
Computational Screening of Energy Materials - DTU Orbit
159
General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from orbit.dtu.dk on: Feb 10, 2022 Computational Screening of Energy Materials Pandey, Mohnish Publication date: 2015 Link back to DTU Orbit Citation (APA): Pandey, M. (2015). Computational Screening of Energy Materials. Technical University of Denmark.
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Computational Screening of Energy Materials - DTU Orbit

General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
Users may download and print one copy of any publication from the public portal for the purpose of private study or research.
You may not further distribute the material or use it for any profit-making activity or commercial gain
You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim.
Downloaded from orbit.dtu.dk on: Feb 10, 2022
Computational Screening of Energy Materials
Pandey, Mohnish
Publication date:2015
Link back to DTU Orbit
Citation (APA):Pandey, M. (2015). Computational Screening of Energy Materials. Technical University of Denmark.

Computational Screeningof Energy Materials
PhD ThesisMohnish Pandey


Computational Screening of Energy MaterialsPhD ThesisJuly 2015
Mohnish [email protected]
Supervisors:Karsten Wedel JacobsenKristian Sommer Thygesen
Center for Atomic-scale Materials DesignDepartment of PhysicsTechnical University of Denmark


Preface
This thesis is submitted for the candidacy of PhD degree in Physics from theTechnical University of Denmark. The work contained in the thesis was carriedout at the Center for Atomic-scale Materials Design (CAMd), Department ofPhysics in the period from August 2012 to July 2015 under the supervision ofProf. Karsten W. Jacobsen and Prof. Kristian S. Thygesen.I cannot thank enough my supervisors Karsten and Kristian for their continu-ous support in my projects. Every bit of discussion I had with them improvedmy understanding of the subject matter. Their immense zeal in the projectsand their unparalleled guidance always kept me motivated and helped me toget over the bottlenecks I faced during my research.Also, I would like to thank Aleksandra, Ivano, Filip, Niels and Thomas forall the suggestions and discussions regarding different projects. Big thanks toChris, Kirsten, Kristian, Martin, Niels and Simon Lamowski for generouslyagreeing to proofread the thesis and for the good times at CAMd. A deepgratitude to Jens Jørgen and Marcin for helping in the code developmentsand troubleshootings and Ole for taking such a good care of Niflheim for itsseamless functioning. A huge thanks to Marianne for making my move toDenmark easy by taking care of all the rigmarole and other administrativematters.Among friends my special thanks to Kristian for his continuous support andinvitations for Indian festival dinners to make me feel home. I would also liketo thank my other colleagues at CAMd - Korina, Manuel, Morten, Per, Simoneand Ulrik for all the fun times.Last but not least my heartfelt thanks to my family members for their continu-ous support and my girlfriend Shrutija for not letting me feel the long-distancerelationship really as long-distance and supporting me in all the ways she could.
Mohnish PandeyKongens Lyngby, July 2015


Abstract
The current energy consumption of the worlds population relies heavily on fos-sil fuels. Unfortunately, the consumption of fossil fuels not only results in theemission of greenhouse gases which have deleterious effect on the envrionmentbut also the fossil fuel reserve is limited. Therefore, it is the need of the hourto search for environmentally benign renewable energy resources. The biggestsource of the renewable energy is our sun and the immense energy it providescan be used to power the whole planet. However, an efficient way to harvestthe solar energy to meet all the energy demand has not been realized yet.
A promising way to utilize the solar energy is the photon assisted watersplitting. The process involves the absorption of sunlight with a semiconduct-ing material (or a photoabsorber) and the generated electron-hole pair can beused to produce hydrogen by splitting the water. However, a single materialcannot accomplish the whole process of the hydrogen evolution. In order doso, a material should be able to absorb the sunlight and generate the electron-hole pairs and evolve hydrogen at the cathode and oxygen at anode using thegenerated electron and hole respectively.
This thesis using first-principle calculations explores materials for the lightabsorption with the bandgap, band edge positions and the stability in aqueousconditions as descriptors. This strategy results in a handful of materials whichcan act as good photoabsorbers for the water splitting reaction. Additionally,strategies to tune the bandgap for different applications is also explored. Tocarry out the cathode reaction, two-dimensional metal dichalcogenides and ox-ides are explored with a suggestion of few potential candidates for the hydrogenevolution reaction.
The thermodynamics of all the above process requires an accurate descrip-tion of the energies with the first-principle calculations. Therefore, along thisline the accuracy and predictability of the Meta-Generalized Gradient Approx-imation functional with Bayesian error estimation is also assessed.


Resumé
Jordens befolkning er i dag fuldstændig afhængig af fossile brændstoffer forat producere den nødvendige energi. Denne afhængighed er meget uforde-lagtig, idet lageret af tilgængelige fossile brændstoffer er stærkt begrænset sam-tidigt med, at afbrændingen af fossile brændstoffer producerer klimaskadeligedrivhusgasser. Det er derfor nødvendigt, at finde miljøsikre vedvarende en-ergikilder. Den største tilgængelige vedvarende energikilde er solen, hvis en-ergiudladning er stor nok til at dække hele vores planets energiforbrug. Dogmangler vi stadigvæk en måde hvorpå solenergien kan høstes effektivt.En lovende metode til at opfange solenergi er foton-assisteret vandspaltning.Denne metode indbefatter et halvleder-materiale, der absorberer en fotonhvilket genererer et elektron-hul par, som kan bruges til at producere brintvia vandspaltning. Det er dog umuligt for et enkelt materiale, at stå for heleden foton-assisterede vandspaltnings proces. For at muliggøre processen erdet nødvendigt både at have et foton-absorberende materiale, der absorberersollyset og genererer elektron-hul parret, et anodemateriale, der faciliterer il-tudvindingsdelen af vandspaltning ved hjælp af det genererede hul, samt etkatodemateriale, som anvender den genererede elektron til at udvikle brint.I denne afhandling anvendes første princip beregninger til at finde foton-absorberende materialer, hvor materialernes båndgab, placering af båndkan-ten samt materialernes stabilitet i vand bruges som deskriptorer. Ved brugaf denne strategi identificeres en håndfuld foton-absorberende materialer, somværende velegnede til brug i foton-assisteret vandspaltning. Derudover under-søges flere muligheder for at optimere et materiales båndgab til brug i forskel-lige sammenhænge. En række todimensionale metaldichalkogener og metalox-ider undersøges til brug som katodematerialer, og flere potentielt brugbarekandidater præsenteres.Det er nødvendigt at bruge metoder, der giver akkurate første princip energier,for korrekt at beskrive termodynamikken i alle de ovenfor nævnte processer.Derfor undersøges præcisionen af funktionalet med Meta-Generaliseret Gradi-ent Approksimation baseret Bayesiansk fejl-estimation.


List of papers
• Heats of Formation of Folids with Error Estimation: The mBEEF Func-tional with and without Fitted Reference Energies M. Pandey and K.W. Jacobsen, Physical Review B 91 (23), 235201 (2015)
• Two-Dimensional Metal Dichalcogenides and Oxides for Hydrogen Evo-lution: A Computational Screening Approach M. Pandey, A. Vojvodic,K. S. Thygesen and K. W. Jacobsen, The Journal of Physical ChemistryLetters 6 (9), 1577-1585 (2015)
• New Light-Harvesting Materials Using Accurate and Efficient BandgapCalculations I. E. Castelli, F. Hüser, M. Pandey, H. Li, K. S. Thygesen,B. Seger, A. Jain, K. A. Persson, G. Ceder and K. W. Jacobsen, Ad-vanced Energy Materials 5 (2) (2015)
• Band-gap Engineering of Functional Perovskites Through Quantum Con-finement and Tunneling I. E. Castelli, M. Pandey, K. S. Thygesen andK. W. Jacobsen, Physical Review B 91 (16), 165309 (2015)


Contents
1 Introduction 1
2 Theory 42.1 Schrödinger Equation . . . . . . . . . . . . . . . . . . . . . . . 4
2.1.1 Adiabatic and Born-Oppenheimer approximation . . . . 52.2 Density Functional Theory: An Introduction . . . . . . . . . . 6
2.2.1 Local (Spin) Density Approximation (L(S)DA) and Gen-eralized Gradient Approximation (GGA) . . . . . . . . 7
2.3 Calculation of Bandgaps with DFT . . . . . . . . . . . . . . . . 82.3.1 A Brief Introduction to the Hybrid Functionals . . . . . 9
2.4 Implementation of DFT in the GPAW (Grid-based ProjectorAugmented Wave) code . . . . . . . . . . . . . . . . . . . . . . 112.4.1 A Brief Introduction to the PAW Method . . . . . . . . 11
3 Heats of Formation of the Solids 133.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133.2 Calculation of the heats of formation without the fitting . . . . 143.3 Calculation of the heats of formation with the fitting . . . . . . 153.4 Outliers in the different predictions . . . . . . . . . . . . . . . . 173.5 True versus predicted error in the mBEEF functional . . . . . . 203.6 Cross validation . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.7 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
4 Hydrogen Evolution from Two-Dimensional Materials 254.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254.2 Details of the atomic structure . . . . . . . . . . . . . . . . . . 264.3 Stability with respect to the standard states . . . . . . . . . . . 294.4 Adsorption of hydrogen on the basal planes . . . . . . . . . . . 29
xi

4.5 Candidates for the HER . . . . . . . . . . . . . . . . . . . . . . 35
5 Materials for Light Absorption 425.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 425.2 Mechanism of photoelectrochemical water splitting . . . . . . . 435.3 Different methods for the bandgap calculation . . . . . . . . . . 445.4 Candidates for photoelectrochemical water splitting . . . . . . 465.5 Bandgap engineering of functional perovskites . . . . . . . . . . 495.6 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
6 Trends in Stability and Bandgaps of Binary Compounds inDifferent Crystal Structures 586.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 586.2 Results and Discussions . . . . . . . . . . . . . . . . . . . . . . 596.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
7 Final Remarks 71
Bibliography 72
Papers 82Paper I . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82Paper II . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94Paper III . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109Paper IV . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138

Chapter 1
Introduction
Chemical fuels are the most widely used energy resource due to their high en-ergy density and ease of availability. Additionally, storing chemical fuels andtransferring them from one place to another is easier e.g. through pipelines.Therefore, all the above factors made society heavily dependent on them for itsenergy consumption which is increasing every year. Eventually, the increasingconsumption of fossil fuels is leading to increased greenhouse gas emissions.For example, the global CO2 emission in 2001 was approximately 24.07 giga-ton/year (Gt/yr) which is projected to increase to 40.3 Gt/yr by 2050 and48.8 Gt/yr by the end of 2100 [1]. An increase in CO2 emission by almosttwo times in the next three decades will pose a serious threat to the environ-ment. Additionally, the availability of the fossil fuels will also become scarce atsome point. Therefore, it is the need of the hour to search for environmentallybenign and abundant renewable energy resources.
Renewable energy sources e.g. wind energy, hydro-electricity, solar thermalconversion, solar electricity, solar fuels etc. may serve as viable alternatives tothe fossil fuels [2]. Among all renewable energy resources, the biggest sourceof the renewable energy is our sun and the immense energy it provides can beused to power the whole planet. However, we are very far from realizing thedream of being completely dependent on the sun for our energy requirements.The challenge lies in utilizing the solar energy in an efficient and economicalway [1, 2]. However, concerted and continuous efforts by theoreticians and ex-perimentalists are being put in order to overcome these challenges. Figure 1.1shows a model of the workflow for the materials design with mutual feedbackof the experimentalists and theoreticians.
1

2
Figure 1.1: Concerted effort of experimentalists and theoreticians. The mu-tual feedback from each other leads to an efficient materials design and un-derstanding of a given physical/chemical process. Image courtesy: SUNCAT(http://suncat.stanford.edu).
Among many possible ways to utilize solar energy one of the most promis-ing ways is to harvest the solar energy for the photon assisted water splitting.The process proceeds via absorption of sunlight with a semiconducting mate-rial and the generated electron-hole pairs can be used to produce hydrogen bysplitting the water [3]. Unfortunately, the process is not as simple as it soundsand the main challenge lies in finding a material which can accomplish thewhole process of hydrogen evolution efficiently. In order to do so, a materialshould be able to absorb the sunlight to generate electron-hole pairs and evolvehydrogen at cathode and oxygen at anode using the generated electron andhole respectively. All these criteria are hard to meet by a single material. Anadditional constraint is also imposed by the abundance and toxicity of differ-ent elements going in the workflow of materials design [4]. Because of all thecomplications involved, even after decades of explorations for a suitable ma-terial for photoelectrochemical watersplitting, the best material has not been

3
found. Additionally, due to limited resources and time a large materials spacemakes it intractable to find a material experimentally which can carry out theabove process. On the other hand, the quantum mechanical calculations onlarge number of materials can be done with relatively less resources and time.Therefore, inputs are required from the quantum mechanical calculations toaccelerate the process of materials design.
This thesis, using first-principle calculations, explores materials for thelight absorption using the bandgap, band edge positions and the stability inaqueous conditions as descriptors. This strategy results in handful of mate-rials which can act as good photoabsorbers for the water splitting reaction.Additionally, strategies to tune the bandgap for different applications is alsoexplored. To carry out the cathode reaction, two-dimensional metal dichalco-genides and oxides are explored with suggestion of few potential candidatesfor the hydrogen evolution reaction.
The thermodynamics of all the above processes requires an accurate de-scription of the energies with first-principle calculations. Therefore, along thisline the accuracy and predictability of the Meta-Generalized Gradient Approx-imation functional with Bayesian error estimation is also assessed.

Chapter 2
Theory
In this chapter a brief description of the electronic structure method is pre-sented. An introduction to the Density Functional Theory (DFT) and theapproximations used for the calculations of the energies and the bandgaps isdiscussed. A condensed overview of the practicalities of the electronic structurecalculations is also presented.
2.1 Schrödinger EquationA complete quantum mechanical description of a system requires the knowl-edge of an abstract object called the wavefunction. In principle, the wave-function can be obtained by solving the time dependent Schrödinger equationwhich can be written as [5]:
i~∂|Ψ〉∂t
= H|Ψ〉, (2.1)
where |Ψ〉 and H are the wavefunction and the Hamiltonian of the systemrespectively. The Hamiltonian holds the information of the total energy ofthe system that is conserved for a time independent potential. Hence, thestationary state solution to the Schrödinger equation will be a product of thetime dependent phase and a time independent part which is nothing but theeigenfunction of the Hamiltonian. Therefore, calculating the stationary stateof the Hamiltonian is central to the time independent description of a system.
Since our interest lies in understanding the physical and chemical propertiesof materials which are governed by the electrons in time independent potential
4

5
in most of the cases, it is relevant to consider the time independent Schrödingerequation. The time independent Schrödinger equation in the position basis canbe written as [6]:
HΨ(r, R) = E(r, R)Ψ(r, R), (2.2)
where E(r, R) is the eigenvalue of the Hamiltonian of the system and r andR represent the electronic and nuclear coordinates. In an expanded form theHamiltonian can be written as:
H = −N∑I=1
~2
2MI∇2I −
n∑i=1
~2
2mi∇2i + e2
2
N∑I=1
N∑J 6=I
ZIZJ|RI −RJ |
(2.3)
+e2
2
n∑i=1
n∑j 6=i
1ri − rj
− e2N∑I=1
n∑i=1
ZI|RI − ri|
,
where first and second term on the right hand side represent the kineticenergy of the nuclei and electrons respectively, third term corresponds tothe nuclear-nuclear Coulomb interaction, fourth term represents the electron-electron Coulomb interaction and the last term is Coulomb interaction betweenthe electrons and nuclei.
Unfortunately, the eigenvalues and eigenfunctions of the full Hamiltonianwith coupled electronic and nuclear degrees of freedom can only be obtainedfor very few simple systems. Therefore, approximations are needed to makethe electronic structure problem tractable.
2.1.1 Adiabatic and Born-Oppenheimer approximationOne of the commonly used approximation to decouple the nuclear and elec-tronic degrees of freedom is the adiabatic approximation. It is based on thefact that the ratio of of the mass of the electrons and nuclei is very small,therefore, the electrons instantaneously adjust their wavefunctions if there is adynamical evolution of the nuclear wavefunctions. In other words, due to thesluggish dynamics of the nuclear wavefunction the electrons are always in astationary state of the Hamiltonian with the instantaneous nuclear potential.The wavefunction of the system within the adiabatic approximation can bewritten as [6]:
Ψ(R, r, t) = Θn(R, t)Φn(R, r), (2.4)
where n denotes the nth adiabatic state of the electrons, Θn(R, t) representsthe nuclear wavefunction and Φn(R, r) denotes the electronic wavefunction.

6
In the above ansatz, the dependence of electronic wavefunction on the nu-clear coordinates gives a correction for the electronic eigenvalues of the orderm/M(which comes from applying the kinetic energy operator of the nuclei onthe electronic wavefunctions). The small correction of the order m/M whenincluded results to the adiabatic approximation and when neglected gives theso called Born-Oppenheimer approximation. The Born-Oppenheimer approx-imation results in an electronic Schrödinger equation Hamiltonian which canbe written as [6]:
he = −n∑i=1
~2
2mi∇2i + e2
2
n∑i=1
n∑j 6=i
1ri − rj
− e2N∑I=1
n∑i=1
ZI|RI − ri|
.
The above approximation simplifies the electronic problem significantly but notsufficiently to make it tractable for complex systems. The complexity mainlyarises from the electron-electron interaction term in the electronic Hamilto-nian. Density functional theory (DFT) which is discussed in the next sectionprovides an elegant way to solve the electronic structure problem of complexelectronic systems.
2.2 Density Functional Theory: An Introduc-tion
The density functional theory came into being from the two theorems by Ho-henberg and Kohn which are [7]:Theorem 1: The electronic density uniquely determines the external poten-tial up to a trivial additive constant.Theorem 2: The ground state energy of an electron system is a universalfunctional of the ground state electronic density.
Above theorems make it possible to map an interacting system to a non-interacting electron system with the same electronic density leading to so calledKohn-Sham equations. The non-interacting electron system is much easier tosolve since the wavefunction of the system factorizes. The mapping signifi-cantly simplifies the electronic structure problem since the electronic densitywhich is dependendent only on three coordinates becomes the central object asopposed to the wavefunction in the Schrödinger equation which has 3N degreesof freedom. The potential which enters the independent particle Hamiltoniancan be derived from the total energy of the system if one knows how the en-ergy depends on the electronic density. Kohn-Sham equation for independent

7
particles can be written as:−1
2∇2 + vext(r) +
∫d3r
n(r)|r− r′| + vxc[n](r)
φi(r) = εiφi(r). (2.5)
The first term denotes the kinetic energy operator, second term is the externalpotential which typically comes from nuclei, third term is the Hartree potentialand vxc[n](r) represents the exchange-correlation potential which arises fromthe antisymmetric and many body nature of the wavefunction. The exchage-correlation potential vxc[n](r) entering the Kohn-Sham equation can be writtenas:
vxc[n](r) = δExcδn(r) . (2.6)
Up to this point no approximations in the Kohn-Sham system has been made,therefore, the formalism in principle is exact. But our ignorance about the ex-act form of Exc demands approximations to calculate the ground state proper-ties of the system hence deviating us from exactness. Fortunately, the approx-imations for the exchange-correlation energy make the quantum mechanicaltreatement of complex materials tractable with a reasonable accuracy. Few ofthe well know approximations are the local density approximation (LDA) [8],the generalized gradient approximation (GGA) [9], and hybrid functionals e.g.HSE06 [10, 11]. A brief overview of the different approximations is given inthe following subsection.
2.2.1 Local (Spin) Density Approximation (L(S)DA) andGeneralized Gradient Approximation (GGA)
The local density approximation is the first approximation employed in thedensity functional theory. It is built using the free electron gas as a modelsystem, and is therefore expected to perform well for systems with reasonablyhomogeneous charge density. Since its inception it has been widely used andhas produced remarkable results. The exchange energy density under theframework of the LDA can be written as:
εX(n(r))LDA = −34
(3π
)1/3n(r)1/3. (2.7)
The correlation part has been derived from quantum Monte Carlo calculationsand can be found in Ref. [6]. Despite being quite succesful LDA occasionallyperforms badly especially for the systems having very inhomogeneous charge

8
density. One might conclude that this behavior arises due to the local natureof the functional. Therefore, a natural way to improve over LDA is to includethe gradients of density in the energy functional. The generalized gradientapproximation provides such a framework to improve over the LDA functionalby an inclusion of the density gradients. The most commonly used functionalunder the GGA framework is known as PBE functional named after its de-velopers [9]. In the PBE functional the exchange energy density of the LDAis augmented by an enhancement factor which depends on the density and itsgradient. The PBE exchange energy can be expressed as:
EGGAX =∫d3rεX(n(r))LDAFX(s), (2.8)
where FX(s) denotes the exchange enhancement factor with s= |∇n(r)]/2kFn(r).One of the crucial property that the enhancement factor should have is thatin the limit of very small s it should behave in a way that the PBE exchangeenergy approaches the exchange energy with the LSDA. Keeping this in mindthe following expression for FX(s) has been proposed:
FX(s) = 1 + κ− κ
1 + µs2/κ. (2.9)
The inclusion of the exchange enhancement factor in the exchange energyshowed significant improvement over the LDA functionals for the systems withsignificantly varying charge density. Since then the PBE functional has beenone of the most widely used functional in electronic structure problems.
2.3 Calculation of Bandgaps with DFTDespite being quite successful in the prediction of ground state properties ofreal materials, Kohn-Sham DFT (KS-DFT) has some drawabacks [12]. Oneof the most commonly known problem with KS-DFT is the systematic un-derestimation of bandgaps [13]. Over the years, numerous studies have beenperformed in order to have an understanding of the bandgap problem and atthe same time finding its solution. A very thorough study to understand thedifferent sources of the errors in the bandgap prediction has been done in theRef. [13]. For example, depending on the convexity (concavity) of the func-tional between the integer particle number, localization (delocalization) leadsto too high (low) bandgap predictions for the periodic systems. Therefore, itwould be desirable to include an additional localization effect in the concave

9
functionals (like LDA) whereas employing a delocalization effect in the convexfunctionals would improve the bandgap predictions.
As explained in Ref. [13] the energy in LDA like functional behave linearlybetween integer points in periodic systems. Therefore one would expect it togive correct bandgaps. But, the linear behavior has wrong slopes due to whichit systematically underestimates the bandgap. To account for the incorrectslopes the correction in the derivative discontinuity can be applied leadingto improved prediction of the bandgap. One such functional is the GLLB-SCfunctional which includes an explicit calculation of the derivative discontinuity.The details of the functional can be found in Ref. [14, 15].
The other method to improve over the LDA/GGA functionals is to incor-porate a fraction of Hartree-Fock exchange (or exact exchange) which has aconvex behavior. Thus, the Hartree-Fock exchange when added in an appro-priate fraction in the LDA exchange gives a reasonable behavior between theinteger points of the particle number. Generally, the LDA/GGA functionalshaving a fraction of exact exchange are called hybrid functionals. Most com-monly used hybrid functionals in condensed matter systems are PBE0 andHSE03/HSE06 [10, 11, 16, 17]. A brief introduction to the HSE functionalwill be provided here since its implementation in GPAW was carried out as apart of this thesis.
2.3.1 A Brief Introduction to the Hybrid FunctionalsThe PBE0 or HSE functionals have 25 % of exact exchange (at least that ishow it started) mixed with 75 % of GGA exchange. The exchange correlationenergy in the PBE0 functional can be written as:
EPBE0xc = 0.25EHFx + 0.75EPBEx + EPBEc . (2.10)
The (1 /|r− r′|) dependence of HF exchange gives rise to a singularity at r =r’( or q = q’ in reciprocal space). Therefore, it is essential to get rid of thesingularity to prevent divergence. Additionally, a very high density of k-pointsis required to resolve the interaction near the singularity.
The singularity problem has been remedied in the HSE functionals by hav-ing an additional term which prevents the exchange term from diverging. TheHSE functional has many commonalities with the PBE0 functionals. How-ever, in the HSE functional the exchange is screened by a screening parameteras opposed to the PBE0 functional which has a bare (or unscreened) exactexchange. The exchange interaction in the HSE is divided into a short range

10
and a long range part using the error function and can be written as:
1r
= erfc(ωr)r
+ erf(ωr)r
. (2.11)
The above expression shows how the splitting of the exchange interaction isachieved. The first term on the right hand side denotes the short range (SR)exact exchange whereas the second terms denotes the long range (LR) ex-change interaction. The strength of the screening is decided by the valueof the parameter ω. The final expression for the exchange energy after thesplitting can be written as:
EHSEx = 0.25EHF,SRx (ω) + 0.25EHF,LRx (ω) + 0.75EPBE,SRx (ω)+EPBE,LRx (ω)− 0.25EPBE,LRx (ω). (2.12)
It turns out that for a range of ω values pertinent for real physical systems, theEHF,LRx (ω) term cancels the −EPBE,LRx (ω) term. Thus the reduced equationfor exchange-correlation energy is:
EHSExc = 0.25EHF,SRx (ω) + 0.75EPBE,SRx (ω) + EPBE,LRx (ω) + EPBEc . (2.13)
From the above equation we can see that the exchange energy has two parts,one is screened HF exchange and the other is screened GGA exchange. Theexpression for screened exact exchange in the plane-wave basis can be writtenas [18]:
Vk(G,G’) = 〈k + G|Vx|k + G’〉
= −4πe2
Ω∑mq
2wqfqm
×∑G”
C∗qm(G’ - G”)Cqm(G - G”)|k -q + G”|2
×(1− e|k -q + G”|2/4ω2). (2.14)
In the above equation we can see that the exchange term does not have asingularity at |k -q + G”| = 0. In the HSE06 functional the optimized valueof the parameter ω is 0.11 a−1
0 (where a0 is the Bohr radius). The currentimplementation of HSE in GPAW is non self-consistent in which the GGA andHF exchange interactions are calculated with PBE calculated ground statedensity and wavefunctions.

11
2.4 Implementation of DFT in the GPAW (Grid-based Projector Augmented Wave) code
The first step in a practical implementation of DFT is choosing a basis forthe expansion of the wavefunctions. There are wide variety of bases and oneis preferred over the other depending on the kind of the calculations. In thecurrent version GPAW has plane wave, grid and linear combination of atomicorbitals (LCAO) as basis sets [19, 20]. In principle, one can solve the all elec-tron problem without making any approximation for the core electrons, butthat is not usually the case. Since for most of the applications the valence elec-trons govern the behavior of materials, its desirable to make approximationsfor the core electrons in order to make the calculations computationally lessdemanding. Many codes use pseudopotential in which the core electrons actas mere spectators and provide an effective potential to the valence electrons[21]. One of the drawbacks of the pseudopotential method is that one com-pletely looses the information of the core electrons which might be required infew cases. In order to circumvent this issue with the pseudopotentials, Blöchlproposed the projector augmented wave (PAW) method [22].
2.4.1 A Brief Introduction to the PAW MethodThe oscillatory behavior of the wavefunctions in the core regions requires largenumber of basis functions for the expansion, therefore, making the calculationscomputationally demanding. In the Blöchl formalism a linear transformationis applied to an auxilliary smooth wavefunction in order to obtain the full allelectron Kohn-Sham (KS) wavefunction. The operation can be written as [23]:
|ψn〉 = T |ψn〉, (2.15)
where |ψn〉 and |ψn〉 are the true and auxilliary wavefunctions respectively.One of the properties required by the transformation operator is that it shouldnot affect the wavefunction outside a given cutoff radius. The above require-ment is due the similar nature of the true wavefunction and the auxilliarywavefunction outside the cutoff radius. Therefore T can be written as:
T = I +∑a
T a, (2.16)
a denotes the atom index and with the expression above the T a does not haveany effect outside the cutoff radius. The true wavefunction inside the augmen-tation sphere can be expanded in terms of the partial waves and the partial

12
waves can be be obtained by the application of the transformation operatoron the auxilliary smooth partial waves. The above steps along with the com-pleteness of the smooth partial waves give the expression of the transformationoperator which then can be used to get the full KS wavefunction. Thus, bythe this approach one always have the access to the full wavefunction.

Chapter 3
Heats of Formation of theSolids
3.1 IntroductionIn the last chapter a brief description about the density functional theory(DFT) was provided with a short introduction to the different functionals andtheir accuracy. In this chapter, one of the application of DFT is looked ati.e. the calculation of heats of formation of the solid compounds with differentfunctionals particularly focussing on the accuracy of their predictions.
The accuracy of the energetics of a thermodynamic process obtained withthe different functionals depends on the fortuituous cancellation of errors.However, if the nature of species on the different side of a reaction differssignificantly then the cancellation of errors may not be complete thus leadingto an inaccurate energetics. For example, one of the most basic reaction is theformation of the solids from the elements in their reference state. In this casethe chemical environment of the solid formed is very different from the chem-ical environment of the elemental phases. In cases like these the cancellationof errors may not be complete thus ending up giving inaccurate results [24].The same reason renders standard LDA/GGA to give the heat of formation ofthe solids deviating from experiments by ∼0.25 eV per atom [25]. Therefore,large errors in the prediction of the heats of formation may not be appropriatein situations like large scale screening of materials where thermodynamic sta-bility is one of the main criterion for the existence of the compounds [26, 27].Hence, in order to get greater accuracy higher level methods are required. On
13

14
the other hand, most of the higher level methods are computationally quiteexpensive and cannot be used for large scale computations.
Recently a method has been proposed by Stevanovic et al. which uses theexperimental heats of formation and DFT total energies to fit the elementalreference energies in order get better prediction for the standard heats of for-mation [28, 25]. In the work by Stevanovic et al. DFT+U [29] has been usedwith non-zero U for the transition metals. However, in our work we find thatthe other functionals like PBE [9], RPBE [30] and TPSS [31, 32, 33, 34, 35]give similar prediction as PBE+U after fitting the reference energies. Sur-prisingly TPSS being a meta-GGA does not improve the prediction and hassimilar error as the standard GGA functionals. But, the recently developedBayesian error estimation meta-GGA functional known as mBEEF improvesthe predictions significantly. Additionally, it provides the uncertainties in theformation energies as well thus giving the information of the trust radius ofthe results. The details of the mBEEF functional can be found in the Ref.[36].
3.2 Calculation of the heats of formation with-out the fitting
The heat of formation of a solid calculated with DFT can be written as:
∆HDFT (Ap1Bp2..) = E(Ap1Bp2..)− Σpiµ0i , (3.1)
where E(Ap1Bp2..) indicates the total energy of Ap1Bp2.. calculated with DFTand the µ0
i denotes the chemical potentials of the elements under standardconditions calculated with DFT. The entropic and zero point corrections havebeen ignored in the expression above.
For the current work, a set of 257 compounds has been selected to com-pare different functionals for the calculation of heats of formation. Compoundshave been selected to ensure that the space of relevant elements is spanned.Figure 3.1 (a), (c), (e), (g) and (i) show the calculated heats of formation ver-sus the experimental values for the different functionals. The figure indicatesthat the RPBE functional deviates the most from the experimental values,which is also expected since the functional parameters have been fitted to giveaccurate adsorption energies which makes it a bit worse for the prediction ofthe bulk properties. Additionally, PBE, PBE+U and TPSS give similar pre-dictions thus TPSS despite being meta-GGA does not perform better thanthe functionals at the GGA level. Therefore, before any fitting of the experi-mental values, mBEEF outperforms other functionals in the predictions with

15
significantly lower mean absolute error (MAE) and standard deviation (σ).It can also be seen that the experimental values are within the uncertaintiespredicted by the mBEEF ensemble.
3.3 Calculation of the heats of formation withthe fitting
As mentioned before, the different chemical environment of the multinary com-pounds and the reference phases leads to an incomplete error cancellation incalculating the energy differences i.e. the heats of formation. This behaviorwas manifested in the predictions in the previous section which was based onthe DFT reference energies of the elemental phases. Fitted elemental refer-ence phase energy (FERE) method [25] solves this problem to some extent byadding corrections to the DFT reference energies. The value of the correctionsis calculated by minimizing the root mean square (RMS) error of the predictedand the experimental values. The FERE heats of formation can be expressedas:
∆HFERE(Ap1Bp2..) = E(Ap1Bp2..)−Σpi(µ0
i + δµ0i ), (3.2)
The only difference between the equation above and the equation (3.1) is theterm δµ0
i which denotes the correction to reference energy of the elementalphase.
As mentioned before, a dataset of 257 compounds has been chosen forexperimental heats of formation, [25, 37] on the other hand, the number ofelements relevant for this work is limited to 62. Therefore, the calculationof the corrections involves solving an overdetermined set of equations whichcan be done by minimizing the RMS error
√∑i(∆Hi
Expt. −∆HiDFT )2. Few
points have to be kept in mind while fitting the reference energies, for example,a reasonable size of the dateset should be taken to avoid over- or under-fittingand the quality of the fit should be validated on a test dataset which hascompounds not used in the fitting procedure.
The calculated heats of formation with the FERE procedure applied to thedifferent functionals is shown in the Figure 3.1 (b), (d), (f), (h) and (j). As canbe seen from the figure, different functionals clearly improve the predictionswhen augmented with the FERE procedure. After the fitting procedure isapplied all the functionals give similar predictions with almost same MAEand σ. It is worth noticing that the mBEEF predictions before the fitting

16
Figure 3.1: (a), (c), (e), (g) and (i) show the calculated heats of formationwith different functionals. The mean absolute error (MAE) and the standarddeviation (σ) of the the difference of the calculated heats of formation andthe experimental values is also shown in the plots. The black line shows theexperimental heat of formation. The figure has been taken from the Paper-1.

17
is not too off from the predictions of the other functionals after the fitting.A possible reason for the better predictions of the mBEEF functional is thefitting of the parameters of the functional to different experimental dataset [36].Additionally, the reduced uncertainties in the Figure 3.1 (j) results from fittingthe ensemble as well to the experimental heats of formation. The individualheats of formation with the mBEEF functional with and without the fitting isshown in the Table 1 of the Paper-1.
3.4 Outliers in the different predictions
The statistical quantity σ indicates that there must be some predictions whichdeviate from the actual value (in the present case, the experimental values)by more than of the order of σ [38] and these predictions are called outliers.A commonly used measure to call a prediction as an outlier is the value of 2σwhich puts 95 % confidence in the results lying within the width of 2σ. Basedon this criterion, outliers selected for different functionals without and withthe FERE are shown in the Table 3.1. and 3.2
Table 3.1 shows that the PBE and RPBE have common outliers to someextent whereas the PBE+U, TPSS and the mBEEF functional have none orvery few common outliers. The feature in the Table 3.1 worth noticing is thatin a few cases all the functionals except mBEEF deviate from the experimentssignificantly, for example, in the PBE, RPBE, PBE+U and TPSS, deviationis as high as 0.85, 0.66, 0.82 and 0.57 eV respectively whereas the maximumdeviation in the mBEEF prediction is 0.41 eV. Therefore, even without theFERE the mBEEF predictions do not significantly deviate from the experi-mental values.
Table 3.2 shows the predictions after the fitting procedure has been applied.As expected the magnitude of the deviation from the experimental heats offormation decreases after employing the fitting. On the other hand, it canalso be seen from the table that the nature of the outliers significantly changesafter the fitting has been applied which is expected in a fitting model since thedatapoints contributing to large errors get penalized more. Additionally, thecommon feature of a large variation in the nature of the outliers before andafter the fitting rules out the possibility of the experimental errors to someextent and rather puts more weight to the limitations of the functionals.

18
Table3.1:O
utliersinthe
calculationswithoutusing
theFER
Eschem
e.The
compoundsexhibiting
deviationsof
thecalculated
heatsof
formation
fromthe
experimentalvalues
bymore
than2σ
havebeen
identifiedas
outliers.The
valuesofσforthe
differentfunctionalsareshow
nin
Fig.3.1.δH
denotesthedifference
betweencalculated
andexperim
entalheatsofform
ation.Table
hasbeen
takenfrom
thePaper-1.
PB
EδHPBE
RP
BE
δHRPBE
PB
E+
UδHPBE
+U
TP
SS
δHTPSS
mB
EE
FδHmBEEF
Al2
O3
0.48A
l2O
30.69
Al2
O3
0.48A
lP0.45
Au
F3
-0.30B
aS-0.52
FeF
20.61
BaS
-0.52B
aI2-0.48
CaF
2-0.35
BaO
-0.47F
eO0.60
BaO
-0.47B
iBr3
-0.51C
dF
2-0.34
FeF
20.61
GaN
0.57C
rS-0.82
CaS
0.48C
u2
Se
0.31F
eO0.49
HfO
20.65
CrF
3-0.47
FeF
20.57
FeS
e0.35
GaS
0.45N
iF2
0.66C
r2O
3-0.75
GaP
0.43G
aN0.33
LaN
0.46-
-G
aN0.42
Ga2
S3
0.44G
a2S3
0.37M
nS
0.60-
-G
a2S3
0.44N
iF2
0.57G
aS0.41
NiF
20.85
--
GaS
0.45P
bB
r2-0.45
GeS
e0.37
--
--
Ge4
O8
0.42S
rBr2
-0.49G
e4O
80.29
--
--
Mn
S-0.48
SrI2
-0.55N
bF
5-0.38
--
--
Mn3
O4
-0.42Z
nS
0.43O
sO4
-0.30-
--
-V
2O
3-0.42
ZrS
20.47
Pb
F2
-0.31-
--
--
--
-S
nO
20.28
--
--
--
--
TiN
-0.30

19
Table3.2:
Outlie
rsin
thecalculations
usingtheFE
RE
sche
me.
The
compo
unds
exhibitin
gde
viations
ofthe
calculated
heatsof
form
ationfro
mtheexpe
rimentalv
alue
sby
morethan
2σha
vebe
enidentifi
edas
outliers.
The
values
ofσfort
hediffe
rent
func
tiona
lsares
hownin
Fig.
3.1.δH
deno
test
hediffe
renc
ebetwe
encalculated
andexpe
rimentalh
eats
ofform
ation.
Tableha
sbe
entakenfro
mthePa
per-1.
PB
EδHFERE
PBE
RP
BE
δHFERE
RPBE
PB
E+
UδHFERE
PBE
+U
TP
SS
δHFERE
TPSS
mB
EE
FδHFERE
mBEEF
Cu
F2
0.22
Cu
F2
0.23
CoS
0.20
BaC
l 20.
24C
aF2
-0.1
8F
eF2
0.33
FeF
20.
27C
o 3O
4-0
.23
CaS
0.21
Cd
F2
-0.1
8F
eSe
-0.1
9M
nO
2-0
.21
CrO
20.
17C
sF-0
.23
Co 3
O4
-0.1
9M
nO
2-0
.25
Nb
F5
-0.3
2F
e 2O
3-0
.17
FeF
20.
29F
e 2O
3-0
.17
Nb
F5
-0.2
9N
i 3S2
-0.1
8G
aF3
0.16
KC
l0.
32F
eF2
0.19
Ni 3
S2
-0.2
1N
iF2
0.38
GeO
20.
20N
bF
5-0
.26
GaP
-0.1
6N
iF2
0.65
Pb
F2
-0.1
8M
gF2
0.19
NiF
20.
37K
F-0
.17
Ru
O4
-0.1
9R
uO
4-0
.33
Nb
F5
-0.2
3R
bI
0.25
Li 3
Sb
0.18
TaF
5-0
.22
TaF
5-0
.24
Sn
O2
0.17
SrS
0.25
MgO
-0.1
5Z
rSi
-0.2
4Z
rSi
-0.2
6T
aF5
-0.1
8S
rI2
-0.2
4M
gF2
0.17
ZrS
20.
26Z
rS2
0.24
TiN
-0.1
9T
lI0.
26M
nO
2-0
.16
--
--
VN
0.26
ZrS
i-0
.24
Nb
F5
-0.2
0-
--
-V
2O
3-0
.36
ZrS
20.
35T
iN-0
.17
--
--
Zn
F2
0.19
--
Zn
F2
0.18
--
--
ZrS
20.
16-
-Z
rS2
0.16

20
3.5 True versus predicted error in the mBEEFfunctional
As previously shown in the Figure 3.1 in most of the cases the experimentalvalues lie within the predicted uncertainties by the mBEEF functional withslight overestimation (large errorbars) of the predicted errors. However, thesize of uncertainties decreased significantly with the FERE. Therefore, in orderto understand the distribution of error before and after the fitting a histogramof the true error (∆HmBEEF - ∆HExpt. and ∆HFERE
mBEEF - ∆HExpt.) dividedby the predicted error (σBEE and σFEREBEE ) is plotted in the Figure 3.2. Thehistogram is a running average calculated as [38]:
P (12 [xi + xi+J ]) ≈ J
N(xi+J − xi), (3.3)
with xi as the statistical quantity plotted in the histogram and an intermediatevalue 20 for the parameter J has been chosen.
If the predicted error matches exactly the true error then one would expectthat the distribution would be a Gaussian of unit width (shown in green inthe figure). However, in the Figure 3.2 this is not the case. As also noticedbefore, the tendency of the mBEEF to overestimate the errors in manifestedin the large peak around zero in (a) which renders the mBEEF to have mostof the experimental values lie within the uncertainties.
However, with the FERE the distribution flattens out and becomes closerto the unit Gaussian implying that the real and the predicted error are close.The tail in the histogram indicates those cases where the predicted error issmaller than the actual error. This is a fairly common feature of the ensembleapproach [39].
3.6 Cross validationAs pointed out before, the fitting model should be such that the data is neitheroverfitted nor underfitted. Therefore, it is of utmost importance that thequality of the fit is tested on a dataset (also called as test set) which is notincluded in the fitting dataset (also called as training set). A good qualityfit should provide a reasonable prediction on a new dataset. A point worthnoticing in the current fitting scheme is that only binary compounds havebeen used in fitting dataset, therefore good predictions are expected for thenew binary compounds. Additionally, reasonable predictions can be expected

21
Figure 3.2: (a) shows the histogram of the true error divided by the predictederror before the fitting (b) shows the histogram of the true error divided by thepredicted error after the fitting. The figure has been taken from the Paper-1.

22
for ternary/tertiary compounds only if their chemical environment does notdiffer significantly from the compounds used in the fitting procedure. Hence,a test set containing a mix of binary and ternary compounds has been selectedfor the validation of the fitting.
Table 3.3 and 3.4 show the heats of formation of the test set without andwith the fitting respectively. The clear decrease in the MAE and σ shows theabsence of overfitting. As expected in any regression scheme, the improvementwith the fitted model is not as much as the improvement seen in the trainingdataset.
In the test set also, the mBEEF predictions without the fitting has thesame quality as the other functionals with the fitting and the improvementwith the fitting is only moderate in the case of the mBEEF. Therefore, areasonable prediction with the mBEEF can be obtained even without usingthe fitting with only negligibly increased computational cost as compared toother GGAs.
3.7 ConclusionThe rapidly growing area of the computational screening of the energy materi-als requiring reasonable predictions of the stability has led forward this work.The synergetic use of the DFT total energies and the experimental heats offormation provides a framework to improve the predictions. Originally, thescheme was developed for the PBE+U functionals but in this work similarimprovements has been seen for the other functionals like PBE, RPBE, TPSSand mBEEF as well.
We see that the recently developed mBEEF functional which has beenoptimized using variety of experimental dataset gives better predictions ascompared to the other functionals. Additionally, the mBEEF functional alsoprovides reasonable estimate of the uncertainties in the predictions, the fea-ture which other functionals used in this work lack. However, the uncertaintiesestimated by the mBEEF ensemble is in general overestimated which can fur-ther be reduced by using the FERE scheme along with the reduction of thetrue error as well.
Despite giving improved results FERE scheme has some drawbacks as well.The corrections are primarily based on nature of the bonding environment inthe training set, therefore, it may not significantly improve the predictions forthe systems differing from the systems used in the training set, for example, inmetal alloys which have significantly different chemical environment than thesemiconductors used in the training set. Therefore, higher level functionals

23
Table 3.3: Heats of formation of test dataset with different functionals withoutthe fitting. All the energies are in eV/atom. Table has been taken from thePaper-1.
Compound ∆HExpt. ∆HPBE ∆HRPBE ∆HPBE+U ∆HTPSS ∆HmBEEF
AgNO3 -0.26 -0.40 -0.31 -0.53 -0.47 -0.60 ± 0.22AlPO4 -2.99 -2.71 -2.58 -2.71 -2.86 -2.97 ± 0.19BeSO4 -2.16 -1.99 -1.83 -1.99 -2.09 -2.19 ± 0.16BiOCl -1.27 -1.26 -1.11 -1.26 -1.62 -1.26 ± 0.16CdSO4 -1.61 -1.42 -1.27 -1.42 -1.53 -1.59 ± 0.17CuCl2 -0.76 -0.51 -0.32 -0.70 -0.80 -0.74 ± 0.21TiBr3 -1.42 -1.24 -1.23 -1.52 -1.71 -1.37 ± 0.08NaClO4 -0.66 -0.54 -0.41 -0.54 -0.67 -0.63 ± 0.15CaSO4 -2.48 -2.24 -2.06 -2.24 -2.37 -2.40 ± 0.17Cs2S -1.24 -1.01 -0.92 -1.01 -1.47 -1.16 ± 0.18CuWO4 -1.91 -1.59 -1.41 -1.76 -1.72 -1.68 ± 0.21PbF4 -1.95 -2.13 -2.05 -2.13 -2.26 -2.32 ± 0.23MgSO4 -2.22 -1.97 -1.79 -1.97 -2.09 -2.16 ± 0.16SrSe -2.00 -2.04 -1.98 -2.04 -2.76 -2.29 ± 0.16NiSO4 -1.51 -1.11 -0.96 -1.35 -1.23 -1.42 ± 0.23FeWO4 -1.99 -1.73 -1.58 -2.01 -1.87 -1.84 ± 0.21GeP -0.11 +0.04 +0.09 +0.04 -0.19 +0.14 ± 0.08VOCl -2.10 -1.79 -1.68 -2.45 -2.07 -2.11 ± 0.24LiBO2 -2.67 -2.42 -2.30 -2.42 -2.57 -2.58 ± 0.17NaBrO3 -0.69 -0.52 -0.41 -0.52 -0.71 -0.60 ± 0.13CoSO4 -1.53 -1.09 -0.95 -1.43 -1.24 -1.40 ± 0.23PbSeO4 -1.05 -0.94 -0.81 -0.94 -1.13 -1.04 ± 0.16Mn2SiO4 -2.56 -1.83 -1.77 -2.58 -2.01 -2.29 ± 0.23ZnSO4 -1.70 -1.37 -1.20 -1.37 -1.47 -1.53 ± 0.16
MAE 0.24 0.35 0.16 0.20 0.12σ 0.28 0.39 0.19 0.26 0.16
are required to improve the description at the electronic structure level andthereby making the FERE scheme unnecessary and the mBEEF functionalseems to be promising in that direction.

24
Table 3.4: Heats of formation of test dataset with different functionals withthe fitting. All the energies are in eV/atom. Table has been taken from thePaper-1.
Compound ∆HExpt. ∆HFEREPBE
∆HFERERPBE
∆HFEREPBE+U ∆HFERE
TPSS∆HFERE
mBEEF
AgNO3 -0.26 -0.58 -0.67 -0.68 -0.45 -0.63 ± 0.16AlPO4 -2.99 -2.95 -2.97 -2.94 -3.02 -3.03 ± 0.07BeSO4 -2.16 -2.22 -2.23 -2.21 -2.19 -2.25 ± 0.11BiOCl -1.27 -1.25 -1.20 -1.23 -1.32 -1.23 ± 0.09CdSO4 -1.61 -1.61 -1.62 -1.60 -1.60 -1.63 ± 0.12CuCl2 -0.76 -0.75 -0.60 -0.84 -0.79 -0.81 ± 0.07TiBr3 -1.42 -1.38 -1.39 -1.61 -1.38 -1.43 ± 0.05NaClO4 -0.66 -0.76 -0.77 -0.73 -0.68 -0.65 ± 0.16CaSO4 -2.48 -2.41 -2.41 -2.41 -2.46 -2.43 ± 0.12Cs2S -1.24 -1.27 -1.24 -1.33 -1.97 -1.23 ± 0.06CuWO4 -1.91 -1.62 -1.60 -1.75 -1.65 -1.71 ± 0.07PbF4 -1.95 -2.19 -2.19 -2.11 -2.24 -2.13 ± 0.08MgSO4 -2.22 -2.22 -2.21 -2.21 -2.20 -2.24 ± 0.10SrSe -2.00 -2.25 -2.26 -2.29 -2.66 -2.29 ± 0.05NiSO4 -1.51 -1.35 -1.36 -1.54 -1.33 -1.50 ± 0.11FeWO4 -1.99 -1.81 -1.81 -1.94 -1.86 -1.89 ± 0.06GeP -0.11 -0.01 +0.03 -0.05 -0.28 -0.02 ± 0.07VOCl -2.10 -1.97 -1.98 -2.41 -2.05 -2.12 ± 0.07LiBO2 -2.67 -2.61 -2.61 -2.58 -2.64 -2.61 ± 0.05NaBrO3 -0.69 -0.74 -0.76 -0.72 -0.69 -0.66 ± 0.11CoSO4 -1.53 -1.30 -1.34 -1.56 -1.31 -1.43 ± 0.11PbSeO4 -1.05 -1.07 -1.09 -1.06 -1.07 -1.08 ± 0.09Mn2SiO4 -2.56 -2.17 -2.19 -2.38 -2.10 -2.25 ± 0.08ZnSO4 -1.70 -1.61 -1.61 -1.61 -1.60 -1.62 ± 0.11
MAE 0.12 0.13 0.11 0.15 0.09σ 0.16 0.17 0.15 0.25 0.14

Chapter 4
Hydrogen Evolution fromTwo-Dimensional Materials
4.1 IntroductionStorage of energy in the form of chemical bonds is one of the most used andefficient way of storing the energy. Transferring energy from one place to otherin form of chemical bonds is easier as compared to the other means such aselectricity. On the other hand, the deteriorating environmental conditions dueto the excess burning of the petroleum fuels needs our attention to look forthe alternative forms of chemical energy not having deleterious effect on theenvironment. One such fuel is hydrogen which can be used in the fuel cells thusinvolving no emission of greenhouse gas whatsoever [40, 41]. However, a cheapand efficient way of producing hydrogen has not been realized yet [42, 43, 44].One of the bottleneck to reduce the cost of hydrogen production is the use ofexpensive catalysts like Platinum a cheaper and efficient alternative of whichhas not been found yet. Recent theoretical and experimental investigations ofthe bulk Ni2P for hydrogen evolution reaction (HER) show promising resultsand hopefully in the future will serve as a viable alternative to Platinum forthe HER [45, 46, 47].
Additionally, over the last few years, two-dimensional (2D) MoS2 has beenexplored for its activity towards the HER with some promising results [48,49, 50, 51, 52]. The initial effort of the MoS2 research was focussed on theedges of the 2H structure of the MoS2 nanoparticle having metallic characteras opposed to the semiconducting states on the basal plane [53, 54]. Unfor-
25

26
tunately, relatively limited number of active sites on the edges gives very lowexchange current density for the HER. However, recent experiments on theother polymorph of the MoS2 and WS2 known as 1T structure demonstratedthe activity of the basal plane for the HER thus giving access to relativelylarger number of active sites [49, 48, 50]. Additionally, the difference in en-ergy of the 1T and 2H phase MoS2 or WS2 decreases as the dimensionalityof the system is reduced from three (bulk) to two (monolayer) thus making itfeasible to synthesize the HER active metastable phase in the 2D form [55].The different activity of the 2H and 1T phase broadens the materials spacefor HER which is the basis of this work. In this work, the basal planes of 100different metal dichalcogenides and oxides have been explored in both the 2Hand 1T structure for the HER. Primarily, criterion of stability of the materialwith respect to the standard reference phases and other competing phases andthe free energy of the hydrogen adsorption on the basal plane has been usedas descriptors for the screening of materials for the HER.
4.2 Details of the atomic structureThe 2H and 1T structures differ by the arrangement of the chalcogen/oxygenatom around the metal atoms. The 2H structure has prismatic arrangement ofthe chalcogen/oxygen atoms around the metal atom whereas in the 1T struc-ture they are octahedrally arranged. The 2H and 1T structures are shownin the Figure 4.1 (a) and (f) respectively. The black square represents theunit cell of the structures. Other structures shown in the 2H and 1T class arethe distorted derivatives of the 2H and 1T structures. The distorted struc-tures have been broadly classified based on their symmetry group which havebeen identified using certain cutoff for the rotations/translations to accountfor the residual forces in the structures. In order to identify the the distortedstructures, atoms are slightly displaced from their symmetric position in abigger unit cell in order to break the symmetry of the structure and then therelaxation is performed.
The above procedure captures all the distortions if any in the 2×2 unit cell.There might be other distortions in the larger unit cell but those cases have notbeen considered here. Fortunately, the charge density wave (CDW) structuresin compounds like TiS2 [56, 57] distorted structure of MoS2, WS2 etc. [58, 59],exhibiting quantum spin Hall effect (QSH) and the distortions in ReS2 [60] arecaptured by the above procedure thus supporting our results. However, thechoice of 0.01 eV/atom for the threshold of energy to differentiate betweenthe symmetrical and the distorted structure categorize TiS2 as symmetrical

27
Figure 4.1: (a), (f) show the 1T undistorted 1T and 2H structures respectively.Yellow spheres represent the chalcogen atoms and the cyan spheres representthe metal atoms. (b) - (e) show the distortions in the 1T structure. The unitcell of the distorted structure is shown with black solid lines and the distortionof the atoms from their ideal symmetric position is shown with black dottedlines. (g) represents the distorted 2H structure with a similar description asabove. The figure has been taken from the Paper-2.

28
structure. But, it turns out that the CDW structure of the TiS2 and thesymmetrical structure are very close in energy having the difference of the orderof 0.005 eV/atom and surprisingly these differences are captured with the aboveprocedure. On the other hand, the adsorption energy of the hydrogen is similaron the symmetrical and the distorted structure in the case of CDW structures,therefore, they have been categorized as symmetrical for consistency due to thethreshold of 0.01 eV/atom. Table 4.1 summarizes the results for the distortedstructures which are classified based on the space group (based on Herman-Maugin notation) of the distorted structure and the size of the reduced unitcell capturing the distortion. Symmetry analysis for the classification hasbeen performed using the tool given in Ref. [61]. The cutoff of of 0.05 Å onthe rotations/translations has been used in order to allow for inaccuracies orresidual forces.
Table 4.1: Classification of different compounds exhibiting distortions based onthe space group (based on Herman-Maugin notation) of the distorted structureand the size of the reduced unit cell capturing the distortions.
Class MX2 Group Unit cell Class MX2 Group Unit cell
2H CoS2 P1 2×2 2H CoSe2 P1 2×22H IrS2 P1 2×2 2H OsS2 P1 2×22H OsSe2 P1 2×2 2H PdS2 P1 2×22H PdSe2 P1 2×2 2H PdTe2 P1 2×22H ReO2 P1 2×2 2H ReS2 P1 2×22H ReSe2 P1 2×2 2H RhS2 P1 2×22H RhSe2 P1 2×2 2H RhTe2 P1 2×22H RuO2 P1 2×2 2H RuS2 P1 2×22H RuSe2 P1 2×2 2H ScS2 P1 2×22H ScSe2 P1 2×21T CoS2 P1 2×2 1T CrS2 P1 2×21T CrSe2 P1 2×2 1T FeS2 P1 2×21T IrS2 P1 2×2 1T IrSe2 P1 2×21T ReO2 P1 2×2 1T ReTe2 P1 2×21T RhS2 P1 2×2 1T RuS2 P1 2×21T RuTe2 P1 2×2 1T MoO2 P1 2×11T MoS2 P1 2×1 1T MoSe2 P1 2×11T MoTe2 P1 2×1 1T OsS2 P1 2×11T OsSe2 P1 2×1 1T OsTe2 P1 2×11T WS2 P1 2×1 1T WSe2 P1 2×11T WTe2 P1 2×1 1T ReS2 P1 2×21T ReSe2 P1 2×2 1T RuSe2 P1 2×21T TaO2 P1 2×2 1T CoSe2 P3m1 2×21T IrTe2 P3m1 2×2 1T NbO2 P3m1 2×21T OsO2 P3m1 2×2 1T RhSe2 P3m1 2×21T RuO2 P3m1 2×2 1T WO2 P3m1 2×2

29
4.3 Stability with respect to the standard statesIn the last chapter, the standard heat of formation of the compounds was dis-cussed. It has to be negative for a compound if the compound has to be stablewith respect to the standard states of the constituent elements. Therefore, asa first step the calculation of the heat of formation of the compounds has beenperformed for all the 2D materials explored here. The heatmap in the Figure4.2 shows the heats of formation of the compounds in the 2H and 1T structureand the difference in energy of the two structures. The figure shows that asignificant fraction of the compounds have positive heats of formation thusunstable [62]. Figure 4.2 (c) shows the difference in energies of the 2H and 1Tstructure. The figure clearly shows that in most of the cases the 2H and 1Tstructures are energetically very close. One of the important implication of thetwo structures having similar energy is that the HER active phase can be syn-thesized and stabilized under normal condition with suitable synthetic routesand the same fact has been realized in the case of MoS2 and WS2 [50, 48].However, an ideal situation would be that the HER active phase is the moststable phase. But, if that is not the case then a small degree of metastabilitywould make it feasible to synthesize the HER active phase. As a side note,since the standard heat of formation by definition is the stability with respectto standard states, the stability with respect to other competing phases mightalso be important, however, stability with respect to the other phases has onlybeen considered for the compounds meeting the criteria for the HER activity.
However, Figure 4.2 only shows the heats of formation of the perfectlysymmetrical 2H and 1T structures. But, as discussed in the last section thepossible distortions have also been explored for all the compounds hence itis crucial to assess the energy difference of the perfectly symmetrical and thedistorted phase of the compounds. Figure 4.3 shows the relative energy ofthe distorted phase with respect to the symmetric phase. The white squarescorresponds to the compounds manifesting massive distortions leading to thestructures not belonging to either of the 2H or 1T class, therefore, they areignored. As can be seen from the figure, a large fraction of compounds do notshow any distortions.
4.4 Adsorption of hydrogen on the basal planesOne of the widely accepted mechanism for the HER is the Volmer-Heyrovskymechanism which is a two step process; the first step is the adsorption of Hon the active site and the second step is the bond formation between the two

30
Figure 4.2: (a), (b) show the standard heats of formation of the compounds inthe 2H and 1T structure respectively. (c) shows the difference of the heats ofthe compounds in the 2H and 1T structure. All the energies are in eV/atom.The figure has been taken from the Paper-2.

31
Figure 4.3: (a) and (b) show the energy of the distorted structures (eV/atom)with respect to the perfectly symmetrical 2H and 1T structures, respectively.The white squares denote massive reconstructions upon relaxation thus leadingto structures not belonging to the 2H and 1T class of structures. All theenergies are in eV/atom. The figure has been taken from the Paper-2.

32
adsorbed hydrogen to evolve the gaseous hydrogen [63, 64]. Schematically, theenergetics of the process is shown in the Figure 4.4.
Figure 4.4: Schematic of the Volmer-Heyrovsky route for the HER.
The product and the reactant are at the same level of energy in the Figure4.4 due to the assumption that the process is at equlibrium under standardconditions thus have zero free energy. Active site in the figure is denoted bythe *. It can be seen from the figure that the intermediate H∗ may lie higheror lower in energy than the product and the reactant. If the intermediate lieshigher in energy than the reactant then the first step will be uphill and if it lieslower in the energy than the reactant then the second process will be uphill.Therefore, based on the thermodynamic argument if the process has a zerobarrier, then the free energy for the adsorption of hydrogen has to be zero[48, 65, 42]. Although the free energy for the hydrogen adsorption provides adescriptor for the HER activity, it does not provide any information about thekinetic barrier for the process but we do not explore the kinetic pathways inthis work.
In order to assess the reactivity of the basal plane the hydrogen adsorptionenergy has been calculated for different active sites on the surface. We find thatthe hydrogen prefers to adsorb on chalcogen/oxygen atoms in tilted positionsin most of the cases and does not prefers to adsorb on the metal site. Ina perfectly symmetric structure all the chalcogen atoms are equivalent thusconsidering just one of them suffices. However, in the distorted structures,the broken symmetry leads to inequivalent chalcogen sites, therefore, all theinequivalent sites have been explored for the hydrogen adsorption and thesite with the lowest adsorption energy has been chosen for further analysis.The coverage of 0.25 monolayer (ML) has been chosen initially and the highercoverage (0.5 ML) is only considered for those structures which bind hydrogentoo strongly (∆Hads
H ≥ −0.8) for 0.25 ML. However, it has been found that athigher coverages the structures massively distort leading to the structures notbelonging to either of the 2H or 1T class, therefore, higher coverages have notbeen considered any further.

