
Crystallographic structure and morphology of bithiophene-fluorene polymer nanocrystals
Upload
independentCategory
view
1download
0
lable at ScienceDirect
Polymer 52 (2011) 3368e3373
Contents lists avai
Polymer
journal homepage: www.elsevier .com/locate/polymer
Crystallographic structure and morphology of bithiophene-fluorene polymernanocrystals
Oliver Werzer a,b, Roland Resel a,*, Boril Chernev c, Harald Plank c,d, Michael M. Rothmann e,Peter Strohriegl e, Gregor Trimmel f,g, Arnaldo Rapallo h, William Porzio h
a Institute of Solid State Physics, Graz University of Technology, Petersgasse 16, A-8010 Graz, AustriabCentre for Organic Electronics, University of Newcastle, AustraliacCentre for Electron Microscopy, Graz, Austriad Institute for Electron Microscopy, Graz University of Technology, Austriae Institute of Macromolecular Chemistry, University of Bayreuth, Germanyf Institute for Chemistry and Technology of Materials, Graz University of Technology, AustriagChristian Doppler Laboratory for Nanocomposite Solar Cells, Graz University of Technology, Austriah Istituto per lo Studio delle Macromolecole, Consiglio Nazionale delle Ricerche, Milano, Italy
a r t i c l e i n f o
Article history:Received 26 January 2011Received in revised form28 April 2011Accepted 29 April 2011Available online 6 May 2011
Keywords:Crystal structureCrystal morphologyF8T2
* Corresponding author. Tel.: þ43 316 873 8476; faE-mail address: [email protected] (R. Resel).
0032-3861/$ e see front matter � 2011 Elsevier Ltd.doi:10.1016/j.polymer.2011.04.063
a b s t r a c t
Nanocrystals of the polymer poly(9,9-dioctylfluorenyl-co-bithiophene) (F8T2) with a molecular weightof 3.2 kg/mol are grown in a para-xylene solution. The typical morphology of the crystals is needle likewith typical widths of 50 nm and lengths of about 200 nm. The crystal structure and morphology arestable up to a temperature of 353 K. The structure solution is obtained by x-ray powder diffraction (XRD)pattern with data modelling by a stochastic global optimization procedure which allows simultaneousindexing and molecular packing determination. Final Rietveld refinement was applied on the mostpromising crystal structure with a ¼ 1.376 nm, b ¼ 3.105 nm, c ¼ 2.690 nm and ß ¼ 109.5� within thespace group C2/c choosing the polymer backbone parallel to the b-axis. The structural motifs of themolecular packing could be identified: aromatic units within a single polymer chain are slightly bentrelative to the chain axis, octyl side chains are aligned along the polymer backbone and aromatic units ofneighbouring molecules display a strong tendency to stack parallel to each other. XRD results of F8T2with a molecular weight of 19 kg/mol reveal the same peak positions compared to the 3.2 kg/molmaterial, showing that both materials crystallise similarly and can be described by the same crystallo-graphic unit cell. The smaller peak intensities together with the broader peak widths, however, show thatthe ability of crystal formation for the 19 kg/mol material is reduced.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Recently poly(9,9-dioctylfluorenyl-co-bithiophene) (F8T2) beco-mes important for the realisation of electronic as well as for opto-electronic applications like thin film transistors, optical emitters,detectors and solar cells [1e6]. Despite the large variety of applica-bility, there is only small knowledge about the packing of thesespecific polymer backbones within the solid state.
Generally, the arrangement of the conjugated molecules withinthe solid state has large influence on the electronic properties [7].Conjugated polymers with branching side chains can arrange inmany different ways relative to neighbouring polymer chains
x: þ43 316 873 8466.
All rights reserved.
whereby the location and density of the alkyl chain attachment sitesas well as the length of the alkyl chains has a decisive influence. Thepacking of the side chains leads to a variety of crystalline statesranging from amorphous to liquid crystalline or highly crystalline.
