in copolymerconcentration, shown in Figure 6, is also related to these factors.Further, the absence of a very sharp change for the micelle togel transition in both concentration- and temperature-dependentrSS studies could be due to the microscopic gelation that startsinitially and then extends throughout the solution with an
Figure 9. Correlation function vs time plots at different P123concentrations recorded at a 90° scattering angle at 303 K. The solidlines represent the fitting curves to the data.
Figure 10. Diffusion coefficient of the faster moving species vs P123concentration obtained at 303 K. Lines are only a guide to the eyes.
5124 J. Phys. Chem. B, Vol. 113, No. 15, 2009 George et al.
increase in the copolymer concentration or the temperature ofthe solution, on approaching the cgc or cgt.
Structural Dynamics of P123 Aggregates by DLS. At thispoint it is worth exploring the structural dynamics of copolymeraggregates by DLS studies as a function of the copolymerconcentration. The intensity correlation function vs time plotsfrom the DLS measurements performed at 303 K and a 90°angle as a function of the P123 concentration are shown inFigure 9. At low P123 concentrations the presence of micellesas the only scattering species is reflected from the single-exponential decay of the correlation function. The nature of thedecay becomes biexponential at higher copolymer concentrationdue to the presence of the bridged network like micellar clusters,as observed in the other block copolymer systems.90-93 Thischange in the nature of the decay of the correlation function isevident from the decay plots for the 5% and 25% P123 solutionsshown in Figure 9. Such a bridged network like micellar clusterat higher copolymer concentrations further substantiates theexistence of the micelle-micelle entanglement, as proposedearlier, and corroborates well with increased rotational time. In30% solutions, where a thermoreversible gel is formed, thecorrelation function undergoes a further change with a significantdecrease in the fraction of the slower moving micellar clusters.To understand the concentration dependence of the correlationfunction plots further, we have calculated the diffusion coef-ficient of the scattering species by analyzing the plots on thebasis of single-exponential and biexponential equations at lowand high copolymer concentrations, respectively. In Figure 10we have shown the variation of the diffusion coefficient of thefaster moving species with the copolymer volume fraction, φ.
The dry copolymer volume fraction value was calculated usingthe specific volume of the copolymer as 0.93.94 Below thegelation concentration the species can be considered as diffusivemicelles. In the gel, in the absence of any micellar diffusion, itmust be associated with some internal motion within the system.The diffusion coefficient increases steadily with the copolymerconcentration up to 25% but shows a significantly faster rateof increase at the gelation concentration. Below the gelationconcentration, the variation in the diffusion coefficient can beexplained on the basis of the hard sphere model with repulsiveintermicellar interaction.10 The sudden increase in the diffusioncoefficient of the faster moving species associated with gelationhas also been observed in other block copolymer micellarsystems.93 In view of the improbable diffusive motion of themicelle in the gel phase, the observation of the correlationfunction of the faster moving species was attributed to thecollective motion of the physically interconnected nodes (“gelmode”).93 The nonlinear nature of Γ vs q2 plots also suggeststhat the scattering species are not diffusive in nature in the gelphase. In the solution phase, however, Γ was found to varylinearly with q2, because of the presence of the diffusive micellesas the scattering species. These DLS results thus highlight thechange in the dynamics of the aggregates present in the aqueousP123 solutions as a function of the P123 concentration up tothe gelation concentration. It should be noted that both motions,the tumbling motion of individual micelles and the overallcollective gel mode of the interconnected micelles in the gelphase, can lead to fluorescence depolarization.
