
EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and...
17
EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation Gu ¨ nther H. S. Richter a,1 , Stephanie Plehm a,1 , Annette Fasan a , Sabine Ro ¨ ssler a , Rebekka Unland b,c , Idriss M. Bennani-Baiti d , Marc Hotfilder c , Diana Lo ¨ wel a , Irene von Luettichau a , Ilona Mossbrugger e , Leticia Quintanilla-Martinez e , Heinrich Kovar d , Martin S. Staege f , Carsten Mu ¨ ller-Tidow b , and Stefan Burdach a,2 a Laboratory for Functional Genomics and Transplantation Biology, Department of Pediatrics, Technische Universita ¨ t Mu ¨ nchen and Pediatric Oncology Center, 81664 Munich, Germany; b Department of Medicine, Hematology, and Oncology, and Interdisciplinary Center for Clinical Research (IZKF), and c Department of Pediatric Hematology and Oncology, University Children’s Hospital, University of Mu ¨ nster, 48149 Mu ¨ nster, Gemany; d Children’s Cancer Research Institute, St. Anna Kinderkrebsforschung, 1090 Vienna, Austria; e Institute of Pathology, Helmholtz Center Munich, German Research Center for Environmental Health, D-85764 Neuherberg, Germany; and f Department of Pediatrics, Martin-Luther-University Halle-Wittenberg, 06097 Halle, Germany Edited by John J. Rossi, Beckman Research Institute of City of Hope, Duarte, CA, and accepted by the Editorial Board February 5, 2009 (received for review October 24, 2008) Ewing tumors (ET) are highly malignant, localized in bone or soft tissue, and are molecularly defined by ews/ets translocations. DNA microarray analysis revealed a relationship of ET to both endothe- lium and fetal neural crest. We identified expression of histone methyltransferase enhancer of Zeste, Drosophila, Homolog 2 (EZH2) to be increased in ET. Suppressive activity of EZH2 maintains stemness in normal and malignant cells. Here, we found EWS/FLI1 bound to the EZH2 promoter in vivo, and induced EZH2 expression in ET and mesenchymal stem cells. Down-regulation of EZH2 by RNA interference in ET suppressed oncogenic transformation by inhibiting clonogenicity in vitro. Similarly, tumor development and metastasis was suppressed in immunodeficient Rag2 / C / mice. EZH2-mediated gene silencing was shown to be dependent on histone deacetylase (HDAC) activity. Subsequent microarray analysis of EZH2 knock down, HDAC-inhibitor treatment and con- firmation in independent assays revealed an undifferentiated phenotype maintained by EZH2 in ET. EZH2 regulated stemness genes such as nerve growth factor receptor (NGFR), as well as genes involved in neuroectodermal and endothelial differentiation (EMP1, EPHB2, GFAP, and GAP43). These data suggest that EZH2 might have a central role in ET pathology by shaping the oncoge- nicity and stem cell phenotype of this tumor. epigenetic regulation Ewing tumor stemness E wing tumors (ET) are highly malignant tumors with an approximate incidence of 3.3/10 6 in children under the age of 15. ET are characterized by early metastases, and metastatic spread is commonly hematogeneous. ET were originally de- scribed by Ewing in 1921 as endothelioma of the bone (1), and we confirmed this endothelial signature by microarray analysis (2). ET are molecularly defined by ews/ets translocations. Trans- location-derived chimeric transcription factors yield transactiva- tion, transformation, and the highly malignant phenotype. In mice, EWS/FLI1 transforms bone marrow derived or mesenchy- mal progenitor cells, and generates tumors (3, 4), which have features of ET. Also, inhibition of EWS/FLI1 expression may allow ET cells to recover the phenotype of their presumed mesenchymal stem cell (MSC) progenitor (5). Multipotent MSCs represent a leading candidate for primary transformation in ET. We revealed a relationship of ET to both endothelial and fetal neural crest-derived cells (2), after having demonstrated neuroectodermal histogenesis of ET in 1985 (6). Based on our recent study, we postulated in 2004 that the ET stem cell is arrested at early mesenchyme development from the neuroec- todermal germ layer, and, thus, the ET stem cell is a neuronal crest-derived stem cell at transition to mesenchymal endothelial development, residing in the bone marrow. However, ectopic EWS/FLI1 expression resulted in a neural phenotype, raising the possibility that transdifferentiation or lineage promiscuity may be an alternative to the MSC histogenetic origin hypothesis of ET (7). We used high density DNA microarrays for the identification of ET specific gene expression profiles compared with 133 normal tissues of diverse origin (normal body atlas, NBA), and identified 37 genes that were highly up-regulated or specifically expressed in ET (2). The histone methyltransferase enhancer of Zeste, Drosophila, Homolog 2 (EZH2), one of these up- regulated genes, is part of the polycomb repressor complex 2 (PRC2), together with embryonic ectoderm development (EED) protein and suppressor of Zeste (SUZ12). Within this complex, EZH2 exhibits methyltransferase activity, and silences target genes by methylating lysine 27 on histone 3 (H3K27). EZH2 is already active at gastrulation, and maintains a stemness expression signature (8). In tumors, it may recruit DNA- methyltransferase to promoter regions, resulting in specific gene switch-off (9). Varambally et al. (10) demonstrated that EZH2 mediated gene silencing could be reverted by histone deacetylase inhibitors (HDACis), because PRC2 complexes interact through EED with HDAC2 to mediate their suppressive activity (11). Here, we analyzed the pathogenetic role of EZH2 in ET in vitro and in vivo. We identified EZH2 to be induced by EWS/FLI1 presumably due to EWS/FLI1 binding to the EZH2 promoter in vivo. Inhibition of EZH2 expression by RNA interference suppressed contact independent growth in vitro, and significantly delayed tumor development and metastasis in mice. Expression profiling and differentiation assays of ET after EZH2 RNA interference and HDAC-inhibitor treatment with Trichostatin A revealed that EZH2 maintains a stemness signature in ET by inhibiting endothelial and neuroectodermal differentiation. Results Histone Methyltransferase EZH2 Is Strongly Up-Regulated in ETs. In a previous microarray analysis, we identified the histone methyl- Author contributions: G.H.S.R., M.H., H.K., and S.B. designed research; G.H.S.R., S.P., A.F., S.R., R.U., I.M.B.-B., D.L., I.M., and M.S.S. performed research; I.v.L. contributed new reagents/analytic tools; S.P., I.M.B.-B., M.H., I.v.L., I.M., L.Q.-M., H.K., M.S.S., and C.M.-T. analyzed data; and G.H.S.R., I.M.B.-B., C.M.-T., and S.B. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. J.J.R. is a guest editor invited by the Editorial Board. Freely available online through the PNAS open access option. Data deposition: The sequence reported in this paper has been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE12692). 1 G.H.S.R. and S.P. contributed equally to this work. 2 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/cgi/content/full/ 0810759106/DCSupplemental. 5324 –5329 PNAS March 31, 2009 vol. 106 no. 13 www.pnas.orgcgidoi10.1073pnas.0810759106
-
Upload
uni-tuebingen1 -
Category
Documents
-
view
3 -
download
0
Transcript of EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and...

EZH2 is a mediator of EWS/FLI1 driven tumorgrowth and metastasis blocking endothelialand neuro-ectodermal differentiationGunther H. S. Richtera,1, Stephanie Plehma,1, Annette Fasana, Sabine Rosslera, Rebekka Unlandb,c,Idriss M. Bennani-Baitid, Marc Hotfilderc, Diana Lowela, Irene von Luettichaua, Ilona Mossbruggere,Leticia Quintanilla-Martineze, Heinrich Kovard, Martin S. Staegef, Carsten Muller-Tidowb, and Stefan Burdacha,2
aLaboratory for Functional Genomics and Transplantation Biology, Department of Pediatrics, Technische Universitat Munchen and Pediatric OncologyCenter, 81664 Munich, Germany; bDepartment of Medicine, Hematology, and Oncology, and Interdisciplinary Center for Clinical Research (IZKF), andcDepartment of Pediatric Hematology and Oncology, University Children’s Hospital, University of Munster, 48149 Munster, Gemany; dChildren’s CancerResearch Institute, St. Anna Kinderkrebsforschung, 1090 Vienna, Austria; eInstitute of Pathology, Helmholtz Center Munich, German Research Center forEnvironmental Health, D-85764 Neuherberg, Germany; and fDepartment of Pediatrics, Martin-Luther-University Halle-Wittenberg, 06097 Halle, Germany
Edited by John J. Rossi, Beckman Research Institute of City of Hope, Duarte, CA, and accepted by the Editorial Board February 5, 2009 (received for reviewOctober 24, 2008)
Ewing tumors (ET) are highly malignant, localized in bone or softtissue, and are molecularly defined by ews/ets translocations. DNAmicroarray analysis revealed a relationship of ET to both endothe-lium and fetal neural crest. We identified expression of histonemethyltransferase enhancer of Zeste, Drosophila, Homolog 2(EZH2) to be increased in ET. Suppressive activity of EZH2 maintainsstemness in normal and malignant cells. Here, we found EWS/FLI1bound to the EZH2 promoter in vivo, and induced EZH2 expressionin ET and mesenchymal stem cells. Down-regulation of EZH2 byRNA interference in ET suppressed oncogenic transformation byinhibiting clonogenicity in vitro. Similarly, tumor development andmetastasis was suppressed in immunodeficient Rag2�/��C
�/�
mice. EZH2-mediated gene silencing was shown to be dependenton histone deacetylase (HDAC) activity. Subsequent microarrayanalysis of EZH2 knock down, HDAC-inhibitor treatment and con-firmation in independent assays revealed an undifferentiatedphenotype maintained by EZH2 in ET. EZH2 regulated stemnessgenes such as nerve growth factor receptor (NGFR), as well asgenes involved in neuroectodermal and endothelial differentiation(EMP1, EPHB2, GFAP, and GAP43). These data suggest that EZH2might have a central role in ET pathology by shaping the oncoge-nicity and stem cell phenotype of this tumor.
epigenetic regulation � Ewing tumor � stemness
Ewing tumors (ET) are highly malignant tumors with anapproximate incidence of 3.3/106 in children under the age of
15. ET are characterized by early metastases, and metastaticspread is commonly hematogeneous. ET were originally de-scribed by Ewing in 1921 as endothelioma of the bone (1), andwe confirmed this endothelial signature by microarray analysis(2). ET are molecularly defined by ews/ets translocations. Trans-location-derived chimeric transcription factors yield transactiva-tion, transformation, and the highly malignant phenotype. Inmice, EWS/FLI1 transforms bone marrow derived or mesenchy-mal progenitor cells, and generates tumors (3, 4), which havefeatures of ET. Also, inhibition of EWS/FLI1 expression mayallow ET cells to recover the phenotype of their presumedmesenchymal stem cell (MSC) progenitor (5). MultipotentMSCs represent a leading candidate for primary transformationin ET. We revealed a relationship of ET to both endothelial andfetal neural crest-derived cells (2), after having demonstratedneuroectodermal histogenesis of ET in 1985 (6). Based on ourrecent study, we postulated in 2004 that the ET stem cell isarrested at early mesenchyme development from the neuroec-todermal germ layer, and, thus, the ET stem cell is a neuronalcrest-derived stem cell at transition to mesenchymal endothelialdevelopment, residing in the bone marrow. However, ectopic
EWS/FLI1 expression resulted in a neural phenotype, raising thepossibility that transdifferentiation or lineage promiscuity may bean alternative to the MSC histogenetic origin hypothesis of ET (7).
We used high density DNA microarrays for the identificationof ET specific gene expression profiles compared with 133normal tissues of diverse origin (normal body atlas, NBA), andidentified 37 genes that were highly up-regulated or specificallyexpressed in ET (2). The histone methyltransferase enhancer ofZeste, Drosophila, Homolog 2 (EZH2), one of these up-regulated genes, is part of the polycomb repressor complex 2(PRC2), together with embryonic ectoderm development(EED) protein and suppressor of Zeste (SUZ12). Within thiscomplex, EZH2 exhibits methyltransferase activity, and silencestarget genes by methylating lysine 27 on histone 3 (H3K27).EZH2 is already active at gastrulation, and maintains a stemnessexpression signature (8). In tumors, it may recruit DNA-methyltransferase to promoter regions, resulting in specific geneswitch-off (9). Varambally et al. (10) demonstrated that EZH2mediated gene silencing could be reverted by histone deacetylaseinhibitors (HDACis), because PRC2 complexes interact throughEED with HDAC2 to mediate their suppressive activity (11).
