
Synthesis and thermal decomposition of GAP–Poly(BAMO) copolymer
13
Synthesis and thermal decomposition of GAPePoly(BAMO) copolymer Sreekumar Pisharath * , How Ghee Ang Energetic Materials Research Centre, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore Received 11 December 2006; received in revised form 5 March 2007; accepted 13 March 2007 Available online 30 March 2007 Abstract An energetic copolymer of glycidyl azide polymer (GAP) and poly(bis(azidomethyl)oxetane (Poly(BAMO)) was synthesized using the Borontrifluorideedimethyl ether complex/diol initiator system. The synthesized copolymer exhibited the characteristics of an energetic thermo- plastic elastomer (ETPE). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to study the thermal decomposition behavior and the results were compared with that of the constituent homopolymers. The main weight loss step in all the polymers coincides with the exothermic dissociation of the azido groups in the side chain. In contrast with the behavior of the homopolymers, the copolymer shows a broad exothermic shoulder peak at 298 C after the main exothermic decomposition peak at 228 C. Kinetic analysis was performed by Vyazovkin’s model-free method, which suggests that the activation energy of the main decomposition step is around 145 kJ/mol and for the second shoulder it is around 220 kJ/mol. Fourier transform infra red (FTIR) spectra of the degradation residues show that the azido groups in the copolymer decompose in two stages at different temperatures which is responsible for the double decomposition behavior. Ó 2007 Elsevier Ltd. All rights reserved. Keywords: Energetic polymer; Thermal decomposition; Model-free kinetics; Thermoplastic elastomer 1. Introduction Azido polymers are unique energetic materials which release heat by decomposition. They are used as energetic binders in propellant and explosive formulations [1]. Inert polymers such as hydroxyl-terminated polybutadiene (HTPB) are being replaced with energetic polymers such as glycidyl azide polymer (GAP) as promising energetic binders. If inert polymers are substituted by energetic polymers as binders, increased performance could be achieved at low oxidizer load- ing. Moreover, low oxidizer content reduces the vulnerability of formulation towards external stimuli. Thus energetic poly- mers as binders in propellant or explosive formulations offer the dual advantage of improved performance and reduced vulnerability. With increasing complexities in the rocket motor designs, commensurate with the performance requirements, structure and processing demands placed on the solid propellants have become more and more strenuous. These requirements have outweighed the ballistic and combustion advantages. In GAP, presence of bulky side group constituents and reduced backbone flexibility render poor low temperature properties. Hence, GAP propellants should be heavily plasticized to meet the structural requirements. GAP based propellants also exhibit low stress and strain capabilities [2]. Copolymerizing crystalline hard segments with soft amor- phous GAP polymer chain is a feasible method to improve the mechanical properties of GAP polymer. It is expected that, the micro-phase separation between the hard and soft seg- ments should provide the copolymers with good mechanical properties. Moreover, the hard segments from each polymer molecule co-crystallize with those from the other molecules to contribute to the three dimensional crosslink density of the elastomer. Therefore, they will behave as crosslinked * Corresponding author. Tel.: þ65 67906409; fax: þ65 67927173. E-mail address: [email protected] (S. Pisharath). 0141-3910/$ - see front matter Ó 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.polymdegradstab.2007.03.016 Polymer Degradation and Stability 92 (2007) 1365e1377 www.elsevier.com/locate/polydegstab
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Synthesis and thermal decomposition of GAP–Poly(BAMO) copolymer

Polymer Degradation and Stability 92 (2007) 1365e1377www.elsevier.com/locate/polydegstab
Synthesis and thermal decomposition ofGAPePoly(BAMO) copolymer
Sreekumar Pisharath*, How Ghee Ang
Energetic Materials Research Centre, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore
Received 11 December 2006; received in revised form 5 March 2007; accepted 13 March 2007
Available online 30 March 2007
Abstract
An energetic copolymer of glycidyl azide polymer (GAP) and poly(bis(azidomethyl)oxetane (Poly(BAMO)) was synthesized using theBorontrifluorideedimethyl ether complex/diol initiator system. The synthesized copolymer exhibited the characteristics of an energetic thermo-plastic elastomer (ETPE). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to study the thermaldecomposition behavior and the results were compared with that of the constituent homopolymers. The main weight loss step in all the polymerscoincides with the exothermic dissociation of the azido groups in the side chain. In contrast with the behavior of the homopolymers, thecopolymer shows a broad exothermic shoulder peak at 298 �C after the main exothermic decomposition peak at 228 �C. Kinetic analysiswas performed by Vyazovkin’s model-free method, which suggests that the activation energy of the main decomposition step is around145 kJ/mol and for the second shoulder it is around 220 kJ/mol. Fourier transform infra red (FTIR) spectra of the degradation residues showthat the azido groups in the copolymer decompose in two stages at different temperatures which is responsible for the double decompositionbehavior.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Energetic polymer; Thermal decomposition; Model-free kinetics; Thermoplastic elastomer
1. Introduction
Azido polymers are unique energetic materials whichrelease heat by decomposition. They are used as energeticbinders in propellant and explosive formulations [1]. Inertpolymers such as hydroxyl-terminated polybutadiene (HTPB)are being replaced with energetic polymers such as glycidylazide polymer (GAP) as promising energetic binders. If inertpolymers are substituted by energetic polymers as binders,increased performance could be achieved at low oxidizer load-ing. Moreover, low oxidizer content reduces the vulnerabilityof formulation towards external stimuli. Thus energetic poly-mers as binders in propellant or explosive formulations offerthe dual advantage of improved performance and reducedvulnerability.
* Corresponding author. Tel.: þ65 67906409; fax: þ65 67927173.
E-mail address: [email protected] (S. Pisharath).
0141-3910/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.polymdegradstab.2007.03.016
With increasing complexities in the rocket motor designs,commensurate with the performance requirements, structureand processing demands placed on the solid propellants havebecome more and more strenuous. These requirements haveoutweighed the ballistic and combustion advantages. InGAP, presence of bulky side group constituents and reducedbackbone flexibility render poor low temperature properties.Hence, GAP propellants should be heavily plasticized tomeet the structural requirements. GAP based propellants alsoexhibit low stress and strain capabilities [2].
Copolymerizing crystalline hard segments with soft amor-phous GAP polymer chain is a feasible method to improvethe mechanical properties of GAP polymer. It is expectedthat, the micro-phase separation between the hard and soft seg-ments should provide the copolymers with good mechanicalproperties. Moreover, the hard segments from each polymermolecule co-crystallize with those from the other moleculesto contribute to the three dimensional crosslink densityof the elastomer. Therefore, they will behave as crosslinked

1366 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
rubber at ambient temperature. Above the melting temperatureof the crystalline hard block, the physical crosslinks betweenthe polymer chains disappear, allowing the polymer to flowlike a thermoplastic. Such materials are known as thermo-plastic elastomers (TPEs). The reversible physical crosslinksfacilitate the recycling of TPEs, thereby avoiding the disposalproblems posed by conventional crosslinked binders.
One of the candidates which could be copolymerized asa crystalline hard segment with GAP is the energetic polymerpoly(bis(azidomethyl)oxetane) (Poly(BAMO)), thereby form-ing an energetic thermoplastic elastomer (ETPE). ETPEcontaining formulations are in the various stages of develop-ment and are being used in missile and propellant developmentdemonstration efforts [3].
