
Colorectal cancers show distinct mutation spectra in members of the canonical WNT signaling pathway...
14
GENES, CHROMOSOMES & CANCER 49:746–759 (2010) Colorectal Cancers Show Distinct Mutation Spectra in Members of the Canonical WNT Signaling Pathway According to Their Anatomical Location and Type of Genetic Instability Cristina Albuquerque, 1 * Ce ´lia Baltazar, 1 Bruno Filipe, 1 Filipa Penha, 1 Teresa Pereira, 2 Ron Smits, 3 Marı ´lia Cravo, 4 Pedro Lage, 4,5 Paulo Fidalgo, 4 Isabel Claro, 4,5 Paula Rodrigues, 5 Isabel Veiga, 6 Jose ´ Silva Ramos, 7 Isabel Fonseca, 2 Carlos Nobre Leita ˜o, 4 and Riccardo Fodde 8 1 Centro de Investigac ¸~ ao de Patobiologia Molecular (CIPM), Instituto Portugu^ es de Oncologia de Lisboa Francisco Gentil, EPE, Lisboa, Portugal 2 Servic ¸o de Anatomia Patolo¤ gica,Instituto Portugu^ es de Oncologia de Lisboa Francisco Gentil, EPE, Lisboa, Portugal 3 Gastroenterology and Hepatology, Erasmus MC University Medical Center,Rotterdam,The Netherlands 4 Servic ¸o de Gastrenterologia, Instituto Portugu^ es de Oncologia de Lisboa Francisco Gentil, EPE, Lisboa, Portugal 5 Cl| nica de Risco Familiar, Instituto Portugu^ es de Oncologia de Lisboa Francisco Gentil, EPE, Lisboa, Portugal 6 Servic ¸o de Gene¤ tica, Instituto Portugu^ es de Oncologia do Porto Francisco Gentil, EPE - Porto, Portugal 7 Servic ¸o de Gastrenterologia, Hospital dos Capuchos, Lisboa, Portugal 8 Department of Pathology, Josephine Nefkens Institute, Erasmus MC,Rotterdam,The Netherlands It is unclear whether the mutation spectra in WNT genes vary among distinct types of colorectal tumors. We have ana- lyzed mutations in specific WNT genes in a cohort of 52 colorectal tumors and performed a meta-analysis of previous studies. Notably, significant differences were found among the mutation spectra. We have previously shown that in familial adenomatous polyposis, APC somatic mutations are selected to provide the ‘‘just-right’’ level of WNT signaling for tumor formation. Here, we found that APC mutations encompassing at least two b-catenin down-regulating motifs (20 a.a. repeats) are significantly more frequent in microsatellite unstable (MSI-H) than in microsatellite stable (MSS) tumors where truncations retaining less than two repeats are more frequent (P ¼ 0.0009). Moreover, in cases where both APC hits are detected, selection for mutations retaining a cumulative number of two 20 a.a. repeats became apparent in MSI-H tumors (P ¼ 0.001). This type of mutations were also more frequent in proximal versus distal colonic tumors, regardless of MSI status (P ¼ 0.0008). Among MSI-H tumors, CTNNB1 mutations were significantly more frequent in HNPCC than in spo- radic lesions (28% versus 6%, P < 10-6) and were preferentially detected in the proximal colon, independently of MSI sta- tus (P ¼ 0.017). In conclusion, the observed spectra of WNT gene mutations in colorectal tumors are likely the result from selection of specific levels of b-catenin signaling, optimal for tumor formation in the context of specific anatomical locations and forms of genetic instability. We suggest that this may underlie the preferential location of MMR deficient tumors in the proximal colon. V V C 2010 Wiley-Liss, Inc. INTRODUCTION The majority of colorectal tumors are thought to be initiated by mutations in the APC (adeno- matous polyposis coli) tumor suppressor gene whose loss of function underlies the transition from the normal polarized intestinal epithelium to dysplastic adenomatous lesion (Fodde et al., 2001). The APC protein encompasses a broad spectrum of functions, ranging from control of the WNT signal transduction pathway, to cell ad- hesion, migration, cell cycle regulation, apoptosis, chromosomal segregation and more recently in cell proliferation and differentiation (Fodde, 2003). Despite its multi-functionality, APC’s main tumor suppressing function resides in its capacity to down-regulate intracellular b-catenin levels along the WNT signal transduction path- way (Fodde et al., 2001). APC, together with the Ser/Thr kinases GSK3b (glycogen synthase ki- nase 3 beta) and CK1 (casein kinase 1), and the scaffold proteins Axin1 and Conductin (Axin2), Supported by: Science and Technology Foundation (FCT), Grant number: POCTI/CBO/47823/2002; Calouste Gulbenkian Foundation. *Correspondence to: Cristina Albuquerque, Centro de Investigac ¸a ˜o de Patobiologia Molecular, Instituto Portugu^ es de Oncologia de Lisboa Francisco Gentil, Rua Prof. Lima Basto 1099-023 Lisboa. E-mail: [email protected] Received 29 November 2009; Accepted 9 April 2010 DOI 10.1002/gcc.20786 Published online 7 May 2010 in Wiley InterScience (www.interscience.wiley.com). V V C 2010 Wiley-Liss, Inc.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Colorectal cancers show distinct mutation spectra in members of the canonical WNT signaling pathway...

GENES, CHROMOSOMES & CANCER 49:746–759 (2010)
Colorectal Cancers Show Distinct Mutation Spectrain Members of the Canonical WNT SignalingPathway According to Their Anatomical Locationand Type of Genetic Instability
Cristina Albuquerque,1* Celia Baltazar,1 Bruno Filipe,1 Filipa Penha,1 Teresa Pereira,2 Ron Smits,3 Marılia Cravo,4
Pedro Lage,4,5 Paulo Fidalgo,4 Isabel Claro,4,5 Paula Rodrigues,5 Isabel Veiga,6 Jose Silva Ramos,7
Isabel Fonseca,2 Carlos Nobre Leitao,4 and Riccardo Fodde8
1Centro de Investigac~ao de Patobiologia Molecular (CIPM),Instituto Portugues de Oncologia de Lisboa Francisco Gentil,EPE,Lisboa,Portugal2Servico de Anatomia Patolo¤ gica,Instituto Portugues de Oncologia de Lisboa Francisco Gentil,EPE,Lisboa,Portugal3Gastroenterologyand Hepatology,Erasmus MCUniversity Medical Center,Rotterdam,The Netherlands4Servico de Gastrenterologia,Instituto Portugues de Oncologia de Lisboa Francisco Gentil,EPE,Lisboa,Portugal5Cl|�nica de Risco Familiar,Instituto Portugues de Oncologia de Lisboa Francisco Gentil,EPE,Lisboa,Portugal6Servico de Gene¤ tica,Instituto Portugues de Oncologia do Porto Francisco Gentil,EPE- Porto,Portugal7Servico de Gastrenterologia,Hospital dos Capuchos,Lisboa,Portugal8Departmentof Pathology,Josephine Nefkens Institute,Erasmus MC,Rotterdam,The Netherlands
It is unclear whether the mutation spectra in WNT genes vary among distinct types of colorectal tumors. We have ana-
lyzed mutations in specific WNT genes in a cohort of 52 colorectal tumors and performed a meta-analysis of previous
studies. Notably, significant differences were found among the mutation spectra. We have previously shown that in familial
adenomatous polyposis, APC somatic mutations are selected to provide the ‘‘just-right’’ level of WNT signaling for tumor
formation. Here, we found that APC mutations encompassing at least two b-catenin down-regulating motifs (20 a.a.
repeats) are significantly more frequent in microsatellite unstable (MSI-H) than in microsatellite stable (MSS) tumors where
truncations retaining less than two repeats are more frequent (P ¼ 0.0009). Moreover, in cases where both APC hits are
detected, selection for mutations retaining a cumulative number of two 20 a.a. repeats became apparent in MSI-H tumors
(P ¼ 0.001). This type of mutations were also more frequent in proximal versus distal colonic tumors, regardless of MSI
status (P ¼ 0.0008). Among MSI-H tumors, CTNNB1 mutations were significantly more frequent in HNPCC than in spo-
radic lesions (28% versus 6%, P < 10-6) and were preferentially detected in the proximal colon, independently of MSI sta-
tus (P ¼ 0.017). In conclusion, the observed spectra of WNT gene mutations in colorectal tumors are likely the result
from selection of specific levels of b-catenin signaling, optimal for tumor formation in the context of specific anatomical
locations and forms of genetic instability. We suggest that this may underlie the preferential location of MMR deficient
tumors in the proximal colon. VVC 2010 Wiley-Liss, Inc.
