
Energy transfer of highly vibrationally excited naphthalene: Collisions with CHF3, CF4, and Kr
7
Energy transfer of highly vibrationally excited biphenyl Hsu Chen Hsu, Yuri Dyakov, and Chi-Kung Ni a Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan Received 15 August 2010; accepted 10 September 2010; published online 3 November 2010 The energy transfer between Kr atoms and highly vibrationally excited, rotationally cold biphenyl in the triplet state was investigated using crossed-beam/time-of-flight mass spectrometer/time-sliced velocity map ion imaging techniques. Compared to the energy transfer of naphthalene, energy transfer of biphenyl shows more forward scattering, less complex formation, larger cross section for vibrational to translational V → T energy transfer, smaller cross section for translational to vibrational and rotational T → VR energy transfer, larger total collisional cross section, and more energy transferred from vibration to translation. Significant increase in the large V → T energy transfer probabilities, termed supercollisions, was observed. The difference in the energy transfer of highly vibrationally excited molecules between rotationally cold naphthalene and rotationally cold biphenyl is very similar to the difference in the energy transfer of highly vibrationally excited molecules between rotationally cold naphthalene and rotationally hot naphthalene. The low-frequency vibrational modes with out-of-plane motion and rotationlike wide-angle motion are attributed to make the energy transfer of biphenyl different from that of naphthalene. © 2010 American Institute of Physics. doi:10.1063/1.3495766 I. INTRODUCTION Collisional energy transfer of molecules containing chemically significant amounts of energy has been studied for several decades. It is one of the important processes in chemistry. In thermal unimolecular reactions, the energies needed for reactions are accumulated via collisions with bath atoms or molecules, whereas in chemical activation, recom- bination reactions, and various photochemical processes, col- lisions can stabilize these highly excited intermediate spe- cies. Lindermann 1 first discovered the importance of molecu- lar energy transfer in thermal unimolecular decomposition in 1921. In Lindermann’s theories, each collision suffered by the activated molecules was considered to be strong. A strong collision is assumed to transfer as much as 30 kcal/ mol or more in a single collision. Although Lindermann’s theory provides a model to explain the pressure dependence of unimolecular reactions, experiments from early unimo- lecular reaction rate coefficients in the low-pressure and fall- off regions shows significantly difference from theoretical prediction. 2 Those experiments indicated that instead of strong collision, average energy transfer in each collision is very small. Energy transfer of highly vibrationally excited diatomic molecule, KBr, has been investigated using crossed-molecular beams. Large amount of energy transfer from vibration to translation was observed. 3–6 On the other hand, new experimental techniques have been developed to directly measure the average energy transfer per collision of highly vibrationally excited polyatomic molecules in the past three decades. 7–15 They confirmed the suggestion from uni- molecular reaction rate coefficient measurements, i.e., the average vibrational energy transfer per collision is generally less than a few kcal/mol and may be less than 0.1 kcal/mol. Despite the wealth of energy-transfer studies of highly vibra- tionally excited molecules, little experimental information about the energy transfer distribution and energy transfer mechanism is available. This is because all these experi- ments have been performed under bulk conditions. Their re- sults are averages of the outcomes of individual collisions over an ensemble of colliding molecules. Detail information about the result of single collision is easily washed. Conse- quently, this is a major bottleneck in understanding the en- ergy transfer of highly vibrationally excited molecules. 16,17 Recently we have studied the energy transfer between rare gas Ar and Kr and highly vibrationally excited mol- ecules azulene, naphthalene, methylnaphthalene using crossed-beam/time-of-flight mass spectrometer/time-sliced velocity map ion imaging techniques. 18–22 Some details of energy transfer distribution and mechanism were obtained. For example, we have demonstrated that at low enough translational collision energies complex with lifetime longer than rotation period can be formed. Although the lifetime of this complex is longer than most of the other collision dura- tions, energy does not transfer from highly vibrationally ex- cited molecules to translational energy. Instead, energy is transferred from translation into vibration and/or rotation of the highly vibrationally excited molecules. Increasing trans- lational collision energy not only increases large amounts of vibration to translation V → T energy transfer, but also en- hances the translation to vibration-rotation T → VR energy transfer. The increase of T → VR energy transfer is much larger than that of V → T energy transfer as the translational collision energy increases. As a result, it does not help to stabilize the highly vibrationally excited molecule. On the other hand, rotation was found to be a gateway to transfer a Also at: Department of Chemistry, National Tsing Hua University, Hsin- chu, 30013 Taiwan. Electronic mail: [email protected]. THE JOURNAL OF CHEMICAL PHYSICS 133, 174315 2010 0021-9606/2010/13317/174315/7/$30.00 © 2010 American Institute of Physics 133, 174315-1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Energy transfer of highly vibrationally excited naphthalene: Collisions with CHF3, CF4, and Kr

Energy transfer of highly vibrationally excited biphenylHsu Chen Hsu, Yuri Dyakov, and Chi-Kung Nia�
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan
�Received 15 August 2010; accepted 10 September 2010; published online 3 November 2010�
The energy transfer between Kr atoms and highly vibrationally excited, rotationally cold biphenylin the triplet state was investigated using crossed-beam/time-of-flight mass spectrometer/time-slicedvelocity map ion imaging techniques. Compared to the energy transfer of naphthalene, energytransfer of biphenyl shows more forward scattering, less complex formation, larger cross section forvibrational to translational �V→T� energy transfer, smaller cross section for translational tovibrational and rotational �T→VR� energy transfer, larger total collisional cross section, and moreenergy transferred from vibration to translation. Significant increase in the large V→T energytransfer probabilities, termed supercollisions, was observed. The difference in the energy transfer ofhighly vibrationally excited molecules between rotationally cold naphthalene and rotationally coldbiphenyl is very similar to the difference in the energy transfer of highly vibrationally excitedmolecules between rotationally cold naphthalene and rotationally hot naphthalene. Thelow-frequency vibrational modes with out-of-plane motion and rotationlike wide-angle motion areattributed to make the energy transfer of biphenyl different from that of naphthalene. © 2010American Institute of Physics. �doi:10.1063/1.3495766�
I. INTRODUCTION
Collisional energy transfer of molecules containingchemically significant amounts of energy has been studiedfor several decades. It is one of the important processes inchemistry. In thermal unimolecular reactions, the energiesneeded for reactions are accumulated via collisions with bathatoms or molecules, whereas in chemical activation, recom-bination reactions, and various photochemical processes, col-lisions can stabilize these highly excited intermediate spe-cies.
