
Culture media - serum, serum free media, cell cytototoxicity & viability

1999, 19(7):4582. Mol. Cell. Biol.
Trinh, Mark W. Majesky and Robert J. SchwartzNarasimhaswamy S. Belaguli, Wei Zhou, Thuy-Hanh T. Binding TargetsTransactivation of Serum Response Factor within the Activation Domain InhibitsResponse Factor: Alternative Splicing Dominant Negative Murine Serum
http://mcb.asm.org/content/19/7/4582Updated information and services can be found at:
These include:
REFERENCEShttp://mcb.asm.org/content/19/7/4582#ref-list-1at:
This article cites 59 articles, 40 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
on M
arch 10, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

MOLECULAR AND CELLULAR BIOLOGY,0270-7306/99/$04.0010
July 1999, p. 4582–4591 Vol. 19, No. 7
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Dominant Negative Murine Serum Response Factor:Alternative Splicing within the Activation Domain InhibitsTransactivation of Serum Response Factor Binding Targets
NARASIMHASWAMY S. BELAGULI,1 WEI ZHOU,1 THUY-HANH T. TRINH,1 MARK W. MAJESKY,1,2
AND ROBERT J. SCHWARTZ1*
Departments of Cell Biology1 and Pathology,2 Baylor College of Medicine, Houston, Texas 77030
Received 30 December 1998/Returned for modification 3 April 1999/Accepted 9 April 1999
Primary transcripts encoding the MADS box superfamily of proteins, such as MEF2 in animals and ZEMain plants, are alternatively spliced, producing several isoformic species. We show here that murine serumresponse factor (SRF) primary RNA transcripts are alternatively spliced at the fifth exon, deleting approxi-mately one-third of the C-terminal activation domain. Among the different muscle types examined, visceralsmooth muscles have a very low ratio of SRFD5 to SRF. Increased levels of SRFD5 correlates well with reducedsmooth muscle contractile gene activity within the elastic aortic arch, suggesting important biological roles fordifferential expression of SRFD5 variant relative to wild-type SRF. SRFD5 forms DNA binding-competenthomodimers and heterodimers. SRFD5 acts as a naturally occurring dominant negative regulatory mutant thatblocks SRF-dependent skeletal a-actin, cardiac a-actin, smooth a-actin, SM22a, and SRF promoter-luciferasereporter activities. Expression of SRFD5 interferes with differentiation of myogenic C2C12 cells and theappearance of skeletal a-actin and myogenin mRNAs. SRFD5 repressed the serum-induced activity of the c-fosserum response element. SRFD5 fused to the yeast Gal4 DNA binding domain displayed low transcriptionalactivity, which was complemented by overexpression of the coactivator ATF6. These results indicate that theabsence of exon 5 might be bypassed through recruitment of transcription factors that interact with extra-exon5 regions in the transcriptional activating domain. The novel alternatively spliced isoform of SRF, SRFD5, mayplay an important regulatory role in modulating SRF-dependent gene expression.
Alternative splicing is a commonly used molecular strategyfor creating diverse gene products from a single genetic locusin most eucaryotic cells. The modular organization of tran-scription factor genes, through exon-encoded structural do-mains, may be conducive for forming a variety of alternativelyspliced isoforms that affect DNA binding avidity and specific-ity, transactivation, subcellular localization, responsiveness tosignaling pathways, and developmental regulation (reviewed inreference 31). For example, alternative splicing within theDNA binding domain of Pax-6 (9) and Wilm’s tumor-associ-ated protein 1 (5, 21) alters their DNA binding specificities.Alternative splicing of exons encoding the transactivating do-mains in the paired family proteins Pax-3 (59), Pax-8 (24), andPax-9 (39), the POU homeodomain family proteins Pit-1 (37),Oct-1 (8), Oct-2 (1), and Brn-3a (38), and the zinc fingertranscription factor GATA-5 (32) results in isoforms possess-ing activation domains with different potencies. Splicing out ofselect exons from the activation domain as in AML1a (56),Oct-2 (29), and CREB family proteins CREB (reviewed inreference 11), Drosophila CREB/CREM (61), and CREM (12)produces isoforms with dominant negative activity. Similarly,several splice variants containing only the DNA binding do-main act as dominant negative isoforms presumably by het-erodimerizing with the wild-type isoforms and competing withthe wild-type isoforms for DNA binding. Thus, additional ge-netic complexity might be further imposed by altering regula-tory processes by the expression of alternatively spliced tran-scription factor isoforms.
Serum response factor (SRF) is a member of an ancientDNA binding protein superfamily whose evolutionarily diver-gent relatives shared a highly conserved DNA binding/dimer-ization domain of 90 amino acids, termed the MADS box(reviewed in reference 51). The complex and novel stratifiedorganization of the MADS box domain recently resolved byX-ray crystallography (43) was probably assembled before thedivergence of plants and animals, since identical MADS boxstructures were present in yeast transcription factors MCM1and ARG80, a large number of homeotic-like plant proteins,and vertebrate SRFs (reviewed in references 46 and 52). All ofthese transcription factors, through their common MADSboxes, bind to virtually the same DNA sequences (reviewed inreferences 46 and 52) and interact with similar kinds of coac-cessory regulatory factors (reviewed in references 52 and 58).SRF-related factors such as RSRF, also described as MEF2,contain the MADS box and the adjacent MEF2 box (44) butprefers binding to divergent MEF2 sites. Dissection of thehuman SRF revealed a modular organization of structural andfunctional domains, such as the central MADS box domain,the N-terminal domain, containing serine 103 phosphorylationsites that facilitate SRF DNA binding activity, and a putativetranscriptional inhibitory domain (22, 47), while the C-termi-nal activation domain binds a diversity of protein factors suchas the RAP74 subunit of TFIIF (23), ATF6 (64), and viral Tax1(13) proteins.
MADS box proteins play a central role for a variety ofregulatory activities. The mammalian MADS box proteins,SRF and MEF2, regulate the SRF-inducible genes and muscle-specific gene expression (19; reviewed in references 40, 45, 52,and 58). The regulation of muscle-specific gene expressionappears to be mediated by the interaction of the SRF MADSbox with striated muscle tissue-restricted factors such as the
* Corresponding author. Mailing address: Department of Cell Biol-ogy, Baylor College of Medicine, One Baylor Plaza, Houston, TX77030. Phone: (713) 798-6649. Fax: (713) 798-7799. E-mail: [email protected].
4582
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

basic helix-loop-helix protein MyoD (18, 50), homeodomainprotein Nkx-2.5 (6), and zinc finger protein GATA-4 (4). Inaddition, high levels of embryonic SRF expression coincidewith the expression of cardiac, skeletal, and smooth musclea-actins (3, 7), noted as early markers for terminal striated andsmooth muscle cell (SMC) differentiation (49). We reportedearlier that the skeletal muscle-enriched expression of SRF isunder the control of a positive autoregulatory loop. Dominantnegative mutants of SRF with point mutations in the DNAbinding domain (SRFpm1) or truncation of the C-terminalactivation domain (SRFDC) repressed SRF promoter activity(3) and myogenesis. Vandromme et al. (60) showed that neu-tralizing SRF activity by microinjection of SRF antibodies, andblocking SRF gene expression through application of SRFantisense oligonucleotides (53) in C2C12 myoblasts, preventsterminal differentiation, thus underscoring the obligate role forSRF in myogenic differentiation and muscle-specific gene ex-pression. These observations were confirmed by the recentanalysis of SRF null mutants, which revealed an absolute de-pendence for SRF for the formation of embryonic myogenicmesoderm (2).
An array of MEF2 proteins are generated by alternativesplicing (27, 33, 34, 35, 62). Furthermore, some of these splic-ing events are cell type restricted, and no specific function forthese isoforms has been ascribed so far. We asked whetherSRF, another closely related MADS box family member, isalso alternatively spliced, and if it is, whether SRF splice vari-ants have biological activity. We show here that murine SRFprimary RNA transcripts are alternatively spliced. An SRFisoform with a deletion in the C-terminal activation domainacts as a naturally occurring dominant negative regulatory mu-tant that interferes with SRF-dependent promoter activity andmuscle cell differentiation.
MATERIALS AND METHODS
Recombinant DNA clones. Luciferase reporter constructs for Gal4-Luc(G5Luc), c-fos serum response element (SRE), cardiac a-actin, skeletal a-actin,SM22a, and 2310 SRF promoters and the expression constructs pCGN, pCGNSRF, pCGNSRFDC, pCGNATF6, GalDB (Gal4 DNA binding domain), andGalSRF 266-508 have been described elsewhere (3, 6, 22, 28). The smootha-actin promoter-luciferase reporter was constructed by cloning a PCR-ampli-fied 2191 to 146 fragment between NheI and XhoI sites of pGL3 basic vector.To construct pCGNSRFD5, pCGNSRF was partially digested with ApalI fol-lowed by digestion to completion with BamHI. ApalI cuts SRF once within exon4. The large ApalI-BamHI fragment containing the pCGN vector backboneincluding the b-globin intron and poly(A) signal sequences and SRF sequencesfrom ATG to nucleotide (nt) 1446 was gel purified. Mouse skeletal muscle cDNAwas amplified with an upstream primer (1317 to 1337) and a downstream primeroverlapping the stop codon which contained an engineered BamHI site. ThePCR-amplified product was digested with ApalI and BamHI and cloned into thelarge ApalI- and BamHI-digested vector fragment. GalSRF 266-508D5 was con-structed by triple ligation of ApalI- and BamHI-digested PCR product containingthe SRFD5 fragment (amino acids 350 to 508) described above, the XbaI-ApalIfragment containing SRF amino acids 266 to 350, and the XbaI and BamHI-cutGal4 expression vector.
