
Expression, Purification, and Biophysical Characterization of the BRCT Domain of Human DNA Ligase...
11
Protein Expression and Purification 21, 401–411 (2001) doi:10.1006/prep.2001.1391, available online at http://www.idealibrary.com on Expression, Purification, and Biophysical Characterization of the BRCT Domain of Human DNA Ligase IIIa Kevin H. Thornton, V. V. Krishnan, Mary G. West, Jennifer Popham, Melissa Ramirez, Michael P. Thelen, and Monique Cosman 1 Molecular and Structural Biology Division, Biology and Biotechnology Research Program, Lawrence Livermore National Laboratory, Livermore, California 94551 Received September 18, 2000, and in revised form December 19, 2000 BRCT 2 domains (BRCA1 C-terminal repeats) are seg- The C-terminal regions of several DNA repair and ments of approximately 100 amino acids in length found cell cycle checkpoint proteins are homologous to the within proteins that function in DNA transcription, re- breast-cancer-associated BRCA-1 protein C-terminal pair, replication, and cell cycle checkpoints (1–3). This region. These regions, known as BRCT domains, have superfamily of domains is structurally similar to the been found to mediate important protein–protein two C-terminal regions of the breast cancer suppresser interactions. We produced the BRCT domain of DNA protein, BRCA1 (4), as deduced from multiple sequence ligase IIIa (L3[86]) for biophysical and structural char- alignments, hydrophobic clustering, and secondary acterization. A glutathione S-transferase (GST) fusion structure predictions. Deletions and mutations within with the L3[86] domain (residues 837–922 of ligase IIIa) these domains of BRCA1 have been identified as a high was expressed in Escherichia coli and purified by glu- risk factor for breast and ovarian cancers (5, 6). BRCT tathione affinity chromatography. The GST fusion pro- domains have also been found to recruit and bind other tein was removed by thrombin digestion and further BRCT domains or unknown protein folds with high af- purification steps. Using this method, 15 N-labeled and finity and specificity. Thus, mutations within these do- 13 C/ 15 N-double-labeled L3[86] proteins were prepared mains might deleteriously affect normal cell functions, to enable a full determination of structure and dynam- such as DNA repair, that rely on protein–protein inter- ics using heteronuclear NMR spectroscopy. To obtain actions. evidence of binding activity to the distal BRCT of the DNA ligase III and XRCC1 are proteins that partici- repair protein XRCC1 (X1BRCTb), as well as to provide pate in the DNA base excision repair pathway, the pro- insight into the interaction between these two BRCT cess that corrects modified DNA bases generated by binding partners, the corresponding BRCT heterocom- plexes were also prepared and studied. Changes in the endogenous cellular metabolic processes and by expo- secondary structures (amount of helix and sheet com- sure to environmental alkylating agents or ionizing ra- ponents) of the two constituents were not observed diation (7). These proteins have been shown to form a upon complex formation. However, the melting tem- 1:1 complex in vitro that is resistant to 2M NaCl (8–10). perature of the complex was significantly higher rela- In addition, xrcc1 mutant Chinese hamster ovary cell tive to the values obtained for the L3[86] or X1BRCTb lines contain significantly reduced amounts of both the proteins alone. This increased thermostability imparted by the interaction between the two BRCT 2 Abbreviations used: BRCT, BRCA1 C-terminal repeats; GST, glu- domains may explain why cells require XRCC1 to main- tathione S-transferase; LB, Luria–Bertani; IPTG, isopropylthioga- tain ligase IIIa activity. q 2001 Academic Press lactopyranoside; DTT, dithiothreitol; S-75, Superdex 75; Ni–NTA, nickel–nitriloacetic acid; bME, b-mercaptoethanol; CD, circular di- chroism; DSC, differential scanning calorimetry; NMR, nuclear mag- netic resonance; HSQC, heteronuclear single quantum correlation; DSS, dimethyl silapentane sulfonate; GuHCl, guanidine hydrochlo- ride; BPTI, basic pancreatic trypsin inhibitor; SEC, size-exclusion 1 To whom correspondence and reprint requests should be ad- dressed. Fax: (925) 424-3130. E-mail: [email protected]. chromatography. 1046-5928/01 $35.00 401 Copyright q 2001 by Academic Press All rights of reproduction in any form reserved.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Expression, Purification, and Biophysical Characterization of the BRCT Domain of Human DNA Ligase...

Protein Expression and Purification 21, 401–411 (2001)doi:10.1006/prep.2001.1391, available online at http://www.idealibrary.com on
Expression, Purification, and Biophysical Characterizationof the BRCT Domain of Human DNA Ligase IIIa
Kevin H. Thornton, V. V. Krishnan, Mary G. West, Jennifer Popham, Melissa Ramirez,Michael P. Thelen, and Monique Cosman1
Molecular and Structural Biology Division, Biology and Biotechnology Research Program,
diation (7). These proteins have been shown to form a1:1 complex in vitro that is resistant to 2M NaCl (8–10).In addition, xrcc1 mutant Chinese hamster ovary cell
Lawrence Livermore National Laboratory, Livermore, Californ
Received September 18, 2000, and in revised form December 19, 2000
The C-terminal regions of several DNA repair andcell cycle checkpoint proteins are homologous to thebreast-cancer-associated BRCA-1 protein C-terminalregion. These regions, known as BRCT domains, havebeen found to mediate important protein–proteininteractions. We produced the BRCT domain of DNAligase IIIa (L3[86]) for biophysical and structural char-acterization. A glutathione S-transferase (GST) fusionwith the L3[86] domain (residues 837–922 of ligase IIIa)was expressed in Escherichia coli and purified by glu-tathione affinity chromatography. The GST fusion pro-tein was removed by thrombin digestion and furtherpurification steps. Using this method, 15N-labeled and13C/15N-double-labeled L3[86] proteins were preparedto enable a full determination of structure and dynam-ics using heteronuclear NMR spectroscopy. To obtainevidence of binding activity to the distal BRCT of therepair protein XRCC1 (X1BRCTb), as well as to provideinsight into the interaction between these two BRCTbinding partners, the corresponding BRCT heterocom-plexes were also prepared and studied. Changes in thesecondary structures (amount of helix and sheet com-ponents) of the two constituents were not observedupon complex formation. However, the melting tem-perature of the complex was significantly higher rela-
tive to the values obtained for the L3[86] or X1BRCTbproteins alone. This increased thermostabilityimparted by the interaction between the two BRCTdomains may explain why cells require XRCC1 to main-tain ligase IIIa activity. q 2001 Academic Press1 To whom correspondence and reprint requests should be ad-dressed. Fax: (925) 424-3130. E-mail: [email protected].
1046-5928/01 $35.00Copyright q 2001 by Academic PressAll rights of reproduction in any form reserved.
ia 94551
BRCT2 domains (BRCA1 C-terminal repeats) are seg-ments of approximately 100 amino acids in length foundwithin proteins that function in DNA transcription, re-pair, replication, and cell cycle checkpoints (1–3). Thissuperfamily of domains is structurally similar to thetwo C-terminal regions of the breast cancer suppresserprotein, BRCA1 (4), as deduced from multiple sequencealignments, hydrophobic clustering, and secondarystructure predictions. Deletions and mutations withinthese domains of BRCA1 have been identified as a highrisk factor for breast and ovarian cancers (5, 6). BRCTdomains have also been found to recruit and bind otherBRCT domains or unknown protein folds with high af-finity and specificity. Thus, mutations within these do-mains might deleteriously affect normal cell functions,such as DNA repair, that rely on protein–protein inter-actions.
