
Cryopreservation of Shoot Tips of Tetraploid Potato ( Solanum tuberosumL.) Clones by Vitrification

Effects of Cryopreservation on the Transcriptome of HumanEmbryonic Stem Cells After Thawing and Culturing
Vilas Wagh & Kesavan Meganathan & Smita Jagtap & John Antonydas Gaspar &
Johannes Winkler & Dimitry Spitkovsky & Jürgen Hescheler & Agapios Sachinidis
Published online: 29 January 2011# Springer Science+Business Media, LLC 2011
Abstract Human embryonic stem cells (hESCs) can bepropagated indefinitely in vitro in an undifferentiatedpluripotent state, can differentiate into derivatives of all threegerm layers and are of considerable interest for applicationsin regenerative medicine. Clinical application of hESCs,however, requires reliable protocols for cryopreservation.Current protocols for cryopreservation of hESCs suffer fromlow recovery rates of hESCs and loss of pluripotency afterthawing. We therefore studied the effects of cryopreservationon the viability, proliferation potential, and the pluripotencystatus of hESCs by combining cellular readouts and tran-scriptomics. We identified biological processes and pathwaysaffected by cryopreservation in order to understand thelimited survival rate of hESCs by comparing transcriptomesof hESCs at different time points after thawing with cells thatdid not undergo cryopreservation. While the transcriptomesof cells post thawing were very similar to those of control non-frozen hESCs for the early time points, we observed increasedexpression of genes involved in apoptosis, embryonicmorphogenesis, ossification, tissue morphogenesis, regenera-tion, vasculature development and cell death at later timepoints. Our data suggest that inhibition of anoikis apoptosisand the stress-induced differentiation pathways are promisingtargets for improving the survival rate and maintainingpluripotency of hESCs after cryopreservation.
Keywords Human embryonic stem cells .
Cryopreservation . Transcriptome . Gene ontology .
Apoptosis . Differentiation . DNA microarrays
Introduction
Human embryonic stem cells (hESCs) are pluripotent cellsisolated from the inner cell mass of pre-implantationembryos [1]. These cells have unlimited proliferativecapacity and can differentiate into all somatic cell types[2–4]. Cryopreservation of hESCs commonly involves useof cryoprotectants such as 10% dimethylsulfoxide (DMSO).These protocols allow large cell numbers to be frozen, butthe survival rate of cells is frequently as low as 5%. Incontrast, vitrification (i.e. rapid freezing to prevent formationof ice crystals) protocols enable high viability (~80%), butare limited to small numbers of cells [5, 6]. Despite itsshortcomings, slow freezing remains the preferred methodfor freezing of bulk amounts of cells.
All current protocols rely on cryopreserving smallclumps of cells to improve survival as hESCs undergomassive cell death after complete dissociation into singlecells due to apoptosis or anoikis (detachment-inducedapoptosis) [7]. Recent reports showed that the use of aselective Rho-associated kinase inhibitor (ROCKi, Y27632[8]) during hESC passaging markedly improves thesurvival of dissociated cells [9–11].
The hESCs are cryopreserved in the presence of DMSOfollowed by slow freezing and storage in liquid nitrogen orthe vapour phase of liquid nitrogen. Most cryopreservationprotocols use DMSO at concentrations from 5 to 20% [12].DMSO not only maintains an osmotic balance between theintra- and extracellular environment [13] but also inhibitsintracellular ice formation during cryopreservation [14].
Electronic supplementary material The online version of this article(doi:10.1007/s12015-011-9230-1) contains supplementary material,which is available to authorized users.
V. Wagh :K. Meganathan : S. Jagtap : J. A. Gaspar : J. Winkler :D. Spitkovsky : J. Hescheler :A. Sachinidis (*)Center of Physiology and Pathophysiology,Institute of Neurophysiology, University of Cologne,Robert-Koch-Str. 39,50931 Cologne, Germanye-mail: [email protected]
Stem Cell Rev and Rep (2011) 7:506–517DOI 10.1007/s12015-011-9230-1
Thawing is rapid, and such methods are very effective for thecryopreservation of murine ESCs as well as primary culturesand lines derived from tumors. When hESCs are cryopre-served using this approach, the viability of recovered cells isextremely low and many of the surviving cells losepluripotency and begin to differentiate [5].
In general, the major steps used in cryopreservation ofmost cell types can be summarized as follows: 1) Addition ofcryoprotective agent to cells prior to cooling, 2) introductionof ice crystal induction in cell suspension at or a few degreesbelow the freezing point of the cell suspension, and eventuallycooling towards a low temperature for the storage, 3) thawingby warming the cell suspension and 4) removal of cryopro-tective agent from cell suspension [15, 16].
Cryoinjury can be due to one or a combination of thefollowing: 1) cytotoxicity of cryoprotective agents [17,18], 2) Osmotic injury caused due to excursion ofcryoprotective agent upon freeze-thawing [19, 20] 3) Intra-cellular ice formation in the cooling process [21–23] 4) re-crystallization of the intracellular ice during the warmingprocess [24–26]. In order to improve the cryopreservationmethodologies for hESCs (and also for iPSCs) it is essential tounderstand the biological processes and pathways affected byfreezing and thawing of hESCs. Therefore we applied thetraditional WiCell cryopreservation protocol and performeddetailed microarray studies on cells post thawing to allowidentification of prominent biological processes and pathwaysaffecting the survival rate of hESCs.
Materials and Methods
hESC Culturing and Freeze-Thawing
H9 hESCs (WiCell, Madison, WI, USA) were culturedand passaged with mechanical dissociation on irradiatedmouse embryonic fibroblasts (MEF) in H9 hESCs growthmedium consisting of- knockout (KO)-DMEM-F12, 20%
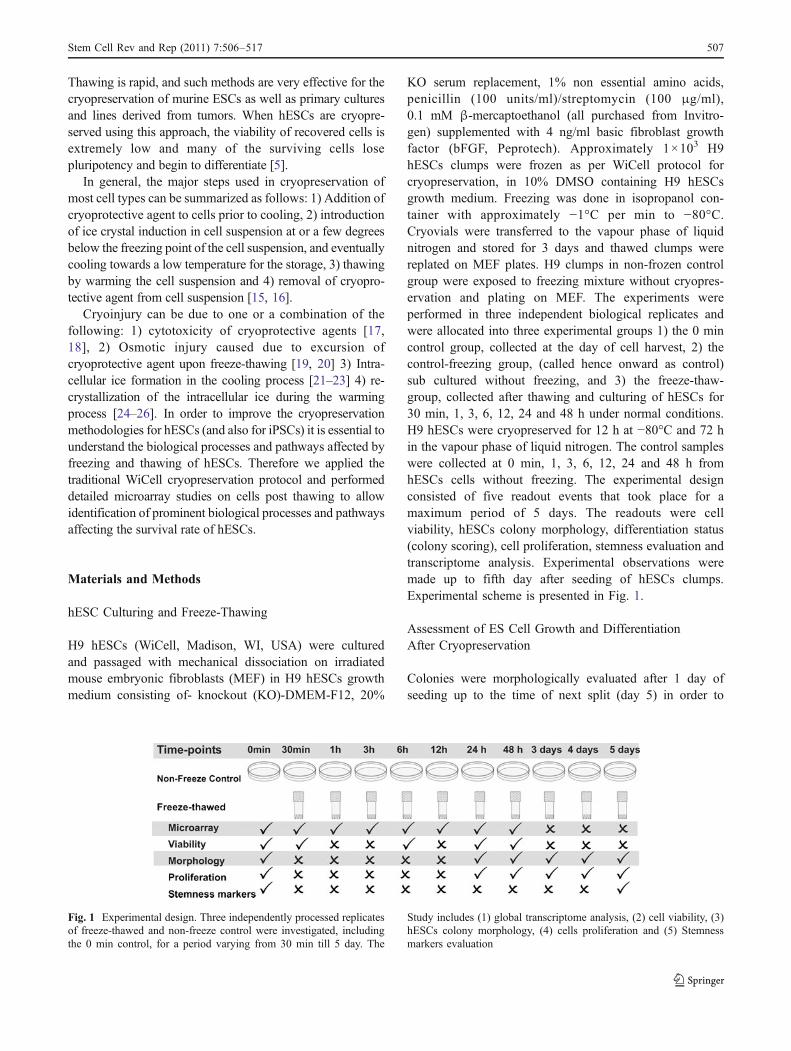
KO serum replacement, 1% non essential amino acids,penicillin (100 units/ml)/streptomycin (100 μg/ml),0.1 mM β-mercaptoethanol (all purchased from Invitro-gen) supplemented with 4 ng/ml basic fibroblast growthfactor (bFGF, Peprotech). Approximately 1×103 H9hESCs clumps were frozen as per WiCell protocol forcryopreservation, in 10% DMSO containing H9 hESCsgrowth medium. Freezing was done in isopropanol con-tainer with approximately −1°C per min to −80°C.Cryovials were transferred to the vapour phase of liquidnitrogen and stored for 3 days and thawed clumps werereplated on MEF plates. H9 clumps in non-frozen controlgroup were exposed to freezing mixture without cryopres-ervation and plating on MEF. The experiments wereperformed in three independent biological replicates andwere allocated into three experimental groups 1) the 0 mincontrol group, collected at the day of cell harvest, 2) thecontrol-freezing group, (called hence onward as control)sub cultured without freezing, and 3) the freeze-thaw-group, collected after thawing and culturing of hESCs for30 min, 1, 3, 6, 12, 24 and 48 h under normal conditions.H9 hESCs were cryopreserved for 12 h at −80°C and 72 hin the vapour phase of liquid nitrogen. The control sampleswere collected at 0 min, 1, 3, 6, 12, 24 and 48 h fromhESCs cells without freezing. The experimental designconsisted of five readout events that took place for amaximum period of 5 days. The readouts were cellviability, hESCs colony morphology, differentiation status(colony scoring), cell proliferation, stemness evaluation andtranscriptome analysis. Experimental observations weremade up to fifth day after seeding of hESCs clumps.Experimental scheme is presented in Fig. 1.
