
Computational determination of the à state absorption spectrum of NH3 and of ND3 using a new...
13
THE JOURNAL OF CHEMICAL PHYSICS 136, 234301 (2012) Computational determination of the ˜ A state absorption spectrum of NH 3 and of ND 3 using a new quasi-diabatic representation of the ˜ X and ˜ A states and full six-dimensional quantum dynamics Xiaolei Zhu, 1, a) JianYi Ma, 2, b) David R. Yarkony, 1, c) and Hua Guo 2, c) 1 Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA 2 Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque, New Mexico 87131, USA (Received 8 April 2012; accepted 18 May 2012; published online 15 June 2012) A recently developed method to represent adiabatic electronic states coupled by conical intersections has been used to construct a full six-dimensional quasi-diabatic representation of the 1 1 A and 2 1 A states of NH 3 . This representation is expected to be appropriate to simulate the photodissociation of ammonia when it is excited to the 2 1 A electronic state. In this work, the electronic structure aspects of this quasi-diabatic representation are analyzed. This representation is then used as the basis for a simulation of the ˜ A ← ˜ X absorption spectrum, dominated by a progression in the v 2 mode, using a full six-dimensional quantum mechanical treatment of the nuclear motion. Results are reported for both NH 3 and ND 3 . This simulation provides the most accurate computational determination of this absorption spectrum reported to date. These results serve to validate the quasi-diabatic representation and set the stage for subsequent studies of vibrationally mediated photodissociation of NH 3 . © 2012 American Institute of Physics.[http://dx.doi.org/10.1063/1.4725496] I. INTRODUCTION It has been almost 80 years since Leifson first reported the ˜ A state absorption of ammonia beginning at 220 nm. 1 The diffuse character of the absorption reflects strong predissoci- ation. In the intervening decades, this state has been probed using a plethora of experimental and theoretical techniques. It is now clear that the photochemistry of the ˜ A state reflects the fact that this state is quasi-bound in the Franck-Condon re- gion and that beyond a small barrier in the dissociation (N–H) coordinate a seam of conical intersections is encountered. Over 50 years after its initial observation, Vaida et al. 2 using a cold supersonic jet, reported a progression in the um- brella mode, v 2 , in the ˜ A band absorption of NH 3 . This pro- gression reflects the change from a trigonal structure in the ˜ X state to a planar structure in the ˜ A state. 3 The lifetimes of the individual levels of the v 2 progression were subsequently measured by several groups. 4–7 Interestingly, however, the an- ticipated progression in the symmetric stretch, the v 1 mode, based on the greater N–H bond length in the ˜ A state when compared to the ˜ X state, 3 was not observed by Vaida et al. More recent vibrationally mediated photodissociation exper- iments have provided access to combinations of levels in the ˜ A state not previously seen. 8–10 The lifetimes of the v 2 levels and the absence of an ob- served progression in v 1 are related to the efficiency of in- tramolecular vibrational energy transfer. The vibrational me- diated photodissociation experiments raised further questions about intramolecular vibrational energy transfer, but these a) Electronic mail: [email protected]. b) Electronic mail: [email protected]. c) Authors to whom correspondence should be addressed. Electronic addresses: [email protected] and [email protected]. experiments also address how the seam of conical intersection impacts the products of the dissociation, NH 2 ( ˜ X 2 B 1 )+H or NH 2 ( ˜ A 2 A 1 )+H. The absence of a progression in v 1 has been attributed to a near 3:1 degeneracy of the v 1 and v 2 modes 11 or a sufficiently low barrier to allow direct dissociation from an excited v 1 mode. 7, 12 This barrier to ˜ A state dissociation has been determined from ab initio calculations and inferred from experimental measurements. Computed barriers include the early work of McCarthy et al. 13, 14 who reported a barrier of 3226 cm −1 and more recent work of Bach et al. 8 and of Li et al. 15 who reported barriers of 2348 cm −1 and 1750 cm −1 , respectively. These results should be compared with the ex- perimental estimate of ∼2100 cm −1 of Henck et al. 7 based on microwave detected, microwave-optical double resonance measurements and supported by the measured lifetimes of Bach et al. 8 obtained from vibrationally mediated photodis- sociation action spectroscopy. The final state resolved dynamics of ammonia photodis- sociation has been investigated experimentally by several groups. 16–26 The production of the ground state NH 2 fragment clearly indicates nonadiabatic coupling between the ˜ X and ˜ A states of ammonia. It is now well established that the nonadia- batic transition is facilitated by a seam of conical intersections encountered after the initial barrier near the Franck-Condon region is traversed. 13–15, 27–30 The photodissociation dynamics following ˜ A state absorption have been the subject of numerous computa- tional studies as well, 31–37 including those of Dixon who determined the ˜ A state lifetimes using three 31 and higher dimensional 32 model potential energy surfaces; those of Bon- hommeau et al. 36 (BVTJ), who simulated the vibrationally mediated photodissociation spectrum measured in Crim’s group, 8–10, 38 using trajectory surface hopping techniques; 34, 39 0021-9606/2012/136(23)/234301/12/$30.00 © 2012 American Institute of Physics 136, 234301-1 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP: 162.129.250.14 On: Sun, 15 Dec 2013 05:44:01
Transcript of Computational determination of the à state absorption spectrum of NH3 and of ND3 using a new...

THE JOURNAL OF CHEMICAL PHYSICS 136, 234301 (2012)
Computational determination of the A state absorption spectrumof NH3 and of ND3 using a new quasi-diabatic representation of theX and A states and full six-dimensional quantum dynamics
Xiaolei Zhu,1,a) JianYi Ma,2,b) David R. Yarkony,1,c) and Hua Guo2,c)1Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA2Department of Chemistry and Chemical Biology, University of New Mexico, Albuquerque,New Mexico 87131, USA
(Received 8 April 2012; accepted 18 May 2012; published online 15 June 2012)
A recently developed method to represent adiabatic electronic states coupled by conical intersectionshas been used to construct a full six-dimensional quasi-diabatic representation of the 11A and 21Astates of NH3. This representation is expected to be appropriate to simulate the photodissociation ofammonia when it is excited to the 21A electronic state. In this work, the electronic structure aspectsof this quasi-diabatic representation are analyzed. This representation is then used as the basis for asimulation of the A ← X absorption spectrum, dominated by a progression in the v2 mode, using afull six-dimensional quantum mechanical treatment of the nuclear motion. Results are reported forboth NH3 and ND3. This simulation provides the most accurate computational determination of thisabsorption spectrum reported to date. These results serve to validate the quasi-diabatic representationand set the stage for subsequent studies of vibrationally mediated photodissociation of NH3. © 2012American Institute of Physics. [http://dx.doi.org/10.1063/1.4725496]
I. INTRODUCTION
It has been almost 80 years since Leifson first reportedthe A state absorption of ammonia beginning at 220 nm.1 Thediffuse character of the absorption reflects strong predissoci-ation. In the intervening decades, this state has been probedusing a plethora of experimental and theoretical techniques.It is now clear that the photochemistry of the A state reflectsthe fact that this state is quasi-bound in the Franck-Condon re-gion and that beyond a small barrier in the dissociation (N–H)coordinate a seam of conical intersections is encountered.
Over 50 years after its initial observation, Vaida et al.2
using a cold supersonic jet, reported a progression in the um-brella mode, v2, in the A band absorption of NH3. This pro-gression reflects the change from a trigonal structure in theX state to a planar structure in the A state.3 The lifetimes ofthe individual levels of the v2 progression were subsequentlymeasured by several groups.4–7 Interestingly, however, the an-ticipated progression in the symmetric stretch, the v1 mode,based on the greater N–H bond length in the A state whencompared to the X state,3 was not observed by Vaida et al.More recent vibrationally mediated photodissociation exper-iments have provided access to combinations of levels in theA state not previously seen.8–10
The lifetimes of the v2 levels and the absence of an ob-served progression in v1 are related to the efficiency of in-tramolecular vibrational energy transfer. The vibrational me-diated photodissociation experiments raised further questionsabout intramolecular vibrational energy transfer, but these
a)Electronic mail: [email protected])Electronic mail: [email protected])Authors to whom correspondence should be addressed. Electronic
addresses: [email protected] and [email protected].
experiments also address how the seam of conical intersectionimpacts the products of the dissociation, NH2(X2B1)+H orNH2(A2A1)+H. The absence of a progression in v1 has beenattributed to a near 3:1 degeneracy of the v1 and v2 modes11
or a sufficiently low barrier to allow direct dissociation froman excited v1 mode.7, 12 This barrier to A state dissociationhas been determined from ab initio calculations and inferredfrom experimental measurements. Computed barriers includethe early work of McCarthy et al.13, 14 who reported a barrierof 3226 cm−1 and more recent work of Bach et al.8 and of Liet al.15 who reported barriers of 2348 cm−1 and 1750 cm−1,respectively. These results should be compared with the ex-perimental estimate of ∼2100 cm−1 of Henck et al.7 basedon microwave detected, microwave-optical double resonancemeasurements and supported by the measured lifetimes ofBach et al.8 obtained from vibrationally mediated photodis-sociation action spectroscopy.