INTRODUCTION
The majority of colorectal tumors are thought
to be initiated by mutations in the APC (adeno-
matous polyposis coli) tumor suppressor gene
whose loss of function underlies the transition
from the normal polarized intestinal epithelium
to dysplastic adenomatous lesion (Fodde et al.,
2001). The APC protein encompasses a broad
spectrum of functions, ranging from control of
the WNT signal transduction pathway, to cell ad-
hesion, migration, cell cycle regulation, apoptosis,
chromosomal segregation and more recently in
cell proliferation and differentiation (Fodde,
2003). Despite its multi-functionality, APC’s
main tumor suppressing function resides in its
capacity to down-regulate intracellular b-cateninlevels along the WNT signal transduction path-
way (Fodde et al., 2001). APC, together with the
Ser/Thr kinases GSK3b (glycogen synthase ki-
nase 3 beta) and CK1 (casein kinase 1), and the
scaffold proteins Axin1 and Conductin (Axin2),
Supported by: Science and Technology Foundation (FCT),Grant number: POCTI/CBO/47823/2002; Calouste GulbenkianFoundation.
*Correspondence to: Cristina Albuquerque, Centro de Investigacaode Patobiologia Molecular, Instituto Portugues de Oncologia deLisboa Francisco Gentil, Rua Prof. Lima Basto 1099-023 Lisboa.E-mail: [email protected]
Received 29 November 2009; Accepted 9 April 2010
DOI 10.1002/gcc.20786
Published online 7 May 2010 inWiley InterScience (www.interscience.wiley.com).
VVC 2010 Wiley-Liss, Inc.

forms the so-called ‘‘destruction complex’’ that
binds to b-catenin and promotes its down-regula-
tion, thus preventing signaling activity to the
nucleus. In the absence of functional APC, b-cat-enin is not efficiently degraded and it translocates
to the nucleus where, together with distinct
members of T-cell family of transcription factors
(TCFs), modulates expression of several Wnt
downstream target genes (Polakis, 2000) (http://
www.stanford.edu/�rnusse/pathways/targets.html).
Virtually all APC mutations result in truncated
proteins that lack all Axin/Conductin binding
motifs and a variable number of 20 a.a. repeats
associated with the down-regulation of intracellu-
lar b-catenin levels (Miyoshi et al., 1992; Powell
et al., 1993; Miyaki et al., 1994; van der Luijt
et al., 1997; Laurent-Puig et al., 1998). Thus,
APC mutations affect the stability and functional-
ity of the destruction complex leading to intracel-
lular accumulation of b-catenin and constitutive
signaling to the nucleus (Korinek et al., 1997).
APC mutations have been found in the major-
ity of MSS (microsatellite stable) tumors and in
20–50% of MSI-High cancers, both from sporadic
and HNPCC (hereditary nonpolyposis colorectal
cancer) patients carrying germline mutations in
MMR genes (Huang et al., 1996, 2004; Homfray
et al., 1998; Rowan et al., 2000). Colorectal
tumors with an intact APC gene, mostly MSI-H,
often carry oncogenic b-catenin (CTNNB1) muta-
tions which make the resulting protein resistant
to proteolytic degradation (Kitaeva et al., 1997;
Mirabelli-Primdahl et al., 1999; Morin et al.,
1997; Muller et al., 1998; Sparks et al., 1998;
Miyaki et al., 1999; Lovig et al., 2002; Johnson
et al., 2005). Moreover, mutations in other WNT
genes like axin (AXIN1) and conductin (AXIN2)have also been reported in MSI tumors and
shown to result in the intracellular accumulation
of b-catenin (Salahshor et al., 1999; Liu et al., 2000;
Satoh et al., 2000; Shitoh et al., 2001; Shimizu
et al., 2002; Suraweera et al., 2006).
Notably, somatic APC mutations in polyps from
patients affected by familial adenomatous polypo-
sis (FAP) are selected based on their residual
b-catenin regulatory activity (Rowan et al., 2000;
Albuquerque et al., 2002a; Crabtree et al., 2003;
Cho et al., 2006). Likewise, the spectra of muta-
tions in WNT genes may follow specific patterns
depending on the sporadic and hereditary nature
of the colorectal tumors, their MSS and MSI sta-
tus, and their location along the GI tract. To
address these issues in a more systematic fashion,
we have performed somatic mutation analyses of
the APC, CTNNB1, and AXIN2 genes in a cohort
of 52 colorectal tumors stratified by their sporadic
or hereditary nature, MSI status, and anatomical
location. In parallel, we performed a meta analy-
sis by employing the mutation analysis data from
colorectal tumors, stratified by MSI status and an-
atomical location, reported to date in the scien-
tific literature (Aoki et al., 1994; Huang et al.,
1996, 2004; Kitaeva et al., 1997; Olschwang et al.,
1997; Yagi et al., 1997; Homfray et al., 1998; Mul-
ler et al., 1998; Otori et al., 1998; Miyaki et al.,
1999; Salahshor et al., 1999; Rowan et al., 2000;
Shitoh et al., 2001; Aust et al., 2002; Lovig et al.,
2002; Shimizu et al., 2002; Smith et al., 2002;
Miyaki et al., 2003; Giaretti et al., 2004; Johnson
et al., 2005; Suraweera et al., 2006).
MATERIALS AND METHODS
Patients and Tumor Samples
Thirty patients with sporadic colorectal cancer
(CRC) and seventeen patients with HNPCC
were included in this study. All sporadic CRC
patients had no known familial history suggestive
of HNPCC or FAP. All HNPCC patients either
belonged to families fulfilling the Amsterdam cri-
teria for HNPCC classification or carry previously
identified germline mutations in DNA mismatch
repair (MMR) genes (Table 2).
Fifty-two colorectal neoplastic lesions were
included: 20 sporadic MSS carcinomas, 10 spo-
radic MSI-H carcinomas, and 12 carcinomas and
10 adenomas from HNPCC patients.
Fresh colorectal adenomas and carcinomas
were obtained from surgical or colectomy speci-
mens from patients who underwent surgery or
colonoscopy in the Portuguese Institute of Oncol-
ogy Francisco Gentil in Lisbon. Sections from
corresponding areas from the specimens submit-
ted for diagnosis were cut in two parts: one was
snap-frozen in liquid nitrogen immediately after
resection until processing for DNA isolation,
while the other was formalin-fixed and paraffin-
embedded for IHC analysis. Dysplasia grading, tu-
mor differentiation, according to the WHO guide-
lines, and the immunohistochemical analysis were
performed by an experienced pathologist (IF).
Moreover, a systematic review of the literature
using Pubmed and the Catalogue of Somatic
Mutations in Cancer of the Sanger Institute was
conducted and a meta-analysis of previous studies
reporting APC, CTNNB1, or AXIN2 mutations was
performed by evaluating the frequency and type
MUTATION SPECTRA IN WNT GENES 747
Genes, Chromosomes & Cancer DOI 10.1002/gcc

of the mutations according to the MSI status,
sporadic or hereditary nature, and anatomical
location along the colon and rectum.
DNA Isolation
Genomic DNA was extracted from each tumor
sample and matched normal colonic mucosa as
previously described (Albuquerque et al., 2002a).
In some cases, total genomic DNA was isolated
from blood by using a commercial kit (Puregene,
Gentra Systems).
Microsatellite Instability Analysis
Microsatellite instability (MSI) was analyzed
using the Bethesda microsatellite markers (Rodri-
guez-Bigas et al., 1997): D2S123, D5S346, D17S250,BAT-25, and BAT-26. Each tumor and paired nor-
mal DNA was amplified by PCR and analyzed by
denaturing polyacrylamide gel electrophoresis as
previously described (Albuquerque et al., 2002b).
Analysis of APC Allelic Loss
Allelic loss at the APC locus was analyzed using
microsatellite markers on chromosome arm 5q
selected from the Human Genome Database
based on their proximity to APC, their degree of
heterozygosity, and size range of the alleles. The
microsatellite markers D5S346, D5S656, D5S421,and D5S1965, were analyzed with the Applied
Biosystems ABI PRISM 310 genetic analyzer
using the GENESCAN software. For each
marker an allelic ratio was calculated by dividing
the intensity of the two alleles in normal control
DNA. This allelic ratio was compared with the
allelic ratio obtained for the two alleles of each
tumor sample. A comparative ratio greater than
1.5 was interpreted as significant, i.e., indicative
of allelic loss.
APC Mutation Analysis
APC mutation analysis was performed using
the protein truncation test (PTT) for exon 15,
and denaturing gradient gel electrophoresis
(DGGE) for exons 1-14. PTT was performed as
previously reported (Albuquerque et al., 2002a).
DGGE was performed as previously described
(Fodde et al., 1992; Olschwang et al., 1993)
except that electrophoresis was performed at
160 V in TAE buffer (40 mM Tris-acetate pH 7.5,
20 mM sodium acetate, 1 mM Na2EDTA) at 60�Cduring 4 hr.
CTNNB1 Mutation Analysis
Genomic DNA from each tumor sample was
amplified by PCR for SSCP (single strand confor-
mational polymorphism) analysis of exon 3 of the
CTNNB1 gene using previously described primers
(Koch et al., 1999). The amplified products were
analyzed in a MDE (mutation detection enhance-
ment) gel (FMC) and visualized by silver stain-
ing. The analysis of large deletions encompassing
exon 3 of the CTNNB1 gene was performed as
previously described (Koch et al., 1999). PCR
products were analyzed in a 1.2% agarose gel,
stained with ethidium bromide.