Lindermann1 first discovered the importance of molecu-lar energy transfer in thermal unimolecular decomposition in1921. In Lindermann’s theories, each collision suffered bythe activated molecules was considered to be strong. Astrong collision is assumed to transfer as much as 30 kcal/mol or more in a single collision. Although Lindermann’stheory provides a model to explain the pressure dependenceof unimolecular reactions, experiments from early unimo-lecular reaction rate coefficients in the low-pressure and fall-off regions shows significantly difference from theoreticalprediction.2 Those experiments indicated that instead ofstrong collision, average energy transfer in each collision isvery small. Energy transfer of highly vibrationally exciteddiatomic molecule, KBr, has been investigated usingcrossed-molecular beams. Large amount of energy transferfrom vibration to translation was observed.3–6 On the otherhand, new experimental techniques have been developed todirectly measure the average energy transfer per collision ofhighly vibrationally excited polyatomic molecules in the pastthree decades.7–15 They confirmed the suggestion from uni-molecular reaction rate coefficient measurements, i.e., the
average vibrational energy transfer per collision is generallyless than a few kcal/mol and may be less than 0.1 kcal/mol.Despite the wealth of energy-transfer studies of highly vibra-tionally excited molecules, little experimental informationabout the energy transfer distribution and energy transfermechanism is available. This is because all these experi-ments have been performed under bulk conditions. Their re-sults are averages of the outcomes of individual collisionsover an ensemble of colliding molecules. Detail informationabout the result of single collision is easily washed. Conse-quently, this is a major bottleneck in understanding the en-ergy transfer of highly vibrationally excited molecules.16,17
Recently we have studied the energy transfer betweenrare gas �Ar and Kr� and highly vibrationally excited mol-ecules �azulene, naphthalene, methylnaphthalene� usingcrossed-beam/time-of-flight mass spectrometer/time-slicedvelocity map ion imaging techniques.18–22 Some details ofenergy transfer distribution and mechanism were obtained.For example, we have demonstrated that at low enoughtranslational collision energies complex with lifetime longerthan rotation period can be formed. Although the lifetime ofthis complex is longer than most of the other collision dura-tions, energy does not transfer from highly vibrationally ex-cited molecules to translational energy. Instead, energy istransferred from translation into vibration and/or rotation ofthe highly vibrationally excited molecules. Increasing trans-lational collision energy not only increases large amounts ofvibration to translation �V→T� energy transfer, but also en-hances the translation to vibration-rotation �T→VR� energytransfer. The increase of T→VR energy transfer is muchlarger than that of V→T energy transfer as the translationalcollision energy increases. As a result, it does not help tostabilize the highly vibrationally excited molecule. On theother hand, rotation was found to be a gateway to transfer
a�Also at: Department of Chemistry, National Tsing Hua University, Hsin-chu, 30013 Taiwan. Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 133, 174315 �2010�
0021-9606/2010/133�17�/174315/7/$30.00 © 2010 American Institute of Physics133, 174315-1

large amount of vibrational energy to translational energy.Increasing the initial rotation energy increases the V→T en-ergy transfer probability, but decreases the T→VR energytransfer probability. Rotation also enhances the large V→Tenergy transfer probabilities, termed supercollisions. Energytransfer of highly vibrationally excited molecules betweenrotationally cold naphthalene and 2-methylnaphthalene doesnot show observable difference, indicating the motion of me-thyl group does not play an important role.
In this work, we report the collisional energy transferbetween Kr and highly vibrationally excited, rotationallycold biphenyl �C6H5C6H5� in the triplet state. Comparison tothe energy transfer of naphthalene shows that low vibrationalfrequency modes of biphenyl, such as out-of-plane motionsand the rotationlike wide-angle motions, greatly enhance thevibration to translation energy transfer. This kind of vibrationmakes the energy transfer of biphenyl very different fromthat of naphthalene.
