
Magnetization reversal studies in structurally tailored cobalt nanowires

Electronic and magnetic structure of bulk cobalt: The �, �, and ε-phasesfrom density functional theory calculations
Víctor Antonio de la Peña O’Shea,1,2,a� Iberio de P. R. Moreira,3 Alberto Roldán,3,4 andFrancesc Illas3
1Thermochemical Processes Group, Instituto Madrileño de Estudios Avanzados en ENERGIA,C/Tulipán s/n 28933, Móstoles, Madrid, Spain2Departament de Química Inorgànica and Institut de Nanociència i Nanotecnologia (IN2UB),Universitat de Barcelona, 08028 Barcelona, Spain3Departament de Química Física and Institut de Química Teòrica i Computacional (IQTCUB),Universitat de Barcelona, C/Martí i Franquès 1, 08028 Barcelona, Spain4Departament de Química Física i Inorgànica, Universitat Rovira i Virgili, C/Marcel lí Domingo s/n,43007 Tarragona, Spain
�Received 21 March 2010; accepted 9 June 2010; published online 8 July 2010�
The geometric, electronic and magnetic properties of the three metallic cobalt phases: hcp���,fcc���, and epsilon��� have been theoretically studied using periodic density functional calculationswith generalized gradient approximation �GGA� and plane wave basis set. These results have beencompared with those obtained with GGA+U approach which have shown a noticeable improvementwith regard to experimental data. For instance, the cohesive energy values predicted by GGA areoverestimated by �25%, whereas GGA+U underestimate them by 14%–17%. On the other hand,magnetic moment values are underestimated in GGA while are overestimated for GGA+U approachby almost the same amount. Besides, the introduction of U parameter gives rise to an electronicredistribution in the d-band structure, which leads to variations in the magnetic properties.Moreover, a higher attention has been paid in the study of the electronic and magnetic properties ofthe �-phase that has not described previously. These studies show that this phase posses specialproperties that could lead to an unusual behavior in magnetic or catalytic applications.© 2010 American Institute of Physics. �doi:10.1063/1.3458691�
I. INTRODUCTION
Cobalt based materials have a great interest in academicand industrial fields due to their chemical, magnetic, andelectronic properties thank to which have potential applica-tions as magnetic data storage1,2 and catalysis3 among others.Metallic cobalt can crystallize in three different crystal struc-tures: Hexagonal closed-packed �hcp� ��-phase�, face-centered cubic �fcc� ��-phase� and primitive cubic phase��-phase�.1,2,4 These three phases possess similar energeticstabilities and, hence, small temperatures or pressure varia-tions give rise to changes in the crystal phase. The similarstability also renders theoretical predictions from first prin-ciples difficult either for bulk or nanoparticles. During thepast years several groups have concentrated their efforts onthe preparation and characterization of nanostructured sys-tems derived from these bulk phases, mainly due to theirinteresting magnetic and catalytic properties. These studieshave shown a strong correlation between crystal structureand magnetic properties in Co-based materials.1,2
As mentioned above, all cobalt metallic phases possessinteresting magnetic properties �in all cases the ferromag-netic structure is the most stable phase�, mainly due to theroom temperature anisotropy of the �-Co �Ref. 5� phases,�-Co,6 and �-Co,7 whose values are: 4.2�106, 2.7�106,and 1.5�106 erg /cm3, respectively. While the highest an-
isotropic magnetic coercivity of the �-Co phase makes itpreferred for magnetic recording applications, the �-Cophase is useful for applications as a soft magnetic materialdue to its symmetrical low coercivity. Although �-Co and�-Co phases can coexist at different conditions, the �-Cophase is more stable at room temperature and ambient pres-sure; while �-Co is a metastable phase formed at tempera-tures above 450 °C.8,9 Both phases are based on a close-packing of atoms although they differ in the stackingsequence of the �111� plane. The low energy required forstacking fault formation gives rise usually to the presence ofboth phases in the same sample. The �-cobalt phase pos-sesses a more complex structure, which was reported by Din-ega et al.10 The synthesis of the �-Co phase has been onlypossible by means of solution-phase chemistry processes.This is a metastable phase and its transformation to �-Co or�-Co structures can be easily achieved by annealing thesample at a suitable temperature.1,10 This �-structure is con-sidered as a soft magnetic material �as �-Co phase� and itsmagnetic properties favor the formation of ordered filmswith applications in magnetic recording. In the past fewyears, �-Co has acquired a great relevance due to the factthat it seems to be a good precursor to obtain �-Co nanopar-ticles for magnetic storage uses. The thermal-magnetic sta-bility determined by the Curie temperature �Tc� of the�-phase has not been reported due to the fact that �-Co �asa�Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 133, 024701 �2010�
0021-9606/2010/133�2�/024701/8/$30.00 © 2010 American Institute of Physics133, 024701-1

�-Co� phase changes its structure to �-Co phase beforeachieving a paramagnetic structure. The Tc for a �-Co phase,under ambient pressure, is 1388 K.11,12
Furthermore, recent studies have confirmed a strong cor-relation between Co phase and catalytic activity. In fact, pre-vious works reported that the behavior of cobalt-based sup-ported catalysts in Fischer–Tropsch synthesis depends on thecharacteristics of the metallic phase �i.e., fcc or hcp�.3,13
These results are also confirmed by preliminary theoreticalstudies14 showing that the Fischer-Tropsch Synthesis �FTS�reaction is structure sensitive.
