
The mitochondrial function was impaired in APP knockout mouse embryo fibroblast cells
Upload
independentCategory
view
1download
0
Published by Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 19 2039–2047
ARTICLE
Conditional tissue-specific expression of the acidα-glucosidase (GAA) gene in the GAA knockout mice: implications for therapyN. Raben*, N. Lu, K. Nagaraju, Y. Rivera, A. Lee, B. Yan, B. Byrne1, P.J. Meikle2, K. Umapathysivam2, J.J. Hopwood2 and P.H. Plotz
Arthritis and Rheumatism Branch, NIAMS, 9000 Rockville Pike, Clinical Center Building 10/9N244, National Institutes of Health, Bethesda, MD 20892, USA, 1Gene Therapy Center and Departments of Medicine, Pediatrics, and Molecular Genetics, University of Florida College of Medicine, Gainesville, FL 32610, USA and 2Department of Chemical Pathology, Women’s and Children’s Hospital, North Adelaide, 5006 Australia
Received June 11, 2001; Revised and Accepted July 13, 2001
Both enzyme replacement and gene therapy of lysosomal storage disorders rely on the receptor-mediateduptake of lysosomal enzymes secreted by cells, and for each lysosomal disorder it is necessary to select thecorrect cell type for recombinant enzyme production or for targeting gene therapy. For example, for thetherapy of Pompe disease, a severe metabolic myopathy and cardiomyopathy caused by deficiency ofacid α-glucosidase (GAA), skeletal muscle seems an obvious choice as a depot organ for local therapy andfor the delivery of the recombinant enzyme into the systemic circulation. Using knockout mice with thisdisease and transgenes containing cDNA for the human enzyme under muscle or liver specific promoterscontrolled by tetracycline, we have demonstrated that the liver provided enzyme far more efficiently. Theachievement of therapeutic levels with skeletal muscle transduction required the entire muscle mass toproduce high levels of enzyme of which little found its way to the plasma, whereas liver, comprising <5% ofbody weight, secreted 100-fold more enzyme, all of which was in the active 110 kDa precursor form. Further-more, using tetracycline regulation, we somatically induced human GAA in the knockout mice, and demonstratedthat the skeletal and cardiac muscle pathology was completely reversible if the treatment was begun early.
INTRODUCTION
Although limited success has been achieved with both enzymeand gene replacement in genetic enzyme deficiencies, it is nowapparent that the particular problems posed by each diseasewill inhibit the spread of success by imitation. In addition tothe problems of vector design, the choice of the optimal loca-tion for the production of an enzyme depends on factors suchas the efficient production of a protein that can be exported,circulated, and taken up and processed by affected cells. Thesame issues arise if the enzyme is made by cells cultured invitro and then injected.
The current state of attempts to treat glycogen storagedisease type II (GSDII) illustrates some of the problems. In thisdisorder caused by the deficiency of acid α-glucosidase(GAA), lysosomal glycogen accumulates in cells. Severedeficiency (Pompe syndrome) manifests as a rapidly progres-sive hypertrophic cardiomyopathy leading to cardiac failureand death before 2 years of age. Partial deficiency results in aprogressive limb-girdle-like myopathy, eventually leading todeath from respiratory insufficiency (1).
The enzyme is synthesized as a catalytically active precursorthat is glycosylated and modified in the Golgi by phosphoryla-tion of mannose residues. Most of the modified precursor goesto the lysosomal compartment, but a portion is secreted, andthis extracellular enzyme can be internalized by other cells.The discovery of this secretion–recapture mechanism viamannose-6-phosphate (M6P) receptors on plasma membrane(2–4) or by an M6P-independent pathway (5) has provided arationale for both cell-mediated and direct enzyme replace-ment therapies for GSDII and other lysosomal disorders (6–8).Clinical trials with recombinant human GAA (rhGAA)produced in Chinese hamster ovary cells or in transgenic rabbitmilk (9–11) have recently been initiated by two groups (12–15),and gene replacement techniques are being explored as well.Skeletal muscle is an attractive site for somatic gene therapydue to its large mass, high vascularity, and large capacity forprotein synthesis, which allows de novo synthesized proteinsaccess to the systemic circulation. Indeed, a number of studieshave demonstrated that skeletal muscle gene therapy canresult in sustained and systemic delivery of therapeutic
*To whom correspondence should be addressed. Tel: +1 301 496 1474; Fax: +1 301 402 0012; Email: [email protected]

2040 Human Molecular Genetics, 2001, Vol. 10, No. 19
proteins (16–20). Furthermore, the idea that easily injectableskeletal muscles of the limbs and trunk which need enzymereplacement might also be a good location for providingenzyme for other affected muscles such as the diaphragm andthe heart is appealing, but the decision about the best locationmust be based on solid experimental grounds. In vitro andex vivo studies using adenovirus- and retrovirus-mediated genetransfer demonstrated that transduced muscle cells were able tosecrete recombinant GAA and provide phenotypic correctionof GAA-deficient muscle cells (21,22), but the results ofin vivo experiments were less convincing; transfer of the GAAgene into skeletal muscle was accompanied by enzymesecretion in some of the studies (23), but not in others (24,25).Recent in vivo observations have demonstrated the benefitsof hepatic-targeted transduction for gene therapy of GSDII(26–28).
The major purpose of the study reported here was to comparethe efficiency of skeletal muscle and liver as locations for genereplacement therapy in GSDII and to determine whether or notestablished disease can be reversed. We have over-expressedhGAA cDNA in skeletal muscle or liver of GAA knockoutmice (–/–) by using tissue-specific promoters, and we haveused a tetracycline-responsive system so that expression of thetransgene can be initiated somatically at different stages ofdisease progression (29,30).
RESULTS
Expression of hGAA in skeletal muscle of knockout mice
Three transgenic hGAA founder lines (F.06, F.53 and F.17)were generated, and each was crossed to GAA–/– mice,resulting in three hGAA/–/– lines. Each of these lines, in turn,was crossed to Mck-T transgenic mice, already on a –/– back-ground (Mck-T/–/–). These double transgenic mice, Mck-T-hGAA/–/–, were designed to constitutively express hGAA inskeletal muscle of knockout animals. Single transgenic miceon either +/– or –/– backgrounds were used as controls. The
single transgenics on a –/– background showed a very low,probably non-specific enzyme activity in all tissues tested(Table 1). The double transgenics derived from each of thethree hGAA founders expressed low, intermediate or highlevels of hGAA in skeletal muscle as reflected by the intensityof green fluorescent protein (GFP) (Fig. 1).
