
Técnicas de microbiología bioquímica y biología molecular ...
Upload
khangminh22Category
view
1download
0
Universidade Federal do Rio Grande do Norte
Centro de Biociências
Programa de Pós-Graduação em Bioquímica
Estudo em complexos fármaco-receptor
utilizando bioquímica quântica
José Xavier de Lima Neto
Natal-RN
2019

José Xavier de Lima Neto
Estudo em complexos fármaco-receptor utilizandobioquímica quântica
Tese de Doutorado apresentada ao Programa
de Pós-Graduação em Bioquímica da UFRN,
como requisito parcial para a obtenção do
Título de Doutor em Bioquímica.
Orientador: Prof. Dr. Eudenilson Lins de
Albuquerque
Coorientador: Prof. Dr. Umberto Laino
Fulco
Natal-RN
2019

Lima Neto, Jose Xavier.
Estudo em complexos fármaco-receptor utilizando bioquímica
quântica / Jose Xavier de Lima Neto. - 2019.
140 f.: il.
Tese (doutorado) - Universidade Federal do Rio Grande do
Norte, Centro de Biociências, Programa de Pós-Graduação em
Bioquímica. Natal, RN, 2019.
Orientador: Prof. Dr. Eudenilson Lins de Albuquerque.
Coorientador: Prof. Dr. Umberto Laino Fulco.
1. GPCRs - Tese. 2. MFCC - Tese. 3. Energia de interação -
Tese. 4. Serotonina - Tese. 5. GABA - Tese. 6. Imunoterapia -
Tese. 7. Bioquímica quântica - Tese. I. Albuquerquer, Eudenilson
Lins de. II. Fulco, Umberto Laino. III. Título.
RN/UF/BCZM CDU 577.33
Universidade Federal do Rio Grande do Norte - UFRNSistema de Bibliotecas - SISBI
Catalogação de Publicação na Fonte. UFRN - Biblioteca Central Zila Mamede
Elaborado por Ana Cristina Cavalcanti Tinôco - CRB-15/262


AGRADECIMENTOS
Agradeço em especial aos deuses imortais; aos meus familiares por todas as horas
dedicadas em minha educação e suporte; ao Prof. Eudenilson Lins de Albuquerque por
seu exemplo como pesquisador e pessoa, sempre disposto a ouvir, aconselhar e auxiliar
da melhor maneira possível; ao Prof. Umberto Laino Fulco por uma década de trabalho
(em especial pela paciência, confiança e sabedoria); ao Prof. Valder Nogueira Freire por
apresentar o método utilizado nesta tese ao nosso laboratório, e pelo exemplo de mente
rápida e intuitiva; ao Prof. Jonas Ivan Nobre Oliveira por todas as conversas e discussões que
tivemos nos últimos dez anos; à Prof. Vanessa de Paula Soares Rachetti, ao sr. Raimundo,
dona Ivonete e aos demais professores e funcionários do Departamento de Biofísica e
Farmacologia; aos meus companheiros de laboratório (em especial a Katyanna Bezerra)
por auxiliar em meu engrandecimento profissional; ao Programa de Pós-Graduação em
Bioquímica pela oportunidade de quatro anos de engrandecimento profissional e pessoal; à
agência CAPES pelo apoio financeiro; e tantos outros, antigos ou recentes, que contribuíram
e motivaram de alguma forma, sabendo ou não, o presente trabalho.

”Não és mais, meu senhor, do que és: um mortal!
Perucas podes ter, com louros aos milhões.
Alçar-te com teus pés nos mais altos tacões,
Serás sempre o que és: um pobre ser mortal!”Trecho retirado do livro "Fausto", Johann Goethe
”Nosso cérebro é o melhor brinquedo já criado:
nele se encontram todos os segredos, inclusive o da felicidade.
A vida é maravilhosa se você não tem medo dela.”Charles Chaplin

Resumo
J. X. Lima Neto. Estudo em complexos fármaco-receptor utilizando bioquímicaquântica. 2019. 140f. Tese (Doutorado em Bioquímica). Universidade Federal do RioGrande do Norte, Natal-RN.
O processo de desenho/desenvolvimento de fármacos vem tornando-se cada vez maisaperfeiçoado ao passo que outras áreas de conhecimento são agregadas ao campo da ciênciafarmacêutica. Dentre essas adições, a introdução da simulação computacional foi semdúvida um dos grandes marcos para o crescimento da área como um todo. Hoje, dois sãoos focos principais no desenvolvimento de fármacos, os receptores acoplados à proteínaG, dentre eles os receptores serotonérgicos e GABAérgicos, e os imunoterapêuticos. Osreceptores 5-HT1B têm sido visados, principalmente, no tratamento de enxaqueca, uma vezque eles atuam no movimento de contração de artérias encefálicas. Em contrapartida, osreceptores que fazem parte da classe C de GPCRs têm sido vistos como potentes alvos deneuromoduladores, mas pouca atenção lhes foi dedicada. Dentre eles, os receptores GABAB
atuam como potentes inibidores do sinal neuronal, levando ao impedimento na liberaçãode vários neurotransmissores e fechamento de canais iônicos no encéfalo. Apesar de suaimportância farmacológica, a única molécula aprovada pelo Food and Drug Administration- FDA para atuar como modulador deste receptor é o baclofeno, sendo utilizado para otratamento de dores neuropáticas. Dentre os imunoterapêuticos, o mais notável tipo demolécula utilizada na clínica são os anticorpos monoclonais que atuam na inibição dainteração entre as proteínas de checkpoint em células T e os ligantes deste receptor nacélula cancerígena, sendo o principal alvo a proteína 1 de morte celular programável (PD-1)e seus ligantes PD-L1 e PD-L2. Neste sentido, este trabalho propõe uma avaliação em nívelquântico para descrever as interações realizadas por compostos que atuam nos receptores5-HT1B, PD-1 e GABAB com o intuito de aprofundar o conhecimento e descrever ospontos-chave para o acoplamento e fixação destes compostos em seus respectivos sítiosde ligação. Para tanto, utilizou-se a estrutura cristalográfica dos três receptores, sendofracionados em resíduos de aminoácido através do métodos de fracionamento molecularcom caps conjugados (MFCC) para posterior cálculo quântico. A partir desse estudo, foipossível predizer a relevância dos aminoácidos que compõem o sítio de ligação para ostrês receptores, dentre eles pode-se citar os resíduos Asp(D)129, Asp(D)352, Asp(D)123,Glu(E)198, Asp(D)204, Phe(F)330, Leu(L)126, Phe(F)351, Ile(I)130, Val(V)201, Val(V)200,Thr(T)355 e Arg(R)114, que fazem parte do sítio de ligação para diidroergotamina em5-HT e os resíduos Ser(S)130, Gly(G)151, Ser(S)153, His(H)170, Tyr(Y)250, Trp(W)278,Glu(E)349, Val(V)201, Ser(S)152, Ser(S)154, Gln(Q)348, Arg(R)168 e Trp(W)65, queformam o ponto de ancoramento de baclofeno, GABA, SCH50911 e 2-hidroxisaclofenoem GABAB, bem como o resíduo de lisina Lys(K)131 de PD-1 exibiu a interação resíduo-resíduo mais forte com os três ligantes estudados, pembrolizumab e nivolumab, assimcomo o ligante natural PD-L1. Estes aminoácidos apresentaram as maiores energias deatração ou repulsão, formando assim o principal ponto de ancoramento para os ligantesque interagem no sítio ortostérico. Utilizando esses dados, tornou-se possível identificar edescrever as regiões funcionais dos ligantes e proteínas, além de permitir uma explicaçãoe diferenciação em nível molecular e energético entre os níveis de afinidade encontradoexperimentalmente para os ligantes estudados.
Palavras-chave: gpcrs, mfcc, energia de interação, serotonina, gaba, imunoterapia.

Abstract
J. X. Lima Neto. Quantum biochemistry study in drug-receptor complexes. 2019.140f. Tese (Doutorado em Bioquímica). Universidade Federal do Rio Grande do Norte,Natal-RN.
The drug development process has been improved by the add of other knowledge areas inthe field of pharmaceutical sciences. Among these improvements, the introduction of thecomputational simulation was, with no doubt, one of the greatest landmarks for the field’sdevelopment. Nowadays, there are two main targets in drug development breakthrough,namely, the G protein-coupled receptors (GPCRs) and the immunotherapeutics. 5-HT1B
receptors have been targeted for the treatment of migraine, since they act in the brainarteries contraction. By the other hand, class C GPCRs are potent neuromodulators,though small attention has been dedicated to them, such as the GABAB receptors. Thesereceptor acts as potent inhibitors of the neuronal signal, leadind to impairments inthe neurotransmitters release and closure of ionic channels. Despite its pharmacologicalrelevance, only baclofen is approved by United States-Food and Drug Administration(US-FDA) as a molecule targeting GABAB, being used to treat neuropathic pain. Forthe class of immunotherapeutics, the most remarkable are the monoclonal antobodies(mAtb) which impair the binding between checkpoint proteins of T cells and their ligandsin cancer cells. The main target for these mAtb is the programmed cell death protein 1(PD-1) and its ligands, PD-L1 and PD-L2. In this sense, this work propose a quantumbiochemistry evaluation of the interactions made by compound thar act in 5-HT1B, PD-1e GABAB receptors aiming to deepen the knowledge and describe the key points for thedocking and stabilization of these compunds within the binding site. Thus, we take intoaccount the crystallographic structure of the three receptors, which were fractionated inamino-acid residues by the molecular fractionation with conjugate caps (MFCC) schemefor posterior quantum (Density Functional Theory - DFT) calculation. By our results, itwas possible to predict the relevance of the amino-acids that compose the binding site fromthe three receptors, including the residues Asp(D)129, Asp(D)352, Asp(D)123, Glu(E)198,Asp(D)204, Phe(F)330, Leu(L)126, Phe(F)351, Ile(I)130, Val(V)201, Val(V)200, Thr(T)355e Arg(R)114, that are part of the binding site for dihydroergotamine-5-HTR complex, theresidues Ser(S)130, Gly(G)151, Ser(S)153, His(H)170, Tyr(Y)250, Trp(W)278, Glu(E)349,Val(V)201, Ser(S)152, Ser(S)154, Gln(Q)348, Arg(R)168 e Trp(W)65, which composethe GABAB’s binding site for GABA, baclofen, SCH50911 and 2-hydroxysaclofen, aswell as the lisine residue Lys(K)131 from PD-1 as the most relevant for the coupling ofpembrolizumab, nivolumab and PD-L1. These residues shown the most intense attractionof repulsion energies, forming the key point for the ligand’s anchoring and stabilization.Through these data, it was possible to identify and describe the most important regionsfor ligands and receptors, as well as explain and differentiate, in a molecular and energeticlevel, the binding affinity experimentally found for these ligands.
Key-words: gpcrs, mfcc, interaction energy, serotonin, gaba, immunotherapy.

LISTA DE ILUSTRAÇÕES
Figura 1.1 – O crescimento dos métodos de escolha em análises in silico no campo daquímica medicinal e especificamente no desenvolvimento de fármacos.Dois conjuntos de termos foram utilizados para a busca: (a) Químicamedicinal e Métodos (vide legenda), e (b) Modelagem molecular e Método. 6
Figura 2.1 – Homologia entre os receptores de serotonina. . . . . . . . . . . . . . . . . 20Figura 2.2 – Estrutura do receptor 5-HT1B composto por sete hélices transmembranares,
duas alças intracelulares e extracelulares (PDB ID: 4IAQ). . . . . . . . . . 23Figura 2.3 – Estado de protonação de DHE. (a) Curva de fração molar variando o pH.
(b) Representação estrutural de diidroergotamina no estado protonado. (c)Superfície de potencial eletrostático. . . . . . . . . . . . . . . . . . . . . . 27
Figura 2.4 – Molécula de diidroergotamina circundada pelas alças e hélices de 5-HT1BR.O bolsão de ligação (r) é representado como um círculo em torno do ligante. 28
Figura 2.5 – Painel gráfico apresentando os resíduos mais relevantes que contribuem para ocomplexo DHE-5-HT1BR. A distância mínima entre cada resíduo e o ligante,bem como a região e os átomos do ligante interagindo com cada resíduono sítio de ligação estão representados. Em preto mostramos os resultadosencontrados para o MFCC e em ciano o EE-MFCC. . . . . . . . . . . . . . 30
Figura 2.6 – Visão detalhada dos resíduos mais importantes envolvidos na ligação deDHE com 5-HT1BR. Potenciais ligações de hidrogênio, cátion-π, e σ-π sãoindicados por linhas de cor azul, laranja e verde, respectivamente. . . . . . 31
Figura 2.7 – Superfície de potencial eletrostático de DHE interagindo com alguns dosaminoácidos mais importantes. . . . . . . . . . . . . . . . . . . . . . . . . 34
Figura 2.8 – Energia de interação total como função da distância do ligante. . . . . . . . 34Figura 2.9 – Resíduos com maior variação energética entre os métodos MFCC e EE-MFCC. 37Figura 3.1 – Complexação de (a) PD-L1, (b) Pembrolizumab (PEM) e (c) Nivolumab
(NIV) à PD-1. Os resíduos do sítio de ligação comum são apresentados.HC e LC representam a cadeia pesada (heavy chain) e cadeia leve (lightchain) do anticorpo monoclonal. . . . . . . . . . . . . . . . . . . . . . 48
Figura 3.2 – Perfil energético para cada resíduo de PD-1 na superfície de reconheci-mento comum.Esta figura representa a soma de todas as energias deinteração de um resíduo de PD-1 com cada aminoácido dos ligantesdentro do raio de 8,0 Å, usando as contantes dielétricas ε20 e ε40. . . . 50
Figura 3.3 – Direita: resíduos que interagem com K131 em cada complexo de PD-1/ligante. Esquerda: painel gráfico mostrando as interações mais rele-vante, considerando as constantes dielétricas deε40 and ε20. (a) Interaçãode K131 com os mais relevantes aminoácidos de PD-L1; (b) o mesmopara o sistema formado com os aminoácidos de PEM; (c) o mesmo parao sistema formado com os aminoácidos de NIV. As linhas tracejadasde cor azul, vermelha, laranja e verde representam ligações de hidro-gênio, interações cátion-π, pontes salinas e ligações de hidrogênio nãoconvencionais, respectivamente. . . . . . . . . . . . . . . . . . . . . . . 52
Figura 4.1 – Estrutura tridimensional dos receptores (a) GABAA e (b) GABAB. . . . . 57

Figura 4.2 – Estrutura tridimensional do domínio N-terminal do heterodímero formadopor GABAB1 e GABAB2. . . . . . . . . . . . . . . . . . . . . . . . . . . 60
Figura 4.3 – Representação estrutural dos ligantes GABA, baclofeno, 2-hidroxisaclofeno eSCH50911 (de cima para baixo) no estado neutro. A superfície de potencialeletrostático e apresentada ao lado direito de cada molécula. . . . . . . . . 64
Figura 4.4 – Energia de interação total como função da distância do ligante. (a) Resultadosforam obtidos a partir do EE-MFCC; (b-c) Resultados foram obtidos a partirdo MFCC utilizando ε10 e ε40, respectivamente. . . . . . . . . . . . . . . . 67
Figura 4.5 – Superposição da energia de interação individual encontrada para cada resíduode GABAB formando um par de interação com SCH50911. (a) EE-MFCC,(b-c) ε10 e ε40, respectivamente. Os resíduos são apresentados começandopor aqueles mais próximos do ligante até àqueles em 9,00 Å. . . . . . . . . 69
Figura 4.6 – Superposição da energia de interação individual encontrada para cada resíduode GABAB formando um par de interação com baclofeno. (a) EE-MFCC,(b-c) ε10 e ε40, respectivamente. Os resíduos são apresentados começandopor aqueles mais próximos do ligante até àqueles em 9,00 Å. . . . . . . . . 70
Figura 4.7 – Superposição da energia de interação individual encontrada para cada resíduode GABAB formando um par de interação com GABA. (a) EE-MFCC, (b-c)ε10 e ε40, respectivamente. Os resíduos são apresentados começando poraqueles mais próximos do ligante até àqueles em 9,00 Å. . . . . . . . . . . 71
Figura 4.8 – Superposição da energia de interação individual encontrada para cada resí-duo de GABAB formando um par de interação com 2-hidroxisaclofeno. (a)EE-MFCC, (b-c) ε10 e ε40, respectivamente. Os resíduos são apresentadoscomeçando por aqueles mais próximos do ligante até àqueles em 9,00 Å. . . 72
Figura 4.9 – Superposição da estrutura tridimensional de GABAB em complexos comGABA, baclofeno, SCH50911 e 2-hidroxisaclofeno. . . . . . . . . . . . . . 74
Figura 4.10–Painel gráfico apresentando os resíduos mais relevantes que contribuem parao complexo (a) SCH50911-GABABR, (b) baclofeno-GABABR, (c) GABA-GABABR e (d) 2-OHS-GABAB. . . . . . . . . . . . . . . . . . . . . . . . 76
Figura 4.11–Visão detalhada dos resíduos mais importantes envolvidos na ligação SCH50911,baclofeno, GABA e 2-OHS (de cima para baixo) com GABAB. Potenciaispontes salinas, ligações de hidrogênio, ligações de hidrogênio não convencio-nais, cátion-π, π-π e σ-π são indicados por linhas de cor vermelho, azul claro,verde, laranja e cinza e ciano escuro, respectivamente. . . . . . . . . . . . . 78
Figura A.1–Figura esquemática apresentando os momentos do MFCC. . . . . . . . . . 137

LISTA DE TABELAS
Tabela 2.1 – Descrição dos 108 resíduos de 5-HT1BR interagindo com DHE. Apresentam-se os grupos funcionais e as distâncias do ligante a cada aminoácido no sítiode ligação (2,0 - 10,0 Å). . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Tabela 2.2 – Energia de ligação e número de contatos das regiões individuais de DHE.Os resultados energéticos são apresentados para os cálculos realizadosutilizando os métodos MFCC e EE-MFCC. . . . . . . . . . . . . . . . 35
Tabela 2.3 – Energia de interação (MFCC e EE-MFCC) por estrutura secundáriado receptor de serotonina. . . . . . . . . . . . . . . . . . . . . . . . . . 36
Tabela 2.4 – Descrição energética dos 108 resíduos de 5-HT1BR interagindo com DHE.Cálculos realizados com o funcional M062X. . . . . . . . . . . . . . . . . . 39
Tabela 2.5 – Descrição energética dos 108 resíduos de 5-HT1BR interagindo com DHE.Cálculos realizados com o funcional B3LYP. . . . . . . . . . . . . . . . . . 40
Tabela 4.1 – Energia total de ligação (EE-MFCC ) e afinidade experimental (IC50)obtida para os quatro complexo, levando em consideração os cálculosrealizados com B97D, M062X, B3LYP, BHANDHLYP, PBE, PBEH1 eHF. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
Tabela 4.2 – Energia total de ligação (ε10) e afinidade experimental (IC50) obtidapara os quatro complexo, levando em consideração os cálculos realizadoscom B97D, M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF. . . . 66
Tabela 4.3 – Energia total de ligação (ε40) e afinidade experimental (IC50) obtidapara os quatro complexo, levando em consideração os cálculos realizadoscom B97D, M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF. . . . 66

LISTA DE ABREVIATURAS E SIGLAS
5-HT 5-Hidroxitriptamina
ADMET Absorção, Distribuição, Metabolismo e Excreção - Toxicidade
CCSD Coupled-Cluster Single Double
CHARMm Chemistry at HARrvard Molecular mechanics
DFT Teoria do Funcional da Densidade; do inglês, Density Functional Theory
DHE Diidroergotamina
DNA Ácido Desoxirribonucleico; do inglês, Deoxyribonucleic Acid
ERG Ergotamina
FDA Administração de Alimentos e Medicamentos; do inglês, Food and DrugAdministration
FMO Fracionamento do orbital molecular; do inglês, Fragment MolecularOrbital
GABA Ácido γ-aminobutírico
GGA Aproximação de gradiente generalizado; do inglês, Generalized-GradientApproximation
GPCR Receptores acoplados à proteína G; do inglês, G Protein-Coupled Re-ceptors
HF Hartree-Fock
HOMO Orbital molecular ocupado de mais alta energia; do inglês, HighestOccupied Molecular Orbital
irAEs Eventos adversos relacionados ao sistema imune; do inglês, ImmuneRelated Adverse Events
LSD Dietilamida do ácido lisérgico; do inglês, Lysergic acid diethylamide
LDA Aproximação de densidade local; do inglês, Local-Density Approximation
LUMO Orbital molecular desocupado de menor energia; do inglês, LowestUnoccupied Molecular Orbital
MFCC Fracionamento molecular com caps conjugados; do inglês, MolecularFractionation with Conjugate Caps
MM Mecânica Molecular (ou mecânica clássica)

MHC Complexo principal de histocompatibilidade; do inglês, Major Histo-compatibility Complex
MP2 Teoria da perturbação de Møller-Plesset [2]
MQ Mecânica Quântica
ONIOM do inglês, Our own N-layered Integrated molecular Orbital and MolecularMechanics
QSAR Relações quantitativas entre estrutura e atividade; do inglês, Quantita-tive Stucture-Activity Relationship
PD-1 Proteína 1 de morte celular programada; do inglês, Programmed celldeath protein 1
PD-L1 Ligante da proteína 1 de morte celular programada; do inglês, Program-med cell Death-Ligand 1
RMSD Raiz do desvio quadrático médio; do inglês, Root Mean Square Deviation
RNM Ressonância Nuclear Magnética
RNA Ácido Ribonucleico; do inglês, Ribonucleic Acid

SUMÁRIO
1 INTRODUÇÃO GERAL . . . . . . . . . . . . . . . . . . . . . . . 11.1 Uma perspectiva histórica sob o desenvolvimento de fármacos . 11.2 Química medicinal e a simulação computacional . . . . . . . . . . 31.3 A modelagem molecular no planejamento de fármacos baseado
na estrutura do receptor . . . . . . . . . . . . . . . . . . . . . . . . 51.4 A mecânica quântica no desenvolvimento de fármacos . . . . . . 91.5 Desenvolvimento de fármacos ligantes de GPCRs e anticorpos
monoclonais e a simulação computacional . . . . . . . . . . . . . . 131.6 Escopo da tese . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2 INTERAÇÃO DIIDROERGOTAMINA-RECEPTOR 5-HT1B
DESCRITA POR BIOQUÍMICA QUÂNTICA . . . . . . . . . . 182.1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182.1.1 Descrição geral dos subtipos de 5-HTR . . . . . . . . . . . . . . . . . . 202.1.2 Receptor 5-HT1B . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212.2 Materiais e Métodos . . . . . . . . . . . . . . . . . . . . . . . . . . . 242.2.1 Dados do complexo DHE-Receptor . . . . . . . . . . . . . . . . . . . . . 242.2.2 Cálculos clássicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242.2.3 Fracionamento molecular com caps conjugados e o efeito da incorporação
eletrostática . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242.2.4 Cálculos DFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252.2.5 Critérios de convergência . . . . . . . . . . . . . . . . . . . . . . . . . . 262.3 Resultados e Discussão . . . . . . . . . . . . . . . . . . . . . . . . . 262.3.1 Estrutura e distribuição de carga de diidroergotamina . . . . . . . . . . 262.3.2 Energia de interação dos fragmentos aminoacídicos . . . . . . . . . . . . 282.3.3 Energia total e convergência . . . . . . . . . . . . . . . . . . . . . . . . 332.3.4 Descrição energética das regiões do ligante . . . . . . . . . . . . . . . . 342.3.5 Descrição energética dos segmentos de 5-HT1BR . . . . . . . . . . . . . 352.3.6 Efeito do esquema de incremento eletrostático . . . . . . . . . . . . . . 362.3.7 Reprodutibilidade utilizando outros métodos de cálculo . . . . . . . . . 382.4 Conclusão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3 DESCREVENDO INIBIDORES DE PROTEÍNAS DE CHECK-
POINT CELULAR POR BIOQUÍMICA QUÂNTICA . . . . . 423.1 A imunoterapia contra o câncer . . . . . . . . . . . . . . . . . . . . 433.2 Materiais e Métodos . . . . . . . . . . . . . . . . . . . . . . . . . . . 473.2.1 Dados do complexo ligante-receptor . . . . . . . . . . . . . . . . . . . . 473.2.2 Cálculos clássicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 473.2.3 Fracionamento molecular com caps conjugados . . . . . . . . . . . . . . 473.2.4 Cálculos DFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 493.3 Resultados e discussão . . . . . . . . . . . . . . . . . . . . . . . . . . 493.4 Conclusão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51

4 ESTUDO IN SILICO DO RECEPTOR GABAB E SEUS AGO-NISTAS/ANTAGONISTAS . . . . . . . . . . . . . . . . . . . . . 54
4.1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 544.1.1 O receptor GABAB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 564.2 Materiais e Métodos . . . . . . . . . . . . . . . . . . . . . . . . . . . 614.2.1 Dados do complexo ligante-Receptor . . . . . . . . . . . . . . . . . . . . 614.2.2 Cálculos clássicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 614.2.3 Fracionamento molecular com caps conjugados e o efeito do ambiente . 614.2.4 Cálculos DFT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 624.2.5 Critérios de convergência . . . . . . . . . . . . . . . . . . . . . . . . . . 634.3 Resultados e discussão . . . . . . . . . . . . . . . . . . . . . . . . . . 634.3.1 Estrutura e distribuição de carga dos ligantes . . . . . . . . . . . . . . . 634.3.2 Energia total, convergência e efeito dos modelos de cargas pontuais e
constante dielétrica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 654.3.3 Energia de interação dos fragmentos aminoacídicos . . . . . . . . . . . . 734.4 Conclusão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
5 CONCLUSÃO FINAL E PERSPECTIVAS . . . . . . . . . . . . 825.1 Conclusão Final . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 825.2 Perspectivas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
REFERÊNCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
APÊNDICES 112
APÊNDICE A – ARTIGOS PUBLICADOS/SUBMETIDOS DU-RANTE O PERÍODO DE DOUTORAMENTO . 113
ANEXOS 129
ANEXO A – FUNDAMENTOS TEÓRICOS DA BIOQUÍMICACOMPUTACIONAL . . . . . . . . . . . . . . . . . . . 130
A.1 Métodos clássicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131A.2 Métodos ab initio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132A.3 Métodos Semi-empíricos . . . . . . . . . . . . . . . . . . . . . . . . 133A.4 Teoria do Funcional da Densidade . . . . . . . . . . . . . . . . . . 133A.5 Fracionamento molecular com caps conjugados - MFCC . . . . 135

1
capítulo 1
INTRODUÇÃO GERAL
O processo de desenho/desenvolvimento de fármacos vem tornando-se cada vezmais aperfeiçoado ao passo que outras áreas de conhecimento são agregadas ao campo dasciências farmacêuticas. Como observado por Drews (2000), o primeiro momento de evoluçãodeste processo, como é conhecido atualmente, se deu com a maturidade e expansão daquímica e com a consolidação da farmacologia como uma disciplina científica, o que só veioa ocorrer no século XIX (DREWS, 2000). O fim do século XX trouxe consigo o advento demétodos experimentais que permitiram a manipulação direta dos modelos de estudo, comoalterações genéticas em células e animais modelo, para o desenvolvimento de avaliações invitro e in vivo, respectivamente, bem como a utilização de métodos computacionais quetornaram menos laborioso o processo de descoberta de novos fármacos. Contudo, deve-senotar que o caminho trilhado até esse momento foi bastante longo. Dessa forma, esta teseé iniciada com uma breve apresentação cronológica sobre a estrada percorrida nestes quasedois séculos, sendo sucedida pelo foco metodológico principal deste trabalho, os métodosin silico, e sua utilização no processo de desenvolvimento de fármacos.
1.1 Uma perspectiva histórica sob o desenvolvimento de fármacos
De acordo com Newman, Cragg e Snader (2000), a descoberta de fármacos iniciou-se quando produtos naturais, principalmente extratos de vegetais, foram utilizados compropósito medicinal pela medicina tradicional (NEWMAN; CRAGG; SNADER, 2000).Posteriormente, com o avanço dos métodos químicos, esses extratos foram refinados elevaram ao isolamento de substância ativas, o que teve um grande impacto na químicamedicinal (ou química farmacêutica, dependendo do autor). Drews afirma que um dosprincipais exemplos deste momento foi a obtenção do analgésico morfina, obtido a partirdo ópio originado da papoula (DREWS, 2000). O surgimento da química analítica nofinal do século XIX tornou possível isolar compostos bioativos, sendo esse um processode grande relevância, que, no começo do século XX, foi acrescido por conceitos novosadvindos da química, como a compreensão da aromaticidade, e que foram importantesna percepção e maturação posterior da teoria dos quimiorreceptores por Paul Ehrlich e,posteriormente, na formulação do conceito de "substâncias receptoras" desenvolvido por J.N. Langley (EHRLICH, 1908; LANGLEY, 1901).
A base do pensamento de Langley era a de que haveriam moléculas receptorasque captariam sinais e gerariam respostas fisiológicas ou farmacológicas. Embora bastantecontroversa na época, sendo visto com maus olhos mesmo por seus alunos que na épocaestudavam esse comportamento em receptores adrenérgicos, a ideia de receptores molecu-lares estimulou a curiosidade daqueles que já trabalhavam com "química medicinal", o quelevou posteriormente ao desenvolvimento de princípios-chave para a "nova farmacologia"que surgia, como as interações ligante-receptor (THOMAS, 2013). Não obstante, a investi-gação de Ehrlich em 1908 pavimentou o caminho para a obtenção dos primeiros fármacossintéticos, bem como para a base dos modelos de triagem de moléculas e os procedimentosde avaliação de novos compostos (PARASCANDOLA, 1981; BOSCH; ROSICH, 2008).
Dando um pequeno salto no tempo, não há como duvidar que a descoberta da

Capítulo 1. Introdução Geral 2
penicilina, em 1928, foi um dos eventos mais marcantes na constituição da indústria far-macêutica. As penicilinas consistem de uma mistura de beta-lactâmicos que compartilhamum núcleo estrutural central e que possuem uma característica de antibiótico, tendo sidobastante útil durante a segunda guerra mundial. Como apresentado acima, a convergênciaentre a característica química de um composto (conhecimento que vem das novas teoriasemergentes na química), sua estrutura molecular e a resposta biológica apresentada apóssua utilização formaram a base para o desenvolvimento de novos compostos análogos àpenicilina após a elucidação de sua estrutura (NEWMAN; CRAGG; SNADER, 2000).Esses análogos logo começaram a ser produzidos e comercializados em larga escala, e abusca por novos antibióticos com estrutura baseada na penicilina foi intensificada.
O período de transição entre as décadas de 1950 e 1960 foi bastante fecundopara o campo da farmacologia, quando vê-se o desfecho de anos de estudo por váriospesquisadores com o artigo publicado por Watson e Crick sobre a estrutura do DNA(FRANKLIN; GOSLING, 1953; WILKINS; STOKES; WILSON, 1953; WATSON; CRICK,1953; PAULING; COREY, 1953), o que serviu de sustentáculo para avanços significativosem técnicas de biologia molecular e celular, incluindo técnicas de DNA recombinante,clonagem e, mais recentemente, os métodos de edição do DNA, que auxiliaram em umarestruturação de toda a indústria farmacêutica (STERNBERG et al., 2014; GERSHELL;ATKINS, 2003; ALBUQUERQUE et al., 2014). Não obstante, o começo da década de1960 viu o nascer de métodos de estudo voltados à análise de relações quantitativas entreestrutura e atividade de compostos, o QSAR (sigla em inglês para a expressão quantitativestucture-activity relationship). Esses métodos aproximaram a estatística, em especial abioestatística, do contexto farmacológico com a comparação entre a composição químicade uma biblioteca de moléculas (um conjunto de centenas ou milhares de compostos) ea atividade em estudo (obtida por análises experimentais), podendo essa atividade sercitotoxicidade, afinidade, potência, eficácia, dentre outras características.
Entretanto, é somente no início da década de 1990 que o desenvolvimento dessasnovas tecnologias uniu-se massivamente ao aperfeiçoamento da química computacional, àemergência dos primeiros bancos de dados de proteínas (uniprot e protein data bank) eao surgimento de métodos em modelagem molecular e bioinformática. A indústria farma-cêutica, juntamente com os avanços na terapia gênica e na compreensão dos mecanismosque causam as doenças, e os resultados de pesquisa do Projeto Genoma Humano, abriramuma infinidade de oportunidades e tornaram possível o desenvolvimento e uso de fármacosvisando especificamente os locais onde as doenças são causadas (LOMBARDINO; III,2004). De fato, o rápido avanço nas ferramentas de modelagem molecular, bem como aaplicação da química combinatória e métodos de triagem de compostos em larga escalatrouxe consigo benefícios imediatos na busca por moléculas "protótipo" (lead compounds)e no processo de otimização dessas. Em pouco tempo, várias bibliotecas de fármacosdesenvolvidos racionalmente, baseados na estrutura química de moléculas já conhecidas,foram criadas e rapidamente triadas na busca de fármacos com melhores respostas, oque diminuiu o tempo gasto entre a formação da biblioteca e a apuração/validação dosresultados (TRIGGLE, 2007; KUBINYI, 2003). Por fim, os dados encontrados eram,e ainda são, utilizados para a otimização do composto em estudo, levando a um ciclointerativo de refinamento, síntese e triagem até a obtenção da propriedade desejada.
Todos esses avanços tecnológicos de ferramentas para o desenvolvimento de fárma-cos levou à crença em um crescimento revolucionário na habilidade de busca de novoscompostos e identificação de novos alvos terapêuticos. Porém, a partir de uma análise dos

Capítulo 1. Introdução Geral 3
medicamentos introduzidos no mercado entre 1990 e 2001, não observam-se vantagenssignificativas desses novos produtos sobre àqueles já no mercado, uma vez que esses com-postos apresentaram maior risco e menor taxa de sobrevida no mercado (MA; ZEMMEL,2002). Assim, há uma divergência entre os pesquisadores sobre a real importância dautilização dessa miscelânea de métodos e áreas de conhecimento no desenvolvimento denovos fármacos, uma vez que houve um aumento significativo na produtividade, mas onúmero de compostos submetidos aos ensaios clínicos e mostrando resultados adequadosnão acompanha esse crescimento (BOOTH; ZEMMEL, 2004).
Apesar do exposto, hoje a bioinformática e outras técnicas in silico tornaram-seimprescindíveis para organizar e gerenciar informações obtidas no processo de descobertae desenvolvimento de fármacos, aumentando a eficiência e a velocidade de análise destasinformações. Neste cenário são observadas algumas estratégias predominantes, sendoas principais aquelas empregadas para estudar as interações dos fármacos com os alvosterapêuticos, apoiadas em técnicas como cristalografia de raios-X, espectrometria de massase ressonância magnética nuclear. Segundo estimativas, estas tecnologias in silico podemreduzir o custo do desenvolvimento de um novo fármaco na ordem de milhões de dólares eno encurtamento de alguns meses de trabalho.
1.2 Química medicinal e a simulação computacional
Mesmo com as tecnologias atuais, o processo de descoberta e desenvolvimento deum novo fármaco potencialmente eficaz e com o mínimo de efeitos adversos é longo eoneroso. A estimativa básica para todo o processo, iniciando na concepção do projetoaté a introdução de um único fármaco no mercado, é de 12-15 anos, com custo estimadona ordem de US$ 500-880 milhões, podendo chegar a US$ 1 bilhão (LOMBARDINO;III, 2004; DIMASI; HANSEN; GRABOWSKI, 2003; ADAMS; BRANTNER, 2006). Nãoobstante, a cada 10.000 compostos considerados promissores em ensaios iniciais, menosde 10 chegam em ensaios clínicos e, sendo bastante otimista, 1 é aprovado pelos órgãosreguladores (NG, 2015).
É nesse contexto que a química medicinal insere-se, contribuindo de modo decisivona descoberta e no desenvolvimento de novos agentes terapêuticos. Por ser uma área deconhecimento completamente multidisciplinar, a química medicinal utiliza-se de diferen-tes abordagens metodológicas no planejamento racional de fármacos. Assim, moléculascandidatas podem ser correlacionadas estruturalmente objetivando planejar análogos combase no tipo de interação ligante-biomacromolécula. Através de pesquisas das relaçõesentre estrutura química e atividade biológica (SAR e/ou QSAR), a estrutura do compostoprotótipo poderá ser otimizada, validada e escolhida para estudos in vitro e posteriormenteselecionadas para ensaios pré-clínicos e clínicos.
A base teórica da química medicinal está intimamente associada à farmacologiamolecular no sentido de compreender cada vez mais as razões moleculares das intera-ções fármaco-receptor (THOMAS, 2013). Como descrito por Cavalli e colaboradores, oconhecimento completo da estrutura molecular é essencial na química medicinal, uma vezque o complexo formado entre moléculas bioativas e os receptores acarreta nas respostasbiológicas, sendo dependente de um mecanismo de reconhecimento molecular, que auxiliana seletividade aos receptores (CAVALLI; CARLONI; RECANATINI, 2006). Na mesmalinha de pensamento, Jing e Han (2010) apresentam a ideia de que um dos principaisobjetivos da pesquisa bioquímica é entender as interações entre a proteína e seus ligantes,

Capítulo 1. Introdução Geral 4
pois isso é o que leva a realização das funções pelos receptores.
Através de ferramentas in silico, tornou-se possível acelerar o processo de desen-volvimento de fármacos por intermédio da predição de propriedades-chave, tais como aabsorção, distribuição, metabolismo e excreção - toxicidade (ADMET ) desses compos-tos. Um composto-protótipo pode ser citotóxico, rapidamente eliminado pelo corpo, sermetabolizado por enzimas antes de atingir seu alvo, ser instável, de difícil síntese, oumesmo de produção inviável economicamente (AUGEN, 2002). Nesse sentido, o processode desenvolvimento de novas moléculas que ligam-se à proteínas alvo tem saído do labora-tório em direção ao computador com bastante intensidade. Modelos gerados por técnicascomputacionais, que servem como bons modelos preditivos de atividade biológica, vemapresentando grande sucesso na predição da estrutura de alvos biológicos, reduzindo, dessaforma, esforços infrutíferos no uso de métodos experimentais, caminhando em conjuntocom as "técnicas de bancada"(ARODOLA; SOLIMAN, 2017).
Nas fases iniciais do processo de desenvolvimento racional de fármacos, geralmente,as moléculas bioativas, ou ligantes, apresentam baixa afinidade, necessitando ser otimizadasem relação a uma série de propriedades (como exemplo, potência, afinidade, seletividade,biodisponibilidade e toxicidade). De acordo com Raha et al. (2007), duas estratégiasbásicas podem ser exploradas no planejamento racional de compostos bioativos (in silico):
• Planejamento baseado na estrutura do ligante (Ligand-Based Drug Design - LBDD)
• Planejamento baseado na estrutura do receptor (Structure-Based Drug Design -SBDD)
No primeiro caso, as interações entre ligante-receptor são levadas em consideraçãoindiretamente, a partir da correlação entre a atividade biológica conhecida experimen-talmente e a estrutura química desses ligantes. Essa correlação se dá por intermédio daseleção de parâmetros físico-químicos que podem ser utilizados como modo de comparaçãocom o efeito biológico, sendo o método mais conhecido o QSAR. Já no segundo caso, asinterações intermoleculares são consideradas explicitamente, necessitando normalmente dautilização de estruturas tridimensionais do alvo molecular para sua realização.
Nota-se, contudo, que em um nível experimental a observação da estrutura molecularde um ligante e de seu receptor é tarefa árdua. São poucos os métodos capazes decaracterizar completamente uma dada estrutura, permitindo sua descrição precisa emtermos espaciais. A cristalografia de raios-X é a técnica mais utilizada para a obtençãodesses dados, embora a ressonância nuclear magnética também tem sido bastante utilizada.Não obstante, ambos os métodos apresentam limitações severas em sua utilização, taiscomo a falta de mobilidade da molécula estudada e o tamanho do sistema (DAVIS;TEAGUE; KLEYWEGT, 2003). Ademais, experimentos podem prover uma caracterizaçãodetalhada da estrutura de macromoléculas biológicas, porém é difícil estudar diretamenteas modificações sofridas por estas moléculas em um pequeno espaço de tempo, assimcomo o mecanismo molecular de processos rápidos, como reações químicas ou o transporteiônico. Portanto, simulações baseadas em princípios físicos têm oferecido o potencial parapreencher os "espaços" não conhecidos, modelando o modo pelo qual estas macromoléculasinteragem, reagem e funcionam (KAMP et al., 2008).
Assim, a modelagem molecular por métodos computacionais surgiu como umaalternativa capaz de estimar a estrutura tridimensional de ligante e receptor, individual-

Capítulo 1. Introdução Geral 5
mente e formando complexos, com um compromisso adequado entre velocidade e precisão(BLUNDELL et al., 2006). Portanto, observa-se a modelagem molecular como um conjuntode ferramentas utilizadas para a construção, edição e visualização, análise e armazenamentode sistemas moleculares complexos.
Uma grande variedade de técnicas de simulação computacional têm sido desenvol-vidas e aplicadas em diversos problemas biomoleculares, como o dobramento de proteínase modificações em suas conformações (DAGGETT, 2006; ELCOCK, 2006), associação deproteínas com pequenas moléculas (MOITESSIER et al., 2004), desenho de drogas baseadoem estrutura (TAFT; SILVA et al., 2008), obtenção da energia livre de ligação (GILSON;ZHOU, 2007), dinâmica de canais iônicos e transporte através da membrana (BECKSTEINet al., 2003) e a modelagem e análise de mecanismos enzimáticos (WARSHEL et al., 2006;KAMP et al., 2008).
Especificamente no desenho de compostos bioativos, a modelagem molecular temse mostrado uma poderosa ferramenta no processo de seleção, identificação, manipula-ção, otimização e caracterização de novos fármacos por sua capacidade de determinarpropriedades termodinâmicas, energéticas e estruturais de complexos receptor-ligante. Ocrescimento no número de estudos utilizando técnicas de modelagem molecular no desenhoracional de fármacos para os últimos dez anos pode ser observado através da figura 1.1,onde percebe-se claramente um aumento na quantidade de artigos publicados, bem comouma preferência por métodos baseados em mecânica clássica, como a dinâmica molecular(YAO et al., 2010).
1.3 A modelagem molecular no planejamento de fármacos baseado na estru-tura do receptor
Existem muitas opções quanto ao método de cálculo a ser aplicado em umadeterminada estratégia de modelagem molecular (BECKER et al., 2001). Neste tópicofaremos uma breve apresentação e explanação sobre o uso frequente desses métodos noâmbito do desenho de fármacos e da química medicinal.
A modelagem por homologia, também conhecida como modelagem comparativa,fornece a estrutura tridimensional de macromoléculas a partir da sequência de aminoácidosdo alvo molecular. Esse método requer a existência de uma estrutura tridimensionalpreviamente obtida por técnicas experimentais de uma proteína homóloga, que possa servircomo template. A base teórica utilizada pela modelagem comparativa é uma das maisacuradas entre as técnicas de predição de estrutura, tendo por alicerce o preceito de que aestrutura de uma proteína pode ser construída a partir de outra evolutivamente similar ecom sequência de aminoácidos homóloga, e que possua estrutura obtida experimentalmente.
Dessa forma, se a identidade de sequência entre a proteína em estudo e aquela queservirá como molde 3D for maior que 30%, os modelos de homologia podem ser construídoscom alta confiabilidade. Quando isto não ocorre, isto é, quando a identidade entre assequências for menor que 30%, a modelagem ab initio pode ser utilizada para a criação domodelo através de uma base mais próxima dos princípios físico-químicos utilizados pelosdemais métodos que serão apresentados posteriormente (SÁNCHEZ et al., 2000; LEACH;LEACH, 2001).
Os passos para realizar uma modelagem por homologia geralmente consistem de:
• Identificação da família à qual pertence a sequência alvo e seleção de um template;

Capítulo 1. Introdução Geral 6
Fonte: Web of Science
Figura 1.1 – O crescimento dos métodos de escolha em análises in silico no campo daquímica medicinal e especificamente no desenvolvimento de fármacos. Doisconjuntos de termos foram utilizados para a busca: (a) Química medicinal eMétodos (vide legenda), e (b) Modelagem molecular e Método.

Capítulo 1. Introdução Geral 7
• Alinhamento da sequência alvo com template;
• Construção do modelo comparativo;
• Avaliação do modelo construído;
Vale ressaltar que um dos fatores mais relevantes para determinar a qualidade damodelagem por homologia é a porcentagem de identidade entre a sequência de aminoácidosda proteína alvo e seu template, pois observa-se uma relação direta entre esses fatores.Dessa forma, quando a identidade está entre 30%-50%, o modelo gerado tende a apresentaraproximadamente 80% de seus átomos a uma distância menor que 3,5 Å da posição esperada.Ademais, quando a identidade é superior a 60%, o modelo aproxima-se, em qualidade,de proteínas obtidas por cristalografia com resolução padrão (aproximadamente 2,0 Å).Não só isso, hoje existem ferramentas que ajudam a acessar a qualidade estereoquímicados modelos, tais como o Molprobity e o Procheck (SALI; KURIYAN, 1999; CHEN etal., 2010; LASKOWSKI et al., 1993; MOSIER; KELLOGG, 2008). Por isso, modelosgerados por modelagem comparativa vem sendo ferramentas bastante úteis para darapoio ao desenho experimental de análises por mutagênese, por exemplo, examinando acapacidade de modulação por inibidores potenciais, bem como na construção de estruturastridimensionais de proteínas que ainda não foram definidas, tais como alguns receptoresacoplados à proteína G.
Modelagem por homologia foi utilizada para construir receptores de serotonina dotipo 5-HT1A e 5-HT2C, dois importantes alvos no tratamento de transtornos mentais eque não possuem estrutura cristalográfica publicada (NOWAK et al., 2006; CÓRDOVA-SINTJAGO et al., 2012). Em 2017, Freyd e colaboradores publicaram a análise de umaestrutura tridimensional completa do receptor GABAB2, uma vez que somente o sítio deligação presente na região N-terminal possui estrutura cristalográfica (FREYD et al., 2017).Juntos, esses trabalhos permitiram uma melhor compreensão de receptores que são alvos(ou potenciais alvos) para fármacos, mas que não possuem uma estrutura tridimensionalbem definida, facilitando o desenho de novas moléculas farmacológicas.
Com a estrutura tridimensional do alvo molecular em mãos, vários métodos dedesenho de fármacos baseado em estrutura podem ser aplicados para inspecionar a interaçãoligante-receptor. Dentre eles, o método de ancoramento (docking) molecular é um dos maisutilizadas devido ao modesto tempo computacional dispendido para realizar acoplamentoentre fármaco e receptor. O docking molecular deve ser utilizado quando a proteína não forco-cristalizada com o ligante que se deseja estudar, ou quando a estrutura tridimensionalé obtida por métodos de homologia. Essa aproximação baseia-se no ajuste espacial deambos, ligante e proteína, buscando as conformações mais favoráveis para a formação docomplexo, dentro de um princípio clássico de obtenção da energia de ligação (potencial)(MENG et al., 2011).
O processo do docking envolve explicitamente predizer a geometria de ligação e asinterações entre ligante e receptor (as poses), bem como estimar a afinidade de ligaçãodo complexo formado (scoring). Dois tipos de docking molecular podem ser realizados naproteína alvo, o docking rígido e o docking flexível. O primeiro caso trata, normalmente,de um processo de complexação onde receptor e ligante são entidades fixas, elaborandoum modelo do tipo chave-fechadura. Contudo, esse termo também pode ser usado para oprocesso onde há a fixação da proteína enquanto o ligante fica livre para se adaptar aosítio e ligação. Já o segundo tipo refere-se ao modelo de estudo onde ambos, proteína e

Capítulo 1. Introdução Geral 8
ligante, podem mover-se livremente para adaptar-se à presença do outro. Independentedo modelo escolhido, os softwares responsáveis por realizar análises de docking molecularutilizam ao menos um dos métodos destes cálculo: métodos estocásticos/aleatórios oumétodos de sistema. Os programas mais conhecidos são o AutoDock, Vina, GOLD e Glide(KONTOYIANNI; MCCLELLAN; SOKOL, 2004; VERDONK et al., 2003; GOODSELL;MORRIS; OLSON, 1996; FRIESNER et al., 2004).
A precisão do docking é determinada por casos-teste, onde complexos proteína-ligante já conhecidos passam por um processo de re-docking, onde o ligante é retirado daestrutura cristalográfica e re-inserido a partir da metodologia de docking que será utilizadano ligante em estudo. O resultado desse re-docking é então obtido e comparado com ocristal através da raiz do desvio quadrático médio (root mean square deviation - RMSD)entre os átomos não-hidrogênio. Para o resultado ser considerado bom, o RMSD entrea pose obtida pelo docking e o cristal deve ser menor que 2,0 Å. Por outro lado, maisestudos têm sido realizados com o intuito de melhorar a robustez deste método, uma vezque falsos positivos são encontrados (LI et al., 2010; PLEWCZYNSKI et al., 2011).
Recentemente, Bartuzi et al. (2017) e Rodríguez, Ranganathan e Carlsson (2015)revisaram os principais avanços na área de docking molecular em receptores acopladosà proteína G, mostrando as diferentes estratégias utilizadas e numerando os maioresproblemas enfrentados, pois assim como outras análises computacionais que serão aquidescritas, o sucesso do docking molecular está intimamente ligado a precisão na escolhados parâmetros de cálculo e nos indicadores de avaliação dos resultados. Rodríguez etal. realizou uma análise por docking de 6,7 milhões de compostos ligados à receptores deadenosina A2A, chegando a um final de 20 possíveis agonistas do receptor que foram testadose comprovados experimentalmente (RODRÍGUEZ et al., 2015a). No ano anterior, o mesmoautor, aproveitando-se da obtenção das primeiras estruturas cristalográficas de receptores5-HT, publicou uma análise de 1,3 milhões de compostos ligados à receptores 5-HT1B e5-HT2B, mostrando que 22 apresentaram atividade seletiva para 5-HT1B (RODRÍGUEZet al., 2014). Sabendo o quão difícil é obter uma estrutura cristalográfica de GPCRs emcomplexo com ligantes não estabilizantes, além de toda a complexidade por trás da síntesede compostos e sua experimentação. Dessa forma, as análises de docking molecular vemfacilitar a busca por moléculas com fins terapêuticos.
É notório que diferentes ligantes podem encaixar-se facilmente nos sítios ortostéricoe alostérico de algumas proteínas, o que torna mais complicada a verificação dos resultadosobtidos por docking. Portanto, métodos complementares devem ser utilizados para validaros estados conformacionais observados. Dinâmica molecular é o principal método utilizadoapós o processo de docking, uma vez que ele permite adicionar um nível maior de flexibili-dade para ligante e receptor em busca de um mínimo energético e, consequentemente, doestado fundamental do complexo. A partir de uma variação térmica ou de pressão, energiaé fornecida para o sistema, que está livre para mover-se. Dessa forma, todas as transiçõesconformacionais relacionadas com o movimento da proteína na presença de diferentesligantes podem ser estudadas, o que não pode ser observado através de estruturas estáticascomo os co-cristais obtidos por cristalografia de raios-x (ICHIKAWA et al., 2016).
A simulação por dinâmica molecular é essencialmente um tipo de modelagemestatística computacional, que considera a evolução temporal de um sistema como sendogovernada pela segunda lei de Newton, permitindo assim estudar uma variedade de proble-mas, tais como dinâmica de fluidos, polímeros, biomoléculas, dentre outros (RAPAPORT,2004). A dinâmica molecular é uma técnica de simulação computacional que, partindo

Capítulo 1. Introdução Geral 9
das equações de movimento clássicas, gera as trajetórias atômicas que relacionam essasinformações com as propriedades macroscópicas para estudar o comportamento dinâmicoe estrutural de sistemas a nível molecular (FRENKEL; SMIT, 2001). No âmbito dasmacromoléculas, a dinâmica molecular é utilizada para estudar variações no movimentode moléculas que dependem do tempo, incluindo o mecanismo de difusão, o dobramentode cadeias moleculares e as alterações estruturais desencadeadas pelo acoplamento defármacos (e seus protótipos) ao sítio de ligação de receptores proteicos. Os programasmais conhecidos são o GROMACS e NAMD (SPOEL et al., 2005; BERENDSEN; SPOEL;DRUNEN, 1995; PHILLIPS et al., 2005).
1.4 A mecânica quântica no desenvolvimento de fármacos
Nos últimos anos, muitos métodos buscando o desenvolvimento de fármacos auxi-liado por computação têm sido desenvolvidos, e vários compostos são avaliados atravésdesses métodos. A ideia por trás deles é gerar padrões farmacofóricos e/ou uma relaçãoquantitativa entre estrutura e atividade (QSAR), ancorar fármacos à um receptor ou cons-truir novos compostos dentro de um sítio de ligação específico (KLEBE, 1995; KUBINYI,1998). Diferentes propriedades moleculares (eletrostáticas, estéricas e hidrofóbicas, porexemplo), bem como ligações de hidrogênio, têm sido utilizadas parar chegar neste objetivo(MESTRES; ROHRER; MAGGIORA, 1997).
Contudo, deve-se ter em mente que os cálculos utilizados por uma grande partedesses estudos baseiam-se em técnicas de mecânica molecular (MM - do inglês, molecularmechanics), derivadas a partir das leis de Newton. Essas operam em um nível molecularacima dos elétrons, e descrevem a energia e forças associadas com estruturas proteicasparticulares por estudar modelos de compostos simples que imitam os grupos químicos emseus componentes aminoacídicos (HUNTER, 2006). Os métodos de mecânica molecularutilizam as leis da física clássica sem considerar explicitamente os efeitos eletrônicose confiam em um campo de força com parâmetros empíricos embutidos. São métodosque possuem a vantagem de ser computacionalmente menos intensivos, rápidos e úteisem computadores com recursos limitados, o que permite sua utilização no estudo demacromoléculas, que são sistemas de centenas ou milhares de átomos. Estes métodospossuem desvantagens, tais como, o fato do campo de força ser aplicável a uma limitadaclasse de moléculas, o de não executar cálculos de propriedades eletrônicas, bem como anecessidade de requerer dados experimentais para a formulação de sua parametrização(JENSEN, 2017).
Quando o algorítimo MM calcula a energia requerida para "esticar ou comprimir"uma ligação química, este aplica uma fórmula similar a lei de Hooke das molas elásticassobre tensão, tratando as moléculas como uma coleção de massas interagindo entre siatravés de forças harmônicas, utilizando as leis clássicas da física para predizer as estruturase as propriedades das moléculas (ARODOLA; SOLIMAN, 2017). Com o intuito de tornaresses algorítimos mais confiáveis, um grande número de aproximações vem sendo aplicadasaos cálculos baseados em mecânica clássica (ou mecânica molecular) para permitir umamaior precisão nos resultados energéticos. Dentre essas, os métodos de mecânica molecularacoplados à resolução da equação de Poisson-Boltzmann (PB) ou o modelo de Borngeneralizado (GB), e a estimativa do deslocamento da área de ação de solventes (SASA,do inglês, solvent accessible surface area), formando os métodos MM/PBSA e MM/GBSA,têm se mostrado promissores (GENHEDEN; RYDE, 2015).