DNA ligase III and XRCC1 are proteins that partici-pate in the DNA base excision repair pathway, the pro-cess that corrects modified DNA bases generated byendogenous cellular metabolic processes and by expo-sure to environmental alkylating agents or ionizing ra-
lines contain significantly reduced amounts of both the
2 Abbreviations used: BRCT, BRCA1 C-terminal repeats; GST, glu-tathione S-transferase; LB, Luria–Bertani; IPTG, isopropylthioga-lactopyranoside; DTT, dithiothreitol; S-75, Superdex 75; Ni–NTA,nickel–nitriloacetic acid; bME, b-mercaptoethanol; CD, circular di-chroism; DSC, differential scanning calorimetry; NMR, nuclear mag-netic resonance; HSQC, heteronuclear single quantum correlation;DSS, dimethyl silapentane sulfonate; GuHCl, guanidine hydrochlo-ride; BPTI, basic pancreatic trypsin inhibitor; SEC, size-exclusionchromatography.
401

402 THORNTO
XRCC1 (11) and the DNA ligase III proteins, suggestingthat the coordinated expression of XRCC1 and ligaseIII may be required to maintain normal levels of DNAligase III activity (12, 13).
Two different forms of DNA ligase III are producedby the same gene and have identical catalytic proper-ties, but they differ as a result of a tissue-specific alter-native splicing mechanism entailing exons coding forthe carboxyl terminus of the protein (14–16). The 96-kDa (862 amino acids) form (ligase IIIb) was shown tooccur only in testes during the latter stages of meioticprophase, whereas the 103-kDa (922 amino acids) form(ligase IIIa) is present at similar levels in several so-matic tissues (16). The C-terminal 148 amino acids ofDNA ligase IIIa are sufficient to bind to the C-terminal96-amino-acid BRCTb domain of XRCC1, whereas the96-kDa ligase IIIb that is 60 residues shorter (at theC-terminal end) does not interact with XRCC1 (16, 17).This observation indicates that the C-terminal 60-amino-acid residues of ligase IIIa are critical for speci-fying the interaction with the XRCC1 BRCTb domain.This region of ligase IIIa comprises most of its BRCT do-main.
XRCC1 contains two BRCT domains, designated hereas X1BRCTa (aa 315–403) and X1BRCTb (aa 538–629),that are separated by an acidic, proline-rich region. Theproximal X1BRCTa domain interacts with poly(ADP-ribose)polymerase and negatively regulates its activityfollowing DNA damage (18), while the BRCT domain ofDNA ligase IIIa, designated here as L3BRCT, interactswith the distal X1BRCTb domain (17). Mutations inthe X1BRCTa domain disable the function of XRCC1in the repair of DNA base damage (11), while alterationsin the X1BRCTb domain disrupt interactions with li-gase IIIa (19) and abolish the XRCC1-dependent repairof DNA strand breaks during the G1 phase of the cellcycle (20).
The X1BRCTb domain isolated as a separate polypep-tide retains the ligase IIIa-binding function (21), andthe crystal structure of this domain (residues 538–629)has been previously determined (http://www.rcsb.org/pdb/ and pdb access code 1cdz) (22). The protein forms ahomodimer containing a four-stranded parallel b-sheetsurrounded by three a-helices (22). Within each mono-meric unit, the order of the strands in the sheet isb2b1b3b4, with a1 and a3 on one side of the sheet anda2 on the other. Here we describe the overexpressionand biophysical characterization of the L3BRCT do-
main and the L3BRCT:X1BRCTb complex, the first stepneeded to understand the properties of these domainsand their association in the complex of XRCC1 andligase IIIa.N ET AL.
EXPERIMENTAL PROCEDURES
Cloning, Overexpression, and Purification of LigaseIIIa BRCT Domains
The cloning and purification of the BRCT domain ofXRCC1 (X1BRCTb, amino acid residues 533–633) hasbeen previously described (21). The X1BRCTb domainobtained contains a Met residue at the N-terminus,as confirmed by mass spectroscopy analysis (SynPep,Dublin, CA). The calculated pI is 4.77, the molecularweight is 12,061 Da, and the extinction coefficient is17,200 M21 cm21 at 280 nm (assuming all cysteines arereduced) (http:/www.expasy.ch).
To obtain soluble ligase IIIa BRCT domain(L3BRCT), two different constructs were made, one thatcould be used to express the domain containing residues843–922 (L3[80]) and another for expressing residues837–922 (L3[86]). The expression vector for L3[86] wasgenerated by amplifying the region containing nucleo-tides 2844–3102 in a DNA ligase IIIa pGSTag expres-sion vector generously provided by A. Tomkinson (16).DNA primers (Genosys) used for amplification by PCRwere forward, 58-TATAGGATCCGCTGATGAGACGCT-GTGCCA-38; and reverse, 58-TATAGAATTCAGCAGG-GAGCTACCAGTCT-38. The underlined nucleotides ofthe forward and reverse primers denote the BamHI andEcoRI sites, respectively, used in cloning the 278-bpproduct. PCR was performed using Platinum Taq poly-merase (Gibco BRL) and the reaction protocol 948C (5min), followed by 25 cycles of 948C denaturation (30 s),558C annealing (45 s), and 728C extension (45 s), andwas completed by a 728C (7 min) extension. The productwas TA cloned using standard techniques into thepCR2.1 vector (Invitrogen). The Escherichia coli strainINVaF8 (Invitrogen) was transformed to ampicillin re-sistance, plasmid DNA was isolated from the trans-formants, and the entire sequence of the insert andflanking regions was verified by DNA sequencing. Theinsert was released by restriction digest with BamHIand EcoRI and ligated into a pGEX-2T vector (Phar-macia) using the BamHI and EcoRI sites in the plasmid.L3[86] was overexpressed as a glutathione S-trans-ferase (GST) fusion protein in E. coli BL21 grown inLB medium, or for nuclear magnetic resonance (NMR)studies, in M9 minimal medium supplemented with 1g/L 15NH4Cl and 5 g/L [13C]glucose. Typically, an over-night culture was diluted 1:100 into LB or M9 mediumwith each 2-L flask containing 500 ml of culture. Thecultures were grown at 378C with shaking at 300 rpmuntil A600 5 0.3–0.4 at which time the heater wasturned off and the shaker allowed to cool to shakertemperature (29–308C). Protein expression was in-
duced at A600 5 0.6 by addition of IPTG to a final concen-tration of 0.4 mM. The bacteria were harvested thefollowing morning by centrifuging at 4000g. Bacteriawere lysed after suspending in 50 mM Tris, pH 8.0, 10
B
CHARACTERIZATION OF THEmM DTT and passing them through a French presstwice at 20,000 psi. After centrifuging the lysate at14.4g for 20 min, the supernatant was clarified usinga Beckman TL ultracentrifuge at 100,000g for 30 min.The supernatant was loaded onto a column containingglutathione–agarose and the column was washed with5 column vol of 50 mM Tris, pH 8.0, 1 mM DTT. Theprotein was eluted with 10 mM glutathione, 50 mMTris, pH 8.0, 1 mM DTT. After quantitation using a Bio-Rad protein microassay, bovine thrombin was added ata ratio of 80 units of thrombin:12 mg of GST-L3[86].The solution was left at room temperature overnightfor complete digestion.