In the low expresser line, the level of GAA activity inskeletal muscle was 5-fold higher than in +/– mice (Table 1),and GAA protein was detected by western analysis in skeletalmuscle, but not in any other organ (Fig. 2A). The increase ofenzyme activity in skeletal muscle was accompanied by a
Figure 1. Expression of GFP in skeletal muscle of the Mck-T-hGAA/–/–mice. (A) Single transgenic control (hGAA/–/–). Three double transgeniclines derived from three independent founders (F.06, F.P53 and F.17) showedlow (B), intermediate (C) or high (D) levels of GAA activity in skeletalmuscle. Muscle cells were examined under a fluorescent microscope usingexcitation light.
Figure 2. Analysis of the low muscle expresser line (F.06). (A) Western blotanalysis. S, spleen; Li, liver; H, heart; M, skeletal muscle. The 76 kDa form ofhGAA is detected only in skeletal muscles from double transgenic mice.Control is an age-matched single transgenic (Mck-T/–/–) littermate. Immuno-detection of vinculin was used for loading control. The gels were loaded with100 µg protein. (B) PAS-stained sections of skeletal muscle, heart, liver andbrain from control (Mck-T/–/–) (a–d) and double transgenic (e–h) mice at4 months of age. Original magnification: ×20.

Human Molecular Genetics, 2001, Vol. 10, No. 19 2041
normalization of glycogen level to wild-type values (Table 2),a finding confirmed by light microscopy (Fig. 2Be). There wasno detectable enzyme activity in plasma, however (Table 1),nor was there any significant increase in the levels of GAAactivity in the distant organs compared with the levels in –/– mice.In fact, the glycogen level in the heart progressively increasedat a rate similar to that in the knockouts. Light microscopyconfirmed these results (Fig. 2Bf–h). Thus, no secretion oruptake occurred with this level of gene expression in skeletalmuscle.
In the intermediate expresser line, the GAA activity inskeletal muscle was ∼80-fold higher than in +/– mice(Table 1). In these mice, hGAA mRNA was detected in bothskeletal muscle and heart (Fig. 3A and B), but not in liver,lung, brain, kidney or spleen, and the lack of GFP expressionin these target organs further supported the northern and RT–PCR data (Fig. 3C, lower panel). Thus, systemic correctioncould be studied in these target organs.
The hGAA protein was detected by western blot in liver,lung and spleen (Fig. 3C, top panel), and increased GAAactivity in these organs reached ∼50, 69 and 80%, respectively,of the values in +/– controls. Low levels of GAA activity couldbe detected in plasma by the enzyme assay (Table 1) andwestern analysis; the 110 kDa precursor was visible afterprolonged exposure (Fig. 3C, top right panel). Morpho-logically, skeletal muscle was completely cleared of glycogen,and the secreted protein provided a phenotypic rescue indistant organs (shown for liver in Fig. 3Db). No protein wasdetected in brain and kidney (Fig. 3C), and glycogen in theseorgans was not reduced (shown for brain in Fig. 3Dc).
In the high expresser line, hGAA was expressed in skeletalmuscle and heart, and the levels of enzyme activity reached1082 ± 142 and 793 ± 85 nmol/h/mg/protein, respectively.These levels were ∼200-fold higher than in +/– mice in skeletalmuscle and ∼90-fold higher in heart. Although glycogencontent in these organs was reduced significantly, morpholog-ically both skeletal and cardiac muscles contained multipleperiodic acid-Schiff (PAS)-positive cells, with some cells ofabnormal shape and smaller size (Fig. 4), suggesting that thehigh GAA levels might be toxic to the cells. For this reason,this line was not used for cross-correction studies.
Expression of hGAA in the liver of knockout mice
Two transgenic Alb-T founder lines (F.21 and F.26) weregenerated, and both were crossed to –/– mice, resulting in twoAlb-T/–/– lines which were then crossed to hGAA/–/– (F.17)to generate double transgenics that constitutively expresshGAA in the liver. The expression of the transgene in the twolines was similar, as shown by the intensity of GFP staining inliver (Fig. 5), and in both lines the hepatic GAA activity was∼25-fold greater than in +/– controls. Results with only oneline (F.21/F.17) are presented here.
hGAA mRNA was detected only in the liver in double trans-genic mice by northern analysis and RT–PCR, but not in skel-etal muscle and heart—the two most affected organs of GSDII(Fig. 6A and B). hGAA protein, however, was detected in allthree organs by western analysis, implying that the enzymewas secreted by liver and taken up by skeletal muscle and heart(Fig. 6C). Indeed, high levels of 110 kDa GAA precursor were
Table 1. GAA activity in double transgenic mice and controls (nmol/h/mg protein)
aSingle transgenics (Mck-T, hGAA and Alb-T) on GAA –/– or +/– were used as controls.
Genotype Muscle Heart Liver Lung Brain Spleen Plasma
–/– (n = 9)a 1.9 ± 0.6 0.9 ± 0.4 2.8 ± 0.4 1.2 ± 0.4 1.45 ± 0.8 2.0 ± 0.6 0
+/– (n = 11)a 5.6 ± 0.5 9.0 ± 0.7 26 ± 3.0 5.8 ± 1.5 39.4 ± 6.6 16.0 ± 2.0 –
Mck-T-hGAA/–/– F.06 (n = 8) 28.9 ± 4.9 1.4 ± 0.4 3.2 ± 0.7 1.6 ± 0.9 0.5 ± 0.4 2.8 ± 0.7 0
Mck-T-hGAA/–/–F.53 (n = 5) 466 ± 90 223 ± 64 13 ± 1.9 4.0 ± 1.96 0.26 ± 0.2 12.8 ± 2.5 4.0 ± 0.7
Alb-T-hGAA/–/– (n = 8) 27.8 ± 5.5 35 ± 9.0 659 ± 47 – 1.5 ± 0.4 – 424 ± 62
Table 2. Glycogen content in double transgenic and control mice (% wet weight)
aDouble transgenics (6 weeks to 8 months); controls (3–8 months). bPreviously reported data (32).
