
Differential Activity of NADPH-Producing Dehydrogenases Renders Rodents Unsuitable Models to Study...
15
Journal of Histochemistry & Cytochemistry 59(5) 489–503 © The Author(s) 2011 Reprints and permission: sagepub.com/journalsPermissions.nav DOI: 10.1369/0022155411400606 http://jhc.sagepub.com The finding of the high frequency of the somatic IDH1 R132 and IDH2 R172 mutations in the isocitrate dehydrogenase (IDH) genes in glioma (Parsons et al. 2008; Bleeker et al. 2009) has recently revolutionized brain tumor research. The IDH2 R172 mutation occurs at a relatively low frequency in glioma (Hartmann et al. 2009; Sonoda et al. 2009; Yan et al. 2009), but the IDH1 R132 mutation occurs in 70% to 80% of secondary glioblastoma (Balss et al. 2008; Bleeker et al. 2009; Hartmann et al. 2009; Ichimura et al. 2009; Nobu- sawa et al. 2009; Parsons et al. 2008; Sanson et al. 2009; Sonoda et al. 2009; Watanabe et al. 2009; Weller et al. 2009; Yan et al. 2009). The IDH1 R132 mutation is an early event in gliomagenesis, and patients with low-grade glioma show even higher frequencies (Parsons et al. 2008; Balss et al. 2008; Bleeker et al. 2009; Ohgaki and Kleihues 2009). The mutation is also associated with a subset of acute myeloid leukemia (Mardis et al. 2009; Chou et al. 2010; Ward et al. 2010), its precursor myelodysplastic syndrome (Andrulis et al. 2010), and thyroid cancer (Murugan et al. 2010). The IDH1 gene encodes for NADP + -dependent IDH1, which is 400606JHC 59 5 10.1369/0022155411400606Atai et al.Journal of Histochemistry & Cytochemistry © The Author(s) 2010 Reprints and permission: sagepub.com/journalsPermissions.nav Received for publication November 11, 2010; accepted January 23, 2011. Corresponding Author: C. J. F.Van Noorden, Department of Cell Biology and Histology, Academic Medical Center, Meibergdreef 15, 1105 AZ Amsterdam, the Netherlands. E-mail: [email protected]. Differential Activity of NADPH-Producing Dehydrogenases Renders Rodents Unsuitable Models to Study IDH1 R132 Mutation Effects in Human Glioblastoma Nadia A. Atai, Nynke A. Renkema-Mills, Joost Bosman, Nadja Schmidt, Denise Rijkeboer, Wikky Tigchelaar, Klazien S. Bosch, Dirk Troost, Ard Jonker, Fonnet E. Bleeker, Hrvoje Miletic, Rolf Bjerkvig, Philip C. De Witt Hamer, and Cornelis J. F.Van Noorden Department of Cell Biology and Histology, Academic Medical Center, University of Amsterdam, the Netherlands (NAA, NARM, JB,NS, DR, WT, KSB, AJ, CJFVN); Nor Lux Neuro-Oncology, Department of Biomedicine, University of Bergen, Norway (HM,RB); Department of Neuropathology, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands (DT); and Neurosurgical Center Amsterdam,VU University Medical Center, Amsterdam, the Netherlands (FEB,PCDWH). Summary The somatic IDH1 R132 mutation in the isocitrate dehydrogenase 1 gene occurs in high frequency in glioma and in lower frequency in acute myeloid leukemia and thyroid cancer but not in other types of cancer. The mutation causes reduced NADPH production capacity in glioblastoma by 40% and is associated with prolonged patient survival. NADPH is a major reducing compound in cells that is essential for detoxification and may be involved in resistance of glioblastoma to treatment. IDH has never been considered important in NADPH production.Therefore, the authors investigated NADPH- producing dehydrogenases using in silico analysis of human cancer gene expression microarray data sets and metabolic mapping of human and rodent tissues to determine the role of IDH in total NADPH production. Expression of most NADPH-producing dehydrogenase genes was not elevated in 34 cancer data sets except for IDH1 in glioma and thyroid cancer, indicating an association with the IDH1 mutation. IDH activity was the main provider of NADPH in human normal brain and glioblastoma, but its role was modest in NADPH production in rodent brain and other tissues. It is concluded that rodents are a poor model to study consequences of the IDH1 R132 mutation in glioblastoma. (J Histochem Cytochem 59:489–503, 2011) Keywords in silico analysis, glioblastoma, metabolic mapping, IDH1, G6PDH, mutation, NADPH, dehydrogenase
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Differential Activity of NADPH-Producing Dehydrogenases Renders Rodents Unsuitable Models to Study...

Journal of Histochemistry & Cytochemistry 59(5) 489 –503© The Author(s) 2011Reprints and permission: sagepub.com/journalsPermissions.navDOI: 10.1369/0022155411400606http://jhc.sagepub.com
The finding of the high frequency of the somatic IDH1R132 and IDH2R172 mutations in the isocitrate dehydrogenase (IDH) genes in glioma (Parsons et al. 2008; Bleeker et al. 2009) has recently revolutionized brain tumor research. The IDH2R172 mutation occurs at a relatively low frequency in glioma (Hartmann et al. 2009; Sonoda et al. 2009; Yan et al. 2009), but the IDH1R132 mutation occurs in 70% to 80% of secondary glioblastoma (Balss et al. 2008; Bleeker et al. 2009; Hartmann et al. 2009; Ichimura et al. 2009; Nobu-sawa et al. 2009; Parsons et al. 2008; Sanson et al. 2009; Sonoda et al. 2009; Watanabe et al. 2009; Weller et al. 2009; Yan et al. 2009). The IDH1R132 mutation is an early event in gliomagenesis, and patients with low-grade glioma show
even higher frequencies (Parsons et al. 2008; Balss et al. 2008; Bleeker et al. 2009; Ohgaki and Kleihues 2009). The mutation is also associated with a subset of acute myeloid leukemia (Mardis et al. 2009; Chou et al. 2010; Ward et al. 2010), its precursor myelodysplastic syndrome (Andrulis et al. 2010), and thyroid cancer (Murugan et al. 2010). The IDH1 gene encodes for NADP+-dependent IDH1, which is
400606 JHC59510.1369/0022155411400606Atai et al.Journal of Histochemistry & Cytochemistry© The Author(s) 2010
Reprints and permission:sagepub.com/journalsPermissions.nav
Received for publication November 11, 2010; accepted January 23, 2011.
Corresponding Author:C. J. F. Van Noorden, Department of Cell Biology and Histology, Academic Medical Center, Meibergdreef 15, 1105 AZ Amsterdam, the Netherlands. E-mail: [email protected].
Differential Activity of NADPH-Producing Dehydrogenases Renders Rodents Unsuitable Models to Study IDH1R132 Mutation Effects in Human Glioblastoma
Nadia A. Atai, Nynke A. Renkema-Mills, Joost Bosman, Nadja Schmidt, Denise Rijkeboer, Wikky Tigchelaar, Klazien S. Bosch, Dirk Troost, Ard Jonker, Fonnet E. Bleeker, Hrvoje Miletic, Rolf Bjerkvig, Philip C. De Witt Hamer, and Cornelis J. F. Van NoordenDepartment of Cell Biology and Histology, Academic Medical Center, University of Amsterdam, the Netherlands (NAA, NARM, JB,NS, DR, WT, KSB, AJ, CJFVN); Nor Lux Neuro-Oncology, Department of Biomedicine, University of Bergen, Norway (HM,RB); Department of Neuropathology, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands (DT); and Neurosurgical Center Amsterdam, VU University Medical Center, Amsterdam, the Netherlands (FEB,PCDWH).
Summary
The somatic IDH1R132 mutation in the isocitrate dehydrogenase 1 gene occurs in high frequency in glioma and in lower frequency in acute myeloid leukemia and thyroid cancer but not in other types of cancer. The mutation causes reduced NADPH production capacity in glioblastoma by 40% and is associated with prolonged patient survival. NADPH is a major reducing compound in cells that is essential for detoxification and may be involved in resistance of glioblastoma to treatment. IDH has never been considered important in NADPH production. Therefore, the authors investigated NADPH-producing dehydrogenases using in silico analysis of human cancer gene expression microarray data sets and metabolic mapping of human and rodent tissues to determine the role of IDH in total NADPH production. Expression of most NADPH-producing dehydrogenase genes was not elevated in 34 cancer data sets except for IDH1 in glioma and thyroid cancer, indicating an association with the IDH1 mutation. IDH activity was the main provider of NADPH in human normal brain and glioblastoma, but its role was modest in NADPH production in rodent brain and other tissues. It is concluded that rodents are a poor model to study consequences of the IDH1R132 mutation in glioblastoma. (J Histochem Cytochem 59:489–503, 2011)
Keywords
in silico analysis, glioblastoma, metabolic mapping, IDH1, G6PDH, mutation, NADPH, dehydrogenase

490 Atai et al.
found in cytoplasm, peroxisomes, and endoplasmic reticu-lum of cells (Geisbrecht and Gould 1999; Margittai and Banhegyi 2008). IDH2 is the second NADP+-dependent IDH localized in mitochondria (Hartmann et al. 2009; Sonoda et al. 2009; Yan et al. 2009). The other three mem-bers of the IDH family are NAD+ dependent, exclusively localized in mitochondria and involved in the Krebs cycle (Ying 2008). NAD+-dependent IDHs have not been found to be mutated in relation with gliomagenesis in particular and cancer in general (Yan et al. 2009).
