
Epidermal growth factor binding sites in human pituitary macroadenomas
Upload
independentCategory
view
1download
0
Epidermal Growth Factor Impairs the Cytochrome C/Caspase-3Apoptotic Pathway Induced by Transforming Growth Factor b
in Rat Fetal Hepatocytes Via a Phosphoinositide3-Kinase–Dependent Pathway
ISABEL FABREGAT,1 BLANCA HERRERA,1 MARGARITA FERNANDEZ,1 ALBERTO M. ALVAREZ,2 ARANZAZU SANCHEZ,1 CESAR RONCERO,1
JUAN-JOSE VENTURA,1 ANGELA M. VALVERDE,1 AND MANUEL BENITO1
Transforming growth factor b (TGF-b)–mediated apopto-sis is one of the major death processes in the liver. We havepreviously shown that epidermal growth factor (EGF) is animportant survival signal for TGF-b–induced apoptosis infetal hepatocytes (Fabregat et al., FEBS Lett 1996;384:14-18). In this work we have studied the intracellular signalingimplicated in the protective effect of EGF. We show herethat EGF activates p42 and p44 mitogen-activated proteinkinases (MAPK). However, mitogen extracellular kinase(MEK) inhibitors do not block the survival effect of EGF.EGF also activates phosphoinositide 3-kinase (PI 3-kinase)and protein kinase B (PKB/AKT) in these cells. The presenceof PI 3-kinase inhibitors blocks the protective effect of EGFon cell viability, DNA fragmentation, and caspase-3 activity.We have found that TGF-b disrupts the mitochondrialtransmembrane potential (DCm) and activates the release ofcytochrome c, this effect being blocked by EGF, via a PI3-kinase–dependent pathway. A detailed study on bcl-2 su-perfamily gene expression shows that TGF-b produces adecrease in the messenger RNA (mRNA) and protein levelsof bcl-xL, an antiapoptotic member of this family, capable ofpreventing cytochrome c release. EGF is able to maintainbcl-xL levels even in the presence of TGF-b. PI 3-kinaseinhibitors completely block the protective effect of EGF onTGF-b–induced bcl-xL down-regulation. We conclude thatPI 3-kinase mediates the survival effect of EGF on TGF-b–induced death by acting upstream from the mitochondrialchanges, i.e., preventing bcl-xL down-regulation, cytochrome c
release, and activation of caspase-3. (HEPATOLOGY 2000;32:528-535.)
Apoptosis is an active cell suicide program characterized bymorphologic changes including membrane blebbing, conden-sation of the nucleoplasm, and degradation of chromosomalDNA. Several concepts have recently emerged with respect tothe role of apoptosis in liver physiology and pathology. First,liver hyperplasia during development or regeneration may beaided by inhibition of apoptosis; second, liver atrophy occursby apoptosis of liver cells in the absence of regeneration; andthird, pathophysiologic processes can trigger the cellular apo-ptotic machinery leading to disease processes (for review seePatel et al.1). Numerous observations suggest that transform-ing growth factor b (TGF-b), an extracellular polypeptideimportant in growth control, development, and differentia-tion (reviewed in Massague2), plays an important biologicalrole mediating hepatocyte death (reviewed in Patel et al.,1
Pitot,3 and Thorgeirsson et al.4). Disruption of the TGF-bpathway at the prereceptor, receptor, or postreceptor levelsoccurs in hepatocellular carcinomas and can cause dysregula-tion of apoptosis.4 In previous works from our group, wefound that TGF-b inhibits growth of fetal hepatocytes, arrest-ing cells in G1 and down-regulating myc expression.5 But, inaddition to these functions, TGF-b, when used at higher con-centrations, induces fetal hepatocyte apoptosis.6 The apopto-tic effect of TGF-b in fetal hepatocytes can be inhibited bytreatment with epidermal growth factor (EGF),7 which sug-gest that a survival signal activated by EGF is impairing theapoptotic signal of TGF-b. However, the molecular level atwhich EGF exerts its action is unknown yet.
Some intracellular signals have been proposed to functionagainst apoptosis induced by TGF-b in different liver cellsystems. Thus, Chen et al.8 found that the suppression of theapoptotic effect of TGF-b in the human hepatoma cells,Hep3B, was dependent on the phosphoinositide 3-kinase (PI3-kinase)/Akt pathway, but not the Ras/mitogen-activatedprotein kinases (MAPK) signaling.8 Activation of PI 3-kinasepathway did not lead to a suppression of Smad (a transcriptionfactor family of proteins mediating TGF-b responses) hetero-oligomerization, or nuclear translocation but blocked TGF-b–induced caspase-3–like activity. Some cytokines, such asinterleukin 6, have been shown to protect TGF-b–inducedapoptotic effect in hepatocytes by a PI 3-kinase and STAT-3dependent pathways.9 It has been also described that a phor-
Abbreviations: TGF-b, transforming growth factor b; EGF, epidermal growth factor;PI 3-kinase, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinase; MEK,mitogen extracellular kinase; PBS, phosphate-buffered saline; EDTA, ethylenediamine-tetraacetic acid; cDNA, complementary DNA; PKB, protein kinase B; ERK, extracellularsignal-regulated kinase; CMXRos, chloromethyl-X-rosamine; mRNA, messenger RNA.
From the 1Departamento de Bioquımica y Biologıa Molecular, Facultad de Farmacia(Centro Mixto CSIC/UCM) and 2Centro de Citometrıa de Flujo y Microscopıa Confocal,Universidad Complutense de Madrid, Madrid, Spain.
Received April 18, 2000; accepted June 9, 2000.Supported by a grant from the Ministerio de Educacion y Cultura (PM97/0052). B.H.
and J.-J.V. were recipients of fellowships from the Ministerio de Educacion y Cultura andA.S. was a recipient of a fellowship from the Comunidad de Madrid.
Address reprint requests to: Isabel Fabregat, Ph.D., Departamento de Bioquımica yBiologıa Molecular, Facultad de Farmacia, Universidad Complutense de Madrid, CiudadUniversitaria, 28040 Madrid, Spain. E-mail: [email protected]; fax: (34)913941779.
Copyright © 2000 by the American Association for the Study of Liver Diseases.0270-9139/00/3203-0013$3.00/0doi:10.1053/jhep.2000.9774
528

bol ester, a selective stimulator of protein kinase C, dimin-ishes the DNA fragmentation and cell death induced byTGF-b in adult hepatocytes.10 Recently, Roberts et al.11 haveshown that EGF-mediated survival effect requires both PI3-kinase and MAPK pathways in adult rat hepatocytes.
We have previously shown that EGF does not regulate pro-tein kinase C activity in fetal hepatocytes.12 Furthermore, inour experimental system, phorbol esters do not protect fetalhepatocytes from the apoptosis induced by TGF-b.13 Theseresults suggest that protein kinase C does not appear to beinvolved in the EGF protective effect. In contrast, EGF acti-vates p42/p44 MAPKs in fetal hepatocytes.12 The possible in-volvement of PI 3-kinase in the signaling elicited by EGF infetal hepatocytes remains unexplored.
