
Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer...
13
Molecular and Cellular Endocrinology 229 (2005) 161–173 Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer cells through differential regulation of c-Myc and cyclin D1 Amanda Mawson a , Angela Lai b , Jason S. Carroll c , C. Marcelo Sergio a , Christopher J. Mitchell d , Boris Sarcevic a,∗ a Cancer Research Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, NSW 2010, Australia b Johnson and Johnson Research Laboratories, Eveleigh, NSW 1430, Australia c Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA 02115, USA d Diabetes and Obesity Research Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, NSW 2010, Australia Received 11 May 2004; received in revised form 14 July 2004; accepted 11 August 2004 Abstract Estrogen and insulin/insulin-like growth factor-I (IGF-I) are major mitogens for breast epithelial cells and when co-administered, syner- gistically induce G 1 –S phase cell cycle progression. We investigated this cooperativity by evaluating if the key cell cycle regulators, c-Myc and cyclin D1, represent points of convergence in the action of these mitogens in MCF-7 breast cancer cells. These studies demonstrated that estrogen significantly increased both c-Myc and cyclin D1 protein, while insulin predominantly increased cyclin D1 levels. This cumulative increase in c-Myc and cyclin D1 contributes to the cooperativity of these mitogens, since ectopic expression of c-Myc or cyclin D1 cooperates with either the estrogen or insulin signaling pathways to increase cell cycle progression. Inhibition of the MAPK or PI3-kinase pathways significantly reduced c-Myc and cyclin D1 protein levels and cell cycle progression. Ectopic expression of cyclin D1 partially overcame this inhibition, while ectopic expression of c-Myc partially overcame MAPK but not PI3-kinase inhibition. Therefore, estrogen and insulin/IGF-1 differentially regulate c-Myc and cyclin D1 to cooperatively stimulate breast cancer cell proliferation. © 2004 Elsevier Ireland Ltd. All rights reserved. Keywords: Estrogen; Insulin; IGF-1; c-Myc; Cyclin D1 1. Introduction Estrogens elicit mitogenic activity which is important for the development of female reproductive tissues, particularly the uterus and mammary gland (Couse et al., 1999) and play a central role in the development and progression of human breast cancer (Colditz, 1998). The mechanisms through which estrogens stimulate cell proliferation are believed to be due to activation of estrogen receptor (ER) transcriptional activity (Couse and Korach, 1999) and potentially through non-transcriptional mechanisms by activation of intracellular signaling pathways such as the MAPK pathway (Migliaccio ∗ Corresponding author. Tel.: +61 2 9295 8330; fax: +61 2 9295 8321. E-mail address: [email protected] (B. Sarcevic). et al., 1996; Castoria et al., 1999). In addition to estrogen, polypeptide growth factors such as IGF-1 also display mitogenic activity on target cells, including human breast epithelial cells (Surmacz et al., 1998). Binding of IGF-1 to IGF-1-IR leads to dimerization of the receptor, activation of its tyrosine kinase activity and phosphorylation of key sub- strate proteins such as insulin receptor substrate proteins-1 (IRS-1) (Myers et al., 1994) and -2 (IRS-2) (He et al., 1996) and Src homolgy collagen (SHC) (Giorgetti et al., 1994). The phosphorylated IRS and SHC substrate proteins in turn recruit different SH2-containing proteins to activate specific intracellular signaling pathways. Thus, phosphorylated IRS-1 activates phosphatidylinositol 3-kinase (PI3-kinase) by recruiting the p85 regulatory subunit of this complex (Jackson et al., 1998), while phosphorylated SHC activates 0303-7207/$ – see front matter © 2004 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.mce.2004.08.002
Transcript of Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in MCF-7 breast cancer...

Molecular and Cellular Endocrinology 229 (2005) 161–173
Estrogen and insulin/IGF-1 cooperatively stimulate cell cycleprogression in MCF-7 breast cancer cells through differential
regulation of c-Myc and cyclin D1
Amanda Mawsona, Angela Laib, Jason S. Carrollc, C. Marcelo Sergioa,Christopher J. Mitchelld, Boris Sarcevica,∗
a Cancer Research Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, NSW 2010, Australiab Johnson and Johnson Research Laboratories, Eveleigh, NSW 1430, Australia
c Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA 02115, USAd Diabetes and Obesity Research Program, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, NSW 2010, Australia
Received 11 May 2004; received in revised form 14 July 2004; accepted 11 August 2004
Abstract
d, syner-g s, c-Myca nstrated thate mulativei ooperatesw pathwayss ame thisi in/IGF-1d©
K
1
ttabwbans
,playast
n ofub-ins-1
turncificted
ase)lex
tes
0d
Estrogen and insulin/insulin-like growth factor-I (IGF-I) are major mitogens for breast epithelial cells and when co-administereistically induce G1–S phase cell cycle progression. We investigated this cooperativity by evaluating if the key cell cycle regulatornd cyclin D1, represent points of convergence in the action of these mitogens in MCF-7 breast cancer cells. These studies demostrogen significantly increased both c-Myc and cyclin D1 protein, while insulin predominantly increased cyclin D1 levels. This cu
ncrease in c-Myc and cyclin D1 contributes to the cooperativity of these mitogens, since ectopic expression of c-Myc or cyclin D1 cith either the estrogen or insulin signaling pathways to increase cell cycle progression. Inhibition of the MAPK or PI3-kinaseignificantly reduced c-Myc and cyclin D1 protein levels and cell cycle progression. Ectopic expression of cyclin D1 partially overcnhibition, while ectopic expression of c-Myc partially overcame MAPK but not PI3-kinase inhibition. Therefore, estrogen and insulifferentially regulate c-Myc and cyclin D1 to cooperatively stimulate breast cancer cell proliferation.2004 Elsevier Ireland Ltd. All rights reserved.
eywords:Estrogen; Insulin; IGF-1; c-Myc; Cyclin D1
. Introduction
Estrogens elicit mitogenic activity which is important forhe development of female reproductive tissues, particularlyhe uterus and mammary gland (Couse et al., 1999) and playcentral role in the development and progression of humanreast cancer (Colditz, 1998). The mechanisms throughhich estrogens stimulate cell proliferation are believed toe due to activation of estrogen receptor (ER) transcriptionalctivity (Couse and Korach, 1999) and potentially throughon-transcriptional mechanisms by activation of intracellularignaling pathways such as the MAPK pathway (Migliaccio
∗ Corresponding author. Tel.: +61 2 9295 8330; fax: +61 2 9295 8321.E-mail address:[email protected] (B. Sarcevic).
et al., 1996; Castoria et al., 1999). In addition to estrogenpolypeptide growth factors such as IGF-1 also dismitogenic activity on target cells, including human breepithelial cells (Surmacz et al., 1998). Binding of IGF-1 toIGF-1-IR leads to dimerization of the receptor, activatioits tyrosine kinase activity and phosphorylation of key sstrate proteins such as insulin receptor substrate prote(IRS-1) (Myers et al., 1994) and -2 (IRS-2) (He et al., 1996)and Src homolgy collagen (SHC) (Giorgetti et al., 1994).The phosphorylated IRS and SHC substrate proteins inrecruit different SH2-containing proteins to activate speintracellular signaling pathways. Thus, phosphorylaIRS-1 activates phosphatidylinositol 3-kinase (PI3-kinby recruiting the p85 regulatory subunit of this comp(Jackson et al., 1998), while phosphorylated SHC activa
303-7207/$ – see front matter © 2004 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.mce.2004.08.002

162 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
the MAPK pathway by recruiting the GRB/SOS adaptorproteins (Giorgetti et al., 1994). Both the PI3-kinase andMAPK pathways are important for IGF-I-stimulated prolif-eration of MCF-7 human breast cancer cells (Jackson et al.,1998).
It is now clear that the estrogen pathway can interactwith the insulin/IGF-1 pathway to elicit synergistic mito-genic responses in breast epithelial cells (van der Burg et al.,1988; Dupont et al., 2000; Lai et al., 2001). Several mecha-nisms have been proposed to underlie this cross-talk. Estro-gen has been reported to increase the expression of polypep-tide growth factors such as IGF-1 and their receptors (IGF-IR)which may then function in an autocrine fashion (Cullen etal., 1989; Lee et al., 1999; Yee and Lee, 2000). Other stud-ies have demonstrated that estrogen stimulates expression ofIRS-1 resulting in enhanced tyrosine phosphorylation fol-lowing IGF-1 stimulation (Lee et al., 1999; Molloy et al.,2000). Furthermore, it has been reported that ligand-boundER may activate the IGF-1 pathway by direct binding andactivation of IGF-IR (Kahlert et al., 2000). Other studieshave suggested that ER may activate IGF-1 signaling path-ways by directly binding to the p85 subunit of PI3-kinaseand the tyrosine kinase Src leading to activation of the PI3-kinase and p21ras/MAPK pathways (Migliaccio et al., 1996;Castoria et al., 2001). In addition to estrogen regulating theI at theI andf an-sa ,2 ran-s K-m ingI eM tim-u t in-h atest 8a .,2
tro-g eryr on.E cellsb ndp ofcIa s-t yc.E ens -io cellp fc ro-
gression (Prall et al., 1998). The mechanism whereby c-Mycstimulates cell cycle progression in breast epithelial cells isnot fully understood, however, recent studies suggest that c-Myc stimulates the expression of numerous genes to elicitits mitogenic effects (Fernandez et al., 2003; Orian et al.,2003). These studies concluded that c-Myc may regulate atleast 10% of all cellular promoters. One major mechanismwhereby c-Myc regulates cell cycle progression appears tobe activation of cyclin E/CDK2 through loss of the CDKinhibitor p21WAF1/Cip1 (Prall et al., 1998). In addition to c-Myc, the involvement of cyclin D/CDKs and cyclin E/CDK2is also pivotal in estrogen- and insulin/IGF-1-mediated G1–Sphase transition. Estrogen and insulin stimulate the expres-sion of cyclin D1 leading to activation of cyclin D1/CDK4(Prall et al., 1997; Lai et al., 2001), while ectopically ex-pressed cyclin D1 mimics the effects of estrogen on cell cy-cle progression (Prall et al., 1998). Conversely, decreasingcyclin D1 with antisense oligonucleotides decreases estro-gen activation of cyclin E/CDK2 (Prall et al., 2001). Thesemitogens also activate cyclin E/CDK2 to promote cell cycleprogression. Insulin increases the expression of cyclin E toactivate CDK2 while estrogen is believed to activate cyclinE/CDK2 by removal of the CDK inhibitor p21WAF1/Cip1 fromthis complex (Prall et al., 1997, 2001; Lai et al., 2001). Ulti-mately, cyclin/CDK complexes promote cell cycle progres-s ates( 998;S
them n es-t cellp esem andc eastc e-p ICI1 n, asd n-s r dif-f hatb olesd ssionl
2
2
c-M as,V td.,N n-t nol-o /42M Akt
GF-1 signaling pathway, recent studies have reported thGF-1 signaling pathway may regulate ER expressionunction. Thus, stimulation with IGF-1 increased ER trcriptional activity in ER-positive breast cancer cells (Lee etl., 1997) or COS-1 cells transfected with ER (Stoica et al.000). The mechanism underlying this increased ER tcriptional activity may be due, at least in part, to MAPediated phosphorylation of serine 118 on ER follow
GF-1 stimulation (Kato et al., 1995). The importance of thAPK and PI3-kinase pathways in IGF-1 and estrogen slated mitogenesis is exemplified by studies showing thaibition of these pathways with specific inhibitors abrog
he mitogenic effects of both IGF-1 (Jackson et al., 199)nd estrogen in human breast cancer cells (Lobenhofer et al000).
