
Detection and subtyping of Actinobacillus pleuropneumoniae strainsby PCR-RFLP analysis of the tbpA...
13
Detection and subtyping of Actinobacillus pleuropneumoniae strains by PCR-RFLP analysis of the tbpA and tbpB genes Víctor A. de la Puente-Redondo a , Noemí García del Blanco a , César B. Gutiérrez-Martín a , Jesús Navas Méndez b , Elías F. Rodríguez Ferri a * a Section of Microbiology and Immunology, Department ofAnimal Health, Faculty of Veterinary Medicine, León, Spain b Department of Molecular Biology (associated with C.I.B.-C.S.I.C), Faculty of Medicine, Santander, Spain Received 1 October 1999; accepted 3 March 2000 Abstract – A PCR-based procedure for detection and serotype identification of Actinobacillus pleuropneumoniae strains was developed and evaluated. The A. pleuropneumoniae tbpA and tbpB genes were used as targets for amplification of DNA fragments, with a pair of specific primers for each gene. Amplification with tbpA primers rendered a 2.8-kb PCR product from all 12 A. pleuropneumoniae reference strains as well as from Actinobacillus suis strain CCM 5586, while amplification of a 1.9-kb PCR product was observed when testing ten Haemophilus parasuis strains of different serovars. Amplification of the tbpB gene from A. pleuropneumoniae serotypes 1, 6, 8 and 12, and A. suis CCM 5586 rendered an identical 1.8-kb fragment, while from A. pleuropneumoniae serotypes 2, 3, 4, 7, 9, 10 and 11, and H. parasuis strains it produced a 1.7-kb fragment. No PCR amplification product was observed when examining strains of 19 other swine pathogens or closely related species. The minimal detection limit for whole-cell A. pleuropneumoniae templates was between 5–50 and 3 × 10 2 –3 × 10 3 CFU when tbpA and tbpB specific primers, respectively, were used. Restriction fragment length polymorphism (RFLP) analysis of the PCR-generated products rendered different patterns, easily allowing us to discriminate between A. pleuropneumoniae, H. parasuis and A. suis and, more importantly, to distinguish ten RFLP A. pleuropneumoniae groups (the highest discrimination reported so far for a PCR assay with A. pleuropneumoniae), in such a way that the only serotypes with profiles identical to each other were 4 to 11 and 7 to 9. Moreover, the PCR-RFLP analysis was assayed in 36 A. pleuropneumoniae field isolates and in porcine samples (lungs and nasal swabs from experimentally infected animals). In both cases the system proved to be very efficient in A. pleuropneumoniae identification and serotype discrimination. © 2000 Éditions scientifiques et médicales Elsevier SAS Actinobacillus pleuropneumoniae / Haemophilus parasuis / tbpA and tbpB genes / PCR-RFLP 1. Introduction Actinobacillus pleuropneumoniae is an encapsu- lated, Gram-negative bacterium of the Pasteurel- laceae family that causes swine pleuropneumo- nia, a frequently fatal and highly contagious respiratory disease encountered worldwide. The disease occurs as acute outbreaks with high fever, coughing, dyspnea, anorexia, ataxia, and severe respiratory distress, or as chronic persis- tent disease. The pathogenesis of this disease has been related to several virulence factors, such as RTX toxins, lipopolysaccharide, capsule, outer membrane proteins and urease [2, 12, 22]. Based on NAD requirements, A. pleuropneu- moniae can be grouped into biotype 1, strains which are NAD-dependent, and biotype 2, strains which are NAD-independent. So far, 12 serotypes have been described within biotype 1, and serotypes 1 and 5 are subdivided into subtypes 1a and 1b, and 5a and 5b, respectively. * Correspondence and reprints: [email protected] Abbreviations: CFU: colony-forming units; PCR: polymerase chain reaction; RFLP: restriction fragment length polymor- phism; NAD: nicotinamide adenine dinucleotide; EDTA: ethylenediaminetetraacetic acid. Res. Microbiol. 151 (2000) 669–681 © 2000 Éditions scientifiques et médicales Elsevier SAS. All rights reserved S0923250800001352/FLA
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Detection and subtyping of Actinobacillus pleuropneumoniae strainsby PCR-RFLP analysis of the tbpA...

Detection and subtyping of Actinobacillus pleuropneumoniae strainsby PCR-RFLP analysis of the tbpA and tbpB genes
Víctor A. de la Puente-Redondoa, Noemí García del Blancoa, César B. Gutiérrez-Martína,Jesús Navas Méndezb, Elías F. Rodríguez Ferria*
aSection of Microbiology and Immunology, Department of Animal Health, Faculty of Veterinary Medicine, León, SpainbDepartment of Molecular Biology (associated with C.I.B.-C.S.I.C), Faculty of Medicine, Santander, Spain
Received 1 October 1999; accepted 3 March 2000
Abstract – A PCR-based procedure for detection and serotype identification of Actinobacillus pleuropneumoniaestrains was developed and evaluated. The A. pleuropneumoniae tbpA and tbpB genes were used as targets foramplification of DNA fragments, with a pair of specific primers for each gene. Amplification with tbpAprimers rendered a 2.8-kb PCR product from all 12 A. pleuropneumoniae reference strains as well as fromActinobacillus suis strain CCM 5586, while amplification of a 1.9-kb PCR product was observed when testingten Haemophilus parasuis strains of different serovars. Amplification of the tbpB gene from A. pleuropneumoniaeserotypes 1, 6, 8 and 12, and A. suis CCM 5586 rendered an identical 1.8-kb fragment, while from A.pleuropneumoniae serotypes 2, 3, 4, 7, 9, 10 and 11, and H. parasuis strains it produced a 1.7-kb fragment. NoPCR amplification product was observed when examining strains of 19 other swine pathogens or closelyrelated species. The minimal detection limit for whole-cell A. pleuropneumoniae templates was between 5–50and 3 × 102–3 × 103 CFU when tbpA and tbpB specific primers, respectively, were used. Restriction fragmentlength polymorphism (RFLP) analysis of the PCR-generated products rendered different patterns, easilyallowing us to discriminate between A. pleuropneumoniae, H. parasuis and A. suis and, more importantly, todistinguish ten RFLP A. pleuropneumoniae groups (the highest discrimination reported so far for a PCR assaywith A. pleuropneumoniae), in such a way that the only serotypes with profiles identical to each other were 4to 11 and 7 to 9. Moreover, the PCR-RFLP analysis was assayed in 36 A. pleuropneumoniae field isolates andin porcine samples (lungs and nasal swabs from experimentally infected animals). In both cases the systemproved to be very efficient in A. pleuropneumoniae identification and serotype discrimination. © 2000 Éditionsscientifiques et médicales Elsevier SAS
Actinobacillus pleuropneumoniae / Haemophilus parasuis / tbpA and tbpB genes / PCR-RFLP
1. Introduction
Actinobacillus pleuropneumoniae is an encapsu-lated, Gram-negative bacterium of the Pasteurel-laceae family that causes swine pleuropneumo-nia, a frequently fatal and highly contagiousrespiratory disease encountered worldwide. The
disease occurs as acute outbreaks with highfever, coughing, dyspnea, anorexia, ataxia, andsevere respiratory distress, or as chronic persis-tent disease. The pathogenesis of this diseasehas been related to several virulence factors,such as RTX toxins, lipopolysaccharide, capsule,outer membrane proteins and urease [2, 12, 22].