Here, we analyzed the pathogenetic role of EZH2 in ET invitro and in vivo. We identified EZH2 to be induced byEWS/FLI1 presumably due to EWS/FLI1 binding to the EZH2promoter in vivo. Inhibition of EZH2 expression by RNAinterference suppressed contact independent growth in vitro,and significantly delayed tumor development and metastasis inmice. Expression profiling and differentiation assays of ETafter EZH2 RNA interference and HDAC-inhibitor treatmentwith Trichostatin A revealed that EZH2 maintains a stemnesssignature in ET by inhibiting endothelial and neuroectodermaldifferentiation.
ResultsHistone Methyltransferase EZH2 Is Strongly Up-Regulated in ETs. In aprevious microarray analysis, we identified the histone methyl-
Author contributions: G.H.S.R., M.H., H.K., and S.B. designed research; G.H.S.R., S.P., A.F.,S.R., R.U., I.M.B.-B., D.L., I.M., and M.S.S. performed research; I.v.L. contributed newreagents/analytic tools; S.P., I.M.B.-B., M.H., I.v.L., I.M., L.Q.-M., H.K., M.S.S., and C.M.-T.analyzed data; and G.H.S.R., I.M.B.-B., C.M.-T., and S.B. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.J.R. is a guest editor invited by the Editorial Board.
Freely available online through the PNAS open access option.
Data deposition: The sequence reported in this paper has been deposited in the GeneExpression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE12692).
1G.H.S.R. and S.P. contributed equally to this work.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0810759106/DCSupplemental.
5324–5329 � PNAS � March 31, 2009 � vol. 106 � no. 13 www.pnas.org�cgi�doi�10.1073�pnas.0810759106
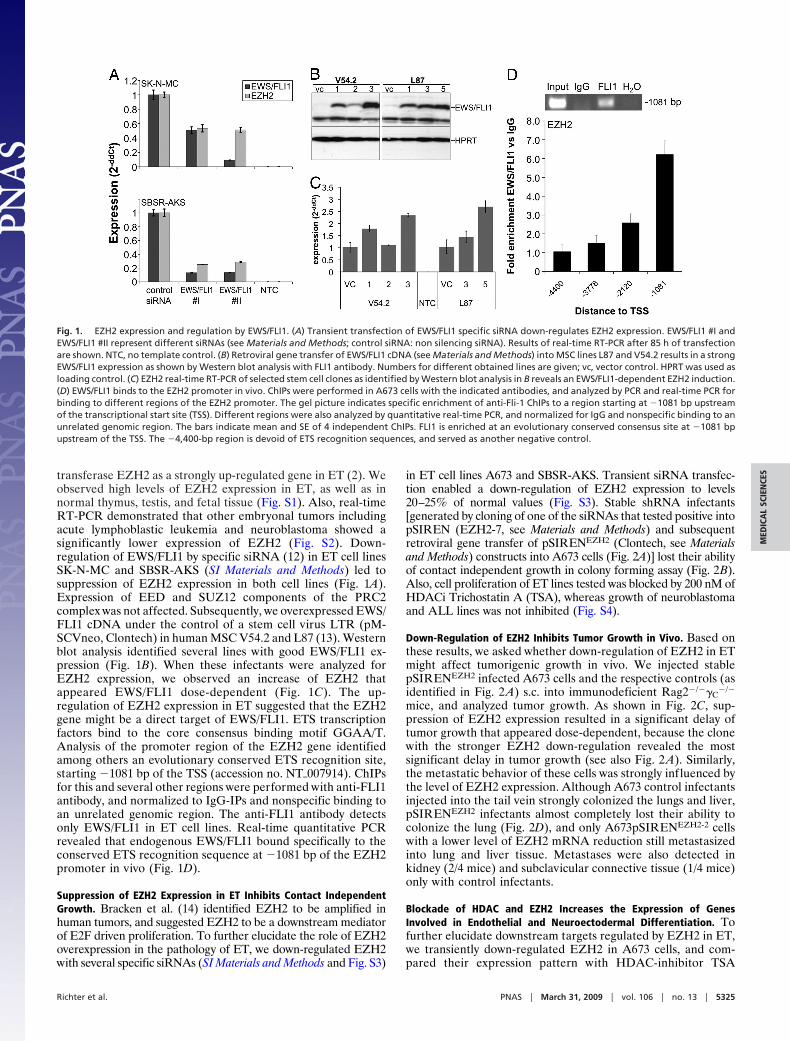
transferase EZH2 as a strongly up-regulated gene in ET (2). Weobserved high levels of EZH2 expression in ET, as well as innormal thymus, testis, and fetal tissue (Fig. S1). Also, real-timeRT-PCR demonstrated that other embryonal tumors includingacute lymphoblastic leukemia and neuroblastoma showed asignificantly lower expression of EZH2 (Fig. S2). Down-regulation of EWS/FLI1 by specific siRNA (12) in ET cell linesSK-N-MC and SBSR-AKS (SI Materials and Methods) led tosuppression of EZH2 expression in both cell lines (Fig. 1A).Expression of EED and SUZ12 components of the PRC2complex was not affected. Subsequently, we overexpressed EWS/FLI1 cDNA under the control of a stem cell virus LTR (pM-SCVneo, Clontech) in human MSC V54.2 and L87 (13). Westernblot analysis identified several lines with good EWS/FLI1 ex-pression (Fig. 1B). When these infectants were analyzed forEZH2 expression, we observed an increase of EZH2 thatappeared EWS/FLI1 dose-dependent (Fig. 1C). The up-regulation of EZH2 expression in ET suggested that the EZH2gene might be a direct target of EWS/FLI1. ETS transcriptionfactors bind to the core consensus binding motif GGAA/T.Analysis of the promoter region of the EZH2 gene identifiedamong others an evolutionary conserved ETS recognition site,starting �1081 bp of the TSS (accession no. NT�007914). ChIPsfor this and several other regions were performed with anti-FLI1antibody, and normalized to IgG-IPs and nonspecific binding toan unrelated genomic region. The anti-FLI1 antibody detectsonly EWS/FLI1 in ET cell lines. Real-time quantitative PCRrevealed that endogenous EWS/FLI1 bound specifically to theconserved ETS recognition sequence at �1081 bp of the EZH2promoter in vivo (Fig. 1D).
Suppression of EZH2 Expression in ET Inhibits Contact IndependentGrowth. Bracken et al. (14) identified EZH2 to be amplified inhuman tumors, and suggested EZH2 to be a downstream mediatorof E2F driven proliferation. To further elucidate the role of EZH2overexpression in the pathology of ET, we down-regulated EZH2with several specific siRNAs (SI Materials and Methods and Fig. S3)
in ET cell lines A673 and SBSR-AKS. Transient siRNA transfec-tion enabled a down-regulation of EZH2 expression to levels20–25% of normal values (Fig. S3). Stable shRNA infectants[generated by cloning of one of the siRNAs that tested positive intopSIREN (EZH2-7, see Materials and Methods) and subsequentretroviral gene transfer of pSIRENEZH2 (Clontech, see Materialsand Methods) constructs into A673 cells (Fig. 2A)] lost their abilityof contact independent growth in colony forming assay (Fig. 2B).Also, cell proliferation of ET lines tested was blocked by 200 nM ofHDACi Trichostatin A (TSA), whereas growth of neuroblastomaand ALL lines was not inhibited (Fig. S4).
Down-Regulation of EZH2 Inhibits Tumor Growth in Vivo. Based onthese results, we asked whether down-regulation of EZH2 in ETmight affect tumorigenic growth in vivo. We injected stablepSIRENEZH2 infected A673 cells and the respective controls (asidentified in Fig. 2 A) s.c. into immunodeficient Rag2�/��C
�/�
mice, and analyzed tumor growth. As shown in Fig. 2C, sup-pression of EZH2 expression resulted in a significant delay oftumor growth that appeared dose-dependent, because the clonewith the stronger EZH2 down-regulation revealed the mostsignificant delay in tumor growth (see also Fig. 2 A). Similarly,the metastatic behavior of these cells was strongly influenced bythe level of EZH2 expression. Although A673 control infectantsinjected into the tail vein strongly colonized the lungs and liver,pSIRENEZH2 infectants almost completely lost their ability tocolonize the lung (Fig. 2D), and only A673pSIRENEZH2-2 cellswith a lower level of EZH2 mRNA reduction still metastasizedinto lung and liver tissue. Metastases were also detected inkidney (2/4 mice) and subclavicular connective tissue (1/4 mice)only with control infectants.
Blockade of HDAC and EZH2 Increases the Expression of GenesInvolved in Endothelial and Neuroectodermal Differentiation. Tofurther elucidate downstream targets regulated by EZH2 in ET,we transiently down-regulated EZH2 in A673 cells, and com-pared their expression pattern with HDAC-inhibitor TSA
Fig. 1. EZH2 expression and regulation by EWS/FLI1. (A) Transient transfection of EWS/FLI1 specific siRNA down-regulates EZH2 expression. EWS/FLI1 #I andEWS/FLI1 #II represent different siRNAs (see Materials and Methods; control siRNA: non silencing siRNA). Results of real-time RT-PCR after 85 h of transfectionare shown. NTC, no template control. (B) Retroviral gene transfer of EWS/FLI1 cDNA (see Materials and Methods) into MSC lines L87 and V54.2 results in a strongEWS/FLI1 expression as shown by Western blot analysis with FLI1 antibody. Numbers for different obtained lines are given; vc, vector control. HPRT was used asloading control. (C) EZH2 real-time RT-PCR of selected stem cell clones as identified by Western blot analysis in B reveals an EWS/FLI1-dependent EZH2 induction.(D) EWS/FLI1 binds to the EZH2 promoter in vivo. ChIPs were performed in A673 cells with the indicated antibodies, and analyzed by PCR and real-time PCR forbinding to different regions of the EZH2 promoter. The gel picture indicates specific enrichment of anti-Fli-1 ChIPs to a region starting at �1081 bp upstreamof the transcriptional start site (TSS). Different regions were also analyzed by quantitative real-time PCR, and normalized for IgG and nonspecific binding to anunrelated genomic region. The bars indicate mean and SE of 4 independent ChIPs. FLI1 is enriched at an evolutionary conserved consensus site at �1081 bpupstream of the TSS. The �4,400-bp region is devoid of ETS recognition sequences, and served as another negative control.