Calculations using density functional theory �DFT�based methods with different exchange correlation potentialssuch as local spin density approximation �LSDA� and gener-alized gradient approximation �GGA� and including explic-itly spin polarization, have been previously reported and ad-dressed the study of the structural and magnetic properties ofmainly �-Co and �-Co phases.15–20 It is well known thatwhile the LSDA method provides a good description formany moderately correlated systems,17,21 it presents someshortcoming when applied to strongly correlated ones, espe-cially in the case of Fe.22 These previous studies and the factthat cobalt systems posses large correlation effects, whatmakes difficult the representation of their magnetic proper-ties. Thus, it is necessary to further explore the capability ofthe GGA method and to investigate alternative methods be-yond the standard LDA or GGA implementations of densityfunctional theory.23 Here, we examine the performance ofmethods that modify the on-site Coulomb repulsion, via adhoc inclusion of a Hubbard effective U term, which haveshown to provide quite accurate description of the electronicstructure and properties of several transition metal and rare-earth systems.24–28 In this work, we present an analysis of theenergetic and magnetic properties associated to the differentcobalt crystal phases as predicted from periodic DFT calcu-lation, using both GGA and GGA+U methods with planewave basis set and focusing mainly in properties of bulk�-Co which have not been previously discussed in the litera-ture.
II. COMPUTATIONAL DETAILS
Spin polarized periodic DFT based calculations with twodifferent exchange-correlation potentials have been carriedout for the �-Co, �-Co, and �-Co phases of bulk metalliccrystalline Co using the VASP program.29,30 The initial unitcells parameter were always constructed from the experi-mental lattice parameters, that were then fully optimized us-ing the procedure described below. For the �-Co, �-Co, and�-Co the unit cell involves 4, 2, and 20 metal atoms, respec-tively. The total energies corresponding to the optimized ge-ometries were calculated using the spin polarized version ofthe PW91 implementation of the GGA exchange correlationfunctional.31,32 To properly describe the magnetic behavior ofall of these cobalt phases an accurate treatment of the elec-tron correlation in the localized d orbital is crucial. Thus, inorder to correct the description of the Co 3d electrons wealso employed the GGA+U approach33 in its rotational in-variant form34 with an effective interaction parameter Ueff
=U-J for Co; note that both U and J represent atomic effec-tive parameters33 although the final results only depend onthe Ueff value. The choice of Ueff is obviously an issue sinceit represents an external input in the calculations; a possibil-ity is obviously to choose Ueff to fit experimental data asdone to simultaneously describe CeO2 and Ce2O3.25 Never-theless, it is important to be aware of the fact that Ueff candepend on the bulk phase studied and even in the calculatedproperty. Thus, Anisimov et al.35 used Ueff=U-J=7.8−0.92=6.88 eV although this corresponds to LaCoO3 at theLDA+U level and, hence, it is larger than the Ueff value forGGA+U.25 A slightly smaller value of Ueff=6.1 has beenused by Wdowik and Parlinski in their study of CoO.36 Avalue of Ueff=4.08 eV has been obtained by Wang et al.performing FLAPW calculations within the GGA functionalon Co-doped SnO2.37 The same authors use Ueff=2.4 eV intheir GGA and GGA+U study of Co under high pressure.38
Zhang et al. have used LDA+U to study Co-doped zinc-blende ZnO.39 These authors explored the effect of Ueff forvalues between 3 and 6 eV and concluded that values largerthan 3 eV do not lead to significant differences. Moreover,Vega et al.40 studied the influence of electron correlation ofthe temperature dependent electronic structure of ferromag-netic fcc cobalt structure using Ueff value of 3,2 eV that wasexplicitly obtained for these systems. In the present work, aneffective UCo value of 3 eV has been used which is close tothe value used for bulk Co �Refs. 38 and 40� and on therange of values above described. The valence one-electronKohn–Sham states have been expanded on a plane wave ba-sis with a cutoff of 415 eV for the kinetic energy and theeffect of the core electrons on the valence electron density,defined by the Co 3d74s2 electrons, has been taken into ac-count through the projector augmented wave �PAW�method41 as implemented by Kresse and Joubert.42 The totalenergy threshold defining self-consistency of the electrondensity was set to 10−4 eV; the convergence criterion forstructural optimization was set to less than 10−3 eV for thedifference of total energy from consecutive geometries en-suring that forces on all atoms are less than 0.3 eV/nm. AGaussian smearing technique with a 0.2 eV width has beenapplied to enhance convergence but all energies presented inthe following have been obtained by extrapolating to zerosmearing �0 K�. Integration in the reciprocal space was car-ried out using the Monkhorst–Pack43 sampling of the Bril-louin zone, after evaluation of several mesh of specialk-points and a special 20�20�20 k-point mesh were usedin other to obtain the desired accuracy in the calculated en-ergies. Charge and spin density analysis have been per-formed by atomic sphere method with a atomic radius of2.460 Å.