Genotype/founder Musclea Heart Diaphragm Tongue
5–8 weeks 4–8 months
Control (–/–) 3.2 ± 0.5 (n = 7) 1.5 ± 0.4 (n = 2) 7.2 ± 1.4 (n = 5) 3.7 ± 0.6 (n = 3) 5.16 ± 0.15 (n = 2)
Wild-type (n = 7)b 0.17 ± 0.08 ND ND 0.03 ± 0.002 –
Mck-T-hGAA/–/– F.06 0.17 ± 0.0 (n = 5) 1.6 ± 0.6 (n = 3) 8.9 ± 2.2 (n = 5) – 5.2 ± 0.12 (n = 2)
Mck-T-hGAA/–/– F.53 0.33 ± 0.08 (n = 8) – 0.13 ± 0.07 (n = 5) 0 (n = 4) –
Alb-T-hGAA/–/– 0.3 ± 0.1 (n = 6) 0.18 ± 0.09 (n = 4) 0.15 ± 0.04 (n = 2) – –
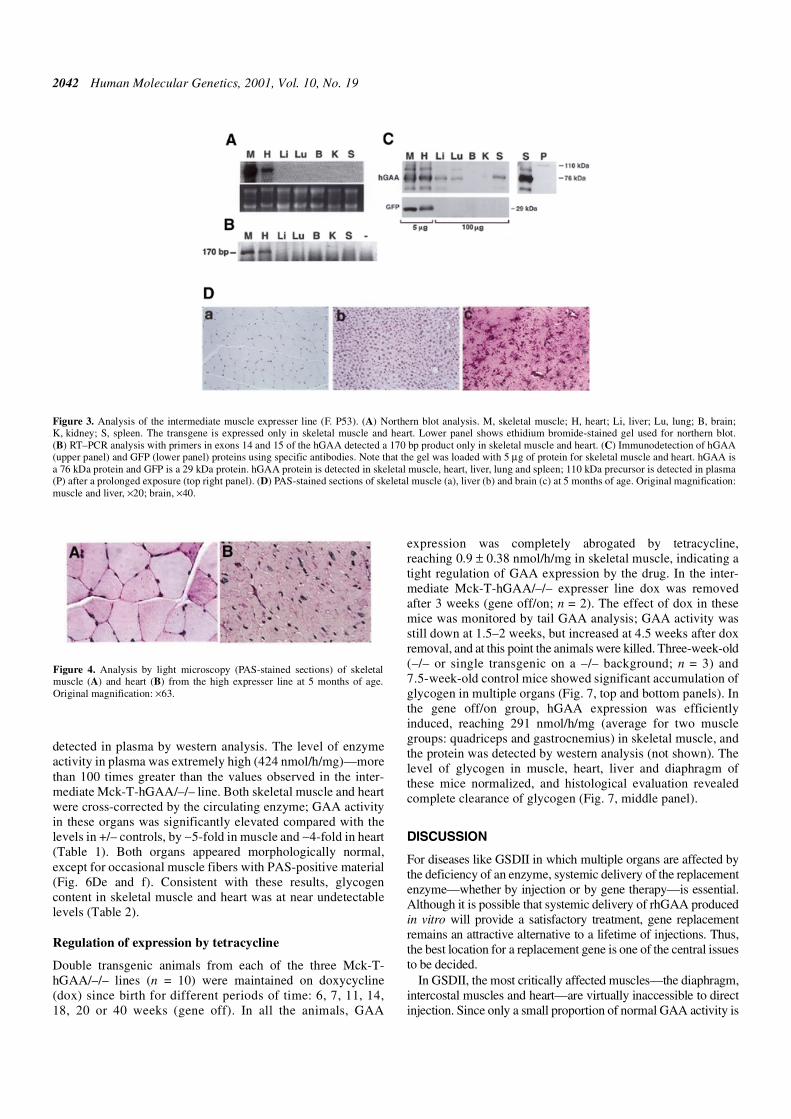
2042 Human Molecular Genetics, 2001, Vol. 10, No. 19
detected in plasma by western analysis. The level of enzymeactivity in plasma was extremely high (424 nmol/h/mg)—morethan 100 times greater than the values observed in the inter-mediate Mck-T-hGAA/–/– line. Both skeletal muscle and heartwere cross-corrected by the circulating enzyme; GAA activityin these organs was significantly elevated compared with thelevels in +/– controls, by ∼5-fold in muscle and ∼4-fold in heart(Table 1). Both organs appeared morphologically normal,except for occasional muscle fibers with PAS-positive material(Fig. 6De and f). Consistent with these results, glycogencontent in skeletal muscle and heart was at near undetectablelevels (Table 2).
