
Crosstalk and signaling switches in mitogen-activated protein kinase cascades
Upload
independentCategory
view
1download
0
Cyclic Nucleotide Phosphodiesterase 3B Is a DownstreamTarget of Protein Kinase B and May Be Involved inRegulation of Effects of Protein Kinase B on ThymidineIncorporation in FDCP2 Cells1
Faiyaz Ahmad,2* Li-Na Cong,† Lena Stenson Holst,‡ Ling-Mei Wang,§ Tova Rahn Landstrom,‡
Jaclyn H. Pierce,§ Michael J. Quon,† Eva Degerman,‡ and Vincent C. Manganiello*
Wild-type (F/B), constitutively active (F/B*), and three kinase-inactive (F/Ba2, F/Bb2, F/Bc2) forms of Akt/protein kinase B(PKB) were permanently overexpressed in FDCP2 cells. In the absence of insulin-like growth factor-1 (IGF-1), activities of PKB,cyclic nucleotide phosphodiesterase 3B (PDE3B), and PDE4 were similar in nontransfected FDCP2 cells, mock-transfected (F/V)cells, and F/B and F/B2 cells. In F/V cells, IGF-1 increased PKB, PDE3B, and PDE4 activities;2-fold. In F/B cells, IGF-1, in awortmannin-sensitive manner, increased PKB activity;10-fold and PDE3B phosphorylation and activity (;4-fold), but increasedPDE4 to the same extent as in F/V cells. In F/B* cells, in the absence of IGF-1, PKB activity was markedly increased (;10-fold)and PDE3B was phosphorylated and activated (3- to 4-fold); wortmannin inhibited these effects. In F/B* cells, IGF-1 had littlefurther effect on PKB and activation/phosphorylation of PDE3B. In F/B2 cells, IGF-1 activated PDE4, not PDE3B, suggesting thatkinase-inactive PKB behaved as a dominant negative with respect to PDE3B activation. Thymidine incorporation was greater inF/B* cells than in F/V cells and was inhibited to a greater extent by PDE3 inhibitors than by rolipram, a PDE4 inhibitor. In F/Bcells, IGF-1-induced phosphorylation of the apoptotic protein BAD was inhibited by the PDE3 inhibitor cilostamide. ActivatedPKB phosphorylated and activated rPDE3B in vitro. These results suggest that PDE3B, not PDE4, is a target of PKB and thatactivated PDE3B may regulate cAMP pools that modulate effects of PKB on thymidine incorporation and BAD phosphorylationin FDCP2 cells. The Journal of Immunology,2000, 164: 4678–4688.
T he serine/threonine protein kinase PKB (protein kinase B)3
(also known as Akt or RAC-PK) is a downstream targetand effector of phosphatidylinositol-3 kinase (PI3-K) ac-
tion, and is thought to mediate many metabolic, mitogenic, andantiapoptotic effects of insulin, IGF-1, IL-3, and other growth fac-tors and cytokines (1–3). Forexample, PKB has been implicatedin insulin-stimulated translocation of GLUT4 transporter andglucose transport in rat adipocytes (4, 5), insulin stimulation ofglycogen synthesis (6, 7), meiotic maturation of frog oocytes(8), and antiapoptotic actions of IGF-1 (9) and IL-3 (10, 11).
There is considerable interest in identifying downstream targetsand effectors of PKB action. Glycogen synthase kinase was thefirst physiological substrate identified (12). Cardiac 6-phospho-fructo-2 kinase, a critical enzyme in the regulation of glycolysis,was also reported to be phosphorylated and activated by PKBin vitro (13). BAD, a member of the Bcl-2 family that promotescell death, is phosphorylated in intact cells and in vitro by PKB.IL-3-induced phosphorylation of BAD is thought to result in itsfunctional inactivation by promoting its binding to 14-3-3 (10).PKB is also thought to promote cell survival by phosphorylatingForkhead family transcription factors, resulting in their associationwith 14-3-3 proteins, retention in the cytosol, and functional inac-tivation (14). In endothelial cells, PKB increases NO productionand release by phosphorylation and activation of endothelial NOsynthase (15).
More recently, in oocytes (8), rat adipocytes (16, 17), and3T3-L1 adipocytes (18), PDE3 isoforms have been suggested tobe downstream targets and effectors of PKB actions. In adipo-cytes, insulin-induced activation of PDE3B, via P13-K- andPKB-dependent signals, is a critical component in the antilipo-lytic action of insulin (19 –21). Activation of PDE3 by PKB isthought to be involved in the resumption of meiosis in quiescentfrog oocytes (8).
FDCP2 promyeloid cells have been utilized to study the mito-genic actions of insulin and IL-4, and to establish the role of in-sulin receptor-substrate proteins (IRS-1, IRS-2) in these processes(22). In these cells, activation of insulin, IGF-1, or IL-4 receptorsgenerates some common signals (23, 24). FDCP2 cells requireIL-3 for growth; IL-4 cannot replace IL-3, but enhances its effects(25). In FDCP2 cells, IL-3 and IL-4 interact with different types of
*Pulmonary/Critical Care Medicine Branch and†Hypertension-Endocrine Branch,National Heart, Lung, and Blood Institute, and§Laboratory of Cellular and MolecularBiology, National Cancer Institute, National Institutes of Health, Bethesda, MD20892; and‡Section for Molecular Signaling, Department of Cell and MolecularBiology, Lund University, Lund, Sweden
Received for publication June 17, 1999. Accepted for publication February 11, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 E.D. was supported by Medical Research Council Grant (Sweden) 3362.2 Address correspondence and reprint requests to Dr. Faiyaz Ahmad, PCCMB, Na-tional Heart, Lung, and Blood Institute, Building 10, Room 5N-307, 10 Center Drive,MSC 1434, Bethesda, MD 20892-1434. E-mail address: [email protected] Abbreviations used in this paper: PKB, protein kinase B; 8-Br-cAMP, 8-bromo-cAMP; CT, carboxyl terminus; GSK-3, glycogen synthase kinase-3; IGF-1, insulin-like growth factor-1; IRS, insulin receptor substrate; MAP, mitogen-activated protein;MBP, myelin basic protein; ERK, extracellular signal-related kinase; MEK, MAP/ERK kinase; MPDE3B, mouse PDE3B; NT, amino terminus; PDE, cyclic nucleotidephosphodiesterase; PDK, phosphoinositide-dependent kinase; PI3-K, phosphatidyl-inositol 3-kinase; PKA, cAMP-dependent protein kinase A; RPDE3B, rat PDE3B; wt,wild-type.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00

receptors and induce tyrosine phosphorylation of differentproteins. For example, IL-4, not IL-3, stimulates tyrosine phos-phorylation of IRS-2 (25, 26). FDCP2 cells also contain PDE4 aswell as PDE3B, and recently we found that in these cells, IL-4activates both PDE3B and PDE4, whereas IL-3 and PMA selec-tively activate PDE4 (26). IL-4, IL-3, and PMA activate PDE4 viamitogen-activated protein kinase (MAP kinase)-dependent signals,whereas, similar to insulin in rat adipocytes (16, 17, 21), IL-4activates PDE3B via MAP kinase-independent signals (26).
The goal of this study was to evaluate the role of PKB in theactivation of PDE3B by IGF-1 in FDCP2 cells and to determinewhether PDE3B was a downstream effector and regulator of somePKB actions. For this purpose, we generated permanently trans-fected FDCP2 cells that overexpress wt (F/B), constitutively active(F/B*), and three kinase-inactive (F/Ba2, F/Bb2, F/Bc2) forms ofPKB (5, 27). In the absence of IGF-1, activities of PKB, PDE3B,and PDE4 were similar in nontransfected FDCP2 cells, cells trans-fected with control vector (F/V), and F/B cells. In F/V cells, IGF-1increased PKB, PDE3B, and PDE4 activities;2-fold. In F/B cells,however, IGF-1, in a wortmannin-sensitive manner, increasedPKB ;10-fold and PDE3B activity (;4-fold) and phosphoryla-tion, but increased PDE4 only;2-fold (as in F/V cells). In F/B*cells, in the absence of IGF-1, PKB was increased;10-fold andPDE3B was activated (3–4-fold) and phosphorylated. Wortman-nin inhibited activation of PKB and phosphorylation/activation ofPDE3B. In F/B* cells, IGF-1 had little further effect on PKB andactivation/phosphorylation of PDE3B, and increased PDE4 to thesame extent as in F/V cells. In cells transfected with kinase-inac-tive forms of PKB, IGF-1 activated PDE4, not PDE3B, suggestingthat kinase-inactive PKB functioned as a dominant negative withrespect to activation of PDE3B. Activated PKB phosphorylatedand activated rPDE3B in vitro. Proliferation of F/B* cells andIGF-1-induced phosphorylation of BAD in F/B cells was moresensitive to inhibition by PDE3 inhibitors than the PDE4 inhibitorrolipram. These and other results are consistent with the idea thatin FDCP2 cells, PDE3B, not PDE4, is a target, if not substrate, ofactivated PKB and a downstream effector of some actions medi-ated by PKB.
Materials and MethodsTransferrin and selenium were purchased from Collaborative Research(Boston, MA); [3H]cAMP, from New England Nuclear (Boston, MA); anti-human PKB-CT, anti-mouse BAD-NT, and anti-rat p70S6 kinase poly-clonal Abs, from Santa Cruz Biotechnology (Santa Cruz, CA); anti-humanextracellular signal-related kinase (ERK1) (MAP kinase) mAb, from Sig-nal Transduction Laboratories (Lexington, KY); anti-rat ERK1-CT poly-clonal (MAP kinase) Ab, from Upstate Biotechnology (Lake Placid, NY);the PI3-K inhibitor wortmannin, from Biomol Research Laboratories (Ply-mouth Meeting, PA); MEK-1 inhibitor PD98059, from New England Bio-labs (Beverly, MA); phosphorus-32 as H3
32PO4 (1000 Ci/mmol) and[g-32P]ATP (3000 Ci/mmol), from ICN Radiochemicals (Costa Mesa,CA); and [3H]thymidine (22 Ci/mmol), from Amersham (ArlingtonHeights, IL). K9 (KKRNRTLTK) and Crosstide (GRPRTSSFAEG) pep-tides were synthesized at the Biomolecular Unit, Lund University (Lund,Sweden). Prestained molecular size markers were from Bio-Rad (Rich-mond, CA). Other materials were obtained as indicated and were of thehighest grade available.
