
Defhc1.1, a homologue of the juvenile myoclonic gene EFHC1, modulates architecture and basal...
10
Defhc1.1, a homologue of the juvenile myoclonic gene EFHC1, modulates architecture and basal activity of the neuromuscular junction in Drosophila Maria Giovanna Rossetto 1, { , Erica Zanarella 1, { , Genny Orso 1 , Michele Scorzeto 2 , Aram Megighian 2 , Vimlesh Kumar 3 , Antonio V. Delgado-Escueta 4,5 and Andrea Daga 1,4, ∗ 1 E. Medea Scientific Institute, Conegliano, Italy, 2 Department of Human Anatomy and Physiology, University of Padova, Padova, Italy, 3 Indian Institute of Science Education and Research (IISER), Bhopal 462023, India, 4 Department of Neurology, The David Geffen School of Medicine, University of California, Los Angeles, CA, USA and 5 Epilepsy Genetics and Genomics labs, Neurology and Research Services of Veterans Affairs Greater Los Angeles Healthcare System, West Los Angeles, CA, USA Received May 17, 2011; Revised and Accepted August 5, 2011 Mutations in the EFHC1 gene have been linked to juvenile myoclonic epilepsy. To understand EFHC1 function in vivo, we generated knockout Drosophila for the fly homolog Defhc1.1. We found that the neuromuscular junction synapse of Defhc1.1 mutants displays an increased number of satellite boutons resulting in increased spontaneous neurotransmitter release. Defhc1.1 binds to microtubules in vitro and overlaps in vivo with axonal and synaptic microtubules. Elimination of Defhc1.1 from synaptic terminals reduces the number of microtubule loops, suggesting that Defhc1.1 is a negative regulator of microtubule dynamics. In fact, pharmacological treatment of Defhc1.1 mutants with vinblastine, an inhibitor of microtubule dynamics, suppresses the satellite bouton phenotype. Furthermore, Defhc1.1 mutants display overgrowth of the den- dritic arbor and Defhc1.1 overexpression reduces dendrite elaboration. These results suggest that Defhc1.1 functions as an inhibitor of neurite growth by finely tuning the microtubule cytoskeleton dynamics and that EFHC1-dependent juvenile myoclonic epilepsy may result from augmented spontaneous neuro- transmitter release due to overgrowth of neuronal processes. INTRODUCTION Among the common idiopathic generalized epilepsies, juven- ile myoclonic epilepsy (JME) is one of the most frequent, accounting for as much as 30% of all epilepsies (1 – 3). Fifteen chromosomal loci have been so far linked to JME (4). At present, however, mutations causing the disease have been identified only in the ion-channel encoding gene GABRA1 (5) and in the non-ion channel gene EFHC1 (6 – 9). EFHC1 encodes a protein that harbors three tandemly repeated DM10 domains of unknown function and a single EF-hand, a calcium-binding motif at the C-terminus. Muta- tions in EFHC1 are to date the most frequent cause of JME (8). Expression of EFHC1 in transfected hippocampal primary neurons produces shorter neurites and fewer branches (9), but loss of function data supporting a function as a negative regulator of neurite growth and/or development are lacking. EFHC1 associates with the centrosome and the mitotic spindle in cultured cells (10) and more recently, EFHC1 has been proposed to be a microtubule-binding protein that regu- lates cell division by controlling mitotic spindle organization and by modulating neuroblast migration (11). Microtubules affect multiple facets of neuronal develop- ment and function, with prominent roles in the growth and maintenance of axons and dendrites (12). The Drosophila neuromuscular junction (NMJ) synapse has been a useful genetic system in which to assay the role of the microtubule † These authors contributed equally to this work. ∗ To whom correspondence should be addressed at: E. Medea Scientific Institute at the University of Padova, Largo Meneghetti 2, 35131 Padova, Italy. Tel: +39 0498275778; Fax: +39 0498275093; Email: [email protected] # The Author 2011. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] Human Molecular Genetics, 2011, Vol. 20, No. 21 4248–4257 doi:10.1093/hmg/ddr352 Advance Access published on August 11, 2011 by guest on June 24, 2016 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Defhc1.1, a homologue of the juvenile myoclonic gene EFHC1, modulates architecture and basal...

Defhc1.1, a homologue of the juvenile myoclonicgene EFHC1, modulates architecture and basalactivity of the neuromuscular junction in Drosophila
Maria Giovanna Rossetto1,{, Erica Zanarella1,{, Genny Orso1, Michele Scorzeto2,
Aram Megighian2, Vimlesh Kumar3, Antonio V. Delgado-Escueta4,5 and Andrea Daga1,4,∗
1E. Medea Scientific Institute, Conegliano, Italy, 2Department of Human Anatomy and Physiology, University of
Padova, Padova, Italy, 3Indian Institute of Science Education and Research (IISER), Bhopal 462023, India,4Department of Neurology, The David Geffen School of Medicine, University of California, Los Angeles, CA, USA and5Epilepsy Genetics and Genomics labs, Neurology and Research Services of Veterans Affairs Greater Los Angeles
Healthcare System, West Los Angeles, CA, USA
Received May 17, 2011; Revised and Accepted August 5, 2011
Mutations in the EFHC1 gene have been linked to juvenile myoclonic epilepsy. To understand EFHC1 functionin vivo, we generated knockout Drosophila for the fly homolog Defhc1.1. We found that the neuromuscularjunction synapse of Defhc1.1 mutants displays an increased number of satellite boutons resulting inincreased spontaneous neurotransmitter release. Defhc1.1 binds to microtubules in vitro and overlaps invivo with axonal and synaptic microtubules. Elimination of Defhc1.1 from synaptic terminals reduces thenumber of microtubule loops, suggesting that Defhc1.1 is a negative regulator of microtubule dynamics. Infact, pharmacological treatment of Defhc1.1 mutants with vinblastine, an inhibitor of microtubule dynamics,suppresses the satellite bouton phenotype. Furthermore, Defhc1.1 mutants display overgrowth of the den-dritic arbor and Defhc1.1 overexpression reduces dendrite elaboration. These results suggest thatDefhc1.1 functions as an inhibitor of neurite growth by finely tuning the microtubule cytoskeleton dynamicsand that EFHC1-dependent juvenile myoclonic epilepsy may result from augmented spontaneous neuro-transmitter release due to overgrowth of neuronal processes.
INTRODUCTION
Among the common idiopathic generalized epilepsies, juven-ile myoclonic epilepsy (JME) is one of the most frequent,accounting for as much as 30% of all epilepsies (1–3).Fifteen chromosomal loci have been so far linked to JME(4). At present, however, mutations causing the diseasehave been identified only in the ion-channel encoding geneGABRA1 (5) and in the non-ion channel gene EFHC1 (6–9). EFHC1 encodes a protein that harbors three tandemlyrepeated DM10 domains of unknown function and a singleEF-hand, a calcium-binding motif at the C-terminus. Muta-tions in EFHC1 are to date the most frequent cause ofJME (8).
