
Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function
36
Review Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function Arthur A. Spector a,b, *, Xiang Fang a , Gary D. Snyder a , Neal L. Weintraub b a Department of Biochemistry, 4-403 Bowen Science Building, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA b Department of Internal Medicine, E-315-A General Hospital, Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA Abstract Epoxyeicosatrienoic acids (EETs), which are synthesized from arachidonic acid by cytochrome P450 epoxygenases, function primarily as autocrine and paracrine effectors in the cardiovascular system and kidney. They modulate ion transport and gene expression, producing vasorelaxation as well as anti- inflammatory and pro-fibrinolytic effects. EETs are incorporated into the sn-2 position of phospholipids and are rapidly mobilized when a cell is treated with a Ca 2+ ionophore, suggesting that they may play a role in phospholipid-mediated signal transduction processes. Soluble epoxide hydrolase (sEH) converts EETs to dihydroxyeicosatrienoic acids (DHETs), and inhibition of sEH is a potential approach for enhancing the biological activity of EETs. EETs also undergo chain-elongation and b-oxidation, and the accumulation of partial b-oxidation products increases when sEH is inhibited. Some functional effects of EETs occur through activation of either the guanine nucleotide binding protein Gas or the Src signal transduction pathways, suggesting that EETs act by binding to membrane receptors. However, other evi- dence indicates that the modulation of gene expression occurs through an intracellular action of EETs. Because of the diversity of biochemical and functional responses produced by EETs, it is doubtful that a single mechanism or signal transduction pathway can account for all of their actions. # 2003 Elsevier Ltd. All rights reserved. Keywords: EET; DHET; Cytochrome P450; Phospholipid; Epoxide hydrolase; b-Oxidation; Chain-elongation; Ion channels; Receptor; EDHF; Inflammation 0163-7827/$ - see front matter # 2003 Elsevier Ltd. All rights reserved. doi:10.1016/S0163-7827(03)00049-3 Progress in Lipid Research 43 (2004) 55–90 www.elsevier.com/locate/plipres * Corresponding author. Tel.: +1-319-335-7913; fax: +1-319-335-9570. E-mail address: [email protected] (A.A. Spector).
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function

Review
Epoxyeicosatrienoic acids (EETs): metabolism andbiochemical function
Arthur A. Spectora,b,*, Xiang Fanga, Gary D. Snydera, Neal L. Weintraubb
aDepartment of Biochemistry, 4-403 Bowen Science Building, Carver College of Medicine,University of Iowa, Iowa City, IA 52242, USA
bDepartment of Internal Medicine, E-315-A General Hospital, Carver College of Medicine,
University of Iowa, Iowa City, IA 52242, USA
www.elsevier.com/locate/plipres
Abstract
Epoxyeicosatrienoic acids (EETs), which are synthesized from arachidonic acid by cytochrome P450epoxygenases, function primarily as autocrine and paracrine effectors in the cardiovascular system andkidney. They modulate ion transport and gene expression, producing vasorelaxation as well as anti-inflammatory and pro-fibrinolytic effects. EETs are incorporated into the sn-2 position of phospholipidsand are rapidly mobilized when a cell is treated with a Ca2+ ionophore, suggesting that they may play arole in phospholipid-mediated signal transduction processes. Soluble epoxide hydrolase (sEH) convertsEETs to dihydroxyeicosatrienoic acids (DHETs), and inhibition of sEH is a potential approach forenhancing the biological activity of EETs. EETs also undergo chain-elongation and b-oxidation, and theaccumulation of partial b-oxidation products increases when sEH is inhibited. Some functional effects ofEETs occur through activation of either the guanine nucleotide binding protein Gas or the Src signaltransduction pathways, suggesting that EETs act by binding to membrane receptors. However, other evi-dence indicates that the modulation of gene expression occurs through an intracellular action of EETs.Because of the diversity of biochemical and functional responses produced by EETs, it is doubtful that asingle mechanism or signal transduction pathway can account for all of their actions.# 2003 Elsevier Ltd. All rights reserved.
Keywords: EET; DHET; Cytochrome P450; Phospholipid; Epoxide hydrolase; b-Oxidation; Chain-elongation; Ionchannels; Receptor; EDHF; Inflammation
0163-7827/$ - see front matter # 2003 Elsevier Ltd. All rights reserved.
doi:10.1016/S0163-7827(03)00049-3
Progress in Lipid Research 43 (2004) 55–90
* Corresponding author. Tel.: +1-319-335-7913; fax: +1-319-335-9570.
E-mail address: [email protected] (A.A. Spector).

Contents
1. Introduction ............................................................................................................................................................. 58
2. EET content of tissue and plasma lipids .................................................................................................................. 60
3. Major pathways of EET metabolism ....................................................................................................................... 613.1. Incorporation into cells ................................................................................................................................... 613.2. Binding to FABP............................................................................................................................................. 613.3. Distribution in cell lipids ................................................................................................................................. 61
3.3.1. Structural effects of EET incorporation into phospholipids ............................................................... 623.3.2. Elimination of EETs from cell lipids................................................................................................... 62
3.4. Conversion to DHETs..................................................................................................................................... 633.4.1. Function of DHETs ............................................................................................................................ 63
3.5. Proposed mechanism for cellular EET metabolism......................................................................................... 63
4. Additional pathways of EET metabolism ................................................................................................................ 644.1. b-Oxidation of EETs ....................................................................................................................................... 64
4.1.1. Function of EET b-oxidation products............................................................................................... 674.1.2. b-Oxidation of 11,12-DHET ............................................................................................................... 67
4.2. Chain-elongation products .............................................................................................................................. 674.3. Fatty acid epoxide derivatives ......................................................................................................................... 68
4.3.1. Metabolism of EETs by cyclooxygenase ............................................................................................. 684.3.2. Formation of epoxy-derivatives by lipoxygenase ................................................................................ 694.3.3. Metabolism of EETs by cytochrome P450 o-oxidase ......................................................................... 704.3.4. Metabolism of EET by glutathione S-transferases.............................................................................. 70
5. Stimulated release of EET from cells ....................................................................................................................... 705.1. EET-mediated vascular relaxation .................................................................................................................. 72
6. o-3 Fatty acid analogues of EETs ........................................................................................................................... 73
7. Soluble epoxide hydrolase (sEH) ............................................................................................................................. 747.1. Distribution of sEH in mammalian tissues ..................................................................................................... 747.2. Molecular biology of sEH ............................................................................................................................... 747.3. Regulation of sEH expression ......................................................................................................................... 757.4. Enzymatic mechanism ..................................................................................................................................... 757.5. sEH and lipid phosphate phosphatase activity................................................................................................ 757.6. Metabolism of EETs by sEH .......................................................................................................................... 767.7. sEH inhibitors ................................................................................................................................................. 767.8. Potential applications of sEH inhibitors ......................................................................................................... 78
7.8.1. Cardiovascular system......................................................................................................................... 787.8.2. Respiratory system .............................................................................................................................. 79
8. Biochemical response to EETs: mechanisms of action............................................................................................. 808.1. The case for EET receptors ............................................................................................................................. 818.2. EETs and ion transport................................................................................................................................... 828.3. EET signaling and G-proteins......................................................................................................................... 838.4. EET signaling through tyrosine kinase-linked cascades .................................................................................. 838.5. EET and cytokine-mediated inflammation...................................................................................................... 858.6. Conclusions regarding EET signaling mechanisms ......................................................................................... 86
Acknowledgements........................................................................................................................................................ 86
References ..................................................................................................................................................................... 86
56 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

Nomenclature
Ang II angiotensin IIBKCa large conductance calcium-activated potassium channelCoA coenzyme ACYP cytochrome P450DCU N,N0-dicyclohexylureaDHET dihydroxyeicosatrienoic acidEDHF endothelium-derived hyperpolarizing factorEDP epoxydocosapentaenoic acidEEQ epoxyeicosaquatraenoic acidEET epoxyeicosatrienoic acidEGF epidermal growth factorEPA eicosapentaenoic acidERK extracellular signal-regulated kinaseFABP fatty acid binding proteinG-protein guanine nucleotide-binding proteinHB-EGF heparin-binding epidermal growth factor-like growth factorHCEC human coronary endothelial cellsHPETE hydroperoxyeicosatetraenoic acidHPLC high-performance liquid chromatographyHSF human skin fibroblastsIC50 half-inhibitory concentrationKATP ATP-sensitive potassium channelmEH microsomal epoxide hydrolaseNF-kB nuclear transcription factor-kBPAEC porcine aortic endothelial cellsPASMC porcine aortic smooth muscle cellsPC phosphatidylcholinePCEC porcine coronary artery endothelial cellsPE phosphatidylethanolaminePG prostaglandinPI phosphatidylinositolPKA protein kinase APPAR peroxisome proliferator-activated receptorsEH soluble epoxide hydrolaseSHR spontaneously hypertensive ratTHETA trihydroxyeicosatrienoic acid
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 57

1. Introduction
Three classes of eicosanoid biomediators are synthesized from arachidonic acid; the cyclooxy-genase, lipoxygenase and cytochrome P450 (CYP) products [1]. The CYP class is subdivided intotwo groups; the epoxyeicosatrienoic acids (EETs) formed by CYP epoxgenases, primarily theCYP2C and 2J isoforms in humans, and the arachidonic acid derivatives that are hydroxylated ator near the o-terminus by CYP o-oxidases, primarily the CYP 4A and 4F isoforms in humans[2,3]. As shown in Fig. 1, CYP epoxygenases produce four EET regioisomers from arachidonicacid; 5,6-, 8,9-, 11,12-, and 14,15-EET.Each CYP epoxygenase isozyme produces all four EET regioisomers, but one or two usually
are the predominant products. For example, 14,15-EET accounts for 41% of the EETs producedby rat heart CYP 2J3 [4], and 11,12-EET accounts for 58% of the EETs produced by rat kidneyCYP 2J23 [5]. These two regioisomers are the predominant EETs produced by many differentcells and tissues: for example, they account for 67–80% of the total EETs produced by five pur-ified and reconstituted rat CYP epoxygenases [2]. Each regioisomer consists of a mixture of R,S-and S,R-enantiomers. As a further complication, the enantiofacial selectivity is different for eachCYP epoxygenase, and the same isozyme can have a different selectivity for epoxidation at eachof the arachidonic acid double bonds [2].The various types of functional effects produced by EETs are listed in Table 1. EETs are
important modulators of cardiovascular function, and they produce these effects by acting pri-marily on the vasculature and in the kidney [6–8]. They are synthesized in blood vessels by CYPepoxygenases contained in endothelial cells [9], and they produce vasodilation in a number ofvascular beds by activating the smooth muscle large conductance Ca2+-activated K+ channels(BKCa) [10]. This hyperpolarizes the smooth muscle and produces vasorelaxation, lowering bloodpressure [6,8,11]. A substantial amount of evidence indicates that EETs may function as endo-thelium-derived hyperpolarizing factors (EDHF) [12], particularly in the coronary circulation[13]. Furthermore, effects of EETs on smooth muscle migration [14], prostaglandin (PG) E2
Fig. 1. EET regioisomers synthesized from arachidonic acid by CYP epoxygenases.
58 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

production [15], Ca2+ influx [16] and aromatase activity [17] have been observed. In the endo-thelium, EETs inhibit cytokine-induced inflammatory responses [18,19], increase Ca2+ entry[20,21], enhance fibrinolysis [22], and stimulate tube formation [23]. Functional effects also occurin other tissues [24–32]. In addition, EET binding to intact cell membranes and to recombinantfatty acid binding protein (FABP) has been observed [31,32].EETs are enzymatically hydrated to the corresponding dihydroxyeicosatrienoic acid (DHET)
by epoxide hydrolases [33,34]. This reaction is illustrated in Fig. 2, which shows the conversion of14,15-EET to 14,15-DHET. DHETs were initially thought to be inactivation products of EETs,but several recent studies indicate that, like EETs, they produce vasodilation and activate smoothmuscle BKCa channels [35–38]. There are two major epoxide hydrolase isozymes in mammaliantissues; soluble epoxide hydrolase (sEH) contained primarily in the cytosol [33,34], and micro-somal epoxide hydrolase (mEH) bound to intracellular membranes [39]. sEH is the main isozymethat acts on the EETs.Two recent findings in the cardiovascular system have heightened interest in EETs. One is that
11,12-EET appears to be a strong candidate for EDHF in the coronary circulation [13]. The otheris that disruption of the sEH gene [40] or inhibition of sEH by N,N0-dicyclohexylurea (DCU), a
Table 1Functional effects of EETs
Process
Regioisomer ReferencesVascular smooth muscle
Activation of BKCa ion channel
All [6,10] Inhibition of migration 11,12-EET [14] Inhibition of PGE2 production 8,9- and 14,15-EET [15] Calcium influx All [16]Inhibition of aromatase activity
11,12- and 14,15-EET [17]Endothelium
Calcium signaling
11,12-EET [20,21] Increased tissue plasminogen activator 11,12-EET [22] Decreased cytokine-induced cell adhesion 11,12-EET [18]Stimulation of tube formation
Not specified [23]Myocardium
Inhibition of sodium ion channel
All [24] Activation of KATP ion channel 8,9- and 11,12-EET [25]Other tissues
Modulation of protein kinase C
14,15-EET [26] Stimulates tyrosine phosphorylation All [27] Epidermal growth factor signaling 14,15-EET [28]Stimulates ADP-ribosylation
14,15-EET [29] Inhibition of platelet cyclooxygenase 8,9- and 14,15-EET [30] Binding to macrophage membrane 14,15-EET [31]Binding to cytosolic fatty acid binding protein
All [32]A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 59