Recent reports have provided evidence that mitochondriaare deeply involved in the regulation of apoptosis.14 The dis-ruption of the mitochondrial transmembrane potential (DCm)has been defined as an early irreversible stage of apoptosis.15
Cytochrome c, which is normally confined in the mitochon-drial intermembrane space, is found in the cytosol of cellsundergoing apoptosis, this process being inhibited by thepresence of Bcl-2 on these organelles.16,17 Cytosolic cyto-chrome c forms an essential part of the vertebrate apopto-some, which is composed of cytochrome c, Apaf-1 and pro-caspase-9. The result is activation of caspase-9, which thenprocesses and activates other caspases, such as caspase-3, toorchestrate the biochemical execution of programmed celldeath.18 Proapoptotic Bcl-2 family proteins, including Baxand Bak, induce cytochrome c release by interacting with themitochondrial permeability transition pores.19,20 In contrast,Bcl-xL, an antiapoptotic member of the Bcl-2 family, is capableof preventing cytochrome c release while also significantlyinhibiting cell death.19,21 Rodrigues et al. have recently shownthat TGF-b decreases mitochondrial transmembrane poten-tial and provokes cytochrome c release in adult hepatocytes.22
In this work we have studied the intracellular signalingimplicated in the protective effect of EGF on different eventsrelated to the apoptosis induced by TGF-b in fetal hepato-cytes. Using specific inhibitors, we have found that PI 3-ki-nase mediates suppression by EGF of DNA fragmentation,caspase-3 activation, cytochrome c release, and disruptionof the mitochondrial transmembrane potential induced byTGF-b. A study on bcl-2 family gene expression shows thatTGF-b down-regulates bcl-xL expression and EGF preventsthis process in a PI 3-kinase–dependent manner.
MATERIALS AND METHODS
Reagents. Human recombinant TGF-b was from Calbiochem (LaJolla, CA) and human recombinant EGF was kindly provided bySerono Laboratories (Madrid, Spain). Collagenase was from Roche(Barcelona, Spain). Fetal and neonatal calf serum and culture mediawere from Imperial Laboratories (Hampshire, UK). Radiochemicalswere from ICN (Irvine, CA). The fluorescent probe chloromethyl-X-rosamine (CMXRos) was from Molecular Probes (Eugene, OR).Antibodies were anti-MAPK (p44/p42) antibody from Upstate Bio-technology (Lake Placid, NY); anti-active MAPK (phospho-p44/phospho-p42) from Promega (Madison, WI); anti-Bcl-x and anti-Baxpolyclonal antibodies from Santa Cruz Biotechnology (Santa Cruz,CA); anti-active AKT from New England Biolabs (Beverly, MA); anti-rat albumin polyclonal antibody from Nordic Immunological Labo-ratories (Tilburg, The Netherlands). Caspase-3 substrate Ac-DEVD-AMC and anti-cytochrome c were from Pharmingen (San Diego,CA). PD098059 (a mitogen extracellular kinase [MEK] inhibitor)and LY294002 (a PI 3-kinase inhibitor) were from Calbiochem.
Other reagents were from Sigma Chemical Co. (St. Louis, MO) orBoehringer (Mannheim, Germany).
Cell Isolation and Culture. Hepatocytes from 20-day-old fetal Wistarrats were isolated as previously described by collagenase disruption(2.5 3 106 cells/fetus)23 and plated on plastic (noncoated) dishes inarginine free medium 199, supplemented with ornithine (200 mmol/L), fetal calf serum (10%), penicillin (120 mg/mL) and streptomycin(100 mg/mL).5 Cells were incubated in 5% CO2, at 37°C for 4 hours,allowing cell attachment to plates. Media was changed at that timeand replaced by one of the same composition except that 10% fetalcalf serum was changed by 2% newborn bovine serum. The presenceof this low concentration of serum (2%) was convenient to maintainthe attachment of cells and progression of culture, but was notenough to induce proliferation. After 18 to 20 hours, the mediumwas again replaced for one of identical composition but in the ab-sence of serum. Two hours later, cells were ready for all the experi-ments described above.
PI 3-Kinase Activity. PI 3-kinase activity was measured by in vitrophosphorylation of phosphatidylinositol. Cells were washed withice-cold phosphate-buffered saline (PBS), solubilized in lysis buffer(10 mmol/L Tris/HCl, 5 mmol/L ethylenediaminetetraacetic acid[EDTA], 50 mmol/L NaCl, 30 mmol/L sodium pyrophosphate, 50mmol/L NaF, 100 mmol/L Na3VO4, 1% Triton X-100, pH 7.6) con-taining leupeptin (10 mg/mL), aprotinin (10 mg/mL) and 1 mmol/Lphenylmethylsulfonyl fluoride. Lysates were clarified by centrifuga-tion at 15,000g for 10 minutes at 4°C, and proteins were immuno-precipitated with a monoclonal antibody anti-Tyr(P)(Py72). The im-munoprecipitates were washed successively in (1) PBS containing1% Triton X-100 and 100 mmol/L Na3VO4 (twice), (2) 100 mmol/LTris (pH 7.5) containing 0.5 mol/L LiCl, 1 mmol/L EDTA and 100mmol/L Na3VO4 (twice), and (3) 25 mmol/L Tris (pH 7.5) containing100 mmol/L NaCl and 1 mmol/L EDTA (twice). To each pellet 25mL of 1 mg/mL L-a-phosphatidylinositol/L-a-phosphatidyl-L-serinesonicated in 25 mmol/L HEPES (pH 7.5) and 1 mmol/L EDTA wereadded. The PI 3-kinase reaction was started by the addition of 100nmol/L (g32P)-adenosine triphosphate (10 mCi) and 300 mmol/Ladenosine triphosphate in 25 mL of 25 mmol/L HEPES pH 7.4, 10mmol/L MgCl2 and 0.5 mmol/L ethylene glycol-bis(b-aminoethylether)-N,N-tetraacetic acid. After 15 minutes at room temperaturethe reaction was stopped by the addition of 500 mL of CHCl3-meth-anol (1:2) in a 1% HCl plus 125 mL chloroform and 125 mL of HCl(10 mmol/L). The samples were centrifuged, and the organic phasewas removed and washed once with 480 mL of methanol-100mmol/L HCl plus 2 mmol/L EDTA (1:1). The organic phase wasextracted, dried in vacuo, and resuspended in chloroform. Sampleswere applied to a silica gel TLC plate (Whatman, Clifton, NJ). TLCplates were developed in propanol-1-acetic acid (2N; 65:35 vol/vol),dried and visualized by autoradiography.
Western Blot Analysis. To detect p42/p44 MAPKs, phospho MAPKsand phospho-AKT, after washing the cells with cold PBS, they werescraped off and lysed at 4°C in a buffer containing 25 mmol/L HEPES(pH 7.5); 0.3 mol/L NaCl; 1.5 mmol/L MgCl2; 0.2 mmol/L EDTA; 0.5mmol/L dithiothreitol; 0.1% Triton X-100; 200 mmol/L b glycero-phosphate; 0.1 mmol/L Na3VO4; 2 mg/mL leupeptin and 1 mmol/Lphenylmethylsulfonyl fluoride. Cells were incubated with buffer for10 minutes at 4°C. Then, samples were clarified by centrifugation at12,000g for 10 minutes at 4°C. To detect total Bcl-x or Bax, superna-tant cells were collected by centrifugation 2,000g for 5 minutes at4°C: attached cells were scraped off in PBS, pelleted by centrifugation4,000g for 10 minutes at 4°C and resuspended in a lysis buffer (25mmol/L HEPES; 2.5 nmol/L EDTA; 0.1% Triton X-100, 1 mmol/Lphenylmethylsulfonyl fluoride, 5 mg/mL leupeptin). Samples weresonicated 30 seconds at 1.5 mA and lysates were clarified by centrif-ugation at 12,000g for 10 minutes. When Bax and Bcl-xL levels wereanalyzed in mitochondrial extracts, the method to isolate mitochon-dria was the same described below (in analysis of cytochrome crelease section). Protein concentration of lysates was determinedusing the Bio-Rad (Hercules, CA) protein assay kit according to themanufacture’s specifications. Following electrophoresis separation
HEPATOLOGY Vol. 32, No. 3, 2000 FABREGAT ET AL. 529

of 50 mg protein/condition in sodium dodecyl sulfate 12% polyacril-amide, gels were transferred by semidry transfer (BioRad Laborato-ries, Richmond, CA) to nitrocellulose membranes (Schleide andSchuell). Immunoblots were blocked in TTBS (10 mmol/L Tris/HCl,150 mmol/L NaCl, pH 7.5, 0.05% Tween 20) containing 5% nonfatdried milk and incubated overnight with the primary antibody (di-luted 1:1,000 in TTBS 0.5% nonfat dried milk). After washing, mem-branes were incubated with peroxide-conjugated secondary antibod-ies (1:5,000 in TTBS 0.5% nonfat dried milk) for 2 hours and the blotwas developed with the ECL system (Amersham, Buckingham-shire, UK).