Richter et al. PNAS � March 31, 2009 � vol. 106 � no. 13 � 5325
MED
ICA
LSC
IEN
CES
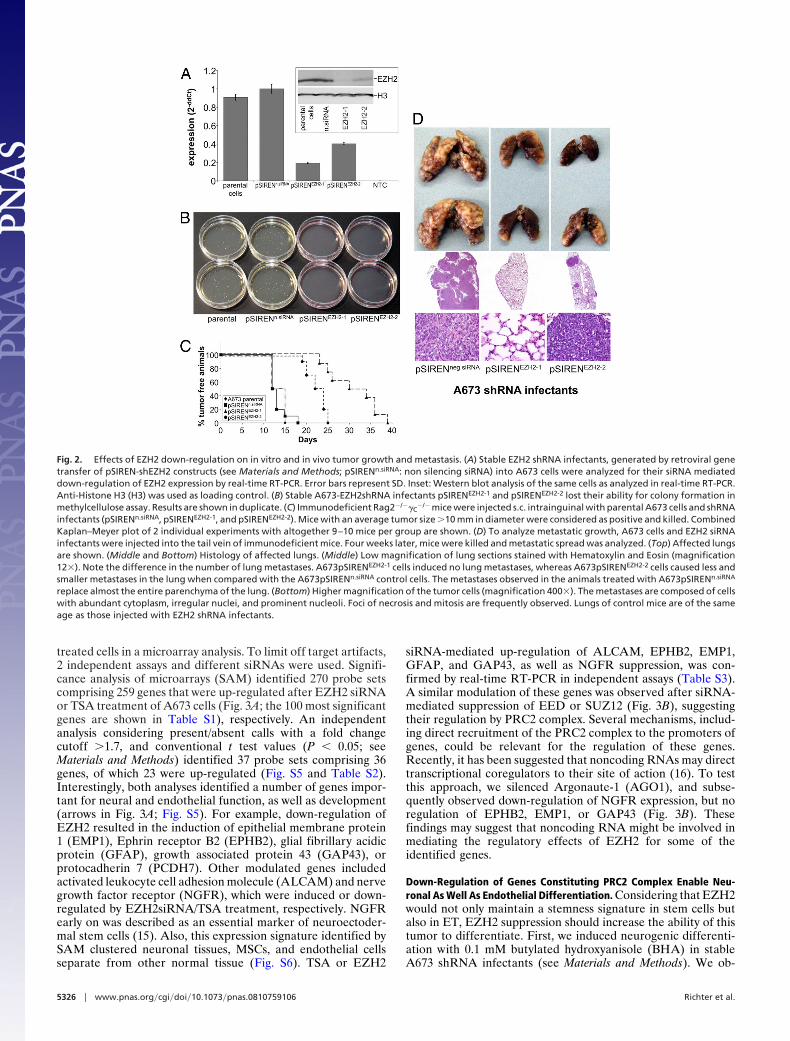
treated cells in a microarray analysis. To limit off target artifacts,2 independent assays and different siRNAs were used. Signifi-cance analysis of microarrays (SAM) identified 270 probe setscomprising 259 genes that were up-regulated after EZH2 siRNAor TSA treatment of A673 cells (Fig. 3A; the 100 most significantgenes are shown in Table S1), respectively. An independentanalysis considering present/absent calls with a fold changecutoff �1.7, and conventional t test values (P � 0.05; seeMaterials and Methods) identified 37 probe sets comprising 36genes, of which 23 were up-regulated (Fig. S5 and Table S2).Interestingly, both analyses identified a number of genes impor-tant for neural and endothelial function, as well as development(arrows in Fig. 3A; Fig. S5). For example, down-regulation ofEZH2 resulted in the induction of epithelial membrane protein1 (EMP1), Ephrin receptor B2 (EPHB2), glial fibrillary acidicprotein (GFAP), growth associated protein 43 (GAP43), orprotocadherin 7 (PCDH7). Other modulated genes includedactivated leukocyte cell adhesion molecule (ALCAM) and nervegrowth factor receptor (NGFR), which were induced or down-regulated by EZH2siRNA/TSA treatment, respectively. NGFRearly on was described as an essential marker of neuroectoder-mal stem cells (15). Also, this expression signature identified bySAM clustered neuronal tissues, MSCs, and endothelial cellsseparate from other normal tissue (Fig. S6). TSA or EZH2
siRNA-mediated up-regulation of ALCAM, EPHB2, EMP1,GFAP, and GAP43, as well as NGFR suppression, was con-firmed by real-time RT-PCR in independent assays (Table S3).A similar modulation of these genes was observed after siRNA-mediated suppression of EED or SUZ12 (Fig. 3B), suggestingtheir regulation by PRC2 complex. Several mechanisms, includ-ing direct recruitment of the PRC2 complex to the promoters ofgenes, could be relevant for the regulation of these genes.Recently, it has been suggested that noncoding RNAs may directtranscriptional coregulators to their site of action (16). To testthis approach, we silenced Argonaute-1 (AGO1), and subse-quently observed down-regulation of NGFR expression, but noregulation of EPHB2, EMP1, or GAP43 (Fig. 3B). Thesefindings may suggest that noncoding RNA might be involved inmediating the regulatory effects of EZH2 for some of theidentified genes.
Down-Regulation of Genes Constituting PRC2 Complex Enable Neu-ronal As Well As Endothelial Differentiation. Considering that EZH2would not only maintain a stemness signature in stem cells butalso in ET, EZH2 suppression should increase the ability of thistumor to differentiate. First, we induced neurogenic differenti-ation with 0.1 mM butylated hydroxyanisole (BHA) in stableA673 shRNA infectants (see Materials and Methods). We ob-
Fig. 2. Effects of EZH2 down-regulation on in vitro and in vivo tumor growth and metastasis. (A) Stable EZH2 shRNA infectants, generated by retroviral genetransfer of pSIREN-shEZH2 constructs (see Materials and Methods; pSIRENn.siRNA: non silencing siRNA) into A673 cells were analyzed for their siRNA mediateddown-regulation of EZH2 expression by real-time RT-PCR. Error bars represent SD. Inset: Western blot analysis of the same cells as analyzed in real-time RT-PCR.Anti-Histone H3 (H3) was used as loading control. (B) Stable A673-EZH2shRNA infectants pSIRENEZH2-1 and pSIRENEZH2-2 lost their ability for colony formation inmethylcellulose assay. Results are shown in duplicate. (C) Immunodeficient Rag2�/��C
�/� mice were injected s.c. intrainguinal with parental A673 cells and shRNAinfectants (pSIRENn.siRNA, pSIRENEZH2-1, and pSIRENEZH2-2). Mice with an average tumor size �10 mm in diameter were considered as positive and killed. CombinedKaplan–Meyer plot of 2 individual experiments with altogether 9–10 mice per group are shown. (D) To analyze metastatic growth, A673 cells and EZH2 siRNAinfectants were injected into the tail vein of immunodeficient mice. Four weeks later, mice were killed and metastatic spread was analyzed. (Top) Affected lungsare shown. (Middle and Bottom) Histology of affected lungs. (Middle) Low magnification of lung sections stained with Hematoxylin and Eosin (magnification12�). Note the difference in the number of lung metastases. A673pSIRENEZH2-1 cells induced no lung metastases, whereas A673pSIRENEZH2-2 cells caused less andsmaller metastases in the lung when compared with the A673pSIRENn.siRNA control cells. The metastases observed in the animals treated with A673pSIRENn.siRNA
replace almost the entire parenchyma of the lung. (Bottom) Higher magnification of the tumor cells (magnification 400�). The metastases are composed of cellswith abundant cytoplasm, irregular nuclei, and prominent nucleoli. Foci of necrosis and mitosis are frequently observed. Lungs of control mice are of the sameage as those injected with EZH2 shRNA infectants.
5326 � www.pnas.org�cgi�doi�10.1073�pnas.0810759106 Richter et al.
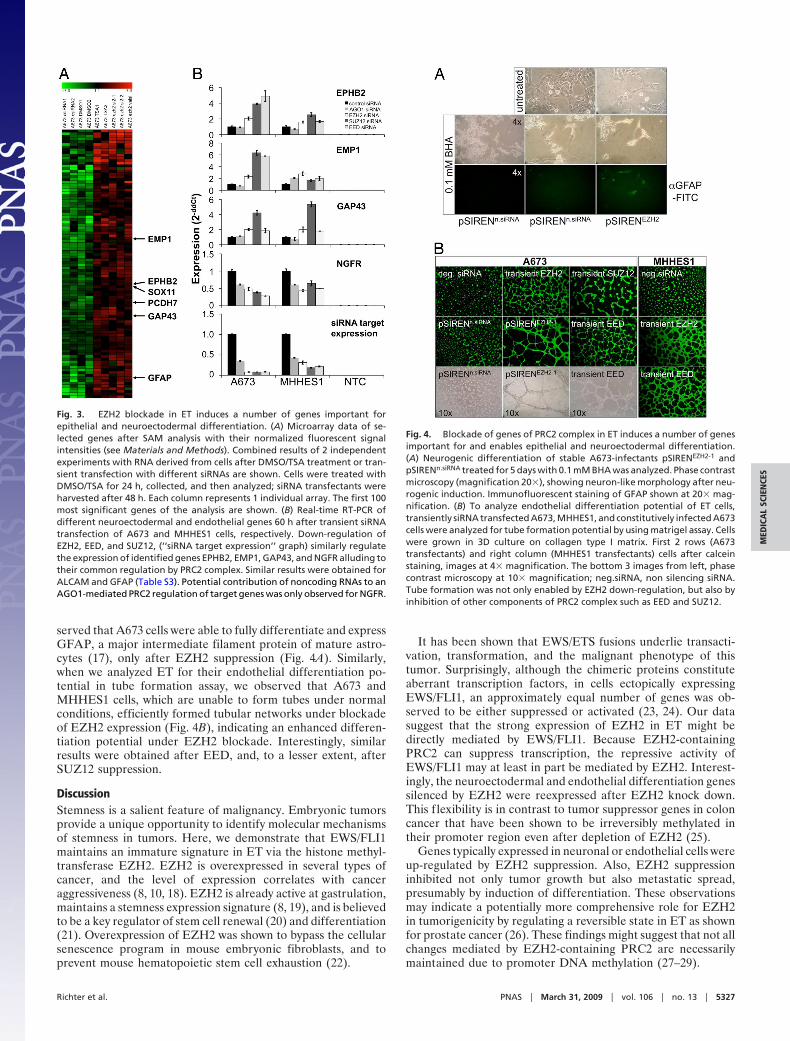
served that A673 cells were able to fully differentiate and expressGFAP, a major intermediate filament protein of mature astro-cytes (17), only after EZH2 suppression (Fig. 4A). Similarly,when we analyzed ET for their endothelial differentiation po-tential in tube formation assay, we observed that A673 andMHHES1 cells, which are unable to form tubes under normalconditions, efficiently formed tubular networks under blockadeof EZH2 expression (Fig. 4B), indicating an enhanced differen-tiation potential under EZH2 blockade. Interestingly, similarresults were obtained after EED, and, to a lesser extent, afterSUZ12 suppression.
DiscussionStemness is a salient feature of malignancy. Embryonic tumorsprovide a unique opportunity to identify molecular mechanismsof stemness in tumors. Here, we demonstrate that EWS/FLI1maintains an immature signature in ET via the histone methyl-transferase EZH2. EZH2 is overexpressed in several types ofcancer, and the level of expression correlates with canceraggressiveness (8, 10, 18). EZH2 is already active at gastrulation,maintains a stemness expression signature (8, 19), and is believedto be a key regulator of stem cell renewal (20) and differentiation(21). Overexpression of EZH2 was shown to bypass the cellularsenescence program in mouse embryonic fibroblasts, and toprevent mouse hematopoietic stem cell exhaustion (22).
It has been shown that EWS/ETS fusions underlie transacti-vation, transformation, and the malignant phenotype of thistumor. Surprisingly, although the chimeric proteins constituteaberrant transcription factors, in cells ectopically expressingEWS/FLI1, an approximately equal number of genes was ob-served to be either suppressed or activated (23, 24). Our datasuggest that the strong expression of EZH2 in ET might bedirectly mediated by EWS/FLI1. Because EZH2-containingPRC2 can suppress transcription, the repressive activity ofEWS/FLI1 may at least in part be mediated by EZH2. Interest-ingly, the neuroectodermal and endothelial differentiation genessilenced by EZH2 were reexpressed after EZH2 knock down.This flexibility is in contrast to tumor suppressor genes in coloncancer that have been shown to be irreversibly methylated intheir promoter region even after depletion of EZH2 (25).
Genes typically expressed in neuronal or endothelial cells wereup-regulated by EZH2 suppression. Also, EZH2 suppressioninhibited not only tumor growth but also metastatic spread,presumably by induction of differentiation. These observationsmay indicate a potentially more comprehensive role for EZH2in tumorigenicity by regulating a reversible state in ET as shownfor prostate cancer (26). These findings might suggest that not allchanges mediated by EZH2-containing PRC2 are necessarilymaintained due to promoter DNA methylation (27–29).
Fig. 3. EZH2 blockade in ET induces a number of genes important forepithelial and neuroectodermal differentiation. (A) Microarray data of se-lected genes after SAM analysis with their normalized fluorescent signalintensities (see Materials and Methods). Combined results of 2 independentexperiments with RNA derived from cells after DMSO/TSA treatment or tran-sient transfection with different siRNAs are shown. Cells were treated withDMSO/TSA for 24 h, collected, and then analyzed; siRNA transfectants wereharvested after 48 h. Each column represents 1 individual array. The first 100most significant genes of the analysis are shown. (B) Real-time RT-PCR ofdifferent neuroectodermal and endothelial genes 60 h after transient siRNAtransfection of A673 and MHHES1 cells, respectively. Down-regulation ofEZH2, EED, and SUZ12, (‘‘siRNA target expression’’ graph) similarly regulatethe expression of identified genes EPHB2, EMP1, GAP43, and NGFR alluding totheir common regulation by PRC2 complex. Similar results were obtained forALCAM and GFAP (Table S3). Potential contribution of noncoding RNAs to anAGO1-mediated PRC2 regulation of target genes was only observed for NGFR.