Regulation of expression by tetracycline
Double transgenic animals from each of the three Mck-T-hGAA/–/– lines (n = 10) were maintained on doxycycline(dox) since birth for different periods of time: 6, 7, 11, 14,18, 20 or 40 weeks (gene off). In all the animals, GAA
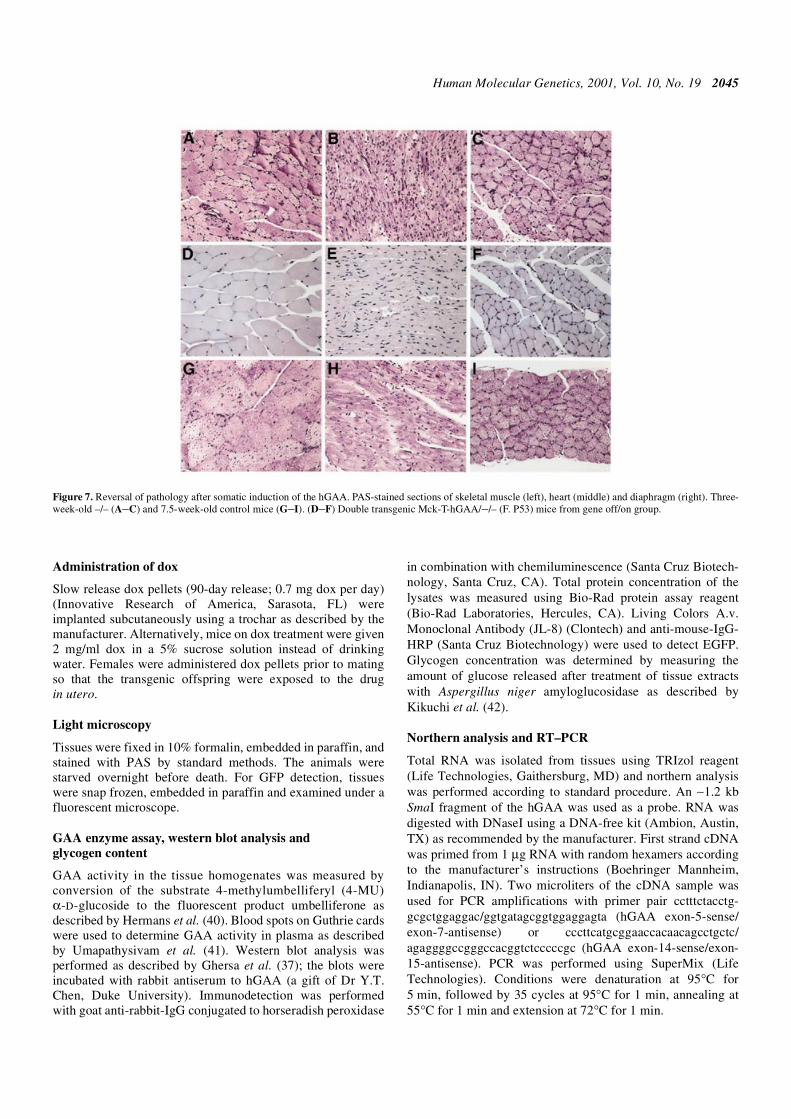
expression was completely abrogated by tetracycline,reaching 0.9 ± 0.38 nmol/h/mg in skeletal muscle, indicating atight regulation of GAA expression by the drug. In the inter-mediate Mck-T-hGAA/–/– expresser line dox was removedafter 3 weeks (gene off/on; n = 2). The effect of dox in thesemice was monitored by tail GAA analysis; GAA activity wasstill down at 1.5–2 weeks, but increased at 4.5 weeks after doxremoval, and at this point the animals were killed. Three-week-old(–/– or single transgenic on a –/– background; n = 3) and7.5-week-old control mice showed significant accumulation ofglycogen in multiple organs (Fig. 7, top and bottom panels). Inthe gene off/on group, hGAA expression was efficientlyinduced, reaching 291 nmol/h/mg (average for two musclegroups: quadriceps and gastrocnemius) in skeletal muscle, andthe protein was detected by western analysis (not shown). Thelevel of glycogen in muscle, heart, liver and diaphragm ofthese mice normalized, and histological evaluation revealedcomplete clearance of glycogen (Fig. 7, middle panel).
DISCUSSION
For diseases like GSDII in which multiple organs are affected bythe deficiency of an enzyme, systemic delivery of the replacementenzyme—whether by injection or by gene therapy—is essential.Although it is possible that systemic delivery of rhGAA producedin vitro will provide a satisfactory treatment, gene replacementremains an attractive alternative to a lifetime of injections. Thus,the best location for a replacement gene is one of the central issuesto be decided.
In GSDII, the most critically affected muscles—the diaphragm,intercostal muscles and heart—are virtually inaccessible to directinjection. Since only a small proportion of normal GAA activity is
Figure 3. Analysis of the intermediate muscle expresser line (F. P53). (A) Northern blot analysis. M, skeletal muscle; H, heart; Li, liver; Lu, lung; B, brain;K, kidney; S, spleen. The transgene is expressed only in skeletal muscle and heart. Lower panel shows ethidium bromide-stained gel used for northern blot.(B) RT–PCR analysis with primers in exons 14 and 15 of the hGAA detected a 170 bp product only in skeletal muscle and heart. (C) Immunodetection of hGAA(upper panel) and GFP (lower panel) proteins using specific antibodies. Note that the gel was loaded with 5 µg of protein for skeletal muscle and heart. hGAA isa 76 kDa protein and GFP is a 29 kDa protein. hGAA protein is detected in skeletal muscle, heart, liver, lung and spleen; 110 kDa precursor is detected in plasma(P) after a prolonged exposure (top right panel). (D) PAS-stained sections of skeletal muscle (a), liver (b) and brain (c) at 5 months of age. Original magnification:muscle and liver, ×20; brain, ×40.
Figure 4. Analysis by light microscopy (PAS-stained sections) of skeletalmuscle (A) and heart (B) from the high expresser line at 5 months of age.Original magnification: ×63.

Human Molecular Genetics, 2001, Vol. 10, No. 19 2043
required to prevent clinical disease, low levels of circulatingenzyme should be sufficient to provide phenotypic correction indistant organs (31). In vitro and ex vivo trials of GAA deliveryfrom transduced cultured cells to cultured mutant cells have beenencouraging. A significant increase in the GAA enzyme level indeficient muscle cells and the phenotypic correction of culturedmuscle cells from a GSDII patient was obtained with a recom-binant adenovirus containing the human gene (22). A high level ofGAA expression was also observed in GAA-deficient myoblaststransduced with a GAA-encoding retrovirus, and those gene-corrected myoblasts provided enzyme to deficient muscle cells byboth secretion and cell fusion (21). The implication of thesestudies is that ex vivo transduced myoblasts from patients could beimplanted into the muscles of affected individuals and would thensecrete the enzyme for uptake by other cells.
The results of the in vivo muscle-mediated GAA delivery,however, have been disappointing. Intracardiac or intramuscularadministration of Ad-GAA into newborn rats resulted in highlevels of GAA expression locally, but the heart, liver, contralateralmuscle and sera showed no increase in GAA activity (24).Similarly, a recombinant Ad-GAA injected into muscle of GAA-deficient quails resulted only in local correction (25). The failureto provide a systemic correction was attributed to a relatively shortduration of the locally expressed transgene.