II. EXPERIMENT
The experimental apparatus includes a pulsed uv laserset at 266 nm, one vacuum ultraviolet �vuv� laser at 157 nm,a differentially pumped crossed-beam vacuum chamber anda time-of-flight mass spectrometer with a time-slicedvelocity-map ion imaging system. The schematic for the ap-paratus is shown in Fig. 1. The details have been described inthe previous studies.19 Only a brief description is describedhere.
The vacuum system that we employed consisted of twosource chambers, a differentially pumped chamber, and amain chamber. In one source chamber, biphenyl molecularbeam was formed as the carrier gas �Ar, 60 psi� flowedthrough a reservoir filled with biphenyl at temperature120 °C and then expanded through a high temperature�180 °C� pulsed nozzle. A Kr atomic beam was created inthe other source chamber by expanding ultrapure Kr at apressure of 200 psi through a pulsed nozzle maintained attemperature of 180 °C. These two beams crossed each other
at a fixed angle �25°� and at a position 4 mm below thecenter of the ion optics. A pulsed uv laser set at 266 nm�25 mJ /cm2� crossed the biphenyl molecular beam 16 mmupstream from the crossing point of the atomic and molecu-lar beams. Biphenyl molecules in the molecular beam wereexcited to the S1 state after absorbing a single 266 nm pho-ton. Highly vibrationally excited biphenyl in the triplet statewas produced after intersystem crossing. Molecules whichabsorbed more than one uv photon either dissociated intofragments quickly �estimated to be within nanosecond� orbecame cations. Fragments have no interference on the ex-perimental measurement because we only probed the parentmass in the time-of-flight mass spectra. Cations created by266 nm multiphoton ionization arrived at the detector about20–30 �s earlier than the cations generated from 157 nmphotoionization. They can be easily discriminated from thearrival time and they have no effects on the measurementeither.
Collision probability between atomic and molecularbeams was only about 5%. Scattered excited biphenyl mol-ecules were ionized by 157 nm laser beam. The 157 nmphoton energy is only large enough to ionize biphenyl in thetriplet state. It is not large enough to ionize biphenyl mol-ecules in the ground state. However, unscattered excited bi-phenyl molecules in the molecular beam were also ionizedby the 157 nm laser beam. These ions having the same ve-locity as the molecular beam were focused by the ion opticsinto a small spot on the microchannel plate �MCP� detector.In order to avoid saturation and possible damage to the de-tector, a stainless-steel pin located 5 cm in front of the de-tector was used to block these ions. Most of the scatteredbiphenyl molecules have velocities different from that of themolecular beam. They were not affected by the stainless-steel pin. Only a small portion of scattered biphenyl mol-ecules in the forward direction was obscured by the stainless-steel pin. The images with Kr atomic beam and the imageswithout Kr beam were taken alternately after every 27 000laser shot accumulation. The final image presented in thiswork, accumulated from a total of 162 000 laser shots, wastaken from the images with Kr beam after the subtraction ofthe images without Kr beam.
We have studied the energy transfer between highly vi-brationally excited naphthalene and Kr.20–22 Here, naphtha-lene under the same experimental conditions as biphenyl wasinvestigated again for comparison.
III. RESULTS AND DISCUSSIONS
A. Generation and detection of highly vibrationallyexcited biphenyl in the triplet state
The electronic spectrum of biphenyl has been investi-gated extensively in vapor,23–28 solution,24,29 and crystalphases.24,28 The electronic absorption spectrum of biphenylis broad and structureless at its lowest energy band due to theexcitation of low-frequency torsional vibrational modes. Theelectronic absorption spectrum in vapor phase at 340 K28
show three bands around 4.6, 5.2, and 6.41 eV associatedwith the �-�� valance transition to the S1, S2, and S3 states,respectively.30 The S1←S0 0–0 transition band is located at
FIG. 1. Schematic for the crossed-beam apparatus.
174315-2 Hsu, Dyakov, and Ni J. Chem. Phys. 133, 174315 �2010�

35 258.0 cm−1 �4.37 eV�, which has been identified by su-personic jet laser spectroscopy.31–34 The lowest triplet statewas measured by high-resolution electron energy-loss spec-troscopy, where biphenyl was deposited on a thin film of Arsolid.35 The spectrum shows a structureless band with onsetaround 3 eV and maximum peak at 3.5 eV. The phosphores-cence spectrum of biphenyl in n-heptane at 77 K shows the0–0 transition from ground state to the lowest excited tripletstate located at 2.84 eV.36,37 Phosphorescence spectra in solidneon host studied by Baca et al. illustrates the T1←S0 00
0
transition at 2.86 eV.38 Both theoretical calculations and ex-perimental evidences confirmed that the geometry of biphe-nyl at the ground state is twisted around the central C–Cbond,39,40 however, it becomes a nearly planer structure atboth singlet �S1�41–43 and triplet states �T1�.43–46 Therefore,the Franck–Condon factor for the 0–0 transition is small. Themain fluorescence parameters are experimentally studied andanalyzed in diluted cyclohexane solution.47–50 The fluores-cence quantum yield is measured to be 0.17�10% withshort decay time �16�5% ns�. The properties of biphenyl intriplet excited state has also been studied in cyclohexanesolution51 proclaiming the intersystem crossing quantumyield from the S1 to T1 state is 0.84. The T1 state has muchlonger lifetime �130 �s� compared to the S1 state lifetime.