Expression of EFHC1 in transfected hippocampal primaryneurons produces shorter neurites and fewer branches (9),but loss of function data supporting a function as a negativeregulator of neurite growth and/or development are lacking.EFHC1 associates with the centrosome and the mitoticspindle in cultured cells (10) and more recently, EFHC1 hasbeen proposed to be a microtubule-binding protein that regu-lates cell division by controlling mitotic spindle organizationand by modulating neuroblast migration (11).
Microtubules affect multiple facets of neuronal develop-ment and function, with prominent roles in the growth andmaintenance of axons and dendrites (12). The Drosophilaneuromuscular junction (NMJ) synapse has been a usefulgenetic system in which to assay the role of the microtubule
†These authors contributed equally to this work.
∗To whom correspondence should be addressed at: E. Medea Scientific Institute at the University of Padova, Largo Meneghetti 2, 35131 Padova, Italy.Tel: +39 0498275778; Fax: +39 0498275093; Email: [email protected]
# The Author 2011. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2011, Vol. 20, No. 21 4248–4257doi:10.1093/hmg/ddr352Advance Access published on August 11, 2011
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

cytoskeleton and model its role in inherited neurologicaldisease. Mutations in the microtubule-interacting proteinFutsch (MAP1b) alter synaptic morphology and VAP-33Aregulate synaptic bouton sprouting through interactions withmicrotubules (13–15). Atypical protein kinase C controlsbouton formation (16) and hereditary spastic paraplegiagenes spastin and spichthyin regulate synaptic structure andgrowth through the control of axonal microtubules (17–20).Furthermore, the Drosophila fragile X-related gene regulatesFutsch thereby modulating synaptic architecture and neuro-transmission strength (21). Microtubules are essential struc-tural components of dendrites, and several genes have beenreported to influence dendrite growth and arbor expansionthrough their action on microtubules (22–24). The importantrole of microtubules in dendritic arbor shape has also beenshown in flies (25). These studies indicate that microtubulesplay key roles in the control of neurite structure and functionand that mutations perturbing microtubule dynamics arecausatively linked with inherited neurological disorders.
Here, we characterize one of the two Drosophila homologsof EFHC1, CG8959, henceforth named Defhc1.1. We gener-ated Defhc1.1 knockout (KO) Drosophila and show thatmutant NMJ synapses display an aberrant increase in satelliteboutons accompanied by an increase in spontaneous neuro-transmitter release. Defhc1.1 binds to microtubules and over-laps in vivo with synaptic microtubules. In the NMJ synapse,disruption of Defhc1.1 function leads to a decrease in thenumber of microtubule loops, whose presence correlateswith halted bouton division, suggesting that Defhc1.1 is anegative regulator of microtubule dynamics. Indeed, the satel-lite bouton phenotype is pharmacologically corrected byadministration of the microtubule dynamics inhibitor vinblastine.We further show that the loss of Defhc1.1 induces overgrowthof the dendritic arbor and in vivo overexpression of Defhc1.1substantially reduces dendrite elaboration. In contrast, overex-pression of the other EFHC1 homolog (CG11048) has noeffect on dendrite arborization. These results indicate thatDefhc1.1 has common functions with the human EFHC1gene and suggests that it functions as an inhibitor of neuritegrowth.
RESULTS
Generation of Defhc1.1 loss of function mutants
The Drosophila genome contains two EFHC1 homologsencoded by the CG8959 and CG11048 genes that we namedDefhc1.1 and Defhc1.2, respectively. The two fly proteinsencoded by these genes have similar homology (52%) anddisplay analogous e-values (CG8959 versus EFHC1 ¼ e286;CG11048 versus EFHC1 ¼ e281) when compared withhuman EFHC1. This study examines Defhc1.1 function inDrosophila. We generated Defhc1.1 KO flies by gene target-ing through ends-out homologous recombination (26). The tar-geting construct consisted of a mini-white marker gene flankedby genomic sequences surrounding the Defhc1.1 open readingframe (ORF) for homologous insertion (Fig. 1A and B). Trans-genic lines carrying the targeting donor construct were gener-ated and homologous recombination between the donor DNAand the endogenous Defhc1.1 locus created a null allele by
replacing the Defhc1.1 ORF with the mini-white transgene(Fig. 1C). Defhc1.1KO lines were identified by genomic PCR(Fig. 1D) and confirmed by reverse transcription polymerasechain reaction (RT–PCR) on total RNA (Fig. 1E). Weraised antibodies against Defhc1.1 and stained different wild-type tissues and developmental stages but were unable todetect a signal. Western blot analysis failed to detect the en-dogenous Defhc1.1 protein in wild-type embryos, larvae oradult lysates. However, the same antibodies reveal transgenicDefhc1.1 upon in vivo overexpression. In situ hybridizationwas also attempted but failed to detect endogenous Defhc1.1transcripts in wild-type animals. These results are in agree-ment with the observation that only a very small number ofexpressed sequence tags (ESTs) corresponding to theDefhc1.1 gene are reported in the databases. Together, thesedata suggest that in wild-type flies Defhc1.1 is expressed atvery low levels.
Defhc1.1 mutants display an increased number of satelliteboutons at the NMJ synapse
Homozygous null mutant Defhc1.1 flies are viable and havenormal lifespan. To uncover potential neuronal phenotypes,we examined the NMJ synapse of third-instar larvae homozy-gous for the KO chromosome. To visualize synaptic morph-ology, we labeled pre-synaptic terminals with anti-horseradish peroxidase (HRP), a neuronal membrane marker,and noted that Defhc1.1 mutant NMJs contained numerous sat-ellite boutons (Fig. 2A–D), small supernumerary boutons pro-truding from the primary terminal axis or stemming fromlarger boutons. We counted satellite boutons of the synapticterminal that innervates muscle 4, suitable for quantitativeanalysis owing to its simpler morphology. When comparedwith controls, a greater than 3-fold increase in satelliteboutons was observed in Defhc1.1 mutants (control ¼ 2.7+0.35, n ¼ 27; Defhcko 8.2+ 0.46, n ¼ 29; P ¼ 3.02e212)(Fig. 2E), although the total number of NMJ boutons, exclud-ing satellite varicosities, was similar in control and mutantlarvae. To ensure that this phenotype was caused by loss ofDefhc1.1 and not by second site mutations or associatedgenetic modifiers, we placed the Defhc1.1KO chromosome intrans to the deficiency Df(1)ED7344 that uncovers theDefhc1.1 locus and found that Defhc1.1ko/Df third-instarlarvae had a number of satellite boutons comparable withthat of the mutant line (9.0+ 0.58, n ¼ 22) (Fig. 2E). Toconfirm that the increase in satellite boutons was due to alack of Defhc1.1 in the nervous system, transgenic Defhc1.1-GFP [UAS-Defhc1.1-green fluorescent protein (GFP)] wasexpressed in the null mutant background with the neuronaldriver elav-Gal4. Neuronal expression of these transgenesrescued the mutant phenotype restoring normal satellitebouton number (3.3+ 0.38, n ¼ 31) (Fig. 2G and H). Theseresults indicate that the development of supernumeraryboutons is the consequence of loss of Defhc1.1 function inthe nervous system and that Defhc1.1 is required presynaptic-ally to negatively regulate bouton formation.