Based on NAD requirements, A. pleuropneu-moniae can be grouped into biotype 1, strainswhich are NAD-dependent, and biotype 2,strains which are NAD-independent. So far, 12serotypes have been described within biotype 1,and serotypes 1 and 5 are subdivided intosubtypes 1a and 1b, and 5a and 5b, respectively.
* Correspondence and reprints: [email protected]: CFU: colony-forming units; PCR: polymerasechain reaction; RFLP: restriction fragment length polymor-phism; NAD: nicotinamide adenine dinucleotide; EDTA:ethylenediaminetetraacetic acid.
Res. Microbiol. 151 (2000) 669–681© 2000 Éditions scientifiques et médicales Elsevier SAS. All rights reservedS0923250800001352/FLA

Serotyping is mainly based on capsular polysac-charide antigens. In addition, the serotypes havedifferent lipopolysaccharide, except for sero-types 1, 9 and 11, serotypes 3, 6 and 8, andserotypes 4 and 7, which share common epitopes[12, 18, 19]. All serotypes can cause severedisease and death; however, virulence variesamong biotypes and serotypes. Indeed, there isevidence that biotype 2 is less virulent thanbiotype 1, and concerning the latter, serotypes1a, 1b, 5a, 5b, 9 and 10 have proven to be morevirulent than the other biotype 1 serotypes [12].Serotyping of the isolates recovered in a givengeographical area becomes essential for controlof swine pleuropneumonia.
Although isolation and culture of A. pleurop-neumoniae is necessary for a definitive diagno-sis, other tests based on immunological or DNAmethods may be more rapid and cheaper forsurveillance programs. In this way, several sero-logical assays have been reported, but theirspecificity varies considerably. More recently,PCR methods have also been developed usingomlA, cpx, cps and aroA genes [8, 14, 15]. In somecases, high specificity and sensitivity resultswere obtained, while in others specificity wasnot complete, as A. pleuropneumoniae also reactedwith other species belonging to the genus Acti-nobacillus [7, 14, 21]. Moreover, none of themwere useful for serotype identification, andtherefore, both a species-specific and a serotype-specific detection is required for an accuratediagnosis of swine pleuropneumonia.
Genes encoding the iron-repressible outer-membrane proteins Tbp1 (tbpA) and Tbp2 (tbpB)in A. pleuropneumoniae, Haemophilus influenzae,Mannheimia (Pasteurella) haemolytica and differ-ent species of the genus Neisseria have beenrecently cloned and sequenced [3, 5, 6, 17]. ThetbpA genes are highly conserved, whereas atleast three different tbpB gene types have beenidentified among the A. pleuropneumoniae sero-types by Southern blot [5]. Based on the con-served nature of the tbpA genes and the hetero-geneity of tbpB genes, the purpose of this studywas to determine whether DNA primers fromthese regions could be used in PCR and furtherrestriction fragment length polymorphism
(RFLP) analysis for identifying A. pleuropneumo-niae strains.
2. Materials and methods
2.1. Bacterial strains and growth conditions
A set of A. pleuropneumoniae reference strainsrepresenting all serotypes of biotype 1 (table I),as well as 36 clinical isolates belonging to dif-ferent serotypes (table II) were used in thisstudy. All the clinical isolates except for strainCM5 (isolated in Canada by Rosendal et al. [20])were recovered from pigs with pleuropneumo-nia from farms located in northeastern andeastern Spain, and they were serotyped bymeans of indirect hemagglutination [11]. In addi-tion, Actinobacillus minor type strain NM305,A. porcinus type strain NM319, A. indolicus typestrain 46KC2, A. rossii NCTC 10801, A. lignieresiiNCTC 4189, A. actinomycetemcomitans NCTC9710, A. equuli NCTC 8529, A. seminis NCTC10851, A. suis CCM 5586 and four clinical iso-lates, Mannheimia (Pasteurella) haemolytica NCTC9712, Pasteurella canis NCTC 11621, P. dagmatisNCTC 11617, P. stomatis NCTC 11623, P. multo-cida subsp. multocida NCTC 10322, P. multocidasubsp. septica NCTC 11619, ten Haemophilusparasuis strains belonging to different serovars(see table I) and 22 clinical isolates (see table II),Klebsiella pneumoniae NCTC 7761, Bordetella bron-chiseptica ATCC 4617, Alcaligenes faecalis ATCC19018, Pseudomonas aeruginosa ATCC 27853, Yers-inia enterocolitica NCTC 10460, Y. ruckeri H.4,Francisella tularensis STU-14, and Escherichia coliHB 101 were also used in the PCR assays. Allstrains except those belonging to the genusActinobacillus, H. parasuis and F. tularensis weregrown on tryptic soy agar. Actinobacillus speciesand H. parasuis were grown on chocolate bloodagar (GC base with 5% defibrinated horse blood)supplemented with 0.025% NAD and 0.1% glu-cose. F. tularensis was grown for 3 days on modi-fied Thayer-Martin agar containing GC mediumbase (36 g/L), hemoglobin (10 g/L) and Vitoxsupplement (2 vials/L; Oxoid Ltd., Hampshire,England). All organisms except Y. ruckeri wereincubated at 37 °C; Y. ruckeri was grown at28 °C.
670 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

2.2. DNA isolation
Two different protocols were used dependingon the sample type. Chromosomal DNAs fromthe reference strains listed in the paragraphabove or in table I were obtained by suspensionof log-phase cells in 10 mM Tris/1 mM EDTA,and incubated at 37 °C for 3 h in 0.5% sodiumdodecyl sulfate (SDS) and 100 µg/mL protein-ase K. The DNA was then purified by repeatedphenol/chloroform extraction, precipitated fromthe aqueous phase by adding 0.6 volumes ofisopropanol and 70% ethanol, dried and resus-pended in sterile water. For sensitivity tests,analyses of A. pleuropneumoniae clinical isolatesand live animal experiments, a short protocolwas used, pelleting the suspension and resus-pending in 200 µL of InstaGene matrix (Biorad).This solution was preheated at 56 °C for 30 minand then boiled for 8 min. After brief centrifu-gation, the supernatants were recovered andstored at –20 °C.
