
Protein kinase C — a family affair
11
Molecular and Cellular Endocrinology, 65 (1989) 1-11 Elsevier Scientific Publishers Ireland, Ltd. MOLCEL 02111 Review Protein kinase C - a family affair Peter J. Parker, Gurdip Kour, Richard M. Marais, Fiona Mitchell, Cathy Pears, Dick Schaap, Silvia Stabel and Catherine Webster Ludwig Institute for Cancer Research, London &y words: Protein kinase C, Effector dependence; Substrate specificity; Signal transduction; (Review) The structural analysis of protein kinase C has led to the identification of a family of related gene products. This family of kinases consists of six unique genes that give rise to at least seven polypeptides. The high degree of conservation and the differential distribution of these mRNAs/proteins suggest that they perform distinct functions in vivo. Characterization of the activities of some of these proteins in vitro shows that there are functional differences with respect to both their regulation and substrate specificity. This indicates that each member of this family may play a unique role in signal transduction. Introduction There have been a number of recent reviews concerning protein kinase C (PKC) (e.g. Ashendel, 1985; Nishizuka, 1986, 1988; Woodgett et al., 1987). These have covered in detail various aspects of the enzymology of this kinase and its role as a signal transducing protein in a plethora of physio- logical responses. It is not the intention of this article to reiterate and update these observations, but rather to focus on the issues that arise from the diversity of the protein kinase C family. As a prelude to discussing the protein kinase C family, it would be pertinent to address the ques- tion of what constitutes a family. There is of course what might be termed a superfamily of protein kinases that from sequence analyses are clearly related (Hanks et al., 1988) and for the most part have been shown to possess protein Address for correspondence: Dr. P.J. Parker, Ludwig In- stitute for Cancer Research, Courtauld Building, 91 Riding House Street, London WlP 8BT, U.K. kinase activity. Within this superfamily there are two readily identifiable branches that correlate with the known target amino acid specificity, namely tyrosine and serine/threonine-specific kinases (Hanks et al., 1988). Useful divisions within for example the serine/threonine-specific protein kinase branch can be made with respect to sequence divergence between the kinase domains of distinct protejn kinases; the family tree derived by these comparisons does to some extent corre- late with the effector dependence of these en- zymes. Ear example the Ca2+/calmodulin-depen- dent enzymes and the cyclic nucleotide-dependent enzymes appear to form distinct groupings within the sequence-related superfamily (Hanks et al., 1988). However, it would be misleading to con- sider these proteins as constituting individual family branches without some understanding of their properties. These observations beg the question ‘To what extent do the PKCs that have been identified through cDNA cloning represent a discrete family of kinases and if they do, how are the limits of the family defined? The following discussion of the PKCs covers some of what is now known about the biochem- 0303-7207/89/$03.50 6 1989 Elsevier Scientific Publishers Ireland, Ltd.
Transcript of Protein kinase C — a family affair

Molecular and Cellular Endocrinology, 65 (1989) 1-11 Elsevier Scientific Publishers Ireland, Ltd.
MOLCEL 02111
Review
Protein kinase C - a family affair
Peter J. Parker, Gurdip Kour, Richard M. Marais, Fiona Mitchell, Cathy Pears, Dick Schaap, Silvia Stabel and Catherine Webster
Ludwig Institute for Cancer Research, London
&y words: Protein kinase C, Effector dependence; Substrate specificity; Signal transduction; (Review)
The structural analysis of protein kinase C has led to the identification of a family of related gene products. This family of kinases consists of six unique genes that give rise to at least seven polypeptides. The high degree of conservation and the differential distribution of these mRNAs/proteins suggest that they perform distinct functions in vivo. Characterization of the activities of some of these proteins in vitro shows that there are functional differences with respect to both their regulation and substrate specificity. This indicates that each member of this family may play a unique role in signal transduction.
Introduction
There have been a number of recent reviews concerning protein kinase C (PKC) (e.g. Ashendel, 1985; Nishizuka, 1986, 1988; Woodgett et al., 1987). These have covered in detail various aspects of the enzymology of this kinase and its role as a signal transducing protein in a plethora of physio- logical responses. It is not the intention of this article to reiterate and update these observations, but rather to focus on the issues that arise from the diversity of the protein kinase C family.
As a prelude to discussing the protein kinase C family, it would be pertinent to address the ques- tion of what constitutes a family. There is of course what might be termed a superfamily of protein kinases that from sequence analyses are clearly related (Hanks et al., 1988) and for the most part have been shown to possess protein
Address for correspondence: Dr. P.J. Parker, Ludwig In-
stitute for Cancer Research, Courtauld Building, 91 Riding House Street, London WlP 8BT, U.K.
kinase activity. Within this superfamily there are two readily identifiable branches that correlate with the known target amino acid specificity, namely tyrosine and serine/threonine-specific kinases (Hanks et al., 1988). Useful divisions within for example the serine/threonine-specific protein kinase branch can be made with respect to sequence divergence between the kinase domains of distinct protejn kinases; the family tree derived by these comparisons does to some extent corre- late with the effector dependence of these en- zymes. Ear example the Ca2+/calmodulin-depen- dent enzymes and the cyclic nucleotide-dependent enzymes appear to form distinct groupings within the sequence-related superfamily (Hanks et al., 1988). However, it would be misleading to con- sider these proteins as constituting individual family branches without some understanding of their properties. These observations beg the question ‘To what extent do the PKCs that have been identified through cDNA cloning represent a discrete family of kinases and if they do, how are the limits of the family defined?
The following discussion of the PKCs covers some of what is now known about the biochem-
0303-7207/89/$03.50 6 1989 Elsevier Scientific Publishers Ireland, Ltd.

2
istry of these proteins and this will in part address the question raised above. To anticipate the answer, it would appear that the PKCs do in fact represent a single family of proteins. However, it should be emphasized that these kinases are not isoenzymes in the sense that lactate dehydro- genase exists as isoenzymes (i.e. different gene products acting to catalyse the same reaction), but rather the PKCs are tied together as a family through their regulatory properties.