Fig. 4. Blockade of genes of PRC2 complex in ET induces a number of genesimportant for and enables epithelial and neuroectodermal differentiation.(A) Neurogenic differentiation of stable A673-infectants pSIRENEZH2-1 andpSIRENn.siRNA treated for 5 days with 0.1 mM BHA was analyzed. Phase contrastmicroscopy (magnification 20�), showing neuron-like morphology after neu-rogenic induction. Immunofluorescent staining of GFAP shown at 20� mag-nification. (B) To analyze endothelial differentiation potential of ET cells,transiently siRNA transfected A673, MHHES1, and constitutively infected A673cells were analyzed for tube formation potential by using matrigel assay. Cellswere grown in 3D culture on collagen type I matrix. First 2 rows (A673transfectants) and right column (MHHES1 transfectants) cells after calceinstaining, images at 4� magnification. The bottom 3 images from left, phasecontrast microscopy at 10� magnification; neg.siRNA, non silencing siRNA.Tube formation was not only enabled by EZH2 down-regulation, but also byinhibition of other components of PRC2 complex such as EED and SUZ12.
Richter et al. PNAS � March 31, 2009 � vol. 106 � no. 13 � 5327
MED
ICA
LSC
IEN
CES

Gene regulation by EZH2 might be direct or indirect. Tofurther elucidate functional involvement of PRC2 complex,suppression of other member components including EED andSUZ12 (20) correspondingly induced genes of neuroectodermaland endothelial signature in ET, and increased their differenti-ation potential. Also, HDAC inhibition by TSA up-regulated asimilar set of genes, possibly delineating EZH2 ability to interactwith HDAC2 via the EED protein (11). Emerging data suggestthat noncoding RNA might be guiding chromatin modifyingcomplexes to epigenetically silenced target genes (16, 30). To testthe relevance of this approach, we suppressed AGO1 that isnecessary for this process (31). We observed suppression ofNGFR expression by �50%, but no change in 3 other analyzedgenes. These findings suggest that for the majority of PRC2suppressed genes in our dataset noncoding RNA direction isunlikely to be the main mechanism.
During the preparation of our manuscript, we noticed 2 recentpublications that investigated the overexpression of EWS/FLI1in human MSCs (32, 33). Both observed that this overexpressiondid not transform MSC and lead to tumorigenicity. The publi-cation by Riggi et al. (33) is in line with our finding thatEWS/FLI1 overexpression induces EZH2 expression, and thatEZH2 may have a role in tumorigenicity of ET. Our findingsindeed go far beyond by demonstrating the role of EZH2 inEWS/FLI1-induced metastasis, and the analysis of the EZH2regulated gene set in ET.
The neuronal expression pattern in the EZH2 siRNA and theHDAC-inhibitor-treated ET cells is in agreement with thepathogenetic hypothesis that EZH2 blocks the (probably EWS/FLI1-induced) neuronal differentiation program of ET. TheMSC/endothelial expression pattern might be indicative ofEZH2-mediated suppression of markers typically expressed inthe ET stem cell. Whether this cell is derived from primordialstreak derived early mesoblasts (e.g., with endothelial differen-tiation capacity, as suggested by James Ewings initial observa-tion; see ref. 1), or whether EWS/FLI1 can induce transdiffer-entiation and lineage promiscuity in more differentiated cellsrequires further investigation. EWS/FLI1 driven overexpressionof EZH2 in ET reveals upstream mechanisms of EZH2 dereg-ulation, which to our knowledge have not been previouslydescribed in malignancies of adulthood.
Overall, our observations indicate that EZH2 expression isrequired in ETs to maintain stemness and metastatic spread.These findings suggest that EZH2 and EZH2-mediated eventsare promising novel targets for ET therapy.
Materials and MethodsMice. Immunodeficient Rag2�/��C
�/� mice on a BALB/c background wereobtained from the Central Institute for Experimental Animals (Kawasaki,Japan), and maintained in our animal facility under pathogen-free conditionsin accordance with the institutional guidelines and approval by local author-ities. Experiments were performed in 6–16 week-old mice.
Cell Lines. ET lines (MHHES1, SK-ES1, SK-N-MC, and TC71), neuroblastoma lines(CHP126, MHHNB11, SH-SY5Y, and SIMA), and pediatric human B cell precur-sor leukemic lines (NALM6, 697, and cALL2) were obtained from the GermanCollection of Microorganisms and Cell Cultures (DSMZ). A673 was from ATCC(LGC Standards). SBSR-AKS is a cell line with a type 1 EWS/FLI1 translocationthat was established in our lab from an extraosseous inguinal metastasis of a17-year old female Ewing patient enrolled in our clinical protocol. MSCs L87and V54.2 were immortalized with SV40 large T-antigen (13). Retroviruspackaging cell line PT67 was from Takara Bio Europe/Clontech.
RNA Interference. For transient RNA interference, cells were transfected withsiRNA. Briefly, 3 � 106 cells were resuspended in a final volume of 12 mLmedium/80 mm Petri dish containing 5 nM siRNA and 36 �L transfectionmedium (HiPerFect, Qiagen), and incubated at 37 °C. To test transfectionefficiency and gene knock-down, RNA was extracted, and gene expressionassessed by real-time RT-PCR. Unspecific induction of an IFN response wasexcluded by monitoring induction of IFN responsive genes G1P2 and IFITM1.
Small interfering RNAs for EWS/FLI1 were synthesized at MWG Biotech, cor-responding to ref. 12, all other siRNAs were purchased from Qiagen. Smallinterfering RNA sequences are provided in SI Materials and Methods.
Constructs and Retroviral Gene Transfer. The cDNA containing the EWS/FLI1-coding region was described previously (2). A BglII fragment was subclonedinto the retrovial vector pMSCVneo (Takara Bio Europe/Clontech). For stablesilencing of EWS/FLI1 or EZH2 expression, oligonucleotides of the short hairpincorresponding to the published EZH2 siRNAs (10), and EWS/FLI1 correspond-ing to the published EWS/FLI1 siRNA, site II (12), were cloned into retroviralvector pSIREN-RetroQ vector (Takara Bio Europe/Clontech) (for detailed se-quences, see SI Materials and Methods). Retroviral constructs were trans-fected by electroporation into PT67 packaging cells, and viral supernatantisolated 48 h after transfection. Infection of target cells was carried out in thepresence of 4 �g/mL polybrene. Stable infectants were isolated after selectionin 600 �g/mL G418 (pMSCVneo) or 2.5 �g/mL puromycin (pSIREN-RetroQ),respectively.
RNA Microarray Analysis. Biotinylated target cRNA was prepared as previouslydescribed (2, 34). A detailed protocol is available at www.affymetrix.com.Samples were hybridized to Affymetrix HG U133A microarrays and analyzedby using Affymetrix software Microarray Suite 5.0 (Affymetrix), and scaled tothe same target intensity of 500. Subsequent analysis was carried out withsignal intensities that were log2 transformed for equal representation of overand under expressed genes, and then median centered to remove biases basedon single expression values. Hierarchical clustering (35) was accomplished byuse of the Genesis software package (36). For the identification of differen-tially expressed genes, we used SAM (37).
Real-Time RT-PCR. Differential gene expression of cDNA or siRNA transfectantswas verified by real-time RT-PCR. Quantitative real-time PCR was performedby use of TaqMan Universal PCR Master Mix (Applied Biosystems), and fluo-rescence detection with an AB 7300 Real-Time PCR System (Applied Biosys-tems). Gene specific primers and probes were obtained as TaqMan GeneExpression Assay sets from Applied Biosystems, which consisted of a FAMdye-labeled TaqMan MGB probe and 2 unlabeled PCR primers; 20� stocksolutions of these reagents were added to the TaqMan Universal PCR MasterMix with cDNA at a final volume of 25 �L. The final concentration of primersand probe was 900 and 250 nM, respectively. A list of used assays is providedin SI Materials and Methods.
Western Blot Analysis. Protein samples were resolved by SDS/PAGE electro-phoresis on 10 or 12% gels, and transferred by semidry blotting onto nitro-cellulose membranes (GE Healthcare). The membranes were blocked to pre-vent nonspecific Ab binding by incubation with specific antibody in Blotto(Pierce) overnight at 4 °C. Membranes were subsequently washed twice inTBST (50 mM Tris�HCl, pH 7.6/0.15 M NaCl/0.05% Tween 20) and then resus-pended in a 1/1,000 dilution of HRP-bovine anti-rabbit IgG in Blotto undergentle agitation for 1 h at room temperature. Membranes were washed twicein TBST and once in TBS, and were finally soaked in ECL� reagent (GEHealthcare) for HRP (60 s), and exposure to autoradiography film for visual-ization of the bands.
ChIP and Quantitative Real-Time PCR. ChIP was carried out as describedpreviously with slight modifications (38). In brief, 3 � 107 A673 cells wereformaldehyde (1% final) fixed for 10 min. After neutralization with glycine,cell pellets were lyzed in RIPA-buffer with protease inhibitors. Samples weresonicated to an average DNA length of 500 bp. ChIP was carried out with 3 �gof antibody either anti-FLI1 (C-19), or anti-IgG (Santa Cruz) added to 0.5 mg ofprecleared chromatin. Quantitative real-time PCR by using Sybr Green (Ap-plied Biosystems) with quantitation performed by the ��Ct-method (38). FLI1data at individual genomic loci were normalized to the IgG control to com-pensate for differences in PCR efficiency, and standardized to 2 nonregulatedgenomic loci outside of the EZH2 locus. Primer sequences are provided in SIMaterials and Methods.
Colony Forming Assay. Cells were seeded in duplicate at a density of 5 � 103
cells per 1.1 mL methylcellulose-based media (R&D Systems) into a 35-mmplate according to the manufacturer’s instruction, and cultured for 14 days at37 °C and 5% CO2 in a humidified atmosphere.
Differentiation Assays. For neuron-like or astrocyte-like cell differentiation,cells were cultured in the presence of 2% dimethyl sulfoxide (DMSO, Sigma-Aldrich) and 0.1 mM BHA (Sigma-Aldrich) for 5 days. The differentiated cells
5328 � www.pnas.org�cgi�doi�10.1073�pnas.0810759106 Richter et al.

were identified after fixation in 4% paraformaldehyde by staining with anantibody directed against glial fibrillary acid protein (GFAP, BD PharMingen),and visualized with a FITC-labeled goat anti-mouse F(ab�)2 fragment (JacksonImmunoResearch Laboratories).
Cellular tube formation was tested by use of a commercial Matrigel Matrixassay (Biocoat, BD Biosciences) according to the manufacturer’s instruction.Briefly, cells were seeded at 50,000 cells per well in a 96-well plate, and grownat 37 °C (5% CO2) in a humidified atmosphere. After 16–18 h, cells werestained with Calcein AM Fluorescent Dye (BD Biosciences) 1 �g/mL for 30 minin the dark. Cells were imaged by fluorescence microscopy by using a NikonEclipse TS 100 with an attached Nikon Coolpix 5400 camera.
In Vivo Experiments. For the analysis of in vivo tumor growth, 2–4 � 106 ET cellsand derivatives were harvested by trypsinization, washed with Dulbecco’s PBS,and injected in a volume of 0.2 mL into immunodeficient Rag2�/��C
�/� mice.To monitor local tumor growth, cells were injected s.c. intrainguinal, andtumor size was determined as described (39). Mice bearing a tumor �10 mmin diameter were considered as positive. To analyze metastatic potentialtumor, cells were injected i.v. into the tail vein. Four weeks later, mice were
killed, and metastatic spread was monitored in individual organs. In allexperiments, tumor and affected tissue after in vivo growth was excised forimmunohistology, and local tumors in addition were analyzed for EZH2activity.
Histology. Organs were fixed in 4% formaldehyde and paraffin embedded; 3to 5 �m thick sections from all tissues were cut and stained with hematoxylinand eosin (H&E). All sections were reviewed by 2 pathologists (I.M. andL.Q-M.).
ACKNOWLEDGMENTS. We thank Ines Volkmer and Colette Zobywalski forexpert technical assistance. L87 and V54.2 MSC lines were kindly provided byPeter Nelson (Medical Policlinic, Ludwig-Maximilians University, Munich). Thiswork was supported by unrestricted Special Grants P31/08//A123/07 from theElse-Kroner-Fresenius Stiftung, and KKF8739175 and 1528/TUM11.16-9c/32269 from the Bayerisches Staatsministerium fur Wissenschaft und Kunst.This work was also supported by the Bundesministerium fur Bildung undForschung (BMBF/DLR) Kompetenznetz Padiatrische Onkologie/TP Immun-und Gentherapie Grant GI9965, Wilhelm-Sander Stiftung Grant 2006.109.1,and Austrian Science Fund (FWF) Grant 18046-B12; and is part of the Trans-lational Sarcoma Research Network supported by the BMBF.
