
The Mycobacterium tuberculosis Rv2540c DNA sequence encodes a bifunctional chorismate synthase

Journal of Structural Biology 174 (2011) 147–155
Contents lists available at ScienceDirect
Journal of Structural Biology
journal homepage: www.elsevier .com/locate /y jsbi
Crystal structure of FabG4 from Mycobacterium tuberculosis revealsthe importance of C-terminal residues in ketoreductase activity
Debajyoti Dutta, Sudipta Bhattacharyya, Somnath Mukherjee, Baisakhee Saha, Amit Kumar Das ⇑Department of Biotechnology, Indian Institute of Technology Kharagpur, Kharagpur 721032, West Bengal, India
a r t i c l e i n f o a b s t r a c t
Article history:Received 8 September 2010Received in revised form 5 November 2010Accepted 8 November 2010Available online 21 November 2010
Keywords:High molecular weight FabGMycobacterium tuberculosisCatalytic triadFlavodoxin type foldShort-chain dehydrogenase/reductase
1047-8477/$ - see front matter � 2010 Elsevier Inc. Adoi:10.1016/j.jsb.2010.11.012
Abbreviations: HMwFabG, high molecular weimolecular weight FabG; KAR, ketoacyl reductase; Snase/reductase.⇑ Corresponding author.
E-mail address: [email protected] (A.K. Da
Rv0242c, also known as FabG4, is a beta-ketoacyl CoA reductase in Mycobacterium tuberculosis. The crys-tal structure of C-terminal truncated FabG4 is solved at 2.5 Å resolution which shows the presence of twodistinct domains, domain I and II. Domain I partially resembles ‘‘flavodoxin type domain’’ and the domainII is a typical ‘‘ketoacyl CoA reductase (KAR) domain’’. The enzyme exhibits ketoacyl CoA reductase activ-ity by reducing acetoacyl CoA to 3-hydroxyacyl CoA in presence of NADH. Conserved catalytic triadSer347, Tyr360, and Lys364 constitute the active site residues of the KAR domain. Presence of the Tyrand the Lys residues in the triad in a particular orientation is imperative for effective catalytic mecha-nism. The importance of loop I and II and the role of the C-terminal residues of KAR domain are high-lighted. Comparative structural analyses clearly demonstrate that loop II is stabilized by hydrophobicinteraction with C-terminal residues to sustain the orientation of Tyr360. Loop I interacts with loop IIvia H-bonding network to restrict the active site residue Lys364 in a catalytically favorable orientation.
� 2010 Elsevier Inc. All rights reserved.
1. Introduction chain only by C22 to C26 (Bloch, 1975, 1977), it meets the basic need
Considering the present global scenario, tuberculosis is stillplaying havoc with mankind. The counts of TB deaths are addedeach day in a significant manner. The situation is even worsenedwith the emergence of multiple drug resistance TB (MDR-TB) andHIV co-infection. The causative agent of TB, Mycobacterium tuber-culosis, possess lipid rich cell envelop which is required for survivalin host. The cell envelop is made up with different unusual lipidmolecules and lipid derivatives like arabinogalactan anchoring my-colic acids, lipoarabinomannan (LAM), lipomannan (LM), etc. My-colic acids and other fatty acid components of lipid moleculesare mostly the products of fatty acid metabolism pathway. Fattyacid metabolism in M. tuberculosis thus deserves attention to iden-tify new drug targets and to design potent drugs against TB.
More than 260 genes of M. tuberculosis genome are particularlyinvolved in metabolism, modification and transport of fatty acid(Cole et al., 1998). Two fatty acid synthesis pathways, FAS-I andFAS-II are reported in Mycobacterium (Takayama et al., 2005).FAS-I which is the de novo synthesis of fatty acid, retains the tradi-tional pathway of a multi-domain enzyme (Fas: Rv2524c).Although the pathway has a limitation to elongate the fatty acyl
ll rights reserved.
ght FabG; LMwFabG, lowDR, short-chain dehydroge-
s).
of fatty acids to the cell. After FAS-I, FAS-II takes over the furtherelongation of its chain length to as long as C70 to C90 (Takayamaet al., 2005). FAS-II pathway is more specialized with disaggregatedenzyme system to prepare the essential components of the cellwall like mycolic acid. Other fatty acid elongation systems are alsoreported in Mycobacterium (Kikuchi and Kusaka, 1982).
The four major enzymes involved in disaggregated fatty acid syn-thesis are termed as FabH (ketoacyl synthase), FabG (ketoacyl reduc-tase), FabA/FabZ (hydroxyacyl dehydratase/isomerase), and FabI(trans 2-enoyl reductase). FabG or b-ketoacyl CoA reductase con-verts keto group to hydroxyl group at the expense of NADPH oxida-tion. A bacterium like Escherichia coli genome is equipped with onlyone copy of FabG gene (Durfee et al., 2008). BLAST results with gen-ome sequences reveal that actinobacteria and some proteobacteriacontain multiple copies of FabG genes and most of the eukaryotescontain more than one copies of ketoreductases distantly relatedto bacteria. M. tuberculosis being an actinobacteria, also contain mul-tiple FabG genes namely Rv0242c (fabG4), Rv1350 (fabG2), Rv1483(fabG1), Rv2002 (fabG3), and Rv2766c (fabG5). Presence of multipleFabG genes in M. tuberculosis genome is speculated to have role invirulence (Banerjee et al., 1998). Requirement of these genes canbe best judged by considering the M. leprae genome where the gen-ome size is massively reduced by eliminating most of the nonfunc-tional genes (Cole et al., 2001). Homolog of fabG3 is absent inleprae whereas those of fabG2 and fabG5 are present as pseudogenes(Marri et al., 2006). Functional copies of fabG1 and fabG4 homologsare present in M. leprae. Multiple sequence alignment of FabG1 to

148 D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155
FabG5 shows that all these enzymes contain GXXXGXG dinucleotidebinding motif and the conserved catalytic triad Ser, Tyr, and Lys.These proteins belong to the short-chain dehydrogenase/reductasefamily. FabG4 is a protein of molecular weight 46.83 kDa whereasmolecular weights of other FabGs are ranging from 27.14 kDa to25.67 kDa. FabG4 shares highest homology with FabG2 (38% in244 residues overlap) followed by FabG1 (33% in 239 residues over-lap) although phylogenetic tree implicates that FabG4 is the closestrelative of FabG1 (unpublished results). FabG4 shares least homol-ogy with FabG3 (31% in 247 residues overlap). Crystal structures ofFabG1 and FabG3 have been solved. FabG1 is essential for very longchain fatty acid synthesis in FAS-II pathway (Marrakchi et al., 2002;Cohen-Gonsaud et al., 2002) and FabG3 is characterized in vitro to bethe NADH dependent 3a,20b-hydroxysteroid dehydrogenase (Yanget al., 2003).