TGF-b–Mediated Cytotoxicity Assay. After cell incubation in absenceor presence of different factors, the medium was discarded, and theremaining viable adherent cells were stained, as described previ-ously5 with crystal violet (0.2% in 2% ethanol) for 20 minutes. Afterthis time, plates were rinsed with tap water, allowed to dry, and 1%sodium dodecyl sulfate was added. The absorbance of each plate wasread photometrically at 560 nm. Remnant viable cells were calcu-lated as percent of absorbance with respect to control cells (incu-bated in the absence of growth factors).
Analysis of Nuclear DNA Content by Flow Cytometry. The ploidy de-termination of hepatocytes was estimated by flow cytometry DNAanalysis, as previously described.6,7 Cells were detached from dishesby addition of 0.25% trypsin-0.02% EDTA, fixed in methanol(220°C) for 1 minute and treated with RNAse (10 mg/mL) for 30minutes at 37°C. The DNA content per cell was then evaluated in aFACScan flow cytometer (Becton-Dickinson, San Jose, CA), afterstaining cells with propidium iodide (0.05 mg/mL) for 15 minutes atroom temperature in the dark. For the computer analysis, only sig-nals from single cells were considered (10,000 cells/assay).
Analysis of Caspase-3 Activity. Cells were scraped off in PBS, col-lected by centrifugation at 2,500g for 5 minutes and lysed at 4°C in 5mmol/L Tris/HCl, pH 8.0, 20 mmol/L EDTA, 0.5% Triton X-100.Lysates were clarified by centrifugation at 13,000g for 10 minutes.Reaction mixture contained 25 mL cellular lysates, 325 mL assaybuffer (20 mmol/L HEPES pH 7.5, 10% glycerol, 2 mmol/L dithio-threitol) and 20 mmol/L caspase-3 substrate (Ac-DEVD-AMC). After2-hour incubation in the dark, enzymatic activity was measured ina Luminescence Spectrophotometer (Perkin Elmer LS-50, Norwalk,CT) (l excitation, 380 nm; l emission, 440 nm). We define a unit ofcaspase-3 activity as the amount of active enzyme necessary to pro-duce an increase in 1 arbitrary unit in the fluorimeter after 2-hourincubation with the reaction mixture. Then, protein concentration ofcell lysates was determined by using the Bio-Rad protein assay kitand final expression of the results is presented as units of caspase-3activity per mg protein.
Analysis of Mitochondrial Transmembrane Potential. The fluorescentprobe chloromethyl-X-rosamine (CMXRos) was used to analyze themitochondrial transmembrane potential by flow cytometry. Fetalhepatocytes were incubated in the presence or in the absence of thedifferent factors and, at different times, cells were detached bytrypsinization and resuspended in PBS. The cellular fluorescenceintensity was measured after 30-minute incubation of the cells with0.1 mmol/L CMXRos. A FACScan flow cytometer (Becton-Dickin-son) was used. For each analysis, 10,000 events were recorded.
Analysis of Cytochrome C Release. Attached cells were scraped off inisotonic isolation buffer (1 mmol/L EDTA; 10 mmol/L HEPES, 250mmol/L sucrose, pH 7.6), collected by centrifugation at 2,500g for5 minutes at 4°C and resuspended in hipotonic isolation buffer (1mmol/L EDTA, 10 mmol/L HEPES, 50 mmol/L sucrose, pH 7.6).Then, cells were incubated at 37°C for 5 minutes and homogenizedunder a teflon pestle (Overhead Stirrer; Wheaton Instruments, Mil-ville, NJ). Hipertonic isolation buffer (1 mmol/L EDTA, 10 mmol/LHEPES, 450 mmol/L sucrose, pH 7.6) was added to balance thebuffer’s tonicity. Samples were centrifuged at 2,000g for 5 minutes at4°C. Supernatants were recovered and centrifuged again at 10,000gfor 10 minutes. Pellet contained the mitochondrial fraction, whichwas resuspended in isotonic isolation buffer, and supernatant thecytosolic protein extract. Cytochrome c was analyzed by Western
blot (as described above) after electrophoresis separation of 50 mg/condition in sodium dodecyl sulfate 15% polyacrilamide gels. Mito-chondrial contamination of the cytosolic protein extracts wasdetermined by analysis of cytochrome c oxidase, measured photo-metrically at 550 nm as previously described.24
RNA Isolation and Northern Blot Analysis. For each assay, total RNAwas extracted from the pooled cells of two 92-mm-diameter dishes,as described by Chomczynski and Sacchi.25 Twenty microgramsRNA per condition were denatured in 50% formamide, 2.2 mol/Lformaldehyde, 20 mmol/L 4 Morpholinepropane sulfonic acid(MOPS), pH 7.0, 6% glycerol at 65°C for 15 minutes, separated bysize on gels containing 0.9% agarose and 0.66 mol/L formaldehydeand blotted on GeneScreenTM membranes (NEN Research Products,Dupont, Boston, MA). Hybridization conditions were previously de-scribed.6 Bcl-xL complementary DNA (cDNA) (a 745-bp fragmentFlag-Bclx in pcDNA3) was kindly provided by Dr. Nunez (Ann Ar-bor, MI) and was labeled with (a-32P) deoxycytidine triphosphate byrandom priming. 18S Ribosomal cDNA was a gift of Dr. Rozengurt(UCLA, Los Angeles, CA) and was labeled with (a-32P)deoxycytidinetriphosphate by nick-translation reaction. Sequential hybridizationwith the different probes was performed.
Statistical Analysis. Differences between means were statisticallycompared by analysis of variance, using Statview 1.03 for Macintosh.Data were compared among them by the Scheffe F test and consid-ered significantly different when P values were less than .05.