The final state resolved dynamics of ammonia photodis-sociation has been investigated experimentally by severalgroups.16–26 The production of the ground state NH2 fragmentclearly indicates nonadiabatic coupling between the X and A
states of ammonia. It is now well established that the nonadia-batic transition is facilitated by a seam of conical intersectionsencountered after the initial barrier near the Franck-Condonregion is traversed.13–15, 27–30
The photodissociation dynamics following A stateabsorption have been the subject of numerous computa-tional studies as well,31–37 including those of Dixon whodetermined the A state lifetimes using three31 and higherdimensional32 model potential energy surfaces; those of Bon-hommeau et al.36 (BVTJ), who simulated the vibrationallymediated photodissociation spectrum measured in Crim’sgroup,8–10, 38 using trajectory surface hopping techniques;34, 39
0021-9606/2012/136(23)/234301/12/$30.00 © 2012 American Institute of Physics136, 234301-1
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-2 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
and those of Lai et. al.35 (LLXG) who computed the lifetimesof the umbrella mode levels using full six-dimensionalquantum dynamics. The results in the LLXG work weresubsequently reproduced semi-quantitatively using the samecoupled potential energy surfaces by Giri et al.40 (GCSW),using the multiconfiguration time-dependent Hartree(MCTDH) method.41, 42 The quantitative differences in bothenergies and widths between the results of LLXG and those ofGCSW are presumably attributable to the approximate natureof aspects of the MCTDH procedure. The studies of BVTJ,LLXG, and GCSW are notable since they employed the samefull six-dimensional approximate diabatic representationof the 1,21A states of NH3 based on ab initio data.15 Theapproximate or quasi-diabatic representation was constructedusing the fourfold way method of Nakamura and Truhlar.43, 44
The use of an approximate diabatic representation is aconsequence of the seam of conical intersections coupling the1,21A adiabatic states. The qualifiers approximate or quasi infront of diabatic, which we shall omit below, remind us thatrigorous diabatic states do not exist for general polyatomicmolecules.45–47
The representation of ab initio determined adiabatic statepotential energy surfaces coupled by conical intersections haslong been a goal of quantum chemistry. Efforts to find dia-batic representations include the vibronic coupling model ofKöppel, Domcke, and Cederbaum;48–56 the perturbation the-ory based work of Mead, Truhlar, and Varandas;57, 58 the di-rect diabatization schemes of Truhlar,43, 44 based on earlierwork of Ruedenberg;59 the regularized diabatization proce-dure of Köppel;60 the block diabatization method of Pacher,Cederbaum, and Köppel;61, 62 the generalized adiabatic an-gle method of Varandas,63, 64 and the Shepard interpolationbased method of Collins, Evenhuis and co-workers.65–68 Ap-proaches based on a combination of the interpolated mov-ing least squares69, 70 and the general dynamically weighted71
state averaged multiconfiguration self-consistent field meth-ods also show promise in this regard. Recently in three pa-pers, denoted P1,72 P2,73 and P374 below, we introduced anapproach, in which a diabatic Hamiltonian, Hd, is constructedwith polynomial expansions of its matrix elements. Hd is ca-pable of representing ab initio energies, gradients, and deriva-tive couplings over a wide range of geometries includingthose for dissociated species and seams of conical intersec-tions. Further since derivative couplings are included in defin-ing the representation it is quantifiably diabatic in a leastsquares sense. However, this method has yet to be tested ina practical calculation.
In the vibrationally mediated photodissociation experi-ments reported by Crim’s group,8–10, 38 a dramatic differencein the NH2 A/X branching fraction was found depending onwhether the X state is excited to v1 = 1 or to v3 = 1, prior todissociation through the A state by a second photon. This dif-ference in the branching fraction is attributed to differences inthe dynamics near the 1,21A conical intersection seam notedabove. The nonadiabatic dynamics study of BVTJ, despitea very careful analysis, was unable to explain the measuredbranching fractions. Further, the line widths of the v2 lev-els computed by LLXG using a full six-dimensional quan-tum treatment, systematically overestimate the measured line
widths of this mode and the spectral intensity distribution waspoorly approximated. The lack of fully satisfactory agreementbetween these careful dynamics studies and the relevant ex-perimental measurements suggests the need to reconsider thedetermination of the coupled diabatic state representation ofthe 1,21A states.
In this work, then a Hd developed using the methods de-scribed in P1–P3 and appropriate for simulating NH3 pho-todissociation will be analyzed. This diabatic representationwill then be validated by comparing the measured line po-sitions, intensities,6 and lifetimes4, 6–8 of the umbrella vibra-tions with computed values based on the coupled diabaticstate description of the 1,21A states of NH3. This simula-tion/analysis of the absorption spectrum will employ the fullsix-dimensional, accurate, fully quantum mechanical treat-ment of the nuclear dynamics of LLXG. The Hd producedin the pioneering work of Li, Valero, and Truhlar15 (LVT)is, to our knowledge, the only other approximate diabatic Hd
available for this system so that comparisons to that work areinevitable.
The computed lifetimes are sensitive to the region of theminimum and the barrier to dissociation on the A state. Theregion of the seam of conical intersections, which is responsi-ble for the branching between X and A states of NH2, is verycarefully described by Hd.73 However, its validation using thefull dimensional quantum dynamics approach is quite costlyand will be the subject of future work.
Section II reviews the form of Hd, its construction andthe full dimensional quantum determination of the positions,intensities, and lifetimes of the v2 levels. Section III de-scribes the electronic structure aspects of the representation.Section IV determines and discusses the positions, intensities,and lifetimes of the v2 levels of the A state. The consequentvalidation of Hd is also discussed. Section V summarizes anddiscusses directions for future studies.
II. THEORETICAL APPROACH
A. Diabatic representations
1. Diabatic states and the reference geometry
The details of constructing Hd have been described in P1and P3. Here, we summarize the key points so as to be able todiscuss the constructed Hd.
The diabatic states !du (q;R) are chosen to agree with the
adiabatic states !a,(ab)I (q;R) at the reference point R0.75 Here,
!a,(ab)I is an adiabatic state, with energy Ea,I,(ab), determined
from ab initio calculations, q denote the electronic coordi-nates and R the Cartesian coordinates of the Nat, nuclei. Thischoice of diabatic representation is permitted as the diabaticstates are defined up to a single arbitrary, geometry indepen-dent, unitary transformation. Since at R0,!d
u is an adiabaticstate, it carries an irreducible representation of the completenuclear permutation inversion (CNPI; Refs. 76–78) group.For ammonia, the CNPI group is isomorphic to D3h. We choseR0 to be a point with D3h point group symmetry so that theCNPI symmetry of the diabatic states at R0 can be determinedfrom the D3h transformation properties of the adiabatic statesat R0.77
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-3 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
Using Hd the electronic Schrödinger equation is given by
[Hd (R) − IEa,J,(m)(R)]dJ = 0. (1)
Here, the superscript (m) indicates that the results come fromthe model Hamiltonian Hd, rather than the ab initio wavefunctions. The diabatic states are constructed from the adi-abatic states, using
!du (r; R) ≡
N state!
J=1
!a,(ab)J (r; R)(d−1)Ju (R)
=N state!
J=1
!a,(ab)J (r; R)du
J (R). (2a)
From Eqs. (1) and (2a) and their derivatives, we find for thederivative couplings
fI,J,(ab)k (R) − f
I,J,(m)k (R) =
!
1≤α<β≤N state
"dI
α (R)dJβ (R)
− dIβ (R)dJ
α (R)#$!d
α (q; R)%%∇k!
dβ (q; R)
&q, (2b)
where
fI,J,(m)k (R) ≡ dI (R)†(∇kHd )dJ (R)
Ea,J,(m)(R) − Ea,I,(m)(R)(2c)
and
fI,J,(ab)k (R) ≡
$!
a,(ab)I (q; R)
%%∇k!a,(ab)J (q; R)
&q. (2d)
Equation (2b) is system of linear equations for the quasi-diabatic state coupling, ⟨!d
α (q; R)|∇k!dβ (q; R)⟩q, in terms of
the residual (the difference between the ab initio and Hd de-termined) couplings. For Nstate = 2, the case considered here,we get the particularly appealing result
f1,2,(a,b)k (R) − f
1,2,(m)k (R) =
$!d
2 (q; R)%%∇k!
d1 (q; R)
&q. (2e)
Thus, the magnitude ∥f a, I, J, (ab)−f a, I, J, (m)∥ is a direct mea-sure of the degree of diabaticity of the representation.