Quite a few crystal structures of FabG are available in PDB. Crys-tal structure of E. coli FabG (PDB: 1Q7B) shows that NADPH is re-quired to orient the catalytic residues specifically Tyr and Lys tothe catalytic conformation (Price et al., 2004). Both the structuresof E. coli FabG (PDB: 1I01, 1Q7B) and M. tuberculosis FabG1 (PDB:1UZN, 1UZL) (Cohen-Gonsaud et al., 2005) also indicate that thepresence of two loop (loops I and II) near the active site are affectedby the presence of the coenzyme which ascertain the proper orien-tation of Tyr and Lys. However, several structures are solved withordered loop I and II where Tyr and Lys reside in catalytic confor-mation irrespective of the presence of coenzyme. It has been pro-posed that Gly, the first residue in the loop II of E. coli and M.tuberculosis FabG is responsible for flexibility and require coen-zyme for stabilization (Poncet-Montange et al., 2007). On the otherhand, FabG structures with Ala, Ser, or Thr in place of Gly do notneed coenzyme for the stabilization of loop II. Bartonella henselaeapo FabG (PDB: 3GRP) where Gly is duly replaced by Thr, on thecontrary, possess disordered loop II. Intriguingly, the C-terminalresidues those are always found in vicinity to the loop II are miss-ing in B. henselae apo FabG, and hence can be regarded as the con-tributing factor for loop II stability. Moreover, the interactionbetween NADP and C-terminal end is also found in Brassica napusFabG (PDB: 1EDO, Fisher et al., 2000) and M. tuberculosis FabG1(Cohen-Gonsaud et al., 2005).
FabG4 exists in a conserved operon and overexpressed whenM. tuberculosis is exposed to the inhibitory conditions of fatty acidsynthesis. It is proposed to involve in some interlinked fatty acidmetabolism pathway (Boshoff et al., 2004). Since FabG proteinsare important for fatty acid metabolism especially in M. tuberculo-sis, we have attempted to solve the structure of FabG4 to addressits structural uniqueness and to delineate the role of C-terminalresidues in its catalytic mechanism. The protein has also been char-acterized in terms of coenzyme specificity and belongs to a groupof high molecular weight ketoacyl reductase.
2. Materials and methods
2.1. Materials
Enzymes used in molecular biology works were from Fermen-tas. Expression vector pQE30 and E. coli M15 (pREP4) cells werefrom QIAGEN, USA. All other chemicals and reagents used werecommercial samples of analytical grade.
2.2. PCR amplification and cloning of fabG4 and its C-terminal deletionmutant
The fabG41–454 was amplified using the primer sets 0242FP1–454
(50 C GAG CTC GTG GCT CCC AAG CGT TCG TC 30) and 0242RP1–454
(50 CCC AAG CTT TCA CGC GCC GAT CAT GGC C 30) containing SacI,
HindIII restriction sites (highlighted) respectively. The fabG417–454
and fabG417–448 were amplified using primer sets 0242FP17–454 (50
CG GGA TCC GGT CCA GGA TCG TTT TTG GC 30), 0242RP17–454 (50
CCC AAG CTT TCA CGC GCC GAT CAT GGC C 30) and 0242FP17–448
(50 C GAG CTC ATG GGT CCA GGA TCG TTT TTG GCC AG 30),0242RP17–448 (50CCC AAG CTT TCA GCC GCA GAC ACG AAT GACGTT G 30) containing BamHI, HindIII and SacI, HindIII restrictionsites (highlighted) respectively. The ORF fabG41–454 correspondsto the full length protein (amino acid sequences 1–454) whereasORF fabG417–454 and fabG417–448 correspond to the proteins contain-ing amino acid residues 17–454 and 17–448, respectively. The first16 amino acids were found to hinder the solubility of the recombi-nant FabG4 protein. The amplified products were purified using gelextraction kit (QIAGEN, USA), digested with restriction enzymesand ligated into the corresponding sites of pQE30 vector that addssix consecutive histidines at the N-terminus. Nucleotide sequencesof the insert genes were determined by automated DNA sequenc-ing to confirm the originality, integrity and absence of PCR-inducedmutation. Recombinant pQE30 vector anchoring fabG417–454 andfabG417–448 were transformed into chemically competent E. coliM15 cells and selected on LB agar plates supplemented with100 lg mL�1 ampicillin and 25 lg mL�1 kanamycin.
2.3. Overexpression and purification of recombinant FabG4
Recombinant M15 cells harboring soluble FabG417–448 orFabG417–454 constructs were grown in 4 L LB media containing100 lg mL�1 ampicillin and 25 lg mL�1 kanamycin at 37 �C untilOD600 reached 0.6. The cells were induced with 0.5 mM isopro-pyl-b-D thiogalactopyranoside (IPTG) and further grown at 37 �Cfor 5 h. All the subsequent steps were performed at 4 �C.