The causal relationship between IDH mutations and gli-omagenesis is only partly understood. Both mutations in the IDH1 and IDH2 genes affect evolutionary-conserved resi-dues (arginines R132 and R172, respectively). The argi-nines are localized in the isocitrate binding site of the NADP+-dependent IDHs (Xu et al. 2004). The mutations inactivate the enzymatic activity of IDH1 and IDH2 (Ichimura et al. 2009; Yan et al. 2009; Bleeker et al. 2010). They cause reduced production of α-ketoglutarate and NADPH from isocitrate and NADP+.
The most important functional consequence of mutated IDH1 is that it converts α-ketoglutarate and NADPH into 2-hydroxyglutarate and NADP+ (Dang et al. 2009). In patients with L-2-hydroxyglutaric aciduria, the accumula-tion of 2-hydroxyglutarate is associated with a higher risk of gliomagenesis (Aghili et al. 2009). Moreover, 2-hydrox-yglutarate may inhibit degradation of hypoxia-inducible factor (HIF) subunit HIF-1α (Gross et al. 2010). HIF-1α can thus form the heterodimer HIF-1, consisting of HIF-1α and HIF-1β, that is transported into the nucleus as transcription factor (Hughes et al. 2010; Pollard and Ratcliffe 2009; Thompson 2009). HIF-1 is the master switch of cellular adaptation to low oxygen levels and induces transcription of more than 100 genes involved in angiogenesis, cell motility, invasion, and anaerobic glycolysis (Bjerkvig et al. 2009; Nobusawa et al. 2009; Atai et al. 2011; Hughes et al. 2010). HIF-1α thus provides a survival kit for glioma cells.
IDH1 can be considered either as a tumor suppressor gene (the mutation causes loss of function by reducing cyto-plasmic α-ketoglutarate levels) or as an oncogene (the mutation causes gain of function by increasing levels of 2-hydroxyglutarate levels and increasing HIF-1α levels). Reduced α-ketoglutarate levels in the cytoplasm due to the IDH1R132 mutation may also reduce degradation of HIF-1α (Zhao et al. 2009). However, α-ketoglutarate levels were found not to be reduced in glioma and acute myeloid leuke-mia with the IDH1R132 mutation (Dang et al. 2009; Gross et al. 2010).
Another clinically important phenomenon of the IDH1R132 mutation is the prolonged survival of glioblas-toma patients with the mutation (Bleeker et al. 2009; Hartmann et al. 2009; Hartmann et al. 2010; Nobusawa et al. 2009; Sanson et al. 2009; Sonoda et al. 2009; Watanabe
et al. 2009; Weller et al. 2009). The IDH1R132 mutation was associated with improved survival of 1 year on average in our set of 98 glioblastoma patients, of whom 18 had the IDH1R132 mutation (Bleeker et al. 2010). We hypothesized in that study that reduced NADPH production in the cyto-plasm of glioma cells is responsible for prolonged survival. We found that the capacity to produce NADPH was reduced by 38% in glioblastoma samples harboring the IDH1R132 mutation, as demonstrated by metabolic mapping (Bleeker et al. 2010). Moreover, the mutated IDH1 consumes NADPH rather than producing it. Thus, the NADPH pro-duction in IDH1-mutated glioblastoma is likely to be even more profoundly decreased. NADPH has a major impact on detoxification. NADPH is among others necessary for the production of reduced glutathione (Koehler and Van Noorden 2003) and reduced thioredoxins (Holmgren and Lu 2010; Biaglow and Miller 2005), formation of active catalase tetramers (Salvemini et al. 1999), and the activity of the members of the cytochrome P450 family (Van Noorden and Butcher 1986, 1991; Koehler and Van Noorden 2003). Oxygen radicals are metabolized by NADPH-dependent systems, and oxygen stress is particularly induced by irradiation and chemotherapy (Ozben 2007). Recently, it was reported that patients with low-grade gli-oma with the mutation responded better to telozolomide treatment (Houillier et al. 2010).
Surprisingly, it appeared that the capacity of IDH to pro-duce NADPH represents 65% of the total NADPH production in the human brain (Bleeker et al. 2010). This is in contrast with the general concept that the irreversible oxidative part of the pentose phosphate pathway by activity of glucose-6-phos-phate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH) is the major NADPH provider in the cytoplasmic compartment of cells, whereas IDH, malate dehy-drogenase (MDH), and hexose-6-phosphate dehdrogenase (H6PDH) play a minor role (Van Noorden 1984; Stoward et al. 1991; Koehler and Van Noorden 2003; Kil et al. 2006; Reitman and Yan 2010). Therefore, we further investigated the relative role in NADPH production by IDH and the other NADPH-producing dehydrogenases in normal human, mouse, and rat tissues and glioblastoma samples using metabolic mapping (Van Noorden 2009, 2010). Furthermore, we analyzed expres-sion of these dehydrogenases in human cancer relative to nor-mal human tissue in silico (Atai et al. 2011; Mir et al. 2010) to establish their potential role in the generation of NADPH in cancer.
Materials and Methods
Glioblastoma SamplesThe activity of NADPH-producing dehydrogenases was localized using metabolic mapping according to Van

NADPH Production and the IDH1 Mutation in Glioblastoma 491
Noorden (2009, 2010) in cryostat sections of samples from glioblastoma patients and non-cancerous brain tissue sam-ples. Twenty-six glioblastoma samples that were classified by the pathologist (DT) according to the World Health Organization (WHO) 2007 classification and five non-cancerous brain tissue samples containing both white and grey matter were used in the study. Tumor samples were included only when at least 80% of the samples consisted of cancer cells, as verified by hematoxylin–eosin staining. Use of patient material fell under the Dutch Code of proper secondary use of human tissue and was waived by the local ethics committee.
All glioblastoma and non-cancerous brain tissue samples were obtained from the tumor bank maintained by the Departments of Neurosurgery and Neuropathology at the Academic Medical Center, Amsterdam, the Netherlands. All samples were snap-frozen in liquid nitrogen in the oper-ating room and stored at –80C until used. The mutational status of the IDH1 and IDH2 genes was determined previ-ously (Bleeker et al. 2009; Bleeker et al. 2010). Thirteen glioblastoma samples mutated in the IDH1 gene (IDH1R132) and 13 glioblastoma samples with wild-type IDH1 and IDH2 genes were selected for further studies described here.
Normal Mouse TissuesThe activity of NADPH-producing dehydrogenases was localized in tissues of five young adult C57 B1/6 mice (Charles Rivers, Someren, the Netherlands) with body weight of 22 to 26 g. The animals were kept under con-stant environmental conditions with a 12-hr dark/12-hr light cycle and free access to food and water. Animals were kept under these conditions for at least 2 weeks. The animals were sacrificed by CO
2 exposure and subse-
quent cervical dislocation. Animal procedures were car-ried out in compliance with Institutional Standards for Human Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee approved the experiments.
Samples of tissues were slowly snap-frozen in small plastic vials in liquid nitrogen and stored at –80C until used as described by Vogels et al. (2009) to ensure preservation of optimum tissue morphology. Cerebrum, cerebellum, spi-nal cord, tongue, small and large intestines, pancreas, liver, and kidney were collected.
Human Glioblastoma in Nude Rat BrainTissues of five glioblastoma tumors were grafted in the brain of eight nude rats (Han: rnu/ruu Rowett) as described previously (Wang et al. 2009). The tumors were allowed to grow for 4 to 5 months, and then brains with tumors were removed and frozen in liquid nitrogen. All procedures and
experiments were approved by the National Animal Research Authority in Norway and conducted according to the European Convention for the Protection of Vertebrates Used for Scientific Purposes.