AXIN2 Mutation Analysis
Specific primers were designed to amplify the
region containing the (G)7, (C)6, and (C)5 tracts
in the AXIN2 gene, where a high incidence of
mutations was previously reported (Liu et al.,
2000). The primers used were as follows: AXIN2
F- 50-AGTCTGCCCGCTCGTCTCCA-30 and
AXIN2 R- 50-GTCAGGGGAGGCATCGCAGG-30.The amplified fragments were electrophoresed in a
7% polyacrilamide gel containing 32.5% formamide
and 6.9 M urea at 48W and visualized by silver
staining.
Sequencing Analysis
All mutations detected in the WNT genes,
were sequenced using the BigDye terminator
cycle sequencing kit (Applied Biosystems) and
analyzed on the ABI PRISM 310 genetic analyzer
(Applied Biosystems) using the SEQUENCING
ANALYSIS software.
Immunohistochemical Analysis
b-catenin, MSH2, MLH1 and MSH6 expres-
sion was evaluated using immunohistochemistry
(IHC) as previously described (Albuquerque
et al., 2002b). Negative and positive controls
were used to score for the presence/absence of
b-catenin expression, and to evaluate its relative
intensity (scored as either negative, weak/focal
(1þ), moderate (2þ) or strong/diffuse (3þ)) and
subcellular location (classified as membrane-
bound, cytoplasmic and nuclear). Controls were
analyzed simultaneously with the tumor samples.
Statistical Analysis
Differences in mutation frequencies among
distinct groups of colorectal tumors, stratified by
748 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

MSI status and anatomical location, were eval-
uated by the Fisher’s exact test.
RESULTS
Mutation analysis of the APC, CTNNB1,
and AXIN2 Genes
Tables 1 and 2 summarize the results of the
mutation, MSI, and immunohistochemistry analyses
of our CRC cohort in relation to tumor location
along the GI tract, histological type, and, in the
case of HNPCC tumors, MMR germline mutation.
We have detected mutations in APC, CTNNB1(b-catenin) or AXIN2 in most colorectal tumors
irrespective of their MSI status and sporadic or
hereditary nature. Overall, mutations in these
genes were found in 85% (17/20) of sporadic
MSS carcinomas, 60% (6/10) of sporadic MSI-H
carcinomas, and in 73% (16/22) of the HNPCC
tumors (both adenomas and carcinomas). Immuno-
histochemistry (IHC) analysis of intracellular
b-catenin accumulation underscored the central
role played by constitutive activation of the
WNT/b-catenin signaling pathway in CRC,
regardless of their MSI status and sporadic or he-
reditary nature. In contrast with its membrane-
bound distribution in normal colonic epithelium,
b-catenin was detected in the cytoplasm and/or in
the nucleus of the vast majority (32/36; 89%) of
the tumors carrying APC, CTNNB1, or AXIN2
TABLE 1. Results of the Mutation Analysis of APC, CTNNB1 and AXIN2 in Sporadic Colorectal Carcinomas Stratifiedfor MSI Status, Anatomical Location, and Histo-Pathological Classification
Carcinomas Histology Location
APC
CTNNB1 AXIN2Point mutation (codon
affected and type of change) Allelic loss
MSS39 Md Ascending N N þ (partial del of exon 3) N15 Md Splenic flexure G1303X C!T N N N61 Md Descending 1300 insA (TAAATAC) N N N47 Md Descending R1114X C!T þ N N51 Muc Descending E1309X G!T NI N N7 Md Sigmoid 903 del2bp N N N27 Md Sigmoid 1309 delAAAGA þ N N29 Wd Sigmoid N N N N37 Md Sigmoid 785 insTC (TCTCTCAT);
Q1367X C!TN N N
55 Wd Sigmoid N N N N57 Md Sigmoid N þ N N59 Md Sigmoid N N N N63 Md Sigmoid R1450X C!T N N N65 Wd Sigmoid E1451X G!T N N N69 Wd Sigmoid E1397X G!T N N N25 Md Rectum E868X G!T N N N31 Md Rectum R232X, C!T N N N33 Wd Rectum R1114X C!T þ N N35 Wd Rectum R232X C!T; 1317
delG (GCTGAA)N N N
53 Wd Rectum IVS 833-6 A!G (splice site) NI N NMSI-H
E-425T Md Caecum N NI S45F N45 Wd Ascending G1477X C!T NI N N97 Pd Ascending N NI N ins G139 Wd, muc Ascending 1495 a) NI N NE-443T Wd Ascending R876X C!T; R1450X C!T NI N N49 Wd, muc Ascending N NI N N5 Wd Descending N NI N N83 UN UN N NI N N3 Wd Rectum N N N NE-429T Wd Rectum R1450X C!T NI N N
Wd, well differentiated; md, moderately differentiated; pd, poor differentiated; muc, mucinous; N, normal; NI, not informative (due to homozigozity
and/or to MSI); þ, positive; ins, insertion; del, deletion; UN, unknown.aNot sequenced due to technical reasons.
MUTATION SPECTRA IN WNT GENES 749
Genes, Chromosomes & Cancer DOI 10.1002/gcc

mutations with IHC patterns ranging from weak/
focal (1þ) to strong/diffuse (3þ) (Table 3). Overall,
intracellular b-catenin accumulation was detected
in the majority of colorectal tumors analyzed,
namely in 100% (20/20) of sporadic MSS tumors,
89% (8/9) of the sporadic MSI-H tumors, and in
60% (6/10) and 78% (7/9) of the HNPCC adenomas
and carcinomas, respectively. Accordingly, a meta-
analysis of previously published data shows that
mutations in WNT genes are detected in the vast
majority of CRC (�90%), even in MSI-H CRC
(Table 4).
However, differences were observed in the fre-
quency and in the type of mutations in the WNT
genes according to the MSI status, sporadic or here-
ditary nature and to the anatomical location.
Association of Somatic WNT Gene Mutations
With MSI and Sporadic/Hereditary Status
Mutation frequencies
Differences in the WNT gene mutation fre-
quencies were found between MSS and MSI-H
tumors and, among the latter, between sporadic
TABLE 2. Results of the Mutation Analysis of APC, CTNNB1 and AXIN2 in HNPCC Tumors in Relation to MMR GermlineMutation and/or Loss of Tumor-Specific Expression, MSI Status, Anatomical Location, and Histological Classification
Tumors Location Histology
MMR germlinemutation or
expression deficiency
APC
CTNNB1 AXIN2
Point mutation(codon and
type of change)Allloss
CarcinomasMSI-H
13 Caecum md MLH1 E721X, T!A N NI N NT7 Caecum md MLH1 IVS1-11 T!A splic. N N S45P N287T Ascending md in course N NI S45F N639 Ascending md MSH2 R236X, C->T N N N N280T1 Hepatic flexure pd, muc MSH2 IVS1277-2 A!C splic. N NI N ins G101 Transverse pd, muc MLH1 R26X, C!T N N S45F N287T1 Transverse wd in course N N S45F N325 Rectum md, muc MSH2 849 insA (AAAAAA);
G1450X C!TNI N N
T16 Rectum UN in course R1114X C!T;1593 insA(AAAAAAA)
NP N N
HO1 UN UN NP R876X C!T;1455 delA(AAAAA)
NP N N
MSSL183T3 Splenic flexure muc MLH1 1976 G!T splic. N N N NL184T4 Splenic flexure muc MLH1 1976 G!T splic. N N N N
AdenomasMSI-H
287A2 Ascending TA, LGD MLH1 1þ <50% N N N N429A1 Transverse TA, LGD MLH1 1þ 90% N NI T41A N429A2 Transverse TA, LGD NP N N T41A NL397A Sigmoid TA, HGD MSH2 R236X, C->T N NI N del GL440A Rectum TA, LGD/HGD MSH2 2þ �20% N NI N N
MSS323A1 Ascending TA, LGD Normal expression G1429X C!T þ N NL447A Ascending TA, LGD/HGD MSH6 3þ �40% 997 delT (AGTTAT);
1494 del16 bpN N N
375A Sigmoid TA, LGD MSH2 2þ �80% R216X C!T þ N NL459A Sigmoid TA, LGD MSH2 2þ �20% R876X C!T;
1310 delAA(AGAAAAG)
N N N
L280A Sigmoid TA, LGD MSH2 - 1182 delA (AAAA);1420 delC(CCCC)
N N N
All loss, allelic loss; wd, well differentiated; md, moderately differentiated; pd, poor differentiated; muc, mucinous; TA, tubular adenoma; LGD, low
grade dysplasia; HGD, high grade dysplasia; N, normal; NI, not informative (due to homozygozity or to MSI); UN, unknown; NP, not performed; þ,
positive; ins, insertion; del, deletion; splic., splice site mutation. L183T3, L183T4 and L180A tumors were considered MMR mutant.