33
The compounds have been grouped based on the nature of the metal atom‘M’ in MX2 i.e. the compounds have been put in the same group if the metalatoms belong to the same group in the periodic table; based on this catego-rization the plots for hydrogen adsorption energies are shown in the Figure4.5.
Figure 4.5: Hydrogen adsorption energies of the individual groups. The com-pounds have been grouped based on the nature of the metal atom ‘M’ in MX2i.e. the compounds have been put in the same group if the metal atoms belongto the same group in the periodic table. The missing data points representmassive reconstruction upon the hydrogen adsorption thus omitted from theplot. All the energies are in eV. The figure has been taken from the Paper-2.
The plot clearly shows that adsorption energies on the 2H and 1T structuresdo not follow any systematic trend in most of the cases, therefore, a simplesystematic analysis cannot be performed to rationalize the different activitiesof the different structures. However, the group containing Cr, Mo and W(group-6) shows an opposite trend as the group containing Ti, Zr and Hf(group-4). In the group-6 the 1T structure binds hydrogen strongly whereasin the group-4 the 2H structure has higher binding energy. Therefore, only thegroup-4 and group-6 have been selected to understand the origin of different

34
reactivity in different structures.Since the strength of the bonding depends on how the adsorbate states
hybridize with the adsorbent states, the position of the center of the p levelof the chalcogen atoms might give a clue about the strength of the bonding[65, 66]. The center of the p band with respect to the Fermi level can becalculated as
εp =∫∞−∞ ρ(ε)εdε∫∞−∞ ρ(ε)dε
(4.1)
The results for the sulphides and selenides of Mo, W, Ti and Zr in both the2H and 1T structures are summarized in the Table 4.2. The table indicatesthat in the case of Mo and W, the 1T structure has higher binding energy(∆HH
ads) for the hydrogen whereas in the case of Ti and Zr hydrogen bindsstrongly in the 2H structure. It can also be seen that the compounds whichhave higher binding energy have the center of the p-level closer to the Fermilevel. For example, in the case of group-6, the center of the p-level in the2H structure lies deeper with respect to the Fermi level as compared to the1T structure whereas in the group-4 the trend is opposite. Thus, it can beconcluded that the position of the p band center is somewhat correlated to thebinding energy.
Table 4.2: Heat of adsorption of hydrogen ∆HHads and the center of the p-band
εp for sulphides and selenides of Mo, W (group-6) and Ti, Zr (group-4) in the2H and 1T structures.
2H εp ∆HHads
1T εp ∆HHads
MoS2 -2.00 1.68 ± 0.07 MoS2 -1.23 0.10 ± 0.13MoSe2 -1.74 1.82 ± 0.13 MoSe2 -1.46 0.64 ± 0.11WS2 -2.32 1.95 ± 0.08 WS2 -1.37 0.23 ± 0.14WSe2 -2.03 2.03 ± 0.14 WSe2 -1.29 0.78 ± 0.15TiS2 -1.02 -0.05 ± 0.13 TiS2 -1.45 0.40 ± 0.09TiSe2 -0.89 0.44 ± 0.12 TiSe2 -1.38 0.90 ± 0.10ZrS2 -0.96 0.11 ± 0.10 ZrS2 -1.42 0.94 ± 0.07ZrSe2 -0.80 0.51 ± 0.10 ZrSe2 -1.34 1.19 ± 0.09
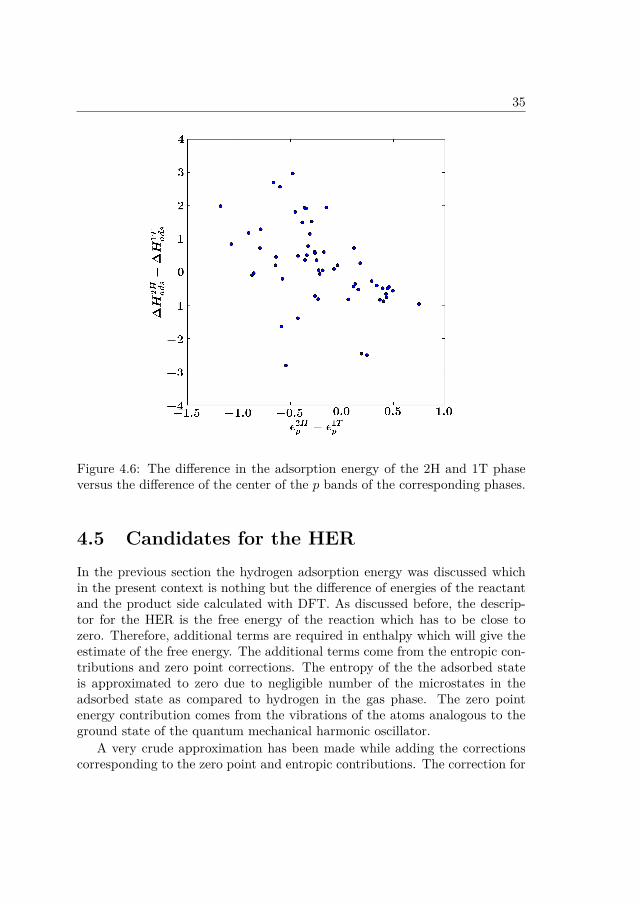
However, as shown in the Figure 4.6 there is hardly any trend when thedifference of the adsorption energies is plotted against the difference of thecenter of the p level for large number of compounds. The absence of any trendcan be attributed to the large variation in the nature of the metal atoms whichmakes it harder to generalize the analysis above for all the groups.
35
Figure 4.6: The difference in the adsorption energy of the 2H and 1T phaseversus the difference of the center of the p bands of the corresponding phases.
4.5 Candidates for the HERIn the previous section the hydrogen adsorption energy was discussed whichin the present context is nothing but the difference of energies of the reactantand the product side calculated with DFT. As discussed before, the descrip-tor for the HER is the free energy of the reaction which has to be close tozero. Therefore, additional terms are required in enthalpy which will give theestimate of the free energy. The additional terms come from the entropic con-tributions and zero point corrections. The entropy of the the adsorbed stateis approximated to zero due to negligible number of the microstates in theadsorbed state as compared to hydrogen in the gas phase. The zero pointenergy contribution comes from the vibrations of the atoms analogous to theground state of the quantum mechanical harmonic oscillator.
A very crude approximation has been made while adding the correctionscorresponding to the zero point and entropic contributions. The correction for

36
the same has been calculated for the 1T-MoS2 and then the same correctionhas been used for the all the other materials. This is not a perfectly validassumption, however, a tolerance of 0.5 eV for the free energy to accountfor the errors introduced due to different approximations at different levelswill likely capture the variability in the zero point and entropic corrections.In the case of 1T-MoS2 as mentioned before the entropic corrections for theadsorbed state has been ignored [54]. The calculated zero point correctionsfor the adsorbed hydrogen comes out as 0.39 eV. The zero point energy of thehydrogen in the gas phase has been taken from the Ref. [67] and is foundto be 0.27 eV and entropy of the gaseous hydrogen has been takes as 0.40 asmentioned in the Ref. [68]. By taking the difference of the corrections in thegas phase and the adsorbed state ∆ZPE comes out as 0.12 eV and -T∆S comesout as 0.40 eV, therefore, ∆ZPE -T∆S comes out to be 0.26 eV (per hydrogenatom). Therefore, the correction of 0.26 eV is added to the adsorption energiesfor all the compounds to have an estimate of the free energy.
As mentioned before, the optimum value of the free energy for the HER is0.0 eV, however, an energy window of 0.5 eV is taken to account for differenteffects like strain, coverage and solvation [48, 69]. Additionally, the estimateof the uncertainties is obtained in the calculation with the BEEF-vdW usingthe ensemble in Ref. [70]. Having the uncertainties along with the energywindow of 0.5 eV helps to calculate the probability for a material to have thefree energy of the hydrogen adsorption to lie within 0.5 eV around zero. Thecalculated probability helps to rank the material in order of their suitabilityfor the HER [71]. The probabilities are calculated as:
P (|∆G| ≤ 0.5) = 1√2πσ2
∫ 0.5+E
−0.5−Eexp
(− E
2
2σ2
)dE. (4.2)
Using the above equation, the ranking of the material meeting the criteria ofhaving the free energy for the HER (including the uncertainties) to lie in therange (-0.5, 0.5) eV is shown in the Figure 4.7. The number of compoundsfulfilling these criterion in the 2H structure in the plot is 23 whereas in the 1Tstructure there are 30 compounds meeting the required criterion.
The plot clearly shows that there are very few compounds which are presentin both the 2H and 1T structure indicating that the chemical properties mightdiffer significantly in different structures of the same compound. Addition-ally, the occurence of the compounds like MoS2 and WS2 in the 1T structurewhich have already been found experimentally as possible HER materials givescredibility to our approach [50, 48].
Up to this point the stability of the compounds have been considered only

37
Figure 4.7: (a) Calculated free energy for the hydrogen adsorption (∆GHads)along with the uncertainties and the probabilities (P(|∆G| ≤ 0.5)) that thecompounds have for the free energy to lie in the range (-0.5, 0.5) eV in the 2Hstructure. Red error bar indicate instability of the compound with respect tothe standard state. (b) Similar plot as (a) for the 1T structure.

38
with respect to the standard states of the elements whereas there might beother potentially competing phases hampering the growth of the 2D materialsfor the HER. Therefore, it is crucial to have a deeper look on the stability ofmaterials potentially suitable for the HER. If the metastability of the candidatematerial comes out to be too large as compared to the competing phase then itsunlikely that the candidate material can be synthesized and stabilized undernormal conditions. Therefore, the stability check for the candidate materialhas been performed with respect to the other competing phases with samestochiometry. Additionally, in the present work we do not explore the stabilityof the compounds in aqueous medium because in some recent works a goodcontrol over the stability of the compounds in water has been achieved bythe use of stabilizing agents [72]. All the competing bulk structures are takenfrom the The Open Quantum Materials Database (OQMD) [73] and then theenergy of candidate material is compared to the energy of the convex hull inorder to have an estimate of the degree of metastability. The data is shown inthe Table 4.3 and 4.4.
∆H in the tables denotes standard heats of formation calculated with theFERE method and ∆Hhull denotes the convex hull [73]. The symbol * insuperscript denotes the cases where the convex hull has been calculated as thelinear combination of the energies of two compounds because no compoundswere present with 1:2 stoichiometry in the database. δHhull denotes the energyof the monolayer with respect to the convex hull. ∆HExpt is the experimentalheat of formation of the compound (if available) lying on the convex hull. Theinitial list of the candidates for the HER is also compared to the predicted2D materials by Lebègue et. al [74]. In order to have an estimate of themetastability of the 2H structure with respect to the 1T structure or vice-versa the difference of the two is also shown as ∆H2H/1T (∆H1T/2H). Finally,the previously discussed probability P(|∆G| ≤ 0.5) is also listed in the table.All the energies mentioned in the table are in eV/atom.
Few important points worth noticing in the table are:• In all the cases the 2H and 1T structure do not differ by more than ∼0.4eV which is similar to the degree of metastability in MoS2 and WS2 inthe 2H and 1T structure and both the compounds can be synthesized inthe stable 2H phase and metastable 1T phase under normal conditions.Similar degree of metastability in other compounds suggest that if theycan be synthesized in one structure then it is likely that they can besynthesized in the other structure as well.
• Few of the HER materials like PdS2, PdTe2 proposed in this work havealso been predicted by Lebègue et. al [74] to exist in the monolayer form.

39
The fact that they lie above the hull by only ∼0.4 eV led us to made achoice of the threshold energy of 0.4 eV for the stability (or the feasibilityof existence) of the monolayer with the respect to the hull. The choiceof the threshold energy helps to narrow down the candidates even more,for example compounds like OsS2, ReO2, OsSe2, ScO2, RuO2 in the 2Hclass of candidates and OsO2 in the 1T class of candidates lying abovethe hull by more than 0.4 eV can be safely discarded. The names of thediscarded compounds are italicized in Table 4.3 and 4.4.
• On comparing the list of the candidates with the list of the predicted 2Dmaterials by Lebègue et. al [74], the compounds common in both thelists are selected. Given the fact that the heurestic approach of Lebègueet. al which is based on the feasibility of cleaving a bulk structure alonga certain direction due to the weak interlayer interaction gives a clue thatthe compounds common in both the list are potentially synthesizable,therefore, potential candidates for the HER. The potential candidatesare marked in bold in the Table 4.3 and 4.4.
In conclusion, this work systematically explores the materials space in thetwo-dimensional 2H and 1T structure for the hydrogen evolution reaction usingthe free energy of hydrogen adsorption as a computational descriptor. Therequirement of the activity on the basal plane ensures the presence of largenumber of active sites as compared to previously explored 2H structure ofthe MoS2 which only has the activity on the edges. A fairly large windowchosen for value of the descriptor provides a flexibility to tune the adsorptionenergy of the hydrogen by different means, for example, strain, environment,doping etc. Additionally, the robust stability analysis of the candidates foundsuitable for the HER provides a list of candidates which do not have very highdegree of metastability with respect to the bulk compounds thus potentiallysynthesizable. The adopted approach also predicts already known MoS2 andWS2 in 1T structure as candidates for HER thus supporting our approach.Finally, the most probable list of candidates is proposed based on work byLebègue et. al. The calculations therefore invite for further investigation ofsome of the best candidates suggested here like PdS2, NbS2, TiS2, TaS2, ZrS2,PdSe2, HfS2 in the 2H structure and CrS2, TaTe2, VTe2, NbS2, CrSe2 in the1T structure in addition to MoS2 and WS2 which are already known.

40
Table 4.3: Relevant energies for analysis of the stabilities of the obtained HERcandidates in the 2H-derived structures. ∆H denotes the calculated standardheat of formation. ∆Hhull denotes the heat of formation of the most stablecompound (i.e. at the convex hull) in the OQMD database[73]. The symbol *in superscript corresponds to the situation where no bulk structure with thecompound composition lies on the convex hull according to the database. Inthat case ∆Hhull is calculated as a linear combination of several structures.δHhull denotes the difference between the two previous columns, i.e. it showshow much the 2D compound lies above or below the convex hull. ∆HExpt
indicates the experimental standard heats of formation as listed in the OQMDdatabase. Lebègue et. al. [74] have analyzed the possibilities for forming 2Dcompounds based on the layered character of the bulk structures and their re-sult is also listed in the Table. ∆H2H/1T is the difference between the energiesin the two (possibly distorted) 2H and 1T structures. Finally P(|∆G| ≤ 0.5 isthe probability that the free energy of hydrogen adsorption lies within 0.5 eVfrom zero
2H-MX2 ∆H ∆Hhull δHhull ∆HExpt. Ref. [74] ∆H2H/1T P(|∆G| ≤ 0.5
RuS2 -0.31 -0.70 0.39 -0.71 No -0.01 1.00NiSe2 -0.21 -0.34 0.13 -0.38 No 0.17 1.00OsS2 0.34 -0.60 0.94 NA No -0.01 1.00ReO2 -0.91 -1.42 0.51 -1.52 No 0.05 1.00TaO2 -2.58 -3.00 0.42 NA No -0.07 1.00PdS2 0.01 -0.31 0.32 -0.28 Yes 0.17 1.00NbS2 -1.21 -1.20 -0.01 NA Yes -0.04 1.00RhS2 -0.11 -0.48 0.37 NA No 0.07 0.99ScS2 -1.46 -1.46 0.00 NA No -0.06 0.99TiS2 -1.23 -1.37 0.14 -1.41 Yes 0.15 0.98TaTe2 -0.32 -0.45 0.13 NA Yes 0.00 0.96TaS2 -1.24 -1.22 -0.02 -1.22 Yes -0.02 0.93IrS2 -0.11 -0.48 0.37 -0.46 No 0.22 0.92RhSe2 -0.17 -0.45 0.28 NA No 0.07 0.92ZrS2 -1.55 -1.73 0.18 -1.99 Yes 0.19 0.91CoS2 -0.33 -0.48 0.15 -0.51 No 0.01 0.90ScSe2 -1.30 -1.25∗ -0.05 NA No -0.01 0.60PdSe2 -0.02 -0.33 0.31 NA Yes 0.22 0.57VS2 -1.16 -1.14 -0.02 NA No -0.02 0.52CrO2 -1.99 -2.15 0.16 -2.01 No 0.03 0.47ScO2 -2.74 -3.17∗ 0.43 NA No 0.05 0.40HfS2 -1.62 -1.80 0.18 NA Yes 0.22 0.26FeS2 -0.54 -0.73 0.19 -0.59 No 0.05 0.06
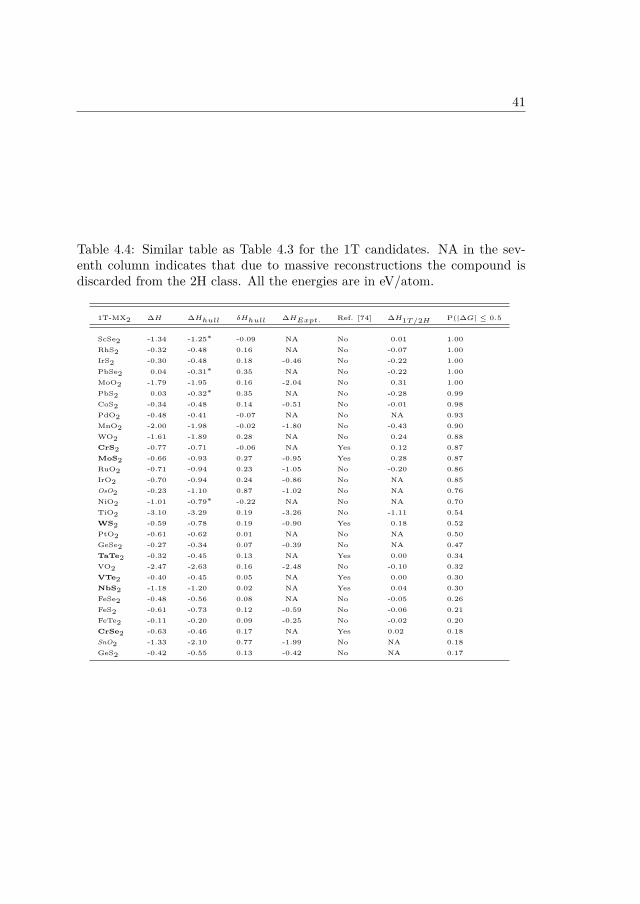
41
Table 4.4: Similar table as Table 4.3 for the 1T candidates. NA in the sev-enth column indicates that due to massive reconstructions the compound isdiscarded from the 2H class. All the energies are in eV/atom.
1T-MX2 ∆H ∆Hhull δHhull ∆HExpt. Ref. [74] ∆H1T/2H P(|∆G| ≤ 0.5
ScSe2 -1.34 -1.25∗ -0.09 NA No 0.01 1.00RhS2 -0.32 -0.48 0.16 NA No -0.07 1.00IrS2 -0.30 -0.48 0.18 -0.46 No -0.22 1.00PbSe2 0.04 -0.31∗ 0.35 NA No -0.22 1.00MoO2 -1.79 -1.95 0.16 -2.04 No 0.31 1.00PbS2 0.03 -0.32∗ 0.35 NA No -0.28 0.99CoS2 -0.34 -0.48 0.14 -0.51 No -0.01 0.98PdO2 -0.48 -0.41 -0.07 NA No NA 0.93MnO2 -2.00 -1.98 -0.02 -1.80 No -0.43 0.90WO2 -1.61 -1.89 0.28 NA No 0.24 0.88CrS2 -0.77 -0.71 -0.06 NA Yes 0.12 0.87MoS2 -0.66 -0.93 0.27 -0.95 Yes 0.28 0.87RuO2 -0.71 -0.94 0.23 -1.05 No -0.20 0.86IrO2 -0.70 -0.94 0.24 -0.86 No NA 0.85OsO2 -0.23 -1.10 0.87 -1.02 No NA 0.76NiO2 -1.01 -0.79∗ -0.22 NA No NA 0.70TiO2 -3.10 -3.29 0.19 -3.26 No -1.11 0.54WS2 -0.59 -0.78 0.19 -0.90 Yes 0.18 0.52PtO2 -0.61 -0.62 0.01 NA No NA 0.50GeSe2 -0.27 -0.34 0.07 -0.39 No NA 0.47TaTe2 -0.32 -0.45 0.13 NA Yes 0.00 0.34VO2 -2.47 -2.63 0.16 -2.48 No -0.10 0.32VTe2 -0.40 -0.45 0.05 NA Yes 0.00 0.30NbS2 -1.18 -1.20 0.02 NA Yes 0.04 0.30FeSe2 -0.48 -0.56 0.08 NA No -0.05 0.26FeS2 -0.61 -0.73 0.12 -0.59 No -0.06 0.21FeTe2 -0.11 -0.20 0.09 -0.25 No -0.02 0.20CrSe2 -0.63 -0.46 0.17 NA Yes 0.02 0.18SnO2 -1.33 -2.10 0.77 -1.99 No NA 0.18GeS2 -0.42 -0.55 0.13 -0.42 No NA 0.17

Chapter 5
Materials for LightAbsorption
5.1 IntroductionIn the last chapter the need to find alternative energy resources has beendiscussed. It was primarily based on the catalytic aspect of the hydrogenproduction. This chapter is mainly focussed on the absorption of sunlightwith semiconducting materials and the absorbed light can be used to producehydrogen by splitting the water. Harvesting the sunlight is crucial becausesolar energy is the most promising alternative resource which can meet thegrowing energy requirement on top of being environmentally benign as com-pared to the fossil fuels. Some of the routes to harness the solar energy aresolar cells, thermoelectrics, photoelectrochemical inter-conversion of chemicalsetc [75, 76, 77, 78, 79, 80, 81, 82, 83]. Photo-electrochemical routes to producechemicals have an added advantage of producing chemicals not just to be usedas fuels but also for other purposes [84].
One of the simplest photoelectrochemical reactions is the splitting of waterinto oxygen and hydrogen. The advent of TiO2 as a material to split thewater into hydrogen and oxygen revolutionized the research in the area ofphotoelectrochemical energy conversion [83]. But the wide bandgap of TiO2limits its performance only to the ultraviolet (UV) region of solar spectrum.Despite continuous effort for more than three decades no abundant and efficientbinary compound has been found to accomplish the task of visible light drivenwater splitting. The limited search space of binary compounds shifted the focus
42

43
to the compounds having more than two elements, especially ternary oxideswhich are quite stable under aqueous condition and irradiation [85, 86, 87].One of the biggest advantages of the ternary/quaternary compounds is thatthere exist lots of possibilities of tuning the bandstructure by choosing differentcombination of elements. A few of the widely studied ternary compounds areoxide perovskites, vanadates, tantalates etc. [26, 88, 85, 86, 87, 89]. Therefore,in order to find multicomponent compounds for efficient light absorption, inthis study a large number of compounds in the Materials Project Database[90, 91] have been explored. Additionally, the possibility of tuning the bandgapby layering of different semiconducting perovskites has also been explored usingBaSnO3 and BaTaO2N as model systems.
5.2 Mechanism of photoelectrochemical watersplitting
The splitting of water using the sunlight is based on the fact that the ab-sorption of sunlight generates electron-hole pairs in the semiconductor. Thegenerated electron-hole pair if have the right energy splits the water to theoxygen and hydrogen. The overall water splitting reaction can be written as[3]:
2H2O→ 2H2 + O2. (5.1)
The oxidation and reduction reactions separately are
2H2O + 4h+ → O2 + 4H+, (5.2)
2H+ + 2e− → H2. (5.3)
By definition of the normal hydrogen electrode (NHE) the free energy of theequation 5.3 is zero. The equation 5.2 however is uphill with a thermody-namic barrier of 1.23 eV [3]. If we ignore the overpotentials involved in thereaction for a moment then the maximum chemical potential of generated elec-tron (equivalently the conduction band minimum (CBM)) should be zero withrespect to the NHE whereas the minimum chemical potential of the generatedhole (equivalently the valence band maximum (VBM)) should be greater than1.23 V. However, due to various overpotentials involved the energy requiredby the electrons and holes to carry out the reaction is higher than the thermo-dynamic voltages mentioned above. A schematic of the photon induced watersplitting reaction described above is shown in the Figure 5.1.

44
As mentioned above the voltage of 1.23 V does not account for any over-potentials or non-equilibrium phenomena. The minimum overpotential asso-ciated with the oxygen evolution reaction (OER ) is ∼0.4 V and with thehydrogen evolution reaction (HER) is 0.1 V [88]. Additionally, the irradia-tion condition disrupts the equlibrium population of the electrons i.e separatequasi-Fermi levels have to be introduced for the conduction and valence bandfor the electron population. This lowers the effective driving force for the re-dox reactions. Typically, the correction due the effect of quasi-Fermi level istaken as ∼0.3 eV [88]. If all the above mentioned effects are added, it turnsout that the semiconductor should have the bandgap of at least 2 eV. In thefollowing sections the results and discussions about the screening of materialsfor the photoelectrochemical water splitting is based on the above mentioneddetails.
Figure 5.1: Schematic of the photon induced water splitting reaction.
5.3 Different methods for the bandgap calcula-tion
As mentioned before, the screening has been performed for the materials avail-able in the Materials Project Database. The Materials Project Database con-tains the subset of compounds present in the experimental database called asThe Inorganic Crystallographic Structures Database (ICSD).[92] The ICSDcontains the compounds which have been synthesized experimentally, there-fore, one can expect that the screened materials from the ICSD will be syn-thesizable under certain experimental conditions. However, the ICSD does

45
not have information about the stability of the materials in aqueous condi-tions which is also explored in this work for the stability of the photocatalystsunder in-situ conditions.
In chapter 1 a few different methods to calculate the bandgap of the semi-conductors were discussed along with their pros and cons. It has also beenpointed out that the calculation of the bandgap in a screening study should bereasonably accurate and at the same time efficient. The previously discussedGLLB-SC functional meets both the criteria in most of the cases. Therefore, inthe present study the bandgap of ∼2400 compounds has been calculated withthe GLLB-SC functional. Additionally, to test the validity of the calculatedbandgap with the GLLB-SC functional the previously discussed hybrid func-tional HSE06 and the many body perturbation theory in different flavors likeG0W0, GW0 and GW have also been used for a few selected materials. Thecomparison is shown in the Figure 5.2. The plot is divided into the high andlow bandgap materials. The figure shows that HSE06 tends to underestimatethe bandgap a bit with respect to GLLB-SC. On the other hand, GW whichis an eigenvalue self-consistent flavor of the GW approximation dovetails verywell with the GLLB-SC bandgaps in the low bandgap region. Additionally,GLLB-SC has a mean absolute error (MAE) of 0.38 eV with respect to GWthus closer to the GW predictions as compared to HSE06 and G0W0 whichhave the MAE of 0.46 and 0.51 eV respectively. GW0, being closest to the GWprediction with a MAE of only 0.29 eV, is highly computationally expensive ascompared to GLLB-SC. Therefore, GLLB-SC being reasonably accurate andan order of magnitude computationally cheaper than the GW approximationserves the purpose of bandgap prediction in a screening study. Table 5.1 sum-marizes the above mentioned mean absolute (signed) error in the bandgap ofthe compounds lying in the low region calculated with different methods withrespect to the other methods.
Table 5.1: Mean absolute (signed) error in eV for the materials in the smallbandgap region in Figure 5.2 using LDA, GLLB-SC, HSE06, G0W0 and GW0and GW
xcref LDA GLLB-SC HSE06 G0W0 GW0 GW
xc
LDA - 1.64 (-1.64) 1.21 (-1.21) 1.08 (-1.08) 1.30 (-1.30) 1.59 (-1.59)GLLB-SC 1.64 (1.64) - 0.61 (0.43) 0.59 (0.56) 0.52 (0.34) 0.38 (0.05)HSE06 1.21 (1.21) 0.61 (-0.43) - 0.25 (0.13) 0.29 (-0.09) 0.46 (-0.38)G0W0 1.08 (1.08) 0.59 (-0.56) 0.25 (-0.13) - 0.22 (-0.22) 0.51 (-0.51)GW0 1.30 (1.30) 0.52 (-0.32) 0.29 (0.09) 0.22 (0.22) - 0.29 (-0.29)GW 1.59 (1.59) 0.38 (-0.05) 0.46 (0.38) 0.51 (0.51) 0.29 (0.29) -

46
Figure 5.2: Bandgaps of few selected materials calculated with different meth-ods versus the GLLB-SC calculated bandgaps. The figure is taken from thePaper-3.
5.4 Candidates for photoelectrochemical watersplitting
A few crucial properties a material should have for an efficient absorption oflight for photoelectrochemical water splitting are:
• Bandgap in the range of visible spectrum because the solar spectrum isdominated by the visible light.
• Proper band edge positions in order to straddle the redox levels of waterin order to have the generated electron and hole at the right chemicalpotential to carry out the reaction.
• Stability of the compound in water because the reaction takes place inan aqueous medium.
Apart from the above mentioned properties, good electron-hole mobility, lowrecombination rates etc. are also required for greater efficiency. However, mod-eling of transport processes, recombination, defect centers etc. is much morecomplicated therefore not considered here for an initial stage of the screening.

47
In order to capture light in the visible range, the bandgap should lie be-tween ∼1.5-3.0 eV. Therefore the calculated bandgap with GLLB-SC shouldlie in this range for efficient absorption of the solar light by the material. Theposition of the band edges in general depend on the surface termination,[93]therefore, in order to calculate the band edges slab calculations have to beperformed. However, the position of the band edges can also be calculatedwith the empirical formula [94]:
EVBM,CBM = (χxAχyBχ
zC)1/(x+y+z) ± Egap/2 + E0, (5.4)
EVBM,CBM is the position of the valence and conduction band edge respectivelyof the compound AxByCz, χ’s are the electronegativity, Egap is the bandgapand E0=-4.5 V is the potential of the NHE with respect to the vacuum leveland the ‘+’, ‘-’ signs correspond to the VBM and CBM respectively. Theabove empirical formula provides band edges which dovetail with the slabmethod and experiments in most of the cases [94] with few exceptions.[95]Finally, the stability in aqueous conditions can be calculated by the Pourbaixdiagram.[96, 97] However, having a very tight threshold of the energy ∆Efor the stability may result in stability diagrams which do not’ agree withexperiments.[96] The reason behind this disagreement is that the stability isnot only the result of thermodynamics but the kinetics as well. On the otherhand, the Pourbaix diagrams do not contain any information about the kineticstherefore a larger energy threshold has to be employed in order to accountfor the phases which are kinetically protected. The sensitivity towards thethreshold can be seen in the Figure 5.3 which is the histogram of the GLLB-SCcalculated bandgap of all the 2400 materials considered in this work. The figureclearly shows that when a very tight threshold of 0 eV is chosen the for stability,most of the compounds turn out to be unstable in water even though many ofthem have been observed to be stable in experimental conditions. However,when a threshold of 1 eV is chosen, a reasonable number of compounds arestable in aqueous conditions. Therefore, a threshold of 1 eV is used to checkfor the stability of the compounds having suitable absorption properties forthe photoelectrochemical water splitting.
The final criteria used for the screening can be summarized as:
• 1.7 ≤ Egap ≤ 3.0 eV (to account the errors in the GLLB-SC calculatedbandgap and overpotentials).
• VBM ≥ 1.6 V and CBM ≤ -0.1 V with respect to the NHE (to accountfor the overpotentials).

48
Figure 5.3: Histogram of the GLLB-SC calculated bandgap of all 2400 mate-rials. Different color denote the stable compounds for different threshold ofenergy chosen for the stability in aqueous condition with pH = 7 and U = 0V with respect to the NHE. The figure is taken from the Paper-3.

49
• ∆E ≤ 1 eV (to account for the metastability and kinetic stabilization)at pH = 7 and -0.4 ≤ V ≤ 2.2 V which is the typical operating voltageof the device.
The materials meeting all the above criteria are selected from the pool of2400 are selected from the pool of ∼2400 and shown in the Figure 5.4. Inthe figure ∆E denotes the degree of stability of the material, the positionof the direct and indirect band edge positions are shown in red and blackrespectively. It is suprising that out of ∼2400 materials only a handful ofmaterials follow all the specified criteria. However, in recent experiments [72]a good control over the stability of semiconductors in water has ben achievedby using protective polymeric layers. Therefore, the size of the materials spacemight be expanded by relaxing the criteria of the stability in aqueous medium.The compounds written in green and underlined in the Figure 5.4 have beenrealized previously for different photoelectrochemical applications, [98, 99, 100,101, 102, 103, 104, 105] and are therefore expected to serve as viable candidatesfor photoelectrochemical water splitting.
5.5 Bandgap engineering of functional perovskitesThe previous section dealt with a specific application of solar light absorptioni.e photoelectrochemical water splitting. However, the other applications likephotovoltaics, transparent conducting oxides etc. require different size of thebandgap.[75, 76] We also saw that different factors limit the suitability ofa material for a given application e.g stability in water, toxicity, cost etc.Therefore, a systematic strategy is required to tune the bandgap of an alreadyexisting material which meets the requirement of toxicity, stability etc.
In this section the possibility of tuning the bandgap by the layering of twoperovskites, namely BaSnO3 and BaTaO2N is explored. The bandgap tuningespecially in perovskites has also been explored in previous works in systemslike SrVO3, SrTiO3 etc.[106, 107, 108, 109, 110, 111] The choice of BaSnO3 andBaTaO2N as a model system here stems from the fact that both the compoundshave been explored recently as light absorbers for the photoelectrochemicalwater splitting applications and have similar lattice constants thus the layeredsystem will not be subjected to a high stress. The protypical structure ofthe layered compound is shown in the Figure 5.5. The α (BaSnO3) and β(BaTaO2N) are stacked along the z-direction while x and y direction have theperiodicity of the cubic perovskite structure and the tuning of the bandgap isexplored by varying the number of layers of α (nα) and the number of layersof β (nβ).

50
Figure 5.4: Materials fulfilling the criteria of the bandgap, band edge positionsand stability in water in neutral pH condition and in the voltage range of -0.4to 2.2 V. ∆E denotes the degree of stability of the material. The positionof the direct and indirect band edge positions are shown in red and blackrespectively. The figure is taken from the Paper-3.

51
Figure 5.5: The structure formed with the layering of BaSnO3 and BaTaO2N.α represents BaSnO3 and β represents BaTaO2N. Stacking is done along thez-direction while x and y direction has the periodicity of the cubic perovskitestructure. The figure is taken from the Paper-4.
The calculation of the bandgap has been performed with the GLLB-SCfunctional and the obtained bandgaps for BaSnO3 and BaTaO2N are 3.33and 1.84 eV which are in good agreement with the measured experimentalbandgap of 3.1 and 1.9 eV for BaSnO3 and BaTaO2N respectively.[112, 113]For comparison, the HSE06 calculated values of the bandgaps are 2.89 and1.71 eV which is slightly lower than the GLLB-SC calculated bandgaps asexpected.
The calculated bandgap of heterostructure for different nα and nβ is shownin the Figure 5.6. The figure shows that the highest bandgap of 2.26 eV isobtained for the αβ stacking and the lowest value of 1.26 eV for α6β6 sequence.The variation of the bandgap by 1 eV for different stacking sequences impliesa high degree of tunability of the bandgap by stacking different layers. Thewide variation in the bandgap can be understood in terms of the quantumconfinement and quantum tunneling effect. The sketch in the Figure 5.7 showshow the local position of the conduction band edge moves downwards uponincreasing the number of α and β layers resulting to decreased confinement.
In order to understand the shift of the local conduction band edge onchanging the number of layers the location and nature of the CBM and VBM

52
Figure 5.6: The bandgap of the heterostructure as a function of nα and nβ .Each rectangle in the plot represents a compound with the stacking sequenceαnβm. The figure is taken from the Paper-4.
states are analyzed. In Figure 5.8 the wavefunctions of the VBM and CBMstates are plotted for αβ and α2β. The figure shows that the VBM mainlyconsists of the N2p orbitals in both αβ and α2β and is located in the β regionof the heterostructure. Additionally, calculations also suggest that the alongwith the character the position of the VBM state also does not change rela-tive to low-lying level for the different structures, therefore, it can be safelyassumed that it is only the nature and position of the conduction band whichis responsible for the bandgap variation in αβn. It can also be seen that in αβthe CBM states are located in the TaON plane and mainly consists of the Tad orbitals. Therefore, in order to see if the same trend is followed in αβn asthe number of β layers is increased the weights of the CBM state is analyzed.The Figure 5.9 shows the planar average (xy plane) of the weights of the CBMstate in the real space. The area of the circle represents the magnitude ofthe weight for the different planes stacked along the z-direction. The boxesrepresent the unit cell and the dotted lines the interface between the α and βlayers. As expected the CBM state is mainly comprised of the Ta d orbitalsand located on the TaON plane. The figure also shows that as the number ofβ layers is increased the CBM states become less confined therefore resulting

53
Figure 5.7: Sketch of the electronic level positions at an interface between αand β for different number of layers. The decreased number of layers resultingto increased confinement moves the local position of the conduction band edgeupwards. The figure is taken from the Paper-4.
Figure 5.8: Wavefunctions of the VBM and CBM states for selected hetere-ostructures. The figure is taken from the Paper-4.

54
to the downshift of the state eventually leading to decrease in the bandgap.
Figure 5.9: Planar average of the weights of the CBM state in the real spaceand the area of the circle represents the magnitude of the average. The unitcell is sketched as rectangles and the dotted lines show the interface betweenthe α and β layers. As expected the CBM state is mainly composed of theTa d orbitals. The bandgaps for different structures is also shown on the top.The figure is taken from the Paper-4.
Until now, the behavior of the heterostructure has only been analyzedwith only one α layer. However, the scenario significantly changes when thenumber of α layers is increased as shown in the Figure 5.10. The figure showsthe similar plot as in Figure 5.9 with the only difference that it has two αlayers as opposed to the Figure 5.9 which has one. As can be seen from thefigure that the CBM is now located mainly in the α part of the heterostructureand primarily consists of the Sn s states. A small weight in the β part of thestructure represents the tunneling effect. However, as expected the tunnelingeffect decreases as the number of β layers is increased and almost diminishesin going from α2β2 to α2β3. Additionally, the weights look similar in allthe structures. Therefore, the weights being similar and the diminishing ofthe tunneling effect results to almost no bandgap change as the number of βlayers ≥ 3.
In the last two kind of heterostructures it is found that keeping the numberof α layers to one has only confinement effect on increasing the number of β

55
Figure 5.10: Similar plot as in 5.9 with the only difference that it has twoα layers as opposed to the Figure 5.9 which has one. The CBM states nowmainly comprise of the Sn s states. The very small weight in the β regionindicates tunneling effect which almost diminished as the number of layers ofβ is increased beyond 2. The figure is taken from the Paper-4.

56
layers and for two α layers it is mainly the tunneling effect responsible for thebandgap variation. Therefore, it can be expected that if the number of α layersis increased while keeping the number of β layers constant the variation of thebandgap would be an interplay between the confinement and the tunnelingeffect. In order to see this effect the Figure 5.11 shows similar plot as Figure5.9 & 5.10 with the difference that the number of β layers is fixed to one whilethe number of α layers is increased. The behavior for αβ and α2β is alreadyshown in the Figure 5.9 & 5.10. In going from α2β to α6β the tunneling as wellas confinement decreses. However, the decreased tunneling has the oppositeeffect as the decreased confinement i.e as the tunneling decreases the bandgapincreases as in Figure 5.10 whereas the decreasing confinement decreases thebandgap as in Figure 5.9. These two competing effects results to a minimain the bandgap for the for a particular number of α layers. The Figure 5.11shows that up to 4 α layers the confinement effect dominates thus leading tothe bandgap reduction in moving from α2β to α4β, however, in going from α4βto α5β the diminishing tunneling dominates over the decreased confinementthus result in an increase in the bandgap. The above analysis for different
Figure 5.11: Similar plot as in 5.9 with the difference that the number of βlayer is fixed to one and the number of α layer is varied. As the number of αlayer is increased beyond α2β the tunneling and confinement effect competewith each other result in decreasing of the bandgap up to α4β and then theincrease of the bandgap. The figure is taken from the Paper-4.

57
heterostructures shows how different effects can be tuned to tailor the bandgapand the variation can be understood with simple physical arguments. Since theanalysis is quite general, therefore, it can be applied to other heterostructuresas well.
5.6 ConclusionIn this chapter a pool of ∼2400 materials is explored which can absorb solarlight for photoelectrochemical water splitting. The criteria imposed for thebandgap, band edge positions and the stability in aqueous solution in neutralpH for a certain potential range gives a handful of candidates which can serveas good photoabsorber for the water splitting reaction. The careful comparisonof the bandgap with different methods involving hybrid functionals and manybody perturbation theory methods assures credibility to the method that isused for the bandgap calculation of a large number of materials. A literaturesurvey for the materials in the list of candidates found which can act as goodphotoabsorbers suggests Ca2PbO4, Cu2PbO2 AgGaO2, AgIn2 and NaBiO3 aspotential candidates.
Additionally, a strategy to engineer the bandgap is also explored via lay-ering of different lattice matched structures. For the model systems exploredi.e BaSnO3 and BaTaO2N the calculations suggest that the variations in thebandgap of the structure can be understood with the simple arguments of con-finement and tunneling effects. This strategy can be applied to other latticematched systems for bandgap engineering.

Chapter 6
Trends in Stability andBandgaps of BinaryCompounds in DifferentCrystal Structures
6.1 IntroductionOne of the simplest class of compounds are binary compounds which onlyhave two constituent elements and different crystal structures e.g wurtzite,zinblende, rocksalt, NiAs and rutile. Often times, same compound exists indifferent crystal structures under different conditions and different structuresmay have significantly different properties. For example, the anatase phase ofTiO2 is found to be photo-catalytically active [83] where as the rutile phaseis not [114] even though both the structures are energetically very close withrutile phase being slightly more stable than anatase phase [115]. Additionally,rocksalt structure of ZnO which is significantly lower in stability as comparedto the native wurtzite structure has been stabilized in MgO matrix [116, 117].Thus, above studies suggest that compounds with different degree of metasta-bility can be synthesized and stabilized at normal conditions which motivatedus to explore different crystal structures of the same compound. In the currentwork we explore four different crystal structures of the binary compounds inAB stoichiometry where A and B are chosen from a set of 44 and 16 elements
58

59
respectively. In the set A, metals which may lead to magnetic compoundshave been ignored due to large degree of uncertainty in their bandgap calcu-lations whereas set B contains non-metals. First principle calculations werecarried out to determine the stability and bandgap of all the compounds. Wesystematically arrange the compounds in tabular form using Pettifor mapswhich involves the ordering of elements based on the their chemical scale fac-tor [118]. We also explore alloys of 32 binary compounds having the band gapin the range of 1.0-3.5 eV inspired by recent experiments on solid solutions forphoto-electrochemical water splitting applications [119, 3]. Clustering analysis[120] of the alloy compounds divides 32 compounds into different groups withcompound belonging to the same group behaving similarly for bandgaps uponalloying, thus giving the freedom too choose one compound over the other fromthe same class if required.
6.2 Results and Discussions
Figure 6.1: Crystal structures explored in the current work. (a) Rocksalt struc-ture (Space Group - Fm3m) (b) Wurtzite structure (Space Group - P63mc)(c) NiAs structure (Space Group - P63/mmc) (d) Zincblende structure (SpaceGroup - F 43m)

60
Table 6.1: Grouping of compounds in Figure 6.3 based on the range of geo-metric mean of chemical scale factor χ
Group Range(√χAχB)
1 0.00 - 1.452 1.45 - 1.753 1.75 - 1.954 1.95 - 2.105 2.10 - 2.306 2.30 - 3.10
Table 6.2: Standard enthalpy ∆H of formation of binary compounds inwurtzite structure having the bandgap in the range of 1.0 - 3.5 eV chosento form ternary wurtzite structures for bandgap engineering of binary com-pounds. ∆HOQMD shows the enthalpy of formation of the with same sto-ichiometry in the minimum energy structure and ∆Hhull represents the en-thalpy above the convex hull calculated from OQMD database. [73] All theenergies are mentioned in eV/atom.
Compound ∆H ∆HOQMD ∆Hhull Compound ∆H ∆HOQMD ∆Hhull
GaN -0.72 -0.59 -0.13 ZnO -1.77 -1.68 -0.09ScN -2.05 -2.04 -0.01 GeO -1.03 -1.94 0.45AgF -0.94 -1.21 0.27 SnO -1.18 -1.58 0.40PbO -0.97 -1.36 0.39 AgI -0.32 -0.38 0.06LaN -1.50 -1.44 -0.06 YN -1.97 -1.81 -0.16InP -0.33 -0.37 0.04 GaP -0.59 -0.55 -0.04AlAs -0.62 -0.49 -0.14 AlP -0.86 -0.76 -0.10AgCl -0.56 -0.67 0.11 AgBr -0.46 -0.55 0.09CdTe -0.37 -0.47 -0.10 AlSb -0.32 -0.17 -0.15CdSe -0.69 -0.64 -0.05 ZnTe -0.56 -0.47 -0.09ZnSe -0.90 -0.72 -0.18 CdS -0.75 -0.81 0.06GeS -0.42 -0.42 0.00 AlI -0.27 -0.05 0.27AlBr -0.58 -0.31 0.34 GeSe -0.33 -0.22 -0.11SnS -0.48 -0.66 0.18 SnSe -0.44 -0.48 0.04PbS -0.41 -0.71 0.30 PbSe -0.38 -0.57 0.19GeTe -0.05 -0.09 0.04 PbTe -0.05 -0.41 0.36
Figure 6.2 shows the ball and stick model of the crystal structures exploredin the present work. Binary compounds crystallizing in NiAs and wurtzitestructures have four atoms in the unit cell with the coordination number ofsix and four respectively, whereas the primitive unit cell of the rocksalt andzincblende structures contains two atoms with a coordination number of sixand four respectively. In the light of recent experiments on the synthesis ofcompounds violating chemical/octet rules under extreme conditions we do notimpose any chemical rules to narrow down the search space in our calculations

61
Figure 6.2: Heatmap of standard enthalpy of formation of compounds in dif-ferent crystal structures. Each square in the heat map represents a compoundswith constituent elements represented by ordinate and abscissa of that square.
62
Figure 6.3: Representation of regions where particular structure has loweststandard enthalpy of formation as compared to other crystal structures. Thecompounds are represented in the same way as in Figure 6.2. Based on geo-metric mean of the chemical scale factor χ [118] the plot is divided into sixdifferent regions where the value of geometric mean increases on moving from‘1’ to ‘6’.

63
Figure 6.4: Band gap of compounds in different crystal structures. The whiteregion in the plots shows the metallic compound i.e. with zero bandgap

64
Figure 6.5: Dendrogram based on difference of average bandgap of the con-stituent compounds in the alloy and calculated bandgap of the alloy. Theconstituent compounds have been selected from the pool of compounds withwurtzite structure and having the bandgap in the range of 1.0-3.5 eV.

65
Figure 6.6: (a) Calculated bandgap of the alloys. The constituent compoundshave wurtzite structures with the bandgap lying in the range of 1.0-3.5 eV.White spaces show alloys with zero bandgap. (b) Shows enthalpy of mixing ofthe alloy compounds with respect to constituent compounds.

66
[121]. In our work we calculate the stability of compounds with respect to thestandard reference states only. Since our interest lies in comparing the trendsin thermodynamic and electronic properties of the compounds in differentcrystal structures with 1:1 stoichimetry, we do not consider the compoundswith the stoichiometry different from 1:1. In the calculations we ignore fewmetals leading to magnetic structures since a reliable approach to calculatethe bandgap of magnetic semiconductors with the GLLB-SC functional hasnot been developed yet.
Figure 6.2 shows the heatmaps of the standard heat of formation of all thecompounds in which the elements are arranged as per the chemical scale χproposed by Pettifor [118]. As can be seen from the figure that the heat of for-mation in all four crystal structures follow the similar pattern which indicatesthat if the compound is stable with respect to the standard states in one crystalstructure it will be stable in the other crystal structure as well. But negativeformation energy does not guarantees that the structure can be stabilized inthat phase which is inhibited by the existence of other competing phases inthe ambient environment. But the control over the ambient condition for thegrowth can be achieved by for example temperature, pressure, surfactants anddoping [122, 123, 124]. Pressure as one of the control mechanism in the growthprocess gives a tool to stabilize structure with different volumes. For example,if the most stable structure has a lower volume (e.g. rocksalt or NiAs) thenapplying tensile stress may favor the higher volume phase (e.g. wurtzite orzincblende) whereas a lower volume phase will be favored under compressivestress if the most stable structure has a higher volume. The above process canbe realized in experiments [116, 125] with the growth on a substrate with differ-ent lattice mismatch thus providing a way to apply the compressive and tensilestress. Thus, above strategies to manipulate crystal structures suggest thatthe compounds can possibily be synthesized and stabilized in different crystalstructures with different volumes. Therefore, in the current work we choosecrystal structures spanning a wide range of volumes with the zincblende andwurzite having higher volumes due to low coordination number as opposed tothe rocksalt and NiAs structures which have larger coordination number thuslower volumes.
In addition to the standard heat of formation as shown in Figure 6.2 onemight also be interested in region where a particular crystal structure has thelowest enthalpy of formation as compared to other crystal structures. Fig-ure 6.3 shows the minimum energy crystal structure for different compounds.As can be inferred from the figure, there are very few isolated regions for agiven crystal structure. Hence, compounds when arranged in Pettifor mapsform clusters having the same most stable crystal structure. Therefore, we

67
group the compounds in Figure 6.3 in six different groups based on the geo-metric mean of the chemical scale factor χ [118]. The range of geometric meanof groups is shown in Table 6.1.
The grouping in Figure 6.3 shows an apparent correlation between thecrystal structure and the geometic mean of the chemical scale factor of thecompounds. For very small values of geometric mean (Group 1) the compoundprefers to have the low volume structure i.e. rocksalt or NiAs whereas at largervalues(Group 6) the compound prefer to have more open structure i.e. wurtziteor zincblende. Few green parts in the Group 1 for compounds like LiF, LiClis the artifact of the calculation since these compounds are known to have therocksalt structure. On the other hand,the difference in energies of the wurtziteand rocksalt structure of these compounds as per our calculations is of theorder of 0.05 eV which is small and can be safely ignored. The region of extremevalues of the geometric mean in Figure 6.3 i.e. Group 1 and 6 are the regionof extreme ionicity of the compounds with very small values indicating largeionicity (more closed structures) whereas very large values showing greatercovalent character (more open structures) [118]. On the other hand, the regionof moderate ionicity i.e. Group 2-5 is not dominated by one crystal structure.Group 2 has large fraction of WZ and NiAs structures, Group 3 ZB structure,Group 4 RS structure and Group 5 WZ and RS structures.
In Figure 6.4 we show the bandgap of all the compounds. The white regionsin the heatmap show the zero bandgap materials by which we can see that outof 704 materials in each group very few turn out be semiconducting with thesmallest number of semiconductors in the NiAs structure and the largest num-ber in the ZB structure. The plot also shows that despite having the similarheat of formation, the wurtzite and zincblende have quite dissimilar trend inbandgaps(especially indirect gap) which is a well known phenomenon [126].Hence, in cases where the WZ and ZB have significantly different bandgaps,stabilizing the structure with the required bandgap can be achieved efficientlydue to the similar heats of formation.
In addition to modifying the structures to tune the bandgap, bandgapengineering can also be achieved by making solid solutions of different semi-conductors. The same has been realized in the experiments in recent workscarried out in Domen’s group [119, 3] in which the mixture of GaN and ZnOhas the bandgap of ∼2.5 eV as opposed to the bandgap of ∼3.4 eV of the con-stituent compounds GaN and ZnO. Thus, these experimental results suggestthat the alloying can be used as a method for the bandgap tailoring. Hence,guided by the above experiments we also explore the alloys of binary com-pounds in the wurtzite structures having the bandgap in the range of 1.0 -3.5 eV. The stability of the alloy with respect to the constituent compounds

68
mixed in an equimolar ratio is described by the enthalpy of mixing as
∆HABCDmix = EABCDtot − EABtot − ECDtot (6.1)
where Eitot is the total energy of the compound ‘i’. Table 6.2 shows the heat offormation of the compounds in the wurtzite structure having the bandgap inthe range of 1.0-3.5 eV, ∆H is the heat of formation calculated in the presentwork, ∆HOQMD is the heat of formation as given in the OQMD (The OpenQuantum Materials Database) database [73] for the structure which has theminimum energy at 1:1 stochiometry and ∆Hhull is the relative energy of thecompound in the current work with respect to the convex hull as given in theOQMD database. Since the enthalpies in OQMD database are based on DFTreference energies whereas we use corrected reference energies as proposed byStevanovic et al. [25], therefore the difference 0.15 eV/atom or less in oursand OQMD calculations will lie within in the error bar due to the differentmethods used for the calculation of the reference energies. Compounds likeAlI, AlBr which are significantly above the convex hull and have no structurewhich is stable at 1:1 stochiometry and also expected to be unstable basedon the valence rule can be ruled out as they may not be possibly synthesizedunder normal/moderate conditions. On the other hand, compounds like GeO,AgF and PbO have different structures other than the wurtzite which arestable and lie below the convex hull. Even though energy of the wurtzitestructure of these compounds lies above the convex hull by ∼0.35 eV/atom(Table 6.1), its likely that they can be stabilized in the wurtzite form sincethey exist in structures which are stable at the same stoichiometry. So, theirsynthesis/stabilization might be possible under moderate conditions.
In order to study the trends we made a crude approximation of the solidsolution with a unit cell of four atoms due large computational time requiredfor a larger unit cell. Based on the previous works, we believe the that theapproximations made will not change the results drastically [127].
The similarity between the compounds forming the mixture in terms of thedeviation of the bandgap of the mixture from the average of the bandgaps ofconstituent compounds has been assessed by the so-called dendrogram plot.Dendrogram is a tree diagram used in the generating hierarchical structuresamong the elements representing the data [120]. In Figure 6.5, we show thedendrogram plot with euclidean metric of the difference of the average bandgapof the constituent compounds and the calculated bandgap of the alloy. Theclustering in a dendrogram is based on the distance measure (dAB) of two

69
components given as
dAB =∑CD
(EgapAB + EgapCD
2 − EgapABCD
)2(6.2)
In the equation above AB corresponds to the compound for which the dis-tance measure has to be calculated and the summation index CD correspondsto other compounds in the set of binary compounds which are combined withAB to form alloys. Based on the distance measure the compounds with cutoffdistance measures will be clustered together. The clustering of compoundslike ZnO and GaN, CdS and CdSe, ZnTe and CdTe, for which the thermo-dynamic/light absorption properties of the solid solutions have already beenexplored experimentally complements our study [119, 128, 129]. As one wouldexpect on the chemical grounds, clustering naturally leads to the grouping ofthe chalcogenides of the Group 12 elements, chalcogenides of Group 14 ele-ments, silver halides and the compounds of the trivalent ions with the Group15 elements. As can be seen from the values close to zero along the blockdiagonal in the dendrogram plot that in most of the cases when the mixturesare formed from compounds in the same group, the resultant bandgaps differonly by ∼0.5 eV from the average of the bandgaps. On the other hand, mix-ing of the compounds from different groups leads to a significant reduction ofthe bandgap with respect to the average of the bandgaps in most of the casesas shown with the red blocks. The detailed description of the above trendwould require electronic structure analysis of every mixture which is beyondthe scope of the current work.
Figure 6.6(a) and (b) show the bandgap and enthalpy of mixing ∆Hmix ofthe mixture as given by equation (6.1). Since, The positive correlation betweenthe bandgap and the stability [130] renders some of the mixtures metallic dueto the large positive heat of mixing. As one would anticipate that the mixingof compounds with very different lattice constants will be energetically un-favorable, the red regions in Figure 6.6(b) manifest the expected trend. Forexample, ZnO, GaN, ScN have similar volumes, so their mixture with the tel-lurides, selenides or compounds of lead which have much larger volumes resultto significantly positive heat of mixing which in turn leads to zero bandgapof those mixtures. Thus, exploring mixtures of compounds with similar lat-tice constant will have a higher probability of giving compounds with a finitebandgap.
Combiningly, Figure 6.5 and 6.6 show that the bandgap of most of thestable mixtures along the block diagonal 1 (bottom left to top right) havebandgap of ∼2.5 eV except the block containing the chalcogenides of zinc and

70
cadmium which has lower bandgap due to low bandgaps of the constituentcompounds. On the other hand, stable compounds along the block diagonal2 (top left to bottom right) are low bandgap mixtures with the bandgap ∼1.5eV. Thus, the dichotomy in the bandgaps allows us to look in a definite regionof the materials space for the required range of bandgaps suitable for theapplication in hand.
6.3 ConclusionIn the current work we have carried out a systematic analysis of the com-pounds in different crystal structures spanning a wide range of volumes andhaving the AB stoichiometry. The correlation between the crystal structureand the chemical scale factor χ gives a rationale to understand the effect ofelectronegativity on the crystal structure of the compounds. In addition tothe binary compounds, detailed analysis of their alloys gives a rationale forthe bandgap engineering via solid solutions of the semiconductors. The anal-ysis carried out in the current work for the binary semiconductors can also begeneralized to the systems containing more than two elements thus providingan elegant route to tailor the bandgap as required by the application in handlike photo-electrochemical water splitting, solar cells.

Chapter 7
Final Remarks
In the previous chapters, challenges involved in the process of materials designhave been looked at. The challenges are not only faced by experimentalists butby theoreticians as well. The bottlenecks in experiments come from the lim-ited resources and time whereas the computational scientists having access topowerful computers are limited by the accuracy of the computational methodsand approximations made to mimic the experiments. But, these limitationsin no way stop us to move forward. Experimental tools are becoming increas-ingly advanced whereas day to day developments in theory and algorithms aremaking computations more and more reliable.
In this work, an attempt to solve some of the materials design problemwith computations and some strategies to face future challenges for the sameare looked at. The assessment of recently developed mBEEF functional for theprediction of the heats of formation is an example for theoretical developmentswhereas the screening of materials for light absorption and hydrogen evolutionreaction is an attempt to solve materials design problem regarding energy.Finally, exploring one of the many methods for the bandgap engineering showshow we can expand the materials space by using the already existing materialsfor different applications.
In all the above problems, we got help from the available experimentaldata whether it was heats of formation, activity for hydrogen evolution orthe photoelectrochemical water splitting. This implies that no matter howfast, cheap and efficient computer simulations become, at the end of the daythe calculated numbers have to agree with experiments. As Feynman oncesaid: “It doesn’t matter how beautiful your theory is, it doesn’t matter howsmart you are. If it doesn’t agree with experiment, it’s wrong.” Therefore,
71

72
the experiments and theory have to go hand in hand to solve materials designproblem in a reliable and efficient way.
One of recent examples of the synergistic effort of the experiments andtheory is machine learning. People are trying to solve materials design prob-lems using machine learning. However, the reliability of machine learning fordifferent fields is different. For example, machine learning in astronomy is rea-sonably reliable because the experimental data is provided only by a very fewtelescopes which are very well tested. On the other hand, machine learningin materials design suffers with a lot of ambiguity, for example, variations inpseudopotentials, different electronic structure codes, plethora of unreliableexperimental data etc. Therefore, I personally feel that the dream of solvingmaterials design problem with machine learning is very far from realizable inthe near future and we still have to resort to more fundamental science thanblindly using computers.

Bibliography
[1] N. S. Lewis and D. G. Nocera. PNAS, 103:15729–15735, 2006.
[2] N. Armaroli and V. Balzani. Angew. Chem. Int. Ed., 46:52–66, 2007.
[3] K. Maeda and K. Domen. Chem. Mater., 22:612–623, 2010.
[4] P. C. K. Vesborg and T. F. Jaramillo. RSC Adv., 2:7933–7947, 2012.
[5] E. Schrödinger. Annalen der Physik, 384:361–376, 1926.
[6] J. Kohanoff. Electronic Structure Calculations for Solids and Molecules.Cambridge University Press, 2006. Cambridge Books Online.
[7] P. Hohenberg and W. Kohn. Phys. Rev., 136:B864, 1964.
[8] W. Kohn and L. J. Sham. Phys. Rev., 140:A1133, 1965.
[9] J. P. Perdew, K. Burke, and M. Ernzerhof. Phys. Rev. Lett., 77:3865,1996.
[10] J. Heyd, G. E. Scuseria, and M. Ernzerhof. J. Chem. Phys., 118:8207,2003.
[11] A. V. Krukau, O. A. Vydrov, A. F. Izmaylov, and G. E. Scuseria. J.Chem. Phys., 125:224106, 2006.
[12] A. J. Cohen, P. Mori-Sánchez, andW. Yang. Science, 321(5890):792–794,2008.
[13] P. Mori-Sánchez, A. J. Cohen, and W. Yang. Phys. Rev. Lett.,100(14):146401, 2008.
[14] O. Gritsenko, R. van Leeuwen, E. van Lenthe, and E. J. Baerends. Phys.Rev. A, 51:1944, 1995.
73

74
[15] M. Kuisma, J. Ojanen, J. Enkovaara, and T. T. Rantala. Phys. Rev. B,82:115106, 2010.
[16] J. P. Perdew, M. Ernzerhof, and K. Burke. J. Chem. Phys., 105:9982,1996.
[17] C. Adamo and V. Barone. J. Chem. Phys., 110:6158, 1999.
[18] J. Paer, M. Marsman, K. Hummer, G. Kresse, and I. C. Gerber. J.Chem. Phys., 124:154709, 2006.
[19] J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak,L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H.Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg,O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A.Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin,M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen,J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen, andK. W. Jacobsen. Journal of Physics: Condensed Matter, 22(25):253202,2010.
[20] J. J. Mortensen, L. B. Hansen, and K. W. Jacobsen. Phys. Rev. B,71:035109, 2005.
[21] D. Vanderbilt. Phys. Rev. B, 41:7892–7895, 1990.
[22] P. E. Blöchl. Phys. Rev. B, 50:17953–17979, 1994.
[23] C. Rostgaard. arXiv preprint arXiv:0910.1921v2 , 2009.
[24] M. Fuchs, J. L. F. Da Silva, C. Stampfl, J. Neugebauer, and M. Scheffler.Phys. Rev. B, 65:245212, 2002.
[25] V. Stevanović, S. Lany, X. Zhang, and A. Zunger. Phys. Rev. B,85:115104, 2012.
[26] I. E. Castelli, T. Olsen, S. Datta, D. D. Landis, S. Dahl, K. S. Thygesen,and K. W. Jacobsen. Energy Environ. Sci., 5:5814–5819, 2012.
[27] X. Zhang, V. Stevanović, M. d’Avezac, S. Lany, and A. Zunger. Phys.Rev. B, 86:014109, 2012.
[28] S. Lany. Phys. Rev. B, 78:245207, 2008.

75
[29] S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys, andA. P. Sutton. Phys. Rev. B, 57:1505, 1998.
[30] B. Hammer, L. B. Hansen, and J. K. Nørskov. Phys. Rev. B, 59:7413,1999.
[31] J. M. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria. Phys.Rev. Lett., 91(14):146401, 2003.
[32] R. Peverati and D. G. Truhlar. Phil. Trans. R. Soc. A, 372:2011, 2014.
[33] L. A. Constantin, E. Fabiano, and F. Della Sala. . Chem. TheoryComput., 9(5):2256–2263, 2013.
[34] J. Sun, J. P. Perdew, and A. Ruzsinszky. Proceedings of the NationalAcademy of Sciences, 112(3):685–689, 2015.
[35] F. Della Sala, E. Fabiano, and L. A. Constantin. Phys. Rev. B,91:035126, 2015.
[36] J. Wellendorff, K. T. Lundgaard, K. W. Jacobsen, and T. Bligaard. J.Chem. Phys., 140:144107, 2014.
[37] Standard thermodynamic properties of chemical substances, 2000. http://www.update.uu.se/~jolkkonen/pdf/CRC_TD.pdf.
[38] W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling.Numerical Recipes in C. Cambridge University Press, 1988.
[39] K. S. Brown S. L. Frederiksen, K. W. Jacobsen and J. P. Sethna. Phys.Rev. Lett., 93(16):165501, 2004.
[40] M. S. Dressselhaus and I. L. Thomas. Nature, 414:332–337, 2001.
[41] G. W. Crabtree, M. S. Dresselhaus, and M. V. Buchanan. Phys. Today,57:39–44, 2004.
[42] J. Greeley, T. F. Jaramillo, J. L. Bonde, I. Chorkendorff, and J. K.Nørskov. Nat. Mater., 5:909–913, 2006.
[43] Y. Okamoto, S. Ida, J. Hyodo, H. Hagiwara, and T. J. Ishihara. J. Am.Chem. Soc., 133:18034–18037, 2011.
[44] S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L-Guetaz,B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave, andV. Artero. Nat. Mater., 11:802–807, 2012.