Metabolic MappingSix-µm-thick cryostat sections were cut of all patient sam-ples and rodent tissue samples at –25C on a HM560 cryo-stat (MICROM, Walldorf, Germany), picked up on glass slides, and stored at –80C until used. By keeping the cabi-net temperature in the cryostat as low as possible, cryostat sectioning of brain could be done despite the high lipid content, as shown by De Witt Hamer et al. (2006). Cryostat sections of these tissues were allowed to dry at room tem-perature for 5 min and were then incubated for the localiza-tion of the activity of G6PDH, 6PGDH, IDH, MDH, and H6PDH, according to Van Noorden and Frederiks (1992). Incubation medium contained 18% polyvinyl alcohol (PVA, weight average Mr 70,000–100,000; Sigma-Aldrich, St. Louis, MO) in 0.1 M phosphate buffer (pH 7.4), 0.32 mM 1-methoxyphenazine methosulphate (Serva, Heidelberg, Germany), 0.8 mM NADP+ (Roche, Mannheim, Germany), 5 mM sodium azide, 5 mM MgCl
2, 5 mM nitro blue tetra-
zolium salt (nitro BT; Sigma-Aldrich), and the respective substrates. For G6PDH, 6PGDH, IDH, MDH, and H6PDH, the following substrates and concentrations were used: 10 mM glucose-6-phosphate (G6P; Serva), 10 mM 6-phosphogluconate (PG; Sigma), 20 mM D,L-isocitrate (Sigma), 100 mM L-malate (Serva), and 10 mM galactose-6-phosphate (Sigma-Aldrich), respectively. The media were freshly prepared just before incubation, and nitro BT was added after being dissolved in a heated mixture of dimethylformamide and ethanol (final dilution of each solvent in the medium was 2% v/v).
For the demonstration of the activity of G6PDH, 6PGDH, IDH, MDH, and H6PDH, sections were incubated at 37C for 10 to 45 min depending on the reaction rate. The incuba-tion was stopped immediately by rinsing the sections in phosphate buffer (0.1 M, pH 5.3, 60C) to remove the vis-cous incubation medium. Afterward, sections were embed-ded in glycerin–gelatin. Experiments were performed in duplicate, and the concentrations of the substrates and coen-zymes in the incubation media were sufficiently high to ensure maximum velocity (V
max) of the enzyme activities
(Stoward and Van Noorden 1991; Van Noorden and Butcher 1991). Control reactions were performed in the absence of substrate (Butcher and Van Noorden 1985).
Image AnalysisThe final reaction product of NADPH-producing dehydro-genase activity (nitro BT-formazan) was analyzed in three areas in each of five sections with the use of quantitative

492 Atai et al.
image analysis, using a Vanox-T photomicroscope with a 10× objective (Olympus, Tokyo, Japan) and a CFW-1312M 1360 × 1024-pixel 10-bit monochrome FireWire camera (Scion, Tucson, AZ) mounted on the front port of the microscope using adapting optics. Sections were illumi-nated with white light that was filtered by a monochromatic filter of 585 nm and an infrared blocking filter to correctly measure the absorbance of both mono- and diformazans (Van Noorden and Butcher 1991; Van Noorden and Frederiks 1992). Absorbance calibration of the images was performed with the use of a calibrated 10-step density tablet (Stouffer, South Bend, IN). After measuring the step tablet, known absorbance values were related to measured gray values using the built-in calibration function of ImageJ, using the Rodbard function. Density calibrated images were recorded in one single run and stored on disk for analysis. The resolution used prevented distributional errors (Chieco et al. 1994). All settings were maintained throughout the recording session and at the end of the ses-sion verified against the step tablet values. Software used for capturing was swf-image, a Scion proprietary camera driver, as an extension to the image-processing application of ImageJ, developed by Rasband (2009). ObjectJ, a plug-in for non-destructive image marking and result linking developed by Vischer and Nastase (2009), was used to indi-cate regions of interest (ROI) that were measured. Using the ObjectJ plug-in, mean absorbance values within the ROI were collected from the test reaction and the control reaction, allowing calculation of specific activity of the NADPH-producing dehydrogenases. Activity was expressed as µmoles NADPH produced per ml of tissue per min (Van Noorden and Frederiks 1992). The use of ObjectJ allows a retrospective quality control of areas measured.
In Silico AnalysisThe genome databases of PubMed (http://www.ncbi.nlm .nih.gov/sites/entrez) were used to collect information on the human NADPH-producing dehydrogenase genes such as Entrez Gene ID, gene name, gene symbol, and gene syn-onyms.
The Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress (http://www.ebi.ac.uk/microar-ray-as/ae/) were searched to retrieve published human micro-array gene expression data sets comparing cancer samples with normal tissue samples. Microarray expression data were retrieved from 34 data sets (after excluding 29 data sets for various reasons such as normal tissue samples consisting of cells in culture; for references, see Table 1) comparing cancer with normal human samples as described previously (Atai et al. 2011; Mir et al. 2010). For all data sets, log
2-transformed
signal intensity measurements were available for each probe in every sample, including probes for the genes investigated.
The original method of spot qualification and data nor-malization was maintained for each data set. To compare between platforms, expression intensity was calculated for each Entrez Gene ID by averaging multiple probe intensi-ties. To retrieve expression data of NADPH-producing dehydrogenases from the data sets, Entrez Gene ID coding for G6PDH, 6PGDH, H6PDH, IDH1, IDH2, ME1, and ME3 (2539, 5226, 3417, 3418, 4199, 10,873, and 9563, respectively) was used. Two expression parameters—namely, frequency of overexpression (the fraction of patients in a data set who showed overexpression of the spe-cific gene in cancer tissue vs normal tissue) and percentile fold change (the level of overexpression of the specific gene determined as a percentile of expression levels of all genes in a particular data set again when comparing cancer tissue and normal tissue)—were used to quantify the frequency and level of gene expression (Atai et al. 2011; Mir et al. 2010). A percentile fold change >0.95 was arbitrarily con-sidered as substantial overexpression, and a frequency of overexpression >30% was arbitrarily considered as frequent overexpression.
Data AnalysisThe R-program (http://www.r-project.org) and Matlab (MatWorks, Natick, MA) were used to calculate the in silico data (expression parameters of NADPH-producing dehydrogenases).
ResultsLocalization of the activity of the major NADPH-producing dehydrogenases IDH and G6PDH in wild-type and IDHR132-mutated glioblastoma is shown in Figure 1. IDH activity is reduced in the mutated glioblastoma. First, an in silico analysis of microarray studies in human cancer versus nor-mal tissue of the seven NADPH-producing dehydrogenases genes was performed to establish the relevance for the NADPH production capacity of the individual dehydroge-nases in cancer in general and in glioma in particular. Table 1 shows that both parameters that we used to analyze the data, percentile fold change and frequency of overexpres-sion, were upregulated to a limited extent only in relation to any type of cancer for each of the seven genes. Remarkably, IDH1 showed the strongest upregulated expression in glio-blastoma, lung carcinoma, and thyroid carcinoma (Table 1). Prostate cancer also showed an elevated percentile fold change, but the frequency of overexpression was small. The two glioma data sets of Bredel et al. (2005) and Kotliarov et al. (2006) both showed upregulation of IDH1 gene expression (Table 2). The percentile fold change is high in all glioma stages in both data sets, but the frequency of overexpression is high only in data set of Bredel et al.