selective sEH inhibitor [41], reduces blood pressure [42], suggesting that sEH is a potentialpharmacological target for hypertension.This review will focus on the metabolism and biochemical mechanisms of action of EETs. The
reader is referred to other recent reviews for in-depth coverage of EET chemistry, physiology andpharmacology [2,3,6–8,11,43,44].
2. EET content of tissue and plasma lipids
Animal and human tissue lipids contain EETs. Rat liver phosphatidylcholine (PC), phosphati-dylethanolamine (PE) and phosphatidylinositol (PI) contain 70, 85 and 106 mmol EET/molphospholipid, respectively [45]. These phosphoglycerides contain 8,9-, 11,12- and 14,15-EET, andall of the EETs are esterified at the sn-2 position of the glycerol moiety. Rabbit kidney contains8,9- and 14,15-EET [46], and human kidney cortex contains 8,9-, 11,12- and 14,15-EET [47]. Theenantiomeric structures of the 11,12- and 14-15-EET present in the human kidney are similar tothose of the EETs produced by the major human CYP epoxygenases [47], leading to the conclu-sion that the EETs in tissue lipids are enzymatic products [45,48]. This may not always be thecase, however, because the amount of EETs esterified in phospholipids of human red blood cellsincreases considerably following incubation with t-butyl hydroperoxide. Therefore, the EETscontained in tissue phospholipids also can arise from lipid peroxidation [49].Rat plasma contains 10.2�0.4 ng/ml EET [48]. The molar ratio of 8,9-, 11,12- and 14,15-EET
in the plasma is 2.8:1:3.4, and more than 90% of the EET is contained in lipoprotein phospholi-pids. The highest concentration is present in low-density lipoproteins, followed by the high- andvery low-density lipoproteins. While it is generally assumed that EETs are produced in the tissueswhere they act, their presence in plasma suggests that they also may be supplied to at least sometissues by the circulation.
Fig. 2. Conversion of EET to DHET by epoxide hydrolase. The reaction is illustrated with 14,15-EET, but the otherEET regioisomers are hydrated in a similar manner.
60 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

3. Major pathways of EET metabolism
Most of the detailed information about EET metabolism has been obtained from incubationsof cultured mammalian cells with chemically synthesized [1-14C]EETs or [5,6,8,9,11,12,14,15-3H]EETs [50,51]. These studies have been done with murine mastocytoma cells [52], porcineand human endothelial cells [50,53,54], porcine arterial smooth muscle cells [15,35,55–57], humanskin fibroblasts (HSF) [58] and rat cerebral astrocytes [59].
3.1. Incorporation into cells
All of the cell types that so far have been tested rapidly take up radiolabelled EETs when theyare added to the culture medium as free fatty acids. It is not known whether the EETs passivelydiffuse through the cell membrane or require a membrane transporter to enter the cell. Theamount taken up by most cells reaches a maximum within 20–60 min. 14,15-EET uptake increa-ses linearly with increasing concentrations up to 5 mM in porcine aortic endothelial cells (PAEC)and up to 20 mM in porcine aortic smooth muscle cells (PASMC) [51,55], indicating that thecapacity of vascular cells to take up EETs is relatively large. The Km for 14,15-EET uptake bymurine mastocytoma cells is 1.1 mM, and the Vmax is 36 pmol/min � 107 cells [52]. Albuminreduces the uptake of 14,15-EET [52], most likely because it binds the EET and reduces theeffective concentration available for uptake. After a 2 h incubation with 2.5 mM EET, the com-parative uptake by PAEC is 5,6-EET>8,9-EET=1 l,12-EET>14,15-EET. The uptakes of 5,6-and 14,15-EET are 40 and 75% less, respectively, than the uptake of arachidonic acid [51]. Thecomparative uptake by PASMC is 8,9- >11,12- >14,15-EET [55]. Similarly, astrocytes take upmore 8,9-EET than 14,15-EET [59].
3.2. Binding to FABP
All of the EET regioisomers bind to cytosolic FABP [32]. Most of the studies have been donewith the heart isoform (H-FABP), but liver FABP (L-FABP) and intestinal FABP (I-FABP) alsobind EETs. The Kd for binding to H-FABP is between 0.4 and 1.7 mM, and the relative values are5,6-EET=11,12-EET<8,9-EET<14,15-EET. These Kd values for EET binding are 7–30 timeshigher than the Kd for arachidonic acid binding, whereas the Kd values for the EETs are 10–40times lower than those for DHET binding. The relatively weak binding of DHETs may explainwhy most of the DHET that is formed by cells is released into the extracellular fluid.These findings suggest that FABP may act as an intracellular transport protein for EETs and
target them to specific organelles or enzymes. Such a function would be consistent with the effectsof FABPs on the intracellular trafficking and utilization of fatty acids [60,61]. However, whetherEETs actually bind to FABP in an intact cell has not yet been determined.
3.3. Distribution in cell lipids
When EETs are taken up by PAEC and PASMC, they are incorporated primarily into phos-pholipids and to a small extent in glycerolipids [51,55]. PC incorporates the largest amount ofEETs in these vascular cells [51,57], whereas PE incorporates the largest amount in mastocytoma
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 61

cells [52]. As compared with other EETs, a relatively large fraction of the 14,15-EET taken up byPAEC and astrocytes is incorporated into PI [51,59], and a protein kinase C signaling mechanismmodulates the incorporation of 14,15-EET in the astrocytes [59]. Studies with acyl coenzyme A(CoA) synthase inhibitors indicate that most of the EET incorporated into phospholipids occursthrough a CoA-dependent mechanism [35,59].High-performance liquid chromatography (HPLC) analysis of cell lipids that were radiolabeled
with EET indicates that most of the incorporated radioactivity remains as EET [51,54,55,58].However, a small amount of the EET is elongated to a 22-carbon product, which is recoveredfrom the cell lipids along with very small amounts of radiolabeled DHET and several un-identified radiolabeled compounds [51,53,56]. Although much less extracellular DHET thanEET ordinarily is taken up [52], bovine aortic endothelial cells can incorporate small amountsof 8,9-, 11,12- and 14,15-DHET into phospholipids [62]. These DHETs are incorporated mainlyinto PC.Treatment of the radiolabeled phospholipids with phospholipase A2 indicates that 80% of the
14,15-EET incorporated into PI by astrocytes is present in the sn-2 position of the glycerol moiety[59]. This is consistent with the finding that endogenous EETs contained in rat liver phospholi-pids are present in the sn-2 position [45]. Likewise, almost all of the 14,15-EET incorporated intoPI and PC by PAEC, and into PI by PASMC, is contained in the sn-1 position [63]. By contrast,70% of the 14,15-EET incorporated into PC by PASMC is present in the sn-2 position. This mayexplain why more 14,15-EET was retained in PASMC than PAEC when these cells were exposedto a Ca2+ ionophore [63].The main phospholipid molecular species that contain EETs in the murine mastocytoma cells
are 16:0/EET in PC, 18:0/EET in PI, and 16:1, 18:0 and 18:2/EET in PE [52]. In addition, alkylether and plasmalogen species that contain EETs are present in the PC and PE fractions.
3.3.1. Structural effects of EET incorporation into phospholipidsIt has been suggested that the presence of phospholipids containing EET in the membrane lipid
bilayer may affect the physical properties of the membrane and the function of proteins embed-ded in the lipid bilayer [2]. However, even if a relatively large amount of EET is present, thenumber of EET acyl chains would be quite small as compared to fatty acyl chains. Therefore, ifsuch structural effects occur, they most likely are localized to only a few lipid domains and affectonly the proteins that interact with these domains. Another possibility is that cellular signaltransduction systems may be perturbed when EETs are present in membrane phospholipids. Thismost likely would involve phospholipase-dependent signaling processes that hydrolyze either PIor PC, the phospholipids that ordinarily incorporate the largest amounts of EET.
3.3.2. Elimination of EETs from cell lipidsMurine mastocytoma cells eliminate newly incorporated [3H] 14,15-EET under basal conditions
with a half-life of 34.9�7 h [52]. The rate of release from the phospholipids is PC>PI>PE. Incomparative studies, the mastocytoma cells were found to eliminate newly incorporated EETmuch faster than arachidonic acid. One possibility is that phospholipids containing esterifiedEETs are better substrates for phospholipases than those containing arachidonic acid in the sn-2position. It is also possible that phospholipids containing the EETs are localized to membranedomains that are more readily accessible to activated phospholipases. Another possibility is that
62 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

phospholipids containing the EETs may be excluded from incorporation into membranes and areinstead targeted to phospholipases for degradation.The retention of EETs in vascular cells is much less than in the neoplastic mastocytoma cells.
PAEC eliminate about 60% of the newly incorporated [1-14C]14,15-EET in 4 h under basal con-ditions. The release is largely from PC and PI [51]. Likewise, PASMC labeled with[3H]11,12-EET eliminate 35% of the uptake in 4 h under basal conditions [56]. Thus, EETsare retained for only a relatively short period of time following their incorporation intovascular cell lipids.
3.4. Conversion to DHETs
As shown in Fig. 2, the main pathway of EET metabolism in many cells is conversion to DHETby epoxide hydrolases, primarily sEH [33,34,44]. PAEC and PASMC convert all of the EETregioisomers tested to DHETs [51,55], whereas rat astrocytes convert 14,15-EET but not 8,9-EETto DHET [59]. The conversion of 14,15-EET to DHET begins rapidly; for example, [3H]14,15-DHET formation can be detected in PASMC cultures after only 3 min of exposure to [3H] 14,15-EET [57]. In PAEC and PASMC cultures, DHET production is not saturated even if the 14,15-EET concentration is as high as 5–10 mM [51,55]. Almost all of the EET that is eliminated fromthe cell lipids of PAEC and PASMC under basal conditions is recovered in the medium as DHET[51,56].
3.4.1. Function of DHETsDHETs are generally thought to be inactivation products of EETs [8,44]. Consistent with this
view, DHETs have diminished activity in many systems as compared with the four correspondingEETs. These include relaxation of the preglomerular vasculature [64], cyclooxygenase inhibitionin PASMC [15], Ca2+ uptake by PASMC [16], inhibition of the rat myocardial Na+ channel [24],activation of the myocardial ATP-sensitive K+(KATP) channel [25], stimulation of ADP-ribosy-lation [29], and relaxation of the bovine coronary artery [65]. On the other hand, DHETs producefunctional effects in several other systems. For example, DHETs inhibit the hydroosmotic actionof arginine vasopressin in the kidney [66], produce relaxation to a similar extent as the corre-sponding EETs in porcine coronary rings preconstricted with the thromboxane mimetic U-46619[35,56,57], dilate canine coronary arterioles preconstricted with endothelin [67], and activate theBKCa channel of rat coronary artery myocytes [37]. Thus, DHETs function as biomediators insome systems and are not merely EET inactivation products in all cases.
3.5. Proposed mechanism for cellular EET metabolism
Fig. 3 illustrates a proposed scheme for EET metabolism based on the currently available data.According to this hypothesis, FABP physically binds unesterified EET in the cytosol and reg-ulates its delivery to target proteins and enzymes. Some of the EET is incorporated into phos-pholipids through a CoA-dependent process and is then slowly eliminated from the phospholipidsby phospholipase-mediated hydrolysis under basal conditions. Some of the unesterified EET isconverted to DHET, and most of the DHET is released into the extracellular fluid. Although notshown in Fig. 3, studies in PAEC, PASMC and porcine coronary artery endothelial cells (PCEC)
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 63