Ultimately the mitogenic pathways stimulated by esen and insulin/IGF-1 impinge on the cell cycle machinesponsible for mediating G1–S phase cell cycle progressistrogens induce mitogenic effects in breast epithelialy stimulating G0/G1 resting cells to enter the cell cycle arogress through the G1–S transition to complete a roundell division (Sutherland et al., 1983; Leung and Potter, 1987).GF-1 also stimulates progression from G1 into S phase (Chennd Rabinovitch, 1990). One of the critical mediators of e
rogen action in G1 phase is the transcription factor c-Mxpression of c-Myc is rapidly induced following estrogtimulation (Dubik et al., 1987) and downregulated followng antiestrogen treatment (Carroll et al., 2002). Antisenseligonucleotides to c-myc inhibit estrogen stimulatedroliferation (Watson et al., 1991), while overexpression o-Myc can mimic the effects of estrogen on cell cycle p
ion by phosphorylation of critical downstream substrWeinberg, 1995; Sarcevic et al., 1997; Zhao et al., 1arcevic et al., 2002).In the present study we sought to further elucidate
olecular mechanisms underlying the cross talk betweerogen and insulin/IGF-1 in stimulating breast cancerroliferation by investigating if pathways stimulated by thitogens cooperate by converging at the level of c-Myc
yclin D1. To perform these studies we used MCF-7 brancer cells, which were arrested in G0 phase by serum drivation and co-treatment with the pure antiestrogen82780 and then stimulated with insulin and/or estrogeescribed previously (Lai et al., 2001). These studies demotrate that estrogen and insulin cooperate through theierential regulation of c-Myc and cyclin D1 levels and toth the MAPK and PI3-kinase pathways play major rownstream of these mitogens by regulating the expre
evels of these key cell cycle regulators.
. Material and methods
.1. Antibodies
The following monoclonal antibodies were used:yc (9E10; American Type Culture Collection, Manass
A) and cyclin D1 (DCS-6; Novacastra Laboratories Lewcastle-upon-Tyne, UK). The following polyclonal a
ibodies were used: c-Myc (C-19; Santa Cruz Biotechgy). p44/42 MAPK (catalogue #9102), phospho p44APK (threonine 202/tyrosine 204) (catalogue #91015),

A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 163
(catalogue #9272) and phospho Akt (serine 473) (catalogue#9270) were all from New England Biolabs, Beverly, CA.The anti-�-Actin antibody (AC-15) was from Sigma.
2.2. Cell culture
The clonal MCF cell lines, mt.102, myc.108 and D1.13were generated following transfection with p�MT (mt.102),p�MTmyc (myc.108) or p�MTcycD1 (D1.13) and main-tained in RPMI 1640 medium as described previously (Prallet al., 1998; Lai et al., 2001). The effects of insulin andestradiol on cell cycle progression in MCF-7 cells were in-vestigated as described previously (Lai et al., 2001). To in-duce ectopic Myc expression in myc.108 cells or ectopiccyclin D1 expression in D1.13 cells, zinc sulphate (Zn2+)was added to a final concentration of 5�M, unless otherwisestated, concurrently with the addition of vehicle or estradioland/or insulin. To evaluate the effects of the MEK inhibitorPD98059 (35�M) and the phosphatidylinositol 3-kinase in-hibitor LY294002 (10�M) (Lobenhofer et al., 2000), thesedrugs were added directly to the media concurrently withthe addition of vehicle or estradiol and/or insulin. Analysisof cell cycle phase distribution by DNA flow cytometry wasperformed as described previously (Lai et al., 2001).
2
alinea endedi MN l1p -d T).T clar-iE ed byS lot-t CLd tions(
3
3ec
di-ae al.,2 CF-7 onistI w-i
et al., 1998), indicating that these molecules could mimicthe proliferative effects of estrogen. However, these stud-ies were performed in the presence of fetal bovine serum(FBS) and therefore the contribution of various polypeptidegrowth factors to this stimulatory effect was unknown. Theimportance of polypeptide growth factors in this context isexemplified by our previous studies in serum-free mediumdemonstrating that estrogen synergizes with insulin/IGF-1 to mediate G1–S phase cell cycle progression (Laiet al., 2001). In the present study we utilized this model toinvestigate if c-Myc and cyclin D1 represent major points ofconvergence in the cooperative action of these mitogens onbreast cancer cell cycle progression.
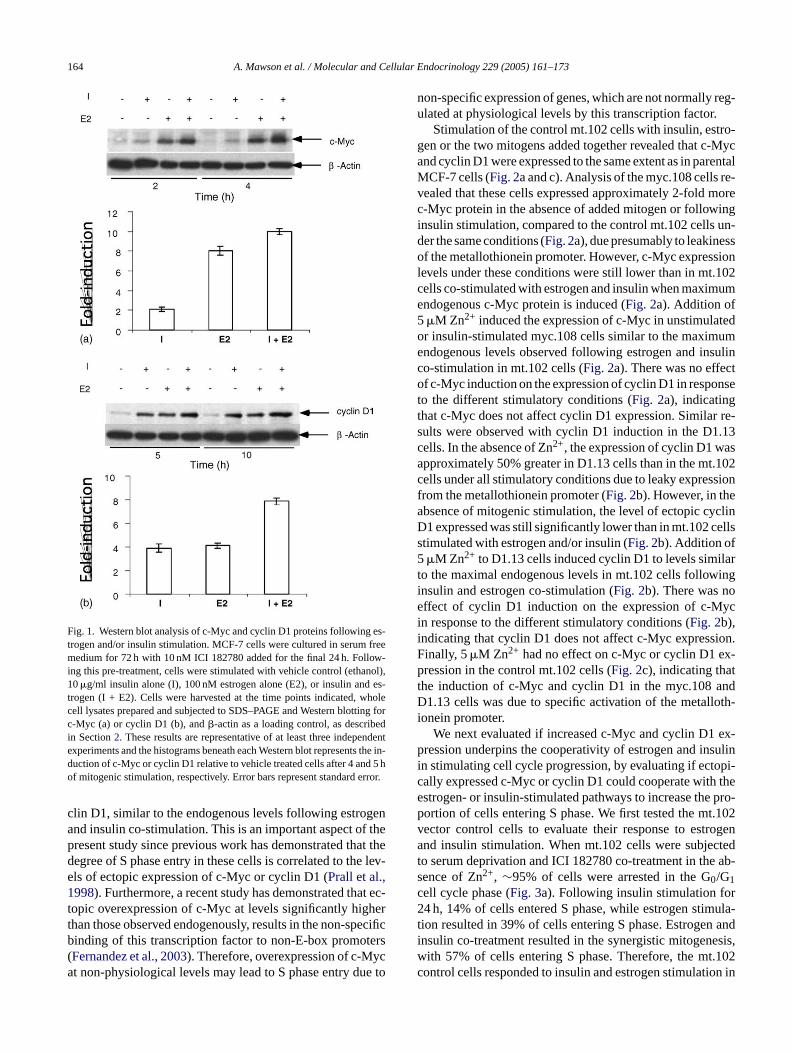
We first analysed the protein expression levels of c-Mycand cyclin D1 following stimulation of serum-deprived andICI 182780 treated MCF-7 cells with either insulin, estro-gen or both mitogens added concurrently (Fig. 1). West-ern blot analysis of the c-Myc protein following stimula-tion by insulin and/or estrogen revealed dramatically dif-ferent degrees of c-Myc expression induced by these mi-togens. Insulin stimulation for 2 or 4 h resulted in a mod-erate∼2-fold increase in c-Myc protein above the levelsdetectable in control unstimulated cells. Conversely, estro-gen significantly increased c-Myc levels to 6- and 8-foldat 2 and 4 h, respectively (Fig. 1). Together, the effects ofe sionw tiale elsrcW n ad-d ee-m2 re-c after1
3e
ationo entp ath-w wayt pon-s lep icallye geno . Top ncerc D1( etal-i on-t trols.W n ofZ cy-
.3. Western blot analysis
Cells were washed with ice-cold phosphate buffered snd then harvested by scraping, centifruged and resusp
n ice-cold lysis buffer (50 mM HEPES, pH 7.5, 150 maCl, 10% (v/v) glycerol, 1% Triton X-100, 1.5 mM MgC2,mM EGTA, 10�g/ml aprotinin, 10�g/ml leupeptin, 1 mMhenylmethylsulfonyl fluoride, 200�M sodium orthovanaate, 10 mM pyrophosphate, 100 mM NaF and 1 mM DThe lysates were incubated on ice for 5 min and then
fied by centrifugation at 15,000× g for 5 min at 4◦C.qual amounts of protein from the lysates were separatDS–PAGE, transferred to nitrocellulose and immunob
ed with the indicated antibody and detected with the Eetection system according to the manufacturers instrucAmersham Pharmacia Biotech).
. Results
.1. Estradiol and Insulin differentially stimulatexpression of c-Myc and cyclin D1 in MCF-7 breastancer cells
Both c-Myc and cyclin D1 are critical downstream metors of estrogen action in MCF-7 breast cancer cells (Prallt al., 1997, 1998, 2001; Lai et al., 2001; Carroll et002). Previous work in our laboratory has shown that Mcells that were growth-arrested by the estrogen antag
CI 182780 could re-initiate cell cycle progression follong ectopic expression of either c-Myc or cyclin D1 (Prall
strogen and insulin co-stimulation on c-Myc expresere additive at both time points. Unlike the differenffects on c-Myc expression, analysis of cyclin D1 levevealed that insulin or estrogen stimulated a∼4-fold in-rease in cyclin D1 protein levels at 5 and 10 h (Fig. 1).hen added together, estrogen and insulin stimulated a
itive ∼8-fold increase in cyclin D1 protein levels, in agrent with similar studies performed previously (Lai et al.,001). The changes in both c-Myc and cyclin D1 levels peded the entry of MCF-7 cells into S phase which began0 h of stimulation in this model system (Lai et al., 2001).
