
First 13C/12C isotopic characterisation of volcanic plume CO2

Journal of Magnetic Resonance 226 (2013) 1–12
Contents lists available at SciVerse ScienceDirect
Journal of Magnetic Resonance
journal homepage: www.elsevier .com/locate / jmr
2H–13C HETCOR MAS NMR for indirect detection of 2H quadrupole patternsand spin–lattice relaxation rates
Xiangyan Shi, Jeffery L. Yarger ⇑, Gregory P. Holland ⇑Department of Chemistry and Biochemistry, Magnetic Resonance Research Center, Arizona State University, Tempe, AZ 85287-1604, United States
a r t i c l e i n f o
Article history:Received 29 August 2012Revised 17 October 2012Available online 1 November 2012
Keywords:Molecular dynamicsTwo-dimensional 2H–13C HETCOR MASNMRSite-specific 2H quadrupole line shapeT1 relaxation time
1090-7807/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.jmr.2012.10.013
⇑ Corresponding authors. Fax: +1 480 965 2747.E-mail addresses: [email protected] (J.L. Yarger)
Holland).
a b s t r a c t
Two-dimensional (2D) cross-polarization magic angle spinning (CP-MAS) 2H–13C heteronuclear correla-tion (HETCOR) experiments were utilized to indirectly detect site-specific deuterium MAS powder pat-terns. The 2H–13C cross-polarization efficiency is orientation-dependent and non-uniform for allcrystallites. This leads to difficulty in extracting the correct 2H MAS quadrupole powder patterns. In orderto obtain accurate deuterium line shapes, 13C spin lock rf field, spin lock rf ramp and CP contact time werecarefully calibrated with the assistance of theoretical simulations. The extracted quadrupole patterns forU-[2H/13C/15N]-alanine indicate that the methyl deuterium undergoes classic, three-site jumping in thefast motion regime (10�8–10�12 s) and the methine deuterium has a rigid deuterium powder pattern.For U-[2H/13C/15N]-phenylalanine, indirectly detected deuterium line shapes illustrate that the aromaticring undergoes 180� flips in the fast motion regime while 2Hb and 2Ha are completely rigid. The exper-imental deuterium line shapes for U-[2H/13C/15N]-proline reflect that 2Hb, 2Hc and 2Hd are subjected tofast, two-site reorientations at an angle of (15 ± 5)�, (30 ± 5)� and (25 ± 10)� respectively. In addition, anapproach that combines a composite inversion pulse with 2H–13C CP-MAS is applied to measure 2H spin–lattice relaxation times in a site-specific, 13C-detected fashion.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction
Deuterium (2H) NMR spectroscopy is of great interest for thestructural biology, polymer and chemistry community. 2H lineshapes and spin–lattice relaxation (T1) times provide rich informa-tion about local molecular dynamics and geometry [1–11]. Staticdeuterium exhibits a broad line shape referred to as the Pake pow-der pattern [12]. When a system undergoes a certain motion likemethyl rotation, a reduced pattern is observed [9,13–18]. Whenmolecular motion rate falls in the range of 104–107 s�1, static deu-terium NMR line shape is dominated by its quadrupolar interactionand provides geometric and local/site-specific dynamic informa-tion for the system [1–18]. With the application of magic anglespinning (MAS), the motional range characterized by 2H line shapeis extended to 102–108 s�1 [19–22]. In the motional timescale of108–1012 s�1, the deuterium line shape becomes independent ofrate and T1 can be utilized for dynamic investigations[1,13,19,23,26]. 2H NMR has been used to investigate dynamicsfor a wide variety of systems such as liquid crystals [20,24–30],partially oriented membranes [31–38] and disordered biopolymers[15–17,39,40]. In most of these studies, site-specific isotopic 2H
ll rights reserved.
, [email protected] (G.P.
labeling was applied for one-dimensional (1D) 2H NMR to avoidassignment difficulties caused by spectral overlap. Selective label-ing is difficult to achieve for most proteins and biopolymers. Forbio-systems, 1D 2H NMR experiments are limited due to the largequadrupolar coupling interaction of deuterium compared with thesmall chemical shift window [19,23,29,41]. Even when performingan experiment under magic angle spinning (MAS), it is difficult toresolve line shapes for different 2H sites in a 1D spectrum [42]. Toovercome this issue, two-dimensional (2D) experiments are re-quired to assist in dispersing the resonances for spin systems inone dimension. The 2H–13C heteronuclear correlation (HETCOR)experiment is suitable for this situation as carbon is accessiblefor most bio-systems and has a large chemical shift range that isstrongly associated with structural information [19,23,29,41].Moreover, with recent advances in preparation of protein nano-crystals/microcrystals, it is now possible to obtain high quality13C solid-state NMR spectra with narrower line widths and betterchemical shift dispersion [43,44]. 2H–13C HETCOR NMR have beensuccessfully used to investigate dynamics information for severalsystems [19,23,29,40,41,45–47]. For example, 2H assignment andmobility information were extracted from 2D 2H–13C heteronu-clear multiple-quantum coherence (HMQC) spectroscopy for4-n-pentyl-40-cyanobiphenyl (5CB) [41] and poly(isoprene) rubber[40]. In addition, by employing deuterium double quantum coher-ence excitation before transferring magnetization to carbon in a 2D

π/2
CPt1
2H
13C
CP
garp
(π/2)x
CPΔ
2H
13C
CP
garp
composite π pulse
A
B
Fig. 1. Pulse sequences for 2H–13C CP-MAS HETCOR experiments. (A) 2D 2H–13C CP-MAS HETCOR pulse sequence. (B) 13C-detected 2H T1 inversion-recovery pulsesequence, the composite p pulse is (p/4)x–(p)y–(p/2)�x–(p)y–(p/4)x. For both pulsesequences, an rf ramp is applied on carbon and 2H GARP decoupling is used duringacquisition at low rf power (�2 kHz).
2 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
experiment, the resolution in deuterium dimension can be signifi-cantly improved [48–50]. An alternative approach is 2D 2H–13Ccross-polarization (CP) experiment. CP NMR between spin-1/2 nu-clei has been theoretically discussed in detail and utilized forchemical shift assignment and signal enhancement for decades[51–55]. The same idea can be applied to spin-1 species such asCP between deuterium and proton or carbon. However, the theoryfor spin-1 nuclei cross polarization is not as straightforward as thespin-1/2 case. This is due to the fact that the quadrupolar interac-tion of spin-1 affects the spin lock significantly making the exper-iments quite challenging to apply in practice [56–60].
Cross-polarization transfer between spin-1 and spin-1/2 wasshown theoretically [56–60] and experimentally [19,23,29,45–47,61–65] to be feasible. Both single-quantum and double-quantum cross polarization can occur during spin lock, dependingon the experimental conditions. When 2H radio frequency (rf) fieldstrength is smaller than quadrupole coupling constant (CQ), the CPmatching condition for each case is different and determined bythe quadrupolar interaction, rf field strength and its transmitteroffset [56–60]. Typically, experimental efforts are put on twoaspects: signal maximization and achieving uniform cross polari-zation transfer for all crystallites [19,23,29,45–47,61–65]. Experi-mental conditions used to achieve the best performance aredifferent for each aspect. In a previous study, the deuteriumspectral assignment was accomplished for 5CB using 2D 2H–13CCP [41]. The experimental conditions were chosen such thatHartmann–Hann matching condition was fulfilled for single-quantum cross-polarization to ensure best signal. However, correctquadrupole line shapes are not necessarily obtained indirectlywhen the CP condition that yields the most intense signal is chosenbecause the cross polarization is non-uniform for all crystallitesunder this condition. In a few recent studies, different experimen-tal strategies were applied to obtain quadrupole line shape fordynamics and geometry investigation, including utilization ofconventional 2H–13C CP [19,23] and modified composite pulsesequences [66,67]. For example, 2H–13C CP experiments were con-ducted for several systems such as a-spectrin [19] SH3 domain,Nac-Val and Nac-Val-Leu-OH [23]. Motionally averaged quadru-pole coupling constants were determined for specific moleculargroups in these systems. Although reasonable dynamics wereproposed for these systems, noticeable differences were observedbetween the experimental and simulated quadrupole patterns[23]. This difference could lead to misinterpretation of geometryand dynamic analysis for some cases [68,69]. Therefore, the 2D2H–13C CP-MAS HETCOR experiments needs to be re-evaluatedfor extracting accurate quadrupole patterns and a set of systemswith known dynamics needs to be tested to determine the validityof this approach.
Deuterium magnetization spin–lattice relaxation is widelyutilized to investigate molecular motion in the timescale of108–1012 s�1, where 2H line shapes are independent of themotional rate [1,13,19,23,26]. 2H T1 is significantly impacted bythe quadrupolar interaction. It is less prone to complications com-pared to 1H and 13C relaxation measurements for investigation ofmolecular motions where multiple relaxation mechanisms arepresent [70,71]. Similar to the case of proton, it is limited todirectly measure deuterium T1 in solid-state NMR due to the lackof spectral resolution. An alternative method is to indirectly detectdeuterium T1 using the 2H–13C CP-MAS approach and exploit theincreased chemical shift dispersion in the 13C dimension. The T1
information is preserved during cross-polarization and can bemeasured by analyzing carbon signal evolution with an inver-sion-recovery experiment in a site-specific manner. This 2Hspin–lattice relaxation time measurement using 2H–13C CPapproach has been successfully applied to several systems inprevious studies [19,23].