In polymer bonded explosive (PBX) formulations, it hasbeen observed that the decomposition characteristics of the en-ergetic binder affect the performance properties of the oxidizer[4e7] making it important to understand the thermal decom-position behavior of the polymer binder. Also, deriving reli-able kinetic data from thermal decomposition process ofpolymer binders will help in the modeling and prediction ofcombustion characteristics of formulations where the polymerbinder is an integral component. In this paper, we report thesynthesis and thermal decomposition kinetics of a GAPePoly(BAMO) copolymer. The thermal decomposition kinetics ofthe homopolymers (GAP and Poly(BAMO)) are also investi-gated for comparison purpose. The chemical structures ofthe studied polymers are presented in Fig. 1.
There have been several literature reports on the studies ofdecomposition of GAP and Poly(BAMO) alone [8e10]. Thecommon decomposition mechanism in these polymers is theexothermic rupture of the azide bonds to produce molecularnitrogen and energy. Secondary decomposition at higher tem-peratures involved rupture of polymer backbone into smallerfragments.
Only limited number of research results has been publishedon the thermal decomposition behavior of ETPE’s. Kimuraand Oyumi [11] compared thermal decomposition of energeticcopolymers of BAMO such as BAMO/NIMMO (whereNIMMO is 3-nitratomethyl-3-methyloxetane) and BAMO/AMMO (where AMMO is 3-azidomethyl-3-methyloxetane).
GAP-Poly (BAMO) Copolymer
HO CH2 C
CH2N3
CH2N3
CH2 O CH2HC O
CH2N3
Hn m
Poly (BAMO)
HO CH2 C CH2 O H
CH2N3
CH2N3
n
GAP
HO CH2
HC
CH2N3
O Hn
Fig. 1. Repeating chemical units of GAP, poly(BAMO) and GAPePoly(BAMO).
It was observed that the heat generated by the decompositionof NIMMO group accelerates the decomposition of the BAMOunit. On the other hand, for the BAMO/AMMO copolymer,the AMMO unit doesn’t affect the thermal decomposition ofBAMO. Reaction pathways or decomposition product analysiswas not reported for the decomposition of the copolymer. Liuet al. [12] studied the thermal characteristics of copolymers oftetrahydrofuran (THF) with BAMO, AMMO and NIMMO. Itwas observed that the decomposition enthalpies were depen-dent on the energetic group contents of the polymers andnot on copolymer types. In another detailed experimentalstudy, Lee et al. [13] studied the pyrolysis of BAMO/AMMO copolymer with and without titanium dioxide (TiO2)using a high power CO2 laser source. Measurement of theevolved decomposition species revealed that the products ofBAMO/AMMO decomposition are similar to those reportedfor BAMO. Addition of TiO2 caused no significant changesin product profiles except for an increase in ammonia (NH3).The product profiles also indicated a simultaneous decomposi-tion of backbone and side chains of the polymer.
There have been no reported investigations on the thermaldecomposition of GAPePoly(BAMO) copolymer whichprompted us for this study. In this work, we use a model-free or iso-conversional method suggested by Vyazovkin andDollimore [14] to investigate the decomposition kinetics.Model-free methods allow the determination of activation en-ergy as a function of extent of decomposition and temperature.These methods are being widely employed to study thermallystimulated processes (e.g., degradation, curing) in polymers[15]. Model-free methods, when applied to propellant formu-lations, help to compare the activation energy dependencies ofthe neat polymer binder with that of a propellant based on thispolymer, allowing the elucidation of the role of the latter [16].
2. Experimental
2.1. Materials
Commercially available materials were used as receivedunless noted otherwise. The solvents were distilled under re-duced pressure over calcium hydride before use.
2.2. Synthesis of monomers
2.2.1. Synthesis of 3,3-bis(chloromethyl)oxetane (BCMO)BCMO was synthesized by the cyclization of pentaerythri-
tol trichlorohydrin using sodium hydroxide as described inRef. [17].
2.2.2. Synthesis of 3,3-bis(azidomethyl)oxetane (BAMO)BAMO was synthesized by the reaction of sodium azide
with BCMO in alkaline medium in the presence of a phasetransfer catalyst, tetrabutyl ammonium bromide as describedin Ref. [18].

1367S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
2.3. Synthesis of polymers
2.3.1. Synthesis of GAPGAP polymer was synthesized in two steps. Scheme 1
outlines the synthesis of GAP. First step was the synthesisof poly(epichlorohydrin) (PECH) of adequate molecularweight. Second step was the conversion of PECH polymerto GAP. The steps involved in polymer synthesis are de-scribed below.
2.3.1.1. Step 1. 1.33 g (0.0144 mol) of butane diol (BDO)dissolved in 75 mL methylene chloride was taken in a three-necked flask fitted with a thermometer and nitrogen inlet.Seven hundred and fifty microliters of BF3edimethyl ethercomplex, was injected into the reaction mixture and stirredat room temperature for 60 min. After cooling the reactionvessel to 0 �C using iceesalt mixture, 46.66 g (0.5044 mol)of epichlorohydrin (ECH) was added drop by drop (at therate of w0.1 mL/min) to the reaction mixture over a periodof 12 h. The mole ratio of BDO to ECH is 1:35. Rate of addi-tion is kept as slow as possible to keep the instantaneous con-centration of ECH in the reaction mixture very low. At nopoint of time, the temperature was allowed to go above 0 �Cduring the reaction.
After the addition of ECH, reaction was continued foranother 12 h at room temperature. Thereafter, reaction wasfrozen by adding distilled water. The organic phase containingPECH was extracted into methylene chloride and washed
several times with distilled water until neutral to pH. Thewashed organic phase was dried over sodium sulphate, filtered,and the solvent evaporated off in vacuum at 40 �C to recover43 g of PECH (89.5% yield).
GPC analysis: Mw¼ 2340 Kg mol�1; Mn¼ 2018 Kg mol�1;polydispersity index¼ 1.16 (against polystyrene standards).
FTIR (KBr): n(eOH)¼ 3450 cm�1; n(CeOeC)¼ 1100 cm�1;n(CeCl)¼ 745 cm�1.
2.3.1.2. Step 2. Twenty eight grams of vacuum dried PECHwas dissolved in 100 mL Dimethyl formamide (DMF) andtaken in a three-necked flask fitted with a thermometer, nitro-gen inlet and water condenser. The reaction mixture washeated to 120 �C, under stirring in an oil bath. Twenty fivegrams of sodium azide was added to the mixture and thereaction continued for another 12 h. Thereafter, reaction wasfrozen by adding distilled water. The unreacted azide andsalted out sodium chloride were filtered off. Organic phasecontaining GAP was extracted into methylene chloride andwashed several times with distilled water until neutral to pH.The washed organic phase was dried over sodium sulphate, fil-tered, and the solvent evaporated off under vacuum. GAP of23 g was obtained (82.14% yield).
GPC analysis: Mw¼ 2490 Kg mol�1; Mn¼ 2178 Kg mol�1;polydispersity index¼ 1.15 (against polystyrene standards).
FTIR (KBr): n(eOH)¼ 3450 cm�1; n(CeN3)¼ 2100 cm�1;n(CeOeC)¼ 1100 cm�1.
CH2 CH2HO CH2 CH2 OH
ClOBF3-Dimethyl ether complex00C, CH2Cl2
(CH2)4 OO CH2 CH OHH2CHC
CH2Cl
HO
(CH2)4 OO CH2 CH OH2CHC
CH2Cl
O CH2 CH OH
CH2Cl
H2CHCHO
CH2Cl
CH2Cl
CH2Cln n
ClO
(CH2)4 OO CH2 CH OH2CHC
CH2N3
O CH2 CH OH
CH2N3
H2CHCHO
CH2N3 CH2N3
n n
NaN3; DMF110°C; 10Hrs
Epichlorohydrin
(1,4-Butane Diol)
Scheme 1. Synthetic scheme for GAP.