Following these results on the structural dynamics of diluteand moderately concentrated copolymer solutions, it is expected
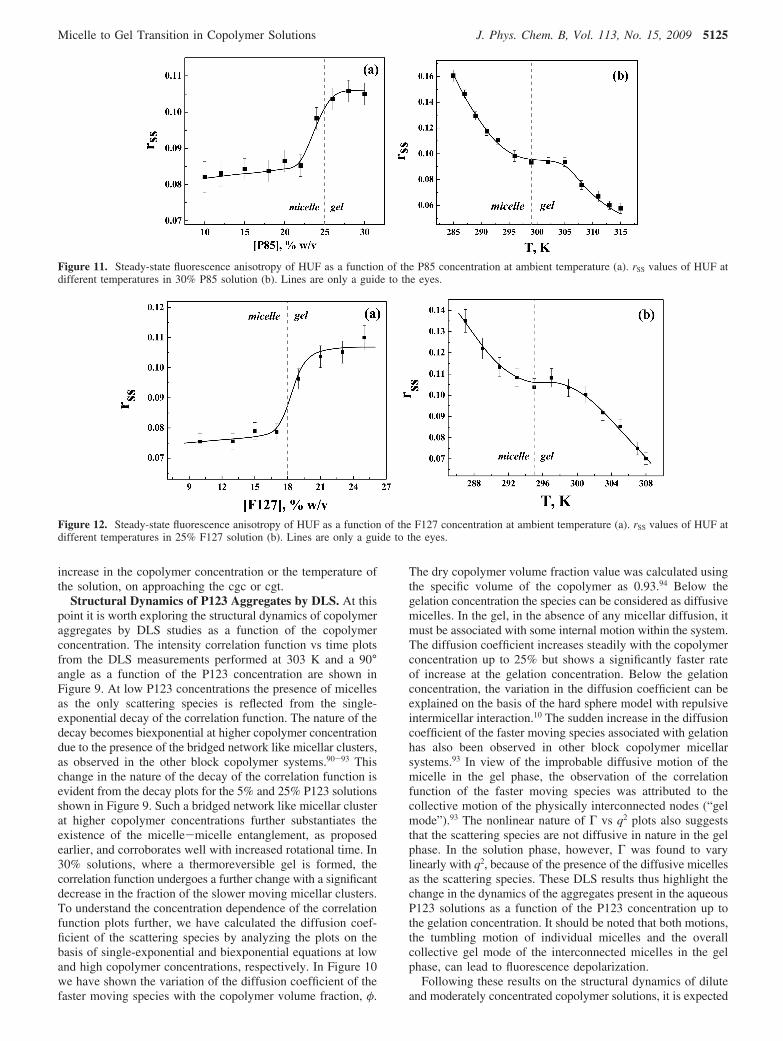
Figure 11. Steady-state fluorescence anisotropy of HUF as a function of the P85 concentration at ambient temperature (a). rSS values of HUF atdifferent temperatures in 30% P85 solution (b). Lines are only a guide to the eyes.
Figure 12. Steady-state fluorescence anisotropy of HUF as a function of the F127 concentration at ambient temperature (a). rSS values of HUF atdifferent temperatures in 25% F127 solution (b). Lines are only a guide to the eyes.
Micelle to Gel Transition in Copolymer Solutions J. Phys. Chem. B, Vol. 113, No. 15, 2009 5125

that the collective motion of the micelles in the gel phase ofP123 will assist the depolarization process for HUF. In thepresent system we propose that the additional depolarizingmotion in the gel phase is not a true translational, tumbling, orcollective motion of the nodes; rather it is a combined complexmotion as a whole of the otherwise physically rigid intercon-nected micelles in the gel phase. It should be further noted thatthe gel mode basically acts against the expected influence of asubstantial decrease in the rotational mobility on the depolar-ization of HUF in the gel phase. In essence we thus observeonly a small increase rather than a large increase in the steady-state and time-resolved anisotropy of HUF.