750 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

TABLE 3. Results of b-Catenin IHC Analysis in Sporadic and HNPCC Tumors
Tumor
Sporadic tumors HNPCC tumors
b-catenin expression b-catenin expression
Nucleus Cytoplasm Membrane Nucleus Cytoplasm Membrane
Carcinomas CarcinomasMSS MSI-H
39 3þ 3þ 1þ 13 – 2þ (50%) 3þ15 2þ 2þ – T7 – 1þ 3þ61 3þ (25%) 3þ 2þ 287T 2þ (50%) 3þ 3þ47 2þ 3þ – 639 – – 3þ51 2þ (rare) 2þ – 280T1 – 1þ 1þ7 2þ 3þ 1þ 101 3þ 3þ –27 – 2þ 2þ 287T1 NP NP NP29 3þ (10%) 3þ 1þ 325 – 2þ 3þ37 3þ (10%) 3þ 1þ T16 – – 3þ55 3þ (<50%) 3þ – HO1 NP NP NP57 3þ (20%) 3þ –59 – 2þ 3þ MSS63 2þ 3þ 1þ L183T3 2þ 3þ –65 3þ (10%) 3þ 2þ L184T4 NP NP NP69 3þ (20%) 2þ –25 3þ (20%) 3þ 1þ Adenomas31 – 2þ – MSI–H33 – 2þ 2þ 287A2 3þ 2þ –35 – 2þ 2þ 429A1 – 1þ 3þ53 3þ 3þ – 429A2 – 2þ 3þ
L397A – 2þ 2þMSI-H L440A – – 3þ
E-425T 3þ (50%) 3þ 2þ45 2þ (20%) 3þ 2þ MSS97 – 2þ 1þ 323A1 – – 3þ139 3þ (30%) 3þ 2þ L447A 1þ (focal) 1þ (focal) 2þ
E-443T – 3þ 1þ L459A – – 3þ49 – – 2þ 375A – 1þ 3þ5 – 1þ 2þ L280A – – 3þ3 3þ 3þ 1þE-429T 3þ 3þ 1þ83 NP NP NP
NP, not performed. b-catenin expression was scored as negative (-); weak (1þ); moderate (2þ) and intense (3þ). The percentages reported next
to a subset of scores indicate the proportion of tumor cells presenting with that particular score.
TABLE 4. Results of the Mutation Analysis of MSI-H Carcinomas, Comprising the Present and Previously Published Studies
Tumors
Mutations
APC CTNNB1 AXIN2 AXIN1
Sporadic MSI-HPresent study 4/10 (40%) 1/10 (10%) 1/10 (10%) –Previous studies (a) 64/144 (44%) 9/161 (6%) 4/14 (28%) 6/26 (23%)
Total 68/154 (44%) 10/171 (6%) 5/24 (20%) 6/26 (23%) � 93% WNT mutationsHNPCC MMR-mutant
Present study 3/12 (25%) 4/12 (33%) 1/12 (8%) –Previous studies (a) 30/72 (42%) 23/85 (27%) – 1/7 (14%)
Total 33/84 (39%) 27/97 (28%) 1/12 (8%) 1/7 (14%) � 89% WNT mutations
(a) Mutation data reported by Huang et al. (1996, 2004), Homfray et al. (1996), Kitaeva et al. (1997), Muller et al. (1998), Miyaki et al. (1999), Sal-
ahshor et al. (1999), Shitoh et al. (2001), Shimizu et al. (2002), Lovig et al. (2002), Johnson et al. (2005) and Suraweera et al. (2006). Despite the
extensive mutational data available in the literature for MSI-H tumors, these studies were selected based on their separate analysis of sporadic
MSI-H and HNPCC MMR-mutant tumors.
MUTATION SPECTRA IN WNT GENES 751
Genes, Chromosomes & Cancer DOI 10.1002/gcc

and HNPCC MMR deficient tumors. As
expected, APC loss of function defects, either
point mutations and/or allelic imbalance, were
significantly more frequent among sporadic MSS
carcinomas (16/20; 80%) than in sporadic MSI-H
carcinomas (4/10; 40%, P ¼ 0.034) and HNPCC
MMR mutant carcinomas and adenomas (4/18;
22%, P ¼ 0.00044). Likewise, CTNNB1 and
AXIN2 mutations were only detected in sporadic
MSI-H and HNPCC tumors, except for one
CTNNB1 deletion detected in 1/20 sporadic MSS
carcinomas. Similarly, whereas APC mutations
were detected in all HNPCC MSS adenomas (4/
4), only one was found among the six MSI-H/
MMR-deficient adenomas where, instead, two
CTNNB1 and one AXIN2 mutations were identi-
fied (Tables 1 and 2).
Among the MSI-H tumors, CTNNB1 mutations
appeared to be more frequent in HNPCC MMR
deficient than in sporadic MSI-H carcinomas (4/
12 versus 1/10). A subsequent CTNNB1 mutation
analysis of a series of paraffin embedded carcino-
mas (7 sporadic MSI-H and 13 HNPCC cases)
confirmed this trend: mutations were found in
14% (1/7) and 31% (4/13) of sporadic MSI-H and
HNPCC carcinomas, respectively (data not
shown). However, APC and AXIN2 mutation fre-
quencies did not appear to differ between spo-
radic and HNPCC MMR deficient carcinomas [4/
10 (40%) and 1/10 (10%) versus 3/12 (25%) and 1/
12 (8%), respectively] (Tables 1 and 2). A meta-
analysis of previous studies merged with the
results presented here confirmed that APC muta-
tion frequency does not differ between HNPCC
MMR deficient and sporadic MSI-H carcinomas
[33/84 (39%) versus 68/154 (44%)]. In contrast,
CTNNB1 mutations are significantly more fre-
quent in the HNPCC MMR-mutant cancers
when compared with sporadic MSI-H carcinomas
[27/97 (28%) versus 10/171 (6%), P < 10�6]
(Table 4). Notably, the frequencies of CTNNB1mutations appear to vary with the disease-causing
MMR gene (Tables 1 and 2): CTNNB1 mutations
were more frequently detected in tumors from
patients carrying germline MLH1 mutations than
in tumors associated to MSH2 germline defects
(4/4 versus 0/4, P ¼ 0.014, Fisher’s exact test).
The latter presented somatic mutations either in
APC (2/4) or in AXIN2 (2/4).
Type of mutations
In polyps from FAP patients, somatic APCmutations are known to be selected based on the
retention of different numbers of b-catenindown-regulating motifs (20 a.a. repeats) in the
corresponding truncated proteins (Rowan et al.,
2000; Albuquerque et al., 2002a; Crabtree et al.,
2003; Cho et al., 2006). A similar selection bias
seems to operate in our cohort of sporadic and
HNPCC colorectal tumors (Table 5 and Fig. 1).
APC mutations located 30 to codon 1397 (resulting
in truncated proteins retaining at least two 20 a.a.
repeats) were significantly more frequent among
sporadic MSI-H and HNPCC MMR-mutant
tumors (8/13; 62%) than in sporadic MSS tumors
(3/17; 18%) (P ¼ 0.016). In the latter group, APCmutations encompassing one or no 20 a.a. repeats
were prevalent (14/17; 82%). A meta-analysis of
previous studies was performed by evaluating
previously reported mutation data with respect to
the resulting number of 20 a.a. repeats. The out-
come of the meta-analysis were then merged
with the results obtained in this study which con-
firmed that APC mutations retaining two or more
20 a.a. repeats were significantly more frequent
among MSI-H tumors, either HNPCC or sporadic
(50/98; 51%), than among MSS tumors (105/311;
34%) (P ¼ 0.0009) (Table 5). Likewise, APCmutations retaining one or none 20 a.a. repeat are
prevalent among MSS tumors (206/311; 66%).
Moreover, the relative incidence of APC muta-
tions retaining one 20 a.a. repeat is clearly lower
than those encompassing 2 of the same motifs
among HNPCC and sporadic MSI-H tumors [18/
98 (18%) versus 50/98 (51%)]. In contrast, among
sporadic MSS tumors these two mutation types
are equally frequent [114/311 (37%) versus 105/
311 (34%), P ¼ 0.0001] (Table 5).