Before ending this section, it is worth pointing out thatthe complex magnetic properties of Co discussed in the in-troduction may require to explicitly introduce relativistic ef-fects. Although this is certainly a possibility, it is importantto be aware of the fact that the complicated relativistic many-body effects are usually included in the Kohn–Sham formal-ism via energy functionals analogous to the exchange-correlation functionals of the nonrelativistic formalism.44
While this is an appealing procedure, there is compelling
024701-2 de la Peña O’Shea et al. J. Chem. Phys. 133, 024701 �2010�
evidence that we are still far from having an accurate univer-sal exchange-correlation functional and that it is likely thatthe error introduced by this functional is larger than the oneoriginated by the neglect of relativistic effects. Consequently,these studies are based on the application of standard spinpolarized DFT and the only additional relativistic correctionsare those entering into the PAW potential to describe theeffect of the core electrons on the valence shells withoutincluding noncollinear magnetism effect even if this is pos-sible through the PAW formalism.45 Consequently, only thespin contribution to the magnetic moment has been included.For systems such as bulk Co with an almost filled d shell thisis a reasonable choice, whereas noncollinear magnetism ismore likely to occur in small magnetic clusters.45 Note alsothat in the absence of spin orbit interaction, where there is nocontribution from orbital to magnetic moment, the spin di-rections are not linked to the crystalline structure and, there-fore, the system is invariant under a general common rota-tion of all spins.
III. RESULTS AND DISCUSSION
First, a brief description of the optimized crystal struc-ture and properties for each of the three Co phases ��, �, and
�� studied as described by GGA and GGA+U methods ispresented. Not surprisingly, the GGA values for the � and �phases are in good agreement with previous works15–19 andhave been discussed recently.20 Therefore, we will omit adirect comparison with these previous studies and will focusonly on the results for the �-phase and on the new GGA+U set of data. The whole set of calculated lattice constants,stability energy per Co atom and per volume unit ��E /V�,cohesion energy �Ecoh�, total magnetic moments ��B� esti-mated from the spin density and Fermi energy �EF� are sum-marized in Table I. These quantities are useful to examine therelative stability of the Co phases although one must bear inmind that the results obtained correspond to 0 K. Tempera-ture effects could be taken into account by appropriate mo-lecular dynamics simulations. However, these have not beenconsidered in the present work.
To carry out the comparison of the optimized resultsobtained for �-Co phase the axial ratio c/a has been used.This value is the same for both GGA and GGA+U methods�1.642 � and is slightly larger than the values found in theliterature �1.622 �.46 This increase in the c /a value could be
TABLE II. Local coordination and nearest-neighbor distances for the twotype of Co atoms in �-cobalt phase.
Atom Near neighbors’ GGA GGA+U Expt.a
Type I Type I �3� 2.295 2.307 2.281Type II �3� 2.432 2.469 2.487Type II �3� 2.557 2.590 2.543Type II �3� 2.590 2.599 2.587
Type II Type I �2� 2.432 2.469 2.487Type I �2� 2.557 2.590 2.543Type I �2� 2.559 2.599 2.587Type II �4� 2.518 2.572 2.544Type II �2� 2.613 2.582 2.580
aReference 10.
A B
C
FIG. 1. Sketch view of different bulk cobalt structures: �a� hcp or �, �b� fccor �, and �c� � �including a detail of the different cobalt position�.
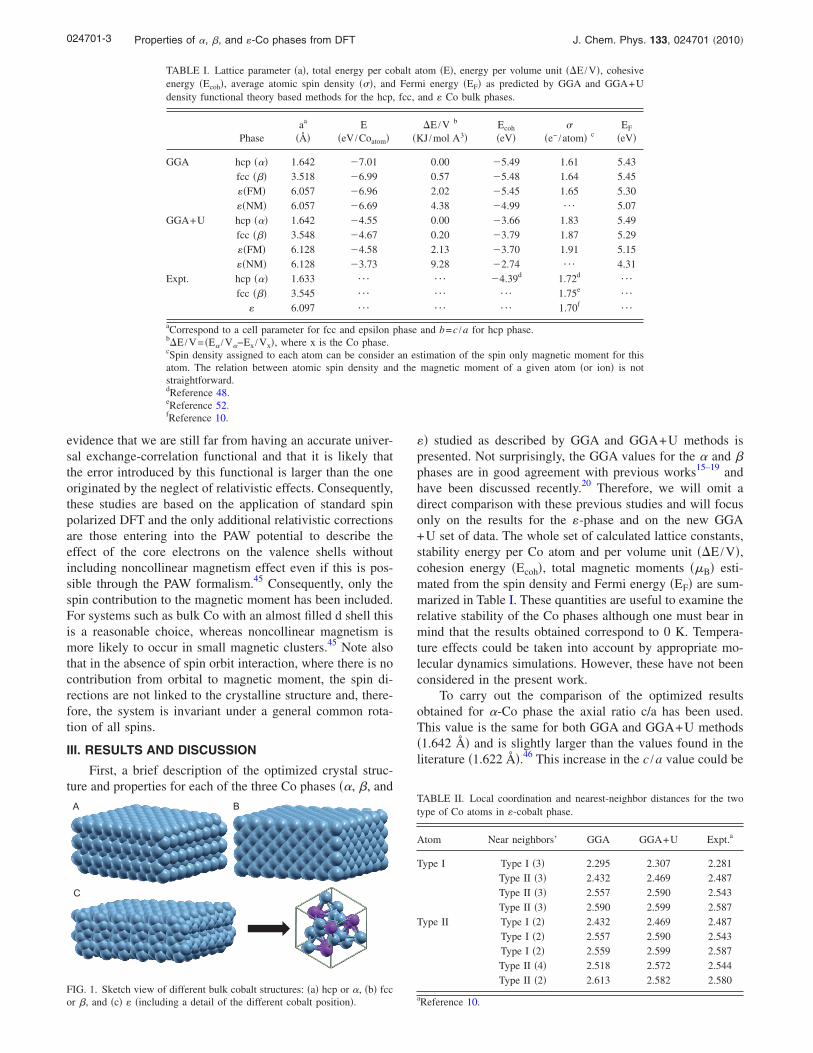
TABLE I. Lattice parameter �a�, total energy per cobalt atom �E�, energy per volume unit ��E /V�, cohesiveenergy �Ecoh�, average atomic spin density ���, and Fermi energy �EF� as predicted by GGA and GGA+Udensity functional theory based methods for the hcp, fcc, and � Co bulk phases.
