
Nanoreinforced biocompatible hydrogels from wood hemicelluloses and cellulose whiskers
Upload
independentCategory
view
0download
0
PAPER www.rsc.org/softmatter | Soft Matter
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
Enzyme-induced graft polymerization for preparation of hydrogels: synergeticeffect of laccase-immobilized-cryogels for pollutants adsorption†
Marina Nieto,a Stefania Nardecchia,a Carmen Peinado,b Fernando Catalina,b Concepci�on Abrusci,c
Mar�ıa C. Guti�errez,a M. Luisa Ferrer*a and Francisco del Monte*a
Received 9th March 2010, Accepted 8th April 2010
DOI: 10.1039/c0sm00079e
The use of polyethylene oxide-polypropylene oxide-polyethylene oxide block-copolymers as a mediator
in the laccase-induced graft polymerization of diacrylic derivate of polyethylene glycols resulted in the
formation of PEG-g-F68 hydrogels. The proper oxygen content in the reaction medium to obtain
reasonable polymerization conversions (i.e., on one hand, laccase needs oxygen as substrate whereas, on
the other, oxygen is a strong inhibitor of radical polymerizations) was achieved by the use of an enzymatic
scavenging system consisting of glucose oxidase and glucose. Eventually, laccase was immobilized within
the resulting PEG-g-F68 hydrogel with full preservation of enzyme activity. Laccases have been used for
bioremediation purposes because of their ability to degrade phenolic compounds. Thus, laccase-
immobilized PEG-g-F68 hydrogels were submitted to the ISISA (ice segregation induced self-assembly)
process for preparation of laccase-immobilized PEG-g-F68 cryogels which exhibited a macroporous
structure where immobilized laccase preserved almost total activity (ca. 90%) for a period exceeding three
months after preparation. Synergy between macroporous structure (deriving from the ISISA process),
amphiphilic domains (deriving from graft copolymer) and activity of the immobilized enzyme provided
outstanding adsorption capabilities to the cryogels (up to 235 mg g�1).
Introduction
Hydrogels are three-dimensional, hydrophilic, polymeric
networks capable of imbibing large amounts of water.1 Among
other applications,2 these networks have been used as adsorbents
to remove organic or heavy metal pollutants from wastewater.3
However, simple hydrophilic materials are not efficient adsor-
bents of organic pollutants and it is necessary for them to also
contain hydrophobic pockets which can accommodate and
retain organic substances. Polymer hydrogels containing
hydrophobic units together with the hydrophilic ones are called
amphiphilic polymer hydrogels or amphiphilic polymer networks
and have been the subject of several reviews.4 These networks
swell to a smaller extent than the corresponding hydrophilic
networks but, in addition, they can also swell in organic solvents.
Thus, hydrogels based on block copolymers containing hydro-
philic and hydrophobic domains are excellent candidates for
adsorption purposes since they can interact effectively with water
(through the hydrophilic domains) and they have as well the
capability (through the hydrophobic domains) to extract and
retain any organic substance eventually dissolved in water. The
specific features of hydrophilic and hydrophobic domains
aInstituto de Ciencia de Materiales de Madrid–ICMM, Consejo Superiorde Investigaciones Cient�ıficas–CSIC, Cantoblanco, 28049 Madrid, SpainbInstituto de Ciencia y Tecnolog�ıa de Pol�ımeros– ICTP, Consejo Superiorde Investigaciones Cient�ıficas–CSIC, C/Juan de la Cierva, 3, 28006Madrid, SpaincDepartamento de Biolog�ıa Molecular, Facultad de Ciencias, UniversidadAut�onoma de Madrid-UAM, Cantoblanco, 28049 Madrid, Spain. E-mail:[email protected]; [email protected]; Fax: +34 91 3720623;Tel: +34 91 334 9033
† Electronic supplementary information (ESI) available: Data resultingfrom fitting experimental adsorption isotherms, SEM and FTIRspectra. See DOI: 10.1039/c0sm00079e
This journal is ª The Royal Society of Chemistry 2010
(e.g. length, the segments arrangement, etc.) will mostly govern
the adsorption performance against a particular solute.5
If one desires to further enhance the adsorption performance of
a certain block-copolymer-based-hydrogel, synergetic moieties
should be included within its structure. Different enzymes, such as
ligninperoxidase, manganese-peroxidase and laccase, have
demonstrated their ability to degrade phenolic compounds6 which
are some of the most recalcitrant pollutants found in wastewaters.
Thus, immobilization of any of these enzymes in the block-
copolymer-based-hydrogels mentioned above could provide
enhanced bioremediation capabilities. However, the challenge
that remains is to immobilize certain unstable enzymes without
compromising their activity and stability.7 The formation of
a hydrogel requires the use of initiators and/or cross-linking
agents (which, ultimately, are incorporated as foreign chemical
functionalities pending from/grafting the polymer chain),8 or
a change of pH, ionic strength, or temperature.9 In every case, the
enzyme can be subjected to suboptimal conditions that may result
in partial or (in the worst scenario) total loss of its catalytic
activity. Enzymatic polymerizations have attracted much atten-
tion lately since they offer mild reaction conditions besides the
occurrence of enzyme immobilization within the formed
hydrogel.10 Suitable enzymes capable of initiating radical reac-
tions comprise peroxidases and laccases.11 The use of laccases
would be interesting for our purposes given that, as mentioned
above, the resulting laccase-immobilized-hydrogels could offer
enhanced bioremediation capabilities. Laccases are especially
efficient for acrylamide polymerizations, but need mediators
(e.g. b-diketones) to polymerize other acrylates.11,12 The oxygen
balance in solution is crucial since, on one hand it is the natural
substrate of laccases while, on the other it is a strong inhibitor of
radical polymerizations.12
Soft Matter, 2010, 6, 3533–3540 | 3533

Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
In this work, we explored the capability of polyethylene oxide-
polypropylene oxide-polyethylene oxide block-copolymers
(e.g. PEO80-PPO27-PEO80, F68) to be used as mediators in
laccase-induced polymerizations. Generation of long-lived mac-
roradicals in oxygen-free media has been widely explored for
PEO and PPO polymers,13 but never via enzymatic. The mono-
mer of choice was a diacrylic derivate of polyethylene glycol
(e.g. PEG550-DA) so that PEG-g-F68 hydrogels were obtained
by grafting of PEG550-DA onto F68 followed by cross-linking
of the acrylic moieties remaining after grafting. Glucose oxidase
(GOX) and glucose (GLU) were also added to obtain the
oxygen-free media required for radical polymerization. Hydro-
gels were thereafter subjected to a unidirectional freezing process
that, by ice-induced-self-assembly-segregation (ISISA) of the
matter homogeneously distributed within the gel and subsequent
high-vacuum sublimation of ice,14 promoted the formation of
macroporous structures while preserving the activity of immo-
bilized biological entities. Freeze-dried hydrogels were called
cryogels from the Greek kryos (krcos, meaning frost of ice).15
The morphology of PEG-g-F68 cryogels was studied by scanning
electron microscopy (SEM). Polymerization conversions were
calculated using the cryogel’s weight after soaking in distilled
water (to wash un-reacted acrylates) divided by their original
weight before washing. FTIR spectroscopy was used to analyze
the composition of cryogels before and after washing. Swelling
experiments were carried out in both water and toluene to
confirm the amphiphilic nature (consisting of hydrophilic and
hydrophobic domains) of washed cryogels. The adsorption of
Brilliant Blue dye Remazol (BBdR) as model of organic
pollutant was used to test the adsorption capability of the cry-
ogels. Adsorption isotherms were carried out with different
samples to assess the synergetic contribution of the activity of the
immobilized laccase besides the macroporous structure and the
amphiphilic nature of the cryogel to the overall adsorption
performance of these materials.