F8T2 is a copolymer with an alternating sequence of a dioctyl-fluorene moiety and two thiophene rings along the polymer chain(see inset of Fig. 2). Besides F8T2, two further prominent conjugatedpolymers are poly(3-hexyl)thiophene (P3HT) and poly(dioctyl-fluorene) (PF8). The polymer P3HT e a polymer formed by thio-phene unitswith regularly attached side chains - shows a crystallinecharacter. The thiophene units form p-p stacking sheets that areseparated by the hexyl chains from each other [8,9]. The lamellaorder results in crystals where even chain folding within stacks wasobserved [10]. The polymer PF8e a polymer consisting of side chainsubstituted dioctyl-fluorene units - shows a variety of crystallo-graphic phases like a-phase, b-phase as well as a nematic liquid

Fig. 1. Atomic force microscopy of a dry drop casted film of the polymer F8T2 with molecular weight of 3.2 kg/mol. On large scales the height image is depicted (left), while a phaseimaging micrograph is given at higher resolution (right).
O. Werzer et al. / Polymer 52 (2011) 3368e3373 3369
crystalline phase [11e14]. All of these phases have in common thata closep-stackingof thephenyl rings is notobserveddue to a twistofadjacent fluorene units resulting in a helix conformation of thepolymer backbone along the polymer axis.
The polymer F8T2 shows a glass transition at 380 K due tomelting of the octyl side chains, a thermal induced crystallinephase formed at a temperature of 393 K [15] and a thermalinduced liquid crystalline phase at a temperature of 463 K [16,17].While all these phases show only small molecular weightdependencies for Mw � 40 kg/mol [18] the nematiceisotropictransition shows a strong molecular weight dependence; highermolecular weight F8T2 melts at higher temperature (e.g. 19 kg/mol melts at 563 K [17] and 3.2 kg/mol 428 K [19]). Due to thechemical similarity of PF8 and F8T2 a similar crystallinearrangement is expected. However the introduction of two thio-phene rings along the polymer backbone is expected to introduceadditional restriction to the polymer e polymer confinement,similar to P3HT in which the sulphur group of adjacent chains aremirrored by 180B. This together with the additional open space,due to the lack of octyl-chains at the thiophene units, allows fordifferent arrangements of the fluorene units.
Within this work F8T2 nanocrystals are investigated by atomicforce microscopy and x-ray diffraction. The molecular packing is
Fig. 2. Specular x-ray diffraction scans of as-prepared 3.2 kg/mol (red) and 19 kg/mol(black) F8T2 films. The green line represents the diffraction pattern of 3.2 kg/mol F8T2after heat treatment at 353 K. The curves are shifted for clarity. (For interpretation ofthe references to colour in this figure legend, the reader is referred to the web versionof this article.)
determined by a combined experimental and theoretical approachrevealing the arrangement of the polymer chains within the unitcell.
2. Materials and methods
F8T2 with a molecular weight of 3.2 kg/mol (Mw/Mn ¼ 1.6) and19 kg/mol were used for the experiment. The 3.2 kg/mol F8T2 wassynthesised following the procedure given in [18]. The 19 kg/molF8T2 polymer was purchased from American Dye Source Ltd. andused without further purification. Nanocrystals of F8T2 were ob-tained by dissolving F8T2 in para-xylene with a concentration of25 mg/ml at a temperature of 353 K by subsequent cooling to roomtemperature. The initial clear solution of the 3.2 kg/mol F8T2material turned turbid andnon transparent after twodays, revealingthe formation of aggregates. Similarly, a turbid solution was ob-tained for the 19 kg/mol material, but after a time period of twoweeks. These turbid solutions are stable up to 353 K, at which thesolutions became clear again. On cooling, the solutions turnedturbid again after the respective time period, revealing the revers-ibility of nanocrystal formation in para-xylene.
Films of F8T2 nanocrystals were prepared by drop casting theturbid solution onto thermally oxidised silicon wafers. The bulksolventwas removed at avacuumof 10�6mbar at room temperature.
Atomic force microscopy (AFM) measurements were performedwith a Dimension3100 microscope equipped with a Hybrid closedloop scan head and a Nanoscope IVa controller (Digital Instru-ments, VEECO). All measurements were done in TappingMode�with different Olympus cantilevers (2e40 N/m) depending on thesample requirements.
Fourier transform infrared absorption spectroscopy (FTIR)measurements were performed using a Bruker Equinox 55 FTIRspectrometer equipped with a Bruker Hyperion 3000 infraredmicroscope and a single - element MCT detector. Thin films weredrop casted on BaF2 support and the transmission spectra werecollected using a �15 Cassegrain objective with spectral resolutionof 4 cm�1 and averaging over 32 interferograms. The area for eachmeasurement was approximately 100 � 100 mm2.