.2. c-Myc and cyclin D1 cooperate with insulin andstradiol to stimulate G1–S phase cell cycle progression
These data suggest that through the differential regulf their expression levels, c-Myc and cyclin D1 represoints of convergence in the estrogen and insulin/IGF-1 pays and that the cumulative contribution of each path
owards the expression of these molecules is in part resible for their cooperative actions on G1–S phase cell cycrogression. To test this hypothesis, we assessed if ectopxpressed c-Myc or cyclin D1 could cooperate with estror insulin stimulation to promote cell cycle progressionerform these studies we utilized clonal MCF-7 breast caell lines in which either c-Myc (clone myc.108) or cyclinclone D1.13) were expressed under the control of the mnducible metallothionein promoter. Clonal MCF-7 cells caining the empty vector (clone mt.102) were used as con
e first performed studies to determine the concentration2+ required to induce expression of ectopic c-Myc or
164 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
Fig. 1. Western blot analysis of c-Myc and cyclin D1 proteins following es-trogen and/or insulin stimulation. MCF-7 cells were cultured in serum freemedium for 72 h with 10 nM ICI 182780 added for the final 24 h. Follow-ing this pre-treatment, cells were stimulated with vehicle control (ethanol),10�g/ml insulin alone (I), 100 nM estrogen alone (E2), or insulin and es-trogen (I + E2). Cells were harvested at the time points indicated, wholecell lysates prepared and subjected to SDS–PAGE and Western blotting forc-Myc (a) or cyclin D1 (b), and�-actin as a loading control, as describedin Section2. These results are representative of at least three independentexperiments and the histograms beneath each Western blot represents the induction of c-Myc or cyclin D1 relative to vehicle treated cells after 4 and 5 hof mitogenic stimulation, respectively. Error bars represent standard error.
clin D1, similar to the endogenous levels following estrogenand insulin co-stimulation. This is an important aspect of thepresent study since previous work has demonstrated that thedegree of S phase entry in these cells is correlated to the lev-els of ectopic expression of c-Myc or cyclin D1 (Prall et al.,1998). Furthermore, a recent study has demonstrated that ec-topic overexpression of c-Myc at levels significantly higherthan those observed endogenously, results in the non-specificbinding of this transcription factor to non-E-box promoters(Fernandez et al., 2003). Therefore, overexpression of c-Mycat non-physiological levels may lead to S phase entry due to
non-specific expression of genes, which are not normally reg-ulated at physiological levels by this transcription factor.
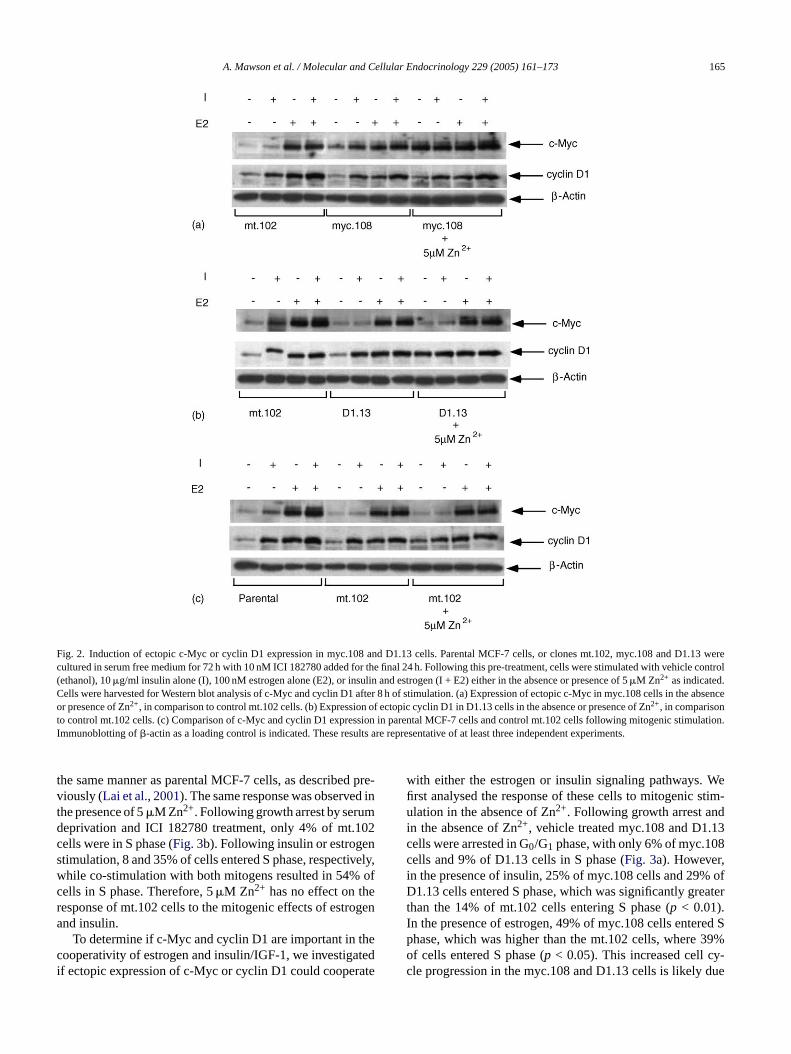
Stimulation of the control mt.102 cells with insulin, estro-gen or the two mitogens added together revealed that c-Mycand cyclin D1 were expressed to the same extent as in parentalMCF-7 cells (Fig. 2a and c). Analysis of the myc.108 cells re-vealed that these cells expressed approximately 2-fold morec-Myc protein in the absence of added mitogen or followinginsulin stimulation, compared to the control mt.102 cells un-der the same conditions (Fig. 2a), due presumably to leakinessof the metallothionein promoter. However, c-Myc expressionlevels under these conditions were still lower than in mt.102cells co-stimulated with estrogen and insulin when maximumendogenous c-Myc protein is induced (Fig. 2a). Addition of5�M Zn2+ induced the expression of c-Myc in unstimulatedor insulin-stimulated myc.108 cells similar to the maximumendogenous levels observed following estrogen and insulinco-stimulation in mt.102 cells (Fig. 2a). There was no effectof c-Myc induction on the expression of cyclin D1 in responseto the different stimulatory conditions (Fig. 2a), indicatingthat c-Myc does not affect cyclin D1 expression. Similar re-sults were observed with cyclin D1 induction in the D1.13cells. In the absence of Zn2+, the expression of cyclin D1 wasapproximately 50% greater in D1.13 cells than in the mt.102cells under all stimulatory conditions due to leaky expressionfa clinD ellss5 lart ingi oe ycii ion.F x-p tt ndD oth-i
ex-p sulini pi-c thee pro-p t.102v ogena ctedt ab-sc r2 ula-t andi esis,w .102c on in
-
rom the metallothionein promoter (Fig. 2b). However, in thebsence of mitogenic stimulation, the level of ectopic cy1 expressed was still significantly lower than in mt.102 ctimulated with estrogen and/or insulin (Fig. 2b). Addition of�M Zn2+ to D1.13 cells induced cyclin D1 to levels simi
o the maximal endogenous levels in mt.102 cells follownsulin and estrogen co-stimulation (Fig. 2b). There was nffect of cyclin D1 induction on the expression of c-M
n response to the different stimulatory conditions (Fig. 2b),ndicating that cyclin D1 does not affect c-Myc expressinally, 5�M Zn2+ had no effect on c-Myc or cyclin D1 eression in the control mt.102 cells (Fig. 2c), indicating tha
he induction of c-Myc and cyclin D1 in the myc.108 a1.13 cells was due to specific activation of the metall
onein promoter.We next evaluated if increased c-Myc and cyclin D1
ression underpins the cooperativity of estrogen and inn stimulating cell cycle progression, by evaluating if ectoally expressed c-Myc or cyclin D1 could cooperate withstrogen- or insulin-stimulated pathways to increase theortion of cells entering S phase. We first tested the mector control cells to evaluate their response to estrnd insulin stimulation. When mt.102 cells were subje
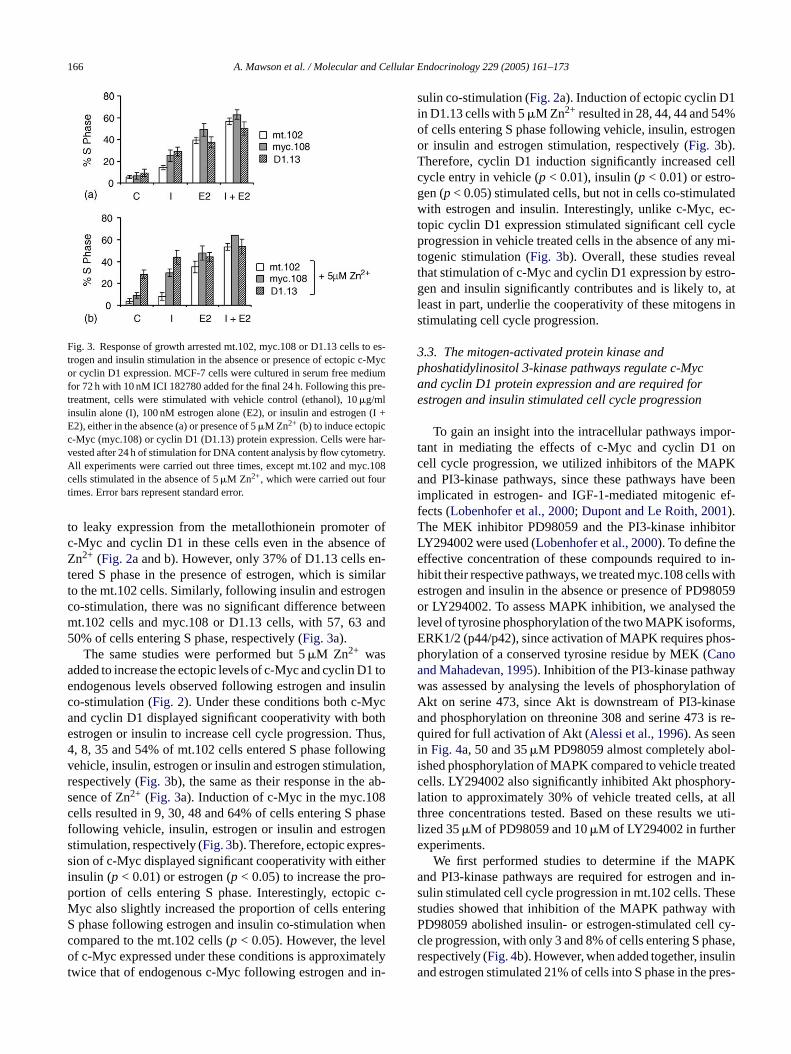
o serum deprivation and ICI 182780 co-treatment in theence of Zn2+, ∼95% of cells were arrested in the G0/G1ell cycle phase (Fig. 3a). Following insulin stimulation fo4 h, 14% of cells entered S phase, while estrogen stim
ion resulted in 39% of cells entering S phase. Estrogennsulin co-treatment resulted in the synergistic mitogenith 57% of cells entering S phase. Therefore, the mtontrol cells responded to insulin and estrogen stimulati
A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 165
Fig. 2. Induction of ectopic c-Myc or cyclin D1 expression in myc.108 and D1.13 cells. Parental MCF-7 cells, or clones mt.102, myc.108 and D1.13 werecultured in serum free medium for 72 h with 10 nM ICI 182780 added for the final 24 h. Following this pre-treatment, cells were stimulated with vehicle control(ethanol), 10�g/ml insulin alone (I), 100 nM estrogen alone (E2), or insulin and estrogen (I + E2) either in the absence or presence of 5�M Zn2+ as indicated.Cells were harvested for Western blot analysis of c-Myc and cyclin D1 after 8 h of stimulation. (a) Expression of ectopic c-Myc in myc.108 cells in the absenceor presence of Zn2+, in comparison to control mt.102 cells. (b) Expression of ectopic cyclin D1 in D1.13 cells in the absence or presence of Zn2+, in comparisonto control mt.102 cells. (c) Comparison of c-Myc and cyclin D1 expression in parental MCF-7 cells and control mt.102 cells following mitogenic stimulation.Immunoblotting of�-actin as a loading control is indicated. These results are representative of at least three independent experiments.