Highly vibrationally excited biphenyl molecules with vi-brational energy of 41.8 kcal/mol can be generated by ab-sorption of one 266 nm photon, corresponding to the excita-tion to the S1 state, followed by intersystem crossing to theT1 state. The ionization potential of biphenyl is about 8.3 eV�191 kcal/mol�.52 Biphenyl in the triplet state can be ionizedusing 157 nm photons �182 kcal/mol�. The photon energy ismuch larger than the ionization threshold of the triplet state5.5 eV �127 kcal/mol� that the ionization cross section isexpected to be not sensitive to the small change of vibra-tional energy in the triplet state. On the other hand, this pho-ton energy is not enough to ionize the biphenyl on the elec-tronic ground state. This allows us to detect the biphenyl onthe triplet state without interference from the molecules onthe ground state. The large ionization cross section differencebetween biphenyl in the ground state and the triplet state at157 nm is shown in Fig. 2. No ion was observed when bi-phenyl molecular beam was not irradiated by 266 nm pho-tons. Only biphenyl excited by 266 nm photons can be ion-
ized by 157 nm laser beam. The 157 photon energy is alsolow enough that the highly vibrationally excited biphenylgenerated in the triplet upon absorption of 266 nm photonswould not dissociate into smaller ionic fragment upon ion-ization by 157 nm photons.53 This allowed us to have mass-selective detection and the measurement would not be inter-fered by multiphoton absorption.
B. Energy transfer distributions
Figure 3 shows the image of scattered biphenyl by Kratoms. The image of scattered naphthalene by Kr atoms isalso shown for comparison. The translational collision ener-gies are 330 and 320 cm−1 for biphenyl and naphthalene,respectively. The rectangular blank area in the upper right-hand corner is the portion of the image blocked by thestainless-steel pin located in front of the MCP detector. Fig-ures 3 also illustrates the respective Newton diagrams, in-cluding the initial naphthalene and biphenyl molecular beamvelocities, initial Kr beam velocities, center-of-mass veloci-ties, relative velocities, and elastic collision circles. The por-tion of image inside the elastic collision circle represents thedecrease of biphenyl or naphthalene velocity in the center ofmass frame after collision by Kr atoms, corresponding to theenergy up, �Eu, translation to vibration and rotation T→VR collisions. The portion of image outside of the circlerepresents the increase of biphenyl or naphthalene velocityafter collision, corresponding to the energy down, �Ed, vi-bration to translation V→T collisions. Notice that the energytransfer for energy down collisions includes the change ofvibrational energy to rotational energy as well as the changeof vibrational energy to translational energy. However, wecan only detect the energy transfer to the translational de-grees of freedom.
If complex is formed during the collision and lifetime ofthe complex is longer than the complex rotation period, theformation of complex can be recognized from the pattern ofimage intensity distribution, i.e., the large intensities in theforward and backward directions with forward-backwardsymmetry. The intensity from the complex in the forwarddirection is overlapped with the large direct forward scatter-ing intensity in this inelastic scattering experiments. There-fore the intensity symmetry is broken. However, the forma-
FIG. 2. Time-of-flight mass spectrum from the photoionization of biphenyland naphthalene molecules by 157 nm photons. The red and green linesrepresent molecules irradiated by 266 nm photons and without irradiation of266 nm photons, respectively.
FIG. 3. The image of scattered naphthalene and biphenyl by Kr atoms.
174315-3 Energy transfer of biphenyl J. Chem. Phys. 133, 174315 �2010�
tion of complex still can be easily identified from thebackward intensity peak. For naphthalene, the intensity ofthe backward peak can be clearly observed and the centralposition of the backward peak is located inside the elasticscattering circle. It indicates the corresponding energy trans-fer is energy up T→VR collisions. On the other hand, thebackward peak in biphenyl scattering is not as strong as thatof naphthalene, indicating the biphenyl-Kr complex forma-tion probability is smaller than that of naphthalene-Kr. How-ever, the position of the backward peak remains inside theelastic circle, suggesting the energy transfer of thebiphenyl-Kr complex is also from translational energy to vi-brational and rotational energy.
The final velocity distribution of scattered biphenyl wasobtained directly from the image intensity distribution. Fromthe conservation of momentum
Mbip � V�bip = MKr � V�Kr �1�
�where Mbip, MKr, V�bip, and V�Kr represents the masses ofbiphenyl, Kr, and the velocity in the center-of-mass frame ofbiphenyl and Kr after collision, respectively� the velocity ofthe corresponding scattered Kr atom after collisions can bedetermined from the measured scattered biphenyl velocity.The amount of energy transfer for each collision thereforecan be calculated from the conservation of energy.