NADPH Production and the IDH1 Mutation in Glioblastoma 493
Table 1. In Silico Analysis of the Expression of NADPH-Producing Dehydrogenases in Cancer versus Normal Tissue Expressed as (A) Percentile Fold Change and (B) Frequency of Overexpression in 34 Data Sets
Normal (n)
Cancer (n) IDH1 IDH2 G6PDH 6PGDH ME1 ME3 H6PDH Reference
(A) Percentile fold changea
Bladder carcinoma 14 13 0.74 0.87 0.85 — 0.4 — 0.23 Dyrskjot et al. 2004 Colon carcinoma 18 18 — 0.86 0.39 0.67 0.88 0.79 — Notterman et al. 2001 Colon carcinoma 5 100 0.51 0.39 0.42 — 0.71 — 0.55 — Esophagus carcinoma 8 8 0.72 0.19 0.11 — 0.05 — 0.37 Kimchi et al. 2005 Gastric carcinoma 8 22 0.53 0.66 0.65 — 0.12 — — Hippo et al. 2002 Gastric carcinoma 23 89 0.18 0.05 — 0.83 0.07 0.09 0.4 Chen et al. 2003 Gastro-intestinal stromal tumor 14 33 0.07 0.47 0.43 0.15 0.75 0.02 0.12 — Glioblastoma 4 31 0.8 0.78 — 0.6 0.11 0.18 0.88 Bredel et al. 2005 Glioblastoma 23 81 0.97 0.67 0.64 — 0.05 — 0.9 Kotliarov et al. 2006 Hepatocellular carcinoma 75 105 — 0.12 — 0.28 0.97 0.33 0.19 Chen et al. 2002 Head and neck carcinoma 3 65 0.6 0.29 — 0.5 0.92 0.34 0.09 Chung et al. 2004 Head and neck carcinoma 22 22 0.84 0.04 — — 0.69 0.08 0.45 Kuriakose et al. 2004 Head and neck carcinoma 4 34 0.16 0.15 — — 0.33 0.19 0.29 Cromer et al. 2004 Leiomyosarcoma 4 9 0.55 0.89 0.06 — 0.74 — — Quade et al. 2004 Lung carcinoma 5 5 0.92 0.31 — — 0.96 0.16 0.43 Wachi et al. 2005 Lung carcinoma 17 154 0.73 0.17 — 0.6 0.74 0.4 0.07 Bhattacharjee et al. 2001 Lung carcinoma 19 34 0.81 0.89 0.9 — 0.59 — — Inamura et al. 2005 Lung carcinoma 9 23 0.97 0.92 0.99 — 0.94 — 0.2 Dehan et al. 2007 Lung carcinoma 10 86 0.86 0.99 — — 0.96 0.94 0.25 Beer et al. 2002 Mamma carcinoma 3 38 — 0.04 — 0.43 — — 0.07 Perou et al. 2000 Mamma carcinoma 7 40 0.34 0.71 0.3 — 0.35 — 0.24 Richardson et al. 2006 Melanoma 7 45 0.41 0.19 0.9 — 0.05 — 0.48 Talantov et al. 2005 Mesothelioma 10 44 0.65 0.84 0.93 — 0.2 — 0.71 Gordon et al. 2005 Ovarium carcinoma 4 27 0.07 0.29 0.6 — 0.94 — — Welsh et al. 2001 Prostate carcinoma 41 62 0.43 0.54 — 0.69 0.07 0.35 0.39 Lapointe et al. 2004 Prostate carcinoma 50 52 0.97 0.97 — — 0.02 0.76 0.13 Singh et al. 2002 Prostate carcinoma 6 13 — 0.61 — 0.81 — 0.5 0.44 Varambally et al. 2005 Prostate carcinoma 19 14 0.47 0.55 0.16 — 0.04 — 0.54 Dhanasekaran et al. 2001 Renal carcinoma 8 9 0.07 0.07 0.64 — 0.58 — 0.81 Lenburg et al. 2003 Renal carcinoma 3 28 — 0.17 — 0.09 0.59 0.19 0.88 Higgins et al. 2003 Seminoma 5 13 0.63 0.91 0.05 — 0.09 0.63 Korkola et al. 2006 Seminoma 14 23 — — — 0.83 0.15 — 0.13 Sperger et al. 2003 Nonseminoma 5 87 0.53 0.9 0.07 — 0.06 — 0.54 Korkola et al. 2006 Thyroid carcinoma 7 7 0.97 0.9 0.62 — 0.04 — 0.59 —(B) Frequency of overexpressionb
Bladder carcinoma 7 7 0.71 0.29 0.00 — 0.00 — 0.00 — Colon carcinoma 14 13 0.00 0.31 0.08 — — 0.00 Dyrskjot et al. 2004 Colon carcinoma 18 18 — 0.00 0.00 0.00 0.00 0.00 — Notterman et al. 2001 Esophagus carcinoma 5 100 0.06 0.13 0.22 — 0.17 — 0.07 — Gastric carcinoma 8 8 0.00 0.00 0.00 — 0.00 — 0.00 Kimchi et al. 2005 Gastric carcinoma 8 22 0.00 0.00 0.05 — 0.00 — — Hippo et al. 2002 Gastro-intestinal stromal tumor 23 89 0.00 0.00 — 0.00 0.00 — 0.00 Chen et al. 2003 Glioblastoma 14 33 0.00 0.00 0.00 0.00 0.00 0.00 0.00 — Glioblastoma 4 31 1.00 0.55 — 0.52 0.00 0.03 0.61 Bredel et al. 2005 Hepatocellular carcinoma 23 81 0.31 0.00 0.02 — 0.00 — 0.02 Kotliarov et al. 2006 Head and neck carcinoma 75 105 — 0.00 — 0.00 0.00 0.00 0.00 Chen et al. 2002 Head and neck carcinoma 3 65 0.15 0.02 — 0.17 0.43 0.27 0.02 Chung et al. 2004 Head and neck carcinoma 22 22 0.00 0.00 — — 0.00 0.00 0.00 Kuriakose et al. 2004 Leiomyosarcoma 4 34 0.03 0.00 — — 0.12 0.00 0.00 Cromer et al. 2004 Lung carcinoma 4 9 0.22 0.44 0.00 — 0.33 — — Quade et al. 2004
(continued)

494 Atai et al.
Table 2. In Silico Analysis of the Expression of NADPH-Producing Dehydrogenases in Stages WHO2, WHO3, and WHO4 (Glioblastoma) of Glioma versus Non-cancerous Brain in the Microarray Data Sets of Bredel et al. (2005) and Kotliarov et al. (2006) Expressed as (A) Percentile Fold Change and (B) Frequency of Overexpression
Dehydrogenase GeneWHO2 (Bredel)
WHO3 (Bredel)
WHO4 (Bredel)
WHO2 (Kotliarov)
WHO3 (Kotliarov)
WHO4 (Kotliarov)
(A) Percentile fold changea
IDH1 0.62 0.76 0.8 0.98 0.98 0.98 IDH2 0.64 0.64 0.78 0.66 0.78 0.67 G6PDH — — — 0.23 0.33 0.64 6PGDH 0.39 0.48 0.6 — — — ME1 0.1 0.09 0.11 0.08 0.04 0.05 ME3 0.17 0.3 0.18 — — — H6PDH 0.8 0.84 0.88 0.79 0.9 0.9(B) Frequency of overexpressionb
IDH1 1.00 0.00 1.00 0.12 0.19 0.31 IDH2 0.00 0.5 0.55 0.00 0.00 0.00 G6PDH — — — 0.00 0.00 0.02 6PGDH 0.22 0.00 0.52 — — — ME1 0.00 0.00 0.00 0.00 0.00 0.00 ME3 0.00 0.00 0.03 — — — H6PDH 0.44 0.5 0.61 0.00 0.00 0.02
aThe numbers in bold are larger then the arbitrarily selected threshold of 0.95, meaning that the gene is among 5% of all genes that are highest upregu-lated in cancer compared to normal tissue in this data set.bThe numbers in bold are larger than the arbitrarily selected threshold of 0.30, meaning that the gene is upregulated in at least 30% of the patients with cancer compared to normal tissue in this data set.
Normal (n)
Cancer (n) IDH1 IDH2 G6PDH 6PGDH ME1 ME3 H6PDH Reference
Lung carcinoma 5 5 0.16 0.00 — — 0.28 — 0.00 Wachi et al. 2005 Lung carcinoma 17 154 0.18 0.00 — 0.15 0.26 0.04 0.03 Bhattacharjee et al. 2001 Lung carcinoma 19 34 0.33 0.47 0.35 — 0.07 — — Inamura et al. 2005 Lung carcinoma 9 23 0.60 0.80 0.60 — 0.80 — 0.00 Dehan et al. 2007 Mamma carcinoma 10 86 0.00 0.02 — — 0.01 0.00 0.00 Beer et al. 2002 Mamma carcinoma 3 38 — 0.00 — 0.18 — — 0.00 Perou et al. 2000 Melanoma 7 40 0.03 0.17 0.03 — 0.00 — 0.00 Richardson et al. 2006 Mesothelioma 7 45 0.00 0.00 0.40 — 0.00 — 0.00 Talantov et al. 2005 Ovarium carcinoma 10 44 0.00 0.00 0.03 — 0.00 — 0.03 Gordon et al. 2005 Prostate carcinoma 4 27 0.04 0.00 0.22 — 0.17 — — Welsh et al. 2001 Prostate carcinoma 41 62 0.00 0.00 — 0.00 0.00 0.00 0.00 Lapointe et al. 2004 Prostate carcinoma 50 52 0.00 0.00 — 0.00 0.03 0.00 Singh et al. 2002 Prostate carcinoma 6 13 — 0.00 — 0.00 — 0.00 0.00 Varambally et al. 2005 Renal carcinoma 19 14 0.00 0.00 0.00 — 0.00 — 0.00 Dhanasekaran et al. 2001 Renal carcinoma 8 9 0.00 0.00 0.11 — 0.00 — 0.00 Lenburg et al. 2003 Seminoma 3 28 — 0.03 — 0.00 0.04 0.03 0.74 Higgins et al. 2003 Seminoma 5 13 0.41 0.95 0.00 — 0.00 — 0.42 Korkola et al. 2006 Nonseminoma 14 23 — — — 0.00 0.00 — 0.00 Sperger et al. 2003 Thyroid carcinoma 5 87 0.00 1.00 0.00 — 0.00 — 0.08 Korkola et al. 2006
The number of samples from non-cancer patients (normal) and cancer patients (cancer) is given for each data set as well as the reference.aThe numbers in bold are larger then the arbitrarily selected threshold of 0.95, meaning that the gene is among 5% of all genes that are highest upregu-lated in cancer compared to normal tissue in this data set.bThe numbers in bold are larger than the arbitrarily selected threshold of 0.30, meaning that the gene is upregulated in at least 30% of the patients with cancer compared to normal tissue in this data set.