2. Diabatic Hamiltonian
a. General form Hd is expanded in terms of basis ma-trices, Bu,v, as
Hd (R) =Nc!
n=1
Vnp(n)(R)Bu(n),v(n), (3)
where Bu,v is an Nstate × Nstate symmetric matrix with a1 in the (u,v), and (v,u) elements and the remaining ele-ments 0. For example, for Nstate = 2 there are three basismatrices
B1,1 ='
1 0
0 0
(
, B1,2 ='
0 1
1 0
(
, and B2,2 ='
0 0
0 1
(
.
The V n, 1 ≤ n ≤ Nc are to be determined by a procedure de-scribed below. The advantage of Hd in the form of Eq. (3)is that it makes the linear dependence of Hd on V j explicitand easy to work with. The polynomials p(n)(R) are symme-try adapted according to the CNPI group. The p(n)(R) are con-structed from basic monomials gl(n)(R) using P µ(n)gl(n)(R)= p(n)(R). P µ is a standard projection operator79 for the µthirreducible representation of the CNPI group
P µ = 1OG
!
x
$(µ)(x)∗x. (4)
Here, OG is the number of group elements, x, whose corre-sponding operator is denoted x. Since only one-dimensionalrepresentations will be required here, given $(µ)(x) the ir-reducible character, and the effect of the group operations,x, on the functions gl, it is straightforward to evaluateP µ(n)gl(n).
The projector used in Eq. (4) depends (only) on theblock of Hd, and different projections are required for dif-ferent blocks. For its contribution to Bu,v to be nonvanishing,a symmetry adapted p(n) should transform as the direct prod-uct µ(n) = ir(u) × ir(v), where ir(u) is the irreducible rep-resentation carried by !d
u . This result, which is also used inRefs. 30 and 15, is rigorous at R0. Its utility is demonstratednumerically in Sec. III.
b. Form of the monomials Since the Hd must describebond breaking, functions that become unbounded for largeinternuclear distances are unacceptable. To address this issue,we use a generalization of an idea introduced by Bowmanand co-workers to describe a single adiabatic potential energysurface.80 Four distinct basic functions of the internal coordi-nates, wi, are used
• Exponential functions w1(ri,j ) = e−s1(ri,j −r(a(i,j ))i,j ), (5a)
• Gaussian functions w2(ri,j ) = e−s2(ri,j −r(b(i,j ))i,j )2
, (5b)
• Reciprocal functions w3(ri,j ) = e−s3(ri,j −r(c(i,j ))i,j )/(ri,j + r
(d(i,j ))i,j ), (5c)
• Dot − cross product functions wi,j,k,l4 = ri,j × ri,k · ri,l/|ri,j ri,kri,lrj,krj,lrk,l|α. (5d)
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-4 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
The dot-cross product functions are due to Collins andco-workers.67 Reciprocal functions as well as the exponentialfunctions in Eq. (5a), are suggested by the work of Bowmanand co-workers.80 The reciprocal functions help describe therepulsive walls. Exponential and reciprocal functions are notlocalized near their origins. Their values are significant for awide range of geometries. Gaussian functions, on the otherhand, are centered on specific origins and their values can bemade to vanish quickly when stepping away from those ori-gins. Gaussian functions are thus local basis functions thatserve to describe the region near their origin. The use of mul-tiple origins provides the flexibility to improve quality of thefit in specific regions.
The gl(R) are constructed as products of the w and havethe following monomial form:
gl(R) =3!
m=1
!
1≤i≤j≤Nat
[wm(ri,j )]α(l)m,i,j
!
(i,j,k,m)
"w
(i,j,k,m)4
#β(l)i,j,k,m
(6)for 1 ≤ l ≤ Ng. Note in Eq. (6), (l) is a label not an exponent,but α
(l)m,i,j and β
(l)i,j,k,m are exponents. Here, (i,j,k,m), again a
label not an exponent, denotes the allowed combinations offour atoms. The order of the monomial gl, o(l), is given by
o(l) =$
m,i<j
α(l)m,i,j +
$
(i,j,k,m)
β(l)i,j,k,m. (7)
The choice of α(l)m,i,j and β
(l)i,j,k,m, which are introduced in
Eq. (6), is in principle arbitrary. Increases in α(n)m,i,j and β
(n)i,j,k,l
are used to reduce the error in the representation of the adi-abatic data, at the expense of a larger Ng. The parameters sj,along with α, r
(a(i,j ))i,j , r
(b(i,j ))i,j , r
(c(i,j ))i,j , and α
(d(i,j ))i,j , are also ar-
bitrary. In this work, they are chosen by trial and error in therange sj > 0, r
(xi,j )i,j > 0 and α > 1/2.
3. Parametrization of Hd
The gl from which the p(n) are constructed are enumer-ated with the help of Table I. Noting that there are three NHand three HH internuclear distances, ri,j, there are 34 func-tions in that table. Denote the order of the jth function in agiven gl as nj. Then the p(n) are constructed from monomi-als, gl, satisfying all the following constraints, on the nj: (i)the total order is lower or equal to 4 (
%34j=1 nj ≤ 4); (ii) the
maximum total order of the NH Gaussian functions is 3; (iii)the maximum total order of the HH Gaussian functions is 3;and (iv) the maximum total order of any reciprocal functionis 1. Also reciprocal functions with 0 shifts in the denomina-tor only appear in 1st order monomials. After projection andelimination of symmetry zeros, we find Nc = 9652.
4. Determination of Hd
The equations used to determine the V and their solu-tion were initially reported in P1 and were revisited in P3.Briefly, the V are determined by the requirement that the en-ergies, energy gradients, and the interstate coupling gradients(= derivative coupling vector times the energy difference)
determined from Hd agree with those determined from theab initio calculations. Data at Npoint = 2536 nuclear con-figurations, producing ∼40 000 equations, were used. Thenuclear configurations were largely generated using ∼2000surface hopping trajectories39, 81 sampled from a Wigner dis-tribution by the requirement that a fixed percentage of trajec-tories, here 99% (1%) remain within the region well defined(approximately defined) by the nuclear configurations. Sincethere are many more equations than Vn, the equations are par-titioned into two groups, those solved in a least squares man-ner and those solved exactly. The equations included in theexact block are not entirely arbitrary. For example, the energydifference at a point of conical intersection must be exactlyzero to ensure that proper intersection adapted coordinates82
can be constructed; the branching plane vectors,83 energy dif-ference gradient, and interstate coupling gradients, also needto be exactly reproduced, so that the residual coupling is finiteat a point of intersection. It is, also, desirable that energies andgradients be reproduced at critical points to ensure the cor-rect topology of Hd. Nuclear configurations whose electronicstructure data are exactly reproduced are called nodes. Notefrom Eq. (2e) that the requirement that the Hd andab initio determined interstate coupling gradients agree, in aleast squares sense or exactly, gives Hd its diabatic character.
5. Quality of the fit
There are two issues concerning the Hd representation: (i)how reliably does the Hd describe the ab initio data and (ii)how reliably does the Hd representation of the NH3 1,21A po-tential energy surfaces and their interactions, describe exper-imentally measured observables. The first question has beenaddressed in P3. The second is the subject of Sec. III. Here,we briefly summarize from P3 how reliably the Hd describesthe ab initio data.
Over the energy range 0–60 000 cm−1, the Hd representa-tion of the 11A (21A) potential energy surface has a mean un-signed [root-mean-square] error of 73 [210] (83 [284]) cm−1.The representation is by construction more accurate in therange 0–50 000 cm−1 where the Hd representation of the 11A(21A) potential energy surface has a mean unsigned [rootmean square] error of 51 [82] (13 [24]) cm−1.
Another assay of the accuracy of the Hd representationinvolves the energy gradients and the derivative couplings.For the preponderance of the points, the energy gradient areaccurate to 1% error and derivative couplings are accurate to10%. The results for the large derivative couplings are par-ticularly gratifying since data in the range 101–108 are wellreproduced, with no point in error by >10%. This is be-cause the large derivative couplings are near points of coni-cal intersection which are nodes in the fitting procedure. Forsmall derivative couplings, which occur in regions of largeenergy separation, agreement decreases, which is not unex-pected since Hd provides a removable approximation to thederivative coupling while the ab initio values contain a smallnonremovable46 part. Up to 50 000 cm−1, the average per-centage error for energy gradients for points with magnitudeof gradient higher than 10−4 is 2.33%. For the full range of
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-5 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
TABLE I. Parameters for gl.