Identification of multiple PKCs
PKC-ci, -p, -y Primary structural analysis of PKC initially led
to the identification of a set of homologous cDNAs (Coussens et al., 1986; Knopf et al., 1986; Ono et al., 1986, 1988a, b; Parker et al., 1986; Housey et al., 1987; Kikkawa et al., 1987a, b; Kubo et al., 1987a, b; Ohno et al., 1987, 1988a). Identification (and isolation) of these cDNAs has come both through protein sequence determination and through transient expression studies. Interestingly, the protein sequences determined for purified brain PKC in different laboratories were variably derived from different PKC isotypes (see Parker et al., 1986 and Knopf et al., 1986). In our labora- tory for example we obtained only protein se- quence for PKC-a (Parker et al., 1986), which in retrospect can be explained on the basis of the purification procedure employed at that time. Nevertheless the sequence homology of these PKCs is such that probes derived from individual peptide sequences can cross-hybridize to distinct PKC cDNAs. Related cDNAs have also been obtained using cloned cDNAs as probes (Ono et al., 1987a, 1988a, b; Ohno et al., 1988b).
The cDNAs initially isolated from various laboratories can be grouped into four PKC iso- types (cw, pi, & and y; see Parker et al., 1989), derived from three distinct genes on human chro-
mosome 17 (cy), chromosome 16 (p) and chro- mosome 19 (y) (Coussens et al., 1986). The pre- dicted sequences for these four cDNAs are shown in Fig. 1. The & and & cDNAs are derived from differential splicing at the 3’ end of the fi gene giving rise to proteins with identical sequence up to residue 621 followed by 52 (pi) and 50 (&) amino acids that show about 50% identity (see Fig. 1). These two /3 3’ sequences are derived from two distinct exons at the 3’ end of the p gene (Coussens et al., 1987; Kubo et al., 1987a, b); the homology would imply that the two exons were derived from a duplication event. There would also appear to be alternative splicing of the (Y and /3 mRNAs outside the coding region as judged by Northern and Southern analysis (see Coussens et al., 1986). By contrast to both (Y and p mRNAs, Northern analysis of brain mRNA with y cDNA probes reveals a single major mRNA of 3.1 kb.
It is evident from the comparison shown in Fig. 1 that these proteins contain four highly conserved regions (C-C,) surrounded by five variable re- gions (Vi-V,). Sequence comparison with other previously identified protein kinases clearly in- dicates that the C-terminal region of the PKCs (C,-V,) corresponds to the catalytic domain (see Parker et al., 1986). This is further substantiated through proteolytic studies that indicate that a catalytically active fragment of PKC-cy is gener- ated by cleavage at a site(s) in the V, region (Young et al., 1988). Within this C-terminal kinase domain there are sequences characteristic of all known protein kinases, such as the GXG- XXG.. . K sequence that forms part of the ATP binding site (Kamps et al., 1984).
There are regions of interest outside the PKC kinase domain, although there is no overall ho- mology to other known proteins. It has been pro- posed that residues 19-36 (in PKC-cw) may con- stitute a pseudosubstrate site (House and Kemp, 1987) that may be responsible for occupying the
Fin. 1. Comparison of PKC-cr. W, -& and -y protein sequences. The predicted amino-acid sequences for PKC-a, -B,. 4, and -y -. -_ _ . . ._ were aligned using the AMPS package (Geoff Barton, Imperial Cancer Research Fund, London, U.K.). The sequences show variable
(V,_,) and conserved (Cl_,) regions. These include the kinase domain (Cs-Vs) that contains sequences (A) characteristic of all the
protein kinase family. The residues in the C, domain that are marked (0) are those in the cysteine repeat unit (see text). The basic residues indicated (W) lie at the primary site of PKC-a cleavage (and consequent activation) by trypsin (Young et al., 1988). The
arrow indicates the point of divergence of the predicted -& and -& sequences (see text).

Vl Cl ________--_--_____-_--------- M?iDVFPAAEPAAPQDVANRFARKGALRQKNVHEVKNHRFIARFFKQPTFCSHCTDFIWGFGK~FQCQVCCE'VVHKRCHE MADPAAGPPPSEGEESTVRFARKGAtRQKNVHEVKNHKFTTFCSHCTDFIWGFGKQGFQCQVCCFHE MADPAAGPPPSEGEESTVRF~~~~~~T~FKQPTFCSHCTDFIWGFGKQGFQCQVCCFWHHE MAGLGPGVGDSEGGPRPL FCRKGALRQKWHEVKSHKFTARFFKQPTFCSHCTDFIWGIGKQGLQCQVCSFWHP.RCHE
00 l eee 00 0 00 0 0 00 0
FVTFSCPGADKGPDTDDPRSKHKFKIHTYGSPTFCDHCGSUYGLIHQGMKCDTCDMMTHKQCVINSLCGMDHTEKRG FVTFSCPGADKGPASDDPRSKIiKE'KIHTYSSPTFCDHCGSLLYGLIHQGMKCDTC SLCGTDHTERRG ~SCPGADKGPASDDPRSKHKFKIHTYSSPTFCDHCGSLLYGLIHQGMKCDTCSLCGTDHTERRG E'VTFECPGAGKGPQTDDPRNKHKF RLHSYSSPTFCDHCGSLLYGLVHQGMKCSCCEMNVHFtRCVRSVP SLCGVDHTERRG . . 00 l e.0 . . 0 . . . l 00 a l 0
v2 c2 -------------
RIYLKAEV TDEKLHVTVRD AKNLIPMDPNGLSDPYVKLKIPDPKNESKQKTKTIRSTLSDKDRR RIYIQAHI DRDVLIVLVRDAKNLVPMDPNGLSDPYVIUUIPDPKSESKQKTKTIKCSLNPEWNETFRFQLKESDKDRR RIYIQAHI DRDVLIVLSJRDAKNLVPMDPNGLSDPYVKLKLIPDPKSESKQKTKTIKCSLNPEWNETFRFQLKESDKDRR RLQLEIRAPTSDEIHVTVGFARNLIPMDPNGLSDPYVKLKLIPDPRNLTKQKTRTVKATLNPVWNETFVFNLKPGDVERR