Micelle to Gel Transition of Aqueous P85 and F127Solutions. The general observation of increased rotationalrigidity upon gelation, the main aim of the present investigation,is unambiguous in both steady-state and time-resolved anisot-ropy studies with the HUF probe. Therefore, for qualitativeexamination of the sol to gel transition of copolymer solutions,simple steady-state anisotropy measurements are equippedenough to provide reasonable insight. To ensure the applicabilityof anisotropy measurements of HUF to indicate the gelationbehavior of copolymer solutions, we have extended our steady-state anisotropy experiments to other triblock copolymers withdifferent EO to PO block length ratios. The EO/PO ratios forP85 and F127 are 1.3 and 3.1, respectively, whereas it is only0.6 for P123. The results of a systematic steady-state investiga-tion employing HUF in P85 and F127 are shown in Figures 11and 12, respectively. The required concentration of triblockcopolymer for the formation of the gel phase generally decreaseswith an increase in the EO/PO ratio.4 The cgc values at roomtemperature for P85 and F127 are reported to be about 25%and 18% (w/v), respectively.4,43,72,95 A comprehensive changein the rSS values due to gelation above 24% (w/v) P85 and 17%(w/v) F127 aqueous solution is indicated in Figures 11a and12a, respectively, and these are in good correspondence withthe reported values. Observed changes in the present systemscan also be visualized with a logic similar to that argued withP123. Thus, the results with P85 and F127 solutions furtherascertain our proposition of using HUF as a suitable indicatorto characterize the micelle to gel transition. In addition to that,temperature-dependent rSS studies, as shown in Figures 11b and12b, also confirm increased rotational restriction above 299 and295 K for 30% (w/v) P85 and 25% (w/v) F127 solutions,respectively. These values closely match with the cgt valuesreported for these triblock copolymers.4,8,43,72,95 Therefore,assimilation of all these results establishes the suitability ofsurfactant probe HUF over the commonly used fluorescenceprobes to realize the micelle to gel transition process.
Conclusions
A fluorescence spectroscopic investigation has been carriedout to realize the micelle to gel transition of aqueous triblockcopolymer P123. It is observed that with proper selection ofthe probe it is possible to clearly realize the micelle to geltransition of block copolymer solutions. Anisotropy results witha normal fluorescent probe (C153) which resides in the coronaregion of the micelle, however, do not exhibit any significantchange due to the micelle to gel transition above the cgc orcgt. The special coumarin probe having a long hydrocarbonchain (HUF) that anchors the probe strongly with the copolymermicelles clearly shows a small but definite increase in theanisotropy above the cgc or cgt. On the basis of the possiblerotational motions responsible for the anisotropy decay inmicellar media, we propose that this increase in anisotropy upon
gelation is probably due to the contribution from the micellarstructural fluctuations to the depolarization of HUF, the effectof which is negligible in the case of normal probe C153. DLSstudies indicate that, in the gel phase, collective motion of thephysically interconnected nodes is much faster than the diffus-sional motion of the micelle itself below the cgc. This collectivemotion in the gel phase is expected to accelerate the fluorescencedepolarization than otherwise expected from the reducedtumbling motion and micellar rotation in the gel phase. A goodcorrelation of the steady-state and time-resolved anisotropyresults indicates the suitability of the present methods forqualitative realization of the sol to gel transition. Extension ofthese studies to other copolymers, such as P85 and F127, furtherconfirms our proposition that surfactant probes are appropriateto investigate the micelle to gel transition process.
Acknowledgment. We acknowledge Dr. G. B. Dutt, RPCDivision, BARC, for many fruitful discussions. Encouragementand support from Dr. T. Mukherjee, Director, Chemistry Group,BARC, and Dr. S. K. Sarkar, Head, RPC Division, BARC, aregratefully acknowledged.
References and Notes
(1) Chu, B. Langmuir 1995, 11, 414.(2) Svingen, R.; Åkerman, B. J. Phys. Chem. B 2004, 108, 2735.(3) Svingen, R.; Alexandridis, P.; Åkerman, B. Langmuir 2002, 18,
8616.(4) Wanka, G.; Hoffmann, H.; Ulbricht, W. Macromolecules 1994, 27,
4145.(5) Alexandridis, P.; Olsson, U.; Lindman, B. Langmuir 1998, 14, 2627.(6) Alexandridis, P.; Hatton, T. A. Colloids Surf., A 1995, 96, 1.(7) Alexandridis, P. Macromolecules 1998, 31, 6935.(8) Alexandridis, P.; Holzwarth, J. F.; Hatton, T. A. Macromolecules
1994, 27, 2414.(9) Mortensen, K.; Pedersen, J. S. Macromolecules 1993, 26, 805.