RNA isolation. Total RNA was isolated from adult mouse tissues and cell linesby using Ultra Spec (Biotecx) according to the manufacturer’s recommendations.
RT-PCR. To analyze SRF RNA processing, total cellular RNA (5 mg) wasdenatured and reverse transcribed with 15 U of Superscript reverse transcrip-tase (RT; Bethesda Research Laboratories) in RT-PCR buffer (Stratagene)containing random primers and deoxynucleoside triphosphates. For negativecontrols, RT was not added. After 1 h of incubation at 42°C, 50 ml of waterwas added, and the RT was heat inactivated. Five microliters of the cDNAwas used for PCR. The 50-ml PCR mixture contained the standard Taqpolymerase buffer with 2.5 mM MgCl2, 0.2 mM deoxynucleoside triphos-phates, and 100 pmol each of upstream (1317 to 1337) and downstream (1889to 1909) primers. The reaction conditions were an initial denaturation at 95°Cfor 10 min, followed by 30 cycles of 1-min denaturation, 1-min annealing at60°C, and 2-min extension at 72°C. A 5-ml aliquot of the reaction product wasfractionated on a 1.5% agarose gel, stained with ethidium bromide, andphotographed. A 0.5-ml aliquot of the RT-PCR product was fractionated ona 1.5% agarose gel and blotted simultaneously onto two pieces of a Zeta-Probe GT membrane (Bio-Rad). One blot was hybridized to a random-
primed SRF cDNA probe, and the other was hybridized to an end-labeledexon 5-specific 25-mer oligonucleotide. Blots were washed under standardconditions and exposed to X-ray film for 10 to 30 min.
For semiquantitative RT-PCR, 32P-end-labeled primers were used, and thereaction was stopped in the linear range (20 to 25 cycles). The PCR productswere resolved on a 5% polyacrylamide gel, dried, and autoradiographed.
Northern blotting. Twenty micrograms of total RNA was denatured, resolvedon a 0.8% formaldehyde-agarose gel, and transferred to a Zeta-Probe GT (Bio-Rad) membrane. The membrane was hybridized to random-primed mouse myo-genin probe at 65°C in 0.25 M Na2HPO4 (pH 7.2)–7% sodium dodecyl sulfate(SDS) and washed at the same temperature in 20 mM Na2HPO4–1% SDS.Subsequently, the membrane was stripped and reprobed with a skeletal a-actin39 untranslated region probe.
RNase protection assays. The antisense probe for RNase protection wassynthesized from XhoI-linearized plasmid pBSSRF 1558-1842, by using T3 RNApolymerase. Radioactive probe (5 3 104 cpm) was hybridized to 40 mg of totalRNA at 50°C overnight in a buffer containing 80% formamide, 10 mM sodiumcitrate (pH 6.4), 300 mM sodium acetate (pH 6.4), and 1 mM EDTA. Unhy-bridized probe and RNA were digested by adding a mixture of RNases A (1 mg)and T1 (20 U) for each sample and incubating the mixtures for 1 h at 37°C.RNase-resistant double-stranded RNA was resolved on a 5% polyacrylamide–8M urea gel. The gel was dried and autoradiographed.
Cell culture and transfections. NIH 3T3 and CV1 cells were maintained inDulbecco modified Eagle medium (DMEM) containing 10% neonatal calf se-rum. 10T1/2 and C2C12 cells were maintained in DMEM containing a mixtureof 10% neonatal calf serum and fetal bovine serum. For serum induction oftransfected cells, 10T1/2 cells in 60-mm-diameter plates were cotransfected with200 ng of SRELuc and 200 ng of pCGNSRF, pCGNSRFD5, pCGNSRFDC, orthe empty vector pCGN along with 200 ng of internal control plasmidpCMVbGal. Following 48 h of serum starvation, cells were induced by theaddition of fetal bovine serum to 20% (final concentration), and cells wereharvested 3 h later. CV1 cells in 60-mm-diameter plates were transfected with 1mg of cardiac a-actin, skeletal a-actin, 3 kbp-Sm22a and 2310 SRF promoter-luciferase reporter constructs along with 150 ng of pCGNSRF, pCGNSRFD5,and pCGNSRFDC. To demonstrate the dose-dependent effect of SRFD5 onsmooth a-actin promoter, 200 ng of smooth a-actin promoter-luciferase con-struct was cotransfected with 150 ng of SRF and various amounts of SRFD5.Cells were harvested 48 h posttransfection, and luciferase activity was measuredaccording to standard methods in a luminometer. When cotransfected withinternal control plasmid pCMVbGal, the luciferase values were normalized tob-galactosidase values.
For stable expression of SRFD5 and SRFpm1, C2C12 myoblasts in 60-mm-diameter plates were transfected with 250 ng of pSV2Neo and indicated plas-mids. A day after transfection, cells were split into 10 100-mm-diameter platesand selected for neomycin resistance. DMEM containing 20% serum and 400 mgof G418 (GIBCO) per ml was changed every 5 days. Neomycin-resistant colonieswere pooled after 2 weeks of selection, expanded, and analyzed.
Whole-cell extracts and electrophoretic gel mobility shift assays (EMSAs).Whole-cell extracts were prepared as described earlier (26). Five micrograms ofcell extract was preincubated with 1 mg of poly(dG-dC) at room temperature for15 min in 20 ml of 13 buffer (50 mM NaCl, 20 mM HEPES-KOH [pH 7.5], 0.1mM EDTA, 0.5 mM dithiothreitol, 10% glycerol). A 50-fold excess of specificand nonspecific competitors and 0.5 ml of antibodies were included in thisreaction. Following the preincubation, 0.02 pmol of end-labeled proximal SRE1probe from the SRF promoter (3) was added, and the mixtures were incubatedat room temperature for a further 15 min. DNA-protein complexes were re-solved on a 5% polyacrylamide gel cast and run in 0.53 Tris-borate-EDTA. Forthe demonstration of SRFD5-SRFDC heterodimers, 2.5 ml of in vitro-translatedSRFD5 was incubated with 2.5 mg of CV1 cell extract overexpressing SRFDC in20 ml of 13 binding buffer.
In vitro transcription and translation and GST pull-down assays. For in vitrotranscription, SRFD5 cloned in pBSSKII(1) was linearized with BamHI andtranscribed with T3 RNA polymerase. Capped SRFD5 and luciferase RNA weretranslated in rabbit reticulocyte lysate (RRL; Promega) in the presence of[35S]methionine according to the manufacturer’s recommendations; 5-ml ali-quots of the programmed lysates were incubated with either glutathione S-transferase (GST) or GST-SRF immobilized on beads. GST pull-down assayswere performed as described earlier (6).
Western blot analysis. NIH 3T3 cell extracts for Western blotting were pre-pared by extracting cells in phosphate-buffered saline containing 1% NonidetP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitors. Solubleprotein (20 mg) was resolved on an SDS–10% polyacrylamide gel, transferred topolyvinylidene difluoride membranes, and probed with SRF immune serum (agift from R. Prywes). Whole-cell extracts prepared from 4-day embryoid bodies(EBs) were probed with a carboxy-terminus peptide epitope antibody (SantaCruz Biotechnology) directed against amino acids 486 to 505 of human SRF.After visualization of the signal by enhanced chemiluminescence the blot wasstripped and reprobed with an exon 5 epitope-specific antibody.
VOL. 19, 1999 SRF GENE REGULATION 4583
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

RESULTS
Alternatively splicing of SRF exon 5. Members of the MADSbox family of proteins such as MEF2 generate isoforms byalternative splicing of the primary transcripts. Some of thesealternatively spliced isoforms are tissue type restricted. Weasked if the primary transcript of SRF also undergoes alterna-tive splicing. Analysis of adult mouse skeletal muscle RNA byRT-PCR using exon 3-specific (nt 1317 to 1337) and exon7-specific (nt 1889 to 1909) primers detected two major am-plified products 592 and 401 nt in length, (Fig. 1A). Based onthe RNA chain lengths, the 592-bp band corresponds to con-stitutively spliced SRF RNA whereas the 401-bp band con-forms to an alternatively spliced form of SRF RNA. Since thelength of exon 5 is 192 bp, it is likely that the smaller amplifiedband of 401 bp arose from removal of exon 5. To confirm thisobservation, Southern blot analysis was performed on the RT-PCR-amplified products. These products, resolved on a 1.5%agarose gel, were transferred bidirectionally onto two mem-branes and probed with an SRF cDNA probe and an exon
5-specific oligonucleotide probe. As shown in Fig. 1B, both592- and 401-bp DNA species hybridized to the SRF cDNAprobe, indicating that both DNA species contain SRF se-quences. However, the exon 5-specific oligonucleotide hybrid-ized only to the 592-bp species, not the 401-bp species, indi-cating that the smaller amplified DNA species is devoid ofexon 5 sequences (Fig. 1C). We confirmed that the 401-bpspecies lacked exon 5 by nucleic acid sequencing. The alterna-tive splicing of exon 4 to exon 6 utilized the authentic splicedonor and acceptor sites. The exon 5 skipping did not alter thereading frame but resulted in substitution of a single aminoacid, glycine for valine at the splice junction. SRFD5 transcriptswere expressed abundantly in brain, cardiac, and skeletal mus-cle and at very low levels in kidney, liver, spleen, and stomach(Fig. 1D).