We have now demonstrated that even when the transgene isconstitutively expressed in the entire muscle mass at levelsexceeding five times those in +/– controls, no secretion by musclecells and no uptake by distant tissues occurred. Since the expres-sion of the transgene in this line is limited to skeletal muscle, otherorgans including heart and brain accumulate glycogen at a ratesimilar to that in the –/– mice. Occasional muscle cells in this linestill contain lysosomal glycogen, indicating that this level ofenzyme does not reliably provide the correction of neighboringcells. A low level of GAA secretion from skeletal muscle wasobserved in mice constitutively expressing 80-fold increasedactivity compared with +/– controls (intermediate expresser line).The high GAA levels in the heart of these mice were a result of thetransgene expression rather than uptake of secreted enzyme, butthe increased GAA activity in liver, lung and spleen reflecteduptake of enzyme secreted by the muscle source.
An extremely high level of GAA expression in skeletal muscle(high expresser line) was not well tolerated, and virtually everycell contained small lysosomes with PAS-positive material.Excess of GAA rather than the transactivator is most likely to beresponsible for the effect since the same Mck-T line was used togenerate three double transgenic lines expressing hGAA inskeletal muscle.
In other lysosomal diseases, in vivo muscle-mediated genetherapy has not been very promising either. AAV-mediated skel-etal muscle gene transfer (human β-glucuronidase) in neonatalmice with mucopolysaccharidosis type VII showed that the secre-tion of the enzyme from an intramuscular source was inefficient(32). Similarly, no enzymatic activity was found in serum orperipheral organs in hexosaminidase A-deficient knockout mice(Tay-Sachs disease) after muscle-mediated gene therapy (33).
The liver, however, proved an efficient organ for productionand secretion of GAA protein into the bloodstream and uptake inperipheral tissues. Comparable levels of transgene expression inboth muscle and liver result in a dramatically different amount ofsecreted protein. In the liver line, only the 110 kDa precursor, butnot the processed forms, was detected in plasma, indicating thatthe liver cells were not damaged. Furthermore, the enzyme wastaken up by skeletal muscle and heart, and targeted to lysosomeswhere it prevented glycogen accumulation. Thus, the liver, whosemass is <5% of body weight, could fully correct all distantaffected tissues except brain, when producing 25-fold moreenzyme than the liver of +/– mice, while the muscle, whose massis ∼40% of body weight, was able to provide correction in distanttissues only when producing 80-fold more enzyme than +/– mice.It is not clear why there is such a difference in the level of secretedGAA from muscle and liver. GAA is normally an intracellularenzyme, and the mechanism by which a small portion of theenzyme is secreted is not well understood. It is possible that thesecretory pathway is not as efficient in skeletal muscle cells as inhepatocytes. Efficient hexosaminidase, and α-galactosidase Asecretion and restoration of enzyme activity was similarlyobserved after liver transduction (33,34), and hepatic targeting ofan Ad vector encoding GAA resulted in systemic correction inknockout mice (26–28).
These findings imply that cultured liver cells could potentiallyprovide a source of the recombinant protein, that both skeletalmuscle and heart have a significant capacity to take up the proteinas long as it is properly processed, and that the expression of GAAin liver at levels lower than we tested may be sufficient to providecross-correction, since the GAA activity in muscle and heart wassignificantly higher than needed to remain phenotypically normal.
Finally, the disease in the knockout mice was reversible. Inmice allowed to develop morphological changes over the first3 weeks of life, accumulated glycogen disappeared completely1 month after the transgene was turned on. The success of thisexperiment should allow the size of the window of opportunity fortherapeutic effect in clinically affected individuals to bedetermined.
Figure 5. Expression of GFP in livers of the Alb-T-hGAA/–/– mice. Liver cells were examined under a fluorescent microscope using excitation light. (A) Singletransgenic control (hGAA/–/–). Two double transgenic lines derived from two independent founders (F. 21 and F. 26) showed similar levels of GFP expression(B and C).

2044 Human Molecular Genetics, 2001, Vol. 10, No. 19
MATERIALS AND METHODS
Generation of transgenic mouse strains
Transgenic strain containing hGAA cDNA. hGAA fragmentwas released from pCDNA3 (a gift from Dr Frank Martiniuk,
New York University Medical Center) by digestion withHindIII/XhoI, and cloned into HindIII/SalI sites of pTRE2(Clontech, Palo Alto, CA) to give rise to pTRE2-hGAA. In thisplasmid, the hGAA is linked to the tetracycline responsiveelement (TRE) to which the tetracycline-controlled transcrip-tional activator (tTA) binds in the absence of tetracycline.Next, the internal ribosome entry site (IRES)-enhanced GFP(EGFP) fragment containing the GFP was released frompIRES2-EGFP (Clontech) by digestion with SmaI/XbaI, andcloned into pTRE2-hGAA to give rise to pTRE2-hGAA-IRES-EGFP. The TRE-hGAA-IRES-EGFP-β-globin poly A frag-ment was released by XhoI/SapI digest, gel purified and usedfor microinjection into FVB embryos. These mice are referredto as hGAA.Transgenic strain containing tTA under the control of liverspecific albumin promoter. The tTA fragment linked to SV40poly A was cloned into the PstI sites of pcDNA2.1 (Invitrogen,San Diego, CA) to give rise to pcDNA2.1-tTA. Next, themouse albumin promoter/enhancer was released frompGEMAlbSVPA (a kind gift from Dr T. Jake Liang, NationalInstitute of Diabetes and Digestive and Kidney Diseases,National Institutes of Health) (35) with ApaI/NotI and clonedinto pcDNA2.1-tTA to generate pcDNA2.1-Alb-tTA. Then a500 bp fragment of the chicken β-globin core insulator (36)was PCR amplified from pNI-CD (a gift from Dr Gary Felsen-feld, National Institute of Diabetes and Digestive and KidneyDiseases, National Institutes of Health) and cloned into bothApaI and SapI sites of the pcDNA2.1-tTA. The insulator-Alb-tTA-SV40 poly A-insulator fragment was released by PacIdigestion, gel purified by electroelution, and used for microin-jection into FVB embryos. These mice are referred to as Alb-T.Transgenic strain containing tTA under the control of musclespecific creatine kinase promoter. Generation of this strain,referred to as Mck-T, was described previously (37).
