
Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in...
12
Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo Vera L. Tarakanova a, ⁎, Eleni Stanitsa a , Steven M. Leonardo a,b , Tarin M. Bigley c , Stephen B. Gauld a,b a Department of Microbiology and Molecular Genetics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USA b Allergy and Clinical Immunology, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USA c Medical Scientist Training Program, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USA abstract article info Article history: Received 14 March 2010 Accepted 24 May 2010 Available online 16 June 2010 Keywords: gammaherpesvirus herpesvirus protein kinase latency H2AX DNA damage response Many herpesvirus-encoded protein kinases facilitate viral lytic replication. Importantly, the role of viral kinases in herpesvirus latency is less clear. Mouse gammaherpesvirus-68 (MHV68)-encoded protein kinase orf36 facilitates lytic replication in part through activation of the host DNA damage response (DDR). Here we show that MHV68 latency was attenuated in the absence of orf36 expression. Unexpectedly, our study uncovered enzymatic activity-independent role of orf36 in the establishment of MHV68 latency following intraperitoneal route of infection. H2AX, an important DDR protein, facilitates MHV68 lytic replication and may be directly phosphorylated by orf36 during lytic infection. In this study, H2AX deficiency, whether systemic or limited to infected cells, attenuated the establishment of MHV68 latency in vivo. Thus, our work reveals viral kinase-dependent regulation of gammaherpesvirus latency and illuminates a novel link between H2AX, a component of a tumor suppressor DDR network, and in vivo latency of a cancer-associated gammaherpesvirus. © 2010 Published by Elsevier Inc. Introduction Cellular protein kinases are critical components of every signaling pathway and represent an elegant tool that regulates many aspects of cell biology. This important cellular tool is usurped by viruses, as all avian and mammalian herpesviruses encode a protein kinase. Herpes- virus protein kinases are separated into H v U s and H v U L families based on amino acid sequence homology (reviewed in Gershburg and Pagano, 2008; Prichard, 2009). Alphaherpesviruses encode members of both protein kinase families, whereas beta- and gammaherpesviruses encode a single H v U L protein kinase. While these enzymes are divergent at the amino acid level, herpesvirus kinases have highly conserved biological roles in viral infection. Herpesvirus protein kinases are virion components that localize to the nucleus and facilitate viral DNA replication and egress (Gershburg et al., 2007; Moffat et al., 1998; Krosky et al., 2003; Wolf et al., 2001; Chang et al., 2009; Gershburg and Pagano, 2008). Protein kinases encoded by gammaherpesviruses target multiple viral and host processes. Orf36 protein kinase encoded by Kaposi's sarcoma-associated herpesvirus (KSHV) phosphorylates Kru- pell-associated box domain-associated protein 1 to facilitate a switch from latency to lytic replication in cell lines (Chang et al., 2009). BGLF4, a kinase encoded by human Epstein–Barr virus (EBV), facilitates expression of select viral genes, viral DNA synthesis, disassembly of the nuclear lamina, inhibition of cellular DNA synthesis, and cellular chromosome condensation (reviewed in Gershburg and Pagano, 2008). Orf36, a kinase encoded by mouse gammaherpesvirus 68 (MHV68), is necessary for the initiation of the DNA damage response (DDR) in lytically infected primary macrophages (Tarakanova et al., 2007). The ability of orf36 to induce DDR is not unique, as expression of BGLF4 is also sufficient to induce DDR (Tarakanova et al., 2007), suggesting that targeting of DDR by herpesvirus kinases may be a conserved biological phenotype. Another conserved biological function is the interaction of MHV68 orf36 and EBV BGLF4 with interferon regulatory factor 3 (IRF3) and subsequent inhibition of its transcriptional activity (Hwang et al., 2009). While many herpesvirus kinases are known to facilitate viral replication, there is a paucity of studies examining the role of these enzymes during viral latency in vivo. Deletion of either orf47 or orf66 varicella-zoster virus (VZV) protein kinase does not have an effect on the establishment of latency in dorsal root ganglia of infected rodents (Sato et al., 2003), suggesting that either VZV kinases do not play a role in viral latency or that the presence of the second herpesvirus kinase is sufficient to functionally compensate for the missing counterpart. Interestingly, infection of mice with US3-deficient herpes simplex virus-2 (HSV-2) leads to an increased number of apoptotic neurons, many of which stain positive for HSV antigens, suggesting that US3 kinase is important for protection against cell death during HSV latency (Asano et al., 2000). MHV68 is a rodent gammaherpesvirus genetically and biologically related to human EBV and KSHV and simian herpesvirus saimiri (HVS) Virology 405 (2010) 50–61 ⁎ Corresponding author. Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USA. Fax: +1 414 955 6535. E-mail address: [email protected] (V.L. Tarakanova). 0042-6822/$ – see front matter © 2010 Published by Elsevier Inc. doi:10.1016/j.virol.2010.05.027 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/yviro
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in...

Virology 405 (2010) 50–61
Contents lists available at ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r.com/ locate /yv i ro
Conserved gammaherpesvirus kinase and histone variant H2AX facilitategammaherpesvirus latency in vivo
Vera L. Tarakanova a,⁎, Eleni Stanitsa a, Steven M. Leonardo a,b, Tarin M. Bigley c, Stephen B. Gauld a,b
a Department of Microbiology and Molecular Genetics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USAb Allergy and Clinical Immunology, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USAc Medical Scientist Training Program, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226, USA
⁎ Corresponding author. Medical College of WisconsMilwaukee, WI 53226, USA. Fax: +1 414 955 6535.
E-mail address: [email protected] (V.L. Tarakanov
0042-6822/$ – see front matter © 2010 Published by Edoi:10.1016/j.virol.2010.05.027
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 March 2010Accepted 24 May 2010Available online 16 June 2010
Keywords:gammaherpesvirusherpesvirus protein kinaselatencyH2AXDNA damage response
Many herpesvirus-encoded protein kinases facilitate viral lytic replication. Importantly, the role of viralkinases in herpesvirus latency is less clear. Mouse gammaherpesvirus-68 (MHV68)-encoded protein kinaseorf36 facilitates lytic replication in part through activation of the host DNA damage response (DDR). Here weshow that MHV68 latency was attenuated in the absence of orf36 expression. Unexpectedly, our studyuncovered enzymatic activity-independent role of orf36 in the establishment of MHV68 latency followingintraperitoneal route of infection. H2AX, an important DDR protein, facilitates MHV68 lytic replication andmay be directly phosphorylated by orf36 during lytic infection. In this study, H2AX deficiency, whethersystemic or limited to infected cells, attenuated the establishment of MHV68 latency in vivo. Thus, our workreveals viral kinase-dependent regulation of gammaherpesvirus latency and illuminates a novel linkbetween H2AX, a component of a tumor suppressor DDR network, and in vivo latency of a cancer-associatedgammaherpesvirus.
in, 8701 Watertown Plank Rd,
a).
lsevier Inc.
© 2010 Published by Elsevier Inc.
Introduction
Cellular protein kinases are critical components of every signalingpathway and represent an elegant tool that regulates many aspects ofcell biology. This important cellular tool is usurped by viruses, as allavian and mammalian herpesviruses encode a protein kinase. Herpes-virus protein kinases are separated into HvUs and HvUL families basedon amino acid sequence homology (reviewed in Gershburg and Pagano,2008; Prichard, 2009). Alphaherpesviruses encode members of bothprotein kinase families, whereas beta- and gammaherpesvirusesencode a single HvUL protein kinase.While these enzymes are divergentat the amino acid level, herpesvirus kinases have highly conservedbiological roles in viral infection. Herpesvirus protein kinases are virioncomponents that localize to the nucleus and facilitate viral DNAreplication and egress (Gershburg et al., 2007; Moffat et al., 1998;Krosky et al., 2003; Wolf et al., 2001; Chang et al., 2009; Gershburg andPagano, 2008). Protein kinases encoded by gammaherpesviruses targetmultiple viral and host processes. Orf36 protein kinase encoded byKaposi's sarcoma-associated herpesvirus (KSHV) phosphorylates Kru-pell-associated box domain-associated protein 1 to facilitate a switchfrom latency to lytic replication in cell lines (Chang et al., 2009). BGLF4,a kinase encoded by human Epstein–Barr virus (EBV), facilitatesexpression of select viral genes, viral DNA synthesis, disassembly of
the nuclear lamina, inhibition of cellular DNA synthesis, and cellularchromosome condensation (reviewed in Gershburg and Pagano, 2008).Orf36, a kinase encoded by mouse gammaherpesvirus 68 (MHV68), isnecessary for the initiation of the DNA damage response (DDR) inlytically infected primary macrophages (Tarakanova et al., 2007). Theability of orf36 to induce DDR is not unique, as expression of BGLF4 isalso sufficient to induce DDR (Tarakanova et al., 2007), suggesting thattargeting of DDR by herpesvirus kinases may be a conserved biologicalphenotype. Another conserved biological function is the interaction ofMHV68 orf36 and EBV BGLF4 with interferon regulatory factor 3 (IRF3)and subsequent inhibition of its transcriptional activity (Hwang et al.,2009).