We characterized the localization and expression of pre-synaptic (Csp, BRP/nc82) and post-synaptic (discs large andglutamate receptor subunits II and III) markers (Supplemen-tary Material, Figs. S1 and S2) in these satellite boutons. As
Human Molecular Genetics, 2011, Vol. 20, No. 21 4249
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

reported for other mutants featuring excess satellite boutons(27,28), we found that all markers assayed were present andnormally localized at supernumerary boutons, suggestingthat these structures have the predicted organization of a func-tional bouton.
We then examined whether gain of Defhc1.1 functionwould also cause defects in synapse formation in vivo. Weanalyzed the NMJ of larvae overexpressingUAS-Defhc1.1-GFP with elav-Gal4 or the motoneuron-specific driver D42-Gal4 and found that the number ofnormal and satellite boutons was not significantly modifiedwhen compared with controls (Fig. 2F). These NMJs displayeda largely normal morphology indicating that increasingDefhc1.1 levels per se does not alter bouton formation.
To determine Defhc1.1 sub-cellular localization, we exam-ined the distribution of transgenic Defhc1.1-GFP expressedin the nervous system. Importantly, Defhc1.1-GFP wasfound along the entire length of the motor nerves andstained robustly all types of synaptic boutons (Supplemen-tary Material, Fig. S3A). GFP fluorescence was alsoobserved throughout the body of the neurons in the larvalventral ganglion (Supplementary Material, Fig. S3B). Thesynaptic localization of the Defhc1.1 protein is consistentwith a role in the local regulation of bouton sprouting. Inaddition, expression of Defhc1.1-GFP with the type IV den-dritic neuron driver ppk-Gal4 shows that Defhc1.1 also
distributes to the dendritic arbor (Supplementary Material,Fig. S3C).
Defhc1.1 mutants show increased spontaneousneurotransmitter release but normal evoked release
The observed synaptic overgrowth prompted us to analyze thefunctional consequences of loss of Defhc1.1 in synaptic trans-mission. We thus measured the basic aspects of synaptic trans-mission in the mutants. Recordings of excitatory junctionalpontentials (EJPs) at the NMJ of third-instar larvae showedthat evoked neurotransmitter release is not altered (43.67+1.4 mV in controls; 47.7+ 2.2 mV in mutants; P ¼ 0.18).However, Defhc1.1 mutants show a significant increase inthe spontaneous neurotransmitter release (or miniature EJPs)(1.24+ 0.03 in controls; 1.62+ 0.17 in mutants; P ¼ 0.02)(Fig. 2 I–L). Importantly, neuronal expression ofDefhc1.1-GFP in the mutant background fully rescued theincreased mEJP (1.24+ 0.08). The increased mEJP frequencyis likely to reflect an increase in the total number of activezones per synapse. Indeed, when Defhc1.1 mutant andcontrol NMJs were immunostained with anti-nc82 antibodythat labels active zones, we found that the total number ofnc82 punctae per synapse was greater. However, when nor-malized to the total bouton volume, the number of nc82punctae was comparable with that of controls (0.41+ 0.04
Figure 1. Generation and characterization of Defhc1.1KO flies. (A) Wild-type Defhc1.1 gene locus. (B) The targeting donor-transgene. In this construct, the whitemini-gene marker is flanked by 3.0 kb and 3.1 kb genomic fragments and these homology arms are further flanked by two I-SceI restriction sites and two FRTsites. (C) The Defhc1.1KO locus, where the Defhc1.1 gene has been entirely replaced by the white mini-gene marker. (D) Genomic PCR demonstrates that theDefhc1.1 locus is present in w1118 control DNA but is not amplified from adult Defhc1.1KO DNA (primer pairs 1–2). In contrast, PCR products amplified usingprimer sets mapping to the inserted mini-white marker and outside the homology arms are found only in Defhc1.1KO DNA (primer pairs 3–4 and 5–6). (E) RT–PCR on total RNA from Defhc1.1KO shows that the endogenous Defhc1.1 transcript is absent while a control transcript (Defhc1.2) is normally amplified. Bothtranscripts are amplified from w1118 control RNA.
4250 Human Molecular Genetics, 2011, Vol. 20, No. 21
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
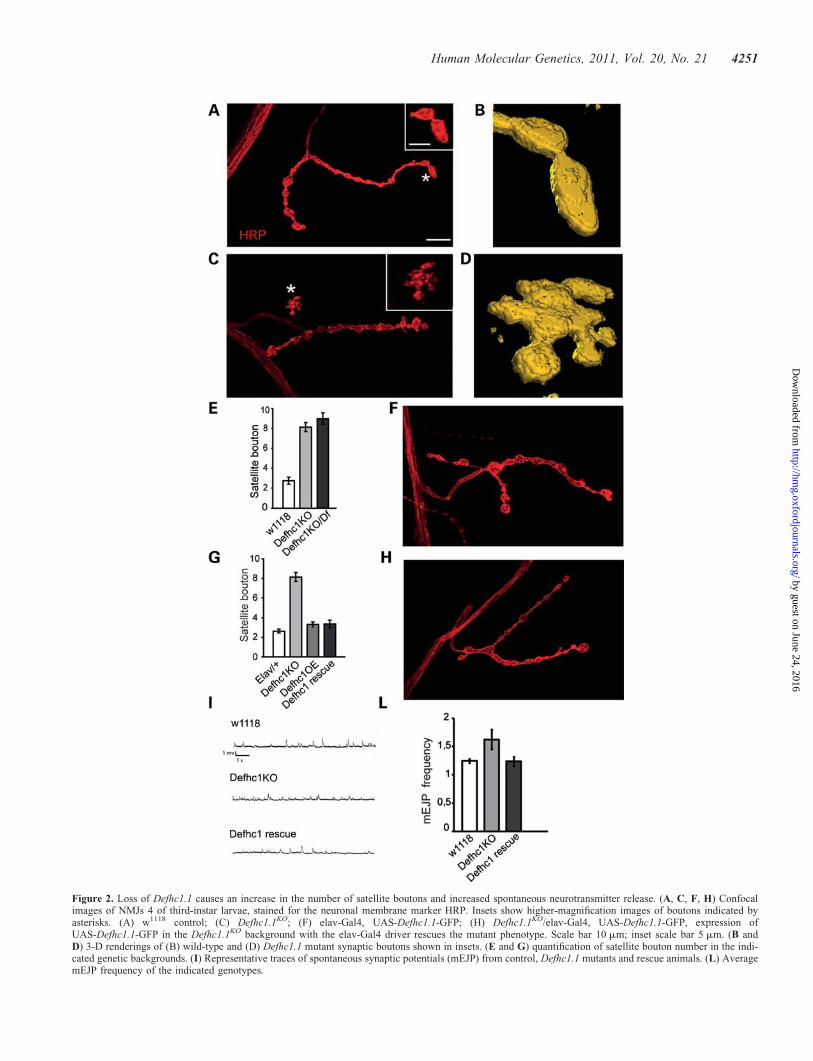
Figure 2. Loss of Defhc1.1 causes an increase in the number of satellite boutons and increased spontaneous neurotransmitter release. (A, C, F, H) Confocalimages of NMJs 4 of third-instar larvae, stained for the neuronal membrane marker HRP. Insets show higher-magnification images of boutons indicated byasterisks. (A) w1118 control; (C) Defhc1.1KO; (F) elav-Gal4, UAS-Defhc1.1-GFP; (H) Defhc1.1KO/elav-Gal4, UAS-Defhc1.1-GFP, expression ofUAS-Defhc1.1-GFP in the Defhc1.1KO background with the elav-Gal4 driver rescues the mutant phenotype. Scale bar 10 mm; inset scale bar 5 mm. (B andD) 3-D renderings of (B) wild-type and (D) Defhc1.1 mutant synaptic boutons shown in insets. (E and G) quantification of satellite bouton number in the indi-cated genetic backgrounds. (I) Representative traces of spontaneous synaptic potentials (mEJP) from control, Defhc1.1 mutants and rescue animals. (L) AveragemEJP frequency of the indicated genotypes.