The cells were harvested by centrifugation at 10,000g for10 min. Resulting cell pellet was resuspended in buffer A (10 mMimidazole, 10 mM Tris, pH 8.0, and 300 mM NaCl) containing pro-tease inhibitor cocktail (0.1 mM each of leupeptin, pepstatin andaprotinin and 0.02 mM phenylmethylsulfonyl fluoride). Cell sus-pension was lysed by MICROSON™ (MISONIX) ultrasonic cell dis-ruptor using 30 s pulse at regular intervals of 60 s. The entireprocedure was repeated twice to ensure complete lysis. Cell debriswas removed by centrifugation at 22,000g for 30 min. Supernatantwas loaded on the Ni–NTA Sepharose high performance affinitymatrix column (GE Healthcare) pre-equilibrated with buffer A.The column was washed with buffer A followed by buffer B(100 mM imidazole, 10 mM Tris, pH 8.0, and 300 mM NaCl). Re-combinant proteins were finally eluted with buffer C (300 mMimidazole, 10 mM Tris, pH 8.0, and 300 mM NaCl).
The eluant was subjected to dialysis against buffer D (25 mMTris, pH 8.0, 50 mM NaCl, and 2 mM dithiothreitol) and the dia-lyzed fraction was loaded on Q Sepharose fast flow column (GEHealthcare) pre-equilibrated with buffer D. This was followed bywashing the column with the same buffer. A linear gradient of NaClwas applied to 1 M and the protein was found to be eluted at 0.5 Mof NaCl on an ÄKTAprime Plus system (GE Healthcare). Peakfractions were pulled together and subjected to size-exclusionchromatography using Superdex 200 prep grade matrix pre-equil-ibrated with buffer E (10 mM Tris, pH 8.0, 50 mM NaCl, and 2 mMdithiothreitol). Fractions of 4 mL were collected with 1 mL min�1
flow rate. Protein concentration was estimated by Bradford’smethod (Bradford, 1976). The purity of the protein was checkedby MALDI ToF analysis.
2.4. Crystallization of FabG417–448
Purified FabG417–448 was concentrated up to 10 mg mL�1 byAmicon concentrators (10 MWCO). Concentrated protein wassubjected to initial crystallization trials using Crystal Screen, Crys-

D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155 149
tal Screen 2, and Index screen from Hampton Research at 298 K bysitting drop vapor diffusion method. Protein of 2 lL was mixedwith of 2 lL of reservoir solution and equilibrated against 100 lLof reservoir solution. Small crystals appeared within 2 days from0.2 M CH3COONH4, 0.1 M Bis-Tris, pH 5.5 and 20% (w/v) PEG3350 at 298 K. Quality of crystals was further improved by varyingthe buffer pH, precipitant, salt, and buffer composition.
2.5. Diffraction data collection and processing
Single crystal of FabG417–448 was cryoprotected using the cryosolution containing 12% (v/v) glycerol in reservoir solution. Thecryoprotected crystal was flash cooled in N2 stream at 100 K. Acomplete diffraction data set was collected using an in house Rig-aku Micromax HF007 rotating anode CuKa X-ray source and Riga-ku Raxis IV++ image plate detector. Crystal to detector distance wasmaintained at 220 mm. Data were processed and scaled using XDS(Kabsch, 1993) and SCALA (Evans, 1993) from the CCP4 suite(CCP4, 1994) respectively, in space group P212121. Final data pro-cessing statistics are summarized in Table 1.
2.6. Phasing, refinement, and model building
Initial phase was obtained by molecular replacement with PHA-SER (McCoy et al., 2007) using two search models of 3-ketoacylCoA reductases of M. tuberculosis (FabG41–454, PDB: 3LLS) and Aqui-fex aeolicus (PDB: 2PNF). Based on Matthew’s coefficient, the num-ber of copies in PHASER was set to two. Solution obtained frommolecular replacement was refined with Refmac5 (Murshudovet al., 1997). NCS was tightly constrained for peptide backboneand medium for the side chains over the first few cycles of re-
Table 1X-ray data collection and refinement statistics
Data collection statistics:Wavelength(Å) 1.5418Resolution (Å) 19.95–2.52 (2.65–2.52)No. of reflections (observed) 195,928 (22,709)No. of reflections (unique) 27,834 (3783)Completeness (%) 99.0 (94.2)Rmerge (%) 10.1 (56.1)Rr.i.m (%) 11.8 (67.0)Rp.i.m (%) 4.4 (26.7)I/r(I) 15.1 (2.9)Redundancy 7.0 (6.0)
Refinement statistics:Space group P212121
Unit cell parameters a = 65.08, b = 80.5, c = 153.95No. of monomers in asymmetric units 2Matthews coefficient (Da Å�3) 2.22Rwork (%) 18.3Rfree (%) 24.0No. of protein atoms 5285No. of water molecules 207Wilson B factors (Å2) 46.3R.m.s.d bond length (Å) 0.016R.m.s.d bond angles (�) 1.652
Ramachandran plotMost favored regions (%) 92Additional allowed region (%) 6.6Generously allowed region (%) 0.7Outliers (%) 0.2
Values in parenthesis are for the highest resolution shells.Rwork ¼
Phkl jjFobsj � jFcalcjj=
PjFobsj where Fobs and Fcalc are the observed and cal-
culated structure factor, respectively.Rfree = same as Rwork, but for 5% of total reflection chosen and omitted fromrefinement.Rr:i:m ¼
Phkl
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiN=ðN � 1Þ
p PijIiðhklÞ � IðhklÞj=
PhklP
iIiðhklÞ.Rp:i:m ¼
Phkl
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi1=ðN � 1Þ
p PijIiðhklÞ � IðhklÞj=
PhklP
i IiðhklÞ.
strained refinement. Alternate cycles of manual model buildingand refinement were carried out in COOT (Emsley and Cowtan,2004) and Refmac5, respectively. The structure was refined to aresolution 2.5 Å, to Rwork 18.3% and Rfree 24%. Stereochemical vali-dation was performed using PROCHECK (Laskowski et al., 1993)and SFCHECK in CCP4 (CCP4, 1994). Refinement statistics and ste-reochemical parameters were summarized in Table 1. Coordinatesand corresponding structure factor amplitudes of FabG417–448 weredeposited in PDB (PDB: 3M1L). Structure sequence alignment wasperformed using ESpript (Gouet et al., 1999). Models were gener-ated using Pymol (http://www.pymol.org). Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/) was used to search forthe similar fold of FabG4. SCOP database (http://scop.mrc-lmb.ca-m.ac.uk/scop) was used to compare the N-terminal fold ofFabG417–448 with the existing a/b fold. Solvent accessible areaand interaction area was calculated by PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html).