Capítulo 1. Introdução Geral 10
De modo geral, métodos de MM são a alternativa mais viável quando reaçõesquímica não devem ser consideradas na simulação (BAO; SURESH, 2003; ARODOLA;SOLIMAN, 2017). Contudo, eles falham em descrever reações que envolvem ligaçõescovalentes ou o reconhecimento em solução, como ocorrem durante as reações biológicas.Torna-se claro que o entendimento das interações de compostos bioativos com componentescelulares deve-se basear na compreensão das partículas fundamentais que controlam esteseventos, tais como os elétrons e o núcleo de átomos que compõem o ligante e seu receptor.Tal entendimento requer informações estruturais das moléculas, bem como energia edistribuição dos elétrons, o que só pode ser fornecido pela mecânica quântica (MQ)(GREEN; JOHNSON; KANG, 1974; HUNTER, 2006; KEE et al., 2009; DIRAC, 1986;COHEN-TANNOUDJI; DIU; LALÖE, 1977). Ademais, o entendimento das interaçõesfármaco-receptor em sistemas biológicos é uma tarefa minuciosa e extremamente complexa,pois envolve uma diversidade de fatores que estão relacionados com a resposta terapêutica,requisitando um nível de detalhamento que a mecânica clássica não pode oferecer.
Melhorias nos hardwares de computadores têm fornecido maior poder computacional,o que, combinado com o desenvolvimento de teorias e algorítimos mais eficientes, têmlevado ao aumento na escala e profundidade das aplicações de modelagem molecular embiologia, em especial nos processos de entrega e desenvolvimento de fármacos, conhecidospelos termos drug delivery e drug discovery, respectivamente (FRIESNER; BEACHY,1998; CHERTOK et al., 2013; OU-YANG et al., 2012).
O incrível sucesso na co-habitação das ciências biológicas e ciências químico-físicastem sido de grande importância para a instalação da Biologia/Bioquímica Quântica comoárea de grande interesse para pesquisadores em todo o mundo (GESTWICKI, 2008). Comonotado desde a criação da teoria, a mecânica quântica pode explicar a manutenção dosseres vivos e seus processos celulares a partir da estabilidade de moléculas e varições deenergia em diferentes estados químicos. Assim, pode-se dizer que os métodos MQ tiveramsucesso onde os métodos MM não conseguiram, uma vez que esses incluem termos comopolarização e posicionamento dos elétrons, essenciais na determinação de interações entremoléculas ligantes, tais como hormônios, neurotransmissores e fármacos, e sítios de ligaçãoem moléculas receptores, tais como proteínas e RNA (ARODOLA; SOLIMAN, 2017).
As sementes da biologia quântica contemporânea foram semeadas por volta de 1930com Erich Hückel, que desenvolveu métodos simplificados baseados na mecânica quânticapara analisar a estrutura de moléculas orgânicas insaturadas, com o intuito de explicaro estado de elétrons em compostos aromáticos (HÜLCKEL, 1931; HÜLCKEL, 1932) ecom Pullman e Pullman (1955), com a analise de hidrocarbonetos carcinogênicos. Porém,somente em 1990 o campo da biologia quântica pôde se estabelecer com o aprimoramentodas aproximações utilizadas pela teoria do funcional da densidade (DFT ).
Baseado na teoria de Hohenberg e Kohn (HOHENBERG; KOHN, 1964), a DFTforneceu uma base para o desenvolvimento de estratégias computacionais objetivandoa obtenção de energia, estrutura e propriedades de átomos e moléculas com um custocomputacional menor que técnicas ab initio tradicionais e semi-empíricos (GEERLINGS;PROFT; LANGENAEKER, 2003; SLATER, 1965). Considerando-se um sistema arbitráriomodelado com N funções de base, o esforço computacional no estudo deste sistema éde N3, utilizando o DFT. Para os métodos Hartree-Fock (HF) há o aumento para N4 ouN5, enquanto para os métodos de perturbação Møller-Plesset (MP2) e de coupled-cluster(CCSD), o custo computacional cresce em ordem de N5 e N6, respectivamente (sendo N otamanho do sistema) (HE et al., 2014). Assim, a DFT se torna bastante útil no estudo de

Capítulo 1. Introdução Geral 11
grandes sistemas moleculares, permitindo uma descrição realística de sistemas orgânicos,inorgânicos, metálicos e semi-condutores (KOCH; HOLTHAUSEN, 2015).
A ideia proposta pela DFT é não mais depender da função de onda do sistemaestudado, mas sim da densidade eletrônica, ρ(~r). Esta densidade mede a probabilidade deencontrar um elétron no ponto de coordenada ~r (KOHN, 1999). Contudo, neste nível, ateoria ainda não consegue determinar com exatidão o termo de troca e correlação, mesmocom a adição da equação Kohn-Shan (KOHN; SHAM, 1965), requerendo uma aproximaçãopara o tratamento das mesmas (KOCH; HOLTHAUSEN, 2015).
A aproximação para descrever as energias de troca e correlação proposta inicialmentefoi a aproximação da densidade local (LDA - do inglês, Local-Density Approximation).Esta é muito utilizada na descrição de sólidos metálicos, porém não é considerada a melhoropção na descrição de ligações de hidrogênio (descrita como a principal interação a guiareventos biológicos) (IRETA; NEUGEBAUER; SCHEFFLER, 2004). Portanto, outrasaproximações foram propostas com o objetivo de descrever melhor os sistemas, dentreestas, as mais comuns são as aproximações de gradiente generalizado (GGA, do inglês,Generalized-Gradient Approximation), um pouco mais complexas que a LDA por envolvero gradiente de densidade (PERDEW; BURKE; ERNZERHOF, 1996).
Contudo, a DFT ainda era falho em descrever energias obtidas a partir das intera-ções dispersivas (como as interações de Van der Waals), por conseguinte as aproximaçõesforam aperfeiçoadas, havendo a inserção de um termo semi-empírico, o que deu origem aoDFT-D (GRIMME, 2006; ORTMANN; SCHMIDT; BECHSTEDT, 2005; TKATCHENKO;SCHEFFLER, 2009).
O crescente aumento na popularidade da DFT deve-se, principalmente, a (1)possibilidade de estudar sistemas moleculares com elevado número de átomos a umcusto computacional relativamente menor e (2) a confiabilidade nos resultados obtidos,comparáveis com cálculos ab initio mais rigorosos. Sua precisão vem sendo testada econfirmada para explicar dados biológicos, como em análises de docking molecular (HU etal., 2013), QSAR (CARVALHO et al., 2014), QM/MM (VALENTIN et al., 2014) e análisesda interação entre ligante e receptor a partir de dados cristalográficos, RNM, dentre outrosmeios (SUÁREZ; SUÁREZ; DÍAZ, 2009; BRNDIAR; STICH, 2012; FRAZÃO et al., 2012).
Em paridade aos avanços nas aproximações de DFT, na década de 1990 houveum aumento no número de estruturas proteicas obtidas por cristalografia de raios-Xe ressonância nuclear magnética (RNM) depositadas no Banco de Dados de Proteína(<http://www.rcsb.org>), o que foi ponto chave para a estruturação da mecânica quânticacomo fonte de pesquisa para explicar fatos biológicos e favoreceu a instalação da biologiaquântica como alvo de estudo no campo da bioquímica (HUNTER, 2006; KARMALI;BLUNDELL; FURNHAM, 2009; BERMAN et al., 2000).
A utilização da bioquímica quântica tem sido adotada por vários trabalhos como objetivo de descrever as interações entre fármaco e proteína, assim como no estudo desistemas de entrega de fármacos (MASERAS; MOROKUMA, 1995; ZHANG; ZHANG,2003; HE; ZHANG, 2005; FRAZÃO et al., 2012). Zhou et al. (2010) enfatiza que métodosquânticos estão se tornando populares no design computacional de fármacos como umaconsequência do alto grau de precisão exigido para estimar as afinidades de ligação relativas.Ou seja, há a adição de informações que não são levadas em consideração durante umcálculo baseado em mecânica clássica. De acordo com Raha et al. (2007), o uso rotineirode métodos quânticos em todas as fases do design de drogas in silico é de fundamental

Capítulo 1. Introdução Geral 12
importância para o avanço do campo. Porém, o bônus em precisão dos métodos quânticoscaminha lado a lado com seu maior ônus, o custo computacional. Atualmente, cálculoseficientes e com boa precisão para macromoléculas ainda apresentam um grande desafiopara o campo da química computacional, sendo o maior problema o escalonamento, isto é,quanto maior o sistema, maior o custo computacional.
Dessa forma, durante muitos anos, os métodos de mecânica quântica foram utilizadossomente em trabalhos visando moléculas pequenas. Dentre esses, pode-se citar a otimizaçãoda estrutura de fármacos (e seus protótipos) antes do processo de docking molecular,o cálculo de propriedades geométricas em cristais de moléculas simples, alterações naestabilidade de compostos e QSAR comparando propriedades físico-químicas, incluindoHOMO, LUMO, gap HOMO-LUMO, mas nada que pudesse ser utilizado diretamente parasustentar o uso dos métodos MQ no desenvolvimento de fármacos baseado em estrutura(KARAMAN; FATTASH; QTAIT, 2013; FILHO et al., 2013; ZANATTA et al., 2014;KARABULUT et al., 2016; FRAZÃO et al., 2016; STROSCIO; DUTTA, 2004).
Como forma de solução para o problema, métodos híbridos que utilizam aproxima-ções de mecânica quântica e mecânica molecular em sistemas biológicos têm se tornadobastante populares, em especial para estudar o mecanismo molecular em reações enzimáti-cas (CHUNG et al., 2015; SENN; THIEL, 2009). Nesta aproximação, emprega-se o métodode cálculo quântico em uma pequena região (como o sítio catalítico e substrato), enquantoa maior parte do sistema é tratado por campos de força.
A utilização de métodos QM/MM (do inglês, quantum mechanics/molecular me-chanics), tem sido bastante difundida no campo da bioquímica quântica, em especial paradescrever o processo de reação enzimática e otimizar estruturas tridimensionais advindas dedocking molecular e dinâmica molecular (ACEVEDO; JORGENSEN, 2009; TVAROŠKA,2015; KHANDELWAL et al., 2005). Particularmente, o uso do QM/MM, conhecido porONIOM (sigla em inglês, our own n-layered integrated molecular orbital and molecularmechanics) em alguns artigos devido a maior utilização desse modelo de QM/MM, foide grande importância para desvendar o mecanismo por trás de incontáveis processosenzimáticos, bem como para apresentar funções biológicas para algumas proteínas queantes não eram conhecidas (KAMP; MULHOLLAND, 2013; LONSDALE; RANAGHAN;MULHOLLAND, 2010; CHUNG et al., 2015).
Outra forma de aplicar os métodos MQ para grandes sistemas baseia-se nas aproxi-mações de escalonamento linear, no qual o sistema é dividido em pequenos subsistemase o cálculo é realizado individualmente em cada um destes (ZHANG; ZHANG, 2003).Muitos esforços têm sido devotados no desenvolvimento de métodos de escalonamentolinear úteis para o cálculo das mas variadas propriedades em grandes sistemas moleculares(GOEDECKER, 1999; STRAIN; SCUSERIA; FRISCH, 1996).
Nas últimas duas décadas, os métodos de fracionamento têm emergido como apro-ximações poderosas para o desenvolvimento de aproximações baseadas no escalonamentolinear utilizando mecânica quântica para tratar grandes sistemas. As aproximações defracionamento partem do pressuposto de que o efeito de perturbação na interação é maiorentre os vizinhos próximos, decaindo rapidamente com o aumento da distância. A partirde uma avaliação química, a macromolécula é dividida em subsistemas (fragmentos), esubsequentemente, a energia total ou outra propriedade molecular do sistema completo,pode ser obtida pela combinação linear dos termos correspondentes de cada fragmento(GORDON et al., 2012).

Capítulo 1. Introdução Geral 13
Atualmente, uma vasta gama de métodos de fracionamento foram propostos,incluindo o método de fracionamento do orbital molecular (FMO - do inglês, fragmentmolecular orbital) (KITAURA et al., 1999; FUKUZAWA et al., 2005), o método defracionamento com caps conjugados (MFCC - do inglês, molecular fractionation withconjugate caps) (HE; ZHANG, 2005; ZHANG et al., 2004), o método de fragmentaçãosistemático (SFM - do inglês, systematic fragmentation method) (DEEV; COLLINS,2005), o método de fragmentação baseado em energia generalizada (GEBF - do inglês,generalized energy-based fragmentation) (LI; LI; FANG, 2005), o método de muitos corposeletrostaticamente embebidos (EE-MB - do inglês, electrostatically embedded many-body)(DAHLKE; TRUHLAR, 2007), dentre outros (GORDON et al., 2012).
Os métodos de fragmentação têm sido utilizados com sucesso para diferentesaplicações em sistemas moleculares, tais como clusters de água, proteínas, e complexosligante-proteína. Desde sua criação, a aproximação MFCC tem se mostrado eficaz em obtera energia de interação entre as três hélices que formam a fibra colágena (RODRIGUES et al.,2013; BEZERRA et al., 2017), entre proteínas e água (ZHANG; CHEN; ZHANG, 2003a),do ligante benzamidina e a β-tripsina (ZHANG et al., 2004), dentre outros trabalhos. Logo,a partir deste método, pode-se quantificar e comparar energias de ligação de compostosbioativos em um mesmo receptor ou em receptores diferentes. Com isso, é possível identificaro composto que mais fortemente interage no sítio de ligação, informação que pode serutilizada para definir o melhor ligante para um determinado sistema. Simultaneamente,através da fragmentação do sistema, é possível observar individualmente as interaçõesformadas entre o complexo ligante-receptor, permitindo identificar as regiões/átomos demaior importância para a resposta biológica, dado que é de grande importância para odesenvolvimento de novos fármacos.
Apesar da precisão desses métodos, poucos trabalhos têm sido publicados utilizandomodelos de GPCRs ou imunoterapêuticos do tipo anticorpos monoclonais, sendo o maisnotável àquele de Heifetz et al. (2015). Neste, os autores obtiveram um conjunto deestruturas cristalográficas de receptores de classe A e utilizaram o FMO para descrevertodas as interações que ocorriam a 4,5 Å de cada ligante. Da mesma forma, apenas oartigo de Tavares et al. (2018) busca apresentar, quanticamente, o modo de interação entreum anticorpo monoclonal com um receptor do tipo PD-1.
1.5 Desenvolvimento de fármacos ligantes de GPCRs e anticorpos monoclo-nais e a simulação computacional
Como apresentado por Imming, Sinning e Meyer (2006), receptores acoplados àproteína G (GPCRs) compõem a superfamília de receptores mais útil sob um pontode vista farmacológico, e essa preferência está refletida na vendas de medicamentos nomercado global, que chegaram ao valor de US$ 890 bilhões entre os anos de 2011 e 2015.Numerosos fatores contribuem para a utilidade dos receptores de proteína G como alvosde fármacos, incluindo sua capazes de se ligar a moléculas pequenas com característicasde fármacos, interação com numerosos tipos de compostos químicos, ação como reguladorde grande parte dos processos fisiológicos e patológicos, e expressão na membrana celular,o que facilita a interação com o meio extracelular (FANG; KENAKIN; LIU, 2015).
Atualmente, a maior tendência no desenvolvimento de fármacos visando GPCRs nãoapenas busca ligantes comuns, que possuam atividade como agonistas ou antagonistas, masprincipalmente agonistas inversos (que conseguem inativar a atividade basal do receptor),

Capítulo 1. Introdução Geral 14
e moduladores alostéricos (moléculas que interagem com o receptor em locais diferentesdo sítio ativo, mas que conseguem modular a resposta fisiológica).
De acordo com Hauser et al., dentre os fármacos aprovados pelo US-Food and DrugAdministration (US-FDA), 475 possuem algum receptor acoplado à proteína G como alvo,ou seja, aproximadamente 34% de todos os fármacos aprovados pelo FDA visam GPCRs(HAUSER et al., 2017). Além disso, nos últimos cinco anos 69 novos compostos foramaprovados e mais 321 estão em fase de triagem clínica, dentre esses apenas 60 visando alvosque não possuíam nenhum fármaco aprovado anteriormente. Percebe-se claramente que osreceptores aminérgicos são alvos de grande parte dos 475 compostos aprovados, contandocom aproximadamente 300, o que pode ser visto como um reflexo da maior visibilidadedestes receptores.
Agonistas e antagonistas de GPCRs são usados no tratamento de doenças emtodos os órgãos, incluindo no sistema nervoso central, sistema cardiovascular, respiratório,metabólico e urogenital. Dentre os receptores que apresentam-se como principais alvosfarmacológicos, os receptores adrenérgicos (20%), histaminérgicos (14%), colinérgicos (12%)e serotonérgicos (12%), são, respectivamente, os mais visados. Além desses, receptoreshormonais também são alvos consideráveis para o mercado farmacêutico, uma vez quesua atividade é desregulada durante problemas endócrinos. Em contrapartida, algunsreceptores de classe B já são alvos validados para fármacos que estão no mercado ou emfase de teste clínico para diabetes, osteoporose, depressão e enxaqueca (BORTOLATO etal., 2014), enquanto os receptores de classe C vem sendo utilizados (ou estão em fase deteste) para hiperparatireoidismo, ansiedade, esquizofrenia, síndrome do X frágil e Parkinson(URWYLER, 2011). Assim, os GPCRs passiveis de ação de fármacos incluem membros decada uma das famílias, com apenas os receptores SMO da classe frizzled e o ADGRG3, dafamília de receptores de adesão, não apresentando nenhum agonista fisiológico conhecido.
Primariamente, fármacos ligantes destes receptores são utilizados em um grandeespectro de doenças. Os receptores de angiotensina II são alvos na prevenção de danosrenais induzidos por diabetes (BARNETT et al., 2004), bem como no tratamento deproblemas cardíacos. Agonistas dos receptores adrenérgicos do tipo α1 são empregados parao tratamento assintomático (antes que os sintomas apareçam) e diminuição da progressãoclínica de hiperplasia na próstata (MCCONNELL et al., 2003). Já os antagonistas dosreceptores do tipo β1 e β2 aumentam a sobrevida de pacientes com insuficiência cardíacae são usados no tratamento de pacientes com hipertensão (SQUIRE; BARNETT, 2000).
Os agonistas de receptores adrenérgicos β2 são utilizados no tratamento de pacientescom asma e doenças pulmonares crônicas. Antagonistas dos receptores de dopamina sãoagentes terapêuticos padrão no tratamento de esquizofrenia, embora alguns representantesdessa classe podem exercer sua atividade em outras subfamílias de receptores, como osreceptores de serotonina (5-HT) (SEEMAN; NIZNIK, 1990). Por outro lado, agonistasdo receptor de dopamina são largamente utilizados para tratar a doença de Parkinson.Inibidores da recaptação de serotonina atuam no tratamento da depressão, da ansiedadee dos transtornos bipolares, bem como agonistas dos subtipos 5-HT1B/1D são utilizadosno tratamento de enxaqueca. Agonistas dos receptores opioides e GABAB são utilizadospara o tratamento de dor crônica e aguda. Assim como alguns outros receptores têm sidoobservados como potenciais alvos contra o câncer, diabetes, obesidade e inflamação (WISE;GEARING; REES, 2002).
Contudo, uma vez que as famílias de receptores compartilham uma estrutura similare, consequentemente, um sítio de ligação que diverge muito pouco, torna-se difícil buscar

Capítulo 1. Introdução Geral 15
um composto que possua habilidade para ligar-se seletivamente (LEE; BOOE; PIOSZAK,2015). Ademais, apenas uma pequena porcentagem dos fármacos aprovados possuem suasvias de sinalização bem descritas. Neste sentido, vê-se que a taxa de sucesso para a triagemclínica entre as fases I, II e III decresce substancialmente, saindo de 70% na primeirafase, para 25% na última. Portanto, faz-se necessário o desenvolvimento de métodos quepermitam a busca de regiões estruturais específicas para cada uma das proteínas com ointuito de diferenciar as características exclusivas de cada receptor (HAUSER et al., 2017).
Descrições moleculares que relacionam interações moleculares com respostas fi-siológicas podem ser guias úteis para identificar moléculas com boas características deeficácia, segurança e uso. Um grande desafio para o desenho/ descoberta de novos fármacose identificar interações moleculares que levem a respostas farmacológicas seguras. Nessecontexto, a apreciação das estruturas cristalográficas disponíveis vem abrindo portas parauma nova era no desenvolvimento de fármacos visando GPCRs. A partir da diferenciaçãoentre a estrutura tridimensional, foi possível delimitar um conjunto de motivos estruturaisque auxiliam na manutenção dos receptores em seu estado ativado ou inibido. Todavia,o número de estruturas cristalográficas disponíveis de GPCRs não compreende 10% dototal de receptores expressos pelo genoma humano, assim como os problemas advindos demodificações na estrutura das proteínas têm limitado a percepção de fatores que favoreçamo desenvolvimento de fármacos capazes de sobrepujar as barreiras encontradas atualmente,tais como baixa seletividade e interação cruzada.
Considerada como o quinto pilar da terapêutica contra o câncer, juntamentecom cirurgia, quimioterapia citotóxica, radiação e a terapia molecular dirigida, a imuno-oncologia tem desempenhado um papel cada vez mais importante no tratamento do câncer.Amplamente definido como o "aproveitamento" do sistema imunológico para direcionaruma resposta aos tumores, a terapia imuno-oncológica é um campo diverso que englobaa aplicação de vacinas, anticorpos monoclonais, terapias celulares, terapias com vírusoncolíticos e imunoterapias não específicas (OISETH; AZIZ, 2017).
Iniciando com a aprovação do primeiro inibidor de pontos de verificação (checkpoint)em 2011, os rápidos avanços na terapia de imuno-oncologia levaram a grandes benefíciosclínicos, com alta taxa de tolerância e efeitos que foram mantidos por um largo períodoem pacientes com neoplasias malignas. Nos últimos anos, os inibidores de checkpointforam aprovados para mais de 15 indicações, e espera-se que este número venha a crescerexponencialmente (EMENS et al., 2016). Até o momento, seis inibidores de checkpoint foramaprovados pelo US-FDA (do inglês, food and drug administration), incluindo um inibidorda proteína 4 associada ao linfócito T citotóxico (CTLA-4), ipilimumab, dois inibidoresda proteína 1 de morte celular programada (PD-1), nivolumab e pembrolizumab, e trêsinibidores da proteína ligante de PD-1 (PD-L1), atezolizumab, avelumab e durvalumab. Eo número de compostos entrando no processo de triagem clínica aumenta constantemente.
Como uma consequência no sucesso encontrado pelos inibidores de pontos deverificação, muitos agentes oncológicos recentemente aprovados receberam designaçõesdo US-FDA que aceleraram o tempo despendido no processo de desenvolvimento. Em2016, o tempo total de revisão pelo órgão governamental era de oito meses, podendo sermuito menor, uma redução de aproximadamente metade do tempo dispendido para outrosfármacos. Análises mostram que o custo total para um estudo de oncologia nos EUA seaproxima de US$ 79 milhões, com um custo médio por paciente de US$ 59.500. Apesardesse investimento financeiro significativo, os resultados clínicos dramáticos observados comagentes de imuno-oncologia estimularam alianças, investimentos e pesquisas (SERTKAYA

Capítulo 1. Introdução Geral 16
et al., 2014).
O rápido desenvolvimento clínico e aprovação de imunoterapias reflete a utilizaçãofrequente de projetos de estudos adaptativos inovadores que registram um grande númerode pacientes, usam coortes de expansão, analisam subgrupos de pacientes e trabalhamem estreita colaboração com a FDA através do processo acelerado de aprovação (EMENSet al., 2016). A aprovação acelerada é baseada em dados que preveem fortes benefíciosclínicos, com a exigência de que dados clínicos confirmatórios adicionais verifiquem osbenefícios reais. Os desafios com essa abordagem podem incluir questões como: a duraçãodo tratamento é ideal para acompanhar as modificações posológicas.
Deve-se notar que uma parcela dos pacientes que fazem uso da imunoterapia têmreportado o desenvolvimento de efeitos adversos, derivados de um nível de toxicidadeimunológica. Não obstante, parte dos usuários de métodos de terapias imuno-oncológicasnão apresentam respostas ao tratamento. Recentemente, foi observado que a proteínaPD-L1 pode existir em forma livre na corrente sanguínea (ZHANG et al., 2015; ALI et al.,2017; COUSIN; SENESCHAL; ITALIANO, 2017).
Por tratar-se de uma área muito recente na terapêutica, e estar sob forte pressãosocial, pouco se conhece sobre o mecanismo de ação e respostas prolongadas dos compostosimunoterapêuticos. Dessa forma, estudos que visem uma melhor compreensão do mecanismode ativação dos receptores relacionados à imunoterapia são altamente benéficos para estecampo de pesquisa.
Apesar de uma vasta gama de estudos em simulação computacional voltados aodesenvolvimento de fármacos para GPCR estarem disponíveis, poucos são àqueles quevisam uma melhor compreensão dos receptores acoplados à proteína G de classes diferentesdos receptores do tipo rodopsina (classe A). Da mesma forma, o número de artigos visandoos receptores imunomodulatórios é ínfimo em comparação à quantidade de possibilidadesna área.
1.6 Escopo da tese
Diante do exposto, observa-se que o campo de estudos em modelagem molecularaplicada ao desenvolvimento de fármacos é amplo e engloba os mais diversos níveis dapesquisa biológica, em especial a área conformacional. Tendo por base o que foi explanadoneste capítulo introdutório e os trabalhos realizados durante os anos de doutoramento,nesta tese foram aplicadas técnicas de modelagem molecular em um nível quântico paraobter as energias de interação internas em diferentes complexos proteína-ligante com oobjetivo de apresentar uma descrição profunda sobre a manutenção dessas estruturas deligação.
A presente tese de doutoramento divide-se atualmente em cinco capítulos, sendoo primeiro esta introdução. No capítulo dois é apresentada uma caracterização deta-lhada do sítio de ligação do receptor de classe A 5-HT1B em complexo com o fármacodiidroergotamina, juntamente com a descrição de alguns conceitos básicos sobre ambasmoléculas, ligante e receptor, assim como a descrição do método de fracionamento MFCC.
No capítulo três são apresentados os resultados para a interação entre os anti-corpos monoclonais pembrolizumab e nivolumab, assim como o ligante natural PD-L1, eo receptor de morte celular programada (PD-1). É feita uma comparação entre os trêsligantes, buscando encontrar o ponto de ancoramento desses na proteína receptora.

Capítulo 1. Introdução Geral 17
No capítulo quatro são apresentados os resultados teóricos para a interação doreceptor de classe C GABAB em complexo com os agonistas ácido γ-aminobutírico (GABA)e baclofeno, assim como é descrita sua interação com os antagonistas 2-hidroxisaclofeno eSCH50911. É feita uma diferenciação ente o modo de ligação de ambas classes de molécula,bem como métodos de cálculo diferentes são utilizados.
No capítulo cinco, apresentam-se as conclusões finais deste trabalho, assim comoas perspectivas de futuros trabalhos originados a partir dessa tese. No Anexo A, há umabreve descrição dos métodos ab initio utilizados nesta tese, enquanto no Apêndice A sãoapresentados os artigos publicados durante o período de doutoramento.

18
capítulo 2
INTERAÇÃO DIIDROERGOTAMINA-RECEPTOR 5-HT1B DESCRITAPOR BIOQUÍMICA QUÂNTICA
2.1 Introdução
Serotonina ou 5-hidroxitriptamina (5-HT) é um dos neurotransmissores mais es-tudados e mais antigos, evolutivamente, tendo sido isolado e sua estrutura apresentadapela primeira vez em 1948 por M. Rapport a partir do composto obtido na correntesanguínea (RAPPORT; GREEN; PAGE, 1948a). Contudo, em 1938, V. Erspamer já haviademonstrado que uma substância desconhecida estimulava a contração intestinal, a qual elechamou de enteramina, sendo posteriormente reconhecida como a molécula de serotonina(ERSPAMER; BORETTI, 1950; ERSPAMER; ASERO, 1952). Pouco depois, em 1953, B.Twarog observou a presença de serotonina no cérebro através da análise de extratos doórgão (TWAROG; PAGE, 1953). Por último, na década de 1990, a função da serotoninacomo causa da contração do músculo liso foi confirmada (GERSHON et al., 1990).
Embora 5-HT tenha sido descoberto no sistema cardiovascular, a função da molé-cula de serotonina como neurotransmissor no sistema nervoso central tem sido amplamenteexplorada e fármacos que afetam os receptores de serotonina (5-HTR) e os transportadoresde serotonina (SERT - do inglês, serotonin transporter), proteínas que transportam aserotonina da fenda sináptica para o neurônio pré-sináptico, têm sido amplamente desen-volvidos e são largamente utilizados para o tratamento de doenças, tais como depressão,esquizofrenia, psicose, náusea, vômito, deficit de cognição e ansiedade (MORRISSETTE;STAHL, 2014; NAVARI, 2014). Nas últimas décadas, a função de 5-HT nos sistemasvascular e respiratório tem sido observada, bem como sua função no controle da respostade outros neurotransmissores, tais como dopamina, acetilcolina, noradrenalina e GABA(FINK; GÖTHERT, 2007). Assim, não é surpresa que mais de 125,000 artigos tenhamsido publicados entre os anos de 2014 e 2015 contendo as palavras chave: serotonina oureceptores de serotonina (MCCORVY; ROTH, 2015).
Células do tipo enterocromafim (ECL), presentes na mucosa intestinal, produzemaproximadamente 90% de toda a serotonina sintetizada no corpo humano (MACHIDA;IIZUKA; HIRAFUJI, 2013). As plaquetas sanguíneas não sintetizam 5-HT, mas possuemSERT, que auxilia na captação e transporte de altas concentrações de serotonina, obtidas apartir do intestino, através do sistema vascular pela corrente sanguínea (BERGER; GRAY;ROTH, 2009). Em contrapartida, estudos recentes vêm mostrando que artérias periféricassão capazes de sintetizar 5-HT através da ação das enzimas triptofano hidroxilase 1 (NIet al., 2008). Na presença de algum dano vascular, uma grande quantidade de 5-HT éliberada das plaquetas no plasma, o que afeta as funções normais de células vasculares domúsculo liso e células endoteliais, tais como contração, proliferação, migração e liberaçãode mediadores vasoativos. Dessa forma, a molécula de serotonina é observada atuando naperistalse através da contração do músculo liso ou despolarização do nervo entérico, alémde estar envolvida nos processos de coagulação sanguínea e vasoconstrição, função quedá nome a molécula ("serum"de sangue, e "tonina", que induz contração) (RAPPORT;GREEN; PAGE, 1948b).

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 19
Embora a maior concentração de serotonina esteja em outras regiões, grande partedas pesquisas em 5-HT são realizadas na área de neuropsiquiatria, em especial no trata-mento de transtornos afetivos, onde continua a ser de extremo interesse o desenvolvimentode fármacos mais eficazes. Compostos que têm como alvo os receptores de serotonina oua própria molécula vem compondo o grupo dos medicamentos mais vendidos na últimadécada, com mais fármacos sendo aprovados para um uso futuro. Esse fato é mais evidenteao falar-se de antidepressivos e antipsicóticos, que juntos somam mais de US$20 bilhõesde dólares em vendas nos Estados Unidos (LINDSLEY, 2013). Ademais, novos compostosvisando as vias de serotonina têm sido aprovados pelo FDA para o tratamento de depressão(CELADA; BORTOLOZZI; ARTIGAS, 2013). Contudo, ainda há a necessidade de fárma-cos novos, mais eficazes, com menos efeitos colaterais associados com atividade cruzada.Por exemplo, agentes para o tratamento da doença de Parkinson foram descontinuadosnos Estados Unidos devido a seu efeito nas vias circulatórias devido a ação em outrosreceptores monoaminérgicos (HORVATH et al., 2004; SETOLA et al., 2005).
5-HT regula suas vias de sinalização através da ativação coordenada de quatorzesubtipos de receptores geneticamente, farmacologicamente e funcionalmente distintos:5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT1F, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT4,5-HT5A, 5-HT5B, 5-HT6 e 5-HT7, e novos receptores têm sido descobertos, como a recenteseparação de 5-HT3 em 5-HT3A e 5-HT3B. Destes, apenas 5-HT3 não é um membro dasuperfamília de receptores acoplados à proteína G de classe A, uma vez que este ageatravés da ativação de seu canal iônico (LUMMIS, 2012). Dessa forma, a molécula deserotonina e seus receptores compõe uma das mais complexas vias de neurotransmissão.A expressão desses receptores no corpo humano é ampla e diversificada, com o RNAmensageiro (RNAm) de 5-HT1, 5-HT2, 5-HT4 e 5-HT7 expressos principalmente em célulasvasculares e endoteliais, enquanto os demais membros só foram observados em células dosistema nervoso central (ULLMER et al., 1995). Uma vez que o objetivo desta tese versaespecificamente sobre os receptores acoplados à proteína G, os dados que seguem estãofocados nos subtipos que fazem parte dessa superfamília. Dessa forma, quando falarmossobre receptores de serotonina, estaremos excluindo àqueles que fazem parte de outrasclasses de proteína.
Como pode-se observar a partir da figura 2.1, a identidade na sequência de ami-noácidos entre os membros da família de receptores de serotonina é baixa. Ademais, essesreceptores podem estar acoplados às vias de sinalização canônicas de proteína G atravésda formação de pares com Gαi/0, Gαq/11, e Gαs, o que permite a esta família de receptoresmodular diferentes vias de sinalização e, consequentemente, influenciar um grande númerode rotas metabólicas (NICHOLS; NICHOLS, 2008). Apesar disso, os treze receptorescompartilham a mesma estrutura tridimensional comum apresentada para os membrosda classe A, com sete hélices α altamente conservadas (TM1-TM7) conectadas por alçasinternas e externas (IL1-3 e EL1-3, respectivamente) (CÓRDOVA-SINTJAGO et al.,2012; SANDERS et al., 2001). Soma-se a isso a semelhança química e estrutural entre oscompostos conhecidos como "monoaminas", tais como serotonina, dopamina, adrenalina,dentre outros. Assim, torna-se imprescindível uma caracterização mais profunda entre ossubtipos de receptor de serotonina que vise diferenciar as nuances presentes nos sítios deligação específicos de cada subtipo. Uma vez que o objetivo central desta tese não entraneste mérito, os mecanismos fisiológicos da molécula não serão expostos nos próximosparágrafos. Contudo, para uma revisão mais completa, ver referência Fink e Göthert(2007).

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 20
Adaptado de Hoyer; Hannon; Martin, 2002.
Figura 2.1 – Homologia entre os receptores de serotonina.
2.1.1 Descrição geral dos subtipos de 5-HTR
Os receptores 5-HT1 e 5-HT5 estão acoplados a Gαi/0, levando a inibição deadenilato ciclase e a uma diminuição nos níveis de AMP cíclico (AMPc) intracelularquando ativados (LIN et al., 2002). Dentre eles, a classe de receptores 5-HT1 possui entre40% e 60% de identidade geral em sua sequência de aminoácidos. Embora 5-HT1E e 5-HT1F
não possuam uma apelação terapêutica e fisiológica forte, o que pode ser uma consequênciada falta de ligantes específicos para marcá-los, 5-HT1A, 5-HT1B e 5-HT1D aparecem emuma grande variedade de tecidos e têm sido alvos de extensa pesquisa farmacológica, emespecial nos campos de comportamento, tais como depressão e ansiedade, e enxaqueca. Emcontrapartida, os receptores 5-HT5 estão entre os menos conhecidos. Contudo, estudos têmmostrado que estes receptores possam ser alvos na terapia contra desordens psiquiátricas(THOMAS, 2006).
Os receptores de serotonina acoplados a Gαq/11 incluem aqueles da família 5-HT2.Esses, tradicionalmente, são relacionados com as vias de Gαq, com a ativação da enzimafosfolipase C, que leva a produção de inositol 3-fosfato (I3F) e diacilglicerol (DAG), ecom aumento nos níveis intracelulares de cálcio (ROTH et al., 1984; ROTH et al., 1998).Contudo, evidências apontam para certo grau de promiscuidade entre receptores do tipo 2,uma vez que já foram relatados casos onde a ativação dos receptores 5-HT2A e 5-HT2C
levaram a inibição de AMPc. No geral, os receptores que compõe 5-HT2 compartilham de46% a 50% na identidade geral em sua sequência de aminoácidos. Não há como questionara relevância terapêutica do receptor 5-HT2A e 5-HT2B. O primeiro é largamente utilizadoem estudos que buscam modular o sistema límbico, enquanto o segundo é um contra-alvo

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 21
primordial na busca por novos terapêuticos, pois, como apresentado por Choi et al., suaação no sistema circulatório é mais proeminente quando comparada com outros subtipos(CHOI et al., 1994). Neste sentido, a ação de novas moléculas em 5-HT2B tem auxiliadono pequeno número de compostos que conseguem chegar ao mercado e se manter, mesmoquando falando de compostos que atuam em receptores adrenérgicos ou dopaminérgicos.
Os receptores 5-HT4, 5-HT6 e 5-HT7 estão acoplados à Gαs, sendo responsáveispela ativação de adenilato ciclase e aumento nos níveis de AMPc, embora certa atividadede Gαq/11 é apresentada pelo primeiro (ROTH et al., 1994; OUADID et al., 1992). Nogeral, as três classes de receptor compartilham entre 20% a 60% com os demais membrosda classe de receptores de serotonina. Pouco se sabe sobre eles, com o receptor 5-HT7
recebendo uma maior atenção devido a alta afinidade que apresenta por ligantes conhecidosde receptores 5-HT1, tais como LSD e ritanserina, indicando um papel no sistema límbicoem desordens afetivas (ROTH et al., 1994).
2.1.2 Receptor 5-HT1B
Os receptores 5-HT1B têm enfrentado um complexo debate durante a história.Eles foram originalmente pensados como um receptor específico de roedores. Contudo,similaridades nos padrões traducionais, funcionais e de distribuição cerebral levaram àopinião de que os receptores de roedores (5-HT1B) e aqueles de não-roedores (5-HT1D)eram homólogos entre as espécies, o que foi comprovado quando ambos foram clonados(HARTIG; BRANCHEK; WEINSHANK, 1992). Parte dessa confusão foi gerada peladificuldade em utilizar radioligantes específicos para cada subtipo, o que mostrava umperfil similar de ligação, além da alta homologia (97%) entre a sequência de aminoácidosdo receptor 5-HT1B humano e aquele de roedores (HOYER; HANNON; MARTIN, 2002).Assim, o antigo receptor 5-HT1D beta foi renomeado como 5-HT1B (HARTIG et al., 1996).
O gene de 5-HT1B em humanos (HTR1B) se encontra no cromossomo 6q13 (com∼390 aminoácidos), sendo expresso nos gânglios basais, estriado e córtex frontal, ondeacredita-se que servem como autoreceptores (HOYER; HANNON; MARTIN, 2002). Emcontrapartida, 5-HT1B pode atuar como heteroreceptor, controlando a liberação de outrosneurotransmissores, tais como dopamina, acetilcolina, glutamato e GABA (PAUWELS,1997). Ademais, 5-HT1B pode ser encontrado nas artérias cerebrais e outros tecidosvasculares, onde os espasmos vasculares têm sido implicados na patogênese da enxaqueca(NILSSON et al., 1999; SCHOENEN, 2014). Similarmente, efeitos periféricos tambémtêm sido descritos, tais como a inibição da liberação de noradrenalina na veia cava. Alémdisso, existem evidências de que o receptor 5-HT1B possa estar envolvido nos mecanismossubjacentes à esquizofrenia, agressão, depressão, deficit de atenção e hiperatividade, eo uso de drogas de abuso (BRAFF et al., 1978; SARI, 2004). Portanto, 5-HT1BR é umalvo atrativo para o desenvolvimento de terapias não apenas para a enxaqueca, mastambém para algumas outras desordens neurológicas (SCHOENEN, 2014; PIETROBON;STRIESSNIG, 2003).
A ativação dos receptores 5-HT1B nas artérias cerebrais por agonistas é respon-sável pela vasoconstricção e tem sido apontado como uma explicação terapêutica para omecanismo de ação de alguns fármacos antimigranosos (GOADSBY, 2005; AMUNDSENet al., 2015; DURHAM; PAPAPETROPOULOS, 2013). Dentre estes, diidroergotamina[DHE; (5’α,10α)-5’-Benzil-12’-hidroxi-2’-metil-3’,6’,18-trioxo-9,10-dihydroergotaman], éum dos fármacos mais prescritos, sendo considerado uma droga clássica para o tratamentode enxaqueca desde 1945 (EDVINSSON; VILLALÓN; MAASSENVANDENBRINK, 2012;

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 22
LABRUIJERE et al., 2015). Vendido sob o nome D.H.E. 45 ou Migranal, DHE é um alca-loide de ergot sintético, ou ergolina, obtido a partir da hidrogenação da dupla ligação entreos átomos C10=C11 de ergotamina (ERG). Este composto atua como um agonista seletivode 5-HT1BR, onde exerce seu efeito anti-migranoso (ROSAS et al., 2013; SILBERSTEIN;MCCRORY, 2003). Contudo, após o desenvolvimento dos medicamentos da classe dostriptanos na década de 1990, houve uma redução em seu emprego clínico devido aos efeitoscolaterais severos relacionados a sua atividade em outros receptores monoaminérgicos,como o receptor D2 de dopamina e β2-adenérgico (ROTH et al., 2007; ROTHMAN et al.,2000). Apesar disso, diidroergotamina continua sendo um dos fármacos anti-miganososmais utilizados no mundo.
A recente determinação da primeira estrutura cristalográfica do receptor 5-HT1B
humano co-cristalizado com os agonistas ergotamina e diidroergotamina foi um primeiropasso em direção a um entendimento profundo do mecanismo de interação de ligantes nosreceptores serotonérgicos (Figura 2.2) (WACKER et al., 2013; WANG et al., 2013a; LIUet al., 2013). Apesar disso, a similaridade estrutural e na sequência de aminoácidos entreos receptores acoplados à proteína G torna difícil a obtenção de agonistas ou antagonistasseletivos para um único receptor (HOYER; HANNON; MARTIN, 2002). Ademais, evi-dências sugerem que diferentes conformações no receptor induzidas por interações nãocovalentes entre ligantes-aminoácidos podem ser responsáveis pelo surgimento de distintasrespostas biológicas no mesmo receptor, o que não pode ser observado pela análise deuma única estrutura cristalográfica (KOBILKA; DEUPI, 2007; ZHANG; STEVENS; XU,2015). Desta forma, técnicas que possam auxiliar na caracterização das interações entreos receptores de serotonina e seus ligantes podem ser bastante úteis no design de novosfármacos mais seletivos aos receptores de serotonina (RAO et al., 2014).
Neste sentido, técnicas computacionais têm sido largamente utilizadas como modelosna busca por novos fármacos com maior especificidade, não apenas visando os receptoresserotonérgicos, mas também para outros GPRCs (RODRÍGUEZ et al., 2014; BELLO et al.,2016). Contudo, os métodos normalmente utilizados, como docking molecular e dinâmicamolecular, oferecem somente um número limitado de informações sobre a interação entreresíduos específicos do receptor e seus diferentes ligantes. Em contrapartida, o uso demecânica quântica (MQ) no desenvolvimento de fármacos in silico tem se tornado bastantepopular nos últimos anos devido a alta acurácia na estimativa da afinidade (relativa) deligação (ZHOU; HUANG; CAFLISCH, 2010; RAHA et al., 2007).
Apesar disso, o alto custo computacional dos métodos MQ para calcular as ener-gias de interação em macromoléculas demanda um balanço entre o tempo de execuçãocomputacional e a acurácia dos resultados. Portanto, métodos de fracionamento têm sidodesenvolvidos com o intuito de tornar o cálculo de propriedades físico-químicas em macro-moléculas computacionalmente menos custoso (GORDON et al., 2012; RAGHAVACHARI;SAHA, 2015). Hoje, vários esquemas baseados na decomposição de proteínas têm sidoaplicados para calcular suas propriedades, incluindo o fracionamento molecular com capsconjugados (ZHANG; ZHANG, 2003) e o esquema de fracionamento do orbital molecular(NAKANO et al., 2002; SRIWILAIJAROEN et al., 2016). Dentre esses, os métodos defracionamento molecular com caps conjugados (MFCC) tem sido largamente utilizados,particularmente para calcular a energia de interação entre fragmentos aminoacídicos eligantes com grande sucesso (ZANATTA et al., 2012; RIBEIRO et al., 2014).
Neste capítulo, é apresentada uma descrição apropriada do perfil energético deinteração entre diidroergotamina e os resíduos individuais do bolsão de ligação de 5-HT1BR

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 23
Figura 2.2 – Estrutura do receptor 5-HT1B composto por sete hélices transmembranares, duasalças intracelulares e extracelulares (PDB ID: 4IAQ).

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 24
através do esquema MFCC dentro do formalismo da teoria do funcional da densidade(DFT) (DA COSTA et al., 2012; JIANG; MA; JIANG, 2006). A contribuição individual dosresíduos envolvidos na ligação entre DHE e o receptor de serotonina foi calculada usandoa estrutura do receptor 5-HT1B co-cristalizado com diidroergotamina (PDB ID: 4IAQ)(WANG et al., 2013a). Para tornar os cálculos energéticos mais fidedignos, um esquemade representação eletrostática foi implementado, sendo aqui chamado de electrostaticallyembedded molecular fractionation with conjugate caps (EE-MFCC). O propósito destainvestigação é delinear a ligação DHE-5-HT1BR através de um perfil energético detalhadoda interação entre os resíduos no sítio de ligação e o fármaco anti-migranoso, sendo essencialnão apenas para prover informações sobre ajustes que possam melhorar a eficácia de novasdrogas específicas para a terapia voltada ao sistema serotonérgico, mas também parapermitir um melhor entendimento sobre as características de ligação em outras GPCRs.Ao final, os resultados obtidos pelo EE-MFCC são comparados com aqueles resultantes doMFCC comum com o intuito de validar o método aplicado.
2.2 Materiais e Métodos
2.2.1 Dados do complexo DHE-Receptor
Para a realização dos cálculos neste estudo utilizou-se a primeira estrutura cris-talográfica do receptor 5-HT1B em complexo com diidroergotamina obtida por difraçãode raios-X e depositada no banco Protein Data Bank (PDB, <www.rcsb.org/pdb>), sobo código 4IAQ e com resolução de 2,80 Å (WANG et al., 2013a). Ademais, o cálculo doestado de protonação em pH fisiológico para ligante e aminoácidos do receptor foi realizadoatravés do uso do software Marvin Sketch, versão 5.5.0.1, e o software PROPKA 3.1,respectivamente (SØNDERGAARD et al., 2011).
2.2.2 Cálculos clássicos
Átomos não elucidados pela difração de raios-X, e portanto ausentes na estruturacristalográfica, como átomos de hidrogênio e cadeias laterais de alguns aminoácidos, foramadicionados e submetidos a otimização clássica em suas geometrias, mantendo os outrosátomos fixos. Esta otimização foi realizado no software Discovery Studio sob os parâmetrosde cálculo do campo de força CHARMm (Chemistry at HARrvard Molecular Mechanics),sendo este parametrizados para todos os átomos, porém com maior especialização paramoléculas orgânicas (MOMANY; RONE, 1992). Para este cálculo os padrões de convergên-cia foram ajustados para 10−5 kcal mol−1 (variação de energia total), 0.001 kcal.mol−1Å−1
(gradiente de desvio médio quadrado) e 10−5Å (deslocamento atômico máximo). Logo apóso processo de otimização, posições e cargas atômicas foram armazenadas para a realizaçãodos passos subsequentes.
2.2.3 Fracionamento molecular com caps conjugados e o efeito da incorporação eletrostá-tica
A energia de interação entre diidroergotamina e os aminoácidos de 5-HT1BR foicalculada através do formalismo do MFCC proposto por Zhang e Zhang (ZHANG; ZHANG,2003). O ligante é apresentado como L e o resíduo interagindo com este como Ri. O capCi−1 (Ci+1) é formado pelos resíduos covalentemente ligados ao grupo amina (carboxila)do resíduo Ri, provendo uma melhor descrição do ambiente eletrônico. Para cada estrutura

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 25
fragmentada, a energia de interação entre ligante-resíduo, EI(L − Ri), é calculada deacordo com:
EIMF CC(L − Ri) = E(L − Ci−1RiCi+1) − E(Ci−1RiCi+1)
− E(L − Ci−1 Ci+1) + E(Ci−1 Ci+1), (2.1)
onde o primeiro termo, o sistema de Ci−1 + Ri + Ci+1 é separado da proteína, e o ligante(L) é adicionado, finalizando o primeiro momento com o cálculo da energia do sistema L,Ci−1, Ri e Ci+1. No segundo momento, o ligante é retirado do cálculo, estando somenteos caps e o resíduo principal (Ci−1, Ri e Ci+1). Posteriormente, o ligante é reintegrado aosistema e o resíduo em estudo é retirado, resultando na interação do ligante com os caps (L,Ci−1 e Ci+1). Ao final, somente a energia dos caps é obtida (Ci−1 e Ci+1). A equação 2.1 foiaplicada por da Costa e colaboradores para estudar a interação entre estatinas e HMG-CoAredutase, e apresenta o que nós intitulamos como MFCC (DA COSTA et al., 2012). Como objetivo de melhorar nossos cálculos, uma forma adaptada do MFCC foi utilizada. Estainclui uma correção eletrostática do ambiente através do uso das cargas atômicas obtidasclassicamente após o processo de otimização para aumentar a similaridade com a proteínainteira, como segue:
EIEE−MF CC(L − Ri) = E(L − CiRiCi∗ + CP C) − E(CiRiCi∗ − CP C)
− E(L − Ci C∗ − CP C) + E(Ci Ci∗ − CP C). (2.2)
Comparando a equação 2.1 com a 2.2, percebe-se que há a inserção do termo CP C .Este representa o conjunto de todos os átomos dos aminoácidos presentes na estruturacristalográfica, sem o resíduo de referência (Ri) e os caps (Ci−1 Ci+1). Nesse modelocada átomo é substituído pela carga atômica correspondente (carga pontual). Similar aocálculo do MFCC com a correção do campo eletrostático adaptado (electrostactic fieldadapted, EFA) aplicado por Jiang et al. (JIANG; MA; JIANG, 2006), cada fragmento foiintroduzido entre todas as cargas pontuais, e as energias de interação foram calculadasincluindo as interações eletrostáticas do ambiente em todos os passos seguindo o esquemade incorporação de cargas pontuais proposto por Hall e Smith (HALL; SMITH, 1984;HALL, 1985; HALL, 1986) (intitulado aqui como EE-MFCC). A presença das cargaseletrostáticas inclui a influência das interações de longo alcance do ambiente a cada energiade interação ligante-resíduo, o que torna o efeito eletrostático do ambiente mais realísticodo que é observado com o uso de outros modelos, como aqueles de solvatação contínua,devido a sua capacidade em reproduzir a heterogeneidade proteica.
Uma descrição completa sobre o método MFCC encontra-se no Anexo A.
2.2.4 Cálculos DFT
Após o fracionamento considerando os esquemas MFCC e EE-MFCC, os cálculosenergéticos para cada resíduo foram realizados através do software Gaussian (G09), dentrodo formalismo da teoria do funcional da densidade utilizando dois funcionais baseados naaproximação do gradiente generalizado (B97D e B3LYP) e um meta-GGA (M062X). Paraexpandir os orbitais de Khon-Sham para todos os elétrons, nós selecionados o conjunto debases 6 − 311 + G(d, p). É importante notar que os cálculos DFT foram realizados para

