
Diagonal 2ph-TDA Green's function simulation of the valence X-ray photoelectron spectra of n-alkane...
20
Chemical Physics 175 (1993) 427-446 North-Holland Diagonal 2ph-TDA Green’s function simulation of the valence X-ray photoelectron spectra of n-alkane compounds: a theoretical search for conformational signatures MichaH Deleuze, Joseph DeIhaIIe Luboratoire de Chimie ThkoriqueAppliquke, Fact&% Universitaires Notre-Dame de la Paix, 6 I rue de Bruxelles, B-5000 Namur, Belgium and Barry T. Pickup Department of Chemistry, the University, Sheffield S3 7HF, UK Received 22 February 1993; in final form 12 May 1993 With the aim of improving and validating theoretical quantum mechanical tools for tracking confonnational signatures in the XPS spectra of n-alkane compounds, the XPS spectra of model structures are simulated by means of direct Green’s function schemes and compared to available experimental records obtained in the gas and solid phases. Calculations are performed using the minimal STP3G and 3-21G basis sets, and se&energy expansions derived at several levels of decoupling approximation: second-order, shifted Born collision (SBC) and diagonal two-particle-hole Tamm-Dankoff approximation (Zph-TDA). The latter scheme is shown to be accurate enough to generate reliable relative line positions and convoluted photoionixation intensities in the inner valence region, which is of fundamental importance for the identification of secondary molecular structures from XPS spectra. Preventing overcounting of self-enetgy diagrams containing a diagonal ladder part is crucial in that respect. As a by- product, this method of simulation is applied to some particular conformations of C9Hmmodeling the topmost layer of polyeth- ylene in crystalline form, enabling the identification of fold structuresat the extreme surface of lamellar crystals of polyethylene. 1. Introduction X-ray photoelectron spectroscopy (XPS or ESCA ) allows the study of the electron energy levels in atoms and molecules in gas, liquid and solid phases. Con- sidering the growing importance in highly demand- ing fields (electronics, catalysis, biotechnology, space) of polymer thin films deposited on various substrates (metal, glasses, etc. ), materials for which classical structural analysis techniques such as IR or NMR are usually inapplicable, the XPS method pro- vides a particularly appropriate tool to probe the sur- faces and interfaces of technologically useful organic materials. Most studies have been devoted to the core- level shifts which supply information on the chemi- cal composition and, in favourable situations, on the primary molecular structure. The valence region has received much less attention, probably because the many XPS valence lines of low intensity to be iden- tified in a small energy interval, typically 50 eV, are more complicated to analyse. Combined with quan- tum mechanical calculations, invaluable direct and indirect information on structural aspects (substitu- tion, conformation) and the chemical bonding of the constitutive compounds can however be obtained from their valence XPS spectra [ 11. In the purpose of finding conformational signatures, recent studies have been conducted at the uncorrelated Koopmans level of approximation on models of polyethylene [ 2 1, syndiotactic propylene [ 3 1, polyoxymethylene [ 41, and polyacrylonitrile [ 5 1. The simulated spec- tra exhibit variations of both band positions and photoionization intensities among the selected con- formations, encouraging efforts to implement corre- 0301-0104/93/S 06.00 0 1993 Elsevier Science Publishers B.V. All rights reserved.
Transcript of Diagonal 2ph-TDA Green's function simulation of the valence X-ray photoelectron spectra of n-alkane...

Chemical Physics 175 (1993) 427-446 North-Holland
Diagonal 2ph-TDA Green’s function simulation of the valence X-ray photoelectron spectra of n-alkane compounds: a theoretical search for conformational signatures
MichaH Deleuze, Joseph DeIhaIIe Luboratoire de Chimie Thkorique Appliquke, Fact&% Universitaires Notre-Dame de la Paix, 6 I rue de Bruxelles, B-5000 Namur, Belgium
and
Barry T. Pickup Department of Chemistry, the University, Sheffield S3 7HF, UK
Received 22 February 1993; in final form 12 May 1993
With the aim of improving and validating theoretical quantum mechanical tools for tracking confonnational signatures in the XPS spectra of n-alkane compounds, the XPS spectra of model structures are simulated by means of direct Green’s function schemes and compared to available experimental records obtained in the gas and solid phases. Calculations are performed using the minimal STP3G and 3-21G basis sets, and se&energy expansions derived at several levels of decoupling approximation: second-order, shifted Born collision (SBC) and diagonal two-particle-hole Tamm-Dankoff approximation (Zph-TDA). The latter scheme is shown to be accurate enough to generate reliable relative line positions and convoluted photoionixation intensities in the inner valence region, which is of fundamental importance for the identification of secondary molecular structures from XPS spectra. Preventing overcounting of self-enetgy diagrams containing a diagonal ladder part is crucial in that respect. As a by- product, this method of simulation is applied to some particular conformations of C9Hm modeling the topmost layer of polyeth- ylene in crystalline form, enabling the identification of fold structures at the extreme surface of lamellar crystals of polyethylene.
1. Introduction
X-ray photoelectron spectroscopy (XPS or ESCA ) allows the study of the electron energy levels in atoms and molecules in gas, liquid and solid phases. Con- sidering the growing importance in highly demand- ing fields (electronics, catalysis, biotechnology, space) of polymer thin films deposited on various substrates (metal, glasses, etc. ), materials for which classical structural analysis techniques such as IR or NMR are usually inapplicable, the XPS method pro- vides a particularly appropriate tool to probe the sur- faces and interfaces of technologically useful organic materials. Most studies have been devoted to the core- level shifts which supply information on the chemi- cal composition and, in favourable situations, on the primary molecular structure. The valence region has
received much less attention, probably because the many XPS valence lines of low intensity to be iden- tified in a small energy interval, typically 50 eV, are more complicated to analyse. Combined with quan- tum mechanical calculations, invaluable direct and indirect information on structural aspects (substitu- tion, conformation) and the chemical bonding of the constitutive compounds can however be obtained from their valence XPS spectra [ 11. In the purpose of finding conformational signatures, recent studies have been conducted at the uncorrelated Koopmans level of approximation on models of polyethylene [ 2 1, syndiotactic propylene [ 3 1, polyoxymethylene [ 41, and polyacrylonitrile [ 5 1. The simulated spec- tra exhibit variations of both band positions and photoionization intensities among the selected con- formations, encouraging efforts to implement corre-
0301-0104/93/S 06.00 0 1993 Elsevier Science Publishers B.V. All rights reserved.

428 M. Deleuze et al. /Chemical Physics 175 (I 993) 427-446
lated schemes reliable enough to ensure conclusions on structural aspects.
At the Koopmans level of approximation [ 6 1, ver- tical ionization potentials (VIP) are obtained, after inversion of sign, as the energies of the occupied HF orbitals. The frozen orbital approximation is as- sumed and correlation effects are neglected. How- ever, the cancellation of the relaxation and correla- tion contributions, often invoked to justify Koopmans’ theorem, can fail severely and may lead to significant errors in the characterization of ioni- zation spectra. Moreover, for accurate simulations, one should take also into account the dispersion of photoionization intensities into shakeup or scatter- ing processes resulting from correlation and relaxa- tion effects. A very sensitive region to such many- body effects is the inner valence region, known to ex- hibit in many cases a strong breakdown of the molec- ular orbital picture [ 7 1. As this region provides the most specific information on structural aspects, most efforts should be concentrated on a better descrip tion of inner-shell photo-ionization events in ex- tended systems.
Particularly well-adapted to the study of interact- ing particle systems,, the one-particle many-body Green’s function [ 8 ] method (MBGF; also referred to as the oneparticle propagator approach) [ 91 has been shown to yield accurate simulation of ioniza- tion and electron attachment spectra for a large vari- ety of molecular systems [ lo]. Compared to more conventional approaches, such as the configuration interaction method, the MBGF theory offers the ad- vantage of a direct, size-consistent and therefore size- extensive [ 11,12 ] description of many-body effects, a requirement which is of major importance when in- vestigating the electronic structure of polymer sys- tems. This approach enables a balanced treatment of correlation and relaxation effects with respect to the multiplicity of excited states but also with the delo- calized character [ 12,13 ] of the molecular orbitals in increasing size systems. In a recent study [ 141, this technique has been applied, using a second-order self- energy expansion, to simulate the XPS valence spec- tra of n-alkane compounds in their all-staggered con- formations or in folded forms modeling the surface of crystalline polyethylene [ 15 1. Assuming an ideal stereoregular polymer structure exhibiting infinite one-dimensional translation symmetry, the band
structure and density of states for polyethylene have either been obtained at the second-order level in elec- tron correlation perturbation [ 16 1. Preliminary sec- ond-order many-body Green’s function (MBGF2 ) investigations for the search of conformational sig- natures have at least been conducted on small cy- cloalkane systems such as cyclopentane and cyclo- hexane [ 17 1. Although significant improvements were obtained compared to the uncorrelated level of description, agreement with experiment was not completely satisfactory.
In extended systems, drastic and successful varia- tions in many-body corrections can be expected after a careful renormalization of the self-energy. In this contribution, we examine the improvements ob- tained in the simulation of XPS valence spectra of extended alkane systems obtained when using the perturbation expansion of the one-particle propaga- tor the perturbation expansion of the one-particle propagator referred [ 18 ] as the diagonal version of the two-particle-hole Tamm-Dankoff approxima- tion ( Zph-TDA) [ 19 1. The diagonal approximation has also been used for the self-energy, thereby imply- ing the neglect of configuration mixing effects in the ionized system. Several reasons have led us to these choices: (i) The minimal size of a chain required to mimic a polymer system is such that it often pre- cludes the use of more rigorous but also costly ap- proaches. (ii) In the particular case of saturated hy- drocarbons, it has been found that the greater part of the photoionization intensity can be attributed [ 201 to main lines which, in regards to the fundamental gap of such systems, larger than 12 eV [ 211, should fall in much lower binding energy regions than their satellite structures of shakeup lines. This large gap, preventing big electronic polarizabilities limits also the electronic correlation and relaxation effects on the ionization processes: in this ideal case, the mixings of the one-hole and two-hole-one-particle contigura- tions, implying nearly complete energy degeneracies of the corresponding electron states, are strongly dis- favored. (iii) Conformational signatures in the XPS spectra of linear alkanes find exclusively their origin in molecular orbital features and can therefore be consistently interpreted [ 15 ] in terms of variations in relative positions and intensities of main pho- toionization lines. As only relative distributions of primary spectral intensity are under the scope, there