Table 1. (continued)

NADPH Production and the IDH1 Mutation in Glioblastoma 495
(2005). The IDH2 gene also shows upregulation but to a lesser extent (Table 2).
Localization of the activity of NADP+-dependent IDH and the dehydrogenases of the pentose phosphate pathway, G6PDH and 6PGDH, in control mouse central nervous sys-tem, tongue epithelium, and liver is shown in Figure 2. The figure shows strong heterogeneity of activity of the dehy-drogenases in all tissues. Image analysis data of the activity of the dehydrogenases in various regions of tissues are shown in Table 3. Remarkably, when we calculated the NADPH production capacity of IDH and the dehydroge-nases of the pentose phosphate pathway, G6PDH and 6PGDH, they showed a similar proportion in each tissue compartment despite the heterogeneity of activity in the tis-sues. In all mouse tissues, G6PDH and 6PGDH together produced more NADPH than IDH1 and IDH2 did together. Particularly, G6PDH is important for NADPH production. This proportion is 25% versus 75% in the cerebellum, 5% versus 95% in the cerebrum, 10% versus 90% in the spinal cord, 10% versus 85% in tongue epithelium, and 50% ver-sus 50% in the liver. H6PDH activity was below the detec-tion limit in all cases and MDH activity as well except for a low MDH activity in tongue (5%). All other mouse organs studied (small and large intestines, pancreas, and kidney) showed similar patterns of NADPH production capacity (data not shown).
In general, our findings show that IDH activity contrib-utes relatively little to the NADPH production in control mouse tissues and particularly in the central nervous sys-tem, whereas the pentose phosphate pathway is responsible for the major part of NADPH production. This is in contrast to the human cerebrum, where IDH activity is responsible for 60% of the NADPH production capacity (Bleeker et al. 2010).
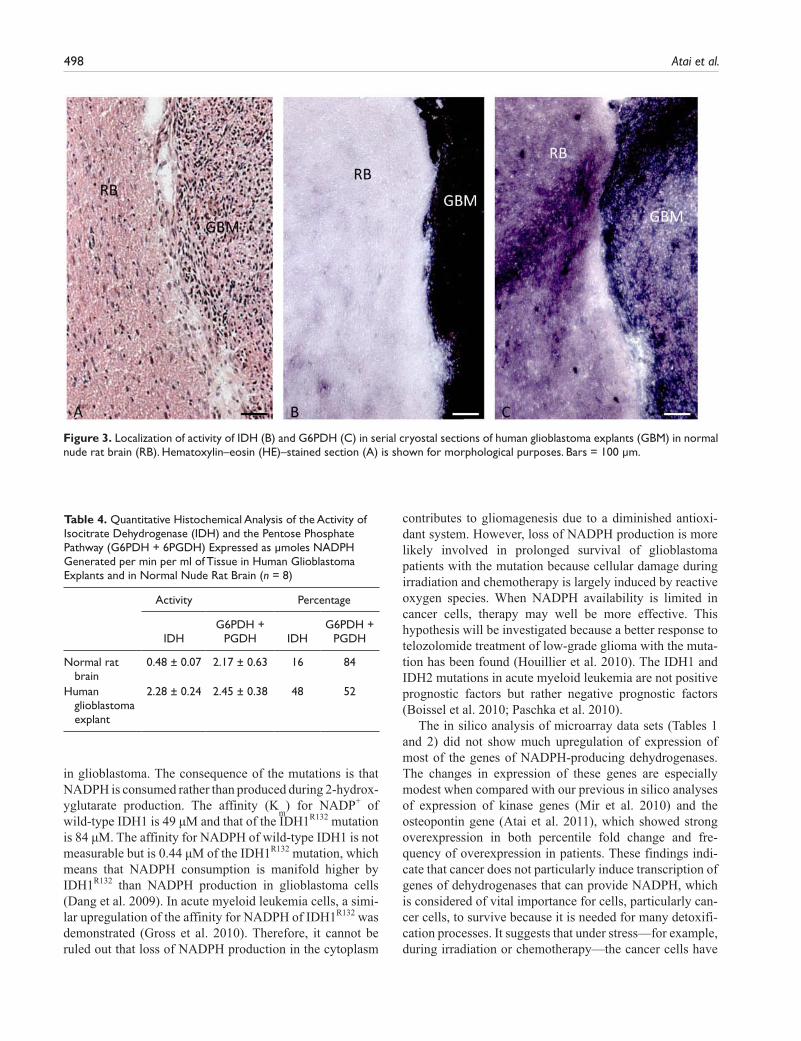
To test the discrepancy that we observed between human and rodent tissues, we determined IDH activity in rat brain in which human glioblastoma explants were grown (Fig. 3; Table 4). Here, IDH activity was responsible for 16% and the pentose phosphate pathway for 84% of the NADPH pro-duction capacity in rat cerebrum and in human glioblastoma for 48% and 52%, respectively, again showing differences in the role of IDH activity in NADPH production between rodents and human.
DiscussionNADPH is an essential compound for detoxification reac-tions in cells. In particular, the cellular antioxidant system relies heavily on NADPH (Koehler and Van Noorden 2003). The general opinion based on rodent models is that the irreversible oxidative part of the pentose phosphate pathway with G6PDH and 6PGDH is the main provider of
Figure 1. Metabolic mapping of the activity of IDH (B, E) and G6PDH (C, F) in serial cryostat sections of human wild-type (A-C) and IDHR132-mutated (D-F) glioblastoma. Hematoxylin–eosin (HE)–stained sections (A, D) are shown for morphological purposes. The amount of blue dye (nitro BT-formazan) reflects IDH or G6PDH activity. v, blood vessel. Bars = 150 µm.

496 Atai et al.
NADPH. However, this may not be the case in humans (Kil et al. 2006) except for erythrocytes in which NADPH is mainly provided by G6PDH (Scott et al. 1991; Peters and Van Noorden 2009).
Indeed, our metabolic mapping study shows a major role for IDH in NADPH production capacity in the normal human brain and glioma (Fig. 1; Bleeker et al. 2010). In contrast, in normal mouse tissues, including the central nervous system, we found that the irreversible oxidative part of the pentose phosphate pathway has the major capacity to generate NADPH, and IDH has only limited capacity (Fig. 2; Table 3). Localization of activity of G6PDH in these mouse organs (Fig. 2) is in good agree-ment with previous metabolic mappings studies (Van Noorden 1984; Biagotti et al. 2000; Biagotti et al. 2002; Biagotti et al. 2005; Ferri et al. 2005). Therefore, we con-clude that the concept that NADPH is mainly provided by the pentose phosphate pathway and not by IDH is based on studies of organisms other than humans. This is further confirmed by the metabolic mapping data on the experi-mental model of human glioblastoma tumors in the brain of nude rats (Fig. 3; Table 4). The relative contribution of IDH to the capacity of NADPH production is much larger in the human glioblastoma explants than in the rat brain. Furthermore, Lee et al. (2002) and Kil et al. (2006, 2007)
demonstrated that IDH is essential in human cells for the provision of NADPH because specific reduction of IDH activity increased oxidative damage, apoptosis, and senes-cence. Silencing of IDH1 expression by siRNA is even lethal in human cancer cells (Abdel-Wahab and Levine 2010), indicating that one wild-type allele must be retained in IDH1 mutant cancer cells. Moreover, homozygously mutated IDH1 glioma has not been found yet. These data show once more that a mouse is not a man and stress that data obtained in rodent models of glioblastoma may not reflect the situation in the glioblastoma patient.
The quantitative analysis of the activity of NADPH-producing dehydrogenases in normal mouse tissues (Table 3) revealed another surprising finding. Despite the strong heterogeneity in activity in the different compartments of a tissue, the relative NADPH production capacity of the dehydrogenases was similar in those tissues. In the cere-brum, 25% was represented by IDH and 75% by G6PDH and 6PGDH; these percentages were 5% and 95% in the cerebellum, 10% and 90% in spinal cord, and so on (Table 3). These data indicate that the regulation of the activity of these enzymes is coordinated per tissue, be it on the tran-scriptional, translational, or posttranslational level. This aspect of the activity of NADPH-producing dehydroge-nases warrants further study.