clearly indicate that very small amounts of DHET are retained intracellularly and can beincorporated into cell phospholipids [51,53,56,62].
4. Additional pathways of EET metabolism
Besides the major pathways illustrated in Fig. 3, EETs can undergo a number of other meta-bolic conversions. These additional reactions, which include partial b-oxidation, chain-elonga-tion, oxidation by the cyclooxygenase, lipoxygenase and CYP o-oxidase enzymes, andconjugation with glutathione are illustrated schematically in Fig. 4.
4.1. �-Oxidation of EETs
The production of radiolabeled b-oxidation products initially was observed during incubationof [3H]11,12-EET with HSF [58]. Fig. 5 illustrates the radiolabeled products detected in the cul-ture medium by reverse-phase HPLC. As opposed to the other cells and tissues that had beenstudied previously, the HSF converted only a relatively small amount of the [3H]11,12-EET toDHET. The majority of the radioactivity was detected in three additional products designated inthe chromatogram as I, II and III.Fig. 6 shows the negative ion chemical ionization mass spectra of the pentafluorobenzyl ester
derivatives of the three metabolites produced by the HSF from 11,12-EET and, for comparison, asimilar mass spectrum of 11,12-EET (Fig. 6A). The metabolites are partial b-oxidation productsof 11,12-EET: I, 9,10-epoxy-�6,12-18:2 (Fig. 6B); II, 7,8-epoxy-�4,10-16:2 (Fig. 6C); III, 5,6-epoxy-�8-14:1 (Fig. 6D).
Fig. 3. Proposed mechanism for cellular metabolism of EETs.
64 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90
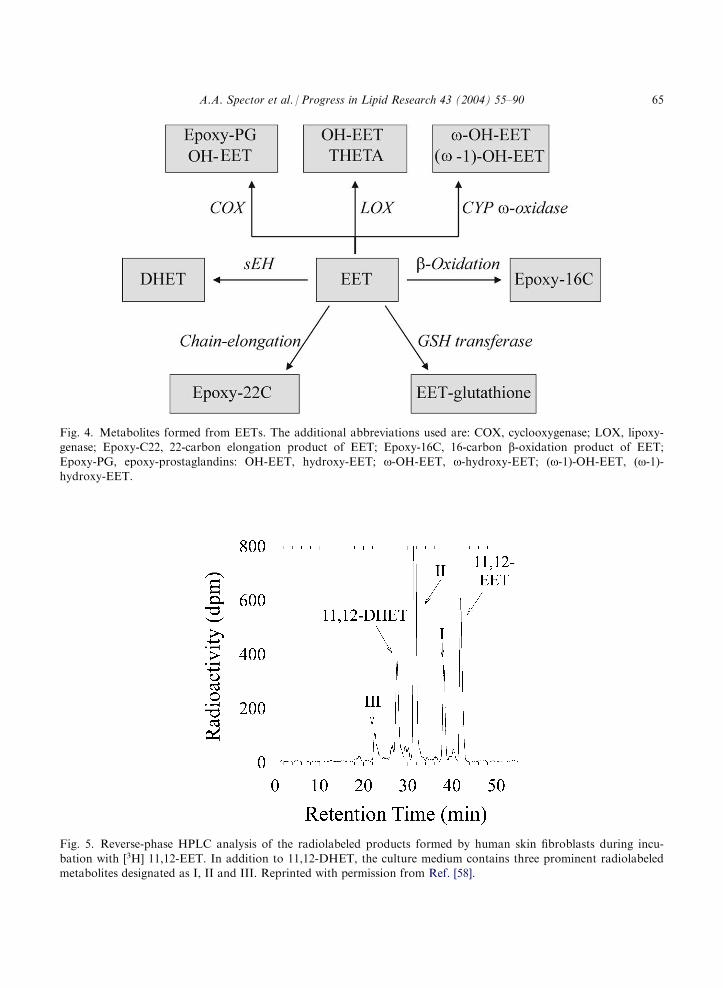
Fig. 4. Metabolites formed from EETs. The additional abbreviations used are: COX, cyclooxygenase; LOX, lipoxy-genase; Epoxy-C22, 22-carbon elongation product of EET; Epoxy-16C, 16-carbon b-oxidation product of EET;Epoxy-PG, epoxy-prostaglandins: OH-EET, hydroxy-EET; o-OH-EET, o-hydroxy-EET; (o-1)-OH-EET, (o-1)-hydroxy-EET.
Fig. 5. Reverse-phase HPLC analysis of the radiolabeled products formed by human skin fibroblasts during incu-
bation with [3H] 11,12-EET. In addition to 11,12-DHET, the culture medium contains three prominent radiolabeledmetabolites designated as I, II and III. Reprinted with permission from Ref. [58].
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 65

Similar radiolabeled metabolites were released into the culture medium when the HSF wereincubated with [3H]14,15-EET, and they also are partial b-oxidation products [58].
14; 15-EET ! 12; 13-epoxy-D6;9-18 :2 ! 10; 11-epoxy-D4;7-16 :2
! 8;9-epoxy-D5-14 :1
In both cases, the main products that accumulate are the 16-carbon metabolites that contain a�4-cis-double bond. Additional studies with mitochondrial and peroxisomal HSF mutants indi-cated that these products are formed by peroxisomal b-oxidation [58]. A radiolabeled metabolite
Fig. 6. Negative ion chemical ionization mass spectra of 11,12-EET and the main b-oxidation metabolites formed byHSF from 11,12-EET. Panel A, 11,12-EET; Panel B, 9,10-epoxy-�6,12-18:2 (metabolite I in Fig. 5); Panel C, 7,8-epoxy-
�4,10-16:2 (metabolite II in Fig. 5); Panel D, 5,6-epoxy-�8,10-14:1 (metabolite III in Fig. 5). Reprinted with permissionfrom Ref. [58].
66 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

with the HPLC properties of a 16-carbon product also was formed by the HSF from [3H]8,9-EET, but this product has not yet been positively identified.Human coronary endothelial cells (HCEC), which convert relatively small amounts of EETs to
DHETs, also produce partial b-oxidation products from [3H]11,12- and [3H]14,15-EET [54].Formation of 14,15-DHET and 10,11-epoxy-16:2 is time- and concentration-dependent. Theestimated Km and Vmax values are 2.2 mM and 1.4 pmol/min for the formation of 14,15-DHET,and 1.08 mM and 1.04 pmol/min for 10,11-epoxy-16:2 [54]. Likewise, human coronary arterysmooth muscle cells and human umbilical vein endothelial cells produce 10,11-epoxy-16:2 from[3H]14,15-EET [54]. By contrast, PCEC, which rapidly convert [3H]14,15-EET to DHET underbasal conditions, produce substantial amounts of chain-shortened EET metabolites only whensEH is inhibited [53,68]. Based on these observations, partial b-oxidation appears to be an alter-native pathway for EET catabolism under ordinary conditions and is prominent only when thesEH activity is inherently low or is inhibited.
4.1.1. Function of EET �-oxidation products10,11-Epoxy-16:2, the 14,15-EET b-oxidation product that accumulates in the extracellular
fluid, relaxes porcine and human coronary microvessels contracted with endothelin [54]. It alsoreduces tumor necrosis factor-a-induced interleukin-8 release from HCEC [54]. The potency of10,11-epoxy-16:2 in these assays is similar to that of 14,15-EET. By contrast, 10,11-epoxy-16:2produced much less relaxation than 14,15-EET in bovine coronary artery rings contracted withU-46619, and the 18- and 14-carbon metabolites also exhibited very little vasoactivity in thissystem [65]. Because of these conflicting observations, more work is needed to determine whetherthe partial b-oxidation products of 11,12- and 14,15-EET have functional effects in the varioustissues that synthesize or are exposed to these EETs.
4.1.2. �-Oxidation of 11,12-DHET11,12-DHET also can undergo partial b-oxidation in PASMC. The 16-carbon intermediate,
7,8-dihydroxy-�4,10-16:2, is the main product that accumulates in the extracellular fluid. This11,12-DHET metabolite relaxes porcine coronary artery rings contracted with U46619 when it isadded in concentrations ranging from 1 to 5 mM [56].
4.2. Chain-elongation products
PCEC convert 14,15-EET to a 22-carbon chain-elongation product, 16,17-epoxy-�7,10,13-22:3[53]. Although most of the 16,17-epoxy-�7,10,13-22:3 is retained in the cell lipids, a small amountis released into the extracellular fluid under some conditions. Based on HPLC retention times,similar chain-elongated products appear to be formed by PASMC from 14,15- 11,12- and 8,9-EET [55,56]. Chemically synthesized 22-carbon EET derivatives dilate constricted canine andporcine coronary microvessels with potencies comparable to that of EETs [69], indicating thatthese products have vasoactive properties. 16,17-Dihydroxy-22:3, the elongation product of14,15-DHET, also has been detected in the cell lipid extract following incubation of PCEC with14,15-EET.When PCEC incubated with [3H]14,15-EET are treated with DCU to selectively inhibit sEH, a
small amount of radiolabeled 14,15-epoxy-�8,11-20:2 is detected in the cell lipids [53]. The
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 67
mechanism through which this dienoic 20-carbon epoxide is formed has not been determined, buta likely possibility is that it is a chain-elongation product of either the 12,13-epoxy-�6,9-18:2 or10,11-epoxy-�4,7-16:2 that are formed from 14,15-EET by PCEC when sEH is inhibited.
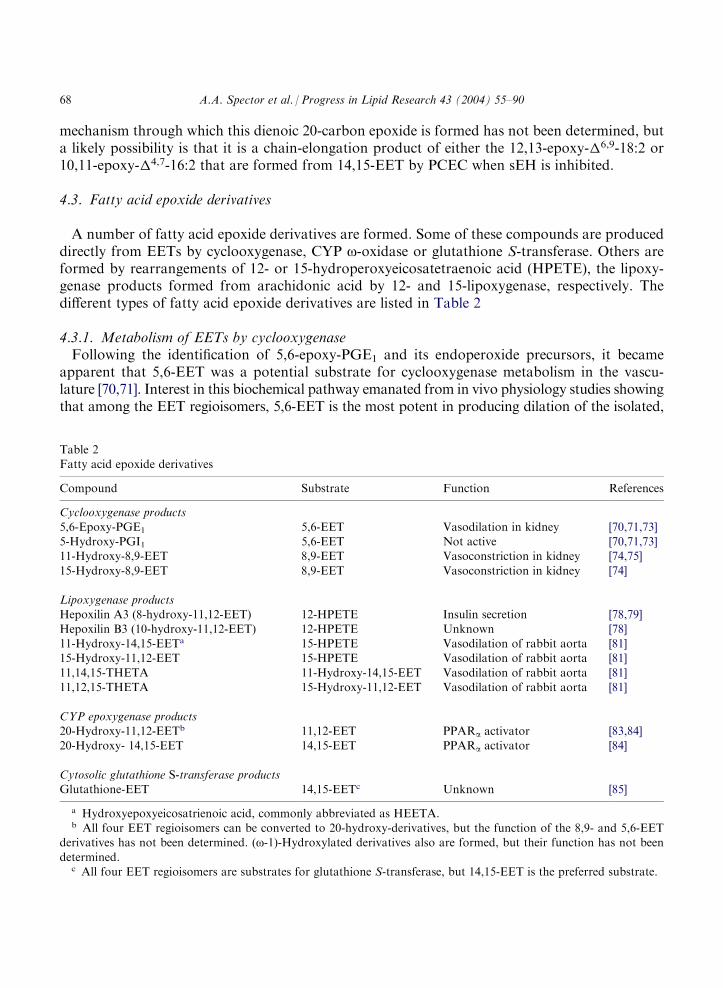
4.3. Fatty acid epoxide derivatives
A number of fatty acid epoxide derivatives are formed. Some of these compounds are produceddirectly from EETs by cyclooxygenase, CYP o-oxidase or glutathione S-transferase. Others areformed by rearrangements of 12- or 15-hydroperoxyeicosatetraenoic acid (HPETE), the lipoxy-genase products formed from arachidonic acid by 12- and 15-lipoxygenase, respectively. Thedifferent types of fatty acid epoxide derivatives are listed in Table 2
4.3.1. Metabolism of EETs by cyclooxygenaseFollowing the identification of 5,6-epoxy-PGE1 and its endoperoxide precursors, it became
apparent that 5,6-EET was a potential substrate for cyclooxygenase metabolism in the vascu-lature [70,71]. Interest in this biochemical pathway emanated from in vivo physiology studies showingthat among the EET regioisomers, 5,6-EET is the most potent in producing dilation of the isolated,
Table 2Fatty acid epoxide derivatives
Compound
Substrate Function ReferencesCyclooxygenase products
5,6-Epoxy-PGE1
5,6-EET Vasodilation in kidney [70,71,73] 5-Hydroxy-PGI1 5,6-EET Not active [70,71,73] 11-Hydroxy-8,9-EET 8,9-EET Vasoconstriction in kidney [74,75]15-Hydroxy-8,9-EET
8,9-EET Vasoconstriction in kidney [74]Lipoxygenase products
Hepoxilin A3 (8-hydroxy-11,12-EET)
12-HPETE Insulin secretion [78,79] Hepoxilin B3 (10-hydroxy-11,12-EET) 12-HPETE Unknown [78] 11-Hydroxy-14,15-EETa 15-HPETE Vasodilation of rabbit aorta [81] 15-Hydroxy-11,12-EET 15-HPETE Vasodilation of rabbit aorta [81]11,14,15-THETA
11-Hydroxy-14,15-EET Vasodilation of rabbit aorta [81] 11,12,15-THETA 15-Hydroxy-11,12-EET Vasodilation of rabbit aorta [81]CYP epoxygenase products
20-Hydroxy-11,12-EETb 11,12-EET PPARa activator [83,84] 20-Hydroxy- 14,15-EET 14,15-EET PPARa activator [84]Cytosolic glutathione S-transferase products
Glutathione-EET 14,15-EETc Unknown [85]a Hydroxyepoxyeicosatrienoic acid, commonly abbreviated as HEETA.b All four EET regioisomers can be converted to 20-hydroxy-derivatives, but the function of the 8,9- and 5,6-EET
derivatives has not been determined. (o-1)-Hydroxylated derivatives also are formed, but their function has not been
determined.c All four EET regioisomers are substrates for glutathione S-transferase, but 14,15-EET is the preferred substrate.
68 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