M. Lkkuze et al. /Chemical Physics I 75 (1993) 42 7-446 429
is no need to address the configuration mixing effects in the ionized system, as it is probably not necessary to introduce ground state correlation effects in a treatment like the extended Zph-TDA scheme [ 22 1. It should also be noted that in the solid phase, the interchain polarization effects on the overall ioniza- tion process should be comparable if not bigger than the corrections to the present results that could be brought by more quantitative Green’s function cal- culations performed over isolated clusters intended to simulate macromolecules. (iv) Finally, the devel- opment of band structure methods based on the one- particle propagator theory provides further motiva- tion for calibrating diagonal schemes with experi- mentally well-characterized systems, such as n- alkanes.
In section 2, the Zph-TDA scheme is presented in its diagonal version and compared to the second-or- der level of approximation. Emphasis is laid to pre- venting overcounting of diagrams containing a lad- der part, by comparing two of the basic formula derived in ref. [ 191. Methodology and model sys- tems are presented in section 3. In the last part, di- agonal 2ph-TDA calculations are performed for in- creasing size linear alkanes, these model oligomers constituting the simplest available series of realistic compounds one can imagine to isolate as building blocks to construct a regular and well-characterized polymer: polyethylene. This is useful in understand- ing the evolution of the one-particle and many-body features when going to the limit of a macromolecule and to validate the method of simulation we intend to apply to related molecular structures. The two di- agonal schemes of renormalization af the self-energy kernel, with or without overcounting of diagrams with a ladder part, are also quantitatively compared.
The method of simulation providing the more re- liable relative distribution of ionization lines in the inner valence region is applied in the last section to the search for conformational signatures in the XPS valence bands of polyethylene. The chosen confor- mations of n-CgHzo are intended to model ideal poly- mer surfaces rich either in folds [ 23 ] or in zig-zag planar chains. Our method of simulation is shown to provide consistent help in solving tricky photoioni- zation intensity questions and to ensure definite con- clusions on structural features.
2. Theory
Designed to preserve the one-particle concept be- yond HF theory, the one-particle propagator is de- fined as an autocorrelation function [ 23 1, providing the probability amplitude of propagation [ 241, de- pending on the time ordering (t,, tz), of an extra par- ticle (or a hole) from the HF spin-orbital & (xi) to the HF spin-orbital x, (&) in the background of the interacting ground state wavefunction 1 !Pg ) :
G&h, h)=i-‘( ~~IT~{ai(t~),~,+(t,)}l%‘> a (1)
Tw is the Wick chronological operator and uk( t) (a,$ (t) ) are standard annihilation (creation) oper- ators for an electron in HF spin-orbital &. They are expressed in the Heisenberg representation:
a~(t)=e~‘u~e-‘H1 2 ak’ =d(O) , (2)
where H is the exact Hamiltonian of the system. Us- ing Moller-Plesset partitioning [ 26 1, it can be written
H=H,+ V, (3)
with Ho the unperturbed HF Hamiltonian and Y a perturbation accounting for the electronic correlation:
+1 C <ijllij>ninj, (5) !!
where (ijll kl) is the standard notation for the anti- symmetrized bielectron integrals over molecular spin- orbitals, e, are the HF energies, and the occupation number nl is 1 if spin-orbital I is occupied, or 0 in the other case.
If the exact Hamiltonian does not depend explic- itly on time, G is time homogeneous and depends only on the difference t= t2- t,. Through Fourier trans- form and spectral resolution, one can derive the one- particle propagator matrix [ 271 in the frequency domain:

430 M. Deleuze et al. /Chemical PhysW 175 (1993) 427-446
where the sums over nr and p run over all the states of the (N- 1 )- and (N+ 1 )-particle system. The im- portance of the one-particle propagator matrix can be readily appreciated from this equation. Poles of G (w) are, after inversion of sign, the exact vertical ioniza- tion potentials and electroattachment energies. Res- idues at the poles are quadratic products of Feyn- man-Dyson amplitudes obtained as partial overlaps between the exact (full CI) wavefunction for the /cth state (k= m or p) of the (M= N+ 1 )-particle system ( !?‘f) and for the ground state of the N-particle sys- tem. Therefore, eq. (6 ) enables a formally exact de- scription of the ionization or electron attachment processes, while partially retaining the one-particle picture associated to the HF molecular spin-orbitals indices i and j.
It does not seem possible at first glance, however, to construct an exact one-particle theory, since the many-electron Hamiltonian contains bi-particle in- teractions. This difftculty can be overcome by intro- ducing an effective frequency-dependent perturba- tion potential, the self-energy Z(o), which results naturally through renormalization [ 25 ] of the one- particle propagator. G(o) can hence be obtained solving the recursive Dyson relation [ 28 ] :
G(o)=G~(~o)+G~(~)X(W)G(~) (7)
with Go(o) the one-particle propagator associated to the HF Hamiltonian
The pole strength related to the ionization of an electron out of the molecular spin-orbital x can be calculated at the diagonal level of approximation for the selfenergy as
(9)
Defined as the residue of 1 / [w-&(o) ] taken at the pole o, it can be equated to the squared overlap I < Yr- ’ I a, I Yg) I ’ between the exact wavefunction
describing the ionized system and the one obtained when annihilating a particle in spin-orbital xc within the exact neutral ground state. Hence, r, represents the fraction of the photoemission intensity associ- ated with a one-electron process, the remaining frac- tion 1 -r, being the intensity dispersed in shake-up or scattering events because of correlation and relax- ation effects.
The self-energy can be expanded in an infinite per- turbation series with respect to the electronic inter- action with the help of diagrammatic techniques [ 29- 331. As the HF energies already include Coulomb and exchange interactions to first order, the expansion of Z (0) starts at second order:
~(o)=I;‘2’(u)+~‘3’(0)+... . (10)
Using the conventions of Abrikosov [ 33 1, Z (co) can be obtained to the nth order in the correlation perturbation by drawing all the time-ordered, topo- logically different and strongly connected diagrams having n point vertices and 2n- 1 Go lines. To sec- ond order, there are two time-ordered self-energy diagrams, whose contributions can be readily evalu- ated using standard diagrammatic rules:
(11)
Much of the physics involved in the ionization process is qualitatively well reproduced using expres- sion ( 11) for the self-energy. A pole of the first term corresponds to the attachment process of an electron to a spin-orbital x, (z) accompanied by a simulta- neous single excitation L-+X (~+x). In the same way, a pole of the second term describes the ionic state with one electron ionized from spin-orbital XI (xb), this process being followed by the excitation fi+x,
(x-xr)- In the diagonal selfenergy approximation (also re-
ferred to as the quasi-particle [ 3 5 ] approximation ) , one removes the coupling between the attachment and ionization sectors of the propagator. The problem of finding the poles of G (w ) is reduced to the resolution of a self-consistent one-particle equation:
W,=&,+ZE(o,). (12)