X-ray diffraction (XRD) experiments were performed witha Bruker D8 Discover diffractometer equipped with a parallelizingpolycapillar optic at the primary side. The radiation is provided bya copper sealed tube (l ¼ 0.154 nm) and is monochromatized bya secondary side graphite monochromator. Temperature depen-dent in-situ x-ray investigations were performed with the DHS 900heating attachment [20] from Anton Paar GmbH (Austria) undera vacuum of 10�3 mbar to reduce sample degradation.

O. Werzer et al. / Polymer 52 (2011) 3368e33733370
The crystal structure determination of the F8T2 crystals wasdealt with by means of the VARICELLA package [21]. The methodallows for searching the cell parameters together with the polymerconformations, orientations and packing, so that a prior indexing ofthe spectrum before the structure solution is not required. Themolecular model is completely flexible (i.e. no constraints areapplied to the stiff molecular degrees of freedom, hence bondlengths and bending angles can vary during the search). Thesampling of the conformational space available to the molecules isguided by two factors, a potential energy provided by the MM2force field as implemented in TINKER [22] and a disagreementfactor between the calculated and the experimental profiles. All theinteractions among the different molecules within the primary celland its periodic images are completely accounted for withina suitable cut-off, during the calculations. Molecular moves areobtained by means of the molecular dynamics, while the cellparameter values are changed in a random fashion. This hybriditerative Monte Carlo procedure starts from any initial crystalstructure and improves the solution according to a global optimi-zation strategy. Input parameters to the method are the spacegroup and the number of independent monomers/molecules.
Themethodwas enrichedwith different featureswith respect toits original formulation, including the possibility to take intoaccount preferred orientations and procedures for better samplingof the octyl side chain conformations. The calculated structuresyielding the best compromise between crystal energy anddisagreement factor between the calculated and the experimentaldiffraction patterns were further optimised by a Rietveld refine-ment procedure. The optimization was performed by minimizingthe weighted profile parameter (Rwp) which writes as:
Rwp ¼
0BB@
Piwi
�Ysimð2qiÞ � Iexpð2qiÞ þ Ybackð2qiÞ
�2
Piwi
�Iexpð2qiÞ
�2
1CCA
1=2
where Ysim is the calculated diffraction pattern, Iexp the experi-mental one, Yback the background subtracted profile and wi anunitary weight applied at each point. The Rietveld optimizationwasperformed with the MATSTUDIO package [23] which accounts forpeak width and asymmetry, crystallite size and lattice strains,orientational parameter, main chain rotation around the b-axis andtranslations along the three cell axes.
3. Experimental results
Fig. 1 (left) shows a typical 10 mm � 10 mm AFM height scan ofa drop casted film prepared from the turbid solution revealinga wavy morphology with maximum height variations of about400 nm. This wide range height variation is attributed to the dropcast process. However, AFM phase images with larger magnifica-tion reveal needle like structures as shown in Fig. 1 (right). Theneedles show a typical width of 45 nm with lengths ranging from100 to 300 nm 2D Fourier analyses of the height and phase imagesdo not reveal a preferred lateral orientation suggesting a randomalignment of the needles at the silica surface.
Crystallographic studies on the F8T2 films were performed byspecular XRD scans, i.e. the scattering vector was kept perpendic-ular to the substrate surface. The XRD pattern for the as-prepared3.2 kg/mol and 19 kg/mol sample are depicted in Fig. 2 andreveal similar diffraction pattern of both samples. Firstly, two broadpeaks with maxima at about 4.9� and 19.7� are noted which belongto the amorphous fraction of F8T2 without crystallographic longrange order of the polymer chains. Secondly, numerous clear andsharp Bragg peaks are present. The diffraction patterns of the two
samples appear at the same peak positions, suggesting that thesame crystalline phase has been formed for the different molecularweights. However, the higher peak intensities and the smaller peakwidth together with the less pronounced amorphous contributionof the 3.2 kg/mol sample compared to the 19 kg/mol indicates thatthe crystallinity in the 3.2 kg/mol sample is higher.