Eint +1
2Mbip � V2
bip +1
2MKr � V2
Kr
= E�int +1
2Mbip � V�2
bip +1
2MKr � V�2
Kr. �2�
Eint and E�int represent the internal energy of biphenyl beforeand after collision, respectively. The internal energy of bi-phenyl before collision depends on the initial internal energyof the biphenyl and the uv photon energy. Due to the super-sonic expansion, biphenyl molecules in the molecular beambefore absorbing uv photons are rotationally and vibra-tionally cold. Therefore, the internal energy before uv exci-tation can be neglected. By combining a molecular beamwith a narrow bandwidth laser, internal energy distribution ofbiphenyl before collision, Eint, is almost equal to photon en-ergy which is quite narrow and well defined. Vbip and VKr
represent the biphenyl and Kr velocity in the center of massframe before collision, respectively. They can be obtainedfrom the velocity of each molecular beam. Energy transfer,defined as �E=E�int−Eint therefore can be obtained from theimage. The same analysis method was used for the collisionsbetween naphthalene and Kr.
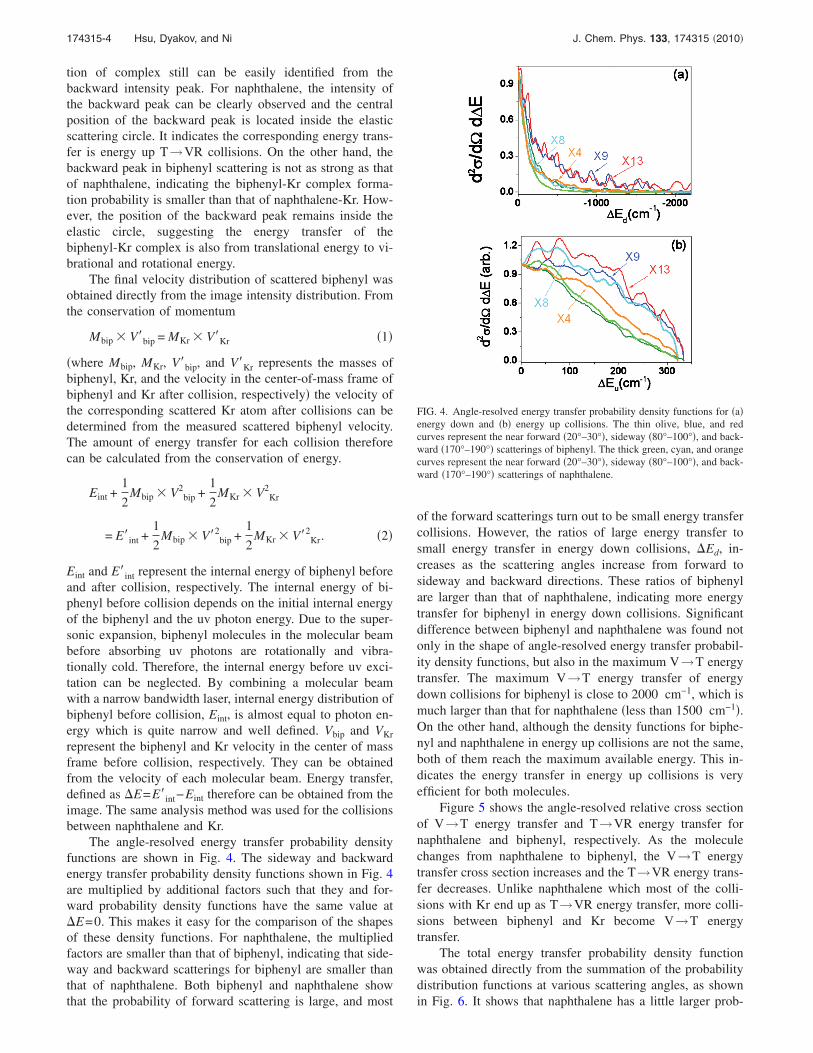
The angle-resolved energy transfer probability densityfunctions are shown in Fig. 4. The sideway and backwardenergy transfer probability density functions shown in Fig. 4are multiplied by additional factors such that they and for-ward probability density functions have the same value at�E=0. This makes it easy for the comparison of the shapesof these density functions. For naphthalene, the multipliedfactors are smaller than that of biphenyl, indicating that side-way and backward scatterings for biphenyl are smaller thanthat of naphthalene. Both biphenyl and naphthalene showthat the probability of forward scattering is large, and most
of the forward scatterings turn out to be small energy transfercollisions. However, the ratios of large energy transfer tosmall energy transfer in energy down collisions, �Ed, in-creases as the scattering angles increase from forward tosideway and backward directions. These ratios of biphenylare larger than that of naphthalene, indicating more energytransfer for biphenyl in energy down collisions. Significantdifference between biphenyl and naphthalene was found notonly in the shape of angle-resolved energy transfer probabil-ity density functions, but also in the maximum V→T energytransfer. The maximum V→T energy transfer of energydown collisions for biphenyl is close to 2000 cm−1, which ismuch larger than that for naphthalene �less than 1500 cm−1�.On the other hand, although the density functions for biphe-nyl and naphthalene in energy up collisions are not the same,both of them reach the maximum available energy. This in-dicates the energy transfer in energy up collisions is veryefficient for both molecules.