Recently, we reported that SRF is expressed predominantlyin mouse brain, skeletal, cardiac, and smooth muscle tissues(3). The relative proportions of the constitutively and alterna-tively spliced SRF RNA species in three different types ofmuscle tissues were assessed by RNase protection assays. Theantisense SRF probe consisted of a cDNA fragment spanningexons 5 through 7. Total RNA prepared from skeletal, cardiac,and visceral smooth muscle tissues protected 284- and 144-ntfragments, representing the constitutively and alternativelyspliced RNA species, respectively (Fig. 2). The relative pro-portion of the constitutively spliced RNA to alternativelyspliced RNA varied for the three muscle types examined, asshown in Fig. 2. Stomach, representing visceral smooth muscle,expressed the highest proportion of constitutively splicedRNA.
SRFD5 is expressed in a rostrocaudal gradient in the neo-natal rat pup aorta. We asked whether SRFD5 was expressedin aortic SMCs and whether the cardiac neural crest derivedaortic arch SMCs differed from mesoderm-derived abdominalaortic SMCs in the expression of SRFD5. As shown in Fig. 3,SRFD5 was expressed in a rostrocaudal gradient, with thehighest level of SRFD5 in aortic arch and the lowest level in the
FIG. 1. Alternative splicing of SRF RNA removes exon 5. The exon-intronorganization of the SRF genome and positions of the primers used for PCR areshown at the top. (A) Ethidium bromide-stained agarose gel showing the RT-PCR products from skeletal muscle (lanes 3 and 4), heart (lanes 5 and 6), brain(lane 7 and 8), and 10T1/2 cells (lane 9). Cloned mSRF cDNA was used as thetemplate in lane 1. Lane 2 contained no input cDNA. RT was omitted duringcDNA synthesis for lanes 4, 6, and 8. Constitutively spliced and alternativelyspliced forms are diagrammatically represented on the left, and sizes of the bandsare shown in base pairs on the right. Southern blots of the gel probed with themSRF cDNA and exon 5 oligonucleotide are shown in panels B and C, respec-tively. End-labeled primers were used for PCR shown in panel D.
FIG. 2. Tissue-restricted expression of alternatively spliced SRFD5 RNA,determined by RNase protection analysis of total cellular RNA isolated fromskeletal muscle (lane 2), heart (lane 3), and stomach (lane 4) with the antisenseSRF probe. The undigested probe was run in lane 1. Protected unspliced andspliced products are diagrammatically represented to the left; sizes of the pro-tected fragments are indicated in base pairs on the right.
4584 BELAGULI ET AL. MOL. CELL. BIOL.
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

abdominal aorta. We further analyzed the expression and dis-tribution of SMC markers such as SM-MHC, SM22a, andSRFD5 in the same aortic segments. Both SM-MHC andSM22a were expressed in a gradient along the aorta, with thelowest level of expression in the elastic aortic arch and thehighest in the contractile abdominal aorta (Fig. 3). Conversely,the highest levels of SRFD5 was in the segments of the aorticarch and was reduced in the segments of abdominal aorta.Thus, there appears to be an inverse relationship between theexpression of these SRF-regulated genes and levels of SRFD5(Fig. 3).
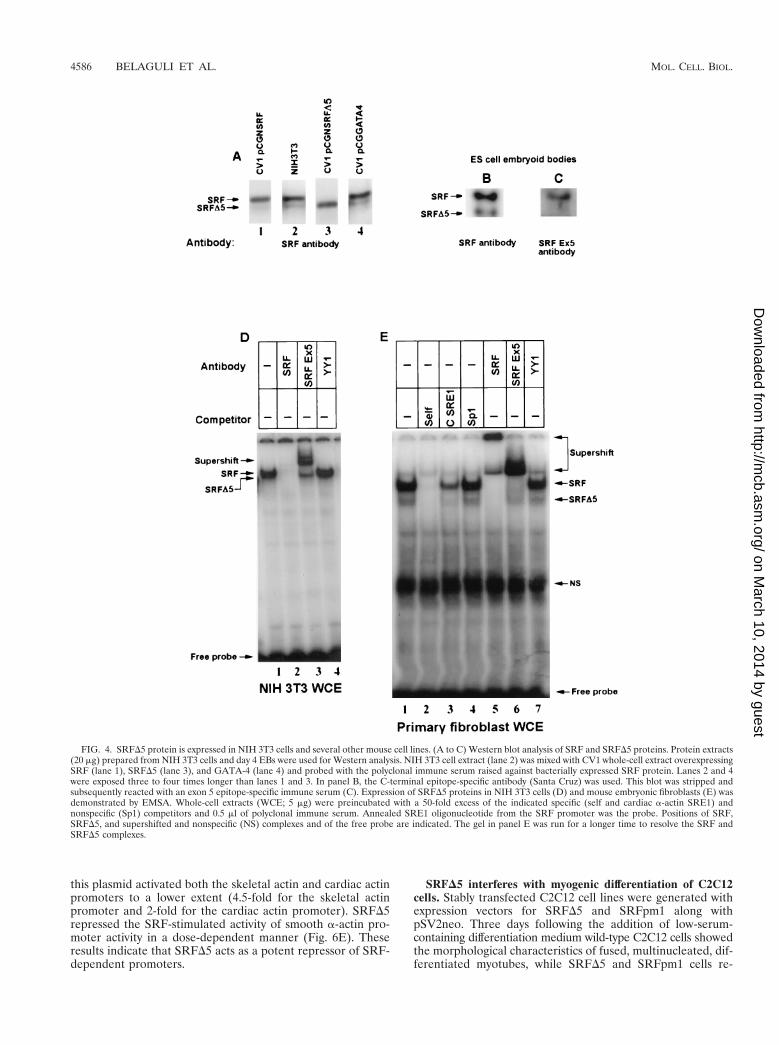
Identification of cellular SRFD5-expressed protein. Weasked whether the alternatively spliced SRF transcript wastranslated into cellular protein. Protein extracts from SRF,SRFD5, and GATA-4 expression vector-transfected CV1 fi-broblasts, which served as ectopically expressed internal stan-dards, were mixed with NIH 3T3 cell extracts, electrophoresed,and then assayed by Western blotting with anti-SRF antibod-ies. As shown in Fig. 4A (lane 2), SRF antiserum reacted with67- and 57-kDa proteins. The 67-kDa band was more intense inthe extracts spiked with SRF-enriched cell extract (Fig. 4A,lane 1). Similarly, the 57-kDa band was more intense when theextract was mixed with the SRFD5-enriched cell extract (Fig.4A, lane 3). The GATA-4 containing extract did not markedlyalter the anti-SRF reactivity (Fig. 4A, lane 4). We also evalu-ated soluble extracts prepared from day 4 EBs with a C-ter-minal epitope (common to both SRF and SRFD5) and theexon 5 epitope-specific peptide antibodies. The C-terminalSRF antibody reacted with two proteins of 67 and 57 kDa.However, stripping and reprobing of this blot with the exon5-specific antibody resulted in immunoreactivity of only the
67-kDa protein (Fig. 4B and C). Thus, based on several criteriaincluding predicted size, comigration with exogenously ex-pressed SRF and SRFD5 proteins, and immune specificity, the67- and 57-kDa proteins were identified as the full-length andalternatively spliced SRFD5 protein isoforms.
EMSAs were used to confirm the presence of two SRFprotein isoforms and to evaluate their DNA binding activities.Incubation of whole-cell extracts prepared from NIH 3T3 cells,CV1 cells, and primary mouse embryo fibroblasts with the SREprobe resulted in two EMSA complexes (Fig. 4D and E), bothof which were competed by a 50-fold molar excess of unlabeledSREs but not by Sp1 sites. Both SRF protein-DNA complexeswere either supershifted and or abolished by SRF antiserumraised against the full-length SRF (Fig. 4D and E), indicatingthe presence of SRF in these complexes. However, the exon 5epitope-specific antibody supershifted the slower-migratingbut not the faster-migrating SRF-DNA complex, suggestingthat only the former contains exon 5-encoded SRF sequences.