Exponential coordinates: w1(ri,j ) = e−s1(ri,j −r
(a(i,j ))i,j )
Coordinate# Atoms s1 r(a(i,j ))i,j Max order
1 N–H 1.5 1.5 42 H–H 0.5 1.0 4
Gaussian coordinates: w2(ri,j ) = e−s2(ri,j −r
(b(i,j ))i,j )2
Coordinate# Atoms s2 r(b(i,j ))i,j Max order
1 N–H 2.3 2.0 32 N–H 1.5 2.53 N–H 1.5 3.04 H–H 0.8 2.5 35 H–H 0.4 3.3
Reciprocal coordinates: w3(ri,j ) = e−s3(ri,j −r
(c(i,j ))i,j )
/!ri,j + r
(d(i,j ))i,j
", Max order = 1.
Coordinate# Atoms s3 r(c(i,j ))i,j r
(d(i,j ))i,j
1 N–H 1.25 1.0 1.02 N–H 1.30 2.0 0.03 H–H 0.5 1.0 1.04 H–H 0.5 1.5 0.0
Scalar triple-product coordinates: wi,j,k,l4 = ri,j × ri,k · ri,l/|ri,j ri,kri,l rj,krj,l rk,l |α , α = 0.55
data, the error in the norms of energy gradients is less that10% except for a limited number of outliers. The outliers arefound in the region of large gradients on repulsive walls, withhigh energies and the errors are only slightly over 10% andall are below 30%. Given their small number, high energies,and size of their error, it is unlikely that they will affect thedynamics.
B. Quantum dynamics
Following LLXG,35 the non-rotating nuclear Hamilto-nian for the NH3 systems is defined in the (2+1) Radau-Jacobi coordinate system,
HQ =2#
i=0
$− 1
2µi
∂2
∂r2i
+ Vi(ri)%
+2#
i=0
j 2i
2µir2i
+&
V (r0, r1, r2, θ1, θ2,ϕ) −2#
i=0
Vi(ri)
'
, (8)
where r1 and r2 are two Radau radial coordinates, r0 is Ja-cobi radial coordinate, µ1, µ2, and µ0 are the correspondingreduced masses, θ1(θ2) is the included angle between −→r1 (−→r2 )and −→r0 , and ϕ is the relative azimuthal angle between −→r1 and−→r2 in body fixed system. j1 and j2 are the angular momentumoperators for r1 and r2, respectively, and j 2
0 = (j1 + j2)2. V isthe six-dimensional potential energy surface, and Vi(ri) arethe reference potential for ri.
The wave function is discretized in a mixed represen-tation. The three radial coordinates were represented by thediscrete variable representation (DVR) or potential-optimizeddiscrete variable representation (PODVR).84 For the angularcoordinates, finite basis representation (FBR) is used. Theparity (p) adapted wave function can thus be expanded on the
FBR basis,
|%⟩p =#
i0i1i2j1j2m
ψpi0i1i2j1j2m
|i0i1i2⟩ |j1j2m,p⟩ , (9)
where ψpi0i1i2j1j2m
is the wave function in the discrete repre-sentation, i0, i1, and i2 denote the indices of three radial co-ordinates, respectively. |j1j2m, p⟩ are the angular FBR basisfunctions defined as parity-adapted products of the sphericalharmonics,
|j1j2m,p⟩ = (2 + 2δm,0)−1/2(|j1m⟩|j2 − m⟩
+p|j1 − m⟩|j2m⟩), p = ±1. (10)
The detailed formulation for how the Hamiltonian acts on ba-sis set can be found in our earlier work.35
To simulate the absorption spectrum, we first calculatedvibrational levels of NH3 on its ground electronic state us-ing the iterative Lanczos algorithm85, 86 with the basis set of5 PODVR for r1 and r2, 21 sine-DVR for r0 ∈ (1.2, 4.5)a0
and j1, j2 = 0 ∼ 24. For ND3, we have used a basis set of5 PODVR for r1 and r2, 25 sine-DVR for r0 ∈ (1.2, 4.5)a0
and j1, j2 = 0 ∼ 30. Five hundred steps of iterations weresufficient to converge the low-lying energy levels.
The absorption spectrum was calculated assuming a Con-don transition, in which the ground state vibrational eigen-functions were placed vertically on the A state. The wavefunctions were transformed into a new grid/basis for the ex-cited state propagation. In particular, we used 21 sine-DVRpoints for three radial coordinates in the region of (1.2, 4.5)a0
and 25 FBR functions for angular coordinates in absorptionspectrum calculation of NH3, 25 sine-DVR points for three ra-dial coordinates in the same region and 30 FBR functions forangular coordinates in calculation of ND3. The evolution ofthe excited state wave packet was performed using the Cheby-shev propagator.87
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-6 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
The A ← X absorption spectrum, S(E), was obtainedfrom the Fourier transform of the Chebyshev auto-correlationfunction Ck ≡ ⟨ψ0 | ψk⟩,88
S(E) = 1πH− sin ϑ
!
k=0
(2 − δk,0) cos(kϑ)Ck, (11)
H− is half-width of the Hamiltonian, ϑ = arccos E is theChebyshev angle, and k is the Chebyshev order. To avoid re-flection at the edge of the grid, a damping function was usedin Chebyshev propagation, and the damping starts at r = 3.1a0
for all three radial coordinates. To obtain a convergent absorp-tion spectrum, 5000 steps of Chebyshev propagation were car-ried out for both NH3 and ND3.
To determine accurately the positions and widths of nar-row resonances, a low-storage filter diagonalization methodwas used.89
III. ANALYSIS OF Hd: ELECTRONIC STRUCTURE
A. Electronic structure: Ab initio treatment
The ab initio electronic structure data used to constructHd were determined from a multireference configuration in-teraction expansion comprised of over 30 × 106 configurationstate functions , and constructed from molecular orbitals ob-tained from a two-state, state averaged multiconfigurationalself-consistent field procedure based on aug-cc-pVTZ baseson N and H with an added 3s Rydberg function on nitrogen.All electronic structure calculations were carried out using theCOLUMBUS electronic structure codes.90–92
B. Electronic structure: Spectroscopic constants
Table II reports the equilibrium structure and harmonicfrequencies of the X1A1 and A1A2 states of NH3, the ex-citation energy, Te, and the ground state dissociation en-ergy, De, and compares those quantities with the availableexperimental data and computational results. Table III re-ports the equivalent quantities for the X2B1 and A2A1 statesof NH2. The formats of Tables II and III follow those ofTables 4 and 5 of LVT (Ref. 15) for ease of comparison.Note that since the structures reported in these tables arenodes in the fit, the ab initio geometries, the values of Te
and De as well as barrier heights reported in these tablesare exactly reproduced by the Hd representation. The ab ini-tio and Hd determined equilibrium and saddle point geome-tries of NH3(X, A) and NH2(X, A) are in very good accordwith the tabulated “best values,” with the largest errors be-ing 0.007 Å and 1.1◦. The ab initio determined Te for NH3
(NH2) is in error = best-calculated by 871 (−205) cm−1
compared to best value of 48 071 (10 978) cm−1. The ab initiodetermined inversion (transition state) barrier on the 11A(21A) potential energy surface is in error by −173 (195)cm−1 compared to the best value of 1784 (2348) cm−1. Theab initio determined dissociation energy, De, of NH3 on theground 11A potential energy surface is in error by 1258 cm−1
compared to a best value of 40 510 cm−1. The dissociation en-ergy on the excited state potential energy surface is in error by
TABLE II. Spectroscopic constants and related data for X1A1 and A1A2states of NH3. Energies in cm−1, distances in Å, and angles in ◦. Ordering ofharmonic frequencies is that of Ref. 95 except for C2v saddle point on 21Apotential energy surface where energy ordering is used. For C2v geometriesH1, H2 are symmetry equivalent.
Hd Ab initio Best
11A stateEquilibrium (C3v−X1A1)re(N–H) 1.0154 1.0154 1.011a
HNH 106.35 106.35 106.75a
Harmonic frequencies3447.08 3456.26 3483.1a
1093.12 1066.24 1052.8a
3581.40 3584.10 3616.0a
1655.33 1674.48 1675.7a
Saddle point (D3h)re(N–H) 0.9979 0.9979 0.9943a
Classical barrier height 1957.2 1957.2 1784.7a
Harmonic frequencies3610.0 3620.1 3640.0a
847.1i 870.0i 837.4ia
3825.2 3830.5 3852.7a
1575.5 1584.0 1585.7a
AsymptoteDe NH3(X) → NH2(X) + H 39 252.1 39 252.1 40 510b
21A state
Equilibrium (D3h−A1A2)re(N–H) 1.0485 1.0485 1.055(8)c
Harmonic frequencies2838.7 2793.7 2870d
780.7 754.10 892d
2949.7 2955.8 3020d
1377.7 1334.3 1110d
Te 47 198.8 47 198.8 48 071d
AsymptoteDe NH3(A) → NH2(A) + H 3237 3236 3416Saddle point (C2v)re(N–H) 1.3055 1.3054 1.298d
re(N–H1) 1.0408 1.0408 1.040d
H2NH1 112.5 112.5 112.4d
HNH1 123.8 123.8 123.8d
Classical barrier height 2152.98 2152.98 2348d
Harmonic frequencies2000.10i 2003.32i508.05 475.29981.30 962.68
1419.51 1435.372995.81 3027.803231.10 3244.42
aReference 93.bReference 19.cReference 96.dReference 8.