2.3. PCR amplification
tbpA and tbpB gene sequences have beenpreviously reported for A. pleuropneumoniae sero-types 1 and 7 [6]. Analysis of their sequencesand their restriction profiles were obtained byusing DNA Strider 1.2 software (DNA andprotein sequence analysis, Service de Biochimieet de Génétique Moléculaire, Département deBiologie Cellulaire et Moléculaire, Direction desSciences de la Vie, CEA, France). PCR amplifi-cation was performed with four primers selectedon the basis of the published nucleotidesequence of the serotype 7 tbpA and tbpB genes(Medline DNA GenBank database accession no.U16017): tbpA33: 5′-AAG CTT GAA ACT AAGGTA CTC TAA-3′; tbpA55: 5′-TTA GCC TTGCTC TTC TTA GCC-3′; tbpB33: 5′-CGT TTTGCA CCA AAG ACA GCG-3′; tbpB55: 5′-ATGCAT TTT AAA CTT AAT CCC-3′. The oligo-nucleotide primers were synthesized and puri-fied by HPLC by Boehringer Mannheim (Ger-
Table I. RFLP patterns of PCR products from the strains listed obtained after digestion with different enzymes.
Serotype RFLP tbpA restriction patterns tbpB restriction patterns
Strains group DraI NlaIV AvaI AsnI TaqI HinpI BstEII NsiI RsaI TaqI AvaII XmnI BglII XbaI
A. pleuropneumoniae ATCC 27088 1 I A A A A A A A A A A A A A AA. pleuropneumoniae ATCC 27089 2 II B B B B B A A nd B B B B B BA. pleuropneumoniae ATCC 27090 3 III A C A B A A A A B B B B B CA. pleuropneumoniae M62 4 IV B B B A B A* A A B B B B B CA. pleuropneumoniae ATCC 33377 5 V A A A A C A A B nd nd nd nd nd ndA. pleuropneumoniae Femφ 6 VI A A A A A A A A C C A C A AA. pleuropneumoniae WF83 7 VII B B B B B A* A A B B B B B CA. pleuropneumoniae 405 8 VIII A* A A A A A A A C C A C A CA. pleuropneumoniae CVJ13261 9 VII B* B B B B A A A B B B B B CA. pleuropneumoniae 13039 10 IX B* C B B B A A A B B C B C CA. pleuropneumoniae 56153 11 IV B B B A B A A A B B B B B CA. pleuropneumoniae 8329 12 X A* A A B A A A A A A A A A AA. suis CCM 5586 – – A* C A C C A A A C D A A* D AH. parasuis H410 2 – C D C D D B B C D B D B C DH. parasuis H409 1 –H. parasuis H411 3 –H. parasuis H412 4 –H. parasuis H 780 6 –H. parasuis H 643 7 –H. parasuis H 553 9 –H. parasuis H 555 10 –H. parasuis H 465 11 –H. parasuis H 790 15 –
* The RFLP of these reference strains did not match exactly with the indicated patterns; they showed very small sizedifferences in the resulting restriction bands. Those differences were difficult to appreciate by visual inspection, so theywere not described as a new subtype but included in the most similar profile. nd: not done.
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 671
many). The reaction mixture for PCRamplification (GeneAmp PCR System 2400, Per-kin Elmer, Germany) consisted of 5 µL ofgenomic DNA, 4 U of Taq DNA polymerase(Bioline, UK), 2.5 mM MgCl2, 10 µL of 10 × PCRamplification buffer (160 mM (NH4)2SO4,670 mM Tris-HCl pH 8.8 at 25 °C, and 0.1%Tween-20), 1 µM each primer, 0.2 mM eachdeoxynucleoside triphosphate, and double-distilled water to a final volume of 100 µL. DNAwas denatured at 94 °C for 5 min, and then atotal of 30 cycles were run under the followingconditions: denaturation at 94 °C for 1 min,annealing at 50 °C for 1 min for tbpA and at45 °C for 1 min for tbpB, and extension at 72 °Cfor 3 min for tbpA and 2 min for tbpB. After thefinal cycle, the reactions were terminated by anextra run at 72 °C for 10 min, and were kept at4 °C until analyzed. Modifications in these PCRconditions (annealing temperature of 40 °C and3.25 mM MgCl2) were performed when testingDNA from A. pleuropneumoniae serotype 5 withtbpB primers. The amplified products were elec-trophoresed in 1% agarose gels in Tris-borate-EDTA buffer containing 0.5 µg of ethidiumbromide/mL.
For testing lung tissue samples, a modifiedPCR protocol was carried out, in which 5 U ofTaq polymerase, 1.85 mM MgCl2 and a denatur-ation temperature of 95 °C for 6 min were used.
2.4. PCR sensitivity
For determination of the PCR sensitivities,serial 10-fold dilutions of 6-h cultures of A. pleu-ropneumoniae strain CM5 (1.1 × 108 to 1.1 × 103
CFU/mL for tbpA and 6.1 × 109 to 6.1 × 104
CFU/mL for tbpB) in DMEM (GibcoBRL, ref.21969-027) were tested. One hundred microli-ters of each dilution were processed as describedabove, and 5 µL of each sample were used astemplates for PCR amplification. Viable cellswere counted by determining the number ofCFU by triplicate plating of the samples ontryptic soy agar supplemented with 0.025%NAD, and counting the colonies after incuba-tion at 37 °C for 24 h. The sensitivity test wascarried out in duplicate.