Experimental part
In a typical batch, F68 (30 mg), PEG550-DA (60 mg), laccase
(0.88 mg, 20 units) and glucose oxidase (GOX, 0.25 mg, 90 units)
were dissolved in a phosphate buffered solution (1.8 mL) also
containing glucose (GLU, 5 mM) for a polymer content of 7 wt%
Table 1 Experimental data used for preparation of graft copolymers at 37 �CF68(2 : 1-7)-lac1N2 which was deaerated under N2 flow. Sample PEG-g-F68(2coming from the use of GOX/Glu. Experimental data used for preparation oare also included
Sample FA68 PEG550DA LACAS
F68(7)-lac1 90 mg – 20UPEG(7)-lac1 – 90 mg 20UPEG-g-F68(2 : 1-7)-lac1 30 mg 60 mg 20UPEG-g-F68(2 : 1-7)-lac1N2 30 mg 60 mg 20UPEG-g-F68(2 : 1-7)-lac1c 30 mg 60 mg 20UPEG-g-F68(2 : 1-7)-lac2 30 mg 60 mg 80UPEG-g-F68(2 : 1-7)-lac3 30 mg 60 mg 200UPEG-g-F68(1 : 1-7)-lac1 45 mg 45 mg 20UPEG-g-F68(3 : 1-7)-lac1 23 mg 69 mg 20UPEG-g-F68(1 : 1-10)-lac1 60 mg 60 mg 20UPEG-g-F68(2 : 1-10)-lac1 40 mg 80 mg 20U
3534 | Soft Matter, 2010, 6, 3533–3540
(see Table 1). The solution was gently stirred for homogeniza-
tion, placed in an insulin syringe and stored 17 h at 37 �C for
gelation. The resulting hydrogel was called PEG-g-F68(2 : 1-7)h-
lac1, where h stands for hydrogel, 2 : 1 stands for PEGDA/F68
weight ratio, 7 for the weight percentage of polymers (F68 +
PEGDA) in solution and lac1 for laccase concentration (20 units)
used for macroradical generation. An identical procedure was
followed for preparation of samples described in Table 1 except
for the use of different PEGDA to F68 weight ratios (1 : 0, 0 : 1,
1 : 1 or 3 : 1), total polymer content (10 wt%) or laccase
concentration (80 units for samples called lac2 and 200 units for
samples called lac3). Moreover, PEG-g-F68(2 : 1-7)-lac1N2
hydrogels were prepared as described above, except for the use of
N2 flow to deaerate the sample instead of using the GOX/GLU
system. Meanwhile, PEG-g-F68(2 : 1-7)-lac1c hydrogels were
prepared using the GOX/GLU system coupled with catalase for
elimination of H2O2 resulting as a by-product of GOX/GLU
system (see Scheme 1). The freshly gelled hydrogels were unidi-
rectionally frozen by dipping the insulin syringes at a constant
rate of 5.9 mm min�1 into a liquid nitrogen bath (77 K). Samples
were freeze-dried using a ThermoSavant Micromodulyo freeze-
drier. The resulting cryogels were monoliths keeping both the
shape and the size of the insulin syringes. Particular cryogels were
called as described above for hydrogels except for the use of c
instead of h superscript (e.g. PEG-g-F68(2 : 1-7)c-lac1 for
a freeze-dried hydrogel prepared with 2 : 1 PEGDA to F68
weight ratio, 7 wt% total polymer content and 20 units of
laccase).
Laccase activity was determined following the oxidation of
ABTS at 420 nm (3 ¼ 3.6 � 104 cm�1 M�1 for the oxidation
product). The reaction mixture contained 100 mL of the enzyme
solution and 900 mL of ABTS (1 mM) in acetate/citrate buffer
(0.05 M, pH 5.0) at 30 �C. One unit of enzyme activity is defined
as the amount of enzyme that oxidizes 1 mmol ABTS in 1 min.
Polymer conversions were determined gravimetrically. Cry-
ogels were immersed in distilled water for 72 h at 25 �C to extract
unreacted monomers and subsequently freeze-dried. Washed
cryogels are called cryogels except for the use of wc instead of c
superscript (e.g. PEG-g-F68(2 : 1-7)wc-lac1 for a washed PEG-g-
F68(2 : 1-7)c-lac1 cryogel). Conversions were calculated (in %)
using the dry gel weight divided by the initial weight of monomer
in the sample. Swelling experiments were carried out by
. GOX/Glu was used as an oxygen scavenger except for sample PEG-g-: 1-7)-lac1c included catalase to address any eventual role played by H2O2
f bare PEG and F68 cryogels (PEG(7)-lac1 and F68(7)-lac1, respectively)
SE GOX GLU CATALASE Conversion
90U 5 mM – No gel90U 5 mM – 56%90U 5 mM – 58%– – – 48%90U 5 mM 250U 57%90U 5 mM – 59%90U 5 mM – 61%90U 5 mM – 50%90U 5 mM – 60%90U 5 mM – 61%90U 5 mM – 63%
This journal is ª The Royal Society of Chemistry 2010

Scheme 1 Use of F68 as mediator in laccase induced polymerization of
PEGDA for the formation of graft copolymers. The ideal oxygen balance
in solution (oxygen is the laccase substrate but it is also a strong inhibitor
of radical polymerizations) was provided by the enzymatic GOX/GLU
system.
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
immersion of PEG-g-F68wc-lac1 cryogels in an excess amount of
either deionized water or toluene at 20 �C. The swollen samples
were weighed at various time intervals. Experiments were con-
ducted in triplicate.