Human Molecular Genetics, 2011, Vol. 20, No. 21 4251
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

for controls and 0.39+ 0.02 for Defhc1.1 mutants). This sug-gests that increased spontaneous neurotransmitter release iscaused by an increase in synapse size. These data indicatethat the aberrant NMJ morphology of Defhc1.1KO mutants cor-relates with abnormal spontaneous neurotransmitter release, adefect potentially relevant to the pathophysiology of epilepsy.
Defhc1.1 associates with microtubules
The regulation of microtubule organization and dynamics iscrucial for axon formation and maintenance and severalstudies that used the Drosophila NMJ as a model have impli-cated microtubules in modulating synaptic growth and boutonbudding (13–16,18–21). Because mammalian EFHC1 hasbeen recently identified as a microtubule-binding protein(11), we tested whether the Drosophila homolog of EFHC1was capable of associating with microtubules. Examinationof Defhc1.1-myc localization in transfected Cos-7 cellsshowed that in resting cells the protein was distributedthroughout the cytoplasm and within the nucleus (Fig. 3A).
However, in cells undergoing mitosis, Defhc1.1 prominentlyoverlapped with the mitotic spindle (Fig. 3B) (10). To testfor binding of Defhc1.1 to microtubules, we performed amicrotubule-binding assay using extracts from cells trans-fected with a Defhc1.1-myc construct, revealing thatDefhc1.1 co-sediments with taxol-stabilized microtubules(Fig. 3C). We also performed microtubule binding in vitrousing bacterially produced Defhc1.1 and purified microtubulesconfirming that binding is direct (Supplementary Material,Fig. S4). To investigate whether Defhc1.1 co-localizes withmicrotubules in vivo, we stained third-instar larvae expressingDefhc1.1-GFP for the microtubule-associated protein Futsch(13). We analyzed motor nerves and synaptic terminals andfound that Defhc1.1-GFP fluorescence largely overlappedwith Futsch immunoreactivity, indicating that in vivoDefhc1.1 co-localizes substantially with microtubules inaxons and synaptic boutons (Fig. 3D and E). These resultsdemonstrate that Defhc1.1 is capable of binding physicallyto microtubules and that in the nervous system it associateswith microtubules within axons.
Figure 3. Defhc1.1 binds to microtubules. Cos-7 cells transfected with Defhc1.1-myc were stained using anti-tubulin (red) and anti-myc (green) antibodies. (A)Resting cell. (B) Cell undergoing mitosis. Scale bar 4 mm. (C) Cos-7 cells were transfected with Defhc1.1-Myc or GFP, lysed and polymerized microtubulespelleted. Western blot analysis shows that Defhc1.1-myc co-sediments with microtubules (top), while GFP remains in the supernatant (bottom). L, lysate; S,supernatant; P, pellet. (D) Segment of motor nerve expressing UAS-Defhc1.1-GFP labeled with Futsch antibody to visualize microtubule bundles. Mergepanel shows that GFP fluorescence widely overlaps with the anti-Futsch signal (Manders’ overlap coefficient ¼ 0.76, co-localization coefficients M1 ¼ 0.92and M2 ¼ 0.99). Co-localization parameters were measured in the boxed region. Scale bar 10 mm. (E) Futsch-labeled (red) synaptic terminal expressingDefhc1.1-GFP (green) shows that these two proteins are largely co-localized within boutons. Scale bar 5 mm. Co-localization parameters for the boutonshown in the inset are: Manders’ overlap coefficient ¼ 0.83, co-localization coefficients Mx ¼ 0.88 and My ¼ 0.98.
4252 Human Molecular Genetics, 2011, Vol. 20, No. 21
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
Defhc1.1 affects microtubule cytoskeleton dynamics
Satellite boutons can be regarded as the site of potential syn-
aptic growth by new bouton sprouting (29). During normal
synaptic growth of the Drosophila NMJ, Futsch is found in
association with loops of bundled microtubules typically
observed within stable boutons. In contrast, dispersion or
destruction of these loops underlies bouton division or sprout-
ing (15). To investigate whether the increase in satellite
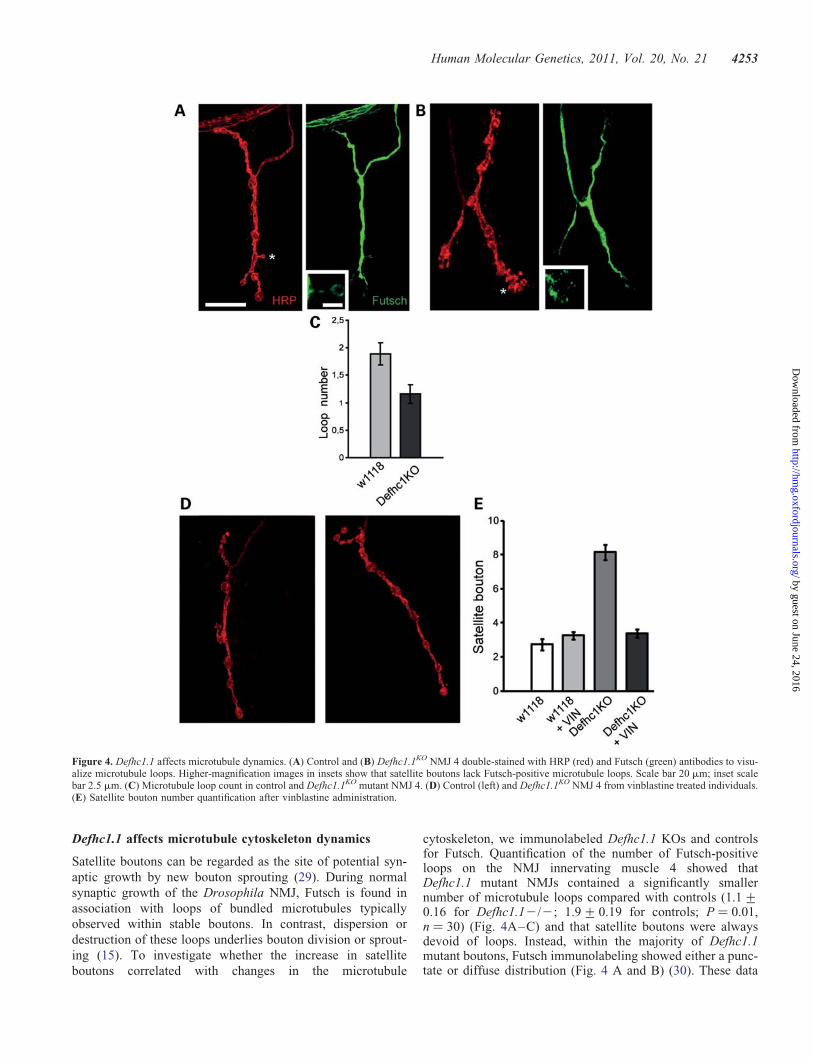
boutons correlated with changes in the microtubule
cytoskeleton, we immunolabeled Defhc1.1 KOs and controlsfor Futsch. Quantification of the number of Futsch-positiveloops on the NMJ innervating muscle 4 showed thatDefhc1.1 mutant NMJs contained a significantly smallernumber of microtubule loops compared with controls (1.1+0.16 for Defhc1.12/2; 1.9+ 0.19 for controls; P ¼ 0.01,n ¼ 30) (Fig. 4A–C) and that satellite boutons were alwaysdevoid of loops. Instead, within the majority of Defhc1.1mutant boutons, Futsch immunolabeling showed either a punc-tate or diffuse distribution (Fig. 4 A and B) (30). These data
Figure 4. Defhc1.1 affects microtubule dynamics. (A) Control and (B) Defhc1.1KO NMJ 4 double-stained with HRP (red) and Futsch (green) antibodies to visu-alize microtubule loops. Higher-magnification images in insets show that satellite boutons lack Futsch-positive microtubule loops. Scale bar 20 mm; inset scalebar 2.5 mm. (C) Microtubule loop count in control and Defhc1.1KO mutant NMJ 4. (D) Control (left) and Defhc1.1KO NMJ 4 from vinblastine treated individuals.(E) Satellite bouton number quantification after vinblastine administration.