Phaseaa
��E
�eV /Coatom��E /V b
�KJ /mol A3�Ecoh
�eV��
�e− /atom� cEF
�eV�
GGA hcp ��� 1.642 7.01 0.00 5.49 1.61 5.43fcc ��� 3.518 6.99 0.57 5.48 1.64 5.45��FM� 6.057 6.96 2.02 5.45 1.65 5.30��NM� 6.057 6.69 4.38 4.99 ¯ 5.07
GGA+U hcp ��� 1.642 4.55 0.00 3.66 1.83 5.49fcc ��� 3.548 4.67 0.20 3.79 1.87 5.29��FM� 6.128 4.58 2.13 3.70 1.91 5.15��NM� 6.128 3.73 9.28 2.74 ¯ 4.31
Expt. hcp ��� 1.633 ¯ ¯ 4.39d 1.72d¯
fcc ��� 3.545 ¯ ¯ ¯ 1.75e¯
� 6.097 ¯ ¯ ¯ 1.70f¯
aCorrespond to a cell parameter for fcc and epsilon phase and b=c /a for hcp phase.b�E /V= �E� /V�−Ex /Vx�, where x is the Co phase.cSpin density assigned to each atom can be consider an estimation of the spin only magnetic moment for thisatom. The relation between atomic spin density and the magnetic moment of a given atom �or ion� is notstraightforward.dReference 48.eReference 52.fReference 10.
024701-3 Properties of �, �, and �-Co phases from DFT J. Chem. Phys. 133, 024701 �2010�

due to the hcp domain that could be deformed to bettermatch the fcc staking. In an ideal hcp structure the b value is1.633 Å and coincides with an fcc structure observed alongthe �111� direction. In the case of �-Co phase GGA andGGA+U calculations predict lattice parameter values of3.518 and 3.548 Å, respectively. Both values are in agree-ment with the experimental one �3.545 Å� �Ref. 47� beingthe GGA+U value closer to experiment. In the case of �-Cophase, 6.057 Å �GGA� and 6.128 Å �GGA+U� values were
obtained for the lattice parameter, which are close to theexperimental value of 6.097 Å.46 The small differences areconsidered within the method error ��2%�. For the �-Cophase, the predicted nearest neighbors distances are 2.478 Å�GGA� and 2.487 Å �GGA+U� that are closer to the experi-mental value of 2.497 Å. Whereas for the for �-Co phase wefind 2.488 Å �GGA� and 2.509 Å �GGA+U�, being the ex-perimental value 2.506 Å. The �-Co phase is a very special
-10 -5 0 5
Densityofstates
E-EF (eV)
-10 -5 0 5
Densityofstates
E-EF (eV)
GGA
GGA+U
A
-10 -5 0 5
Densityofstates
E-EF (eV)
-10 -5 0 5
Densityofstates
E-EF (eV)
GGA
GGA+U
B
-10 -5 0 5
Densityofstates
E-EF (eV)
-10 -5 0 5
Densityofstates
E-EF (eV)
GGA
GGA+U
C
FIG. 2. Total �straight line�, Co-4s �gray pattern�, and Co-3d �dashed pattern� for Density of states calculated using GGA and GGA+U methods for � �a�, ��b�, and � �c� phases. Dashed line indicates the Fermi level. All curves are represented for spin up and down contributions
024701-4 de la Peña O’Shea et al. J. Chem. Phys. 133, 024701 �2010�

case since, compared to the other cobalt phases, exhibits dif-ferent structural properties. The �-Co, described by Dinegaet al.10 posses a cubic structure �space group P4132� with aunit cell similar to that of �-manganese.46 This structure con-tains 20 cobalt atoms, per unit cell, divided in two types:Eight atoms of type I and twelve atoms of type II �Fig. 1�c��.In Table II, the nearest neighbor’s distances for each cobaltatom type are reported.