76
[45] P. Liu and J. A. Rodriguez. J. Am. Chem. Soc., 127:14871–14878, 2005.
[46] E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout,N. S. Lewis, and R. E. Schaak. J. Am. Chem. Soc., 135:9267–9270, 2013.
[47] L. Feng J. Rossmeisl M. H. Hansen, L. A. Stern and X. Hu. Phys. Chem.Chem. Phys., 17:10823–10829, 2015.
[48] D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita,M. Chen, T. Asefa, V. B. Shenoy, G. Eda, and M. Chhowalla . Nat.Mater., 12:850–855, 2013.
[49] M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li, and S. Jin.J. Am. Chem. Soc., 135:10274–10277, 2013.
[50] D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy,G. Eda, and M. Chhowalla. Nano Letters, 13:6222–6227, 2013.
[51] H. Wang, Z. Lu, D. Kong, J. Sun, T. M. Hymel, and Y. Cui. ACS Nano,8:4940–4947, 2014.
[52] T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch, andI. Chorkendorff. Science, 317:100–102, 2007.
[53] M. V. Bollinger, K. W. Jacobsen, and J. K. Nørskov. Phys. Rev. B,67:085410, 2003.
[54] B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen,S. Horsch, I. Chorkendorff, and J. K. Nørskov. J. Am. Chem. Soc.,127:5308–5309, 2005.
[55] A. N. Enyashin, L. Yadgarov, L. Houben, I. Popov, M. Weidenbach,R. Tenne, M. Bar-Sadan, and G. Seifert. J. Phys. Chem. C, 115:24586–24591, 2011.
[56] K. Rossnage. J. Phys.: Condens. Matter, 23:213001, 2011.
[57] K. Dolui and S. Sanvito. arXiv preprint arXiv:1310.1866 , 2013.
[58] L. Fu X. Qian, J. Liu and J. Li. Science, 346:1344–1347, 2014.
[59] M. Kan, J. Y. Wang, X. W. Li, S. H. Zhang, Y. W. Li, Y. Kawazoe,Q. Sun, and P. Jena. J. Phys. Chem. C, 118:1515–1522, 2014.

77
[60] S. Tongay, H. Sahin, C. Ko, A. Luce, W. Fan, K. Liu, J. Zhou, Y.-S.Huang, C.-H. Ho, J. Yan, D. F. Ogletree, S. Aloni, J. Ji, S. Li, J. Li,F. M. Peeters, and J. Wu. Nat. Commun., 5:3252, 2014.
[61] H. T. Stokes and D. M. Hatch. J. Appl. Cryst., 38:237–238, 2005.
[62] C. Ataca, H. Şahin, and S. Ciraci. J. Phys. Chem. C, 116:8983–8999,2012.
[63] S. Trasatti. J. Electroanal. Chem., 39:163, 1972.
[64] J. O’M. Bockris, A. K. N. Reddy, and M. Gamboa-Aldeco. ModernElectrochemistry 2A 2nd ed. Kluwer Academic/Plenum Publishers: NewYork, 1998.
[65] C. Tsai, F. A.-Pedersen, and J. K. Nørskov. Nano Letters, 14:1381–1387,2014.
[66] C. Tsai, J. K. Nørskov K. Chan, and F. A.-Pedersen. J. Phys. Chem.Lett., 5:3884–3889, 2014.
[67] K. K. Irikura. J. Phys. Chem. Ref. Data, 36:389, 2007.
[68]
[69] L. C. Seitz, Z. Chen, A. J. Forman, B. A. Pinaud, J. D. Benck, and T. F.Jaramillo. ChemSusChem, 7:1372, 2014.
[70] J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Lan-dis, J. K. Nørskov, T. Bligaard, and K. W. Jacobsen. Phys. Rev. B,85:235149, 2012.
[71] A. J. Medford, J. Wellendorff, A. Vojvodic, F. Studt, F. A.-Pedersen,K. W. Jacobsen, T. Bligaard, and J. K. Nørkov. Science, 345:197, 2014.
[72] S. Mubeen, J. Lee, N. Singh, M. Moskovitsb, and E. W. McFarland.Energy Environ. Sci., 6:1633, 2013.
[73] J. E. Saal, S. Kirklin, M. Aykol, B. Meredig, and C. Wolverton. JOM,65:1501–1509, 2013.
[74] S. Lebègue, T. Björkman, M. Klintenberg, R. M. Nieminen, andO. Eriksson. Phys. Rev. X, 3:031002, 2013.

78
[75] L. Vayssierres. On Solar Hydrogen and Nanotechnology. John Wileyand Sons (Asia) Pvt. Ltd, 2009.
[76] G.A. Chamberlain. Solar Cells, 8(1):47 – 83, 1983.
[77] M. Grätzel. J. Photochemistry and Photobiology C: PhotochemistryReviews, 4(2):145 – 153, 2003.
[78] M. Ni, M. K.H. Leung, D. Y.C. Leung, and K. Sumathy. Renewable andSustainable Energy Reviews, 11(3):401 – 425, 2007.
[79] A. Kudo and Y. Miseki. Chem. Soc. Rev., 38:253–278, 2009.
[80] K. Sayama Z. Zou, J. Ye and H. Arakawa. Nature, 414:625–627, 2001.
[81] L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend,and J. D. MacKenzie. Science, 293(5532):1119–1122, 2001.
[82] A. Hagfeldt and M. Grätzel. Accounts of Chemical Research, 33(5):269–277, 2000.
[83] A. Fujishima and K. Honda. Nature, 238:37–38, 1972.
[84] B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum, and C. P.Kubiak. Annual Review of Physical Chemistry, 63(1):541–569, 2012.
[85] C. S. Enache, D. Lloyd, M. R. Damen, J. Schoonman, and R. van deKrol. The Journal of Physical Chemistry C, 113(44):19351–19360, 2009.
[86] Y. Liang, T. Tsubota, L. P. A. Mooij, and R. van de Krol. The Journalof Physical Chemistry C, 115(35):17594–17598, 2011.
[87] F. F. Abdi and R. van de Krol. The Journal of Physical Chemistry C,116(17):9398–9404, 2012.
[88] K. S. Thygesen S. Dahl I. Chorkendorff T. F. Jaramillo I. E. Castelli,D. D. Landis and K. W. Jacobsen. Energy Environ. Sci., 5:9034–9043,2012.
[89] M. Oshikiri, M. Boero, J. Ye, Z. Zou, and G. Kido. J. Chem. Phys.,117(15):7313–7318, 2002.
[90] G. Hautier W. Chen W. D. Richards S. Dacek S. Cholia D. Gunter D.Skinner G. Ceder A. Jain, S. P. Ong and K. A. Persson. APL Materials,1:011002, 2013.

79
[91] M. Pandey H. Li K. S. Thygesen B. Seger A. Jain K. A. Persson G.Ceder K. W. Jacobsen I. E. Castelli, F. Hüser. Adv. Energy Mater.,5:1400915, 2015.
[92] A. Belsky, M. Hellenbrandt, V. L. Karen, and P. Luksch. Acta Cryst.,B58:364–369, 2002.
[93] D. S. Ginley W. Tumas V. Stevanovic, S. Lany and A. Zunger. Phys.Chem. Chem. Phys., 16:3706, 2014.
[94] M. A. Butler and D. S. Ginley. J. Electrochem. Soc., 125:228–232, 1978.
[95] Filip A. Rasmussen and Kristian S. Thygesen. J. Phys. Chem. C,119(23):13169–13183, 2015.
[96] K. S. Thygesen I. E. Castelli and K. W. Jacobsen. Top. Catal., 57(1–4):265–272, 2014.
[97] P. Lazic K. A. Persson, B. Waldwick and G. Ceder. Phys. Rev. B,85:235438, 2012.
[98] A. E. Lavat R. P. Diez, E. J. Baran and M. C. Grasselli. J. Phys. Chem.Solids, 56:135, 1995.
[99] K. Rubesova and D. Sykorova. J. Sol-Gel Sci. Technol., 49:228, 2009.
[100] C. L. Teske H. Szillat. Z. Anorg. Allge. Chem., 620:1307, 1994.
[101] R. Nagarajan H. Yanagi, J. Tate and A. W. Sleight. Solid StateCommun., 122:295, 2002.
[102] H. Irie Y. Maruyama and K. Hashimoto. J. Phys. Chem. B, 110:23274,2006.
[103] D. Chen Z. G. Zou S. X. Ouyang, N. Kikugawa and J. H. Ye. J. Phys.Chem. C, 113:1560, 2009.
[104] M. Katagiri T. Kako, Z. G. Zou and J. H. Ye. Chem. Mater., 19:198,2007.
[105] Q. Z. Liu Y. F. Zhu J. J. Liu, S. F. Chen and J. F. Zhang. Chem. Phys.Lett., 572:101, 2013.
[106] H. Kumigashira S. Okamoto S. Aizaki A. Fujimori K. Yoshimatsu,T. Okabe and M. Oshima. Phys. Rev. Lett., 104:147601, 2010.

80
[107] M. Kobayashi K. Horiba T. Yoshida A. Fujimori M. Oshima K. Yoshi-matsu, E. Sakai and H. Kumigashira. Phys. Rev. B, 88:115308, 2013.
[108] H. Kumigashira T. Yoshida A. Fujimori K. Yoshimatsu, K. Horiba andM. Oshima. Science, 333:319, 2011.
[109] S. Kumar S. J. May J. A. Borchers B. B. Maranwille J. Zarestky S. G.E. te Velthuis J. van den Brink T. S. Santos, B. J. Kirby and A. Bhat-tacharya. Phys. Rev. Lett., 107:167202, 2011.
[110] C. R. Freeze C. A. Jackson, J. Y. Zhang and S. Stemmer. Nat. Commun.,5:1, 2014.
[111] B. Ehrlich C. Grote and R. F. Berger. Phys. Rev. B, 90:205202, 2014.
[112] H. W. Eng H. Mizoguchi and P. M. Woodward. Inorg. Chem., 43:1667,2004.
[113] H. Hara J. N. Kondo D. Yamasita, T. Takata and K. Domen. Solid StateIonics, 172:591, 2004.
[114] T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, S. Sutter, andM. Batzill. Scientific Reports, 4:4043, 2014.
[115] M. Lazzeri, A. Vittadini, and A. Selloni. Phys. Rev. B, 63:155409, 2001.
[116] S. W. H. Eijt, J. de Roode, H. Schut, B. J. Kooi, and J. Th. M. DeHosson. App. Phys. Lett., 91:201906, 2007.
[117] A. N. Baranov, O. O. Kurakevych, V. A. Tafeenko, P. S. Sokolov, G. N.Panin, and V. L. Solozhenko. Jour. App. Phys., 107:073519, 2010.
[118] D. G. Pettifor. Jour. Phys. C:Solid State Phys., 19:285–313, 1986.
[119] K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi, andK. Domen. Jour. Am. Chem. Soc., 127:8286–8287, 2005.
[120] I. E. Castelli and K. W. Jacobsen. Modelling Simul. Mater. Sci. Eng.,22:055007, 2014.
[121] W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel,A. O. Lyakhov, E. Stavrou, M. Somayazulu, V. B. Prakapenka, andZ. Konopkova. Science, 342:1502–1505, 2013.
[122] M. Pandey and R. G. S. Pala. Jour. Chem. Phys., 136:044703, 2012.

81
[123] M. Pandey and R. G. S. Pala. Jour. Phys. Chem. C, 117:7643–7647,2013.
[124] P. Gangwar, M. Pandey S. Sivakumar, R. G. S. Pala, andG. Parthasarathy. Cryst. Growth Des., 13:2344–2349, 2013.
[125] H. L. Meyerheim, C. Tusche, A. Ernst, S. Ostanin, I. V. Maznichenko,K. Mohseni, N. Jedrecy, J. Zegenhagen, J. Roy, I. Mertig, andJ. Kirschner. Phys. Rev. Lett., 102:156102, 2009.
[126] C-Y. Yeh, S.-H. Wei, and A. Zunger. Phys. Rev. B, 50(8):2715(R), 1994.
[127] J. N. Hart and N. L. Allan. Adv. Mater., 25:2989–2993, 2013.
[128] V.R. Sidorko and L.V. Goncharuk. J. Alloys Compd., 228:13–15, 1995.
[129] M. A. Hossain, J. R. Jennings, N. Mathews, and Q. Wang. Phys. Chem.Chem. Phys., 14:7154–7161, 2012.
[130] J. Portier, H. S. Hilal, I. Saadeddin, S. J. Hwang, M. A. Subramanian,and G. Campet. Prog. Solid State Ch., 32:207–217, 2004.

Papers
Paper IHeats of formation of solids with error estimation: The mBEEFfunctional with and without fitted reference energiesM. Pandey and K. W. Jacobsen Physical Review B 91 (23), 235201 (2015)
82

PHYSICAL REVIEW B 91, 235201 (2015)
Heats of formation of solids with error estimation: The mBEEF functional with andwithout fitted reference energies
Mohnish Pandey and Karsten W. Jacobsen*
Center for Atomic-scale Materials Design, Department of Physics, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark(Received 30 January 2015; revised manuscript received 12 May 2015; published 3 June 2015)
The need for prediction of accurate electronic binding energies has led to the development of different schemesfor combining density functional calculations, typically at the level of the generalized gradient approximation(GGA), with experimental information. We analyze one such scheme by Stevanovic et al. [Phys. Rev. B85, 115104 (2012)] for predictions of compound enthalpies of formation using fitted elemental-phase referenceenergies. We show that different versions of GGA with or without +U and a meta-GGA (TPSS) lead to comparableaccuracy after fitting the reference energies. Our results also show that the recently developed mBEEF, a Bayesianerror estimation functional, gives comparable accuracy with the other functionals even without the fitting. ThemBEEF functional furthermore supplies an ensemble estimate of the prediction errors in reasonable agreementwith the actual errors. We also show that using the fitting scheme on the mBEEF ensemble leads to improvedaccuracy including realistic error estimation.
DOI: 10.1103/PhysRevB.91.235201 PACS number(s): 71.15.Mb, 31.15.eg, 71.15.Nc
I. INTRODUCTION
In the past two decades, Kohn-Sham density functionaltheory (KS-DFT) based electronic structure calculations [1,2]have greatly enhanced our understanding of the properties ofmaterials. The drastic reduction in the number of degrees offreedom in the electronic structure problem within the KS-DFTframework makes it an efficient tool for quantum mechanicaldescription of materials. The key ingredient in the KS-DFTis an energy functional which depends on the ground-stateelectronic density and the accuracy of calculations dependson the quality of the approximations applied to the functional.Efficient and realistic description of materials requires cal-culations which are not too computationally expensive andreasonably accurate. In the generalized gradient approximationframework (GGA) the PBE functional [3] has been widelyused and has a reasonable trade-off between accuracy andefficiency. Despite being remarkably successful in the past, ithas its limitations as well. For example, the heats of formationpredicted by the PBE functional deviate from experiments by∼0.25 eV/atom [4] which makes it difficult to predict thestabilities of compounds in many cases. It severely plaguesthe process of searching for new materials for differentapplications where stability of the compounds is one of themain criteria [5–7].
Recently, Stevanovic et al. proposed a scheme known as fit-ted elemental reference phase energies (FERE) to improve theprediction of the heats of formation of semiconductors [4,8].Their scheme is based on the idea of using the reference phaseenergies as parameters and calculating these parameters byminimizing the root mean square (rms) error between thecalculated and experimental heats of formation. The schemeuses a mixture of the PBE and PBE with Hubbard-U correction(PBE+U) for the calculation of the heats of formation. Theproposed scheme shows clear improvement when comparingwith the experimental heats of formation of solids. In thepresent work, we carry out similar analysis with a class of GGA
functionals, namely, the PBE without Hubbard-U corrections,PBE with U corrections, and RPBE [9]. We furthermoreexploit the possibilities at the meta-GGA level by includingthe TPSS functional [10–14] as a representative together witha recently developed meta-GGA functional mBEEF [15], aBayesian error estimation functional. One of the advantagesof the mBEEF functional over the other functionals is that itsupplies an error estimate which tells how reliable a particularcalculated energy difference is. The details of the mBEEFfunctional and its comparison to other GGAs and meta-GGAfunctionals in terms of exchange enhancement factors can befound in Ref. [15]. Calculating the heats of formation on atest set of 24 compounds which have not been used in thethe data set for fitting, we show that the mBEEF functionalwithout any fitting is nearly as accurate as the fitted GGAfunctionals and the fitted TPSS functional and includes arealistic error prediction with a small overestimation. Applyingthe FERE scheme to the mBEEF ensemble leads to animproved prediction quality and a corresponding reductionof the predicted error bars. Quantitatively, the rms errors inthe training data set with the PBE, RPBE, PBE+U, TPSS, andmBEEF functionals reduce from 0.22, 0.28, 0.21, 0.21, and0.14 eV per atom to 0.09, 0.09, 0.08, 0.10, and 0.07 eV peratom.
II. COMPUTATIONAL METHODOLOGY
All the calculations in the current work use the GPAWcode [16] with the projector augmented wave (PAW) [17]description of the atoms. We consider the PBE [3], RPBE [9],PBE+U [18], TPSS [10], and mBEEF [15] exchange-correlation functionals. For the PBE+U calculations, as sug-gested by Stevanovic et al., we use the value of U = 3.0 eV forall the transition elements except Ag and Cu for which we usea U value of 5.0 eV. In the calculations involving magnetism,the spin configurations have been taken from the lowest energystructure reported in the experiments. For example, the Fereference state which has the bcc structure has been treatedferromagnetically whereas the iron oxide has been treated
1098-0121/2015/91(23)/235201(9) 235201-1 ©2015 American Physical Society

MOHNISH PANDEY AND KARSTEN W. JACOBSEN PHYSICAL REVIEW B 91, 235201 (2015)
antiferromagnetically as reported in the experiments. We use areal-space description of the wave functions with a grid spacingof 0.18 A. A Fermi temperature of 0.05 eV for the solid phasesis used to enhance convergence. Brillouin zone sampling isdone with a k-point mesh of 33a−1
x × 33a−1y × 33a−1
z with theMonkhorst-Pack [19] sampling scheme. Forces are minimizeddown to 0.05 eV/A for all the relaxations. Uncertainties in theheats of formations with the mBEEF functionals are explicitlycalculated using the ensemble of functionals proposed inRef. [20]. All the experimental heats of formation have beentaken from Refs. [4,21].
III. RESULTS AND DISCUSSIONS
A. Heats of formation with the DFT
The standard heat of formation of a solid calculated withDFT is
H DFT(Ap1Bp2 . . . ) = E(Ap1Bp2 . . . ) − piμ0i , (1)
where E(Ap1Bp2 . . . ) indicates the total energy of Ap1Bp2 . . .
calculated with DFT and the μ0i denotes the chemical potentials
of the elements under standard conditions calculated withDFT. The entropic and zero-point energy corrections have beenignored in the above expression. The calculation of the heatsof formation using the above expression with the PBE, RPBE,TPSS, and PBE+U functionals provide a single number asthe best estimate of the heat of formation. In comparison,the mBEEF functional provides both a best estimate but alsovia the ensemble of functionals an estimation of the error baron the calculated heat of formation. The functionals in themBEEF ensemble differ from each other by the values of theparameters defining the functional [15].
The calculated heats of formation versus the experimentalheats of formation (eV/per atom) of a set of 257 binarycompounds with the PBE, RPBE, PBE+U, TPSS, and mBEEFfunctionals are shown in Fig. 1, panels (a), (c), (e), (g), and (i).The set of compounds we use has about 80% overlap with theset of 252 compounds used by Stevanovic et al. [4] and thefull list of compounds is given in Table I along with the heatsof formation calculated with the mBEEF and the mBEEF withfitting of reference energies. The difference between our dataset and the one of Stevanovic et al. gives rise to somewhatdifferent results in detail but the trends remain the same. In thefigure MAE and σ denote the mean absolute error and standarddeviation with respect to the experimental heats of formation.The observed trend in Figs. 1(a) and 1(c) is a similar behaviorfor the PBE and RPBE functionals with underbinding in mostof the cases with a very few overbinding cases. This behaviorin the GGA functionals arises due to the overbinding of thereference phases and the underbinding in the multinary com-pounds leading to an incomplete cancellation of the errors [22].In Fig. 1(e) the direction of the deviation in the PBE+U heatsof formation is not very systematic, i.e., underbinding in somecases and overbinding in others. This behavior has also beenobserved in Ref. [23]. The predictions do not significantlyimprove with the TPSS functional as shown in Fig. 1(g). TheMAE and rms in the TPSS predictions turn out to be similarto the GGA functionals. An important factor in the calculatederrors is the dissimilar nature of the reactants and the products.Reactions in which both sides have similar compounds are
FIG. 1. (Color online) (a), (c), (e), (g), and (i) show the heatsof formation calculated with the PBE, RPBE, PBE+U, TPSS, andmBEEF functionals, respectively, versus the experimental heats offormation. (b), (d), (f), (h), and (j) show the heats of formationcalculated with PBE, RPBE, PBE+U, TPSS, and mBEEF function-als, respectively, versus the experimental heats of formation aftercorrecting the reference phase energies using the experimental heatsof formation as the training set. MAE and σ in (a)–(j) indicate themean absolute error and standard deviation of the calculated heats offormation with respect to the experimental heats of formation.
235201-2

HEATS OF FORMATION OF SOLIDS WITH ERROR . . . PHYSICAL REVIEW B 91, 235201 (2015)
TABLE I. The heats of formation of the solids used in the training set calculated with the mBEEF (HmBEEF) and mBEEF with the FERE(H FERE
mBEEF). The experimental values (HExpt) are also given for comparison. All the energies are in eV/atom.
Compound HExpt HmBEEF H FEREmBEEF Compound HExpt HmBEEF H FERE
mBEEF
Ag2Se − 0.15 0.02 ± 0.12 − 0.07 ± 0.09 AgCl − 0.66 − 0.56 ± 0.24 − 0.58 ± 0.09AgI − 0.32 − 0.32 ± 0.13 − 0.39 ± 0.08 Ag2O − 0.11 − 0.07 ± 0.20 − 0.14 ± 0.03Ag2O2 − 0.06 − 0.07 ± 0.19 − 0.13 ± 0.04 AlCl3 − 1.82 − 1.74 ± 0.19 − 1.80 ± 0.05AlF3 − 3.90 − 3.93 ± 0.27 − 3.85 ± 0.06 Al2O3 − 3.47 − 3.24 ± 0.24 − 3.41 ± 0.07AlN − 1.65 − 1.55 ± 0.23 − 1.72 ± 0.04 AlAs − 0.61 − 0.66 ± 0.14 − 0.64 ± 0.05AlP − 0.85 − 0.76 ± 0.15 − 0.87 ± 0.06 Al2Se3 − 1.18 − 0.93 ± 0.12 − 1.13 ± 0.04AsI3 − 0.15 − 0.18 ± 0.03 − 0.12 ± 0.04 AuBr − 0.07 − 0.11 ± 0.11 − 0.06 ± 0.03AuCl − 0.18 − 0.27 ± 0.17 − 0.12 ± 0.04 AuI − 0.00 − 0.08 ± 0.11 0.03 ± 0.04Au2O3 − 0.01 − 0.11 ± 0.19 − 0.02 ± 0.05 AuCl3 − 0.17 − 0.39 ± 0.20 − 0.29 ± 0.05AuF3 − 0.94 − 1.24 ± 0.23 − 1.01 ± 0.09 BN − 1.32 − 1.38 ± 0.14 − 1.34 ± 0.04B2O3 − 2.64 − 2.60 ± 0.16 − 2.61 ± 0.05 BaF2 − 4.17 − 4.40 ± 0.28 − 4.25 ± 0.04BaO2 − 2.19 − 2.19 ± 0.23 − 2.22 ± 0.05 BaS − 2.38 − 2.39 ± 0.17 − 2.44 ± 0.04BaCl2 − 2.95 − 2.96 ± 0.23 − 2.94 ± 0.04 BaO − 2.86 − 2.75 ± 0.22 − 2.77 ± 0.04BaBr2 − 2.62 − 2.48 ± 0.13 − 2.59 ± 0.03 BaI2 − 2.10 − 2.02 ± 0.11 − 2.07 ± 0.04BeO − 3.14 − 2.95 ± 0.23 − 3.03 ± 0.05 BeS − 1.21 − 1.18 ± 0.16 − 1.28 ± 0.04BeI2 − 0.67 − 0.64 ± 0.06 − 0.72 ± 0.06 BiBr3 − 0.72 − 0.71 ± 0.08 − 0.81 ± 0.05Bi2O3 − 1.19 − 1.17 ± 0.15 − 1.16 ± 0.07 Bi2S3 − 0.30 − 0.29 ± 0.09 − 0.31 ± 0.07CaF2 − 4.21 − 4.56 ± 0.29 − 4.39 ± 0.07 CaI2 − 1.84 − 1.82 ± 0.10 − 1.84 ± 0.05CaO − 3.29 − 3.25 ± 0.25 − 3.23 ± 0.04 CaS − 2.45 − 2.43 ± 0.19 − 2.43 ± 0.04CaBr2 − 2.36 − 2.26 ± 0.10 − 2.34 ± 0.03 CaCl2 − 2.75 − 2.78 ± 0.22 − 2.72 ± 0.05CaC2 − 0.21 − 0.13 ± 0.12 − 0.21 ± 0.04 CdS − 0.78 − 0.77 ± 0.13 − 0.80 ± 0.05CdF2 − 2.42 − 2.76 ± 0.29 − 2.60 ± 0.08 CdO − 1.34 − 1.20 ± 0.20 − 1.20 ± 0.10CdSe − 0.75 − 0.71 ± 0.10 − 0.74 ± 0.05 CdTe − 0.48 − 0.61 ± 0.10 − 0.51 ± 0.04CdI2 − 0.70 − 0.63 ± 0.07 − 0.66 ± 0.05 CoS − 0.43 − 0.28 ± 0.17 − 0.31 ± 0.08CoSe − 0.32 − 0.27 ± 0.19 − 0.31 ± 0.03 Co3O4 − 1.32 − 1.50 ± 0.38 − 1.51 ± 0.14Co3S4 − 0.53 − 0.45 ± 0.16 − 0.49 ± 0.07 CrS − 0.81 − 0.83 ± 0.16 − 0.77 ± 0.12CrF3 − 3.00 − 3.26 ± 0.34 − 3.04 ± 0.09 CrO2 − 2.07 − 2.06 ± 0.28 − 2.02 ± 0.06Cr2O3 − 2.36 − 2.52 ± 0.33 − 2.46 ± 0.07 CrF4 − 2.58 − 2.72 ± 0.25 − 2.50 ± 0.11CsBr − 2.10 − 1.99 ± 0.10 − 2.10 ± 0.03 CsCl − 2.30 − 2.25 ± 0.18 − 2.27 ± 0.01CsF − 2.87 − 2.98 ± 0.22 − 2.90 ± 0.04 Cu3N 0.18 0.34 ± 0.14 0.12 ± 0.03CuO − 0.82 − 0.58 ± 0.21 − 0.76 ± 0.04 Cu2O − 0.58 − 0.36 ± 0.20 − 0.58 ± 0.05CuF2 − 1.88 − 1.85 ± 0.29 − 1.80 ± 0.13 Cu2Sb − 0.04 0.02 ± 0.05 − 0.07 ± 0.04CuI − 0.35 − 0.15 ± 0.10 − 0.33 ± 0.04 Cu2Se − 0.21 0.10 ± 0.10 − 0.13 ± 0.03Cu3P − 0.17 0.01 ± 0.05 − 0.18 ± 0.09 CuS − 0.28 − 0.17 ± 0.16 − 0.37 ± 0.05Fe2O3 − 1.71 − 1.69 ± 0.26 − 1.89 ± 0.11 FeS − 0.52 − 0.35 ± 0.12 − 0.61 ± 0.04FeF2 − 2.46 − 2.27 ± 0.31 − 2.27 ± 0.10 FeO − 1.41 − 1.15 ± 0.19 − 1.39 ± 0.08FeSe − 0.39 − 0.04 ± 0.11 − 0.30 ± 0.13 GaN − 0.81 − 0.48 ± 0.17 − 0.72 ± 0.06GaP − 0.47 − 0.44 ± 0.08 − 0.63 ± 0.05 GaAs − 0.39 − 0.42 ± 0.08 − 0.47 ± 0.02GaSb − 0.22 − 0.20 ± 0.06 − 0.29 ± 0.03 GaCl3 − 1.36 − 1.25 ± 0.20 − 1.34 ± 0.09GaF3 − 3.01 − 2.96 ± 0.24 − 2.92 ± 0.03 Ga2O3 − 2.26 − 1.99 ± 0.19 − 2.21 ± 0.03Ga2S3 − 1.07 − 0.70 ± 0.11 − 0.95 ± 0.04 Ga2Se3 − 0.85 − 0.59 ± 0.08 − 0.85 ± 0.03GaSe − 0.83 − 0.61 ± 0.06 − 0.91 ± 0.03 GaS − 1.09 − 0.68 ± 0.08 − 0.98 ± 0.04GeTe − 0.10 − 0.04 ± 0.06 − 0.17 ± 0.04 GeS − 0.39 − 0.20 ± 0.08 − 0.46 ± 0.03Ge3O6 − 1.90 − 1.78 ± 0.16 − 1.95 ± 0.07 GeSe − 0.48 − 0.11 ± 0.07 − 0.38 ± 0.04Ge4O8 − 2.00 − 1.71 ± 0.15 − 1.88 ± 0.08 HfN − 1.91 − 1.74 ± 0.14 − 1.94 ± 0.06HfCl4 − 2.05 − 2.07 ± 0.22 − 2.11 ± 0.06 HfO2 − 3.95 − 3.71 ± 0.23 − 3.88 ± 0.05HfF4 − 4.00 − 4.07 ± 0.26 − 3.98 ± 0.04 HgSe − 0.24 − 0.26 ± 0.11 − 0.31 ± 0.02HgTe − 0.22 − 0.32 ± 0.13 − 0.23 ± 0.02 HgS − 0.30 − 0.21 ± 0.13 − 0.26 ± 0.03HgO − 0.47 − 0.36 ± 0.14 − 0.38 ± 0.05 HgI2 − 0.36 − 0.37 ± 0.07 − 0.41 ± 0.02HgCl2 − 0.77 − 0.82 ± 0.21 − 0.79 ± 0.07 InN − 0.10 0.00 ± 0.15 − 0.12 ± 0.03InP − 0.46 − 0.34 ± 0.08 − 0.41 ± 0.04 InTe − 0.50 − 0.32 ± 0.04 − 0.37 ± 0.06InAs − 0.31 − 0.39 ± 0.08 − 0.31 ± 0.03 InS − 0.70 − 0.61 ± 0.13 − 0.78 ± 0.02InSb − 0.16 − 0.27 ± 0.07 − 0.24 ± 0.03 IrO2 − 0.95 − 0.79 ± 0.18 − 0.92 ± 0.03IrCl3 − 0.64 − 0.59 ± 0.17 − 0.64 ± 0.03 IrS2 − 0.48 − 0.36 ± 0.14 − 0.52 ± 0.03K2O − 1.25 − 1.22 ± 0.21 − 1.28 ± 0.05 K2S − 1.31 − 1.24 ± 0.16 − 1.31 ± 0.04K2Se − 1.36 − 1.24 ± 0.14 − 1.31 ± 0.05 KF − 2.94 − 3.19 ± 0.24 − 3.11 ± 0.06KCl − 2.26 − 2.25 ± 0.17 − 2.26 ± 0.02 K2O2 − 1.28 − 1.23 ± 0.22 − 1.28 ± 0.06
235201-3

MOHNISH PANDEY AND KARSTEN W. JACOBSEN PHYSICAL REVIEW B 91, 235201 (2015)
TABLE I. (Continued.)
Compound HExpt HmBEEF H FEREmBEEF Compound HExpt HmBEEF H FERE
mBEEF
K3As − 0.48 − 0.40 ± 0.11 − 0.35 ± 0.06 K2S2 − 1.12 − 1.11 ± 0.14 − 1.18 ± 0.03LaS − 2.36 − 2.14 ± 0.16 − 2.40 ± 0.07 LaN − 1.57 − 1.32 ± 0.18 − 1.53 ± 0.07LaI3 − 1.73 − 1.61 ± 0.09 − 1.76 ± 0.03 LaCl3 − 2.78 − 2.67 ± 0.23 − 2.75 ± 0.06Li2O − 2.07 − 2.00 ± 0.26 − 2.07 ± 0.10 Li2S − 1.52 − 1.51 ± 0.15 − 1.59 ± 0.03Li2Se − 1.45 − 1.37 ± 0.11 − 1.46 ± 0.04 Li3N − 0.43 − 0.45 ± 0.24 − 0.50 ± 0.10Li3Sb − 0.83 − 0.67 ± 0.07 − 0.65 ± 0.09 LiF − 3.19 − 3.37 ± 0.23 − 3.30 ± 0.07Li3Bi − 0.60 − 0.57 ± 0.05 − 0.61 ± 0.09 LiCl − 2.12 − 2.03 ± 0.17 − 2.05 ± 0.11Li2O2 − 1.64 − 1.58 ± 0.21 − 1.64 ± 0.06 MgTe − 1.08 − 1.05 ± 0.09 − 1.06 ± 0.04MgS − 1.79 − 1.60 ± 0.15 − 1.74 ± 0.06 MgSe − 1.52 − 1.41 ± 0.12 − 1.56 ± 0.05MgO − 3.11 − 3.14 ± 0.20 − 3.26 ± 0.06 Mg3Bi2 − 0.32 − 0.23 ± 0.07 − 0.32 ± 0.05MgF2 − 3.88 − 3.79 ± 0.25 − 3.71 ± 0.05 MnS − 1.11 − 1.08 ± 0.23 − 1.03 ± 0.06MnO2 − 1.80 − 1.99 ± 0.28 − 1.96 ± 0.09 Mn3O4 − 2.05 − 2.13 ± 0.28 − 2.07 ± 0.01Mn2O3 − 1.99 − 1.99 ± 0.27 − 1.94 ± 0.02 MoS2 − 0.81 − 0.83 ± 0.08 − 0.86 ± 0.03MoO2 − 2.03 − 1.95 ± 0.18 − 1.96 ± 0.04 MoO3 − 1.93 − 1.93 ± 0.18 − 1.95 ± 0.05NaF − 2.97 − 3.04 ± 0.21 − 2.93 ± 0.05 Na2O − 1.43 − 1.49 ± 0.18 − 1.51 ± 0.10Na2S − 1.26 − 1.24 ± 0.12 − 1.28 ± 0.03 Na2Se − 1.18 − 1.19 ± 0.12 − 1.23 ± 0.04Na2C2 0.06 0.12 ± 0.09 0.04 ± 0.03 NaCl − 2.13 − 2.09 ± 0.17 − 2.07 ± 0.06Na2S2 − 1.03 − 0.97 ± 0.12 − 1.02 ± 0.04 Na2Se2 − 0.97 − 0.95 ± 0.13 − 1.00 ± 0.06Na3Bi − 0.46 − 0.40 ± 0.07 − 0.39 ± 0.06 Na2O2 − 1.32 − 1.26 ± 0.18 − 1.29 ± 0.08NbN − 1.22 − 1.15 ± 0.13 − 1.13 ± 0.04 NbO − 2.10 − 2.06 ± 0.14 − 2.07 ± 0.03Nb2O5 − 2.81 − 2.85 ± 0.21 − 2.87 ± 0.04 NbF5 − 3.13 − 3.52 ± 0.23 − 3.33 ± 0.13NbO2 − 2.75 − 2.76 ± 0.23 − 2.77 ± 0.05 NbCl5 − 1.38 − 1.46 ± 0.22 − 1.42 ± 0.05NiS − 0.43 − 0.17 ± 0.12 − 0.35 ± 0.04 NiSb − 0.34 − 0.36 ± 0.07 − 0.33 ± 0.06NiTe − 0.28 − 0.23 ± 0.06 − 0.28 ± 0.03 NiSe − 0.31 − 0.14 ± 0.09 − 0.32 ± 0.03Ni3S2 − 0.42 − 0.28 ± 0.13 − 0.48 ± 0.03 OsO4 − 0.82 − 1.12 ± 0.19 − 0.83 ± 0.01PI3 − 0.16 − 0.05 ± 0.06 − 0.06 ± 0.05 PCl3 − 0.83 − 0.83 ± 0.21 − 0.76 ± 0.09PBr5 − 0.47 − 0.31 ± 0.04 − 0.42 ± 0.05 PCl5 − 0.77 − 0.74 ± 0.19 − 0.68 ± 0.06PbO − 1.13 − 1.08 ± 0.16 − 1.08 ± 0.04 PbS − 0.52 − 0.52 ± 0.14 − 0.53 ± 0.06PbF2 − 2.29 − 2.60 ± 0.25 − 2.43 ± 0.04 PbO2 − 0.96 − 0.87 ± 0.18 − 0.88 ± 0.05PbCl2 − 1.24 − 1.29 ± 0.19 − 1.24 ± 0.06 PbBr2 − 0.96 − 0.88 ± 0.09 − 0.97 ± 0.03PdO − 0.44 − 0.56 ± 0.27 − 0.52 ± 0.09 Pd4S − 0.14 − 0.14 ± 0.09 − 0.07 ± 0.09PdS − 0.39 − 0.41 ± 0.19 − 0.40 ± 0.07 PdS2 − 0.28 − 0.31 ± 0.14 − 0.33 ± 0.05PtS − 0.42 − 0.43 ± 0.18 − 0.51 ± 0.03 PtS2 − 0.38 − 0.36 ± 0.12 − 0.44 ± 0.06Pt2O2 − 0.37 − 0.19 ± 0.22 − 0.24 ± 0.06 PtO2 − 0.57 − 0.53 ± 0.20 − 0.58 ± 0.04RbF − 2.89 − 2.95 ± 0.22 − 2.87 ± 0.04 RbI − 1.73 − 1.71 ± 0.09 − 1.77 ± 0.05Rb2O − 1.17 − 1.04 ± 0.21 − 1.10 ± 0.05 Rb2S − 1.25 − 1.16 ± 0.15 − 1.23 ± 0.04Rb2O2 − 1.22 − 1.28 ± 0.20 − 1.33 ± 0.05 RbCl − 2.26 − 2.23 ± 0.17 − 2.24 ± 0.02ReO3 − 1.58 − 1.63 ± 0.17 − 1.69 ± 0.06 ReO2 − 1.54 − 1.40 ± 0.17 − 1.46 ± 0.05RhO2 − 0.85 − 0.87 ± 0.23 − 0.87 ± 0.04 RhCl3 − 0.77 − 0.87 ± 0.21 − 0.81 ± 0.03Rh2S3 − 0.54 − 0.53 ± 0.18 − 0.55 ± 0.04 Rh2O3 − 0.84 − 0.80 ± 0.28 − 0.79 ± 0.05RuO2 − 1.05 − 1.11 ± 0.22 − 0.99 ± 0.06 RuBr3 − 0.36 − 0.34 ± 0.08 − 0.35 ± 0.04RuCl3 − 0.53 − 0.74 ± 0.20 − 0.60 ± 0.04 RuO4 − 0.50 − 0.58 ± 0.19 − 0.53 ± 0.19SbF3 − 2.37 − 2.49 ± 0.23 − 2.24 ± 0.07 Sb2O5 − 1.44 − 1.50 ± 0.19 − 1.43 ± 0.06ScAs − 1.39 − 1.53 ± 0.06 − 1.45 ± 0.03 ScF3 − 4.22 − 4.22 ± 0.25 − 4.11 ± 0.06ScCl3 − 2.40 − 2.37 ± 0.21 − 2.39 ± 0.05 SiC − 0.34 − 0.25 ± 0.09 − 0.32 ± 0.07SiO2 − 3.13 − 3.06 ± 0.17 − 3.09 ± 0.06 SiS2 − 0.88 − 0.78 ± 0.06 − 0.83 ± 0.06SiSe2 − 0.61 − 0.50 ± 0.06 − 0.55 ± 0.06 Si3N4 − 1.10 − 1.17 ± 0.14 − 1.15 ± 0.09SnO − 1.48 − 1.20 ± 0.14 − 1.42 ± 0.04 SnS2 − 0.53 − 0.37 ± 0.09 − 0.56 ± 0.03SnSe2 − 0.43 − 0.28 ± 0.06 − 0.48 ± 0.03 SnO2 − 1.97 − 1.73 ± 0.21 − 1.89 ± 0.05SnS − 0.57 − 0.34 ± 0.11 − 0.58 ± 0.03 SnSe − 0.47 − 0.27 ± 0.10 − 0.52 ± 0.04SrO2 − 2.19 − 2.32 ± 0.23 − 2.31 ± 0.06 SrO − 3.07 − 3.08 ± 0.24 − 3.05 ± 0.04SrS − 2.45 − 2.44 ± 0.19 − 2.44 ± 0.04 SrCl2 − 2.86 − 2.88 ± 0.23 − 2.81 ± 0.05SrBr2 − 2.48 − 2.38 ± 0.11 − 2.45 ± 0.03 SrI2 − 1.93 − 1.92 ± 0.10 − 1.93 ± 0.02TaN − 1.30 − 1.14 ± 0.13 − 1.25 ± 0.05 TaSi2 − 0.47 − 0.36 ± 0.17 − 0.43 ± 0.07Ta2O5 − 3.03 − 2.99 ± 0.23 − 3.09 ± 0.06 TaF5 − 3.29 − 3.56 ± 0.23 − 3.41 ± 0.10TaCl5 − 1.48 − 1.51 ± 0.21 − 1.51 ± 0.06 TiS − 1.41 − 1.44 ± 0.10 − 1.37 ± 0.06TiS2 − 1.41 − 1.41 ± 0.12 − 1.38 ± 0.04 TiN − 1.58 − 1.88 ± 0.12 − 1.75 ± 0.03Ti2O3 − 3.15 − 3.14 ± 0.19 − 3.07 ± 0.03 TiAs − 0.78 − 1.02 ± 0.05 − 0.69 ± 0.03
235201-4

HEATS OF FORMATION OF SOLIDS WITH ERROR . . . PHYSICAL REVIEW B 91, 235201 (2015)
TABLE I. (Continued.)
Compound HExpt HmBEEF H FEREmBEEF Compound HExpt HmBEEF H FERE
mBEEF
TiO2 − 3.26 − 3.37 ± 0.21 − 3.32 ± 0.05 TlI − 0.64 − 0.70 ± 0.10 − 0.67 ± 0.04TlBr − 0.90 − 0.95 ± 0.11 − 0.97 ± 0.05 TlCl − 1.06 − 1.16 ± 0.20 − 1.08 ± 0.07Tl2O − 0.62 − 0.68 ± 0.15 − 0.61 ± 0.08 TlF − 1.68 − 1.80 ± 0.21 − 1.62 ± 0.03Tl2S − 0.34 − 0.37 ± 0.12 − 0.32 ± 0.05 VN − 1.13 − 0.99 ± 0.13 − 1.00 ± 0.08V2O3 − 2.53 − 2.58 ± 0.29 − 2.62 ± 0.11 V2O5 − 2.29 − 2.31 ± 0.20 − 2.35 ± 0.06VO2 − 2.47 − 2.48 ± 0.23 − 2.52 ± 0.04 WBr6 − 0.52 − 0.41 ± 0.07 − 0.51 ± 0.03WO3 − 2.04 − 2.08 ± 0.19 − 2.05 ± 0.02 YAs − 1.68 − 1.75 ± 0.08 − 1.68 ± 0.02YCl3 − 2.59 − 2.63 ± 0.22 − 2.67 ± 0.05 YF3 − 4.45 − 4.45 ± 0.27 − 4.36 ± 0.04ZnO − 1.81 − 1.57 ± 0.21 − 1.75 ± 0.04 ZnSe − 0.85 − 0.76 ± 0.12 − 0.97 ± 0.02ZnTe − 0.61 − 0.56 ± 0.08 − 0.64 ± 0.01 ZnS − 1.07 − 0.89 ± 0.14 − 1.10 ± 0.03ZnCl2 − 1.43 − 1.36 ± 0.19 − 1.44 ± 0.05 ZnF2 − 2.64 − 2.50 ± 0.25 − 2.46 ± 0.05ZrO2 − 3.80 − 3.75 ± 0.23 − 3.79 ± 0.05 ZrCl4 − 2.03 − 2.11 ± 0.22 − 2.08 ± 0.03ZrSi − 0.80 − 0.93 ± 0.11 − 0.94 ± 0.05 ZrN − 1.89 − 1.84 ± 0.13 − 1.85 ± 0.06ZrS2 − 1.96 − 1.73 ± 0.14 − 1.79 ± 0.06
shown to give smaller errors when compared to experi-ments [22]. Figure 1(i) shows the calculated heats of formationwith the mBEEF functional with calculated error bars indicatedwith green bars. The calculated values are significantly closerto the experimental values compared to the values obtainedfrom the PBE, RPBE, PBE+U, and TPSS functionals. As canbe seen from the figure the experimental values are within theerror bars predicted by the mBEEF functional.
The mBEEF functional thus seems to be more accuratethan both the GGA functionals and the TPSS which is alsoa meta-GGA functional. However, it should also be notedthat in the construction of the mBEEF functional considerableoptimization to experimental databases was performed. Inthe following we investigate how the scheme suggested byStevanovic et al. [4] helps in improving the predictions for thedifferent functionals.
B. Heats of formation with the FERE
In the previous section we noticed that the limited pre-dictability of the TPSS and the GGA functionals mainly arisesfrom the different nature of the bonding in the multinary phasesand the reference phases. The FERE scheme [4] circumventsthis problem by adding corrections to the reference phaseenergies. The heats of formation calculated with the FEREcan be written as
H FERE(Ap1Bp2 . . . )
= E(Ap1Bp2 . . . ) − pi
(μ0
i + δμ0i
), (2)
where the δμ0i ’s are the corrections added to the reference
phase energies to improve the heats of formation. The valuesof the δμ0
i ’s can be calculated by a linear regression fit byminimizing the root mean square (rms) error between thecalculated (H DFT) and the experimental (H Expt) heats offormation. The size of the training set has to be sufficientlylarge to avoid any overfitting and the quality of the fit must bevalidated on a test set. The linear regression requires that thenumber of parameters in the linear model which need to befitted to the observations be smaller than the number of datapoints; i.e., the system of the equations has to be overdeter-mined. We calculate 62 parameters which correspond to the
corrections to the reference phase energies of 62 elements byusing a training set of 257 compounds with the experimentalheats of formation available. The parameters can be calculatedusing singular value decomposition (SVD) [24] by minimizingthe rms error |HExpt − HDFT|2. The calculated referenceenergies are tabulated in the Supplemental Material [25].
Figure 1, panels (b), (d), (f), (h), and (j), shows the heatsof formation calculated after adding the corrections to thereference phase energies. The comparison with panels (a),(c), (e), (g), and (i) of the figure clearly shows that the MAEand σ are significantly reduced after applying the correctionsto the reference phase energies. Interestingly, all the GGAfunctionals give similar heats of formation after employingthe corrections even though they differ before applying thecorrections. The TPSS functional does not perform any betterthan the GGA functionals after applying the corrections.
As noted the performance of mBEEF before fitting issomewhat better than the GGAs and the TPSS functional andin fact, as we shall see later, it is comparable to the fitted GGAsand the TPSS on a test set. However, for comparison we alsoapply the FERE fitting procedure to the mBEEF functionaland this does naturally lead to an improvement on the trainingset. As mentioned before, we furthermore employ the fittingprocedure on all the functionals in the mBEEF ensembleanticipating a reduction of the error and the fluctuations withinthe ensemble. This is indeed the case. In Fig. 1(j) we can seethat the uncertainties are significantly reduced as compared toFig. 1(i). The reduction in the size of the uncertainties is inagreement with the fact that the fitted mBEEF predictions aremore accurate.
C. Analysis of outliers
The appearance of outliers with and without the fittingfor the PBE, RPBE, and PBE+U functionals may occurfor two reasons: (1) error in the experimental data, and (2)some systems are poorly described with the given functional.The compounds having the deviation of the calculated heatof formation (δH ) from the experimental value by twice ofthe standard deviation (2σ ) are shown in Table II. The tableclearly shows that all the functionals except for PBE and RPBE
235201-5

MOHNISH PANDEY AND KARSTEN W. JACOBSEN PHYSICAL REVIEW B 91, 235201 (2015)
TABLE II. Outliers in the calculations without using the FERE scheme. The compounds exhibiting deviations of the calculated heats offormation from the experimental values by more than 2σ have been identified as outliers. The values of σ for the different functionals areshown in Fig. 1. δH denotes the difference between calculated and experimental heats of formation.
PBE δHPBE RPBE δHRPBE PBE+U δHPBE+U TPSS δHTPSS mBEEF δHmBEEF
Al2O3 0.48 Al2O3 0.69 Al2O3 0.48 AlP 0.45 AuF3 − 0.30BaS − 0.52 FeF2 0.61 BaS − 0.52 BaI2 − 0.48 CaF2 − 0.35BaO − 0.47 FeO 0.60 BaO − 0.47 BiBr3 − 0.51 CdF2 − 0.34FeF2 0.61 GaN 0.57 CrS − 0.82 CaS 0.48 Cu2Se 0.31FeO 0.49 HfO2 0.65 CrF3 − 0.47 FeF2 0.57 FeSe 0.35GaS 0.451 NiF2 0.66 Cr2O3 − 0.75 GaP 0.43 GaN 0.33LaN 0.46 GaN 0.42 Ga2S3 0.44 Ga2S3 0.37MnS 0.60 Ga2S3 0.44 NiF2 0.57 GaS 0.41NiF2 0.85 GaS 0.45 PbBr2 − 0.45 GeSe 0.37
Ge4O8 0.42 SrBr2 − 0.49 Ge4O8 0.29MnS − 0.48 SrI2 − 0.55 NbF5 − 0.38
Mn3O4 − 0.42 ZnS 0.43 OsO4 − 0.30V2O3 − 0.42 ZrS2 0.47 PbF2 − 0.31
SnO2 0.28TiN − 0.30
have none or very few common outliers. For example, thepredictions for barium-containing compounds is a little worseonly in the PBE, PBE+U, and the TPSS whereas the outlierscontaining chromium are present in the PBE+U functionalonly. On the other hand, even if the gallium is present in all thefunctionals it is not the same compound which is an outlier.
Additionally, the outliers present in the mBEEF calcula-tions do not deviate from the experimental value by more than0.41 eV per atom whereas the outliers present in the GGAfunctionals and the TPSS have deviations as high as 0.85and 0.57 eV per atom, respectively. The deviations shownfor the mBEEF functional are relative to a common rms errorσ = 0.14 eV and not based on the ensemble estimated errors.The large variation in outliers with functional seems to indicatethat the appearance of outliers is as might be expected notdue to experimental errors but rather due to limitations of thedifferent functionals.
Table III shows the outliers after the fitting has been applied.We see that the outliers are to a large extent different from theones before the fitting and again they also vary considerably forthe different functionals. This means that we cannot identifyparticular issues with specific systems. The PBE and RPBEfunctionals continue to have significant overlap of outliers afterthe fitting.
D. Statistical analysis of the mBEEF predictions
The error bars predicted by the mBEEF ensemble are inreasonable agreement with the actual errors as can be seenfrom Fig. 1(i). In order to study the quality of the error barprediction in more detail we show in Fig. 2(a) a histogramof the actual error, i.e., the deviation between the mBEEFprediction and the experimental value (HmBEEF − HExpt)divided by the predicted error bar (σBEE). The histogram is a
TABLE III. Outliers in the calculations using the FERE scheme. The compounds exhibiting deviations of the calculated heats of formationfrom the experimental values by more than 2σ have been identified as outliers. The values of σ for the different functionals are shown in Fig. 1.δH denotes the difference between calculated and experimental heats of formation.
PBE δH FEREPBE RPBE δH FERE
RPBE PBE+U δH FEREPBE+U TPSS δH FERE
TPSS mBEEF δH FEREmBEEF
CuF2 0.22 CuF2 0.23 CoS 0.20 BaCl2 0.24 CaF2 − 0.18FeF2 0.33 FeF2 0.27 Co3O4 − 0.23 CaS 0.21 CdF2 − 0.18FeSe − 0.19 MnO2 − 0.21 CrO2 0.17 CsF − 0.23 Co3O4 − 0.19MnO2 − 0.25 NbF5 − 0.32 Fe2O3 − 0.17 FeF2 0.29 Fe2O3 − 0.17NbF5 − 0.29 Ni3S2 − 0.18 GaF3 0.16 KCl 0.32 FeF2 0.19Ni3S2 − 0.21 NiF2 0.38 GeO2 0.20 NbF5 − 0.26 GaP − 0.16NiF2 0.65 PbF2 − 0.18 MgF2 0.19 NiF2 0.37 KF − 0.17RuO4 − 0.19 RuO4 − 0.33 NbF5 − 0.23 RbI 0.25 Li3Sb 0.18TaF5 − 0.22 TaF5 − 0.24 SnO2 0.17 SrS 0.25 MgO − 0.15ZrSi − 0.24 ZrSi − 0.26 TaF5 − 0.18 SrI2 − 0.24 MgF2 0.17ZrS2 0.26 ZrS2 0.24 TiN − 0.19 TlI 0.26 MnO2 − 0.16
VN 0.26 ZrSi − 0.24 NbF5 − 0.20V2O3 − 0.36 ZrS2 0.35 TiN − 0.17ZnF2 0.19 ZnF2 0.18ZrS2 0.16 ZrS2 0.16
235201-6

HEATS OF FORMATION OF SOLIDS WITH ERROR . . . PHYSICAL REVIEW B 91, 235201 (2015)
FIG. 2. (Color online) (a) Shows the probability distribution ofthe calculated error (HmBEEF − HExpt) in the heat of formationdivided by the estimated error (σBEE) from the ensemble of func-tionals. (b) Shows the probability distribution of the calculatederror (H FERE
mBEEF − HExpt) in the heat of formation divided bythe estimated error (σ FERE
BEE ) from the ensemble of functionals aftercorrecting the reference phase energies. The ensemble energies havealso been recalculated employing the fitting eventually giving thenew error estimates σ FERE
BEE . The green plots in (a) and (b) show theGaussian distributions with zero mean and unit standard deviation.
running average calculated as [24]
P
(1
2[xi + xi+J ]
)≈ J
N (xi+J − xi), (3)
with xi being the ratio between actual error and predictederror, and the parameter J = 20. For a perfect statistical errorprediction one could expect that the distribution would beGaussian with a width of 1, which is also shown in the figurefor comparison. The large peak in the histogram around zeroshows that there is some tendency for the error prediction tobe on the large side, but the overall agreement is quite good.
If the FERE fitting procedure is applied to the mBEEFensemble the ratios of real to predicted errors result in thehistogram shown in Fig. 2(b). Both the real (H FERE
mBEEF −HExpt) and the predicted errors (σ FERE
BEE ) are now smaller butthe relative distribution remains fairly close to a Gaussianof unit width. However, now a tail in the histogram appears
indicating that for some systems the predicted error can be 3 or4 times smaller than the actual error. This is a fairly commonfeature of the ensemble approach [26].
E. Cross validation
In any regression process it is necessary to validate thequality of the regression over a set of test data which isnot the part of the training data set. Overfitting, i.e., moreparameters in the model than required to model the data, willlead to poor prediction of the test data set. One of the mostimportant features that a fitting scheme should possess is thepredictability on a completely new data set. One might expectgood predictions on a data set which is similar in nature tothe training data set. For example, in our case, we expect agood predictability for the binary compounds since we useonly binary compounds in the training data set. The fittingprocedure provides corrections for the reference energies of theelements which are independent of the chemical environmentsof the atoms. Therefore, we can expect that if the environmentschange considerably, which can for example be the case forternary or quarternary compounds, the improvement will beless pronounced.
Hence, in the test we not only include the binary compoundsbut the ternary compounds as well. We compose a set of 24binary and ternary compounds where the experimental heatsof formation are available and which are not present in thetraining data. We summarize the results in Table IV. As forthe training set the MAE and σ in general show a significantdecrease with the fitted reference energies indicating that wedo not overfit. However, the improvement is somewhat lessthan for the training set which is also what could be expected.Also for the test set we see that the three functionals PBE,RPBE, and PBE+U reach the same level of accuray afterfitting although PBE+U is considerably better before fitting.The performance of the TPSS functional does not seem to beany better than any of the GGA functionals. In fact the rmserror for TPSS is only slightly reduced after fitting, while theMAE is reduced more. This behavior can be traced to a singlesystem (Cs2S), which is clearly poorly corrected by the fittingscheme. We have not been able to identify why this is thecase. It can be noted that Cs was not included in the databaseconsidered by Stevanovic et al. [4].
The most interesting feature is that the mBEEF functionalalready before fitting is of the same quality as the otherfunctionals after fitting. Furthermore, the improvement of themBEEF results using the fitting is only moderate. This meansthat moving to mBEEF the fitting procedure can be completelyavoided at only a moderate cost in computational time (lessthan a factor to 2) compared to the GGAs.
In compounds such as SrSe and Mn2SiO4 the predictionswith mBEEF remain the same after the fitting procedure;however, the estimated error is significantly reduced leading tolarge real error relative to the predicted uncertainty. It shouldbe noted that it is an inherent limitation in the ensembleerror estimation that fluctuations in the predictions can onlyresult from fluctuations within the defined model space (i.e.,meta-GGA in this case). If errors appear which cannot bedescribed by such fluctuations an underestimation of the errormay result.
235201-7