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 26
cada termo nas Eq. 2.1 e 2.2, isto é, (L − CiRiCi∗), (CiRiCi∗), (L − Ci C∗), (Ci Ci∗),(L − CiRiCi∗ + CP C), (CiRiCi∗ − CP C), (L − Ci C∗ − CP C) e (Ci Ci∗ − CP C).
2.2.5 Critérios de convergência
Para evitar a perda de interações importantes, um estudo de convergência daenergia total de ligações como uma função do raio foi realizada, limitando o número deaminoácidos analisados (DA COSTA et al., 2012). Desta forma, foi adicionada a energiade interação individual dos aminoácidos dentro de esferas imaginárias em um bolsão deinteração, r, tendo por centro o ligante, considerando r = R/2 (R = 1, 2, 3, 4, ... Nn,Nn sendo o próximo número natural na sequência) (DANTAS et al., 2015). Assim, aconvergência é obtida quando a variação de energia entre os raios consecutivos é menorque 10%.
2.3 Resultados e Discussão
GPCR é um dos principais alvos da indústria farmacêutica, contando com aproxi-madamente 30% de todos os fármacos comercializados. Dentre eles, o sistema serotonérgicotem representando uma grande parte dos principais compostos (MCCORVY; ROTH,2015). Uma vez que interações off-target são a principal causa de efeitos colaterais e falhasna triagem clínica em ligantes para o sistema serotonérgico, a identificação de ligantesseletivos é um aspecto crítico no processo de descoberta de novos fármacos.
Neste contexto, é notável que a recente publicação de 5-HT1BR co-cristalizadocom diidroergotamina e ergotamina abriu as portas para uma melhor compreensão dessesreceptores (WANG et al., 2013a). Contudo, existem poucos estudos baseados em mecânicaquântica sobre GPCRs. Dentre eles, simulações usando o fracionamento do orbital moleculardentro do formalismo da teoria da perturbação Møller-Plesset (MP2) foram realizadas emestruturas cristalográficas de receptores acoplados à proteína G, mostrando a relevância dométodos quânticos para desvendar a forma de ligação de seus ligantes à uma distância de4,5 Å do ligante (HEIFETZ et al., 2015). Embora DHE possua alta relevância farmacêutica,nenhum outro estudo de mecânica quântica foi realizado para descrever sua interação com5-HT1B.
2.3.1 Estrutura e distribuição de carga de diidroergotamina
Para iniciar os cálculos, o estado de protonação de diidroergotamina é obtido(Figura 2.3). Como apresentado na figura 2.3a, há uma prevalência do estado protonadode DHE entre os pH de 7.0-7.4 (96.12% - 90.77%). Portanto, a estrutura molecular doligante foi ajustada para a forma carregada (+1) pela adição de um único átomo dehidrogênio ao grupo amina (N6) do ligante (figura 2.3b). Esse resultado está de acordocom a análise da estrutura cristalográfica e configuração absoluta obtida por Herbert(1979), e é um comportamento comum apresentado por serotonina e outras ergolinas, taiscomo ergotamina e dietilamida do ácido lisérgico (LSD), todos estes produzindo efeitosem receptores serotonérgicos (MANTEGANI; BRAMBILLA; VARASI, 1999).
O ligante foi subdividido em quatro regiões para facilitar análises posteriores, onde aregião i representa a porção ergolina (anéis A-D); a região ii é a região da ligação peptídica;a região iii é composta pelos anéis E-G; e a região iv é o anel fenil H. A distribuição dadensidade eletrônica em diidroergotamina no estado carregado é representado na figura 2.3c,
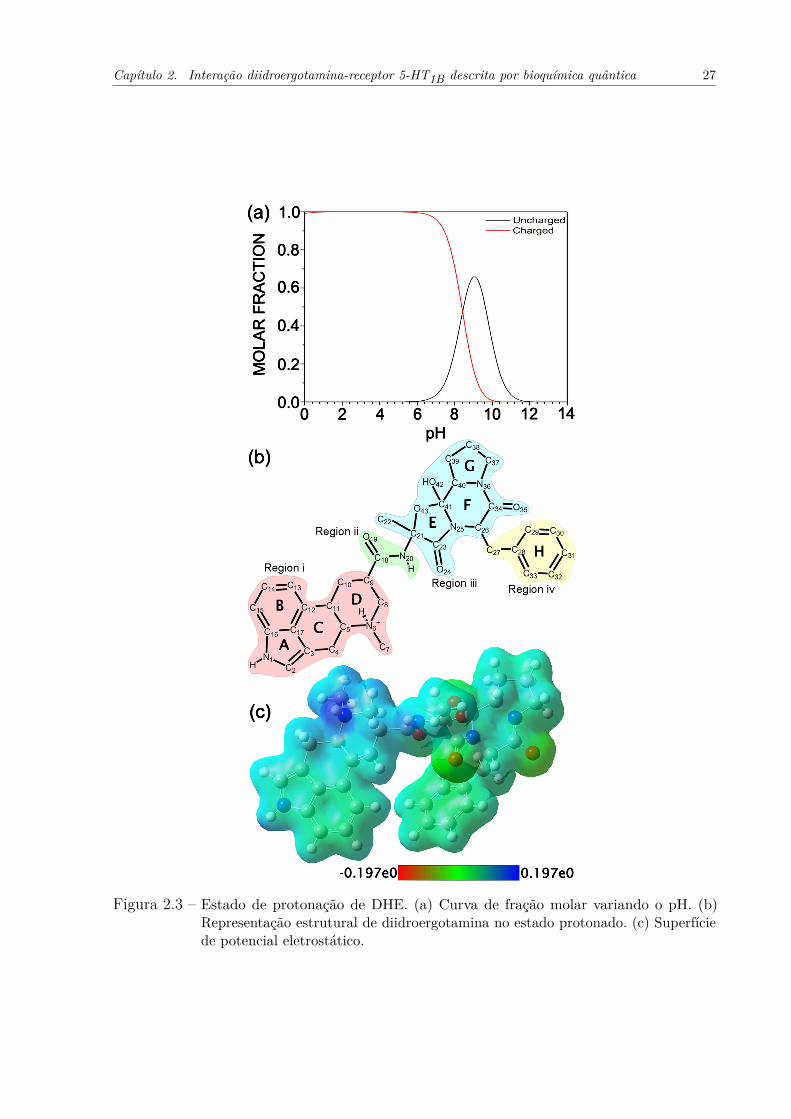
Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 27
Figura 2.3 – Estado de protonação de DHE. (a) Curva de fração molar variando o pH. (b)Representação estrutural de diidroergotamina no estado protonado. (c) Superfíciede potencial eletrostático.
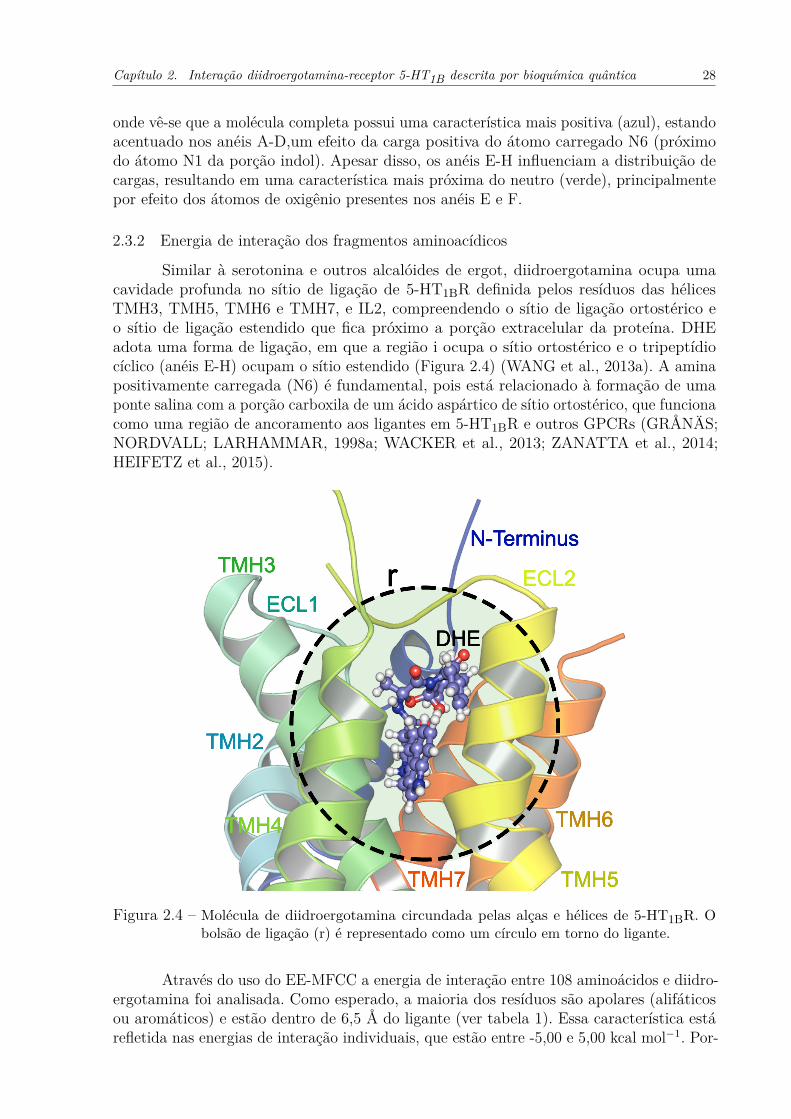
Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 28
onde vê-se que a molécula completa possui uma característica mais positiva (azul), estandoacentuado nos anéis A-D,um efeito da carga positiva do átomo carregado N6 (próximodo átomo N1 da porção indol). Apesar disso, os anéis E-H influenciam a distribuição decargas, resultando em uma característica mais próxima do neutro (verde), principalmentepor efeito dos átomos de oxigênio presentes nos anéis E e F.
2.3.2 Energia de interação dos fragmentos aminoacídicos
Similar à serotonina e outros alcalóides de ergot, diidroergotamina ocupa umacavidade profunda no sítio de ligação de 5-HT1BR definida pelos resíduos das hélicesTMH3, TMH5, TMH6 e TMH7, e IL2, compreendendo o sítio de ligação ortostérico eo sítio de ligação estendido que fica próximo a porção extracelular da proteína. DHEadota uma forma de ligação, em que a região i ocupa o sítio ortostérico e o tripeptídiocíclico (anéis E-H) ocupam o sítio estendido (Figura 2.4) (WANG et al., 2013a). A aminapositivamente carregada (N6) é fundamental, pois está relacionado à formação de umaponte salina com a porção carboxila de um ácido aspártico de sítio ortostérico, que funcionacomo uma região de ancoramento aos ligantes em 5-HT1BR e outros GPCRs (GRÅNÄS;NORDVALL; LARHAMMAR, 1998a; WACKER et al., 2013; ZANATTA et al., 2014;HEIFETZ et al., 2015).
Figura 2.4 – Molécula de diidroergotamina circundada pelas alças e hélices de 5-HT1BR. Obolsão de ligação (r) é representado como um círculo em torno do ligante.
Através do uso do EE-MFCC a energia de interação entre 108 aminoácidos e diidro-ergotamina foi analisada. Como esperado, a maioria dos resíduos são apolares (alifáticosou aromáticos) e estão dentro de 6,5 Å do ligante (ver tabela 1). Essa característica estárefletida nas energias de interação individuais, que estão entre -5,00 e 5,00 kcal mol−1. Por-
Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 29
tanto, as interações com maior energia (entre -6,00 e 6,00 kcal mol−1) foram selecionadose apresentados na figura 2.5.
Como retratado pela figura 2.5, 13 resíduos energeticamente mais significantesforam selecionados após a realização dos cálculos do EE-MFCC, sendo estes: Asp(D)129(-93,92 kcal mol−1, Val(V)201 (-7,38 kcal mol−1), Phe(F)330 (-8,44 kcal mol−1), Phe(F)351(-7,86 kcal mol−1), Leu(L)126 (-7,96 kcal mol−1), Ile(I)130 (-7,43 kcal mol−1), Val(V)200 (-6,69 kcal mol−1), Asp(D)352 (-38,59 kcal mol−1), Thr(T)355 (-6,42 kcal mol−1), Glu(E)198(-20,69 kcal mol−1), Asp(D)204 (-17,32 kcal mol−1) e Arg(R)114 (17,48 kcal mol−1) andAsp(D)123 (-21,44 kcal mol−1). Destes, somente Arg(R)114 apresenta uma energia deinteração positiva, o que reflete a distribuição dos aminoácidos dentro da estrutura proteica,uma vez que este é o único resíduo com carga positiva (grupo guanidina).
Tabela 2.1 – Descrição dos 108 resíduos de 5-HT1BR interagindo com DHE. Apresentam-se osgrupos funcionais e as distâncias do ligante a cada aminoácido no sítio de ligação(2,0 - 10,0 Å).
É bem descrito que ligações de hidrogênio (LH) e pontes salinas (PS) são muitoimportantes para o mecanismo de reconhecimento biológico à ligantes. Em membrosde GPCRs aminérgicos (classe A), sabe-se que a característica mais importante para oreconhecimento de ligantes é uma ponte salina entre o grupo amina carregado do ligantee um ácido aspártico na hélice transmembrana 3 (TMH3), que, em 5-HT1B, é o resíduoAsp(D)129 (GRÅNÄS; NORDVALL; LARHAMMAR, 1998b). A partir da figura 2.6a,pode-se observar que esse é o único resíduo a formar uma ponte salina com i(N6)H. Essainteração é criada entre o seu grupo carboxila e a amina carregada de DHE, em umadistância de 1,94 Å. Ademais, vê-se que há um pequeno grupo de ligações de hidrogênio
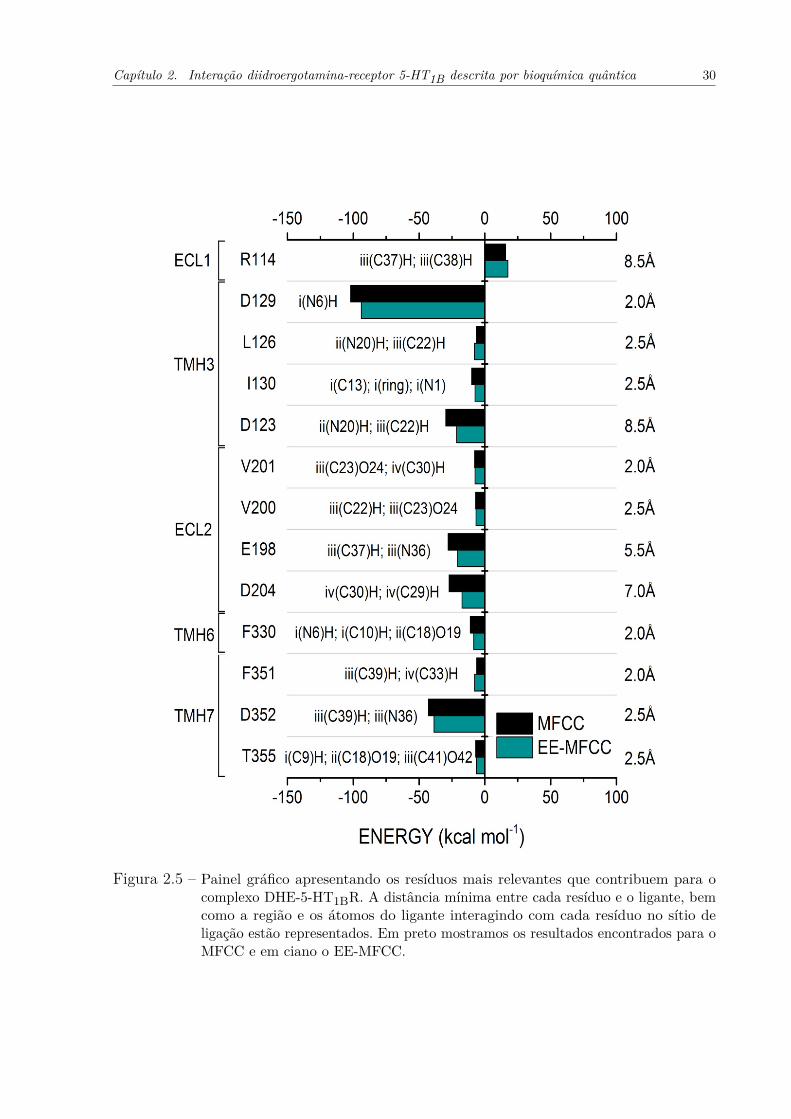
Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 30
Figura 2.5 – Painel gráfico apresentando os resíduos mais relevantes que contribuem para ocomplexo DHE-5-HT1BR. A distância mínima entre cada resíduo e o ligante, bemcomo a região e os átomos do ligante interagindo com cada resíduo no sítio deligação estão representados. Em preto mostramos os resultados encontrados para oMFCC e em ciano o EE-MFCC.
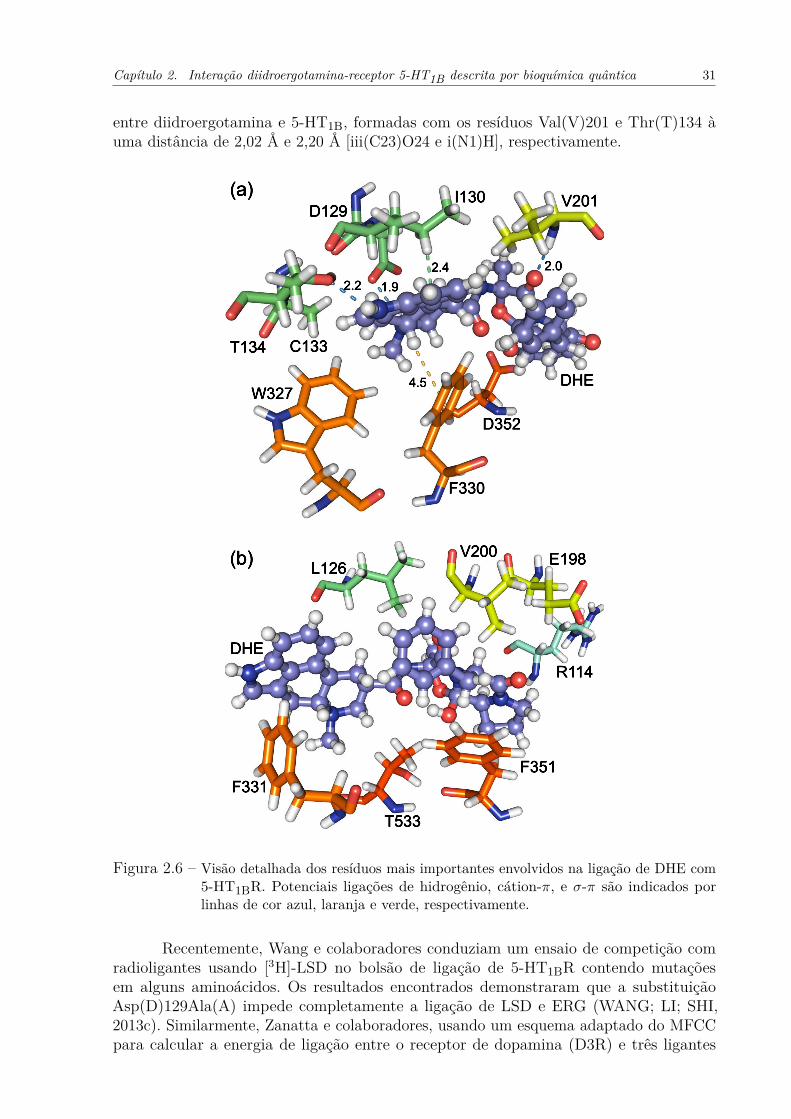
Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 31
entre diidroergotamina e 5-HT1B, formadas com os resíduos Val(V)201 e Thr(T)134 àuma distância de 2,02 Å e 2,20 Å [iii(C23)O24 e i(N1)H], respectivamente.
Figura 2.6 – Visão detalhada dos resíduos mais importantes envolvidos na ligação de DHE com5-HT1BR. Potenciais ligações de hidrogênio, cátion-π, e σ-π são indicados porlinhas de cor azul, laranja e verde, respectivamente.
Recentemente, Wang e colaboradores conduziam um ensaio de competição comradioligantes usando [3H]-LSD no bolsão de ligação de 5-HT1BR contendo mutaçõesem alguns aminoácidos. Os resultados encontrados demonstraram que a substituiçãoAsp(D)129Ala(A) impede completamente a ligação de LSD e ERG (WANG; LI; SHI,2013c). Similarmente, Zanatta e colaboradores, usando um esquema adaptado do MFCCpara calcular a energia de ligação entre o receptor de dopamina (D3R) e três ligantes

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 32
diferentes encontrou que o ácido aspártico na posição 110 (Asp[D]110) foi o resíduocom maior atração para eticloprida, haloperidol e risoperidona (ZANATTA et al., 2012;ZANATTA et al., 2014; ZANATTA et al., 2016). Ademais, Heifetz et al. também mostraramque GPCRs com um ácido aspártico na posição 32 de TMH3 comumente o tem comoo resíduo mais importante, energeticamente (HEIFETZ et al., 2015). Portanto, é de seesperar que o resíduo Asp(D)129 seja observado como um dos aminoácidos com maiorrelevância energética.
Por outro lado, Val(V)201 é um resíduo que compõe a alça extracelular 2 (EL2),formando o sítio de ligação estendido. Através da avaliação de várias estruturas crista-lográficas de receptores acoplados à proteína G, acredita-se que EL2 possui uma funçãode "tampa" que enclausura o composto no sítio de ligação (WHEATLEY et al., 2012).Portanto, espera-se que resíduos da alça extracelular 2 possam ajudar na modulação docomportamento do ligante. Além disso, Val(V)201 está envolvido em contatos hidrofóbicocom iv(C30)H (1.94 Å). O resíduo Thr(T)134 possui um valor energético pouco menor queo valor de referência (-5,84 kcal mol−1), contudo, sua importância deve ser mencionada.Esse resíduo é conservado entre os membros de GPCRs, estando posicionado em umaregião de TMH3 que ocupa uma porção do sítio ortostérico próximo do átomo de nitrogêniono anel indol [i(N1)H], atuando como um aceptor de ligações de hidrogênio. Dentre osGPCRs com estrutura cristalográfica, apenas o receptor de histamina 1 (H1R), 5-HT1BRe 5-HT2BR apresentam uma treonina na mesma posição. Ademais, sua mutação paraalanina reduz a afinidade de ERG, LSD e 5-HT ao receptor, impedindo levemente a ligação(WANG et al., 2013a). Esses resultados estão em acordo com a principal contribuiçãoeletrostática encontrada por Heifetz et al. para o complexo ERG-5-HT1BR (HEIFETZ etal., 2015). Dessa forma, deve-se levar esses dois aminoácidos em consideração durante odesenvolvimento de novos fármacos.
Além das ligações de hidrogênio e pontes salinas, interações hidrofóbicas (IH)apresentam alta relevância para a estabilidade proteica e manutenção de complexos proteína-ligante (PATIL et al., 2010; PACE et al., 2011). No caso de proteínas transmembrana,tais como GPCRs, IHs são importantes fatores para preservar sua estrutura dentro damembrana. Ademais, interações hidrofóbicas ajudam a estabilizar alguns ligantes no sítiode ligação, tendo uma função aditiva à energia total de ligação. Neste estudo, a maioria dosresíduos formam IHs com diidroergotamina, como mencionado anteriormente. Os resíduosPhe(F)330 (TM6) e Ile(I)130 (TM3) estão envolvidos em contatos hidrofóbicos, ligaçõesde hidrogênio não-convencionais, e interações π com a região i de DHE (Figura 7a). Umainteração cátion-π é formada entre o anel aromático de Phe(F)330 e DHE [i(N6)] em umadistância de 4,50 Å, enquanto uma ligação de hidrogênio não-convencional é criada com[[ii(C18)O19]. Enquanto isso, Ile(I)130 apresenta uma σ-π com o anel A de DHE (2,42 Å)e uma LH não-convencional com [i(N1)] em uma distância de 2,91 Å.
Estudos de mutação têm mostrado que esses aminoácidos são muito importan-tes para o reconhecimento de ligantes, uma vez que as mutações Phe(F)330Ala(A) eIle(I)130Ala(A) suprimem completamente a ligação de radioligantes (WANG et al., 2013a).Similarmente, os resultados obtidos pelos cálculos de FMO/MP2 em ERG-5-HT1BR de-monstraram a mesma relevância para estes resíduos, e confirmaram a presença dos termosde dispersão como a principal contribuição energética (HEIFETZ et al., 2015). SegundoChoudhary (1995), Phe(F)330 e seu vizinho Phe(F)331 (TMH6), -4,69 (-4,15) kcal mol−1,estão provavelmente envolvidos no processo de ativação de 5-HT2AR, especialmente porqueeles estão relacionados ao movimento da hélice 6, que foi mostrado por levar à ativação do

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 33
receptor β2 (CHOUDHARY et al., 1995), embora a mutação Phe(F)331Ala(A) não causaum impedimento relevante na ligação de ergolinas.
Juntamente com Phe(F)330, Ile(I)130 e Phe(F)331, Cys(C)133 (TMH3) e Trp(W)327(TMH6) formam uma fenda hidrofóbica que empacota o anel ergolina. A relevância dessesdois resíduos foi observada através da análise de mutação para alanina, que criou umimpedimento completo no receptor, bloqueando a ligação de ergolinas e 5-HT(WANGet al., 2013a). Cys(C)133 é um aminoácido altamente conservado entre os receptores deserotonina, estando presente em todos, com exceção de 5-HT2R e 5-HT4R (MCCORVY;ROTH, 2015). Além disso, uma mutação na serina que ocupa esta posição em 5-HT2ARpara alanina afeta a afinidade de ligantes, tais como 5-HT e triptamina (ALMAULA etal., 1996).
A maioria dos resultados energéticos encontrados para EE-MFCC são aminoácidoscarregados, tais como Asp(D)129, Asp(D)352 (Figura 2.6a), Glu(E)198, Arg(R)114 (Figura2.6b), Asp(D)204 e Asp(D)123. Dentre estes, apenas Asp(D)352 e Asp(D)129 possuem suaimportância apresentadas por estudos experimentais ou teóricos. Ademais, todos eles têmsuas energias de interação relacionadas a um efeito de atração ou repulsão eletrostáticainduzido pela distribuição de cargas positivas do ligante, como indicado por HEIFETZet al. (2015). Arg(R)114, Asp(D)123, Glu(E)198, Asp(D)204 e Asp(D)352 ocupam umaposição próxima da porção extracelular da proteína, interagindo primariamente com asregiões ii(N20), iii(N36) e iv(C29)H em distâncias variadas (ver figura 2.5 e tabela 1). Poroutro lado, Leu(L)126, Val(V)200, Thr(T)355 e Phe(F)351 estão envolvidos em contatoshidrofóbicos e LH fracas com as regiões ii e iii, mostrando sua relevância apresentada porestudos cristalográficos, de mutação e computacionais com 5-HT, LSD e outros fármacosanti-migranosos, tais como ergotamina e sumatriptano (WANG et al., 2013a; RODRÍGUEZet al., 2014; BELLO et al., 2016; MCCORVY; ROTH, 2015; HEIFETZ et al., 2015).
No geral, dentre os aminoácidos analisados neste estudo, percebe-se que as ener-gias de interações seguem a seguinte sequência decrescente: Asp(D)129 > Asp(D)352 >Asp(D)123 > Glu(E)198 > Asp(D)204 > Phe(F)330 > Leu(L)126 > Phe(F)351 > Ile(I)130> Val(V)201 > Val(V)200 > Thr(T)355 > Arg(R)114. Exceto Arg(R)114, Asp(D)123,Glu(E)198 e Asp(D)204, todos esses aminoácidos são consistentes com análises prévias.Ademais, alguns resíduos que não estão dentre os mais anergéticos, têm suas energiasdescritas e interações apresentadas devido a sua relevância. Ao final, mostramos na Figura2.7 a isosuperfície do potencial eletrostático com densidade eletrônica projetada para DHEinteragindo com alguns importantes resíduos do sítio de ligação.
2.3.3 Energia total e convergência
Para avaliar as interações através de métodos quânticos, é importante levar emconsideração cada aminoácido significante que pode influenciar o mecanismo de atração erepulsão. Portanto, em vez de levar em consideração uma área arbitrária, procurou-se umbolsão de interação ótimo (r), em que a variação da energia entre raios sequenciais sejamenor que 10%. Portanto, neste trabalho o bolsão de interação variou de 2,0 Å (-117,91kcal mol−1) à 10,0 Å (-228,46 kcal mol−1) para determinar o melhor valor de r, com aconvergência sendo encontrada no raio de 7,0 Å do ligante (-230,93 kcal mol−1).
De acordo com a figura 2.8, há um aumento na energia de interação entre os raiosde 2,0 Å para 3,0 Å (-216,88 kcal mol−1) devido às energias de interação atrativas dosresíduos próximos a DHE formando ligações de hidrogênio e interações hidrofóbicas. Logo

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 34
Figura 2.7 – Superfície de potencial eletrostático de DHE interagindo com alguns dos aminoáci-dos mais importantes.
após, a energia torna-se menos atrativa no raio de 3,5 Å (-210,87 kcal mol−1), devido aenergia repulsiva do resíduo I137. Uma pequena estabilidade é observada entre os raiosde 4,0, 4,5 e 5,0 Å (-211,36 kcal mol−1, -214,39 kcal mol−1 e -212,26 kcal mol−1), masé desfeita pela energia atrativa do resíduo E198 que aumenta a energia no (r=5,5 Å,-227,90 kcal mol−1). C132 e A135 são responsáveis por reduzir a energia no raio de r=6,0Å (-210,87 kcal mol−1).
Figura 2.8 – Energia de interação total como função da distância do ligante.
2.3.4 Descrição energética das regiões do ligante
Como mostrado por McCorvy e Roth, a estrutura de alguns ligantes serotonérgicoscompartilham similaridades na conformação e composição, o que torna fácil compreender

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 35
o porque os efeitos off-target são frequentes (MCCORVY; ROTH, 2015). Neste contexto,apresentamos os aspectos energéticos e o número de contatos formados pelas regiõesindividuais de diidroergotamina na tabela 2.2. Cada região foi selecionada baseado narelevância estrutural apresentada pela estrutura cristalográfica de DHE (HEBERT, 1979).Energeticamente, as regiões iii e i são a primeira e segunda região mais relevante comvalores de -116,12 e -103,75 kcal mol−1, respectivamente, sendo seguidos pelas regiões ii(-44,26 kcal mol−1) e iv (-36,6 kcal mol−1). Esta sequência muda quando comparamos onúmero de contatos, como segue: região i (59) > região iii (36) > região iv (16) > região ii(4). Este tipo de informação pode ser bastante útil no desenvolvimento de fármacos, umavez que pode mostrar os principais grupos funcionais e regiões do ligante.
Tabela 2.2 – Energia de ligação e número de contatos das regiões individuais de DHE. Osresultados energéticos são apresentados para os cálculos realizados utilizandoos métodos MFCC e EE-MFCC.
2.3.5 Descrição energética dos segmentos de 5-HT1BR
Outra particularidade aqui analisada é a característica energética de cada regiãoindividual de 5-HT1BR. GPCRs exibem uma estrutura tridimensional similar, com setehélices transmembrana unidas por alças internas e externas, contando com 13 motivosestruturais. Tem-se observado que cada um desses motivos, ou segmentos, pode apresentarum movimento diferente após a interação com ligantes, sendo relacionado à seus efeitosnos níveis de ativação da proteína (MCCORVY; ROTH, 2015). Portanto, a energiaindividual de cada aminoácido foi somada e separada por segmento, como apresentadona tabela 4. A maior energia de atração é apresentada por TM3 (-121,57 kcal mol−1)

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 36
seguido por EL2 (-56,60 kcal mol−1), TM7 (-48,88 kcal mol−1), TM6 (-16,59 kcal mol−1),TM5 (-7,94 kcal mol−1), TM4 (-2,42 kcal mol−1) e TM1 (-0,44 kcal mol−1). Ademais, asenergias de interação repulsivas são observadas em EL1 (19,93 kcal mol−1), a região N-terminal (4,25 kcal mol−1) e TM2 (1,80 kcal mol−1). A comparação de algumas estruturascristalográficas mostram que TM1 e TM2 não sofrem grandes mudanças estruturais após ainteração com ligantes, enquanto as hélices TM3, TM6 e TM7 sofrem as maiores mudanças(VENKATAKRISHNAN et al., 2013), o que está em acordo com nossos achados.
Tabela 2.3 – Energia de interação (MFCC e EE-MFCC) por estrutura secundária doreceptor de serotonina.
2.3.6 Efeito do esquema de incremento eletrostático
O método MFCC tem sido largamente utilizado para calcular a energia de ligaçãode complexos biomoleculares (MOTA et al., 2016; RODRIGUES et al., 2013; BARROSO-NETO et al., 2012), uma vez que ele torna possível uma investigação a nível quânticode um grande número de resíduos de aminoácidos em uma proteína com um pequenocusto computacional e grande acurácia (GORDON et al., 2012; GAO et al., 2004). Apesarda boa correlação entre os resultados obtidos pelo MFCC e os resultados experimentais,percebe-se que considerar as contribuições de solvente, íons e outros aminoácidos é muitoimportante nos cálculos moleculares.
De acordo com isso, muitas aproximações têm sido implementadas no esquemado MFCC objetivando tratar o efeito eletrostático do sistema completo em cada passodo fracionamento, tais como o uso de muitos caps (HE; ZHANG, 2006; LIMA NETO etal., 2015), modelos de solvatação contínuos (MARTINS et al., 2016; OURIQUE et al.,2016; MARTINS et al., 2017), dentre outros (ZANATTA et al., 2012; ZANATTA et al.,2014; ZANATTA et al., 2016; WANG et al., 2013b). Além disso, o uso de cargas pontuaisé largamente aplicado para incluir as energias do ambiente eletrostático em métodos defragmentação e cálculos híbridos (SVÄRD; RASMUSON, 2009; LI; LI; JIANG, 2007;SENN; THIEL, 2009), sendo implementadas no MFCC para incluir uma característicade polarização nos fragmentos (JIANG; MA; JIANG, 2006; BRINKMANN; HEIFETS;KANTOROVICH, 2014; OLSEN et al., 2015).
Neste sentido, incluímos o esquema de inclusão eletrostático em nossa análise porMFCC no complexo DHE-5-HT1B para obter a contribuição individual dos aminoácidos no

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 37
sítio de ligação dentro de um raio selecionado, encontrando as interações mais relevantespara o complexo. O emprego dessa forma adaptada do MFCC faz com que o sistematorne-se mais realista através da polarização da densidade eletrônica (OLSEN et al., 2015;JAKOBSEN; KRISTENSEN; JENSEN, 2013).
Para dar-nos a ideia sobre as diferenças entre MFCC e EE-MFCC, comparamosnossos resultados para ambos os métodos por considerar as maiores variações energéticasna figura 2.9, excetuando aqueles mostrados na figura 2.5. A maior variação é encontradapara o resíduo Asp(D)204 (9,82 kcal mol−1), seguido pelo resíduo Tyr(Y)359 (8,1 kcalmol−1), Asp(D)123 (8,19 kcal mol−1) e Asp(D)129 (8,09 kcal mol−1), todos aminoácidossão carregados negativamente, exceto Tyr(Y)359. É importante informar que uma altavariação foi observada em Ile(I)137 (0,74 para 5,88 kcal mol−1) e Ala(A)135 (0,16 para5,10 kcal mol−1). A amina da cadeia principal destes últimos dois resíduos está próximode N1 de diidroergotamina e, como apresentado na figura 2.7, eles se repelem. No caso deTyr(Y)359, também é visto que a cadeia lateral repele a porção i(C7)H3 de DHE.
Figura 2.9 – Resíduos com maior variação energética entre os métodos MFCC e EE-MFCC.
É notável que o uso do esquema de incremento eletrostático trouxe, para a maioriadas energias de interação individuais, uma característica mais positiva quando comparadocom o MFCC convencional, principalmente para os resíduos carregados, onde há umacréscimo de 9,82 kcal mol−1 (Asp[D]204) à 1,75 kcal mol−1 (Arg[R]114), o que está emacordo com a referência Liu et al. (2015). Além disso, ele intensificou o quão atrativo sãoalguns resíduos, tais como Phe(F)351 (-6,23 para -7,86 kcal mol−1) e Leu(L)126 (-6,39para -7,96 kcal mol−1). Apesar disso, na maioria dos casos a ordem de importância destesresíduos é mantida em ambos modelos.
Em um estudo recente realizado por Jakobsen et al., foi mostrado que a polarizaçãocriada por cargas parciais de campos de força comuns, tais como CHAMm e AMBER, não

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 38
são adequados para gerar uma boa descrição eletrostática (JAKOBSEN; KRISTENSEN;JENSEN, 2013). Contudo, os padrões energéticos aqui apresentados têm mostrado queo uso de cargas pontuais é uma aproximação razoável para melhorar a caracterizaçãode complexos ligante-proteína. Isto está em acordo com os resultados encontrados porWang e colaboradores para a energia total de proteínas, calculada através do métodode fracionamento molecular generalizado com caps conjugados usando cargas pontuaisobtidas pelo campo de força Amber94 (WANG et al., 2013b). No geral, os resultados aquiexpostos demonstram que o campo eletrostático usando cargas pontuais é uma formaeficiente para tratar o efeito de polarização no cálculo de fragmentos, tornando a energiade ligação entre resíduo-ligante melhor representada. Contudo, pode-se observar que amaioria dos resíduos não sofre uma alta variação, o que pode ser um efeito do pequenonúmero de grupos polares nos aminoácidos.
2.3.7 Reprodutibilidade utilizando outros métodos de cálculo
Recentemente, Burns e colaboradores (2011) revisaram as propriedades de diversosfuncionais DFT no cálculo de interações não covalentes através da utilização destes nadescrição de um conjunto de estruturas moleculares. Nesse trabalho é apresentada umadiscussão interessante sobre a diferença na formulação matemática e funcional/históricade cada tipo de funcional. Seguindo a classificação adotada por Burns, nós escolhemos trêsfuncionais descritos entre os tipos GGA com correção semi-empírica do termo de dispersão(B97D), meta-GGA (M062X) e GGA hibrido (B3LYP) para a realização deste trabalho.
B97D foi proposto por Grimme e inclui uma correção na descrição dos termosde dispersão (-D) que possui como objetivo melhorar a representação de interações nãocovalentes (GRIMME, 2006), sendo utilizado por Anthony e Grimme em 2012 para calculara energia de interação em alguns complexos proteína-ligante através do esquema MFCC.O funcional M062X foi desenvolvido por Zhao and Truhlar (2008), sendo hoje reconhecidocomo um dos métodos de cálculo mais utilizados no âmbito de otimização de estruturasbiomoleculares, tais como proteínas e complexos proteína-ligante.
Assim como o B97D, M062X é parametrizado para considerar os termos de dis-persão, grande problema encontrado no DFT, mas sua formulação matemática divergeconsideravelmente daquela encontrada no GGA-D e, consequentemente, o resultado finalpode diferir do encontrado nos demais métodos utilizados neste trabalho. Diferente dosfuncionais citados acima, o B3LYP dispensa comentários uma vez que trata-se do primeirofuncional a possuir um nível de correção nos termos de dispersão. Este foi proposto porBecke em 1993 e, desde então, foi largamente empregados nos mais diversos tipos decálculos em moléculas orgânicas e inorgânicas, sendo visto até hoje como método basepara validar os mais diversos tipos de resultados que podem ser obtidos através de cálculosutilizando DFT. Os resultados para os cálculos realizados com M062X e B3LYP podemser observados nas tabelas 4 e 5, respectivamente.
Como pode ser observado comparando as tabelas 1, 4 e 5, o padrão encontrado paraa variação de energia é bastante similar. Contudo, como já esperado, a energia total obtidaa partir de cálculos feitos com M062X e B3LYP é menor do que aquela obtida por B97D.Isso se dá pelo menor recurso da correção dos termos de dispersão, muito importantesem proteínas contendo em sua composição um grande número de resíduos hidrofóbicos,como é o caso dos receptores acoplados à proteína G. Isso pode ser comprovado quandoverificam-se os resultados para aminoácidos como Asp(D)129, que apresenta um valorenergético bastante similar entre B97D e M062X, uma vez que a interação entre este resíduo

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 39
Tabela 2.4 – Descrição energética dos 108 resíduos de 5-HT1BR interagindo com DHE. Cálculosrealizados com o funcional M062X.

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 40
Tabela 2.5 – Descrição energética dos 108 resíduos de 5-HT1BR interagindo com DHE. Cálculosrealizados com o funcional B3LYP.

Capítulo 2. Interação diidroergotamina-receptor 5-HT1B descrita por bioquímica quântica 41
e o ligante se dá principalmente pela formação de uma ponte salina. Em contrapartida,o resíduo Phe(F)331 é parte da fenda hidrofóbica de 5-HT1B, estando empenhado eminterações do tipo hidrofóbica com diidroergotamina.
Em contrapartida, os resultados observados para B3LYP se mostram menores doque aqueles encontrados para os outros dois funcionais. Faz-se interessante notar que osvalores energéticos mais próximo de zero encontrados para esse funcional tornam-o bastanteatrativo para pesquisadores que buscam validação experimental para seus cálculos teóricos,uma vez que as energias encontradas para os demais funcionais são altas em comparaçãoao conhecido experimentalmente. Apesar disso, vê-se claramente que a intensidade dasinterações obtidas para B3LYP enfraqueceu a relevância de alguns aminoácidos bastanteimportantes, como é o caso do Phe(F)331.
2.4 Conclusão
Os receptores serotonérgicos são alvos biológicos atrativos e intensamente exploradosem drug discovery. Dentre eles, o receptor 5-HT1B e seu agonista DHE tem sido largamenteutilizados não somente no tratamento de enxaqueca, mas também para outras disfunçõesneurológicas, o que os tornam importantes alvos na pesquisa terapêutica. Portanto, acompreensão das energias de interação entre ligante-receptor é vital para o sucesso nodesign de fármacos que visem esse receptor.
Para auxiliar neste processo, investigamos a energia de interação deste complexousando cálculos baseados em bioquímica quântica. Não somente utilizamos o MFCCcomum para a realização destes cálculos, mas também incrementamos o método através douso de cargas pontuais, criando um sistema de incremento eletrostático em cada momentodo MFCC.
No geral, nossos resultados foram consistentes com estudos experimentais e teóri-cos anteriores. Os aminoácidos aqui estudados foram selecionados através do critério deconvergência, a partir do qual encontramos uma menor variação na energia total após7,0 Å do ligante. A interação de 108 aminoácidos com DHE foi avaliada, mostrandoque os resíduos Asp(D)129, Asp(D)352, Asp(D)123, Glu(E)198, Asp(D)204, Phe(F)330,Leu(L)126, Phe(F)351, Ile(I)130, Val(V)201, Val(V)200, Thr(T)355 e Arg(R)114 apre-sentam uma maior relevância energética. Também apresentamos a descrição energéticade outros resíduos experimentalmente relevantes, bem como das regiões do ligante e daproteína. Acreditamos que os resultados aqui apresentado são um passo inicial em direçãoao desenvolvimento de novos medicamentos visando não só o tratamento da enxaqueca,mas também outras doenças relacionadas com os receptores serotonérgicos.

42
capítulo 3
DESCREVENDO INIBIDORES DE PROTEÍNAS DE CHECKPOINT
CELULAR POR BIOQUÍMICA QUÂNTICA
O sistema imunológico é composto por um complexo de estruturas biológicas ecélulas especializadas que possuem como função fundamental o reconhecimento e eliminaçãode patógenos. Portanto, faz-se necessário que esse sistema diferencie uma grande variedadede estruturas, principalmente proteicas e glicoproteicas, constituintes do próprio organismo,daquelas oriundas do patógeno. Dessa forma, vê-se que esse sistema é altamente complexo,com vários níveis de regulação celular e tecidual, o que o torna um dos sistemas maisatrativos como fonte de pesquisa, pois pouco se conhece sobre o seu funcionamento, o quetambém ocorre com o sistema nervoso. Ao final, praticamente tudo o que acontece nocorpo de animais passa pelo controle de pelo menos um desses sistemas, e estudos vemmostrando que vegetais e outros organismos vivos apresentam sistemas equivalentes.
Quando um patógeno invade o hospedeiro humano, a resposta contra esse "corpoestranho" é logo montada. Os protagonistas desse intrincado processo de reconhecimento eregulação são vários e largamente diferenciados, indo desde células especializadas até órgãose tecidos. As células hematológicas têm a maior rotatividade de todas as células humanas.Isso requer o reabastecimento dessas populações em uma base constante. Para cumprireste requisito, temos células estaminais hematopoiéticas (CEH), que além de mantera sua própria auto-renovação, dão origem a progenitores linfóides (PL) e progenitoresmielóides (PM). As células PL diferenciam-se em células natural killer (NK), células Be T. As células PM podem se diferenciar em progenitores megacariócitos e eritróides,que são responsáveis pela produção de glóbulos vermelhos e plaquetas, ou progenitor degranulócitos e macrófagos que levam à geração de mastócitos, eosinófilos e células deneutrófilos, bem como monócitos. Os monócitos se diferenciam ainda mais em macrófagose células dendríticas monocíticas (CDM).
Estas células, e a resposta imune como um todo, podem ser dividida em duascategorias de formas de ação: imunidade inata e imunidade adquirida. De forma geral, aimunidade inata é descrita como a resposta imunológica com a qual cada um já nasce, econsiste principalmente de barreiras mecânicas (como a superfície epitelial), à ação decélulas especializadas e proteínas livres ou glicoproteínas transmembrana. Por outro lado aimunidade adquirida refere-se a uma resposta específica e mais complexa do que a respostainata, uma vez que o patógeno deve ser processado e reconhecido pelo corpo, e só então osistema imune cria anticorpos que serão designados especificamente para o alvo. Após aeliminação da infecção, células de memória são criadas, tornando a respostas à um futuroataque mais eficiente (CHAPLIN, 2010; BURNET et al., 1959).
As respostas adaptativas baseiam-se principalmente nos receptores específicos deantígeno expressos nas superfícies dos linfócitos T e B. A produção de anticorpos estárelacionadas com os linfócitos B, enquanto a imunidade mediada por células relaciona-secom os linfócitos T. Monócitos e macrófagos são células efetoras de imunidade e facilitama atividade dos linfócitos T e de modificadores da resposta biológica, como as interleucinas.Mais de setenta atividades biológicas diferentes são mediadas por produtos de linfócitos,monócitos e macrófagos. Assim, pode-se vislumbrar o complexo arranjo de funções mediadas

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 43
pelo sistema imunológico (CORRALES et al., 2017; MEDZHITOV; JR, 1997).
Em humanos, células T, ou linfócitos T, são um tipo de linfócito que possui papelcentral na imunidade mediada por células. A vida de uma célula T inicia no timo, ondecélulas imaturas proliferam e criam um grande repertório de receptores de células T (RCTs)através da recombinação de segmentos dos genes destas RCTs. Um processo de seleçãodessas células é iniciado, onde as escolhidas são posteriormente liberadas como célulasmaduras para circular pela corrente sanguínea, baço e órgãos linfáticos. Estas célulasdiferenciam-se de acordo com sua função em: citotóxicas, auxiliares, memória, naturalkiller, reguladoras ou γ-δ.
Nos locais de ação, essas células são expostas à antígenos externos (no caso deinfecção) ou proteínas do próprio organismos apresentando mutações (no caso de célulasmalignas - como o câncer). Reconhecendo antígenos apresentados pelo complexo principalde histocompatibilidade (MHC) presentes nas células apresentadoras de antígenos (CAA)e ativando cascatas de sinalização, as células T podem levar à uma forte resposta contra oagente patológico e à formação de uma memória imunológica (JANEWAY et al., 2005).
Uma vez que TCRs podem ter especificidade à antígenos do próprio organismo,numerosos pontos de controle regulam a ativação das células T durante uma respostaimune. O ponto inicial é a necessidade de ligação de um TCR ao antígeno apresentadopelo MHC. Contudo, somente isso não é suficiente para a ativação da célula T, sendonecessário a ligação da proteína CD28 da célula T às proteínas B7-1 e B7-2 (CD80 eCD86, respectivamente) presentes em CAA. Após a ativação, a proteína 4 associada aolinfócito T citotóxico (CTLA-4, do inglês - cytotoxic T-lymphocyte-associated protein 4)migra para a membrana celular. CTLA-4 é considerada a líder dos pontos de controlesinibidores da célula T, uma vez que esta inibe células que possam apresentar-se comocélulas T auto-reativas logo no estágio inicial da ativação das células maduras (isto é, desdeo timo). Essa proteína liga-se com maior afinidade à CD80 e CD86 do que CD28, o queleva ao desligamento do complexo CD28-CD80/86, e a inativação da célula T (SHARMA;ALLISON, 2015).
Juntamente com CTLA-4, uma outra proteína tem surgido nos últimos anos comoum importante ponto de controle, a proteína 1 de morte celular programada (PD-1, doinglês - programmed cell death protein 1). PD-1 regula células T previamente ativadasnos últimos estágios da resposta imune, primariamente nos tecidos periféricos. Os ligantesde PD-1 (PD-L1 [B7-H1] e PD-L2 [B7-DC]) são expressos na superfície de CAAs e célulasmalignas, particularmente após haver a liberação de citocinas inflamatórias no local, taiscomo IFNγ. PD-L1 também pode se ligar à CD80 expressos nas células T como umsegundo mecanismo de supressão das células T (BUCHBINDER; DESAI, 2016).
3.1 A imunoterapia contra o câncer
Sabe-se há muitos anos que o sistema imune desempenha um papel importanteno desenvolvimento e controle de neoplasias, uma vez que pacientes imunossuprimidostêm maior risco de desenvolver o câncer, e a regressão espontânea de muitos tipos detumores malignos é um fenômeno raro, mas bem reconhecido, ocorrendo em aproximada-mente 1 em cada 60.000-100.000 casos de câncer (CHALLIS; STAM, 1990; KUCEROVA;CERVINKOVA, 2016). Há aproximadamente 200 anos atrás, os médicos alemães Busch eFehleisen notaram independentemente a regressão de tumores em pacientes com câncerapós infecções acidentais por erisipela (BUSCH, 1868; FEHLEISEN, 1882).

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 44
Foi somente em 1891 que um cirurgião americano, William Coley, observou aregressão de longo prazo de um sarcoma após uma infecção por erisipela, iniciandoum projeto de 43 anos envolvendo a injeção de bactérias inativadas em pacientes comtumores sem possibilidade e retirada cirúrgica, dando inicio ao campo da imunoterapia(COLEY, 1893). Coley reportou um número significativo de regressão e cura em mais de1.000 pacientes, a maior parte com sarcomas, e o método começou a ganhar aceitação enotoriedade (COLEY, 1908).
Com o passar dos anos, esse método foi caindo no esquecimento devido a falhas,pouca capacidade de repetibilidade pelos pares, e o surgimento da radioterapia e quimiote-rapia para tratar câncer. Por mais de 100 anos os princípios de Coley ficaram praticamenteesquecidos (PARISH, 2003), até Morales, Eidinger e Bruce (1976) e Old, Clarke e Bena-cerraf (1959) estabelecerem a eficácia no tratamento de câncer de bexiga com o uso dabactéria Bacilo Calmette-Guérin em modelos animais. Até mesmo infecções virais teriamum efeito supressor de câncer. Em 1904, George Dock descreveu que uma mulher de 42anos com leucemia aguda sofreu remissão temporária após uma suposta infecção cominfluenza em 1896 (LARSON et al., 2015). Ao mesmo tempo, um melhor entendimento dosistema imunológico estava sendo obtido, incluindo descobertas sobre imunidade celular ehumoral. Devido às suas descobertas fundamentais e dedicação ao campo ao longo da vida,Coley e Old têm sido referidos como os "pais da imunoterapia” (OISETH; AZIZ, 2017).
Apesar de possuirmos um complexo sistema imunológico, que é capaz de distinguire eliminar diversos patógenos, ainda há uma lacuna observada no conhecimento atualde seu funcionamento durante algumas patologias, como é o caso do câncer. Uma vezque as células cancerígenas apresentam antígenos específicos que podem ser reconhecidospelas células T, é de se esperar que o sistema de defesa aja na destruição dessas. Contudo,em muitos casos, esse sistema mostra-se incapaz de atuar. Foram James P. Allison eTasuku Honjo, em especial, mas não unicamente, que ajudaram a pavimentaram o caminhopara uma explicação mais consistente sobre esse comportamento (LEACH; KRUMMEL;ALLISON, 1996; ISHIDA et al., 1992; AZUMA et al., 1993).
Esses pesquisadores observaram a expressão de proteínas em algumas célulasespecializadas do sistema imunológico que agem diretamente como pontos de controle(checkpoint) de seu funcionamento (principalmente CTLA-4 e/ou PD-1) (NISHIMURAet al., 1999; KRUMMEL; ALLISON, 1995; HONJO, 2018), e que a modulação dessasproteínas poderia levar à inativação de células T. Poucos anos depois, foi observado quealgumas células cancerígenas expressam ativamente proteínas que servem como ativadoresde checkpoints, além de tecidos tomados pelo tumor maligno apresentarem um ambienteque propicia o não funcionamento das células de defesa, como as células T reguladoras queacumulam-se em tecidos tomados pelo câncer, mas não agem contra ele e podem chegar ainibir outras células (ALLISON, 2018; SHARMA; ALLISON, 2015).
Neste sentido, a imunoterapia contra o câncer está surgindo como uma ferramentabenéfica para o tratamento desse mal, ativando o sistema imunológico para produzirefeitos antitumorais. Recentemente, a imunoterapia do câncer, particularmente a terapiade checkpoint imunológico, progrediu e forneceu novas estratégias para a luta contra essadoença. Esta estratégia alcançou recentemente notável sucesso clínico em pacientes comnumerosos tipos de câncer maligno. Por exemplo, em doentes com melanoma avançado, obloqueio da proteína 4 associada ao linfócito T citotóxico (CTLA-4) através do anticorpoipilimumab e a inibição do receptor 1 de morte celular programada (PD-1) através doanticorpo nivolumab resultou em melhoria geral no tempo de sobrevida do pacientes

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 45
(HODI et al., 2010; TOPALIAN et al., 2014).
Em comparação com as terapias convencionais para o câncer, incluindo radiação equimioterapia, a imunoterapia tem como alvo principal o sistema imunológico ou micro-ambiente tumoral, e pode induzir um efeito sinérgico em terapias combinadas (TANG;QIAO; FU, 2016). Devido ao sucessos da imunoterapia, seis inibidores de checkpoint foramaprovados pelo US-FDA para o combate ao câncer nos últimos anos: ipilimumab (Yervoy;anti-CTLA-4), pembrolizumab (Keytruda; anti-PD-1), nivolumab (Opdivo; anti-PD-1),atezolizumab, avelumab e durvalumabe (anti-PD-L1), e novos fármacos vem sendo testadoscontinuamente.
Os efeitos inibitórios da CTLA-4 ocorrem na fase inicial da resposta imune, interfe-rindo com os sinais necessários para a ativação das células T (TOPALIAN et al., 2012).A ligação da CTLA-4 ao CD80/86 inibe a ativação dos linfócitos T, resultando em umsinal negativo de feedback que diminui a resposta imune. O fármaco ipilimumab induza ativação das células T, desativando a inibição do CTLA-4 e permitindo que o sistemaimunológico seja ativado (MICHOT et al., 2016).
O ipilimumab é atualmente aprovado pela FDA para duas indicações distintasde melanoma (HODI et al., 2010). Demonstrou-se que ele apresenta um efeito sinérgicocom o aumento da iniciação de células T via inibição da CTLA-4 nos linfonodos eatividade citotóxica via inibição da PD-1 no ambiente tumoral (ROBERT et al., 2011). Osefeitos adversos clinicamente significativos incluem enterocolite imunomediada, hepatite eendocrinopatias, com efeitos adversos como fadiga, diarréia, prurido, erupção cutânea ecolite também sendo comuns. A maioria dos eventos relacionados ao sistema imunológicoé reversível com o tratamento com esteróides imunossupressores (ARAGON-SANABRIA;KIM; DONG, 2018).
Por outro lado, com base nas taxas crescentes de resposta ao tratamento, osinibidores de PD-1 e PD-L1 estão sendo aprovados para vários tipos de câncer, incluindomelanoma, câncer de pulmão, câncer urotelial e, mais notavelmente, tumores sólidos commutações que levam instabilidade de microssatélites e reparo incorreto. A aprovação dopembrolizumab em 2015 para os tumores sólidos foi um marco, uma vez que esse foio primeiro momento em que um medicamento foi aprovado com base em um teste debiomarcador para uma característica e não um local anatômico do tumor; isso representauma mudança significativa no tratamento do câncer (KHOJA et al., 2015; KEIR et al.,2008; FRANCISCO; SAGE; SHARPE, 2010).
Os inibidores de PD-L1/inibidores de PD-1 são geralmente bem tolerados; entre-tanto, os pacientes podem sofrer efeitos adversos sérios e possivelmente potencialmentefatais. Embora qualquer sistema orgânico possa ser submetido a uma reação auto-imune,as reações mais comumente observadas são colite, hepatite, endocrinopatias (tireóide esupra-renais), pneumonite e progressão cutânea a síndrome de Stevens-Johnson (GENG etal., 2017; O’DONNELL et al., 2017).
Durante a fase III de testes clínicos, esses fármacos apresentarem bons resultados naremissão do câncer (sem retorno por mais de dois anos). Contudo, a eficácia da imunoterapiacontra o câncer é limitada a apenas alguns pacientes, uma vez que não são todos querespondem a essas manobras imunomoduladoras (TANG; QIAO; FU, 2016). Ademais,dois terços dos pacientes que usaram pelo menos um dos compostos têm mostrado efeitosadversos relacionados com a resposta imune (irAEs), sendo os mais frequentes: fadiga eproblemas dermatológicos (manifestados no início do tratamento), seguidos por problemas

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 46
gastrointestinais, hepatite e problemas endócrinos (principalmente no final do tratamento).Embora presentes nos três fármacos, estes efeitos foram maiores em pacientes que fizeramuso do medicamento Yervoy, o que tem sido relacionado com sua ação anti-CTLA-4 emestágios anteriores ao ataque à célula cancerígena (BEATTY; GLADNEY, 2015).
Dessa forma, fármacos que visam impedir a interação entre PD-1 e seus ligantesvem se mostrando ferramentas chave na terapia contra o câncer. Embora não se conheçaprofundamente seu mecanismo de ação, sabe-se que a ligação de PD-1/PD-L1/L2 levaa interferência na via glicolítica através da proteína Akt, o que acarreta na inibição daproliferação de células T, interferon γ, e fator de necrose tumoral α, bem como na reduçãoda sobrevivência da célula. A expressão de PD-1 é um sinal de uma célula T "exausta",isto é, que já foi muito estimulada, o que ocorre normalmente durante uma infecção oucâncer (NISHINO et al., 2017).
PD-1 (gene Pdcd1) é uma glicoproteína monomérica transmembranar da superfamí-lia das imunoglobulinas composta de aproximadamente 288 aminoácidos, que formam umdomínio imunoglobulina V (IgSF), um domínio de transição, um domínio transmembrana e,por fim, um domínio citoplasmático, que contém dois motivos de sinalização intracelulares:um inibidor e outro ativador. Essa proteína é um membro da família B7-CD28, sendo uminibidor de superfície celular principalmente expresso na fase final de ativação de células TCD4+/CD8+ e natural killer, células apresentadores de antígeno (CAA), incluindo célulasdendríticas, células B e monócitos (ISHIDA et al., 1992; NISHIMURA et al., 1999; IWAIet al., 2017; ZHANG et al., 2004). O domínio extracelular de PD-1 contém um arranjo defolhas-β formado pelas cadeias GFCC’ e ABED, respectivamente (para um revisão verreferência Zak et al. (2017)).
Os ligantes de PD-1, PD-L1 e PD-L2, compartilham >35% similaridade de sequênciae são expressos por diferentes células, embora compartilhem a conformação comum destafamília de proteínas (LATCHMAN et al., 2001). Essa diferença no padrão de expressãoé visto como fator importante para o futuro desenvolvimento de fármacos, uma vez quePD-L1 está presente em células T and B, células dendríticas (DCs), macrófagos e célulasnão hematopoéticas, enquanto PD-L2 é expressa somente em um grupo de células B epode ser induzida em DCs, monócitos e macrófagos, o que tem sido um indicativo da açãode PD-L1 na regulação de outras células de defesa. Some-se a isso, o fato de pacientes queapresentaram remissão do tumor ao utilizarem anti-PD-1 apresentarem comportamentoPD-L1 positivo (GHIOTTO et al., 2010).
A recente publicação das estruturas cristalográficas de PD-1 complexado compembrolizumab, nivolumab e PD-L1 permitiu uma maior compreensão da base estruturalrelacionado ao impedimento da interação entre PD-1-PD-L1. Contudo, não há um trabalhoobjetivando uma comparação entre os três complexos, e, excetuando a própria publicaçãodas estruturas cristalográficas de pembrolizumab e nivolumab, não há ainda a determinaçãodos padrões de interação de cada complexo. Portanto, a observação das interações entreos três complexos, identificando aquelas comuns/diferentes para os 3 complexos, podepermitir o desenvolvimento de novos inibidores de PD-1 e PD-L1 com maior afinidade eespecificidade (ZAK et al., 2015; LEE et al., 2016; TAN et al., 2017).
A superposição das estruturas cristalográficas de PD-1 em complexo com o li-gante natural PD-L1, e com os anticorpos monoclonais pembrolizumab and nivolumabrevelou que esses três ligantes acoplam-se no receptor de forma diferente, embora existauma complementariedade parcial, no que pode ser a região de reconhecimento no sítiode ligação. A figura 3.1 apresenta os onze aminoácidos que formam o sítio de ligação

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 47
comum de PD-1 (Phe[F]63, Val[V]64, Ile[I]126, Ser[S]127, Leu[L]128, Ala[A]129, Phe[F]130,Lys[K]131, Ala[A]132, GLN[Q]133 and Ile[I]134). A relevância de alguns desses resíduosjá foi comprovada experimentalmente e computacionalmente (LÁZÁR-MOLNÁR et al.,2017).
Dentro deste contexto, nosso objetivo neste capítulo é apresentar o primeiro estudocomparativo in silico que visa identificar as energias de interação entre os resíduos deaminoácido do receptor PD-1 com os resíduos dos três ligantes pembrolizumab, nivolumabe PD-L1 com o intuito de mapear o sítio de ligação comum para os três, bem como relataras interações que são responsáveis pela individualidade nas respostas biológicas de cadaum. O esquema de fracionamento molecular com caps conjugados (MFCC) foi usadopara dividir as quatro proteínas em fragmentos aminoacídicos e a energia de interação écalculada a partir da teoria do funcional da densidade (DFT+D).
3.2 Materiais e Métodos
3.2.1 Dados do complexo ligante-receptor
Para a realização dos cálculos neste estudo utilizou-se a estrutura cristalográficado receptor 1 de morte celular programada (PD-1) (região extracelular) em complexo compembrolizumab (PDB ID: 5GGS; 1,99 Å), nivolumab (PDB ID: 5WT9; 2,41 Å) e o PD-L1(PDB ID: 4ZQK; 2.45 Å) obtidas por difração de raios-X e depositadas no banco ProteinData Bank (PDB, <www.rcsb.org/pdb>) (ZAK et al., 2015; LEE et al., 2016; TAN etal., 2017). Ademais, o cálculo do estado de protonação em pH fisiológico para ligantes eaminoácidos foi realizado através do uso do software PROPKA 3.1 (SØNDERGAARD etal., 2011).
3.2.2 Cálculos clássicos
Átomos não elucidados pela difração de raios-X, e portanto ausentes na estruturacristalográfica, como átomos de hidrogênio e cadeias laterais de alguns aminoácidos, foramadicionados e submetidos a otimização clássica em suas geometrias, mantendo os outrosátomos fixos. Com exceção do complexo PD-1/Pembrolizumab, todos os demais passarampor uma otimização de estrutura na cadeia lateral de todos os aminoácidos. Esta otimizaçãofoi realizado sob os parâmetros de cálculo do campo de força CHARMm (Chemistry atHARrvard Molecular Mechanics), sendo este parametrizados para todos os átomos, porémcom maior especialização para moléculas orgânicas (MOMANY; RONE, 1992). Paraeste cálculo os padrões de convergência foram ajustados para 10−5 kcal mol−1 (variaçãode energia total), 0.001 kcal.mol−1Å−1 (gradiente de desvio médio quadrado) e 10−5Å(deslocamento atômico máximo).
3.2.3 Fracionamento molecular com caps conjugados
A energia de interação entre aminoácidos de PD-1 com seus ligantes foi calculadaatravés do formalismo do MFCC proposto por Zhang e Zhang (ZHANG; ZHANG, 2003)e modificado por Rodrigues et al. (2013). Para cada aminoácido de PD-1 na posição Ri,desenhamos uma esfera virtual com raio de 8,0 Å, e avaliamos sua energia de interaçãocom todos os aminoácidos dos ligantes (Rj) que possuem ao menos um átomo dentroda esfera imaginária. Dessa forma, o esquema para computar as energias de interaçãoresíduo-resíduo, EI(Ri − Rj), são calculados como segue:
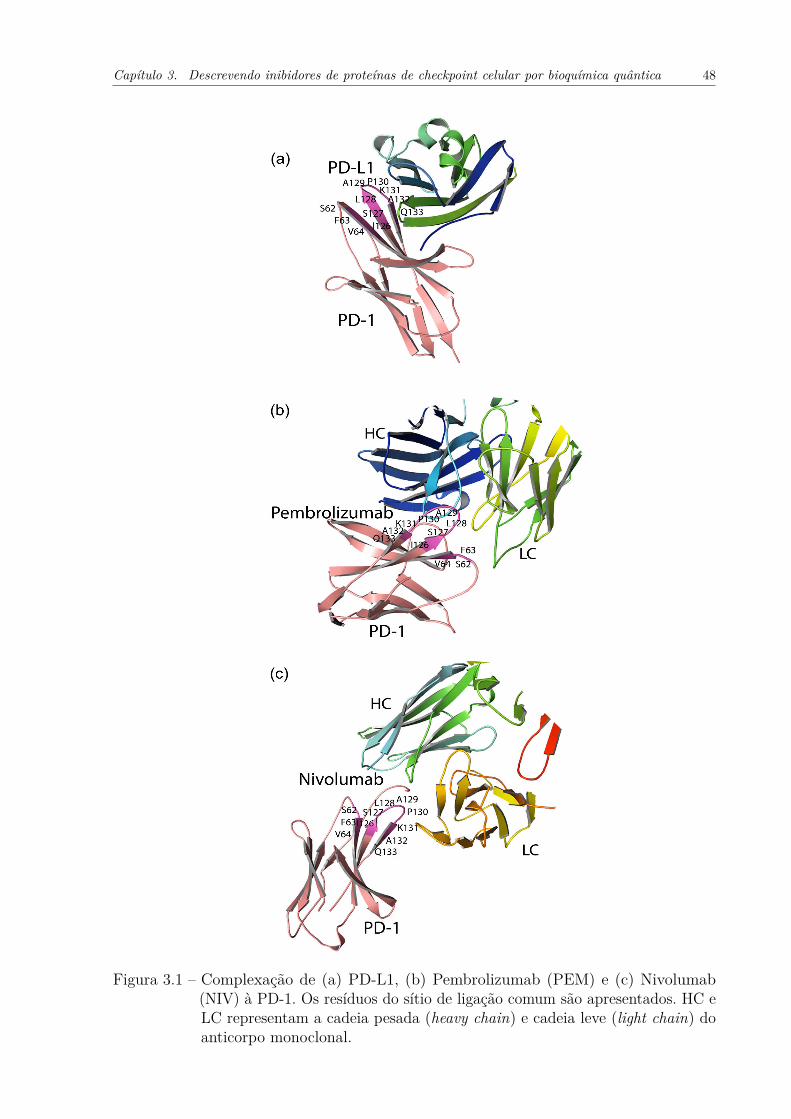
Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 48
Figura 3.1 – Complexação de (a) PD-L1, (b) Pembrolizumab (PEM) e (c) Nivolumab(NIV) à PD-1. Os resíduos do sítio de ligação comum são apresentados. HC eLC representam a cadeia pesada (heavy chain) e cadeia leve (light chain) doanticorpo monoclonal.