The calculated total energy per cobalt atom shows that,for GGA calculations, the �-Co phase is more stable than the�-Co and �-Co phases. However, the GGA+U calculationsshow that the �-Co phase has a lower energy per Co atomthan �-Co and �-Co phases. However, the structural stabilityshould be expressed as energy per cell volume �Ex /Vx,where x is Co-phase� and, in both GGA and GGA+U, thecalculated �E /V �E� /V�−Ex /Vx, where x is Co-phase� val-ues show that the �-Co structure is the most stable, as ex-pected from experimental studies �Table I�.
The cohesive energy �Ecoh� provides an alternative im-portant way to evaluate the stability of the cobalt phases andthe accuracy of the computational method. The Ecoh calcu-lated values in Table I are similar to the experimentalvalue.48 In fact, for all cobalt phases the GGA calculationsfurnish values of the cohesive energy that overestimate theexperimental value by �25%, while the GGA+U methodunderestimate this property by 14%–17%. These data illus-trate that GGA and GGA+U methods, within the form ofone of the most widely used functional such as PW91 pro-vides a rather approximate description of the binding be-tween heavy-element atoms,49 a feature exhibited also byother GGA functionals.50
Next, let us discuss the main features of the electronicstructure of the different Co phases. Upon inclusion of theon-site Hubbard electron correlation correction to GGA pa-rameter �UCo�, the calculated value of the Fermi level �EF�shifts and the variation depends on the cobalt phase. For thefcc and epsilon phases EF increases by 0.16 and 0.15 eV,respectively, whereas for the hcp phase it decreases by 0.06eV. This behavior could be explained thanks to the fact thatthe inclusion of U parameter leads to an expansion of thefilled and semifilled subbands, which could also explain theenergy stabilization previously observed in these phases.
Additional information about the electronic structure ofthese Co bulk phases can be obtained from the total �spin upand down�, Co �4s�, and Co �3d� density of states �DOS�obtained for GGA and GGA+U solutions shown in Fig. 2.
Previous theoretical studies have revealed that the 3d-bandhas a complex structure divided in five nondegeneratesubbands.40 The complete or quasicomplete filled bands aremagnetically inactive, while upper bands provide the mag-netic behavior to this system. Present study shows that allthree phases exhibit related features with concomitant simi-larities in their electronic properties, although a detailedanalysis of the DOS profiles also reveals some differences inthe electronic structure of the different cobalt phases. Thus,in the GGA calculations, the density of states for the d-bandin epsilon phase is broader than in fcc and hcp structures.Besides, the electron distribution density around Fermi levelis slightly higher for epsilon phase than for fcc phase andlower for hcp. These data corroborate the magnetic valuesobtained.
On the other hand, the calculations performed using theHubbard correction lead to similar DOS profiles, although ashift in the energy of the up spin states toward more nega-tives values is observed. Obviously, the inclusion of the Uparameter leads to a variation in the 3d band structure in allcases, giving rise to a redistribution of the density of states inthe subbands, leading to an increase in the spin down densitythat corroborates the enhancement observed in the averageatomic spin density ���. These energy gaps are different foreach structures being higher for epsilon phase.
One of the main properties of these systems is their mag-netic behavior. Experimental studies have shown that all me-tallic cobalt phases have a stable structure which is stronglyferromagnetic at ambient pressure. As previously com-mented, the magnetic properties of � and � phases has beenstudied, corroborating that the hcp and fcc nonmagnetic�NM� phases are less stable than the ferromagnetic �FM�one.15,20 In the case of epsilon phase this kind of calculationshas not been previously evaluated. Our studies show that, asfound for the fcc and hcp structures, the magnetic phase ismore stable than the NM phase �Table I�. Moreover, the sta-bility and cohesion energy differences in both NM and FMare higher in the GGA+U calculation, which indicates thatthe use of the Hubbard algorithm leads to a destabilization ofthe NM phase.
On the other hand, the magnetovolume effect studies inthe �-phase show a stabilization of the NM phase with thevolume decrease. Thus, our calculations show that thesephases lose their magnetic properties into a nonmagneticstructure. Two self-consistent stable structures, with a vol-ume of 214.17 Å3 �GGA� and 204.99 Å3 �GGA+U� wereobtained. Experimental data that corroborate this behavior
TABLE III. Analysis of the atomic spin density for each atom in the �-Co structure as predicted from GGA andGGA+U calculation. The decomposition of the total spin density in atomic components is obtained by properprojection.
�Co
�e− /atom�
AtomGGA GGA+U
4s 4p 3d Tot 4s 4p 3d Tot
1 0.01 0.05 1.67 1.61 0.02 0.08 1.93 1.832 0.01 0.05 1.67 1.61 0.02 0.08 1.93 1.83
024701-5 Properties of �, �, and �-Co phases from DFT J. Chem. Phys. 133, 024701 �2010�

have not been found. However, a similar behavior, for theother Co phases, was previously described fromexperimental51 and theoretical20 studies.