The adsorption isotherms were measured in a Variant Cary
4000 spectrophotometer by taking fixed amounts of adsorbant
(20 mg), which were submerged in 5 mL of a dye solution at
different concentrations (ranging from 50 to 900 mg L�1 for any
of PEG-g-F68-lac0 and PEG-g-F68-lac1 hydrogels/cryogels,
from 50 to 1500 mg L�1 for any of PEG-g-F68-lac2 cryogels and
from 50 to 3500 mg L�1 for any of PEG-g-F68-lac3 cryogels) and
left to adsorb for 72 h at 20 �C. Afterwards, the scaffolds were
removed from the solution and the remaining contaminant
concentration was measured by absorption spectrophotometry.
Experiments were conducted in triplicate. Averaged data was
used for the representation of Langmuir and Freundlich
isotherms. The standard deviation was less than 5%.
Cryogel morphology was studied by SEM using a Zeiss DSM-
950 microscope. Samples were mounted onto the SEM sample
holder and gold sputter coated (for 2 min, to obtain a ca. 5 nm
gold film) prior to SEM observation. The accelerating voltage
was 15–20 kV and the work distance was 14–9 mm. FTIR spectra
were carried out in a FTIR Bruker IFS60v spectrometer.
Fig. 1 SEM micrographs of cross-sectioned PEG-g-F68(2 : 1-7)c-lac1
(a–c) and PEG-g-F68(2 : 1-7)c-lac3 (d). Scale bars are 400 mm in (a),
100 mm in (b) and 40 mm in (c,d).
Results and discussion
The ISISA process is a simple and versatile methodology based
on the unidirectional freezing of a hydrogel by its immersion (at a
rate typically ranging from 0.7 mm min�1 to 9.1 mm min�1) into
a liquid nitrogen bath (at �196 �C).16 Upon freezing, the ice
formation (hexagonal form) causes the hydrogel network
(originally occupying the entire hydrogel volume) to become
segregated and concentrated between adjacent ice crystals.
Freeze-drying resulted in the formation of structures consisting
of micrometre-sized pores corresponding to the empty areas
where ice crystals originally resided. Our interest in the appli-
cation of the ISISA process for the formation of cryogels was
double. First, it was our belief that the achievement of a highly
open microchanneled structure could favour mass transport and
accessibility of small molecules to hydrophilic and hydrophobic
domains distributed throughout the inner structure and thus,
This journal is ª The Royal Society of Chemistry 2010
a potential enhancement to the adsorption performance of these
materials. The second interesting feature of the ISISA process is
that freeze-drying/lyophilization processes are widely used in
biology for preservation and storage of proteins. This feature,
besides the overall biocompatibility of the whole ISISA process
(i.e. it begins from an aqueous suspension and runs in the absence
of further chemical reactions, avoiding potential complications
associated with by-products or purification procedures), can
eventually provide near full preservation of the original activity
of enzymes17 (and even of biological like membrane structures18)
after immobilization within the macroporous structure of the
cryogels. In the particular case of laccase immobilized within
PEG-g-F68 cryogels, almost 90% of activity was preserved over
a period longer than three months (see data below).
With regard to the first issue, the application of the ISISA
process to PEG-g-F68 hydrogels indeed resulted in the formation
of monolithic cryogels exhibiting macropores homogeneously
distributed throughout the whole cross-sectioned structure
(Fig. 1). The structural pattern consisted of well-aligned mac-
ropores in the freezing direction as a consequence of the unidi-
rectional freezing that characterizes the ISISA process
(Fig. S1, ESI†). The macroporous structures of PEG-g-F68
cryogels prepared from different PEGDA:F68 ratios (e.g. 1 : 1,
2 : 1 and 3 : 1), total polymer content (e.g. 7 and 10 wt%) and
laccase concentration were also analyzed (see Fig. 1, 2 and 3) and
no significant differences were observed among the different
samples. SEM micrographs of F68(7)-lac1 and PEG(7)-lac1
cryogels are also depicted in Fig. 4 and Fig. S2, ESI†, for
comparison. Interestingly, F68(7)-lac1 cryogels exhibited much
wider macropores than PEG(7)-lac1 cryogels and wider than any
of the graft copolymer based cryogels mentioned before.
Previous works on cryogels have related the decrease of pore size
to the cross-linking degree of the hydrogels submitted to the
ISISA process.19 Obviously, a lack of cross-linking occurred in
Soft Matter, 2010, 6, 3533–3540 | 3535

Fig. 3 SEM micrographs of cross-sectioned PEG-g-F68(1 : 1-10)c-lac1
(a) and PEG-g-F68(2 : 1-10)c-lac1 (b). Bars are 50 mm.
Fig. 2 SEM micrographs of cross-sectioned PEG-g-F68(1 : 1-7)c-lac1 (a)
and PEG-g-F68(3 : 1-7)c-lac1 (b). Scale bars are 200 mm in (a) and (b), and
20 mm in inset of (a).
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
F68(7)-lac1 because of the absence of acrylate moieties. This
event was confirmed by the determination of the polymer
conversion, that is, the fraction of monomer incorporated into
the hydrogel network (see experimental for details). For this
purpose, cryogels were soaked in distilled water for 72 h (to wash
out unreacted monomers) and, subsequently, freeze-dried and
weighed, and their weight correlated to the initial weight of
monomer in the sample. Thus, F68(7)-lac1 cryogels were
completely dissolved when immersed in distilled water resem-
bling a lack of polymer network formation (null conversion).
Meanwhile, conversions ranging 55 to 65% were obtained for
cross-linked cryogels. Thus, the grafting mechanism that is
conducive to the formation of hydrogels seems to be governed by
the efficient formation of F68 macroradicals (enzymatically
induced) which, thereafter, promote PEGDA polymerization
Fig. 4 SEM micrographs of cross-sectioned F68(7)c-lac1 (a, scale bars
are 500 mm and 20 mm in the inset) and PEG(7)c-lac1 (b, scale bars are
200 mm and 20 mm in the inset).
3536 | Soft Matter, 2010, 6, 3533–3540
and cross-linking (Scheme 1). The above described mechanism
was also valid for the formation of PEG(7)-lac1 hydrogels from
bare PEGDA via enzymatic generation of PEO based macro-
radicals. Actually, the generation of long-lived macroradicals in
oxygen-free media has been demonstrated for both PEO and
PPO based polymers via non-enzymatic processes.13
The conversions reported in Table 1 were slightly below those
previously reported for laccase induced polymerizations (up to
90% for MMA or acrylamide polymerizations carried out at
50–60 �C),11,12 although it is worth noting that, in our case,
temperature was lower (e.g. 37 �C). It is worth noting that the use
of low temperatures may decrease the conversion but it was
crucial for the activity preservation of the immobilized
enzyme.12,20 Furthermore, the macromolecular nature of both
the radicals and the monomers could also play a role in the
achievement of lower conversions than those obtained using
b-diketones as the mediator and methylmethacrylate or acryl-
amide as monomers. In our case, the use of GOX was also crucial
to provide an ideal balance of oxygen concentration in the
solution; enough as substrate for optimum (or near-optimum)
laccase activity but below that which would inhibit radical
polymerization. Similar conversions were obtained in absence of
GOX/GLU only when the solution was thoroughly deaerated
under nitrogen flow (Table 1). Otherwise, hydrogels were not
obtained. It is also worth noting that GLU degradation by GOX
generates low concentrations of H2O2 as a byproduct (Scheme 1).