In the current study, we focus on indirectly detecting accurate,motionally averaged quadrupole patterns through 2H–13C HETCORexperiments with CP-MAS. Experiments were carefully set-up andsuccessfully applied to uniformly 2H/13C/15N-labeled alanine, pro-line and phenylalanine. As will be shown, the typically perceivedoptimal 2H–13C CP condition does not always yield an accurate,indirectly observed 2H MAS quadrupole pattern. Molecular mo-tions and geometry for each group of the amino acid are obtainedbased on quadrupole patterns extracted from 2D experiments andsimulations. In addition, deuterium T1 was determined for eachsite by combining a composite 2H inversion pulse with the2H–13C CP-MAS experiment.
2. Experimental section
2.1. Material
U-[2H4,13C3,15N]-alanine, U-[2H8,13C9,15N]-phenylalanine and U-[2H7,13C5,15N]-proline samples were purchased from CambridgeIsotopes (Andover, MA) and used as received. The powder sampleswere packed in zirconia NMR rotors. The proline sample absorbedwater over time as an isotropic peak was observed in the 2H solid-echo spectrum. Thus, the U-[2H7,13C5,15N]-proline sample wasstored with a calcium sulfate desiccant at room temperature toavoid water absorption and all NMR experiments were conductedon anhydrous proline.
2.2. Solid-state NMR spectroscopy
Solid-state NMR spectra were collected on a Varian VNMRS400 MHz spectrometer equipped with a 3.2 mm MAS probeoperating in 1H–13C–2H triple resonance mode. The 2D 2H–13CCP-MAS HETCOR pulse sequence is depicted in Fig. 1A. A singlequantum deuterium coherence is created by the initial p/2 pulse.After t1 evolution, the single-quantum coherence is transferred to13C by cross-polarization and 13C signal is detected. An rf rampwas applied to 13C during spin lock. The experiment takes advan-

X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12 3
tage of short deuterium spin–lattice relaxation (typically on theorder of millisecond) to compensate for the signal loss during2H–13C CP magnetization transfer. �2 kHz 2H GARP decoupling[72] was utilized during signal acquisition for both experiments.The resolution improvement was �5–20 Hz, depending on thegroup, when 2H decoupling was applied. Due to probe limitations,the available 2H rf field was up to 91 kHz (the pulse length of p/2is 2.75 ls) and this power was used in all 2H–13C CP-MAS experi-ments. 2H rf offset is set such that liquid D2O is on resonance. Therecycle delays for 2D 2H–13C CP-MAS HETCOR experiments were1 s for U-[2H4,13C3,15N]-alanine and U-[2H8,13C9,15N]-phenylalanineand 5 s for U-[2H7,13C5,15N]-proline. To measure deuterium T1, one-dimensional (1D) 2H–13C CP-MAS experiment (Fig. 1B) was imple-mented. Since the applied 2H rf field strength was much smallerthan deuterium CQ in some cases, a composite 2H p pulse [23,73]was applied to provide the necessary bandwidth for inverting theentire 2H powder pattern span. The recycle delays used in T1
measurements were 3 s, 5 s and 60 s for U-[2H4,13C3,15N]-alanine,U-[2H8,13C9,15N]-phenylalanine, and U-[2H7,13C5,15N]-proline,respectively. Experiments were performed under 10 kHz MAS forU-[2H4,13C3,15N]-alanine and U-[2H7,13C5,15N]-proline and 20 kHzMAS for U-[2H8,13C9,15N]-phenylalanine. 20 kHz MAS was chosenfor phenylalanine for better signal to noise in the 2H dimension.The contact time and 13C rf field strength was optimized to achieveuniform as well as efficient cross-polarization for each experimentas described in the following sections. Sample heating due to MASresults in an actual sample temperature of 30 �C for alanine andproline, and 50 �C for phenylalanine. Lead nitrate was used fortemperature calibration [74].
−250−200−150−100−50050100150200250
−250−200−150−100−50050100150200250
2Hβ
2H Frequency (kHz)
2Hα
62.5 kHz
64.5 kHz 13C
2H Frequency (kHz)
A
C
Fig. 2. Deuterium quadrupole line shapes (A and C) and 2H–13C CP matching profiles (Boffset is set such that D2O is on resonance. CP contact time is 1.4 ms. 2Hb quadrupole linejumping model. 2Ha quadrupole line shape is simulated using CQ = 160 kHz, g = 0 and a rShows where the 13C rf field strength equals 2H rf field strength. # Shows where the 13C
2.3. Theoretical simulation
Deuterium MAS and static quadrupole patterns were simulatedusing EXPRESS [75] and SPINEVOLUTION [76] software packages.Methyl and phenyl ring deuterium MAS line shape simulationswere carried out using EXPRESS with the ZCW powder orientationcalculation scheme. Due to the simulation limit on motional time-scale in EXPRESS [75], deuterium MAS quadrupole patterns for ri-gid CD groups were calculated using SPINEVOLUTION with the ASGpowder orientation calculation scheme. Deuterium static quadru-pole line shapes were also simulated with SPINEVOLUTION. Fordeuterium undergoing fast two-site reorientations at small angles,the MAS line shapes were calculated using SPINEVOLUTION withASG powder orientation calculation scheme. A set of 13C–2H CP-MAS experiments for an isolated C–D spin-pair were simulatedin SPINEVOLUTION. The deuterium quadrupolar coupling constant(CQ) and asymmetry parameter (g) used in simulations are detailedin the following discussion sections.
3. Results and discussion
3.1. 2H–13C CP-MAS matching condition
The experimental 2H–13C CP matching profile for methyl andmethine groups of U-[2H4,13C3,15N]-alanine under 10 kHz MAS isshown in Fig. 2. The Fourier transformed 13C spectrum followingcross-polarization transfer from 2H to 13C with different 13C spinlock rf fields is shown. The 2H rf field is 91 kHz which is smaller than
∗
∗
##
13Cβ
13Cα
113.5 kHz13C rf field
rf70.5 kHz 13C rf
B
D
and D) for U-[2H4,13C3,15N]-alanine under 10 kHz MAS. 2H rf field is 91 kHz and itsshape is simulated using CQ = 155 kHz, g = 0 and a methyl deuterium fast three site
igid deuterium model. All simulations are performed with EXPRESS software [75]. �rf field strength is ±10 kHz with respect to the 2H rf field strength .

zHk68zHk5.26
a
b c d
−150 −100 −50 0 50 100 150
A
C
13C rf field strength
2H Frequency (kHz)
−60 −40 −20 0 20 40 60
B
2H Frequency (kHz)
Fig. 3. (A) Ramped 2H–13C CP matching profile for U-[2H4,13C3,15N]-alanine. Eachpair of peaks (Red – Cb, Black – Ca) are from the same experiment performed with aspecific condition. Four conditions are highlighted: a: 67 kHz 13C spin lock field; b:68 kHz 13C spin lock field; c: 69 kHz 13C spin lock field; d: 71 kHz 13C spin lock field;2H rf field is 91 kHz and its offset is set such that D2O is on resonance, CP contacttime is 1.4 ms, 13C rf ramp is 2.5 kHz. Experiments are conducted under 10 kHzMAS. (B) Methyl deuterium quadrupole patterns extracted from 2D 2H–13C CP-MASHETCOR experiment. From left to right: spectra extracted from 2D ramped 2H–13CCP-MAS with conditions a, b, c, d and 1D solid echo spectrum of 2H3-alaninecollected with 100 kHz 2H rf field. (C) Methine deuterium quadrupole patternsextracted from 2D 2H–13C CP-MAS HETCOR experiment, the region from �1 to �7ssb is expanded. From left to right: spectra extracted from 2D ramped 2H–13C CP-MAS under conditions a, b, c, d and simulated deuterium rigid quadrupole pattern.The simulation was conducted using SPINEVOLUTION [76] and a CQ of 160 kHz anda g of 0 were used.