2.7. In silico studies and phylogenetic analysis of FabG4
Amino acid sequences of the proteins hypothetical dehydratase(Rv0241c) and FabG4 were retrieved from TB consortium (http://www.doe-mbi.ucla.edu/TB/) and KEGG database (http://www.genome.jp/dbget-bin/www_bget?mtu). Secondary structure pre-diction was carried out by GOR IV server (Garnier et al., 1996).Domain boundaries were identified by SMART (http://smar-t.embl-heidelberg.de/). Multiple sequence analysis was performedusing ClustlX 2.0.12 (Larkin et al., 2007) whereas the determina-tion of orthologues and paralogues were done using BLAST server(http://blast.ncbi.nlm.nih.gov/Blast.cgi). Phylogenetic tree wasgenerated by ClustalX 2.0.12 and viewed using TreeView (Page,1996).
2.8. FabG4 assay for enzymatic activity
Proteins homogeneously purified after gel filtration chromatog-raphy were subjected to extensive dialysis against assay buffer(50 mM HEPES, pH 7.4). Proteins were assayed for both NADPHand NADH dependent ketoacyl reductase activity. The reductaseactivity was carried out in vitro at 25 �C by monitoring the decreaseof OD340 in kinetic mode as a function of conversion of NADPH toNADP or NADH to NAD in Evolution™ 300 UV–Visible spectropho-tometer (Thermo Fisher scientific). Acetoacetyl CoA is used as elec-tron acceptor and appropriate buffer blank devoid of NADPH/NADH was subtracted from each corresponding spectra. The reac-tion mixture of total volume 0.5 mL contained 0.2 mM of coen-zyme (NADPH/NADH), 0.5 mM acetoacetyl CoA, and 1 lM ofpurified protein. The reaction was initiated by adding coenzymeto the reaction mixture. Dehydrogenase activity was carried outusing the substrate 3-hydroxybutyric acid (Yang et al., 2003).Activity of the protein was measured at pH 5.5, 5.8, and 6 at37 �C using 50 mM MES buffer.
3. Results and discussion
3.1. Expression, purification, and crystallization of FabG4
Recombinant FabG41–454 in M15 (pREP4) cells is found to belocalized as inclusion bodies. Secondary structure prediction indi-cates that the presence of N-terminal flexible loop (1–16) mightbe one of the reasons behind the insolubility of the overexpressedprotein. Hence truncated FabG417–454 and FabG417–448 werecloned, overexpressed in soluble form and purified nearly to homo-geneity (Suppl. Fig. 1). Both the proteins were obtained in dimericforms after size-exclusion chromatography. FabG417–448 was
150 D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155
subjected to crystallization trials. Quality of crystals was signifi-cantly improved by using CH3COONa buffer instead of Bis-Tris buf-fer of the initial condition. Crystal grew to typical dimensions of0.3 � 0.1 � 0.1 mm from 0.2 M CH3COONa, pH 5.6, 15% (w/v) PEG3350 at 298 K in hanging drop vapor diffusion method and dif-fracted up to 2.5 Å.
3.2. Overall structure of FabG417–448
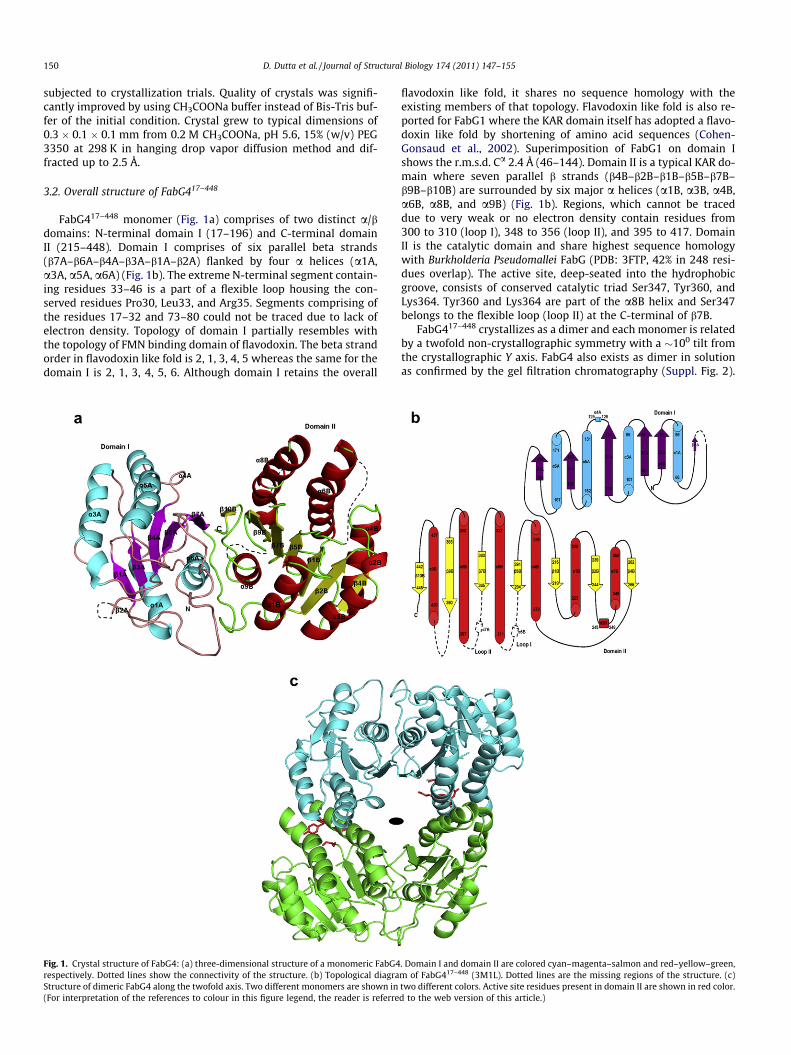
FabG417–448 monomer (Fig. 1a) comprises of two distinct a/bdomains: N-terminal domain I (17–196) and C-terminal domainII (215–448). Domain I comprises of six parallel beta strands(b7A–b6A–b4A–b3A–b1A–b2A) flanked by four a helices (a1A,a3A, a5A, a6A) (Fig. 1b). The extreme N-terminal segment contain-ing residues 33–46 is a part of a flexible loop housing the con-served residues Pro30, Leu33, and Arg35. Segments comprising ofthe residues 17–32 and 73–80 could not be traced due to lack ofelectron density. Topology of domain I partially resembles withthe topology of FMN binding domain of flavodoxin. The beta strandorder in flavodoxin like fold is 2, 1, 3, 4, 5 whereas the same for thedomain I is 2, 1, 3, 4, 5, 6. Although domain I retains the overall
Fig. 1. Crystal structure of FabG4: (a) three-dimensional structure of a monomeric FabG4respectively. Dotted lines show the connectivity of the structure. (b) Topological diagraStructure of dimeric FabG4 along the twofold axis. Two different monomers are shown in(For interpretation of the references to colour in this figure legend, the reader is referre