the same manner as parental MCF-7 cells, as described pre-viously (Lai et al., 2001). The same response was observed inthe presence of 5�M Zn2+. Following growth arrest by serumdeprivation and ICI 182780 treatment, only 4% of mt.102cells were in S phase (Fig. 3b). Following insulin or estrogenstimulation, 8 and 35% of cells entered S phase, respectively,while co-stimulation with both mitogens resulted in 54% ofcells in S phase. Therefore, 5�M Zn2+ has no effect on theresponse of mt.102 cells to the mitogenic effects of estrogenand insulin.
To determine if c-Myc and cyclin D1 are important in thecooperativity of estrogen and insulin/IGF-1, we investigatedif ectopic expression of c-Myc or cyclin D1 could cooperate
with either the estrogen or insulin signaling pathways. Wefirst analysed the response of these cells to mitogenic stim-ulation in the absence of Zn2+. Following growth arrest andin the absence of Zn2+, vehicle treated myc.108 and D1.13cells were arrested in G0/G1 phase, with only 6% of myc.108cells and 9% of D1.13 cells in S phase (Fig. 3a). However,in the presence of insulin, 25% of myc.108 cells and 29% ofD1.13 cells entered S phase, which was significantly greaterthan the 14% of mt.102 cells entering S phase (p < 0.01).In the presence of estrogen, 49% of myc.108 cells entered Sphase, which was higher than the mt.102 cells, where 39%of cells entered S phase (p < 0.05). This increased cell cy-cle progression in the myc.108 and D1.13 cells is likely due
166 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
Fig. 3. Response of growth arrested mt.102, myc.108 or D1.13 cells to es-trogen and insulin stimulation in the absence or presence of ectopic c-Mycor cyclin D1 expression. MCF-7 cells were cultured in serum free mediumfor 72 h with 10 nM ICI 182780 added for the final 24 h. Following this pre-treatment, cells were stimulated with vehicle control (ethanol), 10�g/mlinsulin alone (I), 100 nM estrogen alone (E2), or insulin and estrogen (I +E2), either in the absence (a) or presence of 5�M Zn2+ (b) to induce ectopicc-Myc (myc.108) or cyclin D1 (D1.13) protein expression. Cells were har-vested after 24 h of stimulation for DNA content analysis by flow cytometry.All experiments were carried out three times, except mt.102 and myc.108cells stimulated in the absence of 5�M Zn2+, which were carried out fourtimes. Error bars represent standard error.
to leaky expression from the metallothionein promoter ofc-Myc and cyclin D1 in these cells even in the absence ofZn2+ (Fig. 2a and b). However, only 37% of D1.13 cells en-tered S phase in the presence of estrogen, which is similarto the mt.102 cells. Similarly, following insulin and estrogenco-stimulation, there was no significant difference betweenmt.102 cells and myc.108 or D1.13 cells, with 57, 63 and50% of cells entering S phase, respectively (Fig. 3a).
The same studies were performed but 5�M Zn2+ wasadded to increase the ectopic levels of c-Myc and cyclin D1 toendogenous levels observed following estrogen and insulinco-stimulation (Fig. 2). Under these conditions both c-Mycand cyclin D1 displayed significant cooperativity with bothestrogen or insulin to increase cell cycle progression. Thus,4, 8, 35 and 54% of mt.102 cells entered S phase followingvehicle, insulin, estrogen or insulin and estrogen stimulation,respectively (Fig. 3b), the same as their response in the ab-sence of Zn2+ (Fig. 3a). Induction of c-Myc in the myc.108cells resulted in 9, 30, 48 and 64% of cells entering S phasefollowing vehicle, insulin, estrogen or insulin and estrogenstimulation, respectively (Fig. 3b). Therefore, ectopic expres-sion of c-Myc displayed significant cooperativity with eitherinsulin (p < 0.01) or estrogen (p < 0.05) to increase the pro-portion of cells entering S phase. Interestingly, ectopic c-Myc also slightly increased the proportion of cells enteringS henc elo atelyt in-
sulin co-stimulation (Fig. 2a). Induction of ectopic cyclin D1in D1.13 cells with 5�M Zn2+ resulted in 28, 44, 44 and 54%of cells entering S phase following vehicle, insulin, estrogenor insulin and estrogen stimulation, respectively (Fig. 3b).Therefore, cyclin D1 induction significantly increased cellcycle entry in vehicle (p < 0.01), insulin (p < 0.01) or estro-gen (p< 0.05) stimulated cells, but not in cells co-stimulatedwith estrogen and insulin. Interestingly, unlike c-Myc, ec-topic cyclin D1 expression stimulated significant cell cycleprogression in vehicle treated cells in the absence of any mi-togenic stimulation (Fig. 3b). Overall, these studies revealthat stimulation of c-Myc and cyclin D1 expression by estro-gen and insulin significantly contributes and is likely to, atleast in part, underlie the cooperativity of these mitogens instimulating cell cycle progression.
3.3. The mitogen-activated protein kinase andphoshatidylinositol 3-kinase pathways regulate c-Mycand cyclin D1 protein expression and are required forestrogen and insulin stimulated cell cycle progression
To gain an insight into the intracellular pathways impor-tant in mediating the effects of c-Myc and cyclin D1 oncell cycle progression, we utilized inhibitors of the MAPKand PI3-kinase pathways, since these pathways have beeni ef-fT itorLe o in-h withe 8059o thel s,E os-pa yw on ofA asea is re-qi l-i atedc ry-l t allt e uti-le
PKa d in-s eses ithP l cy-c ase,r lina pres-
phase following estrogen and insulin co-stimulation wompared to the mt.102 cells (p < 0.05). However, the levf c-Myc expressed under these conditions is approxim
wice that of endogenous c-Myc following estrogen and
mplicated in estrogen- and IGF-1-mediated mitogenicects (Lobenhofer et al., 2000; Dupont and Le Roith, 2001).he MEK inhibitor PD98059 and the PI3-kinase inhibY294002 were used (Lobenhofer et al., 2000). To define theffective concentration of these compounds required tibit their respective pathways, we treated myc.108 cellsstrogen and insulin in the absence or presence of PD9r LY294002. To assess MAPK inhibition, we analysed
evel of tyrosine phosphorylation of the two MAPK isoformRK1/2 (p44/p42), since activation of MAPK requires phhorylation of a conserved tyrosine residue by MEK (Canond Mahadevan, 1995). Inhibition of the PI3-kinase pathwaas assessed by analysing the levels of phosphorylatikt on serine 473, since Akt is downstream of PI3-kinnd phosphorylation on threonine 308 and serine 473uired for full activation of Akt (Alessi et al., 1996). As seen
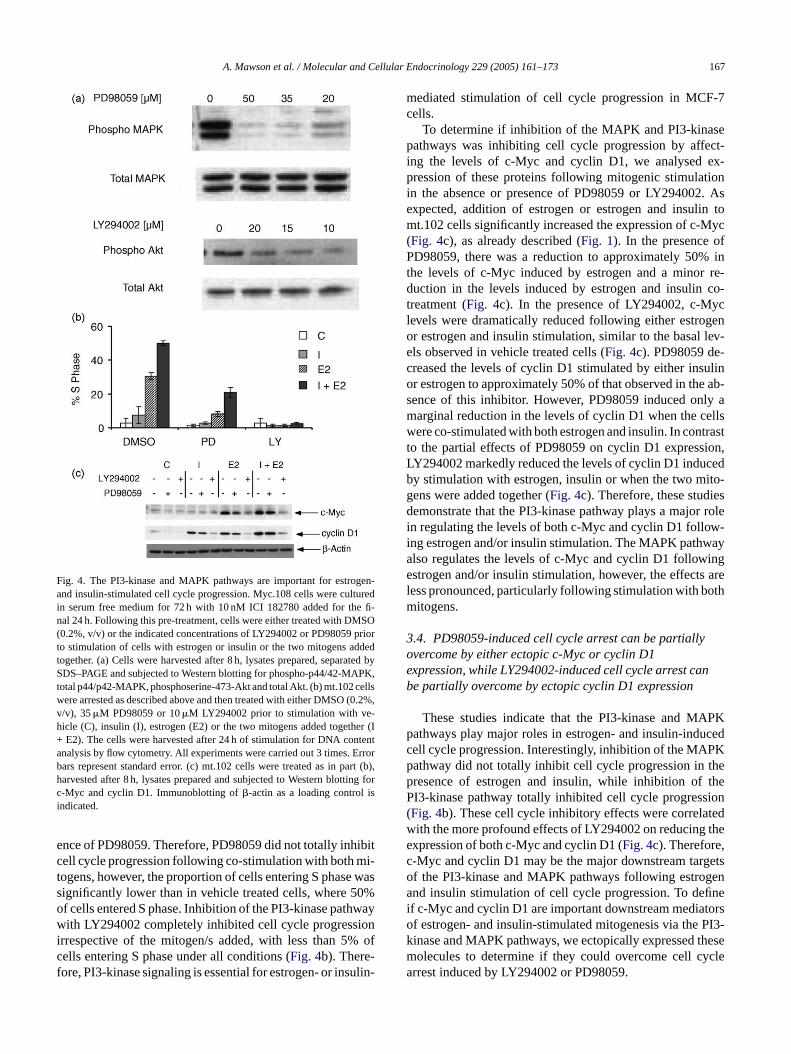
n Fig. 4a, 50 and 35�M PD98059 almost completely aboshed phosphorylation of MAPK compared to vehicle treells. LY294002 also significantly inhibited Akt phosphoation to approximately 30% of vehicle treated cells, ahree concentrations tested. Based on these results wized 35�M of PD98059 and 10�M of LY294002 in furtherxperiments.