The calculated average atomic spin density ��� obtainedfor each phase from GGA and GGA+U are also summarizedin Table I and are in agreement with experimental magneticmoments.52 Nevertheless, the � values obtained from theGGA method are underestimated with respect to experimen-tal results; however, when the Coulomb correction is usedthese values are overestimated. Moreover, the averageatomic spin density of the �-Co phase presents the highestvalue in both GGA and GGA+U calculation. These resultsare corroborated by the examination of spin density valuesover each atom and projected on the 4s, 4p, and 3d states�Tables III–V�, where an occupation increase in the d band isobserved when the GGA+U method is used. These resultsare in agreement with the electronic population observed in
DOS analysis which illustrates that the use of GGA+Umethodology leads to an increase of the number of statesaround of Fermi level �Fig. 2�.
The obtained spin density per atom ��Co� for the differ-ent phases and its decomposition in atomic symmetries issummarized in Tables III–V. For the hcp and fcc phases the�Co values for all atoms are identical with values of 1.61 forGGA and 1.87 for GGA+U for the fcc phase and slightlyhigher values for hcp which are of about 1.64 for GGA and1.87 for GGA+U. These values are quite close to those re-ported in previous studies using similar approaches.18,19 Inthe case of epsilon phase different �Co values are obtained infunction of the atom type. The eight atoms of type I exhibita lower �Co value while for the twelve atoms of type II theatomic spin density is larger �Table V�. Unfortunately, for the� phase comparison with previous studies is not possiblebecause they are inexistent. Note also that in the case of Co
TABLE IV. Analysis of the atomic spin density for each atom in the �-Co structure as predicted from GGA andGGA+U calculation. The decomposition of the total spin density in atomic components is obtained by properprojection.
�Co
�e− /atom�
AtomGGA GGA+U
4s 4p 3d Tot 4s 4p 3d Tot
1 0.01 0.05 1.70 1.64 0.01 0.06 1.95 1.872 0.01 0.05 1.70 1.64 0.01 0.06 1.95 1.873 0.01 0.05 1.70 1.64 0.01 0.06 1.95 1.874 0.01 0.05 1.70 1.64 0.01 0.06 1.95 1.87
TABLE V. Analysis of the atomic spin density for each atom in the �-Co structure as predicted from GGA andGGA+U calculation. The decomposition of the total spin density in atomic components is obtained by properprojection.
�Co
�e− /atom�
AtomGGA GGA+U
4s 4p 3d Tot 4s 4p 3d Tot
1 0.02 0.06 1.67 1.59 0.02 0.08 1.97 1.872 0.02 0.06 1.66 1.59 0.01 0.08 1.97 1.883 0.01 0.06 1.66 1.59 0.02 0.08 1.97 1.884 0.01 0.06 1.64 1.57 0.02 0.08 1.96 1.865 0.02 0.06 1.66 1.59 0.02 0.08 1.97 1.886 0.01 0.06 1.65 1.58 0.02 0.08 1.96 1.877 0.01 0.06 1.66 1.58 0.02 0.08 1.96 1.878 0.01 0.06 1.64 1.57 0.02 0.08 1.95 1.869 0.01 0.05 1.72 1.66 0.01 0.07 2.03 1.95
10 0.01 0.05 1.71 1.65 0.01 0.07 2.00 1.9211 0.01 0.05 1.65 1.59 0.01 0.07 2.01 1.9312 0.01 0.05 1.66 1.60 0.01 0.06 2.01 1.9413 0.01 0.05 1.76 1.70 0.01 0.06 2.03 1.9514 0.01 0.05 1.76 1.70 0.01 0.06 2.02 1.9415 0.01 0.05 1.73 1.68 0.01 0.06 2.01 1.9416 0.01 0.05 1.73 1.67 0.01 0.06 2.01 1.9417 0.01 0.05 1.84 1.78 0.01 0.06 2.02 1.9518 0.01 0.05 1.84 1.78 0.01 0.06 2.02 1.9419 0.01 0.05 1.77 1.72 0.01 0.06 2.01 1.9320 0.01 0.05 1.77 1.71 0.01 0.06 2.01 1.93
024701-6 de la Peña O’Shea et al. J. Chem. Phys. 133, 024701 �2010�

nanoparticles different types of Co atoms will be present atthe surface depending on the crystalline structure of the par-ticle. This fact can lead to different magnetic behavior as afunction of the particle shape. The different behavior of mag-netic and catalytic properties of Co nanoparticles with rela-tion to its shape and phase are actually under study.
Figure 3 shows plots of the electron localization function�ELF� for the different phases as obtained by the GGA andGGA+U methods. In the case of hcp and fcc structures, theELF shows an homogenous electron distribution. The maindifference between these two structures is that hcp phaseshows a radial electron distribution while the fcc phaseshows an evident polarization. However, in the case of �-Cophase a different ELF distribution is observed depending onthe atom type, which one can tentatively relate to the mag-netic behavior of this phase. It is also important to point outthat, for the three phases, the ELF plots in Fig. 3 correspond-ing to calculations with the GGA+U potential, show a morelocalized character than in the case of the GGA calculations,as expected from the well known trend of GGA+U to local-ize the electron density of the levels where the correction isapplied.