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 49
EI(Ri − Rj) = E(CiRiCi∗ − CjRjCj∗) − E(CiRiCi∗ − CjCj∗)
− E(CiCi∗ − CjRjCj∗) + E(CiCi∗ − CjCj∗), (3.1)
onde Ci e Ci∗ (Cj e Cj∗) são respectivamente os resíduos anterior e posterior de Ri (Rj).O termo E(CiRiCi∗ − CjRjCj∗) corresponde a energia total do fragmento composto porambos resíduos, bem como seus caps; E(CiRiCi∗ − CjCj∗) representa a energia total dosistema formado pelos caps e Ri , enquanto em E(CiCi∗ − CjRjCj∗) o resíduo em estudoé substituído, resultando na interação entre os caps e Rj; por fim, E(CiCi∗ − CjCj∗)representa o sistema formado somente pelos caps.
A fim de alcançar a estabilidade estrutural do complexo promovida por interaçõescom a rede de hidratação, todas as moléculas de água formando ligações de hidrogêniocom um resíduo particular foram incluídas para os fragmentos. A análise do cenário deligação baseou-se nos critérios adotados por Bezerra et al. (2017).
3.2.4 Cálculos DFT
Após o fracionamento considerando o esquema MFCC, os cálculos energéticos paracada resíduo foram realizados através do software Gaussian (G09), dentro do formalismoda teoria do funcional da densidade utilizando o funcional baseados na aproximação dogradiente generalizado B97D. Para expandir os orbitais de Khon-Sham para todos oselétrons, nós selecionados o conjunto de bases 6 − 311 + G(d, p). É importante notar queos cálculos DFT foram realizados para cada um dos termos na Eq.3.1.
A representação do ambiente molecular é uma passo essencial para o estudo depropriedades moleculares em biomoléculas. Recentemente, modelos de solvatação implícitostêm sido utilizados atrelados ao esquema do MFCC por Ourique et al. (2016) e Dantaset al. (2015) para calcular a energia de ligação em complexos proteína-ligante, levandoem consideração diferentes valores de constante dielétrica (ε) para representar o efeito doambiente na avaliação das energia eletrostáticas.
Neste trabalho, nós empregamos a aproximação MFCC junto com o modelo CPCM(Conductor-like Polarizable Continuum Model) (COSSI et al., 2003), utilizando dois valoresda constante dielétrica ε=20 e 40 (ε20 e ε40), respectivamente, para tentar aumentar asimilaridade entre o ambiente eletrostático de cada fragmento e àquele encontrado naproteína, além de estimar os efeitos na energia, tais como a polarização do meio promovidopelo solvente.
3.3 Resultados e discussão
Ao realizar um apanhado geral de todos os resíduos de PD-1 que apresentaram aomenos um parceiro de interação nos três ligantes, observa-se que existe uma área de ligaçãocomum em PD-1, compartilhada entre pembrolizumab, nivolumab e PD-L1. Uma vez queessa região pode ser o sítio de reconhecimento para ligantes na proteína, primeiramentefocamos nessa região.
Nota-se que a vasta maioria dos aminoácidos de PD-1 na região comum de ligaçãosão apolares (Phe[F]63, Val[V]64, Ile[I]126, Leu[L]128, Ala[A]129, Ala[A]132, e I[I]134), oque decresce a energia total de ligações, uma vez que a quantidade de interações polares,

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 50
tais como ligações de hidrogênio e pontes salinas, estão reduzidas. Essa afirmação está deacordo com o observado na Figura 3.2, onde a maioria desses mostram energias próximasdo zero para todos os três complexos. Ademais, a maior parte dos pares resíduo-resíduo sãoformados entre os aminoácidos de PD-1 com aqueles da cadeia leve dos fármacos, emboraeles estejam mais distantes da proteína do que aqueles da cadeia pesada (HORITA et al.,2016; NA et al., 2016).
Figura 3.2 – Perfil energético para cada resíduo de PD-1 na superfície de reconhecimentocomum.Esta figura representa a soma de todas as energias de interação deum resíduo de PD-1 com cada aminoácido dos ligantes dentro do raio de 8,0Å, usando as contantes dielétricas ε20 e ε40.
Pode-se observar que para o complexo PD-1/PD-L1, apenas o resíduo Ala(A)132apresenta uma energia de interação repulsiva, com um valor de 0.75 kcal mol−1 (0.86kcal mol−1) for ε40 (ε20). Por outro lado, para o complexo PD-1/PEM, vê-se que doisaminoácidos mostraram um valor repulsivo, enquanto apenas uma repulsão foi encontradapara o complexo PD-1/NIV, nominando: Ser(S)62 (ε40: 10.00 kcal mol−1; ε20: 9.75 kcalmol−1) e Phe(F)63 (ε40: 4.92 kcal mol−1; ε20: 4.93 kcal mol−1) relativo ao primeiro complexo,e Ala(A)129 (ε40: 6.13 kcal mol−1; ε20: 5.49 kcal mol−1) relativo ao último.
Com relação aos anticorpos monoclonais, descobrimos que o principal responsávelpelo resultado energético positivo é a interação com os resíduos de sua cadeia leve.Todosos demais aminácidos avaliados apresentaram energias de interação negativa, com as maisintensas atrações sendo observadas para o resíduo Lys(K)131: em complexo com PD-L1(ε40: -15.02 kcal mol−1; ε20: -18.24 kcal mol−1), com o fármaco PEM (ε40: -29.63 kcalmol−1; ε20: -33.40 kcal mol−1) e com o fármaco NIV (ε40: -37.08 kcal mol−1; ε20: -40.49kcal mol−1).

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 51
Este resíduo de lisina provê uma contribuição muito importante para a formação eestabilização dos complexos como uma consequência de sua cadeia lateral longa, muitomóvel e com o grupo amina carregado, o que cria uma superfície dinâmica capaz de formarligações de hidrogênio, pontes salinas e interações cátion-π, com moléculas de água eresíduos dos ligantes, tais como Asn(N)63P D−L1 (ε40: -1.35 kcal mol−1; ε20: -1.72 kcalmol−1), Gln(Q)66P D−L1 (ε40: -5.21 kcal mol−1; ε20: -5.12 kcal mol−1), Tyr(Y)53P EM (ε40:-4.05 kcal mol−1; ε20: -4.15 kcal mol−1), Glu(E)59P EM (ε40: -9.70 kcal mol−1; ε20: -11.90 kcalmol−1), Met(M)105P EM (ε40: -6.06 kcal mol−1; ε20: -6.62 kcal mol−1), Asp(D)108P EM (ε40:-4.82 kcal mol−1; ε20: -6.56 kcal mol−1), Asp(D)100NIV (ε40: -5.77 kcal mol−1; ε20: -6.58kcal mol−1), Asp(D)101NIV (ε40: -8.24 kcal mol−1; ε20: -10.27 kcal mol−1), Tyr(Y)49NIV
(ε40: -6.62 kcal mol−1; ε20: -6.43 kcal mol−1), Asp(D)50NIV (ε40: -8.82 kcal mol−1; ε20:-10.85 kcal mol−1), como pode ser visto na Figura 3.3.
O resíduo Pro(P)130 mostra-se como o segundo mais atrativo quando complexadocom o fármaco NIV (ε40: -17.65 kcal mol−1; ε20: -18.21 kcal mol−1), enquanto os complexoscom o ligante PD-L1 (ε40: -1.09 kcal mol−1; ε20: -0.92 kcal mol−1) e o fármaco PEM (ε40:-1.63 kcal mol−1; ε20: -1.57 kcal mol−1), apresentam uma pequena energia, que são bastantepróximas uma da outra.
O único resíduo que apresenta um valor energético similar para os três complexos éSer(S)127. Observa-se uma energia de -0.64 kcal mol−1 (-0.69 kcal mol−1) para PD-1 ligadoà PD-L1 considerando uma constante dielétrica ε40 (ε20), -0.48 kcal mol−1 (-0.51 kcalmol−1) and -0.98 kcal mol−1 (-1.17 kcal mol−1), quando PD-L1 está ligado aos fármacosPEM e NIV, respectivamente. Em contrapartida, o resíduo Gln(Q)133 também apresentouuma energia total de interação próxima entre os três complexos, mostrando valores de-2.14 kcal mol−1 (-2.34 kcal mol−1), -1.17 kcal mol−1 (-1.27 kcal mol−1) and -3.03 kcalmol−1 (-3.05 kcal mol−1) para PD-L1, PEM e NIV, respectivamente.
Percebe-se claramente que o hot-spot formado pelos seis resíduos entre Ser(S)62-Leu(L)128 cria uma região de baixa energia de interação, que pode permitir algum grau deflexibilidade e ajude no rearranjo do receptor após a ligação dos antagonistas para evitaros ligantes naturais de se acoplar (FESSAS et al., 2017). Somando as energias de interaçãopara cada par resíduo-resíduo, encontramos a energia de ligação para cada um dos ligantes:-44.34 kcal mol−1 (-47.27 kcal mol−1) para PD-1/PD-L1, considerando a constante dielétricaε40 (ε20). Similarmente, encontramos o valor de -47.54 kcal mol−1 (-51.90 kcal mol−1) paraPD-1/PEM e -65.23 kcal mol−1 (-69.88 kcal mol−1) para o complexo PD-1/NIV.
3.4 Conclusão
Em suma, uma abordagem da bioquímica quântica é usada dentro da estrutura doDFT para estimar as energias de interação entre os resíduos de aminoácidos do receptor 1de morte celular programada (PD-1) e aqueles de seu ligante natural PD-L1, bem comoos dois anticorpos monoclonais aprovados pelo US − FDA, pembrolizumab e nivolumab.O resíduo de lisina K131 exibe a interação resíduo-resíduo mais intensa, um resultadorelacionado com sua capacidade de penetrar-se no sítio de ligação dos ligantes devido asua alta flexibilidade e polaridade, levando a formação de fortes ligações de hidrogêniocom os aminoácidos dos ligantes.
Uma região de baixa energia de interação foi encontrada, o que pode ajudar aoreceptor a adotar uma nova conformação em presença dos inibidores competitivos e impedira ligação de PD-L1. Ademais, o perfil de energia de ligação apresenta dois hot-spots que

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 52
Figura 3.3 – Direita: resíduos que interagem com K131 em cada complexo de PD-1/ligante.Esquerda: painel gráfico mostrando as interações mais relevante, considerandoas constantes dielétricas deε40 and ε20. (a) Interação de K131 com os maisrelevantes aminoácidos de PD-L1; (b) o mesmo para o sistema formadocom os aminoácidos de PEM; (c) o mesmo para o sistema formado com osaminoácidos de NIV. As linhas tracejadas de cor azul, vermelha, laranja everde representam ligações de hidrogênio, interações cátion-π, pontes salinase ligações de hidrogênio não convencionais, respectivamente.

Capítulo 3. Descrevendo inibidores de proteínas de checkpoint celular por bioquímica quântica 53
podem ser utilizados no desenvolvimento de novo compostos.
Nossos resultados confirmam que os métodos computacionais baseados em DFT,utilizados neste trabalho, não são apenas interessantes como ferramenta para desvendar arelevância dos aminoácidos mais importantes, mas também de grande valor para realizaruma avaliação quantitativa de quais são as contribuições relativas de cada resíduo para ainibição competitiva do receptor PD-1. Não há dúvidas sobre a relevância das aproximaçõesutilizadas neste trabalho como ponto de partida na obtenção de novos fármacos anti-cancerígenas.

54
capítulo 4
ESTUDO IN SILICO DO RECEPTOR GABAB E SEUSAGONISTAS/ANTAGONISTAS
4.1 Introdução
A presença de conexões cerebrais funcionais requerer o equilíbrio entre excitaçãoe inibição, e as moléculas neurotransmissoras estão entre os principais responsáveis pelamanutenção dessa homeostase cerebral. Vários aminoácidos são encontrados em altasconcentrações no cérebro agindo como neurotransmissores. Dentre eles, o ácido glutâ-mico (glutamato) é o principal neurotransmissor relacionado com a transmissão sinápticaexcitatória, enquanto o ácido γ-aminobutírico (GABA) é considerado o principal neuro-transmissor presente durante a transmissão sináptica inibitória (KUMAR; KUPPAST,2012). GABA está presente em aproximadamente 30% de todos os neurônios, sendo vistocomo o carro chefe de toda a neurotransmissão inibitória. No sistema nervoso central,os neurônios que liberam GABA são chamados de interneurônios, existindo em todas ascamadas corticais. Ao liberar o neurotransmissor, essas células permitem que as redescerebrais se auto-organizem, sejam mais complexas e aumentem a precisão temporal nodisparo do impulso nervoso, o que, em contrapartida, permite que as redes neurais reajama pequenos estímulos de forma moderada (KOÓS; TEPPER, 1999; MARKRAM et al.,2004).
GABA é produzido pela descarboxilação do ácido glutâmico, que é obtido a partirda transaminação do α-cetoglutarato (advindo do ciclo de Krebs) através da ação daenzima γ-aminobutirato-α-oxoglutarate transaminase (GABA-T) (ROWLEY et al., 2012;SHANK; CAMPBELL, 1982). A enzima glutamato descarboxilase catalisa a reação dedescarboxilação de glutamato em GABA, sendo essa enzima expressa em duas formasque são identificadas apenas em neurônios GABAérgicos e em certos tecidos periféricosconhecidos por sintetizar GABA (PAREDES; ÅGMO, 1992).
Assim como a maioria dos neurotransmissores, GABA é armazenado em vesículasnas células pre-sinápticas até a despolarização da membrana celular por íons cálcio (Ca2+),quando é liberado na fenda sináptica. Posteriormente, o neurotransmissor se difunde pelafenda sináptica e interage com os receptores de GABA (GR) na membrana da célulapós-sináptica. Em seguida, GABA é capturado por proteínas transportadoras presentesna célula pré-sináptica e degradado em succinil semialdeido por GABA transaminasepara regenerar a molécula de glutamato (em presença de α-cetoglutarato), ou em ácidosuccínico, que reentra no ciclo de Krebs (GUASTELLA et al., 1990).
Como apresentado acima, esses neurotransmissores inibem as sinapses pela interaçãocom receptores específicos na membrana plasmática presentes em ambas as células, pré- epós sinápticas. De forma geral, essa interação causa a abertura de canais iônicos, o quepermite a entrada de íons cloro (Cl−) na célula e a saída de íons potássio (K+), assim comoa inibição da entrada de íons cálcio (Ca+) no neurônio (KUMAR et al., 2013). Dependendode qual canal iônico é aberto, o potencial de membrana é hiperpolarizado ou despolarizado,acarretando na diminuição na excitabilidade do neurônio (NUTT; MALIZIA, 2001).
Apesar da função do ácido γ-aminobutírico como neurotransmissor inibitório estar

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 55
hoje bem estabelecida, sua completa aceitação ocorreu a apenas quarenta anos. EmboraGABA tenha sido observado em alguns tecidos biológicos desde 1910, sua presençano sistema nervoso não foi descrito antes de 1950, quando aminoácidos livres foramidentificados no cérebro de mamíferos. Não obstante, a próxima década viu poucos trabalhosque tentaram definir os efeitos desses aminoácidos para a atividade neuronal. Foi apenasno final da década de 1950 que Florey e McLennan (1959) conseguiram extrair do cérebrode mamíferos um composto que eles chamaram de "fator i" (’i’ representa a inibição),o qual eles acreditavam conter GABA. Contudo, análises posteriores apresentaram-secontrárias às expectativas e a posição desse neurotransmissor como agente do sistemanervoso central foi negativa até o final da década de 1960, quando Krnjević e Schwartz(1967) apresentaram provas inequívocas sobre o papel de GABA como neurotransmissorinibitório.
Nos anos seguintes, uma atenção considerável foi dada para definir a naturezados receptores através dos quais GABA gera sua resposta fisiológica, culminando nodescobrimento da estrutura dos receptores ionotrópicos no final da década de 1990 (OLSEN;TOBIN, 1990). Substâncias, tais como anestésicos e, posteriormente, neuroesteróides,apresentaram o efeito de potencializar a resposta de GABA nos receptores ionotrópicos emrelação ao influxo de Cl−, sendo a classe de compostos mais utilizadas como moduladoresde GABA, os benzodiazepínicos, descrita por Haefely et al. (1975) pouco depois. Essesimportantes agentes terapêuticos atuam alostericamente no aumento da frequência comque o canal iônico de GR abra, induzindo um efeito sedativo e ansiolítico, o que proveua base para o desenvolvimento de uma das maiores classes de fármacos utilizados naterapêutica para tratar disfunções do sistema nervoso.
Enquanto as pesquisas em relação ao receptor GABA continuavam, Barber ecolaboradores descreveram uma das enzimas responsáveis pela síntese de GABA na medulaespinhal de mamíferos, indicando que este neurotransmissor não seria encontrado somenteno sistema nervoso central (BARBER et al., 1978). A partir desse estudo, outros o seguiramna tentativa de desenvolver um modelo periférico de ação pré-sináptica de GABA na medulaespinhal, mostrando que GABA também atua em neurônios do sistema simpático periférico,os despolarizando (BOWERY; SMART, 2006). Contudo, na tentativa de caracterizar aresposta de GABA, tornou-se evidente que o efeito do neurotransmissor não foi dependentede Cl− e não pôde ser reproduzido por qualquer outro agonista conhecido do receptor, oque, juntamente com a falha dos antagonistas do receptor GABA em impedir a ação dotransmissor nesses tecidos, só reforçou a suspeita de que um novo receptor estava mediandoos efeitos do neurotransmissor.
Estudos posteriores mostraram β-clorofenil GABA (baclofeno) como um agonistaestereoespecífico deste novo receptor (FILIP; FRANKOWSKA, 2008). Contudo, ele nãodemostrou uma ação similar àquela bem descrita para GABA no sistema nervoso central,apesar de várias tentativas em fazê-lo reconhecer os receptores. Por outro lado, a açãode baclofeno foi idêntica àquele de GABA nos tecidos periféricos, mas essa ação não foiassociado com um aumento na condutância dos íons cloro. Assim, baclofeno tornou-sedisponível no mercado como um fármaco relacionado à ação em um novo tipo de receptorde GABA para o tratamento de espasticidade.
Em pouco tempo ficou claro que esse novo receptor estava presente em outrostecidos periféricos, localizado primariamente em nervos do sistema autônomo, onde suaativação modula a resposta tecidual ao estímulo nervoso. Apesar disso, acreditava-se queo papel de GABA fora do cérebro era provavelmente limitado, e a função do novo receptor

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 56
deveria ser apresentada no sistema nervoso central. Assim, a capacidade de GABA ebaclofeno em inibir a liberação de neurotransmissores causada por K+ no sistema nervosocentral de maneira insensível à bicuculina foi apresentada posteriormente (BOWERY etal., 1980). Por fim, ensaios com radioligantes de [3H]GABA e [3H]baclofeno formaram apeça chave para evidenciar a presença do novo receptor no cérebro de mamíferos (HILL;BOWERY, 1981; BOWERY; HILL; HUDSON, 1983). Este ficou conhecido como GABAB
(GBR) e àquele ionotrópico foi nomeado GABAA (GAR).
Durante os próximos anos, a significância dos receptores GABAB na fisiologiacerebral tornou-se evidente. Caracteristicamente, a ativação dos sítios de GBR são respon-sáveis por mediar uma hiperpolarização lenta no neurônio, em contraste com a respostarápida mediada por receptores GABAA. Ademais, enquanto a resposta do último estárelacionada com os níveis de íon cloro, o primeiro atua devido a um aumento na condutân-cia de íons potássio através da membrana plasmática. Outra característica que distingueambos receptores é a forma de atuação durante a ativação das vias de sinalização, onde osreceptores GABAB apareceram por atuar mediante a ativação da proteína G, que acopla-seà canais de K+ e Ca2+, predominantemente (CHEBIB; JOHNSTON, 1999).
Em uma tentativa de descobrir que conformação de GABA seria responsável porativar os receptores, Johson e colaboradores sintetizaram uma vasta gama de compostosanálogos ao neurotransmissor e notaram que um destes, ácido cis-4-aminocrotônico (CACA)deprimiu o disparo de neurônios espinhais em gatos (JOHNSTON et al., 1975; JOHNSTON,1996; CURTIS et al., 1970). Esse efeito não pôde ser reproduzido por baclofeno, sugerindoa presença de outro receptor, GABAC. Este também se mostrou um receptor de canaliônico, similar aos receptores GABAA, permitindo a entrada de íons cloro. Na figura 4.1apresentamos a estrutura de GABAA e GABAB.
4.1.1 O receptor GABAB
A maior parte da ação tópica de GABA tem sido atribuído à ativação dos receptoresionotrópicos, permeáveis ao íon cloro, GABAA. Esse fato pode ser observado comparandoos estudos entre os três subtipos de receptor. O maior impulso na pesquisa sobre GABAocorreu em 1957 com a descoberta das benzodiazepinas, e posteriormente com o lançamentodo diazepam, em 1963, que rapidamente se tornou um dos medicamentos mais vendidos nosEstados Unidos entre os anos de 1969 e 1982 (STERNBACH, 1979; FROESTL, 2011). Hoje,mais de cinquenta compostos interagindo com o receptor ionotrópico estão no mercadocomo parte de medicamentos ou utilizados como moléculas de estudo, dentre eles temos:haloxazolam, chlordiazepoxide, clonazepam, flumazenil e zopiclon.
Por outro lado, baclofeno foi introduzido no mercado em 1972, sendo utilizado notratamento de espasticidade, distonia e alguns tipos de dor neuropática, e desde entãonenhum outro fármaco que possua os receptores GABAB como alvo foi aprovado peloUS-FDA (BENARROCH, 2012). Apesar disso, GBR ganhou bastante atenção com adescoberta de novos agonistas do receptor, CGP27492 e CGP35024 (FROESTL et al.,1995). Não obstante, foi a partir do desenvolvimento dos primeiros antagonistas, faclofen e2-hidroxisaclofeno, que houve um crescimento notável na pesquisa em torno de GBR, umavez que esses compostos antagonistas mostraram atividade no tratamento de pacientes comproblemas cognitivos e com doença de Alzheimer, com posterior acréscimo de atividadesantidepressivas, contra ansiedade, vício, dentre outras (KERR et al., 1987; KERR et al.,1988; FROESTL et al., 2004). Portanto, não há dúvidas de que há uma necessidade urgentepor novas moléculas que visem a interação com os receptores GABAB.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 57
Adaptado de Freyd et al., 2015.
Figura 4.1 – Estrutura tridimensional dos receptores (a) GABAA e (b) GABAB.
Como apresentado anteriormente, GBR faz parte do grupo de receptores metabo-trópicos acoplados à proteína G de classe C, assim como os receptores metabotrópicos deglutamato. GABAB atua através da proteína Gαi/0, que inibe adenilato ciclase e canaisiônicos (BOWERY, 1993; BOWERY et al., 2002). O GABA liberado na fenda sinápticapode voltar a alimentar os receptores GABAB localizados nos terminais GABAérgicos(homoreceptor) e/ou transbordar para ativar os receptores GABAB hétero-sinápticos nosterminais glutamatérgicos vizinhos. A ativação desses receptores na célula pré-sinápticainibe a liberação de neurotransmissores através de múltiplos alvos, incluindo a ativaçãode canais de cálcio dependentes de voltagem (relacionado a inibição de adenilato ciclase),o bloqueio da condutância ao potássio para desviar o potencial de ação pré-sináptico, ea redução da iniciação vesicular ou interação com a maquinaria de exocitose (MINTZ;BEAN, 1993; THOMPSON; GÄHWILER, 1992; SAKABA; NEHER, 2003; YAMADA etal., 1999).
Através da inibição dos canais de voltagem e, consequentemente, a liberação deneurotransmissores como glutamato, noradrenalina, dopamina, serotonina e acetilcolina,GBR controla vários processos neuronais fundamentais, o que o torna um potencialalvo terapêutico para diversas desordens neurológicas (CHALIFOUX; CARTER, 2011;WALDMEIER; KAUPMANN; URWYLER, 2008; TANIYAMA et al., 1992). Na célulapós-sináptica, a ativação de GBR gera um sinal inibitório lento através da abertura decanais retificadores de potássio, que compensam a entrada do íon através da bomba

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 58
Na+-K+-ATPase, promovendo sua saída e mantendo o gradiente elétrico (GÄHWILER;BROWN, 1985).
A partir da clonagem desses receptores no final da década de 1990, foi identificadaa presença de dois produtos gênicos, as subunidades GABAB1 e GABAB2 (KAUPMANNet al., 1997). O gene de GABAB1 (GABBR1 ) é encontrado no cromossomo 6 (6.p22.1),codificando uma proteína com aproximadamente 900 aminoácidos, sendo que destes, >500 formam sua região extracelular. Por outro lado, o gene para GABAB2 é encontradono cromossomo 9 (9q22.33), codificando uma proteína de mesmo tamanho (∼900 aminoá-cidos), mas com a região extracelular pouco menor (480 aminoácidos). Experimentos derecombinação mostraram que a heterodimerização dessas subunidades é obrigatória para aexpressão na superfície celular, acoplamento com a proteína G e, consequentemente, paraa sinalização dependente de agonistas (JONES et al., 1998; KAUPMANN et al., 1998a;WHITE et al., 1998).
É em GBR1 que existe o sítio de ligação para GABA e demais agonistas/antagonistas,enquanto a subunidade GABAB2 é necessária para o acoplamento à proteína G (ROBBINSet al., 2001). Roedores com duplo knockout nos genes dos receptores GABAB1 (−/−)confirmaram que a montagem heterodimérica é necessária para prover um receptor com-pletamente funcional, apesar de análises em roedores com GABAB2 − /− apresentaremum comportamento atípico na presença de baclofeno (PROSSER et al., 2001; QUÉVA etal., 2003; GASSMANN et al., 2004). Ademais, embora GABAB2 não se ligue a qualqueragonista/antagonista conhecido, sua região extracelular interage diretamente com a re-gião extracelular de GABAB1, aumentando a afinidade de agonistas (MILLIGAN, 2009;GALVEZ et al., 2001; LIU et al., 2004; NOMURA et al., 2008).
Posteriormente, descobriu-se que a subunidade GABAB1 existe em duas isoformas,GABAB1a e GABAB1b, que diferem em seus motivos estruturais e locais de expressão(BENARROCH, 2012; KAUPMANN et al., 1997; BIERMANN et al., 2010). Apesardessas diferenças, ambas isoformas compartilham propriedades farmacológicas e fisiológicassemelhantes, o que pode ter ocultado a sua presença. Enquanto a subunidade GABAB1a épreferencialmente expressa em terminais glutamaérgicos pré-sinápticos e montado com asubunidade GABAB2 para formar os heteroreceptores, as duas isoformas são montadas comGABAB2 para formar autoreceptores em terminais GABAérgicos pré-sinápticos (VIGOTet al., 2006; GUETG et al., 2009). Já no lado pós-sináptico, as duas isoformas estãopresentes, mas GABAB1b provê a maior parte do acoplamento aos canais retificadores depotássio e inibição do potencial de tráfego de cálcio (PÉREZ-GARCI et al., 2006).
Os receptores GABAB são expressos em quase todos os neurônios e células daglia no sistema nervoso central, e sua atividade influencia o sistema neural e estadoscomportamentais. Dessa forma, não é surpresa que esse receptor seja implicado em umagrande variedade de condições neurológicas e psiquiátricas, incluindo epilepsia, ansiedade,depressão, esquizofreina, transtorno compulsivo-obsessivo, vicio e dor (CRUNELLI; EMRI;LERESCHE, 2006). Apesar do envolvimento de GBR em desordens relacionadas a saúdemental, o uso clínico de agonistas desse receptor está limitada, hoje, ao tratamentode narcolepsia, dor neuropática, espasticidade e distonia. Uma razão para isso é que oprincipal efeito de baclofeno, o primeiro agonista e único aprovado para venda no mercado,é o relaxamento muscular, que é um efeito indesejado para indicação no uso em saúdemental (BETTLER et al., 2004; KUMAR et al., 2013). Ademais, esse fármaco apresentabaixa permeabilidade no sistema nervoso central, pequena duração em seu efeito, janelaterapêutica estreita e rápida tolerância do organismo (BROWN et al., 2015). Por outro

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 59
lado, a pesquisa voltada para o uso de antagonistas de GBR tem se mostrado bastantepromissora como forma de modulação da resposta fisiológica em processos de desordensdo sistema nervoso.
Estruturalmente, os receptores GABAB remontam a ideia mais aceita para os recep-tores de Classe C. Cada subunidade do receptor possui aproximadamente 900 aminoácidos,que constituem três domínios distintos: a região extracelular N-terminal (∼500 aminoáci-dos), também conhecida como o domínio Venus flytrap (VFT); o domínio transmembranacaracterístico de todos os receptores acoplados à proteína G; e uma região C-terminalintracelular (FREYD et al., 2017; BROWN et al., 2015). O domínio rico em cisteínas,que liga o N-terminal ao domínio transmembrana nos demais membros dos receptores declasse C está ausente sem sua sequência de aminoácidos (URWYLER, 2011). Dois sítios deligação tem sido caracterizados em receptores GABAB: o sítio de ligação ortostérico, quereconhece GABA e os demais agonsitas/agosnitas presente no N-terminal de GBR (VFT );e um sítio alostérico que tem sido caracterizado como parte do domínio transmembranade GBR2, como pode ser visto na figura 4.1b (BINET et al., 2004; DUPUIS et al., 2006).
Assim como ocorre em outras classes de GPCRs, a base para o conhecimentoestrutural nos receptores GABAB vem a partir da comparação com outros membros damesma classe. Neste caso, o único membro de classe C com estrutura tridimensionaldefinida por métodos experimentais era o receptor metabotrópico de glutamato (mGluR)homodimérico (KUNISHIMA et al., 2000). Essa estrutura cristalográfica do domínio N-terminal de mGluR1 foi obtida a partir de roedores, na ausência e presença do agonistaglutamato, mostrando-se um homodímero unido por pontes dissulfeto que existe em doisestados maiores, um aberto (sem ligante) e outro fechado (com glutamato).
A recente determinação da primeira estrutura cristalográfica do N-terminal hetero-dimérico de GABAB humano co-cristalizado com vários agonistas e antagonistas foi umprimeiro passo em direção a um entendimento profundo do mecanismo de interação deligantes nos receptores GABAérgicos metabotrópicos (Figura 4.2) (GENG et al., 2013).A partir deste estudo, foi apresentado que, similar aos receptores mGlu, a presença dosagonistas induz uma conformação fechada dos lobos L1 e L2 de GABAB1, correspondenteao estado ativo da proteína, enquanto o receptor sem ligante (estado apo) ou ligado aantagonistas manteve-se com os lobos abertos, representando um estado de repouso doreceptor. Em contrapartida, praticamente não foram vistas mudanças em GABAB2 empresença dos ligantes (GENG et al., 2012).
Devido a relevância dos receptores GABAB para a fisiologia do sistema nervosocentral e periférico, e a pouca informação que existe sobre sua estrutura, mecanismo deativação e inibição, além do prover insights sobre os demais membros desta classe dereceptores, um grande número de trabalhos experimentais e computacionais vem sendopublicados no intuito de melhor compreender o funcionamento de GBR (NYPORKOet al., 2015; MELIS; LUMMIS; MOLTENI, 2008; SCHWENK et al., 2016; BERNARD;GUEDIN; HIBERT, 2001; FREYD et al., 2017). Contudo, ainda existe um gap muitogrande nesse conhecimento, o que leva à necessidade de mais estudos sobre esse receptor.
Com este objetivo, nós apresentamos uma descrição apropriada do perfil energé-tico de interação entre os resíduos individuais do bolsão de ligação que compõe domínioN-terminal heterodimérico do receptor GABAB em complexo com quatro ligantes clás-sicos do receptor, dois agonistas (GABA e baclofeno), e dois antagonistas (SCH50911e 2-hidroxisaclofeno), através do esquema MFCC dentro do formalismo da teoria dofuncional da densidade (DFT) (DA COSTA et al., 2012; JIANG; MA; JIANG, 2006).

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 60
Figura 4.2 – Estrutura tridimensional do domínio N-terminal do heterodímero formado porGABAB1 e GABAB2.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 61
A contribuição individual dos resíduos envolvidos na ligação entre agonsita/antagonistae o receptor GABAB foi calculada usando a estrutura do receptor co-cristalizado comGABA, baclofeno, SCH50911 e 2-hidroxisaclofeno (2OHS) (PDB ID: 4MS3, 4MS4, 4MR9e 4MQF, respectivamente). Para tornar os cálculos energéticos mais fidedignos, um es-quema de inclusão eletrostática foi implementado, sendo aqui chamado de EE-MFCC.Com o intuito de comparar o efeito do ambiente causado pela incorporação das cargaspontuais, realizamos cálculos com valores diferentes de constante dielétrica. O propósitodesta investigação é delinear a ligação agonistas/antagonistas-GABAB através de um perfilenergético detalhado da interação entre os resíduos no sítio de ligação e os compostos,sendo essencial não apenas para prover informações sobre ajustes que possam melhorar aeficácia de novas drogas específicas, mas também para permitir uma melhor compreensãosobre as características de ligação em GABAB e outras GPCRs de classe C.
4.2 Materiais e Métodos
4.2.1 Dados do complexo ligante-Receptor
Para a realização dos cálculos neste estudo utilizou-se a primeira estrutura crista-lográfica do receptor GABAB (região extracelular) em complexo com GABA, baclofeno(BACL), SCH50911 e 2-hidroxisaclofeno (2-OHS) obtida por difração de raios-X comresolução de 2,50 Å, 1,90 Å, 2,35 Å e 2,22 Å depositadas no banco de Protein Data Bank(PDB, <www.rcsb.org/pdb>), sob os códigos 4MestaS3, 4MS4 4MR9 e 4MQF (GENG etal., 2013). Ademais, o cálculo do estado de protonação em pH fisiológico para ligante eaminoácidos do receptor foi realizado através do uso do software Marvin Sketch, versão17.24.0, e o software PROPKA 3.1, respectivamente (SØNDERGAARD et al., 2011).
4.2.2 Cálculos clássicos
Átomos não elucidados pela difração de raios-X, e portanto ausentes na estruturacristalográfica, como átomos de hidrogênio e cadeias laterais de alguns aminoácidos,foram adicionados. Cadeias laterais e hidrogênios foram submetidos à otimização clássicaem suas geometrias, mantendo os demais átomos fixos. Esta otimização foi realizadosob os parâmetros de cálculo do campo de força CHARMm (Chemistry at HARrvardMolecular Mechanics), sendo este parametrizados para todos os átomos, porém com maiorespecialização para moléculas orgânicas (MOMANY; RONE, 1992). Para este cálculo ospadrões de convergência foram ajustados para 10−5 kcal mol−1 (variação de energia total),0.001 kcal.mol−1Å−1 (gradiente de desvio médio quadrado) e 10−5Å (deslocamento atômicomáximo). Devido a relevância do monômero GBR2 para o funcionamento de GBR1, umraio de 8,0 Å foi desenhado em torno GBR2 tendo por centro todos os átomos de GBR1.Assim, os átomos de GBR2 que não estavam dentro do raio delimitado foram deletados,ou seja, todos aqueles aminoácidos posicionados a mais de 8,0 Å de GBR1 foram deletadose átomos de hidrogênio foram adicionados aos terminais. Portanto, logo após o processode otimização e subsequente deleção dos átomos do segundo monômero, posições e cargasatômicas de todos os átomos presentes foram armazenadas para a realização dos passossubsequentes.
4.2.3 Fracionamento molecular com caps conjugados e o efeito do ambiente
A energia de interação entre GABA, baclofeno, SCH50911 e 2-hidroxisaclofenoe os aminoácidos do receptor GABAB foi calculada através do formalismo do MFCC

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 62
proposto por Zhang e Zhang (ZHANG; ZHANG, 2003). Nós denominamos o ligantecomo L e o resíduo interagindo com este como Ri. O cap Ci−1 (Ci+1) é formado peloresíduo covalentemente ligado ao grupo amina (carboxila) do resíduo Ri, provendo umamelhor descrição do ambiente eletrônico. Com o objetivo de melhorar nossos cálculos,uma forma adaptada do MFCC foi utilizada. Esta inclui uma correção eletrostática doambiente através do uso das cargas atômicas (CP C) obtidas classicamente após o processode otimização para aumentar a similaridade com a proteína inteira. Para cada estruturafragmentada, a energia de interação entre ligante-resíduo, EIEE−MF CC(L−Ri), é calculadade acordo com:
EIEE−MF CC(L − Ri) = E(L − CiRiCi∗ − CP C) − E(CiRiCi∗ − CP C)
− E(L − Ci C∗ − CP C) + E(Ci Ci∗ − CP C). (4.1)
onde o primeiro termo, o sistema de Ci−1 + Ri + Ci+1 é separado da proteína, e o ligante(L) é adicionado, finalizando o primeiro momento com o cálculo da energia do sistema L,Ci−1, Ri e Ci+1. No segundo momento, o ligante é retirando do cálculo, estando somenteos caps e o resíduo principal (Ci−1, Ri e Ci+1). Posteriormente, o ligante é reintegrado aosistema e o resíduo em estudo é retirado, resultando na interação do ligante com os caps(L, Ci−1 e Ci+1). Ao final, somente a energia dos caps é obtida (Ci−1 e Ci+1).
O termo CP C representa o conjunto de todos os átomos dos aminoácidos presentesna estrutura cristalográfica, sem o resíduo de referência (Ri) e os caps (Ci−1 Ci+1).Nesse modelo cada átomo é substituído pela carga atômica correspondente (carga pontual).Similar ao cálculo do MFCC com a correção do campo eletrostático adaptado (electrostacticfield adapted, EFA) aplicado por Jiang et al. (JIANG; MA; JIANG, 2006), cada fragmentofoi introduzido entre todas as cargas pontuais, e as energias de interação foram calculadasincluindo as interações eletrostáticas do ambiente em todos os passos seguindo o esquemade incorporação de cargas pontuais proposto por Hall e Smith (HALL; SMITH, 1984;HALL, 1985; HALL, 1986) (intitulado aqui como EE-MFCC). A presença das cargaseletrostáticas inclui a influência das interações de longo alcance do ambiente a cada energiade interação ligante-resíduo, o que torna o efeito eletrostático do ambiente mais realísticodo que é observado com o uso de outros modelos, como aqueles de solvatação contínua,devido a sua capacidade em reproduzir a heterogeneidade proteica.
4.2.4 Cálculos DFT
Após o fracionamento considerando o esquema EE-MFCC, os cálculos energéti-cos para cada resíduo foram realizados através do software Gaussian (G09), dentro doformalismo da teoria do funcional da densidade utilizando os funcionais baseados na aproxi-mação do gradiente generalizado B97D, M062X, B3LYP, BHANDHLYP, PBE (PBEPBE)e PBEH1 (PBEH1PBE), bem como o método Hartree-Fock (HF). Para expandir os orbitaisde Khon-Sham para todos os elétrons, nós selecionados o conjunto de bases TZV P . Éimportante notar que os cálculos DFT foram realizados para cada termo na Eq. 4.1, isto é,(L − CiRiCi∗ + CP C), (CiRiCi∗ − CP C), (L − Ci C∗ − CP C) e (Ci Ci∗ − CP C). Como fontede comparação, as cargas pontuais foram retiradas e dois valores de constante dielétricaforam utilizados (ε10 e ε40).

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 63
4.2.5 Critérios de convergência
Para evitar a perda de interações importantes, um estudo de convergência daenergia total de ligações como uma função do raio foi realizada, limitando o número deaminoácidos analisados (DA COSTA et al., 2012). Desta forma, nós adicionamos a energiade interação individual dos aminoácidos dentro de esferas imaginárias em um bolsão deinteração, r, tendo por centro o ligante, considerando r = R/2 (R = 1, 2, 3, 4, ... Nn,Nn sendo o próximo número natural na sequência) (DANTAS et al., 2015). Assim, nósobtemos a convergência quando a variação de energia entre os raios consecutivos é menorque 10%.
4.3 Resultados e discussão
4.3.1 Estrutura e distribuição de carga dos ligantes
Para iniciar os cálculos, o estado de protonação de GABA, baclofeno, SCH50911 e2-hidroxisaclofeno é obtido através do software Marvin Sketch. Como apresentado por C.Chang e colaboradores, baclofeno, o primeiro agonista especifico de GABAB, foi obtidoa partir da estrutura básica do neurotransmissor GABA (CHANG et al., 1982). Emcontrapartida, 2-OHS é um análogo de baclofeno (KERR et al., 1987; KERR et al., 1988).Não obstante, SCH50911 possui a estrutura central de GABA, embora ao grupo aminaátomos de carbono tenham sido adicionados, formando um anel. Dessa forma, espera-seque os quatro ligantes devam possuir uma base estrutural similar, o que pode ser observadoa partir da figura 4.3.
Assim, todos apresentam um grupo negativamente carregado na posição α, e no ladocontrario um grupo amina positivamente carregado compondo uma molécula zwitteriônicade carga total zero. Em baclofeno, essa região de carga negativa é composta por um grupocarboxila, assim como em GABA e SCH50911, enquanto em 2-hidroxisaclofeno um acidosulfônico (H-SO−
3 ) foi adicionado. Como diferença dos demais, baclofeno e 2-hidroxisaclofenopossuem um anel clorofenil-β, o que confere às moléculas uma característica hidrofóbica.Por ultimo, uma hidroxila foi adicionada ao carbono que liga a estrutura central de GABAao anel clorofenil de 2-OHS. Os resultados aqui apresentados em relação a conformação eprotonação dos ligantes estão de acordo com o observado por Chang et al. (1981) e Weber,Craven e McMullan (1983).
Dessa forma, os ligantes foram subdivididos em três regiões para facilitar análisesposteriores, onde a região i representa o ácido-α (carregado negativamente) e o primeirocarbono ligado a este; a região ii é composta pela região de linking entre i e iii; e aregião iii é formada pelo grupo amina e o primeiro carbono ligado a este. A distribuiçãoda densidade eletrônica em GABA, baclofeno, SCH50911 e 2-hidroxisaclofeno no estadozwitteriônico é apresentado (figura 4.3 - direita), onde vê-se que as moléculas possuemuma característica mais positiva (azul), com exceção de GABA, onde observa-se umadistribuição de cargas equivalente entre as três regiões, embora o anel clorofenil apresenteuma característica mais próxima do neutro (verde) e o grupo ácido apresenta uma regiãocom característica negativa.
Essa divisão clara entre a densidade eletrônica serviu de molde para definir as 3regiões dos ligantes. Como já era esperado, a região iii possui uma característica maispositiva, enquanto a região i é apresenta uma maior densidade de elétrons, o que também éobservado para a hidroxila de 2-hidroxisaclofeno. Dessa forma, vê-se que não só a estrutura

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 64
Figura 4.3 – Representação estrutural dos ligantes GABA, baclofeno, 2-hidroxisaclofeno eSCH50911 (de cima para baixo) no estado neutro. A superfície de potencialeletrostático e apresentada ao lado direito de cada molécula.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 65
é bastante similar, mas a distribuição de cargas imita o comportamento entre as moléculas.Contudo, o anel clorofenil de 2-OHS apresenta uma característica mais positiva do que oanel de baclofeno. Por outro lado, no caso do ligante SCH50911, existe uma predominânciade cargas positivas por toda a molécula, sendo levemente modificada pela presença de umoxigênio no anel e, posteriormente, na carboxila.
4.3.2 Energia total, convergência e efeito dos modelos de cargas pontuais e constantedielétrica
Para avaliar as interações através de métodos quânticos, é importante levar emconsideração cada aminoácido significante que pode influenciar o mecanismo de atração erepulsão. O esquema do MFCC em presença de duas constantes dielétricas e de cargaspontuais (EE-MFCC ) foi utilizado para obter a energia total de interação entre os quatroligantes e GABAB. Embora a interação entre pares não seja a forma mais adequada deapresentar a energia total de um sistema, esta é a forma mais utilizada nos dias de hoje,mostrando uma boa correlação entre a analise computacional e experimental (DA COSTAet al., 2012; LIMA NETO et al., 2015). Dessa forma, realizamos a somatória da energiade interação de cada par ligante-resíduo de forma crescente, levando em consideração,inicialmente, aqueles aminoácidos posicionados nas proximidades do ligantes e estendendoessa somatória à resíduos mais distantes, seguindo dois critérios: a afinidade obtidaexperimentalmente, e a convergência da energia de ligação total do sistema.
Segundo Kaupmann e colaboradores, a afinidade de GABA, baclofeno, SCH50911e 2-hidroxisaclofeno para GBR1b segue uma ordem decrescente: SCH50911 > baclofeno >GABA > 2-hidroxisaclofeno (tabelas 4.1-4.3) (KAUPMANN et al., 1998a; KAUPMANNet al., 1998b). Como pode-se observar, dentre todos os sete métodos de cálculo testados(B97D, M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF), dentro dos três modelosde representação do ambiente (carga pontual, ε10 e ε40), o único que conseguiu alcançarenergeticamente, em sua energia total de ligação, a afinidade de ligação experimental foi oB97D. Ademais, isso ocorreu independente do modelo de ambiente utilizado.
Tabela 4.1 – Energia total de ligação (EE-MFCC ) e afinidade experimental (IC50) obtidapara os quatro complexo, levando em consideração os cálculos realizados comB97D, M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF.
Ligante IC50Energia total (kcal mol−1)
(µM) B97D M062X B3LYP BHANDHLYP PBE PBEH1 HF
SCH50911 0,35 -171.73 -185.20 -149.00 -162.35 -151.53 -162.65 -151.06Baclofeno 30,1 -159.67 -159.65 -103.97 -120.01 -117.97 -118.99 -100.13GABA 30,9 -158.00 -164.38 -131.48 -144.37 -134.38 -138.42 -132.722-OHS 49,1 -144.39 -149.23 -111.51 -123.77 -114.23 -118.61 -110.48
Isso pode indicar que, para o modelo de estudo aqui apresentado, o efeito de cargaspontuais ou constante dielétrica não interfere diretamente na energia total do sistema,quando leva-se em consideração a afinidade experimental como ponto de comparação.Contudo, o método de cálculo utilizado para a simulação influencia fortemente no resultadoobservado. Esse mesmo fato foi observado anteriormente por Fouda e Ryde (2016). Aoestudar três complexos enzimáticos diferentes, [NiFe] hidrogenase, glioxalase I e sulfetooxidase, os autores não encontraram diferenças significativas (qualitativamente) entre o

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 66
Tabela 4.2 – Energia total de ligação (ε10) e afinidade experimental (IC50) obtida para osquatro complexo, levando em consideração os cálculos realizados com B97D,M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF.
Ligante IC50Energia total (kcal mol−1)
(µM) B97D M062X B3LYP BHANDHLYP PBE PBEH1 HF
SCH50911 0,35 -80.70 -77.50 -42.64 -51.70 -52.36 -56.07 -35.04Baclofeno 30,1 -69.15 -60.97 -7.24 -19.06 -24.46 -27.87 6.40GABA 30,9 -68.03 -64.61 -33.47 -41.36 -44.36 -46.42 -24.152-OHS 49,1 -63.32 -58.99 -22.58 -31.40 -32.43 -35.96 -14.46
Tabela 4.3 – Energia total de ligação (ε40) e afinidade experimental (IC50) obtida para osquatro complexo, levando em consideração os cálculos realizados com B97D,M062X, B3LYP, BHANDHLYP, PBE, PBEH1 e HF.
Ligante IC50Energia total (kcal mol−1)
(µM) B97D M062X B3LYP BHANDHLYP PBE PBEH1 HF
SCH50911 0,35 -69.08 -65.44 -30.69 -39.41 -40.80 -44.10 -22.37Baclofeno 30,1 -57.86 -49.15 4.36 -7.11 -13.16 -16.15 18.73GABA 30,9 -56.57 -52.77 -21.72 -29.30 -32.96 -34.67 -11.742-OHS 49,1 -54.63 -49.90 -13.61 -22.17 -23.76 -26.97 -5.00
modelo com cargas pontuais e aquele utilizando constante dielétrica, sendo os resultadosencontrados dependentes do funcional utilizado. No entanto, vale a pena mencionar asduas energias totais positivas encontradas para os cálculos utilizando B3LYP (ε40) e HF(ε10 e ε40).
Tendo por base os resultados encontrados para B97D, buscamos verificar o pontode convergência dos sistemas estudados. Como apresentado na figura 4.4, para os doisantagonistas, SCH50911 e 2-hidroxisaclofeno, usando o EE-MFCC, a variação de energianunca chega a diferir do valor esperado (SCH50911 > 2-hidroxisaclofeno). Atingindo aconvergência no raio de 6,5 Å. Por outro lado, não é possível observar o mesmo com-portamento para os dois agonistas (GABA e baclofeno), que invertem sua ordem derelevância energética oito vezes entre os quinte raios calculados. Contudo, essa variaçãoentre os agonistas nos raios finais chega ao máximo de 2,00 kcal mol−1 no raio de 8,00 Å,enquanto entre os rais de 7,50 Å-9,00 Å esse valor não chega a 1,00 kcal mol−1. Isso nãoé um fato isolado, uma vez que estudos em outros sistemas mostraram-se incapazes derepresentar a afinidade experimental quando agonistas são avaliados (LIMA NETO et al.,2015; MARTINS et al., 2013), o que pode ser uma No todo, após o raio de 7,00 Å, a energiatotal não varia significantemente entre os raios calculados. Portanto, consideramos o raiode 7,00 Å como sendo o ponto de convergência da energia de ligação para os complexosem relação à distância do ligante.
Ao observar os resultados para as constantes dielétricas com valores ε10 e ε40,percebe-se uma diferença no comportamento energético dos sistemas em comparaçãoao resultado encontrado para os cálculos utilizando o EE-MFCC. Com um breve olhar,nota-se que após o raio de 6,00 Å (4,00 Å) não há mais uma grande variação na energia deligação entre os ligantes e o receptor para ε10 (ε40). Isto é, após esse ponto não houve mais