1368 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
1H NMR (CDCl3): d¼ 3.7 ppm (3H, eCH2eCHe);3.59 ppm (2H, eCH2N3).
2.3.2. Synthesis of Poly(BAMO)Scheme 2 outlines the synthesis of Poly(BAMO). 0.125 g
(0.00144 mol) of (BDO) in 15 mL methylene chloride was takenin a three-necked flask fitted with a thermometer and nitrogen in-let. Five hundred microlitres of BF3edimethyl ether complex,was injected into the reaction mixture and stirred at room tem-perature for 60 min. After cooling the reaction vessel to 0 �C us-ing iceesalt mixture, 4.65 g (0.0277 mol) of BAMO taken in 9 gof methylene chloride was added to the reaction mixture overa period of 4 h. The solution was allowed to come to room tem-perature and left to react for 24 h. The reaction was quenched bythe addition of saturated brine solution (50 mL). The organicphase was separated and washed with 10% sodium bicarbonatesolution and the solvent was removed by vacuum evaporation.The polymer was precipitated by the addition of methanol anddried under vacuum at 30 �C. Yield was 80%.
GPC analysis: Mw¼ 2179 Kg mol�1; Mn¼ 1653 Kg mol�1;polydispersity index¼ 1.31 (against polystyrene standards).
FTIR (KBr): n(eOH)¼ 3417 cm�1; n(CeN3)¼ 2106 cm�1;n(CeOeC)¼ 1103.2 cm�1.
1H NMR (CDCl3): d¼ 3.36 ppm (4H, (eOCH2)2);3.30 ppm (4H, (eCH2N3)2).
2.3.3. Synthesis of GAPePoly(BAMO) copolymerSynthesis of GAPePoly(BAMO) copolymer was achieved
in two stages as outlined in Scheme 3. First step was the synthe-sis of PECHepoly(BCMO) copolymer of adequate molecularweight. Second step is the conversion of PECHepoly(BCMO)
copolymer to GAPePoly(BAMO) copolymer by conversion ofchloro to azido groups. The steps involved in polymer synthesisare described below.
2.3.3.1. Step 1. Eight grams of pre-synthesized vacuum driedPECH diol was dissolved 30 mL of distilled methylene chlo-ride was taken in a 250 mL three-necked reaction flask equip-ped with a nitrogen inlet, magnetic stirrer, and thermometer.Five hundred microlitres of BF3edimethyl ether complexwas injected into the reaction flask and left to react for 1 hat room temperature. Thereafter, the reaction flask was cooledto 0 �C using iceesalt mixture and calculated quantity offreshly distilled BCMO was added drop by drop througha dropping funnel over a period of 6 h. After the complete ad-dition, the reaction mixture was left to react for 24 h at roomtemperature. The reaction mixture was poured into excess ofmethanol to precipitate out the polymer in 70% yield. Thepolymer was dried in vacuum at 30 �C. Yield¼ 65%.
2.3.3.2. Step 2. Three grams of PECHePoly(BAMO) copoly-mer was reacted with molar excess of sodium azide in 20 mLof DMF in a three-necked flask. The reaction mixture washeated to 120 �C, under stirring in an oil bath. After 24 h,the reaction was frozen by the addition of distilled water.The organic phase containing the copolymer was extractedinto methylene chloride and washed with 10% sodium bicar-bonate solution until neutral to pH. The solution was pouredinto excess of methanol to precipitate out the copolymer.The copolymer was dried at 40 �C under vacuum for 72 h.Yield was 80%.
CH2 CH2HO CH2 CH2 OH
BF3-Dimethyl ether complex
00C, CH2Cl2
(CH2)4 OO CH2 C CH2H2CC
CH2N3
H2C
(CH2)4 OO CH2 CH2CC
CH2N3
H2C
CH2N3
CH2N3
O
N3
N3
BAMO
CH2N3
HO
CH2N3
OH
O
N3
N3
CH2N3 CH2N3
CH2 O H
m m
(1,4,-Butane Diol)
OH
Scheme 2. Synthetic scheme for poly(BAMO).

1369S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
R OO CH2CH2 CH
CH2Cl
O HCHOH
CH2Cl
O
ClCl
blk
PECH Block
BCMO
BF3.OMe2,O0C CH2Cl2
PECH OO CH2CH2 CC
CH2Cl
CH2Cl
CH2 O H
CH2Cl
CH2Cl
CH2OH
blkBCMO Block BCMO Block
m/2 m/2n
m/2 m/2
blk
NaN3, DMF, 110°C
GAP OO CH2CH2 CC
CH2N3
CH2N3
CH2 O H
CH2N3
CH2N3
CH2OH
blkBAMO Block BAMO Block
m/2 m/2n
blk
R=(CH2)4
Scheme 3. Synthetic scheme for GAPePoly(BAMO) copolymer.
GPC analysis: results are provided in Table 1.FTIR (KBr): n(eOH)¼ 3436 cm�1; n(CeN3)¼ 2102.3 cm�1;
n(CeOeC)¼ 1114.8 cm�1.1H NMR (CDCl3): d¼ 3.74 ppm (3H, eCH2eCHe);
3.63 ppm (2H, eCH2N3) (GAP block); 3.50 ppm (4H, (eOCH2)2);3.43 ppm (4H, (eCH2N3)2) (PBAMO block).
2.4. Measurements
GPC analyses of polymers was carried out in a Waters in-strument fitted with a 2414 differential refractive index detec-tor against polystyrene standards using tetrahydrofuran (THF)as eluent at 1 mL/min.
Table 1
Sample Mw (g/mol) Mn (g/mol) Polydispersity
PECH diol 2706 2016 1.34
Copolymer 1 3553 2499 1.4
Copolymer 2 5440 3777 1.44
1H NMR spectra were recorded in a Bruker 400 MHz in-strument using CDCl3 solvent. FTIR spectra were recordedin a Shimadzu FTIR-8300.
Weight loss measurements were carried out in a ShimadzuTGA-50 thermogravimetric analyzer equipped with thermalanalysis software for data analysis. Approximately 5 mg of sam-ples were used for each run. The samples were heated from roomtemperature to 350 �C at a variety of heating rates ranging from0.1 �C/min to 20 �C/min in aluminum crucibles under constantflow of nitrogen (50 mL/min). From a typical TGA experimentat a given heating rate, the fractional weight loss or conversion(a) is computed as ððW0 �WTÞ=ðW0 �WNÞÞ, where W0 is theinitial weight of the sample, WT is the weight at temperatureT, and WN is the final weight of the sample.
DSC measurements were carried out in TA DSC 2010equipment containing thermal analysis software for data inter-pretation. In representative runs, 2e3 mg of samples in sealedaluminum hermetic pans were ramped from room temperatureto 350 �C at the rate of 2 �C/min in a steady flow of nitrogen(50 mL/min).

1370 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
2.5. Kinetic analysis of TGA data
The most straightforward way to evaluate the effectiveactivation energy as a function of decomposition is theFriedman’s method [19]. In this method, no approxima-tions are introduced. However, this is a differential methodwhich produces significant numerical instability andnoise interference. Hence, integral iso-conversional methodswere proposed. For non-isothermal conditions, integral iso-conversional methods were proposed by Ozawa and Flynnand Wall commonly referred to as the OFW method [20].This method uses an oversimplified approximation ofthe temperature integral thereby limiting the accuracy ofthe predictions. To avoid such inaccuracies, Vyazovkinproposed an alternative non-linear iso-conversional method[14].