A more detailed analysis of the tumors from
our series where two independent APC mutations
were detected indicates that HNPCC MMR-mu-
tant and sporadic MSI-H tumors may differ from
sporadic MSS tumors in the type of interdepend-
ence between the two somatic hits. In the
HNPCC/sporadic MSI-H group, allelic losses or
mutations leading to truncated proteins lacking
all 20 a.a. motifs were frequently associated with
mutations encompassing 2 or more 20 a.a. repeats
(5/5 tumors). The latter differs from the results
obtained in sporadic MSS tumors where APCallelic losses or mutations leading to truncated
proteins lacking all 20 a.a. repeats are invariably
found in association with mutations that result in
truncated proteins with one or none 20 a.a. repeat
(4/5 tumors, P ¼ 0.023). By combining our results
with the meta-analysis data, selection for two in-
dependent mutations retaining in total at least
752 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

Figure 1. Schematic representation of the distribution of somaticmutations detected in sporadic MSS, sporadic MSI-H and HNPCC MMR-mutant tumors along the APC protein. APC mutations are depictedaccording to the number of 20 a.a. repeats remaining in the resultingtruncated protein. Mutations retaining two 20 a.a. repeats [orange (orgrey, in black and white version) bars] are significantly more frequent in
sporadic MSI-H and HNPCC MMR-mutant tumors (represented abovethe APC protein) than in sporadic MSS tumors (represented below theAPC protein) (62% versus 18%). In the latter, mutations retaining onlyone 20 a.a. repeat [blue (or black, in black and white version) bars] aremuch more frequent. [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
TABLE 5. Distribution of APC Mutations in Sporadic MSS and Sporadic/HNPCC MSI-H Tumors Accordingto the Number of 20 a.a. Repeats Retained by APC Truncating Mutations
Number of 20 a.a. repeats retainedby each APC truncating mutation
Cumulative number of 20 a.a.repeats retained by APCmutations in tumors withtwo independent hits
Sporadic MSSSporadic/HNPCCMMR-MUTANT
SporadicMSS
Sporadic/HNPCCMSI-H
0 1 �2 0 1 �2 �1 �2 �1 �2
This study Sporadic MSI-H/HNPCCMMR-mutant
8 6 3 5 0 8 4 1 0 5
Olschwanget al. (1997)
Sporadic MSSCRC
36 17 20 – – – 19 20 – –
Huang et al.(1996)
Sporadic MSSand MSI-H
18 18 28 13 4 20 0 6 2 6
Homfrayet al. (1998)
Sporadic MSSand MSI-H andHNPCC MSI-H
4 14 11 4 13 3 2 4 1 8
Rowan et al.(2000)
MSS and MSI-HCRC cell lines
10 7 8 2 0 7 5 14 0 6
Lovig et al.(2002)
Sporadic MSSand MSI-H
16 52 35 1 0 6 3 4 – –
Huang et al.(2004)
HNPCCMSI-H
– – – 5 1 6 – – 0 2
All studies 92 114* 105* 30 18* 50* 33** 49** 3** 27**(30%) (37%) (34%) (31%) (18%) (51%) (40%) (60%) (10%) (90%)
206***(66%)
105***(34%)
48***(49%)
50***(51%)
MSS, microsatellite stable; MSI-H, microsatellite instability; CRC, colorectal carcinomas.
*P ¼ 0.0001; **P ¼ 0.001; ***P ¼ 0.0009.
MUTATION SPECTRA IN WNT GENES 753
Genes, Chromosomes & Cancer DOI 10.1002/gcc
two 20 a.a. repeats was observed in 27/30 (90%)
of the HNPCC and sporadic MSI-H tumors. This
APC mutation spectrum differs from what is
observed among sporadic MSS tumors where the
genotypes resulting in two or more 20 a.a. repeats
or in one or none 20 a.a. repeat are represented
in a 60:40 ratio [49/82 (60%) versus 33/82 (40%),
respectively, (P ¼ 0.001)] (Table 5). This sug-
gests a specific selection bias for truncated APC
proteins encoding for some residual b-catenindown-regulating activity among sporadic MSI-H
and HNPCC MMR deficient tumors (selection
for two or more 20 a.a. repeats), in contrast with
MSS cases.
With respect to the specific type of APC nucle-
otide substitutions, C-T transitions were exclu-
sively observed among HNPCC MMR deficient
and sporadic MSI-H tumors. The latter is likely
to represent a feature of loss of MMR function as
also indicated by the lower incidence of C-T
transitions (10/10 versus 7/11; P ¼ 0.055, Fisher’s
exact test) and the presence of G-T transversions
among MSS tumors (4/11; 36%). As expected,
among all APC point mutations here detected,
1-2 bp insertions/deletions within mono or dinu-
cleotide repeats were more frequently observed
among HNPCC MMR-mutant (5/8; 62%) tumors
when compared with sporadic MSI-H (0/4; P ¼0.07, Fisher’s exact test) and MSS (2/17, 12%; P¼ 0.016, Fisher’s exact test) cases.
Association of Somatic WNT Gene Mutations
With Anatomical Location
Besides the observed differences in the WNT
gene mutation spectra according to MSI status,
we also observed differences according to tumor
location along the colon and rectum, independ-
ently of MSI status. Accordingly, we included in
this analysis only the sporadic and HNPCC MSS
tumors from our cohort and merged them with
the results from the meta-analysis of sporadic
MSS tumors and consecutive series of sporadic
adenomas or carcinomas (among sporadic carcino-
mas, MSI frequency is usually lower than 15%,
thus lowering significantly the bias introduced by
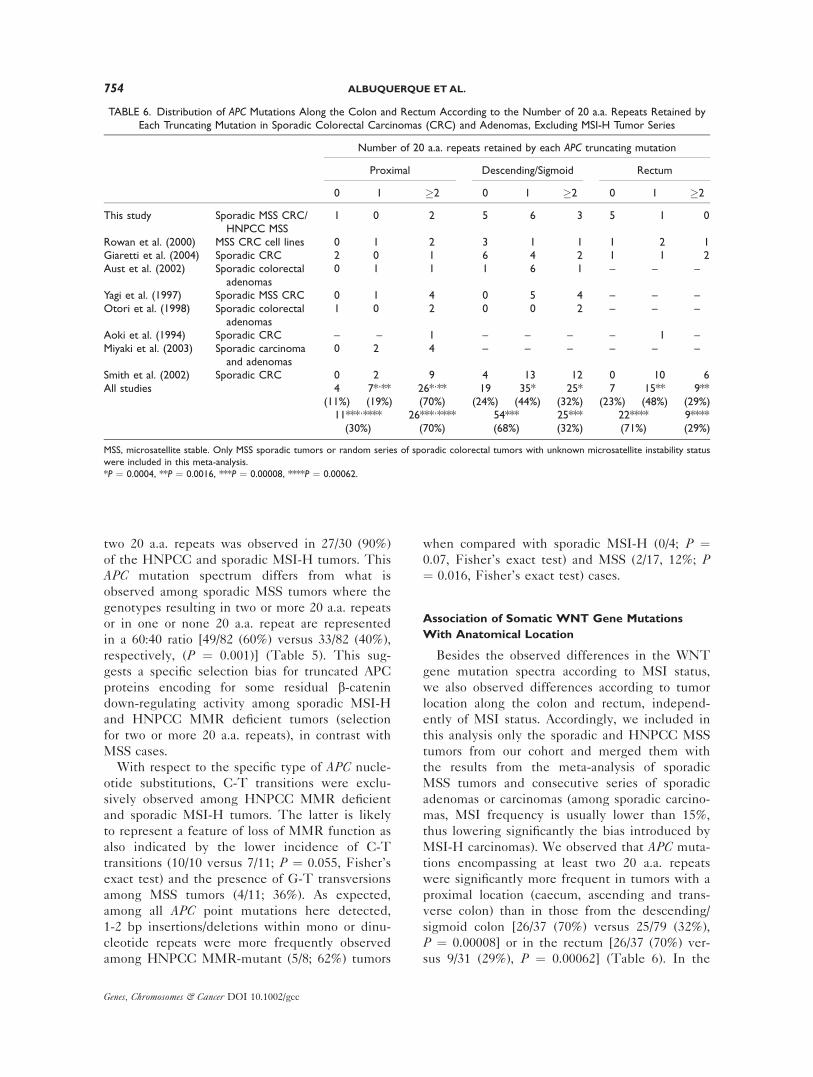
MSI-H carcinomas). We observed that APC muta-
tions encompassing at least two 20 a.a. repeats
were significantly more frequent in tumors with a
proximal location (caecum, ascending and trans-
verse colon) than in those from the descending/
sigmoid colon [26/37 (70%) versus 25/79 (32%),
P ¼ 0.00008] or in the rectum [26/37 (70%) ver-
sus 9/31 (29%), P ¼ 0.00062] (Table 6). In the
TABLE 6. Distribution of APC Mutations Along the Colon and Rectum According to the Number of 20 a.a. Repeats Retained byEach Truncating Mutation in Sporadic Colorectal Carcinomas (CRC) and Adenomas, Excluding MSI-H Tumor Series
Number of 20 a.a. repeats retained by each APC truncating mutation
Proximal Descending/Sigmoid Rectum
0 1 �2 0 1 �2 0 1 �2
This study Sporadic MSS CRC/HNPCC MSS
1 0 2 5 6 3 5 1 0
Rowan et al. (2000) MSS CRC cell lines 0 1 2 3 1 1 1 2 1Giaretti et al. (2004) Sporadic CRC 2 0 1 6 4 2 1 1 2Aust et al. (2002) Sporadic colorectal
adenomas0 1 1 1 6 1 – – –
Yagi et al. (1997) Sporadic MSS CRC 0 1 4 0 5 4 – – –Otori et al. (1998) Sporadic colorectal
adenomas1 0 2 0 0 2 – – –
Aoki et al. (1994) Sporadic CRC – – 1 – – – – 1 –Miyaki et al. (2003) Sporadic carcinoma
and adenomas0 2 4 – – – – – –
Smith et al. (2002) Sporadic CRC 0 2 9 4 13 12 0 10 6All studies 4 7*,** 26*,** 19 35* 25* 7 15** 9**
(11%) (19%) (70%) (24%) (44%) (32%) (23%) (48%) (29%)11***,**** 26***,**** 54*** 25*** 22**** 9****(30%) (70%) (68%) (32%) (71%) (29%)
MSS, microsatellite stable. Only MSS sporadic tumors or random series of sporadic colorectal tumors with unknown microsatellite instability status
were included in this meta-analysis.