Assessment of ES Cell Growth and DifferentiationAfter Cryopreservation
Colonies were morphologically evaluated after 1 day ofseeding up to the time of next split (day 5) in order to
Fig. 1 Experimental design. Three independently processed replicatesof freeze-thawed and non-freeze control were investigated, includingthe 0 min control, for a period varying from 30 min till 5 day. The
Study includes (1) global transcriptome analysis, (2) cell viability, (3)hESCs colony morphology, (4) cells proliferation and (5) Stemnessmarkers evaluation
Stem Cell Rev and Rep (2011) 7:506–517 507

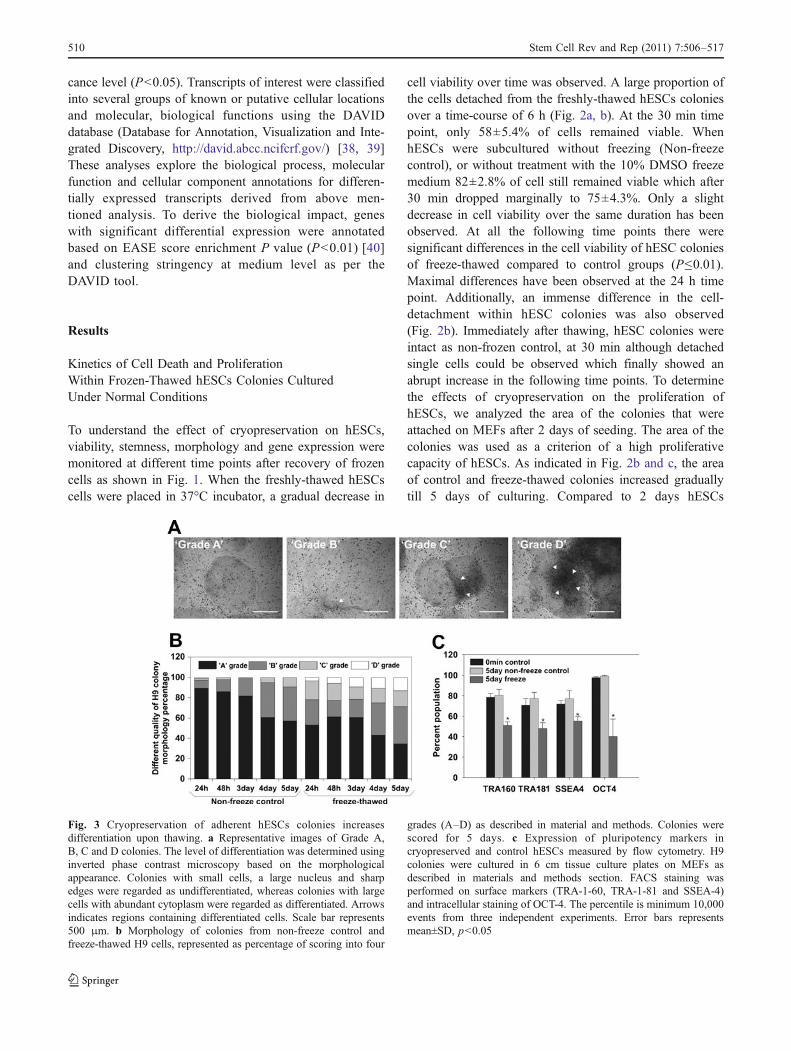
determine the effect of cryopreservation on differentiation.The colonies were assessed by using 2.5× magnification ofan inverted phase contrast microscope (Zeiss Axiovert 25Oberkochen, Germany). The quality of the colonies wasgraded as follows and as described previously [27]: 1)Grade ‘A’, excellent colonies with regular morphology andwell define edges and densely packed cells. The colonieswere tight plus thick multilayer and showed no differenti-ation. 2) Grade ‘B’, good, colonies with a single differen-tiated event. 3) Grade ‘C’, fair, colonies having less than20% differentiated cells and 4) Grade ‘D’, poor, coloniespresenting greater than 20% differentiated morphology andirregular boundaries (Fig. 3a). Both control and freeze-thawed hESCs were revived on irradiated MEFs with theabove mentioned culture medium. Minimum 30 colonieswere pictured captured (DFW-X700, Sony Corporation,Tokyo, Japan) and scored for 1–5 days in all theexperimental sets. The score was defined by three indepen-dent operators in to 4 different phenotypes as mentionedabove. The colonies were monitored for 5 days microscop-ically, and thereafter growing colonies were considered tobe stable with normal growth and expansion.
Cell Viability Using Fluorescein Diacetate/EthidiumBromide (FDA/EB) Staining
Cell viability was determined with FDA/EB staining asdescribed previously [28, 29] with minor modifications. AFDA stock solution was prepared in acetone (5 mg/ml, w/v)and EB stock solution was prepared in water (0.5 mg/ml, w/v). From the stock solution 8 μl FDA and 50 μl EB wereadded to 5 ml of basal media to make 2X staining solution.The stain solution was added to cell suspension fromcontrol and freeze-thawed (0, 30 min, 6, 24, 48 h and5 days) in 1:1 ratio and measured using an invertedfluorescence microscope (Axiocam, Zeiss Axiovert-200,Germany). Live cells hydrolyze FDA and therefore appeargreen whereas dead cells absorb the ethidium bromide andappear red. The percentage of surviving cells was countedas described previously [28] as the ratio between livehESCs after thawing and total number of cells as analyzedusing open-source image-processing toolkit (ImageJ, http://rsbweb.nih.gov/ij/index.html) [30] applying RGB analysisas described previously [31].
Estimation of Area of Colonies and Proliferation Capacity
The proliferation and colony enumeration was performedusing ImageJ-based analysis. High-resolution images fromcontrol and freeze-thawed (1–5 days) hESCs were trainedon convolution based border-detection methods withaccepted quality for images from all time points (Fig. 1).The images were convoluted to high-contrast edge-detected
version of the original image. The contours of colony wereanalyzed with built-in ImageJ function from pre-setdimension and array of data is fed into a spreadsheet. Inorder to enumerate colony numbers, possible clustersformed from H9 colonies were also determined by theAnalyze Particle function, which also provides options forstudying the circularity and size of colonies.