The first and most intense peak is located at 6.88�which corre-
sponds to a real space repeating distance (d-spacing) of 1.28 nm. Ingeneral such a distance is connected with the backboneebackbonedistance separated by the alkyl side chains [24]. Please note that thisdistance is considerably smaller than the length of fully extendedoctyl units, which would result into a d-spacing of at least 2.1 nm, ifinterdigitation of the octyl side chains from neighbouring polymerbackbones would be present. Next to the strong first peak there areseveral additional peaks present revealing a three - dimensionalcrystallographic long range order within the F8T2 films. This is incontrast to P3HT which shows three strong peaks connected to thelamella sheet separation andoneweakpeak connected to intrachainrepeating units across the backbone [8].
As the samples were prepared from solutions, the questionarises if xylene is incorporated within the crystalline structure. IRspectroscopy results (not shown) do not provide an evidence ofincorporated solvent within the vacuum dried thin films. IR peaksbelonging to xylene were only observed right after the drop castingprocess, but on vacuum treatment the amount of xylene (if any)dropped below the detection limit.
For the crystal structure determination of the F8T2 nanocrystalsthe diffraction pattern of the 3.2 kg/mol sample was chosen. Byconsidering the particular chemical species involved, suitable spacegroups were chosen and different searches were performed, whereeither one or twomonomers in the asymmetric unit were included.Within the orthorhombic system, Pbca and P212121 were tested,while P2, P21/c,and C2/c were tried in themonoclinic setting. By far,C2/c emerged as the best choice, allowing for both a close packingof the polymer backbones and reasonable arrangements of the alkylside chains within the cells. All the other groups enforced unreal-istically bent molecular backbone conformations and too highcrystal densities (up to 1.44 g/cm3), and were therefore discarded.Moreover the disagreement factor obtained from the initial calcu-lations for the discarded groups was always much higher than forthe C2/c case. The calculated patternwas further refined against theexperimental diffraction pattern, the result is given in Fig. 3. A finalvalue of Rwp ¼ 0.24 was obtained, which can be considered quiteacceptable, given the evident high level of uncertainty contained inthe experimental XRD data. Such a lack of details in the XRD patternproduces some too close contacts among adjacent macromolecules.In this sense the given structure must be considered as a likelystructural motif of the polymeric crystal. The proposed structurecontains 16 monomer units in the unit cell of a ¼ 1.376 nm,b ¼ 3.105 nm, c ¼ 2.690 nm and beta ¼ 109.5�, and has a crystalmass density of 1.355 g/cm3.
The results reveal a significant preferred orientation (texture)along the (0 0 1) direction. Aside from the amorphous bump cen-tred at 19�, which approximately contributes 50% to the diffractionsignal, evident overlapping of more reflections takes place in theprofile. As a result the majority of peak widths exceeds 0.5�. Hencethe calculation of crystallite size along any crystallographic direc-tion is affected by relevant peak deconvolution problem, except forthe first peak belonging essentially to (0 0 2) and in small part to(�1 1 1) reflections. For this direction, normal to the polymerbackbone, a crystallite dimension of 15 nm has been derived usingline profile analysis [25]. The obtained conformation of one poly-mer backbone is depicted in Fig. 4. The crystallographic unit cell hasthe length of two monomer units, and the repetition of the chainconformation appears due to the space group after two monomer