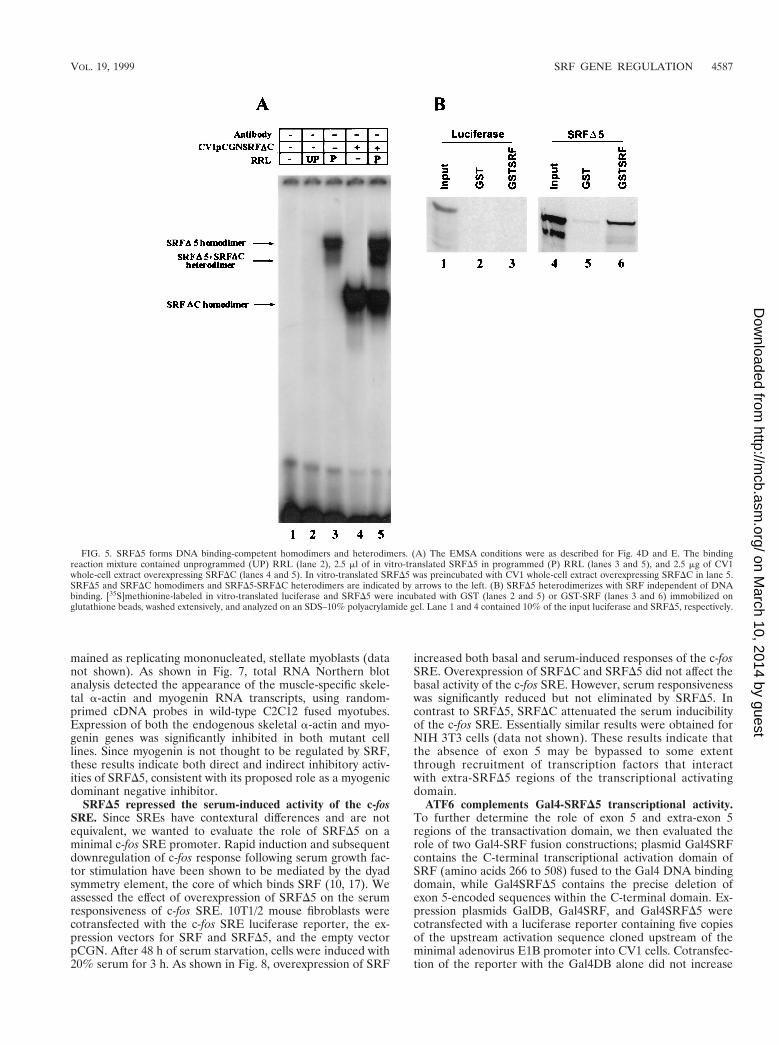
SRFD5 protein binds DNA independently and heterodimer-izes with other SRF species. Although the SRF DNA bindingand dimerization domains overlap the MADS box, sequencesoutside the MADS box have been shown to influence theprotein’s DNA binding activities. We wanted to determinewhether removal of exon 5 (amino acids 384 to 448) perturbsSRFD5 DNA binding and dimerization activities. An EMSAwith in vitro-translated SRFD5 bound the SRE probe with highaffinity, as shown in Fig. 5A, indicating that SRFD5 can bindDNA as a homodimer. Because we could not resolve het-erodimers formed between wild-type SRF and SRFD5, wemixed SRFD5 with truncated SRFDC. SRFDC was recruitedinto a novel complex displaying an intermediate mobility, dem-onstrating the capability of SRFD5 to heterodimerize withother SRF species (Fig. 5A). To directly assess heterodimer-ization of SRFD5 with SRF, we used GST pull-down assays.SRFD5 and luciferase translated in vitro in the presence of35S-labeled methionine were incubated with purified GST orGST-SRF immobilized on glutathione-linked beads. After ex-tensive washing, SRFD5 but not luciferase was retained by theimmobilized GST-SRF, demonstrating that SRFD5 can het-erodimerize with SRF (Fig. 5B); in control assays, we detectedonly background levels of SRFD5 binding to glutathione-linkedbeads.
SRFD5 is a potent repressor of SRE-dependent myogenicpromoters. The activation domain of SRF was mapped previ-ously to the C terminus (22, 26, 30). Earlier studies have shownthat the mutation of the DNA binding domain or deletion ofthe entire transactivating domain of SRF confers a dominantnegative phenotype on the mutant SRF (3, 7, 15, 16). Becauseapproximately one-third of the transactivating region of SRF isencoded by exon 5, we asked if whether SRFD5 also acted asa dominant negative mutant. We evaluated the role of SRFD5on the cardiac, skeletal, smooth a-actin, Sm22a, and SRF genepromoters, because these promoters contain several SREsand promoter activity is highly dependent on SRF binding.Plasmids encoding the wild-type SRF (pCGNSRF), SRFD5(pCGNSRFD5), a C-terminal truncated SRF mutant in whichamino acids 266 to 508 were deleted (pCGNSRFDC), and orthe empty vector (pCGN) were cotransfected with cardiac andskeletal a-actin, Sm22a, and SRF promoter-reporter con-structs into CV1 cells, and reporter activity was assayed 48 hposttransfection. As shown in Fig. 6, all of the promoters wereactivated severalfold by forced expression of SRF; in contrastto wild-type SRF, SRFD5 repressed promoter activity by nearly75 to 90%. As reported previously, SRFDC, in which all of thepreviously mapped transactivation domain was deleted, re-pressed SRF promoter activity by 50% (Fig. 6D). Surprisingly,
FIG. 3. Inverse gradient of expression of SRFD5, SM22a, and SM-MHCalong the aorta. (A) Total RNA isolated from the aortic arch (lane 1) andabdominal aorta (lane 2) analyzed by semiquantitative RT-PCR with the indi-cated end-labeled primers. SRF was amplified for 25 cycles, and SM22a andSM-MHC were amplified for 20 cycles. The linearity of amplification was con-firmed by harvesting the amplified products at 20, 23, 25, 28, and 30 cycles.Constitutively and alternatively spliced isoforms of SRF are diagrammaticallyrepresented to the right. (B) Relative optical densities of bands.
VOL. 19, 1999 SRF GENE REGULATION 4585
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
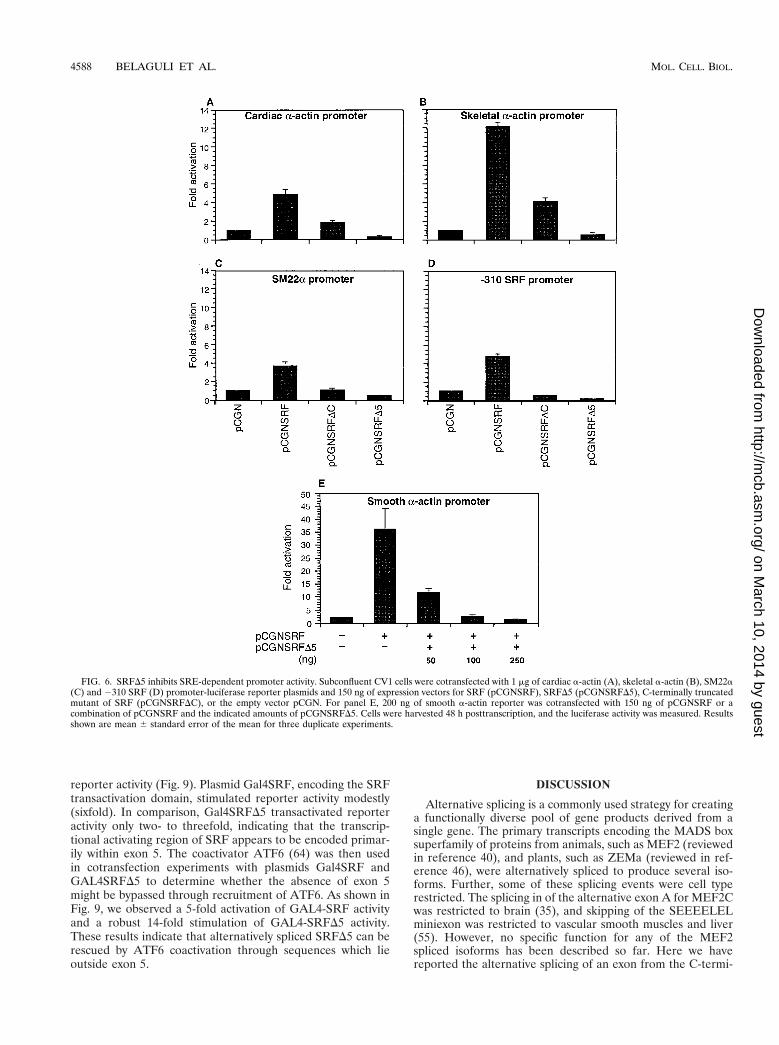
this plasmid activated both the skeletal actin and cardiac actinpromoters to a lower extent (4.5-fold for the skeletal actinpromoter and 2-fold for the cardiac actin promoter). SRFD5repressed the SRF-stimulated activity of smooth a-actin pro-moter activity in a dose-dependent manner (Fig. 6E). Theseresults indicate that SRFD5 acts as a potent repressor of SRF-dependent promoters.