v3 ---________ ___ ----- ii n --------- 8
LSEEIWDWDRTTRNDFMGSLSFGVSEIME35?ASGWYKLLNQEEGEYYNVPIPEGDEEGNVELRQKFE m
LSVEIWDWDLTSRNDFMGSLSFGISELQKASVDGWFIULSQEEGEYFNVPVPPEGSEANEELRQKFE RAKI LSVEIWDWDLTSRNDF'MGSLSFGISEIQKASVDGWFKLLSQEEGEYFNVPVPPEGSEANKEI_XQ~E RAKI LSVEVWDWDRTSRNDFMG?MSFGVSELLKAPVDGWYKLLNQEEGEYYNVPVADADNCNLLQKFEACNYPLELYERVRTGP
c3 -------------~-------_______ GPAGNKVISPSED RRQPSNNLDRVKLTDFNFLMVLGKGSFGKVMLAD RKGTEELYAIKILKKDWIQDDDVECTMVE SQGTKVPEEKTTNTVSKFDNNGNRDRMKLTDFNFLMVLGKGSFG~SE~GTDELYA~IL~WIQDDD~C~ SQGTKVPEEKTTNTVSKFDNNGNRDRMKLTDFNFLMVLGKGSFG~SE~GTDELYA~IL~WIQDDD~C~ SSSPIPSPSPSPTDSKRCFFGASPGEU,HISDFSFLMVLGKGSFGKVMLAE RRGSDELYAIKILKKDVIVQDDDVDCTLVE
AA A A
V4 c5
KRVLALLDK PPFLTQLHSCFQTVDRLYFVMEYVNGGDLMYHIQQVGKFKKPQAVFYAAEISIGLFFLHKRGIIYR KFWTALPGK PPFLTQLHSCFQTi4DRLYFVMEXVNGGDLMYHIQQVGRFKEPHAVFYAAEIAIGLFFLQSKGIIYR KRVIALPGK PPFLTQLHSCFQTMDRLYFVMEYVNGGDLMYHIQQVG~KEPHAVFYAAEIAIGLFFLQSKGIIYR KRVIALGGRGPGGRPHFLTQLHSTFQTF'DIUYFVMEYVTGGDLMYHIQQLGKFKEPHAAFYAAEIAIGLFFLHNQGIIYR
A
DLKLDNVMLDSEGHIKIADF~~~~~~CGTPDPFDGED DLKPDNVMLDSEGHIKIADFGMCKENIWDGVTTKTFCGTPDYIAPEIIAYQPYGKSVDWWAFGVLLYEMLAGQAE'FEGED DLKPDNVMLDSEGHIKIADFGMCKJ~NIWDGVTTKTFCGTPDYIAPEIIAYQPYGKSVDWWAFGVLLYKMLAGQAE'FEGED DLKLDNVMlllAEGHIKITDFGMCKENVFPGSTTRTLCGTPIAYQPYGKSVDWWSFGVLLHPFDGED M A44
EDELFQSIMEHNVAYPKS&%EAVAICKGLMTKHP GKRLGCGPEGERDIKEHAFFRYIDWEKLERKEIQPPYKPKACGR EDELFQSIMEHNVAYPKSMSKEAVAICKGLMTKHPGKBLGCGPEGERDIKEHAFFRYIDWJ3KLEFtKEIQPPYKPKARDKR EEELFQAIMEQTVmPKSLSRICKGFLT~~GSGPDGEPTI~GFF~ID~~E~EI~PF~~CGR
v5 t ____________________~~~~~~~~~~~~~~~---~~~-----___________ GAENFDKFFTGGQPVLTPPDQLVIANIDQSDFEGFSYVNPQFVHPILQSAV NAENFDRJ?FTRHPPVLTPPDNEVIRNIDQSEFEFEGFSFVNSEFLKPEVKS DTSNFDKKFTRQPVKLTPTDKLFIMt-JLDQNEFAGFSYTNPEFVINV SGENFDKl?FTRAAPALTPPDRLVLASIDQAEFQGFTYVNPDFVHPDARSPISPTPVP

(b)
mi* :.::.::+ _:_:_.
::..:
:.... . . I& :. :. ”
.‘.‘. .: “.
.’ ‘.
., ,. .:.:
::. :::. :. : .‘.’
J
cNL_-- . ..O 000 . .
(i RFARKGALROKHUHEUKOHKF
B RFtlRKGALRQKWUHEUKWHKF
I LFCRKERLRQKUUHEUKSHKF
6 TNWRRGAIKUAKIHYIKWHEF
;
PFlKRlJGllURRFl-UHQUNGHKF SIYRRGARRURKLYRRHGHLF
,r
Fig. 2. ((I) A schematic representation of PKC indicating the
interaction of the pseudosubstrate sequence (see text) with the
catalytic domain. It is envisaged that this interaction is lost on
binding of effecters through a conformational event and that
this exposes the catalytic site allowing its function to be
expressed. Activation is also observed following proteolysis;
this again would be predicted to lead to the loss of pseudosub-
strate site interaction. (b) A comparison of the homologous
pseudosubstrate site sequences in the known PKC isotypes.
Identical (0) and conserved residues (0) are shown. The arrow
indicates the ‘target’ residue for which serine substitution
would be predicted to generate a functional substrate. The
differences between these sequences might suggest that there is
specificity with respect to substrates for these different iso-
types; this is evident for PKC-e compared to PKC-cy, -/I,, -&
and -y (see below; Schaap et al., 1989); however, the dif-
ferences are not exclusive.
substrate binding site in the absence of effector molecules (Fig. 2~). It should be noted that this pseudosubstrate site, which is located at the boundary of the V/C, regions (see Fig. l), is fairly well conserved in (Y, B and y (but less so in other PKC isotypes, Fig. 2b; see below). Im- mediately C-terminal to this site is the cysteine-rich C2 region which is characterized by a tandem repeat sequence that centres on a CX,CX,,CX,- CX,CX,C unit. The CX,CX,,CX,C part of the repeat unit shows homology with structures in the
putative DNA binding domain of the steroid re- ceptors (Parker and Ulhich, 1987; Rosenthal et al., 1987). The C, domain also shows homology to c-raf (Ishikawa et al., 1986) and phospholipase A, (Maraganore, 1987). The diversity of functions for these proteins showing limited homology to PKC makes it hard to draw conclusions from these observations with respect to the structure/func- tion of PKC. On the contrary it may prove to be more informative about the related proteins. Thus the amino-terminal domain of c-raf, that is tnm- cated in the transforming v-raf gene (see Rapp et al., 1988), may by analogy with that of PKC act to suppress the kinase activity of this protein in the absence of a suitable effector molecule; there is in this amino-terminal domain of c-raf a region that may constitute a pseudosubstrate site.
The V&,/V, regions of PKC show no par- ticular sequence characteristics that would give clues to their function. We have previously noted the presence of ‘one half of an E-F hand struc- ture in the C, region of PKC-a! (Parker et al., 1986), that we suggested may confer Ca2+ bind- ing; however, this is not conserved in either PKC-P or PKC-y both of which can show Ca2+ depen- dence as discussed below.