Table 3. Quantitative Histochemical Analysis of the Activity of the NADPH-Producing Dehdrogenases IDH, G6PDH, 6PGDH, MDH, and H6PDH (expressed as µmoles NADPH Generated per min per ml of Tissue) in Normal Mouse Tissue (n=5) and Given as a Percentage (%) of the Total NADPH Production Capacity
Activity Percentage
IDH G6PDH 6PGDH IDH G6PDH + 6PGDH
Cerebrum Cortex 0.51 ± 0.25 1.08 ± 0.29 0.30 ± 0.20 27 73 Cortex 0.28 ± 0.16 0.75 ± 0.27 0.30 ± 0.08 21 79 Thalamus 0.23 ± 0.01 0.84 ± 0.04 0.28 ± 0.06 17 83 Corpus callosum 0.58 ± 0.11 1.41 ± 0.20 0.24 ± 0.18 26 74Cerebellum Molecular layer 0.07 ± 0.06 1.79 ± 0.05 0.24 ± 0.06 3 97 Purkinje cells 0.08 ± 0.06 1.38 ± 0.28 0.23 ± 0.03 5 95 Granular layer 0.06 ± 0.04 0.68 ± 0.10 0.16 ± 0.01 7 93 White matter 0.07 ± 0.01 1.31 ± 0.26 0.23 ± 0.07 4 96Spinal cord Gray matter 0.16 ± 0.06 1.14 ± 0.16 0.24 ± 0.03 10 90 White matter 0.13 ± 0.04 0.95 ± 0.18 0.18 ± 0.03 10 90Tongue epithelium Basal cell layer 0.45 ± 0.08 1.00 ± 0.20 0.67 ± 0.19 20 75 Upper cell layer 0.28 ± 0.07 1.80 ± 0.03 1.80 ± 0.05 7 90Colon Bottom crypts 0.50 ± 0.17 0.95 ± 0.31 1.12 ± 0.23 19 81 Upper part crypts 0.18 ± 0.05 0.42 ± 0.18 1.00 ± 0.29 11 89 Surface 0.28 ± 0.09 0.94 ± 0.29 0.92 ± 0.69 13 87Pancreas Endocrine (islets) 1.25 ± 0.09 1.51 ± 0.08 0.26 ± 0.19 41 59
Exocrine 1.72 ± 0.05 1.55 ± 0.05 0.29 ± 0.18 42 58Liver Periportal 0.86 ± 0.06 0.74 ± 0.13 0.14 ± 0.02 49 51 Pericentral 0.70 ± 0.11 0.51 ± 0.17 0.11 ± 0.02 54 46Kidney Corpuscules 0.34 ± 0.13 1.35 ± 0.11 0.08 ± 0.01 19 81 Tubuli 1.53 ± 0.07 1.22 ± 0.09 0.11 ± 0.04 53 47

NADPH Production and the IDH1 Mutation in Glioblastoma 497
The IDH1 and IDH2 gene mutations are linked with gli-omagenesis, acute myeloid leukemia, and thyroid cancer because of 2-hydroxyglutarate production, which is consid-ered an oncometabolite (Dang et al. 2009; Ward et al. 2010; Fu et al. 2010; Gross et al. 2010). However, it was recently found that glioblastoma cells expressing mutant IDH1 show
reduced growth when α-ketoglutarate production via glutaminase is inhibited as well (Seltzer et al. 2010), indi-cating that not only production of the oncometabolite is involved in gliomagenesis but also reduced production of α-ketoglutarate and/or NADPH. We found that the IDH1 mutation reduced the NADPH production capacity as well
Figure 2. Localization of the activity of IDH (B, F, J, N, R), G6PDH (C, G, K, O, S) and 6PGDH (D, H, L, P, T) in serial cryostat sections of mouse cerebrum (A–D), cerebellum (E–H), spinal cord (I–L), tongue (M–P), and liver (Q–T). Hematoxylin–eosin (HE)–stained sections (A, E, I, M, Q) are shown for morphological purposes. The amount of blue dye (nitro BT-formazan) reflects IDH, G6PDH, or 6PGDH activity. Note the strong heterogeneity of activity in the tissues and the specific staining patterns throughout the different structures of the tissues. Cerebrum: 1, gray matter; 2, 3, white matter; 4, archecortex. Cerebellum: 1, molecular layer; 2, Purkinje cells; 3, granular layer; 4, white matter. Spinal cord: 1, gray matter; 2, white matter. Tongue epithelium: 1, basal cell layers; 2, upper cell layers. Liver: 1, periportal area; 2, pericentral area. Note that micrograph O is overexposed because of the intense staining of the epithelium. Bars = 75 µm.
498 Atai et al.
in glioblastoma. The consequence of the mutations is that NADPH is consumed rather than produced during 2-hydrox-yglutarate production. The affinity (K
m) for NADP+ of
wild-type IDH1 is 49 µM and that of the IDH1R132 mutation is 84 µM. The affinity for NADPH of wild-type IDH1 is not measurable but is 0.44 µM of the IDH1R132 mutation, which means that NADPH consumption is manifold higher by IDH1R132 than NADPH production in glioblastoma cells (Dang et al. 2009). In acute myeloid leukemia cells, a simi-lar upregulation of the affinity for NADPH of IDH1R132 was demonstrated (Gross et al. 2010). Therefore, it cannot be ruled out that loss of NADPH production in the cytoplasm
contributes to gliomagenesis due to a diminished antioxi-dant system. However, loss of NADPH production is more likely involved in prolonged survival of glioblastoma patients with the mutation because cellular damage during irradiation and chemotherapy is largely induced by reactive oxygen species. When NADPH availability is limited in cancer cells, therapy may well be more effective. This hypothesis will be investigated because a better response to telozolomide treatment of low-grade glioma with the muta-tion has been found (Houillier et al. 2010). The IDH1 and IDH2 mutations in acute myeloid leukemia are not positive prognostic factors but rather negative prognostic factors (Boissel et al. 2010; Paschka et al. 2010).
The in silico analysis of microarray data sets (Tables 1 and 2) did not show much upregulation of expression of most of the genes of NADPH-producing dehydrogenases. The changes in expression of these genes are especially modest when compared with our previous in silico analyses of expression of kinase genes (Mir et al. 2010) and the osteopontin gene (Atai et al. 2011), which showed strong overexpression in both percentile fold change and fre-quency of overexpression in patients. These findings indi-cate that cancer does not particularly induce transcription of genes of dehydrogenases that can provide NADPH, which is considered of vital importance for cells, particularly can-cer cells, to survive because it is needed for many detoxifi-cation processes. It suggests that under stress—for example, during irradiation or chemotherapy—the cancer cells have
Table 4. Quantitative Histochemical Analysis of the Activity of Isocitrate Dehydrogenase (IDH) and the Pentose Phosphate Pathway (G6PDH + 6PGDH) Expressed as µmoles NADPH Generated per min per ml of Tissue in Human Glioblastoma Explants and in Normal Nude Rat Brain (n = 8)
Activity Percentage
IDHG6PDH +
PGDH IDHG6PDH +
PGDH
Normal rat brain
0.48 ± 0.07 2.17 ± 0.63 16 84
Human glioblastoma explant
2.28 ± 0.24 2.45 ± 0.38 48 52
Figure 3. Localization of activity of IDH (B) and G6PDH (C) in serial cryostal sections of human glioblastoma explants (GBM) in normal nude rat brain (RB). Hematoxylin–eosin (HE)–stained section (A) is shown for morphological purposes. Bars = 100 µm.

NADPH Production and the IDH1 Mutation in Glioblastoma 499
to rely on the NADPH-producing dehydrogenases that are available. This may explain why the IDH1 mutation may have the prolonging effect on patient survival.
It is remarkable that overexpression of IDH1 was observed in glioma (Table 2) and thyroid cancer (Table 1), besides two out of five data sets of lung carcinoma, because the IDH1 mutation has only been found in glioblastoma, thyroid cancer, and acute myeloid leukemia. The relation-ship between IDH2 expression and the mutation in cancer is less clear (Tables 1 and 2). We did not have access to micro-array data sets on acute myeloid leukemia to investigate whether IDH1 and/or IDH2 expression is also elevated in this type of cancer.