(10) Ganguly, R.; Aswal, V. K.; Hassan, P. A.; Gopalakrishnan, I. K.;Yakhmi, J. V. J. Phys. Chem. B 2005, 109, 5653.
(11) Zhang, K.; Khan, A. Macromolecules 1995, 28, 3807.(12) Mortensen, K.; Brown, W.; Norden, B. Phys. ReV. Lett. 1992, 68,
2340.(13) Mortensen, K.; Schwahn, D.; Janssen, S. Phys. ReV. Lett. 1993,
71, 1728.(14) Robbins, M. O.; Kremer, K.; Grest, G. S. J. Chem. Phys. 1988,
88, 3286.(15) Brown, W.; Schillen, K.; Almgren, M.; Hvidt, S.; Bahadur, P.
J. Chem. Phys. 1991, 95, 1850.(16) Malmsten, M.; Lindman, B. Macromolecules 1992, 25, 5440.(17) Cabana, A.; Ait-Kadi, A.; Juhasz, J. J. Colloid Interface Sci. 1997,
190, 307.(18) Glatter, O.; Scherf, G.; Schillen, K.; Brown, W. Macromolecules
1994, 27, 6046.(19) Dutt, G. B. J. Phys. Chem. B 2005, 109, 4923.(20) Grant, C. D.; de Ritter, M. R.; Steege, K. E.; Fadeeva, T. A.;
Castner, E. W., Jr. Langmuir 2005, 21, 1745.(21) Grant, C. D.; Steege, K. E.; Bunagan, M. R.; Castner, E. W. J. J.
Phys. Chem. B 2005, 109, 22273.(22) Jeon, S.; Granick, S.; Kwon, K. C.; Char, K. J. Polym. Sci. 2002,
B40, 2883.(23) Mali, K. S.; Dutt, G. B.; Mukherjee, T. Langmuir 2006, 22, 6837.(24) Mali, K. S.; Dutt, G. B.; Mukherjee, T. J. Chem. Phys. 2006, 124,
054904.(25) Mali, K. S.; Dutt, G. B.; Mukherjee, T. J. Chem. Phys. 2006, 124,
199901.(26) Mali, K. S.; Dutt, G. B.; Mukherjee, T. Langmuir 2007, 23, 1041.(27) Mali, K. S.; Dutt, G. B.; Ganguly, R.; Mukherjee, T. J. Chem. Phys.
2005, 123, 144913.(28) Ghosh, S.; Adhikari, A.; Mandal, U.; Dey, S.; Bhattacharyya, K.
J. Phys. Chem. B 2007, 111, 8775.(29) Ghosh, S.; Dey, S.; Adhikari, A.; Mandal, U.; Bhattacharyya, K.
J. Phys. Chem. B 2007, 111, 7085.(30) Kumbhakar, M. J. Phys. Chem. B 2007, 111, 14250.(31) Kumbhakar, M.; Ganguly, R. J. Phys. Chem. B 2007, 111, 3935.(32) Sen, P.; Ghosh, S.; Sahu, K.; Mondal, S. K.; Roy, D.; Bhattacharyya,
K. J. Chem. Phys. 2006, 124, 204905.(33) Levinger, N. E. Science 2002, 298, 1722.
5126 J. Phys. Chem. B, Vol. 113, No. 15, 2009 George et al.

(34) Kumbhakar, M.; Goel, T.; Nath, S.; Mukherjee, T.; Pal, H. J. Phys.Chem. B 2006, 110, 25646.
(35) Fromherz, P. Biochim. Biophys. Acta 1973, 323, 326.(36) Lukac, S. J. Phys. Chem. 1983, 87, 5045.(37) Lovelock, B.; Grieser, F.; Healy, T. W. J. Phys. Chem. 1985, 89,
501.(38) Drummond, C. J.; Grieser, F. Photochem. Photobiol. 1987, 45, 19.(39) Fernandez, M. S.; Fromherz, P. J. Phys. Chem. 1977, 81, 1755.(40) Kumbhakar, M. J. Phys. Chem. B 2007, 111, 12154.(41) Jansson, J.; Schillen, K.; Olofsson, G.; Silva, R. C. D.; Loh, W. J.