Further purification of L3[86] were carried out usinga Superdex 75 (S-75) size-exclusion column (2.5 3 100cm) attached to a Biologic FPLC (Bio-Rad). The columnwas equilibrated in 50 mM sodium phosphate, pH 7.3,150 mM NaCl, 1 mM DTT and elution of the proteinwas monitored by absorbance at 280 nm. Fractions con-taining L3[86] were combined and concentrated usingan Amicon 8MC microultrafiltration system (YM10membrane). Following the S-75 step L3[86] was againincubated with glutathione–agarose to remove any re-maining GST-tag. The calculated pI of L3[86] is 7.94,the molecular weight is 9,973 Da, and the extinctioncoefficient is 13,940 M21 cm21 at 280 nm (assuming allCys residues are reduced) (23).
For L3[80], the DNA primers used to amplify thesequence by PCR were forward 58TATACATATGCAAA-CAAAGGTATTGCTGGA38 and reverse 58AGTGCGGC-CGCGCAGGGAGCTACCAGTCT38. The ligase IIIcDNA sequence corresponding to residues 843–922 wasligated into pET16b (Novagen). L3[80] protein was notsoluble when expressed alone in the E. coli host. How-ever, transformation of E. coli BL21(DE3) to both kana-mycin and ampicillin resistance with pET29-a(X1BRCTb) (described in (21)) and pET16b(L3[80]),followed by induction with IPTG, resulted in the pro-duction of a soluble L3[80]:X1BRCTb complex. The com-plex was expressed using conditions similar to that de-scribed for L3[86]. However, neither X1BRCTb norL3[80] contained an N-terminal GST or other fusionpartner, but L3[80] contained a C-terminal His-tag (in-cluding residues AAALEHHHHHH). The complex waspurified using the L3[80] His-tag on a Ni–NTA agarosecolumn (Qiagen). To keep the cysteines reduced, 0.1%bME was used instead of DTT prior to size-exclusionchromatography. The resulting L3[80] protein has acalculated pI of 8.77, molecular weight of 10,606 Da,and an extinction coefficient of 13,940 M21 cm21 at 280nm. For the L3[80]:X1BRCTb complex, the calculatedpI is 6.12, the molecular weight is 22,667 Da, and the
extinction coefficient is 31,720 M21 cm21 at 280 nm.The L3[80]:X1BRCTb complex is soluble up to 10 mg/ml but is most stable at lower concentrations for longterm storage.RCT DOMAIN OF LIGASE IIIa 403
SDS–Polyacrylamide Gel Electrophoresis
Protein concentrations were determined using theBio-Rad microassay protocol or by measuring the ab-sorbance at 280 nm. All SDS–PAGE protein sampleswere prepared using a 23 Tris–glycine sample buffer(Novex) with added DTT. The samples were heated for2–3 minutes at 908C prior to electrophoresis on a 4–20%polyacrylamide Tris–glycine gel (Novex) and stainedwith Gelcode blue stain (Pierce). Purity was assessed byanalyzing the stained polyacrylamide gel photographimage, using 1D image analysis software (Eastman Ko-dak Co.) to capture the image and calculate band inten-sities.
Size-Exclusion Chromatography
To estimate the molecular masses, each protein/com-plex sample was mixed with the following standardsand run on the S-75 column: aprotinin (6500 g/M), cyto-chrome C (12,400 g/M), carbonic anhydrase (29,000 g/M), and bovine albumin (66,000 g/M) (Sigma). The col-umn buffer used was 50 mM NaH2PO4, 150 mM NaCl,pH 7.3, or 50 mM KH2PO4, 500 mM KCl, pH 7.3. Thevoid volume (Vo) for the column was 187.5 ml as deter-mined by the elution of blue dextran (MW 5 2,000,000Da). The molecular weights of the proteins were deter-mined from a plot of Ve /Vo 5 log(MW) of the standards,where Ve is the elution volume of the protein and Vo isthe void volume. The resulting correlation coefficient(R2) for the standards was 0.998.
Circular Dichroism (CD)
Spectra were acquired on a Jasco 715 CD spectropo-larimeter using a 0.1-cm cell at 258C unless indicatedotherwise. The system was purged with nitrogen at 50L/min during data collection. Data acquisition parame-ters were as follows: sensitivity, 50 mdeg; resolution,0.5 nm; bandwidth, 1 nm; response, 8 s; scan speed, 20nm/min; and number of accumulations, 4. Backgroundspectra were acquired using the same buffer as usedfor the protein samples. After background subtraction,the spectra were analyzed by a backpropagation neuralnetwork algorithm using the program CircularDichroism Deconvolution (24) (http://bioinformatik.biochemtech.uni-halle.de/cdnn/).
Differential Scanning Calorimetry (DSC)
DSC measurements were performed on a Nano II-differential scanning calorimeter (Calorimetry SciencesCorp.) using a 0.299-ml capillary cell at three atmo-
spheres pressure. Prior to data acquisition, the pro-teins, in 100 mM DTT, were dialyzed (8000 MWCOdispodialyzer, Spectrum Laboratories) three times(1:500) against degassed 50 mM NaH2PO4, 150 mM
8 21 21
conducting the expression in M9 minimal medium sup-
404 THORNTO
NaCl, pH 7.3, at 48C. The dialysate was used for allconditioning and baseline runs as well as in the refer-ence cell. Protein concentrations were 1 mg/ml for allruns. Data analysis was performed using the programCPcalc (Calorimetry Sciences Corp.).
NMR
One dimensional proton and gradient and sensitivityenhanced 15N-1H HSQC (heteronuclear single quantumcorrelation) (25) spectra of 0.6 mM L3[86] dissolved in50 mM NaH2PO4, 150 mM NaCl, 1 mM DTT, pH 6.7,and 5% D2O at 158C were obtained using a 600-MHzVarian INOVA NMR spectrometer. 1H and 15N axeswere referenced with respect to the dimethyl silapen-tane sulfonate (DSS) signal at 158C and indirectly toDSS using the ratio of gs (Hg/Ng), respectively (26).The one dimensional experiment was obtained afterpresaturation of the water resonance (1.5 s with radiofrequency strength of 95 Hz), with an acquisition timeof 0.65 s over a spectral width of 12.5 kHz. In the 1H-15N HSQC experiment, the spectral widths were 7200and 1775 Hz for 1H and 15N dimensions, respectively.The acquisition times along the t2 and t1 dimensionswere 71 and 36 ms, respectively, for each complex point,while States-TPPI was used for quadrature detectionin the indirect dimension. Coherence selection wasachieved by Rance–Kay-type sensitivity enhancement(25) and the recycling delay between the scans was 1.5 swhile the signal was averaged over 64 transients. 15Nhard pulses were applied at a field strength of 10.2 kHz,while the decoupling was achieved by the multipulsesequence GARP1 (27) at 1.5 kHz. Spectra were pro-cessed and analyzed using the program FELIX (MSI,Inc.) and NMRPIPE (28).