flavodoxin like fold, it shares no sequence homology with theexisting members of that topology. Flavodoxin like fold is also re-ported for FabG1 where the KAR domain itself has adopted a flavo-doxin like fold by shortening of amino acid sequences (Cohen-Gonsaud et al., 2002). Superimposition of FabG1 on domain Ishows the r.m.s.d. Ca 2.4 Å (46–144). Domain II is a typical KAR do-main where seven parallel b strands (b4B–b2B–b1B–b5B–b7B–b9B–b10B) are surrounded by six major a helices (a1B, a3B, a4B,a6B, a8B, and a9B) (Fig. 1b). Regions, which cannot be traceddue to very weak or no electron density contain residues from300 to 310 (loop I), 348 to 356 (loop II), and 395 to 417. DomainII is the catalytic domain and share highest sequence homologywith Burkholderia Pseudomallei FabG (PDB: 3FTP, 42% in 248 resi-dues overlap). The active site, deep-seated into the hydrophobicgroove, consists of conserved catalytic triad Ser347, Tyr360, andLys364. Tyr360 and Lys364 are part of the a8B helix and Ser347belongs to the flexible loop (loop II) at the C-terminal of b7B.
FabG417–448 crystallizes as a dimer and each monomer is relatedby a twofold non-crystallographic symmetry with a �100 tilt fromthe crystallographic Y axis. FabG4 also exists as dimer in solutionas confirmed by the gel filtration chromatography (Suppl. Fig. 2).
. Domain I and domain II are colored cyan–magenta–salmon and red–yellow–green,m of FabG417–448 (3M1L). Dotted lines are the missing regions of the structure. (c)two different colors. Active site residues present in domain II are shown in red color.d to the web version of this article.)

D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155 151
As evident from the crystal structure the dimeric interaction is so-lely due to helix–helix interaction (Fig. 1c). Approximately 1514 Å2
of solvent accessible area is consumed by these interactions whichcover roughly 9% of the total solvent accessible area.
Low molecular weight FabGs characterized till date are gener-ally 206–273 residues long covering the ketoacyl reductase domainonly (Fig. 2). In contrast, FabG4 possesses an N-terminal domain(domain I) of 196 residues followed by an entire ketoreductase do-main (domain II). Homologous proteins of FabG4 show that theseproteins are generally of 438–502 amino acid residues. This beingthe very first report of a ketoacyl reductase containing N-terminaldomain, we have designated FabG4 as a high molecular weightFabG (HMwFabG). Exact role of the N-terminal domain I is notknown, however, several conserved residues identified in the spe-cific regions of this domain can be correlated with its function.Helical interface between two monomers are predominated byhydrophobic interaction where the N-terminal domain I of onemonomer interacts with the C-terminal domain II of anothermonomer. Therefore, helices a3A and a5A of domain I interactwith a6B0 and a8B0 of domain II, respectively. Amino acids residueslike Gly143, Ser147, Lys150, and Glu151 are conserved in a5A(131–151) of domain I to provide the largest dimeric interface
Fig. 2. Multiple sequence alignment of FabG4 domain II with different LMwFabG: the stathe active site residues are marked with asterisk. The box indicating the FabG4 residues 2phosphate of adenosine ribose of NADP. The loop is shortened in case of FabG4 and comindicated by arrow mark. C-terminal region of different FabG are shown.
(Fig. 3). The sole hydrogen bonding interaction (3.14 Å) found inthis dimeric interface is due to Ser147 of a5A with conservedThr358 of a8B0. A second region of domain I spanning from 179to 192 is also composed of a number of conserved residues(Fig. 3). This region of N-terminal domain I is nearer to the C-termi-nal of domain II of the same monomer. Thus, relative orientationsof two domains of a single monomer are maintained by hydrogenbonding interactions between the residues Tyr188-Cys447,Gln192-Arg445 within a monomer. Residues Gln192, Arg445,Cys447 are conserved in all HMwFabGs (Fig. 3) which indicate thatthe N-terminal domain is present not only to aid the dimeric inter-action but also to influence the relative orientations of the twodomains.