perfused rabbit kidney, and it is the only isomer whose activity is blocked by cyclooxygenaseinhibitors [72]. Subsequently, the isolated, perfused rabbit kidney was found to convert 5,6-EETto the cyclooxygenase metabolites 5-hydroxy-PGI1 and 5,6-epoxy-PGE1 [73]. Moreover, 5,6-epoxy-PGE1 is equipotent to 5,6-EET as a vasodilator, whereas 5-hydroxy-PGI1 is devoid ofvasodilator activity.8,9-EET also undergoes cyclooxygenase-mediated conversion to 11- and 15-hydroxylation
products; because of the location of the epoxide group, cyclization of 8,9-EET does not occur andit does not form PG endoperoxide compounds [74]. The vasoconstriction produced by 8,9-EET inthe rat kidney is abolished by indomethacin, raising the possibility that the hydroxylated meta-bolites of 8,9-EET formed by cyclooxygenase are biologically active [75]. These and other earlystudies emphasize the potential importance of cyclooxygenase as a pathway of vascular EETmetabolism in vitro and in vivo.Subsequent studies demonstrated a role for cyclooxygenase metabolism in 5,6-EET-mediated
dilation in the intact pulmonary circulation [76]. More recently, all four EET regioisomers werefound to constrict isolated rabbit pulmonary arteries [77]. Interestingly, cyclooxygenase inhibitionblocks responses not only to 5,6-EET, but also to 14,15-EET. Cyclooxygenase metabolites ofEETs were not detected in this study, indicating that the EETs most likely stimulated the releaseof arachidonate, which in turn was metabolized to vasoconstrictor products by cyclooxygenase.
4.3.2. Formation of epoxy-derivatives by lipoxygenaseMetabolism of arachidonate by 12-lipoxygenase yields an unstable hydroperoxy intermediate that
can be converted to hydroxyepoxyeicosatrienoic acids (referred to as hepoxilins) [78]. As the namesuggests, these compounds contain both hydroxyl and epoxide functional groups. There are twohepoxilins, 8-hydroxy- and 10-hydroxy-11,12-epoxyeicosatrienoic acid (8-OH- and 10-OH-11,12-EET). Hepoxilin A3, the 8-hydroxy-isomer, potentiates glucose-dependent insulin secretion in ratpancreatic islets [79]. Hydrolysis of the epoxide group leads to formation of trihydroxy-eicosatrienoic acids (referred to as trioxilins). 15-Lipoxygenase can also produce similar arachidonicacid metabolites. Little is known about the vascular production of these hydroxyepoxy- and trihy-droxy-compounds, which are structurally similar to CYP-derived EETs and DHETs, respectively.One or more products of lipoxygenase metabolism that is distinct from 12-HETE or 15-HETE
produce arachidonic acid-induced dilation of the rabbit aorta [80]. Additional biochemical stud-ies using arachidonic acid as a substrate showed that in the presence of 15-lipoxygenase, aortichomogenates produced 11-hydroxy-14,15-epoxyeicosatrienoic acid (11-OH-14,15-EET), 15-hydroxy-11,12-epoxyeicosatrienoic acid (15-OH-11,12-EET), and their corresponding trihydroxy-eicosatrienoic acid (THETA) derivatives, 11,14,15-THETA and 11,12,15-THETA [81]. Aorticmicrosomes also produce these THETA derivatives from 15-HPETE, the product formed by 15-lipoxygenase from arachidonic acid.Microsomal formation of the epoxide metabolites is blocked by the CYP inhibitor miconazole.
These results were interpreted to suggest that a CYP enzyme is responsible for conversion of15-HPETE to 11-OH-14,15-EET and 15-OH-11,12-EET. This enzymatic reaction appears toinvolve a microsomal CYP enzyme that does not require NADPH as a co-factor, analogous tothe CYP activity of thromboxane synthase [82]. Alternatively, the hydroxy-EET derivativespotentially could be formed through the coordinated activities of a CYP epoxygenase and alipoxygenase.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 69

The enzyme responsible for hydrolysis of the epoxide groups was not identified. Nevertheless,epoxide hydrolysis of the hydroxy-EET derivatives appears to be biologically important becauseTHETAs produce relaxation of rabbit aorta [81]. The extent to which hydroxy-EET derivativesand THETAs are formed and modulate reactivity in other blood vessels remains to be deter-mined.
4.3.3. Metabolism of EETs by cytochrome P450 !-oxidaseo-Hydroxylation of arachidonic acid yields 20-HETE, a compound that has received con-
siderable attention due to its potent biological activity [2,8]. In some vascular beds, 20-HETEappears to act as an EET antagonist and plays an important role as an endogenous vasocon-strictor [7,8]. 11,12-EET can be converted to 20-hydroxy-11,12-EET by renal cortical micro-somes, suggesting that EETs also are potential substrates for CYP o-oxidases in vivo [83]. Adetailed analysis of this pathway was recently reported [84]. All four EET regioisomers are con-verted to their corresponding terminal (o) and subterminal (o-1) hydroxy metabolites by incu-bation with purified rat CYP4A o-hydroxylases. Considerable specificity occurs with regard to o-hydroxylation of the EETs. The rate of conversion depends on the location of the epoxide groupand its position relative to the carboxylate moiety. Surprisingly, the rate of o-hydroxylation of8,9-EET and 11,12-EET exceeds that of arachidonic and lauric acids. Studies with rat livermicrosomes indicate that CYP4A1 and CYP4A2 are the predominant enzymes responsible for o-and o-1-hydroxylation of EETs. Notably, the 20-hydroxy metabolites of 11,12-EET and 14,15-EET bind with high affinity and transactivate peroxisome proliferator-activated receptor-a(PPARa) [84], suggesting that these o-hydroxy compounds may play an important role in reg-ulating fatty acid metabolism.
4.3.4. Metabolism of EET by glutathione S-transferasesCytosolic glutathione S-transferases conjugate EETs with glutathione, a major cellular anti-
oxidant, in a 1:1 ratio [85]. 14,15-EET is a better substrate than 11,12-, 8,9-, or 5,6-EET for thisreaction. The Km for 14,15-EET-glutathione formation by the purified transferase is about 10mM, while the Vmax ranges from 25 to 60 nmol/min/mg [86]. Although conjugation with glu-tathione almost certainly alters the biological activity of the EET, the functional significance ofthis process has not yet been investigated.
5. Stimulated release of EET from cells
Much more EET is released into the culture medium when cells are incubated with a Ca2+
ionophore than under basal conditions. For example, mastocytoma cells loaded with radiolabeled14,15-EET released 50% of the EET into the extracellular fluid in 15 min when they were incu-bated with A23187, a Ca2+ ionophore [52]. Likewise, PCEC incubated with A23187 released 10-times more [3H]14,15-EET than under basal conditions [35,63]. Because most of the intracellularEET is present in phospholipids, the EET release almost certainly is due to the Ca2+ dependentactivation of a phospholipase. The type of phospholipase has not been determined, but the factthat the EETs are present primarily in the sn-2 position suggests that it probably is a phos-pholipase A2.
70 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

Incubation of labeled cells with ionophore A23187 typically is performed for 20–30 min inorder to obtain a sufficient amount of released radioactivity for HPLC analysis. Under theseconditions, about two-thirds of the radioactivity released into the culture medium by [3H]14,15-EET-labeled PCEC is recovered as DHET [35]. If DCU is added to selectively inhibit sEH,however, virtually all of the radioactivity released into the medium remains as EET [53]. Thisdifference is illustrated by the HPLC analysis in Fig. 7, which shows the distribution of theradioactivity released into the medium by cells labeled with [3H]14,15-EET in the absence(Fig. 7A) and presence (Fig. 7B) of sEH inhibition with DCU. Our interpretation of these find-ings is that in cells labeled with [3H]14,15-EET, phospholipase activation initially results in release
Fig. 7. HPLC analysis of the radiolabeled material recovered in the medium following exposure of the endothelialcultures to a calcium ionophore. PCEC labeled with [3H]14,15-EET were incubated with ionophore A23187 for 20 min,and the lipids present into the medium were extracted and analyzed by HPLC. Radiolabeled EET and DHET were
detected in the medium when sEH was not inhibited (Panel A), while only EET was detected when the cultures weretreated with 3 mM DCU (Panel B). Reprinted with permission from Ref. [53].
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 71

of EET from the cells. The EET is then gradually converted to DHET, presumably due toreuptake and hydration by sEH, and the DHET is subsequently released from the cells.
5.1. EET-mediated vascular relaxation
EETs released from the endothelium activate BKCa channels, causing hyperpolarization of thesmooth muscle and vasodilation [6,8,10–13]. The current paradigm, illustrated in Fig. 8, is thatphospholipase activation hydrolyzes arachidonic acid contained in endothelial phospholipids,and the arachidonic acid is subsequently converted to EET by an endothelial CYP epoxygenase[8]. The EET either produces autocrine effects on endothelial function or is released and acts as aparacrine mediator on adjacent cells like vascular smooth muscle.An alternative to this mechanism is suggested by the finding that the vasodilator response to
bradykinin is potentiated in porcine coronary artery rings pretreated with EET [35]. Potentiationof bradykinin-induced relaxation is blocked by triacsin C, an acyl CoA synthase inhibitor thatreduces EET incorporation into endothelial phospholipids. This suggests that the effect of pre-treatment of the rings with EET is exerted on the endothelium, a conclusion that is supported bythe observation that endothelium-independent relaxation is not potentiated in the pretreatedrings. Taken together, these results suggest that pretreatment with EET increases the EET con-tent of endothelial phospholipids and thereby produces a larger EET release in response to bra-dykinin. Because of the increased release of EET, the relaxation response of the smooth muscle isaugmented. Fig. 9 illustrates this alternative mechanism in which EETs are taken up from the
Fig. 8. Mechanism for EET-mediated vasodilation in which the EET is produced from arachidonic acid when theendothelium is activated. According to this paradigm, EET is synthesized by endothelial CYP epoxygenases from thearachidonic acid that is hydrolyzed from endothelial phospholipids as a result of phospholipase activation. The EET is
released from the endothelium and activates the smooth muscle BKCa channel, causing hyperpolarization and vascularrelaxation.
72 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

extracellular fluid by the endothelium, either from the EET contained in the plasma or byreuptake of EET released from the endothelium. Following temporary storage in phospholipids,the EET subsequently is released when a stimulus activates the endothelial phospholipase.
6. o-3 Fatty acid analogues of EET
Eicosapentaenoic acid (EPA, 20:5o-3), the o-3 fatty acid analogue of arachidonic acid, can beconverted to a 17,18-epoxy-derivative by vascular smooth muscle cells [87]. This apparentlyoccurs through the action of either the CYP 4A1 or 4A3 that is expressed in these cells. The17,18-epoxy-derivative of EPA produces greater activation of the rat cerebral artery BKCa chan-nel than 11,12-EET. Likewise, epoxides synthesized chemically by incubation of EPA with m-chloroperoxybenzoic acid [88], called epoxyeicosaquatraenoic acids (EEQ), potently dilate canineand porcine microvessels [69]. Vicinal diols derived from EEQs are detected in the urine ofhumans who ingest o-3 fatty acid supplements [89]. Taken together, these results suggest that theepoxy-derivatives of EPA, like those produced from arachidonic acid, may function as hyperpo-larizing factors, especially when the diet is enriched in o-3 fatty acids [69,87].Five epoxy-derivatives of docosahexaenoic acid (22:6o-3), called epoxydocosapentaenoic acids
(EDP), have been synthesized chemically [90]. The EDPs dilate porcine coronary arteriolescontracted with endothelin. Patch-clamp studies in the inside-out configuration indicate that the
Fig. 9. Alternative mechanism for EET-mediated vasodilation in which preformed EET is released from endothelialphospholipids when the endothelium is activated. The difference in this hypothetical scheme is that preformed EET,
obtained either from the plasma or by recapture of EET released previously from the endothelium, is incorporated andstored in endothelial phospholipids. The stored EET is released when the endothelial phospholipase is activated.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 73