SRFD5 interferes with myogenic differentiation of C2C12cells. Stably transfected C2C12 cell lines were generated withexpression vectors for SRFD5 and SRFpm1 along withpSV2neo. Three days following the addition of low-serum-containing differentiation medium wild-type C2C12 cells showedthe morphological characteristics of fused, multinucleated, dif-ferentiated myotubes, while SRFD5 and SRFpm1 cells re-
FIG. 4. SRFD5 protein is expressed in NIH 3T3 cells and several other mouse cell lines. (A to C) Western blot analysis of SRF and SRFD5 proteins. Protein extracts(20 mg) prepared from NIH 3T3 cells and day 4 EBs were used for Western analysis. NIH 3T3 cell extract (lane 2) was mixed with CV1 whole-cell extract overexpressingSRF (lane 1), SRFD5 (lane 3), and GATA-4 (lane 4) and probed with the polyclonal immune serum raised against bacterially expressed SRF protein. Lanes 2 and 4were exposed three to four times longer than lanes 1 and 3. In panel B, the C-terminal epitope-specific antibody (Santa Cruz) was used. This blot was stripped andsubsequently reacted with an exon 5 epitope-specific immune serum (C). Expression of SRFD5 proteins in NIH 3T3 cells (D) and mouse embryonic fibroblasts (E) wasdemonstrated by EMSA. Whole-cell extracts (WCE; 5 mg) were preincubated with a 50-fold excess of the indicated specific (self and cardiac a-actin SRE1) andnonspecific (Sp1) competitors and 0.5 ml of polyclonal immune serum. Annealed SRE1 oligonucleotide from the SRF promoter was the probe. Positions of SRF,SRFD5, and supershifted and nonspecific (NS) complexes and of the free probe are indicated. The gel in panel E was run for a longer time to resolve the SRF andSRFD5 complexes.
4586 BELAGULI ET AL. MOL. CELL. BIOL.
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
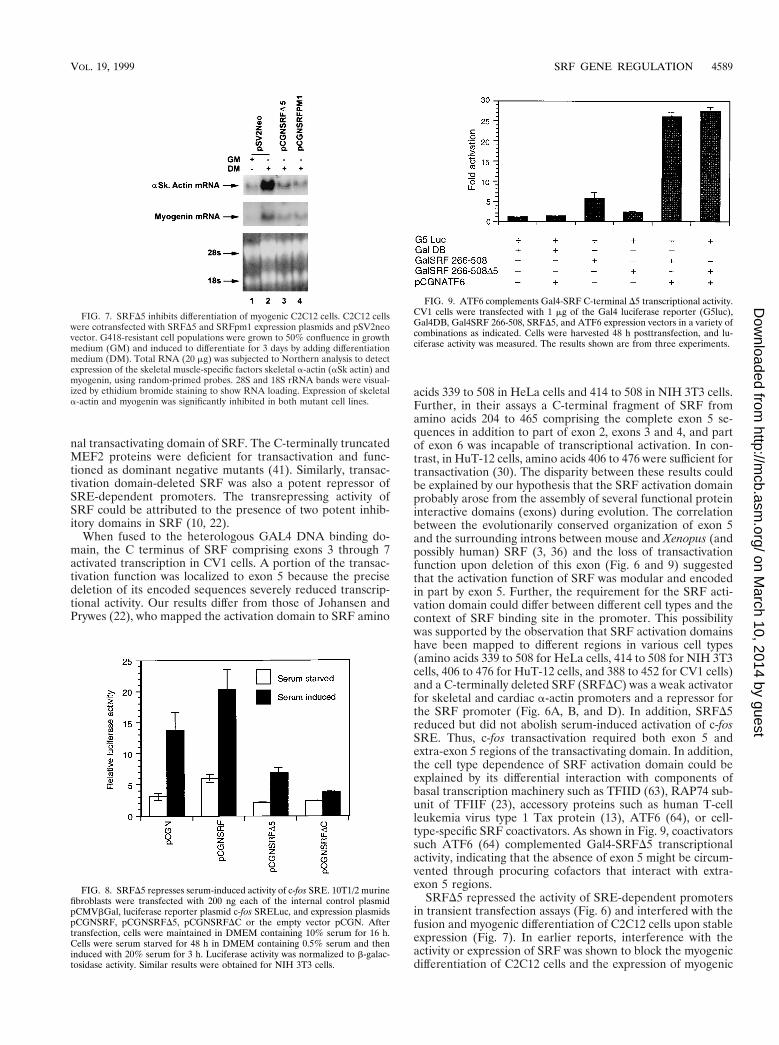
mained as replicating mononucleated, stellate myoblasts (datanot shown). As shown in Fig. 7, total RNA Northern blotanalysis detected the appearance of the muscle-specific skele-tal a-actin and myogenin RNA transcripts, using random-primed cDNA probes in wild-type C2C12 fused myotubes.Expression of both the endogenous skeletal a-actin and myo-genin genes was significantly inhibited in both mutant celllines. Since myogenin is not thought to be regulated by SRF,these results indicate both direct and indirect inhibitory activ-ities of SRFD5, consistent with its proposed role as a myogenicdominant negative inhibitor.
SRFD5 repressed the serum-induced activity of the c-fosSRE. Since SREs have contextural differences and are notequivalent, we wanted to evaluate the role of SRFD5 on aminimal c-fos SRE promoter. Rapid induction and subsequentdownregulation of c-fos response following serum growth fac-tor stimulation have been shown to be mediated by the dyadsymmetry element, the core of which binds SRF (10, 17). Weassessed the effect of overexpression of SRFD5 on the serumresponsiveness of c-fos SRE. 10T1/2 mouse fibroblasts werecotransfected with the c-fos SRE luciferase reporter, the ex-pression vectors for SRF and SRFD5, and the empty vectorpCGN. After 48 h of serum starvation, cells were induced with20% serum for 3 h. As shown in Fig. 8, overexpression of SRF
increased both basal and serum-induced responses of the c-fosSRE. Overexpression of SRFDC and SRFD5 did not affect thebasal activity of the c-fos SRE. However, serum responsivenesswas significantly reduced but not eliminated by SRFD5. Incontrast to SRFD5, SRFDC attenuated the serum inducibilityof the c-fos SRE. Essentially similar results were obtained forNIH 3T3 cells (data not shown). These results indicate thatthe absence of exon 5 may be bypassed to some extentthrough recruitment of transcription factors that interactwith extra-SRFD5 regions of the transcriptional activatingdomain.
ATF6 complements Gal4-SRFD5 transcriptional activity.To further determine the role of exon 5 and extra-exon 5regions of the transactivation domain, we then evaluated therole of two Gal4-SRF fusion constructions; plasmid Gal4SRFcontains the C-terminal transcriptional activation domain ofSRF (amino acids 266 to 508) fused to the Gal4 DNA bindingdomain, while Gal4SRFD5 contains the precise deletion ofexon 5-encoded sequences within the C-terminal domain. Ex-pression plasmids GalDB, Gal4SRF, and Gal4SRFD5 werecotransfected with a luciferase reporter containing five copiesof the upstream activation sequence cloned upstream of theminimal adenovirus E1B promoter into CV1 cells. Cotransfec-tion of the reporter with the Gal4DB alone did not increase
FIG. 5. SRFD5 forms DNA binding-competent homodimers and heterodimers. (A) The EMSA conditions were as described for Fig. 4D and E. The bindingreaction mixture contained unprogrammed (UP) RRL (lane 2), 2.5 ml of in vitro-translated SRFD5 in programmed (P) RRL (lanes 3 and 5), and 2.5 mg of CV1whole-cell extract overexpressing SRFDC (lanes 4 and 5). In vitro-translated SRFD5 was preincubated with CV1 whole-cell extract overexpressing SRFDC in lane 5.SRFD5 and SRFDC homodimers and SRFD5-SRFDC heterodimers are indicated by arrows to the left. (B) SRFD5 heterodimerizes with SRF independent of DNAbinding. [35S]methionine-labeled in vitro-translated luciferase and SRFD5 were incubated with GST (lanes 2 and 5) or GST-SRF (lanes 3 and 6) immobilized onglutathione beads, washed extensively, and analyzed on an SDS–10% polyacrylamide gel. Lane 1 and 4 contained 10% of the input luciferase and SRFD5, respectively.
VOL. 19, 1999 SRF GENE REGULATION 4587
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
reporter activity (Fig. 9). Plasmid Gal4SRF, encoding the SRFtransactivation domain, stimulated reporter activity modestly(sixfold). In comparison, Gal4SRFD5 transactivated reporteractivity only two- to threefold, indicating that the transcrip-tional activating region of SRF appears to be encoded primar-ily within exon 5. The coactivator ATF6 (64) was then usedin cotransfection experiments with plasmids Gal4SRF andGAL4SRFD5 to determine whether the absence of exon 5might be bypassed through recruitment of ATF6. As shown inFig. 9, we observed a 5-fold activation of GAL4-SRF activityand a robust 14-fold stimulation of GAL4-SRFD5 activity.These results indicate that alternatively spliced SRFD5 can berescued by ATF6 coactivation through sequences which lieoutside exon 5.