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 67
Figura 4.4 – Energia de interação total como função da distância do ligante. (a) Resultadosforam obtidos a partir do EE-MFCC; (b-c) Resultados foram obtidos a partir doMFCC utilizando ε10 e ε40, respectivamente.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 68
interações significativas entre os ligantes e os resíduos de aminoácido de GABAB. Dessaforma, para os três modelos testados, os resultado estão de acordo com trabalhos prévios,que descreveram os valores energéticos após 6,0-7,0 Å como de pouca relevância para aenergia total do sistema (RYDE; SÖDERHJELM, 2016; STIERAND; RAREY, 2010).
Com o intuito de comparar os resultados para os quatro sistemas estudados sobmétodos de cálculo diferentes, e tentar identificar quais as diferenças individuais em cadacomplexo, as energias individuais de cada par ligante-resíduo são superpostas e observadasnas figuras 4.5-4.8.
Nota-se que o comportamento energético individual (ligante-resíduo) para cadaum dos complexos estudados é bastante similar, embora com menor intensidade para asconstantes de ε10 e ε40, respectivamente. Esse comportamento reforça o que foi apresentadoanteriormente, onde vê-se que o efeito do modelo de representação do ambiente é pequenoem comparação ao efeito de escolha do método de cálculo quando observa-se o delineamentogeral das energias de interação.
Recentemente, Ribeiro, Lyra e Manzoni (2018) realizaram um cálculo de interaçãoentre a proteína albumina e o ibuprofeno através do esquema do MFCC com cargas pontuaise em vácuo, utilizando os métodos de cálculo HF, B3LYP, CAM-B3LYP e MP2. Os autoresafirmam que o uso das cargas pontuais reduz o erro causado pelos métodos de cálculo,além de tratar as interações de longo alcance com maior acurácia. Com os resultadosaqui apresentados, pode-se dizer que algo similar ocorre para os cálculos realizados comconstante dielétrica, uma vez que, visualmente (sem uma análise estatística), as energiasde interação individuais não parecem diferir muito, qualitativamente, quando comparam-seos métodos de cálculo.
Contudo, precisa-se reforçar a presença da repulsão observada no complexo baclofeno-GABAB para ambas constantes dielétricas (Hartree-Fock - HF), o que não ocorre parao modelo de cargas pontuais, bem como a aproximação entre a energia total de ligaçãode 2-hidroxisaclofeno e a de GABA para ε40, o que não é de se esperar, uma vez que aafinidade obtida experimentalmente difere bastante entre esses dois ligantes.
Em contrapartida, Provorse et al. (2016) utilizaram um modelo de cargas pontuais ede constante dielétrica para a realização de um QM/MM buscando identificar e caracterizaro estado excitado de cromóforos. Esse trabalho nos mostra que a utilização de constantesdielétricas para substituir o modelo de carga pontual não melhora a descrição do sistema,além de aumentar o custo computacional. O que não ocorre no nosso caso, uma vez queos cálculos com constante dielétrica são mais rápidos que aqueles realizados com cargaspontuais.
Por fim, deve-se ressaltar que a maior diferença entre os métodos de cálculo foiobservada para a região entre os quinze primeiros aminoácidos. Essa região é compostapor todos os resíduos dentro de um raio médio de 5,5 Å a partir de cada ligante. Comoobservado anteriormente, é dentro dessa região em que ficam os aminoácidos que formaminterações intermoleculares diretas com os ligantes. Portanto, isso pode indicar que o erroapresentado na energia total desses complexos para os variados métodos de cálculo vêm daformulação desses, uma vez que é bem descrito na literatura que cada método de cálculo,em especial os funcionais DFT, são parametrizados para descrever um tipo especifico decaracterística química de moléculas.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 69
Figura 4.5 – Superposição da energia de interação individual encontrada para cada resíduo deGABAB formando um par de interação com SCH50911. (a) EE-MFCC, (b-c) ε10 eε40, respectivamente. Os resíduos são apresentados começando por aqueles maispróximos do ligante até àqueles em 9,00 Å.
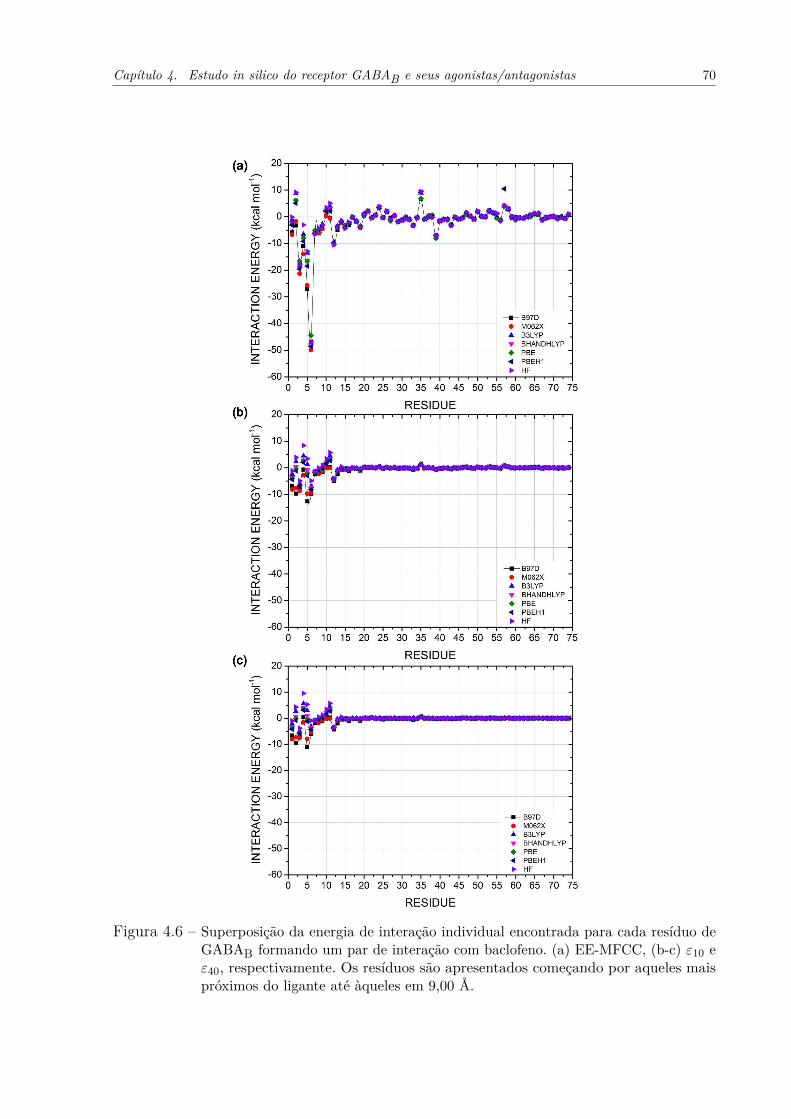
Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 70
Figura 4.6 – Superposição da energia de interação individual encontrada para cada resíduo deGABAB formando um par de interação com baclofeno. (a) EE-MFCC, (b-c) ε10 eε40, respectivamente. Os resíduos são apresentados começando por aqueles maispróximos do ligante até àqueles em 9,00 Å.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 71
Figura 4.7 – Superposição da energia de interação individual encontrada para cada resíduo deGABAB formando um par de interação com GABA. (a) EE-MFCC, (b-c) ε10 eε40, respectivamente. Os resíduos são apresentados começando por aqueles maispróximos do ligante até àqueles em 9,00 Å.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 72
Figura 4.8 – Superposição da energia de interação individual encontrada para cada resíduo deGABAB formando um par de interação com 2-hidroxisaclofeno. (a) EE-MFCC,(b-c) ε10 e ε40, respectivamente. Os resíduos são apresentados começando poraqueles mais próximos do ligante até àqueles em 9,00 Å.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 73
4.3.3 Energia de interação dos fragmentos aminoacídicos
As subunidades GBR1b e GBR2 do receptor GABAB possuem uma arquiteturabilobada similar, com aproximadamente 33% de identidade na sequencia de aminoácidos,e cada subunidade é composta por dois domínios distintos, lobo 1 e lobo 2 (Figura 4.2).Embora similares, as essas duas subunidades possuem arranjos tridimensionais diferentes,com GABAB1b possuindo uma fenda entre seus lobos, onde agonistas e antagonistaspodem ligar-se. Em contrapartida, nenhum ligante foi observado acoplando-se no domínioextracelular de GABAB2, embora compostos alostéricos tenham sido reportados ligando-sea seu domínio transmembrana (BROWN et al., 2015).
Contudo, foi observado que a ação da segunda subunidade aumenta a afinidadede GABAB1 aos ligantes, além de formar o sítio de ancoramento de proteínas efetorasintracelulares (FREYD et al., 2017). Através de uma análise na distancia entre os ligantes eGBR2, vimos que nenhum aminoácido da segunda subunidade do receptor estava presenteem raios menores que 10,0 Å. Portanto, com o intuito de representar a subunidade 2,mas sem aumentar o custo computacional exageradamente, foi decidido trabalhar comuma porção de GBR2. Assim, um limite de 8,0 Å centrado em GBR1b foi feito e todosos aminoácidos de GABAB2 dentro deste raio foram utilizados no estudo como cargaspontuais. Esse raio de corte não representa um valor aleatório, mas é baseado em outrosestudos que mostram este valor como suficiente para representar a interação entre proteínas(BEZERRA et al., 2017; RODRIGUES et al., 2013; TAVARES et al., 2018).
Similar ao que foi observado para GABA, faclofeno e outros análogos do neuro-transmissor, os ligantes aqui estudados ocupam uma cavidade de ligação larga e definidaentre os lobos L1 e L2, que compreende o sítio de ligação ortostérico (Fig. 4.9). As quatromoléculas ocupam uma região similar, com a estrutura básica, formada pela região decarga negativa (i), amina positiva (iii) e carbonos que fazem a ligação entre elas, ocupandouma porção da proteína bem descrita por ser o local de ancoramento de GABA no lobo 1(GENG et al., 2013). Ademais, todos os ligantes utilizados neste estudo formaram umaponte salina entre a amina carregada e um ácido glutâmico, uma característica que foiobservada para outros compostos co-cristalizados com o receptor, indicando que este deveser um ponto importante no reconhecimento e ancoramento de ligantes que visem GABAB
(BROWN et al., 2015).
Com base no que foi apresentado anteriormente, decidimos avaliar a energia deinteração de aproximadamente 65 pares ligante-resíduo para os quatro complexos através douso do EE-MFCC, levando em consideração somente aqueles valores energéticos obtidos apartir do uso do funcional B97D. Mais uma vez, é feita uma comparação entre os resultadosobtidos para o modelo de EE-MFCC e aqueles advindos do modelo de constante dielétrica,buscando identificar diferenças individuais (qualitativamente) entre os aminoácidos maisrelevantes energeticamente, se houverem.
Há uma distribuição similar de aminoácidos hidrofóbicos e hidrofílicos em GABAB,com grande parte daqueles com característica polar presentes em raios menores que 6,0 Å.Como observado acima, todos os ligantes apresentam uma estrutura que atende as duascaracterísticas. Apesar disso, as energias de interação individuais mostraram-se baixas,se comportando normalmente entre -3,0 kcal mol−1 e 3,0 kcal mol−1. Portanto, foramselecionados as interações mais fortes, que apresentaram valores energéticos abaixo de -3,0kcal mol−1 e acima de 3,0 kcal mol−1 e os colocamos na figura 4.10. Vale a pena ressaltar,que a maioria dos aminoácidos que aparecem aqui estão dentro dos quinze citados no

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 74
Figura 4.9 – Superposição da estrutura tridimensional de GABAB em complexos com GABA,baclofeno, SCH50911 e 2-hidroxisaclofeno.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 75
tópico anterior.
Pode-se observar que doze resíduos apresentaram-se neste limite para SCH50911(Fig. 4.10a), e são eles: Ser(S)130 (-24,99 kcal mol−1 para o modelo com cargas pontuais;-12,38 kcal mol−1 para ε10; -11,26 kcal mol−1 para ε40), Ser(S)153 (-22,92 kcal mol−1 parao modelo com cargas pontuais; -13,44 kcal mol−1 para ε10; -12,22 kcal mol−1 para ε40),Glu(E)349 (-52,78 kcal mol−1 para o modelo com cargas pontuais; -12,82 kcal mol−1 paraε10; -9,40 kcal mol−1 para ε40), Gly(G)151 (-4,27 kcal mol−1 para o modelo com cargaspontuais; -1,12 kcal mol−1 para ε10; -0,64 kcal mol−1 para ε40), His(H)170 (-13,61 kcalmol−1 para o modelo com cargas pontuais; -5,39 kcal mol−1 para ε10; -4,40 kcal mol−1
para ε40), Trp(W)65 (4,25 kcal mol−1 para o modelo com cargas pontuais; -6,73 kcal mol−1
para ε10; -6,99 kcal mol−1 para ε40), Trp(W)278 (-10,09 kcal mol−1 para o modelo comcargas pontuais; -6,52 kcal mol−1 para ε10; -6,18 kcal mol−1 para ε40), Cys(C)129 (-8,62kcal mol−1 para o modelo com cargas pontuais; -6,01 kcal mol−1 para ε10; -5,17 kcal mol−1
para ε40), Ser(S)152 (-10,11 kcal mol−1 para o modelo com cargas pontuais; -5,91 kcalmol−1 para ε10; -5,21 kcal mol−1 para ε40), Ser(S)154 (-4,37 kcal mol−1 para o modelocom cargas pontuais; -0,86 kcal mol−1 para ε10; -0,62 kcal mol−1 para ε40), Arg(R)168(-10,12 kcal mol−1 para o modelo com cargas pontuais; -11,04 kcal mol−1 para ε10; -0,65kcal mol−1 para ε40) e Glu(E)309 (-8,32 kcal mol−1 para o modelo com cargas pontuais;-0,84 kcal mol−1 para ε10; -0,29 kcal mol−1 para ε40).
Ao observar-se os resíduos que interagem com baclofeno (Fig. 4.10b), percebe-seque treze aminoácidos foram selecionados: Tyr(Y)250 (-5,80 kcal mol−1 para o modelo comcargas pontuais; -6,88 kcal mol−1 para ε10; -6,65 kcal mol−1 para ε40), Trp(W)65 (-3,26kcal mol−1 para o modelo com cargas pontuais; -9,74 kcal mol−1 para ε10; -9,44 kcal mol−1
para ε40), Ser(S)130 (-17,56 kcal mol−1 para o modelo com cargas pontuais; -7,68 kcalmol−1 para ε10; -6,66 kcal mol−1 para ε40), Ser(S)153 (-10,96 kcal mol−1 para o modelo comcargas pontuais; -0,69 kcal mol−1 para ε10; 0,41 kcal mol−1 para ε40), Trp(W)278 (-26,95kcal mol−1 para o modelo com cargas pontuais; -12,65 kcal mol−1 para ε10; -10,93 kcalmol−1 para ε40), Glu(E)349 (-46,76 kcal mol−1 para o modelo com cargas pontuais; -9,99kcal mol−1 para ε10; -6,16 kcal mol−1 para ε40), Cys(C)129 (-6,10 kcal mol−1 para o modelocom cargas pontuais; -2,44 kcal mol−1 para ε10; -1,83 kcal mol−1 para ε40), Gly(G)151(-5,87 kcal mol−1 para o modelo com cargas pontuais; -1,66 kcal mol−1 para ε10; -1,13 kcalmol−1 para ε40), Val(V)201 (-4,65 kcal mol−1 para o modelo com cargas pontuais; -1,53kcal mol−1 para ε10; -1,13 kcal mol−1 para ε40), Ser(S)152 (-10,13 kcal mol−1 para o modelocom cargas pontuais; -4,98 kcal mol−1 para ε10; -4,29 kcal mol−1 para ε40), His(H)170(-4,94 kcal mol−1 para o modelo com cargas pontuais; -2,45 kcal mol−1 para ε10; -1,97 kcalmol−1 para ε40), Gln(Q)348 (6,73 kcal mol−1 para o modelo com cargas pontuais; 1,16 kcalmol−1 para ε10; 0,41 kcal mol−1 para ε40) e Arg(R)168 (-7,94 kcal mol−1 para o modelocom cargas pontuais; -0,67 kcal mol−1 para ε10; -0,23 kcal mol−1 para ε40).
Na Fig. 4.10c, vê-se que quatorze aminoácidos estão dentro do limite de energia,sendo estes: Ser(S)153 (-19,62 kcal mol−1 para o modelo com cargas pontuais; -9,90 kcalmol−1 para ε10; -8,70 kcal mol−1 para ε40), His(H)170 (-14,60 kcal mol−1 para o modelocom cargas pontuais; -7,36 kcal mol−1 para ε10; -6,24 kcal mol−1 para ε40), Tyr(Y)250(-10,87 kcal mol−1 para o modelo com cargas pontuais; -7,85 kcal mol−1 para ε10; -7,27 kcalmol−1 para ε40), Ser(S)130 (-13,62 kcal mol−1 para o modelo com cargas pontuais; -3,63kcal mol−1 para ε10; -2,61 kcal mol−1 para ε40), Gly(G)151 (-4,39 kcal mol−1 para o modelocom cargas pontuais; -0,54 kcal mol−1 para ε10; -0,13 kcal mol−1 para ε40), Trp(W)278(-25,69 kcal mol−1 para o modelo com cargas pontuais; -11,92 kcal mol−1 para ε10; -10,43
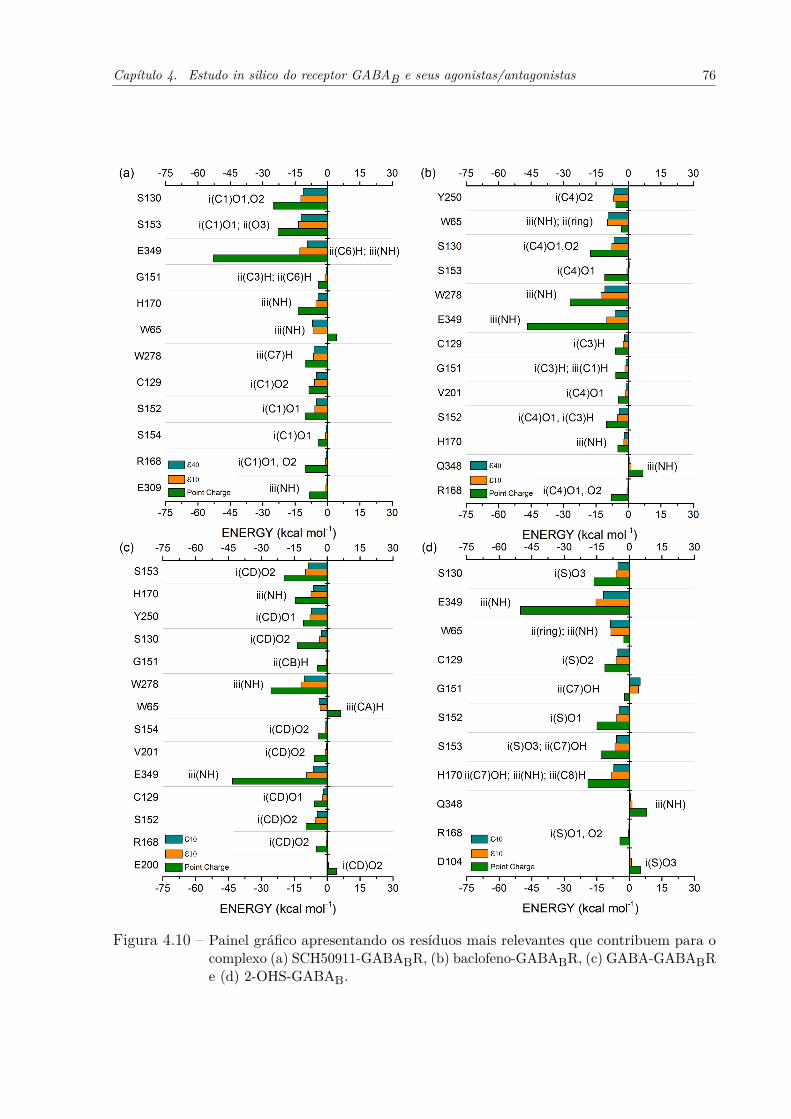
Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 76
Figura 4.10 – Painel gráfico apresentando os resíduos mais relevantes que contribuem para ocomplexo (a) SCH50911-GABABR, (b) baclofeno-GABABR, (c) GABA-GABABRe (d) 2-OHS-GABAB.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 77
kcal mol−1 para ε40), Trp(W)65 (6,13 kcal mol−1 para o modelo com cargas pontuais; -3,25kcal mol−1 para ε10; -3,64 kcal mol−1 para ε40), Ser(S)154 (-4,15 kcal mol−1 para o modelocom cargas pontuais; -0,97 kcal mol−1 para ε10; -0,67 kcal mol−1 para ε40), Val(V)201(-5,90 kcal mol−1 para o modelo com cargas pontuais; -0,94 kcal mol−1 para ε10; -0,30 kcalmol−1 para ε40), Glu(E)349 (-43,34 kcal mol−1 para o modelo com cargas pontuais; -9,34kcal mol−1 para ε10; -6,20 kcal mol−1 para ε40), Cys(C)129 (-5,90 kcal mol−1 para o modelocom cargas pontuais; -2,25 kcal mol−1 para ε10; -1,62 kcal mol−1 para ε40), Ser(S)152 (-9,66kcal mol−1 para o modelo com cargas pontuais; -5,22 kcal mol−1 para ε10; -4,51 kcal mol−1
para ε40), Arg(R)168 (-5,02 kcal mol−1 para o modelo com cargas pontuais; -0,50 kcalmol−1 para ε10; -0,17 kcal mol−1 para ε40) e Glu(E)200 (4,29 kcal mol−1 para o modelocom cargas pontuais; 0,26 kcal mol−1 para ε10; 0,001 kcal mol−1 para ε40).
Em contrapartida, para 2-hidroxisaclofeno somente nove resíduos entraram nestelimite (Fig. 4.10d), sendo eles: Ser(S)130 (-16,26 kcal mol−1 para o modelo com cargaspontuais; -6,09 kcal mol−1 para ε10; -5,25 kcal mol−1 para ε40), Glu(E)349 (-7,94 kcalmol−1 para o modelo com cargas pontuais; -0,67 kcal mol−1 para ε10; -0,23 kcal mol−1 paraε40), HIS170 (-18,77 kcal mol−1), Cys(C)129 (-11,33 kcal mol−1 para o modelo com cargaspontuais; -6,09 kcal mol−1 para ε10; -5,36 kcal mol−1 para ε40), Ser(S)152 (-14,92 kcalmol−1 para o modelo com cargas pontuais; -5,80 kcal mol−1 para ε10; -4,74 kcal mol−1 paraε40), Ser(S)153 (-13,07 kcal mol−1 para o modelo com cargas pontuais; -6,60 kcal mol−1
para ε10; -5,96 kcal mol−1 para ε40), His(H)170 (-19,06 kcal mol−1 para o modelo comcargas pontuais; -8,42 kcal mol−1 para ε10; -7,21 kcal mol−1 para ε40), Gln(Q)348 (7,98kcal mol−1 para o modelo com cargas pontuais; 1,24 kcal mol−1 para ε10; 0,52 kcal mol−1
para ε40), Arg(R)168 (-4,36 kcal mol−1 para o modelo com cargas pontuais; -0,45 kcalmol−1 para ε10; -0,20 kcal mol−1 para ε40) e Asp(D)104 (5,08 kcal mol−1 para o modelocom cargas pontuais; 0,99 kcal mol−1 para ε10; 0,34 kcal mol−1 para ε40). Contudo, doisresíduos apresentaram energia de interação dentro do limite quando constantes dielétricasforma utilizadas: Trp(W)65 (-2,57 kcal mol−1 para o modelo com cargas pontuais; -8,52kcal mol−1 para ε10; -8,73 kcal mol−1 para ε40) e Gly(G)151 (-2,21 kcal mol−1 para omodelo com cargas pontuais; 4,19 kcal mol−1 para ε10; 5,02 kcal mol−1 para ε40).
Como apresentado por Y. Geng e colaboradores (GENG et al., 2013), ao entrarno sítio ortostérico, os agonistas tendem a formar interações que levam ao fechamentodos dois lobos do receptor em torno do ligante, enquanto os antagonistas tendem amanter a estrutura do receptor semelhante àquela encontrada no receptor sem ligantes.Com a aproximação dos lobos, agonistas conseguem realizar mais interações fortes doque antagonistas, o que torna compreensível a diferença observada na Figura 4.10. Essavariação no posicionamento dos aminoácidos pode ser observado na figura 4.11, onde vê-seque os aminoácidos presentes no complexo com baclofeno e GABA estão mais próximos docentro da fenda entre os lobos, o que forma uma estrutura como uma concha fechada emtorno do ligante. Contudo, o mesmo não pode ser visto para o complexo com 2-OHS, umavez que os aminoácidos de LB1 parecem estar um pouco mais distantes do centro da fenda,e aqueles de LB2 não se aproximam o suficiente do antagonista para formar qualquerinteração forte. Uma vez que a única diferença entre os baclofeno e 2-hidroxisaclofenose dá pela presença do grupo sulfônico, a presença de uma distribuição de cargas maispróxima de um valor positivo no anel de 2-hidroxisaclofeno deve estar relacionado com essamaior abertura dos lobos, o que também é observado para SCH50911. Em contrapartida, oacoplamento de SCH50911 é bastante similar àquela de 2-hidroxisaclofeno, embora muitasinterações que são formadas somente com agonistas estão presentes no complexo com esseantagonista.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 78
Figura 4.11 – Visão detalhada dos resíduos mais importantes envolvidos na ligação SCH50911,baclofeno, GABA e 2-OHS (de cima para baixo) com GABAB. Potenciais pontessalinas, ligações de hidrogênio, ligações de hidrogênio não convencionais, cátion-π,π-π e σ-π são indicados por linhas de cor vermelho, azul claro, verde, laranja ecinza e ciano escuro, respectivamente.

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 79
Contudo, existe um padrão de interação que se repete para ambas classes demoléculas. Na figura 4.11 (esquerda), vê-se que o grupo ácido dos ligantes forma ligaçõesde hidrogênio (HB) com Ser(S)153 e Ser(S)130, estabilizando a região (i) de SCH50911,baclofen, GABA e 2-hidroxisaclofeno no sítio ortostérico. Enquanto o primeiro resíduoestá engajado em duas HBs com os oxigênios do grupo ácido de todos os quatro ligantes,Ser(S)153 forma apenas uma HB com i(S)O3 de 2-OHS (2,84 Å), mas também estáenvolvido com a molécula por uma ligação de hidrogênio não convencional com a hidroxilaem ii(C7)OH a uma distancia de 2,49 Å. Por outro lado, Ser(S)130 forma ligações dehidrogênio não convencionais com baclofeno e 2-hidroxisaclofeno.
Já na região contendo a amina positivamente carregada, outro padrão de interaçõesse repete, como pode ser observado nas figuras 4.11. Os aminoácidos His(H)170 e Glu(E)349formam o sítio de ancoramento da porção amina, formando um conjunto de ligações dehidrogênio e pontes salinas. His(H)170 aparece por fazer ligações de hidrogênio com ogrupo iii(NH+
3 ) de SCH50911, baclofen, GABA e 2-hidroxisaclofeno, adicionando umaligação não convencional com iii(C8)H (2,75 Å) de 2-hidroxisaclofeno. Por outro lado,Glu(E)349 forma uma ponte salina com ambos os ligantes na região iii(NH+
3 ) em distânciasimilares, 2,52 para baclofeno e 2,42 Å para 2-hidroxisaclofeno.
Os quatro aminoácidos apresentados acima formam uma região comum paratodos os ligantes, sendo estes agonistas ou antagonistas. Isso foi mostrado por Genget al. (2013), quando estes obtiveram cristais de GABAB co-cristalizado com váriosagonistas e antagonistas. Assim, esses são os únicos pontos em comum entre os ligantes.Energeticamente, vê-se que não houve muita variação quando comparamos os quatrocomplexos. Contudo, a presença de um maior número de interações entre His(H)170 e 2-OHS torna esta ligação mais forte, quando comparado ao agonista. Além desse aminoácido,outra variação importante ocorreu em Ser(S)130. Ao compararmos o posicionamento desseaminoácido nos quatro complexos, percebe-se que este afasta-se do centro da região (i)quando esta é composta pelo ácido sulfônico. Mutações realizadas nesses aminoácidolevaram a uma perda significativa na afinidade e potência de ligantes que interagem comGABAB, mostrando assim a relevância destes (GALVEZ et al., 2000; GALVEZ et al.,1999).
Todos os demais resíduos que serão descritos apresentaram diferenças significativasna forma de interagir com os ligantes. Dentre eles, aqueles que mais divergiram, em termosenergéticos e espaciais, foram Tyr(Y)250 e Trp(W)278, ambos aminoácidos que fazemparte do lobo 2. Como apresentado anteriormente, em presença de agonistas os lobos dodomínio extracelular de GABAB tendem a fechar-se, formando uma estrutura complexa eaproximando os aminoácidos do Lobo 2 dos ligantes. Quando baclofeno ou GABA estãopresentes no sítio de ligação, Tyr(Y)250 aproxima-se da região i(C4)O2 e forma umaligação de hidrogênio com os agonistas. Quando ocorre a ligação do antagonista, estemesmo aminoácido possui como região do ligante mais próxima de si o anel clorofenil de2-hidroxisaclofeno e o grupo carboxila de SCH50911.
Além dessa variação, Trp(W)278 também sofre os efeitos da ligação de 2-OHS eSCH50911. Na presença de baclofeno ou GABA, esse aminoácido forma interações do tipocátion-π entre os anéis que compõe sua cadeia lateral e a amina carregada dos ligantes,que estão a uma distância de 3,6 e 2,9 Å. Contudo, em presença do antagonista há umarotação na cadeia lateral que impede a interação forte com os grupos químicos dos ligantes,resultando em uma energia de interação baixa.
Segundo Geng e colaboradores, mutações realizadas nesses aminoácidos não afetam

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 80
significativamente a ação de antagonistas. Contudo, quando são substituídos por alanina, aresposta à agonistas é reduzida consideravelmente, podendo levar a uma perda da respostado receptor e decréscimo na potência de agonistas. Em contrapartida, foi apresentado quea adaptabilidade conformacional apresentada pela cadeia lateral de Trp(W)278 provê ummecanismo de seleção pelo qual o receptor reconhece ligantes estruturalmente diferentesenquanto mantém a especificidade e afinidade aos ligantes (GENG et al., 2013).
Os aminoácidos GlY151 e Trp(W)65 são os últimos dentre a lista de resíduosenergeticamente importantes que possuem sua relevância representada experimentalmentepor análises de mutação (GENG et al., 2012; GENG et al., 2013). A substituição dessesaminoácidos por alanina levou a uma perda na capacidade de ligação de agonistas eantagonistas, além de uma perda drástica das funções do receptor. Gly(G)151 formaligações de hidrogênio não convencionais com os carbonos da região ii todos os ligantes,embora sua proximidade ao 2-hidroxisaclofeno permite a formação de uma ligação dehidrogênio comum com o grupo hidroxila de ii(C7)OH com distância de 2,04 Å.
Trp(W)65 é o único aminoácido, dentre os mais energéticos, que apresenta umaenergia de interação divergente entre os ligantes. Enquanto uma repulsão foi observadapara SCH50911 e GABA, encontramos uma atração para baclofen e 2-hidroxisaclofeno, oque pode estar relacionado com uma interação do tipo π-π e cátion-π que esse resíduo fazcom os dois últimos. Por outro lado, SCH50911 e GABA só formam interações do tipoσ-π com Trp(W)65.
Por último temos os resíduos Val(V)201, Ser(S)152 e Ser(S)154. Como dito ante-riormente, a relevância desses resíduos não foi apresentada por análises experimentais,portanto, estão com sua importância sendo expostas pela primeira vez neste trabalho.Com exceção de Ser(S)152 ao interagir com 2-OHS, todos estão envolvidos com esseaminoácido por ligações de hidrogênio não convencionais. Val(V)201 forma uma ligação dehidrogênio não convencional com a carboxila do ligantes, com exceção de 2-OHS. Ser(S)152e Ser(S)154 estão circundando o aminoácido Ser(S)153, apresentado anteriormente comoum dos mais importantes para a complexação de agonistas e antagonistas.
No geral, dentre os aminoácidos analisados neste trabalho, foi observado que aenergia de ligação para SCH50911 segue a sequência decrescente (do mais atrativo ao maisrepulsivo): Glu(E)349 > Ser(S)130 > Ser(S)153 > His(H)170 > Arg(R)168 > Ser(S)152> Trp(W)278 > Cys(C)129 > Glu(E)309 > Ser(S)154 > GlY(G)151 > Trp(W)65. Parabaclofeno, a ordem de energia individual seque a ordem: Glu(E)349 > Trp(W)278 >Ser(S)130 > Ser(S)153 > Ser(S)152 > Arg(R)168 > Cys(C)129 > Gly(G)151 > Tyr(Y)250> His(H)170 > Val(V)201 > Trp(W)65 > Gln(Q)348. Observando o complexo com GABA,os resíduos mais importantes são: Glu(E)349 > Trp(W)278 > Ser(S)153 > His(H)170> Ser(S)130 > Tyr(Y)250 > Ser(S)152 > Val(V)201 > Cys(C)129 > Arg(R)168 >Gly(G)151 > Ser(S)154 > Glu(E)200 > Trp(W)65. Para 2-OHS, a energia de interaçãoindividual segue a ordem: Glu(E)349 > His(H)170 > HIS170 > Ser(S)130 > Ser(S)152 >Ser(S)153 > Arg(R)168 > Asp(D)104 > Gln(Q)348 (7,98). Com exceção de Val(V)201,Ser(S)152 e Ser(S)154, todos esses aminoácidos são consistentes com análises publicadasde cristalografia e mutação. Vale a pena ressaltar que todos os resíduos apresentados fazemparte de uma rede intrincada de interações hidrofóbicas com ambos os ligantes, o que vemreforçar a relevância dessas interações para o acoplamento de ligantes no sítio ativo dessaproteína.
Por fim, vale a pena ressaltar que a comparação entre a energia de interação calcu-lada para cada resíduo individual apresentou-se de forma bastante similar para a maioria

Capítulo 4. Estudo in silico do receptor GABAB e seus agonistas/antagonistas 81
dos aminoácidos. Contudo, alguns aminoácidos que formaram ligações de hidrogênio nãoconvencionais, tais como Ser(S)154, Gly(G)151 e Val(V)201, não apresentaram energia deinteração significante para o modelo de constante dielétrica, enquanto apresentaram intera-ções significativas para o modelo com carga pontual. Jakobsen e colaboradores utilizaramesse modelo para obter a estrutura e a densidade de cargas na insulina, mostrando que oresultado com cargas foi mais próximo do esperado a partir de analises experimentais (JA-KOBSEN; KRISTENSEN; JENSEN, 2013). Ademais, Manzoni e colaboradores utilizaramum modelo de cargas pontuais como solvente para as propriedades ópticas e magnéticasde pirimidina e outros compostos, mostrando que o modelo utilizado foi similar ao modelocom átomos explícitos (MANZONI et al., 2010; MANZONI et al., 2011).
4.4 Conclusão
Os receptores GABAérgicos são alvos biológicos atrativos e intensamente exploradosem drug discovery. Dentre eles, o receptor GABAB e seus agonistas/antagonistas têm sidolargamente utilizados na pesquisa sobre modulação do sistema nervoso. Contudo, o únicofármaco aprovado nos dias de hoje é o baclofeno. Portanto, a compreensão das energias deinteração entre ligante-receptor é vital para o sucesso de para o design de fármacos quevisem este receptor.
Para auxiliar neste processo, investigamos a energia de interação dos comple-xos SCH0911-GABAB, baclofeno-GABAB, GABA-GABAB e 2-hidroxisaclofeno-GABAB
usando o método de fracionamento MFCC com os modelos de incremento eletrostático(EE-MFCC) e constante dielétrica sob uma base de cálculos em nível quântico.
No geral, nossos resultados foram consistentes com estudos experimentais e teó-ricos anteriores. Os aminoácidos aqui avaliados foram selecionados através do critériode convergência e afinidade experimental. A interação de aproximadamente 65 ami-noácidos em interação com os quatro ligantes foram estudadas, mostrando que os re-síduos Glu(E)349, Ser(S)130, Ser(S)153, His(H)170, Arg(R)168, Ser(S)152, Trp(W)278,Cys(C)129, Glu(E)309, Ser(S)154, GlY(G)151, Trp(W)65 apresentam uma maior relevânciaenergética. Também apresentamos a descrição energética de outros resíduos experimental-mente relevantes.
Por fim, comparamos os cálculos realizados com sete métodos de cálculos diferentese os três modelos de descrição do ambiente. Encontramos que em termos de energiatotal de ligação, o efeito do método de cálculo é mais importante do que a descrição doambiente. Contudo, ao avaliarmos as energias de interação individuais, algumas diferençasforam encontradas dentro do mesmo método de cálculo. Acreditamos que os resultadosaqui apresentado são um passo inicial em direção não só ao desenvolvimento de novosmedicamentos visando o sistema GABAérgico, mas a uma melhor compreensão dosreceptores acoplados à proteína G.

82
capítulo 5
CONCLUSÃO FINAL E PERSPECTIVAS
5.1 Conclusão Final
Vários processos biológicos não possuem descrição precisa pela falta de conhecimentodo seu mecanismo de ação, o que impede a formulação de fármacos que possam atuarna ativação ou inibição de receptores específicos que podem levar a cura de muitasdoenças. Por muitos anos a indústria farmacêutica se baseou em um conjunto de análisesexperimentais para averiguar a viabilidade de moléculas farmacológicas, demandandouma grande quantidade de tempo e dinheiro. Hoje observa-se que os computadores estãoauxiliando vários momentos da pesquisa farmacêutica, desde a predição de novas moléculasaté a observação de seu comportamento no alvo.
Diferenciar as interações específicas realizadas por cada ligante é essencial paraum melhor entendimento do comportamento do sítio ativo, possuindo grande importânciana compreensão de processos biológicos e no desenvolvimento de medicamentos. Pormeio desta afirmação, percebe-se que uma metodologia que permita a caracterização dasinterações realizadas no sítio de ligação é de extrema importância, pois a diferenciaçãoentre os grupos funcionais que são responsáveis por criar um maior (ou menor) conjuntode ligações favoráveis pode propiciar ajustes na estrutura do ligante que levem a umamelhor eficácia do mesmo.
A partir das informações expostas em todo o corpo desta dissertação, percebe-seque as técnicas computacionais possuem grande relevância para a pesquisa bioquímicae farmacêutica. Deste modo, o presente trabalho vem a reforçar o papel da simulaçãocomputacional em um nível quântico no entendimento da interação de moléculas agonistase antagonistas com representantes das classes A e C dos receptores acoplados à proteínaG, bem como do receptor PD-1, diferenciando, por meio de valores energéticos, os sítiosde interação desses compostos em seus alvos moleculares, com o objetivo de auxílio nodesenvolvimento de fármacos para os sistemas estudados.
Os receptores acoplados à proteína G constituem uma das mais importantes ediversificadas superfamílias de proteínas codificadas pelo genoma humano, com membroque podem ligar-se seletivamente a uma variada gama de moléculas, incluindo íons, aminasbiogênicas, aminoácidos, peptídeos, lipídeos, nucleotídeos e proteínas. Ao serem ativados,estes receptores atuam em conjunto com proteínas citosólicas para traduzir o sinal do meioextracelular para o meio intracelular. Esse sinal é posteriormente amplificado para modularo comportamento fisiológico das células. Dessa forma, estes receptores possuem papelcrucial em vários processos fisiológicos, como neurotransmissão, crescimento, metabolismo ediferenciação celular, secreção e resposta imunológica. Assim, tendo em vista seu alto valorpara o funcionamento dos sistemas biológicos, os receptores acoplados à proteína G temrecebido considerável atenção da indústria farmacêutica, sendo alvo de aproximadamente30-50% de todos os fármacos aprovados e comercializados pelo mundo.
Dentre os receptores acoplados à proteínas G que possuem estrutura tridimensionaldefinida e validação experimental como alvo terapêutico, selecionamos dois modelos dereceptor que poderiam não só clarificar o modo de interação para ligantes da própria

Capítulo 5. Conclusão Final e Perspectivas 83
proteínas, mas que auxiliassem na descrição de outras proteínas da mesma família. Assim,propusemos um trabalho voltado para a descrição das interações chave de receptoresserotonérgicos, um receptor de classe A, e GABAérgicos, receptor de classe C.
Embora a relação comum entre esses receptores e a modulação do sistema nervosopareça quase intuitiva, 5-HT1B e GABAB são alvos terapêuticos utilizados no tratamentode enxaqueca e dores neuropáticas, respectivamente. Em contrapartida, os vários efeitosindesejados advindos da utilização dos fármacos que visam esses receptores torna evidentea necessidade de novos fármacos mais efetivos. Ademais, a similaridade estrutural entreesses receptores e àqueles da mesma classe é grande, o que torna o desenvolvimento defármacos mais difícil para subtipos específicos de receptor.
Não obstante, na última década uma vasta gama de estudos voltados à uma melhorcompreensão do mecanismo molecular relacionado com a correlação entre proteínas decheckpoint imunomodulatórias e a ação de anticorpos monoclonais no combate ao câncertem sido apresentados. Essas proteínas, expressas na superfície de células que agem naresposta imune adquirida, tem sido bastante visadas e tem mostrado respostas animadorasem pacientes que estão passando por esse tipo de terapia.
Dentre os primeiros fármacos aprovados pelo US-FDA, pembrollizumab e nivolu-mab tem sido largamente utilizados, como terapia única, ou combinada com quimioterápi-cos/outros anticorpos monoclonais. O tempo de sobrevida dos pacientes que fazem usodesses compostos aumenta significantemente, o que abriu portas para um amplo desenvol-vimento de novos compostos. Contudo, o número de pacientes mostrando respostas à essasmoléculas ainda é pequeno e alguns efeitos adversos advindos da modulação do sistemanervoso tem sido reportados.
Os métodos empregados no desenho racional de fármacos, em especial os métodosde fragmentação, tem se mostrado bastante eficazes em delinear a estrutura do receptor equantificar a importância de cada interação formada dentro do sítio de ligação. Portanto,neste trabalho, utilizou-se a técnica do fracionamento molecular com caps conjugados, paradeterminar quais os aminoácidos com maior importância na resposta dos dois receptoresescolhidos após a entrada de agonistas ou antagonistas em seu sítio de ligação. Assim,tendo por base as interações formadas com cada um destes aminoácidos, se faz possívelprocurar regiões em que seja possível modificar estas moléculas ligantes e obter uma melhorresposta de agonismo ou antagonismo para o receptor.
Com a fragmentação dos sistemas estudados, foi possível encontrar os aminoácidosmais importantes para a estabilização dos ligantes dentro de seus respectivos sítios deligação. Em ordem de relevância energética, conclui-se que os aminoácidos que possuemas maiores energias de interação são aqueles que participam da rede de interações dehidrogênio, embora interações hidrofóbicas sejam primordiais para o acoplamento de todasos compostos aqui utilizados.
Como apresentado, os membros da classe A dos receptores acoplados à proteína Gpossuem a estrutura característica dessa superfamília, com um domínio transmembranacontendo sete hélices unidas por alças, além de regiões N-terminal e C-terminal curtas.Assim, diferente do que o ocorre nas demais classes, o sítio de ligação à agonistas e antago-nistas está posicionado no interior do domínio transmembrana, diferindo relativamente emdiâmetro de acordo com o tipo de receptor, mas com a mesma característica comumenteobservada nos demais membros desta classe. O mecanismo de ativação desses receptoresé mais simples em relação aos demais, sendo diretamente relacionado ao modo como o

Capítulo 5. Conclusão Final e Perspectivas 84
agonista entra no sítio de ligação e às interações formadas entre este e os aminoácidos doreceptor, podendo variar de acordo com o conjunto de interações formadas.
De modo geral, nossos resultados nos mostraram que diidroergotamina liga-se emum bolsão formado, principalmente, pelos aminoácidos Asp(D)129, Asp(D)352, Asp(D)123,Glu(E)198, Asp(D)204, Phe(F)330, Leu(L)126, Phe(F)351, Ile(I)130, Val(V)201, Val(V)200,The(T)355 e Arg(R)114, que circundam o ligante formando interações fortes que o es-tabilizam. Além disso, apresentamos a ordem de afinidade entre as regões do ligantee a proteína, e entre as estruturas secundárias de 5-HT1B com diidroergotamina. Comexceção de Arg(R)114, Asp(D)123, Glu(E)198 e Asp(D)204, todos os valores energéticosencontrados são consistentes com estudos experimentais e teóricos anteriores, não só parareceptores serotonérgicos, mas também para outros receptores monoaminérgicos.
Ao avaliarmos os resultados para os complexos PD-1/ligantes, observamos que oresíduo de lisina Lys(K)131 exibe a interação resíduo-resíduo mais forte, um resultadorelacionado com sua capacidade de penetrar-se no sítio de ligação dos ligantes devidoa sua alta flexibilidade e polaridade, levando a uma forte ligação de hidrogênio com osaminoácidos dos ligantes. Ademais, uma região de baixa energia de interação foi encontrada,o que pode ajudar ao receptor a adotar uma nova conformação em presença dos inibidorescompetitivos e impede a ligação de PD-L1.
Por outro lado, os receptores de classe C apresentam um sítio de ligação externoao domínio transmembrana, posicionado na região N-terminal em um sítio conhecidocomo Venus Fly Trap. Ademais, esses receptores formam ’dímeros obrigatórios’, o quenão é observado em outros GPCRs. Acredita-se que o mecanismo de ativação dessesreceptores segue uma sequência diferente daquele que ocorre nos demais receptores, ondeos lobos que compõe o domínio extracelular fecham-se à presença de um agonista, o queleva a mudanças conformacionais entre os monômeros e, consequentemente, na formaçãode uma região intracelular favorável a ligação com o parceiro proteico intracelular. Emcontrapartida, a ligação de antagonistas mantém a estrutura geral do domínio extracelularintacta, impedindo o fechamento dos lobos que ocorreria em presença de agonistas.
Neste caso, foi observado que alguns aminoácidos que formam o lobo 1 dessedomínio extracelular compartilham interações entre agonistas e antagonistas, podendo-se predizer que este deve ser o sítio de reconhecimento do receptor para ligantes quetenham GABAB como alvo. Apesar dessa partilha no provável sítio de reconhecimento, osaminoácidos que compõe o lobo 2 de GABAB1 apresentaram uma preferência clara porinteragir com agonistas, criando uma estrutura fechada não vista quando antagonistasentram no sítio de ligação, mesmo quando estruturalmente semelhantes, um provável efeitoda distribuição de cargas que difere, mesmo que levemente, no anel clorofenil dos ligantes.De modo geral, nossos resultados mostraram que os aminoácidos Ser(S)130, Gly(G)151,Ser(S)153, His(H)170, Tyr(Y)250, Trp(W)278, Glu(E)349, Val(V)201, Ser(S)152, Ser(S)154,Gln(Q)348, Arg(R)168 e Trp(W)65 são aqueles que interagem mais fortemente comSCH50911, baclofeno, GABA e 2-hidroxisaclofeno, com os três aminoácidos que apresentamanéis em sua estrutura (Tyr(Y)250, Trp(W)278 e Trp(W)65) mostrando-se mais sensíveisà diferenças entre agonistas e antagonistas. Assim como no caso anterior, a maioria dosvalores energéticos encontrados são consistentes com estudos experimentais e teóricosanteriores.
Além disso, os resultados apresentados para o receptor 5-HT1B mostram que autilização do modelo de incremento eletrostático (electrostatic embedding foi capaz dereduzir a energia total de interação, em comparação ao modelo sem cargas pontuais,

Capítulo 5. Conclusão Final e Perspectivas 85
levando a uma estabilização energética em distâncias confirmadas experimentalmente.Ademais, o uso desse modelo levou a uma boa reprodução computacional para os dadosexperimentais de afinidade em receptores GABAB.
Por fim, os dados aqui obtidos podem ser de grande auxílio no entendimento dos sis-temas serotonérgico, GABAérgico e imuno-oncológico, posteriormente, no desenvolvimentode novos fármacos para esses sistemas, estando de acordo com análises experimentais eacrescentando novos conhecimentos até então não descritos na literatura.
5.2 Perspectivas
Como apresentado anteriormente, a diferenciação entre os subtipos de receptor deserotonina, em especial os receptores 5-HT2B, é um passo importante no desenho de novosfármacos que visem o sistema monoaminérgico, uma vez que vários compostos acabamsendo eliminados em fases clínicas iniciais de avaliação devido a efeitos colaterais severosrelacionados ao mal funcionamento do sistema circulatório.
Desde as primeiras estruturas cristalográficas apresentando a conformação dosreceptores de serotonina serem publicadas até hoje, seis novas estruturas tridimensionaisforam obtidas, excluindo àquela apresentada no capítulo dois deste trabalho. Nestes cincoanos, foram publicadas as estruturas de 5-HT1B com o agonista ergotamina e o antagonistametiotepina, 5-HT2B em complexo com LSD e ergotamina e, recentemente o receptor5-HT2C foi co-cristalografado com ergotamina e ritanserina.
Com essas estruturas em mãos, será possível realizar um estudo comparativovisando caracterizar os pontos chave para a ligação desses compostos em cada subtipo dereceptor. Não só isso, mas a presença de ergotamina em todos os três cristais provê umaoportunidade impar para compreender o modo de interação desta molécula com os demaismembros dos receptores de serotonina.
Não obstante, muito se tem falado sobre a necessidade de dímeros para o fun-cionamento de GABAB. Contudo, trabalhos tem mostrado que essa necessidade não étão aparente em um nível aminoacídico na região extracelular. Portanto, a publicaçãoda estrutura cristalográfica desse dímero com vários agonistas e antagonistas, além daproteína sem ligantes, nos permite avaliar energetica- e estruturalmente as variações nodímero acarretadas pela interação dos ligantes no bolsão de ligação.
Por fim, a avaliação completa da interação entre os fármacos pembrolizumab enivolumab, assim como do ligante natural de PD-L1, em complexo com a proteína PD-1é de grande importância para uma melhor compreensão do funcionamento molecular doreceptor. Ademais, outros complexos já estão disponíveis com PD-L1/fármacos e CTLA-4/fármacos. O que nos abre a porta para o desenvolvimento de uma vasta gama de estudosem imunoterapêuticos.
Destarte, percebe-se que além dos resultados obtidos foram propostos novos tra-balhos que permitirão uma completa descrição do mecanismo funcional dos receptoresaqui apresentados. Esta tese constitui um passo a mais para o completo entendimento desistemas serotonérgico e GABAérgico, bem como de imunoterapias através de anticorposmonoclonais.

86
REFERÊNCIAS
ACEVEDO, O.; JORGENSEN, W. L. Advances in quantum and molecular mechanical(QM/MM) simulations for organic and enzymatic reactions. Accounts of ChemicalResearch, v. 43, p. 142–151, 2009.
ADAMS, C. P.; BRANTNER, V. V. Estimating the cost of new drug development: is itreally $802 million? Health Affairs, v. 25, p. 420–428, 2006.
ALBUQUERQUE, E. L. et al. DNA-based nanobiostructured devices: The role ofquasiperiodicity and correlation effects. Physics Reports, v. 535, p. 139–209, 2014.
ALI, A. et al. Circulating PD-L1 (programmed death-ligand 1) and outcomes in aHER2-positive metastatic breast cancer cohort treated with first-line trastuzumab.Journal of Clinical Oncology, v. 35, p. 1024, 2017.
ALLISON, J. P. Nobel Lecture - Immune Checkpoint Blockade in Cancer Therapy:New insights, opportunities, and prospects for cures. 2018. Disponível em:<https://www.nobelprize.org/prizes/medicine/2018/allison/lecture/>. Acesso em: 19 dez.2018.
ALMAULA, N. et al. Mapping the binding site pocket of the serotonin 5-hydroxytryptamine2a receptor. Ser3.36 (159) provides a second interaction site for theprotonated amine of serotonin but not of lysergic acid diethylamide or bufotenin. Journalof Biological Chemistry, v. 271, p. 14672–14675, 1996.
AMUNDSEN, S. et al. Pharmacological treatment of migraine during pregnancy andbreastfeeding. Nature Reviews Neurology, v. 11, p. 209–219, 2015.
ANTONY, J.; GRIMME, S. Fully ab initio protein-ligand interaction energies withdispersion corrected density functional theory. Journal of Computational Chemistry, v. 33,p. 1730–1739, 2012.
ARAGON-SANABRIA, V.; KIM, G. B.; DONG, C. From cancer immunoediting to newstrategies in cancer immunotherapy: the roles of immune cells and mechanics in oncology.In: Biomechanics in Oncology. Germany: Springer, 2018. p. 113–138.
ARODOLA, O. A.; SOLIMAN, M. E. Quantum mechanics implementation in drug-designworkflows: does it really help? Drug Design, Development and Therapy, v. 11, p. 2551,2017.
ATKINS, P.; PAULA, J. D. Elements of physical chemistry. 5.ed. UK: Oxford UniversityPress, 2013.
AUGEN, J. The evolving role of information technology in the drug discovery process.Drug Discovery Today, v. 7, p. 315–323, 2002.
AZUMA, M. et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature, v. 366,p. 76, 1993.

Referências 87
BAO, G.; SURESH, S. Cell and molecular mechanics of biological materials. NatureMaterials, v. 2, p. 715, 2003.
BARBER, R. P. et al. GABAergic terminals are presynaptic to primary afferent terminalsin the substantia gelatinosa of the rat spinal cord. Brain Research, v. 141, p. 35–55, 1978.
BARNETT, A. H. et al. Angiotensin-receptor blockade versus converting–enzymeinhibition in type 2 diabetes and nephropathy. New England Journal of Medicine, v. 351,p. 1952–1961, 2004.
BARROSO-NETO, I. L. et al. Inactivation of ovine cyclooxygenase-1 by bromoaspirinand aspirin: a quantum chemistry description. The Journal of Physical Chemistry B,v. 116, p. 3270–3279, 2012.
BARTUZI, D. et al. Recent advances and applications of molecular docking to Gprotein-coupled receptors. Molecules, v. 22, p. 340, 2017.
BEATTY, G. L.; GLADNEY, W. L. Immune escape mechanisms as a guide for cancerimmunotherapy. Clinical Cancer Research, v. 21, p. 687–692, 2015.
BECKE, A. D. Density-functional thermochemistry. III. the role of exact exchange. TheJournal of Chemical Physics, v. 98, p. 5648–5652, 1993.
BECKER, O. M. et al. Computational biochemistry and biophysics. New York: MarcelDekker, 2001.
BECKSTEIN, O. et al. Ion channel gating: insights via molecular simulations. FEBSLetters, v. 555, p. 85–90, 2003.
BELLO, F. D. et al. The versatile 2-substituted imidazoline nucleus as a structural motifof ligands directed to the serotonin 5-HT1A receptor. ChemMedChem, v. 11, p. 2287–2298,2016.
BENARROCH, E. E. GABAB receptors structure, functions, and clinical implications.Neurology, v. 78, p. 578–584, 2012.
BERENDSEN, H. J. C.; SPOEL, D. van der; DRUNEN, R. van. GROMACS: amessage-passing parallel molecular dynamics implementation. Computer PhysicsCommunications, v. 91, p. 43–56, 1995.
BERGER, M.; GRAY, J. A.; ROTH, B. L. The expanded biology of serotonin. AnnualReview of Medicine, v. 60, p. 355–366, 2009.
BERMAN, H. M. et al. The protein data bank. Nucleic Acids Research, v. 28, p. 235–242,2000.
BERNARD, P.; GUEDIN, D.; HIBERT, M. Molecular modeling of the GABA/GABAB
receptor complex. Journal of Medicinal Chemistry, v. 44, p. 27–35, 2001.
BETTLER, B. et al. Molecular structure and physiological functions of GABAB receptors.Physiological Reviews, v. 84, p. 835–867, 2004.
BEZERRA, K. S. et al. Quantum binding energy features of the T3-785 collagen-liketriple-helical peptide. RSC Advances, v. 7, p. 2817–2828, 2017.