MOHNISH PANDEY AND KARSTEN W. JACOBSEN PHYSICAL REVIEW B 91, 235201 (2015)
TABLE IV. Heats of formation of the solid compounds calculated with the different functionals with and without employing the fittingprocedure. The set below was not used in the training set for fitting the reference phase energies. All the energies are in eV/atom.
Compound HExpt HPBE HRPBE HPBE+U HTPSS HmBEEF H FEREPBE H FERE
RPBE H FEREPBE+U H FERE
TPSS H FEREmBEEF
AgNO3 − 0.26 − 0.40 − 0.31 − 0.53 − 0.47 −0.60 ± 0.22 − 0.58 − 0.67 − 0.68 − 0.45 −0.63 ± 0.16AlPO4 − 2.99 − 2.71 − 2.58 − 2.71 − 2.86 −2.97 ± 0.19 − 2.95 − 2.97 − 2.94 − 3.02 −3.03 ± 0.07BeSO4 − 2.16 − 1.99 − 1.83 − 1.99 − 2.09 −2.19 ± 0.16 − 2.22 − 2.23 − 2.21 − 2.19 −2.25 ± 0.11BiOCl − 1.27 − 1.26 − 1.11 − 1.26 − 1.62 −1.26 ± 0.16 − 1.25 − 1.20 − 1.23 − 1.32 −1.23 ± 0.09CdSO4 − 1.61 − 1.42 − 1.27 − 1.42 − 1.53 −1.59 ± 0.17 − 1.61 − 1.62 − 1.60 − 1.60 −1.63 ± 0.12CuCl2 − 0.76 − 0.51 − 0.32 − 0.70 − 0.80 −0.74 ± 0.21 − 0.75 − 0.60 − 0.84 − 0.79 −0.81 ± 0.07TiBr3 − 1.42 − 1.24 − 1.23 − 1.52 − 1.71 −1.37 ± 0.08 − 1.38 − 1.39 − 1.61 − 1.38 −1.43 ± 0.05NaClO4 − 0.66 − 0.54 − 0.41 − 0.54 − 0.67 −0.63 ± 0.15 − 0.76 − 0.77 − 0.73 − 0.68 −0.65 ± 0.16CaSO4 − 2.48 − 2.24 − 2.06 − 2.24 − 2.37 −2.40 ± 0.17 − 2.41 − 2.41 − 2.41 − 2.46 −2.43 ± 0.12Cs2S − 1.24 − 1.01 − 0.92 − 1.01 − 1.47 −1.16 ± 0.18 − 1.27 − 1.24 − 1.33 − 1.97 −1.23 ± 0.06CuWO4 − 1.91 − 1.59 − 1.41 − 1.76 − 1.72 −1.68 ± 0.21 − 1.62 − 1.60 − 1.75 − 1.65 −1.71 ± 0.07PbF4 − 1.95 − 2.13 − 2.05 − 2.13 − 2.26 −2.32 ± 0.23 − 2.19 − 2.19 − 2.11 − 2.24 −2.13 ± 0.08MgSO4 − 2.22 − 1.97 − 1.79 − 1.97 − 2.09 −2.16 ± 0.16 − 2.22 − 2.21 − 2.21 − 2.20 −2.24 ± 0.10SrSe − 2.00 − 2.04 − 1.98 − 2.04 − 2.76 −2.29 ± 0.16 − 2.25 − 2.26 − 2.29 − 2.66 −2.29 ± 0.05NiSO4 − 1.51 − 1.11 − 0.96 − 1.35 − 1.23 −1.42 ± 0.23 − 1.35 − 1.36 − 1.54 − 1.33 −1.50 ± 0.11FeWO4 − 1.99 − 1.73 − 1.58 − 2.01 − 1.87 −1.84 ± 0.21 − 1.81 − 1.81 − 1.94 − 1.86 −1.89 ± 0.06GeP − 0.11 +0.04 +0.09 +0.04 − 0.19 +0.14 ± 0.08 − 0.01 +0.03 − 0.05 − 0.28 −0.02 ± 0.07VOCl − 2.10 − 1.79 − 1.68 − 2.45 − 2.07 −2.11 ± 0.24 − 1.97 − 1.98 − 2.41 − 2.05 −2.12 ± 0.07LiBO2 − 2.67 − 2.42 − 2.30 − 2.42 − 2.57 −2.58 ± 0.17 − 2.61 − 2.61 − 2.58 − 2.64 −2.61 ± 0.05NaBrO3 − 0.69 − 0.52 − 0.41 − 0.52 − 0.71 −0.60 ± 0.13 − 0.74 − 0.76 − 0.72 − 0.69 −0.66 ± 0.11CoSO4 − 1.53 − 1.09 − 0.95 − 1.43 − 1.24 −1.40 ± 0.23 − 1.30 − 1.34 − 1.56 − 1.31 −1.43 ± 0.11PbSeO4 − 1.05 − 0.94 − 0.81 − 0.94 − 1.13 −1.04 ± 0.16 − 1.07 − 1.09 − 1.06 − 1.07 −1.08 ± 0.09Mn2SiO4 − 2.56 − 1.83 − 1.77 − 2.58 − 2.01 −2.29 ± 0.23 − 2.17 − 2.19 − 2.38 − 2.10 −2.25 ± 0.08ZnSO4 − 1.70 − 1.37 − 1.20 − 1.37 − 1.47 −1.53 ± 0.16 − 1.61 − 1.61 − 1.61 − 1.60 −1.62 ± 0.11MAE 0.24 0.35 0.16 0.20 0.12 0.12 0.13 0.11 0.15 0.09σ 0.28 0.39 0.19 0.26 0.16 0.16 0.17 0.15 0.25 0.14
IV. CONCLUSION
The need for accurate predictions of material stabilitieshas led to the development of schemes combining DFT totalenergy calculations with experimental information. We haveanalyzed one such scheme for calculation of heats of formationwhich fit the reference energies for elemental systems. Thescheme was developed with the PBE+U functional, but weshow that comparable predictive power is obtained using otherGGAs such as PBE or RPBE or the meta-GGA TPSS. We havefurthermore seen that the mBEEF functional, which is a meta-GGA and which has been extensively optimized to a variety ofexperimental data, leads to much improved estimation of heatsof formation even without applying the fitting procedure. ThemBEEF functional furthermore includes realistic ensembleestimates of the calculated formation energies. Applyingthe fitting scheme to mBEEF leads to a further reductionof the error and narrows the ensemble error estimationaccordingly.
The FERE scheme clearly has its limitations. The correctionof only the binding energies of the reference systems makesmost sense if the character of the bonding differs significantlybetween the material at hand and the reference systems. Thisis for example the case for a metal oxide, in which the bondingmay be quite different from the one in an oxygen moleculeand in the pure metal. However, oxygen can enter in manydifferent ways in different materials and only improving onthe molecular energy cannot be a solution to improved heats offormation in the long run. Moving to more accurate functionalsis therefore a must, and the current work shows that applyinga meta-GGA such as mBEEF already provides a significantimprovement in the prediction of solid heats of formation.
ACKNOWLEDGMENTS
The authors acknowledge CASE (Catalysis for SustainableEnergy) initiative. CASE is funded by the Danish Ministry ofScience, Technology and Innovation.
[1] P. Hohenberg and W. Kohn, Phys. Rev. 136, B864 (1964).[2] W. Kohn and L. J. Sham, Phys. Rev. 140, A1133 (1965).[3] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77,
3865 (1996).[4] V. Stevanovic, S. Lany, X. Zhang, and A. Zunger, Phys. Rev. B
85, 115104 (2012).
[5] I. E. Castelli, T. Olsen, S. Datta, D. D. Landis, S. Dahl, K. S.Thygesen, and K. W. Jacobsen, Energy Environ. Sci. 5, 5814(2012).
[6] A. Jain, G. Hautier, C. J. Moore, S. P. Ong, C. C. Fischer, T.Mueller, K. A. Persson, and G. Ceder, Comput. Mater. Sci. 50,2295 (2011).
235201-8

HEATS OF FORMATION OF SOLIDS WITH ERROR . . . PHYSICAL REVIEW B 91, 235201 (2015)
[7] X. Zhang, V. Stevanovic, M. d’Avezac, S. Lany, and A. Zunger,Phys. Rev. B 86, 014109 (2012).
[8] S. Lany, Phys. Rev. B 78, 245207 (2008).[9] B. Hammer, L. B. Hansen, and J. K. Nørskov, Phys. Rev. B 59,
7413 (1999).[10] J. M. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria,
Phys. Rev. Lett. 91, 146401 (2003).[11] R. Peverati and D. G. Truhlar, Philos. Trans. R. Soc. A 372,
2011 (2014).[12] L. A. Constantin, E. Fabiano, and F. Della Sala, Chem. Theory
Comput. 9, 2256 (2013).[13] J. Sun, J. P. Perdew, and A. Ruzsinszky, Proc. Natl. Acad. Sci.
USA 112, 685 (2015).[14] F. Della Sala, E. Fabiano, and L. A. Constantin, Phys. Rev. B
91, 035126 (2015).[15] J. Wellendorff, K. T. Lundgaard, K. W. Jacobsen, and T.
Bligaard, J. Chem. Phys. 140, 144107 (2014).[16] J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak,
L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen,H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M.Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen,V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange,G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Hakkinen,G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska,
T. T. Rantala, J. Schiøtz, K. S. Thygesen, and K. W. Jacobsen,J. Phys.: Condens. Matter 22, 253202 (2010).
[17] G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 (1999).[18] S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys,
and A. P. Sutton, Phys. Rev. B 57, 1505 (1998).[19] H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188
(1976).[20] J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D.
Landis, J. K. Nørskov, T. Bligaard, and K. W. Jacobsen, Phys.Rev. B 85, 235149 (2012).
[21] Standard thermodynamic properties of chemical substances,http://www.update.uu.se/∼jolkkonen/pdf/CRC_TD.pdf.
[22] M. Fuchs, J. L. F. Da Silva, C. Stampfl, J. Neugebauer, andM. Scheffler, Phys. Rev. B 65, 245212 (2002).
[23] G. Hautier, S. P. Ong, A. Jain, C. J. Moore, and G. Ceder, Phys.Rev. B 85, 155208 (2012).
[24] W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. T.Vetterling, Numerical Recipes in C (Cambridge UniversityPress, Cambridge, 1988).
[25] See Supplemental Material at http://link.aps.org/supplemental/10.1103/PhysRevB.91.235201 for the reference energies calcu-lated with different functionals with and without the fitting.
[26] S. L. Frederiksen, K. W. Jacobsen, K. S. Brown, and J. P. Sethna,Phys. Rev. Lett. 93, 165501 (2004).
235201-9

Supplementary Information: Heats of formation of solids with error estimation: themBEEF functional with and without fitted reference energies.
Mohnish Pandey1 and Karsten W. Jacobsen,11Center for Atomic-scale Materials Design, Department of Physics,
Technical University of Denmark, DK - 2800 Kongens Lyngby, Denmark(Dated: April 14, 2015)
TABLE I: Energies (in eV/atom) of the reference states of elementsbefore and after the fitting
Element µP BE µF EREP BE µRP BE µF ERE
RP BE µP BE+U µF EREP BE+U µT P SS µF ERE
T P SS µmBEEF µF EREmBEEF
Li -1.904 -1.68 -1.796 -1.521 -1.904 -1.685 -2.481 -2.391 -4.62 -4.536Be -3.699 -3.413 -3.412 -3.056 -3.699 -3.406 -4.668 -4.24 -7.528 -7.406B -6.691 -6.612 -6.296 -6.244 -6.691 -6.682 -7.84 -7.646 -11.726 -11.773Na -1.322 -1.116 -1.201 -0.974 -1.322 -1.112 -4.701 -4.492 -15.925 -15.915Mg -1.614 -1.152 -1.422 -0.902 -1.614 -1.154 -5.343 -4.949 -18.121 -17.919Al -3.745 -3.058 -3.489 -2.714 -3.745 -3.103 -7.765 -7.049 -22.134 -21.773Si -5.393 -5.351 -5.094 -5.052 -5.393 -5.299 -9.543 -9.68 -25.89 -25.908K -1.232 -0.977 -1.133 -0.818 -1.232 -0.975 -7.092 -6.937 -32.652 -32.589Ca -1.663 -1.702 -1.473 -1.397 -1.663 -1.65 -8.06 -7.708 -35.485 -35.57Zn -1.201 -0.805 -0.818 -0.405 -1.201 -0.798 -8.592 -8.056 -61.628 -61.3Ga -2.899 -2.324 -2.544 -1.954 -2.899 -2.382 -10.126 -9.52 -66.906 -66.399Ge -4.499 -4.206 -4.161 -3.833 -4.499 -4.229 -11.448 -11.441 -72.126 -71.683As -4.648 -5.026 -4.278 -4.731 -4.648 -4.945 -11.764 -12.12 -75.744 -76.153Rb -0.926 -0.646 -0.814 -0.469 -0.926 -0.618 -8.185 -7.772 -87.101 -87.036Sr -1.677 -1.309 -1.488 -1.003 -1.677 -1.277 -8.095 -8.374 -91.307 -91.402Cd -0.958 -0.858 -0.563 -0.479 -0.958 -0.853 -6.97 -6.766 -130.798 -130.821In -2.793 -2.42 -2.426 -2.043 -2.793 -2.511 -8.594 -8.168 -137.051 -136.79Sn -4.149 -3.924 -3.815 -3.519 -4.149 -3.953 -9.468 -9.515 -142.951 -142.552Sb -4.463 -4.799 -4.093 -4.46 -4.463 -4.66 -9.599 -10.057 -147.587 -147.917Te -3.188 -3.314 -2.834 -3.016 -3.188 -3.268 -8.042 -8.177 -150.633 -150.804Cs -0.813 -0.504 -0.699 -0.333 -0.813 -0.437 -4.551 -3.886 -161.584 -161.519Ba -0.208 -1.365 0.024 -1.049 -0.208 -1.312 -4.588 -4.762 -166.612 -166.602Hg -0.917 -0.902 -0.543 -0.505 -0.917 -0.909 12.263 12.372 -308.363 -308.362Tl -2.569 -2.72 -2.17 -2.399 -2.569 -2.69 11.337 11.435 -316.931 -317.061Pb -3.875 -4.086 -3.49 -3.678 -3.875 -4.037 11.282 10.815 -325.376 -325.429Bi -4.717 -5.16 -4.334 -4.786 -4.717 -5.154 11.223 10.45 -333.537 -333.615Sc -4.667 -4.183 -4.366 -3.763 -3.29 -2.878 -11.312 -11.112 -40.093 -39.857Ti -6.701 -6.818 -6.296 -6.27 -4.303 -4.529 -13.529 -13.779 -44.225 -44.465Y -4.696 -4.104 -4.384 -3.668 -3.289 -2.844 -12.161 -11.813 -97.841 -97.554Zr -7.409 -7.269 -7.007 -6.778 -4.934 -4.981 -14.826 -14.851 -104.147 -104.108Nb -10.387 -10.523 -9.867 -9.918 -6.985 -7.41 -17.614 -17.889 -110.943 -110.977Mo -11.265 -11.707 -10.693 -11.062 -7.716 -8.104 -18.546 -18.813 -115.777 -115.834Ru -9.498 -10.24 -8.839 -9.597 -6.297 -7.733 -16.396 -16.939 -121.956 -122.391Continued on next page

2
Element µP BE µF EREP BE µRP BE µF ERE
RP BE µP BE+U µF EREP BE+U µT P SS µF ERE
T P SS µmBEEF µF EREmBEEF
Rh -7.321 -7.309 -6.677 -6.506 -4.927 -5.661 -14.047 -13.963 -123.897 -123.991Pd -3.924 -4.008 -3.295 -3.329 -3.061 -3.411 -10.189 -10.504 -124.77 -124.889Ag -3.0 -2.887 -2.478 -2.396 -2.708 -2.781 -9.091 -9.053 -128.335 -128.251La -4.631 -3.922 -4.265 -3.488 -3.024 -2.722 -8.023 -7.424 -173.66 -173.225Hf -7.515 -7.06 -7.109 -6.481 -5.042 -4.645 -0.351 -0.011 -260.384 -259.967Ta -9.891 -9.938 -9.399 -9.355 -6.598 -6.798 -2.094 -2.232 -269.497 -269.265Re -11.673 -12.234 -11.081 -11.474 -7.951 -8.59 -2.586 -2.987 -284.828 -284.711Os -11.221 -13.821 -10.593 -13.804 -7.776 -10.239 -1.414 -3.323 -291.189 -292.814Ir -9.401 -9.509 -8.77 -8.704 -6.671 -7.443 0.959 1.13 -296.091 -295.784Pt -6.487 -6.575 -5.834 -5.854 -5.086 -5.641 4.667 4.937 -299.885 -299.82Au -3.251 -3.608 -2.665 -3.115 -2.951 -3.324 8.892 8.668 -303.481 -303.752C -9.224 -9.041 -8.808 -8.669 -9.224 -9.069 -10.548 -10.54 -15.405 -15.242N -8.482 -8.396 -8.301 -7.956 -8.482 -8.257 -10.078 -10.218 -15.89 -15.906O -5.296 -5.071 -5.164 -4.716 -5.296 -5.097 -7.237 -7.227 -14.146 -14.106F -1.982 -1.85 -1.879 -1.656 -1.982 -1.964 -4.51 -4.409 -12.572 -12.793P -5.362 -5.547 -5.054 -5.272 -5.362 -5.45 -10.024 -9.84 -27.769 -27.902S -4.058 -3.904 -3.841 -3.621 -4.058 -3.859 -9.091 -8.903 -28.577 -28.491Cl -1.73 -1.531 -1.635 -1.371 -1.73 -1.581 -6.86 -6.999 -28.461 -28.506Se -3.476 -3.419 -3.236 -3.145 -3.476 -3.373 -10.962 -10.879 -78.331 -78.242Br -1.604 -1.373 -1.395 -1.189 -1.604 -1.413 -8.654 -9.018 -80.263 -80.1I -1.483 -1.382 -1.26 -1.199 -1.483 -1.422 -5.923 -6.25 -153.497 -153.435V -8.538 -8.414 -8.048 -7.84 -5.245 -5.705 -15.485 -15.407 -48.275 -48.239Cr -9.447 -9.001 -8.887 -8.428 -5.931 -7.736 -16.616 -16.204 -51.629 -51.838Mn -9.811 -9.101 -9.271 -8.738 -7.11 -8.23 -17.157 -16.631 -54.674 -54.865Fe -9.077 -8.501 -8.596 -8.011 -7.039 -7.314 -16.46 -15.848 -56.897 -56.463Co -8.376 -8.188 -7.876 -7.533 -6.122 -6.337 -15.79 -15.601 -58.893 -58.915Ni -7.278 -6.915 -6.757 -6.363 -5.539 -5.416 -14.815 -14.428 -60.806 -60.529Cu -3.81 -3.506 -3.307 -2.99 -3.373 -3.243 -11.163 -10.928 -60.624 -60.321W -11.589 -12.593 -11.051 -12.076 -8.108 -9.06 -3.176 -3.858 -278.013 -278.279

94
Paper IITwo-Dimensional Metal Dichalcogenides and Oxides for HydrogenEvolution: A Computational Screening ApproachM. Pandey, A. Vojvodic, K. S. Thygesen and K. W. Jacobsen The Journal ofPhysical Chemistry Letters 6 (9), 1577-1585 (2015)

Two-Dimensional Metal Dichalcogenides and Oxides for HydrogenEvolution: A Computational Screening ApproachMohnish Pandey,† Aleksandra Vojvodic,‡ Kristian S. Thygesen,†,§ and Karsten W. Jacobsen*,†
†Center for Atomic-Scale Materials Design, Department of Physics, Technical University of Denmark, DK-2800 Kongens Lyngby,Denmark‡SUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park,California 94025, United States§Center for Nanostructured Graphene (CNG), Department of Physics, Technical University of Denmark, DK-2800 Kongens Lyngby,Denmark
*S Supporting Information
ABSTRACT: We explore the possibilities of hydrogen evolution by basal planes of2D metal dichalcogenides and oxides in the 2H and 1T class of structures using thehydrogen binding energy as a computational activity descriptor. For some groups ofsystems like the Ti, Zr, and Hf dichalcogenides the hydrogen bonding to the 2Hstructure is stronger than that to the 1T structure, while for the Cr, Mo, and Wdichalcogenides the behavior is opposite. This is rationalized by investigating shifts inthe chalcogenide p levels comparing the two structures. We find that usually for agiven material only at most one of the two phases will be active for the hydrogenevolution reaction; however, in most cases the two phases are very close in formationenergy, opening up the possibility for stabilizing the active phase. The study points tomany new possible 2D HER materials beyond the few that are already known.
Hydrogen holds a crucial place in many chemical synthesesand in energy production;1,2 however, an economical
process for hydrogen production has not been fully realized yet.One of the main challenges lies in finding a cheap catalyst thatcan evolve hydrogen efficiently. Platinum, which is known to beone of the best catalysts for hydrogen evolution, is prohibitivelyexpensive, thus precluding it to be used on large scales. Severalother metals, metal surface alloys and metal oxides, have beenstudied for the same reaction, but unfortunately most of theseare not both efficient and cheap at the same time.3−5 Onlyrecently a few and interesting candidates have been identifiedfor hydrogen evolution reaction (HER), for example, Ni2P.
6,7
Recent promising experiments on 2D metal sulfides haveopened up a new class of materials that could containpromising candidates for HER.8−12 The 2D nature of thesematerials gives additional flexibility of nanostructuring andmanipulating the structures, which is otherwise challenging inthe 3D bulk form. For example, MoS2 exists in both 2H and 1Tphases in monolayer form, whereas the 1T phase isthermodynamically unfavorable in the bulk.13 Despite the factthat the 2H-MoS2 is one of the most studied 2D sulfides forHER, it has active sites on the edges only,14,15 and the limitedactivity is ascribed to the inability of the 2H-MoS2 basal planeto adsorb hydrogen.10 The above limitation has been overcomeby contemporary experiments on 1T-MoS2 and WS2, in whichthe entire sheet has been found to be catalytically active forHER.8−10 The unusual difference between the 2H and 1T
phases thus expands the material space to more structures thatmight be relevant for the given application.In the present work, we explore the HER activity of the basal
planes of 100 dichalcogenides and oxides (MX2) in both the2H and 1T class of structures using the free energy of hydrogenadsorption as a descriptor for the activity of the material.3,16
Rather than assuming the existence of perfectly symmetrical 2Hand 1T structures, we carefully look for deviations of the atomicstructure from the perfectly symmetric 2H and 1T phases andchoose the structure with minimum energy. (We continueusing the terminology 2H and 1T for distorted structures aswell to avoid cluttering of notations.) We choose ‘M’ from a setof 25 elements (shown in the ordinate of Figure 2) and ‘X’from a set of 4 (shown in the abscissa of Figure 2) elements(chalcogens and oxygen). We find a significant difference in thehydrogen adsorption energy of the 2H and 1T phases of a givencompound; on the other hand, the 2H and 1T phases showsimilar thermodynamic stability, thus making it possible tostabilize the structure showing activity toward HER despite thefact that it is not the most stable structure. To find a correlationbetween the adsorption energies and the nature of metal atoms,we group the compounds based on the position of the metalatoms in the periodic table. For the groups showing apparent
Received: February 17, 2015Accepted: April 10, 2015Published: April 10, 2015
Letter
pubs.acs.org/JPCL
© 2015 American Chemical Society 1577 DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585

difference of the 2H and 1T phases for hydrogen adsorption,we show that the relative position of the p level of ‘X’ withrespect to the Fermi level plays a decisive role for hydrogenadsorption. On the basis of the descriptor employed to screenthe materials for HER, we point to many new possible 2D HERmaterials beyond the few that are already known.In the present work, we use GPAW,17 an electronic structure
code based on the projector-augmented wave (PAW)18
formalism. The PBE19 functional is used for the calculationof lattice constants, and the calculated lattice constants havebeen used throughout the work. Structures showing distortionshave been reoptimized, and the recalculated lattice constantsare used. We calculate the heat of formation using the fittedelemental reference phase energies (FERE) scheme employedover the PBE calculated energies, as proposed by Stevanovic etal.20 A grid spacing of 0.18 Å is used to expand the wavefunctions in real space, and a Fermi−Dirac smearing of 0.05 eVis employed to accelerate the convergence. The Brillouin zonefor the smallest unit cell (1 × 1) is sampled using aMonkhorst−Pack21 scheme with a k-point mesh of 18 × 18× 1, and for 2 × 2 unit cells, we use a 9 × 9 × 1 k-point grid. Alloptimizations are carried out using a Quasi-Newton algorithm,and the forces are converged down to 0.05 eV/Å for allrelaxations. Spin-polarized calculations are performed for thecalculation of lattice constants as well as for the adsorptionenergies. Adsorption energies are calculated using the BEEF-vdW functional.22 Uncertainties in adsorption energies areexplicitly calculated using the ensemble of functionals proposedin ref 22. The calculated uncertainties are used to estimate theprobability that a given material will have the free-energydescriptor for HER lying within a given range. The calculatedprobabilities help to rank the different materials based on theirsuitability23 for HER. We add several corrections to thecalculated total energy differences to estimate the adsorption
free energy. The zero point energy corrections to the energiesof all systems are to a first order approximation taken to be thesame as the ones calculated for the 1T-MoS2 monolayerstructure. We get the zero-point correction of the adsorbedhydrogen as 0.39 eV at the standard state. We ignore theentropic corrections for the adsorbed state while calculating thetotal correction as in ref 15. The zero-point energy of the H2molecule has been taken from the ref 24 and is found to be 0.54eV. The entropic correction of 0.40 eV from the gas-phase H2 istaken from ref 25. By taking the difference of the corrections inthe gas phase and the adsorbed state, ΔZPE comes out as 0.12eV and −TΔS comes out as 0.20 eV; therefore, ΔZPE − TΔScomes out to be 0.32 eV.The current work focuses on the 2H and 1T structures and
their distorted derivatives of 2D metal dichalcogenides andoxides some of which have been realized in recent experi-ments.10−12 The structural difference between the 1T and 2Hphases originates from the difference in coordination environ-ment around the metal atom. The 2H phase of MX2 has atrigonal prismatic structure with ‘M’ at the center of the prismand ‘X’ at the vertices, where the 1T phase has an octahedralcoordinated structure with ‘M’ at the center of the octahedronand ‘X’ at the vertices. Figure 1a,f shows the top view of the 2Hand 1T structure, respectively. Figure 1b−e,g representsdistorted derivatives of the 2H and 1T structures, which willbe discussed later. The significant difference in atomic structureof the two phases might lead to differences in theirthermodynamic and electronic properties. The difference inthermodynamic properties will directly influence the relativestability of the two phases, whereas different electronicproperties will have an effect on the chemical reactivity. Todetect the distortions, if any, in the 2H and 1T structures, wefollow the steps: (1) Adsorb the hydrogen in the 2 × 2 unit cellto break the symmetry of the structure and allow the structure
Figure 1. (a) Top view of a 1T monolayer (P32/m1 space group). (b) Monolayer with distortions belonging to the 1T class and P1 spacegroup withunit cell size 2 × 2. (c) Monolayer with distortions belonging to the 1T class and P1 spacegroup with unit cell size 2 × 2. (d) Monolayer withdistortions belonging to the 1T class and P1 spacegroup with unit cell size 2 × 1. (e) Monolayer with distortions belonging to the 1T class and P3m1spacegroup with unit cell size 2 × 2. (f) Top-view of a 2H monolayer (P6 m2 space group). (g) Monolayer with distortions belonging to the 2H classand P1 spacegroup with unicell size 2 × 2. Unit cells have been drawn (black solid lines) to show the size of the unit cell, and a few selected bonds(black broken lines) between metal atoms have been shown to highlight the difference between different structures.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1578

to relax. (2) Remove the hydrogen from the structure obtainedfrom step 1 and relax the structure. (3) If the structure obtainedafter step 2 is the same as the perfectly symmetric structure,then there are no distortions present or else the structure isdistorted. (4) Cases may exist in which step 2 leads to localminima in new structures; therefore, one has to compare theenergy obtained after step 2 and the energy of the perfectlysymmetric structure and choose the one with lower energy.Following the steps previously outlined, the distortions
present under HER conditions can most likely be obtained.Similar distortions in MoS2 have been explored by Kan et al.,26
but we see a wider range of distortions. Therefore, instead ofusing their terminology, we categorize the distortions in a moregeneral way based on the space group and the size of the unitcell.Figure 2a,b shows the calculated standard heats of formation
of the 2D MX2 compounds in the 2H and 1T phases. In
calculating the standard heat of formation, we neglect any zero-point or entropic correction. As can be seen from the Figurethe region of stable compounds is very similar in the twostructures. With very few exceptions, the compound that isstable (unstable) in one structure exhibits stability (instability)in the other structure as well. Figure 2c shows the difference inenthalpies of the different compounds by which we canestimate the extent to which the two phases differthermodynamically. The obtained trend in the relative stabilityof the 2H and 1T phases agrees well with the calculations ofAtaca et al.27 We see that for most of the compounds theenergy difference between the 2H and the 1T phase is smallerthan ∼0.4 eV/atom. Recent experiments on MoS2
10 and WS28
show that the distorted 1T phase despite being energeticallyhigher than the 2H phase by ∼0.3 eV/atom can be stabilized.These experiments thus suggest that the metastable phase of a
2D MX2 compound with a positive heat of formation as high as∼0.3 eV/atom relative to the stable phase can be synthesizedand stabilized under normal conditions using suitable syntheticroutes.28 Thus, the generally small energy differences shown inFigure 2c indicate that the atomic structure of 2D MX2 can betuned, if required, for the application in hand. Therefore, weexplore both the 2H and 1T class of structures of MX2 to findsuitable materials for HER.Figure 3a,b shows the energy of distorted structures with
respect to perfectly symmetric 2H and 1T structures,
respectively. The white squares denote massive reconstructionupon relaxation, thus leading to structures neither belonging tothe 2H or 1T class of structures. We do not investigate thesesystems any further. Upon analyzing the nature of reconstruc-tions in the more moderately distorted structures, it turns outthat the distortions occurring in the 1T structure can becategorized in four different symmetry groups, whereas thedistortions in the 2H structure can be captured by only onegroup. Starting with the structures with slightly displaced atomsfrom their ideal positions in the perfect 2H and 1T structures,that is, without symmetries in a 2 × 2 unit cell, upon relaxation,some compounds in 1T structure gain symmetry in such a waythat all of the symmetry operations can be captured in a 2 × 1unit cell, thus leading to reduction in the size of unit cell. This isnot the case for any of the 2H structures. Therefore, wecategorize the distorted structures based on the space groupsand the unit cell size using the tool described in ref 29. Table 1shows the categorization of the distortions based on the spacegroup and the size of the reduced unit cell. The forces cannotbe brought down to exactly zero during the optimization
Figure 2. (a,b) Heatmap of standard heat of formation (in eV/atom)of compounds in undistorted 2H and 1T structures, respectively. (c)Difference in enthalpies between the 2H and 1T structures (in eV/atom) of different compounds. Each compound is represented by asquare, and the constituent elements are represented by thecorresponding ordinate and abscissa of the square. The difference inenergies is in eV/atom.
Figure 3. (a,b) Energy of the distorted structures (eV/atom) withrespect to the perfectly symmetrical 2H and 1T structures,respectively. The white squares denote massive reconstruction uponrelaxation, thus leading to structures not belonging to the 2H and 1Tclass of structures.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1579

process; therefore, to overcome inaccuracies in the forces, weemploy a cutoff of 0.05 Å on the rotations/translations toidentify the symmetry operations. For six structures where thedifference in energy of the distorted structure and the perfectlysymmetrical structure is <0.01 eV per atom, we categorize theminto the symmetrical structure for the previously mentionedreason. We find that for all of the distorted structures in the 2Hclass, the type of distortion is similar to the one shown inFigure 1g. Therefore, we categorize them in the same class asthose that have the unit cell size of 2 × 2 and the space groupP1. Figure 1b−e shows the four different types of distortionsobserved in the 1T structure. There are subtle differences in allof these four groups. Panel b does not have any symmetry andthus belongs to the P1 group, panel c shows the distortionssimilar to panel b but has an inversion symmetry and thusbelongs to P1 as also observed by Tongay et al. for ReS2.
30 Thedistortions in panel d are such that the structure forms stripeswith periodicity of one unit cell, resulting in a unit cell size of 2× 1. Panel e has the least distortion and inherits most of thesymmetry operations from the symmetric 1T structure andbelongs to the P3m1 space group.Additionally, as previously mentioned, discarding distorted
phases that differ in energy from the perfectly symmetricstructures by <0.01 eV per atom might result in missing someof the charge density wave (CDW) phases, for example, inTiS2.
31,32 In the case of TiS2 we found that for a 12 atom unitcell (2 × 2 unit cell) the distorted and the perfectly symmetricstructure differ by only ∼0.04 eV (∼0.004 eV per atom). Itturns out that due to similar energy differences the CDWphases of other compounds, for example, TaS2, are all discardeddue to the previously mentioned reason. Discarding the CDWphases does not affect our results for the HER, which isdependent only on the energy differences, which are very smallin the previously mentioned cases.In previous works the strength of hydrogen binding on a
catalyst surface has been used as a descriptor for the ability to
Table 1. Categorization of Different Compounds Based onthe Deviation of Their Structures from Perfect 2H or 1TStructures and the Size of the Unit Cella
class MX2 group unit cell class MX2 group unit cell
2H CoS2 P1 2 × 2 2H CoSe2 P1 2 × 22H IrS2 P1 2 × 2 2H OsS2 P1 2 × 22H OsSe2 P1 2 × 2 2H PdS2 P1 2 × 22H PdSe2 P1 2 × 2 2H PdTe2 P1 2 × 22H ReO2 P1 2 × 2 2H ReS2 P1 2 × 22H ReSe2 P1 2 × 2 2H RhS2 P1 2 × 22H RhSe2 P1 2 × 2 2H RhTe2 P1 2 × 22H RuO2 P1 2 × 2 2H RuS2 P1 2 × 22H RuSe2 P1 2 × 2 2H ScS2 P1 2 × 22H ScSe2 P1 2 × 21T CoS2 P1 2 × 2 1T CrS2 P1 2 × 21T CrSe2 P1 2 × 2 1T FeS2 P1 2 × 21T IrS2 P1 2 × 2 1T IrSe2 P1 2 × 21T ReO2 P1 2 × 2 1T ReTe2 P1 2 × 21T RhS2 P1 2 × 2 1T RuS2 P1 2 × 21T RuTe2 P1 2 × 2 1T MoO2 P1 2 × 11T MoS2 P1 2 × 1 1T MoSe2 P1 2 × 11T MoTe2 P1 2 × 1 1T OsS2 P1 2 × 11T OsSe2 P1 2 × 1 1T OsTe2 P1 2 × 11T WS2 P1 2 × 1 1T WSe2 P1 2 × 11T WTe2 P1 2 × 1 1T ReS2 P1 2 × 21T ReSe2 P1 2 × 2 1T RuSe2 P1 2 × 21T TaO2 P1 2 × 2 1T CoSe2 P3m1 2 × 21T IrTe2 P3m1 2 × 2 1T NbO2 P3m1 2 × 21T OsO2 P3m1 2 × 2 1T RhSe2 P3m1 2 × 21T RuO2 P3m1 2 × 2 1T WO2 P3m1 2 × 2
aThe class represents the type of undistorted structure to which thecompound belongs, the group represents the space group of thedistorted structure as per Herman−Maugin notation, and the unit cellrepresents the size of the reduced unit cell with respect to the 1 × 1unit cell of the perfect 2H or 1T structures.
Figure 4. Adsorption energies of the individual groups of compounds. The grouping of the compounds is based on the position of the metal atom ofMX2 in the periodic table. The missing data points in the plots show the instability of those compounds toward hydrogen adsorption; that is, in thesecases hydrogen pulls out the ‘X’ atom from the monolayer and moves far from the surface. All energies shown in the ordinates are in electronvolts.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1580

evolve hydrogen, and it has been found that the optimum valueof the free energy of hydrogen adsorption (ΔGH) on thesurface of the material should be close to zero.3,8,16 The freeenergy of hydrogen adsorption comes out as a descriptor basedon the Volmer−Heyrovsky route for the HER. The stepsinvolved in the Volmer−Heyrovsky process can be writtenas33,34
+ + * → *+ −H e H (1)
* → + *2H H 22 (2)
where * denotes the active site. At zero potential the free-energy difference between H+ + e− and H2 is (by definition)zero, and the intermediate state of adsorbed hydrogen providesan effective barrier for the process, which should be as close tozero as possible. Therefore, to determine the reactivity of thebasal plane, we first calculate the hydrogen adsorption energyon different sites. The adsorption energy is calculated relative tothe hydrogen molecule and the most stable clean substratewithin the given class (1T or 2H). In all of the 1T and 2Hclasses of the MX2 structures, we find that the most favorablehydrogen adsorption site on the basal plane is on top of thechalcogen/oxygen atoms. For distorted structures, dependingon the symmetry, H will bind differently to the differentchalcogen/oxygen atoms. For further analysis we select theadsorption site with the strongest binding. We start with one-fourth (0.25 ML) of a monolayer of coverage (one hydrogenper four chalcogen/metal atom) and select only thecompounds binding hydrogen too strongly (ΔHH
ads ≥ −0.8)for higher coverages. Calculations for higher H adsorptioncoverages reveal massive reconstructions, and the finalstructures do not belong to any of the structure in the 2Hand 1T class; therefore, we choose not to explore the cases ofhigher coverage any further and focus only on one-fourth of amonolayer of coverage in the current work. To establish thetrends in the strength of hydrogen binding, we use the heat ofadsorption (total energy differences) and incorporate zero-point energies and entropic effects only in the stage ofevaluating the suitability of materials for HER.Figure 4 shows the calculated heats of hydrogen adsorption
(ΔHadsH ) on the 2H and 1T basal planes with 0.25 ML coverage
of hydrogen. Upon hydrogen adsorption, not all of the surfacesare stable; therefore, we discard the compounds (missing datapoints) in the plots that are unstable toward hydrogenadsorption, that is, in these cases hydrogen pulls out the ‘X’atom from the monolayer and moves far from the surface or thestructure massively reconstructs and transforms to a structurenot belonging to the 2H or 1T class. As can be seen, the heat ofadsorption varies widely by several electronvolts. An overalltrend is that the bonding strength is increased as theelectronegativity of the chalcogenide is increased. There isclearly no simple relation between the hydrogen bonding to the2H and 1T structures. Depending on the metal andchalcogenide in question the bonding to the 1T class may bestronger or weaker than the bonding to the 2H class.To shed some light on the chemistry behind the different
adsorption energies, we shall focus on only two of the metalgroups that stand out in Figure 4. For the metals Ti, Zr, and Hfthe bonding to the 2H structure is clearly stronger than that forthe 1T, while for the metals Cr, Mo, and W we have anopposite trend. To understand these opposite behaviors weanalyze the density of states (DOS) projected onto the ‘X’ porbital in MX2.
16 Figure 5a−d shows the DOS of pristine
monolayers. The calculated position of the p-band center (ϵp)(obtained as the first moment of the projected density ofstates) with respect to the Fermi level of the pristinemonolayers explains the difference in reactivity of the twogroups. A higher-lying p level indicates possible stronger effectsof hybridization with the hydrogen s state.16 The calculated ϵpfor MoS2 in the 1T structure lies closer to the Fermi level ascompared with the 2H structure, whereas for TiS2, the ϵp forthe 2H structure lies closer to the Fermi level as compared withthe 1T structure. Table 2 shows the adsorption energy (ΔHads
H )and center of p band for compounds selected from the groupsto which MoS2 and TiS2 belong. As can be seen from the Table,other compounds also show the same correlation between ϵpand ΔHads
H . These results show that the nature of the metalatom35 along with the symmetry of the structure has asignificant effect on the reactivity.We calculate for 0.25 ML coverage the heats of adsorption
(ΔHadsH ) including error bars (σ) with the BEEF-vdW functional
to assess the confidence interval of heats of adsorption.22 Asmentioned earlier, we add zero point and entropic correctionsof 0.32 eV in all the heats of adsorption to get the free energy ofadsorption. Here we assume that the corrections will not vary
Figure 5. (a) Density of states (DOS) plot of MoS2 and TiS2 in the2H and 1T structures. MoS2 and TiS2 belong to two different groupsas shown in Figure 4). ϵp denotes the position of the center of the pband with respect to the Fermi level. The shaded region correspondsto occupied states.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1581

much for different compounds, hence we choose the samecorrection as we have calculated for 1T-MoS2 monolayerstructure. Since the optimum value of free energy (ΔGopt) forHER is ∼0.0 eV, we consider the range of free energy from−0.5 to 0.5 eV to take into account the effect of coverage,strain, and so on.8,36 Having an allowable range of free energy,mean adsorption energies along with uncertainties allows us tocalculate the probability (P(|ΔG| ≤ 0.5)) of a material havingfree energy for HER in the given interval. Assuming a Gaussiandistribution of uncertainties around the mean value, E, of theadsorption, probabilities can be calculated as
∫πσ σ
|Δ | ≤ = −− −
+ ⎛⎝⎜
⎞⎠⎟P G
EE( 0.5)
1
2exp
2d
E
E
2 0.5
0.5 2
2(3)
The calculated probabilities will help in narrowing down thematerial space for potential experimental investigation bydiscarding the materials with very small probabilities. In Figure6, we show the compounds in the 2H and 1T class of structuresranked according to the calculated probability measure. TheFigure includes compounds with a probability as low as 0.15.This leads to 21 compounds in the 2H class of structures and26 compounds in the 1T class of structures. For eachcompound, the calculated free energy is shown together withthe error bar from the BEEF−vdW ensemble. We see that
MoS2 and WS2 appear on the list of candidates for the 1Tstructure (although not with the highest probability) in supportof the recent experiments indicating possible hydrogenevolution for these systems.8,10 The only compounds thatappear on both the 2H and 1T lists are NbS2, RhS2, RuS2, IrS2,CoS2, ScSe2, RuO2, and TaTe2, illustrating the fact that thechemical activity is very sensitive to the crystal structure.Having identified possible 2D materials with promising
binding properties for hydrogen, it is appropriate to investigatethe stability of these materials further. There are twopossibilities that may hamper the growth and stability of the2H or 1T phases of the 2D materials found to be active forHER: (1) much higher stability of the competing bulkstructures or the standard states, thus leading to thedissociation of the 2D phase into these compounds, and (2)relative stability of the 2H and 1T phases also matters. Forexample, if the 2H phase of a material is HER-active but ismuch higher in energy than the 1T structure, it is unlikely thatthe material can be synthesized and stabilized in the 2Hstructure. Therefore, the HER-active 2D materials must not lieabove a certain degree of metastability with respect to thecompeting bulk structures or the standard states, and also itshould not be energetically too high with respect to the other2D phase of the material. In the present work, we do notexplore the stability of compounds in water because a recentstudy has shown that with stabilizing agents compounds can bestabilized in water, making this criterion less important.37 Thepresence of water might also have an effect on the adsorptionenergies, but in the current work having a fairly wide window ofthe free energy of adsorption for the candidate materials, weexpect to have allowed for the effect of water.Calculated data to address the previously described issues are
collected in Table 3a,b. The second column of the Table showsthe calculated standard heats of formation, ΔH, for themonolayers (as shown for all the compounds in Figure 2) inthe 2H and 1T classes of structure, respectively. The thirdcolumn ΔHhull is calculated using structural information fromthe OQMD database.38 The OQMD database containsstandard DFT energy calculations for a large selection ofknown compounds from the ICSD database39 plus a number ofstandard structures. In the case of binary systems, this results in
Table 2. Heat of Adsorption of Hydrogen, ΔHadsH , and the
Center of the p Band, ϵp, for Compounds Belonging toDifferent Groups in the 2H and 1T Structuresa
2H ϵp ΔHadsH 1T ϵp ΔHads
H
MoS2 −2.00 1.68 ± 0.07 MoS2 −1.23 0.10 ± 0.13MoSe2 −1.74 1.82 ± 0.13 MoSe2 −1.46 0.64 ± 0.11WS2 −2.32 1.95 ± 0.08 WS2 −1.37 0.23 ± 0.14WSe2 −2.03 2.03 ± 0.14 WSe2 −1.29 0.78 ± 0.15TiS2 −1.02 −0.05 ± 0.13 TiS2 −1.45 0.40 ± 0.09TiSe2 −0.89 0.44 ± 0.12 TiSe2 −1.38 0.90 ± 0.10ZrS2 −0.96 0.11 ± 0.10 ZrS2 −1.42 0.94 ± 0.07ZrSe2 −0.80 0.51 ± 0.10 ZrSe2 −1.34 1.19 ± 0.09
aGrouping of the compounds is performed based on the group of theperiodic table to which the metal atom in MX2 belongs.
Figure 6. (a) 2H compounds having a free energy of hydrogen adsorption (ΔGadsH ) in the range of (−0.5, 0.5) eV along with uncertainties for 0.25
ML coverage and probabilities (P(|ΔG| ≤ 0.5)) as calculated from eq 3. Red error bars indicate that the structure is unstable with respect to thestandard states.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1582

Table 3. (a) Relevant Energies for Analysis of the Stabilities of the Obtained HER Candidates in the 2H-Derived Structuresa
and (b) Similar Table for the 1T Candidatesb
(a)
2H-MX2 ΔH ΔHhull ΔHhull ΔHexpt. ref 40 ΔH2H/1T P(|ΔG| ≤ 0.5)
RuS2 −0.31 −0.70 0.39 −0.71 no −0.01 1.00NiSe2 −0.21 −0.34 0.13 −0.38 no 0.17 1.00OsS2 0.34 −0.60 0.94 NA no −0.01 1.00TaO2 −2.58 −3.00 0.42 NA no −0.07 1.00ReO2 −0.91 −1.42 0.51 −1.52 no 0.05 1.00RhS2 −0.11 −0.48 0.37 NA no 0.07 1.00PdS2 0.01 −0.31 0.32 −0.28 yes 0.17 1.00NbS2 −1.21 −1.20 −0.01 NA yes −0.04 0.98ScS2 −1.46 −1.46 0.00 NA no −0.06 0.96TiS2 −1.23 −1.37 0.14 −1.41 yes 0.15 0.96TaTe2 −0.32 −0.45 0.13 NA yes 0.00 0.89CoS2 −0.33 −0.48 0.15 −0.51 no 0.01 0.86IrS2 −0.11 −0.48 0.37 −0.46 no 0.22 0.84RhSe2 −0.17 −0.45 0.28 NA no 0.07 0.81TaS2 −1.24 −1.22 −0.02 −1.22 yes −0.02 0.78ZrS2 −1.55 −1.73 0.18 −1.99 yes 0.19 0.77ScO2 −2.74 −3.17* 0.43 NA no 0.05 0.49VS2 −1.16 −1.14 −0.02 NA no −0.02 0.46ScSe2 −1.30 −1.25* −0.05 NA no −0.01 0.43CrO2 −1.99 −2.15 0.16 −2.01 no 0.03 0.37PdSe2 −0.02 −0.33 0.31 NA yes 0.22 0.37
(b)
1T-MX2 ΔH ΔHhull ΔHhull ΔHexpt. ref 40 ΔH1T/2H P(|ΔG| ≤ 0.5)
ScSe2 −1.34 −1.25* −0.09 NA no 0.01 1.00MoO2 −1.79 −1.95 0.16 −2.04 no 0.31 1.00RhS2 −0.32 −0.48 0.16 NA no −0.07 1.00IrS2 −0.30 −0.48 0.18 −0.46 no −0.22 0.99PbSe2 0.04 −0.31* 0.35 NA no −0.22 0.99PbS2 0.03 −0.32* 0.35 NA no −0.28 0.98PdO2 −0.48 −0.41 −0.07 NA no NA 0.97WO2 −1.61 −1.89 0.28 NA no 0.24 0.94CoS2 −0.34 −0.48 0.14 −0.51 no −0.01 0.94RuO2 −0.71 −0.94 0.23 −1.05 no −0.20 0.92IrO2 −0.70 −0.94 0.24 −0.86 no NA 0.92MnO2 −2.00 −1.98 −0.02 −1.80 no −0.43 0.87NiO2 −1.01 −0.79* −0.22 NA no NA 0.83CrS2 −0.77 −0.71 −0.06 NA yes 0.12 0.83MoS2 −0.66 −0.93 0.27 −0.95 yes 0.28 0.74OsO2 −0.23 −1.10 0.87 −1.02 no NA 0.65VO2 −2.47 −2.63 0.16 −2.48 no −0.10 0.43TiO2 −3.10 −3.29 0.19 −3.26 no −1.11 0.43GeSe2 −0.27 −0.34 0.07 −0.39 no NA 0.38PtO2 −0.61 −0.62 0.01 NA no NA 0.36WS2 −0.59 −0.78 0.19 −0.90 yes 0.18 0.35VTe2 −0.40 −0.45 0.05 NA yes 0.00 0.27TaTe2 −0.32 −0.45 0.13 NA yes 0.00 0.24FeSe2 −0.48 −0.56 0.08 NA no −0.05 0.23NbS2 −1.18 −1.20 0.02 NA yes 0.04 0.22FeTe2 −0.11 −0.20 0.09 −0.25 no −0.02 0.16
aΔH denotes the calculated standard heat of formation. ΔHhull denotes the heat of formation of the most stable compound (i.e., at the convex hull)in the OQMD database.38 The symbol * in superscript corresponds to the situation, where no bulk structure with the compound composition lies onthe convex hull according to the database. In that case, ΔHhull is calculated as a linear combination of several structures. ΔHhull denotes the differencebetween the two previous columns; that is, it shows how much the 2D compound lies above or below the convex hull. ΔHexpt. indicates theexperimental standard heats of formation as listed in the OQMD database. Lebegue al.40 have analyzed the possibilities for forming 2D compoundsbased on the layered character of the bulk structures and their result is also listed in the Table. ΔH2H/1T is the difference between the energies in thetwo (possibly distorted) 2H and 1T structures. Finally, P(|ΔG| ≤ 0.5) is the probability that the free energy of hydrogen adsorption lies within 0.5 eVfrom zero, as described in Figure 6. All the energies are in eV/atom. bNA in the seventh column indicates that due to massive reconstructions thecompound is discarded from the 2H class. All energies are in eV/atom.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1583

calculations of the most stable structures as a function ofrelative concentration of the two constituents identifying theso-called convex hull of lowest energy structures. For a givenMX2 compound, we extract the structure with the lowestenergy at this 1:2 composition of the M-X phase diagram fromthe database. In most cases a compound with the 1:2composition exists as the most stable one. If that is not thecase we extract the two structures that linearly combine to givethe lowest energy of the convex energy hull at the 1:2composition. We note that all structures are reoptimized andenergies are calculated with the approach we use here. Thefourth column, ΔHhull, shows our calculated heat of formationwith respect to the convex hull. If two structures are used toobtain the energy of the hull, ΔHhull, then it is indicated with anasterisk on the number. For comparison, the experimental heatsof formation for the most stable compounds are shown in thethird column when available in the OQMD database. As can beseen, the calculated heats of formation are in good agreementwith the experimental data with a RMS deviation of only 0.09eV. The fourth column shows the difference between columns1 and 2, that is, how much the energy of the 2D material isabove (or below) the energy at the convex hull. The seventhcolumn in Table 3 shows the heat of formation of the 2H (1T)class of structure with respect to the 1T (2H) class of structuresΔH2H/1T (ΔH1T/2H). We find that the energy differencebetween the catalytically active candidate and its analogue inthe other structure is usually not very large. As previouslymentioned HER-active materials like MoS2 and WS2 in the 1Tphase have a degree of metastability as high as 0.3 eV/atomwith respect to the 2H phase and lie above the hull by ∼0.3 eV/atom; nevertheless, they have been synthesized and stabilizedunder ambient conditions.10 Surprisingly, none of the otherHER-active materials differ from their corresponding 2Danalogue in energy by >0.3 eV/atom. Therefore, in the list ofproposed HER materials, if the material can be synthesized andstabilized in one of the two phases, then it is highly likely that itcan be synthesized and stabilized in the other phase as well.Some of the compounds like PdS2 and PdTe2, which have
been found to be HER-active in the current work, have alsobeen suggested to exist in monolayer form by Lebegue al.40 Ascan be seen from Table 3a,b, PdS2 and PdTe2 lie above the hullby ∼0.35 eV. Therefore, we choose a threshold of 0.4 eV forΔHhull for stability of compounds. The given criteria narrowsthe list of the candidates, specifically OsS2, ReO2, OsSe2, ScO2,and RuO2 in the 2H class of candidates and OsO2 in the 1Tclass of candidates do not fulfill this criteria. The names of thesecompounds are italicized in Table 3a,b. A few monolayers inTable 3a,b have lower energy than the energy of the convexhull. One of the reasons for this behavior might be the existenceof other more stable bulk structures than the ones consideredin the OQMD database, for example, structures obtained bystacking the 2D layers.Additionally, we also compare our findings of 2D materials
for HER with the recent study by Lebegue et al.40 that is basedon predicting the existence of 2D materials from experimentalbulk structures. The exiguous overlap between our results andthe ones by Lebegue al. arises from the fact that ourconclusions are based on thermodynamic arguments obtainedwith ab initio calculations, whereas the work of Lebegue al.relies more on heuristic arguments of the ability of cleaving abulk along a direction of weak bonding. The compounds of theMX2 class proposed in their work are all present in our work,thus supporting our approach. A few compounds in Table 3a,b
are written in bold. We select them based on the work byLebegue et al.40 by looking for “yes” in the column VI in Table3a,b because it is highly likely that they can be synthesized andstabilized with minimal effort.In the current study, we suggest several 2D materials in the
2H and 1T structures as potential candidates for the hydrogenevolution reaction. The activity of the basal plane in all of thediscovered candidates will provide a much larger number ofactive sites as compared with 2D materials like 2H MoS2, whereonly edges are active. Our analysis is using the calculatedadsorption free energy as a well-established descriptor forhydrogen evolution. We furthermore investigate the stability ofthe compounds in some detail by comparing heats of formationof both competing layered phases and bulk structures. Recentexperimental stabilization of different layered phases seem toindicate that fairly large metastability of several tenths of an eV/atom can be overcome by appropriate synthesis routes, makingit likely that many of the suggested compounds could beexperimentally synthesized. It has recently been demonstratedthat the MoS2 and WS2 in the 1T phase can evolve hydrogen,and these systems also appear in our screening, but otheridentified systems should according to the calculations providehigher activity. The calculations therefore invite furtherinvestigation of some of the best candidates suggested here.
ASSOCIATED CONTENT*S Supporting InformationLattice constants and adsorption energy of hydrogen areprovided in the tables. This material is available free of chargevia the Internet at http://pubs.acs.org.
AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] authors declare no competing financial interest.
ACKNOWLEDGMENTSWe acknowledge CASE (Catalysis for Sustainable Energy)initiative of the Danish Ministry of Science for funding theproject. The Center for Nanostructured Graphene is sponsoredby the Danish National Research Foundation, ProjectDNRF58. We thank Jens K. Nørskov and Charlie Tsai fromSUNCAT (Stanford University) for helpful discussions.
REFERENCES(1) Dressselhaus, M. S.; Thomas, I. L. Alternative EnergyTechnologies. Nature 2001, 414, 332−337.(2) Crabtree, G. W.; Dresselhaus, M. S.; Buchanan, M. V. TheHydrogen Economy. Phys. Today 2004, 57, 39−44.(3) Greeley, J.; Jaramillo, T. F.; Bonde, J. L.; Chorkendorff, I.;Nørskov, J. K. Computational High-Throughput Screening ofElectrocatalytic Materials for Hydrogen Evolution. Nat. Mater. 2006,5, 909−913.(4) Okamoto, Y.; Ida, S.; Hyodo, J.; Hagiwara, H.; Ishihara, T. J.Synthesis and Photocatalytic Activity of Rhodium-doped CalciumNiobate Nanosheets for Hydrogen Production from a Water/Methanol System without Cocatalyst Loading. J. Am. Chem. Soc.2011, 133, 18034−18037.(5) Cobo, S.; Heidkamp, J.; Jacques, P.-A.; Fize, J.; Fourmond, V.;Guetaz, L.; Jousselme, B.; Ivanova, V.; Dau, H.; Palacin, S.; et al. AJanus Cobalt-Based Catalytic Material for Electro-Splitting of Water.Nat. Mater. 2012, 11, 802−807.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1584

(6) Liu, P.; Rodriguez, J. A. Catalysts for Hydrogen Evolution fromthe [NiFe] Hydrogenase to the Ni2P (001) Surface: The Importanceof Ensemble Effect. J. Am. Chem. Soc. 2005, 127, 14871−14878.(7) Popczun, E. J.; McKone, J. R.; Read, C. G.; Biacchi, A. J.;Wiltrout, A. M.; Lewis, N. S.; Schaak, R. E. Nanostructured NickelPhosphide as an Electrocatalyst for the Hydrogen Evolution Reaction.J. Am. Chem. Soc. 2013, 135, 9267−9270.(8) Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D. C. B.; Fujita,T.; Chen, M.; Asefa, T.; Shenoy, V. B.; Eda, G.; et al. EnhancedCatalytic Activity in Strained Chemically Exfoliated WS2 Nanosheetsfor Hydrogen Evolution. Nat. Mater. 2013, 12, 850−855.(9) Lukowski, M. A.; Daniel, A. S.; Meng, F.; Forticaux, A.; Li, L.; Jin,S. Enhanced Hydrogen Evolution Catalysis from ChemicallyExfoliated Metallic MoS2 Nanosheets. J. Am. Chem. Soc. 2013, 135,10274−10277.(10) Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.;Shenoy, V. B.; Eda, G.; Chhowalla, M. Conducting MoS2 Nanosheetsas Catalysts for Hydrogen Evolution Reaction. Nano Lett. 2013, 13,6222−6227.(11) Wang, H.; Lu, Z.; Kong, D.; Sun, J.; Hymel, T. M.; Cui, Y.Electrochemical Tuning of MoS2 Nanoparticles on Three-dimensionalSubstrate for Efficient Hydrogen Evolution. ACS Nano 2014, 8, 4940−4947.(12) Jaramillo, T. F.; Jørgensen, K. P.; Bonde, J.; Nielsen, J. H.;Horch, S.; Chorkendorff, I. Identification of Active Edge Sites forElectrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007,317, 100−102.(13) Enyashin, A. N.; Yadgarov, L.; Houben, L.; Popov, I.;Weidenbach, M.; Tenne, R.; Bar-Sadan, M.; Seifert, G. New Routefor Stabilization of 1T-WS2 and MoS2 Phases. J. Phys. Chem. C 2011,115, 24586−24591.(14) Bollinger, M. V.; Jacobsen, K. W.; Nørskov, J. K. Atomic andElectronic Structure of MoS2 Nanoparticles. Phys. Rev. B 2003, 67,085410.(15) Hinnemann, B.; Moses, P. G.; Bonde, J.; Jørgensen, K. P.;Nielsen, J. H.; Horsch, S.; Chorkendorff, I.; Nørskov, J. K. BiomemeticHydrogen Evolution: MoS2 Nanoparticles as Catalyst for HydrogenEvolution. J. Am. Chem. Soc. 2005, 127, 5308−5309.(16) Tsai, C.; A.-Pedersen, F.; Nørskov, J. K. Tuning the MoS2 Edge-Site Activity for Hydrogen Evolution via Support Interactions. NanoLett. 2014, 14, 1381−1387.(17) Enkovaara, J.; Rostgaard, C.; Mortensen, J. J.; Chen, J.; Dulak,M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V.; Hansen, H. A.;et al. Electronic Structure Calculations with GPAW: A real-spaceImplementation of the Projector Augmented-wave Method. J. Phys.:Condens. Matter 2010, 22, 253202.(18) Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to theProjector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758−1775.(19) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized GradientApproximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865.(20) Stevanovic, V.; Lany, S.; Zhang, X.; Zunger, A. CorrectingDensity Functional Ttheory for Accurate Predictions of CompoundEnthalpies of Formation: Fitted Elemental-phase Reference Energies.Phys. Rev. B 2012, 85, 115104.(21) Monkhorst, H. J.; Pack, J. D. Special Points for Brillouin-zoneIntegrations. Phys. Rev. B 1976, 13, 12.(22) Wellendorff, J.; Lundgaard, K. T.; Møgelhøj, A.; Petzold, V.;Landis, D. D.; Nørskov, J. K.; Bligaard, T.; Jacobsen, K. W. DensityFunctionals for Surface Science: Exchange-correlation Model Develop-ment with Bayesian Error Estimation. Phys. Rev. B 2012, 85, 235149.(23) Medford, A. J.; Wellendorff, J.; Vojvodic, A.; Studt, F.; A.-Pedersen, F.; Jacobsen, K. W.; Bligaard, T.; Nørkov, J. K. Assessing theReliability of Calculated Catalytic Ammonia Synthesis Rates. Science2014, 345, 197.(24) Irikura, K. K. Experimental Vibrational Zero-Point Energies:Diatomic Molecules. J. Phys. Chem. Ref. Data 2007, 36, 389.
(25) Linstrom, P. J., Mallard, W. NIST Chemistry WebBook; NISTStandard Reference Database 69; National Institute of Standards andTechnology: Gaithersburg, MD, 1998.(26) Kan, M.; Wang, J. Y.; Li, X. W.; Zhang, S. H.; Li, Y. W.;Kawazoe, Y.; Sun, Q.; Jena, P. Structures and Phase Transition of aMoS2 Monolayer. J. Phys. Chem. C 2014, 118, 1515−1522.(27) Ataca, C.; ahin, H.; Ciraci, S. Stable, Single-Layer MX2Transition-Metal Oxides and Dichalcogenides in a Honeycomb-LikeStructure. J. Phys. Chem. C 2012, 116, 8983−8999.(28) Lin, Y.-C.; Dumcenco, D. O.; Huang, Y.-S.; Suenaga, K. AtomicMechanism of the Semiconducting-to-Metallic Phase Transition inSingle-Layered MoS2. Nat. Nanotechnol. 2014, 9, 391−396.(29) Stokes, H. T.; Hatch, D. M. FINDSYM: Program for Identifyingthe Space Group Symmetry of a Crystal. J. Appl. Crystallogr. 2005, 38,237−238.(30) Tongay, S.; Fan, W.; Kang, J.; Park, J.; Koldemir, U.; Suh, J.;Narang, D. S.; Liu, K.; Ji, J.; Li, J.; et al. Monolayer Behaviour in BulkReS2 Due to Electronic and Vibrational Decoupling. Nat. Commun.2014, 5, 3252.(31) Rossnage, K. On the Origin of Charge-Density Waves in SelectLayered Transition-Metal Dichalcogenides. J. Phys.: Condens. Matter2011, 23, 213001.(32) Dolui, K.; Sanvito, S. Dimensionality Driven Charge DensityWave Instability in TiS2. arXiv 2013, arXiv:1310.1866v1 [cond-mat.mes-hall].(33) Trasatti, S. Work Function, Electronegativity, and Electro-chemical Behaviour of Metals: III. Electrolytic Hydrogen Evolution inAcid Solutions. J. Electroanal. Chem. 1972, 39, 163.(34) Bockris, J. O.; Reddy, A. K. N.; Gamboa-Aldeco, M. ModernElectrochemistry 2A, 2nd ed.; Kluwer Academic/Plenum Publishers:New York, 1998.(35) (a) Tsai, C.; Chan, K.; Nørskov, J. K.; Abild-Pedersen, F.Understanding the Reactivity of Layered Transition-Metal Sulfides: ASingle Electronic Descriptor for Structure and Adsorption. J. Phys.Chem. Lett. 2014, 5, 3884−3889. (b) Tsai, C.; Chan, K.; Nørskov, J.K.; Abild-Pedersen, F. Theoretical Insights into the HydrogenEvolution Activity of Layered Transition Metal Dichalcogenides.Surf. Sci. 2015, 10.1016/j.susc.2015.01.019.(36) Seitz, L. C.; Chen, Z.; Forman, A. J.; Pinaud, B. A.; Benck, J. D.;Jaramillo, T. F. Modeling Practical Performance Limits of Photo-electrochemical Water Splitting Based on the Current State ofMaterials Research. ChemSusChem 2014, 7, 1372.(37) Mubeen, S.; Lee, J.; Singh, N.; Moskovitsb, M.; McFarland, E.W. Stabilizing Inorganic Photoelectrodes for Efficient Solar-to-Chemical Energy Conversion. Energy Environ. Sci. 2013, 6, 1633.(38) Saal, J. E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C.Materials Design and Discovery with High-Throughput DensityFunctional Theory: The Open Quantum Materials Database(OQMD). JOM 2013, 65, 1501−1509.(39) Belsky, A.; Hellenbrandt, M.; Karen, V. L.; Luksch, P. Newdevelopments in the Inorganic Crystal Structure Database (ICSD):Accessibility in Support of Materials Research and Design. ActaCrystallogr. 2002, B58, 364−369.(40) Lebegue, S.; Bjorkman, T.; Klintenberg, M.; Nieminen, R. M.;Eriksson, O. Two-Dimensional Materials from Data Filtering and AbInitio Calculations. Phys. Rev. X 2013, 3, 031002.
The Journal of Physical Chemistry Letters Letter
DOI: 10.1021/acs.jpclett.5b00353J. Phys. Chem. Lett. 2015, 6, 1577−1585
1585