hf. Lkdeuze et al. /Chemical Physics 175 (1993) 427-446 431
The ionization process is now described as the re- moval of a quasi-electron and the creation of a quasi- hole in the occupied space, these electrons and holes being dressed through virtual excitations of the elec- tron system: solving eq. ( 12 ) at the second-order level of approximation allows the excited configurations
(&)-I (L) (xs) and M-i (a)-’ (x,) to interact directly with the one-particle (xc) and one-hole (&) -’ configurations, hence including correlation effects.
Unfortunately, the second-order expansion is in general not adequate for accurate results, although Z(z)(o) has the analytical structure of the exact I;(w) [ 27 1. At that level of approximation, the “internal” polarization effects resulting from the ionization event are evaluated at a level equivalent [ 361 to a summation over states (S.O.S. ) description, which, by its crude character, neglects most of the electron reorganizational effects. The extension to third order will already destroy the spectral structure of Zt2)( o), since quadratic poles will be added by I;(‘) (0). An extension up to a finite order will therefore fail [ 27 ] to describe X(o) if the region in the neighbourhood of its poles is of interest, as it is the case for the inner valence ionization potentials.
The two-particle-hole Tamm-Dankoff approxi- mation (Zph-TDA) scheme leads to a structure con- serving [ 37 ] renormalization of the selfenergy. The free 2p- 1 h and 2h- 1 p energy excitations in eq. ( 11) can be renormalized by summing up to infinite order certain classes of diagrams, enabling the introduction of further electronic interactions. Well-known exam- ples of partial summations of diagrams are the RPA (random phase approximation) and ladder expan- sion of 2 (w). These topological series provide the leading contributions to the self-energy of a particle or a hole propagating in a “high density” electron gas or in “low density” nuclear matter, respectively. For molecular systems, unfortunately, there is no domi- nant class of diagrams [ 27 ] : both the ladder and the RPA series provide large contributions that tend to compensate each other, which renders the evaluation of the self-energy more difficult. In the 2ph-TDA scheme, both kind of contributions are taken into account.
The self-energy is separated into its external ver- tices and the remainder, defining the w-dependent kernel r,
(13)
By definition, r is subject to an expansion in pow- ers of interaction elements, which can be converted into a Dyson-like equation by carrying partial infi- nite summations over the RPA and ladder series of diagrams, as well as all their possible mixing, through the renormalization of the 2plh and 2h-lp re- sponses. A suitable termination [ 191 of this expan- sion yields the following expressions:
l-(o)=[ol -K-C]-‘, (14)
with
(01 -K)M,,,H,,v
=s,,s,,s,,.(U+Ek-&,--E,) ) (15)
c klm,k’l’m,=(fskk’(lmllllm’)+~~mm,(kl)Ilfk()
+6~~(km’Ilmk’))(nkiilrs,-_ifknlnm), (16)
where fi X= 1 -n,. The antisymmetry property of r with respect to the interchange of the indices 1 and m (I’ and m’ ) can be used to restrict the index space ( klm ) to k < 1. The resulting equations are
‘&(o)
= *zrnk ,C,,, (i41~m)r.m,~tmt <I’m’Ilik’> , , ‘3’
(17)
I-(o)= [ol -K-C]-‘, (18)
c Wm,k’l’m’= [(WI~m’>Jkk’
-(l-Pr,)(l-PI.,.)S,,.(k’II(kl’)]
x (n/$,&-&n,nm) . (19)
To avoid the problem of constructing and invert- ing the full inverse in eq. ( 18 ), the off-diagonal ele- ments of C are neglected. This drastic approximation reduces the self-energy to
&j( 0) =
(20)
with
A~~=I2(lmlllm)-(klllkl)-(kmllkm) , (21)

432 M. Deleuze et al. /Chemical Physics 175 (1993) 427-446
where 1~ 1. This diagonal derivation, referred alter- natively as the shifted Born collision (SBC) approx- imation (see ref. [ 18 ] and references therein), has been derived in the framework of both diagrammatic [ 371 and superoperator algebraic techniques [ 38 1. This approximation represents a second-order expression ,tith shifted denominators, which enables a better description of the reorganizational effects on the main ionization processes as the poles of the self- energy correspond now to the first-order values for shake-on and shake-up energies.
lated XPS spectra are contructed from a superposi- tion of peaks centered at the MBGF2 and 2ph-TDA values for electron binding energies, respectively. The peak-shape is represented by a standard linear com- bination of one Lorentzian and one Gaussian, both having the same height and width ( 1.5 eV) over the energy range considered, the peak-height being scaled according to the intensity previously computed.
By expanding the shift out of the denominator, it has been observed [ 18 ] that the SBC expression im- plies the overcounting of diagonal ladder-type dia- grams, a consequence of the fact that the symmetry interchange associated to indices k and 1 (k’ and I’ ) has been removed before restricting the kernel to its diagonal elements. This overcounting can be avoided by considering the unrestricted expressions ( 13) and ( 16) as the starting points for the restriction of the kernel to its diagonal elements. In this approach, the symmetry interchange between indices k and 1 (k’ and I’ ) is properly accounted for, and the correct form of the self-energy in the diagonal Zph-TDA scheme is found. It only differs from the SBC self-energy by a factor 1= l/2 in the energy denominators. A few ap- plications [ 18,391 on molecular systems of rather limited size, like water and formaldehyde, have shown that the shift resulting from the Iz= l/2 factor in the framework of the diagonal Zph-TDA scheme, affect- ing electron binding energies by a few tenths of an electron volt (eV) as compared to those calculated with I= 1, results to better agreement with experiment.
The quasi-particle approximation for the Green’s function has been assumed throughout this work. This approximation leads to results very similar to those obtained when considering the full (non-diagonal) self-energy. For the systems selected here, the devia- tion resulting from the quasi-particle approximation does not exceed 0.04 eV, at the second-order level of expansion of Z( 0). Therefore, no more than a few hundredths of eV of deviation can be expected at the 2ph-TDA level of expansion. Furthermore, the agreement between non-diagonal and quasi-particle results led us to compute the pole strengths r’ in this latter approximation.
3. Methodology and model systems
The calculations have been carried out using, at the ab initio level, the GAUSSIAN 82 seriesof programs [ 4 1 ] and, at the Green’s function level, a homemade program designed to deal with the large systems ad- dressed in this work. This program has been tested on reference molecules with the Sheffteld electron propagator [42,43] (SHEEP) program. The re- quested convergence on the density matrix was fixed to lo-’ and the integral cutoff was fixed to lo-lo hartree. The use of the minimal STO-3G basis set [ 441 was imposed by two factors: on the one hand, the storage of the huge number of bielectron integrals needed for the HF-MBGF calculations on extended molecular systems, and on the other hand, the fact that the relative atomic cross sections used in the Gelius model have been parametrized for atomic populations obtained in a minimal basis set.
Photoionization intensities are computed using the To simulate polyethylene in its well-known zig-zag Gelius model [ 40 ] for molecular cross sections, the planar conformation, finite H-( CH2),-H chains of relative atomic photoionization cross sections used increasing length (n= 1 to 9) and in their all-stag- for CZs, CZp and Hi, being 100, 7.69, 0.00, respec- gered conformations have been considered, the max- tively (in the valence region, core atomic functions imum value of n = 9 being imposed by the high com- do not participate significantly). To describe the in- putational requirements of the Green’s function creasing dispersion of photoionization intensity from calculations. As it has been experimentally shown by main to secondary lines as one runs from the top of Pireaux et al. [ 45,461, the electronic structure of n- the bottom of occupied bands, the Gelius intensities alkanes with only 10 to 13 carbon atoms is represent- are multiplied by the pole strength factor r, Simu- ative of the electronic structure of an infinite real