3.3. Effects of C-terminal deletion from FabG4
In C-terminal truncated FabG417–448 structure (PDB: 3M1L), theelectron density of loop I and loop II of domain II could not be tracedand the active site residues Tyr360 and Lys364 are oriented awayfrom the catalytic conformations in comparison to the structuresof apo LMwFabGs of Pseudomonas aeruginosa (Miller et al., 2006),Plasmodium falciparum (PDB: 2C07; Wickramasinghe et al., 2006),
rting residue numbers of each FabG are shown. Loop I and loop II are indicated and42–244 represents the loop regions of LMwFabG responsible for interacting with 20
posed of hydrophobic amino acids. The residue important for loop II flexibility is

Fig. 3. Multiple sequence alignment of some HMwFabGs from different organisms showing its important regions: helices a3A and a5A involved in dimeric interaction witha6B0 and a8B0 . Some of the conserved residues from 179 to 192 are involved in intramolecular interaction with the residues near C-terminus. Loop I [NAG(I/V)T(R/K)D] isresponsible for Tyr360 orientation. Loop II containing a conserved helix a7B [(I/T/M)XG(I/V/L)AG] directs the Lys364 orientation. Catalytic residues Ser347, Tyr360, andLys364 are marked with asterisk. The C-terminus [CGQXX(I/V/L)GA] region is found to participate in hydrophobic interaction with the loop II a7B helix.
152 D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155
Bacillus anthracis (PDB: 2UVD; Zaccai et al., 2008), Thermus thermo-philus HB8 (PDB: 1ULS) and A. aeolicus VF5 (PDB: 2PNF). Superposi-tion of these structures onto FabG417–448 domain II indicates thatloop I and loop II retain the stable conformations through the inter-action with C-terminal residues. Since FabG417–448 lacks theC-terminal residues (449QAMIGA454), loop I and loop II are disor-dered resulting in Tyr360 and Lys364 to orient away from the cat-alytic conformations. Also structure of FabG41–454 (PDB: 3LLS)shows the presence of active site residues in catalytically active ori-entations which is due to the stable conformations of these twoloops attributed by the presence of C-terminal residues. This clearlydemonstrates the importance of C-terminal residues in conferringenzymatic activity to FabG4 (Fig. 4).
In contrast to the M. tuberculosis FabG4 domain II, FabG1 canmaintain the loop I and loop II in stable conformation only in pres-ence of NADPH and the flexibility of the loop II can be attributed toGly present as the first residue of loop II (Poncet-Montange et al.,2007). Those LMwFabGs that contain Ala/Ser/Thr in place of Gly,require C-terminal residues to maintain the loop I and II in orderedmanner, irrespective of the presence of coenzyme (Fig. 2).
Loop I, joining b5B to a5B, is not a part of active site but hypoth-esized to sequester the coenzyme from the solvent in KAR (Miller
Fig. 4. Comparison of the activity of the two forms of FabG4. (j) points representthe activity of FabG417–454 and (d) represent the activity of FabG417–448.
et al., 2006). In absence of loop I, Tyr360 is rotated 69� away fromthe active site (Fig. 5a and b). Thus the loop I restricts the move-ment of Tyr360 and its presence is required for Tyr360 to sustainthe catalytic orientation. Sequence alignment of differentHMwFabGs has revealed the presence of the signature motifNAG(I/V)T(R/K)D in loop I (Fig. 3). Loop II, joining the b7B witha8B is an active site loop. A part of the loop comprising of the res-idues 348–353 adopts a helical conformation on interacting withthe C-terminal residues and is designated as a7B. In absence ofC-terminal residues the loop becomes disordered allowingLys364 to rotate by 101� away from the active conformation(Fig. 5a and b). a7B is composed of the signature motif (I/T/M)XG(I/V/L)AG (Fig. 3) and engaged in hydrophobic interactionwith C-terminal residues (449QAMIGA454). Interactions betweenloop II and C-terminal residues are also found in case of FabG1.Met243 is involved in hydrophobic interaction with Val141. Struc-tural superimposition of FabG1 (PDB: 1UZN) onto FabG41–454 (PDB:3LLS) shows that in FabG4 the positions of Val141 and Met243 arereplaced with Ile348 and Ala450. Although Ile348 is conserved,Ala450 is not conserved among HMwFabGs. Based on the analysisof FabG417–448 (PDB: 3M1L) and FabG41–454 (PDB: 3LLS) we haveidentified that Ile452 is involved in hydrophobic interaction withIle351 and Ala352. The conserved motif CGQXX(I/V/L)GA (Fig. 3)at the C-terminus shows that the position of Ile452 seems to beconserved and is often occupied by other hydrophobic residues likeVal or Leu in HMwFabGs. Conserved Gly448 and Gly453 probablyact as hinge to allow the free movement of the C-terminus tostrengthen the interaction with loop II. This is the very first reportshowing the structural implications of the C-terminal residues of aketoacyl reductase. Hydrogen bonding interactions and hydropho-bic interactions around the active site residues are shown inFig. 5a.
3.4. Phylogenetic analysis of HMwFabGs
BLAST results have revealed the existence of several homolo-gous genes of FabG4. The corresponding proteins of these genescan be designated as high molecular weight FabG (HMwFabG). Ingeneral, HMwFabG coexist with another dehydratase gene in anoperon. In M. tuberculosis the dehydratase gene (Rv0241c) is over-expressed during the organism’s exposure to the inhibitory condi-tions of fatty acid synthesis (Boshoff et al., 2004). The dehydratasegene is recently shown to introduce unsaturation on the fatty acylbackbone once the hydroxyl group is present on the b carbon

Fig. 5. Orientation of the active site residues is directed by the C-terminal residues. (a) Stereo view of active site residues and its surrounding regions. The green molecule(FabG417–448:3M1L) represents the C-terminal truncated mutant and the magenta molecule (FabG41–454:3LLS) represents the C-terminal intact protein. Loop II is stabilized byhydrophobic interaction with C-terminal residues and forms a short helix a7B in vicinity to the active site. Hydrogen bonding interactions between loop I and loop II arehighlighted. Tyr360 and Lys364 (shown in green) are oriented away from catalytic orientation in absence of the loops. (b) Stereo view of active site residues of C-terminaltruncated mutant in green color (FabG417–448:3M1L) shown in 2Fo � Fc map contoured at 1.0r. FabG41–454 (in magenta) is superimposed on truncated FabG417–448 (in green).(For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155 153
(Sacco et al., 2010). Since FabG4 can incorporate hydroxyl group onthe same b carbon, these two proteins may be related to a singlepathway. This operon is not only conserved in Mycobacteria andclosely related organisms like Rhodococcus, Corynebacterium,Nocardia but also present in some lipid rich proteobacteria. Allthese organisms can synthesize diverse fatty acids and fatty acylderivatives. It has also been hypothesized that in M. tuberculosis,fatty acid metabolism is significantly enriched by the horizontalgene transfer from ancestors of proteobacteria owing to the factthat most actinobacteria is closely related to some of the proteo-bacteria (Kinsella et al., 2003). In the phylogram, HMwFabG isdivided into different clades depending on the order of the organ-ism. Their variation arises mainly due to variability of the N-termi-nal domain where the domain is affected by several insertions anddeletions between b1A and b3A (Suppl. Fig. 3). In contrast, overallsequences of LMwFabGs are conserved and maintain a cleardemarcation from HMwFabGs (Fig. 6).