Referências 88
BIERMANN, B. et al. The sushi domains of GABAB receptors function as axonaltargeting signals. Journal of Neuroscience, v. 30, p. 1385–1394, 2010.
BINET, V. et al. The heptahelical domain of GABAB2 is activated directly by CGP7930,a positive allosteric modulator of the GABAB receptor. Journal of Biological Chemistry,v. 279, p. 29085–29091, 2004.
BLUNDELL, T. L. et al. Structural biology and bioinformatics in drug design:opportunities and challenges for target identification and lead discovery. PhilosophicalTransactions of the Royal Society of London B: Biological Sciences, v. 361, p. 413–423,2006.
BOHÓRQUEZ, H. J. et al. Methods in biocomputational chemistry: a lesson from theamino acids. In: Quantum Biochemistry. New York: John Wiley & Sons, 2010. p. 403–421.
BOOTH, B.; ZEMMEL, R. Prospects for productivity. Nature Reviews Drug Discovery,v. 3, p. 451, 2004.
BORTOLATO, A. et al. Structure of class B GPCRs: new horizons for drug discovery.British Journal of Pharmacology, v. 171, p. 3132–3145, 2014.
BOSCH, F.; ROSICH, L. The contributions of Paul Ehrlich to pharmacology: a tribute onthe occasion of the centenary of his Nobel Prize. Pharmacology, v. 82, p. 171–179, 2008.
BOWERY, N. GABAB receptor pharmacology. Annual Review of Pharmacology andToxicology, v. 33, p. 109–147, 1993.
BOWERY, N. et al. International union of pharmacology. XXXIII. mammalianγ-aminobutyric acid B receptors: structure and function. Pharmacological Reviews, v. 54,p. 247–264, 2002.
BOWERY, N.; HILL, D.; HUDSON, A. Characteristics of GABAB receptor bindingsites on rat whole brain synaptic membranes. British Journal of Pharmacology, v. 78, p.191–206, 1983.
BOWERY, N. et al. (–) baclofen decreases neurotransmitter release in the mammalian cnsby an action at a novel GABA receptor. Nature, v. 283, p. 92, 1980.
BOWERY, N.; SMART, T. GABA and glycine as neurotransmitters: a brief history.British Journal of Pharmacology, v. 147, 2006.
BRAFF, D. et al. Prestimulus effects on human startle reflex in normals and schizophrenics.Psychophysiology, v. 15, p. 339–343, 1978.
BRINKMANN, L.; HEIFETS, E.; KANTOROVICH, L. Density functional calculationsof extended, periodic systems using coulomb corrected molecular fractionation withconjugated caps method (CC-MFCC). Physical Chemistry Chemical Physics, v. 16, p.21252–21270, 2014.
BRNDIAR, J.; STICH, I. van der waals interaction energies of small fragments of P,As, Sb, S, Se, and Te: comparison of complete basis set limit CCSD (T) and DFT withapproximate dispersion. Journal of Chemical Theory and Computation, v. 8, p. 2301–2309,2012.

Referências 89
BROOIJMANS, N.; KUNTZ, I. D. Molecular recognition and docking algorithms. AnnualReview of Biophysics and Biomolecular Structure, v. 32, p. 335–373, 2003.
BROWN, K. M. et al. Activation of the γ-aminobutyric acid type B (GABAB) receptorby agonists and positive allosteric modulators: Miniperspective. Journal of MedicinalChemistry, v. 58, p. 6336–6347, 2015.
BUCHBINDER, E. I.; DESAI, A. CTLA-4 and PD-1 pathways: similarities, differences,and implications of their inhibition. American Journal of Clinical Oncology, v. 39, p. 98,2016.
BURKE, K. Perspective on density functional theory. Journal of Chemical Physics, v. 136,p. 150901, 2012.
BURNET, S. F. M. et al. The clonal selection theory of acquired immunity. Nashville:Vanderbilt University Press, 1959.
BURNS, L. A. et al. Density-functional approaches to noncovalent interactions: acomparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM)theory, and specialized functionals. The Journal of Chemical Physics, v. 134, p. 084107,2011.
BUSCH, W. Aus der sitzung der medicinischen section vom 13 November 1867. BerlinerKlinische Wochenschrift, v. 5, p. 137, 1868.
CARVALHO, L. L. de et al. Molecular features related to HIV integrase inhibitionobtained from structure-and ligand-based approaches. PloS one, v. 9, p. e81301, 2014.
CAVALLI, A.; CARLONI, P.; RECANATINI, M. Target-related applications of firstprinciples quantum chemical methods in drug design. Chemical Reviews, v. 106, p.3497–3519, 2006.
CELADA, P.; BORTOLOZZI, A.; ARTIGAS, F. Serotonin 5-HT1A receptors as targetsfor agents to treat psychiatric disorders: rationale and current status of research. CNSDrugs, v. 27, p. 703–716, 2013.
CHALIFOUX, J. R.; CARTER, A. G. GABAB receptor modulation of voltage-sensitivecalcium channels in spines and dendrites. Journal of Neuroscience, v. 31, p. 4221–4232,2011.
CHALLIS, G. B.; STAM, H. J. The spontaneous regression of cancer: a review of casesfrom 1900 to 1987. Acta Oncologica, v. 29, p. 545–550, 1990.
CHANG, C. et al. The crystal structures of (S) and (R) baclofen and carbamazepine.Acta Crystallographica Section A, v. 37, 1981.
CHANG, C. et al. Structure and absolute configuration of (R)-baclofen monohydrochloride.Acta Crystallographica Section B, v. 38, p. 2065–2067, 1982.
CHAPLIN, D. D. Overview of the immune response. Journal of Allergy and ClinicalImmunology, v. 125, p. S3–S23, 2010.
CHEBIB, M.; JOHNSTON, G. A. The ‘ABC’ of GABA receptors: a brief review. Clinicaland Experimental Pharmacology and Physiology, v. 26, p. 937–940, 1999.

Referências 90
CHEN, V. B. et al. Molprobity: all-atom structure validation for macromolecularcrystallography. Acta Crystallographica Section D, v. 66, p. 12–21, 2010.
CHERTOK, B. et al. Drug delivery interfaces in the 21st century: from science fictionideas to viable technologies. Molecular Pharmaceutics, v. 10, p. 3531–3543, 2013.
CHO, A. E. et al. Quantum mechanical scoring for protein docking. The Journal ofChemical Physics, v. 131, p. 134108, 2009.
CHOI, D.-S. et al. The human serotonin 5-HT2B receptor: Pharmacological link between5-HT2 and 5-HT1D receptors. FEBS Letters, v. 352, p. 393–399, 1994.
CHOUDHARY, M. et al. Differential ergoline and ergopeptine binding to 5-hydroxytryptamine2a receptors: ergolines require an aromatic residue at position 340 forhigh affinity binding. Molecular Pharmacology, v. 47, p. 450–457, 1995.
CHUNG, L. W. et al. The ONIOM method and its applications. Chemical Reviews, v. 115,p. 5678–5796, 2015.
COHEN-TANNOUDJI, C.; DIU, B.; LALÖE, F. Quantum mechanics. New York: JohnWiley & Sons, 1977.
COLEY, W. B. The treatment of malignant tumors by repeated inoculations of erysipelas:with a report of ten original cases. 1. The American Journal of the Medical Sciences,v. 105, p. 487, 1893.
COLEY, W. B. The treatment of sarcoma with the mixed toxins of erysipelas and bacillusprodigiosus. The Boston Medical and Surgical Journal, v. 158, p. 175–182, 1908.
CÓRDOVA-SINTJAGO, T. et al. Human serotonin 5-HT2C G protein-coupled receptorhomology model from the β2 adrenoceptor structure: Ligand docking and mutagenesisstudies. International Journal of Quantum Chemistry, v. 112, p. 140–149, 2012.
CORRALES, L. et al. Innate immune signaling and regulation in cancer immunotherapy.Cell Research, v. 27, p. 96, 2017.
COSSI, M. et al. Energies, structures, and electronic properties of molecules in solutionwith the CPCM solvation model. Journal of Computational Chemistry, v. 24, p. 669–681,2003.
COUSIN, S.; SENESCHAL, J.; ITALIANO, A. Toxicity profiles of immunotherapy.Pharmacology & Therapeutics, v. 181, p. 91–100, 2017.
CRUNELLI, V.; EMRI, Z.; LERESCHE, N. Unravelling the brain targets ofγ-hydroxybutyric acid. Current Opinion in Pharmacology, v. 6, p. 44–52, 2006.
CURTIS, D. et al. GABA, bicuculline and central inhibition. Nature, v. 226, p. 1222, 1970.
DA COSTA, R. F. et al. Explaining statin inhibition effectiveness of HMG-CoA reductaseby quantum biochemistry computations. Physical Chemistry Chemical Physics, v. 14, p.1389–1398, 2012.
DAGGETT, V. Protein folding-simulation. Chemical Reviews, v. 106, p. 1898–1916, 2006.

Referências 91
DAHLKE, E. E.; TRUHLAR, D. G. Electrostatically embedded many-body expansionfor large systems, with applications to water clusters. Journal of Chemical Theory andComputation, v. 3, p. 46–53, 2007.
DANTAS, D. S. et al. Quantum molecular modelling of ibuprofen bound to human serumalbumin. RSC Advances, v. 5, p. 49439–49450, 2015.
DAVIS, A. M.; TEAGUE, S. J.; KLEYWEGT, G. J. Application and limitations of x-raycrystallographic data in structure-based ligand and drug design. Angewandte ChemieInternational Edition, v. 42, p. 2718–2736, 2003.
DEEV, V.; COLLINS, M. A. Approximate ab initio energies by systematic molecularfragmentation. The Journal of Chemical Physics, v. 122, p. 154102, 2005.
DIMASI, J. A.; HANSEN, R. W.; GRABOWSKI, H. G. The price of innovation: newestimates of drug development costs. Journal of Health Economics, v. 22, p. 151–185,2003.
DIRAC, P. A. M. The quantum theory of the electron. Proceedings of the Royal Society A:Mathematical, Physical and Engineering Sciences, v. 117, p. 610–624, 1928.
DIRAC, P. A. M. The principles of quantum mechanics. Oxford: Oxford University Press,1986.
DREWS, J. Drug discovery: A historical perspective. Science, v. 287, p. 1960–1964, 2000.
DUPUIS, D. S. et al. Point mutations in the transmembrane region of GABAB2
facilitate activation by the positive modulator n, n’-dicyclopentyl-2-methylsulfanyl-5-nitro-pyrimidine-4, 6-diamine (GS39783) in the absence of the GABAB1 subunit. MolecularPharmacology, v. 70, p. 2027–2036, 2006.
DURHAM, P.; PAPAPETROPOULOS, S. Biomarkers associated with migraine and theirpotential role in migraine management. Headache: The Journal of Head and Face Pain,v. 53, p. 1262–1277, 2013.
EDVINSSON, L.; VILLALÓN, C. M.; MAASSENVANDENBRINK, A. Basic mechanismsof migraine and its acute treatment. Pharmacology & Therapeutics, v. 136, p. 319–333,2012.
EHRLICH, P. Experimental researches on specific therapeutics. London: H. K. Lewis &Co., 1908.
ELCOCK, A. H. Molecular simulations of cotranslational protein folding: fragmentstabilities, folding cooperativity, and trapping in the ribosome. PLoS ComputationalBiology, v. 2, p. e98, 2006.
EMENS, L. A. et al. Cancer immunotherapy trials: leading a paradigm shift in drugdevelopment. Journal for Immunotherapy of Cancer, v. 4, p. 42, 2016.
ERSPAMER, V.; ASERO, B. Identification of enteramine, the specific hormone of theenterochromaffin cell system, as 5-hydroxytryptamine. Nature, v. 169, p. 800, 1952.
ERSPAMER, V.; BORETTI, G. Identification of enteramine and enteramine-relatedsubstances in extracts of posterior salivary glands ofoctopus vulgaris by paperchromatography. Experientia, v. 6, p. 348–349, 1950.

Referências 92
FANG, Y.; KENAKIN, T.; LIU, C. Orphan GPCRs as emerging drug targets. Frontiersin Pharmacology, v. 6, p. 295, 2015.
FEHLEISEN, F. Ueber die züchtung der erysipelkokken auf künstlichem nährboden undihre übertragbarkeit auf den menschen. Deutsche Medizinische Wochenschrift, v. 8, p.553–554, 1882.
FERMI, E. Un metodo statistico per la determinazione di alcune proprieta dell atomo.Atti della Accademia Nazionale dei Lincei, v. 6, p. 602–607, 1927.
FESSAS, P. et al. A molecular and preclinical comparison of the PD-1-targeted T-cellcheckpoint inhibitors nivolumab and pembrolizumab. Seminars in Oncology, v. 44, p.136–140, 2017.
FILHO, J. G. da S. et al. A comparative density functional theory study of electronicstructure and optical properties of γ-aminobutyric acid and its cocrystals with oxalic andbenzoic acid. Chemical Physics Letters, v. 587, p. 20–24, 2013.
FILIP, M.; FRANKOWSKA, M. GABAB receptors in drug addiction. PharmacologicalReports, v. 60, p. 755, 2008.
FINK, K. B.; GÖTHERT, M. 5-HT receptor regulation of neurotransmitter release.Pharmacological Reviews, v. 59, p. 360–417, 2007.
FLOREY, E.; MCLENNAN, H. The effects of factor i and of gamma-aminobutyric acidon smooth muscle preparations. The Journal of Physiology, v. 145, p. 66–76, 1959.
FOCK, V. Näherungsmethode zur lösung des quantenmechanischen mehrkörperproblems.Zeitschrift für Physik, v. 61, p. 126–148, 1930.
FOUDA, A.; RYDE, U. Does the DFT self-interaction error affect energies calculated inproteins with large QM systems? Journal of Chemical Theory and Computation, v. 12, p.5667–5679, 2016.
FRANCISCO, L. M.; SAGE, P. T.; SHARPE, A. H. The PD-1 pathway in tolerance andautoimmunity. Immunological Reviews, v. 236, p. 219–242, 2010.
FRANKLIN, R. E.; GOSLING, R. G. Molecular configuration in sodium thymonucleate.Nature, v. 171, p. 740–741, 1953.
FRAZÃO, N. F. et al. Four-level levodopa adsorption on C60 fullerene for transdermaland oral administration: a computational study. RSC Advances, v. 2, p. 8306–8322, 2012.
FRAZÃO, N. F. et al. Conformational, optoelectronic and vibrational properties ofthe entacapone molecule: A quantum chemistry study. Journal of Nanoscience andNanotechnology, v. 16, p. 4825–4834, 2016.
FRENKEL, D.; SMIT, B. Understanding molecular simulation: from algorithms toapplications. 2.ed. San Diego: Academic Press, v. 1, 2001.
FREYD, T. et al. Ligand-guided homology modelling of the GABAB2 subunit of theGABAB receptor. PloS one, v. 12, p. e0173889, 2017.

Referências 93
FRIESNER, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1.method and assessment of docking accuracy. Journal of Medicinal Chemistry, v. 47, p.1739–1749, 2004.
FRIESNER, R. A.; BEACHY, M. D. Quantum mechanical calculations on biologicalsystems. Current Opinion in Structural Biology, v. 8, p. 257–262, 1998.
FROESTL, W. An historical perspective on GABAergic drugs. Future MedicinalChemistry, v. 3, p. 163–175, 2011.
FROESTL, W. et al. SGS742: the first GABAB receptor antagonist in clinical trials.Biochemical Pharmacology, v. 68, p. 1479–1487, 2004.
FROESTL, W. et al. Phosphinic acid analogs of GABA. 1. new potent and selectiveGABAB agonists. Journal of Medicinal Chemistry, v. 38, p. 3297–3312, 1995.
FUKUZAWA, K. et al. Ab initio quantum mechanical study of the binding energies ofhuman estrogen receptor α with its ligands: an application of fragment molecular orbitalmethod. Journal of Computational Chemistry, v. 26, p. 1–10, 2005.
GÄHWILER, B.; BROWN, D. A. GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proceedings of the NationalAcademy of Sciences, v. 82, p. 1558–1562, 1985.
GALVEZ, T. et al. Allosteric interactions between GB1 and GB2 subunits are requiredfor optimal GABAB receptor function. The EMBO Journal, v. 20, p. 2152–2159, 2001.
GALVEZ, T. et al. Mutagenesis and modeling of the GABAB receptor extracellulardomain support a venus flytrap mechanism for ligand binding. Journal of BiologicalChemistry, v. 274, p. 13362–13369, 1999.
GALVEZ, T. et al. Mapping the agonist-binding site of GABAB type 1 subunit shedslight on the activation process of GABAB receptors. Journal of Biological Chemistry,v. 275, p. 41166–41174, 2000.
GAO, A. M. et al. An efficient linear scaling method for ab initio calculation of electrondensity of proteins. Chemical Physics Letters, v. 394, p. 293–297, 2004.
GASSMANN, M. et al. Redistribution of GABAB1 protein and atypical GABAB responsesin GABAB2-deficient mice. Journal of Neuroscience, v. 24, p. 6086–6097, 2004.
GEERLINGS, P.; PROFT, F. D.; LANGENAEKER, W. Conceptual density functionaltheory. Chemical Reviews, v. 103, p. 1793–1874, 2003.
GENG, Q. et al. PD-1/PD-L1 inhibitors for immuno-oncology: from antibodies to smallmolecules. Current Pharmaceutical Design, v. 23, p. 6033–6041, 2017.
GENG, Y. et al. Structural mechanism of ligand activation in human GABAB receptor.Nature, v. 504, p. 254, 2013.
GENG, Y. et al. Structure and functional interaction of the extracellular domain ofhuman GABAB receptor GBR2. Nature Neuroscience, v. 15, p. 970, 2012.
GENHEDEN, S.; RYDE, U. The MM/PBSA and MM/GBSA methods to estimateligand-binding affinities. Expert Opinion on Drug Discovery, v. 10, p. 449–461, 2015.

Referências 94
GERSHELL, L. J.; ATKINS, J. H. A brief history of novel drug discovery technologies.Nature Reviews Drug Discovery, v. 2, p. 321, 2003.
GERSHON, M. et al. 5-HT receptor subtypes outside the central nervous system. roles inthe physiology of the gut. Neuropsychopharmacology, v. 3, p. 385–395, 1990.
GESTWICKI, J. E. The interface of chemistry and biology is actually a continuum.Washington: ACS Publications, 2008.
GHIOTTO, M. et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interactionwith PD-1. International Immunology, v. 22, p. 651–660, 2010.
GILSON, M. K.; ZHOU, H.-X. Calculation of protein-ligand binding affinities. AnnualReview of Biophysics and Biomolecular Structure, v. 36, p. 21–42, 2007.
GOADSBY, P. J. Can we develop neurally acting drugs for the treatment of migraine?Nature Reviews Drug Discovery, v. 4, p. 741, 2005.
GOEDECKER, S. Linear scaling electronic structure methods. Reviews of ModernPhysics, v. 71, p. 1085, 1999.
GOODSELL, D. S.; MORRIS, G. M.; OLSON, A. J. Automated docking of flexibleligands: applications of AutoDock. Journal of Molecular Recognition, v. 9, p. 1–5, 1996.
GORDON, M. S. et al. Fragmentation methods: a route to accurate calculations on largesystems. Chemical Reviews, v. 112, p. 632–672, 2012.
GRÅNÄS, C.; NORDVALL, G.; LARHAMMAR, D. Mutagenesis of the human 5-HT1B
receptor: differences from the closely related 5-HT1A receptor and the role of residue F331in signal transduction. Journal of Receptors and Signal Transduction, v. 18, p. 225–241,1998.
GRÅNÄS, C.; NORDVALL, G.; LARHAMMAR, D. Site-directed mutagenesis of thehuman 5-HT1B receptor. European Journal of Pharmacology, v. 349, p. 367–375, 1998.
GREEN, J. P.; JOHNSON, C. L.; KANG, S. Application of quantum chemistry to drugsand their interactions. Annual Review of Pharmacology, v. 14, p. 319–342, 1974.
GRIMME, S. Semiempirical GGA-type density functional constructed with a long-rangedispersion correction. Journal of Computational Chemistry, v. 27, p. 1787–1799, 2006.
GUASTELLA, J. et al. Cloning and expression of a rat brain GABA transporter. Science,v. 249, p. 1303–1306, 1990.
GUETG, N. et al. The GABAB1a isoform mediates heterosynaptic depression athippocampal mossy fiber synapses. Journal of Neuroscience, v. 29, p. 1414–1423, 2009.
HAEFELY, W. et al. Possible involvement of GABA in the central actions ofbenzodiazepines. Advances in Biochemical Psychopharmacology, p. 131–151, 1975.
HALL, G. Atomic charges within molecules. Advances in Atomic and Molecular Physics,v. 20, p. 41–63, 1985.
HALL, G. Point charges and the molecular electrostatic potential. International Reviewsin Physical Chemistry, v. 5, p. 115–120, 1986.

Referências 95
HALL, G.; SMITH, C. Fitting electron densities of molecules. International Journal ofQuantum Chemistry, v. 25, p. 881–890, 1984.
HARTIG, P. R.; BRANCHEK, T. A.; WEINSHANK, R. L. A subfamily of 5-HT1D
receptor genes. Trends in Pharmacological Sciences, v. 13, p. 152–159, 1992.
HARTIG, P. R. et al. Alignment of receptor nomenclature with the human genome:classification of 5-HT1B and 5-HT1D receptor subtypes. Trends in PharmacologicalSciences, v. 17, p. 103–105, 1996.
HARTREE, D. R. The wave mechanics of an atom with a non-coulomb central field. parti. theory and methods. Mathematical Proceedings of the Cambridge Philosophical Society,v. 24, p. 89–110, 1928a.
HARTREE, D. R. The wave mechanics of an atom with a non-coulomb central field. partii. some results and discussion. Mathematical Proceedings of the Cambridge PhilosophicalSociety, v. 24, p. 111–132, 1928b.
HARTREE, D. R. The wave mechanics of an atom with a non-coulomb central field. partiv. further results relating to terms of the optical spectrum. Mathematical Proceedings ofthe Cambridge Philosophical Society, v. 25, p. 310–314, 1929.
HARTREE, D. R. The calculation of atomic structures. Reports on Progress in Physic,v. 113, p. 113–143, 1957.
HATFIELD, M. P. et al. Evaluation of methods to cap molecular fragments in calculatingenergies of interaction in avian pancreatic polypeptide. International Journal of QuantumChemistry, v. 108, p. 1017–1021, 2008.
HAUSER, A. S. et al. Trends in GPCR drug discovery: new agents, targets and indications.Nature Reviews Drug Discovery, v. 16, p. 829, 2017.
HE, X.; ZHANG, J. Z. A new method for direct calculation of total energy of protein.The Journal of Chemical Physics, v. 122, p. 031103, 2005.
HE, X.; ZHANG, J. Z. H. The generalized molecular fractionation with conjugatecaps/molecular mechanics method for direct calculation of protein energy. The Journal ofChemical Physics, v. 124, p. 184703, 2006.
HE, X. et al. Fragment quantum mechanical calculation of proteins and its applications.Accounts of Chemical Research, v. 47, p. 2748–2757, 2014.
HEBERT, H. The crystal structure and absolute configuration of (-)-dihydroergotaminemethanesulfonate monohydrate. Acta Crystallographica Section B, v. 35, p. 2978–2984,1979.
HEIFETZ, A. et al. The fragment molecular orbital method reveals new insight intothe chemical nature of GPCR–ligand interactions. Journal of Chemical Information andModeling, v. 56, p. 159–172, 2015.
HILL, D.; BOWERY, N. 3H-baclofen and 3H-GABA bind to bicuculline-insensitiveGABAB sites in rat brain. Nature, v. 290, p. 149, 1981.

Referências 96
HODI, F. S. et al. Improved survival with ipilimumab in patients with metastaticmelanoma. New England Journal of Medicine, v. 363, p. 711–723, 2010.
HOHENBERG, P.; KOHN, W. Inhomogeneous electron gas. Physical Review, v. 136,p. B864, 1964.
HONJO, T. Nobel Lecture - Serendipities of acquired immunity. 2018. Disponível em:<https://www.nobelprize.org/prizes/medicine/2018/honjo/lecture/>. Acesso em: 19 dez.2018.
HORITA, S. et al. High-resolution crystal structure of the therapeutic antibodypembrolizumab bound to the human PD-1. Scientific Reports, v. 6, p. 35297, 2016.
HORVATH, J. et al. Severe multivalvular heart disease: a new complication of the ergotderivative dopamine agonists. Movement Disorders, v. 19, p. 656–662, 2004.
HOYER, D.; HANNON, J. P.; MARTIN, G. R. Molecular, pharmacological and functionaldiversity of 5-HT receptors. Pharmacology Biochemistry and Behavior, v. 71, p. 533–554,2002.
HU, W. et al. Intercalative interaction of asymmetric copper (II) complex with DNA:experimental, molecular docking, molecular dynamics and TDDFT studies. Journal ofInorganic Biochemistry, v. 127, p. 90–98, 2013.
HÜLCKEL, E. Quantum-theoretical contributions to the benzene problem. i. the electronconfiguration of benzene and related compounds. Zeitschrift für Physik, v. 70, p. 204–286,1931.
HÜLCKEL, E. Quantum theoretical contributions to the problem of aromatic andnon-saturated compounds. The European Physical Journal A, v. 76, p. 628, 1932.
HUNTER, P. A quantum leap in biology: One inscrutable field helps another, as quantumphysics unravels consciousness. EMBO Reports, v. 7, p. 971–974, 2006.
ICHIKAWA, O. et al. G-protein/β-arrestin-linked fluctuating network of G-protein-coupledreceptors for predicting drug efficacy and bias using short-term molecular dynamicssimulation. PloS one, v. 11, p. e0155816, 2016.
IMMING, P.; SINNING, C.; MEYER, A. Drugs, their targets and the nature and numberof drug targets. Nature Reviews Drug Discovery, v. 5, p. 821, 2006.
IRETA, J.; NEUGEBAUER, J.; SCHEFFLER, M. On the accuracy of DFT for describinghydrogen bonds: dependence on the bond directionality. The Journal of PhysicalChemistry A, v. 108, p. 5692–5698, 2004.
ISHIDA, Y. et al. Induced expression of PD-1, a novel member of the immunoglobulingene superfamily, upon programmed cell death. The EMBO Journal, v. 11, p. 3887–3895,1992.
IWAI, Y. et al. Cancer immunotherapies targeting the PD-1 signaling pathway. Journalof Biomedical Science, v. 24, p. 26, 2017.
JAKOBSEN, S.; KRISTENSEN, K.; JENSEN, F. Electrostatic potential of insulin:exploring the limitations of density functional theory and force field methods. Journal ofChemical Theory and Computation, v. 9, p. 3978–3985, 2013.

Referências 97
JANEWAY, C. A. et al. Immunobiology: the immune system in health and disease. 5.ed.New York: Garland Science, 2005.
JASKOLSKI, M.; DAUTER, Z.; WLODAWER, A. A brief history of macromolecularcrystallography, illustrated by a family tree and its nobel fruits. The FEBS journal, v. 281,p. 3985–4009, 2014.
JENSEN, F. Introduction to computational chemistry. 3.ed. New York: John Wiley &Sons, 2017.
JIANG, N.; MA, J.; JIANG, Y. Electrostatic field-adapted molecular fractionation withconjugated caps for energy calculations of charged biomolecules. The Journal of ChemicalPhysics, v. 124, p. 114112, 2006.
JIN, X. et al. A systematic study on RNA NMR chemical shift calculation based on theautomated fragmentation QM/MM approach. RSC Advances, v. 6, p. 108590–108602,2016.
JING, Y.-Q.; HAN, K.-L. Quantum mechanical effect in protein–ligand interaction. ExpertOpinion on Drug Discovery, v. 5, p. 33–49, 2010.
JOHNSTON, G. et al. cis-and trans-4-aminocrotonic acid as GABA analogues ofrestricted conformation. Journal of Neurochemistry, v. 24, p. 157–160, 1975.
JOHNSTON, G. A. GABAC receptors: relatively simple transmitter-gated ion channels?Trends in Pharmacological Sciences, v. 17, p. 319–323, 1996.
JONES, K. A. et al. GABAB receptors function as a heteromeric assembly of the subunitsGABABR1 and GABABR2. Nature, v. 396, p. 674, 1998.
KAMP, M. W.; MULHOLLAND, A. J. Combined quantum mechanics/molecularmechanics (QM/MM) methods in computational enzymology. Biochemistry, v. 52, p.2708–2728, 2013.
KAMP, M. W. et al. Biomolecular simulation and modelling: status, progress andprospects. Journal of The Royal Society Interface, v. 5, p. 173–190, 2008.
KARABULUT, S. et al. A DFT-based QSAR study on inhibition of human dihydrofolatereductase. Journal of Molecular Graphics and Modelling, v. 70, p. 23–29, 2016.
KARAMAN, R.; FATTASH, B.; QTAIT, A. The future of prodrugs–design by quantummechanics methods. Expert Opinion on Drug Delivery, v. 10, p. 713–729, 2013.
KARMALI, A. M.; BLUNDELL, T. L.; FURNHAM, N. Model-building strategies forlow-resolution x-ray crystallographic data. Acta Crystallographica Section D, v. 65, p.121–127, 2009.
KAUPMANN, K. et al. Expression cloning of GABAB receptors uncovers similarity tometabotropic glutamate receptors. Nature, v. 386, p. 239, 1997.
KAUPMANN, K. et al. GABAB-receptor subtypes assemble into functional heteromericcomplexes. Nature, v. 396, p. 683, 1998.

Referências 98
KAUPMANN, K. et al. Human γ-aminobutyric acid type b receptors are differentiallyexpressed and regulate inwardly rectifying K+ channels. Proceedings of the NationalAcademy of Sciences, v. 95, p. 14991–14996, 1998.
KEE, E. A. et al. Aromatic interactions in the binding of ligands to HMG-CoA reductase.The Journal of Physical Chemistry B, v. 113, p. 14810–14815, 2009.
KEIR, M. E. et al. PD-1 and its ligands in tolerance and immunity. Annual Review ofImmunology, v. 26, p. 677–704, 2008.
KERR, D. I. et al. 2-hydroxy-saclofen: an improved antagonist at central and peripheralGABAB receptors. Neuroscience Letters, v. 92, p. 92–96, 1988.
KERR, D. I. et al. Phaclofen: a peripheral and central baclofen antagonist. BrainResearch, v. 405, p. 150–154, 1987.
KHANDELWAL, A. et al. A combination of docking, QM/MM methods, and MDsimulation for binding affinity estimation of metalloprotein ligands. Journal of MedicinalChemistry, v. 48, p. 5437–5447, 2005.
KHOJA, L. et al. Pembrolizumab. Journal for Immunotherapy of Cancer, v. 3, p. 36,2015.
KITAURA, K. et al. Fragment molecular orbital method: an approximate computationalmethod for large molecules. Chemical Physics Letters, v. 313, p. 701–706, 1999.
KLEBE, G. Toward a more efficient handling of conformational flexibility incomputer-assisted modelling of drug molecules. Perspectives in Drug Discovery andDesign, v. 3, p. 85–105, 1995.
KOBILKA, B. K.; DEUPI, X. Conformational complexity of G-protein-coupled receptors.Trends in Pharmacological Sciences, v. 28, p. 397–406, 2007.
KOCH, W.; HOLTHAUSEN, M. C. A chemist’s guide to density functional theory. NewYork: John Wiley & Sons, 2015.
KOHN, W. Nobel lecture: Electronic structure of matter—wave functions and densityfunctionals. Reviews of Modern Physics, v. 71, p. 1253, 1999.
KOHN, W.; SHAM, L. J. Self-consistent equations including exchange and correlationeffects. Physical Review, v. 140, p. A1133, 1965.
KONTOYIANNI, M.; MCCLELLAN, L. M.; SOKOL, G. S. Evaluation of dockingperformance: comparative data on docking algorithms. Journal of Medicinal Chemistry,v. 47, p. 558–565, 2004.
KOÓS, T.; TEPPER, J. M. Inhibitory control of neostriatal projection neurons bygabaergic interneurons. Nature Neuroscience, v. 2, p. 467, 1999.
KRNJEVIĆ, K.; SCHWARTZ, S. The action of γ-aminobutyric acid on cortical neurones.Experimental Brain Research, v. 3, p. 320–336, 1967.
KRUMMEL, M. F.; ALLISON, J. P. CD28 and CTLA-4 have opposing effects on theresponse of T cells to stimulation. Journal of Experimental Medicine, v. 182, p. 459–465,1995.

Referências 99
KUBINYI, H. Combinatorial and computational approaches in structure-based drugdesign. Current Opinion in Drug Discovery and Development, v. 1, p. 16–27, 1998.
KUBINYI, H. Drug research: myths, hype and reality. Nature Reviews Drug Discovery,v. 2, p. 665, 2003.
KUCEROVA, P.; CERVINKOVA, M. Spontaneous regression of tumour and the role ofmicrobial infection–possibilities for cancer treatment. Anti-Cancer Drugs, v. 27, p. 269,2016.
KULIK, H. J. et al. How large should the QM region be in QM/MM calculations? Thecase of catechol o-methyltransferase. The Journal of Physical Chemistry B, v. 120, p.11381–11394, 2016.
KUMAR, K. et al. Therapeutic potential of GABAB receptor ligands in drug addiction,anxiety, depression and other cns disorders. Pharmacology Biochemistry and Behavior,v. 110, p. 174–184, 2013.
KUMAR, M. S.; KUPPAST, I. A review on gamma-aminobutyric acid (GABA) and itsreceptors. International Journal of Pharma & Bio Sciences, v. 3, p. 60–69, 2012.
KUNISHIMA, N. et al. Structural basis of glutamate recognition by a dimericmetabotropic glutamate receptor. Nature, v. 407, p. 971, 2000.
LABRUIJERE, S. et al. Dihydroergotamine and sumatriptan in isolated human coronaryartery, middle meningeal artery and saphenous vein. Cephalalgia, v. 35, p. 182–189, 2015.
LANGLEY, J. N. Observations on the physiological action of extracts of the supra-renalbodies. The Journal of Physiology, v. 27, p. 237–256, 1901.
LARSON, C. et al. Going viral: a review of replication-selective oncolytic adenoviruses.Oncotarget, v. 6, p. 19976, 2015.
LASKOWSKI, R. A. et al. PROCHECK: a program to check the stereochemical qualityof protein structures. Journal of Applied Crystallography, v. 26, p. 283–291, 1993.
LATCHMAN, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation.Nature immunology, v. 2, p. 261, 2001.
LÁZÁR-MOLNÁR, E. et al. Structure-guided development of a high-affinity humanprogrammed cell death-1: Implications for tumor immunotherapy. EBioMedicine, v. 17, p.30–44, 2017.
LEACH, A. R.; LEACH, A. Molecular modelling: principles and applications. 2. ed. NewJersey: Prentice Hall, 2001.
LEACH, D. R.; KRUMMEL, M. F.; ALLISON, J. P. Enhancement of antitumor immunityby CTLA-4 blockade. Science, v. 271, p. 1734–1736, 1996.
LEE, J. Y. et al. Structural basis of checkpoint blockade by monoclonal antibodies incancer immunotherapy. Nature Communications, v. 7, p. 13354, 2016.
LEE, S.-M.; BOOE, J. M.; PIOSZAK, A. A. Structural insights into ligand recognitionand selectivity for classes A, B, and C GPCRs. European Journal of Pharmacology, v. 763,p. 196–205, 2015.

Referências 100
LI, S.; LI, W.; FANG, T. An efficient fragment-based approach for predicting theground-state energies and structures of large molecules. Journal of the American ChemicalSociety, v. 127, p. 7215–7226, 2005.
LI, W.; LI, S.; JIANG, Y. Generalized energy-based fragmentation approach for computingthe ground-state energies and properties of large molecules. The Journal of PhysicalChemistry A, v. 111, p. 2193–2199, 2007.
LI, X. et al. Evaluation of the performance of four molecular docking programs on adiverse set of protein-ligand complexes. Journal of Computational Chemistry, v. 31, p.2109–2125, 2010.
LIMA NETO, J. X. et al. Energetic description of cilengitide bound to integrin. NewJournal of Chemistry, v. 41, p. 11405–11412, 2017.
LIMA NETO, J. X. et al. A quantum biochemistry investigation of willardiine partialagonism in AMPA receptors. Physical Chemistry Chemical Physics, v. 17, p. 13092–13103,2015.
LIN, S. L. et al. Differential coupling of 5-HT1 receptors to g proteins of the Gi family.British Journal of Pharmacology, v. 136, p. 1072–1078, 2002.
LINDSLEY, C. W. The top prescription drugs of 2012 globally: Biologics dominate, butsmall molecule cns drugs hold on to top spots. ACS Chemical Neuroscience, v. 4, p.905–907, 2013.
LIU, J.; HE, X.; ZHANG, J. Z. Structure of liquid water–a dynamical mixture oftetrahedral and ring-and-chain like structures. Physical Chemistry Chemical Physics,v. 19, p. 11931–11936, 2017.
LIU, J. et al. Molecular determinants involved in the allosteric control of agonist affinityin the GABAB receptor by the GABAB2 subunit. Journal of Biological Chemistry, v. 279,p. 15824–15830, 2004.
LIU, J. et al. Calculation of protein–ligand binding affinities based on a fragment quantummechanical method. RSC Advances, v. 5, p. 107020–107030, 2015.
LIU, W. et al. Serial femtosecond crystallography of G protein–coupled receptors. Science,v. 342, p. 1521–1524, 2013.
LOMBARDINO, J. G.; III, J. A. L. A guide to drug discovery: the role of the medicinalchemist in drug discovery—then and now. Nature Reviews Drug Discovery, v. 3, p. 853,2004.
LONSDALE, R.; RANAGHAN, K. E.; MULHOLLAND, A. J. Computational enzymology.Chemical Communications, v. 46, p. 2354–2372, 2010.
LUMMIS, S. C. 5-HT3 receptors. Journal of Biological Chemistry, v. 287, p. 40239–40245,2012.
MA, P.; ZEMMEL, R. Value of novelty? Nature Reviews Drug Discovery, v. 1, p. 1571–572,2002.

Referências 101
MACHIDA, T.; IIZUKA, K.; HIRAFUJI, M. 5-hydroxytryptamine and its receptors insystemic vascular walls. Biological and Pharmaceutical Bulletin, v. 36, p. 1416–1419, 2013.
MANTEGANI, S.; BRAMBILLA, E.; VARASI, M. Ergoline derivatives: receptor affinityand selectivity. Il Farmaco, v. 54, p. 288–296, 1999.
MANZONI, V. et al. Comparison of polarizable continuum model and quantummechanics/molecular mechanics solute electronic polarization: Study of the optical andmagnetic properties of diazines in water. The Journal of Chemical Physics, v. 135, p.144103, 2011.
MANZONI, V. et al. Study of the optical and magnetic properties of pyrimidine in watercombining PCM and QM/MM methodologies. Physical Chemistry Chemical Physics,v. 12, p. 14023–14033, 2010.
MARKRAM, H. et al. Interneurons of the neocortical inhibitory system. Nature ReviewsNeuroscience, v. 5, p. 793, 2004.
MARTINS, A. C. V. et al. An ab initio explanation of the activation and antagonismstrength of an AMPA-sensitive glutamate receptor. RSC Advances, v. 3, p. 14988–14992,2013.
MARTINS, A. C. V. et al. An improved quantum biochemistry description of theglutamate–GluA2 receptor binding within an inhomogeneous dielectric function framework.New Journal of Chemistry, v. 41, p. 6167–6179, 2017.
MARTINS, A. C. V. et al. Modeling of laccase inhibition by formetanate pesticide usingtheoretical approaches. Bioelectrochemistry, v. 108, p. 46–53, 2016.
MASERAS, F.; MOROKUMA, K. IMOMM: A new integrated ab initio + molecularmechanics geometry optimization scheme of equilibrium structures and transition states.Journal of Computational Chemistry, v. 16, p. 1170–1179, 1995.
MCCONNELL, J. D. et al. The long-term effect of doxazosin, finasteride, and combinationtherapy on the clinical progression of benign prostatic hyperplasia. New England Journalof Medicine, v. 349, p. 2387–2398, 2003.
MCCORVY, J. D.; ROTH, B. L. Structure and function of serotonin G protein-coupledreceptors. Pharmacology & Therapeutics, v. 150, p. 129–142, 2015.
MEDZHITOV, R.; JR, C. A. J. Innate immunity: impact on the adaptive immuneresponse. Current Opinion in Immunology, v. 9, p. 4–9, 1997.
MEI, Y.; JI, C.; ZHANG, J. Z. A new quantum method for electrostatic solvation energyof protein. The Journal of Chemical Physics, v. 125, p. 094906, 2006.
MELIS, C.; LUMMIS, S. C.; MOLTENI, C. Molecular dynamics simulations of GABAbinding to the GABAC receptor: the role of Arg104. Biophysical Journal, v. 95, p.4115–4123, 2008.
MENG, X.-Y. et al. Molecular docking: a powerful approach for structure-based drugdiscovery. Current Computer-Aided Drug Design, v. 7, p. 146–157, 2011.

Referências 102
MESTRES, J.; ROHRER, D. C.; MAGGIORA, G. M. MIMIC: A molecular-fieldmatching program. exploiting applicability of molecular similarity approaches. Journal ofComputational Chemistry, v. 18, p. 934–954, 1997.
MICHOT, J. M. et al. Immune-related adverse events with immune checkpoint blockade:a comprehensive review. European Journal of Cancer, v. 54, p. 139–148, 2016.
MILLIGAN, G. G protein-coupled receptor hetero-dimerization: contribution topharmacology and function. British Journal of Pharmacology, v. 158, p. 5–14, 2009.
MINTZ, I. M.; BEAN, B. P. GABAB receptor inhibition of P-type Ca2+ channels incentral neurons. Neuron, v. 10, p. 889–898, 1993.
MOITESSIER, N. et al. Combining pharmacophore search, automated docking, andmolecular dynamics simulations as a novel strategy for flexible docking. proof of concept:docking of arginine- glycine- aspartic acid-like compounds into the αvβ3 binding site.Journal of Medicinal Chemistry, v. 47, p. 4178–4187, 2004.
MOMANY, F. A.; RONE, R. Validation of the general purpose quanta R© 3.2/charmm R©
force field. Journal of Computational Chemistry, v. 13, p. 888–900, 1992.
MORALES, A.; EIDINGER, D.; BRUCE, A. Intracavitary bacillus calmette-guerin in thetreatment of superficial bladder tumors. The Journal of Urology, v. 116, p. 180–182, 1976.
MORRISSETTE, D. A.; STAHL, S. M. Modulating the serotonin system in the treatmentof major depressive disorder. CNS Spectrums, v. 19, p. 54–68, 2014.
MOSIER, P. D.; KELLOGG, G. E. Molecular modeling: Considerations for the designof pharmaceuticals and biopharmaceuticals. In: Biopharmaceutical Drug Design andDevelopment. New York: Humana Press, 2008. p. 267–291.
MOTA, K. B. et al. A quantum biochemistry model of the interaction between theestrogen receptor and the two antagonists used in breast cancer treatment. Computationaland Theoretical Chemistry, v. 1089, p. 21–27, 2016.
NA, Z. et al. Structural basis for blocking PD-1-mediated immune suppression bytherapeutic antibody pembrolizumab. Cell Research, v. 27, p. 147, 2016.
NAKANO, T. et al. Fragment molecular orbital method: use of approximate electrostaticpotential. Chemical Physics Letters, v. 351, p. 475–480, 2002.
NAVARI, R. M. Olanzapine for the prevention and treatment of chronic nausea andchemotherapy-induced nausea and vomiting. European Journal of Pharmacology, v. 722, p.180–186, 2014.
NEWMAN, D. J.; CRAGG, G. M.; SNADER, K. M. The influence of natural productsupon drug discovery. Natural Product Reports, v. 17, p. 215–234, 2000.
NG, R. Drugs: from discovery to approval. 3. ed. New York: John Wiley & Sons, 2015.
NI, W. et al. The existence of a local 5-hydroxytryptaminergic system in peripheralarteries. British Journal of Pharmacology, v. 154, p. 663–674, 2008.
NICHOLS, D. E.; NICHOLS, C. D. Serotonin receptors. Chemical Reviews, v. 108, p.1614–1641, 2008.

Referências 103
NILSSON, T. et al. Contractile 5-HT1B receptors in human cerebral arteries:pharmacological characterization and localization with immunocytochemistry. BritishJournal of Pharmacology, v. 128, p. 1133–1140, 1999.
NISHIMURA, H. et al. Development of lupus-like autoimmune diseases by disruption ofthe PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity, v. 11, p.141–151, 1999.
NISHINO, M. et al. Monitoring immune-checkpoint blockade: response evaluation andbiomarker development. Nature Reviews Clinical Oncology, v. 14, p. 655, 2017.
NOMURA, R. et al. Direct detection of the interaction between recombinant solubleextracellular regions in the heterodimeric metabotropic γ-aminobutyric acid receptor.Journal of Biological Chemistry, v. 283, p. 4665–4673, 2008.
NOWAK, M. et al. Homology modeling of the serotonin 5-HT1A receptor using automateddocking of bioactive compounds with defined geometry. Journal of Medicinal Chemistry,v. 49, p. 205–214, 2006.
NUTT, D. J.; MALIZIA, A. L. New insights into the role of the GABAA–benzodiazepinereceptor in psychiatric disorder. The British Journal of Psychiatry, v. 179, p. 390–396,2001.
NYPORKO, A. Y. et al. 3D reconstruction of a full-size GABAB receptor. Neurophysiology,v. 47, p. 364–375, 2015.
O’DONNELL, J. S. et al. Resistance to PD-1/PD-L1 checkpoint inhibition. CancerTreatment Reviews, v. 52, p. 71–81, 2017.
OISETH, S. J.; AZIZ, M. S. Cancer immunotherapy: a brief review of the history,possibilities, and challenges ahead. Journal of Cancer Metastasis and Treatment, v. 3,p. 251, 2017.
OKADA, O. et al. Molecular dynamics simulations for glutamate-binding and cleft-closingprocesses of the ligand-binding domain of GluR2. Biophysical Chemistry, v. 162, p. 35–44,2012.
OLD, L. J.; CLARKE, D. A.; BENACERRAF, B. Effect of bacillus calmette-guerininfection on transplanted tumours in the mouse. Nature, v. 184, p. 291, 1959.
OLSEN, J. M. H. et al. Accuracy of protein embedding potentials: An analysis in terms ofelectrostatic potentials. Journal of Chemical Theory and Computation, v. 11, p. 1832–1842,2015.
OLSEN, R. W.; TOBIN, A. J. Molecular biology of GABAA receptors. The FASEBJournal, v. 4, p. 1469–1480, 1990.
ORRY, A. J.; ABAGYAN, R. A.; CAVASOTTO, C. N. Structure-based development oftarget-specific compound libraries. Drug Discovery Today, v. 11, p. 261–266, 2006.
ORTMANN, F.; SCHMIDT, W. G.; BECHSTEDT, F. Attracted by long-range electroncorrelation: adenine on graphite. Physical Review Letters, v. 95, p. 186101, 2005.

Referências 104
OU-YANG, S.-s. et al. Computational drug discovery. Acta Pharmacologica Sinica, v. 33,p. 1131, 2012.
OUADID, H. et al. Serotonin increases calcium current in human atrial myocytes via thenewly described 5-hydroxytryptamine–4 receptors. Molecular Pharmacology, v. 41, p.346–351, 1992.
OURIQUE, G. S. et al. A quantum chemistry investigation of a potential inhibitory drugagainst the dengue virus. RSC Advances, v. 6, p. 56562–56570, 2016.
PACE, C. N. et al. Contribution of hydrophobic interactions to protein stability. Journalof Molecular Biology, v. 408, p. 514–528, 2011.
PARASCANDOLA, J. The theoretical basis of Paul Ehrlich’s chemotherapy. Journal ofthe History of Medicine and Allied Sciences, v. 36, p. 19–43, 1981.
PAREDES, R. G.; ÅGMO, A. GABA and behavior: the role of receptor subtypes.Neuroscience & Biobehavioral Reviews, v. 16, p. 145–170, 1992.
PARISH, C. R. Cancer immunotherapy: the past, the present and the future. Immunologyand Cell Biology, v. 81, p. 106–113, 2003.
PATIL, R. et al. Optimized hydrophobic interactions and hydrogen bonding at thetarget-ligand interface leads the pathways of drug-designing. PloS one, v. 5, p. e12029,2010.
PAULING, L.; COREY, R. B. A proposed structure for the nucleic acids. Proceedings ofthe National Academy of Sciences, v. 39, p. 84–97, 1953.
PAUWELS, P. J. 5-HT1B/D receptor antagonists. General Pharmacology: The VascularSystem, v. 29, p. 293–303, 1997.
PERDEW, J. P.; BURKE, K.; ERNZERHOF, M. Generalized gradient approximationmade simple. Physical Review Letters, v. 77, p. 3865, 1996.
PÉREZ-GARCI, E. et al. The GABAB1b isoform mediates long-lasting inhibition ofdendritic ca 2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron, v. 50, p.603–616, 2006.
PHILLIPS, J. C. et al. Scalable molecular dynamics with NAMD. Journal ofComputational Chemistry, v. 26, p. 1781–1802, 2005.
PIETROBON, D.; STRIESSNIG, J. Neurological diseases: Neurobiology of migraine.Nature Reviews Neuroscience, v. 4, p. 386, 2003.
PLATANIA, C. B. M. et al. Homology modeling of dopamine D2 and D3 receptors:molecular dynamics refinement and docking evaluation. PloS one, v. 7, p. e44316, 2012.
PLEWCZYNSKI, D. et al. Can we trust docking results? evaluation of seven commonlyused programs on PDBbind database. Journal of Computational Chemistry, v. 32, p.742–755, 2011.
PROSSER, H. M. et al. Epileptogenesis and enhanced prepulse inhibition inGABAB1-deficient mice. Molecular and Cellular Neuroscience, v. 17, p. 1059–1070, 2001.

Referências 105
PROVORSE, M. R. et al. Convergence of excitation energies in mixed quantum andclassical solvent: Comparison of continuum and point charge models. The Journal ofPhysical Chemistry B, v. 120, p. 12148–12159, 2016.
PULLMAN, A.; PULLMAN, B. Electronic structure and carcinogenic activity of aromaticmolecules new developments. Advances in Cancer Research, v. 3, p. 117–169, 1955.
QUÉVA, C. et al. Effects of GABA agonists on body temperature regulation in GABAB1
-/- mice. British Journal of Pharmacology, v. 140, p. 315–322, 2003.
RAGHAVACHARI, K.; SAHA, A. Accurate composite and fragment-based quantumchemical models for large molecules. Chemical Reviews, v. 115, p. 5643–5677, 2015.
RAHA, K. et al. The role of quantum mechanics in structure-based drug design. DrugDiscovery Today, v. 12, p. 725–731, 2007.
RAO, B. K. et al. Receptor–ligand interaction at 5-HT3 serotonin receptors: a clusterapproach. The Journal of Physical Chemistry A, v. 118, p. 8471–8476, 2014.
RAPAPORT, D. C. The art of molecular dynamics simulation. 2.ed. Cambridge:Cambridge University Press, 2004.
RAPPORT, M. M.; GREEN, A. A.; PAGE, I. H. Crystalline serotonin. Science, v. 108, p.329–330, 1948.
RAPPORT, M. M.; GREEN, A. A.; PAGE, I. H. Partial purification of the vasoconstrictorin beef serum. Journal of Biological Chemistry, v. 174, p. 735–741, 1948.
RIBEIRO, T. C. da S. et al. The quantum biophysics of the isoniazid adduct nadh bindingto its InhA reductase target. New Journal of Chemistry, v. 38, p. 2946–2957, 2014.
RIBEIRO, T. C. da S.; LYRA, M. L.; MANZONI, V. The electrostatic embeddingcontribution to DFT calculations of ligand-amino acid residues interaction. Journal ofMolecular Modeling, v. 24, p. 211, 2018.
ROBBINS, M. J. et al. GABAB2 is essential for g-protein coupling of the GABAB receptorheterodimer. Journal of Neuroscience, v. 21, p. 8043–8052, 2001.
ROBERT, C. et al. Ipilimumab plus dacarbazine for previously untreated metastaticmelanoma. New England Journal of Medicine, v. 364, p. 2517–2526, 2011.
RODRIGUES, C. R. F. et al. Quantum biochemistry study of the T3-785 tropocollagentriple-helical structure. Chemical Physics Letters, v. 559, p. 88–93, 2013.
RODRÍGUEZ, D. et al. Structure-based discovery of selective serotonin 5-HT1B receptorligands. Structure, v. 22, p. 1140–1151, 2014.
RODRÍGUEZ, D. et al. Molecular docking screening using agonist-bound GPCRstructures: probing the A2A adenosine receptor. Journal of Chemical Information andModeling, v. 55, p. 550–563, 2015a.
RODRÍGUEZ, D.; RANGANATHAN, A.; CARLSSON, J. Discovery of GPCR ligands bymolecular docking screening: Novel opportunities provided by crystal structures. CurrentTopics in Medicinal Chemistry, v. 15, p. 2484–2503, 2015.

Referências 106
ROOTHAAN, C. C. J. New developments in molecular orbital theory. Reviews of ModernPhysics, v. 23, p. 69, 1951.
ROSAS, M. B. R. et al. Activation of 5-hydroxytryptamine1B/1D/1F receptors as amechanism of action of antimigraine drugs. Expert Opinion on Pharmacotherapy, v. 14, p.1599–1610, 2013.
ROTH, B. et al. Aortic recognition sites for serotonin (5HT) are coupled to phospholipasec and modulate phosphatidylinositol turnover. Neuropharmacology, v. 23, p. 1223–1225,1984.
ROTH, B. L. et al. Binding of typical and atypical antipsychotic agents to5-hydroxytryptamine–6 and 5-hydroxytryptamine–7 receptors. Journal of Pharmacologyand Experimental Therapeutics, v. 268, p. 1403–1410, 1994.
ROTH, B. L. et al. Drugs and valvular heart disease. The New England Journal ofMedicine, v. 356, p. 6–9, 2007.
ROTH, B. L. et al. 5-hydroxytryptamine2-family receptors (5-hydroxytryptamine2a,5-hydroxytryptamine2b, 5-hydroxytryptamine2c): where structure meets function.Pharmacology & Therapeutics, v. 79, p. 231–257, 1998.
ROTHMAN, R. B. et al. Evidence for possible involvement of 5-HT2B receptors in thecardiac valvulopathy associated with fenfluramine and other serotonergic medications.Circulation, v. 102, p. 2836–2841, 2000.
ROWLEY, N. M. et al. Glutamate and GABA synthesis, release, transport andmetabolism as targets for seizure control. Neurochemistry International, v. 61, p. 546–558,2012.
RYDE, U.; SÖDERHJELM, P. Ligand-binding affinity estimates supported byquantum-mechanical methods. Chemical Reviews, v. 116, p. 5520–5566, 2016.
SAKABA, T.; NEHER, E. Direct modulation of synaptic vesicle priming by GABAB
receptor activation at a glutamatergic synapse. Nature, v. 424, p. 775, 2003.
SALI, A.; KURIYAN, J. Challenges at the frontiers of structural biology. Trends inBiochemical Sciences, v. 24, p. M20–M24, 1999.
SÁNCHEZ, R. et al. Protein structure modeling for structural genomics. Nature Structuraland Molecular Biology, v. 7, p. 986, 2000.
SANDERS, A. R. et al. Genetic diversity of the human serotonin receptor 1B (HTR1B)gene. Genomics, v. 72, p. 1–14, 2001.
SARI, Y. Serotonin1b receptors: from protein to physiological function and behavior.Neuroscience & Biobehavioral Reviews, v. 28, p. 565–582, 2004.
SCHOENEN, J. Migraine and serotonin: The quest for the holy grail goes on. Cephalalgia,v. 34, p. 163–164, 2014.
SCHWENK, J. et al. Modular composition and dynamics of native GABAB receptorsidentified by high-resolution proteomics. Nature Neuroscience, v. 19, p. 233, 2016.