Cell culture and incubations
The murine, IL-3-dependent hemopoietic cell line, FDCP2, was propagatedand maintained in RPMI 1640 medium (Life Technologies, Grand Island,NY) supplemented with 10% FBS, 5% WEHI-3B (American Type CultureCollection, Rockville, MD)-conditioned medium (which contains IL-3),and 2 mM glutamine (25). For most experiments, exponentially growingFDCP2 cells (2–53 105 cells/ml) were collected, centrifuged (5 min,12003 g), washed twice, and cultured overnight in RPMI 1640/10% FBS.Immediately before experiments, cells were washed twice, suspended inserum-free RPMI 1640 medium containing TSB (5mg/ml transferrin, 10
mM selenium, and 1 mg/ml BSA), and incubated (3 ml cells/well,;7 3105 cells/ml) in six-well Costar plates (Cambridge, MA) (25). After 2–3 hat 37°C, IGF-1 or cytokines such as IL-3 were added as indicated. Finally,cells were harvested by centrifugation (12003 g, 5 min), and suspendedand homogenized (10–15 strokes in a Dounce homogenizer (Kontes In-struments, Vineland, NJ)) in lysis buffer A (20 mM Tris, pH 7.5, 150 mMNaCl, 1 mM MgCl2, 1% Nonidet P-40 detergent, 1 mM EDTA, 10 mMNaF, 10 mM sodium pyrophosphate, 1 mM Na3V04, 1 mM PMSF (Sigma,St. Louis, MO), 10mg/ml each of aprotinin, pepstatin, and leupeptin(Boehringer Mannheim, Indianapolis, IN) and 1 mM benzamidine (Sigma),and kept for 30 min at 4°C. Protein was measured using the Bradford assay(Bio-Rad, Richmond, CA) with BSA as standard.
Expression of PKB in FDCP2 cells
The following plasmid vector constructs (4, 27) were transformed intoEscherichia coli, amplified, and purified.pCIS2 (F/V). A vector that generates high levels of expression (27) wasused as parent vector for subsequent constructions.Akt-WT (F/B). A 1.4-kb XbaI/BamHI fragment containing the cDNAfor mouse Akt-1 (28) was blunt ended and ligated in the sense orientationinto theHpaI site in the multiple cloning region of pCIS2.Constitutively active Akt-myrA (F/B*). A BglII/BamHI fragmentcontaining the cDNA for mouse Akt-1 with a myristoylation sequencefrom pp60 c-src (29) fused in-frame with the N terminus of Akt was bluntended and ligated in the sense orientation into theHpaI site in the multiplecloning region of pCIS2.Kinase-inactive Akt (F/B2). A point mutant of Akt-WT with a sub-stitution of alanine for lysine at position 179 in the canonical ATP bindingsite (F/Ba2) was constructed using the mutagenic oligonucleotide-1(59-GC TAC TAT GCC ATG GCG-ATG CTC AAG AAG-G-39) and theMORPH site-specific plasmid DNA mutagenesis kit, according to the man-ufacturer’s instructions (5 Prime3 3 Prime, Boulder, CO). The mutationintroduced aNcoI site and was confirmed by direct sequencing. Substitu-tions of alanine for threonine at position 308 and alanine for serineat position 473 (phosphorylation sites in the regulatory region of Akt)(F/Bb2) were constructed using oligonucleotide-2 (59-CCA CTA TGAAGG CAT TTT GCG GAA CGC CGG-39) and oligonucleotide-3 (59-TTCCCC CAG TTC GCC TAC TCG GCC AGT GGC ACA-39). In addition tointroducing the amino acid changes noted above, the mutagenic oligonu-cleotides also introduced silent mutations that destroyed anXmnI siteand created a newBglI site. The presence of the correct mutations wasconfirmed by direct sequencing. Another kinase-inactive mutant AktAAA(F/Bc2) was created by substitution of alanine for lysine at position 179,alanine for threonine at position 308, and alanine for serine at position 473,as described above.
Expression vectors for the various constructs described above and aplasmid containing the neomycin resistance gene (;20 mg total DNA)were introduced into FDCP2 cells by electroporation; transformants (F/V,F/B, F/B*, and F/Ba,b,c2) were selected by growing cells in 24-well cul-ture plates in medium with G418 (750 mg/L), as previously described (22,24, 30). Six different colonies of each construct were isolated; experimentswere conducted with at least two independent colonies. FDCP2 cells ex-pressing similar amounts of wt (F/B), constitutively active (F/B*), andkinase-inactive PKB (F/Ba2, F/Bb2, F/Bc2) were selected by analyzinglysates (30–40mg protein) from equivalent numbers of cells by SDS-PAGE (Novex, San Diego, CA), and immunoblotting with PKB-CT Ab(Fig. 1). PKB activity was also determined in these lysates.
Incorporation of [3H]thymidine into DNA in FDCP2 cells
Thymidine incorporation was assayed as previously described (22). Cellsin log phase growth were washed twice in PBS and suspended in growthmedium; 33 104 cells were added to each of 24 wells (final volume, 1 ml)containing RPMI 1640 medium with or without conditioned medium con-taining IL-3 (5% WEHI) and the indicated concentrations of PDE inhibi-tors, 8-Br-cAMP, or PGE1. DMSO or ethanol at a final concentration of0.1% or less was added to appropriate wells as vehicle controls. Cells weregrown for 48 h at 37°C. [3H]Thymidine (22 Ci/mmol; Amersham) wasadded to a final concentration of 0.5mCi/ml, and incubation was continuedfor 2 h. Cells were harvested (Skatron Cell Harvester) and lysed on glassmicrofiber filters; unincorporated nucleotide was removed by repeatedwashing with water. Filters were dried and counted in scintillation fluid.Data are presented as mean6 SEM of cpm in samples from triplicateincubations. For cell survival assays, IL-3-depleted cells were seeded at adensity of 33 104/ml in RPMI 1640 containing 10% FBS. In some ex-periments, the ratio of viable to dead cells was determined by trypan blueexclusion.
4679The Journal of Immunology

cAMP PDE assay
Samples of cell lysates (usually 100ml) were incubated for 10 min at 30°Cin a total volume of 0.3 ml containing 50 mM HEPES, pH 7.5, 8.3 mMMgCl2, 0.1 mM EDTA, and 0.1mM [3H]cAMP (25–35,000 cpm) as sub-strate (31). After dephosphorylation of [3H]5-AMP to [3H]adenosine withCrotalus atroxvenom (Sigma), the product was separated from substrateby ion-exchange chromatography (QAE-Sephadex; Pharmacia, Piscat-away, NJ) and quantified by scintillation counting. Lysates were diluted sothat hydrolysis of substrate was usually less than 20%. PDE3 activity is thatfraction of total activity inhibited by 0.3mM cilostamide, a specific PDE3inhibitor, and PDE4 activity, that inhibited by 0.5mM rolipram, a specificPDE4 inhibitor (32). Inhibitor vehicle (DMSO), added in equal quantitiesto samples without inhibitor, did not alter PDE activities.
Immunoprecipitation and immunoblotting
For most experiments, portions of FDCP2 lysates (2–3 mg protein) werefirst cleared by incubation with 1mg of preimmune mouse IgG for 1 h atroom temperature, and then with 60ml of protein G-Sepharose (PharmaciaBiotech, Uppsala, Sweden) for 30 min before centrifugation (28003 g,4°C, 5 min). Cleared cell lysates were incubated with specified Abs for 2 hat room temperature, followed by incubation with fresh protein G-Sepha-rose for 30 min before centrifugation (28003 g, 4°C, 5 min). Immuno-precipitates were washed three times with lysis buffer A containing 0.1%Nonidet P-40, boiled in Laemmli buffer (Bio-Rad, Richmond, CA) (33),and subjected to SDS-PAGE. Proteins were transferred to nitrocellulosemembranes in Tris-glycine buffer (25 mM Tris-base and 192 mM glycineat pH 8.3), containing 20% methanol. Membranes were incubated in blot-ting buffer (150 mM NaCl, 0.05% (v/v) Nonidet P-40, 0.01% NaN3, and 10mM Tris, pH 7.4), containing BSA (50 mg/ml) and OVA (10 mg/ml), for1 h at room temperature with rocking, and then for an additional 2 h in thesame solution containing the appropriate Abs. Membranes were washedthree times (10 min each) in blotting buffer without Abs, followed byincubation with125I-labeled protein A or protein G (2mCi) (AmershamLife Sciences, Arlington Heights, IL) for 1 h atroom temperature, followedby three washes (10 min each) with blotting buffer. Immunoreactive pro-teins were visualized by PhosphorImager analysis (Molecular Dynamics,Sunnyvale, CA).
For some immunoblots, instead of125I-labeled protein A, HRP-labeledsecond Ab (mouse, rat, rabbit, or goat) and enhanced chemiluminescence(ECL) reagents 1 and 2 (Amersham) were used; immunoreactive proteinswere visualized by exposing nitrocellulose filters to Bio-max Kodak filmand developed in a Standard Medical Imaging (Columbia, MD) developer.
Phosphorylation of PDE3B and BAD
PDE3B or BAD proteins were identified in FDCP2 cell lysates by immu-noprecipitation/immunoblotting with anti-PDE3B-NT or anti-PDE3B-CTAbs (rabbit Abs raised against peptides derived from deduced sequences inthe N-terminal (RKDERERDTPAMRSPPP, aa 2–18) and C-terminal(NKLQVDNASLPQADE, aa 1078–1092) portions of rat (R)PDE3B) (34),or anti-BAD-NT Ab, respectively. Immediately before experiments,FDCP2 cells transfected with wt (F/B) or constitutively active (F/B*) PKB,or vector alone (F/V) were washed twice, suspended in serum-free RPMI1640 medium containing TSB, and incubated (3 ml cells,;7 3 105 cells/
ml) in six-well Costar plates (25) for 1–2 h at 37°C. Cells were then in-cubated in RPMI 1640 medium containing 300mM KH2PO4 and 1% BSAwith or without 32PO4 (40 mCi/ml) for 90–100 min at 37°C, and finallywith or without 10 nM IGF-1 for 10 min. PDE3B and BAD were immu-noprecipitated from cleared lysates with anti-PDE3B-NT or anti-BAD-NTAbs, respectively. SDS-PAGE was then conducted, and dried gels werescanned on a PhosphorImager (Molecular Dynamics), as described above.Immunoprecipitates from cells incubated without32PO4 were preparedsimilarly and immunoblotted with anti-PDE3B or anti-BAD Abs. PKBactivity was assessed by phosphorylation of Crosstide in PKBimmunoprecipitates.