hf. Deleuze et al. / Chemical Physics I 75 (I 993) 42 7-446 433
polymer. Hence, the larger systems investigated here would provide a rather correct picture of the con- struction of the corresponding correlated electron band structure in the solid state. All the geometrical parameters in the selected linear chain have been op- timized at the SCF level using the minimal STO-3G basis set.
Two folded structures [ 47 ] of isolated n-&Hz0 molecules have been chosen to model the topmost layer of a particular form of polyethylene, rich in folds emerging from the basal surface of lamellar crystals. This choice implies that interactions between pure hydrocarbon chains have negligible effects, an as- sumption which is supported by the unsuccessful at- tempts of Pireaux et al. in distinguishing crystalline and amorphous polystyrene samples from XPS va- lence spectra. The number of carbon atoms consid- ered comply somehow with the escape depth of photoelectrons in X-ray experiments. The spectra obtained will be also compared to the spectra simu- lated for the n-nonane compound in its all staggered conformation; this particular form models the super- ficial structure of amorphous polyethylene, exhibit- ing zig-zag planar chains lying parallel to the surface. More details in the reasons leading to the choice of the selected model systems is given in a separate con- tribution [ 15 1.
As the leading dynamic many-body effects arise in those regions where electrons propagate with the larger kinetic energies (for instance, in the neigh- bourhood of nuclei, or nearby the nodal surfaces of molecular orbitals), the inclusion of diffuse and po- larization functions in the computational basis is cer- tainly of vital importance in obtaining highly accu- rate results. At first glance, the minimal STO-3G basis set might seem much too limited to yield reliable rel- ative photoionization intensities and line positions in the inner valence region. For the larger systems in- vestigated here, the compensation of the limitation of the STO-3G basis by the contribution of an in- creasing number of atomic functions to the global wavefunction can be invoked as a first justification. In extended systems, this basis is large enough to en- sure a reliable reproduction of the variations ocuring in the relative distribution of primary ionization lines with changes in the molecular conformation. This as- sumption has been assessed in various studies [ l-5 ] conducted at the Koopmans’ level of approximation
and can be also considered at a Green’s function level for systems of low polarizability like saturated hydro- carbons. To assess in a more quantitative way the quality of the STO-3G results, another series of ge- ometry optimizations and Green’s function calcula- tions have been performed on the smallest oligomer systems (n = 1 to 5 ) using the 3-2 1 G basis set [ 48 1. Trends emerging from these calculations for relative line positions are consistent with the trends obtained using a minimal basis set.
4. Results
Our previous studies at the second-order expan- sion of the self-energy have given some confidence in the possibility of solving difficult questions arising from the identification of conformational signatures in photoelectron spectroscopy. With this back- ground, we first analyse the diagonal 2ph-TDA cor- rections to spectra simulated at the Koopmans and MBGF2 levels.
4. I. Diagonal 2ph-TDA study of the valence band formation of linear alkanes; comparison with MBGF2 and SBC results
The diagonal Zph-TDA and MBGF2 spectra of se- lected compounds in the series of the n-alkane oligo- mers are shown in fig. 1. Under each spectrum are represented bars centred at the diagonal 2ph-TDA values for the ionization energies; their height in ar- bitrary units measures the photoionization intensi- ties issued from the model of Gelius and multiplied by the spectroscopic factors r, These spectra should be compared to the simulations displayed at the Koopmans and MBGFZ levels of approximation in ref. [12].
Both spectra reflect the classification of the va- lence molecular orbitals of a C,H,,+, molecule into n inner valence levels in the so-called “C,” region, well separated of the 2n + 1 outer molecular orbitals of dominant C,+H,, character. As shown previ- ously [ 15 I, such a distinction is somewhat arbitrary when dealing with extended alkane molecules. The inner and outer regions differ essentially in their rel- ative intensities, the origin of such a variation arising from the large differences in the diffuse character of

434 M. Dekue et al. /Chmtid My& Ii’5 (I NW) 427-446
100
P e 2 60
60
40
20
-50 -45 -40 -35 -30 -25 -20 -15 -10 -5 0
-50 -45 -40 -35 -30 -25 -20 -15 -10 -5 0 Binding l .rgy (.V)
60.
P e p 60-
60-
-50 -45 -40 -35 -30 -25 -20 -15 -10 -5 0 Binding rnrrgy (et')
Binding .n.rgy (et')
.15 -10 -5 0 Binding m.rgy (&')
-50 -45 -40 -35 -30 -i5 -io -is -io -5 Binding energy (eV;
Fig. 1. Simulated XPS spectra for the selected compounds in the n-alkane series at the Zph-TDA (full line) and MEtGF2 (dotted line) levels of approximation. (a) Methane; (b) ethane; (c) n-propane; (d) n-butane; (e) n-pen-e; (f) n-hexane; (g) n-heptane; and (h) n-octane. The spectrum of the n-nonane compound, in its all-stage& conformation, is given in fig. 7c.

M. Lkkuze et al. /Chemical Physics 175 (1993) 427-446 435
60 60
_;o -45 -40 -55 -jo 35 20 -is -10 -5 Binding .mrgy cm”;
-50 -45 -40 -35 -30 -is -io -is -io -5
Binding enorgy (.“;
Fig. 1 (contimsd).
the CzS and CIP atomic functions and in their behav- iour in the “cusp” region surrounding the nuclei, as shown by the parametric atomic photoionization cross sections used in the Gelius model.
Resolving the one-electron levels in a convoluted spectrum becomes progressively an impossible task as long as the size of the oligomer system increases. The low photoionixation cross sections and the large number of lines in the outer valence region leads to a poorly structured band not only difftcult to analyse, but also to compare with experiment, even in the smaller systems (n-propane). Although inner va- lence levels can be resolved more easily, they exhibit in the larger systems the successive steps in the build- ing up of an unresolved spectral band. As long as the oligomer system extends, an increasing number of electron levels accumulate in the bottom and the top of the inner valence region, of which the two border- ing sharp peaks become progressively the prominent features of the photoelectron spectrum.
Because the relaxation effects, introduced by the hole self-energy, are the leading many-body effects, and because they are enhanced for the innermost electron levels, the inclusion of relaxation effects re- sult in a shift of the ionization potentials towards lower binding energy values, together a contraction of the inner valence band on a reduced energy scale, which induces a smoothing of the structure existing in the Koopmans spectra. This contraction results also in small but significant changes in the convoluted rel- ative photoionization intensities, the latter trend
being partially counterbalanced by the continuous decrease in the pole strength (fig. 2), due to the in- crease in relaxation effects, as long as we move from the top to the bottom of the inner valence band. Ex- cept for the shift and the contraction of the energy scale, Koopmans, MBGF2 and diagonal 2ph-TDA spectra are therefore qualitatively similar, this simi- larity being the final outcome of the combined influ- ences of quite different contributions.
0.96 -0
0.94 - -“o+o o”o
(p
6 0 l 0
0.92 - 0 *a l
0 0
3 l l 0 l t l a 0 0
d O? * 0
.
s 0.99 no 1 C2pregion
Fig. 2. Evolution of the pole strength factor with the inner char- acter of the ionized mokcular orbital at the secondarder (open circles) and diagonal Zph-TDA (closed circles) levels of approximation.

436 M. Lkkze et al. /Chemical Physics 175 (1993) 427-446
As the free 2p1 h and 2h- lp excitation energies are strongly decreased by the inclusion of the direct and exchange scattering contributions, the renormal- ization of the kernel leads to an enhancement of all many-body effects. Modifications in the ionization spectra result after correction of the MBGF2 one- electron energies at the diagonal 2ph-TDA level of approximation in an additional shift towards lower binding energies. Compared to second order, the in- clusion of further electronic correlation and reorgan- izational effects yields also a stronger contraction of the inner valence band, and a larger dispersion of the photoionization intensities from main lines to shake- up processes, as shown by the heavier drop of the val- ues of pole strength associated to the ionization of electrons out of the deeper levels (fig. 2). With the introduction of the second-order correlation and re- laxation corrections, the CZ. bandwidth (full width at half maximum; fwhm) of the n-nonane compound in its all-staggered conformation decreases from 9.6 to 8.2 eV. When using the diagonal Zph-TDA self-en- ergy expansion, this bandwidth decreases ultimately to the value of 7.7 eV, which, considering the approx- imate treatment made in this work and the somewhat arbitrary choice of the spread ( 1.5 eV) of the con- volution function, compares very well to the experi- mental value (7.6 eV) reported [ 49 ] for amorphous polyethylene, exhibiting surfaces rich in long planar zig-zag segments. This point to the consistency of the approach.
In table 1, the STG-3G Green’s function theoreti- cal electron binding energies are compared with the experimental data recorded [ 45,461 in the gas phase and the solid phase by Pireaux and co-workers. In this table, labels of peaks corresponding to the ionization of a single molecular orbital are given in brackets. The maxima of the unresolved peaks corresponding to the ionization of several molecular orbitals were ob- tained through computer convolution fitting, these peaks being characterized by multiple indices. For n- pentane to n-nonane, only inner valence results are displayed.
It can be seen from table 1 that the SBC self-energy expression (with Iz- 1) yields quite unexpectedly closer agreement between the theoretical position of the inner,valence peaks and the experimental data re- corded in the gas phase. Each step in the sophistica- tion of the self-energy expression (MBGFZ, SBC, di-
agonal Zph-TDA) leads to a stronger contraction of bands and increase in the shift of the ionization lines to lower electron binding energies. Although the di- agonal 2ph-TDA formalism changes the Koopmans results in the right direction, it overshoots slightly the experimental ionization potentials. In the outer va- lence region, the situation is even worse, with the more sophisticated self-energy expressions yielding the poorest theoretical values.
The lack of the variational flexibility of the total STO-3G wavefunction can be invoked to explain this disagreement, essentially for the outer valence levels. However, as shown by the comparison of theoretical 3-2 1G MBGF values of ionization potentials with experimental data obtained for the smaller oligo- mers, increasing the basis size does not significantly increase the quality of the diagonal 2ph-TDA results. Moreover, comparing table 1 to table 2 reveals a sig- nificant reduction of the basis set influence on line positions as long as the sophistication of the self-en- ergy expression increases. This reflects a partial can- cellation of errors arising from the limitation of the basis set in the estimation of the Hartree-Fock elec- tron binding energy (e,<O) and of the many-body contributions (&>O) which are both underesti- mated in absolute value with respect to basis saturation.
The underestimation, by about 0.6 + 0.7 eV, of the inner valence ionization potentials is certainly due to a larger part to the lack of ground state electronic cor- relation in the reference ground state wavefunction, which can be introduced only at the third order in the correlation perturbation, mainly by the constant (frequency-independent ) selfenergy diagrams. Be- cause such diagrams do not directly depend on the effective energy (0,) of the ionized electron, their contribution should result in a homogeneous shift, of a few tenths of an electron volt, of all main lines within the same band to higher binding energies, without modifying their relative distribution.
In the solid state, the binding energy of an electron in a given level is lowered compared to the gas phase. Two additive contributions must be taken into ac- count to explain this. ( 1) The first correction is the work function of the solid, defined as the energy dif- ference between the vacuum level, taken as the ref- erence for measurements in the gas phase, and the Fermi level, corresponding to the reference in the