179 cm−1 compared to a best value of 3416 cm−1. The errorsin the Te values and barriers are less than the level spacing inv2 for NH3 A, (See Sec. IV B) while the error in ground stateDe is approximately the spacing of two such levels. The errorsin the Te for NH3 and ground state De for NH3 likely reflectan underestimate of the differential correlation energy of NH3
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-7 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
TABLE III. Spectroscopic constants and related data for X2B1 and A2A1states of NH2. Energies in cm−1, distances in Å, and angles in ◦. Ordering ofharmonic frequencies is that of Ref. 95
Hd Ab initio Best
X2B1 stateEquilibriumre(N–H) 1.0283 1.0283 1.024a
HNH 102.80 102.80 103.4a
Harmonic frequencies2934.62 3350.23 3374.2b
1556.79 1539.57 1523.5b
3427.33 3443.40 3481.2b
A2A1 stateEquilibriumre(N–H) 0.9987 0.9986 1.004c
HNH 145.13 145.10 144c
Harmonic frequencies3796.34 3612.56 3635b
935.75 978.14 964b
4154.31 3924.56 3953b
Te 11 184.01 11 183.1 10 977.8d
aFrom Ref. 97 as cited in LVT.bFrom Ref. 98 as cited in LVT.cReference 95.dFrom LVT.
near its equilibrium geometry. This error is then expected todecrease as the NH3 bond is stretched. Consistent with thisobservation, the error in De for NH3 on the excited state issmall. In the LVT, Hd a correction function was introduced tomake the Hd determined potential energy surfaces reproducethe experimental values of De and Te values exactly. These er-rors in the De and Te values in the Hd described here shouldnot impact the spectral simulations reported in Sec. IV.
The Hd determined harmonic frequencies at the mini-mum and saddle point on the ground state potential energysurface of NH3 are in good accord with the ab initio val-ues. The largest error for an Hd determined frequency is−41 cm−1 compared to a measured value of 1052.8 cm−1
which occurs at the ground state minimum. The ω4 harmonicfrequency at the equilibrium geometry of the A1A′′
2 stateexhibits the greatest deviation from a measured result(1110.9 cm−1) being in error by over −20%. However, itshould be noted that as observed in the Introduction, this min-imum is quite shallow, complicating the determination of har-monic frequencies. For example, the −111 cm−1 error in ω2 isinconsistent with the magnitude of error, <70 cm−1 in the v2
progression discussed in Sec. IV. The harmonic frequenciesfor the asymptotic states of NH2 are in general satisfactorilydescribed.
C. Electronic structure: Conical intersection seam
The seam of conical intersection is considered inTable IV, which reports 26 points on the seam. These pointsare nodes, with the Hd and ab initio determined energy dif-ferences, and g and h constrained to agree. The absolute en-ergies are seen to be in very good agreement (maximum error65 cm−1) even though, with the exception of the minimum
FIG. 1. Three-dimensional plot of adiabatic potential energy surfaces show-ing: (i) minima on ground state and excited states, (ii) saddle points on the11A and 21A potential energy surfaces, and (iii) the minimum energy cross-ing on the 1,21A seam of conical intersection. Energies are indicated in cm−1.The geometry of the NH2 used in this figure is also indicated. The x and y co-ordinates describe the motion of the third H in the bisector plane of the NH2.
energy crossing, they are not so constrained. At all such nu-clear configurations, the 11A and 21A states are degenerateto less than 1 wave number. All points on the seam have atleast a plane of symmetry. This is a consequence of the factthat for planar geometries the 11A and 21A states carry 1A′
and 1A′′ irreducible representations so that the h vector (inter-state coupling vector) must transform as A′′ and there is onlya single a′′ coordinate. The minimum energy conical intersec-tion, the MEX, is approximately 4808 (6154) cm−1 below theA1A′′
2 minimum where the analogous result for LVT is givenin parenthesis. As is the case for the saddle point on the 21Apotential energy surface, the MEX has a single elongated NHbond. Its structure is in good accord with the result of LVT.(See Table IV.)
The electronic structure treatment in this region is notvalidated in the current spectral simulations which do notconsider the NH2(X, A) branching. This analysis will bethe subject of a subsequent study. Figure 1 provides a two-dimensional slice from the 1,21A potential energy surfaces.Key critical points and their energies are indicated althoughonly the approximate geometries of these structures areshown.
IV. ANALYSIS OF Hd: VIBRATIONAL SPECTRA
A. The X 1A1 state
Table V reports the low-lying vibrational levels ofNH3(X) and compares them with the spectroscopic data andthe equivalent theoretical treatment based on the LVT Hd. Itshould be noted that other ground state potential energy sur-faces are available for NH3, for which essentially exact agree-ment with experimental data is possible using the compu-tational treatment discussed in Sec. II B.93, 94 However, the
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-8 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
TABLE IV. Conical intersection seam for 1,21A states of NH3. E1, E2, and !E = E2 − E1 in cm−1 relative to energy of X1A1 state minimum. For C2v
structures, H1 and H2 are symmetry equivalent. The 22 conical intersections with energies <50 000 cm−1 are nodes, with energy difference, g and h fit exactly.(Two such points exhibit small error in ∥g∥, less than 1%.) The ab initio geometries energies are exactly reproduced by Hd. Minimum energy crossing indicatedby a *. Eavg = (E2+E1)/2. ! implies the difference between the Hd determined and the ab initio result.
C2v conical intersectionsrNH1 rNH3 H1NH3 H1NH2 E(avg) !E(avg) E2−E1 !(E2−E1) ∥h∥ ∥g∥
*1.0222 1.9689 125.40 109.20 42 390 0 0.0001 −5.7 × 10−8 0.0523 0.07280.9243 1.9802 126.63 106.74 46 472 −9 0.0017 4.3 × 10−8 0.0536 0.07580.9731 1.9750 126.03 107.95 43 310 −3 0.0002 5.2 × 10−8 0.0529 0.07421.0691 1.9622 124.79 110.43 43 071 −2 0.0001 −4.7 × 10−8 0.0518 0.07191.1626 1.9461 123.49 113.02 47 433 3 0.0072 6.2 × 10−8 0.0508 0.07071.2970 1.9137 121.35 117.30 57 246 −45 0.0001 −3.0 × 10−9 0.049(6) 0.07(11)1.0478 1.9691 125.41 109.18 42 631 −2 0.0014 3.5 × 10−8 0.0523 0.07291.4601 1.3229 156.50 46.99 71 630 −14 0.0001 1.2 × 10−7 0.07(38) 0.0(983)1.3344 1.5875 148.02 63.95 64 028 21 0.0004 1.0 × 10−9 0.07(73) 0.09(58)1.1656 1.7569 138.71 82.58 49 623 −27 0.0013 1.0 × 10−7 0.0730 0.086(4)1.0805 1.8627 131.76 96.49 43 916 4 0.0000 4.2 × 10−8 0.0623 0.07980.9730 2.1167 118.17 123.67 43 944 −22 0.0001 3.0 × 10−8 0.0433 0.06281.0218 1.9782 58.63 117.26 44 599 −1 0.0022 2.6 × 10−8 0.0673 0.0655
Cs intersection pointsrNH1 rNH2 rNH3 H1NH3 H1NH2 Eavg !Eavg E2−E1 !(E2−E1) ∥h∥ ∥g∥1.0225 1.0223 1.9681 127.42 109.14 42 391 0 0.0176 1.4 × 10−8 0.0524 0.07291.0217 1.0217 1.9682 120.34 109.33 42 397 2 0.0449 4.5 × 10−8 0.0525 0.07281.0201 1.0201 1.9662 115.14 109.73 42 420 6 0.0000 −5.1 × 10−8 0.0531 0.07271.0137 1.0137 1.9581 104.36 111.39 42 556 23 0.0004 5.3 × 10−8 0.0553 0.07230.9771 0.9760 1.9190 163.36 122.02 44 824 −65 0.0001 1.9 × 10−8 0.0625 0.069(0)1.0742 0.9684 1.9691 125.41 109.18 43 357 −6 0.0014 2.7 × 10−8 0.0523 0.07301.1271 0.9155 1.9691 125.41 109.18 46 358 −12 0.0022 4.3 × 10−8 0.0524 0.07351.1904 1.0339 2.0693 9.08 101.83 48 452 2 0.7355 −1.7 × 10−7 0.0522 0.03491.0016 1.0009 2.2460 116.87 143.73 48 706 −3 0.0001 −9.3 × 10−8 0.0424 0.03880.9687 0.9651 2.0668 64.13 127.32 45 848 5 0.0005 3.9 × 10−8 0.0674 0.063(6)0.9788 0.8059 1.9972 119.47 108.24 55 786 27 0.0041 3.2 × 10−8 0.053(9) 0.07(70)1.0460 0.9954 1.8319 122.92 89.35 44 825 −4 0.0607 1.0 × 10−7 0.0692 0.08480.9773 0.9593 1.8520 154.96 93.72 45 312 −30 0.0125 6.7 × 10−8 0.0688 0.0831
aMinimum energy crossing from LVT is R(N–H1) = 1.990, R(N–H3) = 1.21, H1NH2 = 110◦, and H3NH2 = 107.9◦.
agreement using the potential energy surface derived fromthe present Hd is quite reasonable, a significant improvementover that of LVT. The improved accuracy will be an impor-tant consideration in future studies of vibrationally mediatedphotodissociation.