PKB assays with Crosstide, K9 peptide, and histone 2Bsubstrates
Immunoprecipitation with anti-PKB-CT Ab (50ml/10 mg IgG) was con-ducted for 2 h atroom temperature or overnight at 4°C, as described above,and washed proteins were suspended in 30–40ml of reaction buffer (20mM HEPES, pH 7.4, 1 mM DTT, 10 mM MnCl2, 10 mM MgCl2, 5 mMATP, 10mCi [g-32P]ATP, 5mM cAMP protein kinase inhibitor) containingeither 13 mg of K9 peptide (KKRNRTLTK), 1 mg of Crosstide(GRPRTSSFAEG), or 2.5mg of histone-2B (Boehringer Mannheim) assubstrate (17, 35, 36). As recently described (26), after incubation for 15min at 30°C, assay reactions with K9 or Crosstide peptides were terminatedby addition of 10ml of 1% BSA, 1 mM ATP, pH 3, and 5ml of 30% TCA.For reaction blanks, the stopping solution was added to immunoprecipi-tated samples before assay buffer. Samples were centrifuged, and 25ml ofsupernatant was applied to phosphocellulose (P81; Whatman, Tewksbury,MA) paper (23 2-cm squares), which were washed three times (5 mineach) with phosphoric acid (7.5 mg/ml), and once with acetone (5 min)before radioassay of32P incorporated into substrate.
With histone 2B as substrate for PKB, reactions were terminated byaddition of Laemmli buffer and boiling. Proteins were separated by SDS-PAGE, the gel was dried, and32P-labeled histone 2B was detected andquantified by PhosphorImager (Molecular Dynamics), as recently de-scribed (26).
MAP kinase and p70S6 kinase assays
Activation of MAP kinase was assessed as recently described (26) by mea-suring phosphorylation of myelin basic protein (MBP) peptide in immu-noprecipitates prepared with anti-ERK1-CT Ab (21ml/10 mg IgG; UpstateBiotechnology). Samples of immunoprecipitates were incubated at 30°Cfor 15 min in 50 mM Tris (pH 7.5), 0.4 mM EGTA, 0.4 mM Na3VO4, 25mM MgCl2, 150mM ATP, 10mCi [g-32P]ATP (10 Ci/mmol), 5mM cAMPprotein kinase inhibitor (Calbiochem, San Diego, CA), and 5mM MBPpeptide (APRTPGGRR) substrate (Upstate Biotechnology), in final vol-ume of 50ml. Reactions were terminated, and phosphocellulose squareswere washed and analyzed, as described for PKB assays.
p70S6 kinase was detected in lysates from F/V and F/B* cells afterseparation of proteins (;30 mg) by SDS-PAGE and immunoblotting withpolyclonal p70S6 kinase Ab. Activated kinase exhibited a lower electro-phoretic mobility than inactive kinase.
Table I. Effects of IGF-1 on PDE3 and PDE4 activities in FDCP2 cellsa
Cells
PDE Activity (pmol cAMP hydrolyzed/min/mg)
2IGF-1 1IGF-1
Total PDE PDE3 PDE4 Total PDE PDE3 PDE4
F (n 5 8) 12.56 1.0 5.76 0.7 6.26 1.1 21.06 2.3 11.06 1.7 10.86 0.5F/V (n 5 9) 13.46 1.3 5.96 0.9 6.66 1.1 24.66 4.7b 12.96 2.9b 11.86 2.5b
F/B (n 5 8) 146 2.6d 6.16 1.4d 7.16 1.5d 4.16 8b 25.56 4.4b 12.76 2.4b
F/Ba2 (n 5 3) 13.86 0.8d 5.96 0.6d 6.16 0.9d 18.96 0.4b 6.26 1.3d 10.96 0.6c
F/Bb2 (n 5 3) 13.46 1.3d 5.96 0.5d 6.56 0.5d 22.16 1.5b 6.06 1.0d 13.26 0.7b
F/Bc2 (n 5 3) 13.46 1.0d 6.06 0.9d 6.66 0.4d 21.06 1.5b 5.86 0.3d 12.36 0.9b
F/B* (n 5 10) 32.56 5.2b 216 3.7b 8.36 2.1c 32.56 1.9b 21.16 1.9b 10.96 1.0c
a Cells were incubated without or with 10 nM IGF-1 for 10 min before total PDE and PDE3 and PDE4 activities in cell lysates were assayed as described inMaterials andMethods.PDE3 activity represents cAMP hydrolysis inhibited by cilostamide (a selective PDE3 inhibitor); PDE4, that activity inhibited by rolipram (a selective PDE4 inhibitor).PDE activities are reported as pmol cAMP hydrolyzed/min/mg (mean6 SEM) from the indicated number (n) of experiments and include data from Figs. 2–4.
b p , 0.001;c p , 0.02;d p . 0.05, compared to activities in F/V cells. PDE activities in nontransfected FDCP2(F) cells were not significantly different from those in F/Vcells.
4680 PDE3B IS A TARGET AND EFFECTOR OF PKB ACTION

Results and DiscussionPDE3 and PDE4 enzymes exhibit high affinity for cAMP (31, 32)and, as shown in Table I, with 0.1mM [3H]cAMP as substrate,account for most of the cAMP hydrolytic activity in FDCP2 celllysates (26). Two PDE3 isoforms, PDE3A and PDE3B, have beenidentified (16); PDE3B mRNA was amplified by RT-PCR fromFDCP2 cell RNA (31) (data not shown). PDE3B protein in FDCP2lysates was identified by immunoprecipitation/immunoblottingwith anti-PDE3B-NT Ab (34) (data not shown).
Characterization of FDCP2 cells transfected with PKBconstructs
Several independently isolated lines were generated from a singletransfection of FDCP2 cells with constructs that expressed vectoralone (F/V) and wt (F/B), constitutively active (F/B*), and threekinase-inactive (F/B a2,b2,c2) forms of Akt/PKB (Fig. 1). Im-munoreactive PKB was much higher in the cells transfected withPKB constructs than in F/V cells (Fig. 1,A andD). In the absenceof IGF-1, PKB activity was similar in F/V and F/B cells, but mark-edly higher (;10-fold) in F/B* cells, and was affected onlyslightly by incubation of these latter cells with IGF-1 (Fig. 1,B andE). Incubation with IGF-1 resulted in;2–3-fold activation of PKBin F/V cells, but;10-fold in F/B cells, to levels comparable withthose in F/B* cells (Fig. 1B). IGF-1 had little effect on PKB ac-tivity in F/Ba2, F/Bb2, F/Bc2 cells, whether assessed as phos-phorylation of histone 2B (data not shown) or of Crosstide by PKBimmunoprecipitates (Fig. 1E).
Role of PKB in IGF-I-induced activation of PDE3B in FDCP2cells
In the absence of IGF-1, activities of PDE3B and PDE4 weresimilar in nontransfected FDCP2 cells, F/V cells, F/B cells, andF/Ba2, F/Bb2, and F/Bc2 cells (Table I). Incubation for 10 minwith 10 nM IGF-1 increased PDE3B and PDE4 activities (#2-fold) in nontransfected FDCP2 cells as well as in F/V cells. As wehave recently reported, IL-4 also increased PDE3B and PDE4 ac-tivities # 2-fold in nontransfected FDCP2 cells (26). In F/B cells,however, IGF-1 increased PDE3B by;4-fold, whereas the in-crease in PDE4 activity was similar to that in F/V cells (#2-fold).In F/Ba2, F/Bb2, and F/Bc2 cells, IGF-1 activated PDE4, notPDE3B, suggesting that kinase-inactive PKB functioned as a dom-
inant negative with regard to activation of PKB (Fig. 1E) andPDE3B (Table I). In the absence of IGF-1, in FDCP2 cells trans-fected with constitutively active PKB (F/B* cells), PDE3B activitywas increased;4-fold and similar to that in F/B cells exposed toIGF-1. PDE3B activity was not increased further by IGF-1, whichincreased PDE4 in F/B* cells to almost the same extent as in F/Bor F/V cells (Table I). These results indicate that PKB is an up-stream regulator of PDE3B, not PDE4, in FDPC2 cells.
As shown in Fig. 1,C andF, MAP kinase was not increased incells transfected with PKB/Akt constructs. IGF-1 activated MAPkinase;2-fold in all cells, including F/B* cells, indicating that,even in cells overexpressing constitutively activated PKB, MAPkinase was regulated appropriately by IGF-1. As shown in Fig. 2,the MEK-1 inhibitor PD98059 did not alter PDE3B (E) or PKB (F)activities in F/B* cells, consistent with the observations that MAPkinase was not elevated in F/B* cells (Figs. 1C and 2D). In non-transfected FDCP2 cells, IL-4-induced activation of PDE3B wasindependent of MAP kinase or p70S6 kinase activity, whereasIL-4-induced activation of PDE4 was MAP kinase dependent andblocked by PD98059 (26). As also shown in Fig. 2A, in F/B* cells(as in other cells transfected with constitutively active PKB (8, 37,38)), p70S6 kinase was activated, evidenced by the shift in itsmobility on SDS-PAGE. Rapamycin blocked activation of p70S6kinase without affecting PKB or PDE3B activity, but did reducePDE4 activity, which was slightly increased in F/B* cells (Table I;Fig. 2B). Although this slight increase in PDE4 activity was notobserved in all experiments with F/B* cells, these results are con-sistent with the findings of Mackenzie et al. (39), which indicatedthat, in 3T3-F442A cells, PDE4A was activated by p70S6 kinase.In untransfected FDCP2 cells, however, effects of IL-3 and IL-4 onPDE4 were blocked by PD98059, not rapamycin (26).
Role of PKB in phosphorylation/activation of PDE3B in FDCP2cells
We next evaluated the effects of IGF-1 and PKB on phosphoryla-tion/activation of PDE3B in32P-labeled FDCP2 cells. As shown inFig. 3, in F/B cells overexpressing wt PKB, neither PDE3B (C) norPKB (D) was activated in the absence of IGF-1, and PDE3B wasnot phosphorylated (A). As shown in Fig. 3, however, after incu-bation of F/B cells with 1 or 10 nM IGF-1 for 10 min, phosphor-ylation of PDE3B was markedly increased (A), as was PDE3B
FIGURE 1. Effects of IGF-1 on PKB and MAP ki-nase activities in transfected FDCP2 cells. FDCP2cells, transfected with empty vector (F/V) or wt PKB(F/B), constitutively active (F/B*), and kinase-inactive(F/Ba2, F/Bb2, F/Bc2) forms of PKB, were incubatedwith or without 10 nM IGF-1 for 10 min.A and D,Proteins (;30mg) in lysates of control and transfectedcells were separated by SDS-PAGE and immunoblot-ted with anti-PKB-CT Ab. PKB activity was assessedby phosphorylation of (B and E) Crosstide (meancpm 6 SEM, n 5 3) or histone 2B (not shown) withPKB immunoprecipitates from lysates (;2.5 mg pro-tein) of control and IGF-1-stimulated cells.C andF,MAP kinase activity was assessed by phosphorylationof MBP peptide (mean cpm6 SEM,n 5 3) with MAPkinase immunoprecipitates from cell lysates (;2.5 mgprotein). In this and all other figures, when not present,SEMs were too small to be displayed graphically.