2.5. RFLP analysis
The RFLP procedure was carried out bydigesting the tbpA PCR-amplified products with
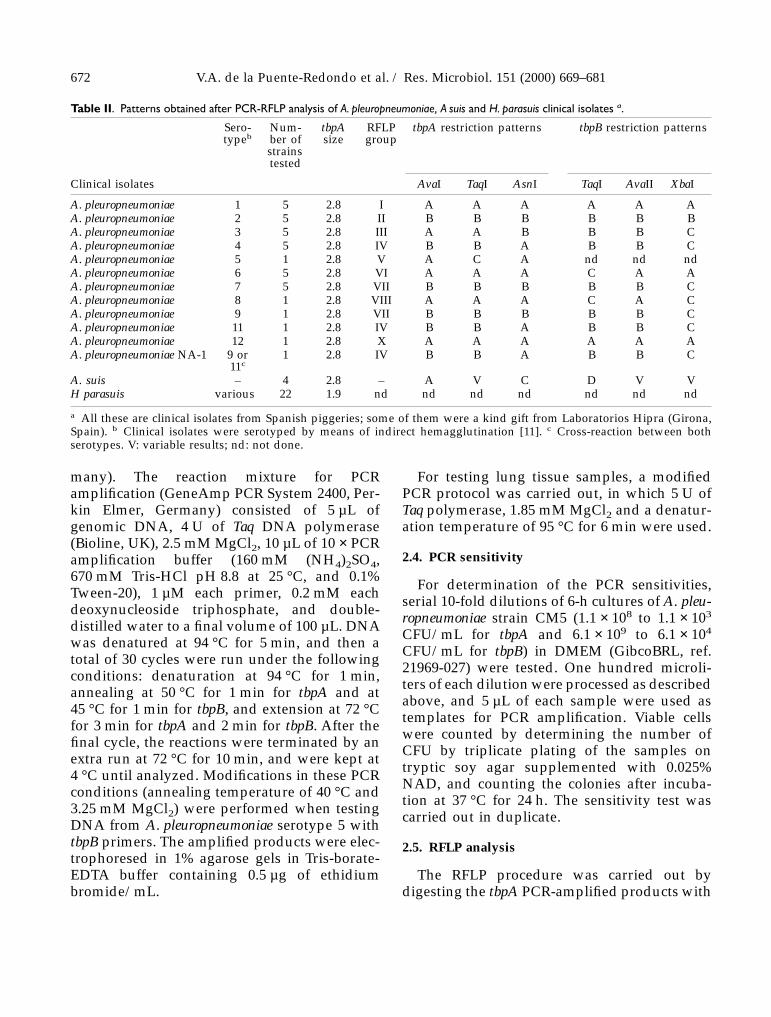
Table II. Patterns obtained after PCR-RFLP analysis of A. pleuropneumoniae, A suis and H. parasuis clinical isolates a.
Sero-typeb
Num-ber ofstrainstested
tbpAsize
RFLPgroup
tbpA restriction patterns tbpB restriction patterns
Clinical isolates AvaI TaqI AsnI TaqI AvaII XbaI
A. pleuropneumoniae 1 5 2.8 I A A A A A AA. pleuropneumoniae 2 5 2.8 II B B B B B BA. pleuropneumoniae 3 5 2.8 III A A B B B CA. pleuropneumoniae 4 5 2.8 IV B B A B B CA. pleuropneumoniae 5 1 2.8 V A C A nd nd ndA. pleuropneumoniae 6 5 2.8 VI A A A C A AA. pleuropneumoniae 7 5 2.8 VII B B B B B CA. pleuropneumoniae 8 1 2.8 VIII A A A C A CA. pleuropneumoniae 9 1 2.8 VII B B B B B CA. pleuropneumoniae 11 1 2.8 IV B B A B B CA. pleuropneumoniae 12 1 2.8 X A A A A A AA. pleuropneumoniae NA-1 9 or
11c1 2.8 IV B B A B B C
A. suis – 4 2.8 – A V C D V VH parasuis various 22 1.9 nd nd nd nd nd nd nd
a All these are clinical isolates from Spanish piggeries; some of them were a kind gift from Laboratorios Hipra (Girona,Spain). b Clinical isolates were serotyped by means of indirect hemagglutination [11]. c Cross-reaction between bothserotypes. V: variable results; nd: not done.
672 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

DraI, NlaIV, AvaI, AsnI, TaqI, HinpI, BstEII andNsiI, and the tbpB-PCR-amplified products withRsaI, TaqI, AvaII, XmnI, BglII and XbaI restrictionendonucleases (New England Biolabs, Beverly,MA). The products were analyzed by agarosegel electrophoresis as described above. For eachenzymatic reaction, letters (A, B, C etc.) wereassigned to each different restriction patternobserved, as shown in table I.
2.6. Pig experiments
Three-week-old large white female pigs werehoused in isolated rooms and were fed non-medicated feed and water ad libitum. The ani-mals had no previous clinical history of A. pleu-ropneumoniae infection and their seronegativityagainst this organism was verified prior tochallenge by using an ELISA [9]. The animaltesting was carried out in accordance with ani-mal protection regulations, and any unneces-sary suffering was avoided. Animals were chal-lenged intranasally with 107 colony-formingunits (CFUs) of each A. pleuropneumoniae sero-type 1, 2 or 4 reference strain (one serotype foreach pig) suspended in DMEM. Another pigremained as control, and received an equivalentvolume of sterile DMEM. During manipula-tions, the animals were tranquilized with azap-erone. The pigs were kept separately during theexperiment. Swabs from each nostril were takenimmediately after death or sacrifice, culturedimmediately on tryptic soy agar supplementedwith 0.025% NAD, and stored at –80 °C forfurther DNA extraction. DNA was extracted byresuspending swabs in sterile phosphate-buffered saline (PBS), pelleted and then resus-pended in InstaGene matrix (Biorad). Subse-quently, samples were heated at 56 °C for 30 minand then boiled for 8 min. After centrifugation,the supernatants were recovered and stored at–20 °C until use in PCR-RFLP assays as detailedabove.
Lung tissue samples were also removed innecropsy, swabs were taken from them asepti-cally, cultured on supplemented tryptic soyagar, and stored at –80 °C. DNA extraction wasconducted as described for nasal swabs. Simul-taneously, approximately 2 × 2 mm lung pieces
were taken from two different sites with lesions,mashed vigorously and vortexed. After cen-trifugation, the supernatant was resuspended inInstaGene matrix (Biorad) as described previ-ously for DNA extraction, used in PCR-RFLPassays, stored at –20 °C and retested some daysafter. For PCR assays, the original protocol wascarried out, but other modified PCR protocols(see above) were also used. Finally, the lungsupernatant was also tested by coagglutinationand immunodiffusion tests. Details of thesetechniques were described earlier [10, 16].
3. Results
3.1. PCR amplification
The two nucleotide primers from the A. pleu-ropneumoniae tbpA gene successfully primed thesynthesis of a 2.8-kb DNA fragment, whichrepresents most of the tbpA gene sequence of thereference strains of the 12 serotypes of A. pleu-ropneumoniae as well as A. suis strain CCM 5586(figure 1B). No quantitative or qualitative differ-ences in the bands rendered by these two Acti-nobacillus species were observed. No PCR ampli-fication product was obtained when theremaining Actinobacillus species or the closelyrelated genera cited in the Materials and meth-ods were used as a source of target DNA (datanot shown), with the exception of the ten H. para-suis strains tested, which all revealed a 1.9-kbband after PCR amplification (figure 1B). Theamplifications which resulted in detectable lev-els of tbpA-primed PCR were achieved when5–50 CFU of A. pleuropneumoniae strain CM5were lysed, on the basis of an average of tworepeated assays.