We first performed studies to determine if the MAnd PI3-kinase pathways are required for estrogen anulin stimulated cell cycle progression in mt.102 cells. Thtudies showed that inhibition of the MAPK pathway wD98059 abolished insulin- or estrogen-stimulated celle progression, with only 3 and 8% of cells entering S phespectively (Fig. 4b). However, when added together, insund estrogen stimulated 21% of cells into S phase in the
A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 167
Fig. 4. The PI3-kinase and MAPK pathways are important for estrogen-and insulin-stimulated cell cycle progression. Myc.108 cells were culturedin serum free medium for 72 h with 10 nM ICI 182780 added for the fi-nal 24 h. Following this pre-treatment, cells were either treated with DMSO(0.2%, v/v) or the indicated concentrations of LY294002 or PD98059 priorto stimulation of cells with estrogen or insulin or the two mitogens addedtogether. (a) Cells were harvested after 8 h, lysates prepared, separated bySDS–PAGE and subjected to Western blotting for phospho-p44/42-MAPK,total p44/p42-MAPK, phosphoserine-473-Akt and total Akt. (b) mt.102 cellswere arrested as described above and then treated with either DMSO (0.2%,v/v), 35�M PD98059 or 10�M LY294002 prior to stimulation with ve-hicle (C), insulin (I), estrogen (E2) or the two mitogens added together (I+ E2). The cells were harvested after 24 h of stimulation for DNA contentanalysis by flow cytometry. All experiments were carried out 3 times. Errorbars represent standard error. (c) mt.102 cells were treated as in part (b),harvested after 8 h, lysates prepared and subjected to Western blotting forc-Myc and cyclin D1. Immunoblotting of�-actin as a loading control isindicated.
ence of PD98059. Therefore, PD98059 did not totally inhibitcell cycle progression following co-stimulation with both mi-togens, however, the proportion of cells entering S phase wassignificantly lower than in vehicle treated cells, where 50%of cells entered S phase. Inhibition of the PI3-kinase pathwaywith LY294002 completely inhibited cell cycle progressionirrespective of the mitogen/s added, with less than 5% ofcells entering S phase under all conditions (Fig. 4b). There-fore, PI3-kinase signaling is essential for estrogen- or insulin-
mediated stimulation of cell cycle progression in MCF-7cells.
To determine if inhibition of the MAPK and PI3-kinasepathways was inhibiting cell cycle progression by affect-ing the levels of c-Myc and cyclin D1, we analysed ex-pression of these proteins following mitogenic stimulationin the absence or presence of PD98059 or LY294002. Asexpected, addition of estrogen or estrogen and insulin tomt.102 cells significantly increased the expression of c-Myc(Fig. 4c), as already described (Fig. 1). In the presence ofPD98059, there was a reduction to approximately 50% inthe levels of c-Myc induced by estrogen and a minor re-duction in the levels induced by estrogen and insulin co-treatment (Fig. 4c). In the presence of LY294002, c-Myclevels were dramatically reduced following either estrogenor estrogen and insulin stimulation, similar to the basal lev-els observed in vehicle treated cells (Fig. 4c). PD98059 de-creased the levels of cyclin D1 stimulated by either insulinor estrogen to approximately 50% of that observed in the ab-sence of this inhibitor. However, PD98059 induced only amarginal reduction in the levels of cyclin D1 when the cellswere co-stimulated with both estrogen and insulin. In contrastto the partial effects of PD98059 on cyclin D1 expression,LY294002 markedly reduced the levels of cyclin D1 inducedby stimulation with estrogen, insulin or when the two mito-g esd r rolei w-i aya inge arel othm
3oe nb
PKp cedc Kp thep theP ion( tedw thee ,c getso gena finei torso PI3-k hesem yclea
ens were added together (Fig. 4c). Therefore, these studiemonstrate that the PI3-kinase pathway plays a majo
n regulating the levels of both c-Myc and cyclin D1 follong estrogen and/or insulin stimulation. The MAPK pathwlso regulates the levels of c-Myc and cyclin D1 followstrogen and/or insulin stimulation, however, the effects
ess pronounced, particularly following stimulation with bitogens.
.4. PD98059-induced cell cycle arrest can be partiallyvercome by either ectopic c-Myc or cyclin D1xpression, while LY294002-induced cell cycle arrest cae partially overcome by ectopic cyclin D1 expression
These studies indicate that the PI3-kinase and MAathways play major roles in estrogen- and insulin-induell cycle progression. Interestingly, inhibition of the MAPathway did not totally inhibit cell cycle progression inresence of estrogen and insulin, while inhibition ofI3-kinase pathway totally inhibited cell cycle progress
Fig. 4b). These cell cycle inhibitory effects were correlaith the more profound effects of LY294002 on reducingxpression of both c-Myc and cyclin D1 (Fig. 4c). Therefore-Myc and cyclin D1 may be the major downstream tarf the PI3-kinase and MAPK pathways following estrond insulin stimulation of cell cycle progression. To de
f c-Myc and cyclin D1 are important downstream mediaf estrogen- and insulin-stimulated mitogenesis via theinase and MAPK pathways, we ectopically expressed tolecules to determine if they could overcome cell crrest induced by LY294002 or PD98059.
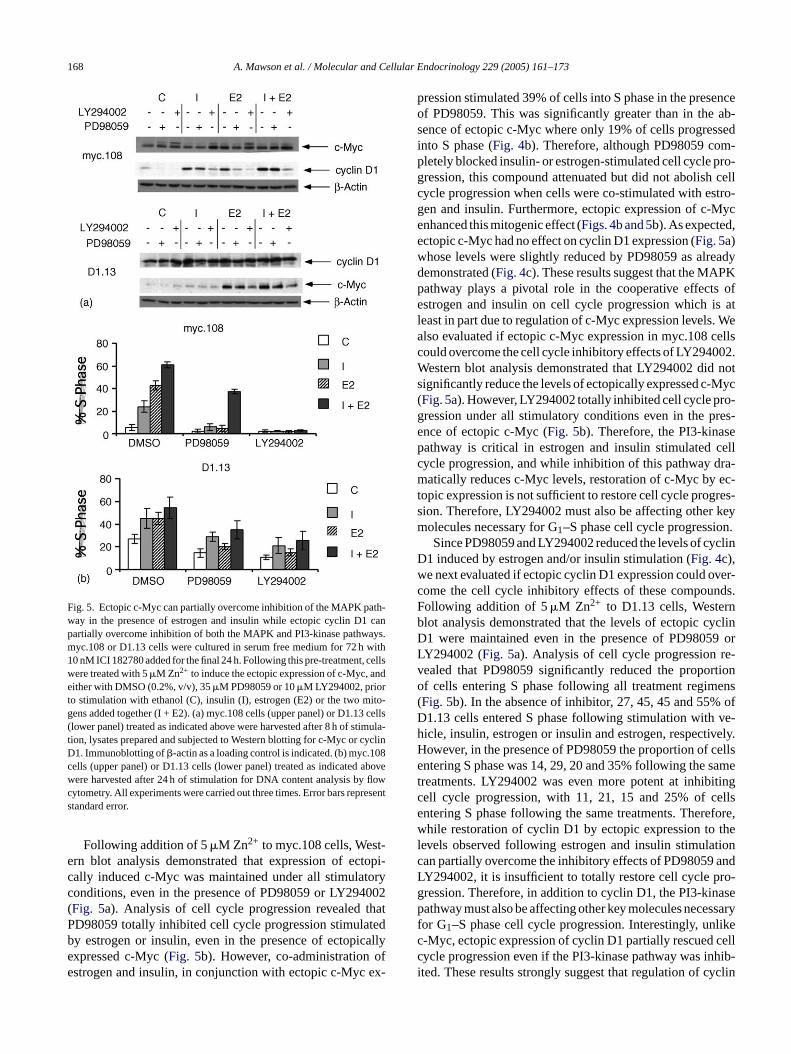
168 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
Fig. 5. Ectopic c-Myc can partially overcome inhibition of the MAPK path-way in the presence of estrogen and insulin while ectopic cyclin D1 canpartially overcome inhibition of both the MAPK and PI3-kinase pathways.myc.108 or D1.13 cells were cultured in serum free medium for 72 h with10 nM ICI 182780 added for the final 24 h. Following this pre-treatment, cellswere treated with 5�M Zn2+ to induce the ectopic expression of c-Myc, andeither with DMSO (0.2%, v/v), 35�M PD98059 or 10�M LY294002, priorto stimulation with ethanol (C), insulin (I), estrogen (E2) or the two mito-gens added together (I + E2). (a) myc.108 cells (upper panel) or D1.13 cells(lower panel) treated as indicated above were harvested after 8 h of stimula-tion, lysates prepared and subjected to Western blotting for c-Myc or cyclinD1. Immunoblotting of�-actin as a loading control is indicated. (b) myc.108cells (upper panel) or D1.13 cells (lower panel) treated as indicated abovewere harvested after 24 h of stimulation for DNA content analysis by flowcytometry. All experiments were carried out three times. Error bars representstandard error.
Following addition of 5�M Zn2+ to myc.108 cells, West-ern blot analysis demonstrated that expression of ectopi-cally induced c-Myc was maintained under all stimulatoryconditions, even in the presence of PD98059 or LY294002(Fig. 5a). Analysis of cell cycle progression revealed thatPD98059 totally inhibited cell cycle progression stimulatedby estrogen or insulin, even in the presence of ectopicallyexpressed c-Myc (Fig. 5b). However, co-administration ofestrogen and insulin, in conjunction with ectopic c-Myc ex-
pression stimulated 39% of cells into S phase in the presenceof PD98059. This was significantly greater than in the ab-sence of ectopic c-Myc where only 19% of cells progressedinto S phase (Fig. 4b). Therefore, although PD98059 com-pletely blocked insulin- or estrogen-stimulated cell cycle pro-gression, this compound attenuated but did not abolish cellcycle progression when cells were co-stimulated with estro-gen and insulin. Furthermore, ectopic expression of c-Mycenhanced this mitogenic effect (Figs. 4b and 5b). As expected,ectopic c-Myc had no effect on cyclin D1 expression (Fig. 5a)whose levels were slightly reduced by PD98059 as alreadydemonstrated (Fig. 4c). These results suggest that the MAPKpathway plays a pivotal role in the cooperative effects ofestrogen and insulin on cell cycle progression which is atleast in part due to regulation of c-Myc expression levels. Wealso evaluated if ectopic c-Myc expression in myc.108 cellscould overcome the cell cycle inhibitory effects of LY294002.Western blot analysis demonstrated that LY294002 did notsignificantly reduce the levels of ectopically expressed c-Myc(Fig. 5a). However, LY294002 totally inhibited cell cycle pro-gression under all stimulatory conditions even in the pres-ence of ectopic c-Myc (Fig. 5b). Therefore, the PI3-kinasepathway is critical in estrogen and insulin stimulated cellcycle progression, and while inhibition of this pathway dra-matically reduces c-Myc levels, restoration of c-Myc by ec-t res-s keym n.