Self-Diffusion Coefficient Measurements
Self-diffusion coefficient measurements of L3[86] dis-solved in 50 mM NaH2PO4,150 mM NaCl, 1 mM DTT,pH 6.7, and 5% D2O at 158C were obtained using aVarian INOVA 600 MHz NMR spectrometer and thebipolar pulsed-field gradient-selective echo dephasingsequence (2931). The experimental parameters were asfollows: acquisition time, 0.328 s; spectral width, 12,500Hz; signal averaging, 256 scans; recycling delay, 3 s;and water-selective pulse, 4 ms. Gradients were variedfrom 1 to 32 Gcm21 in units of 1.0 Gcm21, while theother gradients were applied at a strength of 30 Gcm21
for 1 ms each, yielding a total echo time (t1 1 t2) of14.026 ms. Phase cycling was used to advantageouslyutilize the radiation damping effects for water suppres-sion as previously reported (32). Time domain self-diffu-
sion coefficient data were zero filled once and a cosinebell apodization applied prior to complex Fourier trans-formation. The area under each spectrum from 5 to 21ppm was integrated and a nonlinear least square fit ofN ET AL.
equation (S(q) 5 S(0)exp(2Dsq2(D 2 d/3 2 t/2)) wasused to estimate the self-diffusion coefficients (30). S(q)is the measured integral value as a function of q andS(0) is the value at q 5 0. q is the effective area of thegradient pulse given by (ggzd), where g is the gyromag-
netic ratio of a proton (2.6752 3 10 s T ) and gz andg are the amplitude and duration of the gradient pulse,respectively. Ds is represented in units of m2 s21, whileD and t are delays in seconds employed in the pulse se-quence.RESULTS AND DISCUSSION
Expression and Purification of the Ligase IIIa BRCTDomain, L3[86]To characterize the BRCT domain of ligase IIIa
(L3BRCT) and its interaction with the distal BRCTdomain of XRCC1 (X1BRCTb), these proteins wereoverexpressed in bacteria and purified. This allowed usto study the biophysical properties of isolated L3BRCTand determine how they change upon binding withX1BRCTb. We observed that in the absence of a fusionpartner during expression, the L3[80] protein (residues843–922 of ligase IIIa) was insoluble. The slightlylonger L3[86] protein (residues 837–922), on the otherhand, was soluble when expressed as a GST fusionprotein. After thrombin cleavage, the L3[86] proteinhas two additional nonnative residues (Gly-Ser) at theN-terminus. The purification protocol, consisting of glu-tathione affinity and size exclusion chromatographysteps, yielded 14 mg of highly homogeneous L3[86] from1 liter of bacterial culture. The protein is soluble up to24 mg/ml at pH 7.3 in our preparations, and the finalL3[86] fraction was 98% pure as judged by densitomet-ric evaluation of proteins separated by SDS–gel electro-phoresis (Table 1 and Fig. 1).
For NMR structure and dynamic studies, the 15N-and 15N,13C-labeled L3[86] proteins were prepared by
plemented with 1 g/L 15NH4Cl and either 5 g/L unla-beled glucose or 4 g/L [13C]glucose. In general, the ex-pression yields in M9 medium were approximately halfthat obtained in LB medium.
TABLE 1
Purification of Overexpressed L3[86]
Protein Yield PurificationStep (mg) (%) (fold)
Lysate (soluble) 558 13.2 1GST-affinity (before
thrombin digestion) 83.8 89 6.7
Superdex 75 purified L3(86) 14.1 98 7.4Note. Yield and purification were determined by measuring thequantity of L3[86] in an SDS–polyacrylamide gel using densitometricscanning, compared to the total protein determined in each corres-ponding sample.
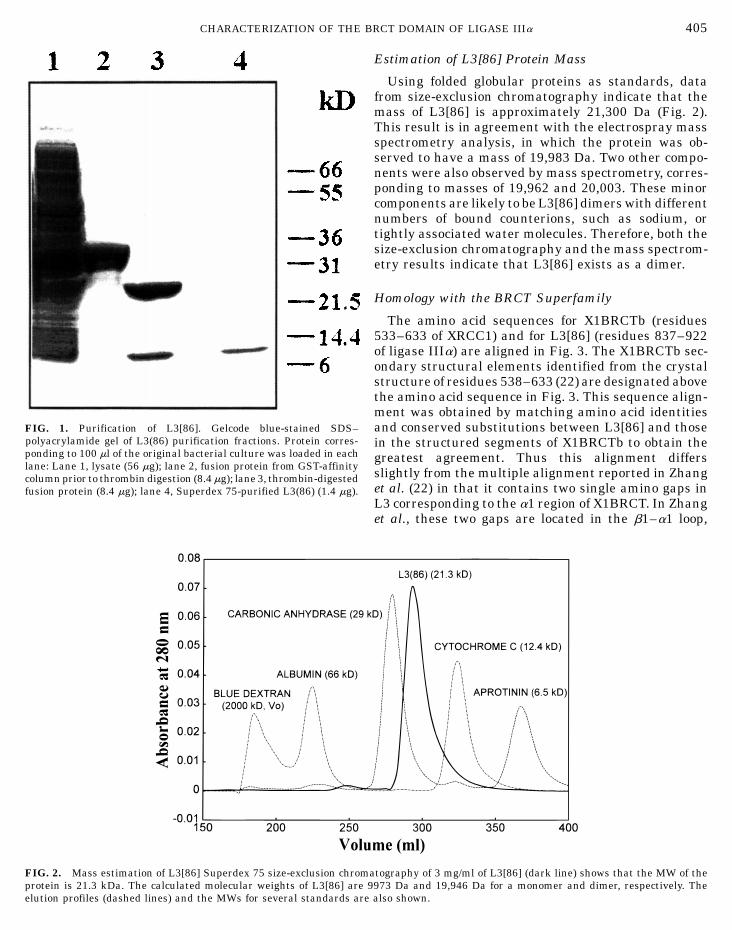
polyacrylamide gel of L3(86) purification fractions. Protein corres-ponding to 100 ml of the original bacterial culture was loaded in eachlane: Lane 1, lysate (56 mg); lane 2, fusion protein from GST-affinity
FIG. 2. Mass estimation of L3[86] Superdex 75 size-exclusion chromprotein is 21.3 kDa. The calculated molecular weights of L3[86] areelution profiles (dashed lines) and the MWs for several standards are
RCT DOMAIN OF LIGASE IIIa 405
Estimation of L3[86] Protein Mass
Using folded globular proteins as standards, datafrom size-exclusion chromatography indicate that themass of L3[86] is approximately 21,300 Da (Fig. 2).This result is in agreement with the electrospray massspectrometry analysis, in which the protein was ob-served to have a mass of 19,983 Da. Two other compo-nents were also observed by mass spectrometry, corres-ponding to masses of 19,962 and 20,003. These minorcomponents are likely to be L3[86] dimers with differentnumbers of bound counterions, such as sodium, ortightly associated water molecules. Therefore, both thesize-exclusion chromatography and the mass spectrom-etry results indicate that L3[86] exists as a dimer.