Phys. Chem. B 2004, 108, 82.(42) Jansson, J.; Schillen, K.; Nilsson, M.; Soderman, O.; Fritz, G.;
Bergmann, A.; Glatter, O. J. Phys. Chem. B 2005, 109, 7073.(43) Ganguly, R.; Aswal, V. K.; Hassan, P. A.; Gopalakrishnan, I. K.;
Kulshreshtha, S. K. J. Phys. Chem. B 2006, 110, 9843.(44) Mandal, U.; Adhikari, A.; Dey, S.; Ghosh, S.; Mondal, S. K.;
Bhattacharyya, K. J. Phys. Chem. B 2007, 111, 5896.(45) Mali, K. S.; Dutt, G. B.; Mukherjee, T. J. Phys. Chem. B 2007,
111, 5878.(46) Mali, K. S.; Dutt, G. B.; Mukherjee, T. J. Chem. Phys. 2007, 127,
154904.(47) Almgren, M.; Stam, J. V.; Lindblad, C.; Li, P.; Stilbs, P.; Bahadur,
P. J. Phys. Chem. 1991, 95, 5677.(48) Hecht, E.; Hoffman, H. Langmuir 1994, 10, 86.(49) Li, Y.; Xu, R.; Bloor, D. M.; Holzwarth, J. F.; Wyn-Jones, E.
Langmuir 2000, 16, 10515.(50) Proietti, N.; Amato, M. E.; Masci, G.; Segre, A. L. Macromolecules
2002, 35, 4365.(51) Lisi, R. D.; Milioto, S.; Muratore, N. Macromolecules 2002, 35,
7067.(52) Ivanova, R.; Alexandridis, P.; Lindman, B. Colloids Surf., A 2001,
41, 183.(53) Couderc-Azouani, S.; Sidhu, J.; Thurn, T.; Xu, R.; Bloor, D. M.;
Penfold, J.; Holzwarth, J. F.; Wyn-Jones, E. Langmuir 2005, 21, 10197.(54) Lipari, G.; Szabo, A. Biophys. J. 1980, 30, 489.(55) Lipari, G.; Szabo, A. J. Chem. Phys. 1981, 75, 2971.(56) Lipari, G.; Szabo, A. J. Am. Chem. Soc. 1982, 104, 4546.(57) Wang, C. C.; Pecora, R. J. Chem. Phys. 1980, 72, 5333.(58) Quitevis, E. L.; Marcus, A. H.; Fayer, M. D. J. Phys. Chem. 1993,
97, 5762.(59) Maiti, N. C.; Krishna, M. M. G.; Britto, P. J.; Periasamy, N. J.
Phys. Chem. B 1997, 101, 11051.(60) Mitra, R. K.; Sinha, S. S.; Pal, S. K. Langmuir 2008, 24, 49.(61) Narayanan, S. S.; Sinha, S. S.; Sarkar, R.; Pal, S. K. J. Phys. Chem.
B 2008, 112, 2859.(62) Kalyansundaram, K. Photochemistry in Microheterogeneous Sys-
tems; Academic: Orlando, FL, 1987.(63) Lakowicz, J. R. Principles of Fluorescence Spectroscopy, 3rd ed.;
Springer: New York, 2006.(64) Brown, J. C.; Pusey, P. N.; Dietz, R. J. J. Chem. Phys. 1975, 62,
1136.(65) Hassan, P. A.; Kulshreshtha, S. K. J. Colloid Interface Sci. 2006,
300, 744.(66) Krylov, S. N.; Dunford, H. B. J. Phys. Chem. 1996, 100, 19719.
(67) Kumbhakar, M.; Nath, S.; Pal, H.; Sapre, A. V.; Mukherjee, T.J. Chem. Phys. 2003, 119, 388.