Genotyping of transgenic lines and breeding strategy
Southern analysis of the founders and F1 progeny from bothhGAA and Alb-T transgenic lines revealed integration of theinjection fragment. Genomic DNA was isolated from tail clips,digested with EcoRI, and genotyping was performed usingstandard procedures. For hGAA, a 733 bp fragment frompIRES2-EGFP vector (BstxI/XbaI digest) was used as a probe,resulting in an ∼2 kb fragment. For Mck-T and Alb-T, an ∼1.5kb fragment from pTet-Off (Clontech) (EcoRI/HindIII digest)was used as a probe, resulting in ∼13 and ∼2.6 kb fragments,respectively. The probes were labeled by the random hexamermethod (Lofstrand, Gaithersburg, MD). Each of the threetransgenic strains was crossed to the knockout mice (–/–) inwhich the GAA gene was disrupted in the middle of exon 6(38,39). Tail DNA was screened by Southern analysis to iden-tify –/– mice, as described previously (38). The resulting linesare referred to as hGAA/–/–, Mck-T/–/– and Alb-T/–/–.
hGAA/–/– mice were mated to either Mck-T/–/– or Alb-T/–/–mice to generate double transgenics expressing hGAA in skeletalmuscle or in liver of the –/– mice (FVB × C57BL6/129 SVj).The resulting lines are referred to as Mck-T-hGAA/–/– andAlb-T-hGAA/–/–.
Figure 6. Analysis of liver expresser line. (A) Northern blot analysis. Li, liver;M, muscle; H, heart. The hGAA transgene is expressed only in liver. Lowerpanel shows ethidium bromide-stained gel used for northern blot. (B) RT–PCRanalysis of cDNA from respective tissues. The primers in exons 5 and 7 of thehGAA detected a 270 bp product only in liver (top); Southern blot analysis witha probe in exon 6 detected a signal only in liver (bottom). (C) Immunodetectionof hGAA (upper panel) and GFP (lower panel) proteins using specific antibod-ies. Note that the gel was loaded with 10 µg of protein for liver. hGAA is a76 kDa protein and GFP is a 29 kDa protein. hGAA protein is detected in liver,muscle and heart; 110 kDa precursor form is easily detectable in plasma (P).Control is a single transgenic (Alb-T/–/–) littermate. (D) PAS-stained sectionsof liver, muscle and heart from control (a–c) and double transgenic Alb-T-hGAA/–/– mice at 3 months of age (d–f). Arrows point to occasional musclecells containing PAS-positive material. Original magnification: ×20.
Human Molecular Genetics, 2001, Vol. 10, No. 19 2045
Administration of dox
Slow release dox pellets (90-day release; 0.7 mg dox per day)(Innovative Research of America, Sarasota, FL) wereimplanted subcutaneously using a trochar as described by themanufacturer. Alternatively, mice on dox treatment were given2 mg/ml dox in a 5% sucrose solution instead of drinkingwater. Females were administered dox pellets prior to matingso that the transgenic offspring were exposed to the drugin utero.
Light microscopy
Tissues were fixed in 10% formalin, embedded in paraffin, andstained with PAS by standard methods. The animals werestarved overnight before death. For GFP detection, tissueswere snap frozen, embedded in paraffin and examined under afluorescent microscope.
GAA enzyme assay, western blot analysis and glycogen content
GAA activity in the tissue homogenates was measured byconversion of the substrate 4-methylumbelliferyl (4-MU)α-D-glucoside to the fluorescent product umbelliferone asdescribed by Hermans et al. (40). Blood spots on Guthrie cardswere used to determine GAA activity in plasma as describedby Umapathysivam et al. (41). Western blot analysis wasperformed as described by Ghersa et al. (37); the blots wereincubated with rabbit antiserum to hGAA (a gift of Dr Y.T.Chen, Duke University). Immunodetection was performedwith goat anti-rabbit-IgG conjugated to horseradish peroxidase
in combination with chemiluminescence (Santa Cruz Biotech-nology, Santa Cruz, CA). Total protein concentration of thelysates was measured using Bio-Rad protein assay reagent(Bio-Rad Laboratories, Hercules, CA). Living Colors A.v.Monoclonal Antibody (JL-8) (Clontech) and anti-mouse-IgG-HRP (Santa Cruz Biotechnology) were used to detect EGFP.Glycogen concentration was determined by measuring theamount of glucose released after treatment of tissue extractswith Aspergillus niger amyloglucosidase as described byKikuchi et al. (42).
Northern analysis and RT–PCR
Total RNA was isolated from tissues using TRIzol reagent(Life Technologies, Gaithersburg, MD) and northern analysiswas performed according to standard procedure. An ∼1.2 kbSmaI fragment of the hGAA was used as a probe. RNA wasdigested with DNaseI using a DNA-free kit (Ambion, Austin,TX) as recommended by the manufacturer. First strand cDNAwas primed from 1 µg RNA with random hexamers accordingto the manufacturer’s instructions (Boehringer Mannheim,Indianapolis, IN). Two microliters of the cDNA sample wasused for PCR amplifications with primer pair cctttctacctg-gcgctggaggac/ggtgatagcggtggaggagta (hGAA exon-5-sense/exon-7-antisense) or cccttcatgcggaaccacaacagcctgctc/agaggggccgggccacggtctcccccgc (hGAA exon-14-sense/exon-15-antisense). PCR was performed using SuperMix (LifeTechnologies). Conditions were denaturation at 95°C for5 min, followed by 35 cycles at 95°C for 1 min, annealing at55°C for 1 min and extension at 72°C for 1 min.
Figure 7. Reversal of pathology after somatic induction of the hGAA. PAS-stained sections of skeletal muscle (left), heart (middle) and diaphragm (right). Three-week-old –/– (A–C) and 7.5-week-old control mice (G–I). (D–F) Double transgenic Mck-T-hGAA/–/– (F. P53) mice from gene off/on group.

2046 Human Molecular Genetics, 2001, Vol. 10, No. 19
Statistical analysis
Data in text and figures are given as means ± standard error.The Student’s t-test was used for comparisons between thegroups. Differences were considered significant at P < 0.005.
REFERENCES
1. Hirschhorn, R. (1995) Glycogen storage disease type II acid α-glucosidase (acid maltase) deficiency. In Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, D. (eds), The Metabolic and Molecular Basis of Inherited Diseases. McGraw-Hill, New York, NY, pp. 2443–2464.