Correction to “Two-Dimensional Metal Dichalcogenides and Oxidesfor Hydrogen Evolution: A Computational Screening Approach”Mohnish Pandey, Aleksandra Vojvodic, Kristian S. Thygesen, and Karsten W. Jacobsen*J. Phys. Chem. Lett. 2015, 6(9), 1577−1585. DOI: 10.1021/acs.jpclett.5b00353
The estimation of the correction to the calculated hydrogenadsorption energy to obtain the free energy of adsorption
is wrong. We state in the present version of the manuscript(pg 1578 column 2) that the zero-point correction for theadsorbed hydrogen is 0.39 eV and that for the H2 moleculeis 0.54 eV. The correct values should be 0.20 and 0.27 eV.This means that the correction to the energy becomes 0.26 eVinstead of the quoted 0.32 eV. The conclusions of the Letter areessentially unchanged because the error is smaller than thecalculated error bars on the individual calculated heats offormation and much smaller than the energy window of 0.5 eV
used for identifying good HER candidates. Furthermore, theestimated correction is just an estimate based on a single system(MoS2) and can be expected to have some variation from systemto system that is not taken into account. However, because of thechange in the correction term Figure 6 and the last column ofTable 3 change in detail. The changes are fairly small, but becausethe probability factor P is used for ordering, some of the systemsare swapped in Figure 6 and Table 3. We have included thefigure and table the way they would look if a correction value of0.26 eV is used instead.
Figure 6.
Table 3
2 H-MX2 ΔH ΔHhulla δHhull ΔHexpt. ref 1 ΔH2H/1T P(|Δ G| ≤ 0.5)
RuS2 −0.31 −0.70 0.39 −0.71 no −0.01 1.00NiSe2 −0.21 −0.34 0.13 −0.38 no 0.17 1.00OsS2 0.34 −0.60 0.94 NA no −0.01 1.00ReO2 −0.91 −1.42 0.51 −1.52 no 0.05 1.00TaO2 −2.58 −3.00 0.42 NA no −0.07 1.00PdS2 0.01 −0.31 0.32 −0.28 yes 0.17 1.00NbS2 −1.21 −1.20 −0.01 NA yes −0.04 1.00RhS2 −0.11 −0.48 0.37 NA no 0.07 0.99ScS2 −1.46 −1.46 0.00 NA no −0.06 0.99TiS2 −1.23 −1.37 0.14 −1.41 yes 0.15 0.98TaTe2 −0.32 −0.45 0.13 NA yes 0.00 0.96TaS2 −1.24 −1.22 −0.02 −1.22 yes −0.02 0.93IrS2 −0.11 −0.48 0.37 −0.46 no 0.22 0.92RhSe2 −0.17 −0.45 0.28 NA no 0.07 0.92ZrS2 −1.55 −1.73 0.18 −1.99 yes 0.19 0.91
Addition/Correction
pubs.acs.org/JPCL
© 2015 American Chemical Society 2669 DOI: 10.1021/acs.jpclett.5b01299J. Phys. Chem. Lett. 2015, 6, 2669−2670

REFERENCES(1) Lebegue, S.; Bjorkman, T.; Klintenberg, M.; Nieminen, R. M.;Eriksson, O. Two-Dimensional Materials from Data Filtering andAb Initio Calculations. Phys. Rev. X 2013, 3, 031002.
2 H-MX2 ΔH ΔHhulla δHhull ΔHexpt. ref 1 ΔH2H/1T P(|Δ G| ≤ 0.5)
CoS2 −0.33 −0.48 0.15 −0.51 no 0.01 0.90ScSe2 −1.30 −1.25* −0.05 NA no −0.01 0.60PdSe2 −0.02 −0.33 0.31 NA yes 0.22 0.57VS2 −1.16 −1.14 −0.02 NA no −0.02 0.52CrO2 −1.99 −2.15 0.16 −2.01 no 0.03 0.47ScO2 −2.74 −3.17* 0.43 NA no 0.05 0.40HfS2 −1.62 −1.80 0.18 NA yes 0.22 0.26FeS2 −0.54 −0.73 0.19 −0.59 no 0.05 0.06
1 T-MX2 ΔH ΔHhulla δHhull ΔHexpt. ref 1 ΔH1T/2H P(|ΔG| ≤ 0.5)
ScSe2 −1.34 −1.25* −0.09 NA no 0.01 1.00RhS2 −0.32 −0.48 0.16 NA no −0.07 1.00IrS2 −0.30 −0.48 0.18 −0.46 no −0.22 1.00PbSe2 0.04 −0.31* 0.35 NA no −0.22 1.00MoO2 −1.79 −1.95 0.16 −2.04 no 0.31 1.00PbS2 0.03 −0.32* 0.35 NA no −0.28 0.99CoS2 −0.34 −0.48 0.14 −0.51 no −0.01 0.98PdO2 −0.48 −0.41 −0.07 NA no NA 0.93MnO2 −2.00 −1.98 −0.02 −1.80 no −0.43 0.90WO2 −1.61 −1.89 0.28 NA no 0.24 0.88CrS2 −0.77 −0.71 −0.06 NA yes 0.12 0.87MoS2 −0.66 −0.93 0.27 −0.95 yes 0.28 0.87RuO2 −0.71 −0.94 0.23 −1.05 no −0.20 0.86IrO2 −0.70 −0.94 0.24 −0.86 no NA 0.85OsO2 −0.23 −1.10 0.87 −1.02 no NA 0.76NiO2 −1.01 −0.79* −0.22 NA no NA 0.70TiO2 −3.10 −3.29 0.19 −3.26 no −1.11 0.54WS2 −0.59 −0.78 0.19 −0.90 yes 0.18 0.52PtO2 −0.61 −0.62 0.01 NA no NA 0.50GeSe2 −0.27 −0.34 0.07 −0.39 no NA 0.47TaTe2 −0.32 −0.45 0.13 NA yes 0.00 0.34VO2 −2.47 −2.63 0.16 −2.48 no −0.10 0.32VTe2 −0.40 −0.45 0.05 NA yes 0.00 0.30NbS2 −1.18 −1.20 0.02 NA yes 0.04 0.30FeSe2 −0.48 −0.56 0.08 NA no −0.05 0.26FeS2 −0.61 −0.73 0.12 −0.59 no −0.06 0.21FeTe2 −0.11 −0.20 0.09 −0.25 no −0.02 0.20CrSe2 −0.63 −0.46 0.17 NA yes 0.02 0.18SnO2 −1.33 −2.10 0.77 −1.99 no NA 0.18GeS2 −0.42 −0.55 0.13 −0.42 no NA 0.17
aThe symbol * in superscript corresponds to the situation where no bulk structure with the compound composition lies on the convex hullaccording to the database. In that case, ΔHhull is calculated as a linear combination of several structures.
Table 3 Continued
The Journal of Physical Chemistry Letters Addition/Correction
DOI: 10.1021/acs.jpclett.5b01299J. Phys. Chem. Lett. 2015, 6, 2669−2670
2670

Supplementary Information: Two-dimensional
Metal Dichalcogenides and Oxides for Hydrogen
Evolution: A Computational Screening Approach
Mohnish Pandey,† Aleksandra Vojvodic,‡ Kristian S. Thygesen,†,¶ and Karsten
W. Jacobsen∗,†
Center for Atomic-scale Materials Design, Department of Physics, Technical University of
Denmark, DK - 2800 Kongens Lyngby, Denmark, SUNCAT Center for Interface Science
and Catalysis, SLAC National Accelerator Laboratory 2575 Sand Hill Road, California
94025, United States, and Center for Nanostructured Graphene (CNG), Department of
Physics, Technical University of Denmark, DK - 2800 Kongens Lyngby, Denmark
E-mail: [email protected]
∗To whom correspondence should be addressed†Center for Atomic-scale Materials Design, Department of Physics, Technical University of Denmark, DK
- 2800 Kongens Lyngby, Denmark‡SUNCAT Center for Interface Science and Catalysis, SLAC National Accelerator Laboratory 2575 Sand
Hill Road, California 94025, United States¶Center for Nanostructured Graphene (CNG), Department of Physics, Technical University of Denmark,
DK - 2800 Kongens Lyngby, Denmark

Table 1: Adsorption energies ∆HHads (in eV) and lattice constants a (in angstrom)
of the compounds with 2H structure and 2×2 unitcell as shown in Figure 4.
2H-MX2 ∆HHads a 2H-MX2 ∆HH
ads a 2H-MX2 ∆HHads a
ScS2 -0.072 7.56 ScSe2 0.205 7.88 CoS2 -0.114 6.45CoSe2 0.425 6.72 RuO2 -0.986 5.83 RuS2 -0.231 6.71RuSe2 0.389 6.94 RuTe2 0.406 7.4 RhS2 -0.425 6.83RhSe2 0.075 7.12 RhTe2 0.727 7.57 PdS2 -0.255 7.81PdSe2 0.22 8.04 ReO2 -0.295 5.6 ReS2 0.897 6.31ReSe2 1.279 6.9 ReTe2 1.653 7.42 OsS2 -0.35 6.77OsSe2 0.409 7.03 OsTe2 1.058 7.5 IrS2 -0.384 6.85IrSe2 0.04 7.11 PtTe2 0.573 7.84 PbO2 -1.778 6.47ScTe2 0.934 7.26 TiS2 -0.05 6.72 TiSe2 0.436 6.98TiTe2 0.683 7.49 VO2 -1.173 5.53 VS2 0.216 6.35VSe2 0.806 6.71 VTe2 1.043 7.2 CrO2 0.259 5.19CrS2 1.228 6.09 CrSe2 1.564 6.41 CrTe2 1.447 6.95MnSe2 1.926 6.72 MnTe2 3.009 7.35 FeO2 -1.436 5.44FeS2 0.347 6.32 FeTe2 1.684 7.15 CoO2 -1.788 5.52CoTe2 0.77 7.25 NiSe2 -0.348 7.03 NiTe2 0.693 7.44ZrS2 0.108 7.14 ZrSe2 0.511 7.4 ZrTe2 0.698 7.84NbS2 -0.038 6.73 NbSe2 0.361 6.94 NbTe2 0.51 7.37MoO2 1.263 5.65 MoS2 1.681 6.35 MoSe2 1.824 6.66MoTe2 1.742 7.09 PdTe2 0.421 8.05 HfS2 0.302 7.08HfSe2 0.643 7.35 HfTe2 0.755 7.82 TaO2 -0.467 5.96TaS2 0.113 6.72 TaSe2 0.498 6.94 TaTe2 0.487 7.39IrTe2 0.022 7.6 WO2 1.842 5.68 WS2 1.95 6.36WSe2 2.033 6.66 WTe2 1.874 7.1 ScO2 -0.829 6.48

Table 2: Adsorption energies ∆HHads (in eV) and lattice constants a (in angstrom)
of the compounds with 1T structure and 2×2 unitcell as shown in Figure 4.
1T-MX2 ∆HHads a 1T-MX2 ∆HH
ads a 1T-MX2 ∆HHads a
CoS2 -0.008 6.41 CoSe2 0.486 6.71 CoTe2 0.647 7.26CrO2 -1.462 5.85 FeO2 -1.196 5.65 FeS2 0.354 6.39FeSe2 0.54 6.76 FeTe2 0.601 7.28 GeO2 1.561 5.82GeS2 0.487 6.87 GeSe2 0.26 7.26 HfO2 2.104 6.46HfS2 1.179 7.3 HfSe2 1.41 7.53 HfTe2 1.318 7.97IrO2 -0.59 6.33 IrS2 -0.137 7.12 IrSe2 0.714 7.43IrTe2 0.662 7.78 MnO2 -0.173 5.81 MnSe2 1.042 6.97MnTe2 2.882 7.48 MoO2 -0.424 5.83 MoS2 0.096 6.34MoSe2 0.643 6.58 MoTe2 0.909 6.98 NbS2 0.376 6.79NbSe2 0.714 6.96 NbTe2 0.47 7.29 NiO2 -0.686 5.71NiS2 0.447 6.74 NiSe2 0.756 7.09 NiTe2 0.994 7.56OsO2 0.115 6.23 OsS2 0.999 6.97 OsTe2 1.223 7.72PbO2 -1.017 6.84 PbSe2 -0.127 8.02 PdO2 -0.515 6.23PdS2 0.37 7.1 PdSe2 0.813 7.46 PdTe2 0.659 8.05PtO2 0.242 6.35 PtS2 0.839 7.14 PtSe2 1.009 7.49PtTe2 0.966 8.03 ReO2 0.997 5.63 ReS2 1.485 6.16ReSe2 1.593 6.33 ReTe2 1.531 6.81 RhO2 -1.285 6.23RhS2 -0.151 7.01 RhSe2 0.406 7.18 RhTe2 0.607 7.59RuO2 -0.563 6.17 RuS2 0.509 6.78 RuSe2 0.862 6.92RuTe2 0.731 7.54 ScO2 -2.259 6.49 ScS2 -0.926 7.48ScSe2 -0.338 7.71 ScTe2 0.57 7.64 SnO2 0.437 6.51SnS2 0.877 7.39 SnSe2 0.778 7.72 SnTe2 0.44 8.24TaO2 0.893 6.14 TaS2 1.041 6.8 TaSe2 0.794 6.99TaTe2 0.327 7.36 TiO2 0.218 6.02 TiS2 0.402 6.89TiSe2 0.901 7.08 TiTe2 1.083 7.51 VO2 -0.857 5.82VS2 0.522 6.35 VSe2 0.69 6.74 VTe2 0.52 7.21WO2 -0.58 5.83 WS2 0.233 6.39 WSe2 0.789 6.62WTe2 0.79 7.0 ZrO2 1.706 6.49 ZrS2 0.941 7.35ZrSe2 1.19 7.58 ZrTe2 1.187 7.96 CoO2 -1.55 5.68PbS2 -0.111 7.68 CrTe2 0.639 7.36 MnS2 0.469 6.75CrS2 -0.089 6.67 CrSe2 0.41 6.87 OsSe2 1.149 7.19

109
Paper IIINew Light-Harvesting Materials Using Accurate and Efficient BandgapCalculationsI. E. Castelli, F. Hüser, M. Pandey, H. Li, K. S. Thygesen, B. Seger, A. Jain,K. A. Persson, G. Ceder and K. W. Jacobsen Advanced Energy Materials 5(2) (2015)

FULL P
APER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim (1 of 7) 1400915wileyonlinelibrary.com
New Light-Harvesting Materials Using Accurate and Effi cient Bandgap Calculations
Ivano E. Castelli ,* Falco Hüser , Mohnish Pandey , Hong Li , Kristian S. Thygesen , Brian Seger , Anubhav Jain , Kristin A. Persson , Gerbrand Ceder , and Karsten W. Jacobsen
the search for stable binary and ternary alloys, [ 1 ] batteries, [ 2 ] carbon capture and storage, [ 3 ] photovoltaics, [ 4,5 ] dye sensitized solar cells, [ 6 ] and water splitting mate-rials [ 7,8 ] has been guided by computational studies. The huge amount of data pro-duced during these studies has been col-lected in several databases, for example, the Materials Project database, [ 9 ] the AFLOWLIB consortium [ 1 ] and the Compu-tational Materials Repository. [ 10 , 11 ]
Experimental data are also collected into databases such as the Inorganic Crystal Structure Database (ICSD) [ 12 ] and the Landolt-Börnstein database [ 13 ] : the former contains around 160 000 crystal structures, the latter collects the electronic, magnetic, thermodynamic properties of 250 000 compounds. The ICSD database is one of the most complete repositories for crystal information. Despite this, the electronic properties are not always available and so
they are not included. One of the tasks for computational condensed matter sci-
entists is to fi ll in the missing information in experimental databases. In this paper, we present the calculations of around 2400 bandgaps of known materials using the GLLB-SC poten-tial by Gritsenko, van Leeuwen, van Lenthe, and Baerends, [ 14 ] (GLLB) adapted by Kuisma et al. [ 15 ] to include the correlation for solids (-SC). The GLLB-SC potential is implemented in the framework of density functional theory (DFT) in the electronic structure code GPAW. [ 16,17 ] The structures under investigation are obtained from the Materials Project database. [ 9 ] As of March 2014, it contains around 50 000 structures optimized with DFT from the ICSD entries. We then compare the bandgaps of 20 compounds calculated with different methods, namely local density approximation (LDA), GLLB-SC, GW approximations (G 0 W 0 , GW 0 , and GW) and the range-separated hybrid func-tional by Heyd, Scuseria, and Ernzerhof (HSE06). At the end, we apply a screening procedure, discussed in detail and used in previous works, [ 7,8 ] to fi nd new light harvesting materials suit-able for water splitting devices.
2. The Calculation of Bandgaps
Experimental databases mostly contain information about the crystal structure of materials. It is more complicated to
Dr. I. E. Castelli, Dr. F. Hüser, M. Pandey, Dr. H. Li, Prof. K. S. Thygesen, Prof. K. W. Jacobsen Center for Atomic-scale Materials Design Department of Physics Technical University of Denmark Kongens Lyngby , DK 2800 , Denmark E-mail: [email protected] Dr. B. Seger Center for Individual Nanoparticle Functionality Department of Physics Technical University of Denmark Kongens Lyngby , DK 2800 , Denmark Dr. A. Jain, Dr. K. A. Persson Computational Research Division Lawrence Berkeley National Laboratory Berkeley , CA 94720 , USA Prof. G. Ceder Massachusetts Institute of Technology Cambridge , MA 02139 , USA
DOI: 10.1002/aenm.201400915
Electronic bandgap calculations are presented for 2400 experimentally known materials from the Materials Project database and the bandgaps, obtained with different types of functionals within density functional theory and (partial) self-consistent GW approximation, are compared for 20 randomly chosen compounds forming an unconventional set of ternary and quaternary materials. It is shown that the computationally cheap GLLB-SC potential gives results in good agreement (around 15%) with the more advanced and demanding eigenvalue-self-consistent GW. This allows for a high-throughput screening of materials for different applications where the bandgaps are used as descriptors for the effi ciency of a photoelectrochemical device. Here, new light harvesting materials are proposed to be used in a one-photon photo-electrochemical device for water splitting by combining the estimation of the bandgaps with the stability analysis using Pourbaix diagrams and with the evaluation of the position of the band edges. Using this methodology, 25 candidate materials are obtained and 5 of them appear to have a realistic pos-sibility of being used as photocatalyst in a one-photon water splitting device.
1. Introduction
High-throughput materials design is becoming more and more important in materials science thanks to theory develop-ments that make computer simulations more reliable, and to an increase in computational resources. During the last decade,
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de

FULL
PAPER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1400915 (2 of 7) wileyonlinelibrary.com
obtain access to information about the electronic structure of compounds. The bandgap is a key discriminating property for a large number of applications, including solar absorbers, thermoelectrics, transparent conductors, contact and buffer layers, etc. In recent works, [ 7,8 ] the bandgap has been used as a descriptor for the effi ciency of a light absorber. In this part, we calculate the electronic bandgaps of experimentally known compounds. All the structures investigated here are available in the Materials Project database [ 9 ] and have been previously optimized using the generalized gradient approximation (GGA) functional by Perdew, Burke, and Ernzerhof (PBE), and GGA PBE+U for some of the structures. [ 18 ] While standard DFT usu-ally gives good result for the optimization of the crystal struc-ture, it fails in the calculation of bandgaps. [ 19 ] The Kohn-Sham bandgaps of semiconductors, given by the minimum energy difference between the bottom of the conduction band and the top of the valence band, are seriously underestimated because of the approximate description of the exchange-correlation functionals, the self-interaction error, [ 20 ] and the missing deriva-tive discontinuity. [ 21 ] Many-body methods, such as the GW approximation, give more reliable results with an increase (at least one order of magnitude) of the computational cost. Hybrid functionals, e.g., PBE0 or HSE06, that incorporate a portion of Hartree-Fock exact exchange, usually give reasonable results for semiconductors, [ 22 ] but fail for metals and wide bandgap insulators. [ 23,24 ] All these methods are expensive to be used in a screening project of several thousands of materials and, in particular the GW approximation, can only be effi ciently used to refi ne the results obtained with computationally cheaper approximations. [ 25 ]
Here, the bandgaps are calculated using the GLLB-SC func-tional, [ 16 ] that is an improved description of the original GLLB functional [ 14 ] adapted for solids. The GLLB functional contains by construction the evaluation of the derivative discontinuity. It is a further approximation to the KLI approximation to the exact exchange optimized effective potential (EXX-OEP). [ 26 ] The fun-damental, or quasi-particle (QP), bandgap is given as the dif-ference in the ionization potential (IP) and the electron affi nity (EA) and thus directly linked to photo-emission and inverse photo-emission measurements. The Kohn-Sham (KS) bandgap differs from the QP gap by the derivative discontinuity, Δ xc :
IP EA .gapQP
gapKSE E xc= − = + Δ (1)
GW, on the other hand, gives directly QP energies. It is impor-tant to keep in mind that the bandgaps obtained from optical measurements can be signifi cantly lower than the QP gaps due to excitonic effects, and one thus speaks of an optical bandgap instead. [ 27 ]
The GLLB-SC functional has been recently tested against other computational methods (mainly non-self-consistent G 0 W 0 ) and experiments for single metal oxides, [ 8 ] for semicon-ductors, [ 28 ] and for perovskite materials for light harvesting. [ 25 ] The GLLB-SC results are expected to be within an error of 0.5 eV. We thus expect that this accuracy is good enough for projects involving thousands of calculations required in a screening study. In addition, with the GLLB-SC is possible to calculate larger crystal structures. For example, recently, the GLLB-SC has been widely used to calculate the bandgaps of 240 organometal halide perovskites [ 29 ] which show very interesting
optical properties for light harvesting and energy conversion. [ 30 ] We note that the GLLB-SC has also given good results for the position of the d -states in noble metals such as silver. [ 31 ]
The Materials Project database is constantly updated and so far we have calculated the bandgaps of around 2400 mate-rials. Those materials have been selected because of their rela-tive simple structure, their stability and because they show a bandgap at the GGA level. Despite its low computational cost, the GLLB-SC functional is at least twice as expensive as a standard GGA calculation [ 32 ] and it is demanding to calculate the bandgaps of large crystal structures of more than 40/50 atoms. Around 6 months has been the computational time required for the bandgap calculations for the 2400 materials using Nifl heim, the supercomputer facility installed at DTU Physics. On a single core machine, the time required would have been around 16.5 years. All the calculated quasi-particle gaps, together with the corresponding ids from the Materials Project and ICSD databases and the chemical formula, are listed in the Supporting Information. In addition, this informa-tion is included and freely available in both the Materials Pro-ject database and the Computational Materials Repository.
The distribution of the bandgaps, calculated with GLLB-SC, of the 2400 materials is shown in Figure 1 (in blue). Even though very large bandgap insulators have been found, the region with a large number of materials correspond to the vis-ible light range, between 1.5 and 3.0 eV. When the stability in water at pH = 7 and at potential 0 V versus normal hydrogen electrode (NHE) is considered by means of Pourbaix dia-grams, [ 33 ] the number of materials that might be stable is signif-icantly reduced. The Pourbaix diagrams give information about the thermodynamics of the reactions, while other factors, such as kinetics and surfaces passivation, are not included. For these and other reasons, here we have considered two energy thresh-olds to defi ne if a material is stable (Δ E = 1 and 0 eV/atom, shown in red and green bars in the fi gure, where Δ E is the total energy difference between the material and the most stable phases in which it can separate). Within the energy threshold of 1 eV/atom, more than 50% of the small bandgap semiconduc-tors are unstable in water, while it seems that all the materials
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de
Figure 1. Histogram of the GLLB-SC bandgaps for all the 2400 calculated materials (in blue). We consider the two energy thresholds 1 eV/atom (in red) and 0 eV/atom (in green) for the stability in water, which is calculated at zero potential ( U = 0 V vs NHE) and neutral pH.

FULL P
APER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim (3 of 7) 1400915wileyonlinelibrary.com
with a gap larger that 10 eV are stable in water. Only around 4% of the materials are stable, when the more strict threshold of 0 eV/atom is used. This may indicate that considering a Δ E larger than zero can help to identify the materials that are experimentally observed to be stable in water.
The electronic structures of 20 materials, randomly picked from the calculated set to cover the full bandgap range and with a reasonable unit cell size, were also calculated using the non-self-consistent G 0 W 0 and the eigenvalue-self-consistent GW 0 and GW as well as the HSE06 hybrid scheme ( Figure 2 ). This unconventional set of structures contains ternary and quater-nary materials including oxides, nitrides, sulfi des, phosphates and chlorides and thus it is a broader set compared to the ones used elsewhere in the literature. [ 34 ]
QP gaps were obtained in the G 0 W 0 approximation in a plane wave representation using LDA wavefunctions and eigen-values as starting point. A detailed description of the imple-mentation of this method in GPAW can be found in ref. [ 28 ] . The initial Kohn-Sham states and energies were calculated in a plane wave basis with kinetic energies up to 600 eV. The same value is used for determining the exact exchange contributions. The G 0 W 0 self-energy was carefully converged with respect to k points, number of bands and plane wave cutoff energy for each material individually. Typically, a (7 × 7 × 7) k -point sam-pling, 100–200 eV energy cutoff and unoccupied bands up to the same energy (a few hundred bands in total) were found to be suffi cient in order to converge band gaps within less than 0.1 eV. Both, the plasmon pole approximation (PPA) by Godby and Needs [ 35 ] and the explicit frequency dependence of the die-lectric function, ε ( ω ), have been used, yielding almost identical results.
It is well known that G 0 W 0 underestimates bandgaps com-pared to experiments and better results can be obtained using (partial) self-consistent GW [ 34 ] where the LDA wavefunctions are kept fi xed while the eigenvalues are updated self-consist-ently. Recently, [ 28 ] it was shown for a set of well known semi-conductors and insulators, that the MAEs for GLLB-SC and
G 0 W 0 with respect to experiments are 0.4 and 0.3 eV, respec-tively, with a tendency of the former to overestimate the gaps, while the latter underestimates them.
Two levels of (partial) self-consistency have been investi-gated: i) in the case of GW 0 , the self-consistency in the eigen-values is performed for the Green’s functions (G) only, whereas ii) in the case of GW, the eigenvalues are updated both in G and in the dielectric matrix of the screened potential ( W ). In general, for the 20 materials described in this section, three or four iterations are necessary to converge band gaps within less than 30 meV and 50 meV for GW 0 and GW, respectively. Due to the high computational costs, the k-point mesh and energy cutoff used for GW 0 and GW are coarser than the ones used for G 0 W 0 . Typically the low convergence criteria of (3 × 3 × 3) k -point sampling and 100 eV energy cutoff are used for GW 0 and GW. The band gaps are then extrapolated to the dense k-point grids and high plane wave cut off, using the difference between the low and high convergence parameters in the G 0 W 0 calculations. For more details about GW 0 and GW, see ref. [ 34 ] and references therein.
Hybrid functional based calculations were performed with the range-separated screened-exchange HSE06 functional. [ 36,37 ] The wavefunctions were expanded in a plane-wave basis with a 700 eV cutoff. We use a Monkhorst-Pack [ 38 ] grid of 33 × ( a x −1 , a y −1 , a z −1 ) k -points, where a x , a y and a z are the lattice constants in x , y and z direction, respectively, and the Γ-point is always included. In the current work, all HSE06 calculations were per-formed non-self-consistently from the PBE ground state density and wavefunctions. Generally, there is good agreement between the non-self-consistent calculations and the self-consistently obtained results [ 24 ] which indicates that self-consistency will not be important in the current work.
For all materials in this study, comparison between the dif-ferent methods is shown by means of the direct Γ point gap, in order to avoid the need for interpolation of the band structure in the case that the minimum of the conduction band is not located at a high symmetry point in the Brillouin zone.
Figure 2 shows the bandgaps for the 20 selected materials calculated with LDA, different levels of the GW approximation, HSE06, and GLLB-SC. Only a few experimental data points are available, and mainly optical measurements which are there-fore not directly comparable with our values. Ideally photoemis-sion and inverse photoemission measurements could be used to compare to our calculated bandgaps, but these are not avail-able for this set of structures.
It is natural to divide the 20 materials into small and wide bandgap semiconductors to give a better evaluation of the signed and mean absolute and relative errors [ 39 ] for the different methods studied here ( Table 1 for the small gap set). Similar data for the wide gaps is reported in the Supporting Informa-tion together with the comparison of band structures calculated with different methods for two compounds.
As expected, for both the groups, LDA seriously underesti-mates the bandgaps. The mean absolute error (MAE) of GLLB-SC with respect to G 0 W 0 and to HSE06 is larger than 0.5 eV for the small bandgaps with a clear tendency for GLLB-SC to over-estimate the bandgaps with respect to HSE06 and to G 0 W 0 as shown by the signed error and Figure 3 a,b. G 0 W 0 and HSE06 are very close, with a MAE of approximately 0.25 eV (G 0 W 0
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de
Figure 2. Bandgaps at Γ-point of 20 structure calculated with LDA (in black), GW approximations with PPA (G 0 W 0 in red, GW 0 in purple and GW in orange), GLLB-SC (in blue), and HSE06 (in green). Both the KS bandgap and the derivative discontinuity are indicated for the GLLB-SC gaps. The materials for which the Γ-point gap corresponds to the bandgap, are indicated with *.

FULL
PAPER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1400915 (4 of 7) wileyonlinelibrary.com
underestimates with respect to HSE06). The GW 0 approxima-tion gives a MAE of around 0.5 eV for the GLLB-SC and slightly less than 0.3 eV for HSE06 and the other two GW levels. The GLLB-SC is the closest to the self-consistent GW with a MAE of 0.38 eV when compared with HSE06 and G 0 W 0 which have a MAE close to 0.5 eV.
GLLB-SC has a mean relative errors (MRE) with respect to GW equal to 15% better that the MRE for HSE06 and G 0 W 0 (16 and 18%, respectively), while GW 0 performs better with an error of 10%. The HSE06 error increases to 23% for the wide bandgap set, as shown in the Supporting Information.
The computational costs required for the methods are very different. G 0 W 0 is one or two orders of magnitude more expen-sive that GLLB-SC which is slightly more expensive than a standard GGA calculation. HSE06 is slightly more expensive than GLLB-SC but still cheaper than G 0 W 0 . The computational cost increases even further for the (partial) self-consistent GW where more iterations are needed.
The bandgaps calculated with GLLB-SC can now be used as a descriptor in a screening study. In the following section, we propose a handful of materials that can be used in a water split-ting device using a high-throughput screening approach.
3. Screening for Water Splitting Materials
The starting point of a screening study is to defi ne the descrip-tors that correlate the microscopic quantities calculated using ab-initio quantum mechanics simulations with the macroscopic properties of a material. [ 40 ] For example, the formation enthalpy of a compound can describe its stability, the bandgap its absorp-tion properties, and so on.
The set of data calculated here can provide the search space for the computational screening of materials for different applications, such as light absorbers (photovoltaics and photo-catalysis), transparent conductors, and thermoelectrics. Here, we illustrate this approach by proposing a handful of mate-rials that can be used to produce energy through photoelec-trochemical splitting of water into oxygen and hydrogen using solar light. In a water splitting device, solar energy is used to divide water into hydrogen and oxygen: the solar light is har-vested by a semiconductor and electron-hole pairs are created. The electrons and holes then reach the surface of the semicon-ductor where, if they are at the right potentials with respect to the redox levels of water, the electrons reduce the protons and the holes oxidize the water. The properties required by a semiconductor to be used in this device are: i) stability, ii) high light absorption, iii) photogenerated charges with appropriate energies. In addition iv) good electron-hole mobility, v) high catalytic activity, vi) non-toxicity, and vii) cost-effectiveness are desirable properties. The screening is based on three criteria: stability, bandgap in the visible light range, and band edges of the semiconductor well positioned versus the redox levels of water. These represent the descriptors for the properties (i–iii), i.e., a stable material with a well positioned bandgap in the vis-ible light range. A more detailed explanation of the water split-ting device can be found in previous works. [ 7,8 ]
Previous studies have described the search for new com-pounds to be used in a water splitting cell both in the perovskite crystal symmetry (cubic, [ 7,8 ] double, [ 41 ] and layered in the Rud-dlesden Popper phase [ 25 ] ) and in the oxynitride and nitride class of materials using a data mining approach. [ 42 ] Here, instead of searching for completely new materials, we consider structures
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de
Table 1. Mean absolute (signed) error, in eV, for the small bandgaps of the materials in Figure 2 calculated using LDA, GLLB-SC, HSE06, G 0 W 0 , GW 0 and GW.
xc ref LDA GLLB-SC HSE06 G 0 W 0 GW 0 GW
xc
LDA – 1.64 (−1.64) 1.21 (−1.21) 1.08 (−1.08) 1.30 (−1.30) 1.59 (−1.59)
GLLB-SC 1.64 (1.64) – 0.61 (0.43) 0.59 (0.56) 0.52 (0.34) 0.38 (0.05)
HSE06 1.21 (1.21) 0.61 (−0.43) – 0.25 (0.13) 0.29 (−0.09) 0.46 (−0.38)
G 0 W 0 1.08 (1.08) 0.59 (−0.56) 0.25 (−0.13) – 0.22 (−0.22) 0.51 (−0.51)
GW 0 1.30 (1.30) 0.52 (−0.34) 0.29 (0.09) 0.22 (0.22) – 0.29 (−0.29)
GW 1.59 (1.59) 0.38 (−0.05) 0.46 (0.38) 0.51 (0.51) 0.29 (0.29) –
Figure 3. a) HSE06, b) G 0 W 0 , c) GW 0 , and d) GW bandgaps as a func-tion of the GLLB-SC gaps. All the methods except GW underestimate the gaps with respect to the GLLB-SC. The signed error of GLLB-SC and GW is 0.05 eV.

FULL P
APER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim (5 of 7) 1400915wileyonlinelibrary.com
already optimized by nature, i.e., known to exist. While no new compounds will be proposed, this scheme has the advantage of the known synthesis procedure so that testing and validation can be prioritized.
Although all the materials studied here are experimen-tally known, i.e., they are stable, or at least metastable, little is known about their stability in contact with water. The corro-sion problem can be investigated using the so-called Pourbaix diagrams, where solid and dissolved substances are combined in a single phase diagram so that the stable species (solid and/or aqueous ion) can be determined, as a function of pH and potential. The total energies of the solid phases, taken from the ICSD and the Materials Project databases, [ 9,12 ] are obtained with DFT (using the RPBE xc-functional [ 43 ] ). Data for the dissolved ions, instead, come from experiments. [ 44,45 ] This method for evaluating stability in water has been already investigated and validated elsewhere. [ 33,46 ]
It is diffi cult to defi ne a single energy threshold under which a material is considered stable because of metastability, reac-tion kinetics, effect and passivation of the surfaces as well as inaccuracy in the calculations and experiments. Here, we con-sider a generous energy threshold of 1 eV/atom. We propose 25 compounds ( Figure 4 ), that also fulfi ll the criteria relating to the bandgap and band edges positions, stable in a potential window corresponding to the working potential of the device (bare redox levels of water plus reaction overpotentials and quasi-Fermi levels, i.e., between −0.4 and 2.2 V) and in neutral pH (pH = 7). Neutral pH is desirable because it is not harmful to environment and not corrosive however the effi ciency of the
device can be improved by operating at very acid or alkaline conditions.
The bandgaps have been calculated with the GLLB-SC func-tional. The bare energy required to split water is 1.23 eV. This energy is not enough to run the water splitting reactions and some overpotentials are needed (0.1 eV for hydrogen evolution and 0.4 eV for oxygen production [ 47 ] ). When the semiconductor is under illumination and electron-hole pairs are created, the electron and hole densities are above their equilibrium values and a single Fermi level cannot describe their populations. The quasi-Fermi levels describe these non-equilibrium popula-tions, located ≈0.25 eV below (above) the conduction (valence) bands for an undoped semiconductor and they correspond to the effective energy of the photogenerated electrons and holes. The minimum bandgap to run the water splitting reaction is at least 2 eV. The maximum realistic effi ciency of a water splitting device is around 7%. [ 48 ] This effi ciency is quite low, especially when compared with the standard technologies for photovol-taics. A higher effi ciency can be obtained using a multiphoton device [ 7,49 ] albeit increasing the technological diffi culties and thus the price of the device. In this work, we focus on the one-photon device emphasizing the simplicity of the device rather than effi ciency. [ 50 ]
There are several methods to obtain the positions of the band edges, [ 51,52 ] all computationally rather demanding and not suited to be used in a screening study. Here, the positions of the band edges have been calculated using an empirical equa-tion based on the geometrical average of the electronegativi-ties in the Mulliken scale of the individual atoms that form the
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de
Figure 4. The most stable materials with potential for one-photon water splitting. The stability in water of each material is calculated as the energy difference between the material and the most stable phases (solid and aqueous) in which it can separate in a potential range between −0.4 and 2.2 V and at pH = 7. The color scale runs from green (i.e., stable) to red (unstable compounds). In the plot, the indirect and direct positions of the valence and conduction band edges (BE) are indicated in black and red as well as the indirect and direct bandgap (BG).

FULL
PAPER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim1400915 (6 of 7) wileyonlinelibrary.com
structure. [ 53 ] For example, the valence (conduction) band edges of ZrS 2 is:
( ) /2 ,VB,CB Zr S2
gap 0E E Eχ χ= ± + (2) where χ Zr and χ S are the electronegativities of Zr and S, E gap the calculated bandgap, and E 0 = −4.5 V the difference between the normal hydrogen electrode (NHE) and vacuum level.
The screening criteria can be summarized as: stability in water: Δ E ≤ 1.0 eV/atom; bandgap: 1.7 ≤ E gap ≤ 3.0 eV; and band edges position: CB edge < −0.1 V vs NHE and VB edge > 1.6 V vs NHE.
Figure 4 shows the 25 stable semiconductors fulfi lling the screening requirements out of the 2400 calculated materials. The fi gure combines the evaluation of the stability using Pour-baix diagrams, calculated at pH = 7 and in a potential range between −0.4 and 2.2 eV, where stable and unstable compounds are indicated in green and red, and the indirect and direct posi-tions of the valence and conduction band edges are drawn with black and red lines, respectively. In particular, oxides tend to be more stable at the oxidative potential, as the O 2p orbitals, that usually form the valence band of oxides, are low in energy and thermodynamically favorable. In general, the problem of sta-bility in water is important but not crucial to the design a new light harvester material. Necessarily, the photoharvester can be protected by transparent protective shields that remove the problem of corrosion due to water and oxygen and hydrogen ions in solution. [ 54 ] On the other hand, the use of a transparent shield increases the manufacturing diffi culties and the total cost of the photodevice.
We performed a literature search for available information of the candidate materials of Figure 4 . In particular, we are inter-ested in data regarding stability in water, light absorption, and industrial applications. Five materials of Figure 4 (green under-lined formula) have a realistic possibility of success as a one-photon photocatalytic water splitting material. Ca 2 PbO 4 has an optical bandgap of approximately 1.8 eV [ 55 ] and it is used as a primer for stainless steel due to its lower toxicity compared to lead oxide. [ 56 ] Cu 2 PbO 2 was originally synthesized by Szillat et al. and they showed the material was insoluble in basic solu-tions. [ 57 ] This compound has an optical bandgap of 1.7 eV and is naturally p-type semiconductor. [ 58 ] α -AgGaO 2 has been shown to have a bandgap of 2.4 eV whereas a bandgap of 2.1–2.2 eV has been found for β-AgGaO 2 . [ 59,60 ] AgInO 2 has a bandgap of 1.9 eV. [ 60 ] AgGaO 2 and AgInO 2 have been successfully tested for photocatalytic degradation of alcohols. [ 59,60 ] NaBiO 3 has a bandgap of 2.6 eV, and has already been used for photocatalytic degradation of pollutants. [ 61 ] Using computational modeling, Liu et al. found a bandgap of 2.2 eV and a valence and conduc-tion band that straddles the water splitting redox reactions. [ 62 ]
Some materials show an experimental bandgap above 3.0 eV and thus are unsuited for an effective water splitting catalyst. For example, BaSnO 3 , which has already been proposed as a light harvester material in previous work [ 7,8 ] in which the cubic perovskites have been investigated, has a bandgap of 3.1–3.3 eV and luminesces at 1.4 eV. [ 63 ] It has been tested for photochem-ical H 2 and O 2 evolution using sacrifi cial donors, however its water splitting activity is inhibited due to defect-assisted recom-bination. [ 64 ] In 2 O 3 has a bandgap near 3.4 eV (however some papers report a bandgap of 2.8 eV [ 65 ] ) and a conduction band
near 0.00 V vs RHE. [ 66 ] It has been used as a photocatalyst [ 67 ] or to enhance the catalytic performances of photocatalysts, such as LaTiO 2 N. [ 68 ] A detailed analysis of all the candidate materials is reported in the Supporting Information.
4. Conclusions
In this work, we have calculated the bandgaps of approximately 2400 known materials, available in the Materials Project data-base, using a recently implemented functional that includes the evaluation of the derivative discontinuity.
As a fi rst step, we compared the bandgaps calculated with the GLLB-SC potential with several levels of the GW approxi-mation and hybrid HSE06 scheme for 20 materials. We showed that the agreement between GLLB-SC and GW is rather good, with a MRE of around 15% better than the agreement between G 0 W 0 (or HSE06) and GW and with a signifi cant savings in the computational cost.
Secondly, we have applied a screening procedure to the set of calculated materials with the goal of fi nding new materials to be used in a one-photon water splitting device. We combined the calculation of the bandgaps with the evaluation of Pourbaix diagrams to estimate the materials’ stability in water with the evaluation of the band edge positions to determine whether the photogenerated charges carry the energy necessary to initiate a water splitting reaction. An a posteriori literature search shows that at least fi ve of them (Ca 2 PbO 4 , Cu 2 PbO 2 , AgGaO 2 , AgInO 2 , and NaBiO 3 ) might be suitable to be used in a water splitting device and require further experimental investigation.
The calculated data may be of relevance for other applica-tions within sustainable energy materials and all the data are made available to the public in the Materials Project database and in the Computational Materials Repository.
Supporting Information Supporting Information is available from the Wiley Online Library or from the author.
Acknowledgments The authors acknowledge support from the Catalysis for Sustainable Energy (CASE) initiative funded by the Danish Ministry of Science, Technology and Innovation, from the Danish National Research Foundation for funding The Center for Individual Nanoparticle Functionality (CINF) (DNRF54) and from the Center on Nanostructuring for the Effi cient Energy Conversion (CNEEC) at Stanford University, an Energy Frontier Research Center founded by the US Department of Energy, Offi ce of Science, Offi ce of Basic Energy Sciences under award number DE-SC0001060. Work at the Lawrence Berkeley National Laboratory was supported by the Assistant Secretary for Energy Effi ciency and Renewable Energy, under Contract No. DE-AC02-05CH11231. The Materials Project work is supported by Department of Energy’s Basic Energy Sciences program under Grant No. EDCBEE.
Received: June 3, 2014 Revised: July 29, 2014
Published online: August 21, 2014
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de

FULL P
APER
© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim (7 of 7) 1400915wileyonlinelibrary.com
[1] S. Curtarolo , W. Setyawan , G. L. Hart , M. Jahnatek , R. V. Chepulskii , R. H. Taylor , S. Wang , J. Xue , K. Yang , O. Levy , M. J. Mehl , H. T. Stokes , D. O. Demchenko , D. Morgan , Comput. Mater. Sci. 2012 , 58 , 218 .
[2] G. Ceder , Y.-M. Chiang , D. R. Sadoway , M. K. Aydinol , Y.-I. Jang , B. Huang , Nature 1998 , 392 , 694 .
[3] L.-C. Lin , A. H. Berger , R. L. Martin , J. Kim , J. A. Swisher , K. Jariwala , C. H. Rycroft , A. S. Bhown , M. W. Deem , M. Haranczyk , B. Smit , Nat. Mater. 2012 , 11 , 633 .
[4] J. Hachmann , R. Olivares-Amaya , S. Atahan-Evrenk , C. Amador-Bedolla , R. S. Sanchez-Carrera , A. Gold-Parker , L. Vogt , A. M. Brockway , A. Aspuru-Guzik , J. Phys. Chem. Lett. 2011 , 2 , 2241 .
[5] M. d’Avezac , J.-W. Luo , T. Chanier , A. Zunger , Phys. Rev. Lett. 2012 , 108 , 027401 .
[6] K. B. Ørnsø , J. M. Garcia-Lastra , K. S. Thygesen , Phys. Chem. Chem. Phys. 2013 , 15 , 19478 .
[7] I. E. Castelli , D. D. Landis , K. S. Thygesen , S. Dahl , I. Chorkendorff , T. F. Jaramillo , K. W. Jacobsen , Energy Environ. Sci. 2012 , 5 , 9034 .
[8] I. E. Castelli , T. Olsen , S. Datta , D. D. Landis , S. Dahl , K. S. Thygesen , K. W. Jacobsen , Energy Environ. Sci. 2012 , 5 , 5814 .
[9] Materials Project - A Materials Genome Approach, http://material-sproject.org/ (accessed August 2014) .
[10] D. D. Landis , J. S. Hummelshoj , S. Nestorov , J. Greeley , M. Dulak , T. Bligaard , J. K. Norskov , K. W. Jacobsen , J. Comput. Sci. Eng. 2012 , 14 , 51 .
[11] Computational Materials Repository, https://cmr.fysik.dtu.dk/ (accessed August 2014) .
[12] ICSDWeb, http://www.fi z-karlsruhe.de/icsd_web.html (accessed July 2014) .
[13] The Landolt-Börnstein Database, http://www.springermaterials.com/docs/index.html (accessed May 2014) .
[14] O. Gritsenko , R. van Leeuwen , E. van Lenthe , E. J. Baerends , Phys. Rev. A 1995 , 51 , 1944 .
[15] M. Kuisma , J. Ojanen , J. Enkovaara , T. T. Rantala , Phys. Rev. B 2010 , 82 , 115106 .
[16] J. J. Mortensen , L. B. Hansen , K. W. Jacobsen , Phys. Rev. B 2005 , 71 , 35109 .
[17] J. Enkovaara , C. Rostgaard , J. J. Mortensen , J. Chen , M. Dulak , L. Ferrighi , J. Gavnholt , C. Glinsvad , V. Haikola , H. A. Hansen , H. H. Kristoffersen , M. Kuisma , A. H. Larsen , L. Lehtovaara , M. Ljungberg , O. Lopez-Acevedo , P. G. Moses , J. Ojanen , T. Olsen , V. Petzold , N. A. Romero , J. Stausholm-Møller , M. Strange , G. A. Tritsaris , M. Vanin , M. Walter , B. Hammer , H. Häkkinen , G. K. H. Madsen , R. M. Nieminen , J. K. Nørskov , M. Puska , T. T. Rantala , J. Schiøtz , K. S. Thygesen , K. W. Jacobsen , J. Phys. Con-dens. Matter 2010 , 22 , 253202 .
[18] A. Jain , S. P. Ong , G. Hautier , W. Chen , W. D. Richards , S. Dacek , S. Cholia , D. Gunter , D. Skinner , G. Ceder , K. A. Persson , APL Mater. 2013 , 1 , 011002 .
[19] J. P. Perdew , Int. J. Quantum Chem. 1985 , 28 , 497 . [20] J. P. Perdew , A. Zunger , Phys. Rev. B 1981 , 23 , 5048 . [21] R. W. Godby , M. Schlüter , L. J. Sham , Phys. Rev. Lett. 1986 , 56 , 2415 . [22] J. Heyd , J. E. Peralta , G. E. Scuseria , R. L. Martin , J. Chem. Phys.
2005 , 123 , 174101 . [23] J. E. Moussa , P. A. Schultz , J. R. Chelikowsky , J. Chem. Phys. 2012 ,
136 , 204117 . [24] J. Paier , M. Marsman , K. Hummer , G. Kresse , I. C. Gerber ,
J. G. Ángyán , J. Chem. Phys. 2006 , 124 , 154709 . [25] I. E. Castelli , J. M. García-Lastra , F. Hüser , K. S. Thygesen ,
K. W. Jacobsen , New J. Phys. 2013 , 15 , 105026 . [26] J. D. Talman , W. F. Shadwick , Phys. Rev. A 1976 , 14 , 36 . [27] G. Onida , L. Reining , A. Rubio , Rev. Mod. Phys. 2002 , 74 , 601 . [28] F. Hüser , T. Olsen , K. S. Thygesen , Phys. Rev. B 2013 , 87 , 235132 . [29] I. E. Castelli , J. M. García-Lastra , K. S. Thygesen , K. W. Jacobsen ,
APL Mater . DOI: 10.1063/1.4893495.
[30] I. Borriello , G. Cantele , D. Ninno , Phys. Rev. B 2008 , 77 , 235214 . [31] J. Yan , K. W. Jacobsen , K. S. Thygesen , Phys. Rev. B 2011 , 84 , 235430 . [32] Typically a k-point mesh fi ner than a GGA calculation is required. For
the calculations presented here, we have used a k-point sampling equal to 35 × a i −1 in each direction, where a i is the lattice constant, with Γ-point are included.
[33] I. E. Castelli , K. S. Thygesen , K. W. Jacobsen , Top. Catal. 2013 , 57 , 1 . [34] M. Shishkin , G. Kresse , Phys. Rev. B 2007 , 75 , 235102 . [35] R. W. Godby , R. J. Needs , Phys. Rev. Lett. 1989 , 62 , 1169 . [36] J. Heyd , G. E. Scuseria , M. Ernzerhof , J. Chem. Phys. 2003 , 118 , 8207 . [37] A. V. Krukau , O. A. Vydrov , A. F. Izmaylov , G. E. Scuseria , J. Chem.
Phys. 2006 , 125 , 224106 . [38] H. J. Monkhorst , J. D. Pack , Phys. Rev. B 1976 , 13 , 5188 . [39] The signed error (SE), is calculated as E En n
SE 1gapxc
gapxcref∑= − , and
the mean absolute error (MAE) as E En nMAE 1
gapxc
gapxcref∑= − , where
Egapxc indicates the bandgaps calculated with the test exchange-cor-
relation (xc) functional, Egapxcref and the bandgaps obtained with the
reference functional. The mean relative error (MRE) is given by
nE E
EnMRE 1 gap
xcgapxcref
gapxcref∑= −
. [40] S. Curtarolo , G. L. W. Hart , M. B. Nardelli , N. Mingo , S. Sanvito ,
O. Levy , Nat. Mater. 2013 , 12 , 191 . [41] I. E. Castelli , K. S. Thygesen , K. W. Jacobsen , MRS Online Proc. Lib.
2013 , 1523 . [42] Y. Wu , P. Lazic , G. Hautier , K. Persson , G. Ceder , Energy Environ. Sci.
2013 , 6 , 157 . [43] B. Hammer , L. B. Hansen , J. K. Nørskov , Phys. Rev. B 1999 , 59 , 7413 . [44] J. W. Johnson , E. H. Oelkers , H. C. Helgeson , Computers Geosci.
1992 , 18 , 899 . [45] M. Pourbaix , Atlas of Electrochemical Equilibria in Aqueous Solutions,
v. 1, Pergamon Press, London 1966 . [46] K. A. Persson , B. Waldwick , P. Lazic , G. Ceder , Phys. Rev. B 2012 , 85 ,
235438 . [47] S. Trasatti , Croat. Chem. Acta , 1990 , 63 , 313 . [48] M. R. Weber , M. J. Dignam , Int. J. Hydrogen Energy 1986 , 11 , 225 . [49] M. Gratzel , Nature 2001 , 414 , 338 . [50] B. A. Pinaud , J. D. Benck , L. C. Seitz , A. J. Forman , Z. Chen ,
T. G. Deutsch , B. D. James , K. N. Baum , G. N. Baum , S. Ardo , H. Wang , E. Millere , T. F. Jaramillo , Energy Environ. Sci. 2013 , 6 , 1983 .
[51] Y. Wu , M. K. Y. Chan , G. Ceder , Phys. Rev. B 2011 , 83 , 235301 . [52] P. G. Moses , C. G. V. de Walle , Appl. Phys. Lett. 2010 , 96 , 021908 . [53] M. A. Butler , D. S. Ginley , J. Electrochem. Soc. 1978 , 125 , 228 . [54] B. Seger , A. B. Laursen , P. C. K. Vesborg , T. Pedersen , O. Hansen ,
S. Dahl , I. Chorkendorff , Angew. Chem. Int. Ed. 2012 , 51 , 9128 . [55] R. P. Diez , E. J. Baran , A. E. Lavat , M. C. Grasselli , J. Phys. Chem.
Solids 1995 , 56 , 135 . [56] K. Rubesova , D. Sykorova , J. Sol-Gel Sci. Technol. 2009 , 49 , 228 . [57] H. Szillat , C. L. Teske , Z. Anorg. Allge. Chem. 1994 , 620 , 1307 . [58] H. Yanagi , J. Tate , R. Nagarajan , A. W. Sleight , Solid State Commun.
2002 , 122 , 295 . [59] Y. Maruyama , H. Irie , K. Hashimoto , J. Phys. Chem. B 2006 , 110 ,
23274 . [60] S. X. Ouyang , N. Kikugawa , D. Chen , Z. G. Zou , J. H. Ye , J. Phys.
Chem. C 2009 , 113 , 1560 . [61] T. Kako , Z. G. Zou , M. Katagiri , J. H. Ye , Chem. Mater. 2007 , 19 , 198 . [62] J. J. Liu , S. F. Chen , Q. Z. Liu , Y. F. Zhu , J. F. Zhang , Chem. Phys.
Lett. 2013 , 572 , 101 . [63] H. Mizoguchi , P. M. Woodward , C. H. Park , D. A. Keszler , J. Am.
Chem. Soc. 2004 , 126 , 9796 . [64] W. Zhang , J. Tang , J. Ye , J. Mater. Res. 2007 , 22 , 1859 . [65] J. Lv , T. Kako , Z. Li , Z. Zou , J. Ye , J. Phys. Chem.C 2010 , 114 , 6157 . [66] E. Y. Wang , L. Hsu , J. Electrochem. Soc. 1978 , 125 , 1328 . [67] A. Kudo , I. Mikami , J. Chem. Soc., Faraday Trans. 1998 , 94 , 2929 . [68] C. M. Leroy , A. E. Maegli , K. Sivula , T. Hisatomi , N. Xanthopoulos ,
E. H. Otal , S. Yoon , A. Weidenkaff , R. Sanjines , M. Gratzel , Chem. Commun. 2012 , 48 , 820 .
Adv. Energy Mater. 2015, 5, 1400915
www.MaterialsViews.comwww.advenergymat.de

Copyright WILEY-VCH Verlag GmbH & Co. KGaA, 69469 Weinheim, Germany, 2014.
Supporting Information
for Adv. Energy Mater., DOI: 10.1002/aenm.201400915
New Light-Harvesting Materials Using Accurate and EfficientBandgap Calculations
Ivano E. Castelli,* Falco Hüser, Mohnish Pandey, Hong Li,Kristian S. Thygesen, Brian Seger, Anubhav Jain, Kristin A.Persson, Gerbrand Ceder, and Karsten W. Jacobsen

New Light Harvesting Materials Using Accurate and EfficientBandgap Calculations - Supplementary Materials InformationIvano E. Castelli,a Falco Huser,a Mohnish Pandey,a Hong Li,a Kristian S. Thygesen,a BrianSeger,b Anubhav Jain,c Kristin Persson,c Gerbrand Ceder,d and Karsten W. Jacobsena
This Supplementary Information is divided in three sec-tions. (i) First, we expand the comparison between the differ-ent methods to calculate the bandgaps, looking at the signed,mean absolute and relative errors for the investigated set of20 materials. In addition, the band structures calculated withGLLB-SC are compared with the eigenvalues obtained fromHSE06 and from the different levels of GW, for the two mate-rials ZrS2 and BaHfN2. (ii) Second, a feasibility study of allthe candidate materials for one-photon water splitting shownin Figure 4, is reported by looking at the literature availableabout these materials. In the manuscript, only the five more re-alistic materials and the materials known to the community aredescribed. (iii) Third, the complete list of calculated bandgapswith their id are reported. These data are also freely availableon the Materials Project database [1] and on the ComputationalMaterials Repository. [2]
Comparison of Different Methods to Calculatethe Bandgaps
Figure 2 in the manuscript, shows the bandgaps for a set ofmaterials calculated with LDA, different levels of the GW ap-proximation, HSE06, and GLLB-SC. The set of materials hasbeen divided into small and wide bandgap semiconductors andonly the analysis of the errors of the small bandgap set hasbeen reported. Here, we expand the analysis also to the widebandgap materials.
Table 1 shows the mean absolute (signed) errors for thewide bandgaps. GLLB-SC is the exchange-correlation func-tional that approximates better the eigenvalue-self-consistentGW with an MAE of 1.54 eV, slightly better than G0W0 (MAEof 1.62 eV). The performance of HSE06 versus GW is con-siderably worst for the wide bandgap set compared with thesmall gap ensemble. In fact, for the small bandgaps, the MAEis 0.46 eV, worse than GLLB-SC and better than G0W0, whilefor the wide gaps, the MAE is 2.38 eV. This is even moreclear from the MRE (Table 2), where the mean relative error
a Center for Atomic-scale Materials Design, Department of Physics, Techni-cal University of Denmark, DK 2800 Kongens Lyngby, Denmark.b Center for Individual Nanoparticle Functionality, Department of Physics,Technical University of Denmark, DK 2800 Kongens Lyngby, Denmark.c Computational Research Division, Lawrence Berkeley National Laboratory,1 Cyclotron Rd, Berkeley, CA 94720, USA.d Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Fig. 1 Band structure, aligned to the valence band, of ZrS2 (id1186) calculated with GLLB-SC (blue lines), HSE06 (greensquares), G0W0 (red dots), GW0 (purple diamonds), and GW(orange triangles).
Fig. 2 Band structure, aligned to the valence band, of BaHfN2 (id10322) calculated with GLLB-SC, HSE06, and different GW levels.
for GLLB-SC and G0W0 with respect to GW is slightly bet-ter for wide gap sets compared to the small set (14.7% and15.1% for the GLLB-SC and 16.1% and 18.0% for the G0W0,respectively), while it is much worst for the wide gaps set withrespect to the small gaps set for HSE06 (22.7% and 16.4%,respectively). As expected for construction, GW0 gives animprovement of the results of G0W0, and gives the best ap-proximation to the GW gaps. Despite this, the computationalcost required by any level of GW is at this stage to high tobe used in a high-throughput screening and cheaper methodsshould be preferred. The materials thus identified can be thenstudied with more accurate methods.
For both HSE06 and GW levels, the computational costs
1

aaaaaaaaaxc
xcrefLDA GLLB-SC HSE06 G0W0 GW0 GW
LDA — 3.75 (−3.75) 2.23 (−2.23) 3.30 (−3.00) 3.57 (−3.57) 4.62 (−4.62)GLLB-SC 3.75 (3.75) — 1.62 (1.51) 1.37 (0.75) 1.39 (0.17) 1.54 (−0.87)HSE06 2.23 (2.23) 1.62 (−1.51) — 0.76 (−0.76) 1.34 (−1.34) 2.38 (−2.38)G0W0 3.00 (3.00) 1.37 (−0.75) 0.76 (0.76) — 0.58 (−0.58) 1.62 (−1.62)GW0 3.57 (3.57) 1.39 (−0.17) 1.34 (1.34) 0.58 (0.58) — 1.04 (−1.04)GW 4.62 (4.62) 1.54 (0.87) 2.38 (2.38) 1.62 (1.62) 1.04 (1.04) —
Table 1 Mean absolute (signed) error, in eV, for the wide bandgaps of the materials in Figure 2 calculated using LDA, GLLB-SC, HSE06,G0W0, GW0 and GW as exchange-correlation functionals.
aaaaaaaaaxc
xcrefLDA GLLB-SC HSE06 G0W0 GW0 GW
LDA — 56.8 (42.3) 50.3 (30.2) 47.9 (35.7) 52.4 (39.4) 56.7(45.7)GLLB-SC 156.9 (73.3) — 27.2 (22.5) 28.0 (18.5) 22.0 (17.0) 15.1 (14.7)HSE06 121.4 (44.6) 20.3 (17.9) — 13.0 (8.1) 12.8 (13.6) 16.4 (22.7)G0W0 103.1 (58.9) 19.5 (16.3) 10.0 (9.5) — 9.0 (6.2) 18.0 (16.1)GW0 124.9 (70.5) 17.9 (17.1) 12.6 (17.0) 10.1 (6.7) — 9.9 (10.6)GW 153.3 (91.0) 15.4 (18.6) 19.7 (31.0) 22.7 (19.4) 11.4 (11.9) —
Table 2 Mean relative error, in %, for the small (wide) bandgaps of the materials in Figure 2 calculated using LDA, GLLB-SC, HSE06,G0W0, GW0 and GW as exchange-correlation functionals.
2

only allow for calculations on rather coarse k-point grids. Anaccurate interpolation of the band structure from these pointsis not possible, since both methods are non-selfconsistent.Nevertheless, we can compare the band structures obtainedwith GLLB-SC with the two last occupied and two first unoc-cupied eigenvalues of some k-points (mainly high-symmetrypoints). For ZrS2 (Figure 1), the band structure and thebandgap calculated with GLLB-SC is very similar to the onesobtained from HSE06 and GW levels. Different levels of GWdiffer from each other only by a constant shift correspond-ing to the difference in the bandgaps. The situation is differ-ent for BaHfN2 (Figure 2). Whereas the valence bands arealmost identical, there are significant changes in the conduc-tion bands. Around the Γ-point, the order of the two lowestconduction bands changes from GLLB-SC through HSE06 toG0W0. For both the cases, an increase of the level of self-consistency, from G0W0 to GW, has only the effect of a con-stant shift of the unoccupied bands equal to the bandgap dif-ference. These two examples shows two cases of excellentmatching and differences between the methods, respectively.
Literature Review of the Candidate Materials
In this section, we list the information found in the literaturefor the candidate materials of Figure 4. In the manuscript,the materials with a realistic possibility of success have beendescribed together with BaSnO3 and In2O3 that are the candi-dates of the list already known to the water splitting commu-nity. We describe now the remaining materials.
• AlAgO2: Sheets et al. have shown that AlAgO2 has anoptical bandgap of 3.6 eV. [3] In an unrelated study pub-lished at almost the same time, Ouyang et al. found abandgap of 2.95 eV. [4] While there is a significant differ-ence in bandgaps, both are too large for optimal absorp-tion from the solar spectrum.
• BaCdO2: this was originally synthesized by von Schner-ing. [5] Very little is known about this material.∗
• Ba3In2O6: initially synthesized by Mader et al. [6], istoxic and harmful [7]. Very little is known about this ma-terial.
• Ba4LiCuO4(CO3)2: originally synthesized by Tams etal.. [8] Very little is known about this material.
• Ba4NaCuO4(CO3)2: this material was initially synthe-sized by Vernooy et al. [9] Very little is known about thismaterial.
• Ba2NaOsO6: this is a Mott-insulator and a ferromag-netic material, [10] which was originally synthesized by
∗Very little is known normally means it has only been synthesized once.
Stitzer et al. [11] The material is black, thus indicating itsbandgap is probably below 1.7 eV, and it is toxic. [7]
• CdIn2O4: originally synthesized by Shannon et al. [12]
This material is typically n-type and can be highly doped.Can be found in either a spinel, inverted spinel, or an or-thorhombic structure. This material has been shown tohave a bandgap of 2.67−3.24 eV. [13]
• Cs2PtBr6: this material was produced only one time, thusthere is little information on it. [14]
• Li2PbO3: there are two forms of this material, [15,16] nei-ther have been investigated thoroughly. It is toxic andharmful. [7]
• LiRhO2: Scheer et al. synthesized a α-LiRhO2. [17] Hob-bie et al. synthesized a black β -LiRhO2. [18] Little in-formation is known on the photochemical properties ofeither of these phases.
• NaBiO2: it was originally synthesized by Schwedes etal. [19] Very little information is known on this material.
• Na2PdCl4: this material is reddish brown, but slightlysoluble in water. [20]
• Na3BiO4: it was originally synthesized by Schwedes etal. [21] Little is known about this material.
• NaCoO2: Takahashi et al. showed that theoretically thebandgap should be 1.3 eV. [22] NaCoO2 can be oxidizedby iodine (redox potential 0.54 V vs NHE), thus it ishighly unlikely that this material will be stable enoughto do water oxidation. [23]
• NaRhO2: originally synthesized by Hobbie et al. [24] Itcan be oxidized by Na2S2O8, thus it is unlikely that itwill be stable during O2 evolution conditions. [25]
• KAg2AsO4: this material has only been synthesized byCurda et al. [26] There is very little information on thismaterial.
• K2PdBr4: this material has a bandgap of around 2 eV, butit is water soluble. [27]
• Sr2FeWO6: this material is black, with a bandgap of0.1 eV. [28]
Calculated Bandgaps
In this section, we report the chemical formulas, the ids, andthe bandgaps of the calculated 2400 materials. The informa-tion reported here are also available electronically in the Ma-terials Project database [1] and in the Computational MaterialsRepository. [2]
3