The capacity to produce NADPH in normal human brain, as well as wild-type and IDH1R132-mutated glioblastoma, is represented for 55% to 65% by IDH, for 30% by the irrevers-ible oxidative pentose phosphate pathway, and for 10% by MDH and H6PDH (Bleeker et al. 2010). Again, this was a sur-prising finding. We expected at least to find a lower contribu-tion of IDH to the total capacity of NADPH production in IDH1R132-mutated glioblastoma. However, the relative contri-butions by the different dehydrogenases remained the same, despite the fact that only IDH activity was decreased signifi-cantly. This discrepancy may well be due to the small number of glioblastoma samples included in the metabolic mapping study (n=13 for both groups), but it may also be a biological phenomenon that is related to the constant contribution of the dehydrogenases to the total NADPH production capacity in the different tissues as discussed above.
In conclusion, the present study reveals that in humans, the relative contribution of IDH activity to NADPH produc-tion capacity in the cerebrum and glioblastoma is largely unlike that in other organisms such as rodents, substantiat-ing that reduction in NADPH production capacity is signifi-cant due to the IDH1R132 mutation in humans but not in rodents. It makes rodents an unsuitable model to study functional consequences of the IDH1R132 mutation in human glioblastoma.
Acknowledgment
The careful preparation of the manuscript by Ms. Monique Arendse is gratefully acknowledged. We are also grateful to Prof. Dr. Alberto Bardelli, Laboratory of Molecular Genetics, University of Torino Medical School, Italy, who enabled the sequencing of the IDH1 and IDH2 genes in the glioblastomas.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the authorship and publication of this article.
Funding
The author(s) received no financial support for the research and authorship of this article.
References
Abdel-Wahab O, Levine RL. 2010. Metabolism and the leukemic stem cell. J Exp Med. 207:677–680.
Aghili M, Zahedi F, Rafiee E. 2009. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J Neurooncol. 91:233–236.
Andrulis M, Capper D, Luft T, Hartmann C, Zentgraf H, Von Deimling A. 2010. Detection of isocitrate dehydrogenase 1 mutation R132H in myelodysplastic syndrome by mutation-specific antibody and direct sequencing. Leukemia Res. 34:1091–1093.
Atai NA, Bosch KS, Bansal M, Lo C, Bosman J, Tigchelaar W, Jonker A, De Witt Hamer PC, Troost D, McCulloch CA, et al. 2011. Osteopontin is upregulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology. 132:39–48.
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, Von Deimling A. 2008. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116:597–602.
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al. 2002. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 8:816–824.
Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. 2001. Classi-fication of human lung carcinomas by mRNA expression pro-filing reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 98:13790–13795.
Biaglow JE, Miller RA. 2005. The thioredoxin reductase/ thioredoxin system. Cancer Biol Ther. 4:6–13.
Biagotti E, Bosch KS, Ninfali P, Frederiks WM, Van Noorden CJF. 2000. Postranslational regulation of glucose-6-phosphate dehydrogenase activity in tongue epithelium. J Histochem Cytochem. 48:971–977.
Biagotti E, Ferri P, Dringen R, Del Grande P, Ninfali P. 2005. Glucose-6-phosphate dehydrogenase and NADPH-consuming enzymes in the rat olfactory bulb. J Neurosci Res. 80:434–441.
Biagotti E, Malatesta M, Capellacci S, Fattoretti P, Gazzanelli G, Ninfali P. 2002. Quantification of G6PD in small and large intestine of rat during aging. Acta Histochem. 104:225–234.
Bjerkvig R, Johansson M, Miletic H, Niclou SP. 2009. Cancer stem cells and angiogenesis. Semin Cancer Biol. 19:279–284.
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, et al. 2010. The prognostic IDH1R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuro-pathol. 119:487–494.
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, et al. 2009. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 30:7–11.

500 Atai et al.
Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N, Bordessoule D, Pautas C, de Revel T, Quesnel B, et al. 2010. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leuke-mia: a study by the acute leukemia French association group. J Clin Oncol. 28:3717–3723.
Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. 2005. Functional network analysis reveals extended gli-omagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 65:8679–8689.
Butcher RG, Van Noorden CJF. 1985. Reaction rate studies of glucose-6-phosphate dehydrogenase activity in sections of rat liver using four tetrazolium salts. Histochem J. 17:993–1008.
Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, et al. 2002. Gene expression patterns in human liver cancers. Mol Biol Cell. 13:1929–1939.
Chen X, Leung SY, Yuen ST, Chu KM, Ji J, Chan AS, Law S, Troyanskaya OG, Wong J, et al. 2003. Variation in gene expression patterns in human gastric cancers. Mol Biol Cell. 14:3208–3215.
Chieco P, Jonker A, Melchiorri C, Vanni G, Van Noorden CJF. 1994. A user’s guide for avoiding errors in absorbance image cytometry: a review with original experimental observations. Histochem J. 26:1–19.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, et al. 2010. Distinct clini-cal and biologic characteristics in adult acute myeloid leuke-mia bearing the isocitrate dehydrogenase 1 mutation. Blood. 115:2749–2754.
Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, Butterfoss D, Xiang D, Zanation A, Yin X, et al. 2004. Molec-ular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 5:489–500.
Cromer A, Carles A, Millon R, Ganguli G, Chalmal F, Lemaire F, Young J, Dembélé D, Thibault C, Muller D, et al. 2004. Identi-fication of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene. 23:2484–2498.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Kennan MC, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 462:739–747.
Dehan E, Ben-Dor A, Liao W, Lipson D, Frimer H, Rienstein S, Simansky D, Krupsky M, Yaron P, Friedman E, et al. 2007. Chromosomal aberrations and gene expression profiles in non–small cell lung cancer. Lung Cancer. 56:175–184.
De Witt Hamer PC, Bleeker FE, Zwinderman AH, Van Noorden CJF. 2006. Can you trust your cryostat? Reproducibility of cryostat section thickness. Microsc Res Techn. 69:835–838.
Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. 2001. Delineation of prognostic biomarks in prostate cancer. Nature. 412:822–826.
Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Lensen JL, Moller K, Orntoft TF. 2004. Gene expression in the urinary bladder: a common carcinoma in situ gene expression sig-nature exists disregarding histopathological classification. Cancer Res. 64:4040–4048.
Ferri P, Biagotti E, Ambrogini P, Santi S, Del Grande P, Ninfali N. 2005. NADPH-consuming enzymes correlate with glucose-6-phosphate dehydrogenase in Purkinje cells: an immunohisto-chemical and enzyme histochemical study of the rat cerebellar cortex. Neurosci Res. 51:185–195.
Fu Y, Huang R, Du J, Yang R, An N, Liang A. 2010. Glioma-derived mutations in IDH: from mechanism to potential ther-apy. Biochem Biophys Res Commun. 397:127–130.
Geisbrecht BV, Gould SJ. 1999. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem. 274:30527–30533.
Gordon GJ, Rockwell GN, Jensen RV, Rheinwald JG, Glickman JN, Aronson JP, Pottorf BJ, Nitz MD, Richards WG, Sugarbaker DJ, et al. 2005. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol. 166: 1827–1840.
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. 2010. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 207:339–344.
Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M Schackert G, Meyermann R, Pietsch T, et al. 2010. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 120:707–718.
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et al. 2009. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 118:469–474.
Higgins JP, Shinghal R, Gill H, Reese JH, Terris M, Cohen RJ, Fero M, Pollack JR, van de Rijn M, Brooks D. 2003. Gene expression patterns in renal cell carcinoma assessed by com-plementary DNA microarray. Am J Pathol. 162:925–932.
Hippo Y, Taniguchi H, Tsutsumi S, Machida N, Chong JM, Fukayama M, Kodama T, Aburatani H. 2002. Global gene expression analysis of gastric cancer by oligonucleotide microarrays. Cancer Res. 62:233–240.
Holmgren A, Lu J. 2010. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Bio-chem Biophys Res Commun. 396:120–124.
Houillier C, Wang X, Kaloshi G, Moktari K, Guillevin R, Laffaire J, Paris S, Boisselier B, Idbaih A, Laigle-Donadey F, et al. 2010.

NADPH Production and the IDH1 Mutation in Glioblastoma 501
IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 75:1560–1566.
Hughes JM, Groot AJ, Van der Groep P, Sersansie R, Vooijs M, Van Diest PJ, Van Noorden CJF, Schlingemann RO, Klaassen I. 2010. Active HIF-1 in the normal human retina. J Histochem Cytochem. 58:247–254.
Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DT, Collins VP. 2009. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-Oncology. 11:341–347.
Inamura K, Fujiwara T, Hoshida Y, Isagawa T, Jones MH, Virtanen C, Shimane M, Satoh Y, Okamura S, Nakagawa K, et al. 2005. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identi-fied by hierarchical clustering and non-negative matrix factor-ization. Oncogene. 24:7105–7113.
Kil IS, Huh TL, Lee YS, Lee YM, Park JW. 2006. Regulation of replicative senescene by NADP+-dependent isocitrate dehy-drogenase. Free Radic Biol Med. 40:110–119.