Nucleotide primers from A. pleuropneumoniaetbpB gene rendered a 1.8-kb amplified band forthe reference strains of A. pleuropneumoniae sero-types 1, 6, 8 and 12, and A. suis CCM 5586, whilethe band was slightly smaller (1.7-kb) for thereference strains of A. pleuropneumoniae sero-types 2, 3, 4, 7, 9, 10 and 11, as well as all theH. parasuis strains tested under standard condi-tions (figure 1A). No amplification product wasobtained when DNA from A. pleuropneumoniae
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 673

serotype 5 and the remaining 19 swine respira-tory pathogens or related species were used astemplate. New trials modifying the PCR condi-tions as described in the Material and methodswere carried out, and then a weak band couldbe observed for A. pleuropneumoniae serotype 5,but also secondary products appeared. For thisreason, this serotype was not included in thetbpB-RFLP analysis. Under these very low strin-gency conditions, barely visible amplificationbands of tbpB could also be seen for A. equuliand A. lignieresii. When tbpB primers were used,the minimal detection limit was determined tobe between 3 × 102 to 3 × 103 CFU of A. pleurop-neumoniae strain CM5.
3.2. PCR-RFLP analysis
The 2.8-kb tbpA amplified fragment from all12 A. pleuropneumoniae serotypes and A. suisCCM 5586 was analyzed by eight restrictionendonucleases (table I and figures 2B, 2C, 3A, 3B,4A and 4C). We found unique RFLP patternswith HinpI and BstEII for all A. pleuropneumo-niae serotypes and A. suis CCM 5586 (figure 2B,C). From the analysis of the restriction profilesobtained with the other six enzymes, sevenRFLP profiles could be observed for the 12
A. pleuropneumoniae serotypes. Concretely,groups of serotypes 1, 6 and 8, 2, 7 and 9, and 4and 11, shared the same restriction patterns forall the digestions, while the remaining serotypes(3, 5, 10 and 12) rendered different profilesdepending on the enzyme used. For A. suis, arestriction pattern completely different fromthose obtained for A. pleuropneumoniae serotypeswas observed when the PCR product wasdigested with AsnI. On the other hand, the1.9-kb tbpA amplified fragment from H. parasuisH410 serovar 2 was also analyzed by restrictionendonucleases, and all the digestions yieldedrestriction patterns easily distinguishable fromthose observed for the Actinobacillus species, ascould be expected because of the smaller size ofthe amplification fragment (table I).
The 1.7-kb or 1.8-kb PCR products fromA. pleuropneumoniae serotypes (except for sero-type 5), A. suis and H. parasuis tbpB gene weredigested with RsaI, TaqI, AvaII, XmnI, BglII, andXbaI endonucleases (table I and figures 2A, 4Band 4C). It is important to note here that sero-type 5 can be identified just with the tbpAamplification product and its RFLP profile,avoiding the poor quality of the bands obtainedfrom tbpB amplification. The analysis of restric-
Figure 1. (A) Results of tbpA amplification from A. pleuropneumoniae serotypes 1–12 (lanes 1–12), A. suis (lane 13) and H. parasuisserovar 2 (lane 14). (B) Results of tbpB probe amplification from different reference strains. The band is 1.8 kb in size fromA. pleuropneumoniae serotypes 1, 6, 8, 12 and A. suis (lanes 1, 5, 7, 11 and 12), and 1.7 kb from A. pleuropneumoniae serotypes 2, 3, 4, 7,9, 10, 11 and H. parasuis serovars 2 and 7 (lanes 2, 3, 4, 6, 8, 9, 10, 13 and 14). M: molecular weight markers: A: MW marker numberX (Boehringer Mannheim); B: HindIII (USB).
674 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

tion patterns rendered allowed grouping A. pleu-ropneumoniae into six groups: the pattern exhib-ited by serotypes 3, 4, 7, 9 and 11; that ofserotypes 1 and 12; that of serotype 2 (thispattern was rather similar to those of serotypes3, 4, 7, 9 and 11); that of serotype 6; that of
serotype 8 and that of serotype 10. A. suisshowed RFLP patterns completely different fromthose obtained for A. pleuropneumoniae serotypesafter TaqI and BglII digestions. As distinguishedfrom that observed for the tbpA gene, the tbpBamplified product from H. parasuis showed
Figure 2. Restriction analysis. (A) tbpB amplification product from A. pleuropneumoniae serotypes 1–4 (lanes 1–4 and 14–17), 6–12(lanes 5–11 and 18–24), A. suis (lanes 12 and 25) and H. parasuis serovar 2 (lanes 13 and 26) restricted with XmnI (lanes 1–13) and BglII(lanes 14–26). (B) tbpA amplification product from A. pleuropneumoniae serotypes 1–12 (lanes 1–12 and 15–26), A. suis (lanes 13 and 27),and H. parasuis serovar 2 (lanes 14 and 28) restricted with HinpI (lanes 1–12) and DraI (lanes 15–26). (C) tbpA amplification product fromA. pleuropneumoniae serotypes 1–12 (lanes 1–12) and A. suis (lane 13) restricted with BstEII. M: MW marker DRIgestIII (AmershamPharmacia Biotech).
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 675

restriction profiles similar to those obtained forsome A. pleuropneumoniae serotypes whendigested with TaqI, XmnI and BglII.
The combination of the results obtained bydigestion of the tbpA- or tbpB-amplified prod-ucts allowed us to readily discriminate most ofthe A. pleuropneumoniae serotypes with theexception of the pairs of serotypes 4-11 and 7-9.From our data, ten RFLP groups can be distin-guished: I (corresponding to serotype 1), II(serotype 2), III (serotype 3), IV (serotypes 4 and11), V (serotype 5), VI (serotype 6), VII (sero-types 7 and 9), VIII (serotypes 8), IX (serotype10) and X (serotype 12).
3.3. Identification of field isolatesby PCR-RFLP analysis
In order to evaluate the usefulness of ourmethod in the identification and typing ofstrains, a total of 62 clinical isolates of the threebacterial species were further analyzed by ourPCR protocol. A 2.8-kb fragment was amplifiedfrom all the A. pleuropneumoniae and A. suis iso-lates when the primers from tbpA gene wereused, and H. parasuis rendered a characteristic1.9-kb fragment. When the PCR products weredigested with endonucleases (table II), the RFLPpatterns generated for all the isolates were
Figure 3. Restriction analysis. (A) tbpA amplification product from A. pleuropneumoniae serotypes 1–12 (lanes 1–12), A. suis (lane 13),and H. parasuis (lane 14) restricted with NlaIV. tbpA amplification product from A. pleuropneumoniae serotypes 1–6 (lanes 15–20)restricted with AvaI. (B) tbpA amplification product from A. pleuropneumoniae serotypes 7–12 (lanes 1–6), A. suis (lane 7), and H. parasuisserovar 2 (lane 8) restricted with AvaI. tpbA amplification product from A. pleuropneumoniae serotypes 1–12 (lanes 9–20) restricted withAsnI. M: MW marker DRIgestIII (Amersham Pharmacia Biotech).