Flow Cytometry
The pluripotency-related markers of control (0 min and5 day) and freeze-thawed (5 day) hESCs were analyzed byflow cytometry using cell surface markers Tra-1-60, Tra-1-81, SSEA-4 and the nuclear marker OCT-4 (also known asPOU5F1). All primary antibodies were PE-conjugatedTRA-1-60 and TRA-1-81 were mouse IgM and SSEA-4were mouse IgG3 (all from Santa Cruz, CA, USA). Oct-4were mouse IgG2a (R&D Systems GmbH, Germany).Briefly, hESC colonies were dissociated into single cellswith 0.25% trypsin-EDTA and washed once with DPBS.Approximately 3×105 cells were stained for detecting thesurface antigen (TRA-1-60, TRA-1-81 and SSEA-4) usinga specific antibody in 50 μl. All staining and washing wasperformed in 0.1% BSA-DPBS solution. Internal stainingwas performed using fixation-permeablization kits (Cytofix/Cytoperm, BD Biosciences, Heidelberg, Germany) as permanufacturers established protocol. As negative controls, thecells were stained with IgG2a/IgG3/IgM isotype-matchedcontrol antibody. Flow cytometric analysis was performedusing BD-FACScan flow cytometer (BD, Biosciences) andacquired data from minimum 10,000 events were analyzedusing BD-CellQuest program.
Microarray Gene Expression Studies
Total RNA was extracted from various time points ofcontrol and freeze-thawed hESCs. The RNA was extractedusing trizol/chloroform and purified with RNeasy minicolumns as recommended by the manufacturer’s instruction(Qiagen, Hilden, Germany). All reagents and instrumenta-tion pertaining to oligonucleotide microarrays were pro-cured from Affymetrix (Affymetrix, Santa Clara, CA, USA,http://www.affymetrix.com). Total RNA (100 ng) was usedfor amplification and in-vitro transcription using theGenechip 3′ IVT Express Kit as per the manufacturer’sinstructions (Affymetrix). The amplified RNA was purifiedwith magnetic beads and 15 μg Biotin-aRNA was frag-mented with fragmentation reagent. 12.5 μg fragmentedaRNAwas hybridized to Affymetrix Human Genome U133plus 2.0 arrays along with hybridization cocktail solutionand then placed in Genechip Hybridization Oven-645(Affymetrix) rotating at 60 rpm at 45°C for 16 h. Afterincubation arrays were washed on Genechip Fluidics
508 Stem Cell Rev and Rep (2011) 7:506–517

Station-450 (Affymetrix) and stained with Affymetrix HWSkit as per manufacturer’s protocols. The chips were scannedwith Affymetrix Gene-Chip Scanner-3000-7G and thequality control matrices were confirmed with AffymetrixGCOS software following the manufacturer’s guidelines.
Identification of Differentially Expressed Genes
Background correction, summarization and normalizationwere done with Robust Multi-array Analysis. The rawdataset was normalized with Quantile normalization [32]method executable with R (Affy)-package [33] carried outat probe feature level. Probe sets which were detected to bepresent were selected and absent were eliminated. MAS5Expression Summary [34] was used to detect present calls.Only 32323 probe sets out of 54613 received present callsas defined by the detection p-value of ≤0.05. Thedifferentially expressed genes were described by a linearmodel implementing R -LIMMA packages (Linear Modelsfor microarray data, [35]). Data sets of differentiallyregulated genes based on cut-off values of 5% error rate(P<0.05, determined by both F-statistics and Moderated t-statistics with Benjamini and Hochberg Multiple Testing
Correction) were filtered out fixing 0 min sample ascommon reference and samples at the other time points asexperimental samples of both frozen and non-frozen takinginto consideration ‘the time’ factor. Beside the abovementioned conditions, fold change value with the thresholdvalue ≥(plus minus) 2.0 were used to filter the significantlyexpressed transcripts. Hierarchical cluster analysis wasperformed with ‘average linkage’ an agglomerative methodusing Cluster tool from Eisen lab [36]. Distances of thesamples were calculated using Spearman Rank correlation.Principal component (PC) analysis was performed using theStats package in R with procomp [6] function. The ‘x’attribute of the procomp object was used to generate 2dimensional scatter plots. The first PC axis accounted for28.6% of the variance in the data set of variable transcriptsand the second accounted for 12.9% [37].
Clustering Analysis of the Differential Expressed Genesand Functional Gene Ontology (GO) Classification
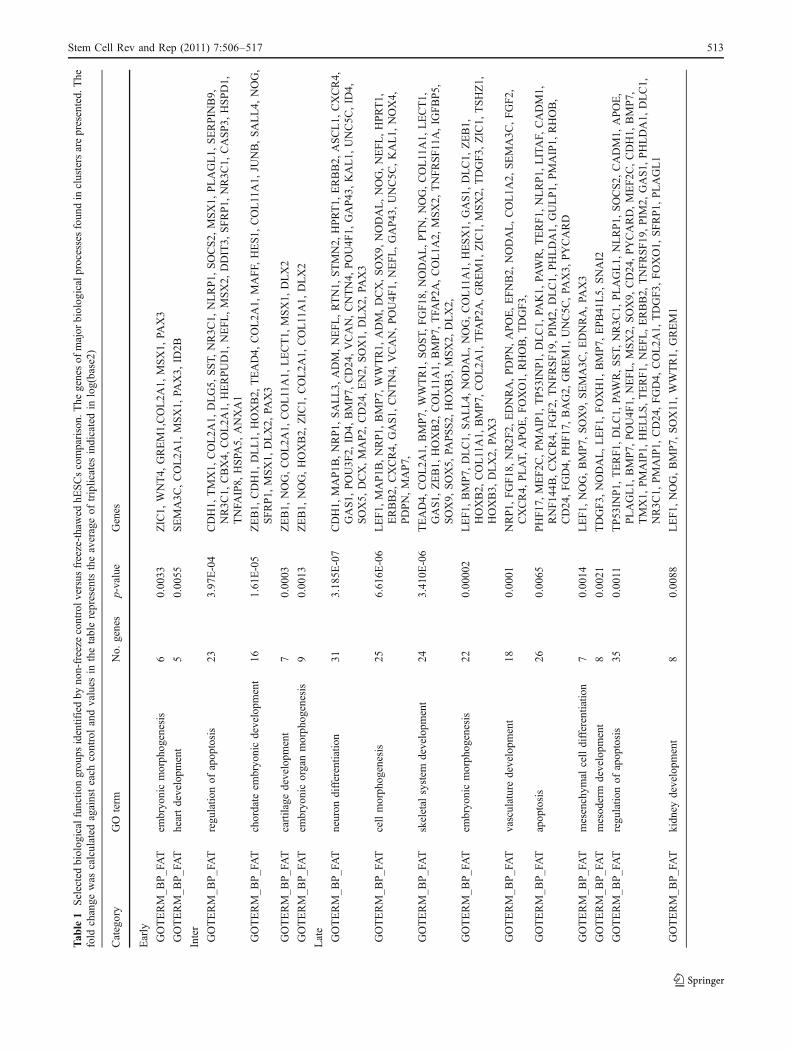
We selected only genes whose expression was altered by atleast 2-folds in response to cryopreservation, verified bystatistical analysis using ‘R’ package at a 0.05% signifi-
Fig. 2 Human ES cells are sensitive to freeze-thaw stress, leading tolow recovery. Few hESC colonies cryopreserved in clumps attachedand grew when replated on MEF plates. Approximately 1×103 H9clumps were frozen as per WiCell protocol for cryopreservation, in10% DMSO with H9 growth medium. a Percentage viability after30 min, 1, 6, 12 and 24 h of growth, and the number of viable colonieson the plate was estimated using FDA/EB staining. b Representative
fluorescence microscopy images presenting FDA/EB staining onfreeze-thawed and control H9 clumps after plating. The live cellsappeared green (FDA) and dead cells appeared with red (EB)fluorescence. Scale bar measures 100 μm. c Colony areas weremeasured every day starting from day 2. Non-frozen hESC coloniesgrew significantly faster than the freeze-thawed cells. Error barsrepresent mean±SD, n=3, *p<0.05.