DISCUSSION
Alternative splicing is a commonly used strategy for creatinga functionally diverse pool of gene products derived from asingle gene. The primary transcripts encoding the MADS boxsuperfamily of proteins from animals, such as MEF2 (reviewedin reference 40), and plants, such as ZEMa (reviewed in ref-erence 46), were alternatively spliced to produce several iso-forms. Further, some of these splicing events were cell typerestricted. The splicing in of the alternative exon A for MEF2Cwas restricted to brain (35), and skipping of the SEEEELELminiexon was restricted to vascular smooth muscles and liver(55). However, no specific function for any of the MEF2spliced isoforms has been described so far. Here we havereported the alternative splicing of an exon from the C-termi-
FIG. 6. SRFD5 inhibits SRE-dependent promoter activity. Subconfluent CV1 cells were cotransfected with 1 mg of cardiac a-actin (A), skeletal a-actin (B), SM22a(C) and 2310 SRF (D) promoter-luciferase reporter plasmids and 150 ng of expression vectors for SRF (pCGNSRF), SRFD5 (pCGNSRFD5), C-terminally truncatedmutant of SRF (pCGNSRFDC), or the empty vector pCGN. For panel E, 200 ng of smooth a-actin reporter was cotransfected with 150 ng of pCGNSRF or acombination of pCGNSRF and the indicated amounts of pCGNSRFD5. Cells were harvested 48 h posttranscription, and the luciferase activity was measured. Resultsshown are mean 6 standard error of the mean for three duplicate experiments.
4588 BELAGULI ET AL. MOL. CELL. BIOL.
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
nal transactivating domain of SRF. The C-terminally truncatedMEF2 proteins were deficient for transactivation and func-tioned as dominant negative mutants (41). Similarly, transac-tivation domain-deleted SRF was also a potent repressor ofSRE-dependent promoters. The transrepressing activity ofSRF could be attributed to the presence of two potent inhib-itory domains in SRF (10, 22).
When fused to the heterologous GAL4 DNA binding do-main, the C terminus of SRF comprising exons 3 through 7activated transcription in CV1 cells. A portion of the transac-tivation function was localized to exon 5 because the precisedeletion of its encoded sequences severely reduced transcrip-tional activity. Our results differ from those of Johansen andPrywes (22), who mapped the activation domain to SRF amino
acids 339 to 508 in HeLa cells and 414 to 508 in NIH 3T3 cells.Further, in their assays a C-terminal fragment of SRF fromamino acids 204 to 465 comprising the complete exon 5 se-quences in addition to part of exon 2, exons 3 and 4, and partof exon 6 was incapable of transcriptional activation. In con-trast, in HuT-12 cells, amino acids 406 to 476 were sufficient fortransactivation (30). The disparity between these results couldbe explained by our hypothesis that the SRF activation domainprobably arose from the assembly of several functional proteininteractive domains (exons) during evolution. The correlationbetween the evolutionarily conserved organization of exon 5and the surrounding introns between mouse and Xenopus (andpossibly human) SRF (3, 36) and the loss of transactivationfunction upon deletion of this exon (Fig. 6 and 9) suggestedthat the activation function of SRF was modular and encodedin part by exon 5. Further, the requirement for the SRF acti-vation domain could differ between different cell types and thecontext of SRF binding site in the promoter. This possibilitywas supported by the observation that SRF activation domainshave been mapped to different regions in various cell types(amino acids 339 to 508 for HeLa cells, 414 to 508 for NIH 3T3cells, 406 to 476 for HuT-12 cells, and 388 to 452 for CV1 cells)and a C-terminally deleted SRF (SRFDC) was a weak activatorfor skeletal and cardiac a-actin promoters and a repressor forthe SRF promoter (Fig. 6A, B, and D). In addition, SRFD5reduced but did not abolish serum-induced activation of c-fosSRE. Thus, c-fos transactivation required both exon 5 andextra-exon 5 regions of the transactivating domain. In addition,the cell type dependence of SRF activation domain could beexplained by its differential interaction with components ofbasal transcription machinery such as TFIID (63), RAP74 sub-unit of TFIIF (23), accessory proteins such as human T-cellleukemia virus type 1 Tax protein (13), ATF6 (64), or cell-type-specific SRF coactivators. As shown in Fig. 9, coactivatorssuch ATF6 (64) complemented Gal4-SRFD5 transcriptionalactivity, indicating that the absence of exon 5 might be circum-vented through procuring cofactors that interact with extra-exon 5 regions.
SRFD5 repressed the activity of SRE-dependent promotersin transient transfection assays (Fig. 6) and interfered with thefusion and myogenic differentiation of C2C12 cells upon stableexpression (Fig. 7). In earlier reports, interference with theactivity or expression of SRF was shown to block the myogenicdifferentiation of C2C12 cells and the expression of myogenic
FIG. 7. SRFD5 inhibits differentiation of myogenic C2C12 cells. C2C12 cellswere cotransfected with SRFD5 and SRFpm1 expression plasmids and pSV2neovector. G418-resistant cell populations were grown to 50% confluence in growthmedium (GM) and induced to differentiate for 3 days by adding differentiationmedium (DM). Total RNA (20 mg) was subjected to Northern analysis to detectexpression of the skeletal muscle-specific factors skeletal a-actin (aSk actin) andmyogenin, using random-primed probes. 28S and 18S rRNA bands were visual-ized by ethidium bromide staining to show RNA loading. Expression of skeletala-actin and myogenin was significantly inhibited in both mutant cell lines.
FIG. 8. SRFD5 represses serum-induced activity of c-fos SRE. 10T1/2 murinefibroblasts were transfected with 200 ng each of the internal control plasmidpCMVbGal, luciferase reporter plasmid c-fos SRELuc, and expression plasmidspCGNSRF, pCGNSRFD5, pCGNSRFDC or the empty vector pCGN. Aftertransfection, cells were maintained in DMEM containing 10% serum for 16 h.Cells were serum starved for 48 h in DMEM containing 0.5% serum and theninduced with 20% serum for 3 h. Luciferase activity was normalized to b-galac-tosidase activity. Similar results were obtained for NIH 3T3 cells.
FIG. 9. ATF6 complements Gal4-SRF C-terminal D5 transcriptional activity.CV1 cells were transfected with 1 mg of the Gal4 luciferase reporter (G5luc),Gal4DB, Gal4SRF 266-508, SRFD5, and ATF6 expression vectors in a variety ofcombinations as indicated. Cells were harvested 48 h posttransfection, and lu-ciferase activity was measured. The results shown are from three experiments.
VOL. 19, 1999 SRF GENE REGULATION 4589
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

regulatory factors such as MyoD and myogenin and muscle-specific markers such as skeletal a-actin and troponin T (7, 16,60). The repression of SRE-dependent promoters by SRFD5appears to be due to the formation of SRF-SRFD5 het-erodimers and SRFD5 homodimers at the SRE (Fig. 5). Inaddition to repression caused by exclusion of SRF binding,SRFD5 homodimers might actively repress transcriptionthrough their repressor domains (10, 22). Two SREs present inthe SRF promoter mediated both the positive autoregulationof the promoter in skeletal myotubes (3) and serum inductionin NIH 3T3 cells (54). Repression of the SRF promoter activityin both cell types is likely to be mediated by binding of SRFD5to the same SREs. Therefore, the SRF promoter is both pos-itively and negatively autoregulated by alternatively splicedproducts of SRF.
The relative proportion of constitutively spliced RNA to thealternatively spliced RNA varied among the tissues examined.SRFD5 RNA was expressed at a lower level in visceral andvascular SMCs (Fig. 2 and 3). Further, SRFD5 was expressed ina rostro-caudal gradient along the aorta, with the highest levelof SRFD5 in the aortic arch and the lowest in the abdominalaorta. The SMC subpopulations within these segments arederived from distinct lineages and differ from each other intheir growth potential and response to transforming growthfactor b (57) and expression of differentiation markers such astropoelastin (14). Interestingly, SM22a and SM-MHC, whichare regulated by SRF, showed an inversely graded expressionto that of SRFD5. We suspect this inverse gradient of SM22aand SM-MHC expression is a consequence of SRFD5-medi-ated repression because overexpression of SRFD5 inhibitedboth endogenous (25) and transfected SM22a promoter activ-ities. SMCs differ from striated muscle cells in that they readilydedifferentiate and reenter cell cycle in response to varioussignals (reviewed in reference 42). Thus, SMCs may requirehigher levels of SRF to be maintained in the nonproliferatingcontractile state. Alternatively, upregulation of SRFD5 couldrepress the SRE-dependent genes and induce the preprolifera-tive synthetic state in these cells. Induction of the syntheticstate is an important step in the pathogenesis of atherosclerosis(reviewed in reference 48). Studies are under way to investi-gate if the signals that induce dedifferentiation of SMCs in-crease the level of SRFD5 RNA and protein.