676 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

Figure 4. Restriction analysis. (A) tbpA amplification product from A. pleuropneumoniae serotypes 1–12 (lanes 1–12), A. suis (lane 13),and H. parasuis serovar 2 (lane 14) restricted with TaqI. (B) tbpB amplification product from A. pleuropneumoniae serotypes 1–4 (lanes1–4), 6–12 (lanes 5–11), A. suis (lane 12), and H. parasuis serovar 2 (lane 13) restricted with TaqI. (C) tbpB amplification product fromA. pleuropneumoniae serotypes 1–4 (lanes 1–4), 6–12 (lanes 5–11), A. suis (lane 12), and H. parasuis serovar 2 (lane 13) restricted withAvaII. tbpB amplification product from A. pleuropneumoniae serotypes 1–4, 6, 7 and 8 (lanes 14–20) restricted with RsaI. (D) tbpBamplification product from A. pleuropneumoniae serotypes 9–12 (lanes 1–4), A. suis (lane 5) and H. parasuis (lane 6) restricted with RsaI.tbpA amplification product from A. pleuropneumoniae serotypes 1–12 (lanes 7–18), A. suis (lane 19), and H. parasuis serovar 2 (lane 20)restricted with NsiI. M: MW marker DRIgestIII (Amersham Pharmacia Biotech).
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 677

consistent with those obtained for the referencestrains belonging to the same serotype.
Indirect hemagglutination had not been ableto separate serotype 9 from serotype 11 for oneof the isolates tested (A. pleuropneumoniae NA-1)because of cross-reactions. By means of PCR-RFLP, this isolate was then classified into groupIV (serotypes 4 and 11), and therefore, thecombination of both methods allowed us todiscard serotypes 4 and 9 and to definitivelyidentify A. pleuropneumoniae NA-1 as belongingto serotype 11.
3.4. Identification of Actinobacilluspleuropneumoniae strains from clinical samples
The three experimentally infected pigs diedbetween 2–6 days after intranasal inoculation.The control pig was sacrificed after the obser-vation period (15 days) and necropsy was per-formed. Negative results were obtained for thedifferent techniques compared with all the typesof samples collected from this animal. A. pleu-ropneumoniae could be isolated in pure culturefrom the lung swabs taken as soon as possibleafter death of the pigs inoculated, but not fromnasal swabs because of the high contaminationobserved in the plates after 24 h of incubation.The unsuccessful attempt to recover A. pleuro-pneumoniae from nostrils might be due to theovergrowth of other highly invasive organisms(mainly belonging to the genus Proteus) rather
than to the real absence of this organism in nasalmucus. On the other hand, coagglutination andimmunodiffusion from the supernatant ofmashed lung samples could not detect A. pleu-ropneumoniae antigen from the serotypes inocu-lated (table III).
When the standard PCR protocol was appliedto lung samples (lung swabs and mashed lungtissues) amplification of the tbpA gene producedan additional weak band that was approxi-mately 1.8 kb in size. In order to avoid this andsome other problems, a modified PCR protocol(as described in the Materials and methodssection) was used and under the new condi-tions, the secondary band was successfully elimi-nated without reducing the intensity of themain band. Therefore, an amplified fragment ofthe expected size was seen for tbpA and tbpBgenes in all lung specimens tested, regardless ofthe manner in which the samples were taken.However, a low DNA stability for the superna-tant obtained from the mashed lungs wasdetected, because after storing for 3 days at–20 °C, some of the samples that were initiallypositive became negative. When the amplifiedproducts were digested by endonucleases, theexpected RFLP profiles according to the sero-type inoculated were seen in all cases (table III).
When nasal swabs were analyzed by PCR-RFLP, positive results were observed in one ofthe three samples taken from each pig inocu-
Table III. Results of the inoculation of some A. pleuropneumoniae serotypes and further detection of these organisms by cultureisolation and PCR-RFLP analysis from different sorts of respiratory samples.
Pig number Serotypeinoculated
(reference strain)
Type of sample Culture isolation PCR-RFLP (number of positivesamples/number of samples taken)
tbpA tbpB
1 1 (ATCC 27088) nasal swabs –a 2 / 3 2 / 3lung swabs + 2 / 2 2 / 2
mashed lungs nd 2 / 2 2 / 22 2 (ATCC 27089) nasal swabs –a 1 / 3 1 / 3
lung swabs + 2 / 2 2 / 2mashed lungs nd 2 / 2 2 / 2
3 4 (M62) nasal swabs –a 1 / 3 1 / 3lung swabs + 2 / 2 2 / 2
mashed lungs nd 2 / 2 2 / 2
a The A. pleuropneumoniae strain inoculated could not be isolated because of the high contamination detected in the plates.nd: not done.
678 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

lated with serotypes 2 and 4, and in two of thosetaken from the animal infected with serotype 1(table III). Neither secondary bands in the PCRof the positive nasal samples or any sort of bandin that of the negative ones could be seen.
4. Discussion
The use of gene amplification by PCR hasmany applications, especially for the rapid iden-tification of bacteria. In this study we were ableto distinguish A. pleuropneumoniae serotypes byPCR amplification of the tbpA and tbpB genesand further RFLP analysis. For the tbpA gene,the pair of primers used did not recognize theother bacteria tested with the exception of A. suisand H. parasuis. Since, for the latter species, adifferent PCR product size was obtained, thistechnique in itself allowed us to distinguishH. parasuis from the complexA. pleuropneumoniae-A. suis without the need touse RFLP analysis. However, we would needmore data to differentiate between A. pleurop-neumoniae and A. suis. Both species can be easilydifferentiated if they have been previously iso-lated in culture, because A. pleuropneumoniaebiotype 1 exhibits NAD-dependent growthwhile A. suis does not. If detection were doneonly by PCR amplification from nasal or lungsamples without cultivation, we would need toperform RFLP analysis of their amplificationproducts for differentiating the two species.When tbpB gene was amplified, A. suis andH. parasuis also gave positive PCR products ofsimilar size as for A. pleuropneumoniae serotypes,and consequently, RFLP analysis became indis-pensable.