The role played by H2O2 in the mechanism depicted in Scheme 1
is still unclear though preliminary studies seem to suggest that its
presence has no influence on PEGDA grafting onto F68. In fact,
the use of an enzymatic oxygen-scavenging system21 based on
coupled GOX and catalase in a buffer containing millimolar
GLU provided quite similar conversions to those where there
was no catalase (see Table 1).
At this stage, we considered that the FTIR spectra of cryogels
before and after washing (e.g. PEG-g-F68c and PEG-g-F68wc,
respectively), and also of the water that was used for washing
them could provide interesting information about cryogel
composition. FTIR spectroscopy confirmed that PEG-g-F68wc
cryogels (i.e. after extraction of unreacted acrylates) were
composed of both PPO segments from F68 (bands at ca.
2950 cm�1 ascribed to CH3 symmetric and antisymmetric
stretching modes)22 and PEO segments from both F68 and
PEGDA (Fig. 5); i.e. pure PEO has strong bands at 844 and
950 cm�1, and a shoulder at 962 cm�1. FTIR spectrum of PEG-
g-F68wc cryogels also confirmed the grafting of PEGDA onto
F68 by the shift of the peak of carbonyl stretching resonance
from 1724 (conjugated with acrylic groups) to 1734 cm�1 (non-
conjugated) upon cross-liking.23 The decrease in the relative
intensity of the peak assigned to C ¼ C vibration (at 1636 cm�1)
versus that of the peak assigned to C ] O stretching (within the
1724–1734 cm�1 range) in PEG-g-F68wc cryogels revealed effi-
cient PEGDA grafting and polymerization onto F68. On the
basis of the data obtained from FTIR spectra, one can conclude
that PEG-g-F68wc cryogels were composed of both F68 and
PEG (thus, of amphiphilic nature), while non-reacted species
mostly corresponded to PEGDA monomers. FTIR spectra of
PEG-g-F68(1 : 1-7)-lac1 and PEG-g-F68(3 : 1-7)-lac1 cryogels
(either before or after washing) exhibited a similar trend
(Fig. S3, ESI†).
This journal is ª The Royal Society of Chemistry 2010
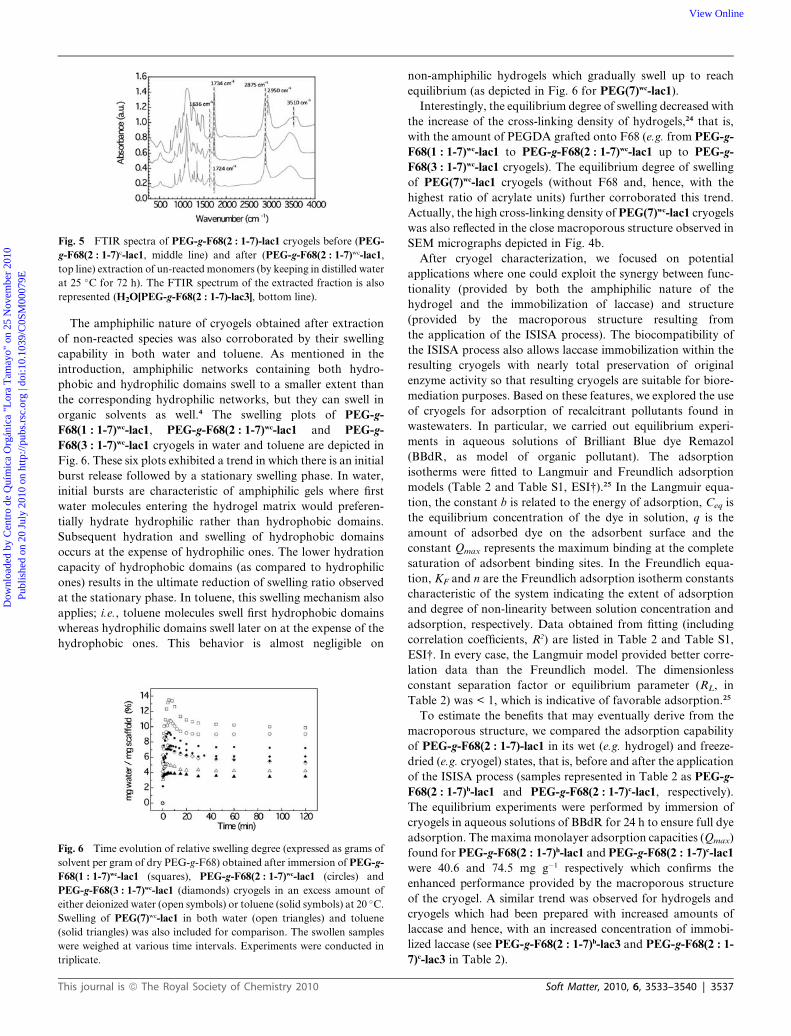
Fig. 5 FTIR spectra of PEG-g-F68(2 : 1-7)-lac1 cryogels before (PEG-
g-F68(2 : 1-7)c-lac1, middle line) and after (PEG-g-F68(2 : 1-7)wc-lac1,
top line) extraction of un-reacted monomers (by keeping in distilled water
at 25 �C for 72 h). The FTIR spectrum of the extracted fraction is also
represented (H2O[PEG-g-F68(2 : 1-7)-lac3], bottom line).
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
The amphiphilic nature of cryogels obtained after extraction
of non-reacted species was also corroborated by their swelling
capability in both water and toluene. As mentioned in the
introduction, amphiphilic networks containing both hydro-
phobic and hydrophilic domains swell to a smaller extent than
the corresponding hydrophilic networks, but they can swell in
organic solvents as well.4 The swelling plots of PEG-g-
F68(1 : 1-7)wc-lac1, PEG-g-F68(2 : 1-7)wc-lac1 and PEG-g-
F68(3 : 1-7)wc-lac1 cryogels in water and toluene are depicted in
Fig. 6. These six plots exhibited a trend in which there is an initial
burst release followed by a stationary swelling phase. In water,
initial bursts are characteristic of amphiphilic gels where first
water molecules entering the hydrogel matrix would preferen-
tially hydrate hydrophilic rather than hydrophobic domains.