PKC-6, -E, -[ Subsequent screening of brain cDNA libraries
has revealed the presence of at least three more PKC transcripts that would appear to be unique gene products (Ono et al., 1987a, 1988a, b; Ohno et al., 1988b; Schaap et al., 1989); these have been termed 4, -E and -{. These three PKCs while showing significant overall homology to -(Y, -fi and -y are evidently less related. As illustrated in Fig. 3 there is colinearity in the kinase domains of 4, -E and -5, but substantial deletions and extensions in the regulatory domains. The C, region is com- pletely missing in all three predicted polypeptides, although the cysteinarich region is retained (it is not clear whether PKC-{ has a second cysteine-rich sequence, since only partial cDNAs have been isolated to date). The divergence amino terminal to the cysteine-rich region includes the putative pseudosubstrate sites (Fig. 2b). The conserved features of this group of PKCs are discussed be- low in the context of their properties.
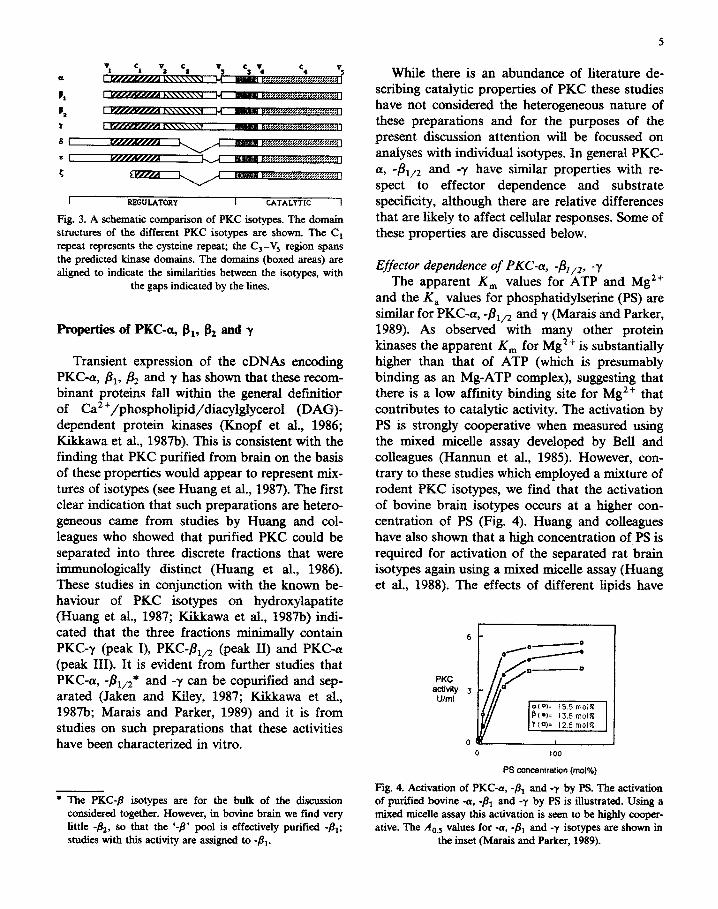
6
t
3
I RRGULATORY I CATALYTIC I
Fig. 3. A schematic comparison of PKC isotypes. The domain structures of tbe different PKC isotypes are shown. The Cr repeat represents the cysteine repeat; the C,-Vs region spans the predicted kinase domains. The domains (boxed areas) are aligned to indicate the similarities between the isotypes, with
the gaps indicated by the lines.
Properties of PKC-a, gt, & and y
Transient expression of the cDNAs encoding PKC-a, & & and y has shown that these recom- binant proteins fall within the general definitior of Ca’~/phospho~pid/diacyl~ycerol (DAG)- dependent protein kinases (Knopf et al., 1986; Kikkawa et al., 1987b). This is consistent with the finding that PKC purified from brain on the basis of these properties would appear to represent mix- tures of isotypes (see Huang et al., 1987). The first clear indication that such prorations are hetero- geneous came from studies by Huang and col- leagues who showed that purified PKC could be separated into three discrete fractions that were immunologically distinct (Huang et al., 1986). These studies in conjunction with the known be- haviour of PKC isotypes on hydroxylapatite (Huang et al., 1987; Kikkawa et al., 1987b) indi- cated that the three fractions minimally contain PKC-y (peak I), PKC-&,, (peak II) and PKC-(U (peak III). It is evident from further studies that PKGa, &* and -y can be copurified and sep- arated (Jaken and Kiley, 1987; Kikkawa et al., 1987b; Marais and Parker, 1989) and it is from studies on such preparations that these activities have been characterized in vitro.
* The PKC-8 isotypes are for the bulk of the discussion considered together. However, in bovine brain we find very little -&, SO that the ‘+I pool is effectively purified -fir; studies with this activity are assigued to -&.
5
While there is an abundance of literature de- scribing catalytic properties of PKC these studies have not considered the heterogeneous nature of these preparations and for the purposes of the present discussion attention will be focussed on analyses with individual isotypes. In general PKC- a, I&,2 and -y have similar properties with re- spect to effector dependence and substrate specificity, although there are relative differences that are likely to affect cellular responses. Some of these properties are discussed below.
Effector dependence of PKC-a, -&,2, -y The apparent K, values for ATP and Mg ’ +
and the K, values for phosphatidylserine (PS) are similar for PKC-a, -&,z and y (Marais and Parker, 1989). As observed with many other protein kinases the apparent K, for Mg*+ is substantially higher than that of ATP (which is presumably binding as an Mg-ATP complex), suggesting that there is a low affinity binding site for Mg2+ that contributes to catalytic activity. The activation by PS is strongly cooperative when measured using the mixed micelle assay developed by Bell and colleagues (Hannun et al., 1985). However, con- trary to these studies which employed a mixture of rodent PKC isotypes, we find that the activation of bovine brain isotypes occurs at a higher con- centration of PS (Fig. 4). Huang and colleagues have also shown that a high concentration of PS is required for activation of the separated rat brain isotypes again using a mixed micelle assay (Huang et al., 1988). The effects of different lipids have
F-KC activity
U/ml a(*)= 15.5 molR
0 100
PS wn~ntmtia~ (mol%)
Fig 4. Activation of PKC-d, $3, and -y by PS. The activation of purifie-d bovine -a, -& and -y by PS is illustrated. Using a mixed micelle assay Uris activation is seen to be highly cooper- ative. The A,, values for -a, -& and -y isotypes are shown in
the inset (Marais and Parker, 1989).