1. Ewing J (1921) Diffuse endothelioma of the bone. Proc NY Pathol Soc 21:17–24.2. Staege MS, et al. (2004) DNA microarrays reveal relationship of Ewing family tumors to
both endothelial and fetal neural crest-derived cells and define novel targets. CancerRes 64:8213–8221.
3. Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA, Ilaria RL, Jr (2005) Expression of theEWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1-dependent, ewing sarcoma-like tumors. Cancer Res 65:8698–8705.
4. Riggi N, et al. (2005) Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res 65:11459–11468.
5. Tirode F, et al. (2007) Mesenchymal stem cell features of Ewing tumors. Cancer Cell11:421–429.
6. Schmidt D, Harms D, Burdach S (1985) Malignant peripheral neuroectodermal tumoursof childhood and adolescence. Virchows Arch A Pathol Anat Histopathol 406:351–365.
7. Hu-Lieskovan S, et al. (2005) EWS-FLI1 fusion protein up-regulates critical genes inneural crest development and is responsible for the observed phenotype of Ewing’sfamily of tumors. Cancer Res 65:4633–4644.
8. Sparmann A, van Lohuizen M (2006) Polycomb silencers control cell fate, developmentand cancer. Nat Rev Cancer 6:846–856.
9. Vire E, et al. (2006) The Polycomb group protein EZH2 directly controls DNA methyl-ation. Nature 439:871–874.
10. Varambally S, et al. (2002) The polycomb group protein EZH2 is involved in progressionof prostate cancer. Nature 419:624–629.
11. van der Vlag J, Otte AP (1999) Transcriptional repression mediated by the humanpolycomb-group protein EED involves histone deacetylation. Nat Genet 23:474–478.
12. Dohjima T, Lee NS, Li H, Ohno T, Rossi JJ (2003) Small interfering RNAs expressed froma Pol III promoter suppress the EWS/Fli-1 transcript in an Ewing sarcoma cell line. MolTher 7:811–816.
13. Moosmann S, Hutter J, Moser C, Krombach F, Huss R (2005) Milieu-adopted in vitro andin vivo differentiation of mesenchymal tissues derived from different adult humanCD34-negative progenitor cell clones. Cells Tissues Organs 179:91–101.
14. Bracken AP, et al. (2003) EZH2 is downstream of the pRB-E2F pathway, essential forproliferation and amplified in cancer. EMBO J 22:5323–5335.
15. Stemple DL, Anderson DJ (1992) Isolation of a stem cell for neurons and glia from themammalian neural crest. Cell 71:973–985.
16. Hawkins PG, Morris KV (2008) RNA and transcriptional modulation of gene expression.Cell Cycle 7:602–607.
17. Reeves SA, Helman LJ, Allison A, Israel MA (1989) Molecular cloning and primarystructure of human glial fibrillary acidic protein. Proc Natl Acad Sci USA 86:5178–5182.
18. Kleer CG, et al. (2003) EZH2 is a marker of aggressive breast cancer and promotesneoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100:11606–11611.
19. Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K (2006) Genome-wide mapping ofPolycomb target genes unravels their roles in cell fate transitions. Genes Dev 20:1123–1136.
20. Lund AH, van Lohuizen M (2004) Polycomb complexes and silencing mechanisms. CurrOpin Cell Biol 16:239–246.
21. Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V (2004) The Polycomb Ezh2methyltransferase regulates muscle gene expression and skeletal muscle differentia-tion. Genes Dev 18:2627–2638.
22. Kamminga LM, et al. (2006) The Polycomb group gene Ezh2 prevents hematopoieticstem cell exhaustion. Blood 107:2170–2179.
23. Arvand A, Welford SM, Teitell MA, Denny CT (2001) The COOH-terminal domain ofFLI-1 is necessary for full tumorigenesis and transcriptional modulation by EWS/FLI-1.Cancer Res 61:5311–5317.
24. Lessnick SL, Dacwag CS, Golub TR (2002) The Ewing’s sarcoma oncoprotein EWS/FLIinduces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell1:393–401.
25. McGarvey KM, Greene E, Fahrner JA, Jenuwein T, Baylin SB (2007) DNA methylationand complete transcriptional silencing of cancer genes persist after depletion of EZH2.Cancer Res 67:5097–5102.
26. Kondo Y, et al. (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylationindependent of promoter DNA methylation. Nat Genet 40:741–750.
27. Ohm JE, et al. (2007) A stem cell-like chromatin pattern may predispose tumorsuppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39:237–242.
28. Schlesinger Y, et al. (2007) Polycomb-mediated methylation on Lys27 of histone H3pre-marks genes for de novo methylation in cancer. Nat Genet 39:232–236.
29. Widschwendter M, et al. (2007) Epigenetic stem cell signature in cancer. Nat Genet39:157–158.
30. Amaral PP, Dinger ME, Mercer TR, Mattick JS (2008) The eukaryotic genome as an RNAmachine. Science 319:1787–1789.
31. Kim DH, Villeneuve LM, Morris KV, Rossi JJ (2006) Argonaute-1 directs siRNA-mediatedtranscriptional gene silencing in human cells. Nat Struct Mol Biol 13:793–797.
32. Miyagawa Y, et al. (2008) Inducible expression of chimeric EWS/ETS proteins confersEwing’s family tumor-like phenotypes to human mesenchymal progenitor cells. MolCell Biol 28:2125–2137.
33. Riggi N, et al. (2008) EWS-FLI-1 expression triggers a Ewing’s sarcoma initiationprogram in primary human mesenchymal stem cells. Cancer Res 68:2176–2185.
34. Hofmann HS, et al. (2005) Matrix metalloproteinase-12 expression correlates with localrecurrence and metastatic disease in non-small cell lung cancer patients. Clin CancerRes 11:1086–1092.
35. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display ofgenome-wide expression patterns. Proc Natl Acad Sci USA 95:14863–14868.
36. Sturn A, Quackenbush J, Trajanoski Z (2002) Genesis: Cluster analysis of microarraydata. Bioinformatics 18:207–208.
37. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to theionizing radiation response. Proc Natl Acad Sci USA 98:5116–5512.
38. Hoemme C, et al. (2008) Chromatin modifications induced by PML-RARalpha represscritical targets in leukemogenesis as analyzed by ChIP-Chip. Blood 111:2887–2895.
39. Qin Z, et al. (1998) B cells inhibit induction of T cell-dependent tumor immunity. NatMed 4:627–630.
Richter et al. PNAS � March 31, 2009 � vol. 106 � no. 13 � 5329
MED
ICA
LSC
IEN
CES

Supporting InformationRichter et al. 10.1073/pnas.0810759106SI Materials and MethodsReagents. Histone deacetylase inhibitor (HDACi) Trichostatin A(TSA) and Polybrene (hexadimethrine bromide) were purchasedfrom SIGMA, Puromycin and G418 were from PAA Labora-tories. Rabbit polyclonal antibodies against C-terminal FLI1,human HPRT, and HRP-conjugated bovine anti-rabbit IgGwere purchased from Santa Cruz Biotechnology. Rabbit poly-clonal antibody against EZH2 was from Cell Signaling, againstH3 was from Upstate Biotechnology.
Small Interfering RNAs Used. AGO1 (Hs�EIF2C1�2�HP) 5�-r-(CCAAGAAUUGUGCAAGUAA)dTdT-3� (sense) 5�-r(UU-ACUUGCACAAUUCUUGG)dAdG-3� (antisense) EWS-FLI1-I 5�-GCUACGGGCAGCAGAACCCUU-3� (sense) and5�-GGGUUCUGCUGCCCGUAGCU-3� (antisense); EWS-FLI1-II 5�-GCAGAACCCUUCUUAUGACUU-3� (sense) and5�-GUCAUAAGAAGGGUUCUGCUUU-3� (antisense);EZH2–2 5�-r(GCAAAUUCUCGGUGUCAAA)dTdT-3�(sense), 5�-r(UUUGACACCGAGAAUUUGC)dTdT-3� (anti-sense), EZH2–7 (Hs�EZH2�7�HP, validated siRNA) 5�-r(CCAUGUUUACAACUAUCAA)dTdT-3� (sense), 5�-r(UUGAUAGUUGUAAACAUGG)dTdT-3� (antisense) andcontrol non silencing siRNA 5�-r(UUCUCCGAACGUGU-CACGU)dTdT-3� (sense), 5�-r(ACGUGACACGUUCG-GAGAA) dTdT-3� (antisense), SUZ12�6�HP validated 5�-r(GCAUAAUGUCAAUAGAUAA)dTdT-3� (sense), 5�-r(UUAUCUAUUGACAUUAUGC)dTdT-3� (antisense),EED�6 5�-r(AAUCCGGUUGUUGCAAUCUUA)dTdT-3�(sense), 5�-r(UAAGAUUGCAACAACCGGA) dTdT-3� (anti-sense).
Short Hairpin RNA Coding Oligonucleotides. EZH2, (1) 5�-GA-TCCGGAGGTTCAGACGAGCTGATTTCAAGAGAAT-CAGCTCGTCTGAACCTCCTTTTTTCTAGAG-3� (sense)and 5�-AATTCTCTAGAAAAAAGGAGGTTCAGAC-GAGCTGATTCTCTTGAAATCAGCTCGTCTGAACC-TCCG-3� (antisense), EWS-FLI1 corresponding to the pub-lished EWS-FLI1 siRNA, site II (2) 5�-GATCCGAGCAGAA-CCCTTCTTATGACTTCAAGAGAGTCATAAGAAGG-GTTCTGCTCTTTTTTCTAGAG-3� (sense) and 5�-AATTC-TCTAGAAAAAAGAGCAGAACCCTTCTTATGACTC-TCTTGAAGTCATAAGAAGGGTTCTGCTCG-3� (anti-sense) and control siRNA 5�-GATCCGTTCTCCGAAC-GTGTCACGTTTCAAGAGAACGTGACACGTTCGGA-GAACTTTTTTCTAGAG-3� (sense) and 5�-AATTCTC-TAGAAAAAAGTTCTCCGAACGTGTCACGTTCTCT-TGAAACGTGACACGTTCGGAGAACG-3�.
Primers and Assays Used for Real-Time PCR. For EWS/FLI1 detec-tion, the following primers 5�-TAGTTACCCACCCCAAACT-GGAT-3� (sense) 5�-GGGCCGTTGCTCTGTATTCTTAC-3�(antisense) and probe 5�-FAM-CAGCTACGGGCAGCA-
GAACCCTTCTT-TAMRA-3� were designed. InventoriedTaqMan Gene Expression Assays (Applied Biosystems) wereused for the genes ALCAM (Hs00233455�m1), AGO1(Hs00201864�m1), EMP1 (Hs00923125�g1), EPHB2(Hs00362096�m1), GAP43 (Hs00176645�m1), EZH2(Hs00544830�m1), G1P2 (Hs00192713�m1), GFAP(Hs00157674�m1), IFITM1 (Hs01652522�g1), NGFR(Hs00182120�m1), EED (Hs00537777�m1), and SUZ12(Hs00248742�m1). Gene expression was normalized to GAPDH(Hs99999905�m1).
Primers Used for Chromatin-IP PCR. Relative to TSS of EZH2:�4400-bp region, 5�-GCACATCAGCCACGCTTCT-3� (sense)and 5�- GGAGCTGAGGGAGCATTTACTG-3� (antisense),�3778-bp region, 5�-ATCCAGCCCCAAGCTGTTT-3� (sense)and 5�-GAACATGAGGTGGTGATAAAAATAAGG-3� (an-tisense), �2120-bp region, 5�-CCAACATTGGAGTGAT-TCAG-3� (sense) and 5�-TCATCAGATGATTTAGCCCA-3�(antisense), �1081-bp region, 5�-GACACGTGCTTAGAAC-TACGAACAG-3� (sense) 5�-TTTGGCTGGCCGAGCTT-3�(antisense).
Cell Proliferation Assay. Cell proliferation was monitored by BrdUincorporation during DNA synthesis by using a BrdU based cellproliferation ELISA (GE Healthcare). Briefly, 5 � 103 cell perwell of 96-well plate were seeded into 200 �L of growth mediumand incubated at various time points. For the final 12 h, BrdUwas added according to the manufacturer’s recommendation.Cells were subsequently spun down, lysed, and DNA denatured.BrdU incorporation was monitored by color development oftetramethyl-benzidine substrate turnover mediated by boundperoxidase-conjugated BrdU-specific Fab fragment, and quan-tified at 450 nm in a MRX microplate reader (Dynex). Alldeterminations were performed in triplicate and measured asthe mean � SEM.