Subsequent hydration and swelling of hydrophobic domains
occurs at the expense of hydrophilic ones. The lower hydration
capacity of hydrophobic domains (as compared to hydrophilic
ones) results in the ultimate reduction of swelling ratio observed
at the stationary phase. In toluene, this swelling mechanism also
applies; i.e., toluene molecules swell first hydrophobic domains
whereas hydrophilic domains swell later on at the expense of the
hydrophobic ones. This behavior is almost negligible on
Fig. 6 Time evolution of relative swelling degree (expressed as grams of
solvent per gram of dry PEG-g-F68) obtained after immersion of PEG-g-
F68(1 : 1-7)wc-lac1 (squares), PEG-g-F68(2 : 1-7)wc-lac1 (circles) and
PEG-g-F68(3 : 1-7)wc-lac1 (diamonds) cryogels in an excess amount of
either deionized water (open symbols) or toluene (solid symbols) at 20 �C.
Swelling of PEG(7)wc-lac1 in both water (open triangles) and toluene
(solid triangles) was also included for comparison. The swollen samples
were weighed at various time intervals. Experiments were conducted in
triplicate.
This journal is ª The Royal Society of Chemistry 2010
non-amphiphilic hydrogels which gradually swell up to reach
equilibrium (as depicted in Fig. 6 for PEG(7)wc-lac1).
Interestingly, the equilibrium degree of swelling decreased with
the increase of the cross-linking density of hydrogels,24 that is,
with the amount of PEGDA grafted onto F68 (e.g. from PEG-g-
F68(1 : 1-7)wc-lac1 to PEG-g-F68(2 : 1-7)wc-lac1 up to PEG-g-
F68(3 : 1-7)wc-lac1 cryogels). The equilibrium degree of swelling
of PEG(7)wc-lac1 cryogels (without F68 and, hence, with the
highest ratio of acrylate units) further corroborated this trend.
Actually, the high cross-linking density of PEG(7)wc-lac1 cryogels
was also reflected in the close macroporous structure observed in
SEM micrographs depicted in Fig. 4b.
After cryogel characterization, we focused on potential
applications where one could exploit the synergy between func-
tionality (provided by both the amphiphilic nature of the
hydrogel and the immobilization of laccase) and structure
(provided by the macroporous structure resulting from
the application of the ISISA process). The biocompatibility of
the ISISA process also allows laccase immobilization within the
resulting cryogels with nearly total preservation of original
enzyme activity so that resulting cryogels are suitable for biore-
mediation purposes. Based on these features, we explored the use
of cryogels for adsorption of recalcitrant pollutants found in
wastewaters. In particular, we carried out equilibrium experi-
ments in aqueous solutions of Brilliant Blue dye Remazol
(BBdR, as model of organic pollutant). The adsorption
isotherms were fitted to Langmuir and Freundlich adsorption
models (Table 2 and Table S1, ESI†).25 In the Langmuir equa-
tion, the constant b is related to the energy of adsorption, Ceq is
the equilibrium concentration of the dye in solution, q is the
amount of adsorbed dye on the adsorbent surface and the
constant Qmax represents the maximum binding at the complete
saturation of adsorbent binding sites. In the Freundlich equa-
tion, KF and n are the Freundlich adsorption isotherm constants
characteristic of the system indicating the extent of adsorption
and degree of non-linearity between solution concentration and
adsorption, respectively. Data obtained from fitting (including
correlation coefficients, R2) are listed in Table 2 and Table S1,
ESI†. In every case, the Langmuir model provided better corre-
lation data than the Freundlich model. The dimensionless
constant separation factor or equilibrium parameter (RL, in
Table 2) was < 1, which is indicative of favorable adsorption.25
To estimate the benefits that may eventually derive from the
macroporous structure, we compared the adsorption capability
of PEG-g-F68(2 : 1-7)-lac1 in its wet (e.g. hydrogel) and freeze-
dried (e.g. cryogel) states, that is, before and after the application
of the ISISA process (samples represented in Table 2 as PEG-g-
F68(2 : 1-7)h-lac1 and PEG-g-F68(2 : 1-7)c-lac1, respectively).
The equilibrium experiments were performed by immersion of
cryogels in aqueous solutions of BBdR for 24 h to ensure full dye
adsorption. The maxima monolayer adsorption capacities (Qmax)
found for PEG-g-F68(2 : 1-7)h-lac1 and PEG-g-F68(2 : 1-7)c-lac1
were 40.6 and 74.5 mg g�1 respectively which confirms the
enhanced performance provided by the macroporous structure
of the cryogel. A similar trend was observed for hydrogels and
cryogels which had been prepared with increased amounts of
laccase and hence, with an increased concentration of immobi-
lized laccase (see PEG-g-F68(2 : 1-7)h-lac3 and PEG-g-F68(2 : 1-
