
Domain Organization and Crystal Structure of the Catalytic Domain of E.coli RluF, a Pseudouridine...
12
Domain Organization and Crystal Structure of the Catalytic Domain of E. coli RluF, a Pseudouridine Synthase that Acts on 23S rRNA S. Sunita 1 , H. Zhenxing 1 , J. Swaathi 1 , Miroslaw Cygler 2 Allan Matte 2 and J. Sivaraman 1 * 1 Department of Biological Sciences, National University of Singapore, 14 Science Drive Singapore, Singapore 117543 2 Biotechnology Research Institute, National Research Council, Canada, 6100 Royalmount Avenue, Montre ´al Que., Canada H4P 2R2 Pseudouridine synthases catalyze the isomerization of uridine to pseudouridine (J) in rRNA and tRNA. The pseudouridine synthase RluF from Escherichia coli (E.C. 4.2.1.70) modifies U2604 in 23S rRNA, and belongs to a large family of pseudouridine synthases present in all kingdoms of life. Here we report the domain architecture and crystal structure of the catalytic domain of E. coli RluF at 2.6 A ˚ resolution. Limited proteolysis, mass spectrometry and N-terminal sequencing indicate that RluF has a distinct domain architecture, with the catalytic domain flanked at the N and C termini by additional domains connected to it by flexible linkers. The structure of the catalytic domain of RluF is similar to those of RsuA and TruB. RluF is a member of the RsuA sequence family of J- synthases, along with RluB and RluE. Structural comparison of RluF with its closest structural homologues, RsuA and TruB, suggests possible functional roles for the N-terminal and C-terminal domains of RluF. q 2006 Elsevier Ltd. All rights reserved. Keywords: pseudouridine synthase; crystal structure; RluF; ribosome; RNA modifying enzyme *Corresponding author Introduction Pseudouridine synthases are present in all king- doms of life and catalyze the isomerization of uridine to pseudouridine (J) in structural RNAs. These enzymes modify specific uridine bases in the rRNA and tRNAs of prokaryotes and eukaryotes, as well as in small nuclear and nucleolar RNAs of eukaryotes. 1–4 Pseudouridine synthesis, along with ribose methylation, is among the most abundant modifications in rRNA. Bacterial pseudouridine synthases are grouped into five sequence families: TruA, TruB, RsuA, RluA 5 and recently TruD. 6 Despite the lack of overall sequence similarity between the families, their three-dimensional struc- tures adopt the same fold, they share a conserved catalytic aspartyl residue and have similar sub- strate-binding clefts, 7–9 indicating a common evol- utionary origin. It has been shown that all pseudouridines in the rRNA of the large ribosomal subunit in organisms from Escherichia coli to human cluster around the peptidyl transferase center (PTC) of the ribosome, 10–14 implying the importance of this modification for the proper function of the ribosome. 15,16 Though the exact role of each pseudouridine residue is not clear, various sugges- tions have been put forward, including a role in the correct processing of rRNA, RNA folding, ribosome tertiary structure or ribosome function. 13,15,17 E. coli rRNA contains 11 pseudouridines, one within 16S rRNA and the remaining ten in 23S rRNA. 11,18,19 The single J516 within the small subunit rRNA is synthesized by the enzyme RsuA. 12 Pseudouridines located in the large subunit rRNA (23 S) are modified by six different pseudour- idine synthases. 19 RluF (formerly YjbC) specifically modifies the uridine at position 2604 and belongs to the RsuA family of J synthases with which it shares 22% sequence identity. 19 Both deletion and site-specific mutagenesis studies confirmed that Asp107, located within the motif GRLD, 19 a conserved sequence found in members of the RsuA family, 20 is the catalytically essential aspartate in RluF. While deletion or mutation of the RluF gene does not affect the growth rate of E. coli, the location 0022-2836/$ - see front matter q 2006 Elsevier Ltd. All rights reserved. Abbreviations used: MS, mass spectrometry; MALDI- TOF, matrix-assisted laser desorption ionization-time of flight; MAD, multiwavelength anomalous dispersion; PDB, Protein Data Bank. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2006.04.019 J. Mol. Biol. (2006) 359, 998–1009
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Domain Organization and Crystal Structure of the Catalytic Domain of E.coli RluF, a Pseudouridine...

00
doi:10.1016/j.jmb.2006.04.019 J. Mol. Biol. (2006) 359, 998–1009
Domain Organization and Crystal Structure of theCatalytic Domain of E. coli RluF, a PseudouridineSynthase that Acts on 23S rRNA
S. Sunita1, H. Zhenxing1, J. Swaathi1, Miroslaw Cygler2
Allan Matte2 and J. Sivaraman1*
1Department of BiologicalSciences, National University ofSingapore, 14 Science DriveSingapore, Singapore 117543
2Biotechnology ResearchInstitute, National ResearchCouncil, Canada, 6100Royalmount Avenue, MontrealQue., Canada H4P 2R2
22-2836/$ - see front matter q 2006 Else
Abbreviations used: MS, mass spTOF, matrix-assisted laser desorptioflight; MAD, multiwavelength anomPDB, Protein Data Bank.
E-mail address of the [email protected]
Pseudouridine synthases catalyze the isomerization of uridine topseudouridine (J) in rRNA and tRNA. The pseudouridine synthaseRluF from Escherichia coli (E.C. 4.2.1.70) modifies U2604 in 23S rRNA, andbelongs to a large family of pseudouridine synthases present in allkingdoms of life. Here we report the domain architecture and crystalstructure of the catalytic domain of E. coli RluF at 2.6 A resolution. Limitedproteolysis, mass spectrometry and N-terminal sequencing indicate thatRluF has a distinct domain architecture, with the catalytic domain flankedat the N and C termini by additional domains connected to it by flexiblelinkers. The structure of the catalytic domain of RluF is similar to those ofRsuA and TruB. RluF is a member of the RsuA sequence family of J-synthases, along with RluB and RluE. Structural comparison of RluF withits closest structural homologues, RsuA and TruB, suggests possiblefunctional roles for the N-terminal and C-terminal domains of RluF.
q 2006 Elsevier Ltd. All rights reserved.