Microarray Data. For comparative gene expression analysis, weprepared a database by combining the following datasets: 12mesenchymal stem cell samples from GSE2248 (3) and GSE6029(4), including bone marrow derived MSC, umbilical cord bloodderived MSC, and MSC derived from differentiated embryonicstem cells; 9 endothelial cell samples from GSE2638 andGSE2639 (5), 36 normal tissues of diverse origin from GSE2361(6). All datasets were downloaded from the GEO database asCEL files, analyzed by using Microarray Suite 5.0, and scaled tothe same target intensity of 500. Subsequent analysis was carriedout with signal intensities that were log2 transformed for equalrepresentation of over and under expressed genes, and thenmedian centered to remove biases based on single expressionvalues. Hierarchical clustering (7) was accomplished by use ofthe Genesis software package (8).
1. Varambally S, et al. (2002) The polycomb group protein EZH2 is involved in progressionof prostate cancer. Nature 419:624–629.
2. Dohjima T, Lee NS, Li H, Ohno T, Rossi JJ (2003) Small interfering RNAs expressed froma Pol III promoter suppress the EWS/Fli-1 transcript in an Ewing sarcoma cell line. MolTher 7:811–816.
3. Barberi T, Willis LM, Socci ND, Studer L (2005) Derivation of multipotent mesenchymalprecursors from human embryonic stem cells. PLoS Med 2:e161.
4. Markov V, et al. (2007) Identification of cord blood-derived mesenchymal stem/stromalcell populations with distinct growth kinetics, differentiation potentials, and geneexpression profiles. Stem Cells Dev 16:53–73.
5. Viemann D, et al. (2006) TNF induces distinct gene expression programs in microvas-cular and macrovascular human endothelial cells. J Leukoc Biol 80:174–185.
6. Ge X, et al. (2005) Interpreting expression profiles of cancers by genome-wide surveyof breadth of expression in normal tissues. Genomics 86:127–141.
7. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display ofgenome-wide expression patterns. Proc Natl Acad Sci USA 95:14863–14868.
8. Sturn A, Quackenbush J, Trajanoski Z (2002) Genesis: cluster analysis of microarraydata. Bioinformatics 18:207–208.
9. Staege MS, et al. (2004) DNA microarrays reveal relationship of Ewing family tumors toboth endothelial and fetal neural crest-derived cells and define novel targets. CancerRes 64:8213–8221.
Richter et al. www.pnas.org/cgi/content/short/0810759106 1 of 11
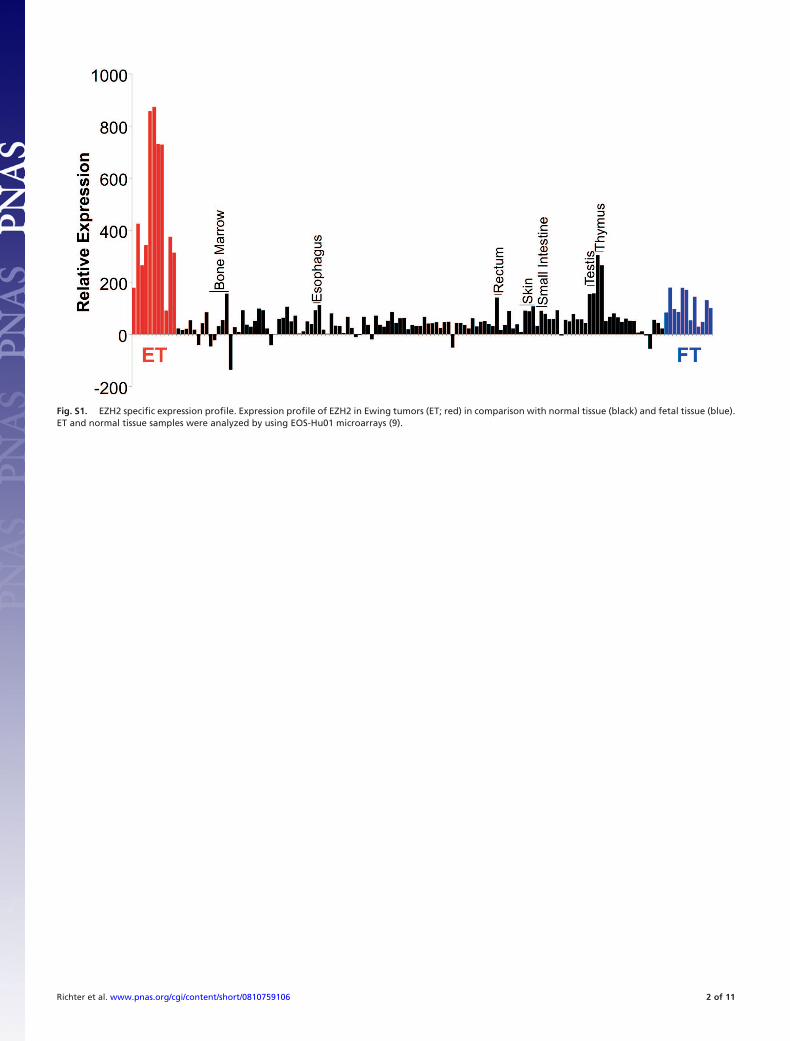
Fig. S1. EZH2 specific expression profile. Expression profile of EZH2 in Ewing tumors (ET; red) in comparison with normal tissue (black) and fetal tissue (blue).ET and normal tissue samples were analyzed by using EOS-Hu01 microarrays (9).
Richter et al. www.pnas.org/cgi/content/short/0810759106 2 of 11
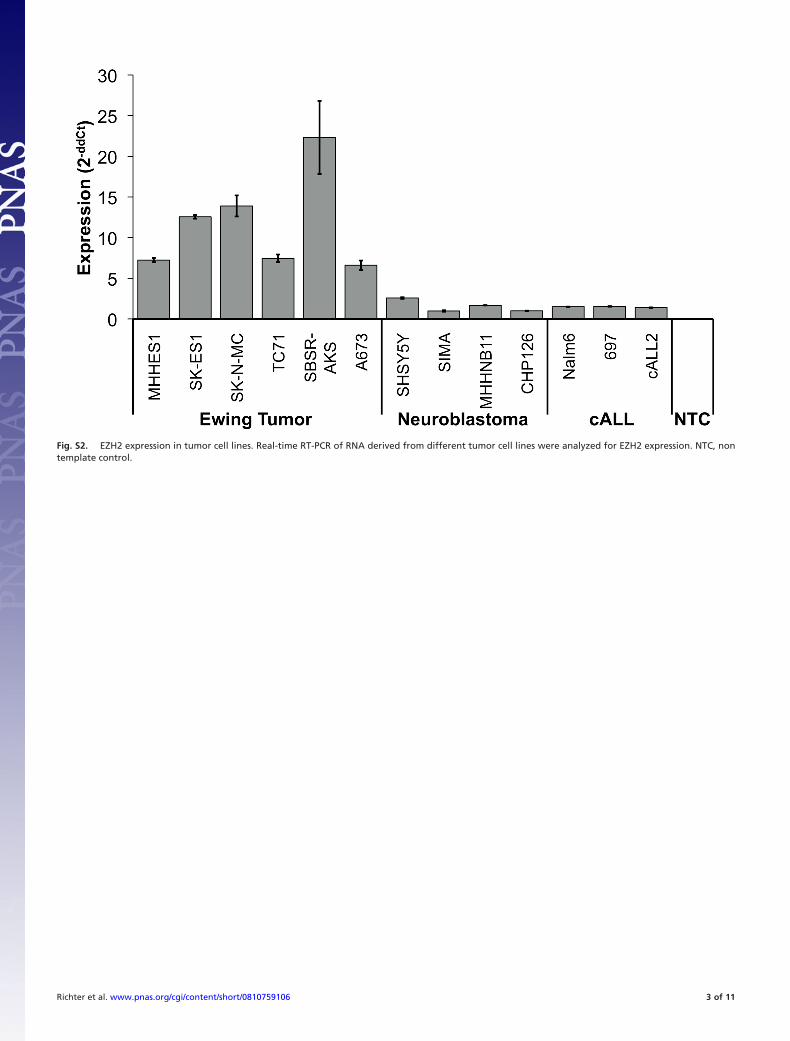
Fig. S2. EZH2 expression in tumor cell lines. Real-time RT-PCR of RNA derived from different tumor cell lines were analyzed for EZH2 expression. NTC, nontemplate control.
Richter et al. www.pnas.org/cgi/content/short/0810759106 3 of 11
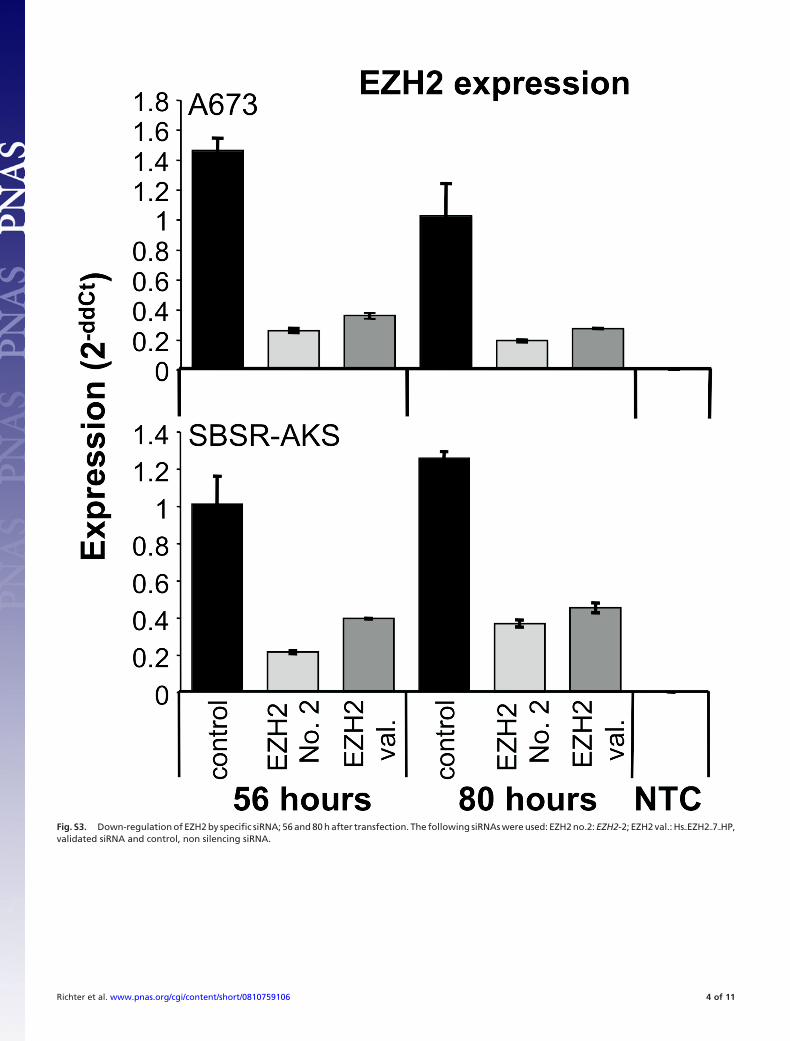
Fig. S3. Down-regulation of EZH2 by specific siRNA; 56 and 80 h after transfection. The following siRNAs were used: EZH2 no.2: EZH2-2; EZH2 val.: Hs�EZH2�7�HP,validated siRNA and control, non silencing siRNA.