While many herpesvirus kinases are known to facilitate viralreplication, there is a paucity of studies examining the role of theseenzymes during viral latency in vivo. Deletion of either orf47 or orf66varicella-zoster virus (VZV) protein kinase does not have an effect onthe establishment of latency in dorsal root ganglia of infected rodents(Sato et al., 2003), suggesting that either VZV kinases do not play arole in viral latency or that the presence of the second herpesviruskinase is sufficient to functionally compensate for the missingcounterpart. Interestingly, infection of micewith US3-deficient herpessimplex virus-2 (HSV-2) leads to an increased number of apoptoticneurons, many of which stain positive for HSV antigens, suggestingthat US3 kinase is important for protection against cell death duringHSV latency (Asano et al., 2000).
MHV68 is a rodent gammaherpesvirus genetically and biologicallyrelated to human EBV and KSHV and simian herpesvirus saimiri (HVS)

51V.L. Tarakanova et al. / Virology 405 (2010) 50–61
(Efstathiou et al., 1990; Virgin et al., 1997). In vivo, MHV68 establisheslatency in a variety of cell types, including macrophages, B cells, anddendritic cells (Flano et al., 2000; Sunil- Chandra et al., 1992; Wecket al., 1999a,b; Willer and Speck, 2003). MHV68-encoded proteinkinase orf36 has two physiologically relevant functions: induction ofDDR during lytic replication in primary macrophages and counterac-tion of type I interferon response via inhibition of IRF3 (Hwang et al.,2009; Tarakanova et al., 2007). Recently, both the expression andkinase activity of MHV68 orf36 were shown to facilitate early spleniclatency following intranasal inoculation (Hwang et al., 2009),suggesting that orf36-mediated induction of DDR and/or inhibitionof IRF-3 may contribute to the establishment of splenic latency in vivo.The type I interferon response controls MHV68 reactivation fromlatency at 28 days post infection (Barton et al., 2005); however, therelationship between DDR and herpesvirus latency is less clear. Whileattenuated DDR skews the choice of HSV life cycle towards latency incell culture systems (Lilley et al., 2005), the interplay between DDRand herpesvirus latency in vivo is not understood.
The DDR is a conserved network of protein–protein interactionsand modifications that orchestrates detection and repair of lesionsin cellular DNA (reviewed in Pandita and Richardson, 2009). Thissignaling network acts as an anti-cancer barrier and is activatedduring an early stage of tumorigenesis in a majority of human cancers(Bartkova et al., 2005; Bartek et al., 2007). Mutations of genesencoding members of the DDR network, if compatible with normaldevelopment, are associated with various degrees of genomeinstability, immune deficiencies, and increased risk of tumorigenesis.H2AX is a core histone 2A (H2A) variant that constitutes 2–25% of allH2A incorporated into nucleosomes (Rogakou et al., 1998). H2AX hasa unique C-terminal amino acid sequence that is rapidly phosphor-ylated early in DDR by ATM, ATR (ATM-related kinase), or DNA-PK tofunction as a scaffold for the assembly of multi-protein complexes atthe DNA lesion. Two independently generated strains of H2AXdeficient mice exhibit increased sensitivity to irradiation, increasedgenomic instability, and defective DNA repair (Bassing et al., 2002,2003; Celeste et al., 2002). H2AX deficient mice display mild immunedefects, including a twofold reduction in the numbers of mature B andT lymphocytes, but no apparent block in the development of thesecell types (Celeste et al., 2002). However, defective class switchrecombination leads to decreased levels of IgA and IgG in H2AXdeficient mice, despite normal B cell proliferation (Celeste et al.,2002).
DDR is actively engaged by many DNA viruses, including herpes-viruses, in lytically infected cells (reviewed in Lilley et al., 2007; Lilleyet al., 2009). Specifically, MHV68 orf36 mediates phosphorylation ofserine 139 of H2AX during lytic infection (Tarakanova et al., 2007).Furthermore, H2AX facilitates MHV68 replication in primary macro-phages (Tarakanova et al., 2007). However, it is not clearwhether H2AXexpression affects MHV68 latency. In this study, we show that MHV68kinase orf36 facilitates viral latency in vivo. Unexpectedly, our studieshave uncovered an enzymatic activity-independent function of orf36during the establishment of viral latency. Furthermore, deficiency ofH2AX, either systemic or limited to infected cells, constrained theestablishment of MHV68 latency in a route of inoculation-specificmanner, suggesting that gammaherpesviruses may usurp componentsof DDR not only during lytic replication, but also during latent infectionin vivo.
Results
Expression and enzymatic activity of MHV68 kinase orf36 are requiredfor the establishment of latency following intranasal inoculation
To define the role of orf36 in the establishment of MHV68 latency,BL6 mice were intranasally inoculated with 104 PFU of wild typeMHV68, a viral mutant incapable of expressing orf36 (N36S), or a
36KN virus that expresses an enzymatically inactive orf36 due to asingle amino acid substitution of lysine 107 (Hwang et al., 2009).Frequencies of MHV68 DNA-positive cells and ex vivo reactivationwere measured in splenocytes and peritoneal exudate cells (PEC)harvested at 16 days post infection as previously described (Wecket al., 1996, 1999b). The frequency of MHV68 genome positive cellswas decreased 16- to 20-fold in splenocytes harvested from miceinfected with orf36 MHV68 mutant viruses as compared to wild type-infected splenocytes (Fig. 1A; 1 in 143 cells positive for wild typeMHV68 vs. 1 in 2297 or 1 in 2917 cells positive for N36S and 36KNmutant virus, respectively). The frequencies of ex vivo reactivationwere also decreased in splenocytes harvested from N36S or 36KN-infected mice as compared to wild type-infected controls (Fig. 1B).The decrease in the frequency of ex vivo reactivation appeared to besimilar in magnitude to the decrease in the frequency of MHV68genome positive cells (Fig. 1A). Therefore, in spite of fewer MHV68positive splenocytes in mice infected with orf36 mutant viruses, thesame proportion of infected splenocytes reactivated ex vivo, suggest-ing that the viral kinase is not required for the efficiency ofreactivation under these experimental conditions. The frequenciesof infected cells and ex vivo reactivation were decreased in PECharvested from mice latently infected with either orf36 mutant(Figs. 1C and D). Thus, following intranasal inoculation, both orf36expression and enzymatic activity facilitated establishment of MHV68latency in the spleen and peritoneum.
Orf36 expression, but not kinase activity, is required for early MHV68latency establishment following intraperitoneal inoculation
Because MHV68 acute replication is attenuated in vivo in theabsence of orf36 (Hwang et al., 2009; Tarakanova et al., 2007), thiscould delay dissemination of orf36 MHV68 mutants to spleen andperitoneum with the subsequent delay in the establishment oflatency. Furthermore, a route of inoculation-dependent spleniclatency phenotype had been described for the MHV68 virus mutantlacking expression of M2 protein (Herskowitz et al., 2005). Therefore,we wanted to determine whether direct inoculation of orf36 MHV68mutants into peritoneum would overcome the defect in theestablishment of latency that was seen following intranasal inocula-tion (Fig. 1). BL6 mice were intraperitoneally infected with 104 PFU ofwild type MHV68 or either of the orf36 mutants described above andsplenocytes and PEC harvested at 16 days post infection. Thefrequency of latently infected splenocytes was decreased approxi-mately 6-fold inmice infectedwith N36Smutant virus as compared towild type infection (1 in 239 splenocytes for wild type virus vs. 1 in1505 splenocytes for N36S mutant, pb0.05; Fig. 2A). Interestingly, thefrequency of splenocytes infected with the 36KN mutant was similarto that measured in the wild type-infected group (1 in 239 for wildtype virus vs. 1 in 464 for 36KN mutant, pN0.05, Fig. 2A). Thefrequency of ex vivo reactivation from splenocytes was significantlydecreased for both orf36 mutants (Fig. 2B). These data suggested thatorf36 expression was required to establish an adequate pool oflatently infected splenocytes and to support efficient MHV68reactivation ex vivo. However, orf36 enzymatic activity was dispens-able for the establishment of a reservoir of latently infectedsplenocytes following intraperitoneal inoculation.