Stem Cell Rev and Rep (2011) 7:506–517 509
cance level (P<0.05). Transcripts of interest were classifiedinto several groups of known or putative cellular locationsand molecular, biological functions using the DAVIDdatabase (Database for Annotation, Visualization and Inte-grated Discovery, http://david.abcc.ncifcrf.gov/) [38, 39]These analyses explore the biological process, molecularfunction and cellular component annotations for differen-tially expressed transcripts derived from above men-tioned analysis. To derive the biological impact, geneswith significant differential expression were annotatedbased on EASE score enrichment P value (P<0.01) [40]and clustering stringency at medium level as per theDAVID tool.
Results
Kinetics of Cell Death and ProliferationWithin Frozen-Thawed hESCs Colonies CulturedUnder Normal Conditions
To understand the effect of cryopreservation on hESCs,viability, stemness, morphology and gene expression weremonitored at different time points after recovery of frozencells as shown in Fig. 1. When the freshly-thawed hESCscells were placed in 37°C incubator, a gradual decrease in
cell viability over time was observed. A large proportion ofthe cells detached from the freshly-thawed hESCs coloniesover a time-course of 6 h (Fig. 2a, b). At the 30 min timepoint, only 58±5.4% of cells remained viable. WhenhESCs were subcultured without freezing (Non-freezecontrol), or without treatment with the 10% DMSO freezemedium 82±2.8% of cell still remained viable which after30 min dropped marginally to 75±4.3%. Only a slightdecrease in cell viability over the same duration has beenobserved. At all the following time points there weresignificant differences in the cell viability of hESC coloniesof freeze-thawed compared to control groups (P≤0.01).Maximal differences have been observed at the 24 h timepoint. Additionally, an immense difference in the cell-detachment within hESC colonies was also observed(Fig. 2b). Immediately after thawing, hESC colonies wereintact as non-frozen control, at 30 min although detachedsingle cells could be observed which finally showed anabrupt increase in the following time points. To determinethe effects of cryopreservation on the proliferation ofhESCs, we analyzed the area of the colonies that wereattached on MEFs after 2 days of seeding. The area of thecolonies was used as a criterion of a high proliferativecapacity of hESCs. As indicated in Fig. 2b and c, the areaof control and freeze-thawed colonies increased graduallytill 5 days of culturing. Compared to 2 days hESCs
Fig. 3 Cryopreservation of adherent hESCs colonies increasesdifferentiation upon thawing. a Representative images of Grade A,B, C and D colonies. The level of differentiation was determined usinginverted phase contrast microscopy based on the morphologicalappearance. Colonies with small cells, a large nucleus and sharpedges were regarded as undifferentiated, whereas colonies with largecells with abundant cytoplasm were regarded as differentiated. Arrowsindicates regions containing differentiated cells. Scale bar represents500 μm. b Morphology of colonies from non-freeze control andfreeze-thawed H9 cells, represented as percentage of scoring into four
grades (A–D) as described in material and methods. Colonies werescored for 5 days. c Expression of pluripotency markers incryopreserved and control hESCs measured by flow cytometry. H9colonies were cultured in 6 cm tissue culture plates on MEFs asdescribed in materials and methods section. FACS staining wasperformed on surface markers (TRA-1-60, TRA-1-81 and SSEA-4)and intracellular staining of OCT-4. The percentile is minimum 10,000events from three independent experiments. Error bars representsmean±SD, p<0.05
510 Stem Cell Rev and Rep (2011) 7:506–517
colonies, approximately a 15 times increase in the area ofthe colonies was observed after 5 days of culturing. Asindicated in Fig. 2c, after 5 days of cultivation on MEFs,the colony areas of the control cell were almost 4 timeshigher compared to those of freeze-thawed. In addition,compared to the 5 days frozen-thawed hESCs colonies, the5 days control colonies had more clear borders consistingof more compact cells.
Colony Morphology of the Cryopreserved hESCAfter Thawing and Culturing and Expressionof Embryonic Stem Cell Markers
Morphology of the colonies from control and frozen-thawed colonies were observed and assessed for theirdifferentiation levels. The percentage of grade -A, -B, -Cand -D morphologies (Fig. 3a) were analyzed from 1 to5 days post thawing (Fig. 3a, 5 days old). Control 1-day oldcolonies shows a highest percent of ‘grade A’ colonies andsubsequently the ‘grade A’ colonies go on decreasing (to60%) and a more percent of ‘grade B’ colonies areobserved towards 5 day (30%). The ‘Grade C’ was closeto 10% while ‘grade D’ colonies were less than 1% after5 days of hESCs culturing (Fig. 3b). On the other hand,frozen-thawed hESC colonies show around 60% of ‘grade A’colonies after 1 day which drastically decreases to about35% after 5 days of cultivation. Similarly, ‘grade B’ alsoshows an increase in the percentage towards day 5 ofcell culturing (35%). Most prominently, the ‘grade C andD’ colonies were found on day 5 (16% and 13%,respectively) of cultivation indicating differentiation ofhESCs (Fig. 3b).
The expression of stemness markers was evaluated for 3different time points: at 0 min, 5 day control and 5 dayfrozen-thawed are shown in Fig. 3c. The undifferentiatedstate of hESCs was determined by pluripotency markerssuch as SSEA-4, TRA-1-60, TRA-1-81 and OCT-4 usingflow cytometry. The 0 min time point hESCs showed >95%
of the cells expressing the nuclear marker OCT-4 and >70%hESCs expressing the surface markers SSEA-4, TRA-1-60and TRA-1-81 (Fig. 3c). More than 95% and 75% of5-days old hESCs express OCT-4 and surface markers,
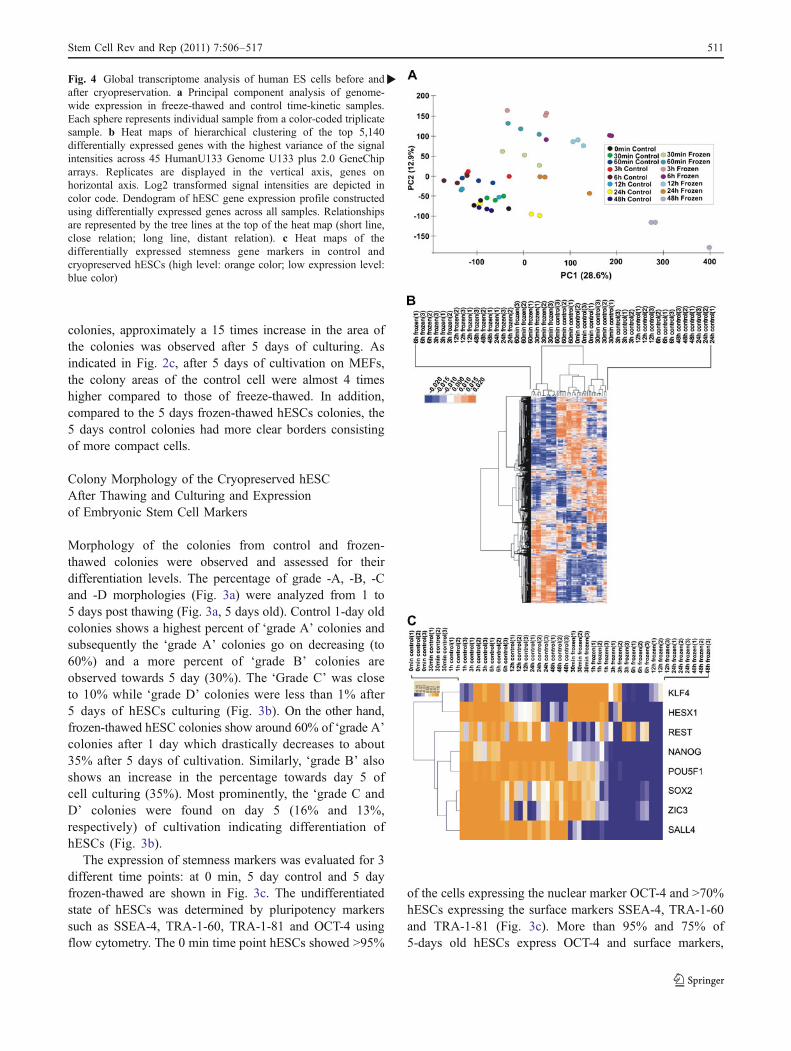
�Fig. 4 Global transcriptome analysis of human ES cells before andafter cryopreservation. a Principal component analysis of genome-wide expression in freeze-thawed and control time-kinetic samples.Each sphere represents individual sample from a color-coded triplicatesample. b Heat maps of hierarchical clustering of the top 5,140differentially expressed genes with the highest variance of the signalintensities across 45 HumanU133 Genome U133 plus 2.0 GeneChiparrays. Replicates are displayed in the vertical axis, genes onhorizontal axis. Log2 transformed signal intensities are depicted incolor code. Dendogram of hESC gene expression profile constructedusing differentially expressed genes across all samples. Relationshipsare represented by the tree lines at the top of the heat map (short line,close relation; long line, distant relation). c Heat maps of thedifferentially expressed stemness gene markers in control andcryopreserved hESCs (high level: orange color; low expression level:blue color)
Stem Cell Rev and Rep (2011) 7:506–517 511

respectively (Fig. 3c). On day 5 of culturing, approximately50% of the frozen-thawed hESCs showed expression of allpluripotency markers implying significant differentiationevents appearing after culturing of the cryopreserved cells(Fig. 3c).