Richter et al. www.pnas.org/cgi/content/short/0810759106 4 of 11
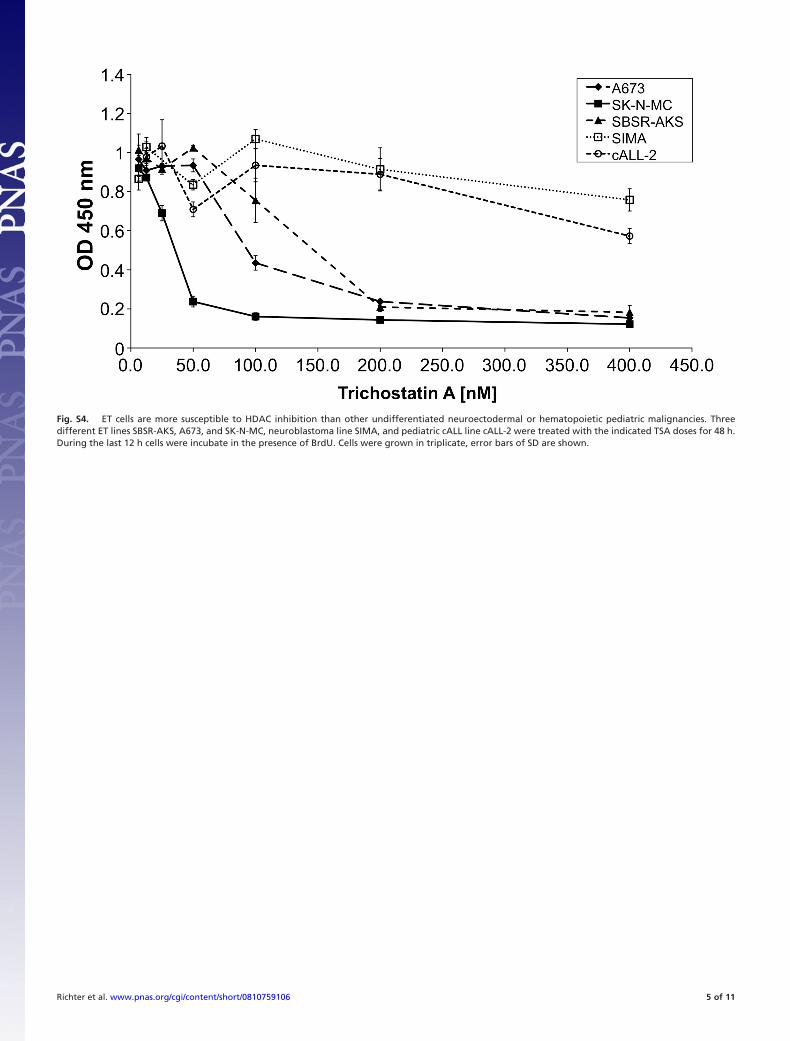
Fig. S4. ET cells are more susceptible to HDAC inhibition than other undifferentiated neuroectodermal or hematopoietic pediatric malignancies. Threedifferent ET lines SBSR-AKS, A673, and SK-N-MC, neuroblastoma line SIMA, and pediatric cALL line cALL-2 were treated with the indicated TSA doses for 48 h.During the last 12 h cells were incubate in the presence of BrdU. Cells were grown in triplicate, error bars of SD are shown.
Richter et al. www.pnas.org/cgi/content/short/0810759106 5 of 11

Fig. S5. Microarray data of differential gene expression after EZH2siRNA/TSA treatment. Selected genes were identified by fold change, P/A calls and P � 0.05and are shown with their normalized fluorescent signal intensities (see Materials and Methods). Combined results of 2 independent assays and different siRNAsused are shown. Each column represents 1 individual array. Arrows mark genes important for neuronal and/or endothelial differentiation (see main article).
Richter et al. www.pnas.org/cgi/content/short/0810759106 6 of 11

Fig. S6. EZH2-suppressed genes are expressed in neuronal tissues, mesenchymal stem cells, and endothelial cells. Expression of genes with significantup-regulation after siRNA mediated suppression of EZH2 in A673 cell were used for cluster analysis (Manhattan distance, complete linkage clustering). For thisend, a panel of datasets from normal tissues, endothelial cells [HMEC, human microvasculature endothelial cells; HUVEC, human macrovasculature (umbilicalcord) endothelial cells], and mesenchymal stem cells (MSC, bone marrow derived MSC; UCB1-MSC and UCB2-MSC, 2 types of umbillical cord blood derived MSC;ES-MSC, ESC-derived MSC) was used (for composition of the database, see Materials and Methods). Raw data were log2 transformed and median centered.
Richter et al. www.pnas.org/cgi/content/short/0810759106 7 of 11
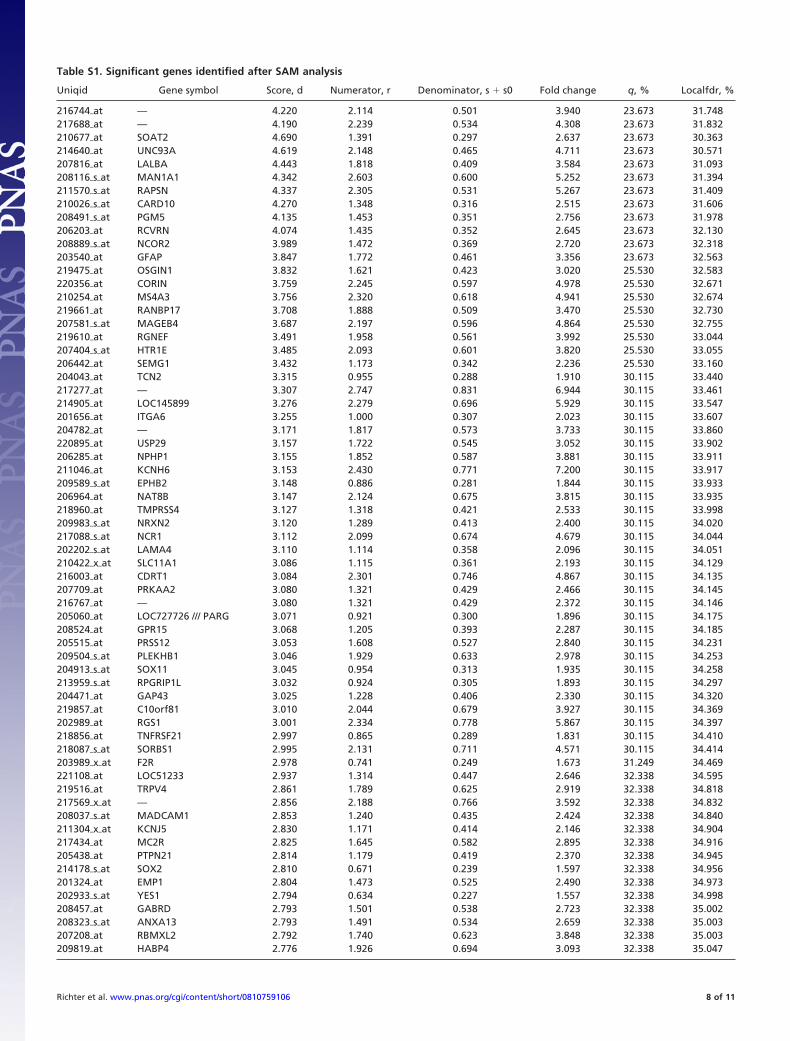
Table S1. Significant genes identified after SAM analysis
Uniqid Gene symbol Score, d Numerator, r Denominator, s � s0 Fold change q, % Localfdr, %
216744�at — 4.220 2.114 0.501 3.940 23.673 31.748217688�at — 4.190 2.239 0.534 4.308 23.673 31.832210677�at SOAT2 4.690 1.391 0.297 2.637 23.673 30.363214640�at UNC93A 4.619 2.148 0.465 4.711 23.673 30.571207816�at LALBA 4.443 1.818 0.409 3.584 23.673 31.093208116�s�at MAN1A1 4.342 2.603 0.600 5.252 23.673 31.394211570�s�at RAPSN 4.337 2.305 0.531 5.267 23.673 31.409210026�s�at CARD10 4.270 1.348 0.316 2.515 23.673 31.606208491�s�at PGM5 4.135 1.453 0.351 2.756 23.673 31.978206203�at RCVRN 4.074 1.435 0.352 2.645 23.673 32.130208889�s�at NCOR2 3.989 1.472 0.369 2.720 23.673 32.318203540�at GFAP 3.847 1.772 0.461 3.356 23.673 32.563219475�at OSGIN1 3.832 1.621 0.423 3.020 25.530 32.583220356�at CORIN 3.759 2.245 0.597 4.978 25.530 32.671210254�at MS4A3 3.756 2.320 0.618 4.941 25.530 32.674219661�at RANBP17 3.708 1.888 0.509 3.470 25.530 32.730207581�s�at MAGEB4 3.687 2.197 0.596 4.864 25.530 32.755219610�at RGNEF 3.491 1.958 0.561 3.992 25.530 33.044207404�s�at HTR1E 3.485 2.093 0.601 3.820 25.530 33.055206442�at SEMG1 3.432 1.173 0.342 2.236 25.530 33.160204043�at TCN2 3.315 0.955 0.288 1.910 30.115 33.440217277�at — 3.307 2.747 0.831 6.944 30.115 33.461214905�at LOC145899 3.276 2.279 0.696 5.929 30.115 33.547201656�at ITGA6 3.255 1.000 0.307 2.023 30.115 33.607204782�at — 3.171 1.817 0.573 3.733 30.115 33.860220895�at USP29 3.157 1.722 0.545 3.052 30.115 33.902206285�at NPHP1 3.155 1.852 0.587 3.881 30.115 33.911211046�at KCNH6 3.153 2.430 0.771 7.200 30.115 33.917209589�s�at EPHB2 3.148 0.886 0.281 1.844 30.115 33.933206964�at NAT8B 3.147 2.124 0.675 3.815 30.115 33.935218960�at TMPRSS4 3.127 1.318 0.421 2.533 30.115 33.998209983�s�at NRXN2 3.120 1.289 0.413 2.400 30.115 34.020217088�s�at NCR1 3.112 2.099 0.674 4.679 30.115 34.044202202�s�at LAMA4 3.110 1.114 0.358 2.096 30.115 34.051210422�x�at SLC11A1 3.086 1.115 0.361 2.193 30.115 34.129216003�at CDRT1 3.084 2.301 0.746 4.867 30.115 34.135207709�at PRKAA2 3.080 1.321 0.429 2.466 30.115 34.145216767�at — 3.080 1.321 0.429 2.372 30.115 34.146205060�at LOC727726 /// PARG 3.071 0.921 0.300 1.896 30.115 34.175208524�at GPR15 3.068 1.205 0.393 2.287 30.115 34.185205515�at PRSS12 3.053 1.608 0.527 2.840 30.115 34.231209504�s�at PLEKHB1 3.046 1.929 0.633 2.978 30.115 34.253204913�s�at SOX11 3.045 0.954 0.313 1.935 30.115 34.258213959�s�at RPGRIP1L 3.032 0.924 0.305 1.893 30.115 34.297204471�at GAP43 3.025 1.228 0.406 2.330 30.115 34.320219857�at C10orf81 3.010 2.044 0.679 3.927 30.115 34.369202989�at RGS1 3.001 2.334 0.778 5.867 30.115 34.397218856�at TNFRSF21 2.997 0.865 0.289 1.831 30.115 34.410218087�s�at SORBS1 2.995 2.131 0.711 4.571 30.115 34.414203989�x�at F2R 2.978 0.741 0.249 1.673 31.249 34.469221108�at LOC51233 2.937 1.314 0.447 2.646 32.338 34.595219516�at TRPV4 2.861 1.789 0.625 2.919 32.338 34.818217569�x�at — 2.856 2.188 0.766 3.592 32.338 34.832208037�s�at MADCAM1 2.853 1.240 0.435 2.424 32.338 34.840211304�x�at KCNJ5 2.830 1.171 0.414 2.146 32.338 34.904217434�at MC2R 2.825 1.645 0.582 2.895 32.338 34.916205438�at PTPN21 2.814 1.179 0.419 2.370 32.338 34.945214178�s�at SOX2 2.810 0.671 0.239 1.597 32.338 34.956201324�at EMP1 2.804 1.473 0.525 2.490 32.338 34.973202933�s�at YES1 2.794 0.634 0.227 1.557 32.338 34.998208457�at GABRD 2.793 1.501 0.538 2.723 32.338 35.002208323�s�at ANXA13 2.793 1.491 0.534 2.659 32.338 35.003207208�at RBMXL2 2.792 1.740 0.623 3.848 32.338 35.003209819�at HABP4 2.776 1.926 0.694 3.093 32.338 35.047
Richter et al. www.pnas.org/cgi/content/short/0810759106 8 of 11

Uniqid Gene symbol Score, d Numerator, r Denominator, s � s0 Fold change q, % Localfdr, %
221186�at — 2.775 1.403 0.505 2.776 32.338 35.048206581�at BNC1 2.774 1.872 0.675 3.426 32.338 35.051217880�at — 2.768 0.666 0.241 1.585 32.338 35.067207160�at IL12A 2.753 0.994 0.361 2.039 32.338 35.105206985�at HSD17B3 2.751 1.864 0.678 3.309 32.338 35.111211925�s�at PLCB1 2.727 2.030 0.744 4.436 32.338 35.172216651�s�at GAD2 2.710 1.577 0.582 2.585 32.338 35.216219899�x�at NDOR1 2.703 1.326 0.491 2.686 32.338 35.234214387�x�at SFTPC 2.689 0.823 0.306 1.774 32.338 35.270209451�at TANK 2.678 0.910 0.340 1.922 32.778 35.300216840�s�at LAMA2 2.674 1.261 0.471 2.420 32.778 35.310215646�s�at VCAN 2.672 2.031 0.760 2.750 32.778 35.314218181�s�at MAP4K4 2.670 0.708 0.265 1.625 32.778 35.320216171�at — 2.666 2.554 0.958 3.200 32.778 35.330210941�at PCDH7 2.664 1.065 0.400 1.981 32.778 35.334205860�x�at FOLH1 2.652 2.100 0.792 4.488 32.778 35.366207426�s�at TNFSF4 2.644 0.707 0.267 1.645 32.778 35.389206322�at SYN3 2.638 1.610 0.610 2.961 32.778 35.405219521�at B3GAT1 2.636 1.942 0.737 4.729 32.778 35.409215767�at ZNF804A 2.635 0.876 0.332 1.799 32.778 35.414210292�s�at PCDH11X /// PCDH11Y 2.632 1.300 0.494 2.472 32.778 35.420220836�at ZNF407 2.629 2.362 0.898 3.733 32.778 35.429204176�at KLHL20 2.626 0.810 0.308 1.751 32.778 35.436215350�at SYNE1 2.619 2.626 1.003 4.800 32.778 35.456205890�s�at UBD 2.603 1.285 0.494 2.528 32.778 35.501216967�at GAP43 2.599 1.221 0.470 2.331 32.778 35.513221697�at MAP1LC3C 2.596 1.347 0.519 2.343 32.778 35.521222342�at — 2.594 2.173 0.838 4.062 32.778 35.525205296�at RBL1 2.592 0.872 0.336 1.782 32.778 35.532210990�s�at LAMA4 2.588 0.880 0.340 1.879 32.778 35.544205485�at RYR1 2.587 2.108 0.815 3.500 32.778 35.546205534�at PCDH7 2.586 1.002 0.387 1.915 32.778 35.549213130�at ZNF473 2.578 0.857 0.333 1.846 32.778 35.572208387�s�at MMP24 2.567 1.995 0.777 3.200 32.778 35.604213479�at NPTX2 2.566 1.518 0.591 2.761 32.778 35.607202627�s�at SERPINE1 2.565 2.104 0.820 3.430 32.778 35.610
The first 100 probe sets are shown.