Human Molecular Genetics, 2011, Vol. 20, No. 21 4253
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

suggest that the lack of Defhc1.1 promotes microtubuledynamic instability within synaptic terminals resulting in over-growth and that Defhc1.1 functions as a negative modulator ofmicrotubule dynamics to prevent this excessive growth.
If additional satellite boutons are the result of increasedmicrotubule dynamics caused by loss of Defhc1.1-dependentinhibition, we reasoned that pharmacological suppression ofmicrotubule dynamics should revert the number of satelliteboutons to wild-type levels. We treated Defhc1.1KO flieswith vinblastine, a microtubule destabilizing drug that in lowdoses suppresses microtubule dynamics in vivo and in vitro(31,32) and in Drosophila has been shown to ameliorate syn-aptic microtubule defects associated with the loss of spastin(17). We found that vinblastine administration suppressed add-itional satellite boutons in Defhc1.1KO NMJs without affectingthe morphology of control synaptic terminals (treatedcontrol ¼ 3.4+ 0.18, n ¼ 29; treated Defhcko 3.3+ 0.16,n ¼ 39) (Fig. 4D and E). These results demonstrate that theloss of Defhc1.1 can be compensated by pharmacological in-hibition of microtubule dynamics and strongly implicateDefhc1.1 in the negative regulation of bouton sproutingthrough the fine inhibition of the microtubule cytoskeletondynamics locally at the synapse.
Defhc1.1 regulates dendritic arbor morphology
Axons and dendrites contain bundles of microtubules that arerequired for the growth and maintenance of these neurites. Wethus examined the phenotypic consequences of loss and gainof Defhc1.1 function on dendrite arborization in Drosophiladendritic neurons. There are four classes of dendritic arboriza-tion neurons, named I–IV in order of increasing dendritic
arbor complexity (33). We concentrated on class IV neuronscharacterized by the most highly branched dendritic arborbecause dendrite outgrowth depends on the microtubule cyto-skeleton (25) and because the elaborate morphology of theirarbor might permit to observe the subtle phenotypic irregular-ities we expected from manipulating Defhc1.1 expressionlevels.
To visualize potential dendritic arbor defects, we used theclass IV-specific promoter ppk-Gal4 (34) to drive theUAS-mCD8::GFP reporter in class-IV neurons and assayeddendritic arbor morphology in third-instar larvae. We drewconcentric circles 100 mm apart centered on the neuron bodyand counted the number of dendrite endings in the outermostcircle as a measure of terminal branching and thus neuritegrowth. We found a small but statistically significant increasein the number of terminal endings in Defhc1.1 mutants whencompared with controls (control ¼ 2.9+ 1.0; Defhc1.1KO ¼8.3+ 2.3; P ¼ 0.04, n ¼ 13) (Fig. 5A, B, D). This result isconsistent with the increased number of satellite boutonsobserved at the NMJ synapse and suggests that the lack ofDefhc1.1 induces terminal dendrite branching and outgrowth,implicating Defhc1.1 in the negative regulation of dendritegrowth. This inhibitory function on dendrite arborizationwas assayed by expressing UAS-Defhc1.1-HA in class-IVdendritic neurons with the ppk-Gal4 driver. Strikingly,ectopic Defhc1.1 expression resulted in a substantial reductionin dendritic arbor elaboration (Fig. 5C). In particular, the totalnumber of dendrite termini was reduced to 50% of that ofcontrols (control ¼ 225+ 8.4; Defhc1.1OE ¼ 107+ 8.9; P ¼5.5e27, n ¼ 7) and total arbor length to 55% (control ¼9644+ 452 mm; Defhc1.1OE ¼ 4921+ 367 mm; P ¼ 3.2e26,n ¼ 7) (Fig. 5E and F). Together, loss- and gain-of-function
Figure 5. Loss and gain of Defhc1.1 function affect dendrite arborization. To visualize dendritic arbor morphology, UAS-mCD8::GFP was expressed with thetype IV dendritic neuron-specific driver ppk-Gal4. Confocal images and corresponding tracings of (A) wild-type, (B) Defhc1.1KO and (C) Defhc1.1 overexpres-sing class IV dendritic neurons. Scale bar 70 mm. (D) Quantification of the number of terminal dendrite endings in w1118 control and Defhc1.1KO individuals.(E–F) Graph bars showing the quantification of the total number of dendrite termini and total arborization length in control and Defhc1.1OE individuals.
4254 Human Molecular Genetics, 2011, Vol. 20, No. 21
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

data provide experimental support for an inhibitory role ofDefhc1.1 in the modulation of dendritic arbor morphology.We also examined whether overexpression of Defhc1.2 hadan effect on dendrite arborization, but found that dendritegrowth was unaffected (Supplementary Material, Figure), pro-viding support to the idea that Defhc1.1 is likely to be theclosest functional homolog of human EFHC1.