Microarray Analysis and Comparison of hESCTranscriptome Before and After Cryopreservation
In order to identify differences in the transcriptomes ofcontrol hESCs and the frozen-thawed group, RNA fromhESCs from the time points 30 min, 1, 3, 6, 12, 24 and 48 hwere analyzed using Affymetrix HG-U133 Plus 2.0 arrays.Expression data were analyzed using R. Monitoring of thedifferences in the full transcriptomes is essential for theidentification of key biological processes and pathwaysinduced by the cryopreservation and thawing process in thehESCs. A comparative analysis was set between thementioned control time points with their respectivefrozen-thawed time points, with an additional comparisonof each group to the 0 min time point as shown in Fig. 1. Toclassify the variance in the data set, a principal componentanalysis (PCA) of the transcriptomes was done (Fig. 4a).The distance between the spheres representing the samplesreflects the degree of similarity of difference between thesamples. The three principal components (PCs) account for50.1% of the total variance in the data set. PC1 (represent-ing 28.6% variance) shows a clear separation of the non-frozen controls and the frozen-thawed samples, with theseparation becoming progressively larger over time (3, 6,12, 24, and 48 h). In PC2 (12.9% of the total variance),differences between the control samples (12, 48, 30 min,1 h) and the frozen-thawed (12, 6, 3, 1 h and 30 min)samples can be observed. Interestingly, no significantvariations are observed in PC2 between the control 24and 12 h compared to the 24 and 48 h frozen-thawed. Thiscan be explained by the fact that intact frozen-thawed
hESCs can be recovered after 24 and 48 h whereas deadcells are eliminated during the 24 and 48 h of culturing.
Heat maps of hierarchical clustering of the top 5,140differentially expressed genes with the highest variance ofthe signal intensities across 45 arrays is shown in Fig. 4b.As indicated there are two main clusters which aresubdivided into more specific sub-clusters. The mainclusters indicate differences existing between control cellsat all the times (including 1 and 30 min frozen-thawed) andthe frozen-thawed samples (6–48 h). Figure 4c, shows theheat map of the differentially expressed genes involved intopluripotency state of the hESCs in control and cryopre-served hESCs. As shown, a downregulation of these geneshas been observed which correlated well with the time ofculturing after thawing of the cryopreserved cells. Down-regulation of the pluripotency markers started at the 3 hfrozen samples and indicated a maximum at 48 h.
The number of the common and the control specificgenes that are upregulated in hESCs is very low at 30 min,show a maximum at 6 h and a minimum within 48 h ofculturing where as the number of the frozen-thawed specificgenes upregulated reaches a maximal plateau within 24 hand dropped down at 48 h (Fig. 5a). As indicated in Fig. 5b,only a few genes are commonly downregulated in controland frozen hESCs over the time of the culturing. Interest-ingly, much more genes were found to be specificallydownregulated in frozen-thawed hESCs to unfrozen controlhESCs. The number of the downregulated genes increasedgradually with the increasing time of culturing. Comparisonof the GO analysis of the dysregulated transcripts (up anddown) between 0 min and to the frozen-thawed time pointsrevealed that the dysregulated genes belong mainly to thebiological process of apoptosis, cell death, embryonicmorphogenesis, cell morphognesis, embryonic organ devel-opment, vascular development, skeletal system develop-ment, bone development, tissue morphogenesis, ossification,chordate embryonic development, cartilage development,
Fig. 5 Common genomic signatures for a comparison between controland freeze-thawed hESCs. The specifically upregulated (a) and down-regulated (b) transcripts and the overlap of detected transcripts between
freeze-thawed and non-freeze thawed groups. List of differentiallyexpressed transcripts were generated by comparison of the respectivesamples with false discovery rate 0.05 (P<0.05, log2 ±1)
512 Stem Cell Rev and Rep (2011) 7:506–517
Tab
le1
Selectedbiolog
icalfunctio
ngrou
psidentifiedby
non-freeze
controlv
ersusfreeze-thawed
hESCscomparison.
The
genesof
major
biolog
icalprocessesfoun
din
clustersarepresented.
The
fold
change
was
calculated
againsteach
controlandvalues
inthetablerepresentstheaverageof
triplicates
indicatedin
log(base2)
Category
GO
term
No.