Figure 5 shows the angle-resolved relative cross sectionof V→T energy transfer and T→VR energy transfer fornaphthalene and biphenyl, respectively. As the moleculechanges from naphthalene to biphenyl, the V→T energytransfer cross section increases and the T→VR energy trans-fer decreases. Unlike naphthalene which most of the colli-sions with Kr end up as T→VR energy transfer, more colli-sions between biphenyl and Kr become V→T energytransfer.
The total energy transfer probability density functionwas obtained directly from the summation of the probabilitydistribution functions at various scattering angles, as shownin Fig. 6. It shows that naphthalene has a little larger prob-
FIG. 4. Angle-resolved energy transfer probability density functions for �a�energy down and �b� energy up collisions. The thin olive, blue, and redcurves represent the near forward �20°–30°�, sideway �80°–100°�, and back-ward �170°–190°� scatterings of biphenyl. The thick green, cyan, and orangecurves represent the near forward �20°–30°�, sideway �80°–100°�, and back-ward �170°–190°� scatterings of naphthalene.
174315-4 Hsu, Dyakov, and Ni J. Chem. Phys. 133, 174315 �2010�

ability at small T→VR energy transfer, and biphenyl has alittle larger probability at large V→T energy transfer. In Fig.6, the total cross section was normalized to be one separatelyfor each molecule so that we can compare the shapes of theenergy transfer probability density functions. In order tocompare the energy transfer efficiency between these twomolecules, the curves in Fig. 6 need to be scaled so that thearea under each curve is proportional to the relative totalcollisional cross sections. The total collisional cross section�coll is related to the ion image intensity Simage, the intensityof rare gas beam Irare, molecular beam intensity, Imole, uvabsorption cross section �266, uv laser intensity I266, and in-tersystem crossing quantum yield �IS, the ionization crosssection of molecule in the triplet state by 157 nm photons,�157, and the 157 nm laser intensity I157
Simage = Irare � Imole � �266 � I266 � �IS � �coll � �157
� I157. �3�
In addition to the measurement of the ion image intensity forthe excited molecules in collisions with rare gases, we alsomeasured the naphthalene and biphenyl ion intensities with-out rare gas atomic beam. The ion intensity measured in suchconditions is simply proportional to the number of moleculesin the triplet state, the ionization cross section at 157 nm, andthe 157 nm laser intensity
Sion = Imole � �266 � I266 � �IS � �157 � I157. �4�
The ratio of the total cross sections for naphthalene and bi-phenyl can be expressed by the following equation:
�coll�bip��coll�nap�
=
Simage�bip�Sion�bip�
Simage�nap�Sion�nap�
. �5�
This equation shows that the uv laser intensity, uv absorptioncross section, intersystem crossing quantum yield, vuv laserintensity, and vuv ionization cross section are all cancelled.We do not need to measure those parameters directly. Weonly need to measure the scattered image intensity and theion intensity without atomic beam. This largely reduces theuncertainty in the measurement. During the experimentalmeasurement, we kept the rare gas atomic beam the same.Meanwhile, we put both naphthalene and biphenyl in themolecular beam so we can measure ion image intensity Simage
and ion intensity Sion for naphthalene and biphenyl alterna-tively in a relatively short period of time in order to reducethe systematic error. The result shows that the ratio of theabsolute total collision cross sections for Kr and naphthaleneto Kr and biphenyl is 1: 1.17�0.1. In Fig. 6, one of thebiphenyl probability curves has been multiplied by 1.17 forcomparison. Although the multiplied factor is not large, itreduces the difference between naphthalene and biphenyl insmall T→VR energy transfer, but enlarges the difference inlarge V→T energy transfer.
C. Energy transfer mechanism
The mechanism of collisional energy transfer between ahighly vibrationally excited polyatomic molecule and a mon-atomic gas has been investigated for benzene-rare-gas sys-tem by carrying out both vibrational close-coupling, infinite-order sudden quantum-scattering computations, and classicaltrajectory calculation.54 It showed that only the three modeswith the smallest frequencies give vibrational relaxationcross sections of appreciable magnitude. These three modesare the out-of-plane vibrations, 16 �frequency 398 cm−1�and 11 �frequency 672 cm−1�, and in-plane vibration 6 �fre-quency 606 cm−1�. The results show that it is the multiquan-tum transitions out of modes with the lowest vibrational fre-quencies that are giving rise to the largest energy transfer,not the smaller quantum transitions out of modes of higherfrequencies. The vibrational relaxation cross sections forbenzene-rare-gas are ��16���6���11�. However, ifthe in-plane vibration 6 was given the same frequency as theout-of-plane vibration 16, the cross sections for 16 are stillmuch larger than those for 6. It suggested that low-frequency vibration modes, particularly if it is an out-of-plane motion are prominent in transferring significantamounts of energy. The other classical trajectory calculationsfor benzene-rare-gas system showed that rotations are gate-way for vibrational energy transfer. Molecules with large ini-tial rotational energy are more efficient in V→T energytransfer than those with small initial rotational energy.55
FIG. 5. The angle-resolved cross section of V→T energy transfer for naph-thalene �orange� and biphenyl �red�, and T→VR energy transfer for naph-thalene �cyan� and biphenyl �blue�, respectively.