6
also been investigated; these studies showed that cardiolipin was as effective as PS in the activation of these isotypes; other lipids were less effective but the relative ability of these lipids did not vary significantly for the isotype preparations (Huang et al., 1988); similar observations have also been made by Asaoka et al. using a pure lipid micelle assay (Asaolca et al., 1988). The two phosphatidyl- alcohols, phosphatidyle~~ol and phosphatidyl- methanol, have also been found to be capable of substitution for PS in the activation of PKC iso- types (using pure lipid micelle preparations). In this case, however, a significant increase in re- sponsiveness was noted for PKC-y compared to the others (Asaoka et al., 1988).
The PKC-(u, -&,2 and -y isotypes are all de- pendent upon DAG or phorbol ester for activa- tion (Huang et al., 1988; Marais and Parker, 1989). However, studies on this dependence are com- plicated by the composition of the phospholipid component; for example Huang and colleagues have found that the K, values for diolein vary by about 2.5-fold if phosphatidylserine is employed in the assay, while the K, values vary by > 20-fold if cardiolipin is employed in place of phosphati- dylserine (Huang et al., 1988). For the bovine isotypes, using phosphatidylse~e in the assay, it has been observed that the K, values for diolein and synthetic short-chain DAGs (frequently employed in cell activation studies) are con- sistently lower (up to 5-fold) for PKC-ru compared to -& or -y; PKC-y was also found to be less sensitive to 12-O-tetradecanoylphorbol Ifacetate (TPA) (Marais and Parker, 1989). The variation in response of the PKC isotypes to DAGs and phorbol esters under different conditions implies that there are real differences between isotypes in the binding of these effecters; the precise pattern of sensitivity that pertains in vivo will be depen- dent upon the composition of the membranes with which the PKCs interact.
There have been a number of reports on the activation of PKC by fatty acids (Murakami et al., 1985, 1986; Sekiguchi et al,, 1987). Of these, one report has addressed the question of specificity with respect to PKC isotypes and has shown that PKC-y is more sensitive to activation by arachi-
. . dome acid than either -& or -(Y (Se&u&i et al., 1987; an increased sensitivity is also found for
bovine PKC-y - R.M.M. and P.J.P., unpub- lished). The nature of the activation by fatty acids is complex, since there is evidence that (on a mixture of PKC isotypes) it acts simply to provide a lipid environment by substituting for PS (Seifert et al., 1988). Furthermore, the biphasic nature of the PKC-y activation (Sekiguchi et al., 1987) is at least in part due to non-specific irreversible (de- naturation?) effects (R.M.M. and P.J.P., unpub- lished). However, reports that fatty acids can activate PKC isotypes in a diacylglycerol-indepen- dent manner (Sekiguchi et al., 1987) and that activation of mixtures of isotypes is stereospecific (Murakami et al., 1985) implies that there may be specific interactions which would have significant implications in vivo with respect to agonist-in- duced activation of PKCs.
The Ca ‘+ dependence o f the PKC isotypes appears to be complex. Data from three laborato- ries provides evidence that there is little difference in the rC, values for Ca2+ with the PKC-(Y, -&/2 and -y isotypes (Jaken and Kiley, 1987; Huang et al., 1988; Marais and Parker, 1989), although we and others have noted a slightly reduced depen- dence for PKC-y (Jaken and Kiley, 1987; Marais and Parker, 1989). There is, however, further data to indicate that PKC-pi,, shows a significant reduction in Ca*+ dependence relative to -cu or -y (Kosaka et al., 1988). This difference may reflect substrate-directed effects since we have also observed a decreased Ca2+ dependence for PKC-& much as described by Nishizuka and colleagues, but only with one particular batch of histone substrate (Marais and Parker, 1989). Some sub- strates can be phosphorylated in a Ca2”-indepen- dent manner (e.g. Nunn and Watson, 1987), fur- ther complicating the issue of Ca2+ dependence. The implication of these obse~ations is that the absolute requirement of Ca2+ in vivo in the activa- tion of PKC isotypes may depend upon the par- ticular substrate under consideration. Whether the Ca2+ concentration is limiting in vivo in any particular situation is a further issue. The pub- lished K, values for Ca2+ for the PKC isotypes (using histone as a substrate in a mixed rnicelle assay) are of the order of 200 nM (Jaken and Kiley, 1987; Marais and Parker, 1989) to 500 nM (Huang et al., 1988). This covers a range from what is probably a resting level to a near-maxi-

mW stimulated level for most cell types. Thus mobilization of Ca2+ would be likely to influence the extent to which these PKC isotypes can be activated in response to diacylglycerol, at least with respect to substrates that are ‘Ca2+-depen- dent’ in their phosphorylation.
Substrate specificity of PKC-a, -/31/z, -y As originally reported by Nishizuka’s labora-
tory (Inoue et al., 1977; Takai et al., 1977), PKC shows a clear preference for the lysine-rich frac- tion with respect to histone substrates and this substrate has been the most commonly used for the purposes of purification and kinetic analysis of these enzymes. However, it is evident that a number of other proteins can be phosphorylated in vitro (see Woodgett et al., 1987), although again for the purposes of this article, discussion will be restricted to protein and peptide substrates that have been assessed with individual isotypes.
For histone Hl (histone 111s) there are some moderate (2-fold) differences in K, for the -LX,
-&,z 3 -y isotypes (Huang et al., 1988; Marais and Parker, 1989); however, these are not consistent between laboratories. Thus PKC-(U has been re- ported to have a K, for histone 2-fold higher than either -&,2 or -y (Huang et al., 1988) but also to have a K, between that of PKC-& and -y (Marais and Parker, 1989). The reasons for these variations are not clear, although the different sources of kinase, histone and the presentation of lipid may cause these discrepancies. Nevertheless the specificity of phosphorylation of histone Hl with respect to phosphopeptide(s) generated has been shown to be the same for all these PKC isotypes (Huang et al., 1988; Marais and Parker, 1989). A similar result has been obtained for myelin basic protein (Huang et al., 1988) and protamine (Marais and Parker, unpublished). With the latter substrate we observed that while the V,, activities for PKC-a, $3, and -y were similar, there was a significant difference in the K, val- ues, with PKC-y showing a K, 3- to 4-fold higher than either -(Y or -& (Marais and Parker, 1989). Similar observations have been made for synthetic peptide substrates where variations in both K,
and v,, were observed indicating that certain primary structures show a limited selectivity for either -(Y, -& or -y (Marais and Parker, 1989).
These variations in K, and V,, for different substrates suggest that these PKC isotypes will phosphorylate different target proteins in vivo as discussed below.