4 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
deuterium CQ of a rigid CD group in amino acids (155–190 kHz)[13–18,28,35,77–79]. The methyl group displays a profile (Fig. 2B)typical for spin-1/2 nuclei CP-MAS in the fast spinning regimewhere the MAS frequency exceeds the heteronuclear dipolar inter-action [53,54]. The matching conditions occur at xI = xS ± nxR
(I = 1/2, S = 1) [53,54]. A more complicated matching profile isobserved for the methine group as shown in Fig. 2D. Strongcross-polarization transfer is observed at xI = 64.5 kHz and70.5 kHz. Unlike the methyl group case, the matching conditionscannot be simply expressed in any useful analytical form as in thecase for spin-1/2. Magnetization transfer efficiency though cross-polarization strongly depends on the orientation of a crystallitewhile the rf field strength is on the order of CQ or smaller [56–60].As a result, cross-polarization dynamics are significantly impactedby the quadrupolar interaction of the system. CQ of methyl deute-rium is motionally averaged to 60 kHz from 160 kHz (Fig. 2A) be-cause of the fast (>108 s�1) methyl group rotation about the C3V
symmetry axis. In this case, the 2H rf is larger than the effectivequadrupolar interaction. Consequently, the influence of quadrupo-lar interaction on CP dynamics is significantly reduced, leading to amatching profile similar to CP between spin-1/2 nuclei [53,54]. TheCP matching behavior is different for the alanine methine group,which has a deuterium CQ of 160 kHz. The orientation-dependentCP efficiency leads to a much more complicated matching profile.Overall, it is hard to find a matching condition satisfying the bestcross-polarization for all groups with different CQ. This situationis improved when all groups in the system posses a similar quadru-polar interaction. For example, the 2H–13C CP matching condition isimpacted differently for groups involved in another classical mo-tion, the 180� aromatic ring flip [24,25,28,32,78,79]. Fig. S1 displaysdeuterium quadrupole line shapes and 2H–13C CP matching profilesfor U-[2H8,13C9,15N]-phenylalanine under 20 kHz MAS (see Supple-mental material). Ca and Cb resonances present the same CP-MASmatching profile within experimental error. This agrees with thefact that 2Ha and 2Hb are both rigid and have the same quadrupolepattern. The matching profile for phenyl ring Cd and Ce resonancesis slightly different. 180� flip of phenyl ring leads to a smaller effec-tive quadrupole coupling constant and results in a quadrupole pat-tern as shown in Fig. S1A and C. This motionally averagedquadrupole pattern is different from the Pake pattern of 2Ha and2Hb, but also covers a broad resonance range. Thus, the differencebetween CP matching profiles for Cd, Ce and Ca, Cb is small. Thepresent investigation is extended to proline that plays an importantrole in protein secondary structure [68,80–83]. The experimentalresults are shown in Fig. S2 for U-[2H7,13C5,15N]-proline (see Sup-plemental material). The dynamics of proline ring can be describedsuch that each deuterium on the ring undergoes fast reorientationbetween two orientations [68,69,80–83]. The previous X-ray andNMR studies have shown that the two orientations deviate by anangle less than 30� [68,69]. Deuterium qudrupole pattern for eachdeuterium on proline ring is close to that for static pattern as shownin Fig. S2A, C and E. As expected, carbon-deuterium groups of pro-line ring possess very close CP-MAS matching profiles because ofthe similar dynamics and similar effective quadrupole couplingsfor the various groups.
In a single-amplitude CP-MAS experiment, it is difficult toestablish efficient matching conditions for all groups with very dif-ferent deuterium quadrupole patterns as in the case of alanine dis-cussed above. This is due to each group possessing a distinct, sharpCP matching condition. Ramped CP has been shown to be effectivein creating flat matching profiles for proton–carbon spin systems[54]. Here, ramped CP is applied to the deuterium–carbon systemand shown to provide a matching condition efficient for differentgroups. Fig. S3 shows CP matching profiles for U-[2H4,13C3,15N]-alanine with different ramp size Dx (see Supplemental material).The application of ramp results in broadened matching profile
and enhanced signal intensity. Consequently, a CP matching condi-tion can be chosen to achieve high signal intensity for both groups.For example, in the case of using a 2.5 kHz rf ramp on 13C, an opti-mal matching condition (marked by an asterisk in Fig. S3) provides94% and 88% of its maximum signal intensity for Ca and Cb respec-tively. Overall, an appropriate ramp can be applied to CP-MASaiming at maximizing signals for all interested groups.
2H–13C CP experiment takes advantage of the short deuteriumspin–lattice relaxation time that compensates for the magnetizationloss during cross-polarization from deuterium to carbon. To illus-trate this, comparisons are made between 2H–13C CP for uniformly2H/13C/15N-labeled alanine/phenylalanine and 1H–13C CP for

13C spin lock rf field strength (kHz)
46 51 56 61 66 71 76 81 86 91 96 101 106 111 116 121 126−100
0100
2 H (kHz)
Fig. 4. Simulated 13C–2H CP spectra for an isolated C–D spin-pair under 10 kHz MAS. 2H rf field strength is fixed as 91 kHz. 13C spin lock field strength varies from 46 kHz to121 kHz with a step of 5 kHz. Simulations were performed using SPINEVOLUTION [76], the deuterium quadrupolar constant used in the simulation is 160 kHz.
56 kHz 13C rf field strength
55.5 kHz 13C rf field strength
X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12 5
uniformly 1H/13C/15N-labeled alanine/phenylalanine. 1H–13C CPprovides 5 and 8 times as much signal as 2H–13C CP per scan for ala-nine Ca and Cb, respectively. However, 2H T1 is 61 ms and 4 ms for2Ha and 2Hb while 1H T1 is 0.55 s for the two groups (see the follow-ing section for detail deuterium T1 analysis). Thus, the signal to noiseper unit time observed in 2H–13C CP is 60% and 150% of that ob-tained in 1H–13C CP for Ca and Cb respectively. Take the CP experi-ment recycle delay limitation into account, 2H–13C CP provides atleast 60% signal to noise per unit time compared to 1H–13C CP. Thus,the short deuterium T1 makes 2H–13C CP nearly comparable to1H–13C CP in terms of signal observed in a certain experimental timeframe. This can be further improved for the case of phenylalanine.2H T1 are 67–101 ms for phenyl ring deuterium and 600 ms formethine and methylene deuterium. T1 times for 1H are much longer,about 3.5–3.7 s for all groups. The detected carbon signal intensityper scan in 1H–13C CP is 3–5.4 times as much as that in 2H–13C CP.Thus, 2H–13C CP provides 70–130% signal of that in 1H–13C CP witha certain experiment time.
55 kHz 13C rf field strength
54.5 kHz 13C rf field strength
54 kHz 13C rf field strength
theoretical C-D line shape
−150 −100 −50 0 50 100 150
2H Frequency (kHz)
Fig. 5. Simulated 13C–2H CP spectra for an isolated C–D spin-pair under 10 kHzMAS. 2H rf field strength is fixed as 91 kHz. 13C spin lock field strength varies from54 kHz to 56 kHz with a step of 0.5 kHz. Simulations were performed usingSPINEVOLUTION [76], the deuterium quadrupolar constant used in the simulation is160 kHz.
3.2. Two-dimensional 2H–13C HETCOR experiment with CP-MAS
Deuterium assignments and quadrupole patterns can be deter-mined from 2D 2H–13C HETCOR experiments with CP-MAS. Inaddition to obtaining maximum signal intensity, extracting thecorrect 2H line shape from indirect dimension of 2D 2H–13C CP-MAS is critical for dynamic investigations. The spin lock fieldstrength and ramp size optimized as discussed above does notensure obtaining correct 2H patterns for all spins in a given sys-tem according to the results of the present study. This is becausethe cross-polarization transfer is non-uniform for all crystallites[56–60]. Most of previous 2D 2H–13C CP-MAS studies neglectthe effect of spin lock field strength on the accuracy of 2H lineshape. Here, part of the CP matching file shown in Fig. S3 is pre-sented in Fig. 3A. 2H–13C CP with 13C rf field strength rangingfrom 62.5 kHz to 78 kHz (the first 25 plots in Fig. 3A) works effi-ciently considering carbon signal intensity. However, most deute-rium line shapes extracted from experiments under theseconditions are quite different and disagree with the simulations.Fig. 3B and C shows 2H quadrupole patterns for alanine methyland methine groups extracted from 2D 2H–13C CP-MAS experi-ments with four different conditions. The 13C spin lock rf field:a – 67 kHz, b – 68 kHz, c – 69 kHz and d – 71 kHz. Conditionsa, b, and c provide methyl deuterium line shape with small differ-ence when compared to the 1D solid-echo spectrum of 2H3-alanine, while condition d results in a line shape with much lessintensity for the centerband. For the methine group, a larger
difference is observed for the four results. Only the patternobtained under condition d is close to the ideal 1D spectra. Thus,the condition providing the correct lineshape is condition d forthe methine group and conditions a, b, c for methyl group. Over-all, the line shape extracted from 2D CP-MAS experiment deviate

6 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
from theory and the difference depend on 13C spin lock rf fieldand 2H quadrupolar interaction of the considered group. Thequadrupole line shape disagreement between the 2D experimentand the ideal 1D spectra cannot be avoided, but only minimizedby carefully choosing experimental conditions. For example, inthe case for methyl group, conditions a, b and c are acceptablefor 2D experiment as their deviation is minimized based on thecurrent results. Despite the deviation from the ideal 1D lineshape, the experimental quadrupole patterns extracted from 2D2H–13C CP-MAS experiment are still able to illustrate that methylundergoes fast rotation and methine is static according to any ofthe four experiment results. Due to the existence of this experi-mental error, careful optimization of CP experimental conditionsare required to distinguish close molecular motions such astwo-site reorientations at small angles as discussed inSection 3.5.3.