Keywords: pseudouridine synthase; crystal structure; RluF; ribosome; RNAmodifying enzyme
*Corresponding authorIntroduction
Pseudouridine synthases are present in all king-doms of life and catalyze the isomerization ofuridine to pseudouridine (J) in structural RNAs.These enzymes modify specific uridine bases in therRNA and tRNAs of prokaryotes and eukaryotes, aswell as in small nuclear and nucleolar RNAs ofeukaryotes.1–4 Pseudouridine synthesis, along withribose methylation, is among the most abundantmodifications in rRNA. Bacterial pseudouridinesynthases are grouped into five sequence families:TruA, TruB, RsuA, RluA5 and recently TruD.6
Despite the lack of overall sequence similaritybetween the families, their three-dimensional struc-tures adopt the same fold, they share a conservedcatalytic aspartyl residue and have similar sub-strate-binding clefts,7–9 indicating a common evol-utionary origin. It has been shown that all
vier Ltd. All rights reserved.
ectrometry; MALDI-n ionization-time ofalous dispersion;
ing author:
pseudouridines in the rRNA of the large ribosomalsubunit in organisms from Escherichia coli to humancluster around the peptidyl transferase center (PTC)of the ribosome,10–14 implying the importance ofthis modification for the proper function of theribosome.15,16 Though the exact role of eachpseudouridine residue is not clear, various sugges-tions have been put forward, including a role in thecorrect processing of rRNA, RNA folding, ribosometertiary structure or ribosome function.13,15,17
E. coli rRNA contains 11 pseudouridines, onewithin 16S rRNA and the remaining ten in 23SrRNA.11,18,19 The single J516 within the smallsubunit rRNA is synthesized by the enzymeRsuA.12 Pseudouridines located in the large subunitrRNA (23 S) are modified by six different pseudour-idine synthases.19 RluF (formerly YjbC) specificallymodifies the uridine at position 2604 and belongs tothe RsuA family of J synthases with which itshares 22% sequence identity.19 Both deletion andsite-specific mutagenesis studies confirmed thatAsp107, located within the motif GRLD,19 aconserved sequence found in members of theRsuA family,20 is the catalytically essential aspartatein RluF. While deletion or mutation of the RluF genedoes not affect the growth rate of E. coli, the location
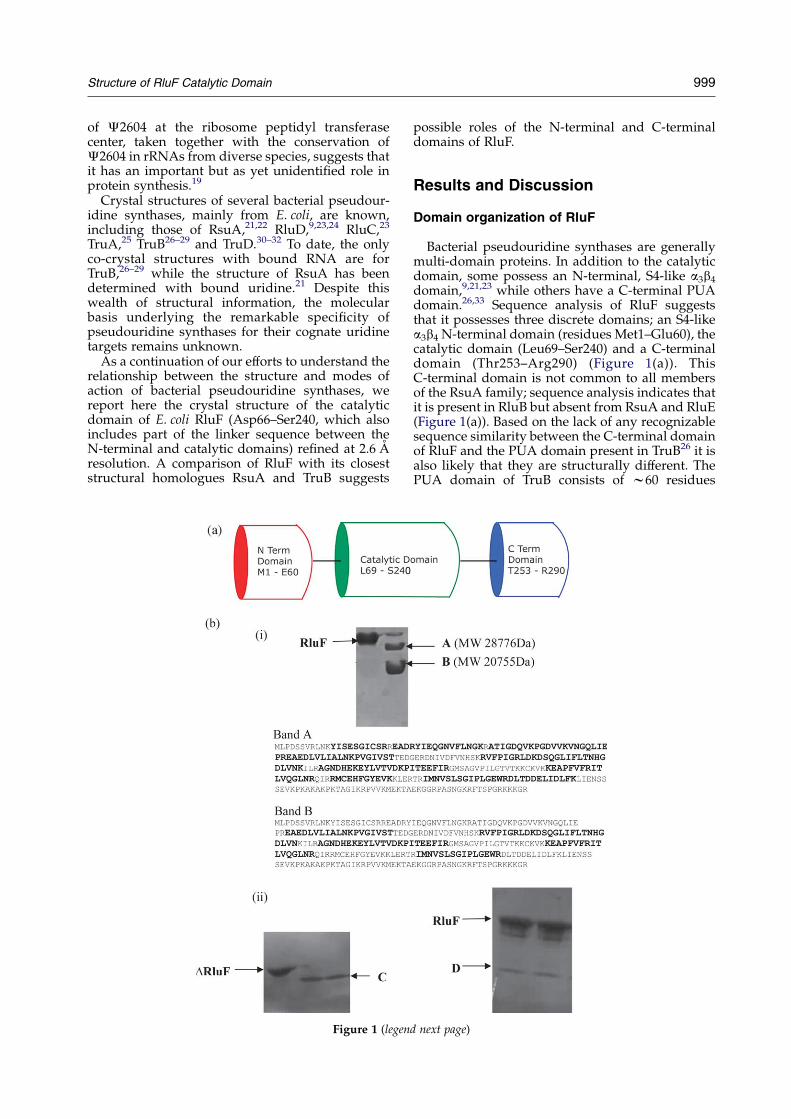
Structure of RluF Catalytic Domain 999
of J2604 at the ribosome peptidyl transferasecenter, taken together with the conservation ofJ2604 in rRNAs from diverse species, suggests thatit has an important but as yet unidentified role inprotein synthesis.19
Crystal structures of several bacterial pseudour-idine synthases, mainly from E. coli, are known,including those of RsuA,21,22 RluD,9,23,24 RluC,23
TruA,25 TruB26–29 and TruD.30–32 To date, the onlyco-crystal structures with bound RNA are forTruB,26–29 while the structure of RsuA has beendetermined with bound uridine.21 Despite thiswealth of structural information, the molecularbasis underlying the remarkable specificity ofpseudouridine synthases for their cognate uridinetargets remains unknown.
As a continuation of our efforts to understand therelationship between the structure and modes ofaction of bacterial pseudouridine synthases, wereport here the crystal structure of the catalyticdomain of E. coli RluF (Asp66–Ser240, which alsoincludes part of the linker sequence between theN-terminal and catalytic domains) refined at 2.6 Aresolution. A comparison of RluF with its closeststructural homologues RsuA and TruB suggests
Figure 1 (legen
possible roles of the N-terminal and C-terminaldomains of RluF.