RESULTS
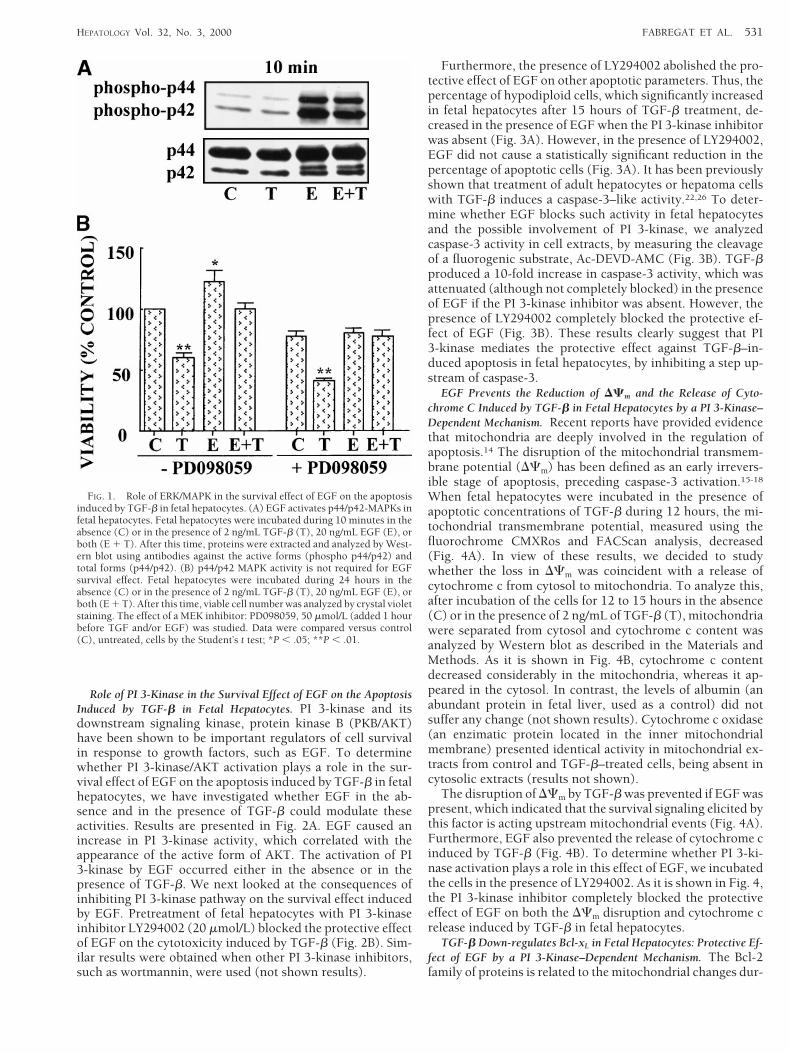
Role of Extracellular Regulated Kinase/MAPK in the Survival Ef-fect of EGF on the Apoptosis Induced by TGF-b in Fetal Hepatocytes.MAPKs are important intermediates in signaling pathwaysthat transduce extracellular signals into intracellular re-sponses and have been implicated in a wide array of physio-logic processes, including cell growth and survival. To deter-mine whether extracellular regulated kinase (ERK)/MAPKactivation plays a role in the survival effect of EGF on theapoptosis induced by TGF-b in fetal hepatocytes, we first ex-amined if EGF in the absence and in the presence of TGF-bcould modulate these activities. Results are presented in Fig.1A. EGF, but not TGF-b, caused a marked increase in theactive forms of p42/p44 MAPKs, which correlated with phos-phorylation of myelin basic protein (data not shown). Theactivation of ERK/MAPK by EGF occurred either in the ab-sence or in the presence of TGF-b. We next looked at theconsequences of blocking specific components of the ERK/MAPK signaling pathway on the survival effect induced byEGF. Pretreatment of fetal hepatocytes with the MEK inhibi-tor PD098059 (50 mmol/L), which completely blocks theERK/MAPK activation caused by EGF in fetal hepatocytes,12
did not affect the protective effect of EGF on the cytotoxicityinduced by TGF-b (Fig. 1B). Similar results were obtainedwhen other parameters related to apoptosis were analyzed.Thus, cell morphology, apoptotic nuclei, and hypodiploidcells did not appear when cells were incubated in the presenceof EGF and TGF-b either in the absence or in the presence ofPD098059 (not shown results). These results suggest thatactivation of the ERK/MAPK pathway is not required for thesurvival effect of EGF. However, pretreatment of fetal hepa-tocytes with PD098059 reduced the ability of EGF to induceproliferation. Thus, when cells were incubated with EGF andMEK inhibitor, cell number did not increase (Fig. 1B) andthere were not changes in (3H)-thymidine incorporation intoDNA and/or cell cycle analysis by flow cytometry (not shownresults).
530 FABREGAT ET AL. HEPATOLOGY September 2000
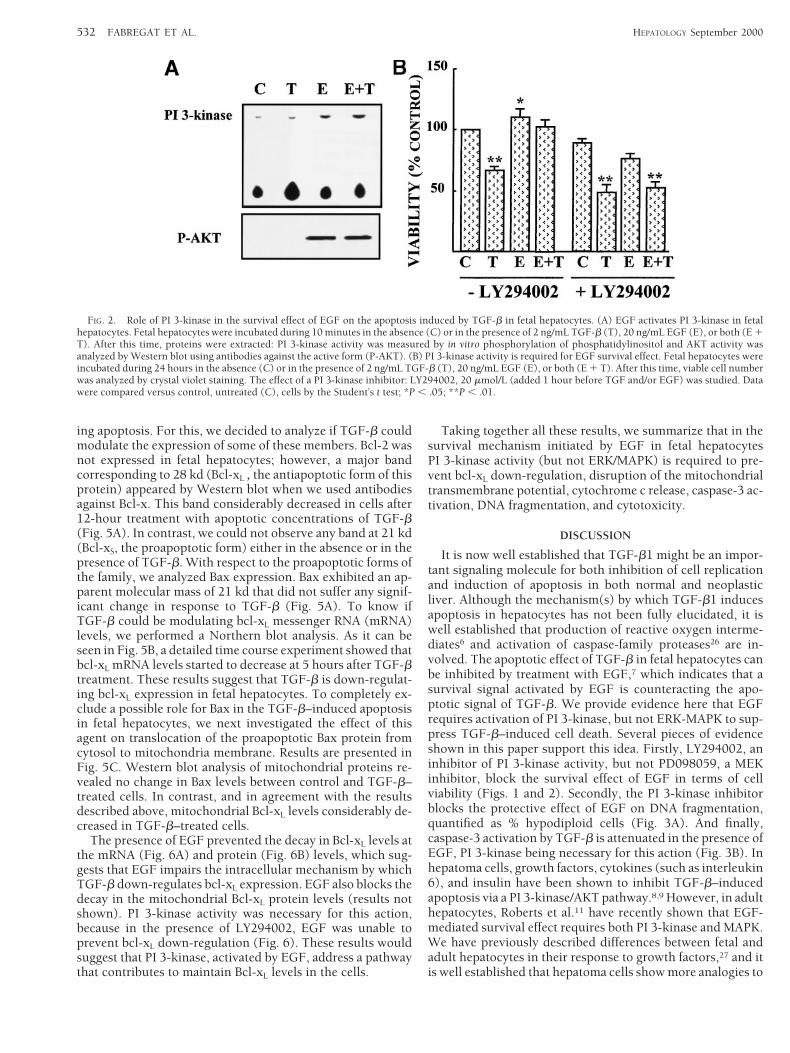
Role of PI 3-Kinase in the Survival Effect of EGF on the ApoptosisInduced by TGF-b in Fetal Hepatocytes. PI 3-kinase and itsdownstream signaling kinase, protein kinase B (PKB/AKT)have been shown to be important regulators of cell survivalin response to growth factors, such as EGF. To determinewhether PI 3-kinase/AKT activation plays a role in the sur-vival effect of EGF on the apoptosis induced by TGF-b in fetalhepatocytes, we have investigated whether EGF in the ab-sence and in the presence of TGF-b could modulate theseactivities. Results are presented in Fig. 2A. EGF caused anincrease in PI 3-kinase activity, which correlated with theappearance of the active form of AKT. The activation of PI3-kinase by EGF occurred either in the absence or in thepresence of TGF-b. We next looked at the consequences ofinhibiting PI 3-kinase pathway on the survival effect inducedby EGF. Pretreatment of fetal hepatocytes with PI 3-kinaseinhibitor LY294002 (20 mmol/L) blocked the protective effectof EGF on the cytotoxicity induced by TGF-b (Fig. 2B). Sim-ilar results were obtained when other PI 3-kinase inhibitors,such as wortmannin, were used (not shown results).