In the peritoneum, the frequency of N36S-infected PEC wasdecreased approximately 4-fold compared to wild type infection (1in 154 vs. 1 in 589 cells; Fig. 2C). On the contrary, the frequency of36KN-infected PEC was indistinguishable from that observed in wildtype MHV68-infected mice (1 in 154 vs. 1 in 151 cells; Fig. 2C). Thefrequency of ex vivo reactivation was decreased approximately 12-fold in PEC infected with N36S mutant as compared to wild type virus(1 in 234 vs. 1 in 2895 cells; Fig. 2D), suggesting that orf36 expressionwas required for both the number of latently infected PEC andefficiency of ex vivo reactivation. The frequency of ex vivo reactivation

Fig. 1. Expression and enzymatic activity of MHV68 kinase orf36 are required for the establishment of latency following intranasal inoculation. BL6 mice were intranasally inoculatedwith 104 PFU of wild type MHV68, N36S viral mutant incapable of expressing orf36 kinase, or 36KN viral mutant expressing an enzymatically inactive orf36. Splenocytes andperitoneal exudate cells (PEC) were harvested at 16 days post infection and pooled from 3 to 5 mice in each experimental group. Frequencies of infected cells (A, C) and ex vivoreactivation (B, D) were determined in indicated experimental groups using limiting dilution assays. Data were pooled from 3 to 4 independent experiments.
Fig. 2. Orf36 expression but not kinase activity is required for early MHV68 latency establishment following intraperitoneal inoculation. BL6 mice were intraperitoneally infectedwith 104 PFU of wild type MHV68, N36S, or 36KN virus mutants. Splenocytes and PEC were harvested and pooled from 3 to 5 mice in each experimental group at 16 days post-infection and frequencies of infected cells (A, C) and ex vivo reactivation (B, D) determined using limiting dilution assays. Data were pooled from 3 independent experiments.
52 V.L. Tarakanova et al. / Virology 405 (2010) 50–61

53V.L. Tarakanova et al. / Virology 405 (2010) 50–61
was only marginally decreased in PEC isolated from 36KN-infectedmice, and this differencewas not statistically significant (1 in 234 cellsfor wild type virus vs. 1 in 492 cells for 36KN, pN0.05; Fig. 2D). Thus,following intraperitoneal inoculation, orf36 expression, but not itsenzymatic activity, was required for efficient latency establishment inthe peritoneum. With the exception of the reactivation phenotype inspleen following intraperitoneal inoculation, our studies revealed aroute of infection-specific role of orf36 enzymatic activity in MHV68latency establishment.
Orf36 does not affect the distribution of latently infected cells betweenCD93-positive and -negative splenic B cell populations
Early MHV68 latency is associated with changes in splenic B cellpopulations, including increases in germinal center B cells, CD138+
plasma cells, and polyclonal B cell activation (Collins et al., 2009;Sangster et al., 2000; Stevenson and Doherty, 1999; Liang et al., 2009;Siegel et al., 2009). Some of these changes are driven by the MHV68-encoded M2 protein (Siegel et al., 2008; Liang et al., 2009). Despitethese obvious changes to the B cell compartment, which B cell subsetsharbor latent virus remains somewhat controversial. Studies haveshown that during early latency the frequency of MHV68 positivecells is similar in IgD+ and IgD− splenic B cells, suggesting that bothnaïve and terminally differentiated B cells harbor latent virus (Willerand Speck, 2003). However, other studies suggest that during earlylatency, MHV68 preferentially resides within B cells positive for thegerminal center markers, such as PNA and GL-7 (Collins et al., 2009;Flano et al., 2000, 2003, 2002; Willer and Speck, 2003). In studies oflong-term latency, latently infected cells are mostly found in the IgD−
B cell compartment (Willer and Speck, 2003). Some of thecontroversial results may have stemmed, in part, from the manipula-tion of CD21 and CD23 on the surface of infected B cells (Collins et al.,2009), making it difficult to interpret some of the data.
Because the frequency of MHV68 positive splenocytes wasdecreased in mice latently infected with orf36 MHV68 mutants, wewanted to determine whether orf36 had a role in the distribution of
Fig. 3. Orf36 does not affect the distribution of latently infected cells between CD93-positivePFU of wild typeMHV68, N36S, or 36KN viral mutant. Splenocytes were harvested and pooledantibodies and analyzed by flow cytometry (A-representative result). (B, C) Splenocytes weusing limiting dilution nested PCR assay. Data were pooled from 3 to 4 independent experi
latently infected cells among splenic B cell populations. We used CD93to differentiate newly emerged transitional B cells frommore mature Bcells, including follicular B cells andmarginal zone B cells (Allman et al.,2001), and investigate whether orf36 plays a role in establishinglatency in these splenic B cell subsets. BL6 mice were intranasallyinoculated with wild type MHV68 or orf36 virus mutants, splenocytesharvested at 16 days post infection and pooled within each group withsubsequent staining with CD93 and B220 (Fig. 3A). Infection did notresult in gross changes to the normal distribution of B220+CD93+ cells,although those receiving the N36S or 36KNMHV68 mutants presentedwith marginal increases in this population based on whole splenocytes(Fig. 3A).
Pooled splenocytes within each experimental group were sortedinto CD93+B220+ (transitional B cells), CD93−B220+ (follicular andmarginal zone B cells), and B220− (non-B-cell) populations and thefrequency of MHV68 positive cells determined by limiting dilutionnested PCR. The frequencies of MHV68 positive cells were similar inCD93-positive (1 in 477 cells) and CD93-negative (1 in 337 cells) Bcell populations isolated from wild type MHV68-infected spleens(Fig. 3B). As expected, a significantly lower frequency of MHV68positive cells was found in B220-negative splenocyte population(Fig. 3B). Although the frequency of MHV68 positive cells was lowerin sorted splenocytes harvested from N36S-infected mice, MHV68genome positive cells had a similar distribution between CD93-positive and -negative B cell subsets (1 in 2822 cells and 1 in 1485cells, respectively; Fig. 3C). Distribution of 36KN-infected splenocyteswas similar to that observed for the N36S-infected group (data notshown). Thus, neither expression nor enzymatic activity of orf36 hadan effect on the distribution of latently infected cells between CD93-positive and -negative B cell subsets.
Orf36 expression and enzymatic activity facilitate MHV68 reactivationduring long-term latency
Between 16 and 42 days post infection, MHV68 transitions fromearly latency to a more stable long-term condition with a decrease
and -negative splenic B cell populations. BL6 mice were intranasally inoculated with 104
from 3 to 5mice/group at 16 days post infection, stainedwith anti-B220 and anti-CD93re sorted into indicated populations and frequency of MHV68 infected cells determinedments for each sorted population.