IV. CONCLUSIONS
The lattice parameter and structural parameters of thefcc, hcp, and � phases of bulk Co have been obtained at theGGA and GGA+U levels of DFT. For the hcp and fccphases, the calculated values are in good agreement with
experimental data and previous GGA calculations. Neverthe-less, the GGA+U values reported in the present work seemto be more accurate and closer to the experimental ones. Thecohesive energy values predicted by these density functionalmethods are also near to the experimental data. However,GGA overestimates this property by �25% and GGA+Uunderestimate it by 14%–17%.
The density of states for the different phases presentssome marked differences. In the GGA calculation, the elec-tron distribution density around Fermi level is slightly higherfor epsilon than for the fcc phase; and lower for the hcp one.On the other hand, the GGA+U calculation shows a shift inthe energy for the up spin states toward more negatives val-ues. This energy shift is higher for epsilon phase and leads toan increase in the average atomic spin density ���.
The study of the magnetic properties shows that thethree cobalt phases exhibit a stable ferromagnetic state.However, the �Co values obtained from GGA method aresmaller than the experimental values while the use of theCoulomb correction overestimates these values. Interestinglyenough, the calculated atomic spin density values for the�-Co phase are the highest. Besides, in the �-Co phase, the�Co values depend on the cobalt atom type, a feature whichfor cobalt nanoparticles can lead to different magnetic behav-ior as a function of the particle shape.
Moreover, the present results show that each cobaltphase, in particular the � one, has inherent structural andelectronic properties that could help to understand the differ-ent behavior in magnetic storage or catalytic application asH2 production or Fischer–Tropsch synthesis.
In summary, density functional calculations within theGGA method allows reaching a rather detailed description ofthe atomic and electronic structure of the �, � phases of bulkCo with experimental values and with previous theoreticalstudies. For these two phases, comparison with experimentaldata indicates that the GGA+U values are more accurate.The precise description of the hcp and fcc Co bulk phasesstrongly suggest that the present description of the � phase;which has not been previously described, is also equally ac-curate.
ACKNOWLEDGMENTS
V.A.P.O. acknowledges financial support from the MI-CINN in the Ramon y Cajal Program. A.R. thanks Univer-sitat Rovira i Virgili, for supporting his pre-doctoral research.Financial support has been provided by Spanish MICINN�Grant Nos. FIS2008-02238, CTQ2008-06549-C02-01, andENE2009-09432� and in part by Generalitat de Catalunya�Grant Nos. 2009SGR1041, 2009SGR462, and XRQTC�.Computational time has been provided by the Centre de Su-percomputació de Catalunya �CESCA� by generous grantsfrom Universitat de Barcelona and CIRIT.
1 V. F. Puntes, K. M. Krishnan, and A. P. Alivisatos, Science 291, 2115�2001�.
2 V.A. de la Peña O’shea, P. Ramírez de la Piscina, N. Homs, G. Aromí,and J. L. G. Fierro, Chem. Mater. 21, 5637 �2009�.
3 V. A. de la Peña O’Shea, J. M. Campos-Martín, and J. L. G. Fierro, Catal.Commun. 5, 635 �2004�.
4 S. Sun and C. B. Murray, J. Appl. Phys. 85, 4325 �1999�.
GGA GGA+U
Type II
Type I
��-Co
��-Co
��-Co
FIG. 3. Schematic representation of the �ELF� for the �-Co hcp; �-Co fccand �-Co epsilon phases as obtained from GGA �left� and GGA+U �right�.
024701-7 Properties of �, �, and �-Co phases from DFT J. Chem. Phys. 133, 024701 �2010�

5 P. Gambardella, S. Rusponi, M. Veronese, D. D. Dhesi, C. Grazioli, A.Dallmeyer, I. Cabria, R. Zeller, P. H. Dederichs, K. Kern, C. Carbone,and H. Brune, Science 300, 1130 �2003�.
6 H. T. Yang, C. M. Shen, Y. K. Su, T. Z. Yang, H. J. Gao, and Y. G. Wang,Appl. Phys. Lett. 82, 4729 �2003�.
7 V. F. Puntes and K. M. Krishnan, IEEE Trans. Magn. 37, 2210 �2001�.8 H. Sato, O. Kitakami, T. Sakurai, Y. Shimada, Y. Otani, and K. Fukami-chi, J. Appl. Phys. 81, 1858 �1997�.
9 O. Kitakami, H. Sato, Y. Shimada, F. Sato, and M. Tanaka, Phys. Rev. B56, 13849 �1997�.
10 D. P. Dinega and M. G. Bawendi, Angew. Chem., Int. Ed. Engl. 38, 1788�1999�.
11 T. Nishizawa and K. Ishida, Bull. Alloy Phase Diagrams 4, 387 �1983�.12 J. B. Staunton and B. L. Gyorffy, Phys. Rev. Lett. 69, 371 �1992�.13 V. A. de la Peña O’Shea, N. Homs, J. L. G. Fierro, and P. Ramírez de la
Piscina, Catal. Today 114, 422 �2006�.14 V.A. de la Peña O’Shea, M.C. Álvarez-Galván J. M. Campos Martín, S.