According to this method, for a set of n experiments carriedout at different heating rates (b), the activation energy at anyparticular value of extent of decomposition (a) can be ob-tained by minimizing the function 4 (Ea):
Xn
i¼1
Xn
js1
IðEa;TaiÞbj
I�Ea;Taj
�bi
ð1Þ
In Eq. (1), the temperature integral is defined as:
IðE;TaÞ ¼ZTa
Ta�Da
exp
��E
RT
�dT ð2Þ
The temperature integral in Eq. (2), will account for stron-ger variation of activation energy with temperature, where Da
is the difference in consecutive values of a chosen for analysis.MATLAB program was developed for the numerical integra-tion of Eq. (2) and minimization of Eq. (1). A function ‘quadv’which uses recursive adaptive Simpson algorithm was used fornumerical integration. Minimization was done using the ‘fmi-nunc’ function for unconstrained problem which uses a quasiNewton line search algorithm.
3. Results
3.1. Synthesis of polymers
The important step in the synthesis of GAP polymer is thesynthesis of PECH diol of adequate molecular weight. Usu-ally, for propellant applications, the molecular weight of theprepolymer used as binders is restricted between 2000 g/moland 3000 g/mol. This is due to the fact that, higher molecularweight of the prepolymer will lead to higher viscosity incur-ring high energy costs in processing operations.
Synthesis of PECH takes place by the cationic ring openingpolymerization of ECH in the presence of BF3edimethyl ethercomplex as initiator and BDO as the co-initiator. In cationicring opening polymerizations, the initiation step is the proton-ation of the monomer by the initiator. Thereafter, protonatedmonomer could either react with BDO or with the non-proton-ated monomer. The reaction proceeds through two propagationmechanisms depending on whether the protonated monomerreacts with BDO (activated monomer mechanism (AMM))or non-protonated monomer (activated chain end mechanism(ACE)). The quality of the polymer obtained depends on thedominance of one mechanism over the other. The operationof AMM will lead to the formation of the preferred lineardiol polymers and the operation of ACE leads to unnecessarylow molecular weight oligomers. These low molecular weightoligomers are mainly constituted of cyclic compounds.Scheme 4 presents the details of the mechanisms of ACEand AMM. From the involved kinetics, in order to enhancethe contribution of AMM, the instantaneous concentration ofthe non-protonated monomer should be kept as low as possible[21]. We have clearly demonstrated this aspect in one of ourearlier papers [22]. By exercising good control over the rateof monomer addition, linear PECH diols with number averagemolecular weight (Mn) of 2040 g/mol and weight averagemolecular weight (Mw) of 2481 g/mol and dispersity¼ 1.21were synthesized.
Like oxiranes, oxetanes such as BCMO and BAMO poly-merize through the cationic polymerization route in the
R OH
ClO
ClOH
RO CH2 CH OH
CH2Cl
HO CH2 CH
CH2Cl
O Cl
AMM
ACE
Linear diol polymer
Cyclic oligomers
Epichlorohydrin
Protonated Epichlorohydrin
HODiol
Scheme 4. Illustrations of AMM and ACE mechanisms involved in cationic ring opening polymerizations. AMM produces linear polymers. ACE produces cyclic
oligomers by backbiting reactions.

1371S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
presence of Lewis acid catalysts. Both AMM and ACEmechanisms are also applicable to the ring opening ofoxetanes too. There have been published reports on the com-parison of reactivities of BCMO and BAMO to undergo ringopening polymerization. It was observed that BAMO andBCMO polymerize through different mechanisms and AMMis more favored with BAMO [23]. Hence, we used BAMOmonomer directly to synthesize Poly(BAMO). Poly(BAMO)with number average molecular weight (Mn) of 1653 g/moland weight average molecular weight (Mw) of 2179 g/moland dispersity¼ 1.31 was synthesized.
In the synthesis of copolymers of PECH and BCMO, weused PECH diol as the macro-initiator along with BF3edimethyl complex as the catalyst. By this route, the PECHmolecule will form the central block and the ring openedBCMO units will be attached to the both ends of PECH moi-ety. We carried out the synthesis of 2 batches of the PECHePoly(BCMO) copolymers namely copolymer 1 and copolymer2 with varying amount of BCMO. An overlay of the GPCchromatograms of the polymers is presented in Fig. 2. Theresult indicates that, with increasing amount of BCMO, theGPC peak of PECH diol shifts to lower retention times (highermolecular weights), meaning that the BCMO units are gettinganchored to the PECH diol block. Results of the GPC analysesare presented in Table 1. It could be observed that the weightaverage molecular weight of PECH diol increases from2706 g/mol to 5440 g/mol with increasing amount of BCMO.
Fig. 3 compares the FTIR spectra of BCMO monomer withthe PECHepoly(BCMO) copolymer. The FTIR peak at980 cm�1 due to the oxetane ring present in the monomer isabsent in the copolymer. It is evident that BCMO undergoesring opening during the polymerization process. We convertedcopolymer 2 to the azido derivative and used it for the thermaldecomposition studies.
Conversion of chloro groups to azido groups is a nucleo-philic substitution reaction. As presented in Fig. 4, the reactioncould be easily monitored by FTIR spectroscopy. The
20 21 22 23 24 25
PECHCopolymer 1Copolymer 2
Retention time (mins)
Increasing
BCMO content
Fig. 2. GPC curves of PECH diol and its copolymers with BCMO. Feed
amounts [PECH diol]:[BCMO], copolymer-1:80:20, copolymer-2:75:25. The
curves shift to lower retention times (higher molecular weights) with increas-
ing BCMO content.
disappearance of the CeCl stretching band in FTIR spectraat 748 cm�1 and appearance of a new stretching band forCeN3 at 2100 cm�1 confirm the substitution reaction. TheGAPePoly(BAMO) copolymer exhibited a weight averagemolecular weight of 5791 g/mol, number average molecularweight of 4136 g/mol and a polydispersity index of 1.4.
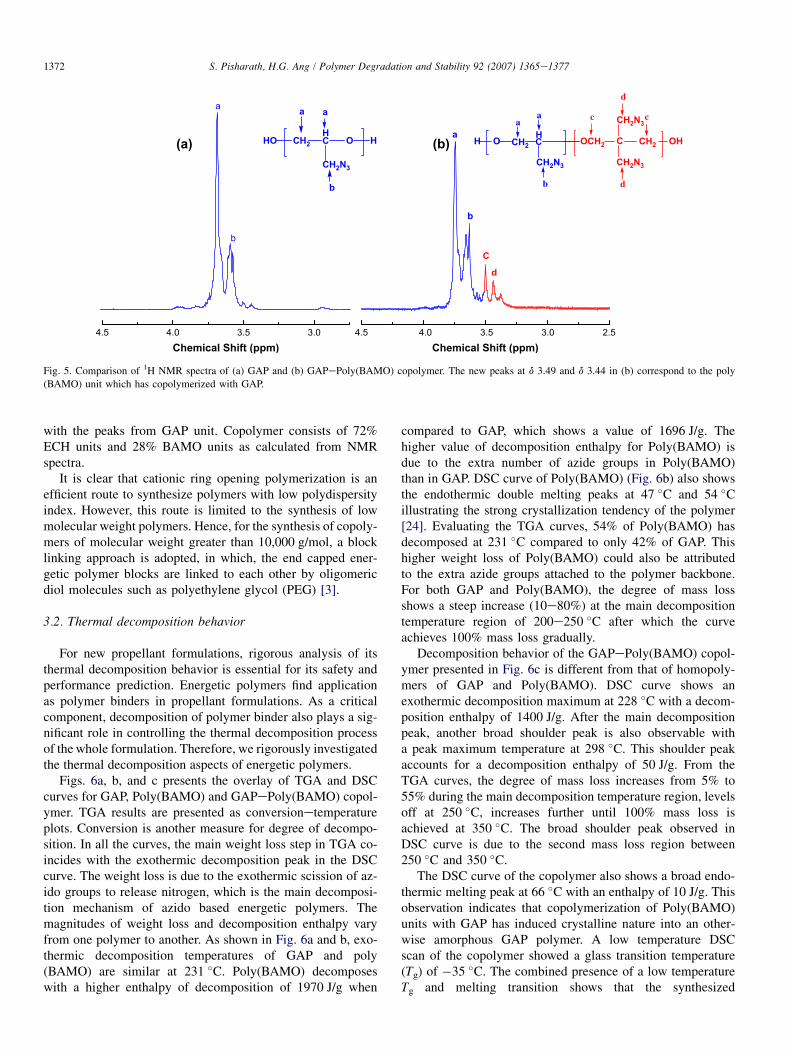
Fig. 5 compares the 1H NMR spectra of GAP with that ofGAPePoly(BAMO). For GAP (Fig. 5a), the spectra show sig-nals due to protons of methylene and methine groups in thepolymer backbone at d 3.6 and that due to the pendant azido-methyl unit at d 3.68. NMR spectra of GAPePoly(BAMO) co-polymer presented in Fig. 5b show two other peaks at d 3.49and d 3.44 corresponding to the Poly(BAMO) unit along
3500 3000 2500 2000 1500 1000
BCMOPECH-Poly(BCMO)
Wavenumber (cm-1)
Oxetane ring at 980 cm-1
C-Cl stretch at 740 cm-1
Fig. 3. Comparison of FTIR spectra of BCMO monomer and PECHepoly
(BCMO) copolymer. The peak due to the oxetane ring at 980 cm�1 is absent
in the copolymer indicating that the oxetane undergoes ring opening in the
presence of PECH diol.