Results and Discussion
Domain organization of RluF
Bacterial pseudouridine synthases are generallymulti-domain proteins. In addition to the catalyticdomain, some possess an N-terminal, S4-like a3b4
domain,9,21,23 while others have a C-terminal PUAdomain.26,33 Sequence analysis of RluF suggeststhat it possesses three discrete domains; an S4-likea3b4 N-terminal domain (residues Met1–Glu60), thecatalytic domain (Leu69–Ser240) and a C-terminaldomain (Thr253–Arg290) (Figure 1(a)). ThisC-terminal domain is not common to all membersof the RsuA family; sequence analysis indicates thatit is present in RluB but absent from RsuA and RluE(Figure 1(a)). Based on the lack of any recognizablesequence similarity between the C-terminal domainof RluF and the PUA domain present in TruB26 it isalso likely that they are structurally different. ThePUA domain of TruB consists of w60 residues
d next page)
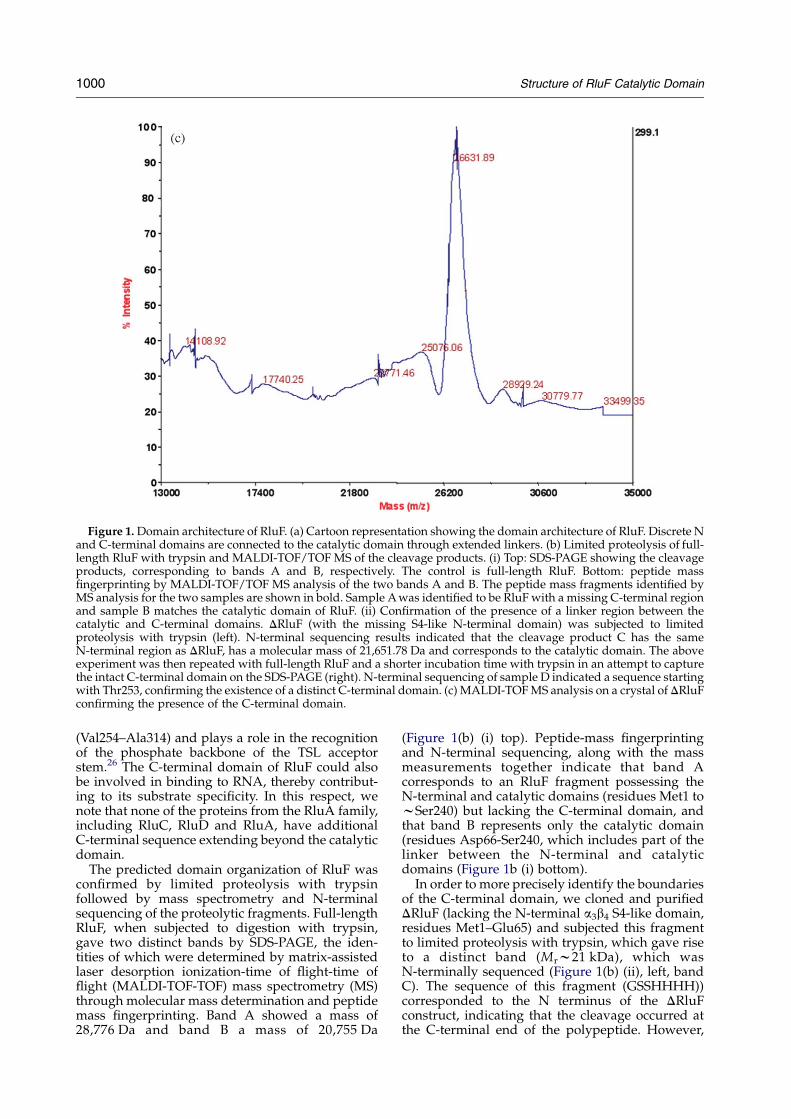
Figure 1. Domain architecture of RluF. (a) Cartoon representation showing the domain architecture of RluF. Discrete Nand C-terminal domains are connected to the catalytic domain through extended linkers. (b) Limited proteolysis of full-length RluF with trypsin and MALDI-TOF/TOF MS of the cleavage products. (i) Top: SDS-PAGE showing the cleavageproducts, corresponding to bands A and B, respectively. The control is full-length RluF. Bottom: peptide massfingerprinting by MALDI-TOF/TOF MS analysis of the two bands A and B. The peptide mass fragments identified byMS analysis for the two samples are shown in bold. Sample A was identified to be RluF with a missing C-terminal regionand sample B matches the catalytic domain of RluF. (ii) Confirmation of the presence of a linker region between thecatalytic and C-terminal domains. DRluF (with the missing S4-like N-terminal domain) was subjected to limitedproteolysis with trypsin (left). N-terminal sequencing results indicated that the cleavage product C has the sameN-terminal region as DRluF, has a molecular mass of 21,651.78 Da and corresponds to the catalytic domain. The aboveexperiment was then repeated with full-length RluF and a shorter incubation time with trypsin in an attempt to capturethe intact C-terminal domain on the SDS-PAGE (right). N-terminal sequencing of sample D indicated a sequence startingwith Thr253, confirming the existence of a distinct C-terminal domain. (c) MALDI-TOF MS analysis on a crystal of DRluFconfirming the presence of the C-terminal domain.
1000 Structure of RluF Catalytic Domain
(Val254–Ala314) and plays a role in the recognitionof the phosphate backbone of the TSL acceptorstem.26 The C-terminal domain of RluF could alsobe involved in binding to RNA, thereby contribut-ing to its substrate specificity. In this respect, wenote that none of the proteins from the RluA family,including RluC, RluD and RluA, have additionalC-terminal sequence extending beyond the catalyticdomain.
The predicted domain organization of RluF wasconfirmed by limited proteolysis with trypsinfollowed by mass spectrometry and N-terminalsequencing of the proteolytic fragments. Full-lengthRluF, when subjected to digestion with trypsin,gave two distinct bands by SDS-PAGE, the iden-tities of which were determined by matrix-assistedlaser desorption ionization-time of flight-time offlight (MALDI-TOF-TOF) mass spectrometry (MS)through molecular mass determination and peptidemass fingerprinting. Band A showed a mass of28,776 Da and band B a mass of 20,755 Da
(Figure 1(b) (i) top). Peptide-mass fingerprintingand N-terminal sequencing, along with the massmeasurements together indicate that band Acorresponds to an RluF fragment possessing theN-terminal and catalytic domains (residues Met1 towSer240) but lacking the C-terminal domain, andthat band B represents only the catalytic domain(residues Asp66-Ser240, which includes part of thelinker between the N-terminal and catalyticdomains (Figure 1b (i) bottom).
In order to more precisely identify the boundariesof the C-terminal domain, we cloned and purifiedDRluF (lacking the N-terminal a3b4 S4-like domain,residues Met1–Glu65) and subjected this fragmentto limited proteolysis with trypsin, which gave riseto a distinct band (Mrw21 kDa), which wasN-terminally sequenced (Figure 1(b) (ii), left, bandC). The sequence of this fragment (GSSHHHH))corresponded to the N terminus of the DRluFconstruct, indicating that the cleavage occurred atthe C-terminal end of the polypeptide. However,

Structure of RluF Catalytic Domain 1001
due to the abundance of tryptic cleavage sites in theC-terminal region and relatively long incubationtimes used, a band corresponding to this domaincould not be readily observed on an SDS-PAGE gel.The above experiment was therefore repeated withfull-length RluF, but with a shorter incubation timewith trypsin. A band with Mrw4.1 kDa wasobserved on the gel, and its N-terminal sequencedetermined to be Thr253-Arg258, corresponding tothe C-terminal domain containing residues Thr253–Arg290 (Figure 1(b) (ii), right, band D). This domainis connected to the catalytic domain by a wtenresidue linker, Glu242–Lys252.
From these studies, we conclude that RluFpossesses a discrete domain organization withdomain boundaries Met1 to Glu60 (N-terminaldomain), Leu69 to Ser240 (catalytic domain) andThr253–Arg290 (C-terminal domain), with thedomains connected via linkers that are readilyaccessible to proteases. The presence of an N-terminalS4-like a3b4 domain in RsuA, RluD and now RluF isfurther supported by sequence alignments andstructural studies of RluD9 and RsuA.21,22
Crystallization of the RluF catalytic domain
While our objective was to crystallize full-lengthRluF, crystals of the full-length enzyme could not beobtained following stringent purification and manycrystallization trials. This inability to crystallize full-length RluF could be attributed to a relatively looseassociation of the domains. Indeed, sequence
Table 1. Crystallographic data and refinement statistics
Data set PeakCell parameters and space group aZ65.60, bZ65.60, cData collectionResolution range (A) 50–2.Wavelength (A) 0.979Observed reflectionsO1 273,25Unique reflections 27,55Completeness (%) 99.8Overall (I/sI) 14.1Rsym (%)a 10.4Refinement and qualityResolution range (A)Rwork (no. of reflections)b
Rfree (no. of reflections)c
rmsd bond lengths (A)rmsd bond angles(deg)Average B-factors (A2)d
Main-chainSide-chains
B-rmsd main-chain (A2)B-rmsd side-chain (A2)C-V sigma-A coordinate error (A)Ramachandran plotMost favored regions (%)Additional allowed regions (%)Generously allowed regions (%)Disallowed regions (%)
a RsymZjIiKhIij/jIij where Ii is the intensity of the ith measuremenb RworkZjFobs–Fcalcj/jFobsj where Fcalc and Fobs are the calculatedc RfreeZas for Rwork, but for 8% of the total reflections chosen at rad Individual B-factor refinement was carried out.