7)c-lac3 in Table 2).
Soft Matter, 2010, 6, 3533–3540 | 3537
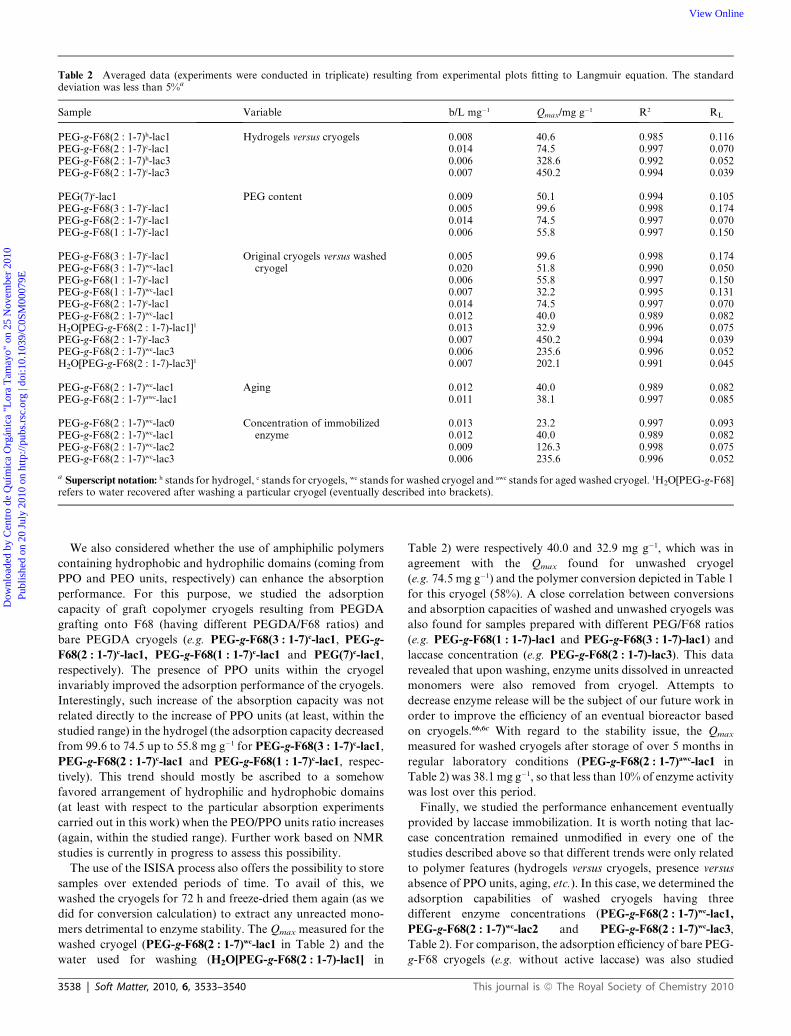
Table 2 Averaged data (experiments were conducted in triplicate) resulting from experimental plots fitting to Langmuir equation. The standarddeviation was less than 5%a
Sample Variable b/L mg�1 Qmax/mg g�1 R2 RL
PEG-g-F68(2 : 1-7)h-lac1 Hydrogels versus cryogels 0.008 40.6 0.985 0.116PEG-g-F68(2 : 1-7)c-lac1 0.014 74.5 0.997 0.070PEG-g-F68(2 : 1-7)h-lac3 0.006 328.6 0.992 0.052PEG-g-F68(2 : 1-7)c-lac3 0.007 450.2 0.994 0.039
PEG(7)c-lac1 PEG content 0.009 50.1 0.994 0.105PEG-g-F68(3 : 1-7)c-lac1 0.005 99.6 0.998 0.174PEG-g-F68(2 : 1-7)c-lac1 0.014 74.5 0.997 0.070PEG-g-F68(1 : 1-7)c-lac1 0.006 55.8 0.997 0.150
PEG-g-F68(3 : 1-7)c-lac1 Original cryogels versus washedcryogel
0.005 99.6 0.998 0.174PEG-g-F68(3 : 1-7)wc-lac1 0.020 51.8 0.990 0.050PEG-g-F68(1 : 1-7)c-lac1 0.006 55.8 0.997 0.150PEG-g-F68(1 : 1-7)wc-lac1 0.007 32.2 0.995 0.131PEG-g-F68(2 : 1-7)c-lac1 0.014 74.5 0.997 0.070PEG-g-F68(2 : 1-7)wc-lac1 0.012 40.0 0.989 0.082H2O[PEG-g-F68(2 : 1-7)-lac1]1 0.013 32.9 0.996 0.075PEG-g-F68(2 : 1-7)c-lac3 0.007 450.2 0.994 0.039PEG-g-F68(2 : 1-7)wc-lac3 0.006 235.6 0.996 0.052H2O[PEG-g-F68(2 : 1-7)-lac3]1 0.007 202.1 0.991 0.045
PEG-g-F68(2 : 1-7)wc-lac1 Aging 0.012 40.0 0.989 0.082PEG-g-F68(2 : 1-7)awc-lac1 0.011 38.1 0.997 0.085
PEG-g-F68(2 : 1-7)wc-lac0 Concentration of immobilizedenzyme
0.013 23.2 0.997 0.093PEG-g-F68(2 : 1-7)wc-lac1 0.012 40.0 0.989 0.082PEG-g-F68(2 : 1-7)wc-lac2 0.009 126.3 0.998 0.075PEG-g-F68(2 : 1-7)wc-lac3 0.006 235.6 0.996 0.052
a Superscript notation: h stands for hydrogel, c stands for cryogels, wc stands for washed cryogel and awc stands for aged washed cryogel. 1H2O[PEG-g-F68]refers to water recovered after washing a particular cryogel (eventually described into brackets).
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
We also considered whether the use of amphiphilic polymers
containing hydrophobic and hydrophilic domains (coming from
PPO and PEO units, respectively) can enhance the absorption
performance. For this purpose, we studied the adsorption
capacity of graft copolymer cryogels resulting from PEGDA
grafting onto F68 (having different PEGDA/F68 ratios) and
bare PEGDA cryogels (e.g. PEG-g-F68(3 : 1-7)c-lac1, PEG-g-
F68(2 : 1-7)c-lac1, PEG-g-F68(1 : 1-7)c-lac1 and PEG(7)c-lac1,
respectively). The presence of PPO units within the cryogel
invariably improved the adsorption performance of the cryogels.
Interestingly, such increase of the absorption capacity was not
related directly to the increase of PPO units (at least, within the
studied range) in the hydrogel (the adsorption capacity decreased
from 99.6 to 74.5 up to 55.8 mg g�1 for PEG-g-F68(3 : 1-7)c-lac1,
PEG-g-F68(2 : 1-7)c-lac1 and PEG-g-F68(1 : 1-7)c-lac1, respec-
tively). This trend should mostly be ascribed to a somehow
favored arrangement of hydrophilic and hydrophobic domains
(at least with respect to the particular absorption experiments
carried out in this work) when the PEO/PPO units ratio increases
(again, within the studied range). Further work based on NMR
studies is currently in progress to assess this possibility.
The use of the ISISA process also offers the possibility to store
samples over extended periods of time. To avail of this, we
washed the cryogels for 72 h and freeze-dried them again (as we
did for conversion calculation) to extract any unreacted mono-
mers detrimental to enzyme stability. The Qmax measured for the
washed cryogel (PEG-g-F68(2 : 1-7)wc-lac1 in Table 2) and the
water used for washing (H2O[PEG-g-F68(2 : 1-7)-lac1] in
3538 | Soft Matter, 2010, 6, 3533–3540
Table 2) were respectively 40.0 and 32.9 mg g�1, which was in
agreement with the Qmax found for unwashed cryogel
(e.g. 74.5 mg g�1) and the polymer conversion depicted in Table 1
for this cryogel (58%). A close correlation between conversions
and absorption capacities of washed and unwashed cryogels was
also found for samples prepared with different PEG/F68 ratios
(e.g. PEG-g-F68(1 : 1-7)-lac1 and PEG-g-F68(3 : 1-7)-lac1) and
laccase concentration (e.g. PEG-g-F68(2 : 1-7)-lac3). This data
revealed that upon washing, enzyme units dissolved in unreacted
monomers were also removed from cryogel. Attempts to
decrease enzyme release will be the subject of our future work in
order to improve the efficiency of an eventual bioreactor based
on cryogels.6b,6c With regard to the stability issue, the Qmax
measured for washed cryogels after storage of over 5 months in
regular laboratory conditions (PEG-g-F68(2 : 1-7)awc-lac1 in
Table 2) was 38.1 mg g�1, so that less than 10% of enzyme activity
was lost over this period.