FIG. 6. Total energy transfer probability density functions for biphenyl �red�and naphthalene �blue�. The areas under these two curves are normalized tobe one, separately. The orange curve represents the red curve multipliedby 1.17.
174315-5 Energy transfer of biphenyl J. Chem. Phys. 133, 174315 �2010�

No theoretical calculations have been performed for thecollisions between biphenyl �or naphthalene� and Kr. How-ever, the energy transfer mechanism of benzene-rare-gas sys-tems investigated from calculations may be applied to biphe-nyl �or naphthalene� and Kr due to the similar molecularstructures, i.e., collisions between an atom and a moleculewith a planar aromatic structure. Table I lists the triplet state
vibrational frequencies of biphenyl and naphthalene. Thenumber of low-frequency vibrational modes for biphenyl islarger than that of naphthalene, especially for the frequencybelow or close 100 cm−1. The motions of these low-frequency vibrational modes happen to be the wide-anglevibration. The vibrational motions of the first three low-frequency vibrations are shown in Fig. 7. The first one is therelative rotation of two aromatic rings along C–C bond axis.The second and the third vibrations are both out-of-planebending motions of two aromatic rings. The motions of thesevibrations are very similar to the rotation of aromatic rings.
In the previous studies of collisions between Kr andnaphthalene, we showed that energy transfer propertieschange significantly as the initial rotational temperature ofnaphthalene changes from �10 to 350 K.21 The changes ofenergy transfer properties due to the change of rotationaltemperature include less complex formation, larger V→Tcross section, smaller T→V cross section, and more super-collisions. These changes happen to be the same as differ-ence between highly vibrationally excited but rotationallycold naphthalene and biphenyl. We suggest that these low-frequency rotationlike vibrations are the most likely factorswhich make the difference between biphenyl and naphtha-lene. They must play an important role in the enhancementof V→T energy transfer and supercollisions. Further theo-retical calculations on this system and similar experimentalexamples will be helpful to investigate the mechanism weproposed.
1 F. A. Lindemann, Trans. Faraday Soc. 17, 598 �1922�.2 D. C. Tardy and B. S. Rabinovitch, Chem. Rev. �Washington, D.C.� 77,369 �1977� and references therein.
3 T. Donohue, M. S. Chou, and G. A. Fisk, Chem. Phys. 2, 271 �1973�.4 F. F. Crim, M. S. Chou, and G. A. Fisk, Chem. Phys. 2, 283 �1973�.5 F. F. Crim, H. B. Bente, and G. A. Fisk, J. Phys. Chem. 78, 2438 �1974�.6 F. F. Crim and G. A. Fisk, Acc. Chem. Res. 10, 73 �1977�.7 H. Hippler, J. Troe, and H. J. Wendelken, J. Chem. Phys. 78, 5351
FIG. 7. Motions of the first three low-frequency vibrations of biphenyl inthe triplet state T1.
TABLE I. Triplet state T1 vibrational frequencies �cm−1�.
Biphenyl Naphthalene
58 790 1374 168 918 318364 794 1416 169 942 3186114 819 1421 289 943 3186192 940 1430 359 952 3193337 950 1471 397 1060 3197355 956 1508 478 1064 3200393 965 1543 483 1136 3213410 980 1555 507 1992 3215414 982 1566 520 1209458 1007 1690 598 1216570 1010 3183 701 1295588 1078 3184 702 1373600 1099 3186 724 1409606 1106 3187 739 1425609 1157 3215 756 1443697 1189 3218 800 1470718 1262 3226 806 1492726 1277 3228 821 1494735 1326 3246 824 1554738 1352 3248 857 1663
174315-6 Hsu, Dyakov, and Ni J. Chem. Phys. 133, 174315 �2010�

�1983�.8 N. Nakashima and Y. Yoshihara, J. Chem. Phys. 79, 2727 �1983�.9 M. J. Rossi and J. R. Barker, J. Chem. Phys. 88, 6219 �1982�.
10 G. V. Hartland, D. Qin, and H. L. Dai, J. Chem. Phys. 100, 7832 �1994�.11 C. A. Michaels, Z. Lin, A. S. Mullin, H. C. Tapalian, and G. W. Flynn, J.
Chem. Phys. 106, 7055 �1997�.12 C. A. Michaels and G. W. Flynn, J. Chem. Phys. 106, 3558 �1997�.13 M. C. Wall and A. S. Mullin, J. Chem. Phys. 108, 9658 �1998�.14 U. Hold, T. Lenzer, K. Luther, K. Reihs, and A. C. Symonds, J. Chem.