2. Reuser, A.J., Kroos, M.A., Ponne, N.J., Wolterman, R.A., Loonen, M.C., Busch, H.F., Visser, W.J. and Bolhuis, P.A. (1984) Uptake and stability of human and bovine acid α-glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp. Cell Res., 155, 178–189.
3. Hoefsloot, L.H., Willemsen, R., Kroos, M., Hoogeveen-Westerveld, M., Hermans, M.M., Van der Ploeg, A.T., Oostra, B.A. and Reuser, A.J.J. (1990) Expression and routeing of human lysosomal α-glucosidase in transiently transfected mammalian cells. Biochem. J., 272, 485–492.
4. Wisselaar, H.A., Kroos, M.A., Hermans, M.M., Van Beeumen, J. and Reuser, A.J.J. (1993) Structural and functional changes of lysosomal acid α-glucosidase during intracellular transport and maturation. J. Biol. Chem., 268, 2223–2231.
5. Tsuji, A., Omura, K. and Suzuki, Y. (1988) Intracellular transport of acid α-glucosidase in human fibroblasts: evidence for involvement of phosphomannosyl receptor-independent system. J. Biochem. (Tokyo), 104, 276–278.
6. Van der Ploeg, A.T., Loonen, M.C.B., Bolhuis, P.A., Busch, H.M.F., Reuser, A.J. and Galjaard, H. (1988) Receptor-mediated uptake of acid α-glucosidase corrects lysosomal glycogen storage in cultured skeletal muscle. Pediatr. Res., 24, 90–94.
7. Van der Ploeg, A.T., Kroos, M.A., Willemsen, R., Brons, H.N.C. and Reuser, A.J. (1991) Intravenous administration of phosphorylated acid α-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice. J. Clin. Invest., 87, 513–518.
8. Neufeld, E.F. (1991) Lysosomal storage diseases. Annu. Rev. Biochem., 60, 257–280.
9. Fuller, M., Van der Ploeg, A.T., Reuser, A.J., Anson, D.S. and Hopwood, J.J. (1995) Isolation and characterization of a recombinant precursor form of lysosomal acid α-glucosidase. Eur. J. Biochem., 234, 903–909.
10. Van Hove, J.L.K., Yang, H.W., Wu, J.Y., Brady, R.O. and Chen, Y.T. (1996) High level production of recombinant human lysosomal acid α-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc. Natl Acad. Sci. USA, 93, 65–70.
11. Bijvoet, A.G.A., Van Hirtum, H., Kroos, M.A., Van de Kamp, E.H., Schoneveld, O., Visser, P., Brakenhoff, J.P., Weggeman, M., van Corven, E.J., Van der Ploeg, A.T. and Reuser, A.J. (1999) Humanacid α-glucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Hum. Mol. Genet., 8, 2145–2153.
12. Van den Hout, H., Reuser, A.J.J., Vulto, A.G., Loonen, M.C.B., Cromme-Dijkhuis, A. and Van der Ploeg, A.T. (2000) Recombinant human α-glucosidase from rabbit milk in Pompe patients. Lancet, 356, 397–398.
13. Chen, Y.T. and Amalfitano, A. (2000) Towards a molecular therapy for glycogen storage disease type II (Pompe disease). Mol. Med. Today, 6, 245–251.
14. Amalfitano, A., Bengur, A.R., Morse, R.P., Majure, J.M., Case, L.E., Veerling, D.L., Mackey, J., Kishnani, P., Smith, W., McVie-Wylie, A. et al. (2001) Recombinant human acid α-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet. Med., 3, 132–138.
15. Van den Hout, J.M., Reuser, A.J., de Klerk, J.B., Arts, W.F., Smeitink, J.A. and Van der Ploeg, AT. (2001) Enzyme therapy for pompe disease with recombinant human alpha-glucosidase from rabbit milk. J. Inherit. Metab. Dis., 24, 266–274.
16. Kessler, P., Pdsakoff, G., Chen, X., McQuiston, S., Colosi, P., Matelis, L., Kurtzman, G. and Byrne, B. (1996) Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl Acad. Sci. USA, 93, 14082–14087.
17. Herzog, R.W., Hagstrom, J.N., Kung, S.H., Tai, S.J., Wilson, J.M., Fisher, K.J. and High, K.A. (1997) Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc. Natl Acad. Sci. USA, 94, 5804–5809.
18. Hasse, G., Kennel, P., Pettmann, B., Vigne, E., Akli, S., Revah, F., Schmalbruch, H. and Kahn, A. (1997) Gene therapy of murine neuron disease using adenoviral vectors for neurotrophic factors. Nat. Med., 3, 429–436.
19. Vincent-Lacaze, N., Snyder, R., Gluzman, R., Bohl, D., Lagarde, C. and Danos, O. (1999) Structure of adeno-associated virus vector DNA following transduction of the skeletal muscle. J. Virol., 73, 1949–1955.
20. Gros, L., Riu, E., Montoliu, L., Ontiveros, M., Lebrigand, L. and Bosch, F. (1999) Insulin production by engineered muscle cells. Hum. Gene Ther., 10, 1207–1217.
21. Zaretsky, J., Candotti, F., Boerkoel, C., Adams, E., Yewdell, J., Blaese, M. and Plotz, P. (1997) Retroviral transfer of acid α-glucosidase cDNA to enzyme-deficient myoblasts results in phenotypic spread of the genotypic correction by both secretion and fusion. Hum. Gene Ther., 8, 1555–1563.
22. Nicolino, M.P., Puech, J.P., Kremer, E.J., Reuser, A.J., Mbebi, C., Verdiere-Sahuque, M., Kahn, A. and Poenaru, L. (1998) Adenovirus-mediated transfer of the acid α-glucosidase gene into fibroblasts, myoblasts and myotubes from patients with glycogen storage disease type II leads to high level expression of enzyme and corrects glycogen accumulation. Hum. Mol. Genet., 7, 1695–1702.
23. Poenaru, L. (2000) Approach to gene therapy of glycogenosis type II (Pompe disease). Mol. Genet. Metab., 70, 163–169.