3.5. Enzymatic activity of FabG4
Enzymatic activity of FabG4 shows that the enzyme is highlyspecific for NADH to reduce acetoacetyl CoA to 3-hydroxyacetylCoA, whereas LMwFabG needs NADPH for the same. Sequencealignment of domain II of FabG4 with other LMwFabG shows thatthe loop that accommodates 20 phosphate of adenosine ribose ofNADP is shortened by two amino acid residues (Fig. 2). AlthoughFabG4 uses NADH as coenzyme to reduce acetoacetyl group tohydroxylacetyl group, the reverse reaction using NAD is notobserved. This clearly indicates that FabG4 is devoid of any dehy-drogenase activity. A comparative catalytic activity between C-ter-minal truncated FabG417–448 with C-terminal intact FabG417–454
has also been verified. As apparent from the structural comparison,the ketoreductase activity of FabG417–448 is reduced by 120-foldcompared to that of FabG417–454 (Fig. 4). FabG4 is also active atacidic pH range 5.5–6.0.

Fig. 6. A phylogram using different FabG protein sequences found in prokaryotes and eukaryote: gi numbers and accession codes are shown in parenthesis. HMwFabG fromorganisms also show a high degree of variation. HMwFabG from actinobacteria are colored green, alpha proteobacteria as red, beta proteobacteria as violet, gammaproteiobacteria as brown, delta proteobacteria as blue and LMwFabGs are in black. LMwFabGs form a separate clade diverged from HMwFabGs. Some actinobacteria are moreclosely related to proteobacteria with respect to HMwFabG. Two Mycobacterial FabGs (FabG1:Rv1483 and FabG4:Rv0242c) are highlighted (shown in box). LMwFabGs arepresent in a separate clade. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
154 D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155
4. Conclusion
FabG4 is a high molecular weight ketoacyl reductase which canspecifically use NADH to reduce 3-ketoacetyl CoA to 3-hydroxyace-tyl CoA. This diverse group of protein is a member of short-chaindehydrogenase/reductase family and is conserved in the genomesof Mycobacteria. Structure of FabG4 shows that the protein is atwo-domain ketoacyl reductase where the domain I is a flavodoxintype domain and the domain II is the actual catalytic domain sim-ilar to that of LMwFabG. The drastic reduction of FabG4 ketoreduc-tase activity is attributed to the lack of six amino acid residues inC-terminus. Due to the absence of C-terminal residues, interactionsstabilizing loop I and loop II are lost rendering their highly disor-dered conformation. As a result, the active site residues Tyr360and Lys364 move away from the catalytically active orientation.
Acknowledgments
We gratefully acknowledge Department of Biotechnology, Gov-ernment of India for financial assistance and for setting up theMacromolecular Crystallography housed at CRF, IIT Kharagpur.Sudipta Bhattacharyya and Somnath Mukherjee acknowledge IITKharagpur and CSIR, Govt. of India for Institutional and individualfellowship, respectively. Mr. Dipankar Manna and Ms. MonolekhaBhattacharya are duly acknowledged for their help during data col-lection and helpful suggestions. The genomic DNA of H37Rv was akind gift from Prof. H. G. Wiker, University of Bergen.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.jsb.2010.11.012.
References
Banerjee, A., Sugantino, M., Sacchettini, J.C., Jacobs Jr., W.R., 1998. The mabA genefrom the inhA operon of Mycobacterium tuberculosis encodes a 3-ketoacylreductase that fails to confer isoniazid resistance. Microbiology 144, 2697–2704.
Bloch, K., 1975. Fatty acid synthases from Mycobacterium phlei. Methods Enzymol.35, 84–90.
Bloch, K., 1977. Control mechanisms for fatty acid synthesis in Mycobacteriumsmegmatis. Adv. Enzymol. Relat. Areas Mol. Biol. 45, 1–84.
Bradford, M.M., 1976. A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye binding.Anal. Biochem. 72, 248–254.
Boshoff, H.I., Myers, T.G., Copp, B.R., McNeil, M.R., Wilson, M.A., Barry 3rd, C.E., 2004.The transcriptional responses of Mycobacterium tuberculosis to inhibitors ofmetabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279,40174–40184.
CCP4, 1994. Collaborative computational project number 4. Acta Crystallogr., D 50,760–763.
Cohen-Gonsaud, M., Ducasse, S., Hoh, F., Zerbib, D., Labesse, G., Quemard, A., 2002.Crystal structure of MabA from Mycobacterium tuberculosis, a reductaseinvolved in long-chain fatty acid biosynthesis. J. Mol. Biol. 320, 249–261.
Cohen-Gonsaud, M., Ducasse-Cabanot, S., Quemard, A., Labesse, G., 2005. Ligand-induced fit in mycobacterial MabA: the sequence-specific C-terminus locks theconformational change. Proteins 60, 392–400.
Cole, S.T., Brosch, R., Parkhill, J., Garnier, T., et al., 1998. Deciphering the biology ofMycobacterium tuberculosis from the complete genome sequence. Nature 393,537–544.
Cole, S.T., Eiglmeier, K., Parkhill, J., James, K.D., et al., 2001. Massive gene decay inthe leprosy bacillus. Nature 409, 1007–1011.