(68) Moller, J. V.; Kragh-Hansen, U. Biochemistry 1975, 14, 2317.(69) Simeonov, A.; Jadhav, A.; Thomas, C. J.; Wang, Y.; Huang, R.;
Southall, N. T.; Shinn, P.; Smith, J.; Austin, C. P.; Auld, D. S.; Inglese, J.J. Med. Chem. 2008, 51, 2363.
(70) Cohen, B.; Huppert, D. J. Phys. Chem. A 2001, 105, 7157.(71) Valeur, B. Molecular Fluorescence; Wiley-VCH: Weinheim,
Germany, 2002.(72) Chu, B.; Zhou, Z. Physical chemistry of polyoxyalkylene block
copolymer surfactants. Nonionic Surfactants; Marcel Dekker: New York,1996; Vol. 60.
(73) Mortensen, K.; Batsberg, W.; Hvidt, S. Macromolecules 2008, 41,1720.
(74) Li, Y.; Xu, R.; Couderc, S.; Bloor, D. M.; Holzwarth, J. F.; Wyn-Jones, E. Langmuir 2001, 17, 5742.
(75) Li, Y.; Xu, R.; Couderc, S.; Bloor, D. M.; Wyn-Jones, E.;Holzwarth, J. F. Langmuir 2001, 17, 183.
(76) Atkins, P. W. Physical Chemistry; Oxford University Press: Oxford,U.K., 1994.
(77) Chapman, C. F.; Fee, R. S.; Maroncelli, M. J. Phys. Chem. 1995,99, 4811.
(78) Maroncelli, M.; Fleming, G. R. J. Chem. Phys. 1987, 86, 6221.(79) Nemkovich, N. A.; Reis, H.; Baumann, W. J. Lumin. 1997, 71,
255.(80) Tsan, P.; Volpon, L.; Besson, F. o.; Lancelin, J.-M. J. Am. Chem.
Soc. 2007, 129, 1968.(81) Dosset, P.; Hus, J.-C.; Blackledge, M.; Marion, D. J. Biomol. NMR
2000, 16, 23.(82) Jarvet, J.; Zdunek, J.; Damberg, P.; Graslund, A. Biochemistry 1997,
36, 8153.(83) Syvitski, R. T.; Burton, I.; Mattatall, N. R.; Douglas, S. E.; Jakeman,
D. L. Biochemistry 2005, 44, 7282.(84) Shirota, H.; Segawa, H. J. Phys. Chem. A 2003, 107, 3719.(85) Shirota, H.; Segawa, H. Chem. Phys. 2004, 306, 43.(86) Ganguly, R.; Aswal, V. K. J. Phys. Chem. B 2008, 112, 7726.(87) Ganguly, R.; Kumbhakar, M.; Aswal, V. K. J. Phys. Chem. B,
revised.(88) Ruthstein, S.; Potapov, A.; Raitsimring, A. M.; Goldfarb, D. J. Phys.
Chem. B 2005, 109, 22843.(89) Soni, S. S.; Brotons, G.; Bellour, M.; Narayanan, T.; Gibaud, A. J.
Phys. Chem. B 2006, 110, 15157.(90) Balsara, N. P.; Tirrel, M.; Lodge, T. P. Macromolecules 1991, 24,
1975.(91) Glotzer, S. C.; Bansil, R.; Gallagher, P. D.; Gyure, M. F.; Sciortino,
F.; Stanley, H. E. Physica A 1993, 201, 482.(92) Raspaud, E.; Lairez, D.; Adam, M.; Carton, J.-P. Macromolecules
1996, 29, 1269.(93) Konak, C.; Fleischer, G.; Tuzar, Z.; Bansil, R. J. Polym. Sci., Part
B: Polym. Phys. 2000, 38, 1312.(94) Nolan, S. L.; Phillips, R. J.; Cotts, P. M.; Dungan, S. R. J. Colloid
Interface Sci. 1997, 191, 291.(95) Malmsten, M.; Lindman, B. Macromolecules 1993, 26, 1282.
JP809826C
Micelle to Gel Transition in Copolymer Solutions J. Phys. Chem. B, Vol. 113, No. 15, 2009 5127
Copyright © 2022 FDOKUMEN




