DISCUSSION
In this study, we functionally characterize the DrosophilaDefhc1.1 gene homologous to EFHC1 whose mutation inhumans is responsible for a form of JME (9). A function forEFHC1 in regulation of cell division and neuronal migrationhas been proposed from studies on the rat developing neocor-tex (11), despite the reported absence of gross anatomicaldefects in Efhc12/2 mouse brains (35). However, a directlink between the defects ensuing loss of EFHC1 inmammals and epileptogenesis is not apparent. Our mutant ana-lysis of Defhc1.1 loss- and gain-of-function alleles in vivo inDrosophila revealed a number of neuronal defects that canbe more promptly reconciled with epileptogenesis.
EFHC1 has been demonstrated to be a microtubule-bindingprotein (11). Likewise, Defhc1.1 co-sediments with polymer-ized microtubules in vitro and co-localizes with axonal micro-tubules in vivo. This observation indicates that the reportedneuronal defects are potentially the result of an impairmentof the neuronal microtubule cytoskeleton dynamics or organ-ization caused by modulating the levels of Defhc1.1 expres-sion. The elaboration of synaptic architecture is welldocumented to be microtubule dependent and, at the Drosoph-ila NMJ, several known regulators of microtubule dynamicshave been shown to modulate the size and complexity of theterminal arbor (13–16,18–21). Spastin and Dmob4/Phoceinmutants, which lack a microtubule-severing protein and aregulator of microtubule dynamics respectively, have elabor-ate supernumerary NMJ boutons (18,19,30). Likewise, lossof Defhc1.1 results in the presence of supernumerary satelliteboutons at the NMJ synapse, suggesting that bouton over-growth could be due to loss of Defhc1.1 activity on microtu-bules. Alterations of microtubule loops architecture havebeen shown to be involved in the process of bouton divisionat the Drosophila NMJ synapse. Microtubule loops havebeen associated with stable synaptic boutons, while the disper-sion of these loops is typical of boutons undergoing division orsprouting (15). Previous studies have shown that mutants char-acterized by supernumerary satellite boutons can display bothmore numerous (29) or less numerous (30) microtubule loops.In both cases, perturbation of microtubule dynamics, whosebalance must be tightly regulated for normal synapticgrowth, underlies these NMJ defects. We found that thenumber of microtubule loops identified by Futsch immunola-beling is reduced in Defhc1.1 mutant NMJs, suggesting thatthe loss of Defhc1.1 is likely to affect microtubule dynamics.We propose that wild-type Defhc1.1 facilitates the stabiliza-tion of microtubules and thereby decreases their dynamic in-stability. Under normal circumstances, this activity wouldforce existing boutons into a more stable mode and limit thesprouting of new boutons. Robust corroboration for this
functional hypothesis comes from the observation that inhib-ition of microtubule dynamics by pharmacological treatmentof Defhc1.1 mutants with vinblastine suppresses extra satellitebouton.
The behavior of growth cone microtubules indicates thatmicrotubule bundling is an important step in the formationof a new axonal segment. Decreased microtubule dynamicslocks microtubules in a non-dynamic array, microtubules donot undergo the subsequent step of bundling and axon elong-ation is halted (31). The parallel behavior of microtubulesobserved at the Drosophila NMJ synapse during boutonsprouting has suggested a fundamental similarity betweenthe mechanisms of synaptic growth and the mechanisms ofgrowth cone motility (15). Loss of mammalian EFHC1 isthought to induce microtubule bundling (11), indicating thatincreased or facilitated bundling could be the mechanismunderlying the synaptic overgrowth seen in response to lossof Defhc1.1 in flies. Lack of Defhc1.1 results in moredynamic and unstable microtubules which would proceedmore easily through bundling, a step thought to be importantfor bouton division and growth. According to this model, thefunction of wild-type Defhc1.1 would be to restrain excessivesynaptic growth by curbing the microtubule dynamic instabil-ity and its positive effects on bundling.
The role of Defhc1.1 is not restricted to the synapse. Lossand gain of Defhc1.1 function also affect the formation ofthe dendritic arbor, where the microtubule cytoskeleton isknown to play a prominent role. Similar to its effect on synap-tic bouton growth, elimination of Defhc1.1 results in an in-crease in terminal branching of dendritic arborizationneurons, suggesting a more general role on neurite growth.Consistent with this result, Defhc1.1 overexpression causes amarked reduction in dendrite branching and complexity.This is in contrast, however, with our observations at theNMJ where ectopic expression of Defhc1.1 had no conse-quences on synaptic elaboration. A possible explanation forthis differential effect could be that the diverse microtubulemilieu within axons and dendrites (12) might render dendriticmicrotubules less tolerant to increased levels of Defhc1.1 thanaxonal microtubules. Thus, our combined data implicateDefhc1.1 as a negative regulator of neurite growth.
The overall subtle phenotypic consequences of loss ofDefhc1.1 are consistent with the absence of obvious defectsin EFHC1 KO mice (35) as well as with the otherwisenormal phenotype of humans with JME (9). Therefore,strong phenotypic consequences would not be predictedfrom the loss of Defhc1.1 function. Accordingly, the modestlyvisible effects of Defhc1.1 elimination on microtubule cyto-skeleton morphology are not surprising. It is known, forexample, that low doses of vinblastine decrease microtubuledynamics and thereby abolish axonal elongation duringgrowth cone advance (31,32), yet do not visibly affect the in-tegrity of the axonal microtubule array. Furthermore, elimin-ation of the other EFHC1 homolog, Defhc1.2, couldexacerbate the phenotypes here described due to genetic re-dundancy.
We also found that the lack of Defhc1.1 results in signifi-cantly increased spontaneous neurotransmitter release suggest-ing a correlation between the abnormal synaptic architectureof Defhc1.1 loss-of-function mutants and defects in synaptic
Human Molecular Genetics, 2011, Vol. 20, No. 21 4255
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

activity. Our observation that Defhc1.1KO has increased spon-taneous release suggests a model in which basal level neuralactivity can contribute to epilepsy. The observation that thedensity of active zones in Defhc1.1 mutant synapses remainsunaltered indicates that the increase in spontaneous fusionevents depends on the increased number of active zones dueto synaptic overgrowth. In conclusion, these results suggestthat EFHC1 may regulate epileptogenesis by subtly increasingthe extent of spontaneous vesicle fusion through a fine regula-tion of the microtubule cytoskeleton and that drugs affectingthe dynamics of microtubules could potentially be tested inthe treatment of this disease.
MATERIALS AND METHODS
Drosophila genetics
Fly culture and transgenesis were performed using standardprocedures. Several transgenic lines for each construct weregenerated and tested. Drosophila strains used: elav-Gal4;D42-Gal4, tubulin-Gal4, UAS-mCD8-GFP (Bloomington);Mef2-Gal4, ppk-Gal4. Targeting of the Defhc1.1 genomiclocus was performed using the ends-out technique asdescribed (26).