3500 3000 2500 2000 1500 1000
C-Cl stretch at 745 cm-1
GAP-Poly(BAMO)
PECH-Poly(BCMO)
Wavenumber (cm-1)
C-N3
stretch at 2100 cm-1
Fig. 4. Comparison of FTIR spectra of PECHepoly(BCMO) copolymer and
GAPePoly(BAMO) copolymer. Appearance of new peak at 2100 cm�1 for
GAPePoly(BAMO) copolymer indicates the substitution of chloro groups in
PECHepoly(BCMO) copolymer with the azido groups.
1372 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
(a)
4.5
(b)
4.0 3.5 3.0 2.5
O CH2
H
H
C
CH2N
3
OCH2
C
CH2N
3
CH2N
3
CH2
OH
aa
b
c c
d
d
Chemical Shift (ppm)
a
b
C
d
4.5 4.0 3.5 3.0
HO CH2
H
C
CH2N
3
O H
a a
b
Chemical Shift (ppm)
a
b
Fig. 5. Comparison of 1H NMR spectra of (a) GAP and (b) GAPePoly(BAMO) copolymer. The new peaks at d 3.49 and d 3.44 in (b) correspond to the poly
(BAMO) unit which has copolymerized with GAP.
with the peaks from GAP unit. Copolymer consists of 72%ECH units and 28% BAMO units as calculated from NMRspectra.
It is clear that cationic ring opening polymerization is anefficient route to synthesize polymers with low polydispersityindex. However, this route is limited to the synthesis of lowmolecular weight polymers. Hence, for the synthesis of copoly-mers of molecular weight greater than 10,000 g/mol, a blocklinking approach is adopted, in which, the end capped ener-getic polymer blocks are linked to each other by oligomericdiol molecules such as polyethylene glycol (PEG) [3].
3.2. Thermal decomposition behavior
For new propellant formulations, rigorous analysis of itsthermal decomposition behavior is essential for its safety andperformance prediction. Energetic polymers find applicationas polymer binders in propellant formulations. As a criticalcomponent, decomposition of polymer binder also plays a sig-nificant role in controlling the thermal decomposition processof the whole formulation. Therefore, we rigorously investigatedthe thermal decomposition aspects of energetic polymers.
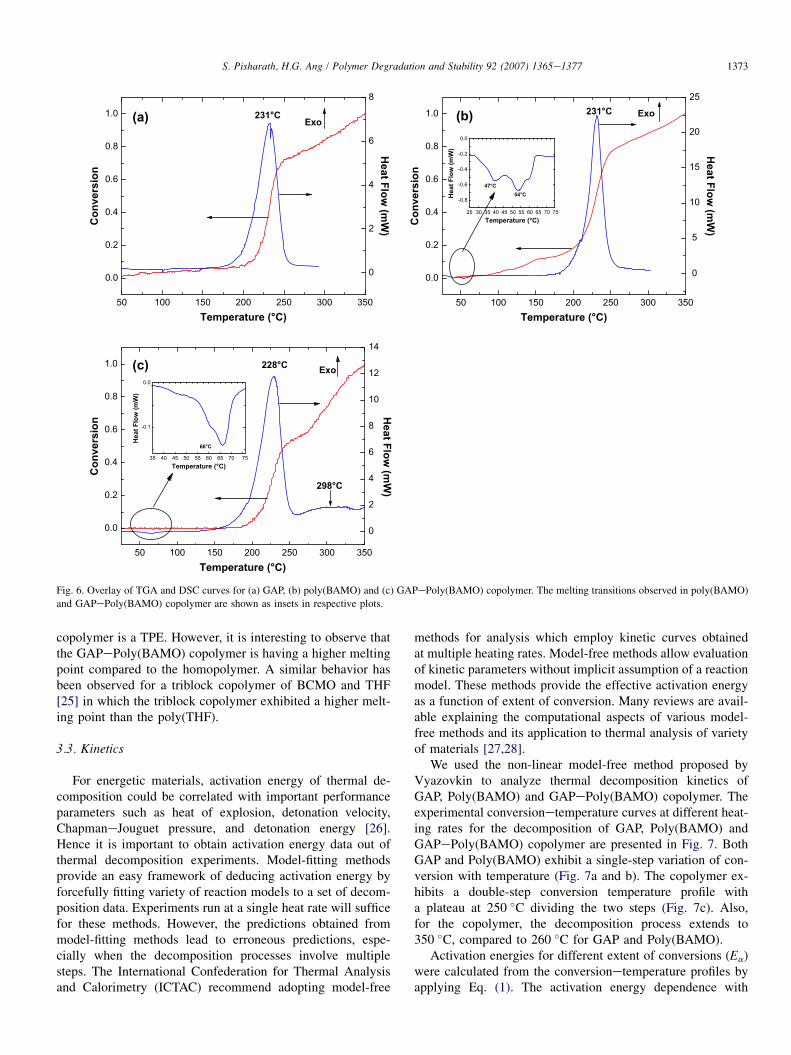
Figs. 6a, b, and c presents the overlay of TGA and DSCcurves for GAP, Poly(BAMO) and GAPePoly(BAMO) copol-ymer. TGA results are presented as conversionetemperatureplots. Conversion is another measure for degree of decompo-sition. In all the curves, the main weight loss step in TGA co-incides with the exothermic decomposition peak in the DSCcurve. The weight loss is due to the exothermic scission of az-ido groups to release nitrogen, which is the main decomposi-tion mechanism of azido based energetic polymers. Themagnitudes of weight loss and decomposition enthalpy varyfrom one polymer to another. As shown in Fig. 6a and b, exo-thermic decomposition temperatures of GAP and poly(BAMO) are similar at 231 �C. Poly(BAMO) decomposeswith a higher enthalpy of decomposition of 1970 J/g when
compared to GAP, which shows a value of 1696 J/g. Thehigher value of decomposition enthalpy for Poly(BAMO) isdue to the extra number of azide groups in Poly(BAMO)than in GAP. DSC curve of Poly(BAMO) (Fig. 6b) also showsthe endothermic double melting peaks at 47 �C and 54 �Cillustrating the strong crystallization tendency of the polymer[24]. Evaluating the TGA curves, 54% of Poly(BAMO) hasdecomposed at 231 �C compared to only 42% of GAP. Thishigher weight loss of Poly(BAMO) could also be attributedto the extra azide groups attached to the polymer backbone.For both GAP and Poly(BAMO), the degree of mass lossshows a steep increase (10e80%) at the main decompositiontemperature region of 200e250 �C after which the curveachieves 100% mass loss gradually.