yclinDw ver-c nds.F nb yclinD 9 orL re-v rtiono ens( ofD ve-h vely.H cellse samet itingc ellse fore,w thel tionc andL ro-g asep ssaryf likec cellc hib-i yclin
opic expression is not sufficient to restore cell cycle progion. Therefore, LY294002 must also be affecting otherolecules necessary for G1–S phase cell cycle progressioSince PD98059 and LY294002 reduced the levels of c
1 induced by estrogen and/or insulin stimulation (Fig. 4c),e next evaluated if ectopic cyclin D1 expression could oome the cell cycle inhibitory effects of these compouollowing addition of 5�M Zn2+ to D1.13 cells, Westerlot analysis demonstrated that the levels of ectopic c1 were maintained even in the presence of PD9805Y294002 (Fig. 5a). Analysis of cell cycle progressionealed that PD98059 significantly reduced the propof cells entering S phase following all treatment regimFig. 5b). In the absence of inhibitor, 27, 45, 45 and 55%1.13 cells entered S phase following stimulation withicle, insulin, estrogen or insulin and estrogen, respectiowever, in the presence of PD98059 the proportion ofntering S phase was 14, 29, 20 and 35% following the
reatments. LY294002 was even more potent at inhibell cycle progression, with 11, 21, 15 and 25% of cntering S phase following the same treatments. Therehile restoration of cyclin D1 by ectopic expression to
evels observed following estrogen and insulin stimulaan partially overcome the inhibitory effects of PD98059Y294002, it is insufficient to totally restore cell cycle pression. Therefore, in addition to cyclin D1, the PI3-kinathway must also be affecting other key molecules nece
or G1–S phase cell cycle progression. Interestingly, un-Myc, ectopic expression of cyclin D1 partially rescuedycle progression even if the PI3-kinase pathway was inted. These results strongly suggest that regulation of c

A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 169
D1 expression levels by the MAPK and PI3-kinase pathwaysfollowing stimulation with estrogen and insulin is critical forG1–S phase cell cycle progression.
4. Discussion
The importance of estrogens in the development of themammary gland and stimulating mitogenic activity in breastcancer cells is now well established. It is also now clear thatthe estrogen-stimulated pathway can synergise with variousgrowth factor stimulated pathways, and the cross talk of thesepathways contributes to the final mitogenic effect on tar-get cells by promoting G1–S phase cell cycle progression.Hence, the downstream molecules important for mediatingG1–S phase cell cycle progression may be concurrently regu-lated by these pathways to elicit the final mitogenic response.Previous studies have implicated pivotal roles for c-Myc andcyclin D1 in estrogen action as outlined in the Introduction,however, it is unclear if the mitogenic effects mediated byc-Myc and cyclin D1 are modulated by or dependent onpolypeptide growth factors. Furthermore, the relative con-tribution of these molecules to estrogen-induced mitogenesisis unclear.
Our results demonstrated differential effects of estrogena clinD andi me-d siono mi-t thes ntiala ratet rel-a ndi nalM er c-M ter.I cy-c velsf thata ssiono rrentw over-e oseo ng oft ze toe thist vels.S wasp ex-p cellc senceo Mycf the
presence of insulin or estrogen, ectopic c-Myc displayed sig-nificant cooperativity in stimulating cell cycle progression.These studies demonstrate several important issues related tothe importance of c-Myc in the mitogenic effects of estrogen.First, they demonstrate that in the absence of mitogenic stim-ulation, ectopic c-Myc cannot promote cell cycle progression,even when expressed at levels equivalent to those observedfollowing estrogen and insulin stimulation. It is possible thatc-Myc overexpressing cells may undergo an extended growthphase prior to cell cycle entry, as c-Myc has been implicatedin cellular growth control by regulating genes involved inprotein synthesis (Schmidt, 1999). However, studies in micehave shown that c-Myc is required for cell cycle progressionand is not required for cellular growth (Trumpp et al., 2001).Previous work has shown that ectopic c-Myc can mimic es-trogen in stimulating cell cycle progression in MCF-7 cells(Prall et al., 1998). However, these studies evaluated the abil-ity of ectopic c-Myc to stimulate S phase entry of MCF-7 cellsarrested with the antiestrogen ICI 182780 in the presenceof FBS, which is replete with polypeptide growth factors.Growth factors present in FBS are likely to be required forc-Myc action, consistent with our results demonstrating thatc-Myc displays significant cooperativity with the insulin sig-naling pathway. Our studies also demonstrate that ectopic c-Myc displayed significantly greater cooperativity with insulint on-s Myce at ec-t iresi Im-p topicc lino yn-e tion.T tar-g ffecto
tro-g thism actormd on-s ion ofc othm firste de-t mi-t yc,e es-s opicc heri n ofc rogena ig-n el ofe ental
nd insulin/IGF-1 on the expression of c-Myc and cy1, suggesting that the synergistic effects of estrogen
nsulin/IGF-1 on cell cycle progression may at least beiated, in part, by the differential and cumulative expresf these proteins. Therefore, the cooperativity of these
ogens is not simply due to the amplified regulation ofame downstream cell cycle molecules but rather differectivation of specific cell cycle molecules which coope
o elicit an increased mitogenic effect. To delineate thetive contribution of c-Myc and cyclin D1 to estrogen- a
nsulin-stimulated cell cycle progression, we utilized cloCF-7 breast cancer cells ectopically expressing eithyc or cyclin D1 under the control of an inducible promo
mportantly, we induced ectopic expression of c-Myc orlin D1 only to the maximum observed endogenous leollowing estrogen and insulin co-stimulation, to ensureny stimulatory effects were not due to gross overexpref these proteins. This is an important aspect of the cuork since a recent study has demonstrated that ectopicxpression of c-Myc at levels significantly higher than thbserved endogenously, results in the non-specific bindi
his transcription factor to non-E-box promoters (Fernandet al., 2003). This may lead to cell cycle progression duexpression of genes which are not normally regulated byranscription factor when expressed at physiological leince our results demonstrated that c-Myc expressionrimarily regulated by estrogen, we determined if ectopicression of c-Myc could mimic the effects of estrogen onycle progression. These studies revealed that in the abf estrogen or insulin signaling, ectopically expressed c-
ailed to stimulate cell cycle progression. However, in
han with estrogen. Since our Western blotting data demtrated that estrogen is predominantly responsible for c-xpression, these results are consistent with the view thopic c-Myc can partially mimic estrogen action but requnsulin/IGF-1 signaling to mediate its mitogenic effects.ortantly, our results also demonstrate that although ec-Myc displayed significant cooperativity with either insur estrogen, this cooperativity was still lower than the srgy observed following estrogen and insulin co-stimulaherefore, in addition to c-Myc, other important estrogenets are responsible for mediating the full stimulatory ef this mitogen.
We also investigated if cyclin D1 can cooperate with esen or insulin to stimulate cell cycle progression, sinceolecule has been implicated in estrogen and growth fitogenesis (Prall et al., 1998; Lai et al., 2001). Unlike theifferential effects on c-Myc expression, our results demtrated that insulin and estrogen stimulated the expressyclin D1 to equivalent levels and co-stimulation with bitogens resulted in an additive level of protein. We
valuated the effects of ectopic cyclin D1 expression toermine if this molecule could mimic the effects of theseogens on cell cycle progression. Interestingly, unlike c-Mctopic cyclin D1 stimulated significant cell cycle progrion in the absence of any mitogenic stimulation. Ectyclin D1 also displayed significant cooperation with eitnsulin or estrogen signaling to increase the proportioells entering S phase. However, in the presence of estnd insulin co-stimulation, ectopic cyclin D1 did not sificantly enhance this effect presumably since the levndogenous cyclin D1 expressed under these experim

170 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
conditions is sufficient and not rate limiting for cell cycleprogression. Although ectopic cyclin D1 displayed signifi-cant cooperativity with either insulin or estrogen, this coop-erativity was still lower than the synergy observed follow-ing estrogen and insulin co-stimulation. Therefore, as withc-Myc, cyclin D1 can only partially mimic the effects of es-trogen and insulin. Interestingly, ectopic cyclin D1 was sig-nificantly more potent than c-Myc in stimulating cell cycleprogression in the absence of any mitogenic stimulation or inconjunction with either mitogen alone. The molecular basisfor this is not fully understood. However, it is now clear thatcyclin D/CDKs have two important functions in regulatingG1–S phase cell cycle progression. One is catalytic, wherebycyclin D binding to CDK4 or CDK6 activates their kinaseactivity to phosphorylate the retinoblastoma gene productpRb, and stimulate G1–S phase progression (Sherr, 1995).An additional function of cyclin D/CDKs is to sequester theCDK inhibitors p21WAF1/Cip1 and p27Kip1 to relieve theirinhibitory action on cyclin E/CDK2, thereby activating thiscyclin/CDK complex in late G1 phase to stimulate progres-sion into S phase (Sherr and Roberts, 1999). Conversely, c-Myc can activate cyclin E/CDK2 by generating complexeslacking p21WAF1/Cip1 (Prall et al., 1998) which is likely dueto a reduction in p21WAF1/Cip1 levels (Lai et al., 2001). In ad-dition, c-Myc has been reported to increase the expression oft DK2( ersd yclinDc llp pRbdW ist ellc exes,w
rpin-n gena andM thati ithP cedt re th-w ofs c,w asek Myc( h-w K-3w mon-s tingc c-M nasep pres-
sion ofc-mycat the transcriptional level while insulin/IGF-1activation of the PI3-kinase and MAPK pathways leads to sta-bilization of c-Myc. Since ectopic expression of c-Myc couldpartially reverse the cell cycle inhibitory effects of PD98059following estrogen and insulin co-stimulation, these resultsdemonstrate that c-Myc regulation by the MAPK pathwayis a critical aspect of cooperation between the estrogen andinsulin. Interestingly, the inability of ectopic c-Myc to over-come cell cycle arrest of PD98059 when MCF-7 cells werestimulated with insulin or estrogen alone indicates that acti-vation of intracellular pathways by both of these mitogens isrequired for cooperation with c-Myc. Unlike the partial res-cue of PD98059-induced cell cycle arrest by ectopic c-Myc inthe presence of estrogen and insulin co-stimulation, ectopicc-Myc was unable to overcome inhibition of the PI3-kinasepathway under any of the conditions tested. Therefore, al-though the PI3-kinase pathway regulates c-Myc expression,this pathway also regulates other key cell cycle moleculeswhich are necessary for cell cycle progression.