4681The Journal of Immunology

activity (3–4-fold) (C) and PKB activity (;6–10-fold) (D). In F/Vcells, IGF-1 increased PDE3B and PDE4 activities;2-fold (Fig.3C). In F/B cells, in contrast to the;4-fold activation of PDE3B,IGF-1 increased PDE4 activity only;2-fold, as in F/V cells (Fig.3C). As shown in Fig. 4, in FDCP2 cells expressing constitutivelyactive PKB (F/B* cells), in the absence of IGF-1, PKB activitywas;10-fold greater than that in F/V cells (Fig. 4D). PDE3B wasphosphorylated (Fig. 4A) and activated (;3- to 4-fold) (Fig. 4C)without change in the amount of immunoreactive PDE3B (Fig.4B). In F/B* cells, IGF-1 had little additional effect on activationof PKB (see Fig. 1) or on phosphorylation/activation of PDE3B(data not shown, Table I).
As also shown in Fig. 3, wortmannin, in a concentration-depen-dent manner, blocked the effects of IGF-1 on PKB (Fig. 3D), phos-phorylation/activation of PDE3B (Fig. 3,A andC), and activationof PDE4 (Fig. 3C). As shown in Fig. 4, in F/B* cells, wortmannin,in a concentration-dependent manner, almost completely blocked(IC50, ;25 nM) the elevated phosphorylation/activation of PDE3B(Fig. 4, A andC) and PKB activity (Fig. 4D).
In nontransfected FDCP2 cells, wortmannin also blocked theactivation of PDE3B and PDE4 by IL-4 (26). Beyond PI3-K, how-ever, IL-4-effects on PDE3B and PDE4 diverged. IL-4-inducedactivation of PDE3B was apparently independent of MAP kinaseand p70S6 kinase and not blocked by PD98059 (MEK-1 inhibitor)or rapamycin (p70S6 kinase inhibitor). Activation of PDE4 byIL-4 was, however, dependent on MAP kinase and blocked byPD98059 (26). In FDCP2 cells expressing wt MEK, IL-3 activatedPDE4, not PDE3B; in FDCP2 cells expressing constitutively ac-tivated MEK, PDE4, not PDE3B, activity was increased in theabsence of IL-3 (26).
Because phosphoinositide-dependent kinases (PDK) are thoughtto phosphorylate and activate PKB directly (1–3), the PI3-K in-hibitor wortmannin, as expected, blocked IGF-1-induced activa-tion of PKB and phosphorylation/activation of PDE3B in F/B cells(Fig. 3). The mutant PKB expressed in F/B* cells, however, is notactivated intrinsically by alteration at the catalytic site, rather itcontains an N-terminal myristoylation sequence that targets it tomembranes, where it is presumably continuously or constitutivelyactivated in response to basal production of phosphoinositides withactivation of PDK isoforms or to a specific pool of active PI3-K.In F/B* cells (Fig. 4), wortmannin could inhibit constitutively ac-tivated PKB by blocking production of phosphoinositides involvedin PKB association with membranes and/or in the activation ofspecific PDK isoforms. Whether activated PKB contributes tofeedback activation of PI3-K or sensitization of PDK to phosphoi-nositides is not known, but other workers have also reported thatwortmannin or LY294002 inhibits constitutively activated PKB(activated by targeting to membranes) in 3T3-L1 adipocytes (38)or HEK293 cells (37).
Taken together, our previously reported data (26) and that pre-sented in this report support the idea that in FDCP2 cells, IGF-1activates PDE3B and PDE4 via PI3-K-dependent signals. Down-stream of PI3-K, PDE3B is phosphorylated/activated by activatedPKB, whereas PDE4 is activated via MEK/MAP kinase signals.
Effects of PKB on phosphorylation/activation of PDE3B in vitro
Because effects of IGF-1 and wortmannin on phosphorylation/ac-tivation of PD3B in F/B or F/B* cells do not prove that PDE3B isa direct substrate for PKB, we tested the ability of PKB to directly
FIGURE 2. Effect of rapamycin and PD98059 onPDE, PKB, MAP kinase, and p70S6K activities inF/V and F/B* cells. FDCP2 cells, transfected withvector alone (F/V) or constitutively active PKB (F/B*cells), were incubated with or without 20 nM rapamy-cin (rap) (A–C) or 10mM PD98059 (PD) (D–F) for 30min. A, Lysates (;30 mg protein) from F/V and F/B*cells were separated on SDS-PAGE and immunoblot-ted with anti-p70S6K Ab.D, MAP kinase activity(mean cpm6 1⁄2 range,n 5 2) in F/V and F/B* cellswas assessed by phosphorylation of MBP peptide withMAP kinase immunoprecipitates from cell lysates(;2.5 mg protein).B andE, Portions of lysates fromcells incubated with or without rapamycin (B) orPD98059 (E) were assayed for total PDE (M) andPDE3 (u) and PDE4 (f) activities (mean cpm6 1⁄2range,n 5 2). C andF, Samples of cell lysates (;2.5mg protein) were immunoprecipitated with anti-PKB-CT Ab; PKB activity (mean cpm6 1⁄2 range,n 5 2) was assessed by phosphorylation of Crosstideby PKB immunoprecipitates.
4682 PDE3B IS A TARGET AND EFFECTOR OF PKB ACTION

phosphorylate and activate recombinant mouse (M)PDE3B. Pre-vious experiments have demonstrated that native RPDE3B wasphosphorylated on serine 302 in intact rat adipocytes (analogous toserine 296 in MPDE3B) incubated with insulin (16), and thatRPDE3B was also phosphorylated in vitro by partially purified ratadipocyte PKB (17) or rPKB (18). Available evidence suggeststhat phosphorylation is not required to maintain, but does increase,basal PDE activity. As shown in Fig. 5, activated PKB, in a time-and concentration-dependent manner, phosphorylated and in-creased the activity of wt rMPDE3B synthesized in Sf-9 insectcells. As shown in Fig. 6, activated PKB did not, however, phos-phorylate or increase the activity of M3BD604, a truncatedrMPDE3B from which the N-terminal 604 aa (including putativephosphorylation sites for PKB and PKA (16, 18)) were removed.The higher sp. act. of M3BD604 compared with that of wtMPDE3B may be related to higher levels of expression or accu-mulation of a larger proportion of active M3BD604 molecules. Itis also possible that the N-terminal portion of PDE3B contains anautoinhibitory domain, the constraints of which are released by
phosphorylation or removal of the N-terminal portion of the mol-ecule. Taken together, these data suggest that PKB not only has arole as an upstream regulator, but also directly acts on PDE3B andcould thereby be an important determinant in control of intracel-lular cyclic nucleotide concentrations.
Effects of PDE inhibitors and cAMP on BAD phosphorylationand thymidine incorporation in F/V and F/B* cells
Activated PKB has been reported to increase cell proliferation/survival (9, 11, 40), and to activate PDE3 and trigger meiosis inXenopusoocytes (8). PKB-catalyzed phosphorylation of BAD maybe involved in the antiapoptotic effects of IL-3 (10). As shown inFig. 7A, in 32P-labeled F/V and F/B cells, but not in F/Bc2 cells,IGF-1 increased phosphorylation of BAD, which was blocked byincubation of F/B cells with 8-Br-cAMP or the PDE3 inhibitorcilostamide (Fig. 7B). Rolipram, a specific PDE4 inhibitor, wasmuch less effective in inhibiting IGF-1-stimulated phosphorylationof BAD (Fig. 7B). These agents did not block IGF-1-induced ac-tivation of PKB (assessed by phosphorylation of Crosstide peptideby immunoprecipitated PKB) (data not shown).
FIGURE 3. Effects of wortmannin on IGF-1-induced PDE3B phosphor-ylation, and PDE and PKB activities in F/V and F/B cells. FDCP2 cells,transfected with vector alone (F/V) or wt PKB (F/B), were incubated with-out or with the indicated concentrations of wortmannin for 1 h in RPMI1640 medium containing 300mM KH2PO4 and TSB, and then with orwithout added32PO4 (40 mCi/ml) for 90 min before addition of the indi-cated concentrations of IGF-1 for 10 min.A, Samples (;2.5 mg protein) oflysates of cells incubated with32PO4 were immunoprecipitated with anti-PDE3B-NT Ab and subjected to SDS-PAGE, and PDE3B bands were vi-sualized by scanning the dried gel on a PhosphorImager (Molecular Dy-namics). Lysates (;2.5 mg protein) of cells incubated without32PO4 wereimmunoprecipitated and immunoblotted with anti-PDE3B Ab (B), or im-munoprecipitated with anti-PKB-CT Ab (D) for assay of PKB activity byphosphorylation of Crosstide (mean cpm6 SEM, n 5 3). C, Samples oflysates of cells incubated without32PO4 were assayed for total PDE (M)and PDE3 (u) and PDE4 (f) activities (mean6 SEM, n 5 3).
FIGURE 4. Effects of wortmannin on PDE3B phosphorylation, andPDE and PKB activities in transfected F/V and F/B* cells. FDCP2 cells,transfected with vector alone (F/V) or constitutively active PKB (F/B*),were incubated without or with the indicated concentrations of wortmanninfor 1 h in RPMI 1640 medium containing 300mM KH2PO4 and TSB, andthen with or without added32PO4 (40mCi/ml) for 90 min at 37°C. Sampleswere prepared as described in the legend to Fig. 3.A, Immunoprecipitated32P-labeled PDE3B.B, PDE3B immunoprecipitated and immunoblottedwith anti-PDE3B-NT Ab.C, Total PDE (M) and PDE3 (u) and PDE4 (f)activities (pmol/min/mg, mean6 SEM (n 5 3)). D, PKB activity (meancpm 6 SEM, n 5 3).