genes
p-value
Genes
Early
GOTERM_B
P_FAT
embryo
nicmorph
ogenesis
60.00
33ZIC1,
WNT4,
GREM1,COL2A
1,MSX1,
PAX3
GOTERM_B
P_FAT
heartdevelopm
ent
50.00
55SEMA3C
,COL2A
1,MSX1,
PAX3,
ID2B
Inter
GOTERM_B
P_FAT
regu
latio
nof
apop
tosis
233.97
E-04
CDH1,
TMX1,
COL2A
1,DLG5,
SST,
NR3C
1,NLRP1,
SOCS2,
MSX1,
PLAGL1,
SERPIN
B9,
NR3C
1,CBX4,
COL2A
1,HERPUD1,
NEFL,MSX2,
DDIT3,
SFRP1,
NR3C
1,CASP3,
HSPD1,
TNFA
IP8,
HSPA
5,ANXA1
GOTERM_B
P_FAT
chordate
embryo
nicdevelopm
ent
161.61
E-05
ZEB1,
CDH1,
DLL1,
HOXB2,
TEAD4,
COL2A
1,MAFF,
HES1,
COL11A1,
JUNB,SALL4,
NOG,
SFRP1,
MSX1,
DLX2,
PAX3
GOTERM_B
P_FAT
cartilage
developm
ent
70.00
03ZEB1,
NOG,COL2A
1,COL11A1,
LECT1,
MSX1,
DLX2
GOTERM_B
P_FAT
embryo
nicorganmorph
ogenesis
90.00
13ZEB1,
NOG,HOXB2,
ZIC1,
COL2A
1,COL11A1,
DLX2
Late
GOTERM_B
P_FAT
neuron
differentiatio
n31
3.18
5E-07
CDH1,
MAP1B
,NRP1,
SALL3,
ADM,NEFL,RTN1,
STMN2,
HPRT1,
ERBB2,
ASCL1,
CXCR4,
GAS1,
POU3F
2,ID
4,BMP7,
CD24
,VCAN,CNTN4,
POU4F
1,GAP43
,KAL1,
UNC5C
,ID
4,SOX5,
DCX,MAP2,
CD24
,EN2,
SOX1,
DLX2,
PAX3
GOTERM_B
P_FAT
cellmorph
ogenesis
256.61
6E-06
LEF1,
MAP1B
,NRP1,
BMP7,
WWTR1,
ADM,DCX,SOX9,
NODAL,NOG,NEFL,HPRT1,
ERBB2,
CXCR4,
GAS1,
CNTN4,
VCAN,POU4F
1,NEFL,GAP43
,UNC5C
,KAL1,
NOX4,
PDPN,MAP7,
GOTERM_B
P_FAT
skeletal
system
developm
ent
243.41
0E-06
TEAD4,
COL2A
1,BMP7,
WWTR1,
SOST,
FGF18
,NODAL,PTN,NOG,COL11A1,
LECT1,
GAS1,
ZEB1,
HOXB2,
COL11A1,
BMP7,
TFA
P2A
,COL1A
2,MSX2,
TNFRSF11A,IG
FBP5,
SOX9,
SOX5,
PAPSS2,
HOXB3,
MSX2,
DLX2,
GOTERM_B
P_FAT
embryo
nicmorph
ogenesis
220.00
002
LEF1,
BMP7,
DLC1,
SALL4,
NODAL,NOG,COL11A1,
HESX1,
GAS1,
DLC1,
ZEB1,
HOXB2,
COL11A1,
BMP7,
COL2A
1,TFA
P2A
,GREM1,
ZIC1,
MSX2,
TDGF3,
ZIC1,
TSHZ1,
HOXB3,
DLX2,
PAX3
GOTERM_B
P_FAT
vasculaturedevelopm
ent
180.00
01NRP1,
FGF18
,NR2F
2,EDNRA,PDPN,APOE,EFNB2,
NODAL,COL1A
2,SEMA3C
,FGF2,
CXCR4,
PLAT,
APOE,FOXO1,
RHOB,TDGF3,
GOTERM_B
P_FAT
apop
tosis
260.00
65PHF17
,MEF2C
,PMAIP1,
TP53
INP1,
DLC1,
PAK1,
PAWR,TERF1,
NLRP1,
LITAF,
CADM1,
RNF14
4B,CXCR4,
FGF2,
TNFRSF19
,PIM
2,DLC1,
PHLDA1,
GULP1,
PMAIP1,
RHOB,
CD24
,FGD4,
PHF17
,BAG2,
GREM1,
UNC5C
,PA
X3,
PYCARD
GOTERM_B
P_FAT
mesenchym
alcelldifferentiatio
n7
0.00
14LEF1,
NOG,BMP7,
SOX9,
SEMA3C
,EDNRA,PA
X3
GOTERM_B
P_FAT
mesod
erm
developm
ent
80.00
21TDGF3,
NODAL,LEF1,
FOXH1,
BMP7,
EPB41
L5,
SNAI2
GOTERM_B
P_FAT
regu
latio
nof
apop
tosis
350.00
11TP53
INP1,
TERF1,
DLC1,
PAWR,SST,
NR3C
1,PLAGL1,
NLRP1,
SOCS2,
CADM1,
APOE,
PLAGL1,
BMP7,
POU4F
1,NEFL,MSX2,
SOX9,
CD24
,PYCARD,MEF2C
,CDH1,
BMP7,
TMX1,
PMAIP1,
HELLS,TERF1,
NEFL,ERBB2,
TNFRSF19
,PIM
2,GAS1,
PHLDA1,
DLC1,
NR3C
1,PMAIP1,
CD24
,FGD4,
COL2A
1,TDGF3,
FOXO1,
SFRP1,
PLAGL1
GOTERM_B
P_FAT
kidn
eydevelopm
ent
80.00
88LEF1,
NOG,BMP7,
SOX11,WWTR1,
GREM1
Stem Cell Rev and Rep (2011) 7:506–517 513

neuronal development and differentiation, mesoderm, heartand kidney development (Table 1). As demonstrated inFig. 6, expression of the genes is regulated in a time-dependent manner and in a regular way.
Discussion
Human embryonic stem cells are considered to be relevantfor therapeutic application of degenerative diseases. How-ever, one of the main problems that remain to be solved isto improve their survival rate and to preserve theirtranscriptomic identity as a criterion of consistency of theirdifferentiation program. Understanding of the biologicalprocesses and pathways involved in these phenomenamight contribute to improved cryopreservation protocolsand thereby the viability and the pluripotency status of thecells after thawing of the cryopreserved hESCs.
Combining a traditional cryopreservation protocol fre-quently used with large scale gene expression profiling weidentify critical time points, biological processes and signaltransduction pathways that might be critical for improvingof culturing of cryopreserved hESC. The phenotypes ofcryopreserved hESCs fully recovered after 5 days ofcultivation. However, the number of colonies was signifi-cantly higher in control groups compared to the cryopre-served hESCs. Furthermore, the colony growth ratefindings indicated an inhibition of the proliferation rateafter thawing of the frozen cells.
Analysis of the transcriptome derived from eight timepoints showed relatively small PC1 and PC2 variancesbetween the control cells reflecting large similarities of theirtranscriptomes. Relatively small differences can beexplained by the fact that hESCs first were dissociatedbefore further culturing. Both processes might require theexpression of a set of genes involved in attachment of thehESCs. Consistent with this hypothesis, the transcriptomeof the 0 min control hESCs which were not detached andthe transcriptomes of the 24 h or 48 h hESCs thatcompletely attached are almost identical (Fig. 4).
Relatively large PC1 and PC2 variances, especially inPC1 have been observed between the frozen-thawed versusunfrozen control cells. Interestingly, the PC1 and PC2variances in the transcriptomes of the different time pointsof the frozen-thawed hESCs increase continuously from30 min to 24 h and declined after 48 h. In this context, anincrease of the number of the up- and down-regulated genesoccurs continuously within 24 h after thawing and culturingand declined within 48 h. These time-dependent increases ofgene expression (up and down) might be explained by thelarge amount of cells dying after freeze-thaw stress but after48 h of culturing cells establish colonies (by attachment onfeeder cells) and grows similar to the control cells.
Obviously, the transcriptome of the 30 min frozen-thawed hESCs fraction which survived the freezing andthawing procedure is very similar to that of the unfrozencells as shown by the PC analysis. In this context, the PC1variance was negligible and the PC2 variance was relativelysmall compared to the control hESCs (1, 3 and 6 h). From theobservation that the 30 min frozen-thawed hESCs yieldedapproximately 25% of the RNA normally isolated from thesame number of control hESCs clumps (SupplementaryFigure 1) we observed that approximately 25% of thehESCs survived the freezing/thawing procedure. Fromthese findings we may conclude that the transcriptome ofthe surviving hESCs is preserved during cryopreservation.Immediately after the post-thaw wash (0 and 30 min), themajority of hESCs within colonies were viable, as assessedby the FDA/EB staining. However, viability declinedrapidly in the consecutive time points. This is in line withprevious publications that observed a rapid decline down to30% viability within 90 min post-thawing due to apoptoticcell death [7]. This observation favors the hypothesis thatphysically induced cellular damage induced by freezingand/or thawing inhibits proper attachment during cultiva-tion resulting in an induction of anoikis apoptotic cell deathpathway despite an almost stable transcriptome. Anoikis isa specific apoptotic pathway induced in cells that fail toattach on the surrounding extracellular matrix (ECM).
The hESCs that escape apoptosis can be recovered after24 and 48 h whereas dead cells are eliminated during the 24and 48 h culturing. This hypothesis is supported by the GOanalysis of the microarray data showing differences inexpression of genes involved in biological process such ascell communication, cell growth and maintenance, celldeath, cell differentiation and cell proliferation. Based onthese findings we suggest that factors such as ECM andanoikis anti-apoptotic agents might improve the survivalrate of the frozen-thawed hESCs.