Homology with the BRCT Superfamily
The amino acid sequences for X1BRCTb (residues533–633 of XRCC1) and for L3[86] (residues 837–922of ligase IIIa) are aligned in Fig. 3. The X1BRCTb sec-ondary structural elements identified from the crystalstructure of residues 538–633 (22) are designated abovethe amino acid sequence in Fig. 3. This sequence align-ment was obtained by matching amino acid identitiesand conserved substitutions between L3[86] and thosein the structured segments of X1BRCTb to obtain thegreatest agreement. Thus this alignment differs
CHARACTERIZATION OF THE B
FIG. 1. Purification of L3[86]. Gelcode blue-stained SDS–
slightly from the multiple alignment reported in Zhang
column prior to thrombin digestion (8.4 mg); lane 3, thrombin-digestedet al. (22) in that it contains two single amino gaps infusion protein (8.4 mg); lane 4, Superdex 75-purified L3(86) (1.4 mg).L3 corresponding to the a1 region of X1BRCT. In Zhanget al., these two gaps are located in the b1–a1 loop,atography of 3 mg/ml of L3[86] (dark line) shows that the MW of the9973 Da and 19,946 Da for a monomer and dimer, respectively. Thealso shown.

FIG. 3. Sequence alignment of X1BRCTb and L3[86]. Consensus amare shown in bold underlined (identities) and italic underlined (conserdesignated by dashes. The secondary structure elements identified fromof X1BRCTb.
which is a highly variable region among the BRCT fam-ily. Positioning the two gaps within the a1-helix yields100% alignment between all the conserved residues(bold underlined, Fig. 3) and suggests that the homolo-gous a1-helix will be two residues shorter in the struc-ture of L3[86] compared to that in the X1BRCTb struc-ture. The L3[86] protein used in our studies is 9 aminoacid residues longer at the N-terminal end and eightresidues shorter at the C-terminal end compared to theX1BRCTb protein used in the crystallographic studies.The alignment also results in a single amino acid gapin the X1BRCTb b2–b3 loop, which is another highlyvariable region, and the same large 10- amino-acid gapin L3[86] as previously reported (22) that encompassesthe entire X1BRCTb a2-helix. The a2-helix inX1BRCTb is the least conserved and the most variableamong the BRCT family members (4). Based on ouralignment (Fig. 3), there is approximately 43% se-quence homology between the X1BRCTb(538–590) andL3[86](846–897) segments and 68% between theX1BRCTb(601–625) and L3[86](898–922) segments.The overall sequence homology (number of underlinedresidues divided by the 75 residues in the most highlyconserved regions) between these X1BRCTb and L3[86]segments is ,51%.
It is noteworthy that while X1BRCTb contains onecysteine residue at position 615, L3[86] contains 3(C842, C912, and C922) (boxed residues, Fig. 3). Thehighly conserved cysteine (C615 in X1BRCTb and C912in L3[86], Fig. 3) is buried in the crystal structure of
X1BRCTb (22) and also in the solution structure ofL3[86] (manuscript in preparation); thus it is not avail-able to participate in disulfide bond formation. The re-maining two L3[86] cysteines would not be expected toino acid residues between L3[86] and its binding partner X1BRCTbved substitutions). Gaps in the alignment between the sequences are
the crystal structure of X1BRCTb (22) are shown above the sequence
form intramolecular disulfide bonds in the folded pro-tein based on structural homology to the X1BRCTbstructure, in which the analogous positions of theL3[86] C842 and C922 residues would place them in theN- and C-terminal ends, respectively, of the X1BRCTbmolecule and thus more than 40 A apart from eachother (22). However, in the absence of reducing agent,the L3[86] C842 and C922 residues might participatein the formation of intermolecular disulfide bonds, butthis would be expected to produce a very complex mix-ture. For example, for nonspecific homodimer forma-tion, three possible configurations are C842A to C842B,C842A to C922B, and C922A to C922B, where A andB designate different monomeric L3[86] molecules. Inaddition, higher order aggregates would also be able toform as only one of these two cysteines would partici-pate in dimerization, while the second cysteine couldinteract with another monomer, dimer or higher orderaggregate to form an additional disulfide bond. Sincethis could lead to undesirable precipitation of the pro-tein, we found it was important to keep reducing agentpresent during purification and data collection. In thepresence of 1 mM DTT, we saw no evidence of a mixtureof higher ordered aggregates, and this has been con-firmed by the NMR structure of the protein. AdditionalNMR data also show that these cysteines do not partici-pate in forming intermolecular disulfide bonds. It hasbeen reported that the 13C NMR chemical shift of eachcysteine’s Cb carbon is extremely sensitive to its redoxstate (33). In reduced cysteine, the observed Cb shifts
406 THORNTON ET AL.
fall within 25.4 to 31.7 ppm, whereas for oxidized cys-teine they are located within 35.2 to 47.2 ppm (33). Ina very few cases, Cb shifts for oxidized and reducedstates can also overlap in a narrow range of 33.0 to 34.0

B
CHARACTERIZATION OF THEppm. However, under the reducing conditions used forour NMR studies, all the cysteines in L3[86] clearlyexhibit Cb chemical shifts (C842, 27.4 ppm; C912, 27.5ppm; and C922, 29.0 ppm) well within the range (25.4to 31.7 ppm) expected for the reduced state.
Secondary Structure Analyses of L3[86]
The far UV CD spectrum of L3[86] (Fig. 4A, top)provides information about the secondary structure ofthis BRCT domain. The percentages of helix, sheet, andcoil present were determined by fitting the far UV CDspectra (190–250 nm) to a theoretical model (24). Usingthis method, L3[86] is predicted to have 20% a-helixand 25% b-sheet. The X-ray structure of X1BRCTb has27.08% helix and 13.54% sheet (22). The smalleramount of helical secondary structure in L3[86] is notsurprising, considering that 10 residues comprising a2that are present in X1BRCTb would most likely not bepresent in the structure of L3BRCT (Fig. 3).
The near UV CD spectrum (250–310 nm, Fig. 4A,bottom) of L3[86] also provides information aboutwhether the aromatic residues are held rigidly in anasymmetric environment, as would be the case in thehydrophobic core of a structured protein, and whetherdisulfide bonds are present in the protein. However,
FIG. 4. (A) CD, DSC, and GuHCl denaturation of L3[86]. The far (1differential scanning calorimetry data, and (C) GuHCl denaturation curat 1.0 mg/ml purified L3(86) in 50 mM NaH2PO4, 150 mM NaCl, pHmg/ml, respectively, in 10 mM NaH2PO4, pH 7.0.