13,14-epoxy-EDP regioisomer is 1000-fold more effective than EETs in activating the BKCa
channel of myocytes from rat small coronary arteries, making it the most potent BKCa activatorso far reported. It is not known, however, if docosahexaenoic acid is converted to 13,14-epoxy-EDP or any of the other EDP regioisomers under physiological conditions.
7. Soluble epoxide hydrolase (sEH)
Epoxide hydrolases (EC 3.3.2.3) are a group of enzymes that convert the epoxide group ofchemical compounds to corresponding diols by the addition of water. The main epoxide hydro-lases have been classified based on the structure of their substrates and their cellular localization.They are cholesterol 5,6-oxide hydrolase, hepoxilin A3 hydrolase, leukotriene A4 hydrolase, sEH(formerly referred to as cytosolic EH), and mEH [91,92]. sEH is the primary enzyme responsiblefor the conversion of EETs to the corresponding DHETs (see Fig. 2). Because of the potentialimportance of this enzyme as a therapeutic target, this section will focus on the properties andfunction of sEH.
7.1. Distribution of sEH in mammalian tissues
sEH activity is widely distributed in mammalian tissues. Those containing high sEH activityinclude liver, kidney, intestine, and vascular tissue. Low levels of sEH are found in testes, lung,brain and spleen [42,51,55,93]. Although sEH is predominately located in the cytosol, sEH enzy-matic activity and protein are also present in the peroxisomes [94–96]. The sEH from liver andkidney of different species, including human, has been purified [97–102]. Native rat liver sEH hasa molecular mass of approximately 120 kDa, consisting of two 61-kDa subunits [99]. Human liversEH has been cloned and expressed in a baculovirus system. The 2101 base clone predicts a 554-residue protein [M(r)=62,640] that contains an apparently weak peroxisomal targeting signal,Ser-Lys-Met, at the carboxyl terminus [100]. The predicted amino acid sequence of the humanenzyme is very similar to that of mouse sEH [102].
7.2. Molecular biology of sEH
The genes encoding mEH and sEH are EPHX1 and EPHX2, respectively. Both EPHX1 andEPHX2 have been cloned and characterized [100,103]. EPHX2 is located in chromosomal region8p21-p12 [104]. It is approximately 45 kb in length and consists of 19 exons with lengths from 27to 265 bp. The translated parts of the exons of EPHX2 correspond to 555 amino acid residues[105], one residue more than predicted by the cDNA expressed in the insect cell system [100].Genetic polymorphisms of human sEH have been identified. Eight variant cDNA loci from 25
human livers have been detected. The coding region contains five silent single nucleotide poly-morphisms and two variant loci resulting in altered protein sequences [Arg287 ! Gln287 (exon 8);Arg403 ! Arg403�404(exon 13)]. Although the two variant alleles retain catalytic activity, the allelecontaing the Arg insertion results in extremely low sEH enzymatic activity [106]. Thirty-six singlenucleotide polymorphisms have been identified in the EPHX2 gene in Japanese individuals, aswell as one insertion/deletion polymorphism in the 50 flanking region [107]. These variants of the
74 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

sEH gene may explain previously reported differences in sEH activity among these individuals[108]. Although increased levels of DHETs are found in the urine of pregnancy-induced hyper-tensive women [109], it is not clear whether sEH gene variants are correlated with these or otherclinical-pathological conditions.
7.3. Regulation of sEH expression
The expression of sEH is up-regulated by activation of PPARa in rodent species. sEH mRNAlevels in mouse liver were increased 8-fold by clofibrate, a hypolipidemic drug that producesperoxisomal proliferation. Increases in sEH mRNA levels are also produced in kidney and heartby clofibrate [93]. In addition, dietary exposure to other peroxisomal proliferating agents, such as2,4-dichlorophenoxyacetic acid, 2,4,5-trichlorophenoxyacetic acid and 2-ethylhexanoic acid,increase sEH activity in mouse liver [110,111]. Testosterone administration also augments sEHactivity in mouse kidney and liver, and testosterone and clofibrate may have additive effects onsEH in these tissues [112].
7.4. Enzymatic mechanism
Epoxide hydrolases are members of the a/b hydrolase fold family. These enzymes hydrolyzetheir substrates in a two-step reaction involving the formation and hydrolysis of a covalent acyl-or alkyl-enzyme intermediate [113–115]. Similar to other a/b hydrolase fold enzymes, the Asp333
and His523 residues that are required for the catalytic activity of sEH are conserved in all epoxidehydrolases. No activity was detectable after the replacement of Asp333 by Ser, and mutation ofHis523 to Gln produced greater than 99% loss of sEH specific activity [102]. Site-directed muta-genesis studies indicate that Asp495 also is required for activity of sEH, and it appears thatAsp333, Asp495 and His523 form the catalytic triad [115]. Furthermore, the crystal structure ofmouse sEH suggests that Tyr381 and Tyr465 can act as acid catalysts, activating the epoxide ringand facilitating the formation of a covalent intermediate between the epoxide and the enzyme.Mutational analyses confirm that both Tyr381 and Tyr465 are required for full catalytic activity ofsEH [116].
7.5. sEH and lipid phosphate phosphatase activity
The mammalian sEH is a homodimer, and each subunit contains carboxyl- and amino-terminaldomains (Fig. 10). The active site for epoxide hydrolysis is located in the carboxyl-terminaldomain; the structure of this domain is similar to that of haloalkane dehalogenase and shares the
Fig. 10. Bifunctional domains of sEH.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 75

a/b hydrolase fold. A vestigial active site is found in the N-terminal domain similar to that ofanother enzyme of halocarbon metabolism, haloacid dehalogenase. The amino-terminal domainof sEH appears to play a critical structural role by stabilizing the dimer in a distinctive con-formation [117]. Furthermore, the amino-terminal domain of sEH has been shown recently tohave phosphatase activity [118].Phosphorylated hydroxy lipids are excellent substrates for the human sEH, particularly the
monophosphate of dihydroxy stearic acid [119]. Rat sEH hydrolyzes 4-nitrophenyl phosphatewith a rate constant of 0.8 s�1 and a Km of 0.24 mM. If the carboxyl-terminal domain is deletedfrom human sEH, the amino-terminal domain still remains active as a phosphatase. This suggeststhat both catalytic sites act independently [118].
7.6. Metabolism of EETs by sEH
The conversion of EET to the corresponding DHET by sEH is regioselective. 14,15-EET is thepreferred substrate as compared with 11,12- and 8,9-EET. Formation of 14,15- and 8,9-DHET bysEH is also stereoselective (4:1 and 7:3 ratio of antipodes, respectively), but hydration of 11,12-EET is not enantioselective. Regioselective and/or enantioselective oxirane water addition occursduring formation of asymmetric 14,15-, and 8,9-DHET, and 14(R),15(S)-, 11(S),12(R)-, and8(S),9(R)-EET are metabolized at substantially higher rates than their antipodes. Conversion ofEETs to methyl esters decreases the rates of epoxide hydration, and catalytic hydrogenation ofEETs also reduces the rates of hydration [33,34].
7.7. sEH inhibitors
Substituted chalcone oxides and trans-3-phenylglycidols are two classes of structurally similar(both are epoxides), potent, and selective sEH inhibitors [120,121]. Fig. 11 (top) shows thestructure of a chalcone oxide. A representative inhibitor of this class is 4-phenylchalcone oxide,which has a half-inhibitory concentration (IC50) of 64 nM for mouse sEH [120]. Inhibition ofsEH by substituted chalcone oxides occurs through electronic stabilization of the covalentenzyme–inhibitor intermediate [122]. Substituted chalcone oxides are poor substrates, and theirinhibitory effects on sEH are transient. These compounds are also relatively unstable, particularlyin the presence of glutathione [122].
trans-3-Phenylglycidol derivatives are enantioselective, slow binding inhibitors of sEH. (2S,3S)-3-(4-Nitrophenyl)glycidol is the most potent of the 3-phenylglycidol series, and the (2S,3S)-enantiomer is about 750-fold more effective as an inhibitor than the (2R,3R)-enantiomer [121].However, even (2S,3S)-3-(4-nitrophenyl)glycidol has an IC50 of 1.6 mM, which is less potent than4-phenylchalcone oxide.A new class of potent, selective and stable sEH inhibitors that are derivatives of urea and car-
bamates recently has been described [41]. DCU is a representative compound of this class (Fig. 11,middle). The IC50 of DCU is 0.09 and 0.16 mM for mouse and human sEH, respectively [41].DCU is poorly soluble in H2O, and this limits its potential usefulness. A series of urea-derivedsEH inhibitors that have equivalent or greater potency than DCU and also have increased H2Osolubility has been developed [123]. For example, N-cyclohexyl-N0-dodecylurea, in which ahydrocarbon chain replaces one of the cyclohexyl groups of DCU, increases the potency of sEH
76 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

inhibition 8- to 16-fold (IC50 of 0.01 and 0.011 mM for mouse and human sEH, respectively)[123]. Addition of a carboxylic acid group to the end of the hydrocarbon chain of N-cyclohexyl-N0-dodecylurea further increases H2O solubility without reducing the potency of sEH inhibition(Fig. 11, bottom).Several metals ions also inhibit epoxide hydrolase activity. Cd2+ and Cu2+ inhibit sEH, while
Hg2+ and Zn2+ inhibit mEH. The IC50 for Cd2+ and Cu2+ is approximately 20 mM. Zn2+ is a
competitive inhibitor of mEH, while Cd2+ and Cu2+ are noncompetitive inhibitors of mouse andhuman sEH [124].It should be noted that the effect of sEH inhibitors is species specific. For example, N-cyclo-
hexyl-N0-dodecylurea and N-cyclohexyl-N0-dodecanoylurea produce 10-fold more potent inhibi-tion of mouse sEH than human sEH [123]. The effect of DCU on mouse sEH is also twice aspotent as on human sEH [41]. Table 3 summarizes the potency and species specificity of repre-sentative sEH inhibitors.A mechanism for the action of the alkylurea inhibitors has been proposed [125]. According to
this mechanism, the urea carbonyl oxygen forms hydrogen bonds with the hydroxyl groups ofTyr381 and Tyr465 of sEH. The urea carbonyl oxygen also interacts with Gln382, which may sta-bilize the partial negative charge on the epoxide oxygen. The carboxylate side chain of Asp333
accepts a hydrogen bond from one of the urea NH-groups in each enzyme–inhibitor complex.
Fig. 11. Representative structures of sEH inhibitors.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 77

Asp333 then interacts with the partial positive charge on the urea NH-group. This is similar to theactivation of a substrate epoxide ring for nucleophilic attack by Asp333 [125].
7.8. Potential applications of sEH inhibitors
sEH inhibitors have a number of beneficial actions that are likely to be useful in treatingdiseases that affect the cardiovascular and respiratory systems.
7.8.1. Cardiovascular systemRecent observations suggest that inhibition of sEH could be a novel approach for the treatment
of hypertension. Although CYP2J expression and EET formation are increased in the kidney ofthe spontaneously hypertensive rat (SHR) [126], sEH expression and, consequently EET hydro-lysis, are also increased. Inhibition of EET hydrolysis with the selective sEH inhibitor DCUreverses the hypertensive phenotype in the SHR [42], presumably by maintaining a high level ofEET in renal tissue. Renal expression of sEH also is increased in rats made hypertensive by theinfusion of angiotensin II (Ang II), and the sEH inhibitor N-cyclohexyl-N0-dodecyl urea decreasesarterial blood pressure in this hypertension model presumably by raising the EET level [127].Likewise, targeted disruption of sEH gene in male mice lowers systolic blood pressure [40].Although four single nucleotide polymorphisms in the EPHX2 gene have been detected in SHRand three produce amino acid substitutions in sEH, these sequence variations do not appear tocontribute to the elevated blood pressure level in the SHR [128].The mechanism through which a sEH inhibitor lowers blood pressure probably involves three
factors, as is illustrated in Fig. 12. One is that EETs, but not DHETs, produce vasodilation in therenal circulation [129, 64]. The enhanced renal microvascular reactivity to Ang II that occurs inhypertension is ameliorated by an analog of 11,12-EET [130], suggesting that an increase inendogenous EET would have a similar effect. Administration of a sEH inhibitor decreases thehydrolysis of EETs and, therefore, maintains a higher endogenous EET level. This reduces renalvasoconstriction and thereby decreases renin-Ang II-aldosterone-dependent hypertension. A sec-ond factor is that sEH inhibition increases the incorporation of EETs into phospholipids, leadingto greater EET mobilization when the cell is stimulated. The increased release of EET potentiatesthe vasorelaxant response [35,53,68]. The third factor is that when sEH is inhibited, increased
Table 3Potency and species specificity of sEH inhibitors
Inhibitor
IC50 (nM) ReferencesMouse sEH
Human sEH4-Phenylchalcone oxide
64–140 200 [120,122] (2S,3S)-3-(4-Nitrophenyl)glycidol 1600 – [121] N,N0-Dicyclohexylurea 90 160 [41]N-Cyclohexyl-N0-dodecylurea
9.8 85.2 [123] N-Cyclohexyl-N0-dodecanoylureaa 11.1 112 [123]a Increased H2O solubility as compared with N-cyclohexyl-N0-dodecylurea.
78 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