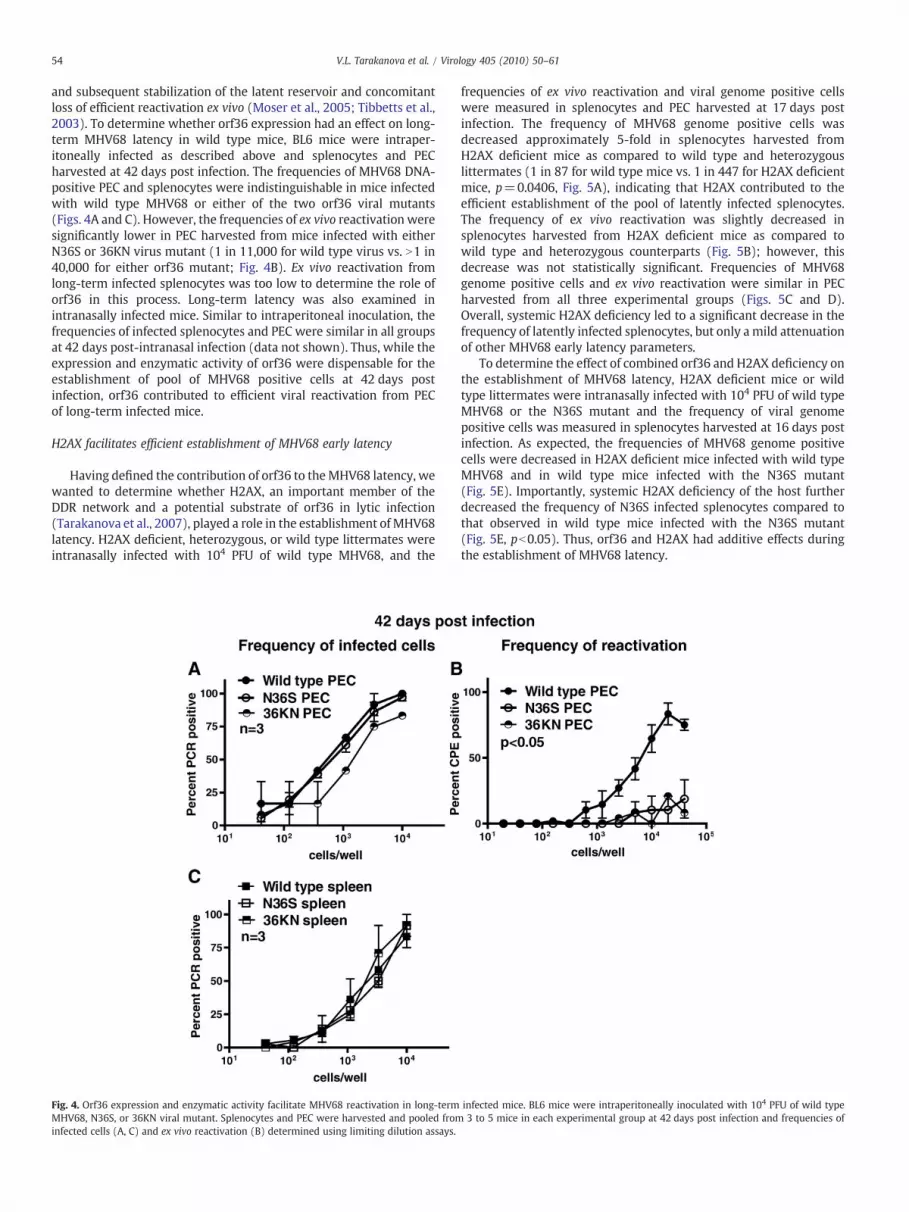
54 V.L. Tarakanova et al. / Virology 405 (2010) 50–61
and subsequent stabilization of the latent reservoir and concomitantloss of efficient reactivation ex vivo (Moser et al., 2005; Tibbetts et al.,2003). To determine whether orf36 expression had an effect on long-term MHV68 latency in wild type mice, BL6 mice were intraper-itoneally infected as described above and splenocytes and PECharvested at 42 days post infection. The frequencies of MHV68 DNA-positive PEC and splenocytes were indistinguishable in mice infectedwith wild type MHV68 or either of the two orf36 viral mutants(Figs. 4A and C). However, the frequencies of ex vivo reactivation weresignificantly lower in PEC harvested from mice infected with eitherN36S or 36KN virus mutant (1 in 11,000 for wild type virus vs. N1 in40,000 for either orf36 mutant; Fig. 4B). Ex vivo reactivation fromlong-term infected splenocytes was too low to determine the role oforf36 in this process. Long-term latency was also examined inintranasally infected mice. Similar to intraperitoneal inoculation, thefrequencies of infected splenocytes and PEC were similar in all groupsat 42 days post-intranasal infection (data not shown). Thus, while theexpression and enzymatic activity of orf36 were dispensable for theestablishment of pool of MHV68 positive cells at 42 days postinfection, orf36 contributed to efficient viral reactivation from PECof long-term infected mice.
H2AX facilitates efficient establishment of MHV68 early latency
Having defined the contribution of orf36 to theMHV68 latency, wewanted to determine whether H2AX, an important member of theDDR network and a potential substrate of orf36 in lytic infection(Tarakanova et al., 2007), played a role in the establishment of MHV68latency. H2AX deficient, heterozygous, or wild type littermates wereintranasally infected with 104 PFU of wild type MHV68, and the
Fig. 4. Orf36 expression and enzymatic activity facilitate MHV68 reactivation in long-termMHV68, N36S, or 36KN viral mutant. Splenocytes and PEC were harvested and pooled frominfected cells (A, C) and ex vivo reactivation (B) determined using limiting dilution assays.
frequencies of ex vivo reactivation and viral genome positive cellswere measured in splenocytes and PEC harvested at 17 days postinfection. The frequency of MHV68 genome positive cells wasdecreased approximately 5-fold in splenocytes harvested fromH2AX deficient mice as compared to wild type and heterozygouslittermates (1 in 87 for wild type mice vs. 1 in 447 for H2AX deficientmice, p=0.0406, Fig. 5A), indicating that H2AX contributed to theefficient establishment of the pool of latently infected splenocytes.The frequency of ex vivo reactivation was slightly decreased insplenocytes harvested from H2AX deficient mice as compared towild type and heterozygous counterparts (Fig. 5B); however, thisdecrease was not statistically significant. Frequencies of MHV68genome positive cells and ex vivo reactivation were similar in PECharvested from all three experimental groups (Figs. 5C and D).Overall, systemic H2AX deficiency led to a significant decrease in thefrequency of latently infected splenocytes, but only a mild attenuationof other MHV68 early latency parameters.
To determine the effect of combined orf36 and H2AX deficiency onthe establishment of MHV68 latency, H2AX deficient mice or wildtype littermates were intranasally infected with 104 PFU of wild typeMHV68 or the N36S mutant and the frequency of viral genomepositive cells was measured in splenocytes harvested at 16 days postinfection. As expected, the frequencies of MHV68 genome positivecells were decreased in H2AX deficient mice infected with wild typeMHV68 and in wild type mice infected with the N36S mutant(Fig. 5E). Importantly, systemic H2AX deficiency of the host furtherdecreased the frequency of N36S infected splenocytes compared tothat observed in wild type mice infected with the N36S mutant(Fig. 5E, pb0.05). Thus, orf36 and H2AX had additive effects duringthe establishment of MHV68 latency.
infected mice. BL6 mice were intraperitoneally inoculated with 104 PFU of wild type3 to 5 mice in each experimental group at 42 days post infection and frequencies of

Fig. 5.H2AX facilitates efficient establishment of MHV68 splenic latency. H2AXwild type, heterozygous (het), or deficient (KO)mice were intranasally infected with 104 PFU of wild-type MHV68 (or N36S viral mutant in E). Splenocytes and PEC were harvested at 16 days post infection and pooled from 3 to 5 mice in each experimental group. Frequencies ofinfected cells (A, C, E) and ex vivo reactivation (B, D) were determined in indicated experimental groups using limiting dilution assays. Data were pooled from 3 to 4 independentexperiments.
55V.L. Tarakanova et al. / Virology 405 (2010) 50–61
Efficient establishment of early MHV68 latency is dependent on thepresence of H2AX within the latently infected cell
An important caveat of MHV68 latency studies in H2AX deficientmice is the presence of the immune deficiencies, such as decrease in Band T cell numbers and defective class switching, identified in thesemice in the absence of pathogen challenge (Celeste et al., 2002). To thebest of our knowledge, there are no reports examining immuneresponses of H2AX deficient mice challenged with a live pathogen.Therefore, it is possible that known immune defects becomeexacerbated or additional immune deficiencies are exposed uponchallenge of H2AX deficient mice with MHV68. Because manyeffectors of immune system, including B and T cells, control MHV68latency (reviewed in Stevenson and Efstathiou, 2005), the parametersof early latency in H2AX deficient mice are likely to combine effects ofH2AX deficiency within the latently infected cells and systemic effectson the immune system. To circumvent this caveat, we analyzed
MHV68 latency establishment in a mouse model where H2AXdeficiency is limited to infected cells.
H2AX deficient mice were generated from a mouse embryonicstem (ES) cell line containing H2AX gene flanked by loxP sites(Bassing et al., 2002). This ES cell line gave rise to the mouse strainwith systemic H2AX deletion (via expression of Cre recombinase in EScells) as well as the H2AX flox/flox mouse strain that allowsconditional deletion of H2AX in vivo (Bassing et al., 2002, 2003). Todetermine whether H2AX deficiency limited to MHV68-infected cellshad an effect on viral latency, H2AX flox/flox mice were intranasallyinfected with 104 PFU of wild type MHV68 or the MHV68-Cre viruscontaining a CMV promoter-driven Cre recombinase transgeneinserted into the 27b-29 locus of the MHV68 (Moser et al., 2005).Importantly, insertion of the Cre transgene has no detectable effectson the ability of MHV68-Cre virus to replicate or establish latency inwild type mice, while allowing efficient deletion of loxP-flankedsequences in mouse models with conditional genetic deficiencies

56 V.L. Tarakanova et al. / Virology 405 (2010) 50–61
(Moser et al., 2005). The frequencies of MHV68 genome positive cellsand ex vivo reactivation were measured in splenocytes and PECcollected at 15–16 days post infection.
The frequency of MHV68 genome positive splenocytes wassignificantly decreased (approximately 6-fold) in H2AX flox/floxmice infected with MHV68-Cre virus as compared to wild type virus-infected controls (1 in 95 cells for wild type virus vs. 1 in 600 cells forMHV68-Cre; Fig. 6A). Concurrent with decrease in the frequency oflatently infected cells, the frequency of ex vivo reactivation was alsodecreased in MHV68-Cre-infected splenocytes (Fig. 6B). The frequen-cies of MHV68 genome positive cells as well as ex vivo reactivationwere decreased in PEC harvested from MHV68-Cre-infected mice(Figs. 6C and D). Thus, specific deletion of H2AX within the infectedcell led to a significant attenuation of early MHV68 latency in thespleen and peritoneum. Notably, at 28 days post infection, thefrequencies of MHV68 positive cells and ex vivo reactivation wereindistinguishable between wild type and MHV68-Cre-infected H2AXflox/flox mice (data not shown).