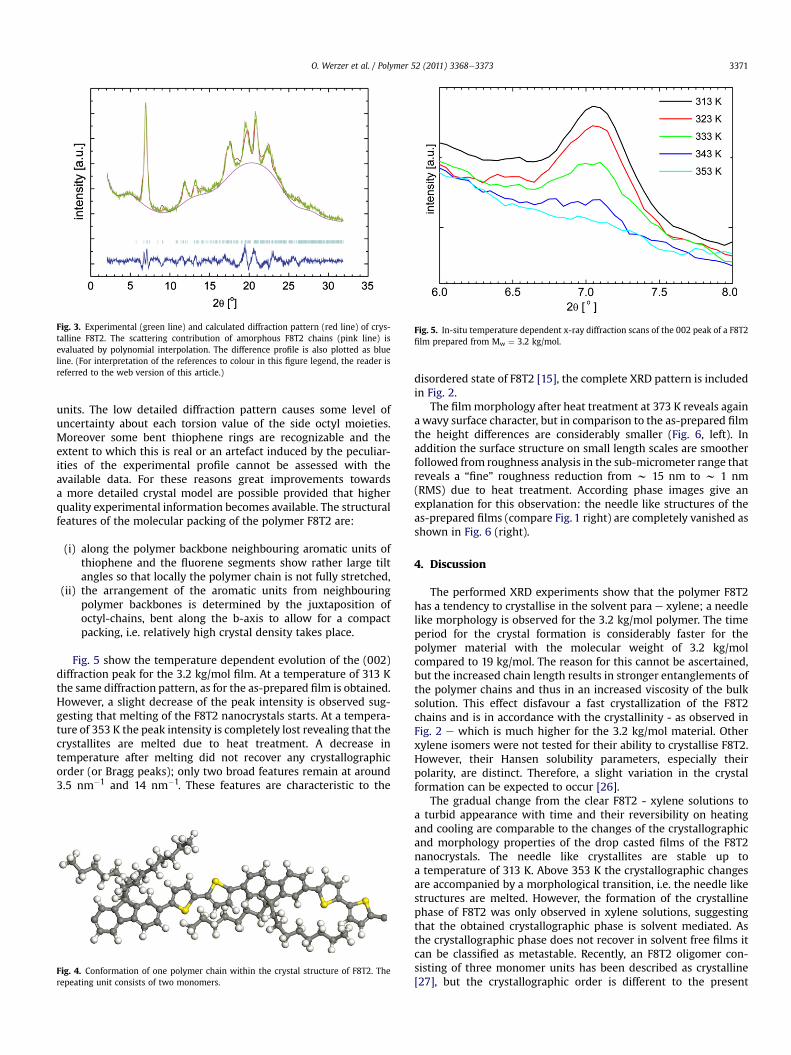
Fig. 5. In-situ temperature dependent x-ray diffraction scans of the 002 peak of a F8T2film prepared from Mw ¼ 3.2 kg/mol.
Fig. 3. Experimental (green line) and calculated diffraction pattern (red line) of crys-talline F8T2. The scattering contribution of amorphous F8T2 chains (pink line) isevaluated by polynomial interpolation. The difference profile is also plotted as blueline. (For interpretation of the references to colour in this figure legend, the reader isreferred to the web version of this article.)
O. Werzer et al. / Polymer 52 (2011) 3368e3373 3371
units. The low detailed diffraction pattern causes some level ofuncertainty about each torsion value of the side octyl moieties.Moreover some bent thiophene rings are recognizable and theextent to which this is real or an artefact induced by the peculiar-ities of the experimental profile cannot be assessed with theavailable data. For these reasons great improvements towardsa more detailed crystal model are possible provided that higherquality experimental information becomes available. The structuralfeatures of the molecular packing of the polymer F8T2 are:
(i) along the polymer backbone neighbouring aromatic units ofthiophene and the fluorene segments show rather large tiltangles so that locally the polymer chain is not fully stretched,
(ii) the arrangement of the aromatic units from neighbouringpolymer backbones is determined by the juxtaposition ofoctyl-chains, bent along the b-axis to allow for a compactpacking, i.e. relatively high crystal density takes place.
Fig. 5 show the temperature dependent evolution of the (002)diffraction peak for the 3.2 kg/mol film. At a temperature of 313 Kthe same diffraction pattern, as for the as-prepared film is obtained.However, a slight decrease of the peak intensity is observed sug-gesting that melting of the F8T2 nanocrystals starts. At a tempera-ture of 353 K the peak intensity is completely lost revealing that thecrystallites are melted due to heat treatment. A decrease intemperature after melting did not recover any crystallographicorder (or Bragg peaks); only two broad features remain at around3.5 nm�1 and 14 nm�1. These features are characteristic to the
Fig. 4. Conformation of one polymer chain within the crystal structure of F8T2. Therepeating unit consists of two monomers.
disordered state of F8T2 [15], the complete XRD pattern is includedin Fig. 2.
The film morphology after heat treatment at 373 K reveals againa wavy surface character, but in comparison to the as-prepared filmthe height differences are considerably smaller (Fig. 6, left). Inaddition the surface structure on small length scales are smootherfollowed from roughness analysis in the sub-micrometer range thatreveals a “fine” roughness reduction from w 15 nm to w 1 nm(RMS) due to heat treatment. According phase images give anexplanation for this observation: the needle like structures of theas-prepared films (compare Fig. 1 right) are completely vanished asshown in Fig. 6 (right).