Molecular biology
The Defhc1.1 and Defhc1.2 full-length cDNAs were obtainedby RT–PCR performed on total head RNA purified withTrizol reagent (Invitrogen). The Defhc1.1 and Defhc1.2cDNAs were subcloned in the pUAST vector in-frame withan HA or GFP tag. The Defhc1.1 cDNA was cloned in thepcDNA3.1 vector in frame with a myc tag. The Defhc1.1 tar-geting construct was generated by cloning PCR-amplified 3 kbgenomic fragments upstream and downstream of the Defhc1.1ORF into the pEndsout2 vector, on both sides of a whitemini-gene.
Immunohistochemistry, immunocytochemistry and in situhybridization
Third-instar larva immunostaining and cell transfection andimmunocytochemistry were carried out following standardprotocols. Antibodies used: mouse anti-CSP (1:100, DSHB),anti-HRP (1:500, Jackson laboratories) rabbit anti-myc(1:200, Santa Cruz Biotechnology), mouse anti-Dlg (1:100,DSHB), mouse anti-synaptotagmin (1:100, DSHB), mouseanti-nc82 (1:20, DSHB), mouse anti-glutamate receptor IIand rabbit anti-glutamate receptor III (1:5000, gift of AaronDiAntonio), mouse anti-acetylated a-Tubulin (1:500,Sigma-Aldrich) and mouse anti-Futsch (1:100, DSHB). Sec-ondary antibodies were Cy5 and Cy3 conjugates fromJackson laboratories and Alexa Fluor 488 conjugates fromInvitrogen. Fluorescent in situ hybridization was performedas described in reference (36).
Quantification of bouton number
Boutons were visualized with anti-CSP and anti-HRPantibodies. Neuromuscolar junctions innervating muscle 4,
abdominal segments 2 and 3, were imaged. Each NMJ wascounted independently by three people blind to the genotype.The bouton number represents the average of the threeindividual counts.
Electrophysiology
Wandering third-instar larvae were dissected in HL3 saline.Excitatory junctional potentials were recorded from muscle6 of hemisegment A2 or A3 by stimulating the nerve at 4–6 mV at 1 Hz. Recordings were collected only from prepara-tions where the resting membrane potential was between260 and 270 mV. mEJP signals were recorded over aperiod of 180 s. Signals were digitized via Digidata 1322Aat a sampling rate of 50 kHz and low pass filtered at 1 kHz.Data were processed and analyzed with pClamp 9 software.Recordings from at least nine animals of each genotypewere used for data analysis.
Microtubule co-sedimentation assay
Cos-7 cells were collected 24 h after transfection and lysed in80 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES)(pH 7.0), 1 mM MgCl2, 0.5 mM ethylene glycol tetraaceticacid (EGTA) and 1.0% Triton X-100 plus protease inhibitorssamples were incubated on ice for 30 min and then centrifuged(610g; 10 min at 48C). The supernatant was collected andincubated with 40 mm paclitaxel and 1 mM GTP in dimethylsulfoxide (DMSO) or else equimolar DMSO alone for30 min at 378C. Samples were layered over a 50% glycerolcushion containing 80 mM PIPES (pH 7.0), 1 mM MgCl2 and0.5 mM EGTA and then centrifuged at 100 000g at 48C for40 min. Lysate, pellet and supernatant fractions were collectedfor western blot analysis. In vitro co-sedimentation wascarried out using the Tubulin Polymerization Assay kit (Cyto-skeleton) according to the manufacturer’s instructions.
Confocal microscopy and images analysis
Images were acquired with a Nikon C1 confocal microscopeand analysed with Volocity (Perkin-Elmer). Co-localizationanalysis was performed with Volocity. Manders’ overlap coef-ficient indicates the percent of signal from both channels thatco-localize; co-localization coefficient Mx and My indicatethe percent of green pixels co-localizing with red pixels andthe percent of red pixels co-localizing with green pixels,respectively. To visualize dendritic arborization neurons, asingle third-instar larva was put in 80% glycerol on a slideand carefully pushed down with a cover slip. Images of den-drites in abdominal segments 4 or 5 were analyzed using Neu-rolucida, Autoneuron and Neuroexplorer softwares (MBFBioscience).
Statistics
The data are presented as mean+SEM. Statistical signifi-cance was determined by using two-tailed, unpaired t-tests.The alpha level for all statistical tests was set at 0.05.
4256 Human Molecular Genetics, 2011, Vol. 20, No. 21
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
ACKNOWLEDGEMENTS
We would like to thank Stefano Casonato for preparing theknockout construct and Andrea Martinuzzi for helpful discus-sion and support.
FUNDING
This study was supported by grants from the Italian Ministryof Health, the Fondazione Telethon and a Veterans Adminis-tration Merit Review Grant.
REFERENCES
1. Delgado-Escueta, A.V. and Enrile-Bacsal, F. (1984) Juvenile myoclonicepilepsy of Janz. Neurology, 34, 285–294.
2. Janz, D. (1985) Epilepsy with impulsive petit mal (juvenile myoclonicepilepsy). Acta Neurol. Scand., 72, 449–459.
3. Delgado-Escueta, A.V., Medina, M.T., Serratosa, J.M., Castroviejo, I.P.,Gee, M.N., Weissbecker, K., Westling, B.W., Fong, C.Y., Alonso, M.E.,Cordova, S. et al. (1999) Mapping and positional cloning of commonidiopathic generalized epilepsies: juvenile myoclonus epilepsy andchildhood absence epilepsy. Adv. Neurol., 79, 351–374.
4. Delgado-Escueta, A.V. (2007) Advances in genetics of juvenilemyoclonic epilepsies. Epilepsy Curr., 7, 61–67.
5. Cossette, P., Liu, L., Brisebois, K., Dong, H., Lortie, A., Vanasse, M.,Saint-Hilaire, J.M., Carmant, L., Verner, A., Lu, W.Y. et al. (2002)Mutation of GABRA1 in an autosomal dominant form of juvenilemyoclonic epilepsy. Nat. Genet., 31, 184–189.
6. Annesi, F., Gambardella, A., Michelucci, R., Bianchi, A., Marini, C.,Canevini, M.P., Capovilla, G., Elia, M., Buti, D., Chifari, R. et al. (2007)Mutational analysis of EFHC1 gene in Italian families with juvenilemyoclonic epilepsy. Epilepsia, 48, 1686–1690.
7. Stogmann, E., Lichtner, P., Baumgartner, C., Bonelli, S., Assem-Hilger,E., Leutmezer, F., Schmied, M., Hotzy, C., Strom, T.M., Meitinger, T.et al. (2006) Idiopathic generalized epilepsy phenotypes associated withdifferent EFHC1 mutations. Neurology, 67, 2029–2031.
8. Medina, M.T., Suzuki, T., Alonso, M.E., Duron, R.M., Martinez-Juarez,I.E., Bailey, J.N., Bai, D., Inoue, Y., Yoshimura, I., Kaneko, S. et al.(2008) Novel mutations in Myoclonin1/EFHC1 in sporadic and familialjuvenile myoclonic epilepsy. Neurology, 70, 2137–2144.
9. Suzuki, T., Delgado-Escueta, A.V., Aguan, K., Alonso, M.E., Shi, J.,Hara, Y., Nishida, M., Numata, T., Medina, M.T., Takeuchi, T. et al.(2004) Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat.Genet., 36, 842–849.