Table 1
M. Deleuze et al. /Chemical Physics 175 (1993) 427-446 431
Experimental f45.461 and theoretical electron binding encrgks (in eV) of the selected alkane compounds (SRI-3G basis set) l )
Molecule Feak Experimental results (XI%) Theoretical results (STO-3G) (MO line )
eaSPha= solid phase Koopmans MBGFZ SBC diagonal Zph-TDA
methane
ethane
propane
n-butane
n-pentane
n-hexane
n-heptane
n-octane
n-nonane
(1) (2)
24.82 23.79 23.02 22.06 14.16 13.52 13.15 12.80
(1) (2) (3) 4-5
26.60 24.97 24.14 23.25 22.15 21.05 20.37 19.68 15.52 14.63 14.20 13.81 12.67 11.68 11.32 11.00
(1) (2) (3) 4-5
(4-5) 6-7 8-9-10
22.93 14.45
23.91 20.42 15.35 12.69
24.60 22.08 19.57 15.64
13.70 12.04
27.54 25.57 24.66 23.87 24.24 22.78 22.11 21.52 20.97 19.76 19.24 18.77 15.44 14.30 13.84 13.36
(16.25-15.41) (15.19-14.34) (14.75-13.93) (14.41-13.59) 14.06 13.16 12.71 12.52 11.96 10.84 10.47 10.20
(1) 24.73(5) 27.94 25.79 24.93 24.23
(2) 23.00( 5) 25.65 23.90 23.20 22.64
(3) 20.81 22.50 21.08 20.55 20.12
(4) 19.11 20.69 19.41 18.86 18.46 5-6-l 15.0 14.76 13.66 13.25 12.97
10-11-12 12.1 11.63 10.43 10.07 9.81
(1) (2) (3)
(: (5)
25.02 23.6( 1) 21.79
(19.875) (18.705)
19.5 18.2 16.6 14.2
28.19 25.93 25.13 24.51 26.52 24.58 23.88 23.36 24.02 22.31 21.80 21.37 21.18 19.78 19.34 19.00
(21.43) (20.03) (19.59) (19.24) (20.56) (19.22) (18.77) (18.42)
(1) (20.0) (28.33) (26.00) (25.25) (24.71)
(2) (19.3) (27.07) (25.00) (24.33) 23.85
(3) 18.0 25.11 23.28 22.70 22.30
(4) 15.8 22.75 21.17 20.69 20.35 5-6 14.4 20.74 19.37 18.97 18.68
(k2 (4) (5) 5-6-l
l-2-3
(3) (4) (5) 6-7-8
19.1 17.9 16.3 14.7 13.7
19.4
27.74 25.62 24.88 24.42 25.89 23.92 24.65 24.21 23.89 22.14 23.36 22.91 21.82 20.30 21.65 21.32 20.68 19.32 18.97 18.71
17.5 15.8 14.0
19.6 18.1 16.8
21.95 25.63 24.99 24.56 26.44 24.38 (23.83) (24.47) 24.78 22.90 22.41 22.09 22.89 21.21 20.79 20.51 20.75 19.32 19.00 18.77
l-2-3 24.5(l)
(4) 22.7( 1)
(5) 21.4( 1)
(6) 20.2( 1) 6-l-8-9 18.8(l) 14.2
28.04 25.66 25.06 24.67 25.46 23.48 23.00 22.69 23.81 22.00 21.57 21.30 22.10 20.49 (20.11) (19.88) 20.68 19.28 18.96 18.75
‘) x-y-z: computer fitted values for peak’s maxima. (x): effective (HF or Dyson) molecular orbitals electron binding energies.

438 M. Dekuze et al. /Chemical Physic!s 175 (1993) 427-446
Table 2 Experimental [45,46] and theoretical electron binding energies (in eV) of the selected alkane compnunds (3-21G basis set) ‘)
Molecule Peak Experimental results (XPS ) Theoretical results (3-2 1G) (MO line)
gas Phasa Koopmans MBGF2 SBC diagonal Zph-TDA
methane (1) 22.93 25.74 23.82 23.14 21.95 (2) 14.45 14.83 13.85 13.56 13.08
ethane (1) 23.91 27.73 25.04 24.21 23.03 (2) 20.42 23.03 21.08 20.49 19.65 (3) 15.35 16.31 14.98 14.58 14.08 4-5 12.69 13.47 12.24 11.93 11.51
propane (1) 24.60 28.61 25.53 24.63 23.48 (2) 22.08 25.24 22.76 22.05 21.20 (3) 19.57 21.89 19.86 19.28 18.60 4-5 15.64 16.34 14.78 14.35 13.85 6-7 13.70 14.78 13.47 13.12 12.75 8-9-10 12.04 11.80 11.45 11.11 10.74
n-butane (1) 24.73( 5) 29.13 25.74 24.84 24.04 (2) 23.00(S) 26.72 23.85 23.11 22.29 (3) 20.81 23.52 21.14 20.53 19.88 (4) 19.11 21.56 19.42 18.87 18.28 5-6-l 15.0 15.63 14.05 13.66 13.27
10-l l-12 12.1 12.53 11.07 10.74 10.41
n-pentane (1) 25.02 29.38 25.85 24.98 24.12 (2) 23.6( 1) 21.64 24.52 23.77 23.00 (3) 21.79 25.07 22.37 21.73 21.09
(4) (19.875) (22.42) (20.12) (19.58) (19.05) (5) (18.705) (21.48) (19.27) (18.73) (18.21)
a1 x-y-z computer fitted values for peak’s maxima. (x): effective (HF or Dyson) molecular orbitals electron binding energies.
solid state. For polyethylene, it has been measured [46] to be between 4.5 and 4.7 eV. (2) In addition to the intramolecular relaxation effects, it is also nec- essary to consider the intermolecular relaxation en- ergy corresponding to a polarization of the neigh- bouring chains in the solid. For polyethylene, Pireaux and Caudano [ 461 propose a value of about 1.2 eV for this contribution.
The Green’s functions formalism provides useful improvements in the simulations displayed at the Koopmans level of approximation, from the outlook of relating XPS spectra to molecular primary and secondary structures. Figs. 3a-3c show correlation diagrams for molecular orbital levels of alkanes taken in the gas phase. The linear correlations between the STO-3G theoretical electron binding energies, ob- tained successively at the Koopmans, MBGFZ, SBC and diagonal Zph-TDA levels of description, and the
experimental electron binding energies provide a slope (respectively 1.268; 1.131; 1.068; 1.009) and an intercept at the origin (respectively - 3.548; - 2.208; - 1.420; -0.635) becoming closer to their ideal values, respectively 1 and 0. Except in the par- ticular case of the Zph-TDA simulations (fig 3c), the Green’s function method leads to linear correlations (figs. 3a and 3b) with experimental data of slightly higher quality than the approach based on Koop mans’ theorem, as shown by the quadratic regression coefficients. Hence the Green’s function method en- ables not only a deeper investigation in the mecha- nisms leading to the construction of a band structure, if offers an advantageous substitute to the pragmatic and empirical procedure of linear contraction and shift in the ab initio energy scale which has been usu- ally used up to now to reproduce the experimental spectra.