4. Discussion
The performed XRD experiments show that the polymer F8T2has a tendency to crystallise in the solvent para e xylene; a needlelike morphology is observed for the 3.2 kg/mol polymer. The timeperiod for the crystal formation is considerably faster for thepolymer material with the molecular weight of 3.2 kg/molcompared to 19 kg/mol. The reason for this cannot be ascertained,but the increased chain length results in stronger entanglements ofthe polymer chains and thus in an increased viscosity of the bulksolution. This effect disfavour a fast crystallization of the F8T2chains and is in accordance with the crystallinity - as observed inFig. 2 e which is much higher for the 3.2 kg/mol material. Otherxylene isomers were not tested for their ability to crystallise F8T2.However, their Hansen solubility parameters, especially theirpolarity, are distinct. Therefore, a slight variation in the crystalformation can be expected to occur [26].
The gradual change from the clear F8T2 - xylene solutions toa turbid appearance with time and their reversibility on heatingand cooling are comparable to the changes of the crystallographicand morphology properties of the drop casted films of the F8T2nanocrystals. The needle like crystallites are stable up toa temperature of 313 K. Above 353 K the crystallographic changesare accompanied by a morphological transition, i.e. the needle likestructures are melted. However, the formation of the crystallinephase of F8T2 was only observed in xylene solutions, suggestingthat the obtained crystallographic phase is solvent mediated. Asthe crystallographic phase does not recover in solvent free films itcan be classified as metastable. Recently, an F8T2 oligomer con-sisting of three monomer units has been described as crystalline[27], but the crystallographic order is different to the present

Fig. 6. Atomic force microscopy of the F8T2 (Mw ¼ 3.2 kg/mol) film after annealing at 373 K: height images (left) and phase images (right).
O. Werzer et al. / Polymer 52 (2011) 3368e33733372
polymer investigation. However, this suggests, that the dioctyl-fluorene e bithiophene moieties have a strong tendency for crys-tallization. In the present case of the polymer with a high numberof repeating units, crystallization is strongly effected by theincreased number of possible polymer chain conformation as wellas polymer backbone twisting along its axis. These entropicaleffects hinder the crystallization and a much less definedarrangement of the polymer chains compared to the oligomerresults. The similarity of the x-ray diffraction pattern shows thatboth molecular weight F8T2 samples are representative for the
Fig. 7. Crystal packing as viewed along mainechain axis b (top) and perspective viewomitted for clarity.
polymer limit; below the oligomer limit a different crystallographicphase is formed [27].
Within our structural motif, a polymer backbone with tilted andslightly bent aromatic units relative to each other together withdirected octyl-chains parallel to the backbone itself, emerges fromthe present structural analysis. Two polymer backbones are alwaysconnected in single columns where the octyl-chains are in thecentre of the columns and the aromatic units within the columnsare closely packed together. This packing involves both thiopheneand fluorene units. The overall packing is constituted by repetition
(bottom), only two neighbouring chains are represented and hydrogen atoms were

O. Werzer et al. / Polymer 52 (2011) 3368e3373 3373
of adjacent column couples of polymer chains developing alongboth a and c axes, separated by side octyl-substituents but also byparallel packing of the aromatic units (see Fig. 7).
These main structural features can be compared to the struc-tural features of the polymeric system PF8. Although a large varietyof the polymer backbone confirmation is concluded for thedifferent phases, neighbouring aromatic units are neither collinearnor coplanar [28]. Moreover in agreement with our results, theanti-parallel alignment of the octyl side-chains is proven for theb-phase of PF8 [13]. But the aromatic units of neighbouring chainsstack parallel to each other, the main feature of the crystal structureP3HT.
5. Conclusion
The formation of crytalline F8T2 in the solvent para e xylene isobserved. XRD and AFMmeasurements of the 3.2 kg/mol F8T2 filmsshow the presence of nanocrystallites with needle likemorphology.On heat treatment the crystalline long range order and the needlelike morphology is lost. The crystal structure of F8T2 is proposedbased on x-ray diffraction studies combined with a recentlydeveloped crystal structure solution method for polymers. It isfound that the crystal structure of F8T2 has structural elementscomparable to poly(octly-fluorene) and poly(hexylthiophene),although the polymer chain conformation are neither colinear norcoplanar. But individual F8T2 polymer chains forms closely packedcolumns and the aromatic units from neighbouring chains arestacked parallel to each other. XRD measurements on the 3.2 kg/mol and the 19 kg/mol F8T2 samples reveal the same peak posi-tions and can be explained by the common crystallographic unitcell of a ¼ 1.376 nm, b ¼ 3.105 nm, c ¼ 2.690 nm and beta ¼ 109.5�.Variation of relative peak intensities are a result of different abilityof crystallization which results in a different preferred orientationsof the crystallites; the small number of repeating units in the 3.2 kg/mol material allows for a faster adaptation of their conformation toneighbouring polymer chains which lead to a more definedcrystallization.