alignment of RluF and RsuA shows that the inter-domain linker region between the N-terminal S4-like a3b4 domain and the catalytic domain is tworesidues longer in RluF than in RsuA. The mobilityof the catalytic domain relative to the S4-likedomain has been documented in RsuA,21,22 andhas proven to be detrimental to crystallization inRluD9 and RluC,34 or results in a domain that isdisordered in the crystal.23,24 Upon purification, afaint band with a molecular mass of w25 kDa, lessthan the full-length enzyme (32.4 kDa), wasobserved by SDS-PAGE (data not shown). Thislower band became more intense as the proteinsample was stored at 4 8C, even in the presence of acocktail of protease inhibitors.
In order to overcome these problems, a newconstruct was made (DRluF) in which the first 65amino acid residues, corresponding to the S4-likea3b4 domain of RluF were deleted. Crystals wereobtained of DRluF, containing the catalytic andC-terminal domains, expressed as a fusion proteinwith a non-cleavable N-terminal His6-tag.
Quality and overall structure
The crystal structure of Se-Met-labeled DRluFwas solved by the multi-wavelength anomalousdispersion (MAD) method.35 DRluF crystallizedwith two molecules in the asymmetric unit, andthe structure was refined to a final R-factor of 0.225(RfreeZ0.287) at 2.6 A resolution with good stereo-chemical parameters (Table 1).
InflectionZ215.71, P43212 aZ65.72, bZ65.72, cZ215.92, P43212
6 50–2.62 0.97944 264,903
5 27,72699.516.211.5
45.0–2.60.225 (25,538)0.287 (2140)
0.0091.6
44.144.21.462.000.52
84.01420
t, and hIi is the mean intensity for that reflection.and observed structure factor amplitudes, respectively.ndom and omitted from refinement.

1002 Structure of RluF Catalytic Domain
Each monomer of DRluF has an a/b architecturewith a central, extended b-sheet core. The moleculecontains 13 b-strands, seven of which form acentral, twisted b-sheet. The order of antiparallelb-strands within the b-sheet is b13Yb1[b5Yb11Yb6[b10Yb9[. This extended b-sheet is twistedalong its longitudinal axis (perpendicular to thestrands), and is flanked on all sides by a total of fivea-helices. The substrate-binding site is located in acleft near the center of the b-sheet and has toaccommodate the RNA substrate. The catalyticresidue, Asp107 (in RluF),19 common to all bacterialpseudouridine synthases, is located in a loopbetween strands b4 and b5 at the edge of the
Figure 2. Structure of DRluF. (a) Ribbon diagram of the DRlub-strands are labeled. (b) Ribbon diagram of the DRluF crystaThe catalytic Asp107 is shown in stick representation. FigureRaster3D.58
RNA-binding cleft. Except for some local differ-ences outside the substrate-binding cleft, the overallfold of DRluF is similar to that found in otherbacterial pseudouridine synthases.9,21
No electron density was observed for the last50 residues of the protein corresponding to theC-terminal domain. To check if this region wasindeed still present in the crystallized protein weanalyzed the protein from dissolved DRluF crystalsby MALDI-TOF MS (Figure 1(c)). The observedmolecular mass of 26,631 Da compared favorablywith the expected molecular mass of 26,558 Da,indicating that the Se-Met labeled protein containedboth the N-terminal His-tag and the intact
F monomer. The N and C termini as well as a-helices andllographic dimer (two monomers of one asymmetric unit).s were prepared using the programs MOLSCRIPT57 and

Structure of RluF Catalytic Domain 1003
C-terminal domain. The lack of electron density forthe C-terminal domain indicates that it is eitherunstructured or is highly mobile within the crystallattice (Figure 2(a)). Indeed, the C-terminal domainhas a basic character, with a calculated pI of 11.9resulting from eight Lys and five Arg residueswithin its sequence. Analysis of the C-terminaldomain sequence using either DisEMBL36 orGlobPlot37 shows a high probability that a signifi-cant portion of the domain is unfolded. Werationalize that this domain becomes folded uponassociation of RluF with its cognate RNA,a property of many ribosomal RNA-binding pro-teins.38,39 Examination of the crystal packing ofRluF molecules shows that sufficient space exists inthe crystal lattice to accommodate the C-terminaldomain.
Oligomerization of RluF and DRluF
The oligomerization state of RluF and the DRluFfragment missing the S4-like a3b4 domain wasinvestigated by dynamic light-scattering (DLS)and gel filtration chromatography. DLS analysis ofpurified, full-length RluF showed a molecular massthat corresponded to that of a dimer, similarly, DLSmeasurements showed that DRluF also exists as anapparent dimer in solution. In a gel filtrationexperiment of DRluF at a concentration of 4 mg/ml, a 7.5-fold lower protein concentration than thatused for crystallization, the enzyme eluted as asingle peak with an apparent molecular mass of50 kDa (26.6 kDa per monomer), indicating thepossibility of dimers in solution. Both dynamiclight-scattering and gel filtration experimentsdepend on the transport/hydrodynamic radius ofthe molecules in solution. If the C-terminal domainis indeed disordered/unfolded, then the possibilityof its contribution to an increased hydrodynamicradius of the molecule cannot be ignored. Toinvestigate this possibility, we have carried outanalytical ultracentrifugation (AUC) sedimentationanalysis. The AUC experiments revealed theexistence of RluF as a monomer in solution with asedimentation co-efficient sZ(1.52G0.07)!10K13,which corresponds to a molecular mass of26,326(G3066) Da with a relative abundance of84(G9)%. We suggest that the observed molecularmass in the dynamic light-scattering andgel filtration experiments may be due to thedisordered/unfolded C-terminal domain of RluF.The observed dimeric arrangement in the crystal,i.e. two molecules in the asymmetric unit of thecrystal, could be a crystallization artifact. With theexception of TruA, the structurally characterizedbacterial pseudouridine synthases have beenshown to exist as monomers. The two monomerswithin the asymmetric unit are related by a 2-foldnon-crystallographic symmetry axis nearly parallelwith the crystallographic c-axis (Figure 2(b)). Thestructures of the two independent monomers in theasymmetric unit are similar, with a root-mean-square (rms) deviation of 0.69 A for all Ca atoms,
when refined without non-crystallographic sym-metry restraints.