H2AX deficiency has no effect on acute MHV68 replication in vivo
MHV68 orf36 facilitates acute MHV68 replication in vivo (Hwanget al., 2009; Tarakanova et al., 2007) To determine whether H2AXdeficiency affects MHV68 acute replication in mouse lungs, H2AXwild type or deficient mice were intranasally inoculated with 104
PFU of wild type MHV68 and lytic virus measured in lungs at 4 dayspost infection, during the peak of viral replication. MHV68 titers weresimilar in lungs of H2AX deficient mice and wild type littermates(Fig. 7A). Similar peak lung titers were also observed upon infection ofH2AX flox/flox mice with wild type MHV68 or MHV68-Cre (Fig. 7B).Thus, neither systemic nor infected cell-specific H2AX deficiency hadan effect on the acute MHV68 replication in the lung.
Fig. 6. Efficient establishment of early MHV68 latency is dependent on the presence of H2AXwPFU of wild type MHV68 or MHV68-Cre. Splenocytes and PEC were harvested at 16 days pinfected cells (A, C) and ex vivo reactivation (B, D) were determined in indicated experiexperiments.
H2AX exerts a route of infection-dependent effect on early MHV68latency
The enzymatic activity of MHV68 orf36 had a route of infection-dependent contribution during the establishment of MHV68 latency(Fig. 2). Because H2AX may be a substrate of orf36 kinase, wewanted to determine whether H2AX may also modulate MHV68latency in a route of infection-dependent manner. H2AX flox/floxmice were intraperitoneally infected with 104 PFU of wild typeMHV68 or MHV68-Cre. Frequencies of viral genome positive cellsand ex vivo reactivation were measured in splenocytes and PECharvested at 16 days post infection. The frequencies of MHV68genome positive cells were similar in splenocytes harvested fromboth experimental groups (1 in 93 cells for wild type virus vs. 1 in202 cells for MHV68-Cre; Fig. 8A). However, a small but statisticallysignificant decrease was observed in the frequency of ex vivoreactivation from MHV68-Cre-infected splenocytes as compared towild type infected group (Fig. 8B), indicating that H2AX expressionwithin latently infected splenocytes facilitated ex vivo reactivation.Similar frequencies of viral genome positive cells were alsoobserved in PEC (1 in 68 for wild type virus vs. 1 in 93 forMHV68-Cre; Fig. 8C). Frequencies of ex vivo reactivation fromlatently infected PEC were indistinguishable between two experi-mental groups (Fig. 8D). Similar data were obtained in H2AX wildtype and deficient mice intraperitoneally inoculated with wild typeMHV68 (data not shown). Thus, we found that, analogous to thephenotype of the 36KN virus mutant, H2AX deficiency had a routeof inoculation-dependent effect on the establishment of MHV68latency.
To assess whether H2AX contributes to long-term MHV68latency, H2AX flox/flox mice were intraperitoneally infected with104 PFU of wild type MHV68 or MHV68-Cre and frequency of viral
ithin the latently infected cell. H2AX flox/floxmice were intranasally infectedwith 104
ost infection and pooled from 3 to 5 mice in each experimental group. Frequencies ofmental groups using limiting dilution assays. Data were pooled from 3 independent

Fig. 7.H2AX deficiency has no effect on acute MHV68 replication in lungs. H2AXwild type or deficient mice (A) or H2AX flox/floxmice (B) were intranasally infected with 104 PFU ofindicated viruses. Lungs were harvested at 4 days post infection, homogenized, and viral titers measured. Each symbol represents an individual mouse.
57V.L. Tarakanova et al. / Virology 405 (2010) 50–61
genome positive splenocytes and PEC determined at 42 dayspost infection. The frequency of MHV68 DNA-positive PEC wasdecreased in mice infected with the MHV68-Cre virus as comparedto wild type virus infection (1 in 1743 vs. 1 in 5451 cells, p=0.03;
Fig. 8. H2AX has a route of infection-dependent effect on MHV68 latency. H2AX flox/flox mSplenocytes and PECwere harvested at 16 days (A–D) or 42 days (E, F) post infection and pooand ex vivo reactivation (B, D) were determined in indicated experimental groups using lim
Fig. 8E). However, the frequencies of MHV68 DNA-positive spleno-cytes were similar in mice infected with either wild type (1 in 1505cells) or MHV68-Cre virus (1 in 2761 cells; Fig. 8F). Thus, deletion ofH2AX within infected cells had long-term effects on MHV68 latency.
ice were intraperitoneally infected with 104 PFU of wild type MHV68 or MHV68-Cre.led from 3 to 5mice in each experimental group. Frequencies of infected cells (A, C, E, F)iting dilution assays. Data were pooled from 3 independent experiments.

58 V.L. Tarakanova et al. / Virology 405 (2010) 50–61
Discussion
This study presents a comprehensive analysis of the role of aconserved gammaherpesvirus kinase in the establishment andmaintenance of viral latency in vivo. Furthermore, this is the firststudy to show that H2AX, a critical component of the host DDRnetwork, modulates gammaherpesvirus latency in vivo. Our experi-ments reveal an interesting correlation between the phenotypes ofMHV68 latency in an H2AX deficient host and latency phenotypesseen in mice infected with MHV68 mutant expressing an enzymat-ically inactive viral kinase (36KN; Table 1). Experiments presentedhere also suggest the possibility that the physiologically relevantfunctions of gammaherpesvirus kinases extend beyond the ability ofthe viral protein to phosphorylate substrates.
The role of orf36 in MHV68 latency
Our studies indicate that orf36 expression and enzymatic activitymodulate both the establishment and long-term maintenance ofMHV68 latency in vivo. The requirement for orf36 during theestablishment of MHV68 latency (Figs. 1 and 2) and MHV68reactivation from long-term infected PEC (Fig. 4B) suggests thatorf36 most likely functions during viral reactivation from latency.Indeed, orf36 is expressed with early–late kinetics in MHV68-infectedfibroblasts (Martinez-Guzman et al., 2003) and facilitatesMHV68 lyticreplication in vitro in a cell type-specific manner (Tarakanova et al.,2007). Furthermore, MHV68 reactivation from latency is likely tofacilitate both the establishment of splenic latency and long-terminfection (Li et al., 2008; Liang et al., 2009; Siegel et al., 2009). Themodulation of latent MHV68 infection by a lytic viral protein is notunprecedented. In fact, MHV68-encoded v-cyclin is one example of alytic viral protein that is required for efficient establishment andmaintenance of MHV68 latency (Van Dyk et al., 2000, 2003; Uptonand Speck, 2006). However, we also cannot rule out that orf36functions in latently infected cells independent of viral reactivation.While orf36 expression was not detected in a tumor cell line latentlyinfected with MHV68 (Martinez-Guzman et al., 2003), the expressionof orf36 during viral latency in vivo has not been formally excluded.Importantly, expression of KSHV orf36 kinase is induced by hypoxia inlatently infected cell lines (Haque et al., 2006), suggesting that certainstimuli may induce viral kinase expression in latently infected cells inthe absence of bona fide reactivation.
In our study, we entertained a possibility that the tropism ofMHV68 for splenic B cell subpopulations would be altered in theabsence of orf36 expression, thus leading to attenuation of latency inthe spleen. Interestingly, we found that the frequencies of infectedcells were similar between the transitional (CD93+B220+) andfollicular and marginal zone B cell populations (CD93−B220+)irrespective of the orf36 status (Figs. 3B and C). While previous
Table 1Summary of in vivo phenotypes of orf36 MHV68 mutants and H2AX deficient host during e
Deficiency of Route of inoculation Fre
Orf36 expression Intraperitoneal SpPE
Orf36 enzymatic activity Intraperitoneal Sp
H2AX within infected cells (H2AX flox/flox) Intraperitoneal Sp
Orf36 expression Intranasal SpPE
Orf36 enzymatic activity Intranasal SpPE
H2AX within infected cells (H2AX flox/flox) Intranasal SpPE
a Compared to wild type virus.b Low-frequency precluded precise quantitation.
studies have shown that the majority of MHV68 infected splenic Bcells during early latency are positive for germinal center markers(Collins et al., 2009; Flano et al., 2000, 2002, 2003; Willer and Speck,2003), our data would suggest that transitional B cells also supportviral latency. This suggests that identification of B cell subsets thatsupport latent infection may be more complicated than previouslyappreciated. This observation warrants future studies of the interac-tion of MHV68 with “immature” B cell populations and locale ofterminally differentiated B cells during the establishment of latency.