Phys. 112, 4076 �2000�.15 S. Hassoon, I. Oref, and C. Steel, J. Chem. Phys. 89, 1743 �1988�.16 I. Oref and D. C. Tardy, Chem. Rev. �Washington, D.C.� 90, 1407
�1990�.17 J. R. Barker, L. M. Yoder, and K. D. King, J. Phys. Chem. A 105, 796
�2001�.18 C. L. Liu, H. C. Hsu, J. J. Lyu, and C. K. Ni, J. Chem. Phys. 123, 131102
�2005�.19 C. L. Liu, H. C. Hsu, J. J. Lyu, and C. K. Ni, J. Chem. Phys. 124, 054302
�2006�.20 C. L. Liu, H. C. Hsu, Y. C. Hsu, and C. K. Ni, J. Chem. Phys. 128,
124320 �2008�.21 C. L. Liu, H. C. Hsu, and C. K. Ni, J. Chem. Phys. 128, 164316 �2008�.22 H. C. Hsu, Ch. L. Liu, Y. C. Hsu, and C. K. Ni, J. Chem. Phys. 129,
044301 �2008�.23 F. Almasy and H. Laemmel, Helv. Chim. Acta 33, 2092 �1950�.24 H. Suzuki, Bull. Chem. Soc. Jpn. 32, 1340 �1959�.25 A. Nakajima, Bull. Chem. Soc. Jpn. 45, 1687 �1972�.26 G. Traverso and J. E. Brunet, Spectrosc. Lett. 13, 657 �1980�.27 N. A. Borisevich, S. M. Kazakov, E. E. Kolesnik, A. V. Kukhto, A. I.
Mit’kovets, D. V. Murtazaliev, and O. V. Khristoforov, J. Appl. Spec-trosc. 68, 447 �2001�.
28 T. G. McLaughlin and L. B. Clark, Chem. Phys. 31, 11 �1978�.29 B. Dick and G. Hohlneicher, Chem. Phys. 94, 131 �1985�.30 C. W. M. Castleton and W. Barford, J. Chem. Phys. 117, 3570 �2002�.31 J. I. Murakami, M. Ito, and K. Kaya, J. Chem. Phys. 74, 6505 �1981�.
32 H. S. Im and E. R. Bernstein, J. Chem. Phys. 88, 7337 �1988�.33 Y. Takei, T. Yamaguchi, Y. Osamura, K. Fuke, and K. Kaya, J. Phys.
Chem. 92, 577 �1988�.34 C. H. Lin, H. Fukii, T. Imasaka, and N. Ishibashi, Anal. Chem. 63, 1433
�1991�.35 P. Swiderek, M. Michaud, G. Hohlneicher, and L. Sanche, Chem. Phys.
Lett. 187, 583 �1991�.36 A. P. Marchetti and D. R. Kearns, J. Am. Chem. Soc. 89, 768 �1967�.37 E. C. Lim and Y. H. Li, J. Chem. Phys. 52, 6416 �1970�.38 A. Baca, R. Rossetti, and L. E. Brus, J. Chem. Phys. 70, 5575 �1979�.39 I. L. Karle and L. O. Brockway, J. Am. Chem. Soc. 66, 1974 �1944�.40 M. P. Johansson and J. Olsen, J. Chem. Theory Comput. 4, 1460 �2008�.41 P. J. Wagner, J. Am. Chem. Soc. 89, 2820 �1967�.42 A. Imamura and R. Hoffmann, J. Am. Chem. Soc. 90, 5379 �1968�.43 M. Rubio, M. Merchan, E. Orti, and B. O. Roos, Chem. Phys. Lett. 234,
373 �1995�.44 G. Buntinx and O. Poizat, J. Chem. Phys. 91, 2153 �1989�.45 F. Negri and M. Z. Zgierski, J. Chem. Phys. 97, 7124 �1992�.46 S. Y. Lee, Bull. Korean Chem. Soc. 19, 93 �1998�.47 N. I. Nijegorodov and W. S. Downey, J. Phys. Chem. 98, 5639 �1994�.48 N. I. Nijegorodov, W. S. Downey, and M. B. Danailov, Spectrochim.
Acta, Part A 56, 783 �2000�.49 R. Mabbs, N. Nijegorodov, and W. S. Downey, Spectrochim. Acta, Part A
59, 1329 �2003�.50 N. Nijegorodov, P. V. C. Luhanga, J. S. Nkoma, and D. P. Winkoun,
Spectrochim. Acta, Part A 64, 1 �2006�.51 X. C. Cai, M. Sakamoto, M. Hara, S. Tojo, K. Kawai, M. Endo, M.
Fujitsuka, and T. Majima, J. Phys. Chem. A 108, 9361 �2004�.52 K. Watanabe, T. Nakayama, and J. Mottl, J. Quant. Spectrosc. Radiat.
Transf. 2, 369 �1962�.53 A. G. Loudon and R. Z. Mazengo, Org. Mass Spectrom. 8, 179 �1974�.54 D. C. Clary, R. G. Gilbert, V. Bernshtein, and I. Oref, Faraday Discuss.
102, 423 �1995�.55 V. Bernshtein and I. Oref, J. Phys. Chem. 106, 7080 �1997�.
174315-7 Energy transfer of biphenyl J. Chem. Phys. 133, 174315 �2010�




