hf. Dekuze et al. /Chemical Physics 175 (1993) 427-446 439
- (a) 28 -
,“, . . . , . . , , . 10 20 22 24
Experimental Binding JZncqics (cl’)
(S~db Y=L268X-3.548 (a2 = 0.989)
+-
a- WRopaac A Bubnc
0 Pentane
n N-
(2) HP-MBGF2 resuh Y = 1.131 x-2.2079 (R2 = 0.996)
;,iztc
0 Ropurc
A Butane
i 0 Pentme
m N-
(3) SBC results : Y=l.O68X-1.4202 (IumO.997)
+Methsne
l m
n Ropanc
A Butane
0 pentanc
8 N-
18, . . . , . . . , . . . , . . . , 10 20 22 24 28
Expwimntd Big Encrgics (eV)
Fii 3. Comlation between !TID3G theoretical results and experimental data recorded in the gas phase.
The diagonal 2ph-TDA values based on STO-3G calculations exhibit a slight but significant drift with respect to the size of the oligomer and the fitted cor- relation line (fig. 3c), which reflects typically a com- pensation to the limitation of the STO-3G basis set by the increasing number of atomic contributions to the global wavefunction. Indeed such a drift seems strongly reduced, if not removed, when diagonal 2ph- TDA calculations are performed using the extended 3-2 1G basis set (fig. 4~). Despite this weak basis set
superposition error, no difference between the rela- tive distribution of lines and peaks obtained for a particular expression of the self-energy, can be really detected from one basis to the other: the slopes of the linear regressions (fig. 4) based on 3-2 1 G results ex- hibit values (in the same order: 1.3 12, 1.113; 1.055; 0.982) that are very close to the corresponding val- ues obtained from STO-3G results.
The above observations hold also if STO-3G re- sults are correlated to experimental data recorded in

440 M. Deleuze et al. /Chemical Physics I 75 (I 993) 427-446
(4) diagond Zph-TDA fesults :
Y=l.W9X-0.6345 (R2 = 0.982)
+ MMe
a-
HRopanc
A Butane
. Fumne
n Nonam
18 20 22 24 26
Expcximdtai BindingEncrgics(eV)
Fig. 3 (codtinud).
the solid phase. The polarization of the neighbouring chains does not affect differently the ionization pro- cess of electrons out of various molecular orbitals. The environmental influence yields simply a homogene- ous shift of the one-electron levels, as reflected by the large similarity of the slopes corresponding to the successive linear regressions displayed in fig. 5 ( 1.3 15; 1.114; 1.078; 1.042) with the related values previ- ously given.
The very nice agreement with experiment of the di- agonal Zph-TDA values for the inner valence ioniza- tion potentials obtained using the minimal STO-3G basis set promote this particular method of simula- tion as the right approach, offering the best compro- mise between the accuracy of results and their com- putational cost, for the purpose of assigning variations in relative line positions and convoluted photoioni- zation intensities upon molecular structure alteration.
4.2. Identification of conformational signatures in the valence XPS spectra of amorphous films and crystalline lameke of polyethylene
AU thermodynamic theories for crystallization agree on the fact that polyethylene crystallizes in mi- crolamellae in which the macromolecular chains emerge at the basal surfaces and fold back to the bulk several times. Although the existence of these folds is indirectly supported by neutron scattering [ 501 or X-
ray diffraction data on crystal grown polyethylene with controlled morphologies [ 5 11, or by the exis- tence of -CHI wagging vibrations at 1346 cm- ’ in the infrared spectra of polyethylene lamellae [ 521, their nature is still a matter of controversy. As un- doubtedly shown by condensation of highly cooled polymer vapors on lamellar crystals of polyethylene [ 521, the outermost part of their surface is relatively well-ordered, and should therefore exhibits a prc- dominant type of folds.
In 1988, simulated XPS valence spectra obtained from Koopmans ionization energies of oligomers modeling the topmost layer of polyethylene, such as the folded (fold A and fold B ) or zig-zag planar (ZZ chain) forms of &Hz0 shown in fig. 6, revealed [ 23 ] a striking conformational dependence in the shape of the peak bordering the inner valence band at its low energy edge. These early theoretical predictions were in qualitative agreement with a recent [54] XPS study of ordered and well-characterized polyethylene surfaces: a sharp and intense peak is indicative of zig- zag planar structures while broadening reveals folded conformations [ 15 1. The expected consistency of the forthcoming analysis is based on the excellent agree- ment observed between the present STG-3G diago- nal 2ph-TDA calculations for linear alkanes in their all-staggered conformation on the one hand, and measured XPS valence bands in gas or amorphous solid phases, on the other hand.

M. Deleuze et al. /Chemical Physics I 75 (1993) 42 7-446 441
(I) Kopmm r?sJlts : Y = 1.312x-3.594 (R2 = 0.990) + Methane
l Erhane
n n A Butane
n Pentanc
(2) HF-MBGFZ results Y=l.l13X-1.814 (R2 = 0.996)
x Methane
OEmane
0 Piup=
A Butane 0 Pentme
18 20 22 24 26
Experimental Binding Bncrgics (ev)
26 . @I
s a (3) SBC resula : 3 24 - Y=l.O%X-1.218
18 I.. . 1.. . I.. . 18 20 22 24 26
25
17
I-
,
10 I
2b 2; 2; - :
Bxpaimental Binding Energks (eV)
Experimental Binding Eacrgks (ev)
:4) dia&md Zph-TDA results :
Y = 0.982 x - 0.433 p2 = 0.995)
+Mcthane
l Ethane
n Ropenc
A Bumne
n Pentnne.
Fig. 4. Correlation between 3-21G them-&d results and experimental data recoded in the gas phase.

M. D&we et al. /Chem&al PhyUcs I75 (1993) 427-446
28 -
p”
R2=0.979
A 0 PcaeIne
R (2)’ n Isuant
26 - A Hcpane
n ckcmc
n N- 24 -
(2) HP-MBGPZ nsulB •I Y=1.1439X+3.2409
22- RZ = 0.981 0 0 Pentane
q Hnane
30 64 (1)Kaqmlarcsults
(1)' Y=1.315X+1.1447
A HepenC 0 cktanc
13 15 17 19 21
26
(3) SBC results : Y = I.078 X + 3.8340 (R2 = 0.984)
. . . , . . . , . - , ,
13 15 17 19 21
Experimental Binding Energies (ev)
24 - (4) diagonal 2ph-TDA rcsuiu :
Y = 1.042 X + 4.08 1
(R2=0.9w
22 0 Pcntane -
q Harane
AHcptam
noaane
20 - n Nonane
13 15 17 19
Fxperimental Binding Enmgies (ev)
21
Fig. 5. Correlation between !TKMG theoretical results and experimental data recorded in the solid phase.