Decomposition behavior of the GAPePoly(BAMO) copol-ymer presented in Fig. 6c is different from that of homopoly-mers of GAP and Poly(BAMO). DSC curve shows anexothermic decomposition maximum at 228 �C with a decom-position enthalpy of 1400 J/g. After the main decompositionpeak, another broad shoulder peak is also observable witha peak maximum temperature at 298 �C. This shoulder peakaccounts for a decomposition enthalpy of 50 J/g. From theTGA curves, the degree of mass loss increases from 5% to55% during the main decomposition temperature region, levelsoff at 250 �C, increases further until 100% mass loss isachieved at 350 �C. The broad shoulder peak observed inDSC curve is due to the second mass loss region between250 �C and 350 �C.
The DSC curve of the copolymer also shows a broad endo-thermic melting peak at 66 �C with an enthalpy of 10 J/g. Thisobservation indicates that copolymerization of Poly(BAMO)units with GAP has induced crystalline nature into an other-wise amorphous GAP polymer. A low temperature DSCscan of the copolymer showed a glass transition temperature(Tg) of �35 �C. The combined presence of a low temperatureTg and melting transition shows that the synthesized
1373S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
25 30 35 40 45 50 55 60 65 70 75
-0.8
-0.6
-0.4
-0.2
0.0
50 100 150 200 250 300 350
0.0
0.2
0.4
0.6
0.8
1.0
54°C
47°C
Temperature (°C)
Co
nversio
n
231°C Exo
0
5
10
15
20
25
Heat F
lo
w (m
W)
Heat F
lo
w (m
W)
Temperature (°C)
(b)
35 40 45 50 55 60 65 70 75
-0.1
0.0
50 100 150 200 250 300 350
0.0
0.2
0.4
0.6
0.8
1.0
66°C
Temperature (°C)
Co
nversio
n
228°C
298°C
Exo
0
2
4
6
8
10
12
14
Heat F
lo
w (m
W)
Temperature (°C)
Heat F
lo
w (m
W)
(c)
50 100 150 200 250 300 350
0.0
0.2
0.4
0.6
0.8
1.0
Temperature (°C)
Co
nversio
n
231°C
0
2
4
6
8
Heat F
lo
w (m
W)
Exo(a)
Fig. 6. Overlay of TGA and DSC curves for (a) GAP, (b) poly(BAMO) and (c) GAPePoly(BAMO) copolymer. The melting transitions observed in poly(BAMO)
and GAPePoly(BAMO) copolymer are shown as insets in respective plots.
copolymer is a TPE. However, it is interesting to observe thatthe GAPePoly(BAMO) copolymer is having a higher meltingpoint compared to the homopolymer. A similar behavior hasbeen observed for a triblock copolymer of BCMO and THF[25] in which the triblock copolymer exhibited a higher melt-ing point than the poly(THF).
3.3. Kinetics
For energetic materials, activation energy of thermal de-composition could be correlated with important performanceparameters such as heat of explosion, detonation velocity,ChapmaneJouguet pressure, and detonation energy [26].Hence it is important to obtain activation energy data out ofthermal decomposition experiments. Model-fitting methodsprovide an easy framework of deducing activation energy byforcefully fitting variety of reaction models to a set of decom-position data. Experiments run at a single heat rate will sufficefor these methods. However, the predictions obtained frommodel-fitting methods lead to erroneous predictions, espe-cially when the decomposition processes involve multiplesteps. The International Confederation for Thermal Analysisand Calorimetry (ICTAC) recommend adopting model-free
methods for analysis which employ kinetic curves obtainedat multiple heating rates. Model-free methods allow evaluationof kinetic parameters without implicit assumption of a reactionmodel. These methods provide the effective activation energyas a function of extent of conversion. Many reviews are avail-able explaining the computational aspects of various model-free methods and its application to thermal analysis of varietyof materials [27,28].
We used the non-linear model-free method proposed byVyazovkin to analyze thermal decomposition kinetics ofGAP, Poly(BAMO) and GAPePoly(BAMO) copolymer. Theexperimental conversionetemperature curves at different heat-ing rates for the decomposition of GAP, Poly(BAMO) andGAPePoly(BAMO) copolymer are presented in Fig. 7. BothGAP and Poly(BAMO) exhibit a single-step variation of con-version with temperature (Fig. 7a and b). The copolymer ex-hibits a double-step conversion temperature profile witha plateau at 250 �C dividing the two steps (Fig. 7c). Also,for the copolymer, the decomposition process extends to350 �C, compared to 260 �C for GAP and Poly(BAMO).
Activation energies for different extent of conversions (Ea)were calculated from the conversionetemperature profiles byapplying Eq. (1). The activation energy dependence with

1374 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
200 210 220 230 240 250 260 270 2800.0
0.2
0.4
0.6
0.8
1.0
0.80°C/min0.50°C/min
Co
nversio
n
Temperature (°C)
0.20°C/min
(a)
170 180 190 200 210 220 230 240 250
0.2
0.4
0.6
0.8
1.0
5.0°C/min
7.50°C/min
Co
nversio
n
Temperature (°C)
2.50°C/min
(b)
180 200 220 240 260 280 300 320 340 3600.0
0.2
0.4
0.6
0.8
1.0
5.0°C/min
1.0°C/min
Co
nversio
n
Temperature (°C)
4.0°C/min
(c)
Fig. 7. Experimental conversionetemperature profiles at various heat rates obtained from TGA results for (a) GAP, (b) poly(BAMO) and (c) GAPePoly(BAMO)
copolymer.
conversion for GAP, Poly(BAMO) and GAPePoly(BAMO)copolymer are presented in Fig. 8. TGA curves of the samplescollected at a heat rate of 5 �C/min are also plotted to show thecorrelation of Ea with temperature. For both the homopoly-mers, activation energy dependence is an increasing functionof conversion (Fig. 8a and b). Two decomposition pathwayscould be clearly evidenced for GAP and Poly(BAMO). Fromthe initial stages of decomposition until a¼ 0.75, Ea remainspractically constant at w170 kJ/mol. This value is consistentwith the reported values of activation energy of azido poly-mers measured by various experimental methods [29,30]. To-wards the end of the decomposition process, Ea increases tow280 kJ/mol for both the polymers. GAP exhibits an abruptincrease, whereas Poly(BAMO) shows a more gradual varia-tion. For the copolymer (Fig. 8c), 4 decomposition stages ofvarying Ea could be identified. During the first mass lossstep (a¼ 0.03e0.36), Ea remains constant at 145 kJ/mol.For the second mass loss step Ea increases from w145 kJ/mol at a¼ 0.36 to w220 kJ/mol at a¼ 0.62. Hereafter, Ea re-mains constant until a¼ 0.78 and further increases tow260 kJ/mol until a¼ 0.98.