3.3. Spin-lock field strength effect on line shape
In the previous section, it was illustrated that deuterium lineshape extracted from 2D HETCOR experiment with 2H–13C CP-MAS is sensitive to 13C spin lock field strength. Even a 1 kHzvariation in 13C rf field can lead to different 2H line shapes. CPefficiency is uneven across the broad deuterium span under a givenset of experimental conditions. Thus, it is necessary to determinehow sensitive the deuterium line shape is to 13C spin lock fieldstrength for an experimental guide. It can be illustrated by theoret-ical calculations of the 1D 13C–2H cross polarization starting with13C transverse magnetization. Here, a series of 13C–2H CP withdifferent 13C field strength for an isolated C–D spin-pair are
−150 −100 −50 0 50 100 150 −
A
100 μs CP contact time
150 μs CP contact time
1400 μs CP contact time
2000 μs CP contact time
2H Frequency (kHz)
Fig. 6. Experimental 2H quadrupole patterns extracted from 2D 2H–13C CP-MAS HETExtracted 2Ha line shapes. (B): Extracted 2Hb line shapes. The MAS frequency is 10 kHz. 2Hspin-lock rf field is 67 kHz.
simulated and presented in Fig. 4. Deuterium quadrupole patternsare obtained under conditions with different 13C spin lock fieldstrengths. Some of the line shapes are significantly distorted. Theseconditions should be avoided in experimental studies. Fig. 5displays five quadrupole line shapes obtained from 13C–2H CPexperiments simulations with very close 13C field strength.Although field strength difference between experiments is only0.5 kHz, non-negligible difference is observed between the results.This would make it difficult to accurately investigate the dynamicsin cases like proline (see below). For accurate dynamics studies, itis necessary to perform theoretical simulations and NMRexperiments on standard samples before conducting 2D 2H–13CCP-MAS experiments on the sample of interest.
3.4. Contact time in 2D 2H–13C HETCOR experiments
In 2H–13C CP-MAS, effective dipolar interaction between deute-rium and carbon is orientation dependent [56–60]. Magnetizationtransfer though cross-polarization is determined by the effectivedipolar interaction. Thus, the contact time needed for efficientcross-polarization is unique for a certain crystallite. In fact, theeffective dipolar interaction becomes smaller for orientations withlarger quadrupolar interactions [60]. To reach cross-polarizationefficiently for all crystallites, long contact times are required.Fig. 6 displays deuterium quadrupole pattern for alanine 2Ha and2Hb extracted from 2D 2H–13C CP-MAS HETCOR experiments withdifferent contact times. With short contact times, 100 ls and150 ls, the line shapes vary significantly from simulation lineshapes. When contact time is increased to 1400 ls and 2000 ls,cross polarization is efficient for all orientations and the obtained
100 −80 −60 −40 −20 0 20 40 60 80 100
2H Frequency (kHz)
B
100 μs CP contact time
150 μs CP contact time
1400 μs CP contact time
2000 μs CP contact time
COR experiments with different CP contact time for U-[2H/13C3/15N]-alanine. (A)rf field strength is 91 kHz and its offset is set such that D2O is on resonance. The 13C
-140-100-60-202060100140
15
20
25
30
35
40
45
50
55
60
10
2H Frequency (kHz)
13C
Che
mic
al S
hift
(pp
m)
−80−60−40−20020406080 −150 −100 −50 0 50 100 150
extracted Dβ quadrupole pattern
extracted Dα quadrupole pattern
simulated three sites jumping quadrupole pattern
simulated CD rigid pattern
CA D
FE
Cβ
Cα
B
13C Chemical Shift (ppm)
2H
Che
mic
al S
hift
(pp
m)
20253035404550
2
3
4
5
6
7
1
α
β
2H Frequency (kHz) 2H Frequency (kHz)
H2NOH
O
α
β
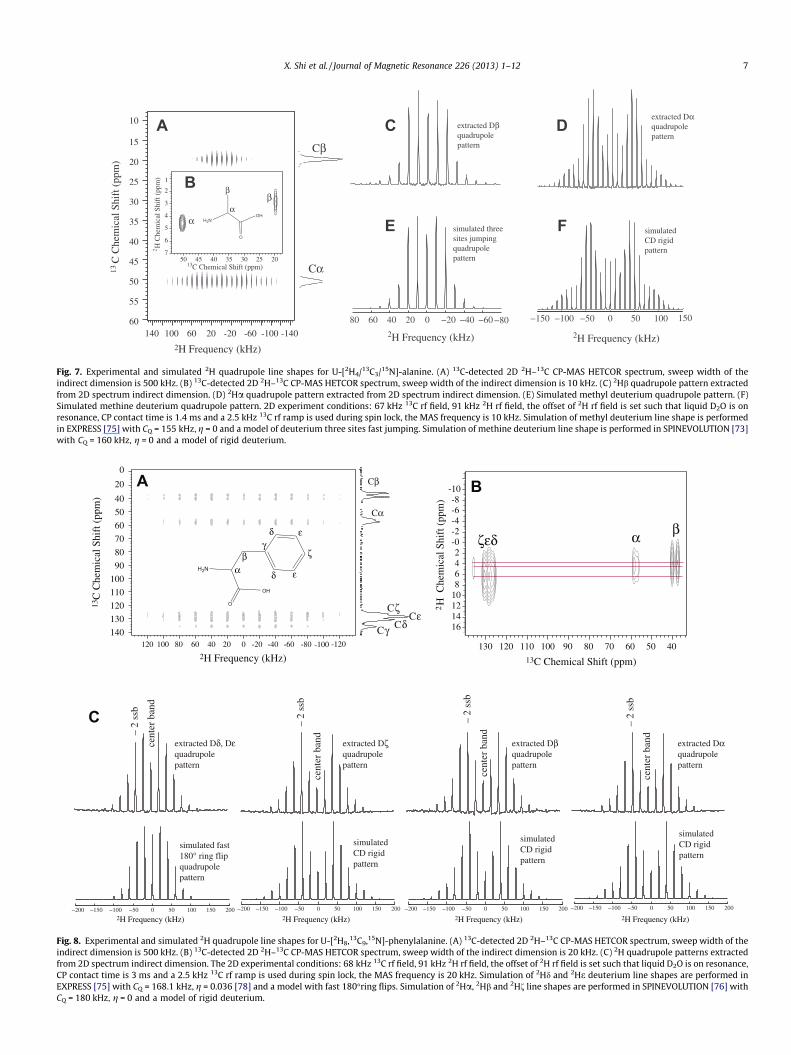
Fig. 7. Experimental and simulated 2H quadrupole line shapes for U-[2H4/13C3/15N]-alanine. (A) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, sweep width of theindirect dimension is 500 kHz. (B) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, sweep width of the indirect dimension is 10 kHz. (C) 2Hb quadrupole pattern extractedfrom 2D spectrum indirect dimension. (D) 2Ha quadrupole pattern extracted from 2D spectrum indirect dimension. (E) Simulated methyl deuterium quadrupole pattern. (F)Simulated methine deuterium quadrupole pattern. 2D experiment conditions: 67 kHz 13C rf field, 91 kHz 2H rf field, the offset of 2H rf field is set such that liquid D2O is onresonance, CP contact time is 1.4 ms and a 2.5 kHz 13C rf ramp is used during spin lock, the MAS frequency is 10 kHz. Simulation of methyl deuterium line shape is performedin EXPRESS [75] with CQ = 155 kHz, g = 0 and a model of deuterium three sites fast jumping. Simulation of methine deuterium line shape is performed in SPINEVOLUTION [73]with CQ = 160 kHz, g = 0 and a model of rigid deuterium.
δ
-120-100-80-60-40-20020406080100120
40
50
60
70
80
90
100
110
120
130
140
20
0
2H Frequency (kHz)
13C
Che
mic
al S
hift
(pp
m)
Cβ
Cα
CδCε
Cγ
13C Chemical Shift (ppm)
2H
Che
mic
al S
hift
(pp
m)
405060708090100110120130
-10-8-6-4-2-02468
10121416
Cζ
ζεδβα
−150 −100 −50 0 50 100 150 200−200 −150 −100 −50 0 50 100 150 200−200−150 −100 −50 0 50 100 150 200−200−150 −100 −50 0 50 100 150 200−200
extracted Dδ, Dε quadrupole pattern
simulated fast 180° ring flip quadrupole pattern
extracted Dζ quadrupole pattern
extracted Dβ quadrupole pattern
extracted Dα quadrupole pattern
simulated CD rigid pattern
simulated CD rigid pattern
simulated CD rigid pattern
A B
C
– 2
ssb
cent
er b
and
– 2
ssb
cent
er b
and
– 2
ssb
cent
er b
and
– 2
ssb
cent
er b
and
2H Frequency (kHz) 2H Frequency (kHz)2H Frequency (kHz)2H Frequency (kHz)
H2N
OH
O
γβ
α ε
ζ
δ ε
Fig. 8. Experimental and simulated 2H quadrupole line shapes for U-[2H8,13C9,15N]-phenylalanine. (A) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, sweep width of theindirect dimension is 500 kHz. (B) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, sweep width of the indirect dimension is 20 kHz. (C) 2H quadrupole patterns extractedfrom 2D spectrum indirect dimension. The 2D experimental conditions: 68 kHz 13C rf field, 91 kHz 2H rf field, the offset of 2H rf field is set such that liquid D2O is on resonance,CP contact time is 3 ms and a 2.5 kHz 13C rf ramp is used during spin lock, the MAS frequency is 20 kHz. Simulation of 2Hd and 2He deuterium line shapes are performed inEXPRESS [75] with CQ = 168.1 kHz, g = 0.036 [78] and a model with fast 180�ring flips. Simulation of 2Ha, 2Hb and 2Hf line shapes are performed in SPINEVOLUTION [76] withCQ = 180 kHz, g = 0 and a model of rigid deuterium.