M. Lkleuze et al. /Chemical Physics 175 (1993) 42 7-446 443
(a)
Fig. 6. Molecular models of (a and b ) folds (respectively fold A and fold B ), and (c ) zig-zag planar n-nonane (ZZ chain).
AU pole strengths decrease asymptotically with re- spect to the size of alkane systems to values larger than 0.9 and 0.8 at respectively the MBGFZ [ 141 and di- agonal 2ph-TDA levels of description. This indicates that the ionization process are well represented by a dominant configuration assumption for the ionized system, and that it is safe to base the search for the origin of the conformational signature on the inner valence molecular orbitals, their energies and pho- toionization cross sections. In this case, the confor- mational dependence can be attributed [ 15 ] both to changes in the contribution of the directional Clp atomic orbitals to the molecular orbitals at the top of the inner valence band and also to variations in the amplitude of methylenic through-space hyper- conjugation.
The theoretical MBGF and Zph-TDA spectra cor- responding to the selected forms of n-nonane are dis- played in fig. 7 and compared to the experimental measurements of Riga and co-workers [ 541 on sur- faces of crystalline lamellae of polyethylene, rich in
folds emerging perpendicularly from the basal plane (figs. 7b), or on surfaces of amorphous polyethylene, exhibiting zig-zag planar chains oriented parallel to the surface (fig. 7~).
Despite the limited-size of the model alkane oligo- mers, the STO-3G diagonal 2ph-TDA simulations lead to relative line positions and photoionization in- tensities in quantitative agreement with experimen- tal records and therefore provide definite contirma- tion about conformational fingerprints in XPS spectra. It is interesting to note the larger resem- blance of the experimental spectrum recorded on la- mellae of polyethylene with the Zph-TDA spectrum simulated for fold B. Both spectra exhibit peaks of rather equal intensity on both size of the inner va- lence band, and two well separated structures in the outer valence region. The Czr bandwidth (fwhm) corresponding to the fold A (and fold B) structures is successively obtainedas 10.8; 9.2; and 8.7 eV ( 10.3; 8.9; and 8.4) eV at respectively the Koopmans, MBGFZ and diagonal Zph-TDA levels of approxi-
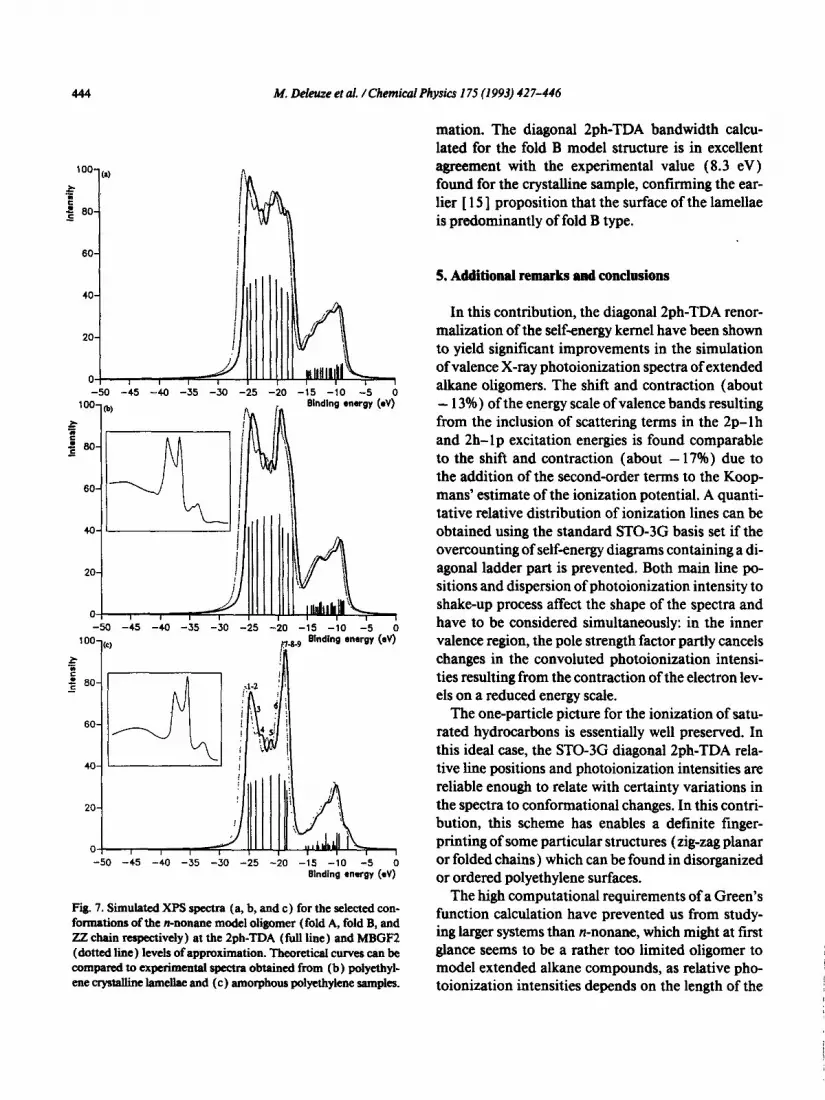
444 M. Lkkuze et al. /Chemical Physics I 7S (1993) 427-446
60 1
40
60-
40-
zo-
.25 - glndlng rnrgy (*I’)
0
-50 -45 -40 -35 -30 -25 -20 -15 -10 -5 0
‘%C, fis-9 Wnding energy (eV)
.e 2 2 60-
60-
20-
o-
-50 -;5 A0 -;5 Go -;5 -;o -is -;o -5 Binding magy (aI’)
Fig. 7. Simulated XPS spectra (a, b, and c) for the selected con- formations of the nnonane model oligomer (fold A, fold B, and ZZ chain respectively) at the Zph-TDA ( fbll line) and MBGF2 (dotted line) levels of approximation. Theoretical curves can be compared to experimental spectra obtained from (b) poiyethyl- ene crystalline lameIke and (c ) amorphous polyethylene samples.
mation. The diagonal Zph-TDA bandwidth calcu- lated for the fold B model structure is in excellent agreement with the experimental value (8.3 eV) found for the crystalline sample, confirming the ear- lier [ 15 ] proposition that the surface of the lamellae is predominantly of fold B type.
5. Additional remarks and conclusions
In this contribution, the diagonal Zph-TDA renor- malization of the self-energy kernel have been shown to yield significant improvements in the simulation of valence X-ray photoionization spectra of extended alkane oligomers. The shift and contraction (about - 13%) of the energy scale of valence bands resulting from the inclusion of scattering terms in the 2p-lh and 2h-lp excitation energies is found comparable to the shift and contraction (about - 17%) due to the addition of the second-order terms to the Koop- mans’ estimate of the ionization potential. A quanti- tative relative distribution of ionization lines can be obtained using the standard STO-3G basis set if the overcounting of self-energy diagrams containing a di- agonal ladder part is prevented. Both main line po- sitions and dispersion of photoionization intensity to shake-up process affect the shape of the spectra and have to be considered simultaneously: in the inner valence region, the pole strength factor partly cancels changes in the convoluted photoionization intensi- ties resulting from the contraction of the electron lev- els on a reduced energy scale.
The one-particle picture for the ionization of satu- rated hydrocarbons is essentially well preserved. In this ideal case, the STO-3G diagonal 2ph-TDA rela- tive line positions and photoionization intensities are reliable enough to relate with certainty variations in the spectra to conformational changes. In this contri- bution, this scheme has enables a definite tinger- printing of some particular structures (zig-zag planar or folded chains) which can be found in disorganized or ordered polyethylene surfaces.
The high computational requirements of a Green’s function calculation have prevented us from study- ing larger systems than n-nonane, which might at first glance seems to be a rather too limited oligomer to model extended alkane compounds, as relative pho- toionization intensities depends on the length of the

M. Deleuze et al. I Chemical Physics 175 (1993) 427-446 445
model system. However, the n-nonane molecule is very close to the smallest oligomer ( 10 to 13 carbon atoms) which is representative of the electronic structure of an infinite real polymer. It should be stressed that the calculations are performed on iso- lated systems, assuming that the interchain interac- tions affect evenly the position and shape of the inner as well as the outer valence levels. The influence of the neighbouring chains should be taken into ac- count, as it might be important to cope with model- ing the escape depth in a less ad hoc way.
Another restriction in the accuracy of our simula- tions results from the computation of photoioniza- tion intensities from the model of Gelius, parame- trized for a minimal basis set and in the framework of a non-correlated approach. Molecular orbital pho- toionization cross sections are needed to modulate the spectroscopic factors r,, which can simply be re- garded as the molecular equivalent of a correlated density of states in extended systems. Hyperconju- gation effects may indeed result [ 15 ] in a strong mu- tual contamination of the CZs and Clp+Hls valence bands at their common border, yielding large varia- tions (about - 20%) in the cross sections associated to the levels at the top of the inner valence band. However, the model of Gelius does not only prevent simulations using larger basis sets than the SIG-3G basis, but its use combined with the inclusion of the pole strength factor may also lead to quantitative er- rors in the computation of the relative photoioniza- tion intensities from one band to another, as some local atomic correlation is introduced twice through the atomic photoionization cross sections. In the long run, photoionization intensities should be calculated using non-parametric techniques implemented in the framework of the Green’s function approach.
Acknowledgement
M. Deleuze is grateful to the FNRS (Belgian Na- tional Fund for Scientific Research) for his Research Assistant position. The authors thank Professor J.M. Andre for his interest in this work. They acknowl- edge also the kind help of Dr. J.G. Fripiat. All calcu- lations reported here have been made on the Namur- Scientific Computing Facility, a result of the cooper- ation between FNRS, IBM-Belgium and the Facultb
Universitaires Notre-Dame de la Paix. The authors acknowledge the support of this project within the framework of the 1991-1992 scientific agreements between the British Council (UK) and the CGRI- Communaute Francaise de Belgique/PNRS (Belgium).
References
[ 1 ] J. DelhaBe and M. Deleuze, J. Mol. Struct. 26 I ( 1992) 187. [2] S. Dellialle, J. Delhalle, C. Demanet and J.M. And& Bull.
Sot. Chim. Bel8.84 (1975) 1071. [ 31 J.J. Pireaux, J. Rica, R. Caudano, J.J. Verbist, J. Delhalle,
S. DeBtaBe, J.M. Andre and Y. Gobillon, Physica Scripta 16 (1977) 329; J. Delhalle, R. Montigny, C. Demanet and J.M. Andre, Theotet. Chim. Acta 50 (1979) 343.
[4] P. Boulanger, R. Lazzaroni, J.J. Verbist and J. Delhalle, Chem. Phys. Letters 129 (1986) 275; P. Boulanger, J. R&a, J.J. Verbist and J. Delhalle, Macromolecules 22 (1989) 173.
[ 51 G. Hennico, J. Delhalle, C. Boiziau and G. IRcayon, J. Chem. Sot. Faraday Trans. 86 ( 1990) 1025.
[6] T.A. Koopmans, Physica 1 (1933) 104. [7] L.S. Cederbaum, J. Schirmer, W. Domcke and W. von
Niessen, Intern. J. Quantum Chem. 14 (1978) 593; L.S. Cederbaum, W. Domcke, J. Schirmer and W. von Niessen, Physica Scripta 21 ( 1980) 481; Advan. Chem. Phys. 65 (1986) 115.
[8] PC. Martin and J. Schwinger, Phys. Rev. 6 (1959) 115; A.J. Layser, Phys. Rev. 129 (1963) 897.
[9] F. Ecker and G. Hohlneicker, Theoret. Chim. Acta 25 (1972) 289; F. Ecker, G. Hohlneicker and W. von Niessen, Mol. Phys. 26 (1973) 1405; B.T. Pickup and 0. Goscinski, Mol. Phys. 26 (1973) 1013.
[ lo] W. von Niessen, J. S&inner and L.S. Cederbaum, Computer Phys. Rept. 1 (1984) 57, and references therein.
[ 111 J.A. Pople, J.S. Binkley and R. Seeger, Intern. J. Quantum Chem. SlO (1976) 1; R.J. Bartlett, Ann. Rev. Phys. Chem. 32 (1981) 359; M. Deleuze, J. Delhalle, B.T. Pickup and J.-L. Calais, Phys. Rev. B 46 ( 1992) 15668.
12 ] M. Deleuze, J. Delhalle and J.M. Andre, Intern. J. Quantum Chem. 41 (1992) 243.
131 M. Deleuze, J. Delhalle and B.T. Pickup, Theoret. Chim. Acta 82 ( 1992) 309.
141 M. Deleuze, J. Delhalle and B.T. Pickup, J. Ekzctron Spectry. Relat. Phenom. 60 ( 1992) 37.
151 M. Deleuze, J.-P. Denis, J. Delhalle and B.T. Pickup, J. Phys. Chem. ( 1993), in press.
[ 161 C.-M. Licgener, Chem. Phys. Letters 167 (1990) 555. [ 171 M. Deleuze et al., unpublished.