Activation energy parameters derived from the model-freemethod support the experimental TGA and DSC results. Ea re-mains practically constant at w170 kJ/mol, for GAP and Pol-y(BAMO) exhibiting single-step decomposition until itincreases to w280 kJ/mol towards the end of decompositionprocess. For the copolymer, double decomposition step inTGA and DSC results could be identified as two regionswith different activation energies in the Ea-dependence plot.First region is between a¼ 0.03 and a¼ 0.36 with Ea ofw145 kJ/mol and the second region with Ea of w220 kJ/mol between a¼ 0.62 and a¼ 0.8. Thus application ofmodel-free techniques to thermal decomposition of materialscould successfully identify the Ea dependence of stepsinvolved.
3.4. Mechanistic considerations
The shape of activation energy dependence helps to shedlight on the kinetic scheme involved in the decomposition pro-cess. An increasing function of Ea dependence as observed inour studies generally denotes the operation of competitive

1375S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
(b)
0.2 0.4 0.6 0.8 1.0
100
150
200
250
300
Activation EnergyTemperature
Conversion
Activatio
n E
nerg
y/kJ m
ol-1
170
180
190
200
210
220
230
Tem
peratu
re (°C
)
(c)
0.0 0.2 0.4 0.6 0.8 1.050
100
150
200
250
300
Region 2
Activation EnergyTemperature
Conversion
Activatio
n E
nerg
y/kJ m
ol-1
Region 1
180
200
220
240
260
280
300
320
340
Tem
peratu
re (°C
)
(a)
0.2 0.4 0.6 0.8 1.050
100
150
200
250
300
350
400
Activation EnergyTemperature
Conversion
Activatio
n E
nerg
y/kJ m
ol-1
200
210
220
230
240
250
260
Tem
peratu
re (°C
)
Fig. 8. Dependence of activation energy on the extent of conversion and the dependence of the extent of conversion on temperature at the heating rate of 5 �C/min
for (a) GAP, (b) poly(BAMO) and (c) GAPePoly(BAMO) copolymer.
reactions during the decomposition [31]. Increasing Ea depen-dence was observed for degradation of linear polymers in theabsence of oxygen. The result was interpreted as competitionbetween the decomposition of individual macromolecules andintermolecular associates formed during the course of the re-action [32].
Mechanism of decomposition of azide polymers has beeninvestigated by employing a variety of experimental tech-niques. Here we attempt to correlate the Ea dependence withthe suggested decomposition mechanism of azido polymers.The established mechanism is the elimination of nitrogenfrom the pendant azide groups of the polymer to form animine. It has also been proposed that the imine intermediateundergoes intermolecular cyclizations to form intermolecularassociates (Scheme 5) [33,34]. We believe that the increasingfunction of Ea dependence is due to the competition betweenthe decomposition of linear GAP polymer and intermolecularassociate formed during the reaction.
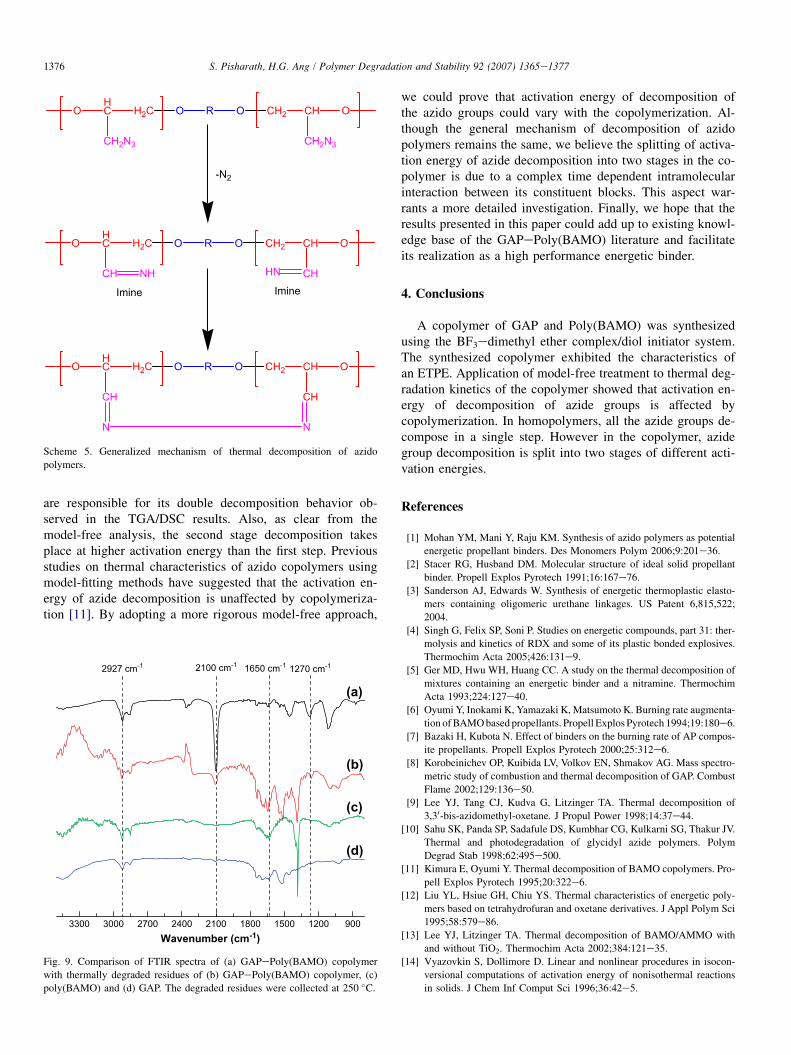
In order to gain more insights into the mechanism of de-composition, we took FTIR spectrum of the decompositionresidues of the polymers at 250 �C (Fig. 9) and comparedwith the spectrum of GAPePoly(BAMO) copolymer beforedecomposition. This temperature was chosen because the
conversionetemperature profile of the copolymer exhibitsa plateau at this temperature. This will enable us to accountfor the energetic factor responsible for the second stage degra-dation observed in the copolymer. Comparing the spectra, itcould be observed that for the degraded copolymer the inten-sity of the characteristic azide band at 2100 cm�1 and1270 cm�1 is reduced considerably after thermal decomposi-tion. These bands are completely absent for the degraded Pol-y(BAMO) and GAP. Also, after the pyrolysis, a new bandappears at 1650 cm�1 for the degraded polymers. This bandcould be readily assigned to the C]N vibration of the imineintermediate formed during the elimination of nitrogen fromthe azide group as shown in Scheme 5. It is noteworthy thatthe band associated with CeH stretching vibrations of thepolymer doesn’t change with pyrolysis suggesting that thepolymer backbone is stable at 250 �C. This observation con-firms that the azide group thermally decomposes before thebackbone does.
FTIR study presented above indicates that some residualazido groups are remaining in the copolymer after the firststage decomposition, whereas for degraded Poly(BAMO)and GAP all the azido groups have completely decomposedat 250 �C itself. These residual azido groups in the copolymer
1376 S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
are responsible for its double decomposition behavior ob-served in the TGA/DSC results. Also, as clear from themodel-free analysis, the second stage decomposition takesplace at higher activation energy than the first step. Previousstudies on thermal characteristics of azido copolymers usingmodel-fitting methods have suggested that the activation en-ergy of azide decomposition is unaffected by copolymeriza-tion [11]. By adopting a more rigorous model-free approach,
3300 3000 2700 2400 2100 1800 1500 1200 900
(d)
(c)
(b)
1270 cm-11650 cm-12100 cm-1
Wavenumber (cm-1
)
2927 cm-1
(a)
Fig. 9. Comparison of FTIR spectra of (a) GAPePoly(BAMO) copolymer
with thermally degraded residues of (b) GAPePoly(BAMO) copolymer, (c)
poly(BAMO) and (d) GAP. The degraded residues were collected at 250 �C.