X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12 7

8 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
line shapes approach the simulated ones. Thus, long CP contacttimes (>1 ms) are needed to obtain correct line shapes from these2D 2H–13C CP-MAS experiments. In addition, one needs to avoidcross-polarization transfer beyond directly bonded 2H–13C spinswhile using long contact times to ensure that the experimentsare site-specific. According to thermodynamic theory of proton tocarbon cross-polarization, proton spin-diffusion leads to energytransfer between protons to achieve quasi-equilibrium tempera-ture for the spin system [84–88]. This would lead to communica-tion between un-bonded proton and carbon spins through spindiffusion and heteronuclear coherence transfer [84–88]. It is muchdifferent for the case of cross-polarization between deuterium andcarbon. The mechanism of deuterium spin-diffusion has been dis-cussed in previous studies [89–92]. Deuterium spin-diffusion coef-ficient is three orders of magnitude weaker than that of proton andsignificantly quenched when the spin quadrupolar interactions aredifferent [89–92]. Therefore, deuterium to carbon coherence trans-fer via heteronuclear coupling following deuterium spin diffusionis limited. In addition, heteronuclear coupling is negligible fornon-directly bonded deuterium–carbon spins compared with thatfor directly bonded spins. Overall, it is reasonable to assume that
−200 −150
-140-120-100-80-60-40-20020406080100120140
20
30
40
50
60
70
80
2H Frequency (kHz)
13C
Che
mic
al S
hift
(pp
m)
Cβ
Cα
Cδ
Cγ
2530354045505560
-6-4-202468
1012
2 H C
hem
ical
Shi
ft (
ppm
)
13C Chemical Shift (ppm)
βα γδ
A
B
C
NH
OH
O
βα
γδ
Fig. 9. Experimental and simulated 2H quadrupole line shapes for U-[2H7,13C5,15N]-proindirect dimension is 500 kHz. (B) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, swfrom 2D spectrum indirect dimension. The 2D experiment conditions: 68 kHz 13C rf field,CP contact time is 2 ms and a 2.5 kHz 13C rf ramp is used during spin lock, the MAS frequ[76] with CQ = 170 kHz, g = 0 [68] and fast two-site reorientation at an angle indicated i
deuterium–carbon cross polarization is limited to directly bondeddeuterium–carbon pairs. Thus, the deuterium quadrupole patternsextracted from 2D 2H–13C HETCOR experiments should be site-specific. From 2D 2H–13C CP-MAS experiment of amino acids withcontact times longer than 1.4 ms, it is shown that no cross polari-zation occurred between non-directly bonded deuterium–carbonas discussed below (Figs. 7B, 8B and 9B). However, in practice, itis recommended to check whether cross-polarization occursbetween non-bonded heteronuclei with a chosen contact time.
3.5. 2D 2H–13C HETCOR experiment for alanine, proline andphenylalanine
Experimental parameters need to be set-up carefully to extractaccurate 2H quadrupole line shape from 2D HETCOR experimentswith 2H–13C CP-MAS for dynamic analysis. Here, we recommendan experimental strategy to assist when conducting experimentson an unknown material of interest. First, a standard sample is cho-sen for experimental parameter optimization. It should contain thesame group or unit as the material of interest. For example, alanineis a preferred standard sample if one is interested in local dynamics
−100 −50 0 50 100 150 200 −200 −150 −100 −50 0 50 100 150 200
CD D
θ θ=40O
θ=15O
θ=10O
static
– 4
ssb
– 4
ssb
– 4
ssb
– 4
ssb
cent
er b
and
cent
er b
and
cent
er b
and
cent
er b
and
2H Frequency (kHz) 2H Frequency (kHz)
D
extracted Dβ quadrupole pattern
extracted Dα quadrupole pattern
extracted Dγ quadrupole pattern
extracted Dδ quadrupole pattern
θ=35O
θ=20O
θ=25O
θ=30O
line. (A) 13C-detected 2D 2H–13C CP-MAS HETCOR spectrum, sweep width of theeep width of the indirect dimension is 10 kHz. (C) 2H quadrupole pattern extracted91 kHz 2H rf field, the offset of 2H rf field is set such that liquid D2O is on resonance,ency is 10 kHz. (D) Simulated deuterium line shapes conducted in SPINEVOLUTIONn the figure.

X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12 9
by characterizing molecular motions of alanine in a protein or ingeneral, when a methyl group is of interest. Secondly, determinea CP rf ramp that provides a relatively flat CP matching profileand best signal enhancement. And then, find several CP matchingconditions, which give strong signals for all molecular groups ofinterest. Note that it is almost impossible to have a matching con-dition maximizing signal intensities for all resonances. The chosenmatching conditions should provide good signal intensity for allgroups instead of favoring one over another. Following this, 2D2H–13C CP-MAS HETCOR experiments on the standard sample areperformed with the chosen matching conditions. The contact timeis set such that it reaches strong signal intensities and no crosspolarization occurred between different groups. In our experience,>1 ms is required for the deuterium–carbon spin system. It is rec-ommended to perform a 2D 2H–13C CP-MAS experiment with ro-tor-synchronized acquisition to ensure the absence of crosspolarization between different groups under certain contact times.Finally, extracted 2H line shapes from the 2D 2H–13C CP-MASexperiments can be compared with the expected, simulated line-shapes. One should then choose the matching condition that yieldsthe quadrupole line shape that best matches simulation for all deu-terium. Then, 2D 2H–13C CP-MAS experiment with this condition isready to be applied to complicated protein and biopolymers. Here,using this strategy, the 2H quadrupole line shapes are determinedfor U-[2H4,13C3,15N]-alanine, U-[2H8,13C9,15N]-phenylalanine andU-[2H7,13C5,15N]-proline.
3.5.1. Alanine site-specific 2H quadrupole patterns2D HETCOR experiments with 2H–13C CP were performed on
U-[2H4,13C3,15N]-alanine under 10 kHz MAS. The experimental con-ditions were optimized using the strategy discussed above. Overall,a 67 kHz 13C spin lock rf field with 2.5 kHz ramp, and 1.4 ms cross-polarization contact time was used. Experimental results arepresented in Fig. 7. Cross-polarization works efficiently for bothmethyl and methine groups. In Fig. 7B, correlation between carbonand deuterium resonances indicate that cross polarization only oc-curred within directly bonded heteronuclear spin pairs. Thus, theextracted deuterium line shapes are site-specific. 2Ha and 2Hbquadrupole patterns extracted from 2H dimension match very wellwith theoretical simulations. As displayed in Fig. 7C and E, theexperimental line shape for methyl deuterium agrees with themodel of three sites jumping in the fast motion regime (10�8–10�12 s) [13–19,23]. For methine group, the determined line shapeis characteristic of a completely rigid deuterium environment(Fig. 7D and F).
3.5.2. Phenylalanine site-specific 2H quadrupole patterns2H line shapes for phenylalanine were determined by 2D HET-
COR experiment using 2H–13C CP. The experiment was conductedunder 20 kHz MAS. The final optimized experimental conditionsare 68 kHz 13C spin lock rf field with a 2.5 kHz ramp, and 3 mscross polarization contact time. Fig. 8 displays the experimental re-sults. The 2D spectrum shown in Fig. 8A indicates cross-polariza-tion magnetization transfer takes place from deuterium to carbonfor all sites. It is noted that both Ca and Cb possess two chemicalshifts that are likely due to crystal packing effects [55]. The ob-tained deuterium quadrupole patterns are site-specific since theoccurrence of cross-polarization is limited to directly bonded het-eronuclear spin pairs. It is verified that no 2H–13C correlation wasobserved between non-bonded deuterium and carbon (Fig. 8B).The only exception is that a weak signal is detected for the quater-nary Cc. This is likely due to the close proximity of four deuteronson the neighboring Cb and Cd sites. Although 2H polarization trans-fer is observed for this quaternary environment, there does not ap-pear to be spin communication between the other deuteronsdirectly bound to carbon. Simulations and experimental deuterium
quadrupole patterns extracted from the indirect dimension of the2D spectrum are presented in Fig. 8C. The reduced 2Hd and 2He lineshapes result from the aromatic ring experiencing 180� flips in fastmolecular motion regime (10�8–10�12 s) [24,25,28,32,78,79]. Thefast 180� flips do not averaged the 2H quadrupole pattern due tothe geometry of the phenyl ring. As expected, a classical deuteriumline shape for a rigid group is observed for 2Ha. Similarly, 2Hb pre-sents a line shape almost identical to that of 2Ha. Thus, methineand methylene deuterium are completely rigid. In summary, 2DHETCOR experiments with 2H–13C CP can be used to extract thesite-specific MAS deuterium line shape for phenylalanine.