Form
ula
MP
IdIC
SDId
Gap
Form
ula
MP
IdIC
SDId
Gap
Form
ula
MP
IdIC
SDId
Gap
CoA
s 227
1542
613
0.1
(0.3
)Sr
2Pb
3082
810
5624
0.1
(0.1
)C
aAgP
1227
710
016
0.1
(0.3
)Sr
MgS
i15
642
4245
70.
1(0
.1)
Rh 2
S 317
173
1534
40.
1(0
.1)
SrM
gGe
1564
342
458
0.1
(0.1
)Ta
AgS
358
2184
910
0.1
(0.4
)K
2MnS
e 287
1665
456
0.1
(0.2
)N
aCuS
e74
3312
155
0.1
(0.1
)V
4As 2
O13
3244
760
781
0.1
(0.1
)L
i 2M
nO3
1898
820
2639
0.1
(0.2
)C
r 4In
CuS
e 820
809
2502
460.
1(0
.1)
YN
iSb
1152
010
5331
0.2
(1.3
)T
l 4Te
3Pb
2074
064
8621
0.2
(0.2
)In
AgT
e 222
386
1044
760.
2(0
.2)
CsM
nBr 3
2304
897
030.
2(0
.3)
ScN
iSb
3432
7669
50.
2(1
.3)
Na 4
CoO
431
593
4196
0.2
(0.2
)Y
SbPt
4964
6495
780.
2(0
.2)
RbM
nBr 3
5055
9697
040.
2(0
.3)
CsM
nI3
5406
0910
042
0.2
(0.3
)C
s 2K
4Fe 2
O5
5412
3465
942
0.2
(0.2
)C
aAgA
s56
1560
4730
0.2
(0.3
)L
i 3L
aSb 2
8405
4962
50.
2(1
.0)
Li 2
AgS
b16
238
5258
90.
2(0
.2)
Cu 2
GeS
315
252
8513
80.
2(0
.2)
Sr2S
n97
865
2722
0.2
(0.2
)C
dSb
1321
5283
10.
2(0
.5)
CuA
gO2
7237
9388
00.
2(0
.2)
CaA
sAu
3927
1079
150.
2(0
.3)
Sb10
455
402
0.2
(0.5
)Z
rSnT
e92
8980
190
0.2
(0.3
)Pb
Au 2
1987
156
261
0.2
(0.4
)L
i 2Z
nGe
1241
117
1498
0.2
(0.4
)C
aSbA
u16
245
5266
20.
2(0
.3)
VPO
418
835
8228
60.
2(0
.2)
CsI
n 354
2339
1028
670.
2(0
.4)
Nb 3
I 827
772
2576
70.
2(0
.3)
MoB
r 323
312
9422
0.2
(0.2
)C
uSe 2
O5
3199
2450
590.
2(0
.3)
MoC
l 322
853
8387
80.
3(0
.3)
YIr
3074
610
4600
0.3
(1.1
)A
s 2R
h15
954
4261
60.
3(0
.4)
Cs 6
Fe2O
554
1385
7313
40.
3(0
.3)
Ba 3
CrN
312
905
1548
020.
3(0
.5)
Sr(A
s 3Pt
2)2
1450
062
518
0.3
(0.4
)B
a(Z
nP) 2
7426
1214
50.
3(0
.8)
SiR
u18
985
209
0.3
(0.5
)H
fSnP
d11
869
1067
730.
3(1
.3)
Sb2R
u20
928
4260
80.
3(0
.6)
BaS
nHg
1181
910
6311
0.3
(0.5
)C
aPA
u10
677
5266
10.
3(0
.3)
Cs 2
CoS
e 287
7067
392
0.3
(0.3
)C
a(C
dSb)
274
3012
151
0.3
(0.4
)L
aP5
1864
9654
50.
3(1
.0)
Na 2
In51
0678
1068
570.
3(0
.4)
CaA
gSb
1121
456
982
0.4
(0.6
)C
4861
7290
0.4
(0.4
)K
3FeO
350
4570
1653
40.
4(0
.4)
LiA
lGe
5920
1520
870.
4(1
.7)
ReT
eS52
2282
721
0.4
(0.8
)T
lTe 3
Pt2
9251
7878
70.
4(0
.6)
HfC
u 2Te
336
5065
5974
0.4
(0.6
)Fe
Si87
163
3541
0.4
(0.4
)Fe
Te2
2065
342
727
0.4
(0.8
)L
iSi
795
1605
380.
4(0
.9)
GeP
tSe
2081
782
20.
5(1
.1)
Te2R
u26
710
6001
0.5
(1.3
)L
iAlS
i31
6115
2086
0.5
(2.2
)P 2
Rh
1595
342
615
0.5
(0.9
)C
dAs 2
471
2051
80.
5(1
.0)
Sr(Z
nAs)
277
7023
248
0.5
(0.5
)L
a 3M
nGaS
750
4891
3841
30.
5(0
.6)
MgB
436
565
3968
0.5
(1.0
)Sb
2Ir
1247
4262
00.
5(0
.8)
TiS 2
2156
6512
170.
5(1
.0)
SrC
uSb
1074
953
339
0.5
(0.9
)A
lVFe
257
7810
7814
0.5
(0.8
)L
iScI
328
835
7333
90.
5(0
.7)
BaB
iO3
2294
215
1894
0.5
(1.1
)A
l 2(F
eSi)
329
110
8366
40.
5(0
.6)
OsS
220
905
6477
500.
6(1
.0)
GaA
s25
3410
7946
0.6
(0.6
)Sb
2Os
2695
6477
570.
6(1
.0)
AuS
e27
9373
700
0.6
(1.5
)R
bGa 3
3149
310
3943
0.6
(0.8
)P
157
1508
730.
6(0
.6)
CoP
214
285
3831
60.
6(0
.7)
Mg 2
Ge
408
1513
890.
6(1
.6)
Cd(
PtO
2)3
8212
3540
70.
6(0
.6)
YB
4Mo
7691
2008
10.
6(0
.7)
LaT
eAs
1038
328
0231
0.6
(0.6
)Sr
(PIr
) 215
074
7353
10.
6(0
.6)
Si3A
g 3(S
nP3)
218
310
5259
50.
6(0
.8)
Si3R
u 222
192
9558
60.
6(0
.6)
BaP
3Pt 2
2837
362
520
0.6
(0.8
)Z
r 3N
i 3Sb
417
926
8799
50.
6(0
.9)
CsC
uBr 3
2733
610
184
0.6
(0.7
)B
a(C
dAs)
282
8130
917
0.6
(0.7
)M
g 2Sn
2343
1581
810.
6(1
.9)
GaA
gTe 2
4899
1561
280.
6(0
.6)
K2C
dPb
5044
9810
041
0.6
(0.7
)In
AuO
219
723
9567
20.
6(1
.5)
Ba(
GeA
s)2
2781
026
417
0.7
(0.9
)M
g 2Si
1367
1637
080.
7(2
.5)
As 2
Ru
766
4257
80.
7(1
.2)
Ca(
ZnA
s)2
9571
1000
640.
7(0
.7)
Al 2
Os
7188
5810
80.
7(1
.5)
ScSb
Pt71
7377
948
0.7
(0.7
)G
aCuT
e 238
3960
0219
0.7
(0.7
)L
iZnS
b99
1942
064
0.7
(0.7
)Y
P 598
5440
9188
0.7
(1.2
)Fe
As 2
2008
4180
50.
7(1
.0)
KA
g 3Se
297
8240
2643
0.7
(0.8
)Sr
(CdA
s)2
7771
2324
90.
7(0
.7)
SrC
uP16
321
5332
30.
7(1
.2)
CaC
uP84
3249
740
0.7
(1.6
)P 3
Au 2
2725
880
580.
7(1
.2)
Ca(
PIr)
211
168
9575
60.
7(0
.7)
CaC
rP2O
719
134
4117
430.
7(0
.7)
SiSb
Pt11
152
4131
940.
7(0
.8)
Ba(
As 3
Pt2)
214
501
6251
90.
7(0
.7)
P 2Ir
1015
544
661
0.8
(1.8
)T
lBiS
e 229
662
4331
40.
8(0
.8)
Si3N
iP4
8311
3945
20.
8(0
.9)
TiS 3
9920
4207
20.
8(0
.8)
KSn
Sb34
8640
816
0.8
(0.8
)K
Sb2
2905
580
945
0.8
(1.1
)C
a(C
dAs)
270
6710
0065
0.8
(0.8
)K
Ga 3
181
1036
490.
8(0
.9)
K2A
g 4S 3
7494
863
0.8
(0.8
)Sr
CaP
b21
166
1720
080.
8(0
.8)
TaTe
4I28
691
6753
30.
8(0
.9)
Te19
1616
900.
8(0
.8)
CaC
uAs
4120
6594
350.
8(0
.9)
CsS
cCl 3
2735
910
474
0.8
(1.1
)T
lPd 2
Se3
7038
7878
60.
8(0
.9)
Sc(A
lC) 3
4798
6604
020.
8(2
.2)
Tl(
TeM
o)3
1609
690
811
0.8
(1.0
)Sn
Te18
8365
2763
0.8
(0.8
)A
gF3
1853
680
477
0.8
(1.0
)A
g 3Te
2Au
5710
6047
880.
8(0
.9)
PtSe
211
1564
9594
0.9
(1.2
)Sr
CaS
n20
726
1720
070.
9(0
.9)
FeSe
276
063
3489
0.9
(1.5
)N
a 2Pd
3O4
2756
261
570.
9(0
.9)
P 2Pd
2826
648
163
0.9
(1.1
)Si
3P2P
t29
157
8494
40.
9(1
.5)
Rb 2
Te5
3100
230
734
0.9
(1.1
)T
l(M
oSe)
334
1153
119
0.9
(1.0
)B
aCaG
e16
252
5268
10.
9(0
.9)
BaC
aSi
1625
352
682
0.9
(0.9
)G
aAgS
e 255
1852
570
0.9
(0.9
)
4

AuS
eBr
2719
928
970.
9(1
.3)
CoP
Se10
368
5306
00.
9(1
.1)
Y3S
b 4A
u 313
654
957
0.9
(1.0
)K
ZnS
b74
3812
161
0.9
(0.9
)B
a 2C
uClO
255
1456
1038
0.9
(1.1
)K
ZnA
s74
2110
459
0.9
(0.9
)B
aCaS
n11
265
5864
10.
9(0
.9)
Na 4
FeO
319
026
2363
50.
9(0
.9)
Ta2C
rO6
3162
951
175
0.9
(0.9
)C
a 2Sb
9925
4213
50.
9(1
.0)
ZrI
323
282
3270
40.
9(1
.0)
La 2
CoI
rO6
1886
279
496
0.9
(0.9
)R
bAg 3
Te2
1048
190
872
1.0
(1.0
)A
g 2B
iO3
2355
841
5958
1.0
(1.0
)Pd
Se2
2418
1703
271.
0(1
.5)
Ca 2
Si25
1715
8275
1.0
(1.0
)T
lBiT
e 227
438
1541
21.
0(1
.0)
CdS
iAs 2
3078
6099
801.
0(1
.0)
Hg 2
GeS
e 431
6759
911
1.0
(1.0
)C
sAg 3
Se2
1623
452
558
1.0
(1.0
)T
lPt 2
Se3
5414
8778
785
1.0
(1.1
)B
i 4R
uI2
5417
7140
6949
1.0
(1.0
)C
a 3G
eO17
193
4133
831.
0(1
.0)
Ag 3
Ge 5
P 617
862
7005
51.
0(1
.2)
Ta2P
d 3Se
818
010
7331
81.
0(1
.2)
Cs 2
SnI 6
2763
622
105
1.0
(1.0
)Pt
I 328
268
4712
01.
0(1
.0)
BaA
s 231
243
4141
391.
0(1
.0)
MnP
448
710
0786
1.0
(1.1
)C
u 2O
361
1721
741.
0(1
.0)
HfS
e 315
622
4207
51.
0(1
.3)
KSn
As
3481
4081
51.
0(1
.0)
Te3A
s 248
454
097
1.0
(1.0
)C
a 2G
e30
444
854
1.0
(1.0
)Sr
(GaT
e 2) 2
6987
4116
61.
0(1
.2)
FeP 2
2002
763
3072
1.0
(1.3
)Ta
CuS
331
0266
0123
1.0
(1.0
)N
bTeI
354
0924
3537
71.
0(1
.1)
SrH
fN2
9383
8253
81.
0(1
.8)
Tl 2
(CdS
b)3
2974
176
500
1.0
(1.0
)Z
rSe 2
2076
1092
911.
1(1
.9)
Mg 3
Sb2
2646
2456
921.
1(1
.9)
PtS
288
6543
791.
1(1
.2)
TlS
e18
3665
2068
1.1
(1.4
)C
a 3Si
O11
649
4133
821.
1(1
.1)
PtO
212
8564
7320
1.1
(1.4
)R
bSc 5
Te8
1333
624
5998
1.1
(1.3
)Si
149
1505
301.
1(3
.1)
As 2
Ir15
649
4257
31.
1(1
.2)
LiA
uI4
5054
6240
6967
1.1
(1.2
)B
i 4R
uBr 2
5417
7240
6951
1.1
(1.1
)C
dP4
7904
2560
51.
1(1
.3)
LiA
s79
4326
472
1.1
(1.3
)B
a 2G
eAs 2
8195
3515
11.
1(1
.3)
Li 3
LaP
284
0749
627
1.1
(1.5
)G
eTe
938
1599
071.
1(1
.2)
GeA
s95
4886
361
1.1
(1.2
)Sc
4C3
1566
142
760
1.1
(1.1
)Si
3Os 2
1660
895
590
1.1
(1.1
)L
aAs 2
5056
4028
0294
1.1
(1.4
)N
a 3In
As 2
2162
230
0139
1.1
(1.1
)N
aNbS
e 279
3926
287
1.1
(1.5
)In
2HgT
e 419
765
2565
21.
1(1
.1)
LaZ
nAsO
5495
8916
3779
1.1
(1.1
)N
bSbR
u50
5297
8392
91.
1(2
.0)
KZ
rCuS
e 393
1880
625
1.1
(1.1
)Sb
2WO
654
1435
7559
51.
1(1
.1)
SiC
u 2Se
315
896
8823
61.
1(1
.1)
ZrT
lCuS
e 370
5082
561
1.1
(1.1
)Ti
Nb 3
O6
2969
928
0002
1.1
(1.3
)Ta
TlS
310
795
4123
851.
1(1
.1)
FeSb
S27
904
2416
11.
1(1
.2)
NaT
iCuS
350
5171
7388
61.
1(1
.1)
SiTe
Pt29
397
4004
871.
1(1
.2)
Li 3
Bi
2322
258
797
1.2
(1.8
)A
s 2O
s24
5542
610
1.2
(1.4
)Sr
2Ge
2576
5392
31.
2(1
.2)
Ca(
MgB
i)2
2920
810
0048
1.2
(1.2
)B
aLaI
430
111
4122
761.
2(1
.2)
ZrS
e 316
8365
2241
1.2
(1.4
)B
a 3P 4
1428
938
322
1.2
(1.2
)C
dTe
406
1616
931.
2(1
.2)
CoA
sS46
2743
221
1.2
(1.4
)A
gF75
9218
008
1.2
(2.6
)L
iNbS
279
3626
284
1.2
(1.9
)B
a 2G
eP2
8194
3515
01.
2(1
.4)
LaR
hO3
5163
1723
531.
2(1
.2)
PdS 2
1368
216
694
1.2
(1.9
)R
b 2N
i 3Se
418
691
7897
61.
2(1
.4)
LiB
iO3
2907
782
277
1.2
(1.3
)Sr
3GeO
3095
041
3385
1.2
(1.2
)Ti
SnPt
3084
710
5799
1.2
(1.9
)L
aZnP
O70
6085
777
1.2
(1.2
)G
a 2H
gTe 4
1633
753
575
1.2
(1.2
)L
aCuT
eS10
288
8801
21.
2(1
.2)
CsZ
rCuS
e 371
5228
0701
1.2
(1.2
)T
l 5Se
2Br
2892
175
960
1.2
(1.2
)N
a 4Fe
O4
1902
259
585
1.2
(1.2
)R
uSe 2
1922
6506
111.
2(1
.4)
InP
2035
164
0192
1.2
(1.2
)Sr
As
1049
8335
31.
2(1
.8)
SrL
aI4
5057
9341
2275
1.2
(1.2
)K
2SnB
i31
486
1076
161.
2(1
.3)
CdS
e26
9162
0439
1.3
(1.3
)Sc
N28
5715
7501
1.3
(2.2
)K
2SnT
e 528
080
3621
51.
3(1
.4)
Ba(
MgB
i)2
2920
910
0049
1.3
(1.3
)T
l 2Te
329
711
4108
951.
3(1
.5)
SrC
aGe
1241
817
2006
1.3
(1.3
)G
eAs 2
1752
423
872
1.3
(1.4
)R
b 2A
s 315
556
4093
811.
3(1
.4)
CdG
eP2
3668
4289
51.
3(1
.3)
MgP
438
442
030
1.3
(1.4
)N
aZnP
4824
1609
551.
3(1
.3)
NbS
eI3
5418
1741
0743
1.3
(1.3
)K
NbS
279
3826
286
1.3
(1.6
)N
aSb
7944
2647
31.
3(1
.4)
Ba(
CdP
) 282
7930
915
1.3
(1.4
)T
l 2Pt
2O7
1380
122
215
1.3
(1.3
)T
l 3G
eTe 3
1721
749
658
1.3
(1.5
)B
a 2T
l 2O
518
327
6322
1.3
(1.3
)Te
2I27
655
107
1.3
(1.3
)N
bTeB
r 328
038
3537
61.
3(1
.4)
P 10A
u 7I
2737
012
162
1.3
(1.4
)B
a(Sc
Te2)
217
501
4163
261.
3(1
.3)
Ta3I
723
238
1421
11.
3(1
.4)
NaN
bS2
7937
2628
51.
3(1
.9)
SnSe
691
1082
931.
3(1
.4)
CsA
uI3
2845
359
269
1.3
(1.3
)L
iCuO
291
5874
978
1.3
(1.6
)N
bFeS
b94
3783
928
1.3
(2.0
)H
fBrN
3030
251
773
1.3
(1.3
)A
g 2C
O3
4691
2810
401.
3(2
.1)
In2H
gS4
2235
656
081
1.3
(1.5
)Sb
(IF 3
) 250
4965
6330
11.
3(1
.3)
SrC
aSi
7084
1720
051.
3(1
.3)
VS 4
5411
5564
770
1.3
(1.4
)C
uTe 2
Cl
3097
164
11.
3(1
.7)
Sr2S
i11
0616
0102
1.3
(1.3
)H
fTlC
uSe 3
9397
8256
31.
3(1
.3)
KC
uSe
7435
1215
71.
3(1
.3)
ZrB
r 323
247
3270
31.
3(1
.4)
Ca 3
(CoO
3)2
1879
224
6282
1.3
(1.3
)Pb
Se20
667
6575
611.
4(1
.4)
TlI
nTe 2
2279
164
0633
1.4
(1.5
)P 2
Os
2319
4260
91.
4(1
.7)
Bi 2
Pt2O
723
341
1611
041.
4(1
.4)
BeP
227
148
2262
1.4
(1.5
)L
i 4N
Cl
2914
984
649
1.4
(2.0
)T
lAgS
e29
238
1007
101.
4(1
.4)
BaN
aBi
3123
541
3810
1.4
(1.4
)Z
rCoB
i31
451
1071
201.
4(2
.1)
Cs 2
Ni 3
Se4
1433
633
891
1.4
(1.6
)Sr
3As 4
1533
910
0110
1.4
(1.6
)C
s 2Pd
I 650
5635
2801
891.
4(1
.5)
Te2M
o60
224
155
1.4
(1.4
)P 2
Pt73
064
7971
1.4
(1.9
)L
iZnN
7575
1679
01.
4(1
.4)
Na 2
SbA
u77
7423
255
1.4
(1.5
)
5

Sr(Z
nP) 2
8276
3091
11.
4(1
.4)
K(M
oS) 3
8116
3075
21.
4(1
.5)
Rb(
MoS
) 381
1730
753
1.4
(1.5
)C
a 3(G
eAs 2
) 218
504
1645
51.
4(1
.5)
Ba(
AgT
e)2
1850
124
6048
1.4
(1.4
)C
s 2Pt
I 623
060
3719
31.
4(1
.4)
RbP
b21
525
4094
361.
4(1
.4)
KPb
2152
640
1436
1.4
(1.4
)C
d 2A
s 3I
2757
782
161.
4(1
.7)
Te2B
r27
648
106
1.4
(1.5
)Z
nCu 2
GeS
464
0815
2752
1.4
(1.4
)In
2HgS
e 420
731
2564
91.
4(1
.4)
TiC
oSb
5967
6249
201.
4(2
.2)
SnSe
266
543
857
1.4
(1.8
)N
bI3O
5462
8541
8088
1.4
(1.4
)K
2Pd 3
Se4
1433
933
894
1.4
(1.5
)C
oAgO
219
178
2461
571.
4(1
.7)
InA
gS2
1983
315
6129
1.4
(1.4
)A
g 2G
eS3
9900
4171
11.
4(1
.5)
RbS
b74
4414
030
1.4
(1.4
)Z
rCl 3
2287
132
702
1.4
(1.6
)R
eCl 3
2317
462
222
1.4
(1.4
)C
a(B
C) 2
1028
988
019
1.5
(1.9
)G
aAgO
211
020
9566
51.
5(2
.5)
Zn(
InTe
2)2
2083
225
650
1.5
(1.5
)Pb
S21
276
6575
601.
5(1
.5)
Rb 2
(NbC
l 3) 3
2297
640
4165
1.5
(1.6
)Z
rCl 2
2316
230
052
1.5
(1.6
)C
lO2
2320
767
663
1.5
(1.5
)R
b 3B
i23
304
5884
91.
5(1
.6)
LiA
g 3O
227
227
4204
1.5
(1.5
)R
bAuB
r 327
300
9577
1.5
(1.6
)N
aAg 3
O2
2730
396
271.
5(1
.5)
Sr3(
InP 2
) 228
324
6133
51.
5(1
.5)
Te4M
oBr
2919
082
245
1.5
(1.5
)Sr
LiB
i51
0112
5880
01.
5(1
.5)
SrP
7931
2626
21.
5(2
.1)
K2T
e 2Pt
8623
4043
21.
5(2
.1)
K2S
e 376
7012
641.
5(1
.7)
CdH
gO2
9146
7484
81.
5(1
.5)
Ba 3
(Si 2
P 3) 2
2788
729
261
1.5
(1.5
)N
a 10C
aSn 1
230
252
2400
061.
5(1
.5)
Nb 2
Se9
5411
0662
538
1.5
(1.5
)L
aS2
1508
4184
051.
5(2
.0)
CuB
iSeO
2311
674
475
1.5
(1.6
)B
i 2Se
O2
5520
9841
1143
1.5
(2.3
)N
aSnP
2952
940
9010
1.5
(2.0
)K
2NiA
s 296
7330
0120
1.5
(1.5
)Ta
SbR
u31
454
1071
231.
5(2
.5)
VC
u 3Se
421
855
6291
481.
5(1
.8)
CdC
u 2G
eS4
1398
226
150
1.5
(1.5
)Y
4OsB
r 428
744
7151
31.
5(1
.5)
Hg 2
As 3
Br
2889
975
169
1.5
(1.7
)T
lInS
220
042
4241
91.
5(2
.0)
CuP
292
765
3601
1.5
(1.6
)C
uSbS
e 220
331
3035
81.
5(1
.6)
CuB
iS2
2298
234
936
1.5
(1.7
)B
aCu 2
SnSe
412
364
1708
571.
5(1
.5)
Ba 2
FeM
oO6
5106
7524
6544
1.5
(1.6
)M
g(B
iO3)
228
447
5000
51.
5(1
.5)
NaZ
rCuS
e 350
5172
7388
81.
5(1
.5)
FeSi
217
1423
408
1.5
(1.5
)N
bI5
3148
725
503
1.5
(1.6
)Te
RhC
l22
945
5685
31.
6(1
.7)
CsA
uCl 3
2348
441
7363
1.6
(1.6
)Y
2Cl 3
2767
823
337
1.6
(1.7
)N
aAuS
e 229
139
8400
41.
6(1
.7)
Ca 3
BiN
3114
910
6320
1.6
(1.6
)Z
nSiA
s 235
9561
1411
1.6
(1.6
)K
Ag 2
AsO
412
015
4097
931.
6(2
.5)
NaS
e15
668
4340
81.
6(1
.6)
TeA
uI50
4668
1661
1.6
(1.7
)N
aTaN
254
7576
382
1.6
(2.0
)M
gAs 4
7623
1079
1.6
(1.6
)Sr
3SbN
7752
1520
521.
6(1
.6)
BaP
378
0823
618
1.6
(1.6
)R
b 2Te
320
9577
994
1.6
(1.7
)Sr
(CdP
) 282
7730
912
1.6
(1.6
)C
a(Z
nP) 2
9569
1000
621.
6(1
.6)
RbA
s10
802
4125
941.
6(1
.6)
K2N
i 3S 4
1722
881
731
1.6
(1.8
)R
b 3Sn
4Au
1740
110
7445
1.6
(1.7
)Pd
I 227
747
2512
01.
6(1
.6)
Na 1
0SrS
n 12
3025
324
0007
1.6
(1.6
)K
CuT
e74
3612
158
1.6
(1.6
)B
aAg 2
GeS
473
9410
040
1.6
(2.4
)Sn
S22
3115
6130
1.6
(1.8
)R
b 2Pd
3Se 4
1434
033
895
1.6
(1.7
)C
uTe 2
Br
3103
667
252
1.6
(1.9
)Sb
2Se 3
2160
1715
691.
6(1
.6)
BaC
uN50
5338
8606
41.
6(1
.6)
KTe
2072
9674
11.
7(1
.7)
Li 3
Sb20
7464
2341
1.7
(3.3
)Y
N21
1416
1075
1.7
(2.0
)N
aInT
e 222
483
2534
61.
7(2
.0)
Ga 2
Te5
2371
3203
41.
7(1
.7)
GeS
e70
063
7866
1.7
(1.7
)A
uClO
2726
581
901.
7(1
.9)
TiB
rN27
849
2739
51.
7(1
.7)
TiN
Cl
2785
027
396
1.7
(1.7
)YA
gTe 2
1290
315
4792
1.7
(1.7
)R
bPt 2
Se3
1479
769
439
1.7
(1.7
)Fe
S 215
2210
9374
1.7
(2.1
)Z
n(G
aTe 2
) 215
777
4488
81.
7(1
.7)
Ga 2
HgS
e 447
3083
712
1.7
(1.7
)B
a(Pd
S 2) 2
5052
3079
930
1.7
(1.9
)Te
AuB
r 854
1134
6312
91.
7(1
.8)
Cs 2
Pd(I
Br 2
) 254
3024
2404
801.
7(1
.8)
SrL
iSb
7756
1722
161.
7(1
.7)
Na 2
AsA
u77
7323
254
1.7
(1.8
)C
a(C
dP) 2
9570
1000
631.
7(1
.7)
Na 5
GeA
s 316
861
3001
871.
7(1
.8)
KN
a 4G
eAs 3
1692
130
0204
1.7
(1.8
)Si
2Os
1712
342
730
1.7
(1.8
)Sr
3(G
eAs 2
) 217
504
1645
41.
7(1
.7)
Ca(
RhO
2)2
1799
817
0597
1.7
(1.7
)Ta
I 2O
2902
780
109
1.7
(1.8
)B
i 4O
730
303
5177
81.
7(1
.9)
CdA
g 4G
e 2S 7
5422
0095
121
1.7
(1.7
)Pt
Br 3
2316
523
127
1.7
(1.7
)T
l 2O
5514
7077
699
1.7
(1.7
)N
aCuO
245
4120
3080
1.7
(2.0
)R
bCu 3
S 210
985
4096
461.
7(1
.8)
LiC
aSb
1626
452
775
1.7
(1.7
)R
eSe 2
5415
8281
813
1.7
(1.7
)K
2Sn 2
Se5
8966
7237
91.
7(2
.0)
TePb
1971
715
3711
1.7
(1.7
)R
uS2
2030
5601
91.
7(2
.1)
KZ
rCuS
393
1780
624
1.7
(1.7
)K
2VC
uSe 4
1522
084
303
1.7
(1.9
)N
aHfC
uSe 3
5054
4840
2578
1.7
(1.7
)B
aLiS
b10
485
2805
741.
8(1
.9)
BaL
iAs
1061
656
445
1.8
(1.8
)B
a(In
Te2)
221
183
4116
81.
8(2
.0)
ZnT
e21
7616
2756
1.8
(1.8
)T
l 2Sn
2S3
2801
133
531
1.8
(1.9
)K
AuS
e 229
138
8400
31.
8(2
.0)
Zr 2
SN2
1158
396
971
1.8
(2.8
)C
d(G
aTe 2
) 213
949
2564
61.
8(1
.8)
KA
gSe
1623
652
586
1.8
(1.8
)C
s 2Pd
3Se 4
5048
5533
892
1.8
(1.9
)W
Br 6
5049
9362
048
1.8
(1.8
)N
aSbS
254
1443
909
1.8
(1.9
)A
uCl 2
5416
5620
1436
1.8
(1.9
)K
YO
284
0949
650
1.8
(1.8
)R
bTe
9064
7317
91.
8(2
.2)
Ca 3
AlA
s 317
186
3272
71.
8(1
.8)
Ga 7
Te10
1838
840
0668
1.8
(1.8
)N
aGe
2965
743
275
1.8
(1.8
)K
2LiI
nAs 2
5054
3140
2147
1.8
(1.9
)B
a 3G
aP3
5417
1540
2177
1.8
(1.8
)B
aCuT
eF13
287
2456
241.
8(1
.8)
VC
u 3S 4
3762
6289
571.
8(2
.1)
Cd 2
SnO
459
6669
297
1.8
(1.8
)T
lPt 2
S 392
7278
784
1.8
(1.8
)K
V(C
uS2)
263
7640
2924
1.8
(1.9
)L
iP95
8810
0465
1.8
(2.0
)R
bCuP
dSe 5
1111
528
1424
1.8
(1.8
)R
b 2VA
gSe 4
1463
550
462
1.8
(2.0
)
6

Sr(I
nTe 2
) 220
397
4116
71.
8(2
.0)
LaC
uSe 2
1179
099
675
1.8
(1.8
)C
uTe 2
I31
037
6725
31.
8(1
.9)
Rb 3
Sb16
319
7799
11.
8(1
.9)
CuR
hO2
1411
629
214
1.8
(1.9
)K
Sb15
3656
529
1.8
(1.8
)N
aPt 2
Se3
2898
778
788
1.8
(1.8
)B
aYA
gTe 3
1033
788
717
1.9
(2.1
)B
P14
7961
5154
1.9
(4.1
)C
d(In
Se2)
222
304
1519
541.
9(1
.9)
Zn(
InSe
2)2
2260
725
647
1.9
(1.9
)In
Se22
691
4147
71.
9(1
.9)
AgB
r23
231
6506
11.
9(3
.2)
Au 2
O3
2725
380
141.
9(2
.1)
Bi(
TeB
r 2) 2
2912
783
806
1.9
(2.0
)C
a 3B
N3
3031
595
814
1.9
(1.9
)N
bCl 4
3104
010
101.
9(1
.9)
Sr3P
414
288
3832
11.
9(2
.0)
Na 2
PtSe
214
588
4042
91.
9(2
.3)
MoS
e 216
3464
4334
1.9
(1.9
)Sc
AgS
e 212
908
1551
151.
9(2
.4)
ScT
lSe 2
1331
341
8475
1.9
(2.1
)Pd
F 413
868
1555
1.9
(1.9
)B
160
1080
261.
9(2
.4)
Rb 3
Ge 4
Au
1783
041
3724
1.9
(2.1
)P 4
Ru
5044
4324
921.
9(2
.1)
TiI 4
5410
1339
820
1.9
(2.0
)L
i 3A
s75
743
987
1.9
(2.9
)B
aSe 2
7547
1635
81.
9(2
.0)
Al 2
HgT
e 479
1025
641
1.9
(1.9
)N
a 3Sb
7956
2688
21.
9(1
.9)
K2A
s 2Pd
8147
3200
91.
9(2
.0)
K2P
382
6233
259
1.9
(1.9
)K
5As 2
Au
8683
4069
81.
9(2
.0)
Cs 2
As 2
Pd88
5769
647
1.9
(2.1
)T
lPd 3
O4
5478
2276
1.9
(2.1
)C
a(M
gSb)
295
6510
0045
1.9
(2.5
)B
a 4Sb
2O97
7440
2284
1.9
(1.9
)B
a(M
gSb)
295
6710
0047
1.9
(2.6
)Z
rSnS
317
324
7371
11.
9(1
.9)
K2S
e 518
609
7238
01.
9(1
.9)
I23
153
6770
41.
9(2
.0)
TeI
2327
366
641
1.9
(1.9
)R
b 5Si
As 3
1749
030
0191
1.9
(2.0
)N
b 2H
g 2O
713
803
2222
61.
9(2
.3)
AlT
lSe 2
9579
1001
301.
9(2
.1)
YZ
nAsO
5460
1116
3780
1.9
(1.9
)L
aTaN
2O55
0514
4111
381.
9(1
.9)
Cs 2
Se3
7449
1409
51.
9(1
.9)
Na 2
AgS
b73
9210
010
1.9
(2.1
)C
uSe 2
Br
2956
741
0004
1.9
(1.9
)H
g 2P 3
Cl
2887
574
771
1.9
(2.4
)C
uSe 2
Cl
3103
868
292
1.9
(2.0
)VA
g 3O
418
889
4174
691.
9(2
.3)
HfT
lCuS
393
9682
562
1.9
(1.9
)C
uSbS
244
6841
8753
1.9
(2.1
)N
aNbN
270
1772
557
1.9
(2.1
)K
3As
1401
826
887
1.9
(2.0
)Fe
MoO
454
1843
4301
31.
9(1
.9)
Cs 2
Ni 3
S 428
486
6301
01.
9(2
.1)
PPdS
e31
2377
772
1.9
(1.9
)N
a 2Pd
S 210
223
7653
32.
0(2
.4)
Rb 2
P 320
7965
4296
2.0
(2.1
)Pd
PbF 4
2080
510
8992
2.0
(2.2
)T
lInS
e 222
232
6405
262.
0(2
.1)
InA
gO2
2266
020
2429
2.0
(2.5
)K
2AgB
i27
549
1156
2.0
(2.2
)T
l 2Pd
Cl 4
2988
989
601
2.0
(2.5
)Sc
TlS
213
312
4184
742.
0(3
.0)
Rb 2
As 2
Pt13
445
1075
292.
0(2
.0)
Cs 2
P 314
652
6518
52.
0(2
.2)
Cd(
RhO
2)2
1410
028
954
2.0
(2.0
)Z
nGeP
245
2463
7511
2.0
(2.1
)G
aTe
5428
1215
3456
2.0
(2.1
)L
i 3P
736
2408
612.
0(3
.3)
KT
lO2
8175
3355
32.
0(2
.2)
K2P
tSe 2
8621
4043
02.
0(2
.5)
Ca 2
Pt3O
887
1065
412
2.0
(2.0
)B
a 2Z
nN2
9307
8037
72.
0(2
.0)
Fe(S
iP) 4
9198
7900
52.
0(2
.1)
K3N
a 2Sn
As 3
1844
740
560
2.0
(2.0
)G
e 19(
PI) 4
2342
063
194
2.0
(2.0
)K
2Te 2
As
2938
030
0184
2.0
(2.1
)K
2SnT
e 329
835
4094
832.
0(2
.0)
Cs 3
Ge 4
Au
5103
4141
3725
2.0
(2.1
)K
2WSe
418
138
5924
22.
0(2
.0)
Ba(
YTe
2)2
1787
290
335
2.0
(2.0
)Ta
(IC
l)2
2868
369
688
2.0
(2.0
)Sc
2CC
l 228
479
5912
42.
0(2
.3)
GaC
uS2
5238
1567
862.
0(2
.0)
BaG
e 221
3915
7612
2.0
(2.0
)C
a 3V
N3
9029
7211
82.
0(2
.0)
Ba 4
LiC
u(C
O5)
215
472
4011
222.
0(2
.5)
WSe
218
2140
752
2.0
(2.0
)R
b(Sb
Se2)
297
9840
2887
2.0
(2.3
)C
o(R
eO4)
231
516
5101
52.
0(2
.0)
SbIr
S86
3041
400
2.0
(2.3
)Z
n(R
hO2)
251
4610
9298
2.0
(2.0
)Sr
2FeW
O6
1926
615
3430
2.0
(2.0
)K
AuB
r 429
694
2800
332.
0(2
.0)
ZnS
e11
9016
2755
2.1
(2.1
)Sr
3(Si
As 2
) 211
677
1645
32.
1(2
.1)
K2N
aInA
s 221
510
3001
372.
1(2
.1)
GeS
2242
6538
962.
1(2
.1)
AlS
b26
2415
1218
2.1
(2.4
)B
i 2SO
227
891
2945
12.
1(2
.4)
Co(
ClO
4)2
3162
133
288
2.1
(2.1
)C
s(Sb
Se2)
233
1241
5580
2.1
(2.4
)O
212
957
1564
812.
1(2
.2)
AlA
gTe 2
1409
228
746
2.1
(2.1
)T
l 2G
eSe 3
1424
235
043
2.1
(2.3
)B
e 2C
1569
6161
852.
1(5
.5)
NaN
bO2
3744
3002
442.
1(2
.1)
Ag 3
SbS 3
4515
6498
62.
1(2
.6)
K2G
eAs 2
8930
7122
22.
1(2
.1)
Al 2
Te5
9254
7894
12.
1(2
.4)
LiB
eAs
9562
1000
042.
1(2
.3)
Ti(S
nO2)
218
288
1632
302.
1(2
.1)
Sr3(
GeP
2)2
1835
141
182
2.1
(2.1
)N
a 4G
e 2Te
528
107
3718
32.
1(2
.1)
PtI 2
2831
960
760
2.1
(2.2
)B
a(Te
P 2) 2
3127
541
2643
2.1
(2.2
)B
e 2Te
7Cl 6
5057
8839
1156
2.1
(2.2
)B
iAuB
r 654
1774
4072
202.
1(2
.1)
Pb3O
454
2494
3625
32.
1(2
.1)
LiB
C92
4478
731
2.1
(2.5
)Sr
TiN
295
1785
770
2.1
(2.1
)In
2S3
2221
615
1645
2.1
(2.1
)C
a 4B
i 2O
5518
7341
6137
2.1
(2.3
)C
s 2C
uF6
2869
265
259
2.1
(2.2
)Pd
SeO
354
5482
2495
042.
1(2
.4)
TaC
u 3Te
492
9580
282
2.1
(2.4
)N
a 2C
uAs
1568
543
937
2.1
(2.3
)C
uBr
2291
330
090
2.1
(2.1
)R
bCuO
274
6715
096
2.1
(2.4
)K
ZnP
7437
1216
02.
1(2
.1)
Na 2
CuP
7639
1153
2.1
(2.4
)K
2VC
uS4
1514
781
414
2.1
(2.4
)R
b 2V
CuS
415
219
8430
22.
1(2
.4)
HfS
nS3
8725
6566
72.
1(2
.1)
Ag 2
PdC
l 428
557
6523
92.
1(2
.2)
Tl 2
Sn(A
sS3)
260
2330
0252
2.1
(2.2
)K
2Pd 3
S 499
1041
885
2.1
(2.2
)K
InTe
219
851
7341
02.
1(2
.3)
AgH
gAsS
362
1565
3762
2.1
(2.3
)N
aP74
4014
009
2.1
(2.1
)Fe
Br 2
2288
040
9571
2.1
(2.1
)B
aCu 2
SnS 4
1795
452
685
2.1
(2.1
)C
r 2A
g 2O
750
4441
2433
2.1
(2.1
)B
aPt 2
S 329
289
2011
462.
1(2
.3)
FeSO
419
254
2409
472.
1(2
.1)
Sc2C
dS4
1095
394
994
2.2
(2.2
)In
6Ge 2
PtO
922
186
1708
972.
2(2
.4)
RbI
nTe 2
2225
575
346
2.2
(2.5
)C
a 2N
Cl
2293
615
3101
2.2
(2.3
)Te
3Cl 2
2762
810
52.
2(2
.3)
K2P
tBr 6
2769
123
771
2.2
(2.2
)K
4CdA
s 229
585
3001
902.
2(2
.3)
NaT
lO2
3056
6449
192.
2(2
.3)
7

Ca 4
Sb2O
1366
016
353
2.2
(2.5
)B
a 2Si
Te4
1444
849
751
2.2
(2.2
)N
aSe 2
1551
440
2584
2.2
(2.3
)A
gAuF
416
060
9007
12.
2(2
.2)
Hg 2
GeO
413
995
2634
02.
2(2
.2)
Na 3
P15
9817
1012
2.2
(2.2
)R
hBr 3
5047
1928
245
2.2
(2.3
)K
Pt2S
351
0130
4006
22.
2(2
.2)
PtS 2
762
6599
632.
2(2
.3)
CsT
e83
6183
351
2.2
(2.2
)N
a 2A
gAs
8411
4900
72.
2(2
.2)
Ba(
AgS
) 285
7950
183
2.2
(2.2
)N
a 3Sb
Se4
8703
6514
12.
2(2
.2)
Ca 2
ZnN
288
1869
049
2.2
(2.3
)Sr
2ZnN
293
0680
376
2.2
(2.2
)B
a 2H
fS4
9321
8065
22.
2(2
.2)
ZrS
399
2142
073
2.2
(2.3
)H
fS3
9922
4207
42.
2(2
.2)
NaL
i 2Sb
5077
5424
42.
2(3
.0)
GaA
gS2
5343
1567
852.
2(2
.2)
Sr5M
o 2N
731
231
4139
322.
2(2
.2)
BaP
1050
4809
3529
52.
2(2
.4)
Rb 5
GeP
317
978
3001
862.
2(2
.2)
Ag 3
PS4
1245
941
6585
2.2
(2.2
)K
Li 2
As
2899
478
938
2.2
(2.2
)H
gAsO
330
284
4112
302.
2(2
.6)
WS 2
224
5601
42.
2(2
.3)
VN
Cl 4
2786
828
128
2.2
(2.2
)B
aHgS
228
007
3264
82.
2(2
.2)
Sr3G
aN3
7191
2812
592.
2(2
.3)
GaS
e19
4343
540
2.2
(2.2
)H
f(Te
2Cl 3
) 229
419
4015
892.
2(2
.2)
HgP
Se3
7293
2565
2.2
(2.2
)B
a 4N
aCu(
CO
5)2
6841
8060
62.
2(2
.7)
K2V
AgS
489
0066
840
2.2
(2.3
)K
(SnS
e 2) 2
2876
972
386
2.2
(2.4
)L
a 2S 3
7475
1515
12.
2(2
.2)
SrTa
NO
255
2454
4111
372.
2(2
.2)
RbC
uSe 4
1836
540
4225
2.2
(2.2
)H
g 2P 3
Br
2887
474
770
2.2
(2.2
)A
gHg 3
SbO
612
362
1707
642.
2(2
.3)
ZrS
211
8656
012
2.3
(3.0
)L
i 2Pb
O3
2245
035
182
2.3
(2.4
)N
aInS
e 222
473
2555
82.
3(2
.5)
K2P
dCl 6
2306
733
709
2.3
(2.3
)C
sSnB
r 327
214
4071
2.3
(2.3
)C
sGeI
328
377
6255
92.
3(2
.3)
PAuS
430
938
4130
092.
3(2
.5)
Hf 3
N4
1166
097
997
2.3
(2.3
)A
g 3A
sS3
4431
6180
62.
3(2
.7)
ZnS
iP2
4763
6481
452.
3(2
.3)
AsS
eI50
5373
2007
992.
3(2
.3)
K2C
d 2O
375
3416
223
2.3
(2.3
)C
aPdF
481
6132
674
2.3
(2.3
)K
3Sb 2
Au 3
9273
7897
72.
3(2
.5)
K5P
2Au
1462
440
700
2.3
(2.4
)N
aAg 3
S 216
992
7319
82.
3(2
.3)
Tl 2
TeS 3
1717
239
1285
2.3
(2.5
)B
iTeI
2296
574
501
2.3
(2.4
)B
a 5P 4
1756
641
3273
2.3
(2.5
)K
GaT
e 217
965
4111
702.
3(2
.3)
Cs 2
VAgS
486
8450
460
2.3
(2.4
)K
Se92
6873
172
2.3
(2.7
)L
iBeP
9915
4203
72.
3(2
.6)
Ag 2
HgI
423
485
1503
432.
3(2
.3)
NbC
u 3Se
440
4362
8479
2.3
(2.7
)R
b 2Sn
As 2
8931
7122
32.
3(2
.4)
K2P
Au
9687
3002
012.
3(2
.3)
AlC
uTe 2
8017
2873
52.
3(2
.3)
Cs 2
SnA
s 289
3471
226
2.3
(2.5
)Sb
I 3C
l 823
536
3266
22.
3(2
.3)
K2T
iS3
2876
672
377
2.3
(2.3
)K
P74
4114
010
2.3
(2.3
)Pd
SO4
2895
279
559
2.3
(2.3
)R
b 2VA
gS4
8901
6684
12.
3(2
.4)
Cd 2
GaA
gS4
6356
1701
082.
3(2
.3)
BaZ
rS3
5407
7123
288
2.3
(2.3
)N
a 2Pt
S 210
246
8721
92.
4(2
.8)
Cs 3
Sb10
378
5324
32.
4(2
.7)
LiI
nTe 2
2078
216
2674
2.4
(2.4
)Z
nO21
3316
3380
2.4
(2.4
)In
Br
2287
055
189
2.4
(2.6
)A
gCl
2292
264
734
2.4
(4.1
)G
a 2Pd
I 830
946
4132
302.
4(2
.5)
Cs 2
TiS 3
3247
6601
772.
4(2
.5)
BaL
iP13
277
4168
902.
4(2
.4)
AlA
gSe 2
1409
128
745
2.4
(2.4
)C
d(G
aSe 2
) 237
7216
3949
2.4
(2.4
)C
s 2Pd
3S4
5102
6826
250
2.4
(2.5
)C
dS67
215
4188
2.4
(2.4
)L
i 2Pd
O2
7608
6119
92.
4(2
.7)
Cs 2
O79
8827
919
2.4
(3.1
)B
a(M
gAs)
282
8030
916
2.4
(2.8
)Si
P 299
9643
098
2.4
(2.4
)K
2NaG
aAs 2
9676
3001
292.
4(2
.4)
Cs 2
Pt3S
e 414
338
3389
32.
4(2
.5)
Tl 2
B2S
e 716
183
4114
682.
4(2
.4)
Na 5
SiA
s 318
139
3001
882.
4(2
.4)
CsG
aTe 2
1768
840
2613
2.4
(2.4
)K
2In 3
AgS
e 621
705
5485
92.
4(2
.4)
Tl 2
Au 4
S 329
898
5123
52.
4(2
.4)
MoS
2Cl 3
5047
1628
062
2.4
(2.4
)In
GaS
e 350
4952
6293
02.
4(2
.4)
AlC
uSe 2
8016
2873
42.
4(2
.4)
BaS
nO3
3163
4313
82.
4(2
.6)
K4H
gAs 2
2948
440
2573
2.4
(2.4
)L
i 2Pd
O2
5457
5019
007
2.4
(2.8
)H
gO12
2440
316
2.4
(2.8
)H
gI2
2319
215
0345
2.4
(2.4
)N
b 2Sn
O6
3324
1638
152.
4(2
.5)
NaA
sS2
5942
6552
192.
4(2
.4)
KN
b(C
uSe 2
) 265
9965
5976
2.4
(2.4
)L
aCuS
248
4141
5078
2.4
(2.4
)T
l 3B
Se3
2849
040
375
2.4
(2.6
)C
rHgO
419
380
4161
472.
4(2
.4)
Cu 3
PS4
3934
4122
402.
4(2
.4)
CsL
a 2C
uSe 4
5058
1593
680
2.4
(2.5
)Sb
2S3
2809
1718
532.
4(2
.5)
NaC
oO2
1892
196
428
2.4
(2.5
)N
a 6P 4
W90
0771
647
2.4
(2.4
)B
aLaC
uTe 3
1706
388
715
2.4
(2.4
)Ta
2FeO
631
755
4012
642.
4(2
.4)
Ca 3
PN11
824
1063
502.
5(2
.5)
GaP
2490
6350
412.
5(2
.7)
K2P
dBr 4
2713
819
822.
5(2
.6)
Ta3N
527
488
1625
32.
5(2
.6)
MgS
iP2
2961
6427
342.
5(2
.5)
Al 2
HgS
e 430
3833
827
2.5
(2.5
)B
aZrN
231
0474
905
2.5
(2.5
)K
Ag 2
PS4
1253
242
0033
2.5
(3.3
)Si
4P4R
u14
983
7900
62.
5(2
.6)
SrL
i 4N
215
845
8741
32.
5(2
.7)
CdP
291
362
336
2.5
(2.6
)C
dSiP
246
6644
260
2.5
(2.5
)R
bAuB
r 451
0267
2602
22.
5(2
.6)
K2P
dO2
5405
8461
582.
5(2
.6)
BaC
dO2
7899
2555
52.
5(2
.6)
K2P
tS2
7928
2625
82.
5(3
.0)
Rb 2
PtS 2
7929
2625
92.
5(2
.9)
Ba(
MgP
) 282
7830
914
2.5
(3.1
)Pt
F 489
4371
579
2.5
(2.5
)R
bTeA
u90
0871
652
2.5
(2.5
)R
b 3Sb
2Au 3
9274
7897
82.
5(2
.7)
AlA
gO2
9631
9566
22.
5(3
.4)
Ti(P
S 3) 2
1366
616
403
2.5
(2.5
)C
a 6G
aN5
1787
533
795
2.5
(2.6
)Sr
3TaA
s 3O
1819
940
9567
2.5
(2.5
)A
gI22
925
1615
792.
5(2
.5)
P 2Pt
O7
2928
220
0871
2.5
(2.5
)N
a 2W
4O13
3253
361
412.
5(3
.0)
RbI
n 3S 5
5426
5459
667
2.5
(2.5
)L
iAlB
1482
0435
336
2.5
(2.9
)R
b 2Si
As 2
6960
6061
72.
5(2
.8)
CuC
l22
914
2398
82.
5(2
.5)
K2S
iAs 2
6984
4042
62.
5(2
.8)
Na 4
HgP
228
591
6726
02.
5(2
.6)
Ba(
AuO
2)2
9297
8032
72.
5(2
.5)
As 2
Se3
909
4405
82.
5(2
.7)
Ba 2
GeS
e 411
902
4141
662.
5(2
.5)
8

Si2H
g 6O
728
712
6912
32.
5(2
.8)
SiA
s18
6315
3457
2.5
(2.6
)In
S20
094
6601
052.
5(2
.7)
Ba 2
PbO
420
098
6654
42.
5(3
.0)
SbSe
I22
996
3129
22.
5(2
.6)
Sc2S
340
165
4308
2.5
(2.5
)C
s 6G
aSb 3
9697
3002
162.
5(2
.5)
TlV
3O8
1899
665
773
2.5
(2.5
)T
lP5
2741
115
021
2.5
(2.6
)W
O3
5104
1765
3651
2.5
(2.5
)C
sYH
gSe 3
1112
328
1440
2.6
(2.6
)C
dIn 2
O4
2104
715
9740
2.6
(2.7
)C
sIn 5
S 822
007
4087
82.
6(2
.7)
Li 3
N22
5115
6902
2.6
(2.7
)N
b(SC
l)2
2736
210
484
2.6
(2.6
)Ir
Br 3
2739
714
212
2.6
(2.7
)A
g 2S
3105
398
454
2.6
(2.8
)Sr
PdF 4
1262
210
8990
2.6
(2.6
)R
b 2Pt
3S4
4030
6495
182.
6(2
.7)
Cs 2
TeI 6
5409
5738
105
2.6
(2.9
)Z
rBrN
5419
1287
797
2.6
(2.6
)N
aRhO
288
3066
280
2.6
(2.7
)T
l 3A
sS3
9791
1002
922.
6(2
.8)
RbS
e90
6373
177
2.6
(2.8
)R
b 3A
sSe 4
1830
540
4080
2.6
(2.6
)N
a 5Sn
P 318
317
3001
892.
6(2
.6)
Sc2P
bSe 4
5428
2615
4528
2.6
(2.7
)Sn
O2
856
1574
492.
6(2
.6)
Cu 2
PbO
229
396
4006
572.
6(2
.8)
Rb 2
Sn2O
378
6324
816
2.6
(2.6
)Sr
5(A
uO4)
228
796
7196
52.
6(2
.6)
CdS
i(C
uS2)
264
4916
924
2.6
(2.6
)Sr
(AuO
2)2
9298
8032
82.
6(2
.7)
NaA
gF4
7386
9903
2.6
(2.7
)A
gSbS
239
2294
647
2.6
(2.7
)A
uCl 3
2764
722
146
2.6
(2.7
)C
a 3A
sN41
9265
7356
2.6
(2.6
)N
aAuO
220
343
4095
472.
6(2
.9)
Ba 3
In2O
620
352
6525
82.
6(3
.1)
KH
gF3
7483
1517
02.
6(2
.6)
SnG
eS3
5045
6377
962.
6(2
.7)
LiR
hO2
1411
529
213
2.6
(2.6
)R
bHgS
bSe 3
6300
9006
72.
6(2
.8)
RbI
n 5S 8
2093
840
877
2.7
(2.8
)A
lAs
2172
6591
402.
7(3
.2)
In2O
322
323
2432
52.
7(2
.7)
Bi 2
S 322
856
2010
662.
7(2
.7)
Br
2315
420
1692
2.7
(2.7
)N
aSi
2402
4099
532.
7(2
.9)
RhC
l 327
770
2576
42.
7(2
.8)
Pd(S
eCl 3
) 228
175
3943
52.
7(2
.8)
PdC
l 229
487
4046
242.
7(2
.8)
Cs 2
PtB
r 630
062
7738
12.
7(2
.7)
P 2Pd
3S8
3006
3536
12.
7(2
.8)
LiM
gAs
1255
810
7954
2.7
(3.2
)K
YTe
216
763
4199
952.
7(3
.6)
K2A
gSb
7643
1155
2.7
(2.8
)A
l 2Z
nTe 4
7908
2563
92.
7(2
.7)
Al 2
CdT
e 479
0925
640
2.7
(2.7
)N
aBeA
s95
7310
0091
2.7
(2.9
)N
a 2B
2Se 7
5004
6586
922.
7(2
.8)
Ca(
MgA
s)2
9564
1000
412.
7(3
.0)
KB
aPSe
418
156
4146
372.
7(2
.7)
IBr
2763
922
120
2.7
(2.8
)K
AuC
l 427
181
2607
2.7
(2.7
)R
b 4Z
r 3Se
1454
2013
2806
362.
7(2
.8)
Na 3
MoN
354
1291
6756
52.
7(2
.7)
NaM
gAs
5962
4179
82.
7(2
.7)
BiT
eCl
2894
479
362
2.7
(2.8
)V
HgO
354
1588
8224
22.
7(2
.8)
InSb
S 321
365
3002
072.
7(2
.8)
CsC
uF4
5409
2135
264
2.7
(2.8
)R
b 2N
bCuS
e 415
222
8430
52.
7(3
.0)
Na 2
ZrS
e 372
1941
2158
2.7
(2.9
)K
2NbC
uSe 4
9003
7332
42.
7(2
.8)
Ca(
AuO
2)2
2898
8338
62.
7(2
.7)
Cs 2
ZrS
e 398
5640
9294
2.7
(2.7
)H
fPbS
322
147
6566
82.
7(2
.7)
AgH
gSI
2314
054
796
2.7
(2.7
)H
g 2W
O4
3254
290
085
2.7
(3.0
)Sr
Cu 2
GeS
418
685
1000
52.
7(2
.8)
K2L
iGaA
s 297
0340
1208
2.7
(2.7
)Z
rFeC
l 628
208
3966
62.
7(2
.7)
BaH
fN2
1032
250
994
2.8
(2.8
)N
aBiO
222
984
1736
242.
8(2
.8)
Ca 3
C3C
l 228
160
3381
82.
8(2
.9)
K4C
dP2
2830
261
084
2.8
(2.9
)T
lAuC
l 428
368
6210
72.
8(2
.8)
Ga 2
TeSe
228
423
6461
72.
8(2
.8)
Cs 2
Au 2
Se3
2919
485
708
2.8
(2.8
)K
3Ag 3
As 2
1420
632
016
2.8
(2.8
)Z
nPdF
613
983
2616
52.
8(2
.8)
RbY
Te2
1676
441
9996
2.8
(3.6
)L
a 4Se
3O4
4412
4191
282.
8(2
.8)
Tl 3
BO
345
8410
196
2.8
(2.8
)G
a 2H
gS4
4809
6722
02.
8(2
.8)
Tl 3
BS 3
5053
9920
2528
2.8
(3.0
)C
l 2O
5054
6640
7768
2.8
(2.8
)R
bBiS
250
5761
5273
52.
8(3
.2)
CsN
bN2
8978
7254
62.
8(2
.8)
Sr3(
AlP
2)2
9843
4091
342.
8(2
.8)
AgA
sS2
1374
018
101
2.8
(2.9
)C
sLaC
dTe 3
1249
117
3316
2.8
(2.8
)K
7TaA
s 418
073
3801
102.
8(2
.9)
InI
2320
255
179
2.8
(2.9
)N
a 2Pd
Cl 4
2935
920
2971
2.8
(2.8
)R
b 2Te
I 628
070
3600
92.
8(3
.0)
Cs 3
Na 2
SnP 3
1770
750
533
2.8
(2.8
)Sr
(InS
e 2) 2
2173
341
555
2.8
(2.8
)N
bCu 3
S 456
2110
8392
2.8
(3.2
)H
gI22
859
1579
812.
8(2
.9)
SnS 2
1170
6509
922.
8(3
.1)
LaB
i 2IO
454
6672
9242
42.
8(3
.2)
Cs(
SbS 2
) 288
9067
976
2.8
(3.0
)T
lSbS
228
230
3549
82.
8(2
.8)
Tl 2
PAuS
495
1085
680
2.8
(2.9
)K
2Sn 2
O3
8624
4046
32.
8(2
.8)
NaA
gO69
8349
752
2.8
(2.9
)L
aSO
2862
668
498
2.8
(3.4
)L
iCuO
5127
4975
52.
8(3
.1)
K3I
nP2
2025
630
0141
2.8
(2.8
)K
Ta(C
uSe 2
) 260
1365
9203
2.8
(2.8
)R
bTa(
CuS
e 2) 2
1192
541
4274
2.8
(2.9
)C
s 2Ti
(CuS
e 2) 2
1048
928
0646
2.8
(3.0
)C
s 2N
bCuS
e 415
223
8430
62.
8(3
.1)
Ba 2
VN
317
696
8017
72.
8(2
.8)
Sr2V
N3
1701
280
176
2.8
(2.8
)C
a 2V
N3
1789
240
9644
2.8
(2.9
)N
aAlT
e 210
163
4470
12.
9(3
.0)
Rb 2
PtC
210
919
9439
52.
9(2
.9)
Sr2P
bO4
2094
416
806
2.9
(3.1
)K
2NaI
nP2
2151
130
0132
2.9
(2.9
)N
a 2Pt
O2
2231
325
018
2.9
(3.2
)C
dO2
2310
1093
392.
9(3
.0)
Na 3
BiO
427
345
1031
92.
9(3
.0)
K2T
eI6
2768
823
649
2.9
(3.1
)K
4BeA
s 229
584
3001
112.
9(2
.9)
ZnG
eN2
2979
1554
632.
9(2
.9)
MgB
9N30
091
2809
382.
9(2
.9)
RbA
u30
373
5842
82.
9(4
.0)
Ga 2
PdB
r 830
945
4132
292.
9(2
.9)
BaP
dF4
1262
310
8991
2.9
(2.9
)Sr
LiP
1327
641
6889
2.9
(3.0
)B
6O13
4665
6231
2.9
(2.9
)A
lP15
5060
9021
2.9
(4.6
)B
aAgT
eF16
742
4193
822.
9(2
.9)
InTe
I50
5356
1007
032.
9(2
.9)
Ag 2
SO4
5625
5235
62.
9(3
.1)
HgF
706
7235
42.
9(3
.7)
AuF
394
280
478
2.9
(3.0
)K
2SiP
282
3536
367
2.9
(3.3
)Sr
4As 2
O82
9933
904
2.9
(2.9
)K
4HgP
287
5367
263
2.9
(3.0
)C
a 4A
s 2O
8789
6820
32.
9(3
.1)
Cs 2
SiP 2
8932
7122
42.
9(3
.4)
NbN
O75
9610
312.
9(3
.0)
Hg 2
NO
458
2461
437
2.9
(2.9
)K
2NaA
lAs 2
9069
7328
02.
9(3
.0)
Cs 2
Pt3S
413
992
2626
62.
9(2
.9)
9