10. de Nijs, L., Lakaye, B., Coumans, B., Leon, C., Ikeda, T.,Delgado-Escueta, A.V., Grisar, T. and Chanas, G. (2006) EFHC1, aprotein mutated in juvenile myoclonic epilepsy, associates with themitotic spindle through its N-terminus. Exp. Cell Res., 312, 2872–2879.
11. de Nijs, L., Leon, C., Nguyen, L., Loturco, J.J., Delgado-Escueta, A.V.,Grisar, T. and Lakaye, B. (2009) EFHC1 interacts with microtubules toregulate cell division and cortical development. Nat. Neurosci., 12,1266–1274.
12. Conde, C. and Caceres, A. (2009) Microtubule assembly, organization anddynamics in axons and dendrites. Nat. Rev. Neurosci., 10, 319–332.
13. Hummel, T., Krukkert, K., Roos, J., Davis, G. and Klambt, C. (2000)Drosophila Futsch/22C10 is a MAP1B-like protein required for dendriticand axonal development. Neuron, 26, 357–370.
14. Pennetta, G., Hiesinger, P.R., Fabian-Fine, R., Meinertzhagen, I.A. andBellen, H.J. (2002) Drosophila VAP-33A directs bouton formation atneuromuscular junctions in a dosage-dependent manner. Neuron, 35,291–306.
15. Roos, J., Hummel, T., Ng, N., Klambt, C. and Davis, G.W. (2000)Drosophila Futsch regulates synaptic microtubule organization and isnecessary for synaptic growth. Neuron, 26, 371–382.
16. Ruiz-Canada, C., Ashley, J., Moeckel-Cole, S., Drier, E., Yin, J. andBudnik, V. (2004) New synaptic bouton formation is disrupted bymisregulation of microtubule stability in aPKC mutants. Neuron, 42, 567–580.
17. Orso, G., Martinuzzi, A., Rossetto, M.G., Sartori, E., Feany, M. and Daga,A. (2005) Disease-related phenotypes in a Drosophila model of hereditaryspastic paraplegia are ameliorated by treatment with vinblastine. J. Clin.
Invest., 115, 3026–3034.18. Sherwood, N.T., Sun, Q., Xue, M., Zhang, B. and Zinn, K. (2004)
Drosophila spastin regulates synaptic microtubule networks and isrequired for normal motor function. PLoS Biol., 2, e429.
19. Trotta, N., Orso, G., Rossetto, M.G., Daga, A. and Broadie, K. (2004) Thehereditary spastic paraplegia gene, spastin, regulates microtubule stabilityto modulate synaptic structure and function. Curr. Biol., 14, 1135–1147.
20. Wang, X., Shaw, W.R., Tsang, H.T., Reid, E. and O’Kane, C.J. (2007)Drosophila spichthyin inhibits BMP signaling and regulates synapticgrowth and axonal microtubules. Nat. Neurosci., 10, 177–185.
21. Zhang, Y.Q., Bailey, A.M., Matthies, H.J., Renden, R.B., Smith, M.A.,Speese, S.D., Rubin, G.M. and Broadie, K. (2001) Drosophila fragileX-related gene regulates the MAP1B homolog Futsch to control synapticstructure and function. Cell, 107, 591–603.
22. Cohen, D., Segal, M. and Reiner, O. (2008) Doublecortin supports thedevelopment of dendritic arbors in primary hippocampal neurons. Dev.
Neurosci., 30, 187–199.23. Huang, J., Furuya, A. and Furuichi, T. (2007) Very-KIND, a KIND
domain containing RasGEF, controls dendrite growth by linking Rassmall GTPases and MAP2. J. Cell Biol., 179, 539–552.
24. Ohkawa, N., Fujitani, K., Tokunaga, E., Furuya, S. and Inokuchi, K.(2007) The microtubule destabilizer stathmin mediates the developmentof dendritic arbors in neuronal cells. J. Cell Sci., 120, 1447–1456.
25. Jinushi-Nakao, S., Arvind, R., Amikura, R., Kinameri, E., Liu, A.W. andMoore, A.W. (2007) Knot/Collier and cut control different aspects ofdendrite cytoskeleton and synergize to define final arbor shape. Neuron,56, 963–978.
26. Gong, W.J. and Golic, K.G. (2003) Ends-out, or replacement, genetargeting in Drosophila. Proc. Natl Acad. Sci. USA, 100, 2556–2561.
27. Dickman, D.K., Lu, Z., Meinertzhagen, I.A. and Schwarz, T.L. (2006)Altered synaptic development and active zone spacing in endocytosismutants. Curr. Biol., 16, 591–598.
28. O’Connor-Giles, K.M., Ho, L.L. and Ganetzky, B. (2008) Nervous wreckinteracts with thickveins and the endocytic machinery to attenuateretrograde BMP signaling during synaptic growth. Neuron, 58, 507–518.
29. Franco, B., Bogdanik, L., Bobinnec, Y., Debec, A., Bockaert, J.,Parmentier, M.L. and Grau, Y. (2004) Shaggy, the homolog of glycogensynthase kinase 3, controls neuromuscular junction growth in Drosophila.J. Neurosci., 24, 6573–6577.
30. Schulte, J., Sepp, K.J., Jorquera, R.A., Wu, C., Song, Y., Hong, P. andLittleton, J.T. (2010) DMob4/Phocein regulates synapse formation, axonaltransport, and microtubule organization. J. Neurosci., 30, 5189–5203.
31. Tanaka, E., Ho, T. and Kirschner, M.W. (1995) The role of microtubuledynamics in growth cone motility and axonal growth. J. Cell Biol., 128,139–155.
32. Tanaka, E.M. and Kirschner, M.W. (1991) Microtubule behavior in thegrowth cones of living neurons during axon elongation. J. Cell Biol., 115,345–363.
33. Grueber, W.B., Jan, L.Y. and Jan, Y.N. (2002) Tiling of the Drosophilaepidermis by multidendritic sensory neurons. Development, 129,2867–2878.
34. Adams, C.M., Anderson, M.G., Motto, D.G., Price, M.P., Johnson, W.A.and Welsh, M.J. (1998) Ripped pocket and pickpocket, novel DrosophilaDEG/ENaC subunits expressed in early development and inmechanosensory neurons. J. Cell Biol., 140, 143–152.
35. Suzuki, T., Miyamoto, H., Nakahari, T., Inoue, I., Suemoto, T., Jiang, B.,Hirota, Y., Itohara, S., Saido, T.C., Tsumoto, T. et al. (2009) Efhc1deficiency causes spontaneous myoclonus and increased seizuresusceptibility. Hum. Mol. Genet., 18, 1099–1109.
36. Lecuyer, E., Parthasarathy, N. and Krause, H.M. (2008) Fluorescent in
situ hybridization protocols in Drosophila embryos and tissues. Methods
Mol. Biol., 420, 289–302.
Human Molecular Genetics, 2011, Vol. 20, No. 21 4257
by guest on June 24, 2016http://hm
g.oxfordjournals.org/D
ownloaded from




