In accordance with findings suggesting an important roleof the ECM signaling on the viability of frozen-thawedhESCs recently it has been reported that adherent culturewere cryopreserved with a marked survival rate afterthawing under matrix embedded adherent culture condi-
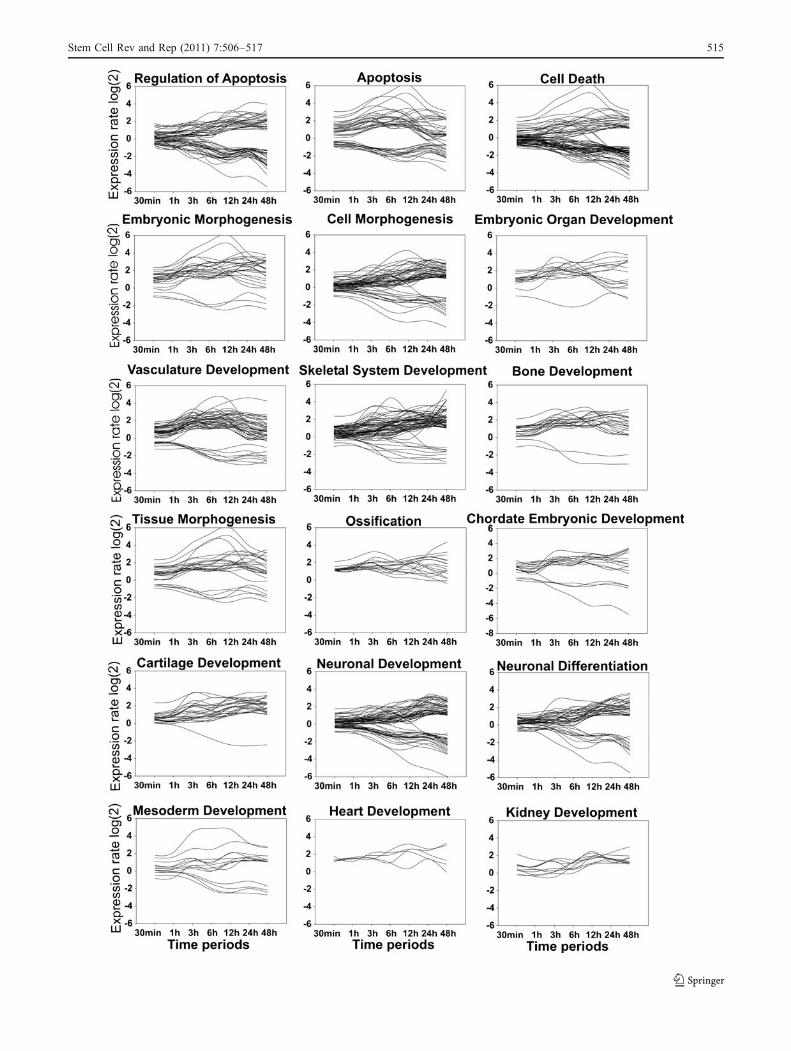
Fig. 6 Gene expression patterns of the genes belonging to the GOsenriched into freezed-thawed hESCs compared to the control hESCsafter different time of culturing. The gene expression data wereobtained from the microarray experiments after isolations of totalRNA from control cells and those exposed to vehicle (DMSO) for30 min, 1-, 6-, 12-, 24 and 48 h before and after cryopreservation.Genes whose expression was altered more than two folds in responseto cryopreservation were included in this analysis. GOs were obtainedusing DAVID analysis. The horizontal axis indicates the time and thevertical axis indicated gene expression ratio in log(2). Genes forselected GO categories are represented. The horizontal axis indicatedthe time-points and vertical axis indicated expression ratio in log(2)
b
514 Stem Cell Rev and Rep (2011) 7:506–517
Stem Cell Rev and Rep (2011) 7:506–517 515

tions [41]. In separate report, it was shown that the overallviability of cryopreserved fibroblasts can be improvedusing caspase-I inhibitor [42]. In the present study, notonly Caspase, but several apoptotic and pro-apoptic GOswere enriched in across the time-points after culturing ofthe freeze-thawed hESCs.
Interestingly, we also demonstrated that several devel-opmental and morphogenesis GO ontology and signalingpathways were deregulated in freeze-thawed hESCs. In asimilar set up of the experiments, it has been shown that thepluripotency maker OCT-4 significantly decreased afterculturing of cryopreserved hESCs for several days suggest-ing differentiation of the cultured cryopreserved cells [43].Accordingly, in our experiments hESCs showed morphol-ogies of differentiation (grade D) after 5 days of culturingpost thawing. Dysregulated genes are participating to themesodermal lineages and neurogenesis. Accordingly, inaddition to the cell death associated GOs we observed thatthe exposure of hESCs to cryopreservation and furtherfreeze-thaw stress increases the expression of almost all genesinvolved in the processes of embryonic morphogenesis,ossification, tissue morphogenesis, regeneration and vascula-ture development. Noticeably, deregulation of the genesparticipating to the cell death associated GOs started after3 h of culturing and showed a maximum after 6 h ofcultivation, and the expression level of those genes tended toreturn to the levels of control (unfrozen) cells after 48 ofcultivation (Fig. 6). All these events might occur by cellularstress caused by the freeze-thaw procedure and/or by thecryoprotectant DMSO.
In summary, our results suggest that anoikis and the ECMmight offer targets for improving the survival rate. Sincecells tend to differentiate after thawing, higher amounts offactors promoting survival of the hESCs might stabilize thecells to remain pluripotent. Moreover, the gene array tran-scriptomic approach in combination with the GO ontologyanalysis offers a quite effective strategy for identification ofcritical biological and molecular events for improvingculturing conditions of cryopreserved hESCs.
Acknowledgement Theworkwas supported by a grant from EuropeanCommission Sixth Framework Programme- CRYSTAL (“Cryo-Bankingof Stem Cells for Human Therapeutic Application”, http://www.crystal-eu.org/, FP6-037261). The authors wish to acknowledge Ms. C.Bottinger, Ms. H. Altenburg and Ms. S. Rohani for the technicalsupport in preparing MEF feeder and cell culturing reagents.
Disclosure The authors declare no potential conflicts of interest.
References
1. Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M.A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem
cell lines derived from human blastocysts. Science, 282(5391),1145–1147.
2. Reubinoff, B. E., Pera, M. F., Fong, C. Y., Trounson, A., &Bongso, A. (2000). Embryonic stem cell lines from humanblastocysts: somatic differentiation in vitro. Nature Biotechnology,18(4), 399–404.
3. Silva, J., & Smith, A. (2008). Capturing pluripotency. Cell, 132(4), 532–536.
4. Odorico, J. S., Kaufman, D. S., & Thomson, J. A. (2001).Multilineage differentiation from human embryonic stem celllines. Stem Cells, 19(3), 193–204.
5. Reubinoff, B. E., Pera, M. F., Vajta, G., & Trounson, A. O.(2001). Effective cryopreservation of human embryonic stem cellsby the open pulled straw vitrification method. Human Reproduc-tion, 16(10), 2187–2194.
6. Zhou, C. Q., Mai, Q. Y., Li, T., & Zhuang, G. L. (2004).Cryopreservation of human embryonic stem cells by vitrification.Chinese Medical Journal, 117(7), 1050–1055.
7. Heng, B. C., Ye, C. P., Liu, H., Toh, W. S., Rufaihah, A. J., Yang,Z., et al. (2006). Loss of viability during freeze-thaw of intact andadherent human embryonic stem cells with conventional slow-cooling protocols is predominantly due to apoptosis rather thancellular necrosis. Journal of Biomedical Science, 13(3), 433–445.
8. Narumiya, S., Ishizaki, T., & Uehata, M. (2000). Use and propertiesof ROCK-specific inhibitor Y-27632. San Diego: Academic.
9. Watanabe, K., Ueno, M., Kamiya, D., Nishiyama, A., Matsumura,M., Wataya, T., et al. (2007). A ROCK inhibitor permits survivalof dissociated human embryonic stem cells. Nature Biotechnology,25(6), 681–686.