In addition to SRFD5, we have detected several other alter-natively spliced isoforms of SRF during differentiation of em-bryonic stem cells into EBs, a process which precisely recapit-ulates the events that occur during early mouse embryogenesis.Based on the SRF cDNA sequence, splice variants lackingindividual exons 4 and 6 and double exons 4-5 and 5-6 arepredicted to express isoforms of SRF with different C termini.Since EBs contain diverse population of cells from differentlineages, different SRF isoforms could serve different functionsamong these cell lineages. Work is under way to characterizethese novel isoforms of SRF.
Several antagonistic protein isoforms derived from the sameprimary transcript by alternative splicing or alternative trans-lation initiation have been reported (reviewed in reference 31).Generation of functionally antagonistic proteins by regulatedalternative splicing of a primary transcript would facilitaterapid termination of the cellular response or switching to thealternate modes of response. An important role for SRFD5 canbe envisioned in silencing the activity of immediate-early genesin differentiated cells and the rapid termination of their re-sponses to mitogens and growth factors. Previous studies havesuggested that both the induction and termination of c-fosresponse are mediated by the SRE (17). In quiescent cells,c-fos expression could be kept repressed by the occupation of
SRE by SRFD5. The growth factor stimuli could result in therapid displacement of SRFD5 with SRF, allowing the expres-sion of c-fos. This could also explain why SRF appeared to beconstitutively bound to SRE irrespective of serum stimulation(20). Termination of the response could again be mediated bydisplacement of SRF by SRFD5. Possibly, the induction ofSRFD5 could also play an important role in the dedifferenti-ation and proliferation of SMCs in wound repair, neovascular-ization, and pathological processes such as atherosclerosis andheart failure. We believe that the novel alternatively splicedisoform of SRF, SRFD5, that we have reported here couldserve as a valuable tool for dissection of the role of SRF in theactivation of immediate-early genes and muscle-specific genesand ultimately play a regulatory role in modulating contractileprotein gene expression that is dependent on SRF.
ACKNOWLEDGMENT
This study was supported by National Institutes of Health grantPO1HL49953.
REFERENCES
1. Annweiler, A., M. Muller-Immergluck, and T. Wirth. 1992. Oct2 transacti-vation from a remote enhancer position requires a B-cell-restricted activity.Mol. Cell. Biol. 12:3107–3116.
2. Arsenian, S., B. Weinhold, M. Oelgeschlager, U. Ruther, and A. Nordheim.1998. Serum response factor is essential for mesoderm formation duringmouse embryogenesis. EMBO J. 17:6289–6299.
3. Belaguli, N. S., L. A. Schildmeyer, and R. J. Schwartz. 1997. Organizationand myogenic restricted expression of the murine serum response factorgene. J. Biol. Chem. 272:18222–18231.
4. Belaguli, N. S., J. L. Sepulveda, V. Nigam, and R. J. Schwartz. Unpublisheddata.
5. Bickmore, W. A., K. Oghene, M. H. Little, A. Seawright, V. VanHeyningen,and N. D. Hastie. 1992. Modulation of DNA binding specificity by alternativesplicing of the Wilms tumor wt1 gene transcript. Science 257:235–238.
6. Chen, C.-Y., and R. J. Schwartz. 1996. Recruitment of the tinman homologNkx-2.5 by serum response factor activates cardiac a-actin gene transcrip-tion. Mol. Cell. Biol. 16:6372–6384.
7. Croissant, J. D., J. H. Kim, G. Eichele, L. Goering, J. Lough, R. Prywes, andR. J. Schwartz. 1996. Avian serum response factor expression restrictedprimarily to muscle cell lineages is required for alpha-actin gene transcrip-tion. Dev. Biol. 177:250–264.
8. Das, G., and W. Herr. 1993. Enhanced activation of the human histone H2Bpromoter by an Oct-1 variant generated by alternative splicing. J. Biol.Chem. 268:25026–25032.
9. Epstein, J. A., T. Glaser, J. Cai, L. Jepeal, D. S. Walton, and R. L. Mass.1994. Two independent and interactive DNA-binding subdomains of thePax6 paired domain are regulated by alternative splicing. Genes Dev.8:2022–2034.
10. Ernst, W. H., R. Janknecht, M. A. Cahill, and A. Nordheim. 1995. Tran-scriptional repression mediated by the serum response factor. FEBS Lett.357:45–49.
11. Foulkes, N. S., and P. Sassone-Corsi. 1992. More is better: activators andrepressors from the same gene. Cell 68:411–414.
12. Foulkes, N. S., B. Mellstrom, E. Benusiglio, and P. Sassone-Corsi. 1992.Developmental switch of CREM function during spermatogenesis: fromantagonist to activator. Nature 355:80–84.
13. Fujii, M., H. Tsuchiya, T. Chuhjo, T. Akizawa, and M. Seiki. 1992. Interac-tion of HTLV-1 Tax1 with p67SRF causes the aberrant induction of cellularimmediate early genes through CArG boxes. Genes Dev. 6:2066–2076.
14. Gadson, P. F., Jr., C. Rossignol, J. McCoy, and T. H. Rosenquist. 1993.Expression of elastin, smooth muscle alpha-actin, and c-jun as a function ofthe embryonic lineage of vascular smooth muscle cells. In Vitro Cell Dev.Biol. Anim. 29A:773–781.
15. Gauthier-Rouviere, C., Q. Q. Cai, N. Lautredou, A. Fernandez, J. M. Blan-chard, and N. J. Lamb. 1993. Expression and purification of the DNA-binding domain of SRF: SRF-DM, a part of a DNA-binding protein whichcan act as a dominant negative mutant in vivo. Exp. Cell Res. 209:208–215.
16. Gauthier-Rouviere, C., M. Vandromme, D. Tuil, N. Lautredou, M. Morris,M. Soulez, A. Kahn, A. Fernandez, and N. J. Lamb. 1996. Expression andactivity of serum response factor is required for expression of the muscle-determining factor MyoD in both dividing and differentiating mouse C2C12myoblasts. Mol. Biol. Cell 7:719–729.
17. Gius, D., X. M. Cao, F. J. Rauscher, D. R. Cohen, T. Curran, and V. P.Sukhatme. 1990. Transcriptional activation and repression by Fos are inde-pendent functions: the C terminus represses immediate-early gene expres-sion via CArG elements. Mol. Cell. Biol. 10:4243–4255.
4590 BELAGULI ET AL. MOL. CELL. BIOL.
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

18. Groisman, R., H. Masutani, M.-P. Leibovitch, P. Robin, I. Soudant, D.Trouche, and A. Harel-Bellan. 1996. Physical interaction between the mito-gen-responsive serum response factor and myogenic basic-helix-loop-helixproteins. J. Biol. Chem. 271:5258–5264.
19. Han, T.-H., and R. Prywes. 1995. Regulatory role of MEF2D in seruminduction of the c-jun promoter. Mol. Cell. Biol. 15:2907–2915.
20. Herrera, R. E., P. E. Shaw, and A. Nordheim. 1989. Occupation of the c-fosserum response element in vivo by a multi-protein complex is unaltered bygrowth factor induction. Nature 340:68–70.
21. Hewitt, S. M., G. C. Fraizer, Y.-J. Wu, F. J. Rauscher III, and G. F. Saun-ders. 1996. Differential function of Wilms’ tumor gene WT1 splice isoformsin transcriptional regulation. J. Biol. Chem. 271:8588–8592.
22. Johansen, F.-E., and R. Prywes. 1993. Identification of transcriptional acti-vation and inhibitory domains in serum response factor (SRF) by usingGAL4-SRF constructs. Mol. Cell. Biol. 18:4640–4647.
23. Jollot, V., M. Demma, and R. Prywes. 1995. Interaction with RAP74 subunitof TFIIF is required for transcriptional activation by serum response factor.Nature 373:632–635.
24. Kozmik, Z., R. Kurzbauer, P. Dorfler, and M. Busslinger. 1993. Alternativesplicing of Pax-8 gene transcripts is developmentally regulated and generatesisoforms with different transactivation properties. Mol. Cell. Biol. 13:6024–6035.
25. Landerholm, T. E., X. R. Dong, J. Lu, N. S. Belaguli, R. J. Schwartz, and M.Majesky. A role for serum response factor in coronary smooth muscledifferentiation from proepicardial cell. Development 126:2053–2062.
26. Lee, T.-C., Y. Shi, and R. J. Schwartz. 1992. Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal alpha-actin tran-scription in embryonic myoblasts. Proc. Natl. Acad. Sci. USA 89:9814–9818.
27. Leifer, D., D. Krainc, Y.-T. Yu, J. McDermott, R. E. Breitbart, J. Heng, R. L.Neve, B. Kosofsky, B. Nadal-Ginard, and B. Lipton. 1992. MEF2C, a MADS/MEF2-family transcription factor expressed in a laminar distribution in ce-rebral cortex. Proc. Natl. Acad. Sci. USA 90:1546–1550.
28. Li, L., Z.-C. Liu, B. Mercer, P. Overbeek, and E. N. Olson. 1997. Evidencefor serum response factor-mediated regulatory networks governing SM22atranscription in smooth, skeletal, and cardiac muscle cells. Dev. Biol. 187:311–321.