Our results indicate that cyclin D1 represents at least oneof the key cell cycle regulators which is downstream of thePI3-kinase pathway and is required in addition to c-Myc tomediate the mitogenic effects of estrogen and insulin. The re-sults with inhibitors of the MAPK and PI3-kinase pathwaysdemonstrated a major role for the PI3-kinase pathway in reg-u clinD clinD thep con-d D1e mon-s asei cy-ca ands ki-n ingte -t nase3 nsi-b ;H Kaet on,i re-q D1i m oft andi
cede withr tro-g ath-w n stim-
he Cdc25A phosphatase which also activates cyclin E/CGalaktionov et al., 1996). The results of this study and othemonstrate that c-Myc does not lead to increases in c1 and activation of CDK4 (Prall et al., 1998). Since bothyclin D1/CDK4 and cyclin E/CDK2 are required for fuhosphorylation and inactivation of substrates such asuring G1–S phase (Resnitzky and Reed, 1995; Lundberg andeinberg, 1998; Harbour et al., 1999), one interpretation
hat cyclin D1 is more potent than c-Myc in stimulating cycle progression since it can activate both kinase complhereas c-Myc can only activate cyclin E/CDK2.Our experiments investigating the mechanism unde
ing the regulation of c-Myc and cyclin D1 levels by estrond insulin demonstrated critical roles of the PI3-kinaseAPK pathways. Previous studies have demonstrated
nhibition of either the PI3-kinase or MAPK pathways wD98059 or LY294002 does not inhibit estrogen-indu
ranscription ofc-mycmRNA in MCF-7 cells (Lobenhofet al., 2000). However, both the MAPK and PI3-kinase paays regulate c-Myc protein stability. Phosphorylationerine 62 by MAPK is required for stabilization of c-Myhile phosphorylation of threonine 58 by glycogen synthinase-3 (GSK-3) is associated with degradation of c-Sears et al., 1999, 2000). Stimulation of the PI3-kinase patay leading to activation of Akt leads to inactivation of GSand thus stabilization of c-Myc (Sears et al., 2000). Ourork is therefore consistent with these studies and detrates a major role for the PI3-kinase pathway in regula-Myc protein levels. The MAPK pathway also affectedyc protein levels but to a lesser extent than the PI3-kiathway. Therefore, estrogen has a major role in the ex
lating both the insulin- and estrogen-induced levels of cy1 and cell cycle progression. Unlike c-Myc, ectopic cy1 was able to partially rescue cell cycle progression inresence of PD98059 or LY294002 under all mitogenicitions tested. The PI3-kinase pathway regulates cyclinxpression at various levels. Previous studies have detrated that treatment of MCF-7 cells with the PI3-kinnhibitor LY294002 inhibits IGF-1-induced increases inlin D1 mRNA and protein (Dufourny et al., 1997, 2000). Inddition, the PI3-kinase pathway regulates the stabilityubcellular localization of cyclin D1. Glycogen synthasease 3� phosphorylates cyclin D1 on threonine 286 lead
o its translocation from the nucleus to the cytoplasm (Diehlt al., 1997, 1998; Hamelers et al., 2002) and increased pro
easomal degradation. Inhibition of glycogen synthase ki� by the PI3-kinase pathway is believed to be respole for stabilization of cyclin D1 protein (Diehl et al., 1998amelers et al., 2002). In addition, studies have linked MAPctivity to cyclin D1 mRNA and protein expression (Lavoiet al., 1996; Balmanno and Cook, 1999). As with c-Myc, ec-
opic cyclin D1 did not fully restore cell cycle progressindicating that other critical targets such as c-Myc areuired for the full mitogenic response. Therefore, cyclin
s a critical mediator of cell cycle progression downstreahe MAPK and PI3-kinase pathways following estrogennsulin stimulation.
Interestingly, both PD98059 and LY294002 redustrogen-stimulated cell cycle progression, associatedeductions in c-Myc and cyclin D1 suggesting that esen may be stimulating the MAPK and PI3-kinase pays. Previous studies have suggested that estrogen ca

A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 171
ulate MAPK activity by stimulating Src family members(Migliaccio et al., 1996; Wong et al., 2002). Other stud-ies have shown that estrogen can stimulate the productionof polypeptide growth factors in breast cancer cells whichcan act in an autocrine fashion to induce MAPK activation(Cullen et al., 1989; Keshamouni et al., 2002). The inter-action between estrogen and polypeptide growth factors iscomplex and it is likely that multiple mechanisms may besimultaneously active.
In conclusion, our studies show that the cooperativity ofestrogen and insulin in stimulating cell cycle progression inMCF-7 breast cancer cells is mediated in part through theirdifferential and cumulative effects on c-Myc and cyclin D1expression, and that the MAPK and PI3-kinase pathways arepivotal to this cooperation. These studies are consistent withprevious work demonstrating that the MAPK and PI3-kinasepathways cooperate with c-Myc to stimulate cell cycle pro-gression (Leone et al., 1997; Kerkhoff and Rapp, 1998; Jonesand Kazlauskas, 2001). Our data suggest that c-Myc and cy-clin D1 are critical points of convergence in estrogen andinsulin/IGF-1 cross-talk, which is required for activation of
F ro-gmccsiPatao
cyclin D1/CDKs and cyclin E/CDK2 to stimulate cell cycleprogression (Fig. 6). Finally, these studies have contributedto our understanding of how increased expression and activ-ity of receptor tyrosine kinase pathways, a common featurein many breast cancers, cooperate with estrogen to stimulatemitogenesis in human breast cancer cells which may haveimplications for breast cancer therapy. Since insulin/IGF-1primarily increases cyclin D1 expression even in the absenceof estrogen signaling, the development of cyclin D1/CDK4inhibitors to augment the antiproliferative effects of antie-strogens such as tamoxifen or for treatment of patients withestrogen receptor negative tumours may represent a new av-enue for the improved clinical management of breast cancer.
Acknowledgements
This work was supported by the National Health and Med-ical Research Council of Australia, The National Breast Can-cer Foundation of Australia and The Cancer Council NSW.
References
Alessi, D., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen,ase
B n isf
C mam-
C cha-CF-
ts of
C D.,of18,
C A.,io,entry
C ulin-der-ysiol.
C hor-r Inst.
ig. 6. Cooperation of c-Myc and cyclin D1 in mediating cell cycle p
ression following estrogen and IGF-1 co-stimulation. The model representsechanisms by which the estrogen and insulin/IGF-1 stimulated pathwaysross-talk to regulate c-Myc and cyclin D1 contributing to the activation ofyclin D1/CDK4 and cyclin E/CDK2 and G1–S phase cell cycle progres-ion. Estrogen induces induction of c-Myc and cyclin D1. IGF-1 primarilynduces cyclin D1 and also stabilizes the levels of c-Myc and cyclin D1 by theI3-kinase and MAPK pathways. Estrogen may also activate the PI3-kinasend MAPK pathways by stimulating the expression of autocrine growth fac-
ors. In addition, estrogen may stimulate the MAPK pathway through c-Srcctivation. Cyclin D1 activates CDK4/6 while c-Myc induces the activationf cyclin E/CDK2 to stimulate maximal G1–S phase cell cycle progression.
C e we417.
C uff,man839.
D thasetion.
D os-the
P., Hemmings, B., 1996. Mechanism of activation of protein kinB by insulin and IGF-1. EMBO J. 15, 6541–6551.
almanno, K., Cook, S., 1999. Sustained MAPK kinase activatiorequired for the expression of cyclin D1, p21Cip1 and a subset oAP-1 proteins in CCL39 cells. Oncogene 18, 3085–3097.
ano, E., Mahadevan, L., 1995. parallel siganal processing amongmalian MAPKs. Trends Biochem. Sci. 20, 117–122.
arroll, J., Swarbrick, A., Musgrove, E., Sutherland, R., 2002. Menisms of growth arrest by c-myc antisense oligonucleotides in M7 breast cancer cells: Implications for the antiproliferative effecantiestrogens. Cancer Res. 62, 3126–3131.
astoria, G., Barone, M., Domenico, M., Bilancio, A., Ametrano,Migliaccio, A., Auricchio, F., 1999. Non-transcriptional actionoestradiol and progestin triggers DNA synthesis. EMBO J.2500–2510.
astoria, G., Migliaccio, A., Bilancio, A., Di Domenico, M., de Falco,Lombardi, M., Fiorentino, R., Varrichio, L., Barone, M., AuricchF., 2001. PI3-kinase in concert with Src promotes the S-phaseof oestradiol-stimulated MCF-7 cells. EMBO J. 20, 6050–6059.
hen, Y., Rabinovitch, P., 1990. Altered cell cycle responses to inslike growth factor I, but not platelet-derived growth factor and epimal growth factor, in senescing human fibroblasts. J. Cell. Ph144, 18–25.
olditz, G., 1998. Relationship between estrogen levels, use ofmone replacement therapy, and breast cancer. J. Natl. Cance90, 814–823.
ouse, J., Korach, K., 1999. Estrogen receptor null mice: what havlearned and where will they lead us? Endocrinol. Rev. 20, 358–
ullen, K., Yee, D., Bates, S., Brunner, N., Clarke, R., Dickson, R., HK., Paik, S., Rosen, N., Valverius, E., 1989. Regulation of hubreast cancer by secreted growth factors. Acta Oncol. 28, 835–
iehl, J., Xheng, M., Roussel, M., Sherr, C., 1998. Glycogen synkinase-3b regulates cyclin D1 proteolysis and subcellular localizaGenes Dev. 12, 3499–3511.
iehl, J.A., Zindy, F., Sherr, C.J., 1997. Inhibition of cyclin D1 phphorylation on threonine-286 prevents its rapid degradation viaubiquitin-proteasome pathway. Genes Dev. 11, 957–972.

172 A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173
Dubik, D., Dembinski, T., Shiu, R., 1987. Stimulation of c-myc oncogeneexpression associated with estogen induced proliferation of humanbreast cancer cells. Cancer Res. 47, 6517–6521.
Dufourny, B., Alblas, J., van Teeffelen, H.A.A.M., van Schaik, F.M.A.,van der Burg, B., Steenbergh, P.H., Sussenbach, J.S., 1997. Mitogenicsignalling of insulin-like growth factor I in MCF-7 human breastcancer cells requires phosphatidylinositol 3-kinase and is independentof mitogen-activated protein kinase. J. Biol. Chem. 272, 31163–31171.
Dufourny, B., van Teeffelen, H., Hamelers, I., Sussenbach, J., Steenbergh,P., 2000. Stabilization of cyclin D1 mRNA via the phosphatidylinositol3-kinase pathway in MCF-7 human breast cancer cells. J. Endocrinol.166, 329–338.
Dupont, J., Karas, M., LeRoith, D., 2000. The potentiation of estrogen oninsulin-like growth factor I action in MCF-7 human breast cancer cellsincludes cell cycle components. J. Biol. Chem. 275, 35893–35901.
Dupont, J., Le Roith, D., 2001. Insulin-like growth factor 1 and oestradiolpromote cell proliferation of MCF-7 breast cancer cells: new insightsinto their synergistic effects. J. Clin. Pathol. Mol. Pathol. 54, 149–154.