Referências 107
SEEMAN, P.; NIZNIK, H. B. Dopamine receptors and transporters in parkinson’s diseaseand schizophrenia. The FASEB Journal, v. 4, p. 2737–2744, 1990.
SENN, H. M.; THIEL, W. QM/MM methods for biomolecular systems. AngewandteChemie International Edition, v. 48, p. 1198–1229, 2009.
SERTKAYA, A. et al. Examination of clinical trial costs and barriers for drug development.Report, US Department of Health and Human Services, Office of the Assistant Secretaryfor Planning and Evaluation, Washington, DC, p. 1–92, 2014.
SETOLA, V. et al. Molecular determinants for the interaction of the valvulopathicanorexigen norfenfluramine with the 5-HT2B receptor. Molecular Pharmacology, v. 68, p.20–33, 2005.
SHANK, R. P.; CAMPBELL, G. L. Glutamine and alpha-ketoglutarate uptake andmetabolism by nerve terminal enriched material from mouse cerebellum. NeurochemicalResearch, v. 7, p. 601–616, 1982.
SHARMA, P.; ALLISON, J. P. The future of immune checkpoint therapy. Science, v. 348,p. 56–61, 2015.
SILBERSTEIN, S. D.; MCCRORY, D. C. Ergotamine and dihydroergotamine: history,pharmacology, and efficacy. Headache: The Journal of Head and Face Pain, v. 43, p.144–166, 2003.
SLATER, J. C. The theory of complex spectra. Physical Review, v. 34, p. 1293, 1929.
SLATER, J. C. Quantum theory of molecules and solids. New York: McGraw-Hill, 1965.
SØNDERGAARD, C. R. et al. Improved treatment of ligands and coupling effects inempirical calculation and rationalization of pKa values. Journal of Chemical Theory andComputation, v. 7, p. 2284–2295, 2011.
SOUSA, S. F.; FERNANDES, P. A.; RAMOS, M. J. Protein–ligand docking: currentstatus and future challenges. Proteins: Structure, Function, and Bioinformatics, v. 65, p.15–26, 2006.
SPOEL, D. V. D. et al. GROMACS: fast, flexible, and free. Journal of ComputationalChemistry, v. 26, p. 1701–1718, 2005.
SQUIRE, I. B.; BARNETT, D. B. The rational use of β-adrenoceptor blockers in thetreatment of heart failure. the changing face of an old therapy. British Journal of ClinicalPharmacology, v. 49, p. 1–9, 2000.
SRIWILAIJAROEN, N. et al. A novel potent and highly specific inhibitor againstinfluenza viral N1–N9 neuraminidases: Insight into neuraminidase–inhibitor interactions.Journal of Medicinal Chemistry, v. 59, p. 4563–4577, 2016.
STERNBACH, L. H. The benzodiazepine story. Journal of Medicinal Chemistry, v. 22,p. 1–7, 1979.
STERNBERG, S. H. et al. DNA interrogation by the CRISPR RNA-guided endonucleaseCas9. Nature, v. 507, p. 62, 2014.

Referências 108
STIERAND, K.; RAREY, M. Drawing the PDB: protein–ligand complexes in twodimensions. ACS Medicinal Chemistry Letters, v. 1, p. 540–545, 2010.
STRAIN, M. C.; SCUSERIA, G. E.; FRISCH, M. J. Achieving linear scaling for theelectronic quantum coulomb problem. Science, v. 271, p. 51–53, 1996.
STROSCIO, M. A.; DUTTA, M. Biological nanostructures and applications ofnanostructures in biology: electrical, mechanical, and optical properties. New York: Kluwer,v. 2, 2004.
SUÁREZ, D.; SUÁREZ, E.; DÍAZ, N. Molecular dynamics and quantum mechanicalcalculations on the mononuclear zinc-β-lactamase from bacillus cereus: Protonation stateof the active site and imipenem binding. Journal of Molecular Structure: THEOCHEM,v. 912, p. 105–112, 2009.
SVÄRD, M.; RASMUSON, Å. C. Force fields and point charges for crystal structuremodeling. Industrial & Engineering Chemistry Research, v. 48, p. 2899–2912, 2009.
TAFT, C. A.; SILVA, V. B. D. et al. Current topics in computer-aided drug design.Journal of Pharmaceutical Sciences, v. 97, p. 1089–1098, 2008.
TAN, S. et al. An unexpected n-terminal loop in PD-1 dominates binding by nivolumab.Nature Communications, v. 8, p. 14369, 2017.
TANG, H.; QIAO, J.; FU, Y.-X. Immunotherapy and tumor microenvironment. CancerLetters, v. 370, p. 85–90, 2016.
TANIYAMA, K. et al. Activation of protein kinase C suppresses the γ-aminobutyric acidB receptor-mediated inhibition of the vesicular release of noradrenaline and acetylcholine.Journal of Neurochemistry, v. 58, p. 1239–1245, 1992.
TAVARES, A. B. M. et al. Inhibition of the checkpoint protein PD-1 by the therapeuticantibody pembrolizumab outlined by quantum chemistry. Scientific Reports, v. 8, p. 1840,2018.
THOMAS, D. R. 5-HT5A receptors as a therapeutic target. Pharmacology & Therapeutics,v. 111, p. 707–714, 2006.
THOMAS, G. Medicinal chemistry: an introduction. 2.ed. New Jersey: John Wiley &Sons, 2013.
THOMAS, L. The calculation of atomic fields. Mathematical Proceedings of the CambridgePhilosophical Society, v. 23, p. 542–548, 1927.
THOMPSON, S. M.; GÄHWILER, B. Comparison of the actions of baclofen at pre-andpostsynaptic receptors in the rat hippocampus in vitro. The Journal of Physiology, v. 451,p. 329–345, 1992.
TKATCHENKO, A.; SCHEFFLER, M. Accurate molecular van der Waals interactionsfrom ground-state electron density and free-atom reference data. Physical Review Letters,v. 102, p. 073005, 2009.
TOPALIAN, S. L. et al. Safety, activity, and immune correlates of anti–PD-1 antibody incancer. New England Journal of Medicine, v. 366, p. 2443–2454, 2012.

Referências 109
TOPALIAN, S. L. et al. Survival, durable tumor remission, and long-term safety inpatients with advanced melanoma receiving nivolumab. Journal of Clinical Oncology,v. 32, p. 1020, 2014.
TRIGGLE, D. J. Drug discovery and delivery in the 21st century. Medical Principles andPractice, v. 16, p. 1–14, 2007.
TVAROŠKA, I. QM/MM methods for studying enzymatic reactions of glycosyltransferases.In: Glycoinformatics. New York: Humana Press, 2015. p. 489–499.
TWAROG, B. M.; PAGE, I. H. Serotonin content of some mammalian tissues and urineand a method for its determination. American Journal of Physiology-Legacy Content,v. 175, p. 157–161, 1953.
ULLMER, C. et al. Expression of serotonin receptor mRNAs in blood vessels. FEBSLetters, v. 370, p. 215–221, 1995.
URWYLER, S. Allosteric modulation of family C G-protein-coupled receptors: frommolecular insights to therapeutic perspectives. Pharmacological Reviews, v. 63, p. 59–126,2011.
VALENTIN, M. D. et al. Evidence for water-mediated triplet–triplet energy transfer inthe photoprotective site of the peridinin–chlorophyll a–protein. Biochimica et BiophysicaActa (BBA)-Bioenergetics, v. 1837, p. 85–97, 2014.
VENKATAKRISHNAN, A. et al. Molecular signatures of G-protein-coupled receptors.Nature, v. 494, p. 185, 2013.
VERDONK, M. L. et al. Improved protein–ligand docking using GOLD. Proteins:Structure, Function, and Bioinformatics, v. 52, p. 609–623, 2003.
VIGOT, R. et al. Differential compartmentalization and distinct functions of GABAB
receptor variants. Neuron, v. 50, p. 589–601, 2006.
WACKER, D. et al. Structural features for functional selectivity at serotonin receptors.Science, v. 340, p. 615–619, 2013.
WALDMEIER, P. C.; KAUPMANN, K.; URWYLER, S. Roles of GABAB receptorsubtypes in presynaptic auto- and heteroreceptor function regulating GABA andglutamate release. Journal of Neural Transmission, v. 115, p. 1401–1411, 2008.
WANG, C. et al. Structural basis for molecular recognition at serotonin receptors. Science,v. 340, p. 610–614, 2013a.
WANG, X. et al. Electrostatically embedded generalized molecular fractionation withconjugate caps method for full quantum mechanical calculation of protein energy. TheJournal of Physical Chemistry A, v. 117, p. 7149–7161, 2013b.
WANG, Y.; LI, Y.; SHI, G. The regulating function of heterotrimeric G proteins in theimmune system. Archivum Immunologiae et Therapiae Experimentalis, v. 61, p. 309–319,2013c.
WARSHEL, A. et al. Electrostatic basis for enzyme catalysis. Chemical Reviews, v. 106, p.3210–3235, 2006.

Referências 110
WATSON, J. D.; CRICK, F. H. Molecular structure of nucleic acids. Nature, v. 171, p.737–738, 1953.
WEBER, H.-P.; CRAVEN, B.; MCMULLAN, R. The neutron structure of and thermalmotion in γ-aminobutyric acid (GABA) at 122 k. Acta Crystallographica Section B, v. 39,p. 360–366, 1983.
WHEATLEY, M. et al. Lifting the lid on GPCRs: the role of extracellular loops. BritishJournal of Pharmacology, v. 165, p. 1688–1703, 2012.
WHITE, J. H. et al. Heterodimerization is required for the formation of a functionalGABAB receptor. Nature, v. 396, p. 679, 1998.
WILKINS, M. H. F.; STOKES, A. R.; WILSON, H. R. Molecular structure of nucleicacids: molecular structure of deoxypentose nucleic acids. Nature, v. 171, p. 738, 1953.
WISE, A.; GEARING, K.; REES, S. Target validation of G-protein coupled receptors.Drug Discovery Today, v. 7, p. 235–246, 2002.
WU, E. L. et al. Quantum and molecular dynamics study for binding of macrocyclicinhibitors to human α-thrombin. Biophysical Journal, v. 92, p. 4244–4253, 2007.
YAMADA, J. et al. GABAB receptor-mediated presynaptic inhibition of glutamatergicand gabaergic transmission in the basolateral amygdala. Neuropharmacology, v. 38, p.1743–1753, 1999.
YAO, J. et al. Novel enoyl-ACP reductase (FabI) potential inhibitors of escherichia colifrom chinese medicine monomers. Bioorganic & Medicinal Chemistry Letters, v. 20, p.56–59, 2010.
ZAK, K. M. et al. Structural biology of the immune checkpoint receptor PD-1 and itsligands PD-L1/PD-L2. Structure, v. 25, p. 1163–1174, 2017.
ZAK, K. M. et al. Structure of the complex of human programmed death 1, PD-1, and itsligand PD-L1. Structure, v. 23, p. 2341–2348, 2015.
ZANATTA, G. et al. Quantum biochemistry description of the human dopamine D3receptor in complex with the selective antagonist eticlopride. Journal of Proteomics andBioinformatics, v. 5, p. 155–162, 2012.
ZANATTA, G. et al. Antipsychotic haloperidol binding to the human dopamine D3receptor: Beyond docking through QM/MM refinement toward the design of improvedschizophrenia medicines. ACS Chemical Neuroscience, v. 5, p. 1041–1054, 2014.
ZANATTA, G. et al. Two binding geometries for risperidone in dopamine D3 receptors:Insights on the fast-off mechanism through docking, quantum biochemistry, and moleculardynamics simulations. ACS Chemical Neuroscience, v. 7, p. 1331–1347, 2016.
ZHANG, D. Quantum mechanical calculation of nanomaterial-ligand interaction energiesby molecular fractionation with conjugated caps method. Scientific Reports, v. 7, p. 44645,2017.
ZHANG, D. W.; CHEN, X.; ZHANG, J. Z. Molecular caps for full quantum mechanicalcomputation of peptide–water interaction energy. Journal of Computational Chemistry,v. 24, p. 1846–1852, 2003a.

Referências 111
ZHANG, D. W. et al. Quantum mechanical map for protein-ligand binding withapplication to β-trypsin/benzamidine complex. The Journal of Chemical Physics, v. 120,p. 1145–1148, 2004.
ZHANG, D. W.; ZHANG, J. Molecular fractionation with conjugate caps for full quantummechanical calculation of protein–molecule interaction energy. The Journal of ChemicalPhysics, v. 119, p. 3599–3605, 2003.
ZHANG, J. et al. Circulating PD-L1 in NSCLC patients and the correlation betweenthe level of PD-L1 expression and the clinical characteristics. Thoracic Cancer, v. 6, p.534–538, 2015.
ZHANG, X. et al. Structural and functional analysis of the costimulatory receptorprogrammed death-1. Immunity, v. 20, p. 337–347, 2004.
ZHANG, X.; STEVENS, R. C.; XU, F. The importance of ligands for G protein-coupledreceptor stability. Trends in Biochemical Sciences, v. 40, p. 79–87, 2015.
ZHAO, Y.; TRUHLAR, D. G. A new local density functional for main-groupthermochemistry, transition metal bonding, thermochemical kinetics, and noncovalentinteractions. Journal of Chemical Physics, v. 125, p. 194101–194118, 2006.
ZHAO, Y.; TRUHLAR, D. G. The M06 suite of density functionals for main groupthermochemistry, thermochemical kinetics, noncovalent interactions, excited states,and transition elements: two new functionals and systematic testing of four M06-classfunctionals and 12 other functionals. Theoretical Chemistry Accounts, v. 120, p. 215–241,2008.
ZHOU, T.; HUANG, D.; CAFLISCH, A. Quantum mechanical methods for drug design.Current Topics in Medicinal Chemistry, v. 10, p. 33–45, 2010.

Apêndices

113
apêndice a
ARTIGOS PUBLICADOS/SUBMETIDOS DURANTE O PERÍODO DEDOUTORAMENTO
É apresentada a seguir uma lista de todos os artigos publicados, submetidos ouescritos pelo autor durante os anos em que esteve matriculado como aluno de doutorado(2015-atual).
1. D. S. Dantas, J. I. N. Oliveira, J. X. Lima Neto, R. F. da Costa, E. M. Bezerra,V. N. Freire, E. W. S. Caetano, U. L. Fulco, E. L. Albuquerque. Quantum molecularmodelling of ibuprofen bound to human serum albumin. RSC Advances, v. 5, p.49439-49450, 2015.
2. J. X Lima Neto, U. L. Fulco, E. L. Albuquerque, G. Corso, E. M. Bezerra, E.W. S. Caetano, R. F. da Costa, V. N Freire. A quantum biochemistry investigationof willardiine partial agonism in AMPA receptors. Physical Chemistry ChemicalPhysics, v. 17, p. 13092-13103, 2015. 1
3. M. L. de Almeida, J. I. N. Oliveira, J. X. Lima Neto, C. E. M. Gomes, U. L. Fulco,E. L. Albuquerque, V. N. Freire, E. W. S. Caetano, F. A. B. F. de Moura, M. L.Lyra. Electronic transport in methylated fragments of DNA. Applied Physics Letters,v. 107, p. 203701, 2015.
4. G. S. Ourique, J. F. Vianna, J. X. Lima Neto, J. I. N. Oliveira, P. W. Mauriz, M.S. Vasconcelos, E. W. S. Caetano, V. N. Freire, E. L. Albuquerque, U. L. Fulco. Aquantum chemistry investigation of a potential inhibitory drug against the denguevirus. RSC Advances, v. 6, p. 56562-56570, 2016.
5. A. J. B. Santos, J. X. Lima Neto, D. H. G. Vieira, L. Dutra-Neto, C. Bellini, N. S.Albuquerque, G. Corso, B. Lobão-Soares. Individual Nest Site Selection in HawksbillTurtles Within and Between Nesting Seasons. Chelonian Conservation and Biology,v. 15, p. 109-114, 2016. 2
6. K. B. Mota, J. X. Lima Neto, A. H. Lima Costa, J. I. N. Oliveira, K. S. Bezerra, E.L. Albuquerque, E. W. S. Caetano, V. N. Freire, U. L. Fulco. A quantum biochemistrymodel of the interaction between the estrogen receptor and the two antagonists usedin breast cancer treatment. Computational and Theoretical Chemistry, v. 1089, p.21-27, 2016.
7. J. X. Lima Neto, K. S. Bezerra, D. N. Manso, K. B. Mota, J. I. N. Oliveira, E. L.Albuquerque, E. W. S. Caetano, V. N. Freire, U. L. Fulco. Energetic description ofcilengitide bound to integrin. New Journal of Chemistry, v. 41, p. 11405-11412, 2017.
8. K. S. Bezerra, J. I. N. Oliveira, J. X. Lima Neto, E. L. Albuquerque, E. W. S.Caetano, V. N. Freire, U. L. Fulco. Quantum binding energy features of the T3-785collagen-like triple-helical peptide. RSC Advances, v. 7, p. 2817-2828, 2017.
1 Trabalho utilizado para obtenção do título de mestre em Ciências Biológicas2 Trabalho realizado durante a iniciação científica sob supervisão do Prof. Gilberto Corso

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 114
9. A. B. M. L. A. Tavares, J. X. Lima Neto, U. L. Fulco, E. L. Albuquerque. Inhibitionof the checkpoint protein PD-1 by the therapeutic antibody pembrolizumab outlinedby quantum chemistry. Scientific Reports, v. 8, p. 1840, 2018.
10. J. X. Lima Neto, V. P. Soares-Rachetti, E. L. Albuquerque, V. Manzoni andU. L. Fulco. Outlining migrainous through dihydroergotamine-serotonin receptorinteractions using quantum biochemistry. New Journal of Chemistry, v. 42, p. 2401-2412, 2018.
11. K. S. Bezerra, J. X. Lima Neto, J. I. N. Oliveira, E. L. Albuquerque, E. W. S.Caetano, V. N. Freire and U. L. Fulco. Computational investigation of the α2β1integrin-collagen triple helix complex interaction. New Journal of Chemistry, v. 42,p. 17115-17125, 2018.
12. A. H. Lima Costa, W. S. Clemente, Jr, K. S. Bezerra, J. X. Lima Neto, E. L.Albuquerque and U. L. Fulco. Computational biochemical investigation of the bindingenergy interactions between an estrogen receptor and its agonists. New Journal ofChemistry, v. 42, p. 19801-19810, 2018.
13. J. X. Lima Neto, A. B. M. L. A. Tavares, U. L. Fulco and E. L. Albuquerque.A Quantum Biochemistry Approach to Investigate Checkpoint Inhibitor Drugs forCancer. New Journal of Chemistry (submetido).
14. J. X. Lima Neto, J. I. N. Oliveira, V. Manzoni, V. P. Soares-Rachetti, E. L. Albu-querque and U. L. Fulco. Exploring GABAB receptor binding site for agonists andantagonists through in silico simulations. ACS Chemical Neuroscience (manuscritoem preparação).
15. J. X. Lima Neto, V. Manzoni, E. L. Albuquerque and U. L. Fulco. Assessing theaccuracy of density functional theory to calculate the protein-ligand binding affinity.Journal of Computational Chemistry (manuscrito em preparação).

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 115
Quantum molecular modelling of ibuprofen boundto human serum albumin†
Diego S. Dantas,a Jonas I. N. Oliveira,a Jose X. Lima Neto,a Roner F. da Costa,b
Eveline M. Bezerra,a Valder N. Freire,c Ewerton W. S. Caetano,d Umberto L. Fulco*a
and Eudenilson L. Albuquerquea
The binding of the nonsteroidal anti-inflammatory drug ibuprofen (IBU) to human serum albumin (HSA) is
investigated using density functional theory (DFT) calculations within a fragmentation strategy.
Crystallographic data for the IBU–HSA supramolecular complex shows that the ligand is confined to a
large cavity at the subdomain IIIA and at the interface between the subdomains IIA and IIB, whose
binding sites are FA3/FA4 and FA6, respectively. The interaction energy between the IBU molecule and
each amino acid residue of these HSA binding pockets was calculated using the Molecular Fractionation
with Conjugate Caps (MFCC) approach employing a dispersion corrected exchange–correlation
functional. Our investigation shows that the total interaction energy of IBU bound to HSA at binding sites
of the fatty acids FA3/FA4 (FA6) converges only for a pocket radius of at least 8.5 A, mainly due to the
action of residues Arg410, Lys414 and Ser489 (Lys351, Ser480 and Leu481) and residues in non-
hydrophobic domains, namely Ile388, Phe395, Phe403, Leu407, Leu430, Val433, and Leu453 (Phe206,
Ala210, Ala213, and Leu327), which is unusual. Our simulations are valuable for a better understanding of
the binding mechanism of IBU to albumin and can lead to the rational design and the development of
novel IBU-derived drugs with improved potency.
1 Introduction
Ibuprofen (IBU) is a well-known nonsteroidal anti-inammatory
agent (NSAIA) widely used and tolerated by patients.1 According
to the World Health Organization (WHO), it is an essential
medicine for a basic health care system when used for acute
pain, fever and palliative care due to its low cost, safety and
efficacy.2 The analgesic and anti-inammatory effects are linked
to one another and are related to non-selective inhibition of
cyclooxygenase-2. In comparison with other NSAIAs, IBU is
relatively low risk for gastrointestinal and hepato-renal events,
and has little prospect of developing renal and cardiovascular
events.3
Regarding its structural features, shown in Fig. 1a, ibuprofen
is a weak acid that is slightly soluble in water and with a exible
chemical structure that can be divided into three regions: the
acidic side chain (region i), the central aryl moiety (region ii),
and the hydrophobic terminal (region iii). Fig. 1b depicts its
electron density projected onto an electrostatic potential iso-
surface. Furthermore, its anti-inammatory activity is usually
associated with the (S)-(+)-isomer and the propionic side chain
due to its lipophilicity and deprotonation energy.4
Once in the bloodstream, around 99% of IBU becomes
strongly bound to human serum albumin (HSA),5 which is a
globular protein abundant in blood plasma (600 M), with a
molecular weight of approximately 66 kDa and a capacity to
bind a large amount of chemical compounds.6,7 Transcribing
from the albumin gene (ALB), located at position q11–22 of
chromosome 4, this single non-glycosylated polypeptide chain
of 585 amino acid residues is formed by three homologous
helical domains (I, II and III), which are divided into six sub-
domains (IA, IB, IIA, IIB, IIIA and IIIB) connected by random
and extensive coils without beta-sheet elements (see Fig. 2).6,8,9
IBU tends to accumulate in appreciable quantities at inamed
compartments where anti-inammatory/analgesic activity
(synovial uids, cerebrospinal uid) is needed.3
A crystallographic study of a HSA–hemin–myristate
complex10 and ve HSA–fatty acid complexes, formed using
saturated medium-chain and long-chain fatty acids,11 revealed
nine distinct binding locations in HSA. Site 1 (fatty acid FA1) is
located in a D-shaped cavity at the center of the four-helix of
subdomain IB. Site 2 (fatty acid FA2) is located between the
subdomains IA and IIA. Sites 3 and 4 (fatty acids FA3 and FA4)
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande do
Norte, 59072-970, Natal, RN, Brazil. E-mail: [email protected] de Fısica, Universidade Federal Rural do Semi-Arido, Caraubas,
59780-000, RN, BrazilcDepartamento de Fısica, Universidade Federal do Ceara, 60455-760, Fortaleza, CE,
BrazildInstituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, 60040-531, Fortaleza,
CE, Brazil
† This document is a collaborative effort of all authors.
Cite this: RSC Adv., 2015, 5, 49439
Received 12th March 2015Accepted 21st May 2015
DOI: 10.1039/c5ra04395f
www.rsc.org/advances
This journal is © The Royal Society of Chemistry 2015 RSC Adv., 2015, 5, 49439–49450 | 49439
RSC Advances
PAPER
Publi
shed
on 0
2 J
une
2015.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
02
/06
/20
15
20
:14
:48
.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 116
13092 | Phys. Chem. Chem. Phys., 2015, 17, 13092--13103 This journal is© the Owner Societies 2015
Cite this:Phys.Chem.Chem.Phys.,
2015, 17, 13092
A quantum biochemistry investigation ofwillardiine partial agonism in AMPA receptors†
Jose X. Lima Neto,a Umberto L. Fulco,*a Eudenilson L. Albuquerque,a
Gilberto Corso,a Eveline M. Bezerra,a Ewerton W. S. Caetano,b Roner F. da Costac
and Valder N. Freired
We employ quantum biochemistry methods based on the Density Functional Theory (DFT) approach to
unveil the detailed binding energy features of willardiines co-crystallized with the AMPA receptor. Our
computational results demonstrate that the total binding energies of fluorine–willardiine (FW), hydrogen–
willardiine (HW), bromine–willardiine (BrW) and iodine–willardiine (IW) to the iGluR2 ligand-pocket
correlate with the agonist binding energies, whose experimental sequential data match our computational
counterpart, excluding the HW case. We find that the main contributions to the total willardiine–iGluR2
binding energy are due to the amino acid residues in decreasing order Glu705 4 Arg485 4 Ser654 4
Tyr450 4 T655. Furthermore, Met708, which is positioned close to the 5-substituent, attracts HW and FW,
but repels BrW and IW. Our results contribute significantly to an improved understanding of the
willardiine–iGluR2 binding mechanisms.
1 Introduction
Disorders in the central nervous system (CNS) affect approximately
1 billion people around the world, leading to more hospitalizations
than any other group of diseases.1 Autism, schizophrenia,
Alzheimer’s and Parkinson’s diseases and epilepsy2–6 have been
ascribed to impairments in the function of ionotropic glutamate
receptors (iGluRs), which are ligand-gated ion channels that undergo
structural changes after activation, culminating with the channel
opening and generating an ion flux through the membrane.7 The
three major subclasses of iGluRs can be differentiated according to
their amino acid sequence and pharmacology as: kainate (GluK1–5),
N-methyl-D-aspartic acid (NMDA; GluN1–3), and a-amino-3-hydroxy-
5-methyl-4-isoxazolepropionic acid (AMPA; GluR1–4),8 being related
to physiological processes such as memory and learning.9
AMPA receptors play a key role in fast synaptic transmission,10
being well distributed throughout the CNS.11 They have a tetrameric
structure organized as a dimer of dimers,12 where each monomer is
composed of an extracellular amino-terminal region (ATD; with
approximately 400 amino acids), a ligand binding domain
(LBD; with approximately 258 amino acids) and a transmembrane
region (TMD; with approximately 169 amino acids),13 whose
molecular structures are depicted in Fig. 1, in which each colour
corresponds to a single chain and the circles in the LBD region
represent the position of the ligand binding site in every subunit.
The iGluR-LBD is formed by polypeptide segments (or lobes) S1
(390–506 amino acids) and S2 (632–775 amino acids),14 which can
be genetically combined and expressed as a soluble protein.15,16
Its structural representation is shown in Fig. 2, which depicts the
monomeric structure of the Ligand Binding Domain (LBD)
coupled to a willardiine molecule (PDB ID: 1MQH). The letters
N and C represent the amino and carboxyl terminal regions,
respectively, that are linked to the Amino Terminal Domain (ATD)
and the Transmembrane Domain (TMD) in the monomer. The
circle marks the region of the protein investigated in this work.
The amino acids in this region are selected by increasing the
binding region radius (r), see Table 1.
Crystallographic and electrophysiological studies have advanced
significantly the structural and functional characterization of
AMPA.17 Isolated structures of the LBD complexed with agonists,
partial agonists and antagonists suggest a correlation between
the degree of lobe closure and channel activation.18–22 When
bound to a full agonist, the lobes were closed by approximately
19.1 to 21.3 degrees, leading to channel activation with high
efficacy levels.14,15,18 In addition, antagonists have shown 2.5 to
9.6 degrees of lobe closure,14 blocking receptor activation. Finally,
partial agonists induce an intermediate closure of the LBD (13.1 to
19 degrees) and the opening of the ion channel.
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande
do Norte, 59072-970, Natal-RN, Brazil. E-mail: [email protected];
Fax: +55-84-32153791; Tel: +55-84-32153793b Instituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, 60040-531,
Fortaleza-CE, BrazilcDepartamento de Fısica, Universidade Federal Rural do Semi-Arido,
59780-000 Caraubas-RN, BrazildDepartamento de Fısica, Universidade Federal do Ceara, 60455-760, Fortaleza-CE,
Brazil
† Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05630b
Received 3rd December 2014,Accepted 13th April 2015
DOI: 10.1039/c4cp05630b
www.rsc.org/pccp
PCCP
PAPER
Publi
shed
on 1
3 A
pri
l 2015.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
06
/05
/20
15
14
:38
:25
.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 117
Electronic transport in methylated fragments of DNA
M. L. de Almeida,1 J. I. N. Oliveira,1 J. X. Lima Neto,1 C. E. M. Gomes,1 U. L. Fulco,1,a)
E. L. Albuquerque,1 V. N. Freire,2 E. W. S. Caetano,3 F. A. B. F. de Moura,4 and M. L. Lyra41Departamento de Biof�ısica e Farmacologia, Universidade Federal do Rio Grande do Norte,59072-970 Natal-RN, Brazil2Departamento de F�ısica, Universidade Federal do Cear�a, 60455-760 Fortaleza, CE, Brazil3Instituto Federal de Educac~ao, Ciencia e Tecnologia do Cear�a, 60040-531 Fortaleza, CE, Brazil4Instituto de F�ısica, Universidade Federal de Alagoas, 57072-900 Macei�o-AL, Brazil
(Received 18 September 2015; accepted 6 November 2015; published online 17 November 2015)
We investigate the electronic transport properties of methylated deoxyribonucleic-acid (DNA)
strands, a biological system in which methyl groups are added to DNA (a major epigenetic modifica-
tion in gene expression), sandwiched between two metallic platinum electrodes. Our theoretical simu-
lations apply an effective Hamiltonian based on a tight-binding model to obtain current-voltage
curves related to the non-methylated/methylated DNA strands. The results suggest potential applica-
tions in the development of novel biosensors for molecular diagnostics.VC 2015 AIP Publishing LLC.
[http://dx.doi.org/10.1063/1.4936099]
Recently, there has been significant interest in the char-
acterization of electronic processes in biomolecules, with a
consequent impact in the development of medical technolo-
gies and therapies.1–4 For instance, investigations of the
compatibility of the interface between electronic devices and
biological systems are nowadays much encouraged, since the
quality of bio-interfaces can play a central role in the pro-
spective success and impact of technologies such as thin film
electronics, in vitro cell culture models, and medical devices
that make use of organic materials in place of conventional
semiconductors.5,6
Natural semiconductors can serve as templates for the
design of synthetic electronic components employing biolog-
ically derived nanomaterials, such as quantum dots and
nanotubes built up from important classes of biomolecules
such as proteins and nucleic acids.7 Their main advantages
are their lower current and power operation, leading to
cheaper and simpler devices, while their main disadvantage
is their low life-time due to degradation.8–10
Among these biological systems, the deoxyribonucleic-
acid (DNA) molecule prevails, not only because it carries the
genetic instruction used for the functioning and reproduction
of all cells in living organisms, but also due to its diversity,
versatility, and amenability as part of a nanoelectronic de-
vice (for a recent review, see Ref. 11).
A gene is a functional unit of heredity, being formed
from DNA segments that represent instructions for protein
synthesis in a living organism. After the Human Genome
Project, we have a nearly complete list of the genes needed
to produce a human being.12 However, gene expression is a
very complex process, relying on a secondary system used
by cells to determine when and where a particular gene in-
formation will be effectively used during development. This
system is overlaid on DNA in the form of epigenetic mecha-
nisms, which refer to heritable and functionally relevant
changes in gene activity, but that do not modify DNA.13
Among them, DNA methylation plays an important role,
being a process by which methyl groups (CH3) are added to
the fifth carbon atom of the cytosine base or the sixth nitro-
gen atom of the adenine base. It is a well-characterized epi-
genetic signaling tool, typically acting to suppress gene
transcription.
Usually, genes are silenced when methylation occurs in
their promoter region, where the transcription process is initi-
ated; however, when the methylation occurs in the gene
body, they may have a positive correlation to their expression
(transcription/translation) process.14 For example, the 5-meth-
ylcytosine-based DNA methylation occurs through the cova-
lent addition of a methyl group at the 5-carbon of the cytosine
heterocyclic aromatic ring by an enzyme called DNA methyl-
transferase resulting in the formation of 5-methylcytosine.15
This is a typical vertebrate DNA methylation pattern, which
occurs predominantly in CpG islands, dinucleotide-rich
regions that possess high relative densities of CpG and are
positioned at the 50 ends of many human genes. Most cell
types display relatively stable DNA methylation patterns, with
70%–80% of all CpGs being methylated.15 This event is asso-
ciated with a number of key processes including embryonic
development, chromosome stability, genomic imprinting, X
chromosome inactivation, suppression of repetitive elements,
and carcinogenesis.16,17
It is the aim of this work to suggest a nanoelectronic
device able to investigate the electron transport properties of
a DNA strand taking into account the contributions of the
5-methylcytosine. Our main purpose is to observe how meth-
ylation of cytosine in a DNA strand modifies its charge trans-
port profiles in comparison to a non-methylated DNA strand.
Both molecules are set in a linear geometry covalently linked
to two platinum electrodes, the source—S and drain—D con-
tacts (see Fig. 1).
We model the non-methylated/methylated DNA strands
adopting a tight-binding Hamiltonian constructed from ab
initio parameters with a single orbital per site, nearest-
neighbor interactions, and neglecting any environment or
complex contacts. Besides, the effects of interatomic matrix
a)Author to whom correspondence should be addressed. Electronic mail:
[email protected]. Tel.: þ-55-84-3215-3419.
0003-6951/2015/107(20)/203701/4/$30.00 VC 2015 AIP Publishing LLC107, 203701-1
APPLIED PHYSICS LETTERS 107, 203701 (2015)
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
189.70.240.105 On: Tue, 17 Nov 2015 16:42:01

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 118
A quantum chemistry investigation of a potentialinhibitory drug against the dengue virus†‡
G. S. Ourique,a J. F. Vianna,a J. X. Lima Neto,a J. I. N. Oliveira,a P. W. Mauriz,b
M. S. Vasconcelos,c E. W. S. Caetano,d V. N. Freire,e E. L. Albuquerquea
and U. L. Fulco*a
We present an in silico study of the interaction energy between NS2B–NS3, a serine protease of the dengue
virus (DENV), and the inhibitor benzoyl-norleucine-Lys-Arg-Arg-aldehyde (Bz-nKRR-H), a crucial step in
the design and development of dengue's antiviral drugs, using quantum chemistry calculations based on
the density functional theory (DFT) at the generalized gradient approximation (GGA). The interaction
energies between the inhibitor Bz-nKRR-H and each amino acid belonging to the binding site was
calculated through the molecular fragmentation with conjugate caps (MFCC) approach employing
a dispersion corrected exchange-correlation functional. Besides the interaction energy, we also
calculated the distances, types of molecular interactions, and the atomic groups involved in the process.
Our results show that the interaction energy of the system reached convergence at 15.0 A, with the
central residues identified in this interaction radius, as well as their attraction/repulsion energies, all of
them being important inputs to improve the effectiveness of antiviral drugs to avoid the dissemination of
the dengue virus.
Introduction
Dengue virus (DENV) belongs to the family Flaviviridae, genus
Flavivirus, which also includes several human pathogens such
as the West Nile virus (WNV), Japanese Encephalitis Virus (JEV),
Yellow Fever Virus (YFV), and Zika Virus (ZIKV), among others.1
It is transmitted by mosquitoes of the species Aedes aegypti and,
less frequently, Aedes albopictus. According to the World Health
Organization (WHO), the incidence of dengue in the world
increased 30 times in the last ve decades.2 Some factors
contributed to this dramatic growing worldwide, such as the
population growth, unplanned urbanization, global warming,
lack of efficient mosquito control, increased air travel and
insufficient public health care facilities. As a result, currently
the disease is present in more than 125 countries, covering
a large amount of the world population.3 A recent estimate
indicates globally 390 million dengue infections per year, of
which 96 million (25% of all cases) manifest clinically with
different degrees of severity of the disease, including 500 000
dengue hemorrhagic fever cases resulting in 25 000 deaths,
mostly children.4 These data clearly point to the urgent need of
a comprehensive understanding of the DENV pathogenesis,
which could lead to the discovery of new control strategies.
Currently, there are four distinct serotypes (DENV-1 to 4)
consolidated by the scientic community and subclassied into
several different phylogenetic clusters or genotypes.5 Recently it
was announced the discovery of a h serotype, DENV-5, found
only in specic regions like the Malaysian and Indonesian
jungles, usually following the sylvatic cycle and not the human
one.6 It is known that the rst four serotypes are genetically
similar and share approximately 65% of their genomes.7
Despite this, although infection with one serotype confers life-
long immunity to that serotype, it does yield only short-term
protection against a secondary infection with a heterologous
serotype. Subsequent infection with a different type increases
the risk of severe complications like the life-threatening dengue
hemorrhagic fever, a rare case characterized by high fever,
damage to lymph and blood vessels, bleeding from the nose and
gums, enlargement of the liver, and failure of the circulatory
system. The symptoms may progress to the dengue shock
syndrome (DSS) with massive bleeding, shock, and ultimately
death, a phenomenon oen attributed to the antibody-
dependent enhancement (ADE).8,9
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande do
Norte, 59072-970, Natal, RN, Brazil. E-mail: [email protected]; Fax: +55-84-
32153791; Tel: +55-84-32153793bDepartamento de Fısica, Instituto Federal de Educaçao, Ciencia e Tecnologia do
Maranhao, 65030-005, Sao Luıs, MA, BrazilcEscola de Ciencia e Tecnologia, Universidade Federal do Rio Grande do Norte, 59072-
970, Natal, RN, BrazildInstituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, 60040-531, Fortaleza,
CE, BrazileDepartamento de Fısica, Universidade Federal do Ceara, 60455-760, Fortaleza, CE,
Brazil
† This document is a collaborative effort of all authors.
‡ Electronic supplementary information (ESI) available. See DOI:
10.1039/c6ra10121f
Cite this: RSC Adv., 2016, 6, 56562
Received 19th April 2016
Accepted 6th June 2016
DOI: 10.1039/c6ra10121f
www.rsc.org/advances
56562 | RSC Adv., 2016, 6, 56562–56570 This journal is © The Royal Society of Chemistry 2016
RSC Advances
PAPER
Publi
shed
on 0
7 J
une
2016.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
13
/06
/20
16
15
:59
:22
.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 119
Chelonian Conservation and Biology, 2016, 15(1): 109–114
� 2016 Chelonian Research Foundation
Individual Nest Site Selection in Hawksbill Turtles Within and Between Nesting Seasons
ARMANDO JOSE BARSANTE SANTOS1,*, JOSE XAVIER LIMA NETO
2,
DANIEL HENRIQUE GIL VIEIRA1, LOURIVAL DUTRA NETO
1, CLAUDIO BELLINI3,
NATALIA DE SOUZA ALBUQUERQUE4, GILBERTO CORSO
2, AND BRUNO LOBAO SOARES2
1Fundacao Pro-Tamar, Fernando de Noronha, Brazil [[email protected]; [email protected]; [email protected]];2Universidade Federal do Rio Grande do Norte, Centro de Biociencias, Departamento de Biofısica e Farmacologia, Natal, Brazil
[[email protected]; [email protected]; [email protected]];3Centro Tamar-Instituto Chico Mendes de Conservacao da Biodiversidade, Parnamirim, Brazil [[email protected]];
4Universidade de Sao Paulo, Instituto de Psicologia, Departamento de Psicologia Experimental, Sao Paulo, Brazil
*Corresponding author
ABSTRACT. – We analyzed 410 nest locations from 150 individual nesting hawksbill turtles
(Eretmochelys imbricata) on the northeastern Brazilian coast during 8 nesting seasons from 2006
to 2014 to evaluate individual nesting preferences. We determined the consistency of nest site
choice within and between nesting seasons for open sand and vegetation nest microhabitats and
also for nest site distances from the current waterline, highest spring tide, vegetation line, and
position along the beach. We found that behavioral consistency within seasons was more robust
than between seasons. This suggests that a decrease in the consistency of nest site choice may be
related to progressive landscape changes in the nesting environment, driving behavioral flexibility
in nesting preferences.
KEY WORDS. – Reptilia; Testudines; Eretmochelys imbricata; hawksbill sea turtle; individual
nesting preferences; nest site choice; behavioral flexibility; preference; sea turtle
Although hawksbill turtles (Eretmochelys imbricata)
spend most of their lifetimes in the sea, they need sandy
beaches to successfully incubate their eggs. The absence of
parental care in this species further makes nest site
selection an important mechanism to maximize fitness
(Resetarits 1996), but choosing an appropriate site is not
without challenges. For example, nests located close to the
sea can provide easy access for hatchlings to reach the
water; however, this location increases the probability of
loss by flood or erosion (Marigatoulis 2005). On the other
hand, nests located farther inland are safer from inundation
but are subject to desiccation, hatchling disorientation,
penetration of roots, and predation (Wood and Bjorndal
2000). Moreover, different microhabitats within the
nesting beach influence the incubation temperature (Kamel
and Mrosovsky 2006a) and therefore hatchling sex ratios
in the nest (Robins 2003).
The study of individual preferences for particular nest
sites (Kamel and Mrosovsky 2004, 2005, 2006b) may
explain the variation in nesting behaviors seen within sea
turtle populations. Sea turtles are interesting taxa to
investigate nest site preferences because they lay several
clutches each nesting season, usually returning to the same
beach area within and between nesting seasons. The study
of such behavior contributes to a better understanding of
the biology of this species and, in addition, can improve
conservation strategies (Kamel and Mrosovsky 2006a).
The aim of this research was to study the individual
nesting preferences of hawksbill turtles along the southern
coastline of Rio Grande do Norte, northeastern Brazil,
during the 2006–2014 nesting seasons. A decrease in the
consistency of nest site choice, caused by changes in nest-
site preferences over time, may reflect behavioral plasticity
as a result of a dynamic nesting environment. As such, our
main goal was to investigate individual nesting profiles
and possible declines in nest site consistency in E.
imbricata using several nest parameters recorded over 8
nesting seasons.
METHODS
Study Site. — The study area is located in
northeastern Brazil in the southern section of the state of
Rio Grande do Norte (Fig. 1). The monitored beach area is
approximately 4.2 km in length, within the municipality of
Tibau do Sul (lat 06.1901218S, long 35.0847208W); this
area consists of Chapadao, Minas, and Sibauma beaches
(Fig. 1). The beach landscape is surrounded by cliffs,
interspersed with dunes, exposing a generally narrow band
of beach, from 0 m (in areas where the cliffs touch the sea)
up to 40 m. The sand is fine grained and of a pale yellow
color. The vegetation is composed primarily of pioneer
herbaceous species, such as Paspalum vaginatum (Poa-
ceae), Blutaparon portulacoides (Amarantaceae), Ipomoea
pes-caprae (Convolvulaceae), and Remirea maritima
(Cyperaceae). A few meters above the pioneer herbaceous
vegetation, the diversity of plants increases, with Chrys-
obalanus icaco (Chrysobalanaceae) and Melocatus (Cac-
taceae) found in dense clusters, as well as a few coconut

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 120
A quantum biochemistry model of the interaction between the estrogen
receptor and the two antagonists used in breast cancer treatment
K.B. Mota a, J.X. Lima Neto a, A.H. Lima Costa a, J.I.N. Oliveira a, K.S. Bezerra a, E.L. Albuquerque a,E.W.S. Caetano b, V.N. Freire c, U.L. Fulco d,⇑
aDepartamento de Biofísica e Farmacologia, Universidade Federal do Rio Grande do Norte, 59072-970 Natal-RN, Brazilb Instituto Federal de Educação, Ciência e Tecnologia do Ceará, 60040-531 Fortaleza-CE, BrazilcDepartamento de Física, Universidade Federal do Ceará, 60455-760 Fortaleza-CE, BrazildDepartamento de Biofísica e Farmacologia, Universidade Federal do Rio Grande do Norte, 59072-970 Lagoa Nova, Natal-RN, Brazil
a r t i c l e i n f o
Article history:
Received 4 April 2016
Received in revised form 3 May 2016
Accepted 8 May 2016
Available online 9 May 2016
Keywords:
Quantum biochemistry
DFT (Density Function Theory)
MFCC (Molecular Fractionation with
Conjugate Caps) scheme
Estrogen receptor
Raloxifene, Tamoxifen
a b s t r a c t
In this work we employed quantum biochemistry methods based on the density functional theory (DFT)
model and the MFCC (Molecular Fractionation with Conjugate Caps) scheme to unveil the detailed
binding energy features of the tissue-selective synthetic agents SERMs (selective estrogen receptor
modulators) 4-hydroxytamoxifen (OHT) and raloxifene (RAL), widely used in the breast cancer treatment,
co-crystallized with the estrogen receptor a (ERa). Our theoretical/computational results demonstrated
that the total binding energies of OHT and RAL to the ERa ligand-pocket correlate with the experimental
binding affinity. Besides, we found that SERMs-OHT binds stronger when compared to SERMs-RAL,
confirming experimental data, whose main contributions to the total SERMs-ERa binding energy are
due to the amino acid residues in decreasing sequence D351 > E542 > D538 > E353 > E423, an important
information to understand their binding mechanisms.
� 2016 Elsevier B.V. All rights reserved.
1. Introduction
Breast cancer is the second most commonmalignant neoplasms
worldwide and the fifth in cause of deaths [1]. Over the years, some
molecular alterations which reflect the biological characteristics of
invasive breast carcinomas have been identified and considered as
molecular markers, such as the estrogen receptors (ERs) [2].
Expressed in approximately 75% of invasive breast tumors [3], ER
belongs to the nuclear receptor (NR) superfamily of ligand-
activated transcription factors, like the glucocorticoid receptor
(GR), androgen receptor (AR), retinoic acid receptor (RAR) and
orphan receptors [4,5]. Through these receptors, NR controls the
transcription of important genes for developmental, reproductive,
neural, skeletal, grow and cardiovascular processes [6,7], trans-
forming them an important drug target.
Similar to other nuclear receptors, estrogen receptors are char-
acterized by presenting a variable N-terminal transactivation
domain, a conserved DNA binding domain (DBD), a variable hinge
region, a conserved ligand binding domain (LBD), and a variable
C-Terminal domain [8]. ER-ligand binding domain (ER-LBD) is
encoded within a region of about 300 amino acids, and its overall
architecture is comprised of helices 1–12 (H1–H12) and two b-
sheet (S1 and S2; see Fig. 1). It possesses the function of ligands
recognition and activation of physiological response [5]. Thus, after
ligand docking, there is an LBD rearrangement to active state with
a shift at the position of helix 12. It is followed by a formation of
(homo or hetero) dimers that recognize specific sequences of the
genomic DNA with the binding of many kinds of co-activators or
co-repressors, which are related to transcriptional activation/
repression, in order to mediate the transcription of various target
genes [9].
Estrogen receptor family is composed by estrogen receptor a(ERa) and estrogen receptor b (ERb), that are expressed in a wide
range of tissues and cell types, sharing some level of sequence
homology [10]. ERa is encoded by ESR1 gene on chromosome 6
and has been found predominantly in bone, testes, epididymis,
prostate, uterus, ovary, liver, mammary gland, adipose tissue,
heart, vascular system and brain. ERb is encoded by ESR2 gene
on chromosome 14 and is present in the bladder, prostate, ovary,
colon, adipose tissue, immune system, heart, vascular system, lung
and brain [11]. In breast cancers cases, it is common to see a
decrease of ERb expression and an increase in proliferative
response associated to the ERa activation [12]. Thus, many ERa
http://dx.doi.org/10.1016/j.comptc.2016.05.006
2210-271X/� 2016 Elsevier B.V. All rights reserved.
⇑ Corresponding author.
E-mail address: [email protected] (U.L. Fulco).
Computational and Theoretical Chemistry 1089 (2016) 21–27
Contents lists available at ScienceDirect
Computational and Theoretical Chemistry
journal homepage: www.elsevier .com/locate /comptc

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 121
This journal is©The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 New J. Chem., 2017, 41, 11405--11412 | 11405
Cite this: NewJ.Chem., 2017,
41, 11405
Energetic description of cilengitide bound tointegrin†‡
Jose X. Lima Neto, a Katyanna S. Bezerra,a Dalila N. Manso,a Kyvia B. Mota,a
Jonas I. N. Oliveira, a Eudenilson L. Albuquerque,a Ewerton W. S. Caetano,b
Valder N. Freirec and Umberto L. Fulco *a
The importance of integrins in several cell types that affect tumour progression has made them an
appealing target for cancer therapy. In particular, the integrin aVb3 plays an important role in angiogenesis
and tumor cell metastasis, besides being evaluated as a target for new therapeutic approaches. By
employing quantum chemistry methods based on Density Functional Theory (DFT) within the Molecular
Fractionation with Conjugate Caps (MFCC) approach, we investigated the binding energy features of
cilengitide interacting with the integrin aVb3 using a CPCM (Conductor-like Polarizable Continuum Model)
scheme to represent the electrostatic environment through a dielectric constant. We have taken
into account all ligand–residue interactions within a radius of 10 Å from the ligand, showing the most
important regions of the ligand and the residues affecting the binding mechanism. Our computational
results give a better understanding of the cilengitide–integrin binding mechanisms, while also being an
efficient alternative towards the development of RGD (R, arginine; G, glycine; D, aspartic acid)-based
drugs, prodrugs, nanoparticles and carriers, leading to new bio-engineering devices for cancer therapy.
Introduction
Cancer is a leading cause of death, accounting for over 8 million
casualties and 14 million cases diagnosed worldwide in 2012,
numbers that are expected to increase in the coming years.1
Mutations in the genome of a cell population form the basis
of tumor development, changing the normal function of
various classes of protein families, such as cytokines, cell surface
receptors, signal transducers and transcription factors, and
disturbing cellular activities that are crucial for the development
and maintenance of multicellular organisms.2
Integrins are heterodimeric a/b transmembrane receptors
which play a central role in cell–cell adhesion and cell–matrix
interaction through binding and association with transmembrane
receptors, proteins and soluble ligands in the extracellular matrix
(ECM).3,4 In addition to adhesive functions, integrins transduce
messages via various signaling pathways and influence the
proliferation and apoptosis of tumor cells, as well as being
responsible for the activation of endothelial cells, which provide
a repertoire of functions needed by cancer cells for migration,
invasion and proliferation.5
In particular, integrins aVb3 and aVb5 are implicated in
tumor neoangiogenesis, cancer development and metastatic
processes, being expressed on malignant tumors and intervening
in adhesion of tumor cells on a variety of extracellular matrix
proteins, thus allowing these cells to migrate during invasion.6
The integrin aVb3 (see Fig. 1) is weakly expressed at low levels in
most normal tissues, while high levels of expression are limited
to bone, the mid-menstrual cycle endometrium, the placenta,
inflammatory sites, invasive tumors, and activated endothelial
cells during angiogenesis where it mediates migration during the
formation of new vessels, which is essential for a sufficient
nutrient supply to the growing tumor.7 Also, high levels of this
integrin receptor in glioblastoma and melanoma cells have been
shown to reinforce tumor proliferation, as well as its spread and
adhesion.8On the other hand, its inhibition by selective antagonists
in preclinical studies has stopped angiogenesis and cell adhesion,
leading to tumor regression in experimental breast cancer
cells and melanoma, besides inducing apoptosis of activated
endothelial cells.9
Integrin aVb3 has been considered an important molecule in
biotechnological approaches within cellular, cancer and anti-
angiogenesis therapies,10,11 and many efforts have been made
to understand it, as well as the molecular basis of the ligand-
binding mechanism. It is assembled from a short cytosolic tail
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande
do Norte, Natal-RN, 59072-970, Brazil. E-mail: [email protected];
Fax: +55-84-32153791; Tel: +55-84-32153793b Instituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, Fortaleza-CE,
60040-531, BrazilcDepartamento de Fısica, Universidade Federal do Ceara, Fortaleza-CE,
60455-760, Brazil
† This document is a collaborative effort of all authors.
‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj02166f
Received 16th June 2017,Accepted 1st September 2017
DOI: 10.1039/c7nj02166f
rsc.li/njc
NJC
PAPER
Publi
shed
on 0
4 S
epte
mber
2017.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
27
/09
/20
17
01
:09
:26
. View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 122
Quantum binding energy features of the T3-785collagen-like triple-helical peptide†‡
Katyanna S. Bezerra,a Jonas I. N. Oliveira,a Jose X. Lima Neto,a
Eudenilson L. Albuquerque,a Ewerton W. S. Caetano,b Valder N. Freirec
and Umberto L. Fulco*a
Collagen-based biomaterials are expected to become a useful matrix substance for various biomedical
applications in the future. By taking advantage of the crystallographic data of the triple-helical peptide
T3-785, a collagen-like peptide whose homotrimeric structure presents large conformational similarity
to the human type III collagen, we present a quantum biochemistry study to unveil its detailed binding
energy features, taking into account the inter-chain interaction energies of 90 amino acid residues
distributed into three interlaced monomers. Our theoretical model is based on the density functional
theory (DFT) formalism within the molecular fragmentation with conjugate caps (MFCC) approach. We
predict the individual relevance (energetically) of the amino acid triplets Pro–Hyp–Gly, Ile–Thr–Gly, Ala–
Arg–Gly and Leu–Gly–Ala, as well as the influence of the N-terminal, central and C-terminal regions,
looking for the integrity of the collagen's triple helix. We found that the amino acid residues comprising
the peptide T3-785 have an interaction energy that depends not only on the chemical nature of the side
chain, but also the surrounding solvent molecules and inter-chain intermolecular interactions. The
energy profile of this collagen-model molecule depicts a character essentially attractive to its
conformational stability, encouraging research focusing on the development and synthesis of artificial
collagen with high stability for bioengineering applications.
Biomaterial science is an expanding area, which encompasses
a wide range of medical knowledge. Collagen, a well-known
protein, is regarded as one of the most useful biomaterial in
the medical, dental and pharmacological elds. Biomedical
applications of collagen require improved methods to synthe-
size its triple helices feature, essential for the development of
articial structures displaying its natural properties. Its excel-
lent biocompatibility and safety due to its biological charac-
teristics, made it the primary resource in medical applications.
Besides, it is the most abundant family of extracellular matrix
proteins expressed in connective tissues of animals.1 Human
beings have at least 45 distinct collagen genes accounting for 28
protein isoforms, amounting about one quarter of the total
protein mass of the human body.2
A single collagen molecule is a structural insoluble brillar
protein used to set up larger collagen aggregates that performs
unique physiological functions in bones, skin, cartilage, liga-
ments and tendons.3 Also, there has been increasing appreciation
of the biological importance of collagen in many cellular process,
such as adhesion, proliferation and migration, matrix
degradation/remodeling, tissue regeneration and homeostasis.4,5
Furthermore, due to a number of biological properties such as
high biocompatibility, rare adverse reactions and weak antige-
nicity,6 collagens have been widely used as a natural material for
diverse biomedical applications, including its clinical use as
biomimetic scaffold and drug delivery system.7 Therefore, the
understanding of how these properties are derived from its
fundamental structural units requires a comprehensive knowl-
edge of the mechanisms underlying its structure and stability.8
Seven types of collagen (I, II, III, V, XI, XXIV and XXVII) are
assembled in stable brils, forming a complex three-
dimensional brous superstructure.9 They are composed of
individual helices of three polypeptide chains/strands twisted
in a right-handed manner and held together by a ladder of
intermolecular backbone hydrogen bonds between adjacent
strands.10 Each chain shows one or more collagenous domain
characterized by a repetition of Xaa–Yaa–Gly motif (or triplet),
where Xaa and Yaa are oen proline and hydroxyproline resi-
dues, respectively. Variations among the collagen family
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande do
Norte, 59072-970, Natal, RN, Brazil. E-mail: [email protected]; Fax: +55-84-
32153791; Tel: +55-84-32153793bInstituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, 60040-531, Fortaleza,
CE, BrazilcDepartamento de Fısica, Universidade Federal do Ceara, 60455-760, Fortaleza, CE,
Brazil
† This document is a collaborative effort of all authors.
‡ Electronic supplementary information (ESI) available. See DOI:
10.1039/c6ra25206k
Cite this: RSC Adv., 2017, 7, 2817
Received 13th October 2016Accepted 10th December 2016
DOI: 10.1039/c6ra25206k
www.rsc.org/advances
This journal is © The Royal Society of Chemistry 2017 RSC Adv., 2017, 7, 2817–2828 | 2817
RSC Advances
PAPER
Open
Acc
ess
Art
icle
. P
ubli
shed
on 1
2 J
anuar
y 2
017.
Dow
nlo
aded
on
13
/01
/20
17
15
:19
:11
.
This
art
icle
is
lice
nse
d u
nder
a C
reat
ive
Com
mons
Att
rib
uti
on
-No
nC
om
mer
cial
3.0
Un
po
rted
Lic
ence
.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 123
1SCIENTIFIC REPORTS | (2018) 8:1840 | DOI:10.1038/s41598-018-20325-0
www.nature.com/scientificreports
Inhibition of the checkpoint protein PD-1 by the therapeutic antibody pembrolizumab outlined by quantum chemistryAna Beatriz M. L. A. Tavares, José X. Lima Neto , Umberto L. Fulco & Eudenilson L. Albuquerque
Much of the recent excitement in the cancer immunotherapy approach has been generated by the recognition that immune checkpoint proteins, like the receptor PD-1, can be blocked by antibody-based drugs with profound effects. Promising clinical data have already been released pointing to the efficiency of the drug pembrolizumab to block the PD-1 pathway, triggering the T-lymphocytes to destroy the cancer cells. Thus, a deep understanding of this drug/receptor complex is essential for the improvement of new drugs targeting the protein PD-1. In this context, by employing quantum chemistry methods based on the Density Functional Theory (DFT), we investigate in silico the binding energy features of the receptor PD-1 in complex with its drug inhibitor. Our computational results give a better understanding of the binding mechanisms, being also an efficient alternative towards the development of antibody-based drugs, pointing to new treatments for cancer therapy.
The World Health Organization (WHO) labels cancer as one of the most pressing health challenges in the world. Statistical data from the National Cancer Institute (NCI) show that one in two (three) men (women) will develop this disease. Cancer is a leading cause of death, accounting for over 8.8 million casualties and 14.1 million cases diagnosed worldwide in 2015, numbers that are expected to increase in the coming years1. It is characterized by an uncontrolled growth of cells in the body, with the potential to invade or spread to its other parts with the formation of metastases. It is induced by mutations in the genome of a cell population, which changes the nor-mal function of various classes of protein families, such as cytokines, cell surface receptors, signal transducers and transcription factor, making it one of the most difficult and complex disease to treat2–4. The more widely the cancer spreads, the harder it becomes to eradicate. In 2013, oncology was ranked on the top therapeutic class by worldwide sales, amounting to $73 billion5. However, though highly expensive, the treatment suffers several fails due to a defense mechanisms developed by some malignant cells, mainly those related to the activation of drug resistance processes6.
Although these mutations provide a selective vantage to populations of cancer cells, they also increase their divergences from the normal one, which can allow the recognition by the immune system cells, such as the T lymphocytes (T cells) and B cells7. Nevertheless, tumors also evolved to deceive immune cells, including the abil-ity to activate co-inhibitory signaling pathways on T cells by immune checkpoint proteins, such as the cytotoxic T-lymphocite protein 4 (CTLA-4) and the programmed cell death protein 1 (PD-1), leading to a state of immune tolerance8,9.
The molecular identification of cancer antigens helped the creation of new approaches for effective therapies, giving rise to a new era of treatment in which our own immune system evade the block created by malignant cells and fights against them. Among these new treatments, the immune checkpoint therapy has been clinically vali-dated as an effective treatment for many cancer types10. The blockade of the ligand-receptor interaction of these immune checkpoint molecules can directly increase the function of the T-cells, whereas it has been shown as a way to release the immune system to unleash anti-tumor immune response11.
The receptor PD-1 (Pdcd1 gene on chromossome 2) is an immune cell-specific surface inhibitor, mainly expressed in the late effector phase on activated CD4+/CD8+ T cells, B cells, monocytes, natural killer T cells,
Departamento de Biofísica e Farmacologia, Universidade Federal do Rio Grande do Norte, 59072-970, Natal, RN, Brazil. Correspondence and requests for materials should be addressed to E.L.A. (email: [email protected])
Received: 2 October 2017
Accepted: 16 January 2018
Published: xx xx xxxx
OPEN