RCT DOMAIN OF LIGASE IIIa 407
were kept in a reduced state using DTT prior to acquir-ing the CD spectra and the sample was continuouspurged with nitrogen gas during acquisition. Therefore,any contribution present in the spectrum from disulfidebonds would be minimal. L3[86] contains five phenylal-anines (F851, F865, F872, F875, F883), two tyrosines(Y857, Y871), and two tryptophans (W908, W910) (Fig3). The presence of signals for these aromatic residuesin the near UV CD spectra of L3[86] confirm that signifi-cant structured regions are present, which is a prereq-uisite for tertiary structure determination by eitherNMR spectroscopy or X-ray diffraction studies.
L3[86] Denaturation Experiments
DSC experiments were carried out on L3[86] to deter-mine its thermal stability (Fig. 4B). Thermal denatur-ation was observed to be irreversible, with L3[86] pre-cipitating out of solution. In spite of the difficulties thispresented in the DSC analysis, the data indicate thatL3[86] undergoes a two-state single unfolding transi-tion with a melting temperature at 49 6 0.18C. TheDSC data confirm the CD secondary structure analysesthat L3[86] has a structured core.
A control experiment was also performed to deter-mine the percent of unfolded and folded protein by ti-trating the protein with different concentrations of the
the contribution of the disulfide chromophore is difficult chemical denaturant guanidine hydrochloride (GuHCl)(Fig. 4C). In agreement with CD and DSC, L3[86] exhib-to distinguish and is often buried under that of the
aromatic phenylalanine, tyrosine, and tryptophan resi- its an unfolding transition at 2 M GuHCl, indicative ofa protein that has tertiary structure in solution.dues. In addition, the cysteine residues in the protein
90–250 nm, top) and near (250–310 nm, bottom) UV CD spectra, (B)ve of L3(86) monitored by far UV CD. DSC experiments were acquired7.3. Far and near UV CD experiments were acquired at 0.1 and 1.0

408 THORNTO
Evidence for L3[86] Purity Determined by NMR
Both one-dimensional 1H and two-dimensional 1H–15N-HSQC NMR spectra of L3[86] exhibit good signaldispersion as expected for homogeneous, structuredproteins (Fig. 5). The spectra show reasonably sharplines, indicating that L3[86] is a good candidate formore detailed structural and dynamic studies, which
will be reported elsewhere. 15N and 15N,13C-labeled pro-FIG. 5. NMR spectra. (A) One-dimensional 1H NMR spectrum of the(25) of 15N-labeled L3[86].
N ET AL.
Homodimer Formation of L3[86] Supported by NMRDiffusion Coefficient Measurements
Since BRCT domains appear to participate in bothhomodimer and heterodimer association interactions,self-diffusion coefficient (Ds) measurements were un-dertaken to further characterize the oligomeric state ofL3[86] under NMR sample conditions (Fig. 6). The Ds
value of a protein, which measures the rate at which
it diffuses through aqueous solutions, is dependent ontein was used to assign all the proton, nitrogen (as the size and shape of the protein. The Ds of L3[86] wasshown for the backbone amide protons and nitrogens determined to be 11.2 6 0.3 3 10211 m2 s21 (Fig. 6), and
this value was compared to that obtained for lysozymein Fig. 5), and carbon chemical shifts in the NMR data.
unlabeled L3[86] and (B) 1H–15N gradient sensitivity enhanced HSQC
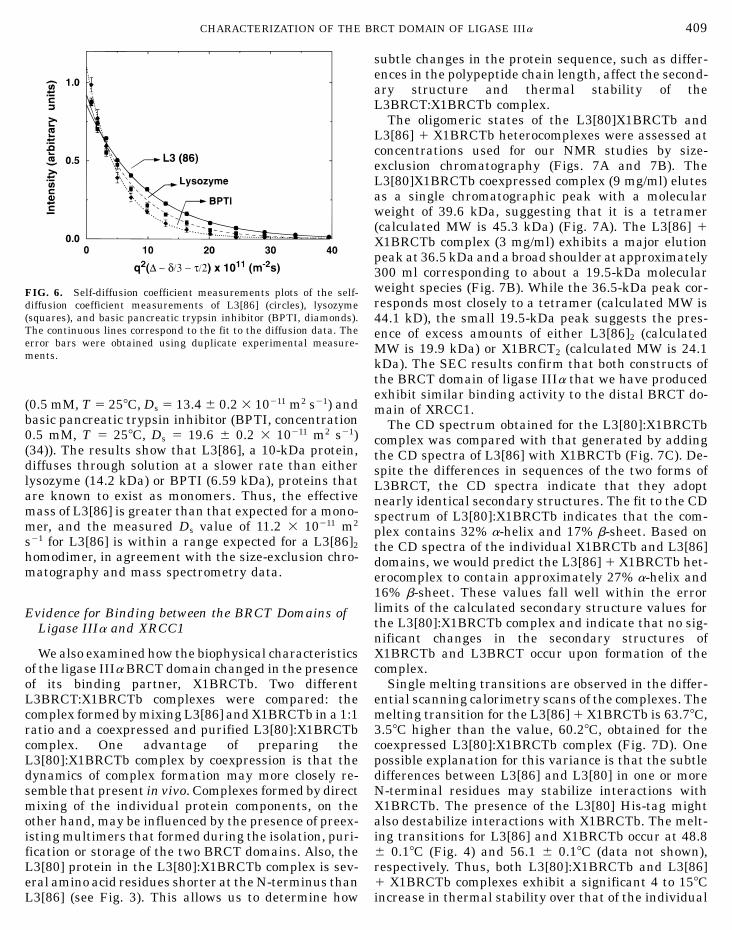
diffusion coefficient measurements of L3[86] (circles), lysozyme
ing transitions for L3[86] and X1BRCTb occur at 48.8
(squares), and basic pancreatic trypsin inhibitor (BPTI, diamonds).The continuous lines correspond to the fit to the diffusion data. Theerror bars were obtained using duplicate experimental measure-ments.
(0.5 mM, T 5 258C, Ds 5 13.4 6 0.2 3 10211 m2 s21) andbasic pancreatic trypsin inhibitor (BPTI, concentration0.5 mM, T 5 258C, Ds 5 19.6 6 0.2 3 10211 m2 s21)(34)). The results show that L3[86], a 10-kDa protein,diffuses through solution at a slower rate than eitherlysozyme (14.2 kDa) or BPTI (6.59 kDa), proteins thatare known to exist as monomers. Thus, the effectivemass of L3[86] is greater than that expected for a mono-mer, and the measured Ds value of 11.2 3 10211 m2
s21 for L3[86] is within a range expected for a L3[86]2
homodimer, in agreement with the size-exclusion chro-matography and mass spectrometry data.
Evidence for Binding between the BRCT Domains ofLigase IIIa and XRCC1
We also examined how the biophysical characteristicsof the ligase IIIa BRCT domain changed in the presenceof its binding partner, X1BRCTb. Two differentL3BRCT:X1BRCTb complexes were compared: thecomplex formed by mixing L3[86] and X1BRCTb in a 1:1ratio and a coexpressed and purified L3[80]:X1BRCTbcomplex. One advantage of preparing theL3[80]:X1BRCTb complex by coexpression is that thedynamics of complex formation may more closely re-semble that present in vivo. Complexes formed by directmixing of the individual protein components, on theother hand, may be influenced by the presence of preex-isting multimers that formed during the isolation, puri-
fication or storage of the two BRCT domains. Also, theL3[80] protein in the L3[80]:X1BRCTb complex is sev-eral amino acid residues shorter at the N-terminus thanL3[86] (see Fig. 3). This allows us to determine howRCT DOMAIN OF LIGASE IIIa 409
subtle changes in the protein sequence, such as differ-ences in the polypeptide chain length, affect the second-ary structure and thermal stability of theL3BRCT:X1BRCTb complex.