B. The A1A′′2 state
The absorption spectrum from the ground vibrationallevel of the X1A1 state is considered in Figs. 2–4. This spec-trum consists of a progression in the v2 mode, in agreementwith experiment. There are three issues to be addressed here,the line positions, the line intensities, and the line widths orlifetimes.
1. Energy levels
Figures 2(a) and 2(b) address the issue of the linepositions. The experimental line positions E exp(n), shiftedHd determined line positions Em
shift(n) ≡ Em(n) − [Em(0)− Eexp(0)] and the energy level error δEm(n) = Eexp(n)− Em
shift(n) are reported. Here, the energy of a level with nquanta in v2 is Em(n) and the superscript denotes the method
used to compute E(n) with m = PW (LVT), if E(n) is deter-mined from the 21A potential energy surface constructed us-ing the Hd in this work (in LVT) or m = exp if experimentalvalues for E(n) are used. Table VI reports the unaltered Em(n).Figures 2(a) and 2(b) report data for NH3 (ND3). Note that theEPW(n) in Table VI reflect a systematic error due principally
TABLE V. Low-lying vibrational levels of NH3(X) obtained using the LVTHd and the present work (PW). Energies in cm−1 relative to correspondingzero-point energy.
v1 vp2 v
l33 v
l44 Expa LVTb PW
0 0+ 00 00 0 0 00 0− 00 00 0.79 0.108 0.450 1+ 00 00 932.43 1093.05 974.360 1− 00 00 968.12 1099.96 997.650 2+ 00 00 1597.47 1966.96 1679.270 0+ 00 11 1626.28 1672.23 1622.550 0− 00 11 1627.37 1672.29 1623.760 2− 00 00 1882.18 2092.07 1918.570 3+ 00 00 2384.15 2653.50 2420.94
aReference 99.bReference 35.
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01
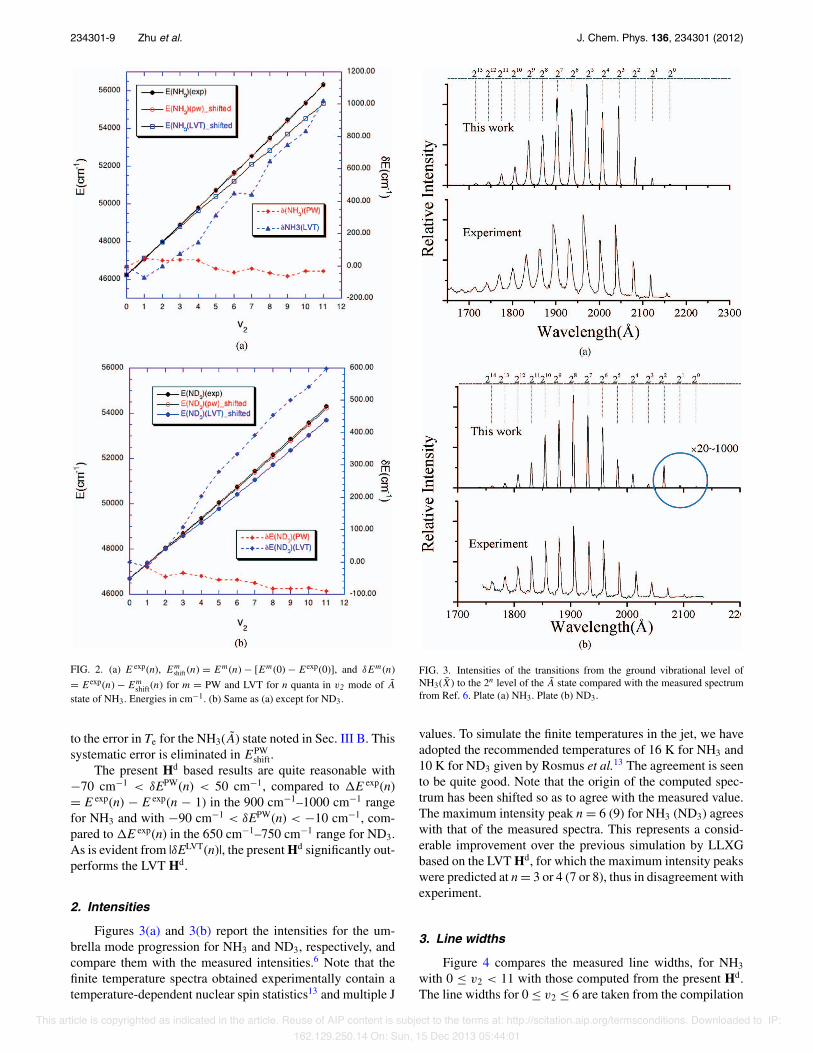
234301-9 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
FIG. 2. (a) E exp(n), Emshift (n) = Em(n) − [Em(0) − Eexp(0)], and δEm(n)
= Eexp(n) − Emshift(n) for m = PW and LVT for n quanta in v2 mode of A
state of NH3. Energies in cm−1. (b) Same as (a) except for ND3.
to the error in Te for the NH3(A) state noted in Sec. III B. Thissystematic error is eliminated in EPW
shift.The present Hd based results are quite reasonable with
−70 cm−1 < δEPW(n) < 50 cm−1, compared to "E exp(n)= E exp(n) − E exp(n − 1) in the 900 cm−1–1000 cm−1 rangefor NH3 and with −90 cm−1 < δEPW(n) < −10 cm−1, com-pared to "E exp(n) in the 650 cm−1–750 cm−1 range for ND3.As is evident from |δELVT(n)|, the present Hd significantly out-performs the LVT Hd.
2. Intensities
Figures 3(a) and 3(b) report the intensities for the um-brella mode progression for NH3 and ND3, respectively, andcompare them with the measured intensities.6 Note that thefinite temperature spectra obtained experimentally contain atemperature-dependent nuclear spin statistics13 and multiple J
FIG. 3. Intensities of the transitions from the ground vibrational level ofNH3(X) to the 2n level of the A state compared with the measured spectrumfrom Ref. 6. Plate (a) NH3. Plate (b) ND3.
values. To simulate the finite temperatures in the jet, we haveadopted the recommended temperatures of 16 K for NH3 and10 K for ND3 given by Rosmus et al.13 The agreement is seento be quite good. Note that the origin of the computed spec-trum has been shifted so as to agree with the measured value.The maximum intensity peak n = 6 (9) for NH3 (ND3) agreeswith that of the measured spectra. This represents a consid-erable improvement over the previous simulation by LLXGbased on the LVT Hd, for which the maximum intensity peakswere predicted at n = 3 or 4 (7 or 8), thus in disagreement withexperiment.
3. Line widths
Figure 4 compares the measured line widths, for NH3
with 0 ≤ v2 < 11 with those computed from the present Hd.The line widths for 0 ≤ v2 ≤ 6 are taken from the compilation
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-10 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
FIG. 4. Calculated line widths of 2n levels of NH3 (plate (a)) and ND3 (plate(b)) using the Hd from the present work (PW) compared with the experimen-tal NH3 values of Ref. 7, 0 ≤ n ≤ 3, and Ref. 4, 3 < n ≤ 6, based on thecompilation of Ref. 8 and the experimental ND3 values of Ref. 7, 0 ≤ n ≤ 2.
in Ref. 8 while the remainder are taken from Ref. 6. For ND3
line widths for the range 0 ≤ v2 ≤ 2, where the experimen-tal data are most consistent,4, 6, 7 are reported.7 The computedresults are seen to be in very good accord with experimen-tal results. Table VI provides a more complete tabulation ofthe line widths. The line widths based on the present Hd areseen to be in good accord with the measured results for NH3
throughout the range of available measurements. The agree-ment with the ND3 results for v2 beyond the range in Fig. 4is somewhat more qualitative, although based on the existingdata the possibility of the experimental line widths being sys-
TABLE VI. Line widths (cm−1) of NH3 from experiment and computationsbased on Hd obtained in the present work (PW) and Hd from LVT, as reportedin Ref. 35. Experimental values taken from Ref. 6 except for v2 ≤ 3 for NH3and v2 ≤ 2 for ND3 where values from Ref. 7, which are more consistentwith the available experimental data are used. The E(n) exhibit a systematicerror owing to the error in Te reported in Table II.