24. Pauly, D.F., Johns, D.C., Matelis, L.A., Lawrence, J.H., Byrne, B.J. and Kessler, P.D. (1998) Complete correction of acid α-glucosidase deficiency in Pompe disease fibroblasts in vitro, and lysosomally targeted expression in neonatal rat cardiac and skeletal muscle. Gene Ther., 5, 473–480.
25. Tsujino, S., Kinoshita, N., Tashiro, T., Ikeda, K., Ichihara, N., Kikuchi, H., Hagiwara, Y., Mizutani, M., Kikuchi, T. and Sakuragawa, N. (1998) Adenovirus-mediated transfer of human acid maltase gene reduces glycogen accumulation in skeletal muscle of Japanese quail with acid maltase deficiency. Hum. Gene Ther., 9, 1609–1616.
26. Amalfitano, A., McVie-Wylie, A.J., Hu, H., Dawson, T., Raben, N., Plotz, P. and Chen, Y.T. (1999) Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid α-glucosidase. Proc. Natl Acad. Sci. USA, 96, 8861–8866.
27. Pauly, D., Fraites, T., Toma, C., Bayes, H., Huie, M., Hirschhorn, R., Plotz, P., Raben, N., Kessler, P. and Byrne, B. (2001) Intracellular transfer of the virally derived precursor form of acid α-glucosidase corrects the enzyme deficiency in inherited cardioskeletal myopathy Pompe disease. Hum. Gene Ther., 12, 527–538.
28. Ding, E.Y., Hodges, B.L., Hu, H., McVie-Wylie, A.J., Serra, D., Migone, F.K., Pressley, D., Chen, Y.T. and Amalfitano, A. (2001) Long-term efficacy after [E1–, polymerase–] adenovirus-mediated transfer of human acid α-glucosidase gene into glycogen storage disease type II knockout mice. Hum. Gene Ther., 12, 955–965.
29. Furth, P.A., Onge, L.S., Boger, H., Gruss, P., Gossen, M., Kistner, A., Bujard, H. and Henninghausen, L. (1994) Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc. Natl Acad. Sci. USA, 91, 9302–9306.
30. Kistner, A., Gossen, M., Zimmermann, F., Jerecic, J., Ullmer, C., Lubbert, H. and Bujard., H. (1996) Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl Acad. Sci. USA, 93, 10933–10938.
31. Reuser, A.J.J., Kroos, M.A., Hermans, M.M., Bijvoet, A.G., Verbeet, M.P., van Diggelen, O.P., Kleijer, W.J. and Van der Ploeg, A.T. (1995) Glycogenosis type II (acid maltase deficiency). Muscle Nerve, 3 (suppl.), S61–S69.
32. Daly, T., Okuyama, T., Vogler, C., Haskins, M., Muzyczka, N. and Sands, M. (1999) Neonatal intramuscular injection with recombinant adeno-associated virus results in prolonged β-glucuronidase expression in situ and correction of liver pathology in mucopolysaccharidosis type VII mice. Hum. Gene Ther., 10, 85–94.
33. Guidotti, J.E., Mignon, A., Haase, G., Caillaud, C., McDonell, N., Kahn, A. and Poenaru, L. (1999) Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum. Mol. Genet., 8, 831–838.
34. Jung, S.C., Han, I.P., Limaye, A., Xu, R., Gelderman, M.P., Zerfas, P., Tirumalai, K., Murray, G.J., During, M.J., Brady, R.O. and Qasba, P.

Human Molecular Genetics, 2001, Vol. 10, No. 19 2047
(2001) Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc. Natl Acad. Sci. USA, 98, 2676–2681.
35. Manickan, E., Satoi, J., Wang, T.C. and Liang, T.J. (2001) Conditional liver specific expression of SV40 antigen leads to regulatable development of hepatic neoplasm in transgenic mice. J. Biol. Chem., 276, 13989–13994.
36. Recillas-Targa, F., Bell, A.C. and Felsenfeld, G. (1999) Positional enhancer-blocking activity of the chicken β-globin insulator in transiently transfected cells. Proc. Natl Acad. Sci. USA, 96, 14354–14359.
37. Ghersa, P., Gobert, R.P., Sattonnet-Roche, P., Richards, C.A., Merlo Pich, E. and Hooft van Huijsduijnen, R. (1998) Highly controlled gene expression using combinations of a tissue-specific promoter, recombinant adenovirus and a tetracycline-regulatable transcription factor. Gene Ther., 5, 1213–1220.
38. Raben, N., Nagaraju, K., Lee, E., Kessler, P., Byrne, B., Lee, L., LaMarca, M., King, C., Ward, J., Sauer, B. and Plotz, P. (1998) Targeted disruption of the acid α-glucosidase gene in mice causes an illness with
critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem., 273, 19086–19092.
39. Raben, N., Nagaraju, K., Lee, E. and Plotz, P. (2000) Modulation of disease severity in mice with targeted disruption of the acid α-glucosidase gene. Neuromusc. Disord., 10, 283–291.
40. Hermans, M.M., Kroos, M.A., van Beeumen, J., Oostra, B.A. and Reuser, A.J.J. (1991) Human lysosomal α-glucosidase. Characterization of the catalytic site. J. Biol. Chem., 266, 13507–13512.
41. Umapathysivam, K., Whittle, A., Ranieri, E., Bindloss, C., Ravenscroft, E., Van Diggelen, O.P., Hopwood, J. and Meikle, P. (2000) Determination of acid α-glucosidase protein: evaluation as a screening marker for Pompe disease and other lysosomal storage disorders. Clin. Chem., 46, 1318–1325.
42. Kikuchi, T., Yang, H.W., Pennybacker, M., Ichihara, N., Mizutani, M., Van Hove, J.L. and Chen, Y.T. (1998) Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase deficient quail. J. Clin. Invest., 101, 827–833.

2048 Human Molecular Genetics, 2001, Vol. 10, No. 19
Copyright © 2022 FDOKUMEN












![Mapping Functional Brain Activation Using [14C]-Iodoantipyrine in Male Serotonin Transporter Knockout Mice](https://static.fdokumen.com/doc/165x107/6323b5f703238a9ff60a8f0e/mapping-functional-brain-activation-using-14c-iodoantipyrine-in-male-serotonin.jpg)