Fernandez, P., Frank, S., Wang, L., Schroeder, M., Liu, S., Greene, J.,Cocito, A., Amati, B., 2003. Genomic targets of the human c-Mycprotein. Genes Dev. 17, 1115–1129.
Galaktionov, K., Chen, X., Beach, D., 1996. Cdc25 cell cycle phosphataseas a target of c-myc. Nature 382, 511–517.
Giorgetti, S., Pelicci, P., Pelicci, G., van Obberghen, E., 1994. Involvementof Src-homology/collagen (SHC) proteins in signaling through theinsulin receptor and the insulin-like growth factor I receptor. Eur. J.Biochem. 223, 195–202.
Hamelers, I., van Schaik, R., Sipkema, J., Sussenbach, J., Steenbergh,P., 2002. Insulin-like growth factor I triggers nuclear accumulationof cyclin D1 in MCF-7S breast cancer cells. J. Biol. Chem. 277,
H 99.tionsCell
H son,the
twoS-2.
J -1 iswthman
J sis re-72.
K ohe,eptor
K saki,. Ac-gen-
K ling.
K -Biol.
L -likeDK2cellsiol.
L 96.
1,
Lee, A., Jackson, J., Gooch, J., Hilsenbeck, S., Coronado-Heinsohn, E.,Osborne, C., Yee, D., 1999. Enhancement of insulin-like growth fac-tor signaling in human breast cancer. Estrogen regulation of insulinreceptor substrate-1 expression in vitro and in vivo. Mol. Endocrinol.13, 787–796.
Lee, A., Weng, C., Jackson, J., Yee, D., 1997. Activation of estrogenreceptor-mediated gene transcription by IGF-1 in human breast cancercells. J. Endocrinol. 152, 39–47.
Leone, G., DeGregori, J., Sears, R., Jakoi, L., Nevins, J., 1997. Myc andRas collaborate in inducing accumulation of active cyclin E/CDK2and E2F. Nature 387, 422–426.
Leung, B., Potter, A., 1987. Mode of estrogen action on cell prolifer-ation in CAMA-1 cells, sensitivity of G1 phase population. J. Cell.Biochem. 34, 213–225.
Lobenhofer, E., Huper, G., Ihlehart, J., Marks, J., 2000. Inhibition ofmitogen-activated protein kinase and phsophatidylinositol 3-kinaseactivity in MCF-7 cells prevents estrogen-induced mitogenesis. CellGrowth Differ. 11, 99–110.
Lundberg, A.S., Weinberg, R.A., 1998. Functional inactivation of theretinoblastoma protein requires sequential modification by at least twodistinct cyclin-cdk complexes. Mol. Cell. Biol. 18, 753–761.
Migliaccio, A., Domenico, M., Castoria, G., de Falco, A., Bontempo,P., Nola, E., Auricchio, F., 1996. Tyrosine kinase/p21ras/MAP-kinasepathway activation by estradiol-receptor complex in MCF-7 cells.EMBO J. 15, 1292–1300.
Molloy, C., May, F., Westley, B., 2000. Insulin receptor substrate-1 ex-pression is regulated by estrogen in the MCF-7 human breast cancercell line. J. Biol. Chem. 275, 12565–12571.
Myers, M.J., Sun, X., White, M., 1994. The IRS-1 signaling system.Trends Biochem. Sci. 19, 289–293.
O , T.,ingrk.
P ol ofin E-
P land,clin99–
P land,ring1 ex-
ciation
R E in69.
S pho--, E-,337.
S gula-ted
S co-
S s Myc
S 000.pro-
S 0.S ative
2.S ole
47645–47652.arbour, J.W., Luo, R.X., Dei Santi, A., Postigo, A.A., Dean, D.D., 19
Cdk phosphorylation triggers sequential intramolecular interacthat progressively block Rb functions as cells move through G1.98, 859–869.
e, W., Craparo, A., Zhu, Y., O’Neill, T., Wang, L., Pierce, J., GustafT., 1996. Interaction of insulin receptor substrate 2 (IRS-2) withinsulin and insulin-like growth factor I receptors. Evidence fordistinct phosphotyrosine-dependent interaction domains within IRJ. Biol. Chem. 271, 11641–11645.
ackson, J., White, M., Yee, D., 1998. Insulin receptor substratethe predominant signaling molecule activated by insulin-like grofactor-I, Insulin, and Interlukin-4 in estrogen receptor-positive hubreast cancer cells. J. Biol. Chem. 273, 9994–10003.
ones, S., Kazlauskas, A., 2001. Growth-factor-dependent mitogenequires two distinct phases of signalling. Nat. Cell Biol. 3, 165–1
ahlert, S., Neudling, S., van Eickels, M., Vetter, H., Meyer, R., GrC., 2000. Estrogen receptor a rapidly activates the IGF-1 recpathway. J. Biol. Chem. 275, 18447–18453.
ato, S., Endoh, H., Masuhiro, Y., Kitamoto, T., Uchiyama, S., SaH., Masushige, S., Gotoh, Y., Nishida, E., Kawashima, H., 1995tivation of the estrogen receptor through phosphorylation by mitoactivated protein kinase. Science 270, 1491–1494.
erkhoff, E., Rapp, U., 1998. Cell cycle targets of Ras/Raf signalOncogene 17, 1457–1462.
eshamouni, V., Mattingly, R., Reddy, K., 2002. Mechanism of 17�-estradiol-induced Erk1/2 activation in breast cancer cells. J.Chem. 277, 22558–22565.
ai, A., Sarcevic, B., Prall, O., Sutherland, R., 2001. Insulin/insulingrowth factor-I and estrogen cooperate to stimulate cyclin E-Cactivation and cell cycle progression in MCF-7 breast cancerthrough differential regulation of cyclin E and p21WAF1/Cip1. J. BChem. 276, 25823–25833.
avoie, J., L’Allemain, G., Brunet, A., Muller, R., Pouyssegur, J., 19Cyclin D1 expression is regulated positively by the p42/p44MAPK
and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 2720608–20616.
rian, A., Steensel, B., Delrow, J., Bussemaker, H., Li, L., SawadoWilliams, E., Loo, L., Cowley, S., Yost, C., 2003. Genomic bindby the Drosophila Myc, Max, Mad/Mnt transcription factor netwoGenes Dev. 17, 1101–1114.
rall, O., Carroll, J., Sutherland, R., 2001. A low abundance ponascent p21WAF1/Cip1 is targeted by estrogen to activate cyclCDK2. J. Biol. Chem. 276, 45433–45442.
rall, O.W.J., Rogan, E.M., Musgrove, E.A., Watts, C.K.W., SutherR.L., 1998. c-Myc or cyclin D1 mimics estrogen effects on cyE-cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 18, 444508.
rall, O.W.J., Sarcevic, B., Musgrove, E.A., Watts, C.K.W., SutherR.L., 1997. Estrogen-induced activation of Cdk4 and Cdk2 duG1–S phase progression is accompanied by increased cyclin Dpression and decreased cyclin-dependent kinase inhibitor assowith cyclin E-Cdk2. J. Biol. Chem. 272, 10882–10894.
esnitzky, D., Reed, S.I., 1995. Different roles for cyclins D1 andregulation of the G1-to-S transition. Mol. Cell. Biol. 15, 3463–34
arcevic, B., Lilischkis, R., Sutherland, R.L., 1997. Differential phosrylation of T-47D human breast cancer cell substrates by D1-, D3and A-type cyclin/CDK complexes. J. Biol. Chem. 272, 33327–33
arcevic, B., Mawson, A., Baker, R.T., Sutherland, R.L., 2002. Retion of the ubiquitin-conjugating enzyme hHR6A by CDK-mediaphosphorylation. EMBO J. 21, 2009–2018.
chmidt, E.V., 1999. The role of c-myc in cellular growth control. Ongene 18, 2988–2996.
ears, R., Leone, G., DeGregori, J., Nevins, J., 1999. ras enhanceprotein stability. Mol. Cell 3, 169–179.
ears, R., Nuckolls, F., Haura, E., Taya, Y., Tamai, K., Nevins, J., 2Multiple Ras-dependent phosphorylation pathways regulate Myctein stability. Genes Dev. 14, 2501–2514.
herr, C.J., 1995. D-type cyclins. Trends Biochem. Sci. 20, 187–19herr, C.J., Roberts, J.M., 1999. CDK inhibitors: positive and neg
regulators of G1-phase progression. Genes Dev. 13, 1501–151toica, A., Saceda, M., Fakhro, A., Joyner, M., Martin, M., 2000. R
of insulin-like growth factor-1 in regulating estrogen receptor-� geneexpression. J. Cell. Bochem. 76, 605–614.

A. Mawson et al. / Molecular and Cellular Endocrinology 229 (2005) 161–173 173
Surmacz, E., Guvakova, M., Nolan, M., Nicosia, R., Sciacca, L., 1998.Type I insulin-like growth factor receptor function in breast cancer.Breast Cancer Res. Treat. 47, 255–267.
Sutherland, R., Reddel, R., Green, M., 1983. Effects of oestrogens on cellproliferation and cell cycle kinetics: a hypothesis on the cell cycleeffects of entioestrogens. Eur. J. Cancer Clin. Oncol. 19, 307–318.
Trumpp, A., Refaeli, Y., Oskarsson, T., Gasser, S., Murphy, M.R.M.G.,Bishop, J.M., 2001. c-Myc regulates mammalian body size by con-trolling cell number but not cell size. Nature 414, 768–773.
van der Burg, B., Rutteman, G., Blankenstein, M., de Laat, S., van Zoe-len, E., 1988. Mitogenic stimulation of human breast cancer cellsin a growth factor-defined medium: synergistic action of insulin andestrogen. J. Cell. Physiol. 134, 101–108.
Watson, P., Pon, R., Shiu, R., 1991. Inhibition of c-myc expression byphosphorothioate antisense oligonucleotide identifies a critical role
for c-myc in the growth of human breast cancer. Cancer Res. 51,3996–4000.
Weinberg, R.A., 1995. The retinoblastoma protein and cell cycle control.Cell 81, 323–330.
Wong, C., McNally, C., Nickbarg, E., Komm, B., Cheskis, B., 2002.Estrogen receptor-interacting protein that modulates its nongenomicactivity-crosstalk with Src/Erk phosphorylation cascade. Proc. Natl.Acad. Sci. 99, 14783–14788.
Yee, D., Lee, A., 2000. Crosstalk between the insulin-like growth factorsand estrogens in breast cancer. J. Mammary Gland Biol. Neoplasia 5,107–115.
Zhao, J., Dynlacht, B., Imai, T., Hori, T., Harlow, E., 1998. Expression ofNPAT, a novel substrate of cyclin E-CDK2, promotes S-phase entry.Genes Dev. 12, 456–461.




