Acknowledgements
Financial support by the Austrian Nano-Initiative (researchproject cluster ISOTEC), the Christian Doppler Research Association
(CDG) and the Austrian Federal Ministry of Economy, Family andYouth (BMWFJ) is gratefully acknowledged.
Appendix. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.polymer.2011.04.063.
References
[1] Sirringhaus H, Wilson RJ, Friend RH, Inbasekaran M, WuW,Woo EP, et al. ApplPhys Lett 2000;77:406.
[2] Fujiwara T, Locklin J, Bao ZN. Appl Phys Lett 2007;90:232108.[3] Noh YY, Zhao N, Caironi M, Sirringhaus H. Nat Nanotech 2007;2:784.[4] Sagane K, Shakutsui M, Tsutsui T, Fujita K. J Photopolymer Sci Technol 2008;
21:193.[5] Levermore PA, Jin R, Wang XH, de Mello JC, Bradley DDC. Adv Funct Mater
2009;19:950.[6] Huang JH, Ho ZY, Kekuda D, Chang Y, Chu CW, Ho KC. Nanotechnology 2009;
20:025202.[7] Nabok D, Puschnig P, Ambrosch-Draxl C, Werzer O, Resel R, Smilgies DM. Phys
Rev B 2007;76:235322.[8] Prosa TJ, Moulton J, Heeger AJ, Winokur MJ. Macromolecules 1999;32:4000.[9] Kline RJ, McGehee MD, Toney MF. Nat Mater 2006;5:222.
[10] Brinkmann M, Rannou P. Macromolecules 2009;42:1125.[11] Kawana S, Durrell M, Lu J, Macdonald JE, Grell M, Bradley DDC, et al. Polymer
2002;43:1907.[12] Chen SH, Chou HL, Su AC, Chen SA. Macromolecules 2004;37:6833.[13] Arif M, Volz C, Guha S. Phys Rev Lett 2006;96:025503.[14] Knaapila M, Stepanyan R, Torkkeli M, Garamus VM, Galbrecht F, Nehls BS,
et al. Phys Rev E 2008;77:051803.[15] Kinder L, Kanicki J, Petroff P. Synth Metals 2004;146:181.[16] Werzer O, Matoy K, Smilgies DM, Rothmann M, Strohriegl P, Resel R. J Appl
Poly Sci 2008;107:1817.[17] Werzer O, Flesch HG, Smilgies DM, Resel R. J Poly Sci B 2009;47:1599.[18] Kinder L, Kanicki J, Swensen J, Petroff P. Proc SPIE 2003;5217:35.[19] Thiem H, Rothmann MM, Strohriegl P. Designed Monomers Polym 2005;8:
619.[20] Resel R, Tamas E, Sonderegger B, Hofbauer P, Keckes J. J Appl Cryst 2003;36:
80.[21] Rapallo AJ. Chem Phys 2009;131:44113.[22] Ren P, Ponder JW. J Phys Chem B 2003;107:5933.[23] MATSTUDIO modeling release 4.0. 9685 Scranton Rd. San Diego CA (USA):
Accelrys Inc.; 2003.[24] Ballauff M, Schmidt GF. Mol Cryst Liq Cryst 1987;147:163.[25] Enzo S, Fagherazzi G, Benedetti A, Polizzi S. J Appl Cryst 1988;21:536.[26] Mulder MHV, Kruitz F, Smolders CA. J Membr Sci 1982;11:349.[27] Li N, Zhang X, Geng Y, Su Z. Polymer 2008;49:4279.[28] Chunwaschirasiri W, Tanto B, Huber DL, Winokur MJ. Phys Rev Lett 2005;94:
107402.
Copyright © 2022 FDOKUMEN




