The role of orf36 enzymatic activity in MHV68 latency
The interpretation of the 36KN latency phenotype is complicatedby the poor understanding of cell types infected with MHV68 duringthe first 10–12 days post infection. Furthermore, trafficking ofMHV68-infected cells during the first 14 days of infection is notclear. Recent studies utilizing luciferase expressing MHV68 mutantsshowed that the kinetics of MHV68 distribution to different organsvary based on the route of inoculation (Hwang et al., 2008; Milhoet al., 2009); however, the mechanism behind this route ofinoculation-dependent viral distribution is not known. It is not clearwhether route of inoculation plays a role in MHV68 pathogenesis. Thelatency phenotype of the orf36.KN mutant is not unprecedented, asthe M2MHV68mutant also exhibits a route of inoculation-dependentphenotype in the establishment of MHV68 latency (Herskowitz et al.,2005). Interestingly, the splenic latency phenotype exhibited by theM2 mutant following intraperitoneal inoculation can be rescued byincreasing the inoculum dose (Herskowitz et al., 2005; Jacoby et al.,2002). This would suggest that a lower inoculum dose may reveal arole for orf36 enzymatic activity in early MHV68 latency following anintraperitoneal inoculation.
While the orf36 substrates have not been extensively character-ized, H2AX and IRF3 are the two physiologically relevant substrates ofthis kinase that play a role during MHV68 lytic replication and theestablishment of latency (Hwang et al., 2009; Tarakanova et al., 2007;and this study). Importantly, combined orf36 and H2AX deficiencyhad an additive effect in the attenuation of MHV68 latency (Fig. 5E),suggesting that orf36 regulates multiple cellular processes (includingDNA damage response and type I interferon signaling) to facilitateviral latency. Importantly, the kinase activity of orf36 was notrequired to inhibit interferon beta reporter activity (Hwang et al.,2009) or establish latency following intraperitoneal inoculation(Fig. 2), suggesting that MHV68 orf36 is a multifunctional viralprotein, with only some of its functions dependent on the enzymaticactivity. This is significant, as the pharmacological approaches thattarget enzymatic activity of herpesvirus kinases (such as maribavirand its derivatives)may only be partially effective, as these agents willlikely fail to inhibit enzymatic activity-independent viral kinasefunctions. Interestingly, the latency phenotypes of the 36KN MHV68
arly MHV68 latency.
quency of infected cellsa Frequency of ex vivo reactivationa
leen: decreased 6-fold Spleen: decreasedb
C: decreased 4-fold PEC: decreased 12-foldleen and PEC: similar to wild type virus Spleen: decreasedb
PEC: similar to wild type virusleen and PEC: similar to wild type virus Spleen: decreasedb
PEC: similar to wild type virusleen: decreased 16- to 20-fold Spleen: decreasedb
C: decreasedb PEC: decreasedb
leen: decreased 16- to 20-fold Spleen: decreasedb
C: decreasedb PEC: decreasedb
leen: decreased 6-fold Spleen: decreasedb
C: decreasedb PEC: decreasedb

59V.L. Tarakanova et al. / Virology 405 (2010) 50–61
mutant seem to mirror the phenotypes of MHV68 latency in theabsence of H2AX, suggesting that phosphorylation of H2AX by orf36may be one of the mechanisms that regulate MHV68 latency andreactivation in vivo.
The role of H2AX in MHV68 latency
In this study, we demonstrate that H2AX facilitates establishmentof MHV68 latency. We utilized mouse strains containing systemic orconditional deletion of H2AX. The advantage of using the H2AX flox/flox system is that specific deletion of H2AX within the infectedcells occurs in the context of a normal immune system. A greaterattenuation of early MHV68 latency observed in H2AX flox/flox miceas compared to mice with global H2AX deficiency (Figs. 5 and 6)suggests that the compromised immune system in H2AX deficientmice may indeed modulate the parameters of MHV68 latency.Alternatively, H2AX may have previously unappreciated effects on Bcell development that could further affect MHV68 latency in micewith systemic deletion of H2AX. The caveat of the flox/flox system isthat the deletion of H2AX requires viral entry and gene expression.Furthermore, the efficiency of Cre-mediated gene excision in thecontext of the MHV68-Cre virus has not been reported. Therefore, it ispossible that the effects of H2AX deficiency within latently infectedcells may be more profound than revealed by our studies due toefficiency and/or timing of Cre-mediated H2AX deletion.
An important question that will be addressed in future studies isthe mechanism by which H2AX functions during MHV68 latency.H2AX, one of the many H2A isoforms, appears to be redundant forcellular chromatin assembly. However, unlike other H2A isoforms,H2AX is a critical member of the DDR network (Yin and Bassing,2008). The unique serine 139 of H2AX is phosphorylated shortly afterdetection of DNA breaks and serves as a scaffold for DNA repairproteins (Yin and Bassing, 2008). Serine 139 of H2AX is phosphor-ylated in an orf36-dependent manner in MHV68 infection and H2AXis required for efficient viral replication in primary macrophages invitro (Tarakanova et al., 2007). It is possible that DDR activation,including H2AX phosphorylation, occurs during viral reactivationfrom latency. The phosphorylation of serine 139 may be the feature ofH2AX required to exert its effects in latently infected cells. Expressionof phosphorylation defective H2AX mutant (S139A) increasessensitivity to DNA damage (Rios-Doria et al., 2009). Furthermore,the phenotype of wild type MHV68 latency in H2AX deficient cellsmirrors the latency phenotype of MHV68 36KNmutant that expressesan enzymatically inactive viral kinase (Table 1). It is tempting tospeculate that, similar to lytic infection, orf36 may mediate H2AXphosphorylation during the establishment of MHV68 latency.
H2AX may function in cis, through its association with MHV68genome, or in trans, by facilitating DDR signaling via cellularchromatin. The association of herpesvirus DNA with histones hasprofound effects on viral gene expression and control of replicationand latency (reviewed in Knipe and Cliffe, 2008). Unfortunately,chromatization of latent MHV68 genome in vivo is poorly understood.Induction of DDR is associated with significant changes in histonemodifications, including acetylation, phosphorylation, and methyla-tion of specific residues that subsequently modulate transcriptionalactivity of the chromatin in the vicinity of the DNA lesion (Pandita andRichardson, 2009; Huertas et al., 2009; van Attikum and Gasser,2009). Therefore, recruitment of H2AX to the viral chromatin andsubsequent activation of DDR may contribute to regulation of MHV68gene expression during reactivation from latency. The role of DDR inherpesvirus reactivation from latency is poorly understood. This issuehas been historically examined in conjunction with radiation orchemotherapy treatment of cancer patients. However, in the clinicalsetting it is frequently impossible to discriminate intrinsic effects ofDDR induction within latently infected cells and the effects of cancer-associated immunosuppression that is further exacerbated by
radiation or chemotherapy. Irradiation upregulates late HSV geneexpression in vitro in cells lytically infected with the gamma 34.5 HSVmutant, but not the wild type virus (Mezhir et al., 2005). Unfortu-nately, the cell-intrinsic effects of DDR induction on herpesvirusreactivation from primary cells are not understood.
In summary, we have shown that the conserved gammaherpes-virus kinase and an important component of DDR network bothregulate gammaherpesvirus latency in vivo. These findings raiseimportant implications for gammaherpesvirus pathogenesis. DDRnetwork functions as a tumor suppressor, and manipulations of thisnetwork during gammaherpesvirus latency may contribute togammaherpesvirus-associated tumorigenesis.
Materials and methods
Animals
C57BL/6 J (BL6) mice were obtained from Jackson Laboratories(Bar Harbor, Maine) and bred at the Medical College of Wisconsin,Milwaukee, WI. H2AX deficient and H2AX flox/flox mice were a kindgift of Dr. Fred Alt (HHMI, Harvard Medical School). All mice werehoused in a specific pathogen-free barrier facility in accordance withfederal and institutional guidelines. Mice were infected between 7and 10 weeks of age, and spleens and peritoneal exudate cells (PEC)were harvested from euthanized mice at indicated days post infectionand processed as previously described (Van Dyk et al., 2000). Allexperimental manipulations of mice were approved by the Institu-tional Animal Care and Use Committee of the Medical College ofWisconsin.
Virus infections
The derivation of orf36 mutant MHV68 viruses and acorresponding wild type control virus was previously described(Hwang et al., 2009; Tarakanova et al., 2007). MHV68-Cre was a kindgift of Dr. Samuel Speck (Emory University School of Medicine). Allviruses were passaged and tittered on NIH 3 T12 cells as previouslydescribed (Weck et al., 1996). Viruses were diluted in Dulbecco'smodified Eagle's medium and 15 or 200 μl per mouse was used forintranasal or intraperitoneal inoculation, respectively. For acutereplication assays, lungs were harvested at 4 days post infection,mechanically disrupted with 1 mm zirconia beads (Biospec products,Bartlesville, OK), briefly centrifuged to remove tissue particles, andsupernatants subjected to plaque assay using NIH 3 T12 cells.