Richter et al. www.pnas.org/cgi/content/short/0810759106 9 of 11
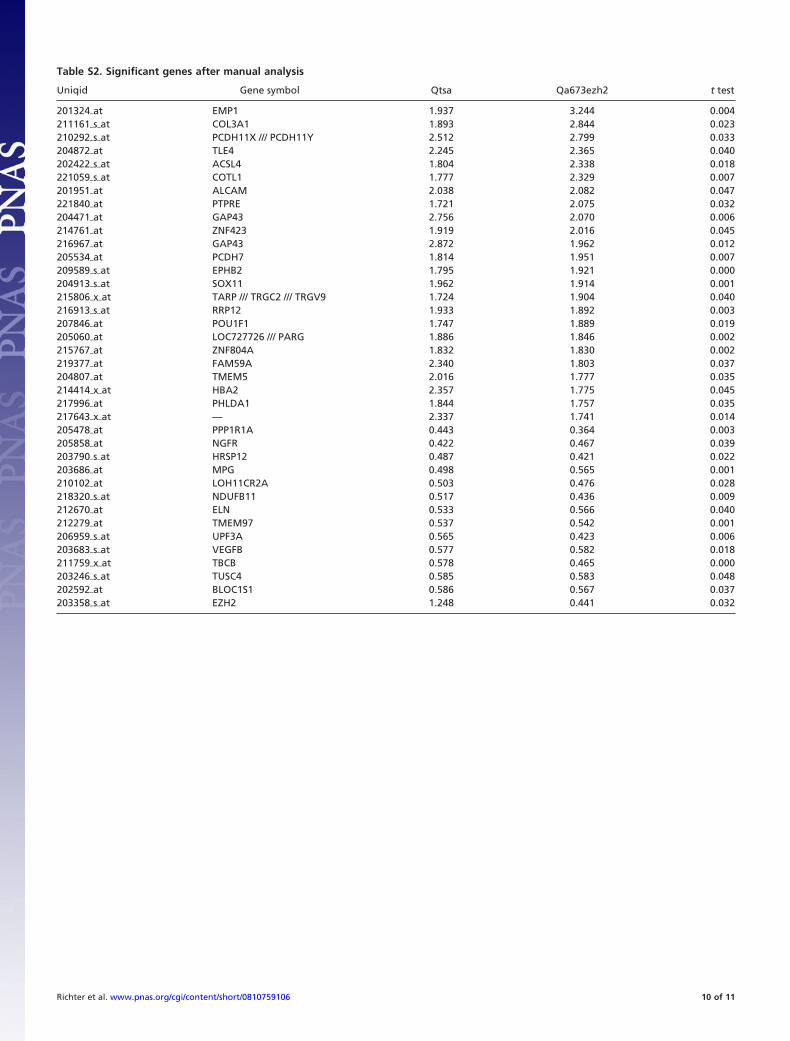
Table S2. Significant genes after manual analysis
Uniqid Gene symbol Qtsa Qa673ezh2 t test
201324�at EMP1 1.937 3.244 0.004211161�s�at COL3A1 1.893 2.844 0.023210292�s�at PCDH11X /// PCDH11Y 2.512 2.799 0.033204872�at TLE4 2.245 2.365 0.040202422�s�at ACSL4 1.804 2.338 0.018221059�s�at COTL1 1.777 2.329 0.007201951�at ALCAM 2.038 2.082 0.047221840�at PTPRE 1.721 2.075 0.032204471�at GAP43 2.756 2.070 0.006214761�at ZNF423 1.919 2.016 0.045216967�at GAP43 2.872 1.962 0.012205534�at PCDH7 1.814 1.951 0.007209589�s�at EPHB2 1.795 1.921 0.000204913�s�at SOX11 1.962 1.914 0.001215806�x�at TARP /// TRGC2 /// TRGV9 1.724 1.904 0.040216913�s�at RRP12 1.933 1.892 0.003207846�at POU1F1 1.747 1.889 0.019205060�at LOC727726 /// PARG 1.886 1.846 0.002215767�at ZNF804A 1.832 1.830 0.002219377�at FAM59A 2.340 1.803 0.037204807�at TMEM5 2.016 1.777 0.035214414�x�at HBA2 2.357 1.775 0.045217996�at PHLDA1 1.844 1.757 0.035217643�x�at — 2.337 1.741 0.014205478�at PPP1R1A 0.443 0.364 0.003205858�at NGFR 0.422 0.467 0.039203790�s�at HRSP12 0.487 0.421 0.022203686�at MPG 0.498 0.565 0.001210102�at LOH11CR2A 0.503 0.476 0.028218320�s�at NDUFB11 0.517 0.436 0.009212670�at ELN 0.533 0.566 0.040212279�at TMEM97 0.537 0.542 0.001206959�s�at UPF3A 0.565 0.423 0.006203683�s�at VEGFB 0.577 0.582 0.018211759�x�at TBCB 0.578 0.465 0.000203246�s�at TUSC4 0.585 0.583 0.048202592�at BLOC1S1 0.586 0.567 0.037203358�s�at EZH2 1.248 0.441 0.032
Richter et al. www.pnas.org/cgi/content/short/0810759106 10 of 11
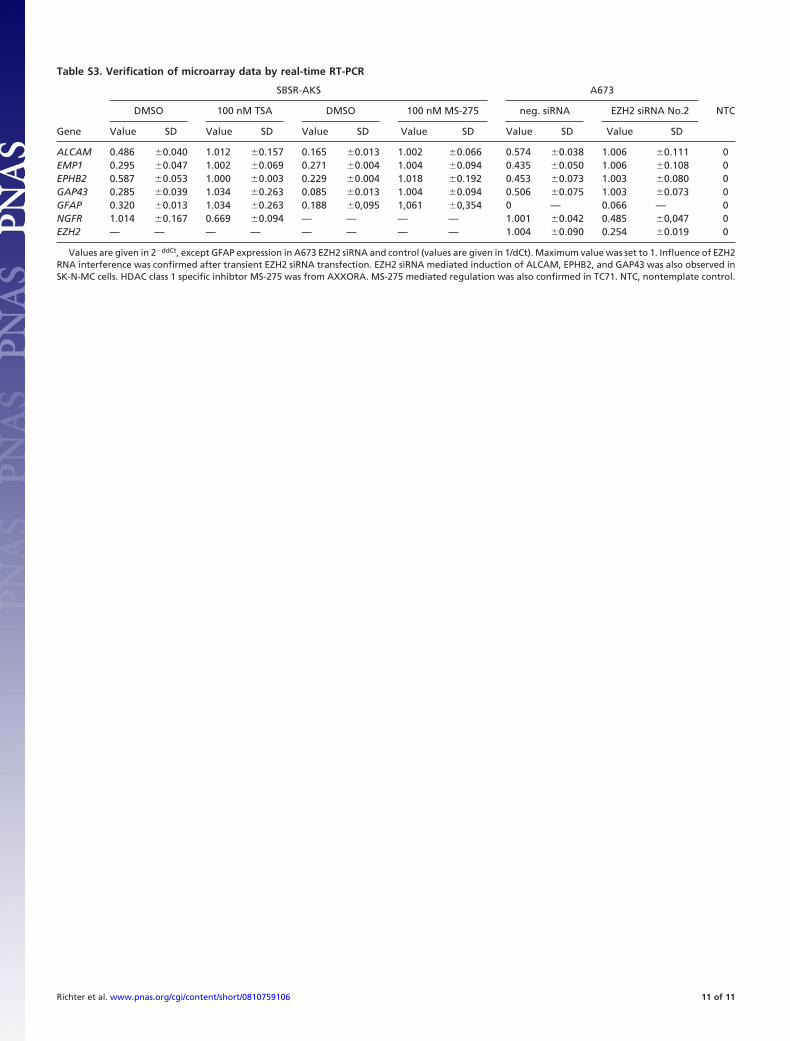
Table S3. Verification of microarray data by real-time RT-PCR
Gene
SBSR-AKS A673
NTCDMSO 100 nM TSA DMSO 100 nM MS-275 neg. siRNA EZH2 siRNA No.2
Value SD Value SD Value SD Value SD Value SD Value SD
ALCAM 0.486 �0.040 1.012 �0.157 0.165 �0.013 1.002 �0.066 0.574 �0.038 1.006 �0.111 0EMP1 0.295 �0.047 1.002 �0.069 0.271 �0.004 1.004 �0.094 0.435 �0.050 1.006 �0.108 0EPHB2 0.587 �0.053 1.000 �0.003 0.229 �0.004 1.018 �0.192 0.453 �0.073 1.003 �0.080 0GAP43 0.285 �0.039 1.034 �0.263 0.085 �0.013 1.004 �0.094 0.506 �0.075 1.003 �0.073 0GFAP 0.320 �0.013 1.034 �0.263 0.188 �0,095 1,061 �0,354 0 — 0.066 — 0NGFR 1.014 �0.167 0.669 �0.094 — — — — 1.001 �0.042 0.485 �0,047 0EZH2 — — — — — — — — 1.004 �0.090 0.254 �0.019 0
Values are given in 2�ddCt, except GFAP expression in A673 EZH2 siRNA and control (values are given in 1/dCt). Maximum value was set to 1. Influence of EZH2RNA interference was confirmed after transient EZH2 siRNA transfection. EZH2 siRNA mediated induction of ALCAM, EPHB2, and GAP43 was also observed inSK-N-MC cells. HDAC class 1 specific inhibtor MS-275 was from AXXORA. MS-275 mediated regulation was also confirmed in TC71. NTC, nontemplate control.
Richter et al. www.pnas.org/cgi/content/short/0810759106 11 of 11



