4683The Journal of Immunology
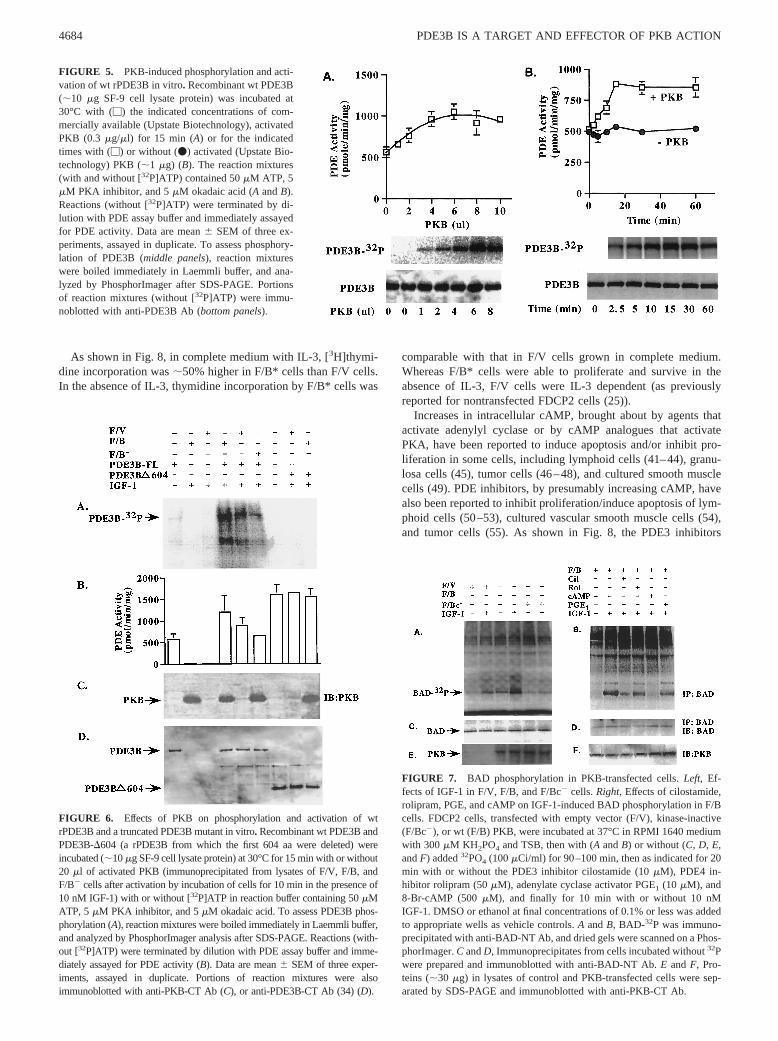
As shown in Fig. 8, in complete medium with IL-3, [3H]thymi-dine incorporation was;50% higher in F/B* cells than F/V cells.In the absence of IL-3, thymidine incorporation by F/B* cells was
comparable with that in F/V cells grown in complete medium.Whereas F/B* cells were able to proliferate and survive in theabsence of IL-3, F/V cells were IL-3 dependent (as previouslyreported for nontransfected FDCP2 cells (25)).
Increases in intracellular cAMP, brought about by agents thatactivate adenylyl cyclase or by cAMP analogues that activatePKA, have been reported to induce apoptosis and/or inhibit pro-liferation in some cells, including lymphoid cells (41–44), granu-losa cells (45), tumor cells (46–48), and cultured smooth musclecells (49). PDE inhibitors, by presumably increasing cAMP, havealso been reported to inhibit proliferation/induce apoptosis of lym-phoid cells (50–53), cultured vascular smooth muscle cells (54),and tumor cells (55). As shown in Fig. 8, the PDE3 inhibitors
FIGURE 5. PKB-induced phosphorylation and acti-vation of wt rPDE3B in vitro. Recombinant wt PDE3B(;10 mg SF-9 cell lysate protein) was incubated at30°C with (M) the indicated concentrations of com-mercially available (Upstate Biotechnology), activatedPKB (0.3 mg/ml) for 15 min (A) or for the indicatedtimes with (M) or without (v) activated (Upstate Bio-technology) PKB (;1 mg) (B). The reaction mixtures(with and without [32P]ATP) contained 50mM ATP, 5mM PKA inhibitor, and 5mM okadaic acid (A andB).Reactions (without [32P]ATP) were terminated by di-lution with PDE assay buffer and immediately assayedfor PDE activity. Data are mean6 SEM of three ex-periments, assayed in duplicate. To assess phosphory-lation of PDE3B (middle panels), reaction mixtureswere boiled immediately in Laemmli buffer, and ana-lyzed by PhosphorImager after SDS-PAGE. Portionsof reaction mixtures (without [32P]ATP) were immu-noblotted with anti-PDE3B Ab (bottom panels).
FIGURE 6. Effects of PKB on phosphorylation and activation of wtrPDE3B and a truncated PDE3B mutant in vitro. Recombinant wt PDE3B andPDE3B-D604 (a rPDE3B from which the first 604 aa were deleted) wereincubated (;10mg SF-9 cell lysate protein) at 30°C for 15 min with or without20 ml of activated PKB (immunoprecipitated from lysates of F/V, F/B, andF/B2 cells after activation by incubation of cells for 10 min in the presence of10 nM IGF-1) with or without [32P]ATP in reaction buffer containing 50mMATP, 5 mM PKA inhibitor, and 5mM okadaic acid. To assess PDE3B phos-phorylation (A), reaction mixtures were boiled immediately in Laemmli buffer,and analyzed by PhosphorImager analysis after SDS-PAGE. Reactions (with-out [32P]ATP) were terminated by dilution with PDE assay buffer and imme-diately assayed for PDE activity (B). Data are mean6 SEM of three exper-iments, assayed in duplicate. Portions of reaction mixtures were alsoimmunoblotted with anti-PKB-CT Ab (C), or anti-PDE3B-CT Ab (34) (D).
FIGURE 7. BAD phosphorylation in PKB-transfected cells.Left, Ef-fects of IGF-1 in F/V, F/B, and F/Bc2 cells.Right, Effects of cilostamide,rolipram, PGE, and cAMP on IGF-1-induced BAD phosphorylation in F/Bcells. FDCP2 cells, transfected with empty vector (F/V), kinase-inactive(F/Bc2), or wt (F/B) PKB, were incubated at 37°C in RPMI 1640 mediumwith 300mM KH2PO4 and TSB, then with (A andB) or without (C, D, E,andF) added32PO4 (100mCi/ml) for 90–100 min, then as indicated for 20min with or without the PDE3 inhibitor cilostamide (10mM), PDE4 in-hibitor rolipram (50mM), adenylate cyclase activator PGE1 (10 mM), and8-Br-cAMP (500 mM), and finally for 10 min with or without 10 nMIGF-1. DMSO or ethanol at final concentrations of 0.1% or less was addedto appropriate wells as vehicle controls.A andB, BAD-32P was immuno-precipitated with anti-BAD-NT Ab, and dried gels were scanned on a Phos-phorImager.C andD, Immunoprecipitates from cells incubated without32Pwere prepared and immunoblotted with anti-BAD-NT Ab.E andF, Pro-teins (;30 mg) in lysates of control and PKB-transfected cells were sep-arated by SDS-PAGE and immunoblotted with anti-PKB-CT Ab.
4684 PDE3B IS A TARGET AND EFFECTOR OF PKB ACTION
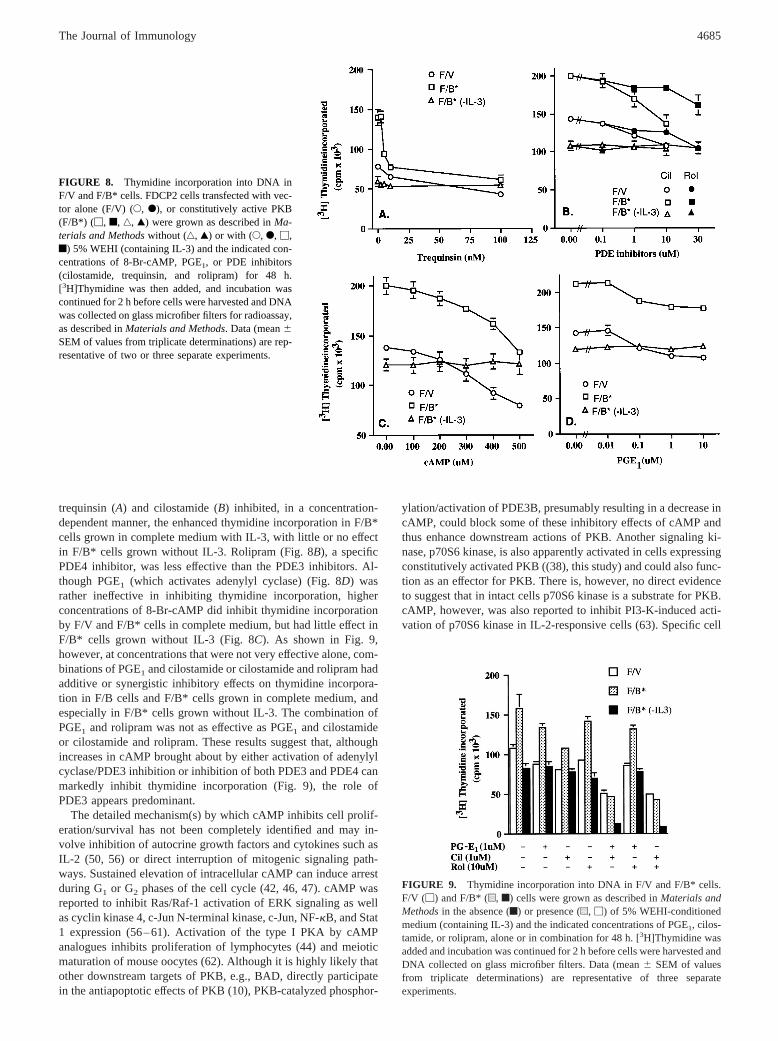
trequinsin (A) and cilostamide (B) inhibited, in a concentration-dependent manner, the enhanced thymidine incorporation in F/B*cells grown in complete medium with IL-3, with little or no effectin F/B* cells grown without IL-3. Rolipram (Fig. 8B), a specificPDE4 inhibitor, was less effective than the PDE3 inhibitors. Al-though PGE1 (which activates adenylyl cyclase) (Fig. 8D) wasrather ineffective in inhibiting thymidine incorporation, higherconcentrations of 8-Br-cAMP did inhibit thymidine incorporationby F/V and F/B* cells in complete medium, but had little effect inF/B* cells grown without IL-3 (Fig. 8C). As shown in Fig. 9,however, at concentrations that were not very effective alone, com-binations of PGE1 and cilostamide or cilostamide and rolipram hadadditive or synergistic inhibitory effects on thymidine incorpora-tion in F/B cells and F/B* cells grown in complete medium, andespecially in F/B* cells grown without IL-3. The combination ofPGE1 and rolipram was not as effective as PGE1 and cilostamideor cilostamide and rolipram. These results suggest that, althoughincreases in cAMP brought about by either activation of adenylylcyclase/PDE3 inhibition or inhibition of both PDE3 and PDE4 canmarkedly inhibit thymidine incorporation (Fig. 9), the role ofPDE3 appears predominant.