R OO CH2 CH O
CH2N3
H2CHC
CH2N3
O
R OO CH2 CH O
CH
H2CHC
CH
O
NH
R OO CH2 CH O
CH
H2CHC
CH
O
N N
HN
-N2
Imine Imine
Scheme 5. Generalized mechanism of thermal decomposition of azido
polymers.
we could prove that activation energy of decomposition ofthe azido groups could vary with the copolymerization. Al-though the general mechanism of decomposition of azidopolymers remains the same, we believe the splitting of activa-tion energy of azide decomposition into two stages in the co-polymer is due to a complex time dependent intramolecularinteraction between its constituent blocks. This aspect war-rants a more detailed investigation. Finally, we hope that theresults presented in this paper could add up to existing knowl-edge base of the GAPePoly(BAMO) literature and facilitateits realization as a high performance energetic binder.
4. Conclusions
A copolymer of GAP and Poly(BAMO) was synthesizedusing the BF3edimethyl ether complex/diol initiator system.The synthesized copolymer exhibited the characteristics ofan ETPE. Application of model-free treatment to thermal deg-radation kinetics of the copolymer showed that activation en-ergy of decomposition of azide groups is affected bycopolymerization. In homopolymers, all the azide groups de-compose in a single step. However in the copolymer, azidegroup decomposition is split into two stages of different acti-vation energies.
References
[1] Mohan YM, Mani Y, Raju KM. Synthesis of azido polymers as potential
energetic propellant binders. Des Monomers Polym 2006;9:201e36.
[2] Stacer RG, Husband DM. Molecular structure of ideal solid propellant
binder. Propell Explos Pyrotech 1991;16:167e76.
[3] Sanderson AJ, Edwards W. Synthesis of energetic thermoplastic elasto-
mers containing oligomeric urethane linkages. US Patent 6,815,522;
2004.
[4] Singh G, Felix SP, Soni P. Studies on energetic compounds, part 31: ther-
molysis and kinetics of RDX and some of its plastic bonded explosives.
Thermochim Acta 2005;426:131e9.
[5] Ger MD, Hwu WH, Huang CC. A study on the thermal decomposition of
mixtures containing an energetic binder and a nitramine. Thermochim
Acta 1993;224:127e40.
[6] Oyumi Y, Inokami K, Yamazaki K, Matsumoto K. Burning rate augmenta-
tion of BAMO based propellants. Propell Explos Pyrotech 1994;19:180e6.
[7] Bazaki H, Kubota N. Effect of binders on the burning rate of AP compos-
ite propellants. Propell Explos Pyrotech 2000;25:312e6.
[8] Korobeinichev OP, Kuibida LV, Volkov EN, Shmakov AG. Mass spectro-
metric study of combustion and thermal decomposition of GAP. Combust
Flame 2002;129:136e50.
[9] Lee YJ, Tang CJ, Kudva G, Litzinger TA. Thermal decomposition of
3,30-bis-azidomethyl-oxetane. J Propul Power 1998;14:37e44.
[10] Sahu SK, Panda SP, Sadafule DS, Kumbhar CG, Kulkarni SG, Thakur JV.
Thermal and photodegradation of glycidyl azide polymers. Polym
Degrad Stab 1998;62:495e500.
[11] Kimura E, Oyumi Y. Thermal decomposition of BAMO copolymers. Pro-
pell Explos Pyrotech 1995;20:322e6.
[12] Liu YL, Hsiue GH, Chiu YS. Thermal characteristics of energetic poly-
mers based on tetrahydrofuran and oxetane derivatives. J Appl Polym Sci
1995;58:579e86.
[13] Lee YJ, Litzinger TA. Thermal decomposition of BAMO/AMMO with
and without TiO2. Thermochim Acta 2002;384:121e35.
[14] Vyazovkin S, Dollimore D. Linear and nonlinear procedures in isocon-
versional computations of activation energy of nonisothermal reactions
in solids. J Chem Inf Comput Sci 1996;36:42e5.

1377S. Pisharath, H.G. Ang / Polymer Degradation and Stability 92 (2007) 1365e1377
[15] Vyazovkin S, Sbirrazzouli N. Isoconversional kinetic analysis of ther-
mally stimulated processes in polymers. Macromol Rapid Commun
2006;27:1515e32.
[16] Sell T, Vyazovkin S, Wight CA. Thermal decomposition kinetics of
PBAN binder and composite solid propellants. Combust Flame
1999;119:174e81.
[17] Stockel RF. Method of preparing 3,3-bis(chloromethyl)oxetane. US Pat-
ent 4,031,110; 1977.
[18] Malik AA, Manser GE, Carson RP, Archibald TG. Solvent free process
for the synthesis of energetic oxetane monomers. US Patent 5,523,424;
1996.
[19] Friedman HL. Kinetics of thermal degradation of char forming plastics
from thermogravimetry application to a phenolic resin. J Polym Sci
Part C 1965;6:183e5.
[20] Flynn JH, Wall LA. A quick direct method for the determination of ac-
tivation energy from thermogravimetric data. Polym Lett 1966;4:323e8.
[21] Kubisa P, Penczek S. Cationic activated monomer polymerization of het-
erocyclic monomers. Prog Polym Sci 1999;24:1409e37.
[22] Pisharath S, Ang HG. Thermal decomposition kinetics of a mixture of
energetic polymer and nitramine oxidizer. Thermochim Acta, in press,
doi:10.1016/j.tca.2007.03.017.
[23] Cheradame H, Andreolety JP, Rousset E. Homopolymerization of 3,3-bi-
s(azidomethyl)oxetane and its copolymerization with 3-chloromethyl-3-
(2,5,8-trioxadecyl) oxetane. Makromol Chem 1991;192:901e18.
[24] Hardenstine KE, Henderson GVS, Sperling LH, Murphy CJ, Manser GE.
Crystallization behavior of poly(3,3-bisethoxymethyl oxetane) and
poly(3,3-bis azidomethyl oxetane). J Polym Sci Part B Polym Phys
1985;23:1597e609.
[25] Hsiue GH, Liu YL, Chiu YS. Tetrahydrofuran and 3,3-bis(chloromethyl)
oxetane triblock copolymers synthesized by two-end living cationic
polymerization. J Polym Sci Part A Polym Chem 1993;31:3371e5.
[26] Zeman S. Modified EvansePolanyieSemenov relationship in the study
of chemical micromechanism governing detonation initiation of individ-
ual energetic materials. Thermochim Acta 2002;384:137e54.
[27] Vyazovkin S. Thermal analysis. Anal Chem 2002;74:2749e62.
[28] Khawam A, Flanagan DR. Basics and applications of solid-state kinetics.
A pharmaceutical perspective. J Pharm Sci 2006;95:472e98.
[29] Puduppakkam KV, Beckstead MW. Combustion modeling of glycidyl
azide polymer with detailed kinetics. Combust Sci Technol
2005;177:1661e97.
[30] Farber M, Harris SP, Srivastava RD. Mass spectrometric kinetic studies
on several azido polymers. Combust Flame 1984;55:203e11.
[31] Vyazovkin S, Wight CA. Kinetics in solids. Annu Rev Phys Chem
1997;48:125e49.
[32] Shlensky OF, Vaynsteyn EF, Matyukhin AA. Dynamic thermal decom-
position of linear polymers and its study by thermo analytical methods.
J Therm Anal 1988;34:645e55.
[33] Ling P, Wight CA. Laser photodissociation and thermal pyrolysis of en-
ergetic polymers. J Phys Chem B 1997;101:2126e31.
[34] Eroglu MS, Guven O. Thermal decomposition of poly(glycidyl azide) as
studied by high temperature FTIR and thermogravimetry. J Appl Polym
Sci 1996;61:201e6.




