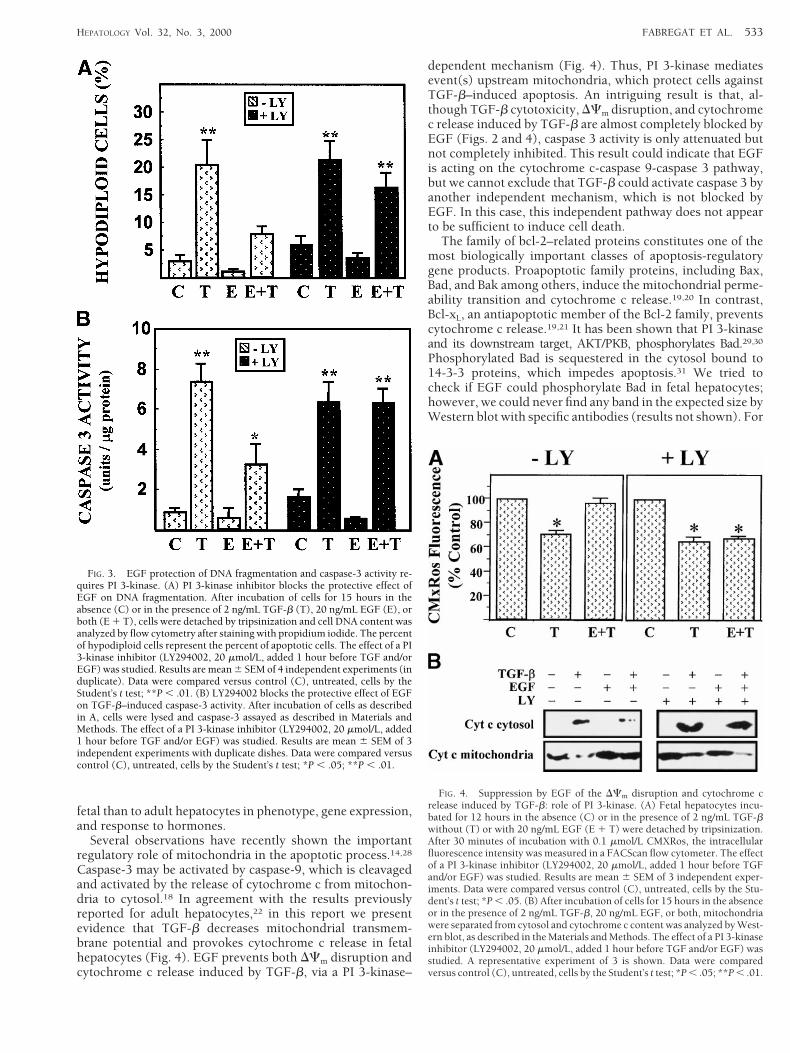
Furthermore, the presence of LY294002 abolished the pro-tective effect of EGF on other apoptotic parameters. Thus, thepercentage of hypodiploid cells, which significantly increasedin fetal hepatocytes after 15 hours of TGF-b treatment, de-creased in the presence of EGF when the PI 3-kinase inhibitorwas absent (Fig. 3A). However, in the presence of LY294002,EGF did not cause a statistically significant reduction in thepercentage of apoptotic cells (Fig. 3A). It has been previouslyshown that treatment of adult hepatocytes or hepatoma cellswith TGF-b induces a caspase-3–like activity.22,26 To deter-mine whether EGF blocks such activity in fetal hepatocytesand the possible involvement of PI 3-kinase, we analyzedcaspase-3 activity in cell extracts, by measuring the cleavageof a fluorogenic substrate, Ac-DEVD-AMC (Fig. 3B). TGF-bproduced a 10-fold increase in caspase-3 activity, which wasattenuated (although not completely blocked) in the presenceof EGF if the PI 3-kinase inhibitor was absent. However, thepresence of LY294002 completely blocked the protective ef-fect of EGF (Fig. 3B). These results clearly suggest that PI3-kinase mediates the protective effect against TGF-b–in-duced apoptosis in fetal hepatocytes, by inhibiting a step up-stream of caspase-3.
EGF Prevents the Reduction of DCm and the Release of Cyto-chrome C Induced by TGF-b in Fetal Hepatocytes by a PI 3-Kinase–Dependent Mechanism. Recent reports have provided evidencethat mitochondria are deeply involved in the regulation ofapoptosis.14 The disruption of the mitochondrial transmem-brane potential (DCm) has been defined as an early irrevers-ible stage of apoptosis, preceding caspase-3 activation.15-18
When fetal hepatocytes were incubated in the presence ofapoptotic concentrations of TGF-b during 12 hours, the mi-tochondrial transmembrane potential, measured using thefluorochrome CMXRos and FACScan analysis, decreased(Fig. 4A). In view of these results, we decided to studywhether the loss in DCm was coincident with a release ofcytochrome c from cytosol to mitochondria. To analyze this,after incubation of the cells for 12 to 15 hours in the absence(C) or in the presence of 2 ng/mL of TGF-b (T), mitochondriawere separated from cytosol and cytochrome c content wasanalyzed by Western blot as described in the Materials andMethods. As it is shown in Fig. 4B, cytochrome c contentdecreased considerably in the mitochondria, whereas it ap-peared in the cytosol. In contrast, the levels of albumin (anabundant protein in fetal liver, used as a control) did notsuffer any change (not shown results). Cytochrome c oxidase(an enzimatic protein located in the inner mitochondrialmembrane) presented identical activity in mitochondrial ex-tracts from control and TGF-b–treated cells, being absent incytosolic extracts (results not shown).
The disruption of DCm by TGF-b was prevented if EGF waspresent, which indicated that the survival signaling elicited bythis factor is acting upstream mitochondrial events (Fig. 4A).Furthermore, EGF also prevented the release of cytochrome cinduced by TGF-b (Fig. 4B). To determine whether PI 3-ki-nase activation plays a role in this effect of EGF, we incubatedthe cells in the presence of LY294002. As it is shown in Fig. 4,the PI 3-kinase inhibitor completely blocked the protectiveeffect of EGF on both the DCm disruption and cytochrome crelease induced by TGF-b in fetal hepatocytes.
TGF-b Down-regulates Bcl-xL in Fetal Hepatocytes: Protective Ef-fect of EGF by a PI 3-Kinase–Dependent Mechanism. The Bcl-2family of proteins is related to the mitochondrial changes dur-
FIG. 1. Role of ERK/MAPK in the survival effect of EGF on the apoptosisinduced by TGF-b in fetal hepatocytes. (A) EGF activates p44/p42-MAPKs infetal hepatocytes. Fetal hepatocytes were incubated during 10 minutes in theabsence (C) or in the presence of 2 ng/mL TGF-b (T), 20 ng/mL EGF (E), orboth (E 1 T). After this time, proteins were extracted and analyzed by West-ern blot using antibodies against the active forms (phospho p44/p42) andtotal forms (p44/p42). (B) p44/p42 MAPK activity is not required for EGFsurvival effect. Fetal hepatocytes were incubated during 24 hours in theabsence (C) or in the presence of 2 ng/mL TGF-b (T), 20 ng/mL EGF (E), orboth (E 1 T). After this time, viable cell number was analyzed by crystal violetstaining. The effect of a MEK inhibitor: PD098059, 50 mmol/L (added 1 hourbefore TGF and/or EGF) was studied. Data were compared versus control(C), untreated, cells by the Student’s t test; *P , .05; **P , .01.
HEPATOLOGY Vol. 32, No. 3, 2000 FABREGAT ET AL. 531
ing apoptosis. For this, we decided to analyze if TGF-b couldmodulate the expression of some of these members. Bcl-2 wasnot expressed in fetal hepatocytes; however, a major bandcorresponding to 28 kd (Bcl-xL , the antiapoptotic form of thisprotein) appeared by Western blot when we used antibodiesagainst Bcl-x. This band considerably decreased in cells after12-hour treatment with apoptotic concentrations of TGF-b(Fig. 5A). In contrast, we could not observe any band at 21 kd(Bcl-xS, the proapoptotic form) either in the absence or in thepresence of TGF-b. With respect to the proapoptotic forms ofthe family, we analyzed Bax expression. Bax exhibited an ap-parent molecular mass of 21 kd that did not suffer any signif-icant change in response to TGF-b (Fig. 5A). To know ifTGF-b could be modulating bcl-xL messenger RNA (mRNA)levels, we performed a Northern blot analysis. As it can beseen in Fig. 5B, a detailed time course experiment showed thatbcl-xL mRNA levels started to decrease at 5 hours after TGF-btreatment. These results suggest that TGF-b is down-regulat-ing bcl-xL expression in fetal hepatocytes. To completely ex-clude a possible role for Bax in the TGF-b–induced apoptosisin fetal hepatocytes, we next investigated the effect of thisagent on translocation of the proapoptotic Bax protein fromcytosol to mitochondria membrane. Results are presented inFig. 5C. Western blot analysis of mitochondrial proteins re-vealed no change in Bax levels between control and TGF-b–treated cells. In contrast, and in agreement with the resultsdescribed above, mitochondrial Bcl-xL levels considerably de-creased in TGF-b–treated cells.