amounts of EET are converted to chain-shortened epoxy fatty acids through b-oxidation. Epoxy-16:2, the main chain-shortened epoxy fatty acid that accumulates in the medium under theseconditions [53], causes potent vasodilation [54].In addition to regulating blood pressure, the sEH inhibitor DCU inhibits human vascular
smooth muscle cell proliferation in a dose-dependent manner. This effect of DCU is associatedwith a decrease in the level of cyclin Dl, a cell cycle regulatory protein that activates the cyclin-dependant kinases. EETs mimic the proliferation suppressive activity of DCU [131]. However,there is no direct evidence as to whether the inhibitory effect of DCU on smooth muscleproliferation is due to inhibition of EET hydrolysis or to an unrelated action.
7.8.2. Respiratory systemLinoleic acid can be converted to leukotoxin, a linoleic acid epoxide produced by CYP in leu-
kocytes [132,133]. Similar to EETs, leukotoxin is converted to leukotoxin-diol by sEH [134]. Thisis illustrated in Fig. 13. Leukotoxin has been associated with multiple organ failure and the adultrespiratory distress syndrome. Early studies found that the cytotoxic effects of leukotoxin occuronly in the presence of epoxide hydrolase [135], and this observation was recently confirmed invivo in a study showing that inhibition of sEH by 4-phenylchalcone oxide decreased leukotoxin-induced mortality [136]. The cytotoxic effects of leukotoxin-diol include mitochrondrial dysfunc-tion, eosinophilic exudates, alveolar edema, and hemorrhage [135–139]. These observations indi-cate that the conversion of leukotoxin to leukotoxin-diol by sEH is the process responsible for the
Fig. 12. Proposed mechanisms that contribute to the regulation of blood pressure by sEH inhibitors.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 79

cytotoxic actions. Therefore, inhibition of sEH has a potentially beneficial effect for the treatmentof leukotoxin-induced adult respiratory distress syndrome.
8. Biochemical response to EETs: mechanisms of action
Considerable progress has been made during the last two decades in determining the functionaleffects and metabolism of EETs [2,7,8,11,44]. In contrast, much less is known about the bio-chemical mechanisms through which EETs produce their responses. Because the physiologicalactions vary widely, depending upon the EET regioisomer and the specific cell type (see Table 1),a single biochemical response mechanism probably does not exist. Prostaglandins and throm-boxanes act through a family of structurally similar plasma membrane receptors linked to specificbiochemical cascades [140]. The potency and functional effects of EETs suggests that they alsomight act through a similar membrane receptor mechanism, and the presence of EET-receptorisoforms could account for the variety of biochemical responses and regioisomeric specificities.Although evidence for EET membrane receptors coupled to specific signaling cascades is growing
Fig. 13. Pathway for leukotoxin production from linoleic acid, and formation of its cytotoxic diol metabolite.
80 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

[11,141], the possibility that EETs mediate some of their actions on cells by having a direct effecton intracellular mechanisms cannot be excluded.
8.1. The case for EET receptors
The earliest indication for the presence of an EET receptor was the finding that EETs are spe-cifically bound to the plasma membrane of human U937 transformed monocytes [141]. Specific,high-affinity binding sites for 14,15-EET were detected with a Kd of 13.8 nM. The binding of14(R),15(S)-EET was displaced more effectively by 14(R),15(S)-EET than 14(S),15(R)-EET,indicating stereospecificity. Consistent with this finding, functional stereospecificity is beingobserved with increasing frequency as EET stereoisomers become more readily available [142–144]. Similar results were obtained with guinea pig mononuclear cells [145]. The binding of14(R),15(S)-EET to the U937 and guinea pig mononuclear cells was disrupted by mild proteasetreatment [141], providing further evidence that a membrane protein binds the EET. Additionaldata suggested that in both cases, the putative receptor was linked to a cyclic AMP and proteinkinase A (PKA) signal transduction pathway that subsequently down-regulates the receptor[141,145].Although these results are compelling, they remain open to question for several reasons. A
receptor has not been purified or cloned from either of these cells. Furthermore, 14(R),15(S)-EETbinding to the guinea pig mononuclear cells is about 20-times weaker than to U937 cells, sug-gesting that the binding proteins may be different in each case. Cell membranes contain fatty acidtransporters [146], and the possibility that one of these proteins rather than a selective EETreceptor might be responsible for the binding cannot be excluded. Another problem is that thedata regarding linkage to the cyclic AMP and PKA signal transduction pathway is based onaddition of dibutyryl cyclic AMP and inhibitors and, therefore, is indirect. Finally, recentstudies have shown that o-hydroxylated 11,12- and 14,15-EET derivatives formed from thecorresponding EETs in the microsomes activate PPARa [84], suggesting that EET-inducedregulation of gene expression occurs through an intracellular mechanism. While an intracel-lular mechanism is a reasonable hypothesis for EET-mediated gene expression, it seems lesslikely for processes like EET-stimulated ion fluxes that occur at the plasma membrane[6,10,16,20,21,24,25]. Therefore, efforts to obtain additional evidence for a plasma membraneEET receptor are continuing.A recent approach utilized incubation of rat aortic smooth muscle cells with 14,15-EET
attached to silica beads through a sulfanilamide linkage [17]. The EET-sulfanilamide derivative isstable, and the attachment to the silica bead restricts the EET to the exterior surface of the cell.Data obtained with this tethered EET preparation demonstrated that a biological response couldoccur through 14,15-EET association with the plasma membrane. The 14,15-EET–bead complexinhibited aromatase activity to the same extent as 14,15-EET and the 14,15-EET-sulfanilamidederivative. Because the EET in the bead complex was restricted to the cell surface, the inhibitoryeffect could not be due to EET entry into the cells.Additional results indicate that EETs stimulate cyclic AMP production by adenylyl cyclase in
guinea pig cerebral microvascular cells and mammalian heart cells [147,148], or by receptor-mediated tyrosine kinase cascades in cultured renal epithelial cells (LLCPKc14) and humanendothelial cells [27,149]. These biochemical responses are processes that are generally associated
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 81

with ligands binding to plasma membrane receptors. When considered together with the bindingdata and tethered EET data [17,31,141,145], these findings provide strong circumstantial evidencefor the existence of plasma membrane EET receptors.
8.2. EETs and ion transport
The involvement of EETs in ion homeostasis was first demonstrated by their ability to release45Ca2+ from aortic smooth muscle microsomes [150]. All of the EET regioisomers were foundsubsequently to produce a transient increase in intracellular Ca2+ concentration in porcine aorticsmooth muscle cells by stimulating Ca influx through L-type Ca2+ channels [16]. Likewise, both5,6-EET and 11,12-EET increase Ca2+ entry into guinea pig ventricular myocytes [151]. EETsalso stimulate the release of Ca2+ from intracellular stores in glomerular mesangial cells [152].Ca2+ mobilization from intracellular stores or influx from the extracellular fluid has been shownrepeatedly to either precede or occur concurrently with EET-stimulated biological responses[16,151–155], indicating that an increase in the cytoplasmic Ca2+ concentration may be a centralcomponent of the cellular response to EETs. As opposed to these findings, 11,12-EET was foundto decrease the open probability of porcine L-type Ca2+ channels reconstituted in a planar lipidbilayer [152]. Inhibition also occurred when phosphatidylcholine that contains 11,12-EET in thesn-2 position was added to the lipid bilayer, suggesting that the inhibitory effect probably resultedfrom either direct contact between the EET and the channel or perturbation of the lipid domainsurrounding the channel. These disparate results suggest that the effect of EETs on Ca2+
channels may depend on the functional state of the cell [44].EETs activate K+ transport, and this is the basis for their action as an EDHF [6,7,10,12,13].
They increase the open state probability of the BKCa channel [156], causing hyperpolarization ofthe smooth muscle and vasorelaxation. Activation of the channel may be triggered by an EET-mediated increase in intracellular Ca2+. Release of Ca2+ from intracellular stores or opening ofmembrane Ca2+ channels in close proximity to the BKCa channel (calcium sparks) can increasethe localized Ca2+ concentration to as high as 30 mM, an amount sufficient to activate the BKCa
channel [157]. However, activation also occurs when EET is added to either a cell-attached orcell-free patch clamp, where the Ca2+ concentration is held constant [10,12]. Therefore, BKCa
channel activation by EET is not dependent on opening of a membrane Ca2+ channel or anincrease in intracellular Ca2+ concentration. These patch clamp results suggest that EETs havethe potential to activate the BKCa channel through direct interaction with a channel protein or anassociated membrane protein, or by interacting with the membrane lipid domains that surroundsthe channels. It remains to be determined whether a calcium spark, physical interaction with thechannel, or physical interaction with an associated membrane protein is the process that mediatesEET-induced activation of BKCa channels under physiological conditions.Investigation of the effect of EETs on Na+ movement across the plasma membrane has not
been extensive. While Na+/H+ exchange is stimulated by EETs in rat glomerular mesangial cellsby approximately 40–60%, presumably as a pH balancing response [154], the open state prob-ability of Na+ channels is reduced in rat ventricular myocardium by as much as 70% by 5 mMEET [24]. The inhibition is similar to that produced by local anesthetics such as lidocaine. Inneither case is there enough evidence presented to permit speculation as to a mechanism by whichEETs affect Na+ movements.
82 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

8.3. EET signaling and G-proteins
A membrane-associated protein that appears to be involved in EET-mediated activation ofBKCa channels is the guanine nucleotide-binding protein (G-protein) Gas [158]. EETs activate theGas subunit of a heterotrimeric G-protein by ADP-ribosylation [159]. In general, when G-pro-teins participate in transduction of extracellular signals, they are coupled with a plasma mem-brane receptor to initiate a signal cascade [160]. The class of receptors that typically function withG-proteins are the G-protein coupled receptors that have seven transmembrane domains [160].By analogy, the involvement of Gas in activation of the BKCa channel suggests that EETs initiatea signal cascade by binding to a G-protein coupled receptor in some types of cells [159]. A similarmechanism linked to a signal cascade involving cyclic AMP and PKA was proposed to explainthe down regulation of EET binding to human U937 and guinea pig mononuclear cells [141,145],and it is consistent with the mechanism of action of other prostanoids [161,162]. The increase intissue plasminogen activator expression in cultured endothelial cells produced by 11,12-EET alsois associated with activation of Gas and the production of cyclic AMP [22], providing furthersupport for this type of mechanism.Fig. 14 illustrates this hypothetical model for EET-mediated functional responses through
activation of Gas. As an extension of this mechanism, we suggest that activation of PKA may bea branch point in transduction of the EET signal. We propose that one branch reaches thenucleus and mediates EET-dependent effects on gene expression, and the other activates theBKCa channel by phosphorylation of a serine residue of the channel a-subunit [163].
8.4. EET signaling through tyrosine kinase-linked cascades
A tyrosine kinase cascade also may mediate EET-stimulated biological responses. Tyrosinekinase activity in porcine aortic endothelial cell lysates increased as much as 3-fold within a fewminutes after addition of as little as 100 nM 11,12-EET [164]. An important implication of theseresults is that the EET-induced increase in tyrosine kinase activity does not require changes inCa2+ concentration or K+ channel activation. However, these findings do not indicate whetherthe tyrosine kinase that was activated was soluble or membrane associated.Additional studies indicated that both 11,12-EET and 14,15-EET stimulated tyrosine kinase
activity in cultured LLCPKc14 cells. Phosphorylation of proteins of several different molecularsizes occurred in the intact cell [27,149], including the epidermal growth factor (EGF) receptor,the regulatory subunit of PI 3-kinase, the src homologous and collagen-like protein (SHC), andextracellular signal-regulated kinase (ERK)1 and ERK2. c-Src activity also was increased up to3-fold within 1–3 min after addition of either 14,15-EET or the EET derivative, 14,15-EET-sulfonamide, to the culture medium. It is likely that this entire series of protein phosphorylations,which describes a typical tyrosine kinase-signaling cascade, is mediated by the activation of theEGF receptor [165].No evidence has been presented so far to show that EET is bound to or even closely associated
with the EGF receptor [27,149,166], and it is possible that the activation of this pathway occursthrough activation of a metalloproteinase [167]. Metalloproteinases cleave an endogenous mem-brane protein, heparin-binding EGF-like growth factor (HB-EGF), and thereby release EGF-likegrowth factor, a ligand that binds to the EGF receptor [167]. The activation of the EGF receptor
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 83
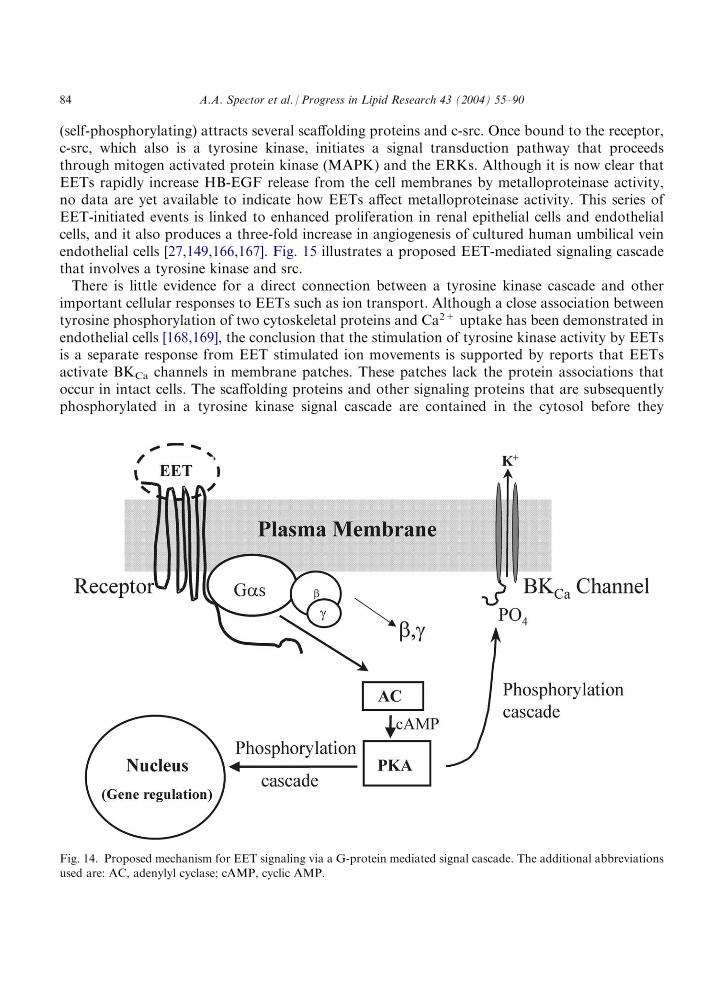
(self-phosphorylating) attracts several scaffolding proteins and c-src. Once bound to the receptor,c-src, which also is a tyrosine kinase, initiates a signal transduction pathway that proceedsthrough mitogen activated protein kinase (MAPK) and the ERKs. Although it is now clear thatEETs rapidly increase HB-EGF release from the cell membranes by metalloproteinase activity,no data are yet available to indicate how EETs affect metalloproteinase activity. This series ofEET-initiated events is linked to enhanced proliferation in renal epithelial cells and endothelialcells, and it also produces a three-fold increase in angiogenesis of cultured human umbilical veinendothelial cells [27,149,166,167]. Fig. 15 illustrates a proposed EET-mediated signaling cascadethat involves a tyrosine kinase and src.There is little evidence for a direct connection between a tyrosine kinase cascade and other
important cellular responses to EETs such as ion transport. Although a close association betweentyrosine phosphorylation of two cytoskeletal proteins and Ca2+ uptake has been demonstrated inendothelial cells [168,169], the conclusion that the stimulation of tyrosine kinase activity by EETsis a separate response from EET stimulated ion movements is supported by reports that EETsactivate BKCa channels in membrane patches. These patches lack the protein associations thatoccur in intact cells. The scaffolding proteins and other signaling proteins that are subsequentlyphosphorylated in a tyrosine kinase signal cascade are contained in the cytosol before they
Fig. 14. Proposed mechanism for EET signaling via a G-protein mediated signal cascade. The additional abbreviationsused are: AC, adenylyl cyclase; cAMP, cyclic AMP.
84 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