29. Lillycrop, K. A., and D. S. Latchman. 1992. Alternative splicing of the Oct-2transcription factor is differentially regulated in B cells and neuronal cellsand results in protein isoforms with opposite effects on the activity of oc-tamer/TAATGARAT-containing promoters. J. Biol. Chem. 267:24960–24966.
30. Liu, S. H., J. T. Ma, A. Y. Yueh, S. P. Lees-Miller, C. W. Anderson, and S. Y.Ng. 1993. The carboxyl-terminal transactivation domain of human serumresponse factor contains DNA-activated protein kinase phosphorylationsites. J. Biol. Chem. 268:21147–21154.
31. Lopez, J. A. 1995. Developmental role of transcription factor isoforms gen-erated by alternative splicing. Dev. Biol. 172:396–411.
32. Macneil, C., B. Ayres, A. C. Laverriere, and J. E. Burch. 1997. Transcripts forfunctionally distinct isoforms of chicken GATA-5 are differentially expressedfrom alternative first exons. J. Biol. Chem. 272:8396–8401.
33. Martin, J. F., J. J. Schwarz, and E. N. Olson. 1993. Myocyte enhancer factor(MEF) 2C: a tissue-restricted member of the MEF-2 family of transcriptionfactors. Proc. Natl. Acad. Sci. USA 90:5282–5286.
34. Martin, J. F., J. M. Miano, C. M. Hustad, N. G. Copeland, N. A. Jenkins, andE. N. Olson. 1994. A Mef2 gene that generates a muscle-specific isoform viaalternative mRNA splicing. Mol. Cell. Biol. 14:1647–1656.
35. McDermott, J. C., M. C. Cordoso, Y.-T. Yu, V. Andres, D. Leifer, D. Krainc,S. A. Lipton, and B. Nadal-Ginard. 1993. HMEF2C gene encodes skeletalmuscle- and brain-specific transcription factors. Mol. Cell. Biol. 13:2564–2577.
36. Mohun, T. J., A. E. Chambers, N. Towers, and M. V. Taylor. 1991. Expres-sion of genes encoding the transcription factor SRF during early develop-ment of Xenopus laevis: identification of a CArG box-binding activity asSRF. EMBO J. 10:933–940.
37. Morris, A. E., B. Kloss, R. E. McChesney, C. Bancroft, and L. A. Chasin.1992. An alternatively spliced Pit-1 isoform altered in its ability to transac-tivate. Nucleic Acids Res. 20:1355–1361.
38. Morris, P. J., T. Theil, C. J. A. Ring, K. A. Lillycrop, T. Moroy, and D. S.Latchman. 1994. The opposite and antagonistic effects of the closely relatedPOU family transcription factors on the activity of a target promoter aredependent upon differences in the POU domain. Mol. Cell. Biol. 14:6907–6914.
39. Nornes, S., I. Mikkola, S. Krauss, M. Delghandi, M. Perander, and T.Johansen. 1996. Zebrafish Pax9 encodes two proteins with distinct C-termi-nal transactivating domains of different potency negatively regulated byadjacent N-terminal sequences. J. Biol. Chem. 271:26914–26923.
40. Olson, E. N., M. Perry, and R. A. Schulz. 1995. Regulation of muscle
differentiation by the MEF2 family of MADS box transcription factors. Dev.Biol. 172:2–14.
41. Ornatsky, O. I., J. J. Andreucci, and J. C. McDermott. 1997. A dominant-negative form of transcription factor MEF2 inhibits myogenesis. J. Biol.Chem. 272:33271–33278.
42. Owens, G. K. 1995. Regulation of differentiation of vascular smooth musclecells. Physiol. Rev. 75:487–517.
43. Pellegrini, L., S. Tans, and T. J. Richmond. 1995. Structure of serum re-sponse factor core bound to DNA. Nature 376:490–498.
44. Pollock, R., and R. Treisman. 1991. Human SRF-related proteins: DNA-binding properties and potential regulatory targets. Genes Dev. 5:2327–2341.
45. Reecy, J., N. S. Belaguli, and R. J. Schwartz. 1998. SRF/homeobox proteininteractions, p. 273–290. In R. Harvey and N. Rosenthal (ed.), Heart devel-opment. Academic Press, San Diego, Calif.
46. Reichmann, J. L., and E. M. Meyerowitz. 1997. MADS domain proteins inplant development J. Biol. Chem. 378:1079–1101.
47. Rivera, V. M., C. K. Miranti, R. P. Misra, D. D. Ginty, R.-H. Chen, J. Blenis,and M. E. Greenberg. 1993. A growth factor-induced kinase phosphorylatesthe serum response factor at a site that regulates its DNA-binding activity.Mol. Cell. Biol. 13:6260–6273.
48. Ross, R. 1993. Atherosclerosis: a defence mechanism gone awry. Am. J.Pathol. 143:987–1002.
49. Ruzicka, D. L., and R. J. Schwartz. 1988. Sequential activation of alpha-actingenes during avian cardiogenesis: vascular smooth muscle alpha-actin genetranscripts mark the onset of cardiomyocyte differentiation. J. Cell Biol.107:2575–2586.
50. Sartorelli, V., K. Webster, and L. Kedes. 1990. Muscle-specific expression ofthe cardiac a-actin gene requires MyoD1, CArG-box binding factor, andSP1. Genes Dev. 4:1811–1822.
51. Schwarz-Sommer, Z., P. Huijser, W. Nacken, H. Seadler, and H. Sommer.1990. Genetic control of flower development by homeotic genes in Antir-rhinum majus. Science 250:931–936.
52. Shore, P., and A. D. Sharrocks. 1995. The MADS-box family of transcriptionfactors. Eur. J. Biochem. 229:1–13.
53. Soulez, M., C. Gauthier-Rouviere, P. Chafey, D. Hentzen, M. Vandromme,N. Lautredou, N. Lamb, N. Kahn, and D. Tuil. 1996. Growth and differen-tiation of C2 myogenic cells are dependent on serum response factor. Mol.Cell. Biol. 16:6065–6074.
54. Spencer, J. A., and R. P. Misra. 1996. Expression of the serum responsefactor gene is regulated by serum response factor binding sites. J. Biol.Chem. 271:16535–16543.
55. Suzuki, E., K. M. Guo, M. Kolman, Y.-T. Yu, and K. Walsh. 1995. Seruminduction of MEF2/RSRF in vascular myocytes is mediated at the level oftranslation. Mol. Cell. Biol. 15:3415–3423.
56. Tanaka, T., K. Tanaka, S. Ogawa, M. Kurokawa, K. Mitani, J. Nishida, Y.Shibata, Y. Yazaki, and H. Hirai. 1995. An acute myeloid leukemia gene,AML1, regulates hemopoietic myeloid cell differentiation and transcrip-tional activation antagonistically by two alternatively spliced forms. EMBO J.14:341–350.
57. Topouzis, S., and M. Majesky. 1996. Smooth muscle lineage diversity in thechick embryo. Two types of aortic smooth muscle cell differ in growth andreceptor-mediated transcriptional responses to transforming growth factor-beta. Dev. Biol. 178:430–445.
58. Treisman, R. 1995. Journey to the surface of the cell: Fos regulation and theSRE. EMBO J. 14:4905–4913.
59. Tsukamoto, K., Y. Nakamura, and N. Niikawa. 1994. Isolation of two iso-forms of the PAX3 gene transcripts and their tissue-specific alternativeexpression in human adult tissues. Hum. Genet. 93:270–274.
60. Vandromme, M., C. Gauthier-Rouviere, G. Carnac, N. Lamb, and A. Fer-nandez. 1992. Serum response factor p67SRF is expressed and requiredduring myogenic differentiation of both mouse C2 and rat L6 muscle celllines. J. Cell Biol. 118:1489–1500.
61. Yin, J. C. P., J. S. Wallach, E. L. Wilder, J. Klingensmith, D. Dang, N.Perrimon, H. Zhou, T. Tully, and W. G. Quinn. 1995. A Drosophila CREB/CREM homolog encodes multiple isoforms, including a cyclic AMP-depen-dent protein kinase-responsive transcriptional activator and antagonist. Mol.Cell. Biol. 15:5123–5130.
62. Yu, Y.-T., R. E. Breitbart, L. B. Smoot, Y. Lee, V. Mahdavi, and B. Nadal-Ginard. 1992. Human myocyte-specific enhancer factor 2 comprises a groupof tissue-restricted MADS box transcription factors. Genes Dev. 6:1783–1798.
63. Zhu, H., A. L. Roy, R. G. Roeder, and R. Prywes. 1991. Serum responsefactor affects preinitiation complex formation by TFIID in vitro. New Biol.3:455–464.
64. Zhu, C., F.-E. Johansen, and R. Prywes. 1997. Interaction of ATF6 andserum response factor. Mol. Cell. Biol. 17:4957–4966.
VOL. 19, 1999 SRF GENE REGULATION 4591
on March 10, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