Finally, we studied the performance enhancement eventually
provided by laccase immobilization. It is worth noting that lac-
case concentration remained unmodified in every one of the
studies described above so that different trends were only related
to polymer features (hydrogels versus cryogels, presence versus
absence of PPO units, aging, etc.). In this case, we determined the
adsorption capabilities of washed cryogels having three
different enzyme concentrations (PEG-g-F68(2 : 1-7)wc-lac1,
PEG-g-F68(2 : 1-7)wc-lac2 and PEG-g-F68(2 : 1-7)wc-lac3,
Table 2). For comparison, the adsorption efficiency of bare PEG-
g-F68 cryogels (e.g. without active laccase) was also studied
This journal is ª The Royal Society of Chemistry 2010

Fig. 7 Experimental (symbols), Langmuir (solid line) and Freundlich
(dash line) isotherms for adsorption of Brilliant Blue dye Remazol on
PEG-g-F68(2 : 1-7)wc-lac0 (bottom half solid circles), PEG-g-
F68(2 : 1-7)wc-lac1 (solid circles), PEG-g-F68(2 : 1-7)wc-lac2 (open circles)
and PEG-g-F68(2 : 1-7)wc-lac3 (bottom half open circles). Lac0 stands for
0 units, lac1 stands for 20 units lac2 stands for 80 units and lac3 for 200
units. Langmuir and Freundlinch equations, and data resulting from
Langmuir equation fitting are also included.
Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
(e.g. PEG-g-F68(2 : 1-7)wc-lac0). Since the block copolymer
based cryogels used in this study cannot be obtained in absence
of laccase, PEG-g-F68(2 : 1-7)wc-lac0 was obtained by submis-
sion of one of the PEG-g-F68 cryogels having immobilized lac-
case (e.g. PEG-g-F68(2 : 1-7)wc-lac1) to a thermal treatment
consisting of 60 �C for 24 h for laccase inactivation.12,20 Fig. 7
shows the adsorption isotherms fitted Langmuir and Freundlich
adsorption models as well as the data resulting from fitting (also
depicted in Table 2 and Table S1, ESI†). The Qmax found for
PEG-g-F68(2 : 1-7)wc-lac0 was 23.2 mg g�1 and increased linearly
with the concentration of immobilized laccase (from 40.0 mg g�1
for PEG-g-F68(2 : 1-7)wc-lac1, to 126.3 mg g�1 for PEG-g-
F68(2 : 1-7)wc-lac2, up to 235.6 mg g�1 for PEG-g-F68(2 : 1-7)wc-
lac3, see Fig. 7). Such enhancement of the adsorption capacity
confirms the favorable synergetic effect between the amphiphilic
polymer and the immobilized enzyme for adsorption purposes.
As mentioned above, synergy must result from the ability of
laccase to degrade phenolic compounds.26 Thus, BBdR adsorbed
on the hydrated block-copolymer based cryogel would, after
adsorption, be degraded by immobilized laccase. Dye degrada-
tion would result in partial renewal of adsorption domains at
block-copolymer based hydrogel allowing for further BBdR
adsorption. It is worth noting that byproducts of enzymatic
BBdR degradation remained adsorbed (they were not detected in
solution by UV-Vis spectroscopy, see Fig. S4, ESI†). Considering
that the Langmuir isotherm assumes a monolayer coverage and
uniform activity distribution on the adsorbent surface, the
enhancement of the adsorption capacity must be explained in
terms of availability of adsorption domains for BBdR byprod-
ucts (due their size, charge and/or hydrophilic-hydrophobic
nature), which were not initially available for original BBdR.27
Conclusions
Summarizing, PEG-g-F68 hydrogels have been obtained via (1)
laccase-generation of long-lived macroradicals in PEO-PPO
segments of F68, and (2) macroradical induced grafting of
PEG550DA onto F68. The use of GOX/GLU provided the
proper balance of oxygen in the reaction medium (i.e., on one
This journal is ª The Royal Society of Chemistry 2010
hand, laccase needs oxygen as a substrate while, on the other,
oxygen is a strong inhibitor of radical polymerizations) to obtain
reasonable polymerization conversions. Eventually, laccase was
immobilized within the resulting PEG-g-F68 hydrogel with full
preservation of enzyme activity (90% of activity was still
preserved three months after preparation). Laccase-immobilized
PEG-g-F68 hydrogels were submitted to the ISISA process for
preparation of laccase-immobilized PEG-g-F68 cryogels, which
exhibited outstanding adsorption capabilities (up to 235 mg g�1).
This eminent suitability of cryogels for adsorption of recalcitrant
pollutants found in wastewaters resided in the synergetic effect of
(1) the macroporous structure that allow easy mass transport to
adsorption sites, (2) the amphiphilic nature of the polymer that
provides hydrophilic and hydrophobic domains for pollutants
adsorption, and (3) the immobilized enzyme that degrades
adsorbed pollutant thus allowing further adsorption on renewed
sites.
Acknowledgements
This work was supported by MICINN (MAT2009-10214,
MAT2009-09671 and PET2008-0168-01) and CSIC
(200660F011). M. N. and S. N. thank CSIC for a research
contract and a PhD fellowship, respectively. C. A. thanks
MICINN for a Ramon y Cajal contract. We thank F. Pinto for
assistance with SEM.
References
1 (a) D. J. Lloyd, Colloid Chemistry. ed. J. Alexander, The ChemicalCatalogue Co., New York, 1926, vol. 1, p. 767; (b) P. Flory,Faraday Discuss. Chem. Soc., 1974, 57, 7; (c) L. A. Estroff andA. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217.
2 (a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879;(b) A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12.
3 (a) Y. S. Ho and G. McKay, Process Biochem., 1999, 34, 451–465;(b) B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5,3452–3460; (c) Y. Chujo, K. Sada and T. Saegusa, Macromolecules,1993, 26, 6315–6319; (d) H. Kasxg€oz, S. €Ozg€um€u�s and M. Orbay,Polymer, 2003, 44, 1785–1793.
4 C. S. Patrickios and T. K. Georgiou, Curr. Opin. Colloid InterfaceSci., 2003, 8, 76–85.
5 V. Bekiari and P. Lianos, Chem. Mater., 2006, 18, 4142–4146.6 (a) G. Palmieri, P. Giardina and G. Sannia, Biotechnol. Prog., 2005,
21, 1436–1441; (b) G. Palmieri, P. Giardina and G. Sannia, EnzymeMicrob. Technol., 2005, 36, 17–24; (c) V. Arantes andA. M. F. Milagres, Enzyme Microb. Technol., 2007, 42, 17–22.
7 Q. Wang, Z. Yang, Y. Gao, W. Ge, L. Wanga and B. Xu, Soft Matter,2008, 4, 550–553.
8 A. Barbetta, M. Massimi, L. C. Devirgiliis and M. Dentini,Biomacromolecules, 2006, 7, 3059–3068.
9 (a) L. Mart�ın, M. Alonso, M. M€oller, J. C. Rodr�ıguez-Cabello andP. Mela, Soft Matter, 2009, 5, 1591–1593; (b) R. E. Sallach, W. Cui,J. Wen, A. Martinez, V. P. Conticello and E. L. Chaikof,Biomaterials, 2009, 30, 409–422.