*P ¼ 0.0004, **P ¼ 0.0016, ***P ¼ 0.00008, ****P ¼ 0.00062.
754 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

descending/sigmoid colon and in the rectum,
truncating proteins encompassing either one or
none of the 20 a.a. repeats were more frequent
[54/79 (68%) and 22/31 (71%), respectively], with a
preference for one 20 a.a. repeat with respect to
none [35/79 (44%) versus 19/79 (24%) in the de-
scending/sigmoid colon and 15/31 (48%) versus 7/
31 (23%) in the rectum]. The analysis of the com-
bination of both APC hits with respect to anatomi-
cal location was not possible because of the lack of
information concerning tumor location in the ma-
jority of studies where both hits were identified.
The incidence of CTNNB1 mutations among spo-
radic MSI-H and HNPCC MMR deficient groups
also varied with the location of the tumor along
the colorectal tract (Tables 1 and 2). We detected
CTNNB1 mutations in 7/16 (44%) of the proximal
tumors but in none of the 10 distal or rectal tumors
(P ¼ 0.017).
DISCUSSION
It is generally accepted that constitutional acti-
vation of the WNT signaling pathway is a rate-
limiting step for colorectal tumor formation (Pola-
kis, 2000; Fodde et al., 2001; Fodde, 2003).
Although several studies have previously reported
mutational analysis of WNT genes in sporadic
MSS, MSI-H, and HNPCC tumors, most did not
address HNPCC and sporadic MSI-H tumors as
distinct entities and the relative role of the ana-
tomical location of the tumors. Here, we have
carried out a thorough mutation analysis of the
APC, CTNNB1, and AXIN2 genes in a cohort of
52 colorectal tumors comprehensive of sporadic
MSS and MSI-H carcinomas, and of HNPCC
adenomas and carcinomas, together with the IHC
analysis of b-catenin and the main MMR genes.
Overall, mutations in APC, CTNNB1, or AXIN2genes were found in the vast majority of tumors
of the present cohort, thus underscoring the cen-
tral role of the WNT signaling pathway not only
in MSS tumors but also in both hereditary and
sporadic MMR-deficient colorectal tumors, as also
validated by the presence of b-catenin intracellu-
lar accumulation in most HNPCC (13/19; 68%)
and sporadic MSI-H (8/9; 89%) tumors (Table 3).
In tumors characterized by intracellular b-cateninaccumulation, but without APC, CTNNB1 or
AXIN2 mutations, alternative (epi)genetic defects
in additional WNT genes not analyzed in our
study are likely to underlie tumor formation and
progression.
In parallel with the above mentioned mutation
data, we used the considerable body of mutation
analysis in colorectal tumors, stratified by MSI
status, hereditary nature and by anatomical loca-
tion, reported to date in the scientific literature.
As shown in Table 4, the overall incidence of
mutations in WNT genes in either sporadic or
HNPCC MSI-H groups adds up to approximately
90% of the total cases studied. This high fre-
quency among sporadic MSI-H tumors is similar
to that of MLH1 hypermethylation and BRAF on-
cogenic mutations (Higuchi and Jass, 2004).
Hence, in sporadic MSI-H tumors, genetic and
epigenetic mutation events at these genes are
likely to coexist in the same tumors. Accordingly,
in our cohort we detected the V600E BRAFmutation in 4/10 sporadic MSI-H tumors (data
not shown), two of which carry APC and AXIN2mutations, respectively. The same is true for the
G13D and G12D K-RAS oncogenic mutations
found in 3/10 sporadic MSI-H tumors (data not
shown), two of which also carry APC mutations.
MLH1 hypermethylation was also detected in
8/10 tumors, five of which carry APC (three
cases), CTNNB1 and AXIN2 mutations.
Notably, in addition to the high contribution of
WNT gene mutations in CRC irrespective of
MSI status, the WNT mutation spectra revealed
significant differences with respect to the fre-
quency and type of mutations according to the
type of genetic instability, hereditary nature and
tumor location. The analysis of the WNT genes
mutation spectra, combining the meta-analysis
with our results, confirmed that, whereas sporadic
MSS tumors mainly carry APC defects (80%),
HNPCC and sporadic MSI-H lesions revealed a
more heterogeneous spectrum with CTNNB1 and
AXIN2 mutations and with a lower frequency of
APC mutations (40%). However, the latter two
groups differ in the relative frequency of
CTNNB1 mutations with a significantly increased
incidence in HNPCC MMR deficient than in
sporadic MSI-H tumors (28% versus 6%, respec-
tively) (Table 4).
A more detailed analysis of the WNT genes
mutation spectra shows that the distribution of
APC mutations also differs between MSS and
MSI-H tumors. This observation is only partly
explained by the non-random distribution of short
repeats along the APC coding region, known to
represent preferential mutation targets in MMR-
deficient cells. Of note, we found a significant
increase in the incidence of mutations leading to
truncating proteins retaining two 20 a.a. repeats
MUTATION SPECTRA IN WNT GENES 755
Genes, Chromosomes & Cancer DOI 10.1002/gcc

(51%) among the MSI-H sporadic/HNPCC cases
when compared with mutations encompassing
one or none repeat (18% and 31%, respectively).
This was in sharp contrast with MSS tumors
where truncating proteins encompassing none,
one or two 20 a.a. repeats are more equally dis-
tributed (30%, 37% and 34%, respectively) (P ¼0.0009) (Table 5). The difference between trun-
cating proteins retaining two and one 20 a.a.
repeat (respectively, 51% versus 18% in the spo-
radic/HNPCC MSI-H group and 34% versus 37%
in the MSS group, P ¼ 0.0001) further highlights
selection for two, instead of one, 20 a.a. repeats
in the MSI-H group. Moreover, this difference in
APC mutation spectra between sporadic MSS and
sporadic/HNPCC MSI-H tumors is confirmed
when looking at the interdependence between
two somatic APC hits in the same tumor. Accord-
ingly, the preference for two 20 a.a. repeats as a
result of the combination of both APC hits is evi-
dent in the sporadic/HNPCC MSI-H group (90%
versus 10%), when compared with the MSS cases
(60% versus 40%, P ¼ 0.001) (Table 5).
Notably, truncating mutations retaining two 20
a.a. repeats are also more frequent in proximal
than in descending/sigmoid tumors (70% versus
32%, P ¼ 0.00008) (Table 6). The higher fre-
quency of this type of APC mutations among
proximal colonic tumors could result from the
preferential proximal location of sporadic MSI-H
and HNPCC tumors and from the non-random
distribution of mono and dinucleotide repeats,
more prone to MMR errors, in the proximity of
codons 1,400–1,500. However, this should not
play any major confounding role in our data set
as we included only MSS tumors in addition to
sporadic adenomas where MSI is rarely detected,
and sporadic carcinomas where the frequency of
MSI-H cases is approximately 15%. Accordingly,
CTNNB1 mutations were significantly associated
with a proximal location among MSI-H tumors
from the present cohort (P ¼ 0.017) which further
suggests that distinct WNT gene mutation spec-
tra are selected along the different segments of
the colon. A schematic representation of the col-
orectum showing the most frequent types of
WNT gene mutations in the proximal versus de-
scending/sigmoid colon, according to the nature
of the tumors (MSS, sporadic MSI-H and
HNPCC), is depicted in Figure 2.
The observed nonrandom distribution of APCtruncated alleles retaining different numbers of
20 a.a. repeats, and thus encoding different levels
of b-catenin signaling along the colon and rec-
tum, is reminiscent of the APC mutation spec-
trum in FAP adenomas where APC mutation are
selected to retain a total of one or two 20 a.a.
repeats (Albuquerque et al., 2002a). This correla-
tion between anatomical location and the spectra
of APC somatic mutations was also observed in
FAP adenomas where APC germline mutations
deleting all 20 a.a. repeats were associated with
somatic mutations retaining one or two 20 a.a.
repeats in the descending/sigmoid and in the
proximal colon, respectively. Germline defects
encompassing one repeat were associated with
second hits with no or one repeat, respectively, in
the descending/sigmoid and in the proximal colon
(Albuquerque et al., manuscript in preparation).