The presence of EGF prevented the decay in Bcl-xL levels atthe mRNA (Fig. 6A) and protein (Fig. 6B) levels, which sug-gests that EGF impairs the intracellular mechanism by whichTGF-b down-regulates bcl-xL expression. EGF also blocks thedecay in the mitochondrial Bcl-xL protein levels (results notshown). PI 3-kinase activity was necessary for this action,because in the presence of LY294002, EGF was unable toprevent bcl-xL down-regulation (Fig. 6). These results wouldsuggest that PI 3-kinase, activated by EGF, address a pathwaythat contributes to maintain Bcl-xL levels in the cells.
Taking together all these results, we summarize that in thesurvival mechanism initiated by EGF in fetal hepatocytesPI 3-kinase activity (but not ERK/MAPK) is required to pre-vent bcl-xL down-regulation, disruption of the mitochondrialtransmembrane potential, cytochrome c release, caspase-3 ac-tivation, DNA fragmentation, and cytotoxicity.
DISCUSSION
It is now well established that TGF-b1 might be an impor-tant signaling molecule for both inhibition of cell replicationand induction of apoptosis in both normal and neoplasticliver. Although the mechanism(s) by which TGF-b1 inducesapoptosis in hepatocytes has not been fully elucidated, it iswell established that production of reactive oxygen interme-diates6 and activation of caspase-family proteases26 are in-volved. The apoptotic effect of TGF-b in fetal hepatocytes canbe inhibited by treatment with EGF,7 which indicates that asurvival signal activated by EGF is counteracting the apo-ptotic signal of TGF-b. We provide evidence here that EGFrequires activation of PI 3-kinase, but not ERK-MAPK to sup-press TGF-b–induced cell death. Several pieces of evidenceshown in this paper support this idea. Firstly, LY294002, aninhibitor of PI 3-kinase activity, but not PD098059, a MEKinhibitor, block the survival effect of EGF in terms of cellviability (Figs. 1 and 2). Secondly, the PI 3-kinase inhibitorblocks the protective effect of EGF on DNA fragmentation,quantified as % hypodiploid cells (Fig. 3A). And finally,caspase-3 activation by TGF-b is attenuated in the presence ofEGF, PI 3-kinase being necessary for this action (Fig. 3B). Inhepatoma cells, growth factors, cytokines (such as interleukin6), and insulin have been shown to inhibit TGF-b–inducedapoptosis via a PI 3-kinase/AKT pathway.8,9 However, in adulthepatocytes, Roberts et al.11 have recently shown that EGF-mediated survival effect requires both PI 3-kinase and MAPK.We have previously described differences between fetal andadult hepatocytes in their response to growth factors,27 and itis well established that hepatoma cells show more analogies to
FIG. 2. Role of PI 3-kinase in the survival effect of EGF on the apoptosis induced by TGF-b in fetal hepatocytes. (A) EGF activates PI 3-kinase in fetalhepatocytes. Fetal hepatocytes were incubated during 10 minutes in the absence (C) or in the presence of 2 ng/mL TGF-b (T), 20 ng/mL EGF (E), or both (E 1T). After this time, proteins were extracted: PI 3-kinase activity was measured by in vitro phosphorylation of phosphatidylinositol and AKT activity wasanalyzed by Western blot using antibodies against the active form (P-AKT). (B) PI 3-kinase activity is required for EGF survival effect. Fetal hepatocytes wereincubated during 24 hours in the absence (C) or in the presence of 2 ng/mL TGF-b (T), 20 ng/mL EGF (E), or both (E 1 T). After this time, viable cell numberwas analyzed by crystal violet staining. The effect of a PI 3-kinase inhibitor: LY294002, 20 mmol/L (added 1 hour before TGF and/or EGF) was studied. Datawere compared versus control, untreated (C), cells by the Student’s t test; *P , .05; **P , .01.
532 FABREGAT ET AL. HEPATOLOGY September 2000
fetal than to adult hepatocytes in phenotype, gene expression,and response to hormones.
Several observations have recently shown the importantregulatory role of mitochondria in the apoptotic process.14,28
Caspase-3 may be activated by caspase-9, which is cleavagedand activated by the release of cytochrome c from mitochon-dria to cytosol.18 In agreement with the results previouslyreported for adult hepatocytes,22 in this report we presentevidence that TGF-b decreases mitochondrial transmem-brane potential and provokes cytochrome c release in fetalhepatocytes (Fig. 4). EGF prevents both DCm disruption andcytochrome c release induced by TGF-b, via a PI 3-kinase–
dependent mechanism (Fig. 4). Thus, PI 3-kinase mediatesevent(s) upstream mitochondria, which protect cells againstTGF-b–induced apoptosis. An intriguing result is that, al-though TGF-b cytotoxicity, DCm disruption, and cytochromec release induced by TGF-b are almost completely blocked byEGF (Figs. 2 and 4), caspase 3 activity is only attenuated butnot completely inhibited. This result could indicate that EGFis acting on the cytochrome c-caspase 9-caspase 3 pathway,but we cannot exclude that TGF-b could activate caspase 3 byanother independent mechanism, which is not blocked byEGF. In this case, this independent pathway does not appearto be sufficient to induce cell death.
The family of bcl-2–related proteins constitutes one of themost biologically important classes of apoptosis-regulatorygene products. Proapoptotic family proteins, including Bax,Bad, and Bak among others, induce the mitochondrial perme-ability transition and cytochrome c release.19,20 In contrast,Bcl-xL, an antiapoptotic member of the Bcl-2 family, preventscytochrome c release.19,21 It has been shown that PI 3-kinaseand its downstream target, AKT/PKB, phosphorylates Bad.29,30
Phosphorylated Bad is sequestered in the cytosol bound to14-3-3 proteins, which impedes apoptosis.31 We tried tocheck if EGF could phosphorylate Bad in fetal hepatocytes;however, we could never find any band in the expected size byWestern blot with specific antibodies (results not shown). For
FIG. 3. EGF protection of DNA fragmentation and caspase-3 activity re-quires PI 3-kinase. (A) PI 3-kinase inhibitor blocks the protective effect ofEGF on DNA fragmentation. After incubation of cells for 15 hours in theabsence (C) or in the presence of 2 ng/mL TGF-b (T), 20 ng/mL EGF (E), orboth (E 1 T), cells were detached by tripsinization and cell DNA content wasanalyzed by flow cytometry after staining with propidium iodide. The percentof hypodiploid cells represent the percent of apoptotic cells. The effect of a PI3-kinase inhibitor (LY294002, 20 mmol/L, added 1 hour before TGF and/orEGF) was studied. Results are mean 6 SEM of 4 independent experiments (induplicate). Data were compared versus control (C), untreated, cells by theStudent’s t test; **P , .01. (B) LY294002 blocks the protective effect of EGFon TGF-b–induced caspase-3 activity. After incubation of cells as describedin A, cells were lysed and caspase-3 assayed as described in Materials andMethods. The effect of a PI 3-kinase inhibitor (LY294002, 20 mmol/L, added1 hour before TGF and/or EGF) was studied. Results are mean 6 SEM of 3independent experiments with duplicate dishes. Data were compared versuscontrol (C), untreated, cells by the Student’s t test; *P , .05; **P , .01.