Gonzalez F. Illas, and J. L. G. Fierro, International Symposium on Ca-talysis for Clean Energy and Sustainable Chemistry, Madrid, 2008.
15 E. G. Moroni, G. Kresse, and J. Hafner, Phys. Rev. B 56, 15629 �1997�.16 F. Cleri and V. Rosato, Phys. Rev. B 48, 22 �1993�.17 T. C. Leung, C. T. Chan, and B. N. Harnon, Phys. Rev. B 44, 2923
�1991�.18 M. Körling and J. Häglund, Phys. Rev. B 45, 13293 �1992�.19 J.-H. Cho and M. Scheffler, Phys. Rev. B 53, 10685 �1996�.20 S. F. Matar, A. Houari, and M. A. Belkhir, Phys. Rev. B 75, 245109
�2007�.21 R. O. Jones and O. Gunnarsson, Rev. Mod. Phys. 61, 689 �1989�.22 C. S. Wang, B. M. Klein, and H. Krakauer, Phys. Rev. Lett. 54, 1852
�1985�.23 A. Grechnev, I. Di Marco, M. I. Katsnelson, A. I. Lichtenstein, J. Wills,
and O. Eriksson, Phys. Rev. B 76, 035107 �2007�.24 L. Wang, T. Maxisch, and G. Ceder, Phys. Rev. B 73, 195107 �2006�.25 C. Loschen, J. Carrasco, K. M. Neyman, and F. Illas, Phys. Rev. B 75,
035115 �2007�.26 H. J. Kulik, M. Cococcioni, D. A. Scherlis, and N. Marzari, Phys. Rev.
Lett. 97, 103001 �2006�.27 P. Rivero, C. Loschen, I. P. R. Moreira, and F. Illas, J. Comput. Chem.
30, 2316 �2009�.28 C. Loschen, A. Migani, S. T. Bromley, F. Illas, and K. M. Neyman, Phys.
Chem. Chem. Phys. 10, 5730 �2008�.29 G. Kresse and J. Furthmüller, Comput. Mater. Sci. 6, 15 �1996�.30 G. Kresse and J. Hafner, Phys. Rev. B 47, 558 �1993�.31 J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson,
D. J. Singh, and C. Fiolhais, Phys. Rev. B 46, 6671 �1992�.32 J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson,
D. J. Singh, and C. Fiolhais, Phys. Rev. B 48, 4978 �1993�.33 V. I. Anisimov, F. Aryasetiawan, and A. I. Lichtenstein, J. Phys.: Con-
dens. Matter 9, 767 �1997�.34 S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys, and A. P.
Sutto, Phys. Rev. B 57, 1505 �1998�.35 M. A. Korotin, S. Yu. Ezhov, I. V. Solovyev, V. I. Anisimov, D. I. Khom-
skii, and G. A. Sawatzky, Phys. Rev. B 54, 5309 �1996�.36 U. D. Wdowik and K. Parlinski, J. Phys.: Condens. Matter 21, 125601
�2009�.37 H. Wang, Y. Yan, Y. Sh. Mohammed, X. Du, K. Li, and H. Jin, J. Magn.
Magn. Mater. 321, 3114 �2009�.38 Y. Shoaib Mohammed, Y. Yan, H. Wang, K. Li, and X. Du, Magn. Magn.
Mater. 322, 653 �2010�.39 J. Zhang, K. L. Yao, Z. L. Liu, and G. Y. Gao, Physica B 405, 1447
�2010�.40 A. Vega and W. Nolting, Phys. Status Solidi B 193, 177 �1996�.41 P. E. Blöchl, Phys. Rev. B 50, 17953 �1994�.42 G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 �1999�.43 H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188 �1976�.44 A. H. MacDonaldt and S. H. Vosko, J. Phys. C 12, 2977 �1979�.45 D. Hobbs, G. Kresse, and J. Hafner, Phys. Rev. B 62, 11556 �2000�.46 See x-ray powder JCPDS diffraction files 5.0727.47 See x-ray powder JCPDS diffraction files 15.806.48 C. Kittel, Introduction to Solid State Physics �Wiley, New York, 2005�.49 A. Roldán, F. Viñes, F. Illas, J. M. Ricart, and K. M. Neyman, Theor.
Chem. Acc. 120, 565 �2008�.50 K. M. Neyman, G. N. Vayssilov, and N. Rösch, J. Organomet. Chem.
689, 4384 �2004�.51 C. S. Yoo, H. Cynn, P. Soderling, and V. Iota, Phys. Rev. Lett. 84, 4132
�2000�.52 Landolt-Börnstein Numerical Data and Functional Relationships in Sci-
ence and Technology, edited by K. H. Hellwege and O. Madelung�Springer, New York, 1986�, Vol. 19/a.
024701-8 de la Peña O’Shea et al. J. Chem. Phys. 133, 024701 �2010�
Copyright © 2022 FDOKUMEN




