Comparison of PKC-j3, and -p2 The preceding section has dealt for the most
part with PKC-/3,/P, (but see note above) since there have been no reports of the chromatographic separation of PKC-8, and -fi2 (these two isotypes coelute on hydroxylapatite). Thus the only com- parison of these two activities has been carried out using recombinant proteins purified from COS cells expressing the relevant cDNA construct (Ono et al., 1987b). This work has shown that there are no detectable differences in the activities of these kinases with respect to their regulation (Ono et al., 1987b), an observation which is entirely consistent with the identity of their regulatory domains (see above). More detailed analyses of these proteins are currently in progress employing purified pro- teins derived from the baculovirus expression sys- tem. The large (mg) amounts of PKC that can be produced in this expression system (Pate1 and Stabel, 1989) have been extremely useful not only in the analysis of these PKC isotypes but also in the characterization of the novel PKCs that have only been identified through cDNA cloning (see below).
Properties of PKC-6, -8, -5
PKC-6, -E and -l have to date been identified only through cDNA cloning (Ono et al., 1987a, 1988a, b; Ohno et al., 1988b; Schaap et al., 1989) and their activities have been investigated through transient expression of cDNA constructs (Ohno et al., 1988b; Ono et al., 1988a, b; Schaap et al., 1989). Thus we have described that transient ex- pression of the murine cDNA for -E increased phorbol ester binding activity in COS cells much as is seen for other PKCs (e.g. Knopf et al., 1986). On extraction and fractionation, however, this binding activity did not appear to have an associ- ated histone kinase activity, although the kinase activity of -E could be detected using a synthetic peptide based upon the -E pseudosubstrate site sequence (Fig. 2; Schaap et al., 1989). Thus PKC-E has a quite distinct substrate specificity compared

8
TABLE 1
RELATIVE SUBSTRATE SPECIFICITY OF PKC-a AND PKC-e
Peak I and control refer to the activities detected on ion-ex- change c~mato~aphy of COS cell extracts foIlowing trans- fection of PKC-e or a control vector respectively (see Schaap et al., 1989). The assays were performed either in the presence (+) or absence (-) of PS/TPA. Substrates were assayed at a final concentration of 1.25 mg/ml. Values are normahsed to an activity of 1.0 for the conventional PKC substrate histone 111s.
Substrate Peak I (-E) Control (-a)
+ - + -
Peptide-e 64 2 3.3 i 0.3 Histone IIS co.3 n < 0.3 n Histone IIAS c 0.3 n CO.3 n Histone 111s 1.0 < 0.3 1.0 < 0.3 Histone VS CO.3 n 0.3 n Histone VIS CO.3 n eO.3 n Histone VIIS X0.3 n 0.3 n Poly Arg-Ser 11 10 2.1 2.7 Poly Lys-Ser 40 25 2.0 2.3 Protamine sulphate 95 42 3.3 2.3 Protamine base 2 3 1.3 1.0 a-Casein GO.3 n < 0.3 n
n. not determined.
to PKC-a, -&,2 and -y as illustrated in Table 1. This observation has been confirmed and the anal- ysis extended with purified recombinant PKC-E expressed in insect cells (D.S. and P.J.P., sub- mitted). These findings are consistent with the data from Olmo et al., who ~~~eplet~ PKC-ar from COS cells in order to demonstrate the very low histone kinase activity expressed by PKC-e (Ohno et al., 1988b). As discussed previ- ously (Schaap et al., 1989) the activity described by Nishizuka and colleagues (Ono et al., 1988a, b) is likely to have been due to a proteolytic frag- ment of PKGE. It is not clear from the parallel studies on PKCd @no et al., 1988a, b) whether the described activity is that of the holoenzyme or of a 4 proteolytic fragment. To date no expres- sion studies have been carried out with PKC-[ due to the partial nature of the clones isolated (Ono et al., 1988a, b; C.W. and P.J.P., unpublished).
One other notable feature with respect to the properties of PICC-E is that both the phorbol bind- ing and kinase activities of -e are independent of
Ca2+ (Schaap et al., 1989). This is provocative with respect to the domain structure of the PKCs. Thus the deletion of the C, domain in PKC-6, -8, -[ (Fig. 3) may be associated with a loss in Ca2+ dependence; it will be possible to test this through domain exchanges between PKCs. In the same vein, the conserved lipid/phorbol binding activity of the regulatory domains may be associated with the conservation of the cysteine-rich region in these proteins (Fig. 3) and this may therefore constitute the binding domain(s); direct evidence that this may indeed be the case has come from the expression of deleted PKC-& constructs (Knopf et al., 1988).
A ~tion~ization for the PKC family
In the introduction to this review the question of whether or not these PKC-related genes con- stituted a family was posed. A comparison of the properties of PKCs -ty, -&,z, -y, -E as outlined above suggests that these proteins do indeed make up a family. It has been the convention to classify serine/threonine kinases into families on the basis of their regulation and the foregoing summary would indicate that the PKCs make up a family of lipid-dependent, diacyl~ycerol-activated protein kinases. Whether it is useful to subdivide the members up into Ca2+-dependent and Ca2+-inde- pendent PKCs is an issue that may be more clearly resolved when evidence is obtained as to the requirement for Ca2+ mobilization in vivo in the activation of the in~~du~ isotypes.
While we would argue that these proteins con- stitute a family, the biochemical analyses sum- marized above indicate that there are functional differences between members of this kinase family, as might be expected from the high degree of conservation of these isotypes from five mam- malian species (for a comparison see Parker et al., 1989). These functional variations are concerned both with the potency with which the physiologi- cal activators will act and the potential targets for the activated kinase activities. Thus it is antic- ipated that these isotypes do not function inter- changeably but rather where coexpressed they act in parallel to transduce specific cellular responses.
The relatively subtle differences in the activa- tion of PKCs -OL, -&,2 and -y by diacylglycerols

9
may permit the selective activation of one isotype under situations where the extent of diacylglycerol accumulation is limited. There is evidence that the extent/rate of diacylglycerol production can be limiting, for example the potentiat~g effect of the diglyceride (DG)-kinase inhibitor R59022 in the activation of neutrophils (Dell et al., 1988). Whether the diverse phosphatidylinositol 43 -bisphosphate (PIP&specific phospholipase Cs that have been described (Katan et al., 1988; Stahl et al, 1988; Suh et al., 1988a, b) are coupled to different receptor pathways to produce di- acylglycerol at different rates remains to be de- termined; however, it is evident from the rapid metabolism of diacylglycerol that the rate of in- ositol lipid hydrolysis will greatly influence the extent of accumulation. The possibility exists therefore that specific receptors may either activate one or more of the PKC isotypes through differen- tial coupling pathways. The consequences of activation will also vary through the differences in the efficiencies with which these isotypes will phosphorylate target proteins. The very evident difference in specificity of PKC-e with respect to
PKCs -a, -8r,2, -y (Schaap et al., 1989) is readily incorporated into a scheme of parallel distinct pathways (see Fig. 5). The more subtle variations in the specificities of PKC-cy, -&,z and -y observed in vitro are less obviously accommodated into such a scheme, but it should be noted that through the action of phosphatases the phosphorylation of proteins in vivo is dynamic; the relatively modest differences between these PKCs detected in vitro under initial rate conditions will therefore be exaggerated in vivo.