D. Dutta et al. / Journal of Structural Biology 174 (2011) 147–155 155
Durfee, T., Nelson, R., Baldwin, S., Plunkett 3rd, G., Burland, V., Mau, B., Petrosino,J.F., Qin, X., Muzny, D.M., Ayele, M., Gibbs, R.A., Csörgo, B., Pósfai, G., Weinstock,G.M., Blattner, F.R., 2008. The complete genome sequence of Escherichia coliDH10B: insights into the biology of a laboratory workhorse. J. Bacteriol. 190,2597–2606.
Emsley, P., Cowtan, K., 2004. Coot: model-building tools for molecular graphics.Acta Crystallogr., D 60, 2126–2132.
Evans, P.R., 1993. In: Sawyer, L., Isaacs, N., Bailey, S. (Eds.), Proceedings of the CCP4Study Weekend. Data Collection and Processing, Warrington, DaresburyLaboratory, pp. 114–122.
Fisher, M., Kroon, J.T., Martindale, W., Stuitje, A.R., Slabas, A.R., Rafferty, J.B., 2000.The X-ray structure of Brassica napus beta-keto acyl carrier protein reductaseand its implications for substrate binding and catalysis. Structure 8, 339–347.
Garnier, J., Gibrat, J.F., Robson, B., 1996. GOR method for predicting proteinsecondary structure from amino acid sequence. Methods Enzymol. 266, 540–553.
Gouet, P., Courcelle, E., Stuaet, D.I., Metoz, F., 1999. Espript: multiple sequencealignments in PostScript. Bioinformatics 15, 305–308.
Kabsch, W., 1993. Automatic processing of rotation diffraction data from crystals ofinitially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800.
Kikuchi, S., Kusaka, T., 1982. New malonyl-CoA-dependent fatty acid elongati onsystem in Mycobacterium smegmatis. J. Biochem. (Tokyo) 92, 839–844.
Kinsella, R.J., Fitzpatrick, D.A., Creevey, C.J., McInerney, J.O., 2003. Fatty acidbiosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptiveevolution, and gene duplication. Proc. Natl. Acad. Sci. USA 100, 10320–10325.
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam,H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J.,Higgins, D.G., 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23,2947–2948.
Laskowski, R.A., MacArthur, M.W., Moss, D.S., Thornton, J.M., 1993. PROCHECK: aprogram to check the stereochemical quality of protein structures. J. Appl.Crystallogr. 26, 283–291.
Marrakchi, H., Ducasse, S., Labesse, G., Margeat, E., Montrozier, H., Emorine, L.,Charpentier, X., Lanéelle, G., Quémard, A., 2002. Biochemical and structuralstudies of the Mycobacterium tuberculosis MabA (FabG1) protein involved in thefatty acid elongation system, FAS-II. Microbiology 148, 951–960.
Marri, P.R., Bannantine, J.P., Golding, G.B., 2006. Comparative genomics of metabolicpathways in Mycobacterium species: gene duplication, gene decay and lateralgene transfer. FEMS Microbiol. Rev. 30, 906–925.
McCoy, A.J., Grosse-Kunstleve, R.W., Adams, P.D., Winn, M.D., Storoni, L.C., Read, R.J.,2007. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674.
Miller, D.J., Zhang, Y.M., Rock, C.O., White, S.W., 2006. Structure of RhlG, an essentialbeta-ketoacyl reductase in the rhamnolipid biosynthetic pathway ofPseudomonas aeruginosa. J. Biol. Chem. 281, 18025–18032.
Murshudov, G.N., Vagin, A.A., Dodson, E.J., 1997. Refinement of macromolecularstructures by the maximum-likelihood method. Acta Crystallogr., D 53, 240–255.
Page, R.D.M., 1996. TreeView: an application to display phylogenetic trees onpersonal computers. Bioinformatics 12, 357–358.
Poncet-Montange, G., Ducasse-Cabanot, S., Quemard, A., Labesse, G., Cohen-Gonsaud, M., 2007. Lack of dynamics in the MabA active site kills the enzymeactivity: practical consequences for drug-design studies. Acta Crystallogr., D 63,923–925.
Price, A.C., Zhang, Y.M., Rock, C.O., White, S.W., 2004. Cofactor-inducedconformational rearrangements establish a catalytically competent active siteand a proton relay conduit in FabG. Structure 12, 417–428.
Sacco, E., Slama, N., Bäckbro, K., Parish, T., Laval, F., Daffé, M., Eynard, N., Quémard,A., 2010. Revisiting the assignment of Rv0241c to fatty acid synthase type II ofMycobacterium tuberculosis. J. Bacteriol. 192, 4037–4044.
Takayama, K., Wang, C., Besra, G.S., 2005. Pathway to synthesis and processing ofmycolic acids in Mycobacterium tuberculosis. Clin. Microb. Rev. 18, 81–101.
Wickramasinghe, S.R., Inglis, K.A., Urch, J.E., Muller, S., van Aalten, D.M.F., Fairlamb,A.H., 2006. Kinetic, inhibition and structural studies on 3-oxoacyl-ACPreductase from Plasmodium falciparum, a key enzyme in fatty acidbiosynthesis. Biochem. J. 393, 447–457.
Yang, J.K., Park, M.S., Waldo, G.S., Suh, S.W., 2003. Directed evolution approach to astructural genomics project: Rv2002 from Mycobacterium tuberculosis. Proc.Natl. Acad. Sci. USA 100, 455–460.
Zaccai, N.R., Carter, L.G., Berrow, N.S., Sainsbury, S., Nettleship, J.E., Walter, T.S.,Harlos, K., Owens, R.J., Wilson, K.S., Stuart, D.I., Esnouf, R.M., 2008. Crystalstructure of a 3-oxoacyl-(acylcarrier protein) reductase (BA3989) from Bacillusanthracis at 2.4-A resolution. Proteins 70, 562–567.
Copyright © 2022 FDOKUMEN




