Previous reports have dealt with the develop-ment of PCR assays for A. pleuropneumoniaespecies-specific detection. Gram et al. [7] andSirois et al. [21], similarly to our study, alsodetected amplification products for other Acti-nobacillus species, and notably A. lignieresii [7,21], a species related to A. pleuropneumoniae atthe DNA level, but which has never been iso-lated from swine [4]. This cross-amplificationhas been further avoided in other PCR assays,
which were found to be completely specific forA. pleuropneumoniae [8].
The sensitivity of our PCR assay with thetbpA gene was higher than that described inprevious A. pleuropneumoniae studies, that is, 102
[15] or 103 CFU [7], but it correlated with thosevalues when the tbpB gene was used. However,in another report, considerably lower detectionlimits (about 12 CFU of A. pleuropneumoniae)were observed [14]. The relatively high detec-tion level found in our study (especially whenthe tbpB gene was tested) might be due toincomplete release of DNA from the cells,because of the simplicity of the sample prepa-ration and lysing steps used. However, thesefeatures made our PCR fast and simple tohandle even though they might have adverseeffects on the detection level. In addition, it hasbeen stated that too low detection limits are notdesirable for diagnostic purposes [7].
According to the PCR-RFLP results obtainedfor the two genes tested, digestion with AvaI,TaqI, AsnI, NsiI, AvaII and XbaI endonucleaseswould be sufficient for differentiation of A. pleu-ropneumoniae serotypes, except for pairs of sero-types 4-11 and 7-9. These pairs could easily bediscriminated by PCR amplification of the toxingene apxIA [1]. The PCR method described hereused as probes DNA sequences larger thanthose previously described, which proved to beextremely useful for our diagnostic purposes,because it increased the quality of the RFLPprofiles obtained: easily observable fragments,ranging from 0.5–2.5 kb, were adequate for sepa-rating A. pleuropneumonaie serotypes withoutapplying to smaller fragments below 0.5 kb.
Hennessy et al. [13] reported an arbitrarilyprimed PCR assay for typing of A. pleuropneu-moniae, in which unique profiles that specificallydistinguished each serotype were obtained, butthis method has the disadvantages of requiringpure bacterial samples, it is highly susceptibleto contamination and results are poorly repeti-tive. Based on omlA gene nucleotide sequencesof the 12 A. pleuropneumoniae serotypes, Gramand Ahrens [8] had previously divided theminto four clusters: that composed of serotypes 1,9, 11 and 12; that of serotype 2; that of serotypes
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 679

3, 4, 6, 7 and 8; and that of serotypes 5 and 10. Loet al. [15] described the first use of primers toamplify conserved and serotype-specific capsu-lar DNA regions to simultaneously identifyA. pleuropneumoniae and the serotype, but theywere only able to distinguish serotype 5 fromother serotypes. More recently, based on thearoA gene sequence, Hernanz et al. [14] havebeen able to separate the 12 serotypes of biotype1 into only three groups: that including sero-types 1, 4, 5, 9, 11 and 12; that includingserotypes 2, 3, 6, 7, 8 and A. equuli; and thatincluding serotype 10. Although the latter studyhas proven to be highly sensitive for bacterialstrains, its ability to detect A. pleuropneumoniaeserotypes directly from clinical samples was notcorroborated. However, the results obtainedwith our PCR-RFLP protocol clearly show ahigher discriminating capacity among serotypes,because at least eight of the 12 serotypes couldbe separated. Therefore, the PCR-RFLP assayhere described is at present one of the mostpromising tools for accurate DNA-based typingof A. pleuropneumoniae strains.
Our PCR-RFLP protocol was also used tocharacterize 50 A. pleuropneumoniae, A. suis andH. parasuis field isolates. A. pleuropneumoniaeisolates included the serotypes most frequentlyrecovered in Spain [11]. The test seems accuratewith regard to the typing of these isolates and,therefore, very suitable for the definitive dis-crimination of isolates (like A. pleuropneumoniaeNA-1) showing cross-reactions by conventionalserotyping methods. However, to make thisprotocol more reliable, a higher number of fieldisolates should be tested.
As concerns results obtained from experi-mental inoculation of pigs with A. pleuropneu-moniae serotypes 1, 2 and 4, the high sensitivityof our PCR-RFLP protocol would seem to beconfirmed by: the successful amplification oftbpA and tbpB genes irrespective of the type ofporcine sample tested; the failure to detectA. pleuropneumoniae antigen from mashed lungswith coagglutination and immunodiffusion tests(the former method was previously reported tobe a sensitive procedure for detecting this organ-ism from lung samples [16]); and the unsuccess-
ful culture isolation of strains inoculated fromnasal swabs. However, the study was carriedout on a limited number of experimentallyinfected animals and therefore further studieswill be necessary to better evaluate the useful-ness of this test, using both naturally and experi-mentally infected pigs.
The high sensitivity of other PCR methods forthe detection of A. pleuropneumoniae on mixedbacterial cultures from tonsils, a sample ascontaminated as nasal swabs in our study, hasalready been reported [7, 8]. The low DNAstability shown in our study by the supernatantextracted from mashed lungs might be explainedby the DNA extraction method used, whichmay not have been efficient enough in eliminat-ing both bacterial and tissue endonucleases.With respect to this, Lo et al. [15] reportedproblems of DNA degradation in the cps generegion, which did not allow the amplification ofthe 1.1-kb cps product in a multiplex PCR car-ried out from frozen lung tissue samples ofswine infected with A. pleuropneumoniae sero-type 5.
In conclusion, PCR amplification of the tbpAand tbpB genes is a useful tool for A. pleuropneu-moniae identification, and it is considerablyenhanced when it is combined with RFLP analy-sis by means of AvaI, TaqI, AsnI, NsiI, AvaII andXbaI endonucleases, which enable discrimina-tion of all 12 serotypes with the exception ofpairs of serotypes 4-11 and 7-9. In addition, ourPCR-RFLP protocol seems to be a quick andeffective method for detecting and typing fieldisolates as well as strains directly from porcinesamples, even if they are highly contaminated.This one might be a useful method for detectingand typing of A. pleuropneumoniae from thenasopharynx of live animals, without slaughter-ing them.
Acknowledgments
We thank Dr P. Kielstein for providing uswith H. parasuis reference strains, LaboratoriosHipra (Girona, Spain) for providing us withH. parasuis field isolates and E. Keitch López forassistance with the computer-generated images.