The preceding comments on the possible paral- lel pathways operating through different PKCs have so far been confined to their role down- stream of inositol lipid degradation, as originally proposed by Nishizuka and colleagues (Takai et al., 1979). However, the agonist-induced produc- tion of diacylglycerol could, in theory, come from phospholipase C (PLC) hydrolysis of any cellular lipid. Thus there is evidence that phosphati- dylcho~e hydrolysis through a PLC may also be receptor coupled (e.g. Bocckino et al., 1985; Irving and Exton, 1987). The question that therefore arises, is whether the DAG that is produced in the absence of inositol1,4,Misphosphate (IP,) is suf-
Fig. 5. Signal transduction and PKC isotypes. The scheme
ilhtstrates the ‘classical’ pho~~tidy~ositol pathway with the
IP, head group being responsible for Ca2+ release from the
endoplasmic reticulum and diacylglycerol for the activation of
all the PKC isotypes. Tbis activation will induce the phos-
phorylation of a spectrum of target proteins, the nature of which will be dependent upon the pattern of &type expression
(see text). In parallel is shown a distinct pathway of di-
acylglycerol production (e.g. phospholipase C hydrolysis of
phosphatidylcholine) which in the absence of any CL?+ release would be predicted to lead to the full activation of only a
subset of the PKC isotypes (see text).
ficient to activate one or other PKC. The answer to this depends upon the issue of the Ca2+ depen- dence for the -1y, -&,2, -y, and -E isotypes. As discussed above the K, for Ca2+ for the -LY, -j3,,2 and -y isotypes lies within the physiological limits of cytosolic free Ca2+ and as such the degree of activation may be influenced by the mobilization of Ca2+. Thus it could be argued that in the absence of IP3 production -(Y, -&,z and -y may not be activated above some critical threshold level required to drive the phospho~la~on of target proteins, but by contrast the Ca2+ indepen- dence of PKC-e (and 6?, l?) would permit activa- tion of this isotype in the absence of Ca2+ mobih- zation. In a sense the very existence of the inde- pendent activities (E, S?, {?) would suggest that these polypeptides have been conserved to in- crease the options available to cells in the way they respond to different agonists. The variations in the substrate specificity of these kinases further diversify the potential for response (Fig. 5).

10
Perspectives
The various properties of the PKC isotypes and some of the possible physiological consequences are outlined above and shown schematically in Fig. 5, but do these hypothetical schemes have any bearing on reality? It is now necessary to de- termine how the in vitro behaviour of these pro- teins relates to their performance in vivo. This will be much helped by the availability of cDNAs that will permit the expression and/or overexpression of various combinations of normal or mutated PKCs. It is anticipated that novel approaches to the monitoring of the activation of individual iso- types will also prove important (Mitchell et al., 1989). Ultimately the goal will be to define the roles that this family of proteins play in different physiological (and pathological) contexts and this will come from a detailed description of the sub- strates for these kinases and the situations in which they become activated.
References
Asaoka, Y., Kikkawa, U., Sekiguchi, K., Shearman, MS.,
Kosaka, Y., Nakano, Y., Satoh, T. and Nishizuki, Y. (1988)
FEBS Lett. 231, 221-224.
Ashendel, C.L. (1985) B&him. Biophys. Acta 822, 219-242.
Bocckino, S.B., Blackmore, P.F. and Exton, J.H. (1985) J. Biol.
Chem. 260, 14201-14207.
Coussens, L., Parker, P.J., Rhee, L., Yang-Feng, T.L., Chen, E.,
Waterfield, M.D., Francke, U. and Ulhich, A. (1986) Sci-
ence 233, 859-866.
Coussens, L., Rhee, L., Parker, P.J. and Ullrich, A. (1987)
DNA 6, 389-394.
Dell, K.R., Walsh, M.P. and Severson, D.I. (1988) Biochem. J.
254, 455-462.
Hanks, S.K., Quin, A.M. and Hunter, T. (1988) Science 241,
42-52.
Hannun, Y.A., Loomis, C.R. and Bell, R.M. (1985) J. Biol.
Chem. 260,10039-10043.
Hoshijima, M., Kikuchi, A., Tanimoto, T., Kaibuchi, K. and
Takai, Y. (1986) Cancer Res. 46, 3000-3004.
House, C. and Kemp, B.E. (1987) Science 238, 1726-1728.
Housey, G.M., O’Brian, C.A., Johnson, M.D., Kirschmeier, P.
and Weinstein, LB. (1987) Proc. Natl. Acad. Sci. U.S.A. 84,
1065-1069.
Huang, K.-P. and Huang, F.L. (1986) B&hem. Biophys. Res.
Commun. 139, 320-326.
Huang, K.-P., Nakabayashi, H. and Huang, F.L. (1986) Proc.
Natl. Acad. Sci. U.S.A. 83, 8535-8539..
Huang, F.L., Yoshida, Y., Nakabayashi, H., Knopf, J., Young, W.S. and Huang, K.-P. (1987) B&hem. Biophys. Res.
Commun. 149, 946-952.
Huang, K.-P., Huang, F.L., Nakabayashi, H. and Yoshida. Y.
(1988) J. Biol. Chem. 263, 14839-14845.
Ido, M., Sekiguchi, K., Kikkawa, U. and Nishizuka, Y. (1987)
FEBS Lett. 219, 215-218.
Inoue, M., Kistimoto, A., Takai, Y. and Nishizuka, Y. (1977) J.
Biol. Chem. 252, 7610-7616.
Irving, H.R. and Exton, J.H. (1987) J. Biol. Chem. 262,
3440-3443.
Ishikawa, F., Takaku, F., Nagao, M. and Sugimura, T. (1986)
Jpn. J. Cancer Res. 77, 1183-1187.