FIG. 4. Suppression by EGF of the DCm disruption and cytochrome crelease induced by TGF-b: role of PI 3-kinase. (A) Fetal hepatocytes incu-bated for 12 hours in the absence (C) or in the presence of 2 ng/mL TGF-bwithout (T) or with 20 ng/mL EGF (E 1 T) were detached by tripsinization.After 30 minutes of incubation with 0.1 mmol/L CMXRos, the intracellularfluorescence intensity was measured in a FACScan flow cytometer. The effectof a PI 3-kinase inhibitor (LY294002, 20 mmol/L, added 1 hour before TGFand/or EGF) was studied. Results are mean 6 SEM of 3 independent exper-iments. Data were compared versus control (C), untreated, cells by the Stu-dent’s t test; *P , .05. (B) After incubation of cells for 15 hours in the absenceor in the presence of 2 ng/mL TGF-b, 20 ng/mL EGF, or both, mitochondriawere separated from cytosol and cytochrome c content was analyzed by West-ern blot, as described in the Materials and Methods. The effect of a PI 3-kinaseinhibitor (LY294002, 20 mmol/L, added 1 hour before TGF and/or EGF) wasstudied. A representative experiment of 3 is shown. Data were comparedversus control (C), untreated, cells by the Student’s t test; *P , .05; **P , .01.
HEPATOLOGY Vol. 32, No. 3, 2000 FABREGAT ET AL. 533

this, we decided to study the possible effects of TGF-b andEGF on bcl-2 family gene expression. We show here thatTGF-b induces in fetal hepatocytes a decrease in the proteinand mRNA levels of Bcl-xL (Fig. 5). In contrast, there is nochange in the expression and/or translocation to mitochon-dria of Bax, a proapoptotic member of the same family that hasbeen previously reported to increase its levels in response toTGF-b in other liver cell systems.32 Although Bax expressiondoes not appear to be affected, the mitochondrial ratio Bcl-xL/Bax clearly decreases (Fig. 5C). It has been recently reportedthat TGF-b can down-regulate Bcl-xL protein levels insome hepatoma cells (i.e., HuH7, McA-RH777, or McA-RH8994)33,34 and mouse hepatocytes.35 However, it is not ageneral mechanism, because other hepatoma cells (i.e.,Hep3B) do not respond to TGF-b down-regulating Bcl-xL.36
We also show here that EGF maintains bcl-xL mRNA andprotein levels (Fig. 6), preventing the effect of TGF-b. Therole of EGF on bcl-xL expression is unclear at this moment.Although it has been described that EGF increases bcl-xL
mRNA and protein levels in human keratinocytes,37,38 it doesnot appear to modulate bcl-xL expression in hepatoma cells.34
In fact, we cannot observe any regulation on bcl-xL expressionby EGF itself, but it blocks the down-regulatory effect ofTGF-b (Fig. 6). The maintenance of bcl-xL levels could ex-plain the inhibition of cytochrome c release and the stability ofmitochondrial transmembrane potential promoted by EGF(Fig. 4). When we analyze the implication of PI 3-kinase in
this protective effect, we observe that in the presence of the PI3-kinase inhibitor, EGF is not capable of maintaining bcl-xL
mRNA (Fig. 6A) and protein (Fig. 6B) levels. Thus, theseresults clearly show that PI 3-kinase activated by EGF medi-ates the maintenance of bcl-xL levels and the ratio Bcl-xL/Baxin fetal hepatocytes, even if TGF-b is present. A role for bcl-xL
in the regulation of apoptosis by EGF and TGF-b in mammaryepithelial cells has been previously proposed.39 It has beenalso recently suggested that PI 3-kinase may mediate the up-regulation of Bcl-xL expression by IGF-1 in Baf-3 cells.40 How-ever, this report proposes this idea for liver cells. Further workwill be necessary to completely understand the molecularmechanism of this effect.
It is apparent that failure to positively regulate apoptosishas emerged as one of the critical steps toward hepatocarci-nogenesis.3,4,41,42 Several lines of evidence suggest that growthfactors, which activate PI 3-kinase/AKT pathway, play an im-portant role in this process. The results presented in this re-port provide strong evidence that in the survival mechanisminitiated by EGF in fetal hepatocytes, PI 3-kinase activity (butnot ERK/MAPK) is required to prevent bcl-xL down-regula-tion, impairing the activation of cytochrome c/caspase-3 apo-ptotic pathway. The ability to define the antiapoptotic poten-tial of signaling pathways, like that for PI 3-kinase in fetalhepatocytes, should allow enhanced efficiency in the treat-ment of hepatocellular carcinoma. It is possible that inhibi-tion of the PI 3-kinase stimulatory pathway will further sen-sitize tumor cells to apoptosis induced by therapeutic drugs oreven by TGF-b, whose expression is very high during livercarcinogenesis42 (at least in those cases in which TGF-b re-ceptors were not affected). Additional studies will be neces-sary to further define the PI 3-kinase/AKT pathway as a ther-apeutic target for treatment of hepatocellular carcinoma.
FIG. 5. Effect of TGF-b on Bcl-xL and Bax expression. (A) After 12-hourincubation of cells without (C) or with 2 ng/mL TGF-b (T), proteins wereextracted and analyzed the levels of Bcl-xL and Bax by Western blot. A repre-sentative experiment of at least 5 is shown. (B) Time course analysis of theeffect of TGF-b on bcl-xL mRNA levels. Twenty micrograms total RNA, ex-tracted from the pooled cells of two 92-mm-diameter dishes, were denaturedand separated by size on formaldehyde and blotted on GeneScreenTM mem-branes. Serial hybridizations with bcl-xL cDNA and 18S ribosomal cDNAwere performed as described in the Materials and Methods. A representativeexperiment of 2 is shown. (C) After 12-hour incubation of cells without (C)or with 2 ng/mL TGF-b (T), mitochondria were separated from cytosol andthe levels of Bcl-xL and Bax were analyzed by Western blot, as described in theMaterials and Methods. A representative experiment of 3 is shown.
FIG. 6. EGF suppresses the down-regulation of bcl-xL expression induced byTGF-b via a PI 3-kinase pathway. (A) After 6-hour incubation of cells without orwith 2 ng/mL TGF-b, and/or 20 ng/mL EGF (as indicated in the figure) 20 mg totalRNA was extracted from the pooled cells of two 92-mm-diameter dishes, dena-tured, separated by size on formaldehyde gels, and blotted on GeneScreenTMmembranes. Serialhybridizationswithbcl-xL cDNAand18SribosomalcDNAwereperformed. The effect of a PI 3-kinase inhibitor (LY294002, 20 mmol/L, added 1hour before TGF and/or EGF) was studied. A representative experiment of 3 isshown. (B) After 12-hour incubation of cells under the same conditions describedin A, proteins were extracted and analyzed the levels of Bcl-xL and Albumin byWestern blot, as described in the Materials and Methods. A representative experi-ment of 4 is shown.
534 FABREGAT ET AL. HEPATOLOGY September 2000

Acknowledgment: The authors thank Drs. G. Nunez (AnnArbor, MI) and E. Rozengurt (UCLA, Los Angeles, CA) forproviding plasmids and A. Vazquez for expert assistance withthe flow cytometer. We also thank Drs. J. Gil and A. Lopez-Rivas for helpful discussions.
REFERENCES
1. Patel T, Roberts LR, Jones BA, Gores GJ. Dysregulation of apoptosis as amechanism of liver disease: an overview. Semin Liver Dis 1998;18:105-114.
2. Massague J. TGF-b signal transduction. Annu Rev Biochem 1998;67:753-791.
3. Pitot HC. Hepatocyte death in hepatocarcinogenesis. HEPATOLOGY 1998;28:1-5.
4. Thorgeirsson SS, Teramoto T, Factor V. Regulation of apoptosis in hep-atocellular carcinoma. Semin Liver Dis 1998;18:115-122.
5. Sanchez A, Alvarez AM, Benito M, Fabregat I. Transforming growth fac-tor beta modulates growth and differentiation of fetal hepatocytes inprimary culture. J Cell Physiol 1995;165:398-405.