10. Koyanagi, M., Takahashi, J., Arakawa, Y., Doi, D., Fukuda, H.,Hayashi, H., et al. (2008). Inhibition of the Rho/ROCK pathwayreduces apoptosis during transplantation of embryonic stem cell-derived neural precursors. Journal of Neuroscience Research, 86(2), 270–280.
11. Li, X. Y., Meng, G. L., Krawetz, R., Liu, S. Y., & Rancourt, D. E.(2008). The ROCK inhibitor Y-27632 enhances the survival rateof human embryonic stem cells following cryopreservation. StemCells and Development, 17(6), 1079–1085.
12. McLellan, M. R., & Day, J. G. (1995). Cryopreservation andfreeze-drying protocols. Introduction. Methods in MolecularBiology, 38, 1–5.
13. Sum, A. K., & de Pablo, J. J. (2003). Molecular simulation studyon the influence of dimethylsulfoxide on the structure ofphospholipid bilayers. Biophysical Journal, 85(6), 3636–3645.
14. Karlsson, J. O. M., Cravalho, E. G., Rinkes, I. H. M. B., Tompkins, R.G., Yarmush,M. L., & Toner,M. (1993). Nucleation and growth of icecrystals inside cultured-hepatocytes during freezing in the presence ofdimethyl-sulfoxide. Biophysical Journal, 65(6), 2524–2536.
15. Gao, D. Y., Chang, Q., Liu, C., Farris, K., Harvey, K., Mcgann, L.E., et al. (1998). Fundamental cryobiology of human hematopoieticprogenitor cells I: Osmotic characteristics and volume distribution.Cryobiology, 36(1), 40–48.
16. Hubel, A. (1997). Parameters of cell freezing: implications for thecryopreservation of stem cells. Transfusion Medicine Reviews, 11(3), 224–233.
17. Schneider, U., & Mazur, P. (1984). Osmotic consequences ofcryoprotectant permeability and its relation to the survival offrozen-thawed embryos. Theriogenology, 21(1), 68–79.
18. Muldrew, K., & Mcgann, L. E. (1994). The osmotic rupturehypothesis of intracellular freezing-injury. Biophysical Journal, 66(2), 532–541.
19. Mazur, P., & Schneider, U. (1986). Osmotic responses ofpreimplantation mouse and bovine embryos and their cryobiologicalimplications. Cell Biophysics, 8(4), 259–285.
20. Gao, D. Y., Liu, J., Liu, C., Mcgann, L. E., Watson, P. F.,Kleinhans, F. W., et al. (1995). Prevention of osmotic injury to
516 Stem Cell Rev and Rep (2011) 7:506–517

human spermatozoa during addition and removal of glycerol.Human Reproduction, 10(5), 1109–1122.
21. Fujikawa, S. (1980). Freeze-fracture and etching studies onmembrane damage on human-erythrocytes caused by formationof intracellular ice. Cryobiology, 17(4), 351–362.
22. Mazur, P., Leibo, S. P., & Chu, E. H. Y. (1972). 2-Factorhypothesis of freezing injury—evidence from chinese-hamstertissue-culture cells. Experimental Cell Research, 71(2), 345–355.
23. Mazur, P. (1984). Freezing of living cells—mechanisms andimplications. American Journal of Physiology, 247(3), C125–C142.
24. Mazur, P., & Cole, K. W. (1989). Roles of unfrozen fraction, saltconcentration, and changes in cell-volume in the survival offrozen human-erythrocytes. Cryobiology, 26(1), 1–29.
25. Mazur, P. (1990). Equilibrium, quasi-equilibrium, and nonequi-librium freezing of mammalian embryos. Cell Biophysics, 17(1),53–92.
26. Trump, B. F., Young, D. E., Arnold, E. A., & Stowell, R. E.(1965) Effects of freezing and thawing on structure chemicalconstitution and function of cytoplasmic structures. FederationProceedings 24(2S15), S144–S168.
27. Baharvand, H., Ashtiani, S. K., Taee, A., Massumi, M., Valojerdi,M. R., Yazdi, P. E., et al. (2006). Generation of new humanembryonic stem cell lines with diploid and triploid karyotypes.Development Growth & Differentiation, 48(2), 117–128.
28. Ehrhart, F., Schulz, J. C., Katsen-Globa, A., Shirley, S. G., Reuter,D., Bach, F., et al. (2009). A comparative study of freezing singlecells and spheroids: towards a new model system for optimizingfreezing protocols for cryobanking of human tumours. Cryobiology,58(2), 119–127.
29. Kvach, J. T., Munguia, G., & Strand, S. H. (1984). Staining tissue-derived mycobacterium-leprae with fluorescein diacetate andethidium-bromide. International Journal of Leprosy and OtherMycobacterial Diseases, 52(2), 176–182.
30. Abramoff, M. D., Magelhaes, P. J., & Ram, S. J. (2004). Imageprocessing with ImageJ. Biophotonics International, 11, 36–42.
31. Vrekoussis, T., Chaniotis, V., Navrozoglou, I., Dousias, V., Pavlakis,K., Stathopoulos, E. N., et al. (2009). Image analysis of breast cancerimmunohistochemistry-stained sections using ImageJ: an RGB-based model. Anticancer Research, 29(12), 4995–4998.
32. Bolstad, B. M., Irizarry, R. A., Astrand, M., & Speed, T. P. (2003).A comparison of normalization methods for high density oligonu-cleotide array data based on variance and bias. Bioinformatics, 19(2), 185–193.
33. Gautier, L., Cope, L., Bolstad, B. M., & Irizarry, R. A. (2004).affy—analysis of Affymetrix GeneChip data at the probe level.Bioinformatics, 20(3), 307–315.
34. Pepper, S. D., Saunders, E. K., Edwards, L. E., Wilson, C. L., &Miller, C. J. (2007) The utility of MAS5 expression summary anddetection call algorithms. Bmc Bioinformatics 8.
35. Smyth, G. K. (2004) Linear models and empirical bayes methodsfor assessing differential expression in microarray experiments.Statistical applications in genetics and molecular biology 3,Article3.
36. Eisen, M. B., Spellman, P. T., Brown, P. O., & Botstein, D.(1998). Cluster analysis and display of genome-wide expressionpatterns. Proceedings of the National Academy of Sciences of theUnited States of America, 95(25), 14863–14868.
37. Mardia, K. V., Kent, J. T., & Bibby, J. M. (1979). Multivariateanalysis. London: Academic.
38. Huang, D. W., Sherman, B. T., & Lempicki, R. A. (2009).Systematic and integrative analysis of large gene lists usingDAVID bioinformatics resources. Nature Protocols, 4(1), 44–57.
39. Dennis, G., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W.,Lane, H. C. et al. (2003) DAVID: Database for annotation,visualization, and integrated discovery. Genome Biology 4(9).
40. Hosack, D. A., Dennis, G., Jr., Sherman, B. T., Lane, H. C., &Lempicki, R. A. (2003). Identifying biological themes within listsof genes with EASE. Genome Biology, 4(10), R70.
41. Ji, L., de Pablo, J. J., & Palecek, S. P. (2004). Cryopreservation ofadherent human embryonic stem cells. Biotechnology andBioengineering, 88(3), 299–312.
42. Baust, J. M., Van Buskirk, R., & Baust, J. G. (2000). Cellviability improves following inhibition of cryopreservation-induced apoptosis. Vitro Cellular & Developmental Biology-Animal, 36(4), 262–270.
43. Katkov, I. I., Kim, M. S., Bajpai, R., Altman, Y. S., Mercola, M.,Loring, J. F., et al. (2006). Cryopreservation by slow cooling withDMSO diminished production of Oct-4 pluripotency marker inhuman embryonic stem cells. Cryobiology, 53(2), 194–205.
Stem Cell Rev and Rep (2011) 7:506–517 517
Copyright © 2022 FDOKUMEN




