K4T
a 2S 1
118
664
4106
722.
9(2
.9)
TlI
328
329
6134
92.
9(3
.0)
K3S
nSe 3
5418
7541
0863
2.9
(3.0
)R
b 2S 3
7446
1409
22.
9(3
.0)
K2C
uP84
4661
082
2.9
(2.9
)K
2CuA
s15
684
4393
62.
9(2
.9)
K2A
gP97
7840
2572
2.9
(2.9
)Sr
CuS
eF21
228
1727
982.
9(2
.9)
BaS
nS2
1218
125
872.
9(3
.0)
SbSI
2304
128
305
2.9
(3.0
)R
eNC
lF5
2309
833
542
2.9
(2.9
)B
aYA
gSe 3
6647
1042
372.
9(3
.1)
LiI
nSe 2
2018
756
532
2.9
(2.9
)B
a(A
lTe 2
) 228
505
4116
52.
9(2
.9)
GeA
sSe
5053
6010
0828
2.9
(2.9
)Sr
4PdO
650
5606
8813
52.
9(2
.9)
Na 2
LiG
aAs 2
9722
4021
112.
9(2
.9)
YSF
1008
687
130
3.0
(3.0
)K
SbS 2
1170
360
138
3.0
(3.1
)C
a 2Pb
O4
2113
736
629
3.0
(3.2
)A
s(B
rF2)
328
159
3381
13.
0(3
.1)
Pd(S
Cl 3
) 228
174
3943
43.
0(3
.1)
Sn2O
F 529
590
4093
933.
0(3
.6)
Na 3
AgO
235
2724
817
3.0
(3.1
)N
aSrP
1327
541
6887
3.0
(3.0
)T
l 3V
S 455
1365
5086
3.0
(3.0
)T
l 2Pt
Cl 6
5046
9526
710
3.0
(3.0
)A
uBr
5053
6620
0287
3.0
(3.0
)C
a 4P 2
O53
8040
2952
3.0
(3.2
)L
aSF
5394
9335
03.
0(3
.0)
CsB
iS2
5413
7872
975
3.0
(3.1
)K
2B2S
e 754
2637
6586
933.
0(3
.1)
SiP
2798
8714
93.
0(3
.0)
GaN
805
1538
903.
0(3
.0)
Zr(
PS3)
282
0335
298
3.0
(3.2
)Sr
4P2O
8298
3390
33.
0(3
.1)
Cs 2
NbA
gSe 4
1463
750
464
3.0
(3.1
)B
aLa 2
ZnS
516
452
9371
13.
0(3
.1)
NaB
iO3
2305
491
776
3.0
(3.0
)B
iIO
2298
724
610
3.0
(3.3
)A
l 2Pd
Cl 8
5045
4715
595
3.0
(3.0
)K
3Cu 3
As 2
1420
532
015
3.0
(3.1
)C
sCu 3
O2
5533
0341
3342
3.0
(3.1
)H
g 3(T
eBr)
227
853
2740
23.
0(3
.0)
PPbS
e 320
316
6555
653.
0(3
.1)
NaN
b(C
uS2)
261
8184
301
3.0
(3.0
)K
4ZnP
211
719
6726
13.
1(3
.1)
PBr 5
2287
415
559
3.1
(3.2
)K
2PdC
l 422
956
2752
23.
1(3
.1)
PtC
l 223
290
9866
83.
1(3
.1)
IrC
l 327
666
2317
13.
1(3
.2)
KT
lO27
716
1570
3.1
(3.5
)K
3Al 2
As 3
2834
760
950
3.1
(3.1
)C
dPdF
613
984
2616
63.
1(3
.1)
Tl 2
SiSe
314
241
3504
23.
1(3
.3)
Y2M
gSe 4
1580
376
052
3.1
(3.1
)M
g 3P 2
2514
6427
253.
1(3
.1)
Rb 2
GeS
e 397
9440
2719
3.1
(3.1
)Sn
I 227
194
2831
3.1
(3.2
)Z
rP2S
731
014
5070
13.
1(3
.2)
K3C
u 3P 2
7439
1216
33.
1(3
.2)
YB
i 2B
rO4
5532
4392
418
3.1
(3.4
)Y
Bi 2
IO4
5518
1692
432
3.1
(3.5
)H
gPS 3
2717
825
643.
1(3
.2)
V2O
551
0583
1579
883.
1(3
.3)
SbSB
r22
971
6180
23.
1(3
.2)
Sr2G
eSe 4
3029
341
3023
3.1
(3.1
)K
2NaG
aP2
9666
3001
123.
1(3
.2)
Rb 2
Ti(C
uS2)
271
2928
0644
3.1
(3.3
)Se
Br
5409
4337
017
3.1
(3.2
)K
AgF
473
8772
715
3.1
(3.2
)N
bAgO
312
725
5564
93.
1(3
.6)
HgS
O4
3228
1003
163.
1(3
.4)
Tl 3
SbS 4
1536
010
0849
3.1
(3.1
)H
g(A
uF4)
229
170
8541
43.
2(3
.2)
PtPb
F 620
458
4057
3.2
(3.5
)Pb
F 320
652
2346
73.
2(3
.2)
Rb 2
PbO
321
489
1413
3.2
(3.2
)In
GaO
322
606
3033
93.
2(3
.4)
K2S
eBr 6
2303
636
228
3.2
(3.5
)B
eTe
252
6164
393.
2(4
.9)
InO
F27
175
2521
3.2
(3.2
)C
a(A
uF6)
228
153
3931
53.
2(3
.2)
Cd(
AuF
4)2
2916
985
413
3.2
(3.2
)P 2
PdO
629
274
2006
773.
2(3
.3)
AsC
l 530
106
4121
033.
2(3
.2)
K3G
eSe 3
1443
547
112
3.2
(3.2
)B
a(B
eN) 2
1292
741
5304
3.2
(3.2
)C
aTe
1519
6196
183.
2(4
.2)
Ba 2
PdF 6
5056
2188
802
3.2
(3.2
)C
s 2Pt
C2
5058
2594
397
3.2
(3.2
)A
sS54
2810
1532
813.
2(3
.2)
BaS
268
442
134
3.2
(3.2
)M
gPdF
679
2126
163
3.2
(3.2
)H
gF2
8177
3361
43.
2(3
.2)
Rb 2
B2S
e 716
184
4114
693.
2(3
.3)
Zn(
AuF
4)2
1751
265
286
3.2
(3.3
)B
a 2Sn
Se3F
217
805
1713
443.
2(3
.2)
RbP
Se6
1794
517
0334
3.2
(3.2
)N
a 3G
eSe 3
1841
161
400
3.2
(3.2
)R
bI3
2832
861
348
3.2
(3.3
)C
s 7A
u 5O
251
0075
9582
13.
2(3
.3)
KB
i(W
O4)
251
0667
4191
143.
2(3
.3)
Rb 2
WS 4
1704
828
1586
3.2
(3.2
)A
sSe 3
(ClF
2)3
2357
575
450
3.2
(3.3
)C
uBS 2
1295
415
6413
3.2
(3.2
)N
aV(O
F)2
1895
375
418
3.2
(3.6
)L
aCuS
eO55
2488
9675
83.
2(3
.2)
YB
i 2C
lO4
5526
0492
405
3.2
(3.4
)B
a 2B
iSbO
623
127
1429
3.2
(4.0
)V
2CdO
655
0436
1592
63.
2(3
.2)
Rb 2
TaC
uSe 4
1192
441
4272
3.2
(3.4
)N
aCuO
1458
049
756
3.2
(3.3
)H
g 2P 2
S 727
171
2490
3.2
(3.5
)H
g 3(T
eCl)
250
4706
2740
13.
2(3
.2)
K2T
aCuS
e 489
7272
427
3.2
(3.3
)K
Sb(P
Se3)
271
2390
152
3.2
(3.3
)K
PSe 6
1862
517
0333
3.2
(3.2
)N
a 4Sn
Se4
2876
872
384
3.2
(3.2
)C
s 2A
gSbS
451
0710
5536
73.
2(3
.2)
TaT
l(C
uS2)
298
1540
2636
3.2
(3.2
)Sb
Br 5
F 654
1259
6905
63.
2(3
.2)
Ca 4
PdO
610
299
8813
43.
2(3
.2)
Mg(
InS 2
) 220
493
5309
63.
3(3
.3)
Li 4
PbO
422
170
3835
03.
3(3
.3)
KL
i 6B
iO6
2358
272
840
3.3
(3.3
)IC
l 327
729
2471
43.
3(3
.3)
LiC
aN31
468
1073
043.
3(3
.3)
Li 2
ZrN
232
1678
790
3.3
(4.0
)Sr
2LiR
eO6
1252
541
8991
3.3
(3.3
)C
d 2Si
O4
4530
5053
13.
3(3
.4)
GeI
250
4756
2317
63.
3(3
.6)
Ga 2
S 353
934
647
3.3
(3.3
)R
b 2C
d(PS
e 3) 2
5418
9750
959
3.3
(3.4
)A
u 2S
947
6122
823.
3(3
.3)
CdP
S 353
2879
556
3.3
(3.7
)A
g 2Se
O3
1691
378
388
3.3
(3.4
)C
sI3
2287
627
252
3.3
(3.3
)C
a 2PI
2304
060
683.
3(3
.6)
TaTe
Br9
2971
641
0949
3.3
(3.4
)C
s 2Pd
Cl 4
3031
495
812
3.3
(3.3
)K
2GeS
e 396
9230
0233
3.3
(3.4
)Sr
(CuO
) 213
900
2500
23.
3(3
.3)
GaC
uI4
2940
340
0817
3.3
(3.3
)C
sCuO
5410
3737
079
3.3
(3.3
)Ta
Cu 3
S 410
748
5333
53.
3(3
.7)
Ba(
GaS
e 2) 2
7841
2438
63.
3(3
.4)
Hg 3
(SeC
l)2
2785
127
400
3.3
(3.3
)K
2SnS
e 396
9330
0234
3.3
(3.3
)M
gCrO
419
120
1811
73.
3(3
.4)
PbO
2044
264
7260
3.3
(3.7
)K
2PA
uS4
9509
8567
93.
3(3
.3)
YC
uS2
1053
392
458
3.3
(3.3
)Y
CuP
bS3
5428
0215
2555
3.3
(3.5
)In
I 250
5387
2016
073.
3(3
.3)
TlB
r 250
4527
1421
63.
3(3
.3)
Pt(S
Cl 4
) 228
721
6601
23.
3(3
.3)
Zn(
SbO
3)2
3188
3040
93.
3(3
.3)
K3V
5O14
5407
8724
068
3.3
(3.4
)
10

BaT
e10
0061
6165
3.4
(3.7
)C
sAuO
1054
840
9553
3.4
(3.4
)K
2Mg(
PSe 3
) 211
643
4131
683.
4(3
.4)
Rb 2
PtC
l 623
350
2902
83.
4(3
.4)
Rb 2
TeB
r 623
383
4952
13.
4(3
.6)
GaT
eCl
2744
915
582
3.4
(3.4
)B
e 4Te
O7
2760
813
223.
4(3
.4)
PI2
2944
320
3216
3.4
(3.5
)R
uS3C
l 829
568
4101
123.
4(3
.4)
Cd(
GaO
2)2
3443
1597
393.
4(3
.4)
GaS
2507
6546
853.
4(3
.5)
KS
1287
4340
63.
4(3
.7)
SiC
7631
1561
903.
4(4
.5)
K3S
iTe 3
7657
1238
3.4
(3.6
)K
PS3
8267
3327
83.
4(3
.4)
RbC
aAs
9845
4091
773.
4(3
.5)
Na 3
AlP
251
2240
2081
3.4
(3.5
)M
g(Sc
S 2) 2
1430
737
423
3.4
(3.4
)L
i 2Pd
F 613
985
2616
73.
4(3
.4)
Sr2L
iReN
410
556
4114
543.
4(3
.4)
Cs 2
WS 4
1736
124
9347
3.4
(3.4
)R
bSnI
329
405
4009
343.
4(3
.4)
CsS
nI3
2738
114
070
3.4
(3.5
)T
l 6Si
2O7
2722
842
303.
4(3
.4)
Ca 4
GeN
429
808
2802
513.
4(3
.4)
CsI
2Br
5046
4123
120
3.4
(3.4
)G
e 3N
413
852
2367
23.
4(3
.4)
SbPb
ClO
223
138
3615
93.
4(3
.4)
Rb 2
SnSe
391
4574
844
3.4
(3.4
)B
a 3B
PO3
9712
4020
173.
4(3
.6)
Cs 2
HgI
428
421
6311
03.
4(3
.4)
RbC
uO86
6540
159
3.4
(3.4
)H
gMoO
419
363
2533
3.4
(3.6
)K
2W2O
719
037
6728
43.
4(3
.9)
CaC
rO4
1921
583
387
3.4
(3.4
)B
a 3B
AsO
397
9340
2682
3.4
(3.5
)L
i 10B
rN3
2898
978
819
3.4
(3.7
)H
g 2P 2
O7
5049
8159
280
3.4
(3.8
)K
2TeS
e 350
4959
6301
23.
4(3
.5)
Al 2
Se3
1167
414
373
3.5
(3.5
)K
2Pb 2
O3
2069
414
123.
5(3
.5)
BiS
Cl
2331
810
0173
3.5
(3.5
)C
s 2Te
Br 6
2340
527
695
3.5
(3.7
)A
uI27
725
2461
93.
5(3
.6)
Tl 2
TeB
r 631
076
9912
73.
5(3
.6)
Al 2
CdS
e 431
5951
422
3.5
(3.5
)L
iAuF
412
263
9908
3.5
(3.5
)N
aTl 2
RhF
614
037
2734
03.
5(3
.5)
CsA
gO14
579
4975
43.
5(3
.5)
Sr2S
b 2O
741
0377
062
3.5
(3.8
)Z
nPtF
682
5637
444
3.5
(3.5
)R
bAgO
8603
4975
33.
5(3
.5)
Na 3
AsS
e 386
8650
491
3.5
(3.5
)R
b 3B
As 2
9718
4020
823.
5(3
.6)
RbC
aSb
9846
4091
783.
5(3
.6)
K4B
eP2
9872
3001
103.
5(3
.5)
Hg 3
(SF)
275
8016
927
3.5
(3.6
)R
bAuS
e97
3140
2190
3.5
(3.6
)T
lBS 2
8946
7159
33.
5(3
.6)
RbA
uO10
547
4095
523.
5(3
.5)
K2N
bAgS
415
214
8429
23.
5(3
.6)
RbS
bS2
1536
620
0263
3.5
(3.5
)Sr
3(G
aN2)
216
945
1704
413.
5(3
.5)
K2S
517
146
4467
53.
5(3
.5)
Tl 3
B3S
1017
823
7308
63.
5(3
.5)
AsI
323
218
5656
73.
5(3
.6)
KT
lBr 4
2804
835
418
3.5
(3.5
)L
a 3A
gSnS
754
2888
4173
163.
5(3
.5)
SrC
uSF
1244
415
7433
3.5
(3.5
)B
a(C
uO) 2
7374
9456
3.5
(3.5
)Sb
I 323
281
5656
93.
5(3
.5)
NaA
uF4
7388
9905
3.5
(3.5
)K
CuO
1429
637
325
3.5
(3.6
)Sr
Te19
5865
2879
3.5
(4.2
)Pb
CN
219
727
4109
153.
5(3
.8)
NaI
nS2
2028
964
0036
3.5
(3.7
)Sc
PS4
6999
6755
93.
5(3
.6)
BiV
O4
6131
7265
3651
3.5
(3.5
)C
aPSe
311
007
4127
653.
6(3
.7)
ZnS
1069
516
2754
3.6
(3.6
)Sc
AgO
211
022
9566
83.
6(4
.0)
CsB
r 2F
2865
069
124
3.6
(3.7
)K
2PtC
l 422
934
3701
43.
6(3
.6)
BiS
Br
2332
431
389
3.6
(3.6
)C
sAu
2667
1509
713.
6(4
.1)
TlF
326
3218
029
3.6
(3.6
)G
a 2Te
S 227
255
8028
3.6
(3.6
)M
gPSe
330
943
4131
653.
6(3
.6)
KA
uF6
1244
241
5874
3.6
(3.6
)K
2ZnT
e 212
535
4200
883.
6(3
.7)
La 2
TeO
245
4789
557
3.6
(3.9
)K
2PdF
450
4853
3388
83.
6(3
.7)
Rb 2
Zn 3
O4
5055
0140
754
3.6
(3.6
)L
iInO
254
8863
9886
3.6
(4.2
)A
l 2H
gS4
7906
2563
53.
6(3
.6)
Tl 4
SiS 4
8479
5917
03.
6(3
.8)
KA
gO86
6040
154
3.6
(3.6
)L
aSeF
7738
2101
03.
6(3
.6)
RbA
u 3Se
293
8582
541
3.6
(3.8
)K
AuS
e98
8140
759
3.6
(3.7
)N
aS2
1218
025
863.
6(3
.6)
Ba 2
LiR
eN4
1055
541
1453
3.6
(3.6
)R
b 2S 5
1691
110
0321
3.6
(3.6
)B
a 3(A
lN2)
217
133
4105
783.
6(3
.7)
Na 3
SiTe
317
291
1557
93.
6(3
.6)
Mg(
AuF
4)2
1755
565
287
3.6
(3.6
)K
4SnS
e 418
132
7238
53.
6(3
.6)
LiG
aI3
2934
520
2642
3.6
(3.6
)Z
rI4
5411
1262
243
3.6
(3.6
)H
gBr
2317
715
7980
3.6
(3.9
)N
ClO
5057
2741
1511
3.6
(3.7
)Sr
SnO
328
7916
1783
3.6
(3.7
)B
a 2C
aMoO
619
403
4531
73.
6(3
.7)
As 2
PbO
620
015
8106
33.
6(4
.0)
V2C
d 2O
718
740
6208
13.
6(3
.8)
NbA
gO3
5537
5564
63.
6(3
.6)
Na 3
SbS 4
1016
744
707
3.7
(3.7
)B
aPSe
311
008
4127
683.
7(3
.8)
CsY
CdS
e 311
116
2814
333.
7(3
.7)
SrM
g 2N
210
550
4108
263.
7(3
.7)
KIn
P 2S 7
2258
391
755
3.7
(3.7
)PC
l 523
228
2666
13.
7(3
.7)
NC
lO6
2777
425
817
3.7
(3.7
)R
b 2N
aRhF
614
038
2734
13.
7(3
.7)
K2N
aRhF
614
039
2734
23.
7(3
.7)
Cd(
GaS
2)2
4452
6198
723.
7(3
.7)
InB
rO50
4656
2405
93.
7(3
.8)
CsP
S 350
4838
3327
73.
7(3
.7)
AlA
gS2
5782
6046
983.
7(3
.7)
Al 2
ZnS
e 479
0725
636
3.7
(3.7
)B
a 4L
i(Sb
O4)
379
7120
2200
3.7
(3.8
)C
dPtF
658
5878
906
3.7
(3.7
)Sr
3ScR
hO6
1824
751
137
3.7
(3.7
)K
AlT
e 218
347
4111
713.
7(3
.7)
LaI
327
979
3159
63.
7(3
.8)
NbA
lCl 8
2835
862
029
3.7
(3.7
)R
b 2C
dO2
2836
462
054
3.7
(3.7
)N
a 4Se
O5
2987
141
1401
3.7
(3.8
)K
2Zn 3
O4
5049
3662
146
3.7
(3.7
)B
a 2Sn
S 454
1832
4203
63.
7(3
.7)
AgN
O3
5521
8537
43.
7(4
.0)
Cs 2
HfI
629
398
2013
083.
7(3
.8)
SrPS
e 371
9841
2766
3.7
(3.7
)H
g 2SO
474
6115
005
3.7
(4.3
)V
2Zn 2
O7
1970
728
863.
7(3
.8)
PbW
O4
5407
1981
13.
7(3
.7)
OsO
3F2
2884
573
732
3.7
(3.7
)K
2TeB
r 622
963
6511
53.
8(3
.9)
BiP
bClO
223
084
3186
03.
8(3
.8)
Ba 2
YB
iO6
2313
765
555
3.8
(3.8
)T
lCl 2
2720
540
313.
8(3
.8)
SBr
2809
937
020
3.8
(3.9
)B
iClF
828
194
3955
53.
8(3
.8)
B6P
2839
562
748
3.8
(4.0
)K
BiO
230
988
1623
93.
8(3
.8)
Ba 2
ZnG
e 2S 6
O17
244
1417
43.
8(3
.8)
CsB
r 350
4635
2213
03.
8(3
.8)
KA
gCO
354
1966
4094
843.
8(3
.8)
SrC
rO4
5428
8516
0793
3.8
(3.8
)
11

B6A
s62
410
7916
3.8
(3.8
)C
sRb 2
PdF 5
8202
3528
63.
8(3
.8)
RbS
9062
7317
63.
8(3
.9)
K3B
P 296
6430
0104
3.8
(3.9
)K
3Nb 3
(BO
6)2
1524
885
091
3.8
(3.8
)K
3AsS
e 318
594
5049
23.
8(3
.8)
TlI
2285
855
193
3.8
(3.9
)PI
Cl 6
2782
426
594
3.8
(3.8
)K
2CdO
227
742
2500
43.
8(3
.8)
CdI
228
248
3811
63.
8(3
.9)
LiI
nI4
5410
0136
599
3.8
(3.8
)A
lCuS
249
7942
124
3.8
(3.8
)K
2NaA
lP2
9068
7327
93.
8(3
.9)
Sb2O
321
3664
7390
3.8
(3.8
)C
dMoO
419
039
8445
53.
8(3
.9)
Rb 2
TaC
uS4
1192
341
4271
3.8
(4.0
)B
a 2B
i 2O
528
670
6986
43.
8(3
.8)
AlC
uO2
3748
6084
43.
8(4
.5)
Sr3C
dPtO
610
392
2805
183.
8(3
.9)
KSb
O2
1041
741
1214
3.9
(3.9
)C
sGeC
l 322
988
6255
73.
9(3
.9)
MoP
bO4
2505
456
111
3.9
(4.0
)A
lIC
l 627
935
2651
03.
9(3
.9)
Cs 2
PtC
l 429
216
1001
553.
9(3
.9)
BrN
O3
2952
640
7765
3.9
(3.9
)Sr
TiO
346
5117
0091
3.9
(3.9
)L
iBiO
250
4893
4602
23.
9(3
.9)
KA
uF4
5309
1032
73.
9(3
.9)
TaT
l 3Se
410
644
5243
13.
9(3
.9)
Sr2S
nS3F
217
676
1713
463.
9(3
.9)
Ba 2
CdS
318
309
6665
43.
9(3
.9)
PI3
2752
931
13.
9(3
.9)
Na 3
TlO
229
454
2020
283.
9(4
.1)
Sr(I
nS2)
221
781
3001
763.
9(3
.9)
HgP
b 2(C
lO) 2
5468
6274
973
3.9
(4.3
)C
d(A
sO3)
271
2828
0576
3.9
(4.2
)M
gV2O
650
4510
1039
13.
9(3
.9)
Rb 2
TiC
l 627
827
2668
93.
9(3
.9)
SbC
l 523
176
2503
633.
9(3
.9)
Rb 2
Cr 2
O7
1965
815
034
3.9
(3.9
)H
f(PS
3)2
1444
447
228
3.9
(4.0
)M
g 2V
2O7
3250
023
213.
9(3
.9)
Sr(B
eN) 2
1191
941
3356
4.0
(4.1
)C
sAgB
r 223
454
1503
014.
0(4
.3)
CsS
2926
620
0474
4.0
(4.2
)G
aI3
3095
441
3457
4.0
(4.0
)R
bAuF
434
1933
952
4.0
(4.0
)A
l 2Z
nS4
4842
7627
84.
0(4
.0)
Rb 2
PbO
250
4598
2267
4.0
(4.0
)G
eS2
5426
1365
4304
4.0
(4.1
)C
aMg 2
N2
5795
4111
754.
0(4
.0)
Ba 2
ZnS
355
8765
3999
4.0
(4.0
)R
b 3B
P 297
2040
2084
4.0
(4.1
)L
i 2Pt
F 613
986
2616
84.
0(4
.0)
SrT
lPS 4
1709
024
9346
4.0
(4.0
)M
g 2G
eS4
1744
123
525
4.0
(4.1
)Sr
2NbN
317
701
5102
84.
0(4
.1)
RbL
iZn 2
O3
1842
267
145
4.0
(4.0
)C
sIC
l 222
990
1426
04.
0(4
.1)
SCl 2
2812
838
351
4.0
(4.0
)K
3Nb 3
Si2O
1317
059
1593
84.
0(4
.0)
RbA
uS90
1071
654
4.0
(4.2
)A
sPO
514
367
3664
94.
0(4
.1)
Ba(
InS 2
) 221
943
4602
04.
0(4
.0)
RbL
aSe 2
7176
2810
674.
0(4
.4)
Sc2S
O2
7288
2450
4.0
(4.4
)C
sCu 3
S 277
8623
326
4.0
(4.1
)C
aV2O
632
526
2106
44.
0(4
.1)
Tl 3
VO
432
479
8066
74.
0(4
.0)
K2L
iAlP
264
5077
275
4.0
(4.2
)Sc
VO
419
247
7807
34.
0(4
.0)
RbL
iCrO
418
741
7254
84.
0(4
.1)
BaC
rO4
1966
262
560
4.0
(4.0
)L
i 2G
ePbS
419
896
2810
114.
0(4
.1)
CsC
u 2B
r 323
017
4961
34.
0(4
.0)
Sr2G
eS4
4578
2538
14.
0(4
.1)
VC
l 3O
5048
2036
214
4.0
(4.0
)B
a(A
uF4)
228
719
6528
94.
0(4
.0)
InPS
420
790
1699
4.1
(4.2
)Si
Se2
2214
265
1863
4.1
(4.1
)T
lBr
2287
561
532
4.1
(4.1
)B
rF3
2329
731
689
4.1
(4.3
)K
Sn2B
r 523
541
1519
834.
1(4
.3)
Tl 2
TeC
l 627
833
2670
54.
1(4
.3)
Bi 3
ClO
429
558
4093
304.
1(4
.3)
KTa
O3
3614
5644
04.
1(4
.6)
NaG
e 2N
314
433
4710
04.
1(4
.2)
Nb 2
O5
1595
6537
374.
1(4
.6)
K3G
eS3
1443
463
6776
4.1
(4.1
)B
a 2Si
Se4
1444
749
750
4.1
(4.1
)K
2PtF
638
2187
360
4.1
(4.1
)L
iAlT
e 245
8616
2672
4.1
(4.1
)T
l 2Si
S 381
9035
041
4.1
(4.2
)N
a 5R
eO6
8253
3838
24.
1(4
.1)
Ca 2
Ta2O
714
026
2712
14.
1(4
.1)
Rb 2
TaA
gS4
1521
784
295
4.1
(4.1
)Sr
3(A
lN2)
217
129
7482
44.
1(4
.1)
Ba 2
Nb 6
Te2O
2116
998
4051
074.
1(4
.1)
Ba 2
TaN
317
400
7450
34.
1(4
.1)
TiT
l 2O
317
986
6105
4.1
(4.1
)B
i 3B
rO4
2944
720
1691
4.1
(4.2
)G
aPS 4
3097
926
134.
1(4
.1)
Li 6
MoN
488
0466
095
4.1
(4.2
)B
aZnS
O54
8469
1712
394.
1(4
.1)
KA
uS70
7720
2178
4.1
(4.2
)L
i 4M
oO5
1911
740
270
4.1
(4.1
)B
a 2In
O3F
1995
681
876
4.1
(4.6
)Sr
4Ti 3
O10
3121
334
630
4.1
(4.3
)N
b 2C
d 2O
754
7216
1921
4.1
(4.3
)R
bSbO
210
418
4112
164.
2(4
.3)
CaI
n 2O
422
766
5239
04.
2(4
.2)
Rb 2
TeC
l 622
975
2902
74.
2(4
.3)
BiB
rO23
072
2914
44.
2(4
.5)
KA
s 4IO
623
126
6520
74.
2(4
.6)
BPS
427
724
2461
84.
2(4
.6)
Rb 2
SeC
l 627
829
2669
24.
2(4
.4)
Ga 4
GeO
829
455
2020
444.
2(4
.2)
RbB
iO2
2952
140
7208
4.2
(4.2
)Sn
Br 2
2986
241
1177
4.2
(4.3
)Sr
3(B
S 3) 2
3023
941
2879
4.2
(4.3
)Sb
S 2N
F 612
368
1708
714.
2(4
.2)
ZnP
bF6
1361
015
107
4.2
(4.2
)B
aSe
1253
6161
244.
2(4
.4)
TiO
239
016
1908
4.2
(4.5
)YA
gO2
5100
3495
674
4.2
(4.4
)Ta
TeC
l954
1877
4109
474.
2(4
.3)
Rb 2
PtF 6
8192
3510
84.
2(4
.2)
CaP
tF6
8255
3744
34.
2(4
.2)
Li 2
O2
841
1521
834.
2(4
.2)
ZnO
284
8460
763
4.2
(4.3
)B
aLa 2
PtO
588
0968
794
4.2
(4.2
)G
a 2O
388
683
645
4.2
(4.2
)C
sAu 3
Se2
9386
8254
24.
2(4
.4)
NbB
O4
8615
6320
24.
2(4
.2)
K3S
bS4
9781
4026
424.
2(4
.2)
Cs 2
TaA
gS4
1521
884
296
4.2
(4.2
)B
a 2Sn
S 3F 2
1791
817
1343
4.2
(4.2
)K
3AlT
e 318
378
3001
684.
2(4
.4)
PbI 2
2288
377
325
4.2
(4.3
)R
bPbI
323
517
6067
4.2
(4.2
)K
BiP
2S7
2357
274
019
4.2
(4.2
)B
iI3
2284
936
182
4.2
(4.2
)Te
6Br 2
O11
2925
120
0007
4.2
(4.2
)C
a 3PI
327
272
9026
4.2
(4.2
)C
d 4O
F 650
5174
7403
14.
2(4
.2)
K2T
eS3
5055
5628
0023
4.2
(4.2
)L
aBN
250
5493
4105
984.
2(4
.3)
Li 4
GeS
451
0033
9564
94.
2(4
.3)
CdC
N2
1096
995
264
4.2
(4.2
)Sr
3BPO
397
0240
1207
4.2
(4.4
)Y
2NC
l 328
580
6582
94.
2(4
.7)
Na 3
PS4
2878
272
860
4.2
(4.2
)K
2SrT
a 2O
771
4893
492
4.2
(4.5
)C
aSe
1415
6195
724.
2(5
.1)
ReA
gO4
7094
2800
864.
2(4
.2)
LiV
O3
1944
023
477
4.2
(4.2
)Te
O2
2125
1593
264.
2(4
.2)
WO
F 454
0636
1039
34.
2(4
.2)
NaY
S 210
226
7654
34.
3(5
.1)
12

Na 4
ReN
310
419
4112
434.
3(4
.4)
NaS
r 4(B
N2)
310
811
9257
74.
3(5
.0)
TlC
l23
167
2910
74.
3(4
.3)
ClF
2950
440
6442
4.3
(4.3
)C
a 3A
sBr 3
5044
8842
64.
3(4
.4)
K3B
rO50
4857
3392
04.
3(4
.3)
SrZ
nO2
5637
2555
64.
3(4
.3)
K3I
O28
171
3651
24.
3(4
.3)
K4I
2O9
2776
425
694
4.3
(4.3
)N
a 3A
uO2
2836
562
066
4.3
(4.3
)K
GaI
429
402
4008
164.
3(4
.3)
CsP
bI3
5408
3927
979
4.3
(4.3
)B
aHgO
239
1583
411
4.3
(4.9
)Sc
CuO
236
4215
1929
4.3
(4.7
)Z
nWO
418
918
1626
834.
3(4
.3)
CdW
O4
1938
787
937
4.3
(4.4
)N
aGeS
bO5
6526
3968
94.
3(4
.3)
K3A
uSe 2
1557
140
2000
4.3
(4.3
)M
gTe
1039
6428
834.
4(4
.4)
CsB
rF22
935
8402
04.
4(4
.4)
CsP
bCl 3
2303
729
072
4.4
(4.4
)L
i 2Te
2530
6423
994.
4(5
.2)
KT
lCl 4
2738
514
105
4.4
(4.4
)B
iF5
2774
325
023
4.4
(4.6
)C
s 2Se
Cl 6
2783
026
693
4.4
(4.5
)N
a 4Sn
S 429
628
4203
54.
4(4
.4)
CsN
O2
3288
5032
84.
4(4
.5)
ZrS
O35
1931
721
4.4
(5.0
)L
i 3A
uS2
1599
928
0535
4.4
(4.4
)B
eSe
1541
6164
194.
4(5
.9)
K2B
2S7
4351
6578
094.
4(4
.5)
Na 5
InS 4
5054
1430
0175
4.4
(4.4
)N
aInO
251
7534
600
4.4
(4.6
)Z
rNC
l54
2791
1514
684.
4(4
.4)
Zn(
GaO
2)2
5794
8111
34.
4(4
.5)
Na 3
AsS
358
3010
364.
4(4
.4)
Cs 2
PtF 6
9262
7895
54.
4(4
.4)
Na 3
GaS
e 317
148
3001
644.
4(4
.4)
Sr2T
aN3
1751
896
126
4.4
(4.5
)N
a 5A
sO5
1781
641
1721
4.4
(4.5
)K
3TaS
418
148
5935
54.
4(4
.4)
RbI
nI4
2819
836
601
4.4
(4.4
)T
lCdB
r 328
219
3980
84.
4(4
.4)
Ge 2
S 3B
r 254
0792
2437
04.
4(4
.5)
InG
aBr 4
5412
8369
652
4.4
(4.5
)C
aHgO
270
4180
717
4.4
(5.0
)K
4Br 2
O28
627
6850
54.
4(4
.4)
HgC
l22
897
1579
794.
4(4
.6)
Sr4L
i(B
N2)
397
2340
2173
4.4
(5.1
)V
InO
425
113
1551
624.
4(4
.4)
LiS
bO3
4995
2030
324.
4(4
.4)
KV
O3
1881
544
528
4.4
(4.4
)Y
6WO
1219
005
9203
74.
4(4
.5)
KC
uS28
270
4900
84.
4(4
.4)
Li 5
ReN
438
3892
468
4.4
(4.4
)R
b 2Sn
O2
2793
124
805
4.5
(4.5
)Sn
2OF 2
2748
094
84.
5(4
.6)
InC
lO27
702
2405
84.
5(4
.7)
NO
227
8920
1142
4.5
(4.5
)C
a 3A
sCl 3
2806
936
002
4.5
(4.7
)Ti
(SeO
3)2
2926
020
0203
4.5
(4.5
)C
sBiO
229
506
4065
644.
5(4
.5)
Sr2S
nS4
3029
441
3024
4.5
(4.5
)Pb
F 434
178
895
4.5
(4.7
)Sr
Se27
5853
949
4.5
(5.0
)G
e 2N
2O41
8720
0849
4.5
(4.6
)R
b 2H
gO2
5072
6627
64.
5(4
.5)
P 2S 5
5417
8840
9061
4.5
(4.6
)T
l 6Se
I 428
517
4052
04.
5(4
.5)
MgG
eN2
7798
2350
24.
5(4
.5)
KN
aTe
8755
6727
64.
5(4
.5)
KB
aPS 4
1708
841
4639
4.5
(4.5
)I 2
O5
2326
127
516
4.5
(4.5
)R
b 4In
2S5
2767
023
253
4.5
(4.5
)B
aNb 2
O6
2815
039
272
4.5
(4.8
)L
i 4Ta
N3
5057
9541
2585
4.5
(4.7
)L
a 2Se
O2
7233
2580
44.
5(4
.9)
SnF 3
8289
3378
64.
5(4
.8)
CsS
bO2
5102
7359
329
4.5
(4.5
)H
gBr 2
2329
230
290
4.5
(4.5
)R
b 2H
gF4
8962
7235
24.
5(5
.6)
TiB
r 427
634
2210
34.
5(4
.5)
LiC
a 4(B
N2)
367
9940
0339
4.5
(5.1
)N
aVC
dO4
1944
915
1411
4.5
(4.5
)G
aCuC
l 450
5410
3001
034.
5(4
.5)
LaV
O4
1916
241
1083
4.5
(4.5
)K
2HgS
228
859
7450
04.
5(4
.5)
Hg 3
(BO
3)2
4710
4096
884.
5(4
.6)
BaT
i(B
O3)
211
659
9797
24.
6(4
.6)
BiC
lO22
939
2914
34.
6(5
.2)
Na 2
O2
2340
1092
764.
6(4
.9)
K2S
nCl 6
2349
960
584.
6(4
.6)
Tl 2
SnC
l 627
832
2670
04.
6(4
.7)
Na 2
Te27
8465
6377
4.6
(4.6
)L
iGaB
r 328
327
6133
84.
6(4
.6)
KL
aSiS
412
924
4145
444.
6(4
.6)
BaS
1500
6160
594.
6(4
.7)
CsN
aTe
5339
1075
694.
6(4
.6)
CsA
gCl 2
5427
7215
0299
4.6
(4.9
)K
2HgO
258
6066
275
4.6
(4.6
)C
aGeN
278
0123
523
4.6
(4.6
)N
aLiT
e87
5467
274
4.6
(4.6
)B
aTeO
354
3110
106
4.6
(4.6
)N
aAlS
e 217
060
3001
734.
6(4
.6)
Rb 3
TaS 4
1722
051
115
4.6
(4.6
)T
lSnF
717
649
7889
94.
6(4
.6)
LiI
nGeO
417
854
6222
94.
6(4
.6)
Na 2
Si2S
e 518
562
4900
14.
6(4
.7)
KIO
323
487
2019
84.
6(4
.6)
SBr 2
O28
407
6297
24.
6(4
.7)
Cs 2
AgI
354
0881
3065
84.
6(4
.6)
Cs 2
HgF
489
6372
353
4.6
(5.6
)Ta
Tl 3
S 475
6216
571
4.6
(4.6
)K
NaV
2O6
1943
215
3450
4.6
(4.6
)C
sCu 2
Cl 3
2306
549
612
4.6
(4.6
)N
aVO
319
083
2103
4.6
(4.6
)Sr
2ZnW
O6
1928
272
811
4.6
(4.7
)N
a 3A
gS2
9634
2018
004.
6(4
.8)
Tl 6
SI4
2793
829
265
4.6
(4.6
)L
iZnP
S 411
175
9578
54.
7(5
.0)
Cl 2
2284
820
1696
4.7
(4.8
)G
aBr 2
2838
462
665
4.7
(4.7
)K
LaS
215
781
4494
24.
7(4
.9)
BaZ
nO2
4236
2581
24.
7(4
.7)
NaN
bO3
4514
2815
24.
7(4
.7)
CsO
7896
2552
94.
7(4
.8)
BaT
iO3
5020
1004
644.
7(5
.0)
Sr(B
S 2) 2
8947
7159
44.
7(4
.7)
RbL
aS2
9361
8139
44.
7(5
.0)
BI 3
2318
928
328
4.7
(4.9
)T
lIO
322
981
6210
64.
7(4
.7)
Ba 3
(GaS
3)2
2930
220
1421
4.7
(4.7
)K
Ag(
NO
3)2
1842
910
0711
4.7
(4.7
)R
bGeI
O6
5496
9773
613
4.7
(4.8
)H
fNC
l54
1911
8779
54.
7(4
.7)
Sr2C
aWO
618
742
3646
04.
7(4
.7)
Ba 2
CaW
O6
1918
224
6117
4.7
(4.8
)Sr
2MgW
O6
1942
015
5310
4.7
(4.7
)B
a 3N
b 2Z
nO9
7249
1570
444.
7(4
.9)
TaPC
l 10
5046
8426
097
4.7
(4.7
)R
b 2Sn
Cl 6
2305
929
026
4.8
(4.8
)SO
227
726
2464
54.
8(5
.2)
Li 5
Br 2
N29
025
7883
64.
8(4
.9)
Sr(B
iO2)
229
048
8066
84.
8(4
.8)
TeSe
O4
2929
920
1413
4.8
(4.9
)B
a(B
S 2) 2
3012
641
2516
4.8
(4.9
)T
l 2Z
nI4
3053
237
099
4.8
(4.9
)A
lBiB
r 631
268
4142
624.
8(4
.8)
KB
S 215
012
7961
44.
8(4
.8)
CaS
1672
6195
404.
8(5
.5)
KL
iTe
4495
6788
74.
8(4
.8)
SCl
5048
2437
016
4.8
(4.9
)A
l 2C
dS4
5928
2563
44.
8(4
.8)
InPO
475
6616
618
4.8
(5.0
)H
fSO
7787
2332
74.
8(5
.5)
CsA
u 3S 2
9384
8254
04.
8(5
.0)
Sr2T
a 2O
712
286
601
4.8
(4.8
)M
gSe 2
O5
1677
140
2917
4.8
(4.8
)B
a 3Te
2O9
1700
910
0797
4.8
(4.8
)
13

NbB
iO4
2341
374
338
4.8
(4.9
)Ta
Cl 4
F27
854
2741
34.
8(4
.8)
KT
lF4
2720
940
464.
8(4
.8)
TiSO
550
5270
8062
74.
8(4
.8)
RbC
dBr 3
5045
2980
84.
8(4
.9)
K3T
a 3(B
O6)
298
7020
1143
4.8
(4.9
)B
a 2Sn
O4
3359
8185
04.
8(5
.1)
Cd 2
B2O
572
1028
1357
4.8
(4.8
)M
gWO
418
875
6790
54.
8(4
.9)
Tl 2
MoO
418
938
2806
084.
8(4
.8)
Sr2C
dWO
619
014
2456
834.
8(4
.9)
Ca 3
V2O
854
2076
4122
734.
8(4
.9)
SrTe
O4
4274
6050
74.
8(4
.8)
Ca(
BeN
) 211
918
4133
574.
9(5
.0)
PbB
rF23
008
1550
114.
9(4
.9)
KA
s 4B
rO6
2308
365
206
4.9
(5.1
)N
a 3B
S 329
976
4116
084.
9(5
.0)
Na 2
Se12
6654
281
4.9
(4.9
)T
l 3A
sO4
1557
340
7561
4.9
(5.1
)M
gSe
1303
115
9398
4.9
(4.9
)B
aO13
4261
6005
4.9
(4.9
)R
b 2O
1394
7790
64.
9(5
.3)
LiG
aS2
3647
9691
44.
9(4
.9)
CdC
O3
4385
1567
554.
9(5
.7)
OsO
454
0783
2380
34.
9(4
.9)
K2Z
nO2
8187
3460
34.
9(4
.9)
CsN
aSe
8658
4132
24.
9(5
.0)
ZnS
e 2O
518
373
2355
4.9
(4.9
)M
gTi 2
O5
2823
237
232
4.9
(4.9
)R
bIO
327
193
2825
4.9
(4.9
)B
aSbC
lO2
5455
0020
0962
4.9
(4.9
)In
BO
370
2775
254
4.9
(5.3
)L
aCuO
220
072
1810
24.
9(5
.0)
KG
eNO
6955
6000
24.
9(4
.9)
KN
aSe
2859
567
278
4.9
(4.9
)C
aMoO
419
330
4097
854.
9(4
.9)
P 3N
519
5495
782
4.9
(5.6
)Sr
S10
8724
9177
5.0
(5.4
)R
b 2Se
1132
736
584
5.0
(5.2
)K
Sn2C
l 523
539
1519
825.
0(5
.0)
RbB
S 215
013
7961
55.
0(5
.0)
Tl 3
PO4
5709
6078
05.
0(5
.2)
SrG
a 4O
718
253
1031
65.
0(5
.0)
Ba 2
Ga 2
S 518
421
3825
65.
0(5
.0)
Sr2I
nI5
2350
415
2021
5.0
(5.0
)C
sIO
328
295
6100
95.
0(5
.0)
CsS
nCl 3
2739
414
199
5.0
(5.0
)C
s 2Z
n 3S 4
5056
3353
242
5.0
(5.0
)R
bLiS
e92
5078
878
5.0
(5.0
)N
a 3Sb
O4
7404
1032
05.
0(5
.0)
NaL
iSe
2860
367
359
5.0
(5.0
)L
i 2W
O4
1926
010
445.
0(5
.1)
CdS
e 2O
591
7875
230
5.0
(5.2
)L
i 6W
N4
3503
1536
205.
0(5
.0)
La 2
Sn2O
740
8615
3813
5.0
(5.0
)A
l 6C
d 4Te
O12
9535
8615
55.
0(5
.0)
Ta2O
510
390
2803
965.
1(5
.3)
PbSe
O3
2071
698
376
5.1
(5.1
)R
b 3In
S 322
303
2325
25.
1(5
.1)
AsB
r 323
317
2457
95.
1(5
.3)
CdB
r 227
934
2578
25.
1(5
.1)
BiB
O3
3125
041
3621
5.1
(5.1
)Ti
CdO
313
641
1598
95.
1(5
.1)
BeS
422
6164
135.
1(7
.5)
MgG
eO3
4819
2016
645.
1(5
.2)
Rb 2
ZnI
450
5748
5642
55.
1(5
.1)
Cs 2
Sn(G
eO3)
354
0707
1903
15.
1(5
.2)
CsC
dBr 3
5418
9987
261
5.1
(5.1
)T
lNO
359
1575
253
5.1
(5.2
)C
d 4P 6
SN12
8921
7101
95.
1(5
.1)
Sr2B
N2F
1023
450
843
5.1
(5.1
)K
3Ta 3
Si2O
1316
855
1593
75.
1(5
.1)
K3B
S 329
975
4116
075.
1(5
.2)
Pb2O
F 227
355
1041
65.
1(5
.3)
Ba 3
Zn 6
Si4T
eO20
5430
3441
6231
5.1
(5.1
)Sn
3BrF
523
452
2000
315.
1(5
.1)
BaB
iIO
255
1243
9751
15.
1(5
.3)
NbI
nO4
9595
1006
955.
1(5
.3)
Sr3V
2O8
1938
673
258
5.1
(5.2
)Y
2Sn 2
O7
3370
1601
155.
1(5
.1)
KIn
Br 4
2293
247
141
5.2
(5.2
)R
bPb 2
Br 5
2304
326
663
5.2
(5.4
)A
l 2S 3
2654
3002
135.
2(5
.2)
K2T
e17
4764
1376
5.2
(5.2
)C
aInB
r 350
5024
5413
85.
2(5
.4)
RbO
7895
2552
85.
2(5
.3)
KL
iSe
8756
6727
75.
2(5
.3)
CaT
iGeO
517
784
1717
955.
2(5
.2)
K3P
S 417
989
2459
95.
2(5
.2)
Cd 2
P 2O
727
686
2354
25.
2(5
.5)
Sr3T
e 4O
1130
026
5679
85.
2(5
.3)
Ta2Z
nO6
1776
536
289
5.2
(5.2
)B
a 2Y
NbO
672
5117
2407
5.2
(5.2
)N
aCaV
O4
1930
232
573
5.2
(5.2
)H
gCl 2
2285
576
648
5.2
(5.2
)L
iAlS
e 271
1728
0225
5.2
(5.2
)C
aPS 3
9789
4051
925.
2(5
.2)
MgM
oO4
1904
720
418
5.2
(5.3
)L
iAl(
MoO
4)2
1920
916
175
5.2
(5.2
)N
a 4W
O5
1933
485
063
5.2
(5.2
)R
bLa(
WO
4)2
1968
027
737
5.2
(5.2
)Sr
(AlS
e 2) 2
8422
4973
25.
2(5
.2)
Li 4
TeO
548
0420
2326
5.2
(5.3
)R
bTlF
427
210
4047
5.2
(5.3
)C
dSeO
391
8675
273
5.2
(5.2
)B
aPS 3
1100
641
2764
5.3
(5.3
)A
gClO
422
993
9629
5.3
(6.1
)N
a 3B
iO3
2791
423
347
5.3
(5.3
)Pb
Br 2
2807
736
170
5.3
(5.4
)L
iBeN
2946
340
2341
5.3
(5.4
)L
iNO
381
8067
981
5.3
(5.6
)N
a 3Sb
O3
8076
2334
65.
3(5
.3)
Rb 3
PS4
1686
340
9818
5.3
(5.3
)Z
n(IO
3)2
2336
054
086
5.3
(5.3
)T
lCdC
l 328
218
3980
75.
3(5
.3)
Ga 1
0GeP
b 3O
2050
4827
3717
85.
3(5
.4)
La 3
TaO
751
0513
5565
55.
3(5
.3)
Ca(
SbO
3)2
9125
7453
95.
3(5
.6)
Na 4
Br 2
O28
599
6728
35.
3(5
.3)
KSb
O3
5477
9233
546
5.3
(5.4
)B
a 3V
2O8
1936
541
8460
5.3
(5.4
)Sr
PS3
9788
4051
915.
3(5
.3)
P 4N
3Cl 1
128
792
7191
35.
3(5
.3)
ScT
l(M
oO4)
219
450
2503
405.
3(5
.3)
SnF 2
7457
1419
55.
3(6
.0)
SnC
l 229
179
8197
95.
3(5
.3)
Sr2B
N2C
l23
131
4063
925.
4(5
.6)
Li 2
Se22
8664
2355
5.4
(5.8
)L
iBiF
627
419
1511
95.
4(5
.4)
Na 3
PS3O
1173
898
651
5.4
(5.4
)L
iNbO
337
3141
5526
5.4
(5.4
)N
aNO
345
3142
0041
5.4
(5.5
)C
sAl(
MoO
4)2
5421
1628
0947
5.4
(5.4
)Sb
OF
7609
1901
95.
4(5
.6)
Li 3
BS 3
5614
3801
045.
4(5
.4)
K2S
e84
2660
440
5.4
(5.5
)C
dSO
484
5960
571
5.4
(5.7
)B
a(Sb
O3)
291
2774
541
5.4
(5.5
)B
a 2Z
nO3
1791
136
659
5.4
(5.4
)C
d(IO
3)2
2764
013
975.
4(5
.5)
TlB
O2
2824
436
404
5.4
(5.4
)Sr
Ta2O
617
715
3970
65.
4(5
.4)
PBr 2
N23
457
2727
15.
4(5
.4)
K3V
O4
1905
241
385.
4(5
.4)
Ca 2
SnO
447
4717
3626
5.4
(5.4
)A
lTl(
MoO
4)2
1873
325
0339
5.4
(5.4
)Sr
MoO
418
834
2458
025.
4(5
.4)
KA
l(M
oO4)
219
352
2801
85.
4(5
.4)
Na 4
SnO
496
5520
2818
5.4
(5.4
)R
bNbO
332
8320
0854
5.4
(5.4
)Pb
SeO
422
342
4092
15.
5(5
.6)
AsC
l 2F 3
2344
433
884
5.5
(5.5
)Sr
O2
2697
6474
745.
5(5
.7)
PBr 3
2725
780
525.
5(5
.5)
CsB
iF6
2742
215
122
5.5
(5.6
)Te
2O3F
229
185
8216
25.
5(5
.5)
Cs 3
As 5
O9
3030
041
3151
5.5
(5.6
)M
gS13
1565
9124
5.5
(6.3
)Si
S 216
0227
205
5.5
(5.8
)
14

BaS
iN2
3777
1702
695.
5(5
.6)
La 2
SO2
4511
2601
455.
5(6
.0)
SrSi
N2
4549
1702
705.
5(5
.6)
TaB
O4
4624
4024
045.
5(5
.5)
Rb 2
CdC
l 450
5668
5116
85.
5(5
.9)
Tl 2
CO
354
3045
2412
455.
5(5
.6)
SeO
272
659
712
5.5
(5.9
)M
g 3Te
O6
3118
2361
15.
5(5
.7)
Rb 2
S80
4129
208
5.5
(5.8
)K
2O97
164
1282
5.5
(5.9
)K
NO
351
5828
077
5.5
(5.5
)L
iTa 3
O8
7638
493
5.5
(5.6
)N
a 4I 2
O22
937
6711
25.
5(5
.5)
K3B
iO3
2952
440
7293
5.5
(5.5
)Ta
BiO
430
900
9742
35.
5(5
.6)
NO
F50
5726
4115
105.
5(5
.6)
BaS
n(G
eO3)
354
0635
1038
45.
5(5
.6)
BaB
iClO
255
2806
7953
25.
5(5
.5)
SrB
iBrO
255
2234
9750
95.
5(5
.6)
Na 3
ClO
2860
267
319
5.5
(5.5
)C
s 2L
iVO
454
1190
4021
95.
5(5
.5)
Cs 2
NaV
O4
1944
765
963
5.5
(5.6
)Sr
2YN
bO6
6019
1636
645.
5(5
.5)
Rb 2
LiV
O4
1912
340
218
5.5
(5.5
)R
bLa(
MoO
4)2
1968
742
700
5.5
(5.6
)Sb
Br 3
2739
914
217
5.5
(5.7
)N
aInB
r 428
569
6546
25.
5(5
.5)
BaO
211
0580
750
5.6
(5.6
)B
iOF
2307
424
096
5.6
(5.6
)N
a 2O
2352
6449
175.
6(5
.6)
BrF
527
987
3169
05.
6(5
.6)
BaA
l 2Sb
2O7
1288
515
4362
5.6
(5.8
)Y
2SO
212
894
1545
825.
6(5
.7)
CsG
aS2
5038
4188
85.
6(5
.7)
YN
bO4
5387
2033
55.
6(5
.8)
CaN
667
641
2259
5.6
(5.6
)R
bNaS
8799
6848
95.
6(5
.7)
Li 3
SbO
457
6927
9592
5.6
(5.6
)K
SeO
2F92
0578
398
5.6
(5.6
)In
2P2O
717
100
4128
555.
6(5
.6)
LiG
aCl 3
2934
420
2641
5.6
(5.7
)Sr
2CuB
rO2
5525
3765
470
5.6
(5.7
)N
a 2Z
nGeO
464
0276
314
5.6
(5.7
)N
aSr 3
NbO
663
6141
8494
5.6
(5.7
)G
a 2Pb
O4
2049
680
129
5.7
(5.8
)B
iCl 3
2290
841
179
5.7
(5.9
)G
e 3(B
iO3)
423
560
2013
565.
7(5
.8)
MgS
eO4
1263
010
9070
5.7
(5.8
)Sn
ClF
5045
1964
75.
7(5
.7)
CaW
O4
5105
6315
5792
5.7
(5.7
)N
aReO
455
5852
337
5.7
(5.7
)M
g 2A
s 2O
756
1823
548
5.7
(5.7
)B
aAl 4
S 782
5833
237
5.7
(5.8
)B
aTiO
F 416
915
7274
05.
7(5
.7)
LiZ
nAsO
418
048
8618
45.
7(5
.7)
Be 3
N2
1833
741
2667
5.7
(5.7
)R
b 4C
dBr 6
2831
560
625
5.7
(5.7
)L
iGaB
r 428
326
6133
75.
7(5
.7)
CaN
b 2O
617
101
1520
85.
7(5
.7)
Bi 3
B5O
1223
549
4128
315.
7(5
.9)
CsN
aS69
7341
323
5.7
(5.7
)Sr
BiC
lO2
5472
4484
636
5.7
(5.7
)C
sTa(
BO
3)2
9309
8042
35.
7(5
.9)
SbA
sO3
2810
937
187
5.7
(5.8
)N
a 3M
o(O
F)3
1875
397
453
5.7
(5.8
)A
lCuC
l 428
020
3505
05.
7(5
.8)
SbC
lF8
2731
498
995.
7(5
.7)
TiT
l 2F 6
1040
241
0802
5.8
(5.9
)Pb
BrC
l22
997
4588
15.
8(5
.9)
CaH
223
713
1559
875.
8(6
.6)
BaH
223
715
1559
895.
8(6
.3)
K2N
aAlH
624
412
1528
915.
8(5
.8)
LiN
326
5915
5166
5.8
(6.2
)Z
nCN
229
826
2805
235.
8(6
.1)
AlI
330
930
3912
475.
8(5
.8)
Ba(
IO3)
230
991
2327
65.
8(5
.9)
Ca(
AsO
3)2
4555
7737
95.
8(6
.1)
RbI
n(M
oO4)
250
4506
1018
65.
8(5
.8)
Na 2
S64
865
6376
5.8
(5.8
)T
lF72
090
996
5.8
(5.9
)L
i 2Z
nGeO
481
8434
362
5.8
(5.8
)Sn
B4O
713
252
2492
065.
8(6
.3)
Pb2C
O4
5057
0291
714
5.8
(5.9
)K
NaS
5049
3862
658
5.8
(5.8
)B
aPbF
619
799
2552
15.
8(5
.8)
ScB
rO54
6279
1707
745.
8(5
.8)
BaM
oO4
1927
616
1851
5.8
(5.8
)R
bTaO
330
3323
015.
8(5
.8)
AsC
lF8
2792
623
775
5.9
(5.9
)N
aGaO
233
3836
652
5.9
(5.9
)K
2TiO
313
133
1622
165.
9(5
.9)
Mg 2
PN3
3933
5022
45.
9(6
.6)
CsM
gI3
5055
8687
262
5.9
(6.0
)C
aBiC
lO2
5530
2584
635
5.9
(6.0
)A
s 2O
315
8165
5563
5.9
(6.0
)R
bLiS
8751
6725
45.
9(5
.9)
TaIn
O4
8979
7256
95.
9(6
.0)
LiG
aO2
5854
9308
75.
9(5
.9)
Li 3
BN
250
0115
5129
5.9
(6.2
)R
b 3G
aO3
1374
422
705.
9(5
.9)
Na 3
ScB
r 629
417
4013
355.
9(5
.9)
K4Z
r 5O
1227
377
1402
45.
9(5
.9)
Sr3G
a 4O
930
158
5154
65.
9(5
.9)
NaG
aBr 4
5050
8469
650
5.9
(5.9
)G
aAsO
439
9641
949
5.9
(5.9
)K
2S10
2264
1321
6.0
(6.1
)K
NaL
aNbO
510
942
9474
36.
0(6
.0)
SrN
621
3141
2255
6.0
(6.0
)C
dCl 2
2288
130
255
6.0
(6.0
)SC
l 2O
2840
662
971
6.0
(6.0
)Z
n 3S 2
O9
3098
615
280
6.0
(6.0
)Sr
SeO
333
9524
0888
6.0
(6.0
)Sr
2ZnG
e 2O
717
392
3915
96.
0(6
.0)
GeO
273
359
639
6.0
(6.1
)K
2TeO
387
2465
640
6.0
(6.2
)L
i 16T
a 2N
8O14
871
7169
66.
0(6
.0)
NaI
O3
2298
929
098
6.0
(6.1
)L
i 2Te
O3
2723
143
176.
0(6
.2)
Na 3
In2(
PO4)
317
349
8477
66.
0(6
.1)
SbC
l 322
872
8258
6.0
(6.1
)C
sPbF
320
282
9343
86.
0(6
.0)
KA
lAs 2
O7
9230
7971
16.
0(6
.1)
CsS
bClF
323
576
2002
966.
0(6
.0)
Li 2
SnO
335
4021
053
6.0
(6.1
)Z
n(R
eO4)
210
326
5101
76.
1(6
.1)
Li 2
S11
5354
396
6.1
(7.0
)L
aBr 3
2326
331
581
6.1
(6.1
)K
CdC
l 323
442
3135
76.
1(6
.2)
CdF
224
125
0165
6.1
(6.6
)B
a 2Y
TaO
612
385
1711
766.
1(6
.1)
Ba 2
LaT
aO6
1305
516
0169
6.1
(6.3
)L
i 2Ti
GeO
513
182
2502
976.
1(6
.2)
BaZ
rO3
3834
9746
26.
1(6
.2)
KSO
476
2254
024
6.1
(6.2
)B
aI2
2326
015
707
6.1
(6.1
)Ta
2Zn 3
O8
2825
146
001
6.1
(6.1
)N
a 3N
bO4
2724
761
166.
1(6
.1)
LiA
lGeO
416
947
6723
86.
1(6
.1)
HfT
e 3O
818
352
9078
6.1
(6.1
)R
bMgH
323
738
1591
766.
1(6
.3)
Sr2I
2O55
1203
4098
966.
1(6
.1)
K2M
oO4
1891
415
0842
6.1
(6.2
)Pb
CO
319
893
5610
16.
1(6
.2)
BaC
N2
2889
875
041
6.1
(6.1
)K
BrO
322
958
3366
36.
2(6
.2)
PbC
lF22
964
3028
76.
2(6
.2)
SrH
223
714
1559
886.
2(6
.9)
BeA
lH5
2371
915
6311
6.2
(6.2
)R
b 2Z
rCl 6
2783
126
694
6.2
(6.2
)Sr
4I6O
2991
028
0585
6.2
(6.2
)K
ReO
447
5772
503
6.2
(6.2
)L
aNbO
452
9581
618
6.2
(6.3
)N
a 2C
N2
5419
8941
1341
6.2
(6.3
)N
aLiS
8452
6109
16.
2(6
.2)
Na 5
TaO
589
5772
297
6.2
(6.2
)K
MgH
323
737
1591
756.
2(7
.9)
SrW
O4
1916
315
5793
6.2
(6.2
)M
gPbF
619
734
1510
66.
2(6
.2)
La 3
Ga 5
SnO
1467
8855
416
6.2
(6.2
)
15