10 (a) J. S. Dordick, Trends Biotechnol., 1992, 10, 287–29; (b) A. Singhand D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 211–224;(c) S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353.
11 (a) D. Teixeira, T. Lalot, M. Brigodiot and E. Mar�echal,Macromolecules, 1999, 32, 70–72; (b) R. Ikeda, H. Tanaka,H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1998, 19,423–425; (c) T. Tsujimoto, H. Uyama and S. Kobayashi, Macromol.Biosci., 2001, 1, 228–232; (d) B. Kalra and R. A. Gross, GreenChem., 2002, 4, 174–178.
12 F. Hollmann, Y. Gumulya, C. T€olle, A. Liese and O. Thum,Macromolecules, 2008, 41, 8520–8524.
13 (a) J. Yamauchi, A. Yamaoka, K. Ikemoto and T. Matsui, J. Appl.Polym. Sci., 1991, 43, 1197–1203; (b) L. Bromberg, J. Phys. Chem. B,
Soft Matter, 2010, 6, 3533–3540 | 3539

Dow
nloa
ded
by C
entr
o de
Quí
mic
a O
rgán
ica
"Lor
a T
amay
o" o
n 25
Nov
embe
r 20
10Pu
blis
hed
on 2
0 Ju
ly 2
010
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C0S
M00
079E
View Online
1998, 102, 10736–10744; (c) L. Bromberg, J. Phys. Chem. B, 1998, 102,1956–1963; (d) L. Bromberg, Langmuir, 1998, 14, 5806–5812;(e) Y. Zhang and Y. M. Lam, J. Colloid Interface Sci., 2007, 306,398–404; (f) S.-P. Zhao, L.-M. Zhang, D. Ma, C. Yang and L. Yan,J. Phys. Chem. B, 2006, 110, 16503–16507.
14 (a) S. R. Mukai, H. Nishihara and H. Tamon, Chem. Commun., 2004,874; (b) H. Nishihara, S. R. Mukai and H. Tamon, Carbon, 2004, 42,899; (c) Zhang, I. Hussain, M. Brust, M. F. Butler, S. P. Rannard andA. I. Cooper, Nat. Mater., 2005, 4, 787; (d) S. Deville, E. Saiz,R. K. Nalla and A. P. Tomsia, Science, 2006, 311, 515; (e) S. Bandiand D. A. Schiraldi, Macromolecules, 2006, 39, 6537–654;(f) M. C. Gutierrez, M. J. Hortig€uela, R. Jimenez, J. M. Amarilla,M. L. Ferrer and F. del Monte, J. Phys. Chem. C, 2007, 111, 5557;(g) A. Abarrategui, M. C. Gutierrez, M. J. Hortig€uela, V. Ramos,J. L. Lopez-Lacomba, M. L. Ferrer and F. del Monte, Biomaterials,2008, 29, 94; (h) Q. Shi, H. Liang, D. Feng, J. Wang andG. D. Stucky, J. Am. Chem. Soc., 2008, 130, 5034; (i) F. Khan,D. Walsh, A. J. Patil, A. W. Perriman and S. Mann, Soft Matter,2009, 5, 3081–3085.
15 M. B. Dainiak, A. Kumar, I. Yu. Galaev and B. Mattiasson, Proc.Natl. Acad. Sci. U. S. A., 2006, 103, 849–854.
16 (a) M. C. Gutierrez, Z. Y. Garcia-Carvajal, M. Jobbagy, L. Yuste,F. Rojo, M. L. Ferrer and F. del Monte, Adv. Funct. Mater., 2007,17, 3505–3513; (b) M. C. Gutierrez, M. L. Ferrer and F. del Monte,Chem. Mater., 2008, 20, 634–648.
17 M. C. Gutierrez, M. Jobbagy, N. Rapun, M. L. Ferrer and F. delMonte, Adv. Mater., 2006, 18, 1137.
18 (a) M. L. Ferrer, R. Esquembre, I. Ortega, C. R. Mateo and F. delMonte, Chem. Mater., 2006, 18, 554; (b) M. C. Gutierrez,
3540 | Soft Matter, 2010, 6, 3533–3540
Z. Y. Garcia-Carvajal, M. Jobbagy, F. Catalina, C. Abrusci,L. Yuste, F. Rojo, M. L. Ferrer and F. del Monte, Chem. Mater.,2007, 19, 1968; (c) M. C. Gutierrez, Z. Y. Garcia-Carvajal,M. J. Hortig€uela, L. Yuste, F. Rojo, M. L. Ferrer and F. delMonte, J. Mater. Chem., 2007, 17, 2992.
19 X. Wu, Y. Liu, X. Li, P. Wen, Y. Zhang, Y. Long, X. Wang, Y. Guo,F. Xing and J. Gao, Acta Biomater., 2010, 6, 1167–1177.
20 A. Rek�uc, J. Bryjak, K. Szymanska and A. B. Jarzebski, ProcessBiochem., 2009, 44, 191–198.
21 S. C. Blanchard, H. D. Kim, R. L. GonzalezJr., J. D. Puglisi andS. Chu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12893–12898.
22 (a) C. Guo, H. Liu, J. Wang and J. Chen, J. Colloid Interface Sci.,1999, 209, 368–373; (b) C.-H. Choi, J.-H. Jung, T.-S. Hwang andC.-S. Leezz, Macromol. Res., 2009, 17, 163–167.
23 (a) Y. Wu, F.-B. Che and J.-H. Chen, J. Appl. Polym. Sci., 2008, 110,1118–1128; (b) D. Biswal and J. Z. Hilt, Polymer, 2006, 47, 7355–7360.
24 (a) D. S. Achilleos, T. K. Georgiou and C. S. Patrickios,Biomacromolecules, 2006, 7, 3396–3405; (b) F. M. Goycoolea,A. Heras, I. Aranaz, G. Galed, M. E. Fern�andez-Valle andW. Arg€uelles-Monal, Macromol. Biosci., 2003, 3, 612–619.
25 See, for instance: K. Vijayaraghavan, S. W. Won, J. Mao andY.-S. Yun, Chem. Eng. J., 2008, 145, 1–6.
26 P.-P. Champagne and J. A. Ramsay, Appl. Microbiol. Biotechnol.,2007, 77, 819–823.
27 (a) L. Pereira, A. V. Coelho, C. A. Viegas, C. Ganachaud, G. Iacazio,T. Tron, M. P. Robalo and L. O. Martinsa, Adv. Synth. Catal., 2009,351, 1857–1865; (b) Y. Sugano, Y. Matsushima, K. Tsuchiya,H. Aoki, M. Hirai and M. Shoda, Biodegradation, 2009, 20,433–44.
This journal is ª The Royal Society of Chemistry 2010
Copyright © 2022 FDOKUMEN




