Comparison of the sequences of the C-terminaldomains of RluF and TruB shows that they differ inlength and sequence, suggesting that they mayrepresent two unique types of domains inJ-synthases, each with its own specificity forsubstrate recognition. As both domains are attachedto the catalytic domain through extended linkers,they are evidently mobile with respect to thecatalytic domain. In the proposed model of full-length RluF, the N and C-terminal domains arelocated at opposite ends of the catalytic domain. Asthese domains are not immediately adjacent tothe RNA substrate-binding cleft, they may notdirectly participate in binding RNA structure nearthe catalytic site, but instead, may contribute tothe recognition of RNA structure distant from theactive site, which would be important for theexquisite specificity exhibited by this enzyme.
Sequence and structural similarity
Pairwise sequence alignments show that RluF has31% sequence identity with RluB, 22% with RsuA and21% with RluE. The most important region is thehighly conserved active site motif (X)RLD (motifII5,41) containing the essential catalytic aspartate42
(Figure 3).Comparison of the structure of DRluF with those
of other pseudouridine synthases was performedusing the program sPDBviewer.43 Pairwise super-positions showed that the catalytic domain of RluFis structurally similar to the correspondingdomain found in RsuA (PDB 1KSK, 1.4 A, 140 Ca
pairs), TruB (PDB 1K8W, 1.6 A for 101 Ca pairs),RluD (PDB 1PRZ, 1.7 A for 93 Ca pairs), RluC (PDB1V9K, 1.0 A for 49 Ca pairs) and TruA (PDB 1DJO,1.6 A for 80 Ca pairs). In all cases, the central b-sheetis the most structurally conserved feature, whilewith RsuA, some a-helical regions also sharestructural similarity. Among these proteins, themost structurally similar to RluF were found to beRsuA and TruB. The structure-based sequencealignment of RluF with RsuA and TruB is summar-ized in Figure 3(b). Sixteen invariant side-chains areobserved in the structure-based alignment of thethree structures, including a number of residues inthe active site region (Leu72, Lys74, Pro75, Gly77,Gly104, Leu106, Asp107, Gly113, Lys135, Tyr137,Val139, Gly184, Gly197, Val200, Leu203 andArg205). His43 of TruB, a highly sequence-con-served but non-essential residue in catalysis,44 isnot structurally conserved in RluF (Figure 5).
RNA binding and recognition
A cleft 9 A wide, 22 A long and 14 A deep suitablefor binding the rRNA substrate is located in themiddle of the catalytic domain. The shape andsurface electrostatic potential of the molecular sur-face calculated using GRASP40 revealed adepression in the center of the cleft with a positively

Figure 3. (a) Sequence alignment of the S4-like N-terminal domain of members of the RsuA family done usingClustalW.59,60 (b) Alignment of the catalytic domains. Structure-based sequence alignment was performed for the topthree sequences titled in red (RsuA_ec and TruB_ec) and blue (RluF_ec) using the program O.52 The secondary structuralelements and the sequence numbering shown in blue are for RluF_ec (E. coli). For RluF_sh (Shigella flexneri), RluF_sa(Salmonella typhi), RluB_ec (E. coli) and RluE_ec (E. coli), only sequence-based alignment was carried out. The threesequence and structurally conserved motifs (motifs 1, 2 and 3)5 are indicated by blue dotted lines. The active siteaspartate (D107 for RluF) is shown by a blue asterisk. The residues involved in binding to the substrate are denoted byred asterisks, identified by the structural superposition of DRluF with TSL-bound TruB. (c) Sequence alignment of the C-terminal domains of RluF and RluB done using ClustalW. These Figures were created by using the program ESPript.61
1004 Structure of RluF Catalytic Domain
charged region inside the depression that couldprovide a suitable site for rRNA binding, similar tothat observed in RluD.9 (Figure 4). Superposition ofDRluF with the TruB–TSL co-crystal structure26
shows that the T-stem loop readily fits within thesubstrate-binding cleft of RluF without stericclashes. A total of 41 possible hydrogen bondingcontacts (!3.2 A) are formed between the super-imposed TSL and DRluF, 13 of which are with thetarget fluorouridine. Though the substrate is differ-ent for RluF, similar interactions can be expected.The walls of the putative RNA-binding cleft containseveral positively charged residues, includingLys125, Lys135, Arg187, and Arg190. The conservedcatalytic Asp107 is located at the entrance of the cleftwith its side-chain directed towards the solvent.
In E. coli 23 S rRNA (PDB 1PNU), U2604 islocated at the bottom of a cleft towards one side,with the base buried within the cleft. It would benecessary to “flip out” this base in order to bringabout the modification, probably in a way similar toU55 in the TruB–TSL complex.26,28
Experimental Procedures
Expression and purification
The full-length rluF gene, as well as an N-terminal, 65-residue truncation (DRluF) were amplified from E. coliO157:H7 EDL933 genomic DNA45 using oligonucleotideprimers (IDT, Coralville, IA, USA) and cloned into the

Figure 5. Stereo Ca superposition of DRluF (red), RsuAsuperposition was generated using the program O,52 using tchain of the active site Asp of DRluF and bound UMP of RsuAmodels are 1.6 A for 155 Ca atoms for RsuA and 1.9 A for 11program MOLSCRIPT.57
Figure 4. Molecular surface of the DRluF (monomer)depicting the positively charged, putative RNA-bindingcleft (blue) prepared using the program GRASP.40 Theactive site aspartate (Asp107) is located within thisdepression as indicated.
Structure of RluF Catalytic Domain 1005
plasmid pF04, a derivative of pET15b (Novagen) contain-ing an N-terminal, non-cleavable His6 tag. Plasmid DNAwas transformed into E. coli BL21(DE3) and grown in 1 lof LB broth at 37 8C until the A600 nm reached 0.5–0.6. Theculture was induced with 100 mM IPTG and continued togrow at 25 8C overnight. Cells were harvested bycentrifugation (9000g; 20 min, 4 8C) and resuspended in40 ml of lysis buffer (50 mM Tris–HCl (pH 7.5), 400 mMNaCl, 5% (v/v) glycerol, 10 mM imidazole, 0.5% (v/v)Triton X-100, 10 mM b-mercaptoethanol and one tablet ofCompletee protease inhibitor cocktail (Roche Diagnos-tics)). Selenomethionine-substituted RluF was preparedby growing cells under conditions of endogenousmethionine synthesis inhibition in M9 medium, asdescribed.46 Incorporation of SeMet in the protein wasverified by MALDI-TOF MS.
Purification of full-length RluF and DRluF wasperformed in two steps using DEAE Sepharose FastFlow (Amersham BioSciences) followed by affinitychromatography with Ni-NTA agarose (Qiagen). Thenon-cleavable His6-tag protein was eluted with 350 mMimidazole following a series of wash steps. The elutedprotein was dialyzed overnight against buffer (20 mMTris–HCl (pH 7.5), 250 mM NaCl, 5% glycerol, 10 mMDTT, 0.1 mM EDTA) to remove imidazole and furtherpurified on a FPLC Hiload 16/60 Superdex75 gelfiltration column using an AKTA FPLC UPC-900 system(Amersham Biosciences), followed by concentrationof the protein by ultrafiltration to 40 mg/ml. Formolecular mass determination, the gel filtration columnwas calibrated with low molecular mass standards fromBio-Rad.