The oligomeric states of the L3[80]X1BRCTb andL3[86] 1 X1BRCTb heterocomplexes were assessed atconcentrations used for our NMR studies by size-exclusion chromatography (Figs. 7A and 7B). TheL3[80]X1BRCTb coexpressed complex (9 mg/ml) elutesas a single chromatographic peak with a molecularweight of 39.6 kDa, suggesting that it is a tetramer(calculated MW is 45.3 kDa) (Fig. 7A). The L3[86] 1X1BRCTb complex (3 mg/ml) exhibits a major elutionpeak at 36.5 kDa and a broad shoulder at approximately300 ml corresponding to about a 19.5-kDa molecularweight species (Fig. 7B). While the 36.5-kDa peak cor-responds most closely to a tetramer (calculated MW is44.1 kD), the small 19.5-kDa peak suggests the pres-ence of excess amounts of either L3[86]2 (calculatedMW is 19.9 kDa) or X1BRCT2 (calculated MW is 24.1kDa). The SEC results confirm that both constructs ofthe BRCT domain of ligase IIIa that we have producedexhibit similar binding activity to the distal BRCT do-main of XRCC1.
The CD spectrum obtained for the L3[80]:X1BRCTbcomplex was compared with that generated by addingthe CD spectra of L3[86] with X1BRCTb (Fig. 7C). De-spite the differences in sequences of the two forms ofL3BRCT, the CD spectra indicate that they adoptnearly identical secondary structures. The fit to the CDspectrum of L3[80]:X1BRCTb indicates that the com-plex contains 32% a-helix and 17% b-sheet. Based onthe CD spectra of the individual X1BRCTb and L3[86]domains, we would predict the L3[86] 1 X1BRCTb het-erocomplex to contain approximately 27% a-helix and16% b-sheet. These values fall well within the errorlimits of the calculated secondary structure values forthe L3[80]:X1BRCTb complex and indicate that no sig-nificant changes in the secondary structures ofX1BRCTb and L3BRCT occur upon formation of thecomplex.
Single melting transitions are observed in the differ-ential scanning calorimetry scans of the complexes. Themelting transition for the L3[86] 1 X1BRCTb is 63.78C,3.58C higher than the value, 60.28C, obtained for thecoexpressed L3[80]:X1BRCTb complex (Fig. 7D). Onepossible explanation for this variance is that the subtledifferences between L3[86] and L3[80] in one or moreN-terminal residues may stabilize interactions withX1BRCTb. The presence of the L3[80] His-tag mightalso destabilize interactions with X1BRCTb. The melt-
CHARACTERIZATION OF THE B
FIG. 6. Self-diffusion coefficient measurements plots of the self-
6 0.18C (Fig. 4) and 56.1 6 0.18C (data not shown),respectively. Thus, both L3[80]:X1BRCTb and L3[86]1 X1BRCTb complexes exhibit a significant 4 to 158Cincrease in thermal stability over that of the individual

8w
FIG. 7. Complexes. (A) Size-exclusion chromatography (SEC) of L3[ml, 36.5 kDa. The arrow indicates the presence of a minor componentof 0.1 mg/ml L3[80]X1BRCTb (solid line) compared to that obtainedX1BRCTb and dividing by 2 (dashed line). (D) DSC spectrum of L3[8and X1BRCTb (dashed line).
410 THORNTON ET AL.
components L3[86] and X1BRCTb. This result providesdirect experimental evidence that L3BRCT andX1BRCTb interact with one another to form a stablecomplex.
CONCLUSION
Using BRCT domains produced by overexpression inE. coli, we have demonstrated that the ligase IIIa BRCTdomain, L3[86], is autonomously folded and that it con-sists of both helical and b-sheet segments. Size-exclu-sion chromatography and NMR diffusion coefficientstudies show that L3[86] exists as a homodimer in solu-tion. We have prepared 15N- and 15N,13C-labeled L3[86]protein and the NMR data acquired are of sufficientquality to determine the structure and dynamics of theprotein in the solution state. Circular dichroism studiesshow that neither L3[86] nor X1BRCTb undergo a sig-nificant change in secondary structure upon binding toone another. However, calorimetry studies show thatL3[86] is stabilized by binding to the X1BRCTb domain,as evidenced by the 158C increase in the melting transi-
tion of the complex relative to that of L3[86] alone. Theincreased thermostability of the heterocomplex may ex-plain, in part, why the presence of XRCC1 is critical inmaintaining ligase IIIa levels and activity in vivo.0]:X1BRCTb at 9 mg/ml, 39.6 kDa; and (B) L3[86] 1 X1BRCT 3 mg/ith a molecular weight of approximately 19.5 kDa. (C) CD spectrumfrom adding the CD spectrum of 0.1 mg/ml L3[86] and 0.1 mg/ml
0]:X1BRCTb (solid line) compared to that of a 1:1 mixture of L3[86]
ACKNOWLEDGMENTS
The authors are grateful to Dr. Alan Tomkinson for the GSTag-ligase IIIa expression vector and to Dr. Rod Balhorn for critical read-ing and comments on the manuscript. This work was performed under
the auspices of the U.S. Department of Energy by the LawrenceLivermore National Laboratory under Contract W-7405-ENG-48,supported by the Lawrence Livermore National Laboratory LDRDDirector’s Initiative Grant 99-LW-031 to M.C., and partly funded byNIH Grant CA76116 to M.P.T.REFERENCES
1. Bork, P., Hofmann, K., Bucher, P., Neuwald, A. F., Altschul, S.F., and Koonin, E. V. (1997) A superfamily of conserved domainsin DNA damage responsive cell cycle checkpoint proteins. FASEBJ. 11, 68–76.
2. Callebaut, I., and Mornon, J. P. (1997) From BRCA1 to RAP1:A widespread BRCT module closely associated with DNA repair.FEBS Lett. 400, 25–30.
3. Huyton, T., Bates, P. A., Zhang, X., Sternberg, M. J. R., andFreemont, P. S. (2000) The BRCA1 C-terminal domain: Structureand function. Mutat. Res. 460, 319–332.
4. Koonin, E. V., Altschul, S. F., and Bork, P. (1996) Functionalmotifs. Nature Genet. 13, 266–268.
5. Futreal, P. A., Liu, Q. Y., Shattuckeidens, D., Cochran, C., Harsh-
man, K., Tavtigian, S., Bennett, L. M., Haugenstrano, A.,Swensen, J., Miki, Y., Eddington, K., McClure, M., Frye, C., Weav-erfeldhaus, J., Ding, W., et al. (1994) BRCA1 mutations in pri-mary breast and ovarian carcinomas. Science 266, 120–122.6. Friedman, L. S., Ostermeyer, E. A., Szabo, C. I., Dowd, P., Lynch,

CHARACTERIZATION OF THE B
E. D., Rowell, S. E., and King, M. C. (1994) Confirmation ofBRCA1 analysis of germline mutations linked to breast and ovar-ian cancer in ten families. Nature Genet. 8, 399–404.