It has been described that truncated APC is
required for optimal cell proliferation and to modu-
late the transcriptional activity of b-catenin in a
cell cycle dependent manner (Schneikert and Beh-
rens 2006; Schneikert et al., 2007). Therefore, the
selection for truncated proteins encompassing a dif-
ferent number of 20 a.a. repeats between the prox-
imal and distal colon may reflect the requirement
for distinct b-catenin down-regulation activities.
Recently, Buchert et al. (2010) identified dif-
ferences in the b-catenin signaling threshold lev-
els that determine physiological and pathological
outcomes during embryogenesis, intestinal and
hepatic homeostasis. Similarly, in the human gas-
trointestinal tract an optimal signaling level will
ensure cell homeostasis. Therefore, APC trun-
cated proteins appear to be selected based on the
capacity to overcome the threshold level above
which this homeostasis is disturbed, thus allowing
clonal expansion and neoplastic transformation.
The absence of 20 a.a. repeats as a result of the
two APC mutational events, i.e., too high signal-
ing levels, is selected against, presumably due to
the induction of apoptosis. In agreement with
this, it has been reported that b-catenin overex-
pression and high levels of WNT transcriptional
activity are associated to high apoptotic rates
(Kim et al., 2000; Bordonaro et al., 2008)
As the level of b-catenin signaling appears to
be selected in cancer in a tissue-specific fashion,
as a function of the APC mutations, providing the
‘‘just right’’ level of WNT signaling for tumor for-
mation (Albuquerque et al., 2002a; Groves et al.,
2002; Cho et al., 2006; Gaspar et al., 2009;
Buchert et al., 2010), we suggest that the optimal
level of b-catenin signaling for tumor formation
appears to differ not only between MSS and
MSI-H tumors, sporadic and hereditary, but also
between tumors located in the proximal and
756 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

Figure 2. Schematic representation of WNT mutation spectrathroughout the colon and rectum in MSS, sporadic MSI-H andHNPCC MMR-mutant tumors. For each of these three groups oftumors, the frequency of somatic mutations in the WNT genes (APC,CTNNB1, and AXIN2) and the frequency of APC truncating mutationsretaining 1/0 or 2/3 20 a.a. repeats are represented by vertical andhorizontal graphic bars, respectively. In the colorectum image, each
mutation/type of mutation is depicted in the proximal or in the distalside of colon according to its prevalence in each of these two seg-ments. Mutations/types of mutations limited by a square and writtenin bold-type letter are the most prevalent whereas the remainingones are detected less frequently (�10% of the tumors). [Color fig-ure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
MUTATION SPECTRA IN WNT GENES 757
Genes, Chromosomes & Cancer DOI 10.1002/gcc

distal colon. At the MSI level, this selection for
distinct levels of b-catenin signaling may be asso-
ciated with differences in apoptosis resistance,
known to be increased in MMR deficient com-
pared with proficient tumors. As for tumor loca-
tion, the different embryological origin and
histological features between proximal and distal
colon may reflect specific and distinct WNT sig-
naling threshold levels to trigger tumor initiation.
Accordingly, the decrease of absorptive cells and
the increase of goblet cells from proximal to dis-
tal colon reflect differences in cell differentiation
previously proposed to explain tumor regionality
in the mouse intestine (Haigis et al., 2004). Like-
wise, the specific differentiation pattern of the
proximal colon should lead to a specific threshold
level for which the just-right signaling for tumor
formation is most likely provided by two or three
20 a.a. repeats (mutations in codons 1400-1500).
This and the increased probability of MMR
errors in mono and dinucleotide repeats in the
proximity of codons 1,400–1,500, may compre-
hensively result in the observed preferential loca-
tion of MMR deficient tumors in the proximal
colon. More recently, it has been also reported
that the proximal and distal colon have distinct
and specific gene expression profiles (Glebov
et al., 2003) and follow different tumorigenic
pathways (Sugai et al., 2006), which further sup-
ports the selection for specific b-catenin signaling
levels in these two regions.
In conclusion, alterations in the WNT pathway
genes APC, CTNNB1, and AXIN2 are found in
the vast majority of colorectal tumors. However,
the frequency and the type of mutations, namely
the number of 20 a.a. repeats retained by the
truncated APC gene mutations, significantly differ
among MSS and MSI-H, sporadic and hereditary,
and proximal and distal colonic tumors. We sug-
gest that, as it was shown for adenomas in FAP
patients carrying APC germline mutations, the
observed distinct patterns of WNT gene muta-
tions in sporadic and HNPCC colorectal tumors
may result from selection for the ‘‘just right’’
level of b-catenin signaling optimal for tumor for-
mation depending on their anatomical location
(proximal or distal colon) and the type of genetic
instability (MSI-H or MSS).
REFERENCES
Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P,Slors FJ, Leitao CN, Fodde R, Smits R. 2002a. The ‘just-right’signaling model: APC somatic mutations are selected based ona specific level of activation of the beta-catenin signaling cas-cade. Hum Mol Genet 11:1549–1560.
Albuquerque C, Cravo M, Cruz C, Lage P, Chaves P, Fidalgo P,Suspiro A, Nobre Leitao C. 2002b. Genetic characterization ofpatients with multiple colonic polyps. J Med Genet 39:297–302.
Aoki T, Takeda S, Yanagisawa A, Kato Y, Ajioka Y, Watanabe H,Kudo S, Nakamura Y. 1994. APC and p53 mutations in de novocolorectal adenocarcinomas. Hum Mutat 3:342–346.
Aust DE, Terdiman JP, Willenbucher RF, Chang CG, Molinaro-Clark A, Baretton GB, Loehrs U, Waldman FM. 2002. TheAPC/beta-catenin pathway in ulcerative colitis-related colorectalcarcinomas: a mutational analysis. Cancer 94:1421–1427.
Bordonaro M, Lazarova DL, Sartorelli AC. 2008. Hyperinductionof Wnt activity: A new paradigm for the treatment of colorectalcancer? Oncol Res 17:1–9.
Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, SamuelMS, Jarnicki AG, Winbanks CE, Newton IP, Meniel VS,Suzuki H, Stacker SA, Nathke IS, Tosh D, Huelsken J, ClarkeAR, Heath JK, Sansom OJ, Ernst M. 2010. Genetic dissectionof differential signaling threshold requirements for the Wnt/beta-catenin pathway in vivo. PLoS Genet 6:e1000816.
Cho KH, Baek S, Sung MH. 2006. Wnt pathway mutationsselected by optimal beta-catenin signaling for tumorigenesis.FEBS Lett 580:3665–3670.
Crabtree M, Sieber OM, Lipton L, Hodgson SV, Lamlum H,Thomas HJ, Neale K, Phillips RK, Heinimann K, TomlinsonIP. 2003. Refining the relation between ‘first hits’ and ‘secondhits’ at the APC locus: The ‘loose fit’ model and evidence fordifferences in somatic mutation spectra among patients. Onco-gene 22:4257–4265.
Fodde R. 2003. The multiple functions of tumor suppressors: It’sall in APC. Nat Cell Bio 5:190–192.
Fodde R, van der Luijt R, Wijnen J, Tops C, van der Klift H, vanLeeuwen-Cornelisse I, Griffioen G, Vasen H, Khan PM. 1992.Eight novel inactivating germ line mutations at the APC geneidentified by denaturing gradient gel electrophoresis. Genomics13:1162–1168.
Fodde R, Smits R, Clevers H. 2001. APC, signal transductionand genetic instability in colorectal cancer. Nat Rev Cancer1:55–67.
Gaspar C, Franken P, Molenaar L, Breukel C, van der Valk M,Smits R, Fodde R. 2009. A targeted constitutive mutation inthe APC tumor suppressor gene underlies mammary but not in-testinal tumorigenesis. PLoS Genet 5:e1000547.
Giaretti W, Venesio T, Prevosto C, Lombardo F, Ceccarelli J,Molinu S, Risio M. 2004. Chromosomal instability and APCgene mutations in human sporadic colorectal adenomas.J Pathol 204:193–199.
Groves C, Lamlum H, Crabtree M, Williamson J, Taylor C, BassS, Cuthbert-Heavens D, Hodgson S, Phillips R, Tomlinson I.2002. Mutation cluster region, association between germlineand somatic mutations and genotype-phenotype correlation inupper gastrointestinal familial adenomatous polyposis. Am JPathol 160:2055–2061.
Glebov OK, Rodriguez LM, Nakahara K, Jenkins J, Cliatt J,Humbyrd CJ, DeNobille J, Soballe P, Simon R, Wright G,Lynch P, Patterson S, Lynch H, Gallinger S, Buchbinder A,Gordon G, Hawk E, Kirsch IR. 2003. Distinguishing right fromleft colon by the pattern of gene expression. Cancer EpidemiolBiomark Prev 12:755–762.
Haigis KM, Hoff PD, White A, Shoemaker AR, Halberg RB,Dove WF. 2004. Tumor regionality in the mouse intestinereflects the mechanism of loss of Apc function. Proc Natl AcadSci USA 101:9769–9773.