NH3
Band E(PW) E(exp) !(PW) !(exp) !(LVT)
00 45 426 46 222 35.3 34 103.921 46 309 47 057 23.1 29 58.822 47 202 47 964 36.7 44 67.423 48 109 48 869 72.0 94 121.224 49 019 49 783 139.4 132 235.125 49 918 50 730 201.3 178 394.426 50 819 51 656 238.9 228 369.727 51 729 52 543 248.3 269 367.128 52 654 53 496 238.5 253 280.829 53 596 54 454 225.9 271 213.4210 54 551 55 380 222.3 263 146.1211 55 514 56 342 229.0 277 108.5212 56 482 57 300 243.8 279 16.9213 57 454 58 285 263.4 293 24.5
ND3
Band E(PW) E(exp) !(PW) !(exp) !(LVT)00 46 025 46 701 2.4 4.2 11.21 46 676 47 369 1.1 1.5 5.822 47 330 48 052 16.8 15.4 33.723 47 988 48 697 29.6 61.8 97.524 48 655 49 375 32.6 52.5 93.925 49 331 50 062 31.2 56.5 79.926 50 017 50 748 29.0 59.1 69.527 50 710 51 451 27.8 60.4 62.428 51 410 52 168 28.4 60.4 60.329 52 116 52 874 34.0 69.3 64.8210 52 826 53 582 44.6 73.7 73.9211 53 540 54 306 61.0 79.6 82.2212 54 258 55 054 83.6 101.7 81.8213 54 977 55 751 113.2 103.0 71.5
tematically too large for v2 ≥ 3, cannot be ruled out. Againthe performance for the LVT Hd whose results are also re-ported in Table VI are seen to be less satisfactory. The mostlikely source of the difference in the line widths between thepresent Hd and that developed by LVT is the difference inthe classical barrier height which is 2153 cm−1 in the presentwork and 1774 cm−1 in that of LVT.
V. SUMMARY AND CONCLUSION
A method for determining a coupled diabatic state repre-sentation of adiabatic states coupled by conical intersectionshas been previously introduced. The method is notable for itsuse of derivative coupling and energy gradient informationas well as energies to construct the diabatic representationand its combination of least squares and interpolative tech-niques. The incorporation of energy gradient and derivativecoupling information reduces the number of points at whichthe electronic structure data must be determined. The deriva-tive coupling information also enables the representation tobe quantifiably diabatic in a least squares sense. However, a
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-11 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
representation produced by this approach has yet to be em-ployed in a fully quantum mechanical nuclear dynamics sim-ulation, which provides a stringent test of the quality of therepresentation.
Concomitantly, there have been a considerable number ofcomputational and experimental studies of the photodissocia-tion of the A state of NH3 which exhibits both adiabatic andnonadiabatic product channels. Of particular relevance herewas a full six-dimensional quantum mechanical study of theabsorption from the ground vibrational level of the X stateto the A state by LLXG. These calculations were based on apreviously reported two-state diabatic Hamiltonian. However,the agreement with experiment was modest at best. Therefore,in this work we report an analysis of the ground state absorp-tion spectrum, line positions, intensities, and lifetimes, us-ing the full six-dimensional quantum dynamical treatment ofLLXG and a Hd constructed using the algorithm discussed inSec. II. Results for both NH3 and ND3 were reported. Agree-ment with the measured spectrum, the line positions, intensi-ties, and lifetimes is quite good.
This work focused on the adiabatic aspects of the absorp-tion spectrum, that is, it ignores the non-adiabatic transitionsand final state of NH2 produced. Determination of the branch-ing ratio of NH2(X) to NH2(A), as, for example, determinedin vibrationally mediated photodissociation, requires a nona-diabatic dynamics simulation. This can be accomplished us-ing a slightly refined extension of the current Hd. However,using the exact quantum techniques employed here the nona-diabatic simulation is much more time consuming and will bereported in a subsequent study.
ACKNOWLEDGMENTS
X.Z. and D.R.Y., who are responsible for the constructionof Hd, acknowledge the support of the National Science Foun-dation Grant No. CHE-1010644 to D.R.Y. J.Y.M. and H.G.,who performed the six-dimensional quantum mechanical cal-culations, acknowledge the support of the National ScienceFoundation Grant No. CHE-0910828 to H.G.
1S. W. Leifson, Astrophys. J. 63, 73 (1933).2V. Vaida, W. Hess, and J. L. Roebber, J. Phys. Chem. 88, 3397 (1984).3A. D. Walsh and P. A. Warsop, Trans. Faraday Soc. 57, 345 (1961).4L. D. Ziegler, J. Chem. Phys. 82, 664 (1985).5M. N. R. Ashfold, C. L. Bennett, and R. N. Dixon, Faraday Discuss. Chem.Soc. 82, 163 (1986).
6V. Vaida, M. I. McCarthy, P. C. Engelking, P. Rosmus, H.-J. Werner, andP. Botschwina, J. Chem. Phys. 86, 6669 (1987).
7S. A. Henck, M. A. Mason, W.-B. Yan, K. K. Lehmann, and S. L. Coy,J. Chem. Phys. 102, 4783 (1995).
8A. Bach, J. M. Hutchison, R. J. Holiday, and F. F. Crim, J. Chem. Phys.116, 9315 (2002).
9M. L. Hause, Y. H. Yoon, and F. F. Crim, J. Chem. Phys. 125, 174309(2006).
10A. Bach, J. M. Hutchison, R. J. Holiday, and F. F. Crim, J. Chem. Phys.116, 4955 (2002).
11W. R. Harshbarger, J. Chem. Phys. 53, 903 (1970).12S. L. Tang and D. G. Imre, Chem. Phys. Lett. 144, 6 (1988).13P. Rosmus, P. Botschwina, H.-J. Werner, V. Vaida, P. C. Engelking, and
M. I. McCarthy, J. Chem. Phys. 86, 6677 (1987).14M. I. McCarthy, P. Rosmus, H.-J. Werner, P. Botschwina, and V. Vaida,
J. Chem. Phys. 86, 6693 (1987).15Z. H. Li, R. Valero, and D. G. Truhlar, Theor. Chem. Acc. 118, 9 (2007).
16J. Biesner, L. Schnieder, G. Ahlers, X. Xie, K. H. Welge, M. N. R. Ashfold,and R. N. Dixon, J. Chem. Phys. 88, 3607 (1988).
17J. Biesner, L. Schneider, G. Ahlers, X. Xie, K. H. Welge, M. N. R. Ashfold,and R. N. Dixon, J. Chem. Phys. 91, 2901 (1989).
18E. L. Woodbridge, M. N. R. Ashfold, and S. R. Leone, J. Chem. Phys. 94,4195 (1991).
19D. H. Mordaunt, M. N. R. Ashfold, and R. N. Dixon, J. Chem. Phys. 104,6472 (1996).
20D. H. Mordaunt, M. N. R. Ashfold, and R. N. Dixon, J. Chem. Phys. 104,6460 (1996).
21D. H. Mordaunt, M. N. R. Ashfold, R. N. Dixon, P. Löffler, L. Schneider,and K. H. Welge, J. Chem. Phys. 108, 519 (1998).
22R. A. Loomis and M. I. Lester, Annu. Rev. Phys. Chem. 48, 643 (1997).23J. P. Reid, R. A. Loomis, and S. R. Leone, J. Chem. Phys. 112, 3181 (2000).24J. P. Reid, R. A. Loomis, and S. R. Leone, Chem. Phys. Lett. 324, 240
(2000).25J. P. Reid, R. A. Loomis, and S. R. Leone, J. Phys. Chem. A 104, 10139
(2000).26K. L. Wells, G. Perriam, and V. G. Stavros, J. Chem. Phys. 130, 074308
(2009).27R. Runau, S. D. Peyerimhoff, and R. J. Buenker, J. Mol. Spectrosc. 68, 253
(1977).28U. Manz, E.-A. Reinsch, P. Rosmus, H.-J. Werner, and S. V. O’Neil,
J. Chem. Soc. Faraday Trans. 87, 1809 (1991).29D. R. Yarkony, J. Chem. Phys. 121, 628 (2004).30S. Nangia and D. G. Truhlar, J. Chem. Phys. 124, 124309 (2006).31R. N. Dixon, Chem. Phys. Lett. 147, 377 (1988).32R. N. Dixon, Mol. Phys. 88, 949 (1996).33R. N. Dixon and T. W. R. Hancock, J. Phys. Chem. A 101, 7567 (1997).34D. Bonhommeau and D. G. Truhlar, J. Chem. Phys. 129, 014302 (2008).35W. Lai, S. Y. Lin, D. Xie, and H. Guo, J. Chem. Phys. 129, 154311 (2008).36D. Bonhommeau, R. Valero, D. G. Truhlar, and A. W. Jasper, J. Chem.