680 V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681

The useful comments of Dr M. Mulks, MichiganState University, Department of Microbiology,are gratefully acknowledged. This work wassupported by grants AGF 96-0489, AGF 99-0196and 1FD97-1371-CO2-01 from the ‘ProgramaNacional de Investigación y Desarrollo Ganad-ero’, CICYT, Spain. V.A.D.R. was a recipient of apredoctoral fellowship from the ‘Ministerio deEducación y Ciencia’, Spain.
References
[1] Beck M., van den Bosch J.F., Jongenelen I.M.C.A., Loeffen P.L.W.,Nielsen R., Nicolet J., Frey J., RTX toxin genotypes and phenotypesin Actinobacillus pleuropneumoniae field strains, J. Clin. Microbiol. 32(1994) 2749–2754.
[2] Bossé J.T., MacInnes J.I., Genetic and biochemical analyses of Acti-nobacillus pleuropneumoniae urease, Infect. Immun. 65 (1997)4389–4394.
[3] Charland N., D’Silva C.G., Dumont R.A., Niven D.F., Contact-dependent acquisition of transferrin-bound iron by two strains ofHaemophilus parasuis, Can. J. Microbiol. 41 (1995) 70–74.
[4] Dewhirst F.E., Paster B.J., Olsen I., Fraser G.J., Phylogeny of 54representative strains of species in the family Pasteurellaceae asdetermined by comparison of 16S rRNA sequences, J. Bacteriol.174 (1992) 2002–2013.
[5] Gerlach G.F., Klashinsky S., Anderson C., Poter A.A., Willson P.J.,Characterization of two genes encoding distinct transferrin-bindingproteins in different Actinobacillus pleuropneumoniae isolates, Infect.Immun. 60 (1992) 3253–3261.
[6] González G.C., Yu R.H., Rosteck P.R., Schryvers A.B., Sequence,genetic analysis, and expression of Actinobacillus pleuropneumoniaetransferrin receptor genes, Microbiology 141 (1995) 2405–2416.
[7] Gram T., Ahrens P., Nielsen J.P., Evaluation of a PCR for detection ofActinobacillus pleuropneumoniae in mixed bacterial cultures fromtonsils, Vet. Microbiol. 51 (1996) 95–104.
[8] Gram T., Ahrens P., Improved diagnostic PCR assay for Actinobacilluspleuropneumoniae based on the nucleotide sequence of an outermembrane lipoprotein, J. Clin. Microbiol. 36 (1998) 443–448.
[9] Gutiérrez C.B., Tascón R.I., Vázquez Boland J.A., RodríguezFerri E.F., Cross-reactivity between Actinobacillus pleuropneumoniaeserotypes comparing different antigens and serological tests, Res.Vet. Sci. 50 (1991) 308–310.
[10] Gutiérrez C.B., Rodríguez Barbosa J.I., González O.R., Tascón R.I.,Rodríguez Ferri E.F., Viability of Actinobacillus pleuropneumoniae infrozen pig lung samples and comparison of different methods ofdirect diagnosis in fresh samples, Comp. Immun. Microbiol. Infect.Dis. 15 (1992) 89–95.
[11] Gutiérrez C.B., Rodríguez Barbosa J.I., Tascón R.I., Costa L.,Riera P., Rodríguez Ferri E.F., Serological characterisation and anti-microbial susceptibility of Actinobacillus pleuropneumoniae strainsisolated from pigs in Spain, Vet. Rec. 137 (1995) 62–64.
[12] Haesebrouck F., Chiers K., van Overbeke I., Ducatelle R., Actinoba-cillus pleuropneumoniae infections in pigs: the role of virulencefactors in pathogenesis and protection, Vet. Microbiol. 58 (1997)239–249.
[13] Hennessy K.J., Iandolo J.J., Fenwick B.W., Serotype identification ofActinobacillus pleuropneumoniae by arbitrarily primed polymerasechain reaction, J. Clin. Microbiol. 31 (1993) 1155–1159.
[14] Hernanz C., Cascón A., Sánchez M., Yugueros J., Suárez S., Naha-rro G., Molecular cloning and sequencing of the aroA gene fromActinobacillus pleuropneumoniae and its use in a PCR assay for rapididentification, J. Clin. Microbiol. 37 (1999) 1575–1578.
[15] Lo T.M., Ward C.K., Inzana T.J., Detection and identification ofActinobacillus pleuropneumoniae serotype 5 by multiplex PCR, J. Clin.Microbiol. 36 (1998) 1704–1710.
[16] Mittal K.R., Higgins R., Larivière S., Detection of type-specificantigens in the lungs of Haemophilus pleuropneumoniae-infected pigsby coagglutination test, J. Clin. Microbiol. 18 (1983) 1355–1357.
[17] Ogunnariwo J.A., Woo T.K.W., Lo R.Y.C., González G.C.,Schyvers A.B., Characterization of the Pasteurella haemolytica trans-ferrin receptor genes and the recombinant receptor proteins,Microb. Pathog. 23 (1997) 273–284.
[18] Rodríguez Barbosa J.I., Gutiérrez Martín C.B., Tascón R.I., Suárez J.,Rodríguez Ferri E.F., Evidence obtained with monoclonal antibodiesthat O antigen is the major antigen responsible for the cross-reactivities between serotypes 4 and 7 of Actinobacillus (Haemophi-lus) pleuropneumoniae, Clin. Diagn. Lab. Immunol. 2 (1995) 563–568.
[19] Rodríguez Barbosa J.I., Gutiérrez Martín C.B., Tascón R.I.,González O.R., Mittal K.R., Rodríguez Ferri E.F., Characterization ofmonoclonal antibodies that recognize common epitopes locatedon O antigen of lipopolysaccharide of serotypes 1, 9 and 11 ofActinobacillus pleuropneumoniae, FEMS Immunol. Med. Microbiol. 16(1996) 173–181.
[20] Rosendal S., Carpenter D.S., Mitchell W.R., Wilson M.R., Vaccina-tion against pleuropneumonia of pigs caused by Haemophilus pleu-ropneumoniae, Can. Vet. J. 22 (1981) 34–35.
[21] Sirois M., Lemire E.G., Levesque R.C., Construction of a DNAprobe and detection of Actinobacillus pleuropneumoniae by usingpolymerase chain reaction, J. Clin. Microbiol. 29 (1991) 1183–1187.
[22] Tascón R.I., Vázquez-Boland J.A., Gutiérrez-Martín C.B., Rodríguez-Barbosa J.I., Rodríguez-Ferri E.F., Virulence factors of the swinepathogen Actinobacillus pleuropneumoniae, Microbiologia SEM 12(1996) 171–184.
V.A. de la Puente-Redondo et al. / Res. Microbiol. 151 (2000) 669–681 681




