Higuchi T, Jass JR. 2004. My approach to serrated polyps of thecolorectum. J Clin Pathol 57:682–686.
Homfray TF, Cottrell SE, Ilyas M, Rowan A, Talbot IC, BodmerWF, Tomlinson IP. 1998. Defects in mismatch repair occur af-ter APC mutations in the pathogenesis of sporadic colorectaltumors. Hum Mutat 11:114–120.
Huang J, Papadopoulos N, McKinley AJ, Farrington SM, CurtisLJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P,Kinzler KW, Vogelstein B, Dunlop MG. 1996. APC mutationsin colorectal tumors with mismatch repair deficiency. Proc NatlAcad Sci USA 93:9049–9054.
Huang J, Zheng S, Jin S-H, Zhang S-Z. 2004. Somatic mutationsof APC gene in carcinomas from hereditary non-polyposis colo-rectal cancer patients. World J Gastroenterol 10:834–836.
Johnson V, Volikos E, Halford SE, Eftekhar Sadat ET, Popat S,Talbot I, Truninger K, Martin J, Jass J, Houlston R, Atkin W,Tomlinson IP, Silver AR. 2005. Exon 3 beta-catenin mutations
758 ALBUQUERQUE ETAL.
Genes, Chromosomes & Cancer DOI 10.1002/gcc

are specifically associated with colorectal carcinomas in heredi-tary non-polyposis colorectal cancer syndrome. Gut 54:264–267.
Kim K, Pang KM, Evans M, Hay ED. 2000. Overexpression ofbeta-catenin induces apoptosis independent of its transactiva-tion function with LEF-1 or the involvement of major G1 cellcycle regulators. Mol Biol Cell 11:3509–3523.
Kitaeva MN, Grogan L, Williams JP, Dimond E, Nakahara K,Hausner P, DeNobile JW, Soballe PW, Kirsch IR. 1997. Muta-tions in beta-catenin are uncommon in colorectal cancer occur-ring in occasional replication error-positive tumors. Cancer Res57:4478–4481.
Koch A, Denkhaus D, Albrecht S, Leuschner I, Schweinitz D,Pietsch T. 1999. Childhood hepatoblastomas frequently carry amutated degradation targeting box of the beta-catenin gene.Cancer Res 59:269–273.
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kin-zler KW, Vogelstein B, Clevers H. 1997. Constitutive transcrip-tional activation by a beta-catenin-Tcf complex in APC-/- coloncarcinoma. Science 275:1784–1787.
Laurent-Puig P, Beroud C, Soussi T. 1998. APC gene: Databaseof germline and somatic mutations in human tumors and celllines. Nucleic Acids Res 26:269–270.
Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, KrishnadathKK, Halling KC, Cunningham JM, Boardman LA, Qian C,Christensen E, Schmidt SS, Roche PC, Smith DI, ThibodeauSN. 2000. Mutations in AXIN2 cause colorectal cancer with de-fective mismatch repair by activating beta-catenin/TCF signal-ing. Nat Genet 26:146–147.
Løvig T, Meling GI, Diep CB, Thorstensen L, Norheim Ander-sen S, Lothe RA, Rognum TO. 2002. APC and CTNNB1mutations in a large series of sporadic colorectal carcinomasstratified by the microsatellite instability status. Scand J Gastro-enterol 10:1184–1193.
Mirabelli-Primdahl L, Gryfe R, Kim H, Millar A, Luceri C, DaleD, Holowaty E, Bapat B, Gallinger S, Redston M. 1999. Beta-catenin mutations are specific for colorectal carcinomas withmicrosatellite instability but occur in endometrial carcinomasirrespective of mutator pathway. Cancer Res 59:3346–3351.
Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T,Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M,Maeda Y, Iwama T, Mishima Y, Mori T, Koike M. 1994. Char-acteristics of somatic mutation of the adenomatous polyposiscoli gene in colorectal tumors. Cancer Res 54:3011–3020.
Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y,Koike M, Shitara N, Iwama T, Kuroki T. 1999. Frequent muta-tion of beta-catenin and APC genes in primary colorectaltumors from patients with hereditary nonpolyposis colorectalcancer. Cancer Res 59:4506–4509.
Miyaki M, Iijima T, Ohue M, Kita Y, Hishima T, Kuroki T,Iwama T, Mori T. 2003. A novel case with germline p53 genemutation having concurrent multiple primary colon tumors. Gut52:304–306.
Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S,Aoki T, Miki Y, Mori T, Nakamura Y. 1992. Somatic mutationsof the APC gene in colorectal tumors: Mutation cluster regionin the APC gene. Hum Mol Genet 1:229–233.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, VogelsteinB, Kinzler KW. 1997. Activation of beta-catenin-Tcf signalingin colon cancer by mutations in beta-catenin or APC. Science275:1787–1790.
Muller O, Nimmrich I, Finke U, Friedl W, Hoffmann I. 1998. Abeta-catenin mutation in a sporadic colorectal tumor of theRER phenotype and absence of beta-catenin germline muta-tions in FAP patients. Genes Chromosomes Cancer 22:37–41.
Olschwang S, Laurent-Puig P, Groden J, White R, Thomas G.1993. Germ-line mutations in the first 14 exons of the adenoma-tous polyposis coli (APC) gene. Am J Hum Genet 52:273–279.
Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, De RyckeY, Li YJ, Muzeau F, Girodet J, Salmon RJ, Thomas G. 1997.Alternative genetic pathways in colorectal carcinogenesis. ProcNatl Acad Sci USA 94:12122–12127.
Otori K, Konishi M, Sugiyama K, Hasebe T, Shimoda T, Kiku-chi-Yanoshita R, Mukai K, Fukushima S, Miyaki M, Esumi H.1998. Infrequent somatic mutation of the adenomatous polypo-sis coli gene in aberrant crypt foci of human colon tissue. Can-cer 83:896–900.
Polakis P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837–1851.
Powell SM, Petersen GM, Krush AJ, Booker S, Jen J, GiardielloFM, Hamilton SR, Vogelstein B, Kinzler KW. 1993. Moleculardiagnosis of familial adenomatous polyposis. N Engl J Med329:1982–1987.
Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR,Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, Srivastava S.1997. A National Cancer Institute Workshop on Hereditary Nonpo-lyposis Colorectal Cancer Syndrome: Meeting highlights andBethesda guidelines. J Natl Cancer Inst 89:1758–1762.
Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopou-lou A, Bicknell D, Bodmer WF, Tomlinson IP. 2000. APCmutations in sporadic colorectal tumors: A mutational ‘‘hotspot’’and interdependence of the ‘‘two hits’’. Proc Natl Acad SciUSA 97:3352–3357.
Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G,Lindblom A. 1999. Colorectal cancer with and without microsa-tellite instability involves different genes. Genes ChromosomesCancer 26:247–252.
Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T,Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, ImaokaS, Murata M, Shimano T, Yamaoka Y, Nakamura Y. 2000.AXIN1 mutations in hepatocellular carcinomas, and growth sup-pression in cancer cells by virus-mediated transfer of AXIN1.Nat Genet 24:245–250.
Schneikert J and Behrens J. 2006. Truncated APC is requiredfor cell proliferation and DNA replication. Int J Cancer 119:74–79.
Schneikert J, Grohmann A, Behrens J. 2007. Truncated APC regu-lates the transcriptional activity of b-catenin in a cell cycle de-pendent manner. Hum Mol Genet 16:199–209.
Shimizu Y, Ikeda S, Fujimori M, Kodama S, Nakahara M, Oka-jima M, Asahara T. 2002. Frequent alterations in the Wnt sig-naling pathway in colorectal cancer with microsatelliteinstability. Genes Chromosomes Cancer 33:73–81.
Shitoh K, Furukawa T, Kojima M, Konishi F, Miyaki M, TsukamotoT, Nagai H. 2001. Frequent activation of the beta-catenin-Tcf sig-naling pathway in nonfamilial colorectal carcinomas with microsa-tellite instability. Genes Chromosomes Cancer 30:32–37.
Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, CoxheadJ, Garner RC, Steele RJ, Wolf CR. 2002. Mutations in APC,Kirsten-ras, and p53–alternative genetic pathways to colorectalcancer. Proc Natl Acad Sci USA 99:9433–9438.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. 1998. Mutationalanalysis of the APC/beta-catenin/Tcf pathway in colorectal can-cer. Cancer Res 58:1130–1134.
Sugai T, Habano W, Jiao YF, Tsukahara M, Takeda Y, Otsuka K,Nakamura S. 2006. Analysis of molecular alterations in left- andright-sided colorectal carcinomas reveals distinct pathways ofcarcinogenesis: Proposal for new molecular profile of colorectalcarcinomas. J Mol Diagn 8:193–201.
Suraweera N, Robinson J, Volikos E, Guenther T, Talbot I, Tom-linson I, Silver A. 2006. Mutations within Wnt pathway genesin sporadic colorectal cancers and cell lines. Int J Cancer119:1837–1842.
vander Luijt RB, Khan PM, Vasen HF, Tops CM, van Leeuwen-Cornelisse