(PDB 1KSK, blue) and TruB (PDB 1K8W, green). Thehe catalytic Asp residues as the starting point. The side-are shown. The rmsd between DRluF and the superposed
2 Ca atoms for TruB. This Figure was prepared using the

Figure 6. Sedimentation velocity data for DRluF. Thedata were analyzed by the software ULTRASCAN 7.3.Initial sedimentation profile was analyzed by the vanHolde–Weischet method to identify the homogeneity ofthe solution.
1006 Structure of RluF Catalytic Domain
Dynamic light-scattering
Dynamic light-scattering experiments on purifiedRluF/DRluF were performed using a DynaProe (ProteinSolutionse) dynamic light-scattering instrument. Thequality of the protein samples was monitored duringthe various stages of concentration in order to avoidaggregation. Experiments were carried out at 20 8C. ThePolydIndx was lower than 0.1 for all protein samples atvarious concentrations.
Limited proteolysis and N-terminal sequencing
A sample of purified RluF or DRluF (1 mg/ml) wasincubated at 20 8C with trypsin (1 mg/ml) at a ratio of100:1 (w/w) for 30–60 min. Digestion products wereseparated by 12.5% (w/v) SDS-PAGE. The protein wasthen transferred to a PVDF membrane by using atransblot apparatus (Biorad) at 200 mA for 1 h andsubjected to automated Edman degradation.
MALDI-TOF MS and MS-MS analysis
Molecular mass determination was done with the aid ofa Voyager STR MALDI-TOF mass spectrometer (AppliedBiosystems). For MS/MS analysis, sample digestion,desalting and concentration steps were carried out byusing the Montagew In-Gel digestion Kits (MilliporeCorp.) Protein spots were analyzed using an AppliedBiosystems 4700 Proteomics Analyzer MALDI-TOF/TOF(Applied Biosystems, Framigham, MA, USA). Dataprocessing and interpretation was carried out using theGPS explorer Software (Applied Biosystems) and data-base searching was done using the MASCOT program(Matrix Science Ltd., London, UK). The NCBI databasewas used for combined MS and MS/MS search.
Analytical ultracentrifugation
Samples for analytical ultracentrifugation were dilutedin buffer (50 mM Hepes (pH 7.5), 250 mM NaCl, 5% (v/v)glycerol, 5 mM BME and 0.1 mM EDTA) to obtain anabsorbance (A280) of 1.0, 0.5 and 0.25 at 280 nm. Finaldilutions were 1.0, 0.4 and !0.2, the last dilution wasdiscarded from the analysis. Sedimentation velocity datawere analyzed by the software ULTRASCAN 7.3 (BorriesDemeler, University of Texas System, San Antonio,Texas). Initial sedimentation profile was analyzed by thevan Holde–Weischet method to identify homogeneity/heterogeneity of the solution. Coefficient of sedimentation(s) and buoyant molecular weight (MW) were obtainedwith C(s) analysis with global frictional ratio (f/fo)floating to convergence (Figure 6).
Crystallization
Crystallization trials were carried out at 20 8C byhanging-drop vapor-diffusion using crystallizationscreens from Hampton Research (Aliso Viejo, CA, USA)and by the microbatch method using JB crystallizationscreens (Jena Biosciences, Jena, Germany). Initial crystalsof DRluF was obtained in JB Screen 6, condition D2containing 3 M ammonium sulfate, 1% (v/v) 2-methyl-2,4-pentanediol (MPD). The best native crystals wereobtained by mixing 0.5 ml of protein in buffer with 0.5 mlcrystallization solution under paraffin oil in a microbatchplate. Rod-shaped crystals with the smallest dimensionmeasuring w0.07 mm were obtained over the course of
four days. Crystals belonged to space group P3221 withaZbZ163.9 A and cZ59.6 A and diffracted up to 3.0 Aresolution.
For crystallization of selenomethionyl-DRluF, the pro-tein was concentrated to 30 mg/ml, using a reservoircontaining 2 M ammonium sulfate and 200 mM KSCN,by the hanging-drop vapor-diffusion method by mixing1 ml of protein with 2 ml of buffer (20 mM Tris–HCl(pH 7.5), 250 mM NaCl, 5% glycerol, 10 mM DTT,0.1 mM EDTA) and 3 ml of reservoir solution. Crystalsmeasuring w0.11 mm in length grew over the courseof two days, and belonged to space group P43212 withaZbZ65.7 A and cZ215.9 A and contained two mol-ecules in the asymmetric unit. The Matthews coefficient is2.2 A3/Da,47 giving a solvent content of 44%.
Data collection, structure solution and refinement
The best cryoprotectant was identified to be a 1:1mixture of mineral oil and paraffin oil. Crystals werecryoprotected in the above solution and flash cooled in aN2 cold stream at 100 K. X-ray diffraction data for nativeDRluF crystals were collected to a resolution of 3.0 Ausing an R-axis IVCC image plate detector mounted on aRU-H3RHB rotating anode generator (Rigaku Corp.,Tokyo, Japan). Attempts to solve the structure bymolecular replacement, using the program AMoRe48
with the structure of E. coli RsuA as the search modelwere unsuccessful. Subsequently, the structure wasdetermined using crystals of SeMet-labeled protein bythe multiwavelength anomalous dispersion (MAD)method.35 X-ray diffraction data were collected at beam-line X25, National Synchrotron Light Source, BrookhavenNational Laboratory using a Q315 CCD detector (AreaDetector Systems Corp., Poway, CA, USA). Two data setswere collected at wavelengths corresponding to the peakand the inflection point of the Se K edge. Data wereprocessed and scaled to a resolution of 2.6 A using theprogram HKL2000.49 Six of the ten expected Se sites werelocated using the program SOLVE.50 The missing Se sitesturned out to be located in the disordered C-terminaldomain. The phases were further improved using Sharp(v3.0.15).51 Density modification was performed with the

Figure 7. Simulated-annealing FoKFc omit map in the active-site region of DRluF. Active site Asp107 and all atomswithin 3.5 A of Asp107 were omitted prior to refinement. The map is contoured at a level of 3.0s. This Figure wasprepared using the program Bobscript.62
Structure of RluF Catalytic Domain 1007
program Sharp (v3.0.15) and improved the overall figureof merit to 0.68. The initial model containing w120residues of each monomer was built automatically withthe program ARP/wARP.52 This model was completed bymanual fitting using the program O53 and refined againstthe Se inflection point dataset using the program CNS54
interspersed with several rounds of manual refitting. Allmeasured reflections were used in the refinement. TheC-terminal domain was disordered in the crystal and thefinal model contains only residues from Asp66 to Ser240for each monomer, corresponding to the catalytic domain.The N-terminal His6-tag and the linker were alsodisordered. There are 125 solvent molecules included inthe model. The final R-factor is 0.225 (RfreeZ0.287) at2.6 A resolution (Table 1). Statistics for the Ramachandranplot from an analysis using PROCHECK55 showed nooutliers in the Ramachandran plot and 84% of non-glycine residues are in the most-favored region (Table 1).An example of the electron density from a simulatedannealing omit map in the active-site region is presentedin Figure 7.