3.5.3. Proline site-specific 2H quadrupole patternsProline deuterium quadrupole patterns were extracted from 2D
HETCOR experiment utilizing 2H–13C CP under 10 kHz MAS. Theexperiments were performed using optimized condition: 68 kHz13C spin lock rf filed with a 2.5 kHz ramp, and 2 ms cross polariza-tion contact time. Spectra are displayed in Fig. 9. Signals were ob-served for all carbons on the five-member ring after crosspolarization magnetization transfer from 2H to 13C as shown inFig. 9A. Similar to alanine and phenylalanine, cross-polarizationonly occurs between directly bonded deuterium and carbon pairsand provide site-specific deuterium quadrupole patterns. This iswell illustrated by the correlation spectra shown in Fig. 9B. Deute-rium quadrupole patterns for the four sites on proline ring arepresented in Fig. 9C. All the line shapes are very close to rigid quad-rupole patterns. Comparison of the line shapes indicates that 2Hcare the most mobile deuterons and 2Ha is the most rigid one. Thisobservation agrees with previous studies [68,69,80–83]. Structureand dynamics studies show that proline ring cannot execute largeamplitude internal motions [68,69,80–83]. The flexibility varieswith sites on the ring: Cc > Cb > Cd > Ca. The dynamics of proline2Hb, 2Hc and 2Hd can be described as fast two-site reorientationsat small angles [68,69]. Two deuterium solid-state NMR studies re-ported reorientation angles for these sites. Torchia showed that2Hb, 2Hc and 2Hd reorient rapidly (>108 s�1) through angles of17�, 20� and 12� for L-[D7]-proline at 20 �C [69]. Sarkar and cowork-ers determined the fast reorientation angles for 2Hb, 2Hc and 2Hdare 22.1�, 29.0� and 10.8� for DL-[2H6]-proline at 20 �C [68].Fig. 9D displays simulated 2H quadrupole MAS patterns for fasttwo-site reorientation at different angles. Comparing experimentalline shape shown in Fig. 9C with simulated ones, a range of reori-entation angles could be obtained for each site. Here, it is con-cluded that 2Hb, 2Hc and 2Hd have a reorientation angle of(15 ± 5)�, (30 ± 5)� and (25 ± 10)�, respectively. The results fromthe 2D 2H–13C CP-MAS HETCOR experiment roughly agree withprevious results [68,69]. The obtained two-site reorientation anglerange illustrates the small amplitude mobility and relative flexibil-ity for each site very well for proline. Thus, 2D HETCOR experimentwith 2H–13C CP successfully provides site-specific proline deute-rium quadrupole patterns, which can be further applied to biopoly-mers and proteins.
3.6. T1 indirect measurement with 2H–13C HETCOR experiments
Deuterium is a powerful tool to characterize dynamics in the108–1012 s�1 timescale, where line shape is independent of mo-tional rate by measuring 2H spin–lattice relaxation times[1,13,19–23,26]. Indirect deuterium T1 measurements by 2H–13CHERCOR experiments with CP-MAS can be used to overcome thepoor resonance resolution in directly detected 2H experiments.More importantly, 2H–13C HETCOR using CP provides correlationslimited to directly bonded heteronuclear pairs due to negligibledeuterium spin diffusion and heteronuclear coupling betweenun-bonded spins as discussed above [84–92]. This enables one toobtain T1 information site-specifically. It should be noted that

−0. 1 0. 1 0. 3 0. 5 0. 7 0. 9 1. 1 1. 3 1. 5 1. 7 1. 9 2. 1 2. 3 2. 5 2. 7 2. 9 3. 1
−120
−100
−80
−60
−40
−20
0
20
40
60
Dα: T1= 61 ms
Dβ: T1= 4 ms
Δ (s)
Sign
al I
nten
sity
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58 60 62−50
−40
−30
−20
−10
0
10
20
30
40
50
60
Δ (s)
Sign
al I
nten
sity
Dα: T1= 7.63 s
Dδ: T1= 7.64 s
Dβ: T1= 7.87 s
Dγ: T1= 7.79 s
Δ (s)
Sign
al I
nten
sity
0 0. 5 1 1. 5 2 2. 5 3 3. 5 4 4. 5 5
−2
−1.5
−1
−0.5
0
0.5
1
Dε: T1= 97 ms
Dδ: T1= 67 ms
Dβ: T1= 605 ms
Dα: T1= 602 ms
Dζ: T1= 110 ms
A
B
C
Fig. 10. 13C-detected 2H T1 recovery curves for (A) U-[2H4/13C3/15N]-alanine, (B) U-[2H8,13C9,15N]-phenylalanine and (C) U-[2H7,13C5,15N]-proline. The T1 for each deuterium isindicated in the figure.
10 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
2H–13C HETCOR experiments only provide average values for theorientation dependent deuterium T1 [91,93–95]. In the currentwork, T1 measurements were performed on alanine, phenylalanineand proline using the sequence shown in Fig. 1B. Deuterium T1
information is preserved during spin lock and indirectly deter-mined by carbon signal evolution behavior. The experimental con-ditions are the same as those in 2D 2H–13C CP experimentsdiscussed in Section 3.5. Fitted T1 inversion-recovery curves forthe three samples are presented in Fig. 10. For alanine, T1 for 2Haand 2Hb is 61 ms and 4 ms, respectively (Fig. 10A). The very short2Hb results from fast methyl rotation. Deuterium on phenylalaninering have shorter T1 than 2Ha and 2Hb: 97 ms, 67 ms and 101 msfor 2He, 2Hd and 2H1 (Fig. 10B). It is a consequence of the phenyl-alanine 180� ring flip dynamics. In contact, 2Ha and 2Hb are rela-
tively rigid with T1’s of 602 ms and 605 ms as shown in Fig. 10B.All deuterium on proline ring posses close T1’s of 7.63 s, 7.87 s,7.79 s and 7.64 s for 2Ha 2Hb, 2Hc and 2Hd (Fig. 10C). The T1 reflectsthat the proline ring motion is restricted to a small amplitude.
4. Conclusion
Deuterium line shape and spin–lattice T1 relaxation rates can becharacterized by 2H–13C CP-MAS HETCOR experiments for molecu-lar dynamic and geometry studies. The 2H–13C CP is significantlyimpacted by the deuterium quadrupole interaction and dependenton crystallite orientation. Thus, line shapes extracted from a 2D2H–13C CP-MAS HETCOR experiments are very sensitive to spin lock

X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12 11
rf field strength as well as cross-polarization contact time. Theobtained line shapes can show a large deviation from theoreticalsimulations if experimental conditions are not chosen carefully.Here, a strategy is proposed to set-up 2D 2H–13C CP-MAS experi-ments correctly. Using this strategy, site-specific deuterium quad-rupole patterns were determined for alanine, phenylalanine andproline. The extracted quadrupole patterns illustrate the motionsfor each group and agree with theoretical simulations very well.In addition, site-specific deuterium T1 is measured indirectlythough 2H–13C CP HETCOR experiments for the three amino acids.In summary, the present article shows improved determination ofdeuterium quadrupole line shapes using 2H–13C CP-MAS HETCORexperiments compared to previous studies. The 2H–13C CP-MASHETCOR experiments can be applied to uniformly labeled bio-systems for local/site-specific molecular motion and geometryinvestigations if the experimental conditions are carefully deter-mined with the described approach.
Acknowledgments
This research was supported by grants from the Department ofDefense Air Force Office of Scientific Research (AFOSR) underAward No. FA9550-10-1-0275 and the National ScienceFoundation, Division of Materials Research under Award No.DMR-0805197. We thank Dr. Brian Cherry for help with NMRinstrumentation, student training and scientific discussion.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.jmr.2012.10.013.
References
[1] L.W. Jelinski, Solid state deuterium NMR studies of polymer chain dynamics,Ann. Rev. Mater. Sci. 15 (1985) 359–377.
[2] T.M. Barbara, Deuterium quadrupole echo NMR spectroscopy. I. Effects ofchemical exchange during single and composite pulses, J. Magn. Reson. 69(1986) 311–330.
[3] A.D. Ronemus, R.L. Vold, R.R. Vold, Deuterium quadrupole echo NMRspectroscopy. II. Artifact suppression, J. Magn. Reson. 70 (1986) 416–426.
[4] M.S. Greenfield, A.D. Ronemus, R.L. Vold, R.R. Vold, Deuterium quadrupole echoNMR spectroscopy. III. Practical aspects of lineshape calculations for multiaxisrotational process, J. Magn. Reson. 72 (1987) 89–107.
[5] N.J. Heaton, R.R. Vold, R.L. Vold, Deuterium quadrupole echo NMRspectroscopy. IV. Inversion of full width deuterium powder patterns, J. Magn.Reson. 77 (1988) 572–576.
[6] H.W. Spiess, Pulsed deuteron NMR investigation of structure and dynamics ofsolid polymers, J. Mol. Struct. 111 (1983) 119–133.
[7] L.E. Kay, NMR studies of protein structure and dynamics, J. Magn. Reson. 173(2005) 193–207.
[8] S.E. Ashbrook, Recent advances in solid-state NMR spectroscopy ofquadrupolar nuclei, Phys. Chem. Chem. Phys. 11 (2009) 6875–6905.
[9] J.P. Jacobsen, Determination of deuterium quadrupole coupling constants inmethyl groups by high-resolution NMR spectroscopy, J. Magn. Reson. 28(1977) 191–201.
[10] J. Hirschinger, A.D. English, Differentiation of models of rapid molecularmotion by 2H NMR lineshape simulations, J. Magn. Reson. 85 (1989) 542–553.