AsC
l 323
280
3513
36.
3(6
.6)
PbC
l 223
291
2773
66.
3(6
.5)
CsS
rN9
2922
810
0461
6.3
(6.4
)K
AsO
230
298
4131
496.
3(6
.4)
MgS
eO3
1227
149
46.
3(6
.5)
CaT
aAlO
515
733
5071
86.
3(6
.3)
BaN
617
0765
3607
6.3
(6.3
)R
bReO
440
3552
339
6.3
(6.3
)K
LiS
8430
4722
96.
3(6
.3)
TaA
lO4
1433
333
885
6.3
(6.5
)C
a 4A
l 6Te
O12
1531
286
156
6.3
(6.3
)M
gI2
2320
552
279
6.3
(6.4
)C
sMgH
323
751
1622
606.
3(6
.6)
MgS
n(B
O3)
211
715
2826
66.
3(6
.4)
ScA
sO4
5461
2515
5920
6.3
(6.3
)R
b 2M
oO4
1921
224
904
6.3
(6.3
)Z
n 4B
6O13
4812
1002
906.
3(6
.4)
NaA
lI4
2939
540
0521
6.3
(6.3
)C
a 2B
N2F
1023
350
842
6.3
(6.3
)Si
3(B
iO3)
423
331
2678
76.
4(6
.5)
SrC
N2
1231
759
860
6.4
(7.1
)R
b 3A
lO3
1495
174
969
6.4
(6.4
)N
aGe 2
(PO
4)3
5429
1916
4019
6.4
(6.5
)A
lN66
116
3953
6.4
(6.4
)G
eF2
7595
1803
06.
4(6
.8)
Na 2
GeO
357
8416
226.
4(6
.6)
CsC
dF3
8399
4958
26.
4(7
.3)
Ba 3
NaT
aO6
8961
7233
16.
4(6
.4)
YTa
O4
5377
3842
06.
4(6
.7)
Rb 6
Ge 2
O7
1822
473
551
6.4
(6.4
)B
aSeO
369
8954
156
6.4
(6.5
)Sr
Sn(B
O3)
280
0028
267
6.4
(6.8
)K
2CN
210
408
4110
946.
5(6
.5)
PCl 3
2323
027
798
6.5
(6.6
)K
2ZnB
r 423
535
7293
16.
5(6
.5)
ZrO
228
5815
7403
6.5
(6.5
)A
l 2Z
nO4
2908
1632
686.
5(6
.5)
RbA
sO2
3029
941
3150
6.5
(6.5
)Sb
PO4
3439
6297
76.
5(6
.8)
MgS
iN2
3677
9073
16.
5(6
.8)
ZrG
eO4
8042
2926
26.
5(6
.6)
NaS
c(G
eO3)
280
5422
503
6.5
(6.5
)Z
n 4P 6
SN12
1583
376
440
6.5
(6.7
)Sr
GeO
317
464
5930
36.
5(6
.6)
CaG
eO3
1776
140
3086
6.5
(6.6
)Sr
CdP
2O7
1769
372
672
6.5
(6.5
)C
a 3Sc
2(G
eO4)
321
989
2021
56.
5(6
.5)
Na 3
AlH
623
705
1546
736.
5(6
.5)
RbB
rO3
2887
274
768
6.5
(6.5
)N
a 2Z
nSiO
463
9134
565
6.5
(6.6
)G
eP2O
728
883
7487
66.
5(6
.5)
Rb 2
TiO
354
0378
842
6.5
(6.5
)N
aN3
2200
377
323
6.6
(7.0
)B
Br 3
2322
517
3374
6.6
(6.8
)M
gO2
2589
4173
26.
6(6
.7)
SeO
F 227
367
1211
06.
6(6
.6)
SeF 4
2917
285
451
6.6
(6.6
)Sr
2YSb
O6
1287
815
4033
6.6
(6.7
)C
aCN
241
2441
8451
6.6
(7.3
)YA
sO4
8058
2451
36.
6(6
.6)
Na 2
SeO
451
4115
0706
6.6
(6.7
)T
lZnP
O4
1816
874
811
6.6
(6.7
)N
aScC
l 429
432
4022
736.
6(6
.7)
Zr 4
Cd(
PO4)
654
1299
6682
06.
6(6
.6)
Na 4
GeO
429
7062
595
6.6
(6.6
)K
SO2F
6560
9306
06.
6(6
.6)
SnF 4
2706
7889
46.
6(6
.8)
KC
dF3
9628
2013
296.
6(6
.6)
BaW
O4
1904
815
5512
6.6
(6.6
)B
aBA
sO5
9784
4044
396.
6(6
.6)
Mg(
BeN
) 211
917
4133
586.
7(7
.3)
ScC
l 323
309
3823
56.
7(6
.7)
K2N
aTlF
613
798
2211
46.
7(6
.7)
RbS
cO2
7650
1270
6.7
(7.1
)T
lSbF
683
4836
264
6.7
(6.9
)K
Na 2
BO
382
6333
261
6.7
(6.9
)R
bNa 2
BO
388
7267
525
6.7
(6.9
)B
aSeO
412
010
4098
106.
7(6
.8)
BaG
eO3
1386
323
925
6.7
(6.7
)L
i 2Z
nSiO
417
288
8237
6.7
(6.7
)N
aMgH
323
730
1582
516.
7(6
.7)
Sc2(
SeO
3)3
3106
598
624
6.7
(6.8
)N
a 2L
i 3G
aO4
5409
4537
071
6.7
(6.7
)S(
ClO
) 228
405
6297
06.
7(6
.7)
HfC
l 429
422
4020
546.
7(6
.7)
RbS
O2F
6384
9306
86.
7(6
.7)
KC
SN65
1167
582
6.7
(6.7
)Sr
ZrO
343
8715
9454
6.7
(6.8
)H
fGeO
497
5520
2080
6.7
(6.7
)C
aPbF
620
463
2552
26.
8(6
.8)
KH
SeO
324
433
2086
26.
8(6
.9)
CaI
230
031
5228
06.
8(7
.2)
Mg 3
NF 3
7604
1832
06.
8(6
.8)
RbL
aO2
7972
2733
16.
8(6
.8)
KN
382
715
5168
6.8
(6.8
)N
a 2Se
O3
5416
2809
416.
8(6
.8)
Mg 4
Ta2O
917
481
6530
16.
8(6
.8)
KA
sOF 4
1753
990
276.
8(6
.8)
K2L
i 3G
aO4
1777
435
290
6.8
(6.8
)C
aZn 2
(PO
4)2
1830
820
2571
6.8
(6.9
)N
aI23
268
6150
26.
8(6
.8)
K2B
eO2
2791
523
633
6.8
(6.9
)B
a 4I 6
O29
909
2805
846.
8(6
.8)
Na 3
BO
351
0256
1351
6.8
(6.9
)C
sBrO
328
873
7476
96.
8(6
.8)
NaC
SN66
3395
46.
8(6
.8)
KL
i 6Ta
O6
9059
7315
96.
8(6
.9)
TlS
bF4
2928
620
1084
6.8
(6.8
)Z
nCl 2
2290
915
916
6.9
(6.9
)K
4CdC
l 623
392
6075
36.
9(7
.0)
SrO
2472
2491
786.
9(7
.0)
As 2
SO6
2723
042
976.
9(7
.0)
NaC
dF3
4360
1638
676.
9(6
.9)
KL
aO2
7958
2700
16.
9(6
.9)
Rb 2
Li 2
GeO
484
5061
087
6.9
(7.0
)N
aSi 2
N3
8973
7246
66.
9(6
.9)
K2S
eO4
5226
2018
826.
9(6
.9)
Ba 1
0P6S
O24
1699
041
0785
6.9
(6.9
)R
b 2Si
3SnO
917
382
1902
86.
9(7
.2)
K3N
a(Se
O4)
217
839
7346
36.
9(6
.9)
BaS
i 3Sn
O9
1850
210
385
6.9
(7.1
)Z
nF2
1873
5398
16.
9(7
.0)
K2W
O4
1878
015
0840
6.9
(7.0
)Sb
F 318
8030
411
6.9
(7.2
)K
3AlO
391
5774
968
6.9
(6.9
)Sr
3GaO
4F65
0950
735
6.9
(7.0
)Z
nSO
451
2671
018
6.9
(7.0
)Pb
SO4
2229
815
4273
7.0
(7.2
)B
a(B
rO3)
250
5008
4028
77.
0(7
.1)
RbN
374
315
5169
7.0
(7.0
)N
aScO
279
1425
729
7.0
(7.7
)C
s 2L
i 2G
eO4
8313
3653
27.
0(7
.0)
NaY
GeO
417
543
8549
77.
0(7
.1)
Cs 6
Si2O
718
315
4116
657.
0(7
.0)
Na 3
Ca 2
TaO
618
480
2802
847.
0(7
.0)
BaL
a 2B
eO5
1841
465
292
7.0
(7.0
)R
b 4C
dCl 6
2293
015
1893
7.0
(7.0
)L
iI22
899
4142
447.
0(7
.0)
PNC
l 223
375
1092
727.
0(7
.0)
Tl 2
SnF 6
1040
141
0801
7.0
(7.0
)L
aGeB
O5
1995
739
262
7.0
(7.0
)H
fO2
352
1731
587.
0(7
.0)
AlG
aCl 4
5411
1162
232
7.0
(7.0
)L
aAlO
329
2015
3830
7.1
(7.1
)K
2ZrG
e 2O
716
871
8884
37.
1(7
.1)
RbA
lO2
1407
028
373
7.1
(7.1
)R
b 2L
iAsO
414
363
3664
47.
1(7
.2)
KSc
O2
8188
3495
87.
1(7
.5)
CsN
a 2B
O3
8871
6752
47.
1(7
.1)
Li 2
CN
296
1020
0369
7.1
(7.2
)N
a 4H
f 2(G
eO4)
314
526
6580
77.
1(7
.1)
Zr 3
Tl 2
OF 1
217
315
4800
37.
1(7
.1)
RbZ
nPO
418
463
7177
87.
1(7
.1)
Rb 3
PbC
l 529
883
8957
07.
1(7
.1)
LiI
nP2O
784
9160
935
7.1
(7.1
)C
dS2O
717
586
6306
77.
1(7
.1)
Y2C
(NO
) 254
6864
2453
327.
1(7
.1)
CsS
O2F
6368
9307
87.
1(7
.2)
CaM
gAsO
4F70
3556
862
7.1
(7.1
)
16

La 2
MgG
eO6
1158
597
016
7.2
(7.3
)N
aBrO
323
339
2883
17.
2(7
.2)
TaA
gF6
2999
341
1796
7.2
(7.2
)Si
2CN
430
161
9354
47.
2(7
.2)
BA
sO4
3277
4134
387.
2(7
.6)
ZnS
nF6
1390
325
012
7.2
(7.2
)L
i 4G
eO4
4558
6517
77.
2(7
.2)
LiS
cO2
5840
3612
47.
2(7
.2)
Na 4
SiO
475
0062
594
7.2
(7.3
)C
s 2N
aAsO
483
1436
533
7.2
(7.2
)C
s 2Z
rO3
8759
6734
57.
2(7
.2)
Cs 2
Li 2
TiO
482
9433
810
7.2
(7.3
)Si
3N4
988
1700
067.
2(7
.4)
Ba 3
(AsO
4)2
9783
4044
387.
2(7
.2)
CsZ
nPO
418
673
8545
77.
2(7
.2)
Sr10
P 6SO
2417
447
4107
837.
2(7
.2)
IF5
2325
760
217.
2(7
.2)
TlT
eF5
2990
390
619
7.2
(7.4
)C
a 2H
f 7O
1 627
221
4136
7.2
(7.2
)C
sNa 3
Li 1
2(G
eO4)
417
125
7909
87.
2(7
.3)
NaS
bF4
2926
920
0573
7.2
(7.2
)C
aO26
0516
3628
7.3
(7.6
)L
i 2C
aGeO
476
1119
024
7.3
(7.3
)Sr
Zn 2
(PO
4)2
8810
6888
17.
3(7
.3)
SrG
a 2(S
iO4)
214
235
3441
47.
3(7
.3)
KL
i 3G
eO4
1800
238
324
7.3
(7.3
)K
I22
898
5224
47.
3(7
.3)
RbI
2290
361
536
7.3
(7.3
)B
aIF
2295
111
287.
3(7
.3)
K4B
e 3O
528
158
3380
87.
3(7
.3)
LiG
aSiO
418
147
6512
57.
3(7
.3)
Na 4
TiO
414
726
6962
17.
3(7
.3)
NaL
i 2A
sO4
9066
7320
07.
3(7
.4)
LaA
sO4
4917
1559
177.
3(7
.3)
LaB
rO23
023
8433
67.
4(7
.4)
PbF 2
315
5398
47.
4(7
.5)
CdS
nF6
1390
725
017
7.4
(7.4
)A
lTlF
437
5120
2458
7.4
(7.5
)B
a 3N
b 2O
854
2201
9519
37.
4(7
.4)
K2Z
rO3
1844
916
264
7.4
(7.4
)K
ZnP
O4
1859
188
955
7.4
(7.4
)N
bF5
1868
726
647
7.4
(7.4
)Sr
IF23
046
1592
807.
4(7
.4)
Zr 2
P 2O
927
132
1922
7.4
(7.5
)L
i 3A
sO4
9197
7592
77.
4(7
.5)
SrH
fO3
3378
1629
067.
4(7
.4)
BiF
323
237
9015
7.5
(7.6
)A
lBr 3
2328
839
768
7.5
(7.5
)C
sMgB
r 329
750
8726
07.
5(7
.5)
Li 2
TiF 6
7603
1831
37.
5(7
.5)
BaT
iF6
8291
3378
97.
5(7
.5)
InB
F 485
8650
218
7.5
(7.5
)R
b 2L
iGaF
614
638
5046
87.
5(7
.5)
RbG
aCl 4
3023
140
9650
7.5
(7.5
)N
aZnF
337
9572
320
7.5
(7.5
)K
2Pb(
SO4)
221
099
7689
17.
6(7
.8)
LaC
l 322
896
3157
47.
6(7
.6)
CsL
iBr 2
2305
724
5979
7.6
(7.9
)C
sAsF
457
0754
852
7.6
(7.6
)A
sPO
481
4031
879
7.6
(7.6
)C
sK2S
c(PO
4)2
8564
6178
77.
6(7
.7)
ScB
O3
8697
6501
07.
6(7
.6)
LiG
eBO
488
7367
535
7.6
(7.8
)B
N98
435
538
7.6
(7.9
)N
aAlO
292
1279
404
7.6
(7.6
)L
iYO
270
2045
511
7.6
(7.6
)N
a 3Y
Br 6
2908
082
355
7.6
(7.7
)Sc
OF
4661
1005
647.
7(7
.7)
Ba 2
Zn 7
F 18
5418
2740
925
7.7
(7.7
)A
lAsO
478
4924
512
7.7
(7.7
)K
6Si 2
O7
3099
017
064
7.7
(7.7
)In
F 369
4938
306
7.7
(7.7
)N
a 2M
gSiO
464
0615
619
7.7
(7.7
)C
aAl 4
O7
4867
4451
97.
7(7
.7)
La 2
Hf 2
O7
1253
315
3815
7.7
(7.7
)N
aPN
210
572
4118
187.
8(7
.8)
BaZ
nCl 4
2337
341
0193
7.8
(7.8
)B
aBr 2
2745
615
706
7.8
(7.8
)N
a 4B
2O5
2756
410
061
7.8
(7.8
)Sr
Be 3
O4
2779
126
179
7.8
(7.8
)G
aPO
435
1815
5446
7.8
(7.8
)L
iBiF
450
5032
6540
47.
8(8
.1)
Sc2S
i 2O
755
9416
214
7.8
(7.9
)Z
n(PO
3)2
8230
3604
07.
8(7
.8)
K3S
c(PO
4)2
1703
561
786
7.8
(7.8
)N
a 3Ta
F 817
245
2661
17.
8(7
.8)
LaC
lO23
025
8433
07.
9(7
.9)
MgB
r 230
034
5236
67.
9(7
.9)
CsA
lO2
1406
928
372
7.9
(7.9
)Si
2N2O
4497
4155
757.
9(7
.9)
Na 2
SiO
345
3374
640
7.9
(8.1
)Z
rSiO
448
2015
8108
7.9
(8.1
)L
iInF
488
9266
693
7.9
(7.9
)Sc
2(SO
4)3
1680
041
1221
7.9
(7.9
)K
2Li 1
4Zr 3
O14
1720
865
445
7.9
(7.9
)Sr
CO
338
2256
099
7.9
(7.9
)L
i 2N
bF6
9752
2017
557.
9(7
.9)
Y2B
e 2G
eO7
5410
4039
122
7.9
(7.9
)C
sNbF
611
980
4124
448.
0(8
.1)
NaB
r22
916
6167
28.
0(8
.0)
BC
l 323
184
2786
98.
0(8
.1)
Li 6
MgB
r 829
008
7327
58.
0(8
.0)
ScPO
442
0020
1132
8.0
(8.2
)N
aBO
238
8915
967
8.0
(8.0
)K
NbF
675
7116
729
8.0
(8.0
)B
a 2L
aC3O
9F18
111
2500
598.
0(8
.2)
Ca 4
LaB
3O10
6076
2500
128.
0(8
.0)
CaB
r 222
888
1422
08.
1(8
.1)
CsB
r22
906
4429
08.
1(8
.1)
RbL
iBr 2
2823
730
719
8.1
(8.1
)C
sCaB
r 330
056
7724
28.
1(8
.2)
SrL
iBO
310
814
9284
28.
1(8
.3)
Sr3S
c(B
O3)
317
562
7533
98.
1(8
.2)
NaT
eF5
2935
520
2879
8.1
(8.1
)N
a 2M
g(C
O3)
260
2610
0482
8.1
(8.1
)M
gO12
6516
1607
8.1
(8.1
)N
a 2C
O3
2238
181
003
8.2
(8.2
)L
iAlO
234
2730
249
8.2
(8.2
)Sr
(ClO
3)2
5049
9161
157
8.2
(8.3
)B
eSiN
279
1325
704
8.2
(8.4
)Z
nP4O
1115
438
3002
298.
2(8
.2)
Sr2S
iO4
1851
036
041
8.2
(8.2
)L
aOF
7100
7642
78.
2(8
.4)
Ba 2
Mg(
BO
3)2
9259
7598
68.
2(8
.2)
LiB
r23
259
2798
28.
3(8
.3)
NaC
NO
5465
0027
138
8.3
(8.5
)M
g 2B
O3F
7995
2809
68.
3(8
.3)
Na 3
Sc2(
PO4)
316
956
6540
68.
3(8
.3)
Ge(
SF6)
228
291
6007
98.
3(8
.3)
ZrP
bF6
7310
4051
8.3
(8.3
)R
bBr
2286
718
017
8.4
(8.4
)K
Br
2325
118
015
8.4
(8.4
)A
sF3
2802
735
132
8.4
(8.5
)Y
ClO
5047
8631
667
8.4
(8.4
)M
gCO
353
4915
6763
8.4
(8.9
)A
sF5
8723
6547
78.
4(8
.4)
Ba 2
SiO
417
612
2847
68.
4(8
.4)
BaH
f(PO
4)2
5455
4824
5690
8.4
(8.5
)B
aSiO
373
3962
458.
4(8
.5)
BaL
iBO
364
9992
843
8.4
(8.5
)Y
OF
3637
3062
38.
5(8
.5)
BaZ
nF4
3881
4029
268.
5(8
.5)
CaC
O3
3953
1584
728.
5(8
.5)
Tl 2
SiF 6
5033
5229
28.
5(8
.5)
KA
sF6
7569
1666
38.
5(8
.5)
K2Z
nF4
9583
1002
988.
5(9
.4)
KA
lSiO
494
8083
449
8.5
(8.5
)C
sMgP
O4
1832
926
0423
8.5
(8.5
)B
aBeS
iO4
5507
5186
792
8.5
(8.6
)K
2CO
339
6366
943
8.5
(8.5
)Sr
BrF
2302
415
5008
8.6
(8.6
)Y
Cl 3
2745
515
684
8.6
(8.6
)L
i 4Si
O4
1173
798
615
8.6
(8.8
)G
aF3
588
4095
078.
6(8
.6)
RbN
a 3L
i 12(
SiO
4)4
1724
074
865
8.6
(8.6
)C
sKN
a 2L
i 12(
SiO
4)4
1771
874
864
8.6
(8.7
)B
a 3In
2F12
2827
448
182
8.6
(8.6
)C
aMg(
CO
3)2
6459
1715
088.
6(8
.8)
Sr2M
gSi 2
O7
6564
1553
008.
6(8
.8)
17

Ba 2
MgS
i 2O
793
3881
117
8.6
(8.7
)T
lClO
430
530
3356
48.
7(8
.7)
HfS
iO4
4609
5911
18.
7(8
.8)
Y2S
i 2O
756
5228
1313
8.7
(8.7
)K
CaC
O3F
6867
1546
858.
7(8
.7)
BaB
rF23
070
3539
38.
8(8
.8)
Cs 2
NaY
Cl 6
2312
024
5353
8.8
(8.8
)L
i 2O
5530
9010
8886
8.8
(8.8
)L
i 2Si
O3
5012
1004
028.
8(9
.0)
RbM
gCl 3
5044
5940
368.
8(8
.8)
La 2
SO6
4078
6682
48.
8(9
.0)
BaA
l 2B
2O7
9844
4091
718.
8(8
.8)
Li 2
CaS
iO4
7610
1902
38.
9(9
.3)
Mg 3
(BO
3)2
5005
3138
58.
9(9
.2)
Rb 2
SrP 2
O7
1435
439
506
8.9
(8.9
)M
gP4O
1115
437
3002
148.
9(8
.9)
LiA
lSiO
418
220
9270
88.
9(9
.0)
BaC
l 223
199
1570
59.
0(9
.0)
BeB
r 230
139
9258
49.
0(9
.2)
Na 2
BeB
2O5
1673
724
9341
9.0
(9.0
)P 2
O5
2452
7737
79.
0(9
.0)
RbS
bF6
9821
4080
719.
0(9
.0)
Cs 2
SrP 2
O7
1435
539
507
9.0
(9.0
)M
g 2B
2O5
1825
681
229
9.0
(9.1
)B
aSnF
682
9033
788
9.0
(9.0
)C
sLiC
l 223
364
2459
729.
1(9
.3)
Na 2
S 2O
731
269
4130
499.
1(9
.2)
SrA
l 2B
2O7
1593
989
423
9.1
(9.1
)C
sSbF
696
3620
1886
9.1
(9.1
)R
b 2Z
r 3O
F 12
1708
595
848
9.1
(9.2
)K
SrPO
417
975
8359
89.
1(9
.1)
RbB
aPO
417
832
7200
19.
1(9
.1)
KH
CO
323
724
1571
669.
1(9
.1)
SrC
l 223
209
2896
49.
2(9
.5)
K2M
gCl 4
2720
740
359.
2(9
.4)
LiB
O2
3635
2008
919.
2(9
.2)
Mg 3
(PO
4)2
1439
631
005
9.2
(9.2
)R
b 2B
4O7
1698
085
093
9.2
(9.2
)A
lH12
(ClO
2)3
2374
322
071
9.2
(9.2
)N
a 2C
a(PO
3)4
5415
2281
393
9.2
(9.2
)N
aAlP
2O7
1677
740
0462
9.2
(9.2
)C
a 4C
l 6O
2332
641
8946
9.2
(9.2
)K
2Be 2
PbF 8
7385
9902
9.2
(9.3
)A
l 2O
311
4316
1060
9.3
(9.3
)C
sCl
2286
544
289
9.3
(9.3
)C
sMgC
l 323
004
5416
79.
3(9
.3)
AlC
lO27
863
2781
29.
3(9
.3)
Ba 3
(PO
4)2
3857
1508
669.
3(9
.4)
NaA
lPO
4F86
7840
522
9.3
(9.3
)C
sBeP
O4
1539
520
2604
9.3
(9.3
)K
Na 2
(PO
3)3
1788
923
469.
3(9
.3)
K2Z
r 3O
F 12
1788
828
1350
9.3
(9.3
)N
a 2B
4O7
1794
120
409.
3(9
.3)
Al 2
O3
5525
5843
732
9.3
(9.3
)L
aSiB
O5
7051
8339
89.
3(9
.3)
Sr3(
PO4)
246
3215
0869
9.4
(9.5
)R
bLiC
l 250
4888
3621
69.
4(9
.4)
SrZ
nF4
5078
3136
79.
4(9
.4)
BaB
e 2Si
2O7
1279
715
1563
9.4
(9.6
)M
g(PO
3)2
1862
042
809.
4(9
.5)
BaM
gP2O
718
343
3939
89.
4(9
.4)
SrM
gP2O
718
469
2807
829.
4(9
.4)
YAl 3
(BO
3)4
6062
9196
29.
4(9
.5)
RbP
O3
4135
7473
69.
4(9
.4)
Na 6
S 2C
lO8F
2365
726
914
9.5
(9.5
)M
gSO
475
7216
759
9.5
(9.5
)N
aAlC
l 423
363
3527
89.
5(9
.5)
SiC
l 428
391
6227
99.
5(9
.5)
Ba(
AlC
l 4) 2
5057
5556
735
9.5
(9.5
)N
a 2Sn
F 688
1168
980
9.5
(9.5
)Sr
BPO
564
8677
519
9.5
(9.5
)N
aCl
2286
224
0598
9.6
(9.6
)R
bCl
2329
516
2798
9.6
(9.6
)B
aGeF
614
006
2661
49.
6(9
.6)
Na 2
SO4
4770
8150
69.
6(9
.6)
KA
l(SO
4)2
7645
6017
09.
6(9
.6)
LiC
lO4
3030
141
3238
9.6
(9.7
)C
s 2Sn
F 672
9728
19.
6(9
.6)
CaC
l 222
904
2464
179.
7(9
.7)
NaC
lO4
2296
820
0405
9.7
(9.9
)L
i 2G
eF6
7791
2340
69.
7(9
.7)
Li 2
BeS
iO4
8070
2830
79.
7(9
.9)
LiA
lCl 4
2298
335
275
9.7
(9.7
)C
sAlC
l 427
260
8118
9.7
(9.7
)Si
O2
6945
1626
159.
7(9
.7)
K3N
a(SO
4)2
2245
727
658
9.8
(9.9
)K
Cl
2319
322
156
9.8
(9.8
)B
aClF
2343
235
487
9.8
(9.9
)K
ClO
423
526
3511
19.
8(9
.8)
Al 4
(B2O
5)3
5105
0954
854
9.8
(10.
0)Y
PO4
5132
5611
39.
8(9
.8)
RbC
lO4
2843
363
363
9.8
(9.8
)H
fF4
3103
366
008
9.8
(9.9
)T
lBF 4
5101
3630
0222
9.8
(9.8
)M
gCl 2
2321
026
157
9.9
(9.9
)L
i 3PO
413
725
1025
79.
9(9
.9)
K2Z
rF6
5450
2503
719.
9(9
.9)
CaS
nF6
8224
3571
39.
9(9
.9)
NaZ
r 2Z
nF11
1514
181
223
10.0
(10.
1)G
eF4
9816
2025
5810
.0(1
0.2)
SrSO
452
8592
608
10.0
(10.
1)R
b 2H
f 3O
F 12
1725
695
847
10.0
(10.
0)B
aZr 2
F 10
5054
0020
2530
10.0
(10.
0)L
i 3B
7O12
1682
868
475
10.0
(10.
0)K
LiS
O4
6800
8882
910
.0(1
0.0)
LaP
O4
3962
2106
610
.0(1
0.0)
CaS
O4
4406
5610
710
.1(1
0.5)
SF6
8560
6336
210
.1(1
0.1)
SiO
254
7211
1622
4610
.1(1
0.1)
AlP
O4
7848
2451
110
.2(1
0.2)
LiC
l22
905
2798
110
.3(1
0.3)
K2G
eF6
1416
830
310
10.3
(10.
3)L
iSiB
O4
8874
6753
610
.4(1
0.8)
CsF
1784
5383
210
.5(1
0.8)
Rb 2
GeF
688
1268
982
10.5
(10.
5)C
s 2G
eF6
8217
3554
710
.6(1
0.6)
RbH
2OF
2370
041
5010
10.7
(10.
7)C
sBe 2
BO
3F2
5533
4220
000
10.7
(10.
7)K
2HfF
614
128
2951
410
.8(1
0.9)
B2O
330
636
066
10.8
(10.
9)C
s 2N
aAlF
665
2893
459
10.8
(10.
8)B
eCl 2
2326
731
696
10.9
(11.
0)Sc
F 310
694
7707
111
.0(1
1.4)
KR
b 2G
aF6
1319
041
6605
11.0
(11.
0)L
i 2Z
rF6
4002
1679
311
.0(1
1.1)
KB
e 2B
O3F
268
7015
5156
11.0
(11.
0)B
eO25
4216
3819
11.1
(11.
1)L
iCaG
aF6
1282
915
2300
11.1
(11.
2)Si
H4
2373
915
9307
11.1
(11.
1)K
Be 3
ZnF
918
509
1802
211
.2(1
1.2)
Cs 2
ZrF
679
0325
598
11.2
(11.
2)R
bF11
718
5382
811
.3(1
1.4)
BeS
O4
5055
6844
801
11.4
(11.
5)Sr
B4O
755
4095
300
11.4
(11.
7)C
sCO
F 314
734
6997
111
.4(1
1.4)
Li 2
CaH
fF8
1657
795
248
11.4
(11.
4)C
O2
2006
622
398
11.5
(11.
5)B
PO4
3589
1503
8011
.6(1
1.6)
Cs 2
CaF
415
157
8261
611
.7(1
1.8)
Rb 2
MgF
488
6169
681
12.1
(12.
2)C
s 2H
fF6
1394
825
600
12.2
(12.
2)K
F46
353
824
12.2
(12.
2)R
bCaF
336
5420
1253
12.3
(12.
4)K
PF6
4608
5625
812
.4(1
2.4)
NaF
682
5384
012
.4(1
2.4)
BaF
210
2953
980
12.5
(12.
6)K
2MgF
431
212
3351
912
.5(1
2.6)
CsC
aF3
7104
4530
912
.5(1
2.5)
MgF
218
1015
9315
12.5
(12.
5)PF
585
1162
554
12.6
(12.
6)K
MgF
334
4894
089
12.7
(12.
9)R
b 3N
aBe 2
F 813
630
1576
412
.7(1
2.8)
SrF 2
981
4140
212
.7(1
2.7)
Cs 2
NaA
l 3F 1
212
309
1578
412
.7(1
2.7)
CaF
227
4165
6448
12.8
(13.
0)K
AlF
429
1020
1949
12.8
(12.
8)L
iPF 6
9143
7483
012
.8(1
2.8)
K2N
aAlF
665
8660
2712
.8(1
2.8)
NaP
F 610
474
9061
512
.9(1
2.9)
18

BaS
iF6
5588
2015
1012
.9(1
2.9)
K2A
lF5
9486
8186
412
.9(1
2.9)
CaA
lF5
8836
6956
313
.0(1
3.0)
SrA
lF5
1655
741
1509
13.0
(13.
0)Y
F 324
1626
595
13.1
(13.
1)L
aF3
905
2397
213
.1(1
3.1)
NaC
aAlF
617
944
8054
213
.2(1
3.2)
Rb 2
SiF 6
1049
238
547
13.3
(13.
3)A
lF3
468
3830
513
.3(1
3.3)
K2S
iF6
3042
3854
613
.4(1
3.4)
LiY
F 437
0096
727
13.4
(13.
4)C
s 2Si
F 640
4738
548
13.4
(13.
4)N
a 2L
iBe 2
F 712
240
9430
13.5
(13.
6)L
iCaA
lF6
6134
1503
3313
.6(1
3.6)
SrB
eF4
9726
4043
9613
.7(1
3.7)
SiF 4
1818
4814
714
.0(1
4.4)
LiF
1138
5383
914
.6(1
4.6)
BeF
215
951
4149
214
.6(1
4.8)
CF 4
1167
6578
616
.0(1
6.1)
Tabl
e3
Lis
toft
hein
dire
ct(d
irec
t)ba
ndga
psso
rted
fori
ncre
asin
gga
p,in
eV,o
fthe
calc
ulat
ed24
50st
ruct
ures
.The
chem
ical
form
ulas
,the
Mat
eria
lsPr
ojec
tand
ICSD
Ids
are
also
incl
uded
.
19

REFERENCES REFERENCES
References
[1] Materials Project - A Materials Genome Approach,http://materialsproject.org/.
[2] Computational Materials Repository, https://wiki.fysik.dtu.dk/cmr/ (Documentation) ,and https://cmr.fysik.dtu.dk/ (Database).
[3] W. C. Sheets, E. S. Stampler, M. I. Bertoni, M. Sasaki,T. J. Marks, T. O. Mason, K. R. Poeppelmeier, InorganicChemistry 47, 2696 (2008).
[4] S. Ouyang, Z. Li, Z. Ouyang, T. Yu, J. Ye, Z. Zou, Jour-nal of Physical Chemistry C 112, 3134 (2008).
[5] H. G. Schnering, Zeitschrift Fur Anorganische Und All-gemeine Chemie 314, 144 (1962).
[6] K. Mader, H. Mullerbuschbaum, Zeitschrift Fur Anor-ganische Und Allgemeine Chemie 559, 89 (1988).
[7] The Landolt-Bornstein Database, http://www.springermaterials.com/docs/index.html.
[8] G. Tams, H. Mullerbuschbaum, Zeitschrift Fur Natur-forschung Section B-a Journal of Chemical Sciences 50,56 (1995).
[9] P. D. Vernooy, A. M. Stacy, Journal of Solid State Chem-istry 95, 270 (1991).
[10] A. S. Erickson, S. Misra, G. J. Miller, R. R. Gupta,Z. Schlesinger, W. A. Harrison, J. M. Kim, I. R. Fisher,Physical Review Letters 99 (2007).
[11] K. E. Stitzer, M. D. Smith, H. C. zur Loye, Solid StateSciences 4, 311 (2002).
[12] R. D. Shannon, J. L. Gillson, R. J. Bouchard, Journal ofPhysics and Chemistry of Solids 38, 877 (1977).
[13] T. Pisarkiewicz, K. Zakrzewska, E. Leja, Thin SolidFilms 153, 479 (1987).
[14] G. Ferrari, L. Coghi, Gazzetta Chimica Italiana 71, 440(1941).
[15] B. Brazel, R. Hoppe, Zeitschrift Fur NaturforschungSection B-a Journal of Chemical Sciences 37, 1369(1982).
[16] B. Brazel, R. Hoppe, Zeitschrift Fur NaturforschungSection B-a Journal of Chemical Sciences 38, 661(1983).
[17] J. J. Scheer, A. E. Vanarkel, R. D. Heyding, CanadianJournal of Chemistry-Revue Canadienne De Chimie 33,683 (1955).
[18] K. Hobbie, R. Hoppe, Zeitschrift Fur Anorganische UndAllgemeine Chemie 535, 20 (1986).
[19] B. Schwedes, R. Hoppe, Zeitschrift Fur AnorganischeUnd Allgemeine Chemie 391, 313 (1972).
[20] D. Choueiry, E.-i. Negishi, Handbook of Organopalla-dium Chemistry for Organic Synthesis (2002), ISBN 0-471-31506-0.
[21] B. Schwedes, R. Hoppe, Zeitschrift Fur AnorganischeUnd Allgemeine Chemie 393, 136 (1972).
[22] Y. Takahashi, Y. Gotoh, J. Akimoto, Journal of SolidState Chemistry 172, 22 (2003).
[23] S. Miyazaki, S. Kikkawa, M. Koizumi, Synthetic Metals6, 211 (1983).
[24] K. Hobbie, R. Hoppe, Zeitschrift Fur Anorganische UndAllgemeine Chemie 565, 106 (1988).
[25] S. Park, K. Kang, W. Si, W. S. Yoon, Y. Lee, A. R. Mood-enbaugh, L. H. Lewis, T. Vogt, Solid State Communica-tions 135, 51 (2005).
[26] J. Curda, E. M. Peters, W. Klein, M. Jansen, ZeitschriftFur Kristallographie-New Crystal Structures 219, 345(2004).
[27] Y. Pelletier, C. Reber, Inorganic Chemistry 36, 721(1997).
[28] N. E. Massa, J. A. Alonso, M. J. Martinez-Lope, M. T.Casais, Physical Review B 72 (2005).
20

138
Paper IVBand-gap engineering of functional perovskites through quantumconfinement and tunnelingI. E. Castelli, M. Pandey, K. S. Thygesen and K. W. Jacobsen Physical ReviewB 91 (16), 165309 (2015)

PHYSICAL REVIEW B 91, 165309 (2015)
Band-gap engineering of functional perovskites through quantum confinement and tunneling
Ivano E. Castelli,* Mohnish Pandey, Kristian S. Thygesen, and Karsten W. JacobsenCenter for Atomic-scale Materials Design, Department of Physics, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark
(Received 30 December 2014; revised manuscript received 25 March 2015; published 24 April 2015)
An optimal band gap that allows for a high solar-to-fuel energy conversion efficiency is one of the key factors toachieve sustainability. We investigate computationally the band gaps and optical spectra of functional perovskitescomposed of layers of the two cubic perovskite semiconductors BaSnO3 and BaTaO2N. Starting from an indirectgap of around 3.3 eV for BaSnO3 and a direct gap of 1.8 eV for BaTaO2N, different layerings can be used todesign a direct gap of the functional perovskite between 2.3 and 1.2 eV. The variations of the band gap can beunderstood in terms of quantum confinement and tunneling. We also calculate the light absorption of the differentheterostructures and demonstrate a large sensitivity to the detailed layering.
DOI: 10.1103/PhysRevB.91.165309 PACS number(s): 68.35.bg, 73.21.Ac, 73.20.−r, 78.20.−e
I. INTRODUCTION
Functional oxides form a fascinating class of materialsexhibiting a large range of phenomena and with great potentialfor technological applications. Some of their properties includehigh-temperature superconductivity, multiferroic and half-metallic behavior, thermoelectric, magnetocaloric, and photo-conductivity effects, transport phenomena, and catalytic prop-erties [1]. The oxides in the perovskite structure constitute aninteresting subclass with high stability and new underexploredpossibilities for producing layered heterostructures with atom-ically well-defined interfaces. Effects of quantum confinementin atomically layered perovskites have been discussed inseveral different heterosystems [2]. Yoshimatsu et al. [3–5]have studied quantum wells of the metal SrVO3 embeddedin an insulator, SrTiO3, with photoemission, demonstratingthat modifications of the electronic structure develop belowsix layers of SrVO3 and that for a single layer a substantialgap appears. Other studies include confinement effects onthe magnetic structure of LaMnO3/SrMnO3 superlattices [6]and recent investigations of how non-Fermi-liquid behaviorappears when a SrTiO3 quantum well embedded in SmTiO3
is sufficiently thin [7]. More recently, Grote et al. [8] haveinvestigated how to tune the band gap of tin- and lead-halideperovskites through effects of atomic layering and quantumconfinement.
In the present work, we investigate the band gaps and thelight-absorption properties of functional perovskites obtainedby stacking cubic perovskite planes, with general formulaABO3, in one direction (say, the z axis) while the other twodirections preserve the cubic symmetry, as shown in Fig. 1. Thepossibilities for producing such structures are numerous, butlittle is known about the potential for systematic, quantitativecontrol of their properties. We show that a large variation of theband gap can be obtained and that the size of the band gap fora particular stacking sequence can be understood in terms ofconfinement and tunneling behavior. Using these ingredients,an engineering of the band gap can be pursued to tune the gap
*[email protected]. Present address: Theory and Simulation ofMaterials (THEOS) and National Center for Computational Designand Discovery of Novel Materials (MARVEL), Ecole PolytechniqueFederale de Lausanne, CH-1015 Lausanne, Switzerland.
to a desired window. This approach could potentially be usedto achieve high efficiencies in light-harvesting devices.
More specifically, we consider combinations of the twocubic perovskite semiconductors BaSnO3 and BaTaO2N,indicated with α and β in Fig. 1, respectively [9]. The choiceof these two materials as building blocks is based on the factthat both BaSnO3 and BaTaO2N have been previously selectedas good materials for light harvesting and photocatalytic watersplitting [10,11] and their crystal lattices are rather similar,with the consequence that the obtained layered structure [12]will not be subjected to high stress.
All of the calculations presented in this work are performedin the framework of density functional theory (DFT) usingthe electronic structure code GPAW [13,14]. Due to the well-known problem of standard DFT with the underestimationof the band gaps, the gaps have been calculated using theGLLB-SC potential by Gritsenko, van Leeuwen, van Lenthe,and Baerends (GLLB) [15], modified by Kuisma et al. [16]to include the correlation for solids (-SC). This potentialhas been shown to provide realistic estimates of band gapswhen compared with other more advanced computationalmethods and experiments for a range of semiconductors andinsulators including oxides without too strong correlationeffects [10,17–19]. One reason for the favorable comparisonis the addition to the DFT Kohn-Sham gap of the so-calledderivative discontinuity, which is explicitly calculated in theGLLB-SC approach. We have furthermore performed hybridcalculations using the functional proposed by Heyd, Scuseria,and Ernzerhof (HSE06) [20,21] as a comparison for a subsetof the layered materials investigated in this work.
II. BAND GAPS
The compounds that we study here are all obtained bystacking nα layers of α with nβ layers of β, where 1 nα(β) 6, and then repeating this unit periodically. The latticeparameter is taken equal to the average value of the latticesof α and β (4.1 A [22]). BaSnO3 and BaTaO2N have beenfrozen in their perfect cubic perovskite symmetry, i.e., withoutany distortion. Even though distortions usually have largeeffects on the band gaps, BaSnO3 and BaTaO2N have a highcubicity so that the changes in the band gaps are expected tobe small. Keeping the structures frozen in the cubic symmetryfurthermore allows us to analyze the changes in the electronic
1098-0121/2015/91(16)/165309(6) 165309-1 ©2015 American Physical Society

CASTELLI, PANDEY, THYGESEN, AND JACOBSEN PHYSICAL REVIEW B 91, 165309 (2015)
FIG. 1. (Color online) Unit cell of the αβ structure. The cubicperovskite planes are stacked in the z direction, while the x and y
directions maintain the usual periodicity of a cubic perovskite. α
indicates the BaSnO3 perovskite, and β the BaTaO2N.
properties of the materials due only to the different stackings,regardless of any changes caused by structure relaxation.
Using GLLB-SC, BaSnO3 shows an indirect band gapbetween the and R points of 3.33 eV, while BaTaO2Nis found to have a direct band gap at of 1.84 eV. Thiscompares favorably with experiments where the optical gapshave been measured, through diffuse reflectance spectra, to 3.1and 1.9 eV for BaSnO3 [23] and BaTaO2N [24], respectively.HSE calculations slightly underestimate the gaps (2.89 eV forBaSnO3 and 1.71 eV for BaTaO2N).
Figure 2 reports the band gaps for the 36 αnαβnβ
structuresas a function of the number of α and β planes. The gaps varyconsiderably spanning a region of 1 eV, illustrating the highdegree of tunability of the band gap. The simplest combinationwith only one layer of α and β in the heterostructure gives thewidest gap with a value of 2.26 eV, not too far from the averageof the band gaps of the two constituent cubic perovskites. (Forcomparison, the HSE method gives again a slightly lower valueof 2.04 eV.) More complex combinations exhibit reduced band
FIG. 2. (Color online) Calculated band gaps as a function of thenumber of α (BaSnO3) and β (BaTaO2N) layers. Each rectangle inthe plot represents a layered periodic structure with sequence αnβm.
FIG. 3. (Color online) Sketch of the electronic level positions atan interface between layers of BaSnO3 and BaTaO2N. When the layerthickness is reduced, the local position of the conduction-band edgemoves up due to confinement.
gaps depending on their composition. As we shall show in thefollowing, the significant complicated variation of the bandgap shown in Fig. 2 can essentially be understood in terms ofelectronic confinement and tunneling effects.
In Fig. 3, we sketch how the local band edges are positionedrelative to each other for different layer thicknesses. For thethickest layer structure, α6β6, we have the smallest band gapof 1.26 eV. The state at the valence-band maximum (VBM)is composed mainly of N2p orbitals with a minor contributionfrom the O2p orbitals and is located in the β part of the material.In fact, all of the mixed compositions have a direct band gapat the point, with the VBM state of this character locatedin the β part of the material. The character of the VBM statecan, for example, be seen in Figs. 4(a) and 4(c) for the αβ
and α2β structures, respectively. Not only is the character ofthe VBM state the same for all structures, but the calculationsalso indicate that it does not move much relative to a low-lyingatomic state in BaTaO2N, and we shall therefore regard thislevel as fixed in the following and ascribe the variations tochanges in the conduction band.
FIG. 4. (Color online) Wave functions of states at the valence-band maximum (VBM) and the conduction-band minimum (CBM)for some combinations of α and β layers.
165309-2

BAND-GAP ENGINEERING OF FUNCTIONAL . . . PHYSICAL REVIEW B 91, 165309 (2015)
FIG. 5. (Color online) The figure illustrates the weights of theCBM state in real space. The vertical axes are along the stackingdirection of the material and the areas of the circles indicate theweights of the CBM state for a particular atomic xy plane. The boxesshow the extent of the supercell in the direction of the stacking (z),and the dashed lines mark the interfaces between the α and β layers.Above the figure, the calculated band gaps for the different stackingsequences are denoted. The CBM state is mainly composed of Ta d
orbitals, as shown in Fig. 4(b).
To understand the variation of the conduction-band min-imum in Fig. 3, we first consider the compounds with theformula αβ
nβ, where the band gap decreases as a function of
the number of β layers. The CBM states for these systemsare located only on the TaON plane, as shown in Fig. 5, andgenerated by the Ta5d orbitals, as plotted in Fig. 4(b) for theαβ case. The variation of the band gap as a function of nβ
is a result of quantum confinement. The empty states in thesingle α layer are shifted up out of reach, and the CBM statein β becomes less confined with the increase of the numberof β layers in the αβ
nβstructures, as can be seen in Fig. 5.
The reduction of the confinement results in a down-shift of theCBM level and thus a reduced band gap, as also sketched inFig. 3.
The situation is radically different for all the combinationsα
nαβ
nβ, with nα 2. Now the CBM state is not located in β,
but in α. It is located mainly on the Sn5s orbitals, as shown inFig. 4(d). If, for example, we consider the compounds α2βn,the CBM state is localized in the α2 layers and essentiallylooks the same, as seen in Fig. 6. The band gap is thereforealso largely unchanged for n 3. For n = 2, a small reductionrelative to the situation with n 3 is seen and this reductionbecomes even larger for n = 1 (see Fig. 2). We ascribe thisreduction to quantum tunneling through the thin β layers. Ascan be seen in Fig. 6, the CBM states decay into the β layersand the tunneling coupling, for small thicknesses, will resultin a lowering of the CBM level and thus a decrease of the bandgap.
The interplay between quantum confinement and tunnelingis seen most clearly for the nβ = 1 systems (Fig. 7). Ignoringthe αβ structure that has a different nature for the CBM levelwith respect to the other systems, the CBM state becomes
FIG. 6. (Color online) Weight of the CBM state in each xy planeof the nα = 2 structure. The CBM state is now composed of Sn s
states, with some tunneling through the TaON plane [Fig. 4(d)]. Thetunneling progressively reduces with the increase of β layers.
less confined with the increase of the number of α layers,with the consequence of a decrease in the band gap. Andas we have seen, the band gap is further reduced becauseof tunneling through the single β TaON layer. However,for larger thicknesses of the α layer, the tunneling effect isreduced because the now less-confined CBM state has loweramplitude at the interface. This interplay between confinementand tunneling leads to the increase of the band gap betweennα = 4 and nα = 5.
As we have seen, the variation of the band gaps for thedifferent periodic αnβm compounds can be understood fromthe confinement effects shown in the level diagram in Fig. 3together with additional tunneling effects if β1 or β2 layers arepresent. Does this lesson apply to more complicated sequences
FIG. 7. (Color online) Weight of the CBM state in each xy planeof the nβ = 1 structure. The character of the CBM state changesdrastically with nα: TaON is responsible for the CBM state for thenα = 1 structure, while for nα > 1, it is SnO2 [Figs. 4(b) and 4(d)].The tunneling across the TaON plane has an effect until nα = 4.
165309-3

CASTELLI, PANDEY, THYGESEN, AND JACOBSEN PHYSICAL REVIEW B 91, 165309 (2015)
of layers? Some test calculations seem to indicate so. Acompound with the periodic repetition of αβαβnβ
reproducesexactly the same gaps as the αβnβ
. This is to be expected sincefrom the level diagram in Fig. 3 we should expect the CBMstate to be located in the βnβ
layer with little tunneling throughthe α layers. Another example is the systems with sequenceαβαnα
β, which exhibit a small increase (up to about 0.2 eV) ofthe band gap for nα 2 as compared to the αnα
β compounds.This can be understood in terms of reduced tunneling becausethe αnα
layers are now separated by βαβ instead of a single β
layer reducing the tunneling effect.
III. OPTICAL PROPERTIES
A number of technological applications such as photo-voltaics or photocatalysis depend on the availability of efficientabsorbers of light in the visible spectrum. This requires anappropriate band gap of the material, and band-gap tuning istherefore a key issue. However, the band gap does not by itselfprovide any information about the magnitude of the matrixelements responsible for light absorption. For symmetryreasons, the light absorption can be dipole allowed or forbiddenand—in particular for heterostructures—the transitions maytake place between states with different degree of spatialoverlap, giving rise to large variations of the absorptionstrengths.
To address this issue, we perform linear response cal-culations [25], using the adiabatic local density approxima-tion (ALDA), and determine the optical absorption of theinvestigated systems focusing for simplicity on the systemswith nα = 1 or nβ = 1 [26]. The optical absorption spec-trum is calculated using time-dependent density functionaltheory (TDDFT) from the density response function χ .The response function evaluated at point r to first orderin a time-dependent perturbation of frequency ω applied atpoint r′ is χ (r,r′,ω) = δn(r,ω)/δVext(r′,ω), where δn is theinduced density under the perturbation caused by the externalpotential Vext.
The microscopic dielectric matrix is defined as
ε−1GG′(q,ω) = δGG′ + 4π
|q + G|2 χGG′(q,ω), (1)
where G and G′ are reciprocal lattice vectors, and q is awave vector of the first Brillouin zone. The optical absorptionspectrum is given by Imε(q → 0,ω), where ε(q → 0,ω) =
1ε−1
00 (q→0,ω).
Figure 8 shows the optical absorption for the nα = 1systems. In the plot, we distinguish between the case wherethe light is polarized along the xy and the z directions. Thexy plane, in fact, maintains the cubic symmetry, while thestacking of the layers takes place in the z direction. For thesecompounds, the CBM and VBM states are located in the sameregion of space, namely, in the TaON layers, and thus theabsorption starts at the direct band gap and is quite intense,especially for polarizations in the xy direction. The situationis different for the nβ = 1 systems (Fig. 9), where the VBMstate is located in the TaON layer, while the CBM state hasmost weight on the BaO2 layers. The absorption here startsat much higher energies than the band gap (except for the αβ
compound, in black in the figure, which has the VBM and
FIG. 8. (Color online) Calculated optical absorption for the nα =1 systems. The direct band gaps are indicated with verticalarrows.
CBM states located in the same region). The first transitionwith appreciable weight is between two TaON states in the β
layer, and the absorption curves are therefore fairly similar,independent of the band gap.
Table I reports the efficiencies of the two sequences. Theefficiency is calculated as the percentage of the collectedphotons of the global solar spectrum at AM1.5 [27]. As alsoshown in Figs. 8 and 9, the efficiency is higher for lightpolarized in the xy direction than along z. αβ2 and α2β arealmost comparable because the higher absorption properties ofthe former are balanced by the lower band gap of the latter, andthe two systems collect almost the same amount of photons.The efficiency of the αβnβ
sequence always increases with thenumber of layers, while the one of αnαβ decreases even thoughthe gap closes.
The calculations thus indicate that the absorption crosssection at the α − β interface is quite limited and that the
FIG. 9. (Color online) Calculated optical absorption for the nβ =1 systems. The direct band gaps are indicated with vertical arrows.
165309-4

BAND-GAP ENGINEERING OF FUNCTIONAL . . . PHYSICAL REVIEW B 91, 165309 (2015)
TABLE I. Gap (in eV) and photon-absorption efficiency η (in%, for light polarized in the xy and z directions) of the sequencesαβnβ
and αnαβ , calculated for a thickness of 10−7 m. The efficiencycalculated for the pure α and β cubic perovskites is included forcomparison.
Gap ηxy ηz Gap ηxy ηz
α 3.33 0.1 0.1 β 1.84 14.9 6.4αβ 2.26 4.3 0.9 αβ 2.26 4.3 0.9αβ2 2.10 7.0 1.5 α2β 1.57 5.3 2.1αβ3 2.04 8.9 2.4 α3β 1.41 4.3 2.0αβ4 2.00 10.0 3.0 α4β 1.27 4.1 2.0αβ5 1.98 10.7 3.5 α5β 1.40 2.8 1.5αβ6 1.97 11.3 3.8 α6β 1.35 2.6 1.5
band-edge states have to be localized in the same layers toobtain efficient absorption.
IV. CONCLUSIONS
In this work, we have investigated the electronic propertiesof perovskite heterostructures obtained by stacking BaSnO3
and BaTaO2N layers. The band gap is seen to be tunableover the wide range of around 1 eV and the variationcan be understood in terms of quantum confinement andtunneling. Confinement leads to up-shifts of the conduction-band minimum and thus to increase of the band gap, whiletunneling effects reduce the confinement and lead to lowerband gap. The tunneling effects are seen to decay over a fewperovskite unit cells. The systems studied here are close tocubic and with similar lattice constants, but in general band-gap formation in layered perovskites can be expected to dependsensitively also on strain and lattice distortions/reconstructions[28].
The calculated optical absorption spectra for the het-erostructures indicate that high absorption is only obtained
if the VBM and CBM states are localized in the same spatialregion. The design of heterostructures for efficient visible-lightabsorption therefore requires not only appropriate bandgaps, but also tailored band-edge states with proper spatialoverlap.
The stacking of BaSnO3 and BaTaO2N layers that we havedescribed here is a type-II heterojunction with the conductionband of BaSnO3 above the valence band of BaTaO2N. A type-Iheterojunction can be designed using different perovskites.One example is LaAlO3 (as α) and LaTiO2N (as β), with acalculated indirect band gap between and R points of 6.11and a direct gap at the point of 1.49 eV, respectively, andwhere the band edges of LaTiO2N are placed in between theedges of LaAlO3. Preliminary results show that due to the largeband gap of α, there is no tunneling through the α layer andthere is already a full confinement of the β layers with a singleα [29]. In addition, the band gaps of the layered combinationsare direct, with the VBM formed by N2p orbitals and the CBMcomposed of Ti3d . Also in this case, the stacking has the effectof placing the VBM and CBM closer together spatially. Thisfact might increase the absorption properties of the materialsand, together with the possibility of tuning the band gap usingquantum confinement and tunneling, can be used to designnovel light-harvesting heterojunctions.
ACKNOWLEDGMENTS
The authors acknowledge support from the Catalysis forSustainable Energy (CASE) initiative funded by the DanishMinistry of Science, Technology and Innovation, and fromthe Center on Nanostructuring for the Efficient Energy Con-version (CNEEC) at Stanford University, an Energy FrontierResearch Center founded by the U.S. Department of Energy,Office of Science, Office of Basic Energy Sciences underAward No. DE-SC0001060. K.S.T. acknowledges supportfrom the Danish Council for Independent Research SapereAude Program through Grant No. 11-1051390. The Center forNanostructured Graphene is sponsored by the Danish NationalResearch Foundation, Project No. DNRF58.
[1] F. M. Granozio, G. Koster, and G. Rijnders, Mater. Res. Bull.38, 1017 (2013).
[2] S. Stemmer and A. J. Millis, Mater. Res. Bull. 38, 1032(2013).
[3] K. Yoshimatsu, T. Okabe, H. Kumigashira, S. Okamoto,S. Aizaki, A. Fujimori, and M. Oshima, Phys. Rev. Lett. 104,147601 (2010).
[4] K. Yoshimatsu, E. Sakai, M. Kobayashi, K. Horiba, T. Yoshida,A. Fujimori, M. Oshima, and H. Kumigashira, Phys. Rev. B 88,115308 (2013).
[5] K. Yoshimatsu, K. Horiba, H. Kumigashira, T. Yoshida,A. Fujimori, and M. Oshima, Science 333, 319 (2011).
[6] T. S. Santos, B. J. Kirby, S. Kumar, S. J. May, J. A. Borchers,B. B. Maranville, J. Zarestky, S. G. E. te Velthuis, J. vanden Brink, and A. Bhattacharya, Phys. Rev. Lett. 107, 167202(2011).
[7] C. A. Jackson, J. Y. Zhang, C. R. Freeze, and S. Stemmer, Nat.Commun. 5, 1 (2014).
[8] C. Grote, B. Ehrlich, and R. F. Berger, Phys. Rev. B 90, 205202(2014).
[9] We consider, for simplicity, only the simplest ordered structureof the nitrogen atoms. Some check calculations within the unitcells we use indicate that exchanging O and N atoms so thatthe number of O and N neighbors of the Ta atoms stays fixedonly gives minor changes to both the stability and the band gap(changes less than 0.1 eV). If the O and N coordination of theTa atoms is changed, the stability is significantly decreased.
[10] I. E. Castelli, T. Olsen, S. Datta, D. D. Landis, S. Dahl, K. S.Thygesen, and K. W. Jacobsen, Energy Environ. Sci. 5, 5814(2012).
[11] I. E. Castelli, D. D. Landis, K. S. Thygesen, S. Dahl,I. Chorkendorff, T. F. Jaramillo, and K. W. Jacobsen, EnergyEnviron. Sci. 5, 9034 (2012).
[12] Layered perovskite usually is the name given to ABO3 per-ovskites separated by some motifs. Some examples are theRuddlesden-Popper and the Dion-Jacobson phases. In this work,
165309-5

CASTELLI, PANDEY, THYGESEN, AND JACOBSEN PHYSICAL REVIEW B 91, 165309 (2015)
with layered perovskite, we indicate layers of cubic perovskitesstacked in the z direction.
[13] J. J. Mortensen, L. B. Hansen, and K. W. Jacobsen, Phys. Rev.B 71, 035109 (2005).
[14] J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dulak,L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen,H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara,M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen,T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller,M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer,H. Hakkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov,M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen, and K. W.Jacobsen, J. Phys. Condens. Matter 22, 253202 (2010).
[15] O. Gritsenko, R. van Leeuwen, E. van Lenthe, and E. J.Baerends, Phys. Rev. A 51, 1944 (1995).
[16] M. Kuisma, J. Ojanen, J. Enkovaara, and T. T. Rantala, Phys.Rev. B 82, 115106 (2010).
[17] F. Huser, T. Olsen, and K. S. Thygesen, Phys. Rev. B 87, 235132(2013).
[18] I. E. Castelli, J. M. Garcıa-Lastra, F. Huser, K. S. Thygesen, andK. W. Jacobsen, New J. Phys. 15, 105026 (2013).
[19] I. E. Castelli, F. Huser, M. Pandey, H. Li, K. S. Thygesen,B. Seger, A. Jain, K. A. Persson, G. Ceder, and K. W. Jacobsen,Adv. Energy Mater. 5, 1400915 (2015).
[20] J. Heyd, G. E. Scuseria, and M. Ernzerhof, J. Chem. Phys. 118,8207 (2003).
[21] A. V. Krukau, O. A. Vydrov, A. F. Izmaylov, and G. E. Scuseria,J. Chem. Phys. 125, 224106 (2006).
[22] The lattice parameters of α and β calculated with the PBEsolfunctional [30] are 4.12 and 4.08 A, respectively, in very goodagreement with the experimental values (4.11 A for both α [31]and β [32]).
[23] H. Mizoguchi, H. W. Eng, and P. M. Woodward, Inorg. Chem.43, 1667 (2004).
[24] D. Yamasita, T. Takata, M. Hara, J. N. Kondo, and K. Domen,Solid State Ionics 172, 591 (2004).
[25] J. Yan, J. J. Mortensen, K. W. Jacobsen, and K. S. Thygesen,Phys. Rev. B 83, 245122 (2011).
[26] TDDFT-ALDA does not include excitonic effects. However, theexciton binding energy Eexc can be estimated using the Wannier-Mott model,
Eexc = Rμ
m
1
ε2M
,
where R = 13.606 eV, 1μ
= 1μ∗
c+ 1
μ∗v, with μ∗
c and μ∗v the
hole and electron effective masses at the CBM and VBM,respectively, and εM is the macroscopic dielectric constant.Based on this, we estimate the exciton binding energies forBaSnO3 and BaTaO2N to be of the order of 0.3 and 0.1 eV,respectively, and about 0.2 eV for the αβ sequence.
[27] The efficiency η is given by
η = 1
ntot
∫ ∞
gapphabs(E)nph(E)dE,
where ntot is the total number of photons from the sun measuredat AM1.5, phabs(E) is the photon absorptivity of the material,and nph(E) is the number of sun photons as a function ofenergy E, in eV. The photon absorptivity depends on the theabsorption coefficient α(E) and on the depth of the materialalong the absorption direction L: phabs(E) = 1 − e−α(E)L. Theabsorption coefficient is α(E) = 2Ek(E)
c, where and c are the
Planck constant and the speed of light, respectively, and k
is obtained from the absorption spectrum as k2 = 12 (−Reε +√
Re2ε + Im2ε).[28] H. Li, I. E. Castelli, K. S. Thygesen, and K. W. Jacobsen, Phys.
Rev. B 91, 045204 (2015).[29] The calculated gaps for this combination, as well as for the
BaSnO3-BaTaO2N, are available in the Computational MaterialsRepository at https://cmr.fysik.dtu.dk/.
[30] J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E.Scuseria, L. A. Constantin, X. Zhou, and K. Burke, Phys. Rev.Lett. 100, 136406 (2008).
[31] H. Mizoguchi, P. M. Woodward, C.-H. Park, and D. A. Keszler,J. Am. Chem. Soc. 126, 9796 (2004).
[32] F. Pors, R. Marchand, Y. Laurent, P. Bacher, and G. Roult, Mater.Res. Bull. 23, 1447 (1988).
165309-6




