Kil IS, Kim SY, Lee SJ, Park JW. 2007. Small interfering RNA-mediated silencing of mitochondrial NADP+-dependent iso-citrate dehydrogenase enhances the sensitivity of HeLa cells toward tumor necrosis factor-α and anticancer drugs. Free Radic Biol Med. 43:1197–1207.
Kimchi ET, Posner MC, Park JO, Darga TE, Kocherginsky M, Karrison T, Hart J. 2005. Progression of Barrett’s metaplasia to adenocarcinoma is associated with the suppression of the transcriptional programs of epidermal differentiation. Cancer Res. 65:3146–3154.
Koehler A, Van Noorden CJF. 2003. Reduced nicotinamide adenine dinucleotide phosphate and the higher incidence of pollution-induced liver cancer in female flounder. Environ Toxicol Chem. 22:2703–2710.
Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, Bosl GJ, Chaganti RS. 2006. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 66:820–827.
Kotliarov Y, Steed ME, Christopher N, Walling J, Su Q, Center A, Heiss J, Rosenblum M, Mikkelsen T, Zenklusen JC, et al. 2006. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res. 66:9428–9436.
Kuriakose MA, Chen WT, He ZM, Sikora AG, Zhang P, Zhang ZY, Qiu WL, Hsu DF, McMunn-Coffran C, Brown SM, et al. 2004. Selection and validation of differentially expressed genes in head and neck cancer. Cell Mol Life Sci. 61:1372–1383.
Lapointe J, Li C, Higgins JP, Van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, et al. 2004. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 101:811–816.
Lee AM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. 2002. Cytosolic NADP+-dependent isocitrate dehydrogenase status
modulates oxidative damage to cells. Free Radic Biol Med. 32:1185–1196.
Lenburg ME, Liou LS, Gerry NP, Framton GM, Cohen HT, Christman MF. 2003. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric anal-ysis of microarray data. BMC Cancer. 3:31.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. 2009. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 361:1058–1066.
Margittai E, Banhegyi G. 2008. Isocitrate dehydrogenase: a NADPH-generating enzyme in the lumen of the endoplasmic reticulum. Arch Biochem Biophys. 471:184–190.
Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AAG, Zwinderman AH, Geerts D, Kaspers GJ, et al. 2010. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 18:244–257.
Murugan AK, Bojdani E, Xing M. 2010. Identification and func-tional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 393:555–559.
Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. 2009. IDH1 mutations as molecular signature and predictive factor of sec-ondary glioblastomas. Clin Cancer Res. 15:6002–6007.
Notterman DA, Alon U, Sierk AJ, Levine AJ. 2001. Transcrip-tional gene expression profiles of colorectal adenoma, adeno-carcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 61:3124–3130.
Ohgaki H, Kleihues. 2009. Genetic alterations and signaling path-ways in the evolution of gliomas. Cancer Sci. 100:2235–2241.
Ozben T. 2007. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci. 96:2181–2196.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. 2008. An inte-grated genomic analysis of human glioblastoma multiforme. Science. 321:1807–1812.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K, et al. 2010. IDH1 and IDH2 mutations are frequent genetic altera-tions in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 28:3636–3643.
Perou CM, Sørlie T, Eisen MB, Van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. 2000. Molecular portraits of human breast tumours. Nature. 406:747–752.
Peters AL, Van Noorden CJF. 2009. Glucose-6-phosphate dehy-drogenase deficiency and malaria: cytochemical detection of heterozygous G6PD deficiency in women. J Histochem Cyto-chem. 57:1003–1011.
Pollard PJ, Ratcliffe PJ. 2009. Puzzling patterns of predisposition. Science. 324:192–194.

502 Atai et al.
Quade BJ, Wang TY, Sornberger K, Dal CP, Mutter GL, Morton CC. 2004. Molecular pathogenesis of uterine smooth muscle tumors from transcriptional profiling. Genes Chromosomes Cancer. 40:97–108.
Rasband WS. 2009. ImageJ. http://rsbweb.nih.gov/ij.Reitman ZJ, Yan H. 2010. Isocitrate dehydrogenase 1 and 2 muta-
tions in cancer: altenations at a crossroads of cellular metabo-lism. J Natl Cancer Inst. 102:932–941.
Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. 2006. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 9:121–132.
Salvemini F, Franze A, Iervolino A, Filosa S, Salzano S, Ursini MV. 1999. Enhanced glutathione levels and oxidoresistance mediated by increased glucose-6-phosphate dehydrogenase expression. J Biol Chem. 274:2750–2757.
Sanson M, Marie Y, Paris S, Idbaith A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, et al. 2009. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 27:4150–4154.
Scott MD, Zuo L, Lubin BH, Chiu DYT. 1991. NADPH, not glu-tathione status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood. 77:2059–2064.
Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. 2010. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70:8981–8987.
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP, et al. 2002. Gene expression correlates of clinical prostate cancer behav-iour. Cancer Cell. 1:203–209.
Sonoda Y, Kumabe T, Nakamura T, Saits R, Kanamori M, Yamashita Y, Suzuki H, Tominaga T. 2009. Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci. 100:1996–1998.
Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, Brooks JD, Andrews PW, Brown PO, Thomson JA. 2003. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci U S A. 100:13350–13355.
Stoward PJ, Meijer AEFH, Seidler E, Wohlrab F. 1991. Dehydro-genases. In Stoward PJ, Pearse AGE, eds. Histochemistry. Vol 3, 4th ed. Edinburgh, UK: Churchill Livingstone.
Stoward PJ, Van Noorden CJF. 1991. Histochemical methods for dehydrogenases. In Stoward PJ, Pearse AGE, eds. Histochemistry. Vol 3, 4th ed. Edinburgh, UK: Churchill Livingstone.
Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. 2005. Novel genes associated with malig-nant melanoma but not benign melanocytic lesions. Clin Can-cer Res. 11:7234–42.
Thompson CB. 2009. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 360:813–815.
Van Noorden CJF. 1984. Histochemistry and cytochemistry of glucose-6-phosphate dehydrogenase. Progr Histochem Cyto-chem. 15(4):1–85.
Van Noorden CJF. 2009. Metabolic mapping by enzyme histo-chemistry in living animals, tissues and cells. J Physiol Phar-macol. 60(Suppl 4):125–129.
Van Noorden CJF. 2010. Imaging enzymes at work: metabolic mapping by enzyme histochemistry. J Histochem Cytochem. 58:481–497.
Van Noorden CJF, Butcher RG. 1986. A quantitative histochemi-cal study of NADPH-ferrihemoprotein reductase activity. His-tochem J. 18:364–370.
Van Noorden CJF, Butcher RG. 1991. Quantitative enzyme histo-chemistry. In Stoward PJ, Pearse AGE, eds. Histochemistry. Vol 3, 4th ed. Edinburgh, UK: Churchill Livingstone.
Van Noorden CJF, Frederiks WM. 1992. Enzyme histochemistry: a laboratory manual of current methods. Oxford, UK: Oxford Science Publications.
Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, et al. 2005. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 8:393–406.
Vischer N, Nastase S. 2009. ObjectJ. http://simon.bio.uva.nl/.Vogels IMC, Hoeben KA, Van Noorden CJF. 2009. Rapid com-
bined light and electron microscopy on large frozen biological samples. J Microsc. 235:252–258.
Wachi S, Yoneda K, Wu R. 2005. Interactome-transcriptome analy-sis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 21:4205–4208.
Wang J, Miletic H, Sakariassen PØ, Huszthy PC, Jacobsen H, Brekkå N, Li X, Zhao P, Mørk S, Chekenya M, et al. 2009. A reproducible brain tumor model established from human glio-blastoma biopsies. BMC Cancer. 9:465.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. 2010. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17: 225–234.
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. 2009. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153.
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M, Tonn JC, et al. 2009. Molecular predictors of progression-free and over-all survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German glioma network. J Clin Oncol. 27:5743–5750.
Welsh JB, Zarrinkar PP, Sapinoso LM, Kern SG, Behling CA, Monk BJ, Lockhart DJ, Burger RA, Hampton GM. 2001. Analysis of gene expression profiles in normal and neoplastic

NADPH Production and the IDH1 Mutation in Glioblastoma 503
ovarian tissue samples identifies candidate molecular mark-ers of epithelial ovarian cancer. Proc Natl Acad Sci U S A. 98:1176–1181.
Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. 2004. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 279:33946–33957.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos J, Batinic-Haberle I, Jones S, Riggins GJ, et al.
2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 360:765–773.
Ying W. 2008. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological conse-quences. Antioxid Redox Signal. 10:179–206.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peny Y, et al. 2009. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324:261–265.




