associate with an activated receptor. Thus, the tyrosine kinase-dependent EET pathway seems tobe separate from the mechanism that controls ion movements in EET-stimulated cells. Even so,there are at least a few reports showing that EET-mediated EGF signaling can be linked throughPI and protein kinase C activation to an increase in intracellular free Ca2+, a frequent responseto EETs [16,148,152,165,170–174].
8.5. EET and cytokine-mediated inflammation
EETs also function as anti-inflammatory mediators through autocrine effects on endothelialcells [18,19]. In particular, addition of 11,12-EET or overexpression of CYP2J2 decreases cyto-kine-induced endothelial cell adhesion molecule expression, reducing leukocyte adhesion to thevascular wall [18]. Nuclear transcription factor-kB (NF-kB) is a key component responsible forcytokine-mediated inflammation. NF-kB is inactive when bound to IkB, an inhibitory proteinthat is degraded by IkB kinase [175]. EETs inhibit IkB kinase, preventing degradation of IkB.This maintains NF-kB in the inactive state and inhibits NF-kB-mediated gene transcription [19].In addition, EETs induce polymorphonuclear leukocyte aggregation, and this also decreasesleukocyte adhesion to endothelial cells [176].
Fig. 15. Outline of a proposed tyrosine kinase pathway for EET signaling. The additional abbreviation used is: PI3-K,phosphatidylinositol 3-kinase.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 85

Inhibitors of CYP epoxygenases augment lipopolysaccharide- and interleukin-1b-induced feverin rat and mouse models [177,178], suggesting a role for EETs or their metabolites in the controlof temperature homeostasis. Furthermore, administration of EETs attenuates fever induced bylipopolysaccharide- and interleukin-1b [179,180]. The antipyretic effect may occur because 8,9-and 14,15-EET reduce the production of PGE2 [15], an endogenous pyrogen that contributes tocytokine-induced fever.
8.6. Conclusions regarding EET signaling mechanisms
Because EETs elicit multifunctional responses that are distributed across a wide variety of tis-sues, they probably act through more than one biochemical mechanism. Evidence is growing fora plasma membrane EET receptor or family of receptors that mediate some of the functionalresponses. It is possible that a signaling cascade initiated by a putative receptor, throughbranching and complex interactions with other signaling pathways, could produce many of thefunctions attributed to EETs. However, EETs are incorporated into cell phospholipids, andmetabolites that are generated intracellularly from EETs interact with nuclear receptors to reg-ulate gene expression. Therefore, an intracellular mechanism of action probably also exists, and itis doubtful that a single mechanism or signal transduction pathway will be able to account for thediversity of functional responses that are produced by EETs.
Acknowledgements
The work from our laboratory described in this article was supported by the National Institutesof Health grants HL72845, HL49264 and HL62984, and by American Heart Association grants0060413Z and 0230096N.
References
[1] Brash AR. J Clin Invest 2001;107:1339–45.[2] Capdevila JH, Falck JR, Harris RC. J Lipid Res 2000;41:163–81.[3] Capdevila JH, Falck JR. Biochem Biophys Res Commun 2001;285:571–6.[4] Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, et al. J Biol Chem 1997;272:12551–9.
[5] Karara A, Makita K, Jacobson HR, Falck JR, Guengerich FP, DuBois RN, et al. J Biol Chem 1993;268:13565–70.
[6] Harder DR, Campbell WB, Roman RJ. J Vasc Res 1995;32:79–92.
[7] Campbell WB, Harder DR. Circ Res 1999;84:484–8.[8] Roman RJ. Physiol Rev 2002;82:131–85.[9] Rosolowsky M, Campbell WB. Biochim Biophys Acta 1996;1299:267–77.
[10] Hu S, Kim HS. Eur J Pharmacol 1993;230:215–21.[11] Kroetz DL, Zeldin DC. Curr Opin Lipidol 2002;13:273–84.[12] Campbell WB, Gebremedhun D, Pratt PF, Harder DR. Circ Res 1996;78:877–84.
[13] Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, et al. Nature 1999;401:493–7.[14] Sun J, Sui XX, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Circ Res 2002;90:1020–7.
86 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

[15] Fang X, Moore SA, Stoll LL, Rich G, Kaduce TL, Weintraub NL, et al. Am J Physiol 1998;275:H2113–21.
[16] Fang X, Weintraub NL, Stoll LL, Spector AA. Hypertension 1999;34:1242–6.[17] Snyder GD, Krishna UM, Falck JR, Spector AA. Am J Physiol 2002;283:H1936–42.[18] Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Science 1999;285:1276–9.
[19] Campbell WB. Trends Pharmacol Sci 2000;21:125–7.[20] Graier WF, Simecek S, Sturek M. J Physiol 1995;482:259–74.[21] Mombouli J-V, Holzmann S, Kostner GM, Graier WF. J Cardiovas Pharmacol 1999;33:779–84.
[22] Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, et al. J Biol Chem 2001;276:15983–9.[23] Munzenmaier DH, Harder DR. Am J Physiol 2000;278:H1163–7.[24] Lee H-C, Lu T, Weintraub NL, VanRollins M, Spector AA, Shibata EF. J Physiol 1999;519(1):153–68.[25] Lu T, Hoshi T, Weintraub NL, Spector AA, Lee H-C. J Physiol 2001;537(3):811–27.
[26] Peri KG, Almazan G, Varma DR, Chemtob S. Biochim Biophys Res Commun 1998;244:96–101.[27] Chen J-C, Falck JR, Reddy KM, Capdevila JH, Harris RC. J Biol Chem 1998;273:29254–61.[28] Chen J-C, Wang D-W, Falck JR, Capdevila JH, Harris RC. J Biol Chem 1999;274:4764–9.
[29] Seki K, Hirai A, Noda M, Tamura Y, Kato I, Yoshida S. Biochem J 1992;281:185–90.[30] Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF. J Biol Chem 1986;261:15334–8.[31] Wong PY-K, Lin K-T, Yan Y-T, Ahern D, Iles J, Shen SY, et al. J Lipid Mediators 1993;6:199–208.
[32] Widstrom RL, Norris AW, Spector AA. Biochemistry 2001;40:1070–6.[33] Zeldin D, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, et al. J Biol Chem 1993;268:6402–7.[34] Zeldin DC,Wei S, Falck JR. Hammock BD, Snapper JR, Capdevila JH Arch Biochem Biophys 1995;316:443–51.
[35] Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA. Circ Res 1997;81:258–67.[36] Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Circ Res 1998;8:932–9.[37] Lu T, Katakam PVG, VanRollins M, Weintraub M, Spector AA, Lee H-C. J Physiol 2001;534(3):651–67.[38] Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li PL. Am J Physiol 2002;282:H1656–64.
[39] Morisseau C, Newman JW, Dowdy DL, Goodrow MH, Hammock BD. Chem Res Toxicol 2001;14:409–15.[40] Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. J Biol Chem 2000;275:40504–10.[41] Morisseau C, Goodrow MH, Dowdy DL, Zheng J, Greene JF, Sanborn IR, et al. Proc Natl Acad Sci USA
1999;96:8849–54.[42] Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. Circ Res 2000;87:992–8.[43] Oliw EH. Prog Lipid Res 1994;33:329–54.
[44] Zeldin DC. J Biol Chem 2001;276:36059–62.[45] Karara A, Dishman E, Falck JR, Capdevila JH. J Biol Chem 1991;266:7561–9.[46] Falck JR, Schueler VJ, Jacobson HR, Siddhanta AK, Pramanik B, Capdevila J. J Lipid Res 1987;28:840–6.
[47] Karara A, Dishman B, Jacobson H, Falck JR, Capdevila JH. FEBS Lett 1990;268:227–30.[48] Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. Biochem Biophys Res Commun 1992;182:1320–5.[49] Nakamura T, Bratton DL, Murphy RC. J Mass Spectrom 1997;32:888–96.[50] Falck JR, Yadagiri P, Capdevila J. Methods Enzymol 1990;187:357–64.
[51] VanRollins M, Kaduce TL, Knapp HR, Spector AA. J Lipid Res 1993;34:1931–42.[52] Bernstrom K, Kayganich K, Murphy RC, Fitzpatrick FA. J Biol Chem 1992;267:3686–90.[53] Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, et al. J Biol Chem 2001;276:14867–
74.[54] Fang X, Weintraub NL, Oltman CL, Stoll LL, Kaduce TL, Harmon S, et al. Am J Physiol 2002;283:H2306–14.[55] Fang X, VanRollins M, Kaduce TL, Spector AA. J Lipid Res 1995;36:1236–46.
[56] Fang X, Kaduce TL, Weintraub NL, VanRollins M, Spector AA. Circ Res 1996;79:784–93.[57] Fang X, Kaduce TL, Weintraub NL, Spector AA. Prostagl Leukot Essent Fatty Acids 1997;57:367–71.[58] Fang X, Kaduce TL, VanRollins M, Weintraub NL, Spector AA. J Lipid Res 2000;41:66–74.[59] Shrivachar AC, Willoughby KA, Ellis EF. J Neurochem 1995;65:338–46.
[60] Glatz JFC, Storch J. Curr Opin Lipidol 2001;12:267–75.[61] Veerkamp JH, Zimmerman AW. J Mol Neurosci 2001;16:133–42.[62] VanRollins M, Kaduce TL, Fang X, Knapp HR, Spector AA. J Biol Chem 1996;271:14001–9.
[63] Fang X, Weintraub NL, Spector AA. Prostagl Other Lipid Mediat 2003;71:33–42.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 87