The detailed mechanism(s) by which cAMP inhibits cell prolif-eration/survival has not been completely identified and may in-volve inhibition of autocrine growth factors and cytokines such asIL-2 (50, 56) or direct interruption of mitogenic signaling path-ways. Sustained elevation of intracellular cAMP can induce arrestduring G1 or G2 phases of the cell cycle (42, 46, 47). cAMP wasreported to inhibit Ras/Raf-1 activation of ERK signaling as wellas cyclin kinase 4, c-Jun N-terminal kinase, c-Jun, NF-kB, and Stat1 expression (56–61). Activation of the type I PKA by cAMPanalogues inhibits proliferation of lymphocytes (44) and meioticmaturation of mouse oocytes (62). Although it is highly likely thatother downstream targets of PKB, e.g., BAD, directly participatein the antiapoptotic effects of PKB (10), PKB-catalyzed phosphor-
ylation/activation of PDE3B, presumably resulting in a decrease incAMP, could block some of these inhibitory effects of cAMP andthus enhance downstream actions of PKB. Another signaling ki-nase, p70S6 kinase, is also apparently activated in cells expressingconstitutively activated PKB ((38), this study) and could also func-tion as an effector for PKB. There is, however, no direct evidenceto suggest that in intact cells p70S6 kinase is a substrate for PKB.cAMP, however, was also reported to inhibit PI3-K-induced acti-vation of p70S6 kinase in IL-2-responsive cells (63). Specific cell
FIGURE 8. Thymidine incorporation into DNA inF/V and F/B* cells. FDCP2 cells transfected with vec-tor alone (F/V) (E, F), or constitutively active PKB(F/B*) (M, f, ‚, Œ) were grown as described inMa-terials and Methodswithout (‚, Œ) or with (E, F, M,f) 5% WEHI (containing IL-3) and the indicated con-centrations of 8-Br-cAMP, PGE1, or PDE inhibitors(cilostamide, trequinsin, and rolipram) for 48 h.[3H]Thymidine was then added, and incubation wascontinued for 2 h before cells were harvested and DNAwas collected on glass microfiber filters for radioassay,as described inMaterials and Methods. Data (mean6SEM of values from triplicate determinations) are rep-resentative of two or three separate experiments.
FIGURE 9. Thymidine incorporation into DNA in F/V and F/B* cells.F/V (M) and F/B* (u, f) cells were grown as described inMaterials andMethodsin the absence (f) or presence (u, M) of 5% WEHI-conditionedmedium (containing IL-3) and the indicated concentrations of PGE1, cilos-tamide, or rolipram, alone or in combination for 48 h. [3H]Thymidine wasadded and incubation was continued for 2 h before cells were harvested andDNA collected on glass microfiber filters. Data (mean6 SEM of valuesfrom triplicate determinations) are representative of three separateexperiments.
4685The Journal of Immunology

context may be important in the function of PDE3B and cAMP inthe action of PKB on cell proliferation/survival, since, in somecells, cAMP actually promotes growth (48, 64) and prevents ordelays apoptosis (65).
The proapoptotic protein BAD, a member of the Bcl-2 proteinfamily, promotes apoptosis by heterodimerization with the survivalfactors Bcl-2 or Bcl-xL. Bcl or Bcl-xL suppresses apoptosis, atleast in part, by preventing release of cytochromec from mito-chondria, and thus blocking activation of caspase proteases (66).Survival factors, including IGF-1, IL-3, nerve growth factor, andother cytokines, induce phosphorylation of BAD on critical serineresidues, leading to its interaction with and sequestration by 14-3-3proteins, its dissociation from the BAD/Bcl complex, and subse-quent translocation from mitochondria to the cytosol; free Bcl pro-teins act as suppressors of apoptosis (66).
Although it is clear that PKB-induced phosphorylation of BADcan play an important role in mechanisms that regulate cell sur-vival/proliferation (10, 67), in some cells other signaling pathwaysinvolving Raf-1 and PKA regulate phosphorylation of BAD (68),and PKB-independent pathways involving MEK and MAP kinasessuppress apoptosis (68–70). Furthermore, in lymphocytes, activa-tion of the TCR can induce apoptosis via Ca21-induced activationof the phosphatase calcineurin (PP2b) (71), resulting in dephos-phorylation of BAD, its heterodimerization with Bcl proteins, andinitiation of apoptosis (72).
Recently, Minishall et al. (73) demonstrated that IGF-1 and IL-4increased expression of Bcl-2 and survival of FDCP2 cells viaPI3-K-dependent pathways. In that study, effects of IGF-1 on PKBand BAD phosphorylation, or of cAMP on the antiapoptotic ac-tions of IGF-1 were not assessed. Our results indicate that inFDCP2 cells, IGF-1 activates PDE3 and PDE4 via PI3-K-depen-dent pathways (29). Downstream of PI3-K, PDE3 is activated byPKB-dependent and PDE4 by MEK/MAP kinase-dependent (29)signals. In this context, our findings are consistent with the ideathat in FDCP2 cells, both the extent and duration of increasedintracellular cAMP may be important in regulation of BAD phos-phorylation and thymidine incorporation, and that PDEs (especial-ly PDE3B) can influence these parameters and, consequently, cellsurvival. It is possible that in FDCP2 cells, increased cAMP andactivation of PKA could directly or indirectly block association of[32P]BAD with 14-3-3 protein, leading to dephosphorylation ofBAD, perhaps by PP2B (calcineurin), and thus allowing its asso-ciation with Bcl proteins and stimulation of apoptosis (71, 72). Therelationship, if any, between effects of cAMP on Bcl-2 expression,BAD phosphorylation, and cell survival of FDCP2 cells has notbeen established. Such information could help elucidate mecha-nisms responsible for the proapoptotic and antiapoptotic actions ofcAMP in different cells. It would be of further interest to learnwhether cAMP and PDE3 inhibitors could block proliferation/sur-vival of ovarian, breast, and pancreatic carcinoma cells in whichPKB/Akt is apparently amplified (74, 75).
It is important to identify substrates for PKB. BAD (10), GSK-3(12), phosphofructokinase (13), and endothelial NO synthase (15)were phosphorylated in vitro by PKB, with accompanying changesin activities of GSK-3 and phosphofructokinase. All four containan RXRXXS motif with arginine residues at n-3 and n-5 of thephosphorylated serine. The RXRXXS motif, however, also appar-ently served as a substrate for p70S6 kinase and MAP kinase-activated protein kinase-1 (13). Incubation of intact rat adiopcyteswith insulin resulted in phosphorylation of serine 302 in endoge-nous rat adipocyte PDE3B (analogous to serine 296 in MPDE3B)(76), which is in the sequence KMFRRPS, i.e., different from thatin GSK-3 (12, 13). Although in their recently published reportKitamura et al. (18) did not identify sites phosphorylated in en-
dogenous MPDE3B in 3T3L1 adipocytes, they did report thatserine 276 (within an RXRXXS motif) in rMPDE3B is phosphor-ylated by activated PKB. Current studies in our laboratories areattempting to reconcile these apparent differences and identify thesite(s) involved in phosphorylation/activation of PDE3B by PKBin vitro and intact cells.
Insulin, IGF-1, or IL-4, each of which utilizes IRS proteins toinitiate at least some of its receptor signaling cascades, activatesPDE3 in adipocytes (16, 17, 21), FDCP2 cells, pancreaticb cells(77), hepatocytes (78), andXenopusoocytes (8, 79, 80). In thesecells, some effects of the polypeptides are counterregulatory tothose of cAMP, which increases lipolysis (19, 20, 81), inhibits cellproliferation/survival (41–55), stimulates insulin secretion (77)and glycogenolysis (82), and blocks meiosis (8), respectively. It is,therefore, tempting to speculate that in these cells, activation ofPDE3, which would presumably reduce cAMP and PKA, may playan important role in the counterregulatory effects of the polypep-tides, i.e., inhibiting lipolysis (16, 19–21), enhancing cell prolif-eration, inhibiting insulin secretion (77) and glycogenolysis (82),and stimulating meiosis (8, 79, 80), respectively (Fig. 10). From abroader perspective, these and other studies are consistent with theidea that PDEs comprise a complex group of structurally relatedand highly regulated enzymes that are critical, if not essential, ininfluencing specificity, compartmentation, and overall regulationof cyclic nucleotide-mediated processes (16, 83–85).
AcknowledgmentsWe thank Dr. Martha Vaughan (National Heart, Lung, and Blood Institutefor her suggestions and critical reading of our manuscript, and Carol Koshfor excellent secretarial assistance.
References1. Alessi, D. R., and P. Cohen. 1998. Mechanism and activation of protein kinase B.
Curr. Opin. Genet. Dev. 8:55.2. Franke, T. F., D. R. Kaplan, and L. C. Cantley. 1997. PI3K: downstream AKT ion
blocks apoptosis.Cell 88:435.3. Hemmings, B. A. 1997. Akt signaling: linking membrane events to life and death
decisions.Science 275:628.4. Cong, L.-N., H. Chen, Y. Li, L. Zhou, M. A. McGibbon, S. I. Taylor, and
M. J. Quon. 1997. Physiological role of Akt in insulin-stimulated translocation ofGLUT-4 in transfected rat adipose cells.Mol. Endocrinol. 11:1881.
5. Kohn, A., S. A. Simmons, M. J. Birnbaum, and R. A. Roth. 1996. Expression ofconstitutively active Akt Ser/Thr kinase in 3T3–L1 adipocytes stimulates glucoseuptake and glucose transporter 4 translocation.J. Biol. Chem. 271:31372.
6. Cross, D., P. Watt, M. Shaw, J. van der Kay, C. Downes, J. Holiday, andP. Cohen. 1997. Insulin activates protein kinase B, inhibits glycogen synthasekinase-3 and activates glycogen synthase by rapamycin-insensitive pathways inskeletal muscle and adipose tissue.FEBS Lett. 406:211.