Protein Data Bank accession code
Coordinates and structure factors have been depositedwith RCSB Protein Data Bank56 with code 2GML.
Acknowledgements
We thank Pietro Iannuzzi for assistance incloning, and Drs Anand Saxena and Michael Beckerfor assistance in data collection. Data for this studywere measured at beamlines X12C and X25 ofthe National Synchrotron Light Source. Financial
support comes principally from the Office ofBiological and Environmental Research and ofBasic Energy Sciences of the US Department ofEnergy, and the National Center for ResearchResources of the National Institutes of Health.This research was supported, in part, by a grantfrom the Canadian Institutes of Health Research(200103GSP-90094-GMX-CFAA-19924) to M.C. J.S.acknowledges full research support from theAcademic Research Fund, National University ofSingapore (NUS). We thank Mr Jean-Claude Lavoiefor conducting the analytical ultracentrifugationexperiments. We also thank the Protein andProteomics Centre, Department of BiologicalSciences, NUS for providing mass spectrometryfacilities.
References
1. Maden, B. E. H. (1990). The numerous modifiednucleotides in eukaryotic ribosomal RNA. Nucl. AcidsRes. Mol. Biol. 39, 241–303.
2. Sprinzl, M., Horn, C., Brown, M., Ioudovitch, A. &Steinberg, S. (1998). Compilation of tRNA sequencesand sequences of tRNA genes. Nucl. Acids Res. 26,148–153.
3. Massenet, S., Mougin, A. & Branlant, C. (1998). Posttranscriptional modifications in the U small nuclearRNAs. In Modification and Editing of RNA (Grosjean,H. & Benne, R., eds), pp. 201–227, ASM Press,Washington, DC.
4. Ofengand, J. & Fournier, M. (1998). The pseudour-idine residues of rRNA: number, location, biosyn-thesis, and function. In Modification and Editing of RNA

1008 Structure of RluF Catalytic Domain
(Grosjean, H. & Benne, R., eds), pp. 229–254, ASMPress, Washington, DC.
5. Koonin, E. V. (1996). Pseudouridine synthases: fourfamilies of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTPdeaminases. Nucl. Acids Res. 24, 2411–2415.
6. Kaya, Y. & Ofengand, J. (2003). A novel unanticipatedtype of pseudouridine synthase with homologs inbacteria, archaea and eukarya. RNA, 9, 711–721.
7. Mueller, E. G. (2002). Chips off the old block. NatureStruct. Biol. 9, 320–322.
8. Ferre-D’Amare, A. R. (2003). RNA-modifyingenzymes. Curr. Opin. Struct. Biol. 13, 49–55.
9. Sivaraman, J., Iannuzzi, P., Cygler, M. & Matte, A.(2004). Crystal structure of the RluD pseudouridinesynthase catalytic module, an enzyme that modifies23 S rRNA and is essential for normal cell growth ofEscherichia coli. J. Mol. Biol. 335, 87–101.
10. Bakin, A. & Ofengand, J. (1993). Four newly locatedpseudouridylate residues in Escherichia coli 23 Sribosomal RNA are all at the peptidyltransferasecenter: analysis by the application of a new sequen-cing technique. Biochemistry, 32, 9754–9782.
11. Bakin, A., Lane, B. G. & Ofengand, J. (1994).Clustering of pseudouridine residues around thepeptidyltransferase center of yeast cytoplasmic andmitochondrial ribosomes. Biochemistry, 33,13475–13483.
12. Wrzesinski, J., Bakin, A., Nurse, K., Lane, B. &Ofengand, J. (1995). Purification, cloning and proper-ties of the 16 S RNA 516 synthase from Escherichia coli.Biochemistry, 34, 8904–8913.
13. Ofengand, J., Bakin, A., Wrzesinski, J., Nurse, K. &Lane, B. G. (1995). The pseudouridine residues ofribosomal RNA. Biochem. Cell Biol. 73, 915–924.
14. Ofengand, J. & Bakin, A. (1997). Mapping tonucleotide resolution of pseudouridine residues inlarge subunit ribosomal RNAs from representativeeukaryotes, prokaryotes, archaebacteria, mitochon-dria and chloroplasts. J. Mol. Biol. 266, 246–268.
15. King, T. H., Liu, B., McCully, R. R. & Fournier, M. J.(2003). Ribosome structure and activity are alteredin cells lacking snoNPs that form pseudouridinesin the peptidyl transferase center. Mol. Cell, 11,425–435.
16. Gutgsell, N. S., Deutscher, M. P. & Ofengand, J. (2005).The pseudouridine synthase RluD is required fornormal ribosome assembly and function in Escherichiacoli. RNA, 11, 1141–1152.
17. Ofengand, J., Malhorta, A., Remme, J., Gutgsell, N. S.,Del Campo, M., Jean-Charles, S. et al. (2001).Pseudouridines and pseudouridine synthases of theribosome. Cold Spring Harbor Symp. Quant. Biol. 66,147–159.
18. Bakin, A., Kowalak, J. A., McCloskey, J. A. &Ofengand, J. (1994). The single pseudouridine residuein Escherichia coli 16 S RNA is located at position 516.Nucl. Acids Res. 22, 3681–3684.
19. Del Campo, M., Kaya, Y. & Ofengand, J. (2001).Identification and site of action of the remaining fourputative pseudouridine synthases in Escherichia coli.RNA, 7, 1603–1615.
20. Conrad, J., Niu, L., Rudd, K., Lane, B. G. & Ofengand,J. (1999). 16 S ribosomal RNA pseudouridine synthaseRsuA of Escherichia coli: deletion, mutation of theconserved Asp102 residue, and sequence comparisonamong all other pseudouridine synthases. RNA, 5,751–763.
21. Sivaraman, J., Sauve, V., Larocque, R., Stura, E. A.,Schrag, J. D., Cygler, M. & Matte, A. (2002). Structureof the 16 S rRNA pseudouridine synthase RsuAbound to uracil and UMP. Nature Struct. Biol. 9,353–358.
22. Matte, A., Louie, G. V., Sivaraman, J., Cygler, M. &Burley, S. K. (2005). Structure of the pseudouridinesynthase RsuA from Haemophilus influenzae. ActaCrystallog. sect. F, 61, 350–354.
23. Mizutani, K., Machida, Y., Unzai, S., Park, S-Y. &Tame, J. R. H. (2004). Crystal structures of the catalyticdomains of pseudouridine synthases RluC and RluDfrom Escherichia coli. Biochemistry, 43, 4454–4463.
24. Del Campo, M., Ofengand, J. & Malhotra, A. (2004).Crystal structure of the catalytic domain of RluD, theonly rRNA pseudouridine synthase required fornormal growth of Escherichia coli. RNA, 10, 231–239.
25. Foster, P. G., Huang, L., Santi, D. & Stroud, R. M.(2000). The structural basis for tRNA recognition andpseudouridine formation by pseudouridine synthaseI. Nature Struct. Biol. 7, 23–27.