Phys. 130, 234303 (2009).37W. Lai, S. Y. Lin, D. Xie, and H. Guo, J. Phys. Chem. A 114, 3121 (2010).38A. Bach, J. M. Hutchison, R. J. Holiday, and F. F. Crim, J. Chem. Phys.
118, 7144 (2003).39J. C. Tully, J. Chem. Phys. 93, 1061 (1990).40K. Giri, E. Chapman, C. S. Sanz, and G. Worth, J. Chem. Phys. 135, 044311
(2011).41M. Beck, A. Jackle, G. A. Worth, and H.-D. Meyer, Phys. Rep. 324,
1 (2000).42G. A. Worth, H.-D. Meyer, H. Köppel, L. S. Cederbaum, and I. Burghardt,
Int. Rev. Phys. Chem. 27, 569 (2008).43H. Nakamura and D. G. Truhlar, J. Chem. Phys 117, 5576 (2002).44H. Nakamura and D. G. Truhlar, J. Chem. Phys. 118, 6816 (2003).45M. Baer, Chem. Phys. 15, 49 (1976).46C. A. Mead and D. G. Truhlar, J. Chem. Phys. 77, 6090 (1982).47M. Baer, Phys. Rep. 358, 75 (2002).48H. Köppel, W. Domcke, and L. S. Cederbaum, Adv. Chem. Phys. 57, 59
(1984).49H. Köppel, W. Domcke, and L. S. Cederbaum, in Conical Intersections,
edited by W. Domcke, D. R. Yarkony, and H. Köppel (World Scientific,New Jersey, 2004), Vol. 15, p. 323.
50S. Mahapatra, G. A. Worth, H.-D. Meyer, L. S. Cederbaum, and H. Köppel,J. Phys. Chem. A 105, 5567 (2001).
51S. Mahapatra, V. Vallet, C. Woywod, H. Köppel, and W. Domcke, Chem.Phys. 304, 17 (2004).
52A. Viel and W. Eisfeld, J. Chem. Phys. 120, 4603 (2004).53W. Eisfeld and A. Viel, J. Chem. Phys. 122, 204317 (2005).54T. Ichino, A. J. Gianola, W. C. Lineberger, and J. F. Stanton, J. Chem. Phys.
125, 084312 (2006).55B. N. Papas, M. S. Schuurman, and D. R. Yarkony, J. Chem. Phys. 129,
124104 (2008).56D. Opalka and W. Domcke, J. Chem. Phys. 132, 154108 (2010).57T. C. Thompson, G. Izmirlian, S. J. Lemon, D. G. Truhlar, and C. A. Mead,
J. Chem. Phys 82, 5597 (1985).58A. J. C. Varandas, F. B. Brown, C. A. Mead, D. G. Truhlar, and N. C. Blais,
J. Chem. Phys. 86, 6258 (1987).59G. J. Atchity and K. Ruedenberg, Theor. Chem. Acc. 97, 47 (1997).60H. Köppel, J. Gronki, and S. Mahapatra, J. Chem. Phys. 115, 2377 (2001).61T. Pacher, L. S. Cederbaum, and H. Köppel, J. Chem. Phys. 95, 6668
(1991).62T. Pacher, L. S. Cederbaum, and H. Köppel, J. Chem. Phys. 89, 7367
(1988).
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

234301-12 Zhu et al. J. Chem. Phys. 136, 234301 (2012)
63V. C. Mota and A. J. C. Varandas, J. Phys. Chem. A 112, 3768 (2008).64V. C. Mota, P. J. S. B. Caridade, and A. J. C. Varandas, Int. J. Quantum
Chem. 111, 3776 (2011).65C. R. Evenhuis and M. A. Collins, J. Chem. Phys. 121, 2515 (2004).66C. R. Evenhuis, X. Lin, D. H. Zhang, D. R. Yarkony, and M. A. Collins,
J. Chem. Phys. 123, 134110 (2005).67O. Godsi, C. R. Evenhuis, and M. A. Collins, J. Chem. Phys. 125, 104105
(2006).68C. Evenhuis and T. J. Martínez, J. Chem. Phys. 135, 224110 (2011).69R. Dawes, A. Passalacqua, T. D. Sewell, A. F. Wagner, M. Minkoff, and D.
L. Thompson, J. Chem. Phys. 130, 144107 (2009).70R. Dawes, D. L. Thompson, A. F. Wagner, and M. Minkoff, J. Chem. Phys.
128, 84107 (2009).71R. Dawes, A. W. Jasper, C. Tao, C. Richmond, C. Mukarakate, S. H. Kable,
and S. A. Reid, J. Phys. Chem. Lett. 1, 641 (2010).72X. Zhu and D. R. Yarkony, J. Chem. Phys. 132, 104101 (2010).73X. Zhu and D. R. Yarkony, Mol. Phys. 108(19), 2611 (2010).74X. Zhu and D. R. Yarkony, J. Chem. Phys. 136, 174110 (2012).75D. R. Yarkony, J. Phys. Chem. A101, 4263 (1997).76H. C. Longuet-Higgins, Mol. Phys. 6, 445 (1963).77P. R. Bunker, Molecular Symmetry and Spectroscopy (Academic, New
York, 1979).78J. T. Hougen, J. Mol. Spectrosc. 256, 170 (2009).79M. Tinkham, Group Theory and Quantum Mechanics (McGraw-Hill, New
York, 1964).80B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem. 28, 577 (2009).81J. C. Tully, in Modern Theoretical Chemistry, edited by W. H. Miller
(Plenum, New York, 1976), Vol. 2, p. 217.82G. J. Atchity, S. S. Xantheas, and K. Ruedenberg, J. Chem. Phys. 95, 1862
(1991).
83D. R. Yarkony, Acc. Chem. Res. 31, 511 (1998).84J. C. Light and T. Carrington, Jr., Adv. Chem. Phys. 114, 263 (2000).85C. Lanczos, J. Res. Natl. Bur. Stand. 49, 33 (1952).86H. Guo, Rev. Comput. Chem. 25, 285 (2007).87H. Guo, Int. Rev. Phys. Chem. 31, 1 (2012).88H. Guo, J. Chem. Phys. 108, 2466 (1998).89R. Chen and H. Guo, J. Chem. Phys. 111, 464 (1999).90H. Lischka, R. Shepard, I. Shavitt, R. Pitzer, M. Dallos, T. Müller, P. G.
Szalay, F. B. Brown, R. Alhrichs, H. J. Böhm, A. Chang, D. C. Comeau,R. Gdanitz, H. Dachsel, C. Erhard, M. Ernzerhof, P. Höchtl, S. Irle,G. Kedziora, T. Kovar, V. Parasuk, M. Pepper, P. Scharf, H. Schiffer,M. Schindler, M. Schüler, and J.-G. Zhao, COLUMBUS, An ab initio elec-tronic structure program, 2003.
91H. Lischka, M. Dallos, P. Szalay, D. R. Yarkony, and R. Shepard, J. Chem.Phys. 120, 7322 (2004).
92M. Dallos, H. Lischka, P. Szalay, R. Shepard, and D. R. Yarkony, J. Chem.Phys. 120, 7330 (2004).
93X. Huang, D. W. Schwenke, and T. J. Lee, J. Chem. Phys. 134, 044320(2011).
94S. N. Yurchenko, R. J. Barber, J. Tennyson, W. Thiel, and P. Jensen, J. Mol.Spectrosc. 268, 123 (2011).
95G. Herzberg, Electronic Spectra and Electronic Structure of PolyatomicMolecules (Van Nostrand Reinhold, New York, 1966).
96S. A. Henck, M. A. Mason, W.-B. Yan, K. K. Lehmann, and S. L. Coy,J. Chem. Phys. 102, 4772 (1995).
97I. Morino and K. J. Kawaguchi, Mol. Spectrosc. 182, 428 (1997).98W. Gabriel, G. Chambaud, P. Rosmus, S. Carter, and N. C. Handy, Mol.
Phys. 81, 1445 (1994).99S. N. Yurchenko, J. Zheng, H. Lin, P. Jensen, and W. Thiel, J. Chem. Phys.
123, 134308 (2005).
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:162.129.250.14 On: Sun, 15 Dec 2013 05:44:01

Copyright and Permission Notice:
Reprinted with permission from J. Chem. Phys. 136, 234301 (2012),
Copyright 2012, AIP Publishing LLC. This article may be downloaded for
personal use only. Any other use requires prior permission of the author and
the AIP Publishing LLC




