6. Sanchez A, Alvarez AM, Benito M, Fabregat I. Apoptosis induced bytransforming growth factor-beta in fetal hepatocyte primary cultures:involvement of reactive oxygen intermediates. J Biol Chem 1996;271:7416-7422.
7. Fabregat I, Sanchez A, Alvarez AM, Nakamura T, Benito M. Epidermalgrowth factor, but not hepatocyte growth factor, suppresses the apoptosisinduced by transforming growth factor-beta in fetal hepatocytes in pri-mary culture. FEBS Lett 1996;384:14-18.
8. Chen R-H, Su Y-H, Chuang RLC, Chang T-Y. Suppression of transform-ing growth factor-b-induced apoptosis through a phosphatidylinositol3-kinase/Akt-dependent pathway. Oncogene 1998;17:1959-1968.
9. Chen R-H, Chang M-C, Su Y-H, Tsai Y-T, Kuo M-L. Interleukin-6 inhib-its transforming growth factor-b-induced apoptosis through the phos-phatidylinositol 3-kinase/Akt and signal transducers and activators oftranscription pathways. J Biol Chem 1999;274:23013-23019.
10. Diez-Fernandez C, Sanz N, Wolf A, Cascales M. Effect of phorbol ester(PMA) on antioxidant enzyme expression in TGF-beta 1-induced apo-ptosis in primary cultures of hepatocytes. Biofactors 1998;8:65-71.
11. Roberts RA, James NH, Cosulich SC. The role of protein kinase B andmitogen activated protein kinase in epidermal growth factor and tumornecrosis factor a-mediated rat hepatocyte survival and apoptosis. HEPA-TOLOGY 2000;31:420-427.
12. Roncero C, Ventura JJ, Sanchez A, Bois-Joyeux B, Mesa ML, ThomassinH, Danan JL, et al. Phorbol esters down-regulate alpha-fetoprotein geneexpression without affecting growth in fetal hepatocytes in primary cul-ture. Biochim Biophys Acta 1998;1402:151-164.
13. Sanchez A, Alvarez AM, Benito M, Fabregat I. Cycloheximide preventsapoptosis, reactive oxygen species production, and glutathione depletioninduced by transforming growth factor beta in fetal rat hepatocytes inprimary culture. HEPATOLOGY 1997;26:935-943.
14. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309-1312.
15. Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX,Kroemer G. Reduction in mitochondrial potential constitutes an earlyirreversible step of programmed lymphocyte death in vivo. J Exp Med1995;181:1661-1672.
16. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, et al.Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochon-dria blocked. Science 1997:275:1129-1132.
17. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release ofcytochrome c from mitochondria: a primary site for Bcl-2 regulation ofapoptosis. Science 1997;275:1132-1136.
18. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES,Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479-489.
19. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate therelease of apoptogenic cytochrome c by the mitochondrial channelVDAC. Nature 1999;399:483-487.
20. Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HLA,Prevost M-C, et al. Bax and adenine nucleotide translocator cooperate inthe mitochondrial control of apoptosis. Science 1998;281:2027-2031.
21. Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR.Bax-induced caspase activation and apoptosis via cytochrome c releasefrom mitochondria is inhibitable by Bcl-xL. J Biol Chem 1999;274:2225-2233.
22. Rodrigues CM, Ma X, Linehan-Stieers C, Fan G, Kren BT, Steer CJ.Ursodeoxycholic acid prevents cytochrome c release in apoptosis by in-hibiting mitochondrial membrane depolarization and channel forma-tion. Cell Death Differ 1999;6:842-854.
23. Roncero C, Lorenzo M, Fabregat I, Benito M. Rates of lipogenesis in fetalhepatocytes in suspension and in primary culture: hormonal effects. Bio-chim Biophys Acta 1989;1012:320-324.
24. Lowerson SA, Taylor L, Briggs HL, Turnbull DM. Measurement of theactivity of individual respiratory chain complexes in isolated fibroblastmitochondria. Anal Biochem 1992;205:372-374.
25. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acidguanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem1987;162:156-159.
26. Chen RH, Chang TY. Involvement of caspase family proteases in trans-forming growth factor-beta-induced apoptosis. Cell Growth Differ 1997;8:821-827.
27. De Juan C, Benito M, Alvarez A, Fabregat I. Differential proliferativeresponse of cultured fetal and regenerating hepatocytes to growth factorsand hormones. Exp Cell Res 1992;202:495-500.
28. Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/liferegulator in apoptosis and necrosis. Annu Rev Physiol 1998;60:619-642.
29. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Aktphosphorylation of BAD couples survival signals to the cell-intrinsicdeath machinery. Cell 1997;91:231-241.
30. Peso L, Gonzalez-Garcıa M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science1997;278:687-689.
31. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylationof death agonist BAD in response to survival factors results in binding to14-3-3 not BCL-XL. Cell 1996;87:619-628.
32. Teramoto T, Kiss A, Thorgeirsson SS. Induction of p53 and Bax duringTGF-beta 1 initiated apoptosis in rat liver epithelial cells. Biochem Bio-phys Res Commun 1998;251:56-60.
33. Yamamoto M, Fukuda K, Miura N, Suzuki R, Kido T, Komatsu Y. Inhi-bition by dexamethasone of transforming growth factor beta1-inducedapoptosis in rat hepatoma cells: a possible association with Bcl-xL induc-tion. HEPATOLOGY 1998;27:959-966.
34. Shima Y, Nakao Y, Nakashima T, Kawakami A, Nakata K, Hamasaki K,Kato Y, et al. Activation of caspase-8 in transforming growth factor-b-induced apoptosis of human hepatoma cells. HEPATOLOGY 1999;30:1215-1222.
35. Christensen JG, Gonzales AJ, Cattley RC, Goldsworthy TL. Regulation ofapoptosis in mouse hepatocytes and alteration of apoptosis by nongeno-toxic carcinogens. Cell Growth Differ 1998;9:815-825.
36. Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The transforminggrowth factor b1-inducible transcription factor, TIEG1, mediates apo-ptosis through oxidative stress. HEPATOLOGY 1999;30:1490-1497.
37. Rodeck U, Jost M, DuHadaway J, Kari C, Jensen PJ, Risse B, Ewert DL.Regulation of Bcl-xL expression in human keratinocytes by cell-substra-tum adhesion and the epidermal growth factor receptor. Proc Natl AcadSci U S A 1997;94:5067-5072.
38. Stoll SW, Benedict M, Mitra R, Hiniker A, Elder JT, Nunez G. EGFreceptor signaling inhibits keratinocyte apoptosis: evidence for media-tion by Bcl-xL. Oncogene 1998;16:1493-1499.
39. Nass SJ, Li M, Amundadottir LT, Furth PA, Dickson RB. Role of Bcl-xL inthe regulation of apoptosis by EGF and TGF-b1 in c-myc overexpressingmammary epithelial cells. Biochem Biophys Res Commun 1996;227:248-256.
40. Leverrier Y, Thomas J, Mathieu AL, Low W, Blanquier B, Marvel J. Role ofPI3-kinase mediated by IL-3 or IGF-1 in Baf-3 cells. Cell Death Differ1999;6:290-296.
41. Grasl-Kraupp B, Ruttkay-Nedecky B, Mullauer L, Taper H, Huber W,Bursch W, Schulte-Hermann R. Inherent increase of apoptosis in livertumors: implications for carcinogenesis and tumor regression. HEPATOL-OGY 1997;25:906-912.
42. Schulte-Hermann R, Bursch W, Low-Baselli A, Wagner A, Grasl-KrauppB. Apoptosis in the liver and its role in hepatocarcinogenesis. Cell BiolToxicol 1997;13:339-348.
HEPATOLOGY Vol. 32, No. 3, 2000 FABREGAT ET AL. 535
Copyright © 2022 FDOKUMEN




