26. Hoang, C. & Ferre-D’Amare, A. R. (2001). Cocrystalstructure of a tRNA j55 pseudouridine synthase:nucleotide flipping by an RNA-modifying enzyme.Cell, 71, 929–939.
27. Chaudhuri, B. N., Chan, S., Perry, L. J. & Yeates, T. O.(2004). Crystal structure of the apo forms of 55 tRNApseudouridine synthase from Mycobacterium tubercu-losis. J. Biol. Chem. 279, 24585–24591.
28. Pan, H., Agarwalla, S., Moustakas, D. T., Finer-Moore,J. & Stroud, R. M. (2003). Structure of tRNApseudouridine synthase TruB and its RNA complex:RNA recognition through a combination of rigiddocking and induced fit. Proc. Natl Acad. Sci. USA,100, 12648–12653.
29. Phannachet, K. & Huang, R. H. (2004). Conformation-al change of pseudouridine 55 synthase upon itsassociation with RNA substrate. Nucl. Acids Res. 32,1422–1429.
30. Kaya, Y., Del Campo, M., Ofengand, J. & Malhotra, A.(2004). Crystal structure of TruD, a novel pseudour-idine synthase with a new protein fold. J. Biol. Chem.279, 18107–18110.
31. Hoang, C. & Ferre-D’Amare, A. R. (2004). Crystalstructure of the highly divergent pseudouridinesynthase TruD reveals a circular permutation of aconserved fold. RNA, 10, 1026–1033.
32. Ericsson, U. B., Nordlund, P. & Hallberg, M. (2004). X-ray structure of tRNA pseudouridine synthase TruDreveals an inserted domain with a novel fold. FEBSLetters, 565, 59–64.
33. Aravind, L. & Koonin, E. V. (1999). Novel predictedRNA-binding domains associated with the trans-lation machinery. J. Mol. Evol. 48, 291–302.
34. Corollo, D., Blair-Johnson, M., Fiedler, T., Sun, D.,Wang, L., Ofengand, J. & Fenna, R. (1999). Crystal-lization and characterization of a fragment ofpseudouridine synthase RluC from Escherichia coli.Acta Crystallog. sect. D, 55, 302–304.
35. Hendrickson, W. A., Horton, J. R. & LeMaster, D. M.(1990). Selenomethionyl proteins produced for anal-ysis by multiwavelength anomalous diffraction(MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 9, 1665–1672.
36. Linding, R., Jensen, L. J., Diella, F., Bork, P., Gibson,T. J. & Russell, R. B. (2003). Protein disorderprediction: implications for structural proteomics.Structure, 11, 1453–1459.

Structure of RluF Catalytic Domain 1009
37. Linding, R., Russell, R. B., Neduva, V. & Gibson, T. J.(2003). GlobPlot: exploring protein sequences forglobularity and disorder. Nucl. Acids Res. 31,3701–3708.
38. Klein, D. J., Moore, P. B. & Steitz, T. A. (2004). The rolesof ribosomal proteins in the structure, assembly, andevolution of the large ribosomal subunit. J. Mol. Biol.340, 141–177.
39. Bateman, A., Birney, E., Cerruti, L., Durbin, R.,Etwiller, L., Eddy, S. R. et al. (2002). The Pfam proteinfamilies database. Nucl. Acids Res. 30, 276–280.
40. Nichols, A., Sharp, K. A. & Honig, B. (1991). Proteinfolding and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281–296.
41. Gustafsson, C., Reid, R., Greene, P. J. & Santi, D. V.(1996). Identification of new RNA modifying enzymesby iterative genome search using known modifyingenzymes as probes. Nucl. Acids Res. 24, 3756–3762.
42. Huang, L., Pookanjanatavip, M., Gu, X. & Santi, D. V.(1998). A conserved aspartate of tRNA pseudouridinesynthase is essential for activity and a probablenucleophilic catalyst. Biochemistry, 37, 344–351.
43. Guex, N. & Peitsch, M. C. (1997). Swiss-Model and theSwiss-Pdb viewer: an environment for comparativeprotein modelling. Electrophoresis, 18, 2714–2723.
44. Hamilton, C. S., Spedaliere, C. J., Ginter, J. M.,Johnston, M. V. & Mueller, E. G. (2005). The roles ofthe essential Asp-48 and highly conserved His-43elucidated by the pH dependence of the pseudour-idine synthase TruB. Arch. Biochem. Biophys. 433,322–334.
45. Perna, N. T., Plunkett, G., 3rd, Burland, V., Mau, B.,Glasner, J. D. & Blattner, F. R. (2001). Genomesequence of enterohaemorrhagic Escherichia coliO157:H7. Nature, 409, 529–533.
46. Doublie, S. (1997). Preparation of selenomethionylproteins for phase determination. Methods Enzymol.276, 523–530.
47. Matthews, B. W. (1968). Solvent content of proteincrystals. J. Mol. Biol. 33, 491–497.
48. Navaza, J. (2001). Implementation of molecularreplacement in AMoRe. Acta Crystallog. sect. D, 57,1367–1372.
49. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode.Methods Enzymol. 276, 307–326.
50. Terwilliger, T. C. & Berendzen, J. (1999). AutomatedMAD and MIR structure solution. Acta Crystallog. sect.D, 55, 849–861.
51. Bricogne, G., Vonrheim, C., Flensburg, C., Schiltz, M.& Paciorek, W. (2003). Generation, representation andflow of phase information in structure determination:recent developments in and around SHARP 2.0. ActaCrystallog. sect. D, 59, 2023–2030.
52. Perrakis, A., Morris, R. & Lamzin, V. S. (1999).Automated protein model building combined withiterative structure refinement. Nature Struct. Biol. 6,458–463.
53. Jones, T. A., Zou, J-Y., Cowan, S. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallog. sect A, 47,100–119.
54. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography and NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905–921.
55. Laskowski, R. A., McArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallog. 26, 282–291.
56. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G.,Bhat, T. N., Weissig, H. et al. (2000). The Protein DataBank. Nucl. Acids Res. 28, 235–242.
57. Kraulis, J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of protein struc-tures. J. Appl. Crystallog. 24, 946–950.
58. Merritt, E. A. & Bacon, D. J. (1997). Raster3D:photorealistic molecular graphics. Methods Enzymol.277, 505–524.
59. Chenna, R., Sugawara, H., Koike, T., Lopez, R.,Gibson, T. J., Higgins, D. G. & Thompson, J. D.(2003). Multiple sequence alignment with the Clustalseries of programs. Nucl. Acids Res. 31, 3497–3500.
60. Huang, X. & Miller, W. (1991). A time-efficient linearspace local similarity algorithm. Advan. Appl. Math.12, 337–357.
61. Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999).ESPript: multiple sequence alignments in PostScript.Bioinformatics, 15, 305–308.
62. Esnouf, R. M. (1999). Further additions to MolscriptVersion 1.4 including reading and contouring ofelectron-density maps. Acta Crystallog. sect. D, 55,938–940.
Edited by J. Doudna
(Received 2 November 2005; received in revised form 27 March 2006; accepted 5 April 2006)Available online 25 April 2006




