7. Parikh, S. S., Mol, C. D., and Tainer, J. A. (1997) Base excisionrepair enzyme family portrait: Integrating the structure andchemistry of an entire DNA repair pathway. Structure 5, 1543–1550.
8. Caldecott, K. W., McKeown, C. K., Tucker, J. D., Ljungquist, S.,and Thompson, L. H. (1994) An interaction between the mamma-lian DNA repair protein XRCC1 and DNA ligase-III. Mol. Cell.Biol. 14, 68–76.
9. Caldecott, K. W., Tucker, J. D., Stanker, L. H., and Thompson,L. H. (1995) Characterization of the XRCC1–DNA ligase III com-plex in vitro and its absence from mutant hamster cells. NucleicAcids Res 23, 4836–4843.
10. Ljungquist, S., Kenne, K., Olsson, L., and Sandstrom, M. (1994)Altered DNA ligase III activity in the CHO EM9 mutant. Mutat.Res. 314, 177–186.
11. Shen, M. R., Zdzienicka, M. Z., Mohrenweiser, H., Thompson, L.H., and Thelen, M. P. (1998) Mutations in hamster single-strandbreak repair gene XRCC1 causing defective DNA repair. NucleicAcids Res. 26, 1032–1037.
12. Cappelli, E., Taylor, R., Cevasco, M., Abbondandolo, A., Caldecott,K., and Frosina, G. (1997) Involvement of XRCC1 and DNA ligaseIII gene products in DNA base excision repair. J. Biol. Chem.272, 23970–23975.
13. Wilson, S. H., and Singhal, R. K. (1998) Mammalian DNA repairand the cellular DNA polymerases in “DNA Damage and Repair:DNA Repair in Higher Eukaryotes,” pp. 161–180, Humana Press,Totowa, NJ.
14. Chen, J., Tomkinson, A. E., Ramos, W., Mackey, Z. B., Danehower,S., Walter, C. A., Schultz, R. A., Besterman, J. M., and Husain,I. (1995) Mammalian DNA ligase III: Molecular cloning, chromo-somal localization, and expression in spermatocytes undergoingmeiotic recombination. Mol. Cell. Biol. 15, 5412–5422.
15. Wei, Y. F., Robins, P., Carter, K., Caldecott, K., Pappin, D. J. C.,Yu, G. L., Wang, R. P., Shell, B. K., Nash, R. A., Schar, P., Barnes,D. E., Haseltine, W. A., and Lindahl, T. (1995) Molecular cloning
and expression of human cDNAs encoding a novel DNA ligaseIV and DNA ligase III, an enzyme active in DNA repair andrecombination. Mol. Cell. Biol. 15, 3206–3216.16. Mackey, Z. B., Ramos, W., Levin, D. S., Walter, C. A., McCarrey,J. R., and Tomkinson, A. E. (1997) An alternative splicing eventwhich occurs in mouse pachytene spermatocytes generates a formof DNA ligase III with distinct biochemical properties that mayfunction in meiotic recombination. Mol. Cell. Biol. 17, 989–998.
17. Nash, R. A., Caldecott, K. W., Barnes, D. E., and Lindahl, T.(1997) XRCC1 protein interacts with one of two distinct formsof DNA ligase III. Biochemistry 36, 5207–5211.
18. Masson, M., Niedergang, C., Schreiber, V., Muller, S., Menissier-deMurcia, J., and deMurcia, G. (1998) XRCC1 is specifically asso-ciated with poly(ADP-ribose) polymerase and negatively regu-lates its activity following DNA damage. Mol. Cell. Biol. 18, 3563–3571.
RCT DOMAIN OF LIGASE IIIa 411
19. Taylor, R. M., Wickstead, B., Cronin, S., and Caldecott, K. W.(1998) Role of a BRCT domain in the interaction of DNA ligaseIII-a with the DNA repair protein XRCC1. Curr. Biol. 8, 877–880.
20. Taylor, R. M., Moore, D. J., Whitehouse, J., Johnson, P., andCaldecott, K. W. (2000) A cell cycle-specific requirement for theXRCC1 BRCT II domain during mammalian DNA strand breakrepair. Mol. Cell. Biol. 20, 735–740.
21. Thornton, K., Forstner, M., Shen, M. R., West, M. G., Rupp,B., and Thelen, M. P. (1999) Purification, characterization, andcrystallization of the distal BRCT domain of the human XRCC1DNA repair protein. Protein Express. Purif. 16, 236–242.
22. Zhang, X. D., Morera, S., Bates, P. A., Whitehead, P. C., Coffer,A. I., Hainbucher, K., Nash, R. A., Sternberg, M. J. E., Lindahl,T., and Freemont, P. S. (1998) Structure of an XRCC1 BRCTdomain: A new protein–protein interaction module. EMBO J.17, 6404–6411.
23. Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995)How to measure and predict the molar absorption coefficient ofa protein. Protein Sci. 4, 2411–2423.
24. Bohm, G., Muhr, R., and Jaenicke, R. (1992) Quantitative analy-sis of protein far UV circular dichroism spectra by neural net-works. Protein Eng. 5, 191–195.
25. Kay, L. E., Keifer, P., and Saarinen, T. (1992) Pure absorptiongradient enhanced heteronuclear single quantum correlationspectroscopy with improved sensitivity. J. Am. Chem. Soc. 114,10663–10665.
26. Wishart, D. S., Bigam, C. G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J. L., and Sykes, B. D. (1995) H-1, C-13 and N-15 chemical shift referencing in biomolecular NMR. J.Biomol. NMR 6, 135–140.
27. Shaka, A. J., Barker, P. B., and Freeman, R. (1985) Computeroptimized decoupling scheme for wide-band applications and low-level operation. J. Magn. Res. 64, 547–552.
28. Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., andBax, A. (1995) NMRPipe—A multidimensional spectral pro-cessing system based on UNIX pipes. J. Biomol. NMR 6, 277–293.
29. Bax, A., and Subramanian, S. (1986) J. Magn. Reson. 67,565–569.
30. Wu, D. H., Chen, A. D., and Johnson, C. S. (1995) An improveddiffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 115, 260–264.
31. Mescher, M., Tannus, A., Johnson, M. O., and Garwood, M. (1996)Solvent suppression using selective echo dephasing. J. Magn.Reson. Ser. A 123, 226–229.
32. Krishnan, V. V., Thornton, K. H., and Cosman, M. (1999) Animproved experimental scheme to measure self-diffusion coeffi-cients of biomolecules with an advantageous use of radiationdamping. Chem. Phys. Lett. 302, 317–323.
33. Sharma, D., and Rajarathnam, K. (2000) C-13 NMR chemicalshifts can predict disulfide bond formation. J. Biomol. NMR18, 165–171.
34. Teller, D. C., Swanson, E., and de Haen, C. (1979) The trans-lational friction coefficient of proteins. Methods Enzymol. 61,103–124.




