7. Ueki, K., R. Yamamoto-Honda, Y. Kaburagi, T. Yamauchi, K. Tobe,B. Bugering, P. Coffer, I. Komuro, Y. Akanuma, Y. Yazaki, and T. Kadowaki.1998. Potential role of protein kinase B in insulin-induced glucose transport,glycogen synthesis and protein synthesis.J. Biol. Chem. 273:5315.
8. Andersen, C., R. Roth, and M. Conti. 1998. Protein kinase B/Akt induces re-sumption of meiosis inXenopusoocytes.J. Biol. Chem. 273:18705.
FIGURE 10. Postulated role for PKB-induced activation of PDE3 ineffects of growth factors such as insulin and IGF-1 on several cAMP-mediated processes.
4686 PDE3B IS A TARGET AND EFFECTOR OF PKB ACTION

9. Kulek, G., A. Klippel, and M. J. Weber. 1997. Antiapoptotic signaling by theinsulin-like growth factor receptor 1, phosphatidylinositol 3-kinase and Akt.Mol.Cell. Biol. 17:1595.
10. Del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997.Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt.Science 278:687.
11. Songyang, Z., D. Baltimore, L. Cantley, D. Kaplan, and T. Franke. 1997. Inter-leukin-3 dependent survival by the Akt protein kinase.Proc. Natl. Acad. Sci. USA91:11345.
12. Cross, D., D. R. Alessi, P. Cohen, M. Andjelkovich, and B. Hemmings. 1995.Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B.Nature 378:785.
13. Deprez, J., D. Vertommen, D. Alessi, L. Hue, and M. Rider. 1997. Phosphory-lation and activation of heart 6-phosphofructokinase by protein kinase B andother kinases of the insulin signaling cascades.J. Biol. Chem. 272:17269.
14. Brunet, A., A. Bonni, M. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson,K. C. Arden, J. Blenis, and M. Greenberg. 1999. Akt promotes cell survival byphosphorylating and inhibiting a forkhead transcription factor.Cell 96:857.
15. Fulton, D., J.-P. Gratton, T. J. McCabe, J. Fontana, Y. Fujio, K. Walsh,T. F. Franke, A. Papapetropoulos, and W. C. Sessa. 1999. Regulation of endo-thelium-derived nitric oxide production by the protein kinase Akt.Nature 399:597.
16. Degerman, E., P. Belfrage, and V. C. Manganiello. 1997. Structure, localization,and regulation of cGMP-inhibited phosphodiesterase (PDE3).J. Biol. Chem. 272:6823.
17. Wijkander, J., T. Rahn Landstrom, V. C. Manganiello, P. Belfrage, andE. Degerman. 1998. Insulin induced phosphorylation and activation of phospho-diesterase 3B in rat adipocytes: possible role for protein kinase B but not mito-gen-activated protein kinase or p70S6 kinase.Endocrinology 139:219.
18. Kitamura, T., Y. Kitamura, S. Kuroda, Y. Hino, M. Ando, K. Kotani, H. Konishi,H. Matsuzaki, U. Kikkawa, W. Ogawa, and M. Kasuga. 1999. Insulin-inducedphosphorylation and activation of cyclic nucleotide phosphodiesterase 3B byserine-threonine kinase Akt.Mol. Cell. Biol. 19:6286.
19. Belfrage, P., J. Donner, H. Eriksson, and P. Stralfors. 1986. Mechanisms forcontrol of lipolysis by insulin and growth hormone. InMechanisms of InsulinAction. P. Belfrage, J. Donner, and P. Stralfors, eds. Elsevier, Amsterdam, pp.323–340.
20. Eriksson, H., M. Ridderstrale, E. Degerman, D. Ekholm, C. Smith,V. C. Manganiello, and P. Belfrage. 1995. Evidence for the role of the cGMP-inhibited phosphodiesterase in the antilipolytic action of insulin.Biochim. Bio-phys. Acta 1266:101.
21. Rahn, T., M. Ridderstrale, H. Tornqvist, V. C. Manganiello, G. Fredrickson,P. Belfrage, and E. Degerman. 1994. Essential role of phosphatidylinositol-3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibitedcAMP phosphodiesterase in rat adipocytes using the selective inhibitor wortman-nin. FEBS Lett. 350:314.
22. Wang, L. M., M. G. Myers, X. J. Sun, S. A. Aaronson, M. White, and J. H. Pierce.1993. IRS-1 is essential for insulin stimulated and IL-4 stimulated mitogenesis inhematopoietic cells.Science 261:1591.
23. Sun, X.-J., S. Pons, L.-M. Wang, Y. Zhang, L. Yenush, D. Burks, M. Myers, Jr.,E. Glastein, N. Copeland, N. Jenkins, J. Pierce, and M. White. 1997. The IRS-2gene on murine chromosome 8 encodes a unique signalling adapter for insulinand cytokine action.Mol. Endocrinol. 11:251.
24. Wang, L. M., A. D. Keegan, W. Q. Li, G. E. Lienhard, J. S. Gutkind, M. G. Myer,X. J. Sun, M. F. White, S. A. Aaronson, W. E. Paul, and J. H. Pierce. 1993.Common elements in interleukin-4 and insulin signaling pathways in factor de-pendent hematopoietic cells.Proc. Natl. Acad. Sci. USA 90:4032.
25. Wang, L.-M., A. D. Keegan, W. E. Paul, M. A. Heideran, J. S. Gutkind, andJ. H. Pierce. 1992. IL-4 activates a distinct signal transduction cascade from IL-3in factor-dependent myeloid cells.EMBO J. 11:4899.
26. Ahmad, F., G. Gao, L.-M. Wang, T. Rahn-Landstrom, E. Degerman, J. Pierce,and V. C. Manganiello. 1999. Interleukin-3 and interleukin-4 activate cyclic nu-cleotide phosphodiesterases 3 (PDE3) and 4 (PDE4) by different mechanisms inFDCP2 cells.J. Immunol. 162:4864.
27. Quon, M. J., M. J. Zarnowski, M. Guerremillo, M. D. Sierra, S. I. Taylor, andS. W. Cushman. 1993. Transfection of DNA into isolated rat adipose cells byelectroporation: evaluation of promoter activity in transfected adipose cells whichare highly responsive to insulin after one day in culture.Biochem. Biophys. Res.Commun. 194:338.
28. Datta, K., A. Bellacosa, T. O. Chan, and P. N. Tsichlis. 1996. Akt is a direct targetof the phosphatidylinositol 3-kinase: activation by growth factors, v-src and v-Ha-ras, in Sf9 and mammalian cells.J. Biol. Chem. 271:30835.
29. Klippel, A., C. Reinhard, M. W. Kavanaugh, G. Apell, M. A. Escobedo, andL. T. Williams. 1996. Membrane localization of phosphatidylinositol 3-kinase issufficient to activate multiple signal-transducing kinase pathways.Mol. Cell. Biol.16:4117.
30. Pierce, J., M. Ruggiero, T. Fleming, P. DiFiore, J. Greenberger, L. Varticovski,J. Schlessinger, G. Rovera, and S. Aaronson. 1988. Signal transduction throughthe EGF receptor transfected in IL-3 dependent hematopoietic cells.Science 239:628.
31. Ekholm, D., B. Hemmer, G. Gao, M. Vergelli, R. Martin, and V. C. Manganiello.1997. Differential expression of cyclic nucleotide phosphodiesterase 3 and 4 ac-tivities in human T cell clones specific for myelin basic protein.J. Immunol.159:1520.
32. Beavo, J., and D. Reifsnyder. 1990. Primary sequence of cyclic nucleotide phos-phodiesterase isoenzymes and the design of selective inhibitors.Trends Biochem.Sci. 11:150.
33. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of thehead of bacteriophage T4.Nature 277:680.
34. Leroy, M.-J., E. Degerman, M. Taira, T. Murata, L.-H. Wang, M. Movsesian,E. Meacci, and V. C. Manganiello. 1996. Characterization of two recombinantPDE3 (cGMP-inhibited cyclic nucleotide phosphodiesterase) isoforms, RcGIP1and HcGIP2, expressed in NIH 3006 murine fibroblasts and Sf9 insect cells.Biochemistry 35:10194.
35. Moule, S. K., G. Welsh, N. Edgell, E. Foulstone, C. Proud, and R. Denton. 1997.Regulation of protein kinase B and glycogen synthase kinase-3 by insulin andb-adrenergic agents in rat epididymal fat cells.J. Biol. Chem. 272:7713.
36. Wijkander, J., L. S. Holst, T. Rahn, S. Resjo, I. Castan, V. C. Manganiello,P. Belfrage, and E. Degerman. 1997. Regulation of protein kinase B in rat adi-pocytes by insulin, vanadate, and peroxovanadate: membrane translocation inresponse to peroxovanadate.J. Biol. Chem. 272:21520.
37. Andjelkovic, M., D. Alessi, R. Meier, A. Fernandez, N. Lamb, M. Frech, P. Cron,P. Cohen, J. Lucocq, and B. A. Hemmings. 1997. Role of translocation in acti-vation and function of protein kinase B.J. Biol. Chem. 272:31515.
38. Kohn, A. D., A. Barthel, K. S. Kovacina, A. Boge, B. Wallach, S. A. Summers,M. J. Birnbaum, P. H. Scott, J. C. Lawrence, and R. A. Roth. 1998. Constructionand characterization of a constitutively active version of the serine/threoninekinase Akt.J. Biol. Chem. 273:11937.
39. Mackenzie, S. J., S. Yarwood, A. Peden, G. Bolger, R. Vernon, and M. Houslay.1998. Stimulation of p70S6 kinase via a growth hormone-controlled phosphati-dyl-inositol 3-kinase pathway leads to activation of a PDE4A cyclic AMP-spe-cific phosphodiesterase in 3T3-442A preadipocytes.Proc. Natl. Acad. Sci. USA95:3549.
40. Ahmad, N., H. L. Grimes, A. Bellacosa, T. Chan, and P. Tsichlis. 1997. Trans-duction of interleukin-2 antiapoptotic and proliferative signals by Akt proteinkinase.Proc. Natl. Acad. Sci. USA 94:3627.
41. Brown, D., and R. Phipps. 1996. Bcl-2 expression inhibits prostaglandin E2-mediated apoptosis in B-cell lymphomas.J. Immunol. 157:1359.
42. Lalli, E., P. Sassone-Corsi, and R. Ceredig. 1996. Block of T-lymphocyte dif-ferentiation by activation of the cAMP-dependent signal transduction pathway.EMBO J. 15:528.