Jaken, S. and Kiley, S.C. (1987) Proc. Natl. Acad. Sci. U.S.A.
84,4418-4422.
Kamps, M.P., Taylor, S.S. and Sefton, B.M. (1984) Nature 310,
589-592.
Katan, M., Kriz, R.W., Totty, N., Philp, R., Medrum, E.,
Aldape, R.A., Knopf, J. and Parker, P.J. (1988) Cell 54,
171-177.
Kikkawa, U., Ogita, K., Ono, Y., Asaoka, Y., Shearman, M.S.,
Fujii, T., Sekiguchi, K., Igarashi, K. and Nishizuka, Y.
(1987a) FEBS Lett. 223, 212-216.
Kikkawa, U., Ono, Y., Ogita, K., Fujii, T., Asaoka, Y., Seki-
guchi, K., Kosaka, Y., Igarashi, K. and Nishizuka, Y.
(1987b) FEBS Lett. 217, 227-231.
Knopf, J.L., Lee, M.-H., Sultzman, L.A., Kriz, R.W., Loomis,
CR., Hewick, R.M. and Bell, R.M. (1986) Cell 46,491-502.
Kosaka, Y., Ogita, K., Ase, K., Nomura, H., Kikkawa, U. and
Nishizuka, Y. (1988) Biochem. Biophys. Res. Commun.
151, 973-981.
Kubo, K., Ohno, S. and Suzuki, K. (1987a) Nucleic Acid Res.
15, 7179-7180.
Kubo, K., Ohno, S. and Suzuki, K. (1987b) FEBS Lett. 223,
138-142.
Maraganore, J.M. (1987) Trends Biochem. Sci. 12, 176-177.
Marais, R.M. and Parker, P.J. (1989) Eur. J. B&hem. (in
press).
Mitchell, F., Marais, R.M. and Parker, P.J. (1989) B&hem. J.
(in press).
Murakami, K. and Routtenberg, A. (1985) FEBS Lett. 192,
189-193.
Murakami, K., Chan, S. and Routtenberg, A. (1986) J. Biol.
Chem. 261, 15424-15429.
Nishizuka, Y. (1986) Science 233, 305-312.
Nishizuka, Y. (1988) Nature 334, 661-665.
Nunn, D.L. and Watson, S.P. (1987) Biochem. J. 234, 809-813.
Ohno, S., Kawasaki, H., Imajoh, S. and Suzuki, K. (1987)
Nature 325, 161-166.
Ohno, S., Kawasaki, H., Konno, Y., Inagaki, M., Hidaka, H.
and Suzuki, K. (1988a) Biochemistry 27, 2083-2087.
Ohno, S., Akita, Y., Konno, Y., Imajoh, S. and Suzuki, K.
(1988b) Cell 53, 731-741.
Ono, Y., Kurokawa, T., Fujii, T., Kawahara, K., Igarishi, K.,
Kikkawa, U., Ogita, K. and Nishizuka, Y. (1986) FEBS
Lett. 206, 347-352.
Ono, Y., Fujii, T., Ogita, K., Kikkawa, U., Igarashi, K. and
Nishizuka, Y. (1987a) FEBS Lett. 226,125-128.
Ono, Y., Kikkawa, U., Ogita, K., Fujii, T., Kurokawa, T.,
Asaoka, Y., Sekiguchi, K., Ase, K., Igarashi, K. and
Nishizuka, Y. (1987b) Science 236, 1116-1120.

11
Ono, Y., Fujii, T., Ogita, K., Kikkawa, U., Igarashi, K. and Nishizuka, Y. (1988a) J. Biol. Chem. 263,6927-6932.
Ono, Y., Fujii, T., Igarashi, K., Kikkawa, U., Ogita, K. and Nishizuki, Y. (1988b) Nucleic Acid Res. 16, 5199-5200.
Parker, PJ. and Ulhich, A. (1987) in Hormones and Cell Regulation, Vol. 11 (Dumont, J.E. and Nunez, J. eds.), pp. 41-48, Colloque Inserrn/John Libbey Eurotext, Vol. 153.
Parker, P.J., Coussens, L., Totty, N., Rhee, L., Young, S., Chen, E., Stabel, S., Waterfield, M.D. and Ullrich, A. (1986) Science 233, 853-859.
Parker, P.J., Ulhich, A. and Stabel, S. (1989) in Genes and Signal Transduction in Multistage Carcinogenesis, Ch. 13 (Colbum, N., ed.), pp. 263-278, Marcel Delcker, New York.
Patel, G. and Stabel, S. (1989) Cell. Signal. (in press). Rapp, U.R., Cleveland, J.L. and Bonner, T.I. (1988) in
Handbook of Oncogenes, Ch. 14 (Reddy, P., Curran, T. and Skalka, A., eds.), pp. 213-253, Elsevier, Amsterdam.
Rosenthal, A., Rhee, L., Yadegari, R., Paro, R., Ulhich, A. and Goeddd, D.V. (1987) EMBO J. 6,433-441.
Schaap, D., Parker, P.J., Bristol, A., Kriz, R. and Knopf, J. (1989) FEBS Lett. 243, 351-357.
Seifert, R., Schachtele, C., Rosenthal, W. and Schultz, G. (1988) B&hem. Biophys. Res. Commun. 154,20-26.
Stahl, M.L., Ferenz, C.R., Kelleher, K.S., Kriz, R.W. and Knopf, J.L. (1988) Nature 332, 269-272.
Suh, P.-G., Ryu, S.H., Moon, K.H., Suh, H.W. and Rhee, S.G. (1988a) Cell 54, 161-169.
Suh, P.-G., Ryu, S.H., Muon, K.H., Suh, H.W. and Rhee, S.G. (1988b) Proc. Natl. Acad. Sci. U.S.A. 85, 5419-5423.
Takai, Y., Kishimoto, A., Inoue, M. and Nishixuka, Y. (1977) J. Biol. Chem. 252, 7603-7609.
Takai, Y., Kishimoto, A., Kikkawa, U., Mori, T. and Nishizuka, Y. (1979) Biochem. Biophys. Res. Commun. 91,1218-1224.
Woodgett, J.R., Hunter, T. and Gould, K.L. (1987) in Cell Membranes: Methods and Reviews, Vol. 3 @son, E.L., Frazier, W.A. and Glaser, L., eds.), pp. 215-340, Plenum, New York.
Young, S., Rothbard, J. and Parker, P. (1988) Eur. J. B&hem. 173, 247-252.




