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 124
This journal is©The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 New J. Chem., 2018, 42, 2401--2412 | 2401
Cite this: NewJ.Chem., 2018,
42, 2401
Outlining migrainous throughdihydroergotamine–serotonin receptorinteractions using quantum biochemistry†
Jose X. Lima Neto, a Vanessa P. Soares-Rachetti,a Eudenilson L. Albuquerque,a
Vinicius Manzonib and Umberto L. Fulco *a
Since the early days of migrainous research, serotonin receptors (5-HTR) have been considered a major
target of drugs, and the 5-HT1B and 5-HT1D agonists have been among the most marketed ones for the
treatment of migraine attacks. Besides, the 5-HT1BR agonist is also involved in the mechanisms
underlying many neurological dysfunctions, making it a critical target for the rational development of
drugs. In this context, by taking advantage of its first crystallographic structure co-crystallized with
dihydroergotamine (DHE), one of the oldest and largely used antimigraine drugs, a quantum bio-
chemistry study based on the electrostatically embedded molecular fractionation with conjugate caps
(EE-MFCC) scheme within the density functional theory (DFT) formalism is performed to unveil this
complex’s detailed binding energy. In the EE-MFCC model, each fragment is embedded within the
electrostatic effect of the full protein, in such a way that all atoms are replaced by their corresponding
atomic charges, excluding the reference residues. Our results reveal not only the pivotal role played by
the amino-acid residue D129 in the dihydroergotamine–5-HT1BR total binding energy, but also the
importance of the residues D352, D123, E198, D204, F330, L126, F351, I130, V201, V200, T355 and R114
in this complex. The total binding energy reaches convergence after a pocket radius r = 7.0 Å, with a
value around �230.93 kcal mol�1. Furthermore, we predict the relevance (energetically) of the DHE
regions, as well as the influence of each protein segment to DHE–serotonin receptor binding. Finally, for
completeness, we compared our results obtained by using the EE-MFCC approach with those
considering the ordinary MFCC scheme. We believe that our work is a first step using in silico quantum
biochemical design as a means to influence the discovery of new drugs to treat migraine and other
diseases related to the 5-HT1BR agonist.
Introduction
Migraine is a complex condition with a wide variety of symp-
toms, the main feature usually being a painful headache,
affecting up to one billion people worldwide.1 There is currently
no cure for migraine, although it may be related to a mix of
environmental and genetic factors, and the treatments avail-
able are varied and differ from person to person. A migraine
attack can be very frightening with severe throbbing pain or a
pulsing sensation, usually on just one side of the head, often
accompanied by nausea, vomiting, and extreme sensitivity to
light, sound and smell. It usually comes on gradually and is
generally made worse by physical activity. It is a severe and
disabling neurological disorder that typically begins in adoles-
cence or early adulthood, showing higher incidence among
women in reproductive years, peaking in their fourth decade of
life.2 It usually lasts from four hours to three days, having an
enormous impact on the work, family and social lives of the
sufferers, and significantly diminishes their quality of life. Up
to one-third of them have an aura, a spectacular and sometimes
frightening focal neurological disturbance manifesting as visual,
sensory or motor symptoms, which signals that the headache will
soon occur. Migraine ranks as the third most common (costly)
disabling neurological disease (disorder).3 It costs in the United
States (Europe) nearly US$12 (h67) billion annually,4 being almost
h585 per patient in Western Europe.5,6 Despite that, unfortu-
nately, migraine remains a poorly understood disease that is
often undiagnosed and undertreated.
The pathophysiology of migraine is still unclear. In recent
decades, important strides have been made to understand it,
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande
do Norte, 59072-970, Natal-RN, Brazil. E-mail: [email protected];
Fax: +55 (84) 3215-3791; Tel: +55 (84) 3215-3793b Instituto de Fısica, Universidade Federal de Alagoas, 57072-970, Maceio-AL, Brazil
† This document is a collaborative effort of all authors.
Received 22nd September 2017,Accepted 27th December 2017
DOI: 10.1039/c7nj03645k
rsc.li/njc
NJC
PAPER
Publi
shed
on 0
3 J
anuar
y 2
018.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
29
/03
/20
18
18
:05
:58
.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 125
This journal is©The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 New J. Chem., 2018, 42, 17115--17125 | 17115
Cite this: NewJ.Chem., 2018,
42, 17115
Computational investigation of the a2b1integrin–collagen triple helix complexinteraction†
K. S. Bezerra,a J. X. Lima Neto, a J. I. N. Oliveira, a E. L. Albuquerque, a
E. W. S. Caetano, b V. N. Freirec and U. L. Fulco *a
The greatest difficulty for the development of drugs targeting the a2b1 integrin, a transmembrane receptor
that facilitates cell-extracellular matrix (ECM) adhesion, is the current understanding of its interaction with
ligands, such as the collagen molecule – one of the most complex cell adhesion systems. In this sense, this
work performs a quantum biochemistry analysis of the interaction between the a2b1 I-domain integrin and a
collagen structure containing the GFOGER (Glycine–Phenylalanine–Hydroxyproline–Glycine–Glutamate–
Arginine) sequence. Our study was carried out by using the Molecular Fractional with Conjugate Caps
(MFCC) method within Density Functional Theory (DFT) with generalized gradient approximations (GGA), and
Grimme’s long-range dispersion correction. Our results confirm the importance of the amino-acids residues
Thr221, Asp219, Asp254 and Glu256 (Arg12, Glu33, Arg34, Glu55 and Arg56) present in the MIDAS – Metal
Ion Dependent Adhesion Site – region (GFOGER motif) of the integrin–collagen interaction. Besides, we
depicted the relevance of each strand (A, B and C chains) in the triple-helical collagen structure, helping the
understanding of the events involving the interaction integrin–collagen.
Introduction
The control of cellular interactions in multicellular organisms
is fundamental for many biological processes, such as embryo-
genesis, immune responses, tissue integrity and homeostasis,
to cite just a few.1 Several interactions occurring among cell–
cell and cell–matrix components are performed by integrating
specific ligands with cell adhesion receptors. Among them, the
most known are the members of the family of integrins,2 type I
heterodimeric transmembrane glycoproteins, which function
as themajormetazoan receptor for cell adhesion and transmission
of bidirectional signals. They play a central role in physical support
functions for signal transduction, actin cytoskeleton assembly,
gene expression and cell functions, including cell adhesion,
migration proliferation, differentiation, and apoptosis.3
Integrins are composed of two subunits, namely a (containing
B750 residues) and b (with B1000 residues), not covalently
associated, jointly forming twenty-four distinct heterodimeric
structures with different binding and distribution properties in
various tissues.4 These subunits show a large extracellular domain
folded into N-terminal globular portions, which are combined
to create the ligand binding surface. This globular portion is
followed by a type I transmembrane domain, a single-stranded
structure of about 25–29 amino-acids residues forming a
coiled a-helix, and by a short C-terminal cytoplasmic tail region
(10–70 amino-acids) which binds to cytoskeletal elements
through cytoplasmic adapter proteins.5,6 They can also bind
to a number of proteins of the extracellular matrix (ECM), being
found in the surface of other cells during different physio-
logical contexts, making them a target for therapies against a
large number of diseases, such as thrombosis, infection,
immune system disorders, osteoporosis and cancer.
Among their heterodimeric structures, the a2b1 integrin, a
collagen receptor, has been involved in platelet adhesion,
epithelial differentiation, morphogenesis, cancer, wound healing,
angiogenesis, inflammation and immunity.7 In vitro studies have
shown that the deletion of the a2 structure causes a reduction in
platelet response, blocking the binding to type I collagen, as well as
the destabilization of thrombus formation. Patients either with
reduced levels of a2b1 integrin expression on platelets, or with the
presence of auto-antibodies, had platelet activation impaired by
collagen but not by other molecules.8 Studies related to metastasis
in tumor cells have revealed that the overexpression of the a2b1
integrin may be related to the acceleration of metastasis in
aDepartamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande
do Norte, 59072-970, Natal-RN, Brazil. E-mail: [email protected];
Fax: +-55-84-32153791; Tel: +-55-84-32153793b Instituto Federal de Educaçao, Ciencia e Tecnologia do Ceara, 60040-531,
Fortaleza-CE, BrazilcDepartamento de Fısica, Universidade Federal do Ceara, 60455-760, Fortaleza-CE,
Brazil
† This document is a collaborative effort of all authors.
Received 15th August 2018,
Accepted 14th September 2018
DOI: 10.1039/c8nj04175j
rsc.li/njc
NJC
PAPER
Publi
shed
on 1
9 S
epte
mber
2018.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
10
/19
/20
18
1:4
1:1
8 A
M.
View Article OnlineView Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 126
This journal is©The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 New J. Chem., 2018, 42, 19801--19810 | 19801
Cite this: NewJ.Chem., 2018,
42, 19801
Computational biochemical investigation of thebinding energy interactions between an estrogenreceptor and its agonists†
Aranthya H. Lima Costa, Washington S. Clemente Jr, Katyanna S. Bezerra,Jose X. Lima Neto, Eudenilson L. Albuquerque and Umberto L. Fulco *‡
In this work we describe the profiles of the estrogen receptor ERa, the most common type of hormone
receptor on breast cells, in terms of its total binding energy in the presence of its full agonist ligands,
endogenous 17b-estradiol and synthetic diethylstilbestrol, which are responsible for a large percentage
of all breast cancers. The X-ray crystallographic structures of ERa in complexes with both agonists are
considered, and the electrostatically embedded molecular fractionation with conjugate caps (EE-MFCC)
scheme is applied within a dispersion-corrected density functional theory (DFT-D) framework, taking
into account the B97D/cc-pVTZ level of theory. Our quantum chemical computational results show that
the total binding energy of both agonists to the ERa ligand-pocket, with radius varying from 2.0 to
12.5 Å, is well correlated with the experimental agonist binding affinity. In addition, we found that the
most significant contributions to the total binding energy of the receptor–endogenous (synthetic) ligand
complexes are mainly due to 12 (14) amino acid residues. Finally, we predict the energetic relevance
of the estradiol and diethylstilbestrol regions, as well as the influence of each protein segment on
ERa–agonist binding. This information is important for helping to decide whether or not the cancer is
likely to respond to either endocrine therapy or other clinical treatments.
Introduction
Breast cancer is a serious health concern worldwide; it is the
second most common cause of malignant neoplasms and the
fifth leading cause of death.1,2 Although there is no reliable way
to prevent breast cancer, a knowledge of risk factors, like
heredity, obesity and prolonged hormone therapy, may help
the medical community to take preventive measures to reduce
the likelihood of developing the disease.3,4 Previous studies
have suggested that some of these risk factors are associated
with the expression levels of estrogen receptors (ERs), proges-
terone receptors (PRs) and human epidermal growth factor
receptor 2 (HER2) in tumors, making targeting these proteins
a powerful strategy for traditional therapy.5
There is a strong pharmaceutical interest in understanding
ERs, which are expressed in approximately 75% of invasive
breast tumors and are crucial for the development of malignant
cells.6 They belong to the superfamily of nuclear receptors
(NRs) and share structural features with members of the steroid
receptor family. They are composed of a variable amino-
terminal domain, a conserved DNA-binding domain (DBD) and
a carboxy-terminal ligand binding domain (LBD) linked by a
flexible hinge peptide.7 In addition to forming the ligand-
binding pocket, the LBD comprises dimerization surfaces and
sites for co-regulator interactions. Upon ligand binding, the DBD
is responsible for the recognition of estrogen response elements
and ER dimerization.8 The ER–ligand binding domain (ER-LBD)
is encoded within a region of about 300 amino acids organized
as twelve a-helices (H1–H12) and two b-sheets (S1–S2). Here,
a-helices 3, 6, 8, 11 and 12 form the hydrophobic pocket for
ligands; a-helix 12 is also essential for transactivation of gene
expression.9
In mammals, estrogen receptors are expressed as two iso-
forms, ERa and ERb, which share some level of sequence
homology and are observed in a wide range of tissues and cell
types.10 Their activation by estrogens and their hyperactivity
have been linked to the genesis of breast cancer, cardiovascular
diseases, osteoporosis and obesity.11 Besides, the agonist action
of 17b-estradiol (E2) at ERa has been found to induce chromo-
somal changes that result in the neoplastic transformation of
breast epithelial cells.12,13 Notwithstanding their potential side-
effects and the risks associated with a long-term use, synthetic
Departamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande do
Norte, 59072-970, Natal-RN, Brazil. E-mail: [email protected];
Fax: +55-84-32153791; Tel: +55-84-32153793
† Electronic supplementary information (ESI) available. See DOI: 10.1039/
c8nj03521k
‡ This document is a collaborative effort by all the authors.
Received 14th July 2018,Accepted 15th October 2018
DOI: 10.1039/c8nj03521k
rsc.li/njc
NJC
PAPER
Publi
shed
on 1
8 O
ctober
2018.
Dow
nlo
aded
by U
niv
ersi
dad
e F
eder
al d
o R
io G
ran
de
do
No
rte
on
12
/5/2
01
8 1
0:3
0:2
9 P
M. View Article Online
View Journal | View Issue

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 127
A Quantum Biochemistry Approach to Investigate
Checkpoint Inhibitor Drugs for Cancer‡
Ana Beatriz M. L. A. Tavares,a José X. Lima Neto,b Umberto L. Fulcob and Eudenilson
L. Albuquerque∗b
Targeted immunotherapy has reignited enthusiasm for the
development of checkpoint inhibitors drugs for cancer,
noticeably improving the overall survival of cancer patients.
In this sense, the inhibition of the programmed cell death
receptor-1 (PD-1)/programmed cell death ligand-1 (PD-L1)
pathway by the US FDA-approved monoclonal antibodies
Pembrolizumab and Nivolumab, the leading drugs of this
therapeutic revolution, was recognized as an important
issue for clinical research. Although the drug’s immune-
related binding interactions to PD-1 is slightly different,
they competitively hinder the natural ligand PD-L1. Besides,
their binding energy features undoubtedly need to be better
understood to assure their safe use as new medicines in
clinical practice. In this context, by employing quantum
chemistry methods based on the Density Functional Theory
(DFT), we investigate in silico the coupling profiles of the
receptor PD-1 in complex with its natural ligand PDL-1 and
these two drugs inhibitors, looking for a better understand-
ing of their binding interactions, essential to validate their
importance on the PD-1/PDL-1 pathway blockade for the
treatment of malignant melanomas.
Cancer immunotherapy, a treatment by which the patient’s own
immune system recognize and destroy tumour cells, has shown
great outcomes in the cancer treatment1,2. A number of different
immunotherapeutic approaches have been investigated, depict-
ing the inhibition of checkpoint proteins as a leading strategy for
evade the blockade created by tumour environment against com-
monly used therapies3. In this sense, immunomodulatory anti-
bodies that directly enhance the function of the lymphocyte T (T-
a Hospital das Clínicas, Universidade Federal de Pernambuco, 50.670-901, Recife-PE,
Brazil.b Departamento de Biofísica e Farmacologia, Universidade Federal do Rio Grande do
Norte, 59072-970, Natal-RN, Brazil. Fax: +-55-84-3215-3791; Tel: +-55-84-3215-
3793; E-mail: [email protected]
‡ This document is a collaborative effort of all authors.
cell) by avoiding the activation of its negative regulators (named
molecular checkpoints), are considered the major breakthrough in
cancer immunotherapy4,5. Among these checkpoint proteins, in-
hibitors that block the programmed cell death receptor 1 (PD-1)
pathway showed a powerful clinical potential since the PD-1 re-
ceptors expressed on the T-cells play a pivotal role in the down
regulation of the immune system, and their ligands are overex-
pressed in malignant cells6,7. The receptor PD-1 (Pdcd1 gene
on chromosome 2), a member of the B7-CD28 family, is an im-
mune cell-specific surface inhibitor, mainly expressed in the late
effector phase on activated CD4+/CD8+ T-cells, B-cells, mono-
cytes, natural killer T-cells, and antigen-presenting cells (APC),
including dendritic cells8. It is a 55-kDa monomeric type I sur-
face transmembrane glycoprotein of the Ig superfamily, account-
ing for 288 amino-acids, displaying four domains including a sin-
gle V-set immunoglobulin superfamily (IgSF) domain, a stalk, a
transmembrane domain and a cytoplasmic domain, which con-
tains two tyrosine-based immunoreceptor signalling motifs: the
inhibitory motif (ITIM) and the switch one (ITSM)9.
In spite of that, though clinical results using blocking agents
against the human cell surface receptor PD-1 and its ligants PD-
L1 and PD-L2 point to unprecedented rates of long lasting anti-
tumors activity in patients with different types of cancer, some
of them present resistance to the blockage of the receptor PD-
1 and overstimulation for the immune responses10. Therefore,
the quick design of new drugs is essential and requires a pre-
cise knowledge not only of its biochemical structure, but also its
binding mechanism with the receptor PD-1, leading to a better
understanding of how this complex system influences the cancer
tumor and its microenvironment. Monoclonal antibodies, like the
US FDA-approved drugs Pembrolizumab (PEM) and Nivolumab
(NIV), can block this binding and boost the immune response,
leading to an efficient treatment of some cancer diseases.
Due to its relevance for immune system maintenance, as well
as the multiple evidence indicating the success of monoclonal an-
tibody therapy against PD-1/PD-L1 pathway, a number of crystal-
1–4 | 1

APÊNDICE A. Artigos publicados/submetidos durante o período de doutoramento 128
Exploring GABAB receptor binding site for agonists and antagonists through in silico
simulations
Jose X. Lima Neto, Jonas I. N. Oliveira, Vanessa P.
Soares-Rachetti, Eudenilson L. Albuquerque, and Umberto L. Fulco∗
Departamento de Biofısica e Farmacologia, Universidade Federal do Rio Grande do Norte, 59072-970, Natal-RN, Brazil.
Vincius ManzoniInstituto de Fsica, Universidade Federal do Alagoas, 59072-970, Natal-RN, Brazil.
(Dated: January 7, 2019)
Keywords: MFCC, DFT, GABAB , baclofen, GABA, 2-hydroxysaclofen, SCH50911
INTRODUCTION
γ-Aminobutyric acid (GABA) is the main inhibitoryneurotransmitter in mammalian central nervous system(CNS), where it activates the ligand-gated ion chan-nels GABAA/C and the GABAB receptors, a class C Gprotein-coupled receptor (GPCR) [1]. GABAB receptors(GBRs) modulate the neuronal excitability by inhibit-ing calcium channels and adenylyl cyclase, and activatingthe inwardly rectifying potassium channels, thus regulat-ing the neurotransmitter release (including GABA, sero-tonin, dopamine and noradrenaline) and the neuronalmembrane hyperpolarization processes [2, 3]. Accord-ingly, the GABAB receptors plays a key role in manypathways throughout the brain. Thus, the pharmacolog-ical manipulation of GBRs has been shown to be pivotalfor the treatment of a wide variety of neurological dis-eases and psychiatric disorders, such as anxiety, spastic-ity, epilepsy, autism spectrum disorders, drug addiction,alcoholism, neuropathic pain, Parkinson and Alzheimer[4–6].
Despite the high relevance of GABAB , the only drugapproved by the food and drug administration (US-FDA)and currently used in the clinic is baclofen. Baclofen, alipophilic analogue of GABA, is considered the drug ofchoice to treat muscle spasticity in patients with multiplesclerosis, cerebral palsy and spinal cord injury [7]. How-ever, the side-effects related to baclofen limit its poten-tial applications in other neurological conditions. Hence,a number of ligands based on GABA structure have beendeveloped with broad range of binding affinity values andselectivity, showing to be useful in the treatment of pain,addiction, social deficits and other neurological diseases[8]. On the other hand, there is limited information aboutthe therapeutic properties of GABAB antagonists, suchas phaclofen, saclofen, SCH50911 and CGP54626A, al-though preclinical behavioural models of depression andcognitive disorders have shown promising results [9].
Understand the structural mechanisms that rule therelationship between structure and function of GABAB
∗ Corresponding author, e-mail: [email protected]; Tel:
+-55-84-32153793; Fax: +-55-84-32153791
is a crucial step for the development of new drugs withless side-effects and higher affinity. GBR is a func-tional heterodimer formed by the union of GABAB1
(GB1)/GABAB2 (GB2) subunits, with GABAB1 show-ing two isoforms, GABAB1a and GABAB1b. Similar toother class C GPCRs, including metabotropic glutamatereceptors (mGluRs), each subunit consists of three dis-tinct domains: the extracellular domain (N-terminal re-gion), which is also known as Venus fly trap (VFT), thetransmembrane domain, and the intracellular domain (C-terminal region) [10, 11]. GABAB subunits carry distinctfunctions for GABAergic signalling, where GABAB1 isresponsible for ligand-binding, while GABAB2 is ac-countable for G protein activation. In this sense, themost described activation mechanism for GBR suggesta direct transference of structural rearrangements fromGB1 to GB2 [12].
Currently, much information about GBR’s structurehas been obtained by comparing it with metabotropicglutamate receptor (mGluRs), since the crystallographicstructure of the last was early obtained [13]. However,some authors have showed the likely of severe differencesbetween these receptors, which could impair a good de-scription of GABAB features [1, 14]. Besides, there is lessstudies trying to provide a deeper structural character-ization of class C GPCRs, mainly GABAB , when com-pared with other GPCR’s classes [15, 16]. In this sense,the recent determination of the first high-resolution crys-tal structure of the human VFT-GABAB1b/GABAB2 re-ceptor co-crystallized with several ligands (agonists andantagonists) was a first step toward a deeper understand-ing of the ligand binding mechanism in GABAB [17]. Ithas provided the opportunity for structure-based drugresearch through in silico simulations, which have beenquite helpful in guiding the development of most selectivecompounds [18].
Therefore, to achieve a deeper understanding of themolecular mechanism of action for agonists and antag-onists in GABAB binding site, and to aid the discov-ery of novel drugs targeting the receptor, we determinethe binding energy profile, as well as the interaction be-tween the individual residues at the GBR binding pocketand ligands through quantum biochemistry techniquesbased on the MFCC scheme within the Density Func-

Anexos

130
anexo a
FUNDAMENTOS TEÓRICOS DA BIOQUÍMICA COMPUTACIONAL
No decorrer deste capítulo será descrito o modelo teórico empregado nesta tese.Aqui será abordado de forma sucinta a teoria do funcional da densidade (DFT), tendo sidoesta aplicada na obtenção das energias de interação entre os sistemas biológicos estudados.
A bioquímica computacional abrange um conjunto de ferramentas, que se valemde conhecimentos físicos, químicos, biológicos, matemáticos, estatísticos, e cujo objetivoé simular, avaliar e interpretar informações de sistemas biológicos complexos necessáriaspara a resolução de problemas bioquímicos. Através de softwares que processam cálculosbaseados nas equações da física clássica e quântica, busca-se a representação das estruturasmoleculares, bem como a simulação de seu comportamento em ambiente fisiológico, deforma a obter-se informações sobre sua geometria (comprimentos de ligação, ângulosde ligação, ângulos de torção), energia (calor de formação, energia de ativação, etc.),propriedades eletrônicas (momento de dipolo, potencial de ionização, afinidade eletrônica),e propriedades espectroscópicas (modos de vibração), entre outras (volume, área desuperfície, difusão, viscosidade, etc.).
Este campo vem crescendo muito nas últimas décadas devido aos grandes avançosdos hardwares de computadores e ao desenvolvimento eficiente de softwares de simulação,permitindo que outros campos da ciência também sejam agraciados com estas ferramentapara complementar seus estudos (ATKINS; PAULA, 2013).
Uma importante abordagem da bioquímica computacional é o estudo da interaçãoentre proteínas receptoras ou enzimas e seus ligantes. Neste sentido, identificar possíveissítios de ligação e quantificar a energia de interação entre fármaco e suas proteínas-alvo, alémde determinar orientações e conformações de moléculas de interesse farmacológico estãoentre as áreas nas quais a bioquímica computacional pode contribuir para a compreensãode fenômenos biológicos e o planejamento de novos fármacos. Deste modo, a análise deestruturas obtidas por técnicas de difração de raios-X, RMN ou geradas por modelagemmolecular podem acelerar o processo de descobrimento de fármacos (RAHA et al., 2007;ZHOU; HUANG; CAFLISCH, 2010; JASKOLSKI; DAUTER; WLODAWER, 2014).
O método a ser escolhido durante uma análise através da bioquímica computacionaldependerá da complexidade do sistema, do tipo de informação que se deseja obter e dapoder computacional disponível. Quanto maior o rigor teórico, maior a precisão do resultadoe maior o custo computacional. Como consequência, simulações de sistemas contendo umgrande número de átomos normalmente empregam métodos clássicos de análise, os quaispermitem maior velocidade de cálculo, contudo perdendo em precisão. Exemplos destasaplicações são encontrados em algoritmos de ancoramento molecular (docking, em inglês),onde a geometria de ligação de peptídeos ou pequenas moléculas em proteínas são obtidasem um curto espaço de tempo (ORRY; ABAGYAN; CAVASOTTO, 2006; BROOIJMANS;KUNTZ, 2003; SOUSA; FERNANDES; RAMOS, 2006), e de dinâmica molecular clássica(OKADA et al., 2012; PLATANIA et al., 2012), onde são realizadas simulações, desdegrandes proteínas a pequenas moléculas, em meio líquido explicito durante uma curtafração de tempo. Por outro lado, apesar de limitados a sistemas com reduzido número deátomos, os métodos quânticos, por incluírem a informação eletrônica em seus cálculos,

ANEXO A. Fundamentos teóricos da bioquímica computacional 131
possuem a capacidade de apresentar grande precisão em seus resultados (CHO et al., 2009)e tem sido de grande importância para o desenvolvimento in silico de fármacos (RAHA etal., 2007; BOHÓRQUEZ et al., 2010), além de mostrar-se úteis na estimativa (relativa)das energias de interação/ligação entre proteína e ligantes e na análise estrutural, óptica eeletrônica de pequenos cristais, entre outras aplicações.
O desafio central para os cálculos em química teórica é a solução da Equação deonda de Schrödinger, HΨ=EΨ, em que E é a energia eletrônica e Ψ é a função de ondamulti-eletrônica, que é a função das coordenadas de todos os elétrons e do núcleo (ATKINS;PAULA, 2013). Contudo, excluindo-se o átomo de hidrogênio, essa equação é insolúveldevido a sua complexidade. Assim, algumas restrições matemáticas são aplicadas paraobter a solução do problema.
Atualmente há uma grande variedade de métodos de cálculos computacionais, osquais são caracterizados pela presença de diferentes níveis de teoria, além de variadosmodos de aplicação. Em grandes sistemas, onde o objetivo principal é buscar a geometriaótima ou simplesmente como um input, os métodos clássicos, embora menos precisos,fornecem uma fonte confiável. Em contrapartida, os métodos quânticos, que podem serclassificados como semi-empíricos, ab initio e o método da teoria do funcional da densidade(DFT - do inglês, Density Functinal Theory), representam uma fonte mais confiável, masque possui restrições fortes com relação ao tamanho do sistema. Neste tópico será descritorapidamente os método utilizados nesta tese. Como introdução ao DFT, apresentamosos métodos de Hartree-Fock e os métodos semi-empíricos, sendo dado um maior enfoquepara a teoria do funcional da densidade.
Os métodos ab initio, aqui representado pelo Hartee-Fock e MP2, são capazes defornecer resultados bem precisos para moléculas com poucos átomos e elétrons, porém sãocomputacionalmente inviáveis para moléculas maiores. Em contraste, os métodos semiem-píricos e o DFT podem ser utilizados para a realização de cálculos em moléculas maiorescom maior facilidade, sacrificando, contudo, os resultados em termos de confiabilidade.
Assim, a escolha do método para a resolução de um problema químico não é umatarefa simples. A precisão química associada com o método e o custo computacionaldeve ser levado em consideração. De fato, não há uma única metodologia existente paraser aplicada para todas as moléculas (ORTOLAN, 2014). No entanto, a promessa que aquímica computacional tem para melhorar a habilidade de predizer propriedades físicas equímicas de uma ampla variedade de moléculas é o suficiente para impulsionar ainda maiso desenvolvimento de estruturas eletrônicas.
A.1 Métodos clássicos
Os métodos clássicos, também referidos como métodos de mecânica molecularfazem uso de uma expressão algébrica simplificada durante o cálculo das propriedades deum sistema, dispensando o formalismo da mecânica quântica e o cálculo da função de ondaou da densidade total dos elétrons. Este tem sido um dos principais métodos utilizadospela bioquímicos computacionais devido a sua capacidade de simular grandes moléculas,como proteínas, membrana, DNA, entre outras.
Neste método, a expressão da energia consiste em uma equação clássica, capazde descrever o sistema em termos de energia associada ao alongamento, flexão e rotaçãode ligações, e forças intermoleculares, como as interações de van der Waals e ligações dehidrogênio. Para a resolução dessas equações, são utilizadas bases de dados conhecidas como

ANEXO A. Fundamentos teóricos da bioquímica computacional 132
campo de força, cujo conjunto de parâmetros foi obtido através de dados experimentais oude cálculos ab initio. A energia potencial (U) de um sistema, é determinada segundo asequações:
U = Ecovalente + Enãocovalente (A.1)
Ecovalente = Eligação + Eangular + Ediedral (A.2)
Enãocovalente = Eeletrostática + EvanderW aals (A.3)
onde Ecovalente é a energia envolvida nas ligações covalentes e a formação da estruturaespacial das moléculas, Eligação é a energia de ligação, Eangular é a energia envolvida nadefinição do ângulo entre três átomos sequenciais, Ediedral energia envolvida na formaçãode diedros. O termo Enãocovalente remete a energia de interação intermolecular e divide-seem: Eeletrostática, que trata da energia referente as interação eletrostáticas (computadonormalmente através das leis de Coulomb) e, por fim, Evan der W aals retrata a energiade interações de van der Waals comumente obtida através do potencial de Lenard-Jones(CRAMER, 2010).
Contudo, apesar destes métodos serem úteis para sistemas complexos, calculandogeometrias e energias, eles não fornecem informações sobre a estrutura eletrônica dasmoléculas estudadas, limitando assim a informação obtida.
A.2 Métodos ab initio
O termo ab initio vem do latim e significa "do inicio", implicando que nenhumparâmetro é empregado. Assim, esse método utiliza a solução numérica da equação de ondaindependente do tempo e não relativística de Schrödinger, fazendo uso da aproximação deBorn-Oppenheimer, a qual separa o movimento dos elétrons dos núcleos, devido a estespossuírem massa muito menor, e se movimentarem muito mais rapidamente que o núcleo.
Neste método de cálculo, aplicam-se aproximações bem definidas, as quais podemser sistematicamente melhoradas até o resultado convergente dos cálculos realizados. Estesmétodos não utilizam dados experimentais, exceto constantes físicas fundamentais.
D. R. Hartree propôs em 1927 um método auto consistente para a solução numéricada equação de onda de Schrödinger, o qual relaciona a interação do elétron com o núcleo ecom os demais elétrons do sistema. Este método é dito auto consistente, conhecido comoSCF (do inglês, Self Consistent Feld), pois resolve as equações propostas por Hartreeescolhendo um conjunto inicial de funções, observa o potencial obtido e com eles calcula asnovas funções de onda, as quais são utilizadas para obter um novo potencial médio, o qualé novamente utilizado para resolver novas funções de onda até que os valores da energiafinal não mais variem em cada etapa.
O método proposto possuía o inconveniente de falhar em satisfazer o principio daassimetria, o qual consiste que a função de onda total que descreve um sistema de férmions

ANEXO A. Fundamentos teóricos da bioquímica computacional 133
deve ser antissimétrica na troca de qualquer conjunto de coordenadas de spin espaciais.Por conjunto de coordenadas de spin espacial, entende-se que os férmions não tem apenastrês graus de liberdade espaciais, mas também uma coordenada intrínseca de spin.
Menos de um ano após a publicação dos trabalhos de Hartree (HARTREE, 1928a;HARTREE, 1928b; HARTREE, 1929; HARTREE, 1957), Slater apresentou uma forma maissimples de representar a a função de onda, satisfazendo aos princípios da indistinguibilidadee da assimetria, conhecido simplesmente como determinante de Slater (SLATER, 1929).
Utilizando o principio variacional, o qual afirma que o valor médio do operadorhamiltoniano de um sistema será sempre maior ou igual a energia real deste sistema, Fock,um ano após a publicação de Slater, aplicou o método de Hartree na forma de determinantede Slater e deduziu as equações conhecidas hoje como HF (ou Hartree-Fock), o qual aprincipio foi chamada pelo próprio Fock como método de Hartree generalizado (FOCK,1930).
Em seguida, deduziu-se as equações de HF para o átomo de sódio, sendo que osprimeiros resultados obtidos a partir da solução numérica foi apresentada apenas quatroanos após a obtenção destas equações.
A resolução do método de HF também depende do campo auto consistente, queconsiste em partir de uma função de onda teste, submete-la ao operador de Fock, comparara função de onda emergente com a anterior e testar a convergência da resposta.
Neste método há a divisão dos orbitais moleculares (OM) em dois grupos, os quaisforam definidos como alfa e beta. Esses orbitais moleculares são ocupados separadamentepor elétrons com apenas um determinado spin (ROOTHAAN, 1951). Para sistemas comcamadas fechadas, o spin eletrônico não precisa ser considerado nas equações, já que ospin dos elétrons emparelhados nos OM se anulam mutualmente, porém para um sistemacom elétrons desemparelhados, o número de orbitais alfa ocupados é uma unidade maiorque o número de orbitais beta.
A.3 Métodos Semi-empíricos
A equação de Schrödinger é uma equação diferencial e sua resolução envolve aavaliação de um grande número de integrais. No caso de cálculos com métodos ab initio, onúmero de integrais cresce aproximadamente com a quarta potência do número de funçõesbase, chegando a alguns milhões mesmo para moléculas pequenas. Nos métodos quânticossemi-empíricos (SE), a negligência de um grande número dessas integrais foi a soluçãoadotada para economizar tempo de máquina e também reduzir a quantidade de memórianecessária nos cálculos.
Os método semiempíricos, assim como o método Hartree-Fock, utilizam o procedi-mento SCF, contudo, ao contrário do método HF, os métodos SE não resolvem as matrizesde Fock, apelando pra dados espectroscópicos ou propriedades físicas, como energia deionização, ou então utilizando uma série de regras para definir certas integrais com valorigual a zero.
A.4 Teoria do Funcional da Densidade
A teoria do funcional da densidade, DFT (da sigla inglesa Density FunctionalTheory), tem causado um impacto sem precedentes na aplicação da mecânica quântica

ANEXO A. Fundamentos teóricos da bioquímica computacional 134
(COHEN et al., 2012), sendo que ao longo dos últimos 20 anos, foi uma das ferramentasmais utilizadas na investigação e solução de problemas nas áreas de atuação da química(BURKE, 2012).
O uso da densidade eletrônica (ρ) como variável básica na descrição de um sistemaeletrônico remonta a proposta apresentada por Thomas (1927) e Fermi (1927). Os doispesquisadores, trabalhando de forma independente, empregaram um modelo estatísticopara aproximar a distribuição dos elétrons nos átomos, o qual ficou conhecido como modelode Thomas-Fermi, que posteriormente foi acrescido do termo da energia de troca por Dirac(DIRAC, 1928). Apesar da baixa qualidade das previsões para sistemas reais, este modeloé o precursor da moderna Teoria do Funcional da Densidade.
Em 1964, o norte-americano Walter Kohn publicou, juntamente com o seu alunoPierre Hohenberg, um artigo onde apresentavam uma reformulação da mecânica quânticabaseada, não em funções de onda, mas em densidade eletrônica, ρ(~r). Esta densidade medea probabilidade de encontrar-se um elétron no ponto de coordenada ~r (HOHENBERG;KOHN, 1964).
Neste trabalho, é demonstrado através de dois teoremas, que a partir da densidadeeletrônica do sistema no estado fundamental é possível obter a energia do estado funda-mental de maneira exata. Contudo, ainda não era possível determinar na prática o ρ(~r), oque foi conseguido no ano posterior com o auxílio de Lu Sham (KOHN; SHAM, 1965),onde o problema dos muitos corpos interagentes foi substituído por um problema auxiliarde partículas independentes.
Este procedimento (ou aproximação), conduziu ao cálculo exato de propriedades desistemas de muitos corpos, usando o método das partículas independentes; na prática, issotem permitido gerar formulações aproximadas com notável sucesso (PERDEW; BURKE;ERNZERHOF, 1996; GRIMME, 2006; ZHAO; TRUHLAR, 2006). Assim como os métodosde auto consistência, a aproximação de Kohn-Sham envolve partículas independentes, mascom uma densidade de partículas interagentes.
A aproximação de Kohn-Sham tem por objetivo substituir as interações complicadasde um sistema de muitos corpos, por um sistema auxiliar que pode ser resolvido muitofacilmente. Esta aproximação assume que a densidade do estado fundamental do sistemaoriginal interagente é igual a de algum sistema não-interagente. Isso conduz a equaçõesde partículas independentes, que descrevem um sistema não interagente, considerando-opassível de solução.
Contudo ainda se faz necessário incluir os termos de muitos corpos no Hamiltonianopara que esse sistema de partículas independentes possa descrever perfeitamente o problemareal. Assim, estes termos são implementados através do funcional de troca-correlação dadensidade, que é um dos termos do Hamiltoniano não interagente. Deste modo, a solução daequação nos fornece a densidade do estado fundamental e a energia do sistema interagenteoriginal com uma precisão limitada, sendo isso uma consequência das aproximações feitasno funcional de troca e correlação.
As duas principais aproximações utilizadas na construção dos funcionais de troca-correlação são as aproximações de densidade local (LDA - do inglês, Local Density Ap-proximation) e a de gradiente generalizado (GGA - do inglês, Generalized GradientApproximation). Nos parágrafos subsequentes será feita uma rápida descrição da estruturadestas duas aproximações e dos conjuntos de bases.

ANEXO A. Fundamentos teóricos da bioquímica computacional 135
A base da LDA está em considerar a energia de troca-correlação, Exc, para umsistema de densidade ρ(~r), como sendo a energia de troca-correlação para um gás deelétrons uniforme com a mesma densidade, que é conhecida de forma precisa. Presupõe-seque a densidade varia suavemente nas proximidades de ~r, ou seja, a energia de troca-correlação de um elétron em um dado ponto depende da densidade eletrônica nesse ponto,em vez de depender da densidade eletrônica em todos os pontos do espaço.
Contudo, métodos baseados em LDA não oferecem a precisão desejada em sistemasque apresentam inomogeneidade significativa na densidade eletrônica, levando a errosde forma não sistemática. Um avanço no sentido de resolver essas pendências consistemem introduzir a dependência com o gradiente da densidade na expressão do funcional detroca-correlação (GGA).
Neste formalismo, o funcional EGGAxc é geralmente dividido em duas partes, uma
contendo os termos do funcional de troca, e a outra o funcional de correlação. Existemtambém os funcionais híbridos, que são formados a partir de uma combinação de parte dotermo de troca do HF no funcional de troca da DFT (a partir de dados experimentais parasistemas moleculares conhecidos, contendo parâmetros ajustáveis). O uso desses funcionaisfaz com que alguns autores questionem a DFT como sendo uma teoria ab initio.
No manual do software Gaussian, pode-se obter uma lista com parte dos funcionaisde troca-correlação utilizados. Dentre eles, pode-se citar que os mais utilizados para ossistemas biológicos são:
• PBE - funcional de Perdew, Burke e Ernzerhof (1996)
• PW91 - funcional de correlação obtido por Perdew e Wang (1991)
• PWC - funcional de correlação obtido por Perdew e Wang (1992)
• B3LYP - termo de troca exato obtido por Becke (1993)
• Família M06 - são em sua maioria híbridos (M06, M062X, M06HF) ou puros (M06L),obtidos por Zhao e Truhlar (a parir de 2006)
Quando comparado com funcionais LDA, o uso de funcionais GGA apresentamelhoras consideráveis em descrever ligações entre moléculas sem representar um aumentoproibitivo no custo computacional. Contudo a descrição de ligações fracas permanecemproblemáticas, sendo necessária a utilização de métodos de correção da dispersão, comoos obtidos por Grimme e Ortmann et al. (GRIMME, 2006; ORTMANN; BECHSTEDT;SCHMIDT, 2006; GRIMME; EHRLICH; GOERIGK, 2011).
A.5 Fracionamento molecular com caps conjugados - MFCC
O método de fracionamento molecular com caps conjugados foi desenvolvido porZhang e Zhang no ano de 2003, tendo por base o pressuposto químico de que quanto maisdistante, menos o meio influencia na descrição das propriedades locais de uma molécula(ZHANG; ZHANG, 2003). Esta mesma linha de pensamento levou ao desenvolvimento dométodo de fragmentação do orbital molecular (FMO) por K. Kitaura em 1999 (KITAURAet al., 1999), um dos primeiros métodos de fragmentação molecular, e a diferenciação entrea região quântica e clássica utilizada em métodos híbridos (ex.: QM/MM) (KULIK et al.,2016).

ANEXO A. Fundamentos teóricos da bioquímica computacional 136
A estrutura geral do MFCC está apresentada na figura 4.7, onde vê-se que amolécula, representada por uma sequência primária repetira (onde n-1 e n representamos resíduos anteriores e posteriores, respectivamente). Em um modelo proteico, cadaresíduo individual é recortado da proteína em sua ligação peptídica e capas são adicionadasposteriormente com o intuito de fechar a valência que fora aberta com o fracionamento daproteína e imitar o ambiente eletrostático do local onde o fragmento estava (WU et al.,2007). Originalmente, as capas utilizadas foram fragmentos de CH3 e NH2 adicionadosàs regiões N e C-terminal, respectivamente. Pouco depois, M. Hatfield e colaboradoresdesenvolveram um estudo procurando identificar qual seria a composição das capas quemelhor representaria o meio, chegando conclusão de que quanto mais informação foradicionada, melhor a representação do ambiente eletrostático (HATFIELD et al., 2008).
Ao ser comparados com diversos cálculos em sistemas completos, utilizando variadosníveis de cálculo, o MFCC se mostrou eficaz em reproduzir os demais resultados, o quevem levando sua aplicação em vários sistemas, como biomoléculas e nanomateriais, paraobter um grande número de propriedades, incluindo energia total para estabilidade depeptídeos, ressonância nuclear magnética e a estabilidade de nanomateriais carbonados emcomplexo com íons e moléculas de água, sendo mais utilizado para o cálculo da energia deinteração entre complexos proteína-ligante (LIMA NETO et al., 2017; ZHANG, 2017; JINet al., 2016; LIU; HE; ZHANG, 2017), seguindo a equação:
EIMF CC(L − Ri) = E(L − CiRiCi∗) − E(CiRiCi∗)
− E(L − Ci Ci∗) + E(Ci Ci∗), (A.4)
onde o primeiro termo, o sistema de Ci + Ri + Ci∗ é separado da proteína, e o ligante (L)é adicionado, finalizando o primeiro momento com o cálculo da energia do sistema L, Ci,Ri e Ci∗. No segundo momento, o ligante é retirando do cálculo, estando somente os capse o resíduo principal (Ci, Ri e Ci∗). Posteriormente, o ligante é reintegrado ao sistema e oresíduo em estudo é retirado, resultando na interação do ligante com os caps (L, Ci e Ci∗).Ao final, somente a energia dos caps é obtida (Ci e Ci∗).
Similarmente ao que ocorreu com os demais métodos de fracionamento molecular,as últimas duas décadas viram um crescente desenvolvimento de aproximações que tentamaproximar o ambiente molecular em cada passo do processo de fragmentação. Lima Neto ecolaboradores apresentaram um modelo onde dez caps foram utilizados, Li e colaboradorespropuseram um método de correção energético baseado no MFCC (EC-MFCC) e queposteriormente foi nomeado fragmentação baseada na energia generalizada (GEBF), paracitar apenas alguns (LIMA NETO et al., 2015; LI; LI; FANG, 2005; LI; LI; JIANG, 2007).
O primeiro artigo publicado por nosso grupo utilizando o MFCC foi um estudosobre o modo de interação de estatinas em complexo com HMG-CoA redutase (DA COSTAet al., 2012), sendo seguido por BARROSO-NETO et al. e ZANATTA et al., com os doisprimeiros fazendo parte da tese de doutorado de R. Costa. Cada um desses trabalhos focouem um ponto diferente do uso do MFCC. DA COSTA et al. foi o primeiro trabalho autilizar o aminoácido inteiro como cap, formando um sistema onde o aminoácido anterior eposterior foram tratados no sistema, além de de sugerir a somatória da energia individualde cada resíduo por raio estudado para formar a energia total de ligação no sistemaestudado. BARROSO-NETO et al. utilizou o pressuposto de que após 6,5 Å as interaçõesintermoleculares tornam-se desprezíveis para calcular a energia de interação individual

ANEXO A. Fundamentos teóricos da bioquímica computacional 137
Adaptado de Zanatta et al. (2012)
Figura A.1 – Figura esquemática apresentando os momentos do MFCC.

ANEXO A. Fundamentos teóricos da bioquímica computacional 138
do complexo formado pela enzima ciclooxigenase 1 com bromoaspirina, diferente do casoanterior onde a quantidade de aminoácidos foi delimitada pela convergência do sistema.Já ZANATTA et al., utilizaram um sistema de blindagem para representar as barreirasfísicas e energéticas em um sistema formado pelo receptor de dopamina D3 e eticloprida.
A base por trás do método de blindagem utilizado por ZANATTA et al. vem domodelo de cap generalizado proposto por X. He e J. Zhang em 2006 (GMFCC ) (HE;ZHANG, 2006). Neste, dois aminoácidos são adicionados ao cálculo como caps extras, quenão estão ligados covalentemente ao resíduo de estudo. Esses aminoácidos são escolhidos apartir de um critério que pode ser considerado até certo ponto aleatório: dois aminoácidosque estejam dentro de um raio de 4,0 Å do resíduo principal e na mesma distância entre si.No caso do método de blindagem, todos os aminoácidos que estejam formando uma barreirafísica entre ligante e resíduo são adicionados ao cálculo, sendo a energia de interação entreligante e resíduo é obtida através da equação:
EI(L − Ri) = E(L − CiRiCi∗ − CjRjCj∗) − E(CiRiCi∗ − CjRjCj∗)
− E(CiCi∗ − CjCj∗) + E(CiCi∗ − CjCj∗), (A.5)
onde CjRjCj∗ representam o aminoácido que interfere na ligação do resíduo principal(Rj) com seus caps (CjCj∗). Para uma descrição visual do modelo, ver figura 2 do artigo(ZANATTA et al., 2012).
Na mesma época, Rodrigues e colaboradores desenvolviam um modelo derivadodo MFFC para trabalhar com modelos de interação entre proteína-proteína, sendo oprimeiro trabalho publicado com um estudo profundo da estabilidade nas três hélices deum colágeno sintético (RODRIGUES et al., 2013; BEZERRA et al., 2017). Enquanto isso,Dantas e colaboradores utilizavam o método de blindagem juntamente com um modelo desolvatação implícita derivado do GMFCC-CPCM desenvolvido pelo grupo de J. Zhang(DANTAS et al., 2015; MEI; JI; ZHANG, 2006).
Alguns autores vem apresentando os modelos que utilizam cargas pontuais pararepresentar o ambiente eletrostático como modelos mais fidedignos do que àqueles quefazem uso de modelos de solvatação implícita. Assim, X. Wang propôs um modelo ondeas cargas pontuais do sistema são incorporadas ao cálculos do GMFCC, sendo portantochamado de MFCC eletrostaticamente embebido (EE-MFCC ) (WANG et al., 2013b).Diferente do que ocorria anteriormente, a utilização das cargas pontuais não incorria emproblemas com relação ao limite no número de átomos do sistema, como ocorre duranteo uso de modelos de muitos caps (ex.: blindagem), bem como o sistema não é força aapresentar uma polarização que não condiz com o valor real, como ocorre nos modelos desolvatação contínua implícita.
O uso de cargas pontuais para aprimorar aprimorar o cálculo de propriedades emsistemas biológicos baseado em métodos de fracionamento vem sendo observado desde osseus primórdios, uma vez que esse modelo confere certo nível de polarização aos fragmentos.JIANG; MA; JIANG apresentaram um método de cálculo semelhante ao EE-MFCC aquiproposto, bem como LI; LI; JIANG e BRINKMANN; HEIFETS; KANTOROVICH. Ambosos trabalhos mostrando a precisão do uso de cargas pontuais ao MFCC. Não obstante, omodelo de cargas pontuais é largamente utilizado em cálculos híbridos (ex.: QM/MM),como forma de aproximar no cálculo quântico a posição e energia da região que está sendotratada classicamente, dessa forma comunicando as duas regiões (SENN; THIEL, 2009).

ANEXO A. Fundamentos teóricos da bioquímica computacional 139
Outro uso bastante comum para o uso das cargas pontuais ocorre no cálculo depropriedades de pequenas moléculas, em especial como forma simplificada de representaro efeito de solventes, uma vez que o tempo computacional gasto para calcular o efeito dosolvente a partir de átomos explícitos é muito maior do que utilizando as cargas individuaisdestes no seu lugar. Jakobsen e colaboradores utilizaram esse modelo para obter a estruturae a densidade de cargas na insulina, mostrando que o resultado com cargas foi mais próximodo esperado a partir de analises experimentais (JAKOBSEN; KRISTENSEN; JENSEN,2013). Manzoni e colaboradores utilizaram um modelo de cargas pontuais como solventepara as propriedades ópticas e magnéticas de pirimidina e outros compostos, mostrandoque o modelo utilizado foi similar ao modelo com átomos explícitos (MANZONI et al.,2010; MANZONI et al., 2011).
De forma simplista, pode-se representar as cargas pontuais a partir de um breveolhar no tópico de mecânica clássica acima. Uma vez que a mecânica molecular comumentenão faz referencia aos elétrons que compõe o átomo, a carga do átomo para a resoluçãodos termos de Coulomb é vista como um "ponto de carga", isto e, uma carga que possui ascoordenadas cartesianas bem definidas no espaço. O termo eletrostático obtido a partir daequação de Coulomb diz que duas cargas, com posições definidas, são atraídas ou repelidascom uma intensidade relacionada diretamente com a distancia entre elas. Assim, quantomenor a distancia, maior o efeito eletrostático. Esse ponto de carga com posição definidano espaço é a carga pontual.
Diferente dos modelos de solvatação implícitos, onde os átomos explícitos sãonormalmente circundados por um meio polarizável, onde cargas são adicionadas de formaa permitir que ambos, solvente e soluto, possam encontrar equilíbrio, as cargas pontuaisatuam de forma direta no calculo dos termos eletrostáticos. Dessa forma, surge umadificuldade para o uso desses modelos, pois seu efeito sobre a polarização do "soluto"não étao efetivo (em termos visíveis) quanto aquele do modelo de solvatação continua, emboracom maior similaridade ao sistema real.
Recentemente, Liu e colaboradores publicaram um trabalho procurando unir ométodo EE-GMFCC com o GMFCC-CPCM baseando-se na ideia proposta pelo métodoclássico MM/P(G)BSA (LIU et al., 2015). Nesse método, a energia de cada termo éfragmentada e calculada através de um modelo diferente. O termo MM (mecânica molecular)é utilizado para obter os termos energéticos comuns (ver mecânica clássica). Já os termosPB ou GB sao referentes a energia da solvatação, sendo obtidas através da resoluçãodas equações de Poisson-Boltzmann (PB) ou de Born generalizado (GB). Por ultimo, écalculado o efeito do deslocamento do solvente a partir da movimentação do soluto durantea dinâmica molecular (SA). Assim foi feito com o EE-GMFCC-CPCM. Durante o calculodo EE-GMFCC, a energia total do sistema é obtida, e posteriormente é realizado umcalculo usando modelos de solvatação implícitos para obter o termo de polarização dosistema, similar ao P(G)B. Ao final, ambas energias são somadas, resultando na energiatotal do sistema.
No presente trabalho, as energias de interação foram calculadas utilizando umaadaptação do esquema EE-GMFCC. Neste caso, não utilizamos os caps adicionais, tratandoo sistema contendo todos os átomos das proteínas como cargas pontuais em cada termodo cálculo energético. Isto é, em todos os termos, a energia dos átomos dos aminoácidosimplícitos são inseridas através de suas respectivas cargas atômicas parciais, considerando-se, dessa forma, o backgroung de cargas e sua contribuição eletrostática na energia deinteração entre cada fragmento. Portanto, a expressão utilizada para calcular essa energia

ANEXO A. Fundamentos teóricos da bioquímica computacional 140
é dada pela equação:
EIEE−MF CC(L − Ri) = E(L − CiRiCi∗ + CP C) − E(CiRiCi∗ − CP C)
− E(L − Ci C∗ − CP C) + E(Ci Ci∗ − CP C). (A.6)
onde o termo CP C representa todo o montante de cargas atreladas aos átomos explícitos. Osistema de Ci + Ri + Ci∗ é separado da proteína, e o ligante (L) é adicionado, finalizandoo primeiro momento com o cálculo da energia do sistema L, Ci, Ri e Ci∗. No segundomomento, o ligante é retirando do cálculo, estando somente os caps e o resíduo principal(Ci, Ri e Ci∗). Posteriormente, o ligante é reintegrado ao sistema e o resíduo em estudo éretirado, resultando na interação do ligante com os caps (L, Ci e Ci∗). Ao final, somente aenergia dos caps é obtida (Ci e Ci∗).
Copyright © 2022 FDOKUMEN




















![[ALACIP] NOBRE, Fábio. A Participação Estadunidense no Conflito Colombiano sob a Ótica dos Complexos Regionais de Segurança- Penetration ou Overlay](https://static.fdokumen.com/doc/165x107/631a2a98bb40f9952b01e216/alacip-nobre-fabio-a-participacao-estadunidense-no-conflito-colombiano-sob.jpg)