446 M. Dekuzeetal./ChcmicalPhys~u 175(1993)427-446
[ 181 Y. Ghm and G. Born, Advan. Quantum Chem. 13 ( 198 1)
[19]::SchirmerandL.S.Cederbaum,J.Phys.B1I (1978) 1889. [ 201 L.S. Cederbaum, W. Domcke, J. Schirmer, W. von Niessen,
G.H.F. Diercksen and W.P. Kramer, J. Chem. Phys. 69 (1978) 1591.
[21] T.J. Fabish, CRC Crit. Rev. Solid State Mater. Sci. (Dec. 1979); J. Mort and G. Pfister, Electronic properties of polymers (Wiley, New York, 1982).
[22] 0. Walter and J. Schinner, J. Phys. B 14 (1981) 3805. [23] J. Delhalle, S. Delhalle and J. Riga, J. Chem. Sot. Faraday
Trans. 83 (I 987) 503. [24] D.N. Zubarev, Sov. Phys. Uspekhi 3 (1960) 320. [25] G.Y. Csanak and H.S. Taylor, Advan. At. Mol. Phys. 7
(1971) 287. [ 261 C. Mdler and M.S. Plesset, Phys. Rev. 46 ( 1934) 618. 1271 L.S. Cederbaum and W. Domcke, Advan. Chem. Phys. 36
(1977) 205. [28] F.J. Dyson, Phys. Rev. 75 (1949) 486,1746. [ 291 N.H. March, W.H. Young and S. Sampatar, The many-body
problem in quantum mechanics (Cambridge Univ. Press, Cambridge, 1967).
[ 301 A.D. Mattuck, A guide to Feynman diagrams in the many- body problem (McGraw-Hill, New York, 1967).
[31] D.J. Thouless, The quantum mechanics of many-body system (Academic Press, New York, 196 1) .
[ 321 A.L. Fetter and J.D. Walecka, Quantum theory of many- particle systems (McGraw-Hill, New York, 197 1) .
[ 331 A. Szabo and N.S. Ostlund, Modem quantum chemistry ( McMilian, New York, 1982 ) .
[34] A.A. Abrikosov, L.P. Gorkov and I.E. Dzyaloshinski, Methods of quantum field theory in statistical physics (Prentice Hall, Englewood Cliffs, 1963).
[ 351 S.T. Pantelides, D.J. Mickish and A.B. Kunz, Phys. Rev. B IO (1974) 2602.
(361 M. Deleuze, P. Horeczky, J. Delhalle and B.T. Pickup, Intern. J. Quantum Chem. S26 ( 1992) 3 1.
1371 L.S. Cederbaum, J. Chem. Phys. 62 (1975) 2160. [ 381 G.D. Purvis and Y. ohm, J. Chem. Phys. 60 ( 1974) 4063;
62 (1975) 2045. [39] G. Born and Y. (Shm, Chem. Phys. Letters 61 (1979) 307. [40] U. Gehus, J. Electron Spectry. Relat. Phenom. 5 (1974) 985. [41] M.J. Frisch, J.S. Binkley, H.B. Schlegel, K. Raghavachari,
R.L. Martin, J.J.P. Stewart, F.W. Bobrowicz, D.J. DeFrees, R. Seeger, R.A. Whiteside, D.J. Fox, E.M. Fleuder and J.A.
Pople, GAUSSIAN 82, release C (Carnegie-Mellon University, Pittsburgh, PA, 1984).
[42] J. Baker, Chem. Phys. 79 (1983) 117; Chem. Phys. Letters 101 (1983) 136; Chem. Phys. 79 (1984) 2693; J. Chem. Phys. 80 (1985) 117; Intern. J. Quantum Chem. 27 (1983) 145.
[43] J. Baker,M. Deleuze,D.H. MosleyandB.T. Pickup,SHEEP, Sheffield Electron Propagator Package (University of Sheffield, Sheflield, UK, 1980-1992).
[44] W.J. Hehre, L. Radom, P. von R. Schleyer and J.A. Pople, Ab initio molecular orbital theory (Wiley, New York, 1986).
[45] J.J. Pireaux and R. Caudano, Am. J. Phys. 52 (1984) 821; J.J. Pireaux, S. Svensson, E. Basilier, P.-A. Malmqvist, U. Gehus, R. Caudano and K Siegbahn, Phys. Rev. A 14 (1976) 2133; J.J. Pireaux, R. Caudano, S. Svensson, E. Basilier, P.-A. Malmqvist, U. Gelius and K. Siegbahn, J. Phys. (Paris) 38 (1977) 1213;
[ 461 J.J. Pireaux and R. Caudano, Phys. Rev. B 15 ( 1977) 2242. [47] V. Petraccone, G. Allegra and P. Corradini, J. Polym. Sci.
C 38 (1972) 419. [48] T.H. Dunning and P.J. Hoy, in: Modem theoretical
chemistry, Vol. 3, ed. H.F. Schaefer III (Plenum Press, New York, 1977) pp. l-28.
[49] J. Delhalle, J.M. Andre, S. Delhahe, J.J. Pireaux, R. Caudano and J.J. Verbist, J. Chem. Phys. 60 (1974) 595.
[ 501 D.M. Sadler and A. Keller, Macromolecules 10 (1977) 1128; Science 203 (1979) 263; J.M. Guenet, Macromolecules 13 ( 1980) 387; Polymer 22 (1981) 313; M. Stamm, E.W. Fischer, M. Dettenmaier and P. Convert, Faraday Discussions Chem. Sot. 68 (1979) 263.
[51] A. Keller, Polymer 3 (1962) 393; Faraday Discussions Chem. Sot. 68 (1979) 145; J.J. Point, M.Ch. Colet and M. Dosiere, J. Polym. Sci. Polymer Phys. 24 ( 1986) 345; M. Do&e, in: Handbook of polymer science and technology, ed. N.P. Chereminisoff (Dekker, New York, 1989) pp. 347-436.
[ 52 ] S.J. Spells, S.J. Organ, A. Keller and G. Zerbi, Polymer 28 (1987) 697.
1531 J.C. Wittmann andB. Lotz, J. Polym. Sci. 23 (1985) 205. [ 541 J. Delhalle, J.-P. Denis, M. Deleuze and J. Riga, Chem. Phys.
Letters, to be published.




