[64] Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. J Am Soc Nephrol 1996;7:2364–70.
[65] Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, et al. Am J Physiol 2003;284:H337–49.
[66] Hirt DL, Capdevila J, Falck JR, Breyer MD, Jacobson HR. J Clin Invest 1989;84:1805–12.
[67] Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Circ Res 1998;83:932–9.[68] Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA. Am J Physiol 1999;277:H2098–
108.
[69] Zhang Y, Oltman CL, Lu T, Lee HC, Dellsperger KC, VanRollins M. Am J Physiol 2001;280:H2430–40.[70] Oliw EH. J Biol Chem 1984;259:2716–21.[71] Oliw EH. FEBS Lett 1984;172:279–93.[72] Carroll MA, Garcia MP, Falck JR, McGiff JC. J Pharmacol Exp Ther 1992;260:104–9.
[73] Carroll MA, Balazy M, Margiotta P, Falck JR, McGiff JC. J Biol Chem 1993;268:12260–6.[74] Zhang V, Prakash C, Yamashita K, Blair IA. Biochem Biophys Res Commun 1992;183:138–43.[75] Katoh T, Takahashi K, Capdevila J, Karara A, Falck JR, Jacobson HR, Badr KF. Am J Physiol 1991;
261:F578–86.[76] Stephenson AH, Sprague RS, Lonigro AJ. Am J Physiol 1998;275:H100–9.[77] Zhu D, Bousamara II M, Zeldin DC, Falck JR, Townsley M, Harder DR, et al. Am J Physiol 2000;278:L335–
43.[78] Pace-Asciak CR, Granstrom E, Samuelsson B. J Biol Chem 1983;258:6835–40.[79] Pace-Asciak CR, Martin JR. Prostagl Leukot Med 1984;16:173–80.
[80] Pfister SL, Spitzbarth N, Edgemond W, Campbell WB. Am J Physiol 1996;270:H1021–30.[81] Pfister SL, Spitzbarth N, Nithipatikom K, Edgemond WS, Falck JIR, Campbell WB. J Biol Chem 1998;
273:30879–87.[82] Weiss RH, Arnold JL, Estabrook RW. Arch Biochem Biophys 1987;252:334–8.
[83] Ma YH, Schwartzman ML, Roman RJ. Am J Physiol 1994;267:R579–89.[84] Cowart LA, Wei S, Hsu MH, Johnson EF, Krishna MU, Falck JR, et al. J Biol Chem 2002;277:35105–12.[85] Spearman ME, Prough RA, Estabrook RW, Falck JR, Manna S, Leibman KC, et al. Arch Biochem Biophys
1985;242:225–30.[86] Murphy RC, Zarini S. Prostaglandins Other Lipid Mediat 2002;68/69:471–82.[87] Lauterbach B, Barbosa-Sicard E,WangMH, Honeck H, Kargel E, Theuer I, et al. Hypertension 2002;39:609–13.
[88] VanRollins M. Lipids 1990;25:481–90.[89] Knapp HR, Miller AJ, Lawson JA. Prostaglandins 1991;42:47–54.[90] Ye D, Zhang D, Oltman C, Dellsperger K, Lee H-C, VanRollins M. J Pharmacol Exp Ther 2002;303:768–76.
[91] Guengerich FP. Biochem Toxicol 1981;4:5–30.[92] Fretland AJ, Omiecinski CJ. Chem Biol Interact 2000;129:41–59.[93] Johansson C, Stark A, Sandberg M, Ek B, Rask L, Meijer J. Arch Toxicol 1995;70:61–3.[94] Hollinshead M, Meijer J. Eur J Cell Biol 1988;46:394–402.
[95] Eriksson AM, Zetterqvist MA, Lundgren B, Andersson K, Beije B, DePierre JW. Eur J Biochem 1991;198:471–6.[96] Mullen RT, Trelease RN, Duerk H, Arand M, Hammock BD, Oesch F, et al. FEBS Lett 1999;445:301–5.[97] Waechter F, Merdes M, Bien F, Staubli W, Bentley P. Eur J Biochem 1982;125:457–61.
[98] Schladt L, Thomas H, Hartmann R, Oesch F. Eur J Biochem 1988;176:715–23.[99] Schladt L, Hartmann R, Worner W, Thomas H, Oesch F. Eur J Biochem 1988;176:31–7.[100] Beetham JK, Tian T, Hammock BD. Arch Biochem Biophys 1993;305:197.
[101] Beetham JK, Grant D, Arand M, Garbarino J, Kiyosue T, Pinot F, et al. DNA and Cell Biol 1995;14:61–71.[102] Pinot F, Grant DF, Beetham JK, Parker AG, Borhan B, Landt S, et al. J Biol Chem 1995;270:7968–74.[103] Grant DF, Storms DH, Hammock BD. J Biol Chem 1993;268:17628–33.[104] Larsson C, White I, Johansson C, Stark A, Meijer J. Hum Genet 1995;95:356–8.
[105] Sandberg M, Meijer J. Biochem Biophys Res Commun 1996;221:333–9.[106] Sandberg M, Hassett C, Adman ET, Meijer J, Omiecinski CJ. J Biol Chem 2000;275:28873–81.[107] Saito S, Iida A, Sekine A, Eguchi C, Miura Y, Nakamura Y. J Hum Genet 2001;46:325–9.
[108] Vesell ES. Ann Genet 1991;34:167–72.
88 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

[109] Catella F, Lawson JA, Fitzgerald DJ, FitzGerald GA. Proc Natl Acad Sci USA 1990;87:5893–7.
[110] Lundgren B, Meijer J, DePierre JW. Biochem Pharmacol 1987;36:815–21.[111] Lundgren B, Meijer J, Birberg W, Pilotti A, Depierre JW. Chem Biol Interact 1988;68:219–40.[112] Pinot F, Grant DF, Spearow JL, Parker AG, Hammock BD. Biochem Pharmacol 1995;50:501–8.
[113] Hammock BD, Pinot F, Beetham JK, Grant DF, Arand ME, Oesch F. Biochem Biophys Res Commun 1994;198:850–6.
[114] Borhan B, Jones AD, Pinot F, Grant DF, Kurth MJ, Hammock BD. J Biol Chem 1995;270:26923–30.
[115] Arand M, Wagner H, Oesch F. J Biol Chem 1996;271:4223–9.[116] Yamada T, Morisseau C, Maxwell JE, Argiriadi MA, Christianson DW, Hammock BD. J Biol Chem 2000;
275:23082–8.[117] Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Proc Natl Acad Sci USA 1999;96:10637–42.
[118] Cronin A, Mowbray S, Durk H, Homburg S, Fleming I, Fisslthaler B, et al. Proc Natl Acad Sci USA 2003;100:1552–7.
[119] Newman JW, Morisseau C, Harris TR, Hammock BD. Proc Natl Acad Sci USA 2003;100:1558–63.
[120] Mullin CA, Hammock BD. Arch Biochem Biophys 1982;216:423–39.[121] Dietze EC, Kuwano B, Casas J, Hammock BD. Biochem Pharmacol 1991;42:1163–75.[122] Morisseau C, Du G, Newman IW, Hammock BD. Arch Biochem Biophys 1998;356:214–28.
[123] Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Biochem Pharmacol2002;63:1599–608.
[124] Draper AJ, Hammock BD. Toxicol Sci 1999;52:26–32.
[125] Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW. J Biol Chem 2000;275:15265–70.
[126] Yu Z, Ruse LM, Adler P, Graham L, Ma J, Zeldin DC, Kroetz DL. Mol Pharmacol 2000;57:1011–20.[127] Imig ID, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Hypertension 2002;39:690–4.
[128] Fornage M, Hinojos CA, Nurowska BW, Boerwinkle E, Hammock BD, Morisseau CH, et al. Hypertension2002;40:485–90.
[129] Capdevila JH, Wei S, Yan J, Karara A, Jacobson HR, Falck JR, et al. J Biol Chem 1992;267:21720–6.
[130] Imig JD, Zhao X, Falck JR, Wei S. Capdevila JR J Hypertens 2001;19:983–92.[131] Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Proc Natl Acad Sci USA
2002;99:2222–7.
[132] Ozawa T, Sugiyama S, Hayakawa M, Taki F, Hanaki Y. Biochem Biophys Res Commun 1988;152:1310–8.[133] Moghaddam M, Motoba K, Borhan B, Pinot F, Hammock BD. Biochim Biophys Acta 1996;1290:327–39.[134] Greene JF, Williamson KC, Newman JW, Morisseau C, Hammock BD. Arch Biochem Biophys 2000;376:420–
32.[135] Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Nat Med 1997;3:562–6.[136] Zheng J, Plopper CG, Lakritz J, Storms DH, Hammock BD. Am J Respir Cell Mol Biol 2001;25:434–8.[137] Greene JF, Newman JW, Williamson KC, Hammock BD. Chem Res Toxicol 2000;13:217–26.
[138] Moran JH, Mon T, Hendrickson TL, Mitchell LA, Grant DF. Chem Res Toxicol 2001;14:431–7.[139] Mitchell LA, Moran JH, Grant DF. Toxicol Lett 2002;126:187–96.[140] Funk CD. Science 2001;294:1871–5.
[141] Wong PY, Lai PS, Shen SY, Belosludtsev YY, Falck JR. J Lipid Mediat Cell Signal 1997;16:155–69.[142] Sakairi Y, Jacobson HR, Noland TD, Capdevila JH, Falck JR, Breyer MD. Am J Physiol 1995;268:F931–9.[143] Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, et al. Am J Physiol 1996;270:F822–
32.[144] Pascual JM, McKenzie A, Yankaskas JR, Falck JR, Zeldin DC. J Pharmacol Exp Ther 1998;286:772–9.[145] Wong PY, Lai PS, Falck JR. Prostaglandins Other Lipid Mediat 2000;62:321–33.[146] Stahl A, Gimeno RE, Tartaglia LA, Lodish HF. Trends Endocrinol Metab 2001;12:266–73.
[147] Leffler CW, Fedinec AL. Am J Physiol 1997;273:H333–8.[148] Xiao YF, Huang L, Morgan JP. J Physiol 1988;508:33777–92.[149] Chen J, Capdevila JH, Zeldin DC, Rosenberg RL. Mol Pharmacol 1999;55:288–95.
[150] Kutsky P, Falck JR, Weiss GB, Manna S, Chacos N, Capdevila J. Prostaglandins 1983;26:13–21.
A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90 89

[151] Moffat MP, Ward CA, Bend IR, Mock T, Farhangkhoee P, Kannazyn M. Am J Physiol 1993;264:H1154–60.
[152] Force T, Hyman G, Hajjar R, Sellmayer A, Bonventre JV. J Biol Chem 1991;266:4295–302.[153] Hoebel BG, Graier WF. Eur J Pharmacol 1998;346:115–7.[154] Harris RC, Homma T, Jacobson HR, Capdevila J. J Cell Physiol 1990;144:429–37.
[155] Furstenau M, Lohn M, Ried C, Lull FC, Haller H, Gollasch M. J Hypertension 2000;18:1212–5.[156] Li PL, Campbell WB. Circ Res 1997;80:877–84.[157] Perez GJ, Bonev AD, Nelson MT. Am J Physiol 2001;281:C1769–75.
[158] Li PL, Campbell WB. Circ Res 1997;80:877–84.[159] Li PL, Chen CL, Bortell R, Campbell WB. Circ Res 1999;85:349–56.[160] Pierce KL, Premont RT, Lefkowitz RI. Nat Rev Mol Cell Biol 2002;9:639–50.[161] Breyer MD, Breyer RM. Annu Rev Physiol 2001;63:579–605.
[162] Ashby B. Receptor 1994;4:31–42.[163] Zhou XB, Arntz C, Kamm S, Motejlek K, Sausbier U, Wang GX, et al. J Biol Chem 2001;276:43239–45.[164] Hoebel BG, Graier WF. Eur J Pharmacol 1998;346:115–7.
[165] Chen JK, Capdevilla J, Harris RC. J Biol Chem 2000;276:13789–92.[166] Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. FASBB J 2003;17:770–2.[167] Chen JK, Capdevila J, Hams RC. Proc Natl Acad Sci USA 2002;99:6029–34.
[168] Fleming I, Fisslthaler B, Busse R. J Biol Chem 1996;271:11009–15.[169] Fleming I, Fisslthaler B, Busse R. Circ Res 1995;76:522–9.[170] Snyder G, Lattanzio F, Yadagiri P, Falck JR, Capdevila J. Biochem Biophys Res Commun 1986;139:1188–94.
[171] Thibonnier M, Bayer AL, Laethem CL, Koop DR, Simonson MS. Am J Physiol 1993;265:E108–14.[172] Mombouli IV, Holzmann S, Kostner GM, Graier WF. J Cardiovasc Pharmacol 1999;33:779–84.[173] Rzigalinski BA, Willoughby KA, Hoffman SW, Falck IR, Ellis EF. J Biol Chem 1999;274:175–82.[174] Hoebel BG, Kostner GM, Graier WF. Br J Pharmacol 1997;121:1579–88.
[175] Karin M. J Biol Chem 1999;274:27339–42.[176] Pratt PF. Rosolowsky M, Campbell WB Life Sci 2002;70:2521–33.[177] Nakashima T, Yoshida Y, Miyata S, Kiyohara T. Neurosci Lett 2001;310:141–4.
[178] Kozak W, Mayfield KP, Kozak A, Kluger MJ. J Therm Biol 2000;25:45–50.[179] Nakashima T, Harada Y, Miyata J, Kiyohara T. Am J Physiol 1996;271:R1274–79.[180] Kozak W, Kluger MJ, Kozak A, Wachulec M, Dokiadny K. Am J Physiol 2000;279:R455–60.
90 A.A. Spector et al. / Progress in Lipid Research 43 (2004) 55–90

















![Effects of Fatty Acids on Benzo[a]pyrene Uptake and Metabolism in Human Lung Adenocarcinoma A549 Cells](https://static.fdokumen.com/doc/165x107/63334a02a290d455630a124b/effects-of-fatty-acids-on-benzoapyrene-uptake-and-metabolism-in-human-lung-adenocarcinoma.jpg)