43. McConkey, D., S. Orrenius, and M. Jondal. 1990. Agents that elevate cAMPstimulate DNA fragmentation in thymocytes.J. Immunol. 145:1227.
44. Shalhegg, B., B. Landmark, S. Doskeland, V. Hansson, T. Lea, and T. Johnson.1992. Cyclic AMP-dependent protein kinase type I mediates the inhibitory effectsof cAMP on cell replication in T-lymphocytes.J. Biol. Chem. 267:15707.
45. Aharoni, D., A. Dantes, M. Oren, and A. Amsterdam. 1995. cAMP-mediatedsignals as determinants for apoptosis in primary granulosa cells.Exp. Cell Res.218:271.
46. Friedman, D. L., R. Johnson, and C. Zeilig. 1976. The role of cyclic nucleotidesin the cell cycle.Adv. Cyclic Nucleotide Res. 7:69.
47. Pastan, I. H., G. S. Johnson, and W. B. Anderson. 1974. Role of cyclic nucleo-tides in growth control.Adv. Cancer Res. 19:303.
48. Roger, P. P., S. Reuse, C. Maenhaut, and J. E. Dumont. 1995. Multiple facets ofthe modulation of growth by cAMP.Vitam. Horm. 51:59.
49. Tomlinson, P., J. Wilson, and A. Stewart. 1995. Salbutamol inhibits the prolif-eration of human airway smooth muscle cells grown in culture, relationship toelevated cAMP levels.Biochem. Pharmacol. 49:1809.
50. Giembycz, M., C. Corrigan, J. Seybold, R. Newton, and P. Barnes. 1996. Iden-tification of phosphodiesterases 3, 4, and 7 in human CD41 and CD81 T-lym-phocytes: role in regulating proliferation and the synthesis of interleukin-2.Br. J. Pharmacol. 118:1945.
51. Jiang, X., J. Li, M. Paskind, and D. Epstein. 1996. Inhibition of calmodulin-dependent phosphodiesterase induces apoptosis in human leukemic cells.Proc.Natl. Acad. Sci. USA 93:11236.
52. Kim, D.-H., and A. Lerner. 1998. Type 4 adenosine monophosphate phosphodi-esterase as a therapeutic target in chronic lymphocytic leukemia.Blood 92:2484.
53. Robicsek, S., D. Blanchard, J. Djeu, J. Krzanowski, A. Szentwvanyi, andJ. Polson. 1991. Multiple high-affinity cAMP phosphodiesterases in human T-lymphocytes.Biochem. Pharmacol. 42:869.
54. Souness, J., G. Hassal, and D. Parrot. 1992. Inhibition of pig aortic smooth mus-cle DNA synthesis by selective type III and type IV cAMP phosphodiesteraseinhibitors.Biochem. Pharmacol. 48:827.
55. Drees, M., R. Zimmerman, and G. Eisenbard. 1993. 3959-Cyclic nucleotide phos-phodiesterases in tumor cells as potential targets for tumor growth inhibition.Cancer Res. 53:3058.
56. Chen, D., and E. Rothenberg. 1994. Interleukin-2 transcription factors as molec-ular targets of cAMP inhibition: delayed inhibition-kinetics and combinatorialtranscription roles.J. Exp. Med. 179:931.
57. Cook, S. J., and F. McCormick. 1993. Inhibition by cAMP of Ras-dependentactivation of Raf.Science 262:1069.
58. Ivashkev, L., E. Schmitt, and A. Castro. 1996. Inhibition of transcription factorStat 1 activity in mononuclear cell cultures and T-cells by the cyclic AMP sig-naling pathway.J. Immunol. 157:1415.
59. Kato, J., M. Matsuoka, K. Polyak, J. Massague, and C. J. Sherr. 1994. cAMP-induced G1 phase arrest mediated by an inhibitor of cyclin-dependent kinase 4.Cell 79:487.
60. Rao, G., and M. Runge. 1996. Cyclic AMP inhibition of thrombin-inducedgrowth in vascular smooth muscle cells correlates with decreased JNK1 activityand c-Jun expression.J. Biol. Chem. 271:20805.
61. Tamir, A., Y. Granot, and N. Isakov. 1996. Inhibition of T-lymphocyte activationis associated with down-regulation of two parallel mitogen-activated protein ki-nase pathways, the extracellular signal-related kinase and c-Jun N-terminal ki-nase.J. Immunol. 157:1514.
4687The Journal of Immunology

62. Downs, S. M., and M. Hunzicker-Dunn. 1995. Differential regulation of oocytematuration and cumulus expansion in the mouse oocyte-cumulus complex bysite-selective analogs of cyclic adenosine monophosphate.Dev. Biol. 172:72.
63. Monfar, M., K. Lemon, T. Grammar, L. Cheatham, L. Chung, C. Vlakos, andJ. Blenis. 1995. Activation of pp70/85 S6 kinase in IL-2-responsive cells is me-diated by phosphatidylinositol 3-kinase and inhibited by cAMP.Mol. Cell. Biol.15:326.
64. Pirson, I., K. Coulonval, F. Lamy, and J. E. Dumont. 1996. C-Myc expression iscontrolled by the mitogenic cAMP-cascade in thymocytes.J. Cell. Physiol. 168:59.
65. Parvathenani, L. K., E. S. Buescher, E. Chacon-Cruz, and S. Beebe. 1998. Type1 cAMP-dependent protein kinase delays apoptosis in human neutrophils at a siteupstream of caspase-3.J. Biol. Chem. 273:6736.
66. Franke, T., and L. Cantley. 1997. A BAD kinase makes good.Nature 390:116.67. Datta, S. R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and
M. E. Greenberg. 1997. Akt phosphorylation of BAD couples survival signals tothe cell-intrinsic death machinery.Cell 91:231.
68. Harada, H., B. Becknell, M. Wilm, M. Mann, L. Huang, S. S. Taylor, J. D. Scott,and S. J. Korsmeyer. 1999. Phosphorylation and inactivation of BAD by themitochondria-anchored protein kinase A.Mol. Cell 3:413.
69. Parrizas, M., A. Saltiel, and D. LeRoith. 1997. Insulin-like growth factor 1 in-hibits apoptosis using the phosphatidyl 39-kinase and mitogen-activated proteinkinase pathways.J. Biol. Chem. 272:154.
70. Scheid, M. D., and V. Duorino. 1998. Dissociation of cytokine-induced phos-phorylation of BAD and activation of PKB/Akt: involvement of MEK upstreamof BAD phosphorylation.Proc. Natl. Acad. Sci. USA 95:7439.
71. Klee, C. B., H. Ren, and X. Wang. 1998. Regulation of the calmodulin-stimulatedphosphatase, calcineurin.J. Biol. Chem. 273:13367.
72. Wang, H.-G., N. Patham, I. Ethell, S. Krajewski, Y. Yamaguchi, F. Shibaski,F. McKeon, T. Bobo, T. Franke, and J. C. Reed. 1999. Ca21-induced apoptosisthrough calcineurin dephosphorylation of BAD.Science 284:339.
73. Minishall, C., S. Arkins, R. Dantzer, G. C. Freund, and K. W. Kelley. 1999.Phosphatidyl-3-kinase, but not S6-kinase, is required for insulin-like growth fac-tor-1 and IL-4 to maintain expression of Bcl-2 and promote survival of myeloidprogenitors.J. Immunol. 162:4542.
74. Cheng, J., A. Goodwin, A. Bellacosa, T. Taguchi, T. Franke, T. Hamilton,P. Tsichles, and J. Testa. 1992. AKT2, a putative protooncogene encoding asubfamily of protein serine kinases is amplified in human ovarian carcinomas.Proc. Natl. Acad. Sci. USA 89:9267.
75. Cheng, J., B. Ruggeri, W. Klein, G. Sonoda, D. Altomare, D. Watson, andJ. Testa. 1996. Amplification of AKT2 in human pancreatic-cancer cells andinhibition of AKT2 expression and tumorigenicity by antisense RNA.Proc. Natl.Acad. Sci. USA 93:3636.
76. Rahn, T., L. Ronnstrand, M.-J. Leroy, C. Wernstedt, H. Tornqvist,V. C. Manganiello, P. Belfrage, and E. Degerman. 1996. Identification of the sitein the cGMP-inhibited phosphodiesterase phosphorylated in adipocytes in re-sponse to insulin and isoproterenol.J. Biol. Chem. 271:11575.
77. Zhao, A., H. Zhao, J. Teague, W. Fujimoto, and J. Beavo. 1997. Attenuation ofinsulin secretion by IGF-1 is mediated through activation of phosphodiesterase3B. Proc. Natl. Acad. Sci. USA 94:3223.
78. Pyne, N., M. Cooper, and M. Houslay. 1987. The insulin- and glucagon-stimu-lated “dense vesicle” high affinity cAMP phosphodiesterase from rat liver.Bio-chem. J. 242:33.
79. Sadler, S. 1991. Type III phosphodiesterase plays a necessary role in the growthpromoting actions of insulin, IGF-1 and Hp21 ras in Xenopus laevisoocytes.Mol.Endocrinol. 5:1939.
80. Sadler, S., and J. Maller. 1987. In vivo regulation of cAMP phosphodiesterase inXenopusoocytes: stimulation by insulin and IGF-1.J. Biol. Chem. 262:10644.
81. Londos, C., R. Honor, and C. S. Dhillon. 1985. cAMP-dependent protein kinaseand lipolysis in rat adipocytes III.J. Biol. Chem. 260:15781.
82. Beebe, S. J., J. Redmon, P. Blackmore, and J. Corbin. 1985. Discriminativeinsulin antagonism of stimulatory effects of various cAMP analogs on adipocytelipolysis and hepatic glycogenolysis.J. Biol. Chem. 260:15781.
83. Conti, M., G. Nemoz, C. Sette, and E. Vicini. 1995. Recent progress in under-standing regulation of phosphodiesterases.Endocr. Rev. 16:370.
84. Houslay, M., and G. Milligan. 1997. Tailoring cAMP responses through isoformmultiplicity. Trends Biochem. Sci. 22:217.
85. Torphy, T. 1998. Phosphodiesterase isoenzymes: molecular targets for novel an-tiasthma agents.Am. J. Respir. Crit. Care Med. 157:351.
4688 PDE3B IS A TARGET AND EFFECTOR OF PKB ACTION
Copyright © 2022 FDOKUMEN




