Limiting-dilution assays
The frequency of cells reactivating MHV68 ex vivo and thefrequency of cells harboring viral genome were determined aspreviously described (Weck et al., 1996). Briefly, to determine thefrequency of cells reactivating virus ex vivo, serial twofold dilutions ofsplenocytes or PEC suspensions harvested from infected mice wereplated onto monolayers of mouse embryonic fibroblasts (MEF) at 24replicates per dilution. In order to control for any pre-formedinfectious virus, twofold serial dilutions of mechanically disruptedsplenocytes or PEC were plated as above. MHV68 was allowed toreactivate from splenocytes or PEC, and virus was further amplifiedwithin the same well via subsequent replication in MEF. Cytopa-thic effect was scored at 21 days post-plating in all replicates anddilutions. Because primary MEF were used to amplify the virus, thesensitivity of limiting dilution reactivation assay was a single plaqueforming unit of MHV68. Because the endpoint of viral amplification inMEF was measured, the limiting dilution reactivation assay was notsusceptible to variability of titers released from primary cells uponviral reactivation ex vivo.

60 V.L. Tarakanova et al. / Virology 405 (2010) 50–61
To determine the frequency of cells harboring MHV68 genome,splenocyte or PEC suspensions were plated in serial threefolddilutions in a background of uninfected NIH 3 T12 cells (12 replicatesper dilution), digested with proteinase K, and subjected to nested PCRusing primers against orf72 sequences. Data were subjected tononlinear regression analysis (GraphPad Prism software, San Diego,CA), and Poisson distribution was used to determine the frequenciesof reactivating or MHV68 genome positive cells.
Flow cytometry and cell sorting
Spleens frommock-, wild type-, N36S-, and 36KN-infected C57BL/6mice were homogenized, red blood cells were lysed using RBC lysisbuffer (Sigma-Aldrich, Saint Louis, MO), and Fc receptors were blockedaccording tomanufacturer's recommendations (24G2, BDPharmingen).Cells were then stained with antibodies against B220-AF647 (RA3-6B2)and CD93-PE (AA4.1- eBiosciences). Cells were sorted on the BDFACSAria into 3 separate populations B220−CD93−, B220+CD93+, andB220+CD93− with an average sort purity of greater than 96%, 91%, and94%, respectively.
Statistical analyses
Data from limiting dilution assays were subjected to nonlinearregression analysis (GraphPad Prism software, San Diego, CA). Basedon Poisson distribution, the intersection of linear regression curvewith 63.2% was used to determine the frequency of reactivating orMHV68 genome positive cells. ANOVA or paired Student t-test wereused to determine statistical significance in multi- or two-groupexperiments, respectively (GraphPad Prism software, San Diego, CA).
Acknowledgments
We are grateful to Frederick Alt for his generous gift of H2AXdeficient and flox/flox mouse strains and to Samuel Speck for theMHV68-Cre virus. An expert technical and managerial support wasprovided by Fei Chin Tsan. We also thank Amy Hudson's, WilliamJackson's, and Scott Terhune's laboratories for helpful discussions.
This work was supported by the Advancing Healthier Wisconsinand the Medical College of Wisconsin Cancer Center (V.L.T.) andby The Medical College of Wisconsin/The Children's ResearchInstitute (S.B.G.). The funding sources had no involvement in thepublished studies.
References
Allman, D., Lindsley, R.C., DeMuth, W., Rudd, K., Shinton, S.A., Hardy, R.R., 2001.Resolution of three nonproliferative immature splenic B cell subsets revealsmultiple selection points during peripheral B cell maturation. J. Immunol. 167,6834–6840.
Asano, S., Honda, T., Goshima, F., Nishiyama, Y., Sugiura, Y., 2000. US3 protein kinase ofherpes simplex virus protects primary afferent neurons from virus-inducedapoptosis in ICR mice. Neurosci. Lett. 294, 105–108.
Bartek, J., Lukas, J., Bartkova, J., 2007. DNA damage response as an anti-cancer barrier:damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 6,2344–2347.
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested,M., Nesland, J.M., Lukas, C., Orntoft, T., Lukas, J., Bartek, J., 2005. DNA damageresponse as a candidate anti-cancer barrier in early human tumorigenesis. Nature434, 864–870.
Barton, E.S., Lutzke, M.L., Rochford, R., Virgin, H.W., 2005. Alpha/beta interferonsregulate murine gammaherpesvirus latent gene expression and reactivation fromlatency. J. Virol. 79, 14149–14160.
Bassing, C.H., Chua, K.F., Sekiguchi, J., Suh, H., Whitlow, S.R., Fleming, J.C., Monroe, B.C.,Ciccone, D.N., Yan, C., Vlasakova, K., Livingston, D.M., Ferguson, D.O., Scully, R., Alt, F.W., 2002. Increased ionizing radiation sensitivity and genomic instability in theabsence of histone H2AX. Proc. Natl. Acad. Sci. U. S. A. 99, 8173–8178.
Bassing, C.H., Suh, H., Ferguson, D.O., Chua, K.F., Manis, J., Eckersdorff, M., Gleason, M.,Bronson, R., Lee, C., Alt, F.W., 2003. Histone H2AX: a dosage-dependent suppressorof oncogenic translocations and tumors. Cell 114, 359–370.
Celeste, A., Petersen, S., Romanienko, P.J., Fernandez-Capetillo, O., Chen, H.T.,Sedelnikova, O.A., Reina-San-Martin, B., Coppola, V., Meffre, E., Difilippantonio, M.
J., Redon, C., Pilch, D.R., Olaru, A., Eckhaus, M., Camerini-Otero, R.D., Tessarollo, L.,Livak, F., Manova, K., Bonner, W.M., Nussenzweig, M.C., Nussenzweig, A., 2002.Genomic instability in mice lacking histone H2AX. Science 296, 922–927.
Chang, P.C., Fitzgerald, L.D., Van Geelen, A., Izumiya, Y., Ellison, T.J., Wang, D.H., Ann, D.K., Luciw, P.A., Kung, H.J., 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and itsmodulation by the viral protein kinase. Cancer Res. 69, 5681–5689.
Collins, C.M., Boss, J.M., Speck, S.H., 2009. Identification of infected B-cell populations byusing a recombinant murine gammaherpesvirus 68 expressing a fluorescentprotein. J. Virol. 83, 6484–6493.
Efstathiou, S., Ho, Y.M., Hall, S., Styles, C.J., Scott, S.D., Gompels, U.A., 1990. Murineherpesvirus 68 is genetically related to the gammaherpesviruses Epstein–Barr virusand herpesvirus saimiri. J. Gen. Virol. 71, 1365–1372.
Flano, E., Husain, S.M., Sample, J.T., Woodland, D.L., Blackman, M.A., 2000. Latent murinegamma-herpesvirus infection is established in activated B cells, dendritic cells, andmacrophages. J. Immunol. 165, 1074–1081.
Flano, E., Kim, I.J., Woodland, D.L., Blackman, M.A., 2002. gamma-Herpesvirus latency ispreferentially maintained in splenic germinal center and memory B cells. J. Exp.Med. 196, 1363–1372.
Flano, E., Kim, I.J., Moore, J., Woodland, D.L., Blackman, M.A., 2003. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets duringpersistent infection in mice. J. Immunol. 170, 3828–3834.
Gershburg, E., Pagano, J.S., 2008. Conserved herpesvirus protein kinases. Biochim.Biophys. Acta 1784, 203–212.
Gershburg, E., Raffa, S., Torrisi, M.R., Pagano, J.S., 2007. Epstein–Barr virus-encodedprotein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 81,5407–5412.
Haque, M., Wang, V., Davis, D.A., Zheng, Z.M., Yarchoan, R., 2006. Genetic organizationand hypoxic activation of the Kaposi's sarcoma-associated herpesvirus ORF34-37gene cluster. J. Virol. 80, 7037–7051.
Herskowitz, J., Jacoby,M.A., Speck, S.H., 2005. Themurine gammaherpesvirus 68 M2gene isrequired for efficient reactivation from latently infected B cells. J. Virol. 79, 2261–2273.
Huertas, D., Sendra, R., Munoz, P., 2009. Chromatin dynamics coupled to DNA repair.Epigenetics 4, 31–42.
Hwang, S., Wu, T.T., Tong, L.M., Kim, K.S., Martinez-Guzman, D., Colantonio, A.D.,Uittenbogaart, C.H., Sun, R., 2008. Persistent gammaherpesvirus replication anddynamic interaction with the host in vivo. J. Virol. 82, 12498–12509.
Hwang, S., Kim, K.S., Flano, E., Wu, T.T., Tong, L.M., Park, A.N., Song, M.J., Sanchez, D.J.,O'Connell, R.M., Cheng, G., Sun, R., 2009. Conserved herpesviral kinase promotesviral persistence by inhibiting the IRF-3-mediated type I interferon response. CellHost Microbe 5, 166–178.