[11] B. Bose-Basu, J. Zajicek, G. Bondo, S. Zhao, M. Kubsch, I. Carmichael, A.S.Serianni, Deuterium nuclear spin–lattice relaxation times and quadrupolarcoupling constants in isotopically labeled saccharides, J. Magn. Reson. (2000)207–216.
[12] G.E. Pake, Nuclear resonance absorption in hydrated crystals: fine structure ofthe proton line, J. Chem. Phys. 16 (1948) 327–336.
[13] L.S. Batchelder, C.H. Niu, D.A. Torchia, Methyl reorientation in polycrystallineamino acids and peptides: A 2H NMR spin–lattice relaxation study, J. Am.Chem. Soc. 105 (1983) 2228–2231.
[14] K. Beshah, E.T. Olejniczak, R.G. Griffin, Deuterium NMR study of methyl groupdynamics in L-alanine, J. Chem. Phys. 86 (1987) 4730–4736.
[15] A.H. Simmons, C.A. Michal, L.W. Jelinski, Molecular orientation and two-component nature of the crystalline fraction of spider dragline silk, Science271 (1996) 84–87.
[16] Z. Yang, O. Liivak, A. Seidel, G. LaVerde, D.B. Zax, L.W. Jelinski, Supercontractionand backbone dynamics in spider silk: 13C and 2H NMR Studies, J. Am. Chem.Soc. 122 (2000) 9019–9025.
[17] L.W. Jelinski, C.E. Sullivan, D.A. Torchia, 2H NMR study of molecular motion incollagen fibrils, Nature 284 (1980) 531–534.
[18] T. Weidner, N.F. Breen, K. Li, G.P. Drobny, D.G. Castner, Sum frequencygeneration and solid-state NMR study of the structure, orientation, anddynamics of polystyrene-adsorbed peptides, Proc. Natl. Acad. Sci. 107 (2010)13288–13293.
[19] M. Hologne, K. Faelber, A. Diehl, B. Reif, Characterization of dynamics ofperdeuterated proteins by MAS solid-state NMR, J. Am. Chem. Soc. 127 (2005)11208–11209.
[20] E.B. Brouwer, G.D. Enright, J.A. Ripmeester, Solid-state NMR and diffraction of atunable p-tert-Butylcalix[4]arene�Guest structure, J. Am. Chem. Soc. 119(1997) 5404–5412.
[21] R.J. Schadt, E.J. Cain, A.D. English, Simulation of one-dimensional 2H NMR lineshapes, J. Phys. Chem. 97 (1993) 8387–8392.
[22] M. Hologne, J. Hirschinger, Molecular dynamics as studied by static-powderand magic-angle spinning 2H NMR, Solid State Nucl. Magn. Reson. 26 (2004) 1–10.
[23] M. Hologne, Z. Chen, B. Reif, Characterization of dynamic processes usingdeuterium in uniformly 2H,13C,15N enriched peptides by MAS solid-state NMR,J. Magn. Reson. 179 (2006) 20–28.
[24] H. Zhang, R.G. Bryant, Bound ligand motion in crystalline carboxypeptidase A,Biophys. J. 72 (1997) 363–372.
[25] Y. Hiyama, J.V. Silverton, D.A. Torchia, J.T. Gerig, S.J. Hammond, Molecularstructure and dynamics of crystalline p-Fluoro-D, L-phenylalanine. Acombined X-ray/NMR investigation, J. Am. Chem. Soc. 108 (1986) 2715–2723.
[26] J.W. Mack, D.A. Torchia, A 2H NMR study of the molecular dynamics of solidcyclopentane, J. Phys. Chem. 95 (1991) 4207–4213.
[27] Y. Suzuki, M. Sato, K. Takanohashi, T. Ida, M. Mizuno, Deuterium NMR study ofmolecular dynamics and phase transition in acetonitrile crystal, J. Phys. Chem.A 112 (2008) 13481–13486.
[28] D.M. Rice, R.J. Wittebort, R.G. Griffin, E. Meirovitch, E.R. Stimson, Y.C.Meinwald, J.H. Freed, H.A. Scheraga, Rotational jumps of the tyrosine sidechain in crystalline enkephalin. 2H NMR line shapes for aromatic ring motionin solids, J. Am. Chem. Soc. 103 (1981) 7707–7710.
[29] C. Auger, A. Lesage, S. Caldarelli, P. Hodgkinson, L. Emsley, Deuterium–carbonNMR correlation spectroscopy in oriented materials, J. Am. Chem. Soc. 119(1997) 12000–12001.
[30] K. Beshah, R.G. Griffine, Deuterium quadrupole echo NMR study of methylgroup dynamics in N-Acetyl-DL-(c-d6)-valine, J. Magn. Reson. 84 (1989) 268–274.
[31] W. Ying, S.E. Irvine, R.A. Beekman, D.J. Siminovitch, S.O. Smith, Deuterium NMRreveals helix packing interactions in phospholamban, J. Am. Chem. Soc. 122(2000) 11125–11128.
[32] G.C. Leo, L.A. Colnago, K.G. Valentine, S.J. Opella, Dynamics of fd coat protein inlipid bilayers, Biochemistry 26 (1987) 854–862.
[33] R.G. Griffin, Solid state nuclear magnetic resonance of lipid bilayers, MethodsEnzymol. 72 (1981) 109–174.
[34] C.R. Sanders, B.J. Hare, K.P. Howard, J.H. Prestegard, Magnetically-orientedphospholipid micelles as a tool for the study of membrane-associatedmolecules, Prog. NMR Spectrosc. 26 (1994) 421–444.
[35] R.A. Kinsey, A. Kintanar, E. Oldfield, Dynamics of amino acid side chains inmembrane proteins by high field solid state deuterium nuclear magneticresonance spectroscopy, J. Biol. Chem. 256 (1981) 9028–9036.
[36] R.S. Prosser, S.A. Hunt, J.A. DiNatale, R.R. Vold, Magnetically aligned membranemodel systems with positive order parameter: switching the sign of Szz withparamagnetic ions, J. Am. Chem. Soc. 118 (1996) 269–270.
[37] J. Seelig, P.M. Macdonald, Phospholipids and proteins in biological membranes.2H NMR as a method to study structure, dynamics, and interactions, Acc.Chem. Res. 20 (1987) 221–228.
[38] H. McConnell, A. Radhakrishnan, Theory of the deuterium of sterol-phospholipid membranes, Proc. Natl. Acad. Sci. 103 (2006) 1184–1189.
[39] D.A. Torchia, Solid state NMR studies of molecular motion in collagen fibrils,Methods Enzymol. 82 (1982) 174–186.
[40] H. Mengezaz, S. Hediger, L. Emsley, D. Sandström, Segmental mobility inpoly(isoprene) rubber studied by deuterium–carbon NMR correlationspectroscopy, Polym. Bull. 46 (2001) 183–190.
[41] D. Sandström, H. Zimmermann, Deuterium–carbon multiple-quantum NMR inliquid crystals: polarization transfer and off-magic-angle spinning, Mol. Phys.100 (2002) 1935–1940.
[42] A.E. Aliev, S.E. Mann, A.S. Rahman, P.F. McMillan, F. Corà, D. Iuga, C.E. Hughes,D.M. Harris, High-resolution solid-state 2H NMR spectroscopy of polymorphsof glycine, J. Phys. Chem. A 115 (2011) 12201–12211.
[43] T.I. Igumenova, A.E. McDermott, K.W. Zilm, R.W. Martin, E.K. Paulson, A.J.Wand, Assignments of carbon NMR resonances for microcrystalline ubiquitin,J. Am. Chem. Soc. 126 (2004) 6720–6727.
[44] R.W. Martin, K.W. Zilm, Preparation of protein nanocrystals and theircharacterization by solid state NMR, J. Magn. Reson. 165 (2003) 162–174.
[45] P. Jonsen, A.C. Olivieri, S.F. Tanner, 13C CP/MAS NMR of a 13C–2Hresidual dipolar coupled pair, Solid State Nucl. Magn. Reson. 7 (1996)121–125.
[46] C. Auger, A. Lesage, S. Caldarelli, P. Hodgkinson, L. Emsley, Assignment andmeasurement of deuterium quadrupolar couplings in liquid crystals bydeuterium–carbon NMR correlation spectroscopy, J. Phys. Chem. 102 (1998)3718–3728.

12 X. Shi et al. / Journal of Magnetic Resonance 226 (2013) 1–12
[47] D. Sandstrom, M. Hong, K. Schmidt-Rohr, Identification and mobility ofdeuterated residues in peptides and proteins by 2H–13C solid-state NMR,Chem. Phys. Lett. 300 (1999) 213–220.
[48] S. Vega, T.W. Shattuck, A. Pines, Fourier-transform double-quantum NMR insolid, Phys. Rev. Lett. 37 (1976) 43–46.
[49] V. Agarwal, K. Faelber, P. Schmieder, B. Reif, High-resolution double-quantumdeuterium magic angle spinning solid-state NMR spectroscopy ofperdeuterated proteins, J. Am. Chem. Soc. 131 (2009) 2–3.
[50] D. Lalli, P. Schanda, A. Chowdhury, J. Retel, M. Hiller, V.A. Higman, L. Handel, V.Agarwal, B. Reif, B. van Rossum, Ü. Akbey, H. Oschkinat, Three-dimensionaldeuterium–carbon correlation experiments for high-resolution solid-stateMAS NMR spectroscopy of large proteins, J. Biomol. NMR 51 (2011) 477–485.
[51] S.R. Hartmann, E.L. Hann, Nuclear double resonance in the rotating frame,Phys. Rev. 128 (1962) 2042–2053.
[52] A. Pines, Proton-enhanced NMR of dilute spins in solids, J. Chem. Phys. 59(1973) 569–590.
[53] B.H. Meier, Cross polarization under fast magic angle spinning:thermodynamical consideration, Chem. Phys. Lett. 188 (1992) 201–207.
[54] G. Metz, X. Wu, S.O. Smith, Ramped-amplitude cross polarization in magic-angle-spinning NMR, J. Magn. Reson. A 110 (1994) 219–227.
[55] C.S. Yannoni, High-resolution NMR in solids: the CPMAS experiment, Acc.Chem. Res. 15 (1982) 201–208.
[56] S. Vega, T.W. Shattuck, A. Pines, Double-quantum cross-polarization NMR insolids, Phys. Rev. A 22 (1980) 638–661.
[57] L. Müller, Proton-deuterium polarization transfer in magic angle spinningpolycrystalline solids in the rotation frame, Chem. Phys. 61 (1981) 235–248.
[58] P. Hodgkinson, C. Auger, L. Emsley, Deuterium to carbon cross-polarization inliquid crystals, J. Chem. Phys. 109 (1998) 1873–1884.
[59] D. Marks, N. Zumbulyadis, S. Vega, Deuterium cross-polarization magic-anglespinning, J. Magn. Reson. A 122 (1996) 16–36.
[60] K. Gopalakrishnan, G. Bodenhausen, Cross polarization from spins I = 1/2 tospins S = 1 in nuclear magnetic resonance with magic angle sample spinning, J.Chem. Phys. 124 (2006) 194311.
[61] N. Zumbulyadis, J.M. O’Reilly, Intermolecular proton-deuterium polarizationtransfer in magic angle spinning NMR spectra: a new spectroscopic tool forinterfaces, J. Am. Chem. Soc. 115 (1993) 4407–4408.
[62] N. Zumbulyadis, M.R. Landry, T.P. Russell, Interphase mixing in symmetricdiblock copolymers determined by proton-deuterium CP/MAS NMR,Macromolecules 29 (1996) 2201–2204.
[63] D.F. Shantz, R.F. Lobo, 2H–{1H} CPMAS NMR of guest–host species in zeolites:an experimental study, J. Phys. Chem. B 103 (1999) 592–5927.
[64] W. Kolodziejski, A. Corma, 2H–1H cross-polarization in deuterated glycine,Solid State Nucl. Magn. 7 (1996) 67–72.
[65] N. Zumbulyadis, C.J.T. Landry, T.E. Long, Determination of polymer miscibilityby proton-deuterium CP/MAS NMR spectroscopy, Macromolecules 26 (1993)2647–2648.
[66] D. Wei, U. Akbey, B. Paaske, H. Oschkinat, B. Reif, M. Bjerring, N.C. Nielsen,Optimal 2H rf pulses and 2H–13C cross-polarization methods for solid-state 2HMAS NMR of perdeuterated proteins, J. Phys. Chem. Lett. 2 (2011) 1289–1294.
[67] S. Jayanthi, K.V. Ramanathan, Deuterium–carbon correlation in static orientedsystems using DAPT – a windowless multiple pulse sequence, Chem. Phys.Lett. 487 (2010) 122–128.
[68] S.K. Sarkar, P.E. Young, D.A. Torchia, Ring dynamics of DL-proline and DL-proline hydrochloride in the solid state: a 2H nuclear magnetic resonancestudy, J. Am. Chem. Soc. 108 (1986). 6549-6464.
[69] D.A. Torchia, Solid state NMR studies of protein internal dynamics, Ann. Rev.Biophys. Bioeng. 13 (1984) 125–144.
[70] R. Freeman, H.D.W. Hill, B.L. Tomlinson, L.D. Hall, Dipolar contribution to NMRspin–lattice relaxation of protons, J. Chem. Phys. 61 (1974) 4466–4473.
[71] V.A. Jarymowycz, M.J. Stone, Fast time scale dynamics of protein backbones:NMR relaxation methods, applications, and functional consequences, Chem.Rev. 106 (2006) 1624–1671.
[72] A.J. Shaka, P.B. Barker, R. Freeman, Computer-optimized decoupling schemefor wideband applications and low-level operation, J. Magn. Reson. 64 (1985)547–552.
[73] M.H. Levitt, Composite pulses, Prog. Magn. Reson. 18 (1986) 61–122.[74] A. Bielecki, D.P. Burum, Temperature dependence of 207Pb MAS spectra of solid
lead nitrate. An accurate, sensitive thermometer for variable-temperatureMAS, J. Magn. Reson. 116 (1995) 215–220.
[75] R.L. Vold, G.L. Hoatson, Effects of jump dynamics on solid state nuclearmagnetic resonance line shapes and spin relaxation times, J. Magn. Reson. 198(2009) 57–72.
[76] M. Veshtort, R.G. Griffin, SPINEVOLUTION: a powerful tool for the simulation ofsolid and liquid state NMR experiments, J. Magn. Reson. 178 (2006) 248–282.
[77] P.L. Olympia, Deuteron quadrupole coupling constants in deuterocarbons, J.Chem. Phys. 51 (1969) 1610–1614.
[78] C.M. Gall, J.A. Diverdi, S.J. Opella, Phenylalanine ring dynamics by solid-state2H NMR, J. Am. Chem. Soc. 103 (1981) 5039–5043.
[79] D.M. Rice, Y.C. Meinwald, H.A. Scheraga, R.G. Griffin, Tyrosyl motion inpeptides: 2H NMR line shapes and spin–lattice relaxation, J. Am. Chem. Soc.109 (1987) 1636–1640.
[80] R.E. London, On the interpretation of 13C spin–lattice relaxation resulting fromring puckering in proline, J. Am. Chem. Soc. 100 (1978) 2675–2685.
[81] S.K. Sarkar, D.A. Tochia, K.D. Kopple, D.L. VanderHart, Study of proline peptidebond conformation and ring dynamics in crystalline cyclic peptides using 13CMAS NMR, J. Am. Chem. Soc. 106 (1984) 3328–3331.
[82] E.C. Kostansek, W.N. Lipscomb, W.E. Thiessen, Crystal structure andconformation of the cyclic hexapeptide cyclo-(Gly-L-Pro-D-Ala)2, J. Am.Chem. Soc. 101 (1979) 834–837.
[83] Y. Mitsui, M. Tsuboi, Y. Iitaka, The crystal structure of dl-proline hydrochloride,Acta Cryst. B25 (1969) 2182–2192.
[84] N. Bloembergen, On the interaction of nuclear spins in a crystalline lattice,Physica XV (1949) 386–426.
[85] X. Wang, S. Zhang, X. Wu, Two-stage feature of Harimann-Hahn crossrelaxation in magic-angle sample spinning, Phys. Rev. B 37 (1988) 9827–9829.
[86] W. Kolodziejski, J. Klinowski, Kinetics of cross-polarization in solid-state NMR:a guide for chemists, Chem. Rev. 102 (2002) 613–628.
[87] M.H. Levitt, D. Suter, R.R. Ernst, Spin dynamics and thermodynamics in solid-state NMR cross polarization, J. Chem. Phys. 84 (1986) 4243–4255.
[88] C. Tripon, M. Aluas, X. Filip, C. Filip, Polarization transfer from remote protonsin 13C CP/MAS, J. Magn. Reson. 183 (2006) 68–76.
[89] R. Eckman, A. Pines, R. Tycko, D.P. Weitekamp, Spin diffusion betweeninequivalent quadrupolar nuclei by double-quantum flip-flpos, Chem. Phys.Lett. 99 (1983) 35–40.
[90] G. Diezemann, Dipolar interactions in deuteron spin systems. I. Spin diffusion,J. Chem. Phys. 103 (1995) 6368–6384.
[91] W. Schajor, N. Pislewski, H. Zimmermann, U. Haeberlen, Spin diffusion andspin–lattice relaxation of deuterons in solids, Chem. Phys. Lett. 76 (1980) 409–412.
[92] N. Zumbulyadis, A simple model for deuterium cross-polarization magic-anglespinning nuclear magnetic resonance at the interphases of amorphousmaterials, Solid State Nucl. Magn. Reson. 5 (1995) 3–7.
[93] J. Tang, L. Sterna, A. Pines, Anisotropic spin–lattice relaxation of deuteratedhexamethylbenzene, J. Magn. Reson. 41 (1980) 389–394.
[94] C. Ye, R. Eckman, A. Pines, Spin diffusion and orientation dependence ofdeuterium Tl in solids, J. Magn. Reson. 55 (1983) 334–337.
[95] R.L. Vold, G.L. Hoatson, T.Y. Tse, Effects of slow motion on deuteron relaxationtime anisotropy, Chem. Phys. Lett. 263 (1996) 271–275.
Copyright © 2022 FDOKUMEN




