Jacoby, M.A., Virgin, H.W., Speck, S.H., 2002. Disruption of the M2 gene of murinegammaherpesvirus 68 alters splenic latency following intranasal, but notintraperitoneal, inoculation. J. Virol. 76, 1790–1801.
Knipe, D.M., Cliffe, A., 2008. Chromatin control of herpes simplex virus lytic and latentinfection. Nat. Rev. Microbiol. 6, 211–221.
Krosky, P.M., Baek, M.C., Coen, D.M., 2003. The human cytomegalovirus UL97 proteinkinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 77,905–914.
Li, H., Ikuta, K., Sixbey, J.W., Tibbetts, S.A., 2008. A replication-defective {gamma}-herpesvirus efficiently establishes long-term latency inmacrophages but not B cellsin vivo. J. Virol. 82 (17), 8500–8508.
Liang, X., Collins, C.M., Mendel, J.B., Iwakoshi, N.N., Speck, S.H., 2009. Gammaherpes-virus-driven plasma cell differentiation regulates virus reactivation from latentlyinfected B lymphocytes. PLoS Pathog. 5, e1000677.
Lilley, C.E., Carson, C.T., Muotri, A.R., Gage, F.H., Weitzman, M.D., 2005. DNA repairproteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A.102, 5844–5849.
Lilley, C.E., Schwartz, R.A., Weitzman, M.D., 2007. Using or abusing: viruses and thecellular DNA damage response. Trends Microbiol. 15, 119–126.
Lilley, C.E., Chaurushiya, M.S., Weitzman, M.D., 2009. Chromatin at the intersection ofviral infection and DNA damage. Biochim. Biophys. Acta 1799 (3–4), 319–327.
Martinez-Guzman, D., Rickabaugh, T., Wu, T.T., Brown, H., Cole, S., Song, M.J., Tong, L.,Sun, R., 2003. Transcription program of murine gammaherpesvirus 68. J. Virol. 77,10488–10503.
Mezhir, J.J., Advani, S.J., Smith, K.D., Darga, T.E., Poon, A.P., SCHMIDT, H., Posner, M.C.,Roizman, B., Weichselbaum, R.R., 2005. Ionizing radiation activates late herpessimplex virus 1 promoters via the p38 pathway in tumors treated with oncolyticviruses. Cancer Res. 65, 9479–9484.
Milho, R., Smith, C.M., Marques, S., Alenquer, M., May, J.S., Gillet, L., Gaspar, M.,Efstathiou, S., Simas, J.P., Stevenson, P.G., 2009. In vivo imaging of muridherpesvirus-4 infection. J. Gen. Virol. 90, 21–32.
Moffat, J.F., Zerboni, L., Sommer, M.H., Heineman, T.C., Cohen, J.I., Kaneshima, H., Arvin,A.M., 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virusdetermine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl.Acad. Sci. U. S. A. 95, 11969–11974.
Moser, J.M., Upton, J.W., Allen III, R.D., Wilson, C.B., Speck, S.H., 2005. Role of B-cellproliferation in the establishment of gammaherpesvirus latency. J. Virol. 79,9480–9491.
Pandita, T.K., Richardson, C., 2009. Chromatin remodeling finds its place in the DNAdouble-strand break response. Nucleic Acids Res. 37, 1363–1377.
Prichard, M.N., 2009. Function of human cytomegalovirus UL97 kinase in viral infectionand its inhibition by maribavir. Rev. Med. Virol. 19, 215–229.
Rios-Doria, J., Velkova, A., Dapic, V., Galan-Caridad, J.M., Dapic, V., Carvalho, M.A.,Melendez, J., Monteiro, A.N., 2009. Ectopic expression of histone H2AX mutantsreveals a role for its post-translational modifications. Cancer Biol. Ther. 8, 422–434.

61V.L. Tarakanova et al. / Virology 405 (2010) 50–61
Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S., Bonner, W.M., 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem.273, 5858–5868.
Sangster, M.Y., Topham, D.J., D'Costa, S., Cardin, R.D., Marion, T.N., Myers, L.K., Doherty,P.C., 2000. Analysis of the virus-specific and nonspecific B cell response to apersistent B-lymphotropic gammaherpesvirus. J. Immunol. 164, 1820–1828.
Sato, H., Pesnicak, L., Cohen, J.I., 2003. Varicella-zoster virus ORF47 protein kinase,which is required for replication in human T cells, and ORF66 protein kinase, whichis expressed during latency, are dispensable for establishment of latency. J. Virol.77, 11180–11185.
Siegel, A.M., Herskowitz, J.H., Speck, S.H., 2008. The MHV68 M2 protein drives IL-10dependent B cell proliferation and differentiation. Plos Pathog. 4.
Siegel, A.M., Rangaswamy,U.S., Napier, R.J., Speck, S.H., 2009. Blimp-1 dependent plasmacell differentiation is required for efficient maintenance of murine gammaherpes-virus latency and antiviral antibody responses. J. Virol. 84 (2), 674–685.
Stevenson, P.G., Doherty, P.C., 1999. Non-antigen-specific B-cell activation followingmurine gammaherpesvirus infection is CD4 independent in vitro but CD4dependent in vivo. J. Virol. 73, 1075–1079.
Stevenson, P.G., Efstathiou, S., 2005. Immune mechanisms in murine gammaherpes-virus-68 infection. Viral Immunol. 18, 445–456.
Sunil- Chandra, N.P., Efstathiou, S., Nash, A.A., 1992. Murine gammaherpesvirus 68establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73,3275–3279.
Tarakanova, V.L., Leung-Pineda, V., Hwang, S., Yang, C.-W., Matatall, K., Basson, M., Sun,R., Piwnica-Worms, H., Sleckman, B.P., Virgin, H.W., 2007. Gamma-herpesviruskinase actively initiates a DNA damage response by inducing phosphorylation ofH2AX to foster viral replication. Cell Host Microbe 1, 275–286.
Tibbetts, S.A., Loh, J., van, B.V., McClellan, J.S., Jacoby, M.A., Kapadia, S.B., Speck, S.H.,Virgin, H.W., 2003. Establishment and maintenance of gammaherpesviruslatency are independent of infective dose and route of infection. J. Virol. 77,7696–7701.
Upton, J.W., Speck, S.H., 2006. Evidence for CDK-dependent and CDK-independentfunctions of the murine gammaherpesvirus 68 v-cyclin. J. Virol. 80, 11946–11959.
van Attikum, H., Gasser, S.M., 2009. Crosstalk between histone modifications during theDNA damage response. Trends Cell Biol. 19, 207–217.
Van Dyk, L.F., Virgin, H.W., Speck, S.H., 2000. Themurine gammaherpesvirus 68 v-cyclinis a critical regulator of reactivation from latency. J. Virol. 74, 7451–7461.
Van Dyk, L.F., Virgin, H.W., Speck, S.H., 2003. Maintenance of gammaherpesvirus latencyrequires viral cyclin in the absence of B lymphocytes. J. Virol. 77, 5118–5126.
Virgin, H.W., Latreille, P., Wamsley, P., Hallsworth, K., Weck, K.E., Dal Canto, A.J., Speck,S.H., 1997. Complete sequence and genomic analysis of murine gammaherpesvirus68. J. Virol. 71, 5894–5904.
Wang, J.T., Doong, S.L., Teng, S.C., Lee, C.P., Tsai, C.H., Chen, M.R., 2009. Epstein–Barrvirus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway.J. Virol. 83, 1856–1869.
Weck, K.E., Barkon, M.L., Yoo, L.I., Speck, S.H., Virgin, H.W., 1996. Mature B cells arerequired for acute splenic infection, but not for establishment of latency, by murinegammaherpesvirus 68. J. Virol. 70, 6775–6780.
Weck, K.E., Kim, S.S., Virgin, H.W., Speck, S.H., 1999a. B cells regulate murinegammaherpesvirus 68 latency. J. Virol. 73, 4651–4661.
Weck, K.E., Kim, S.S., Virgin, H.W., Speck, S.H., 1999b. Macrophages are the majorreservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73,3273–3283.
Willer, D.O., Speck, S.H., 2003. Long-term latent murine Gammaherpesvirus 68infection is preferentially found within the surface immunoglobulin D-negativesubset of splenic B cells in vivo. J. Virol. 77, 8310–8321.
Wolf, D.G., Courcelle, C.T., Prichard, M.N., Mocarski, E.S., 2001. Distinct and separateroles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis andencapsidation. Proc. Natl. Acad. Sci. U. S. A. 98, 1895–1900.
Yin, B., Bassing, C.H., 2008. The sticky business of histone H2AX in V(D)J recombination,maintenance of genomic stability, and suppression of lymphoma. Immunol. Res. 42,29–40.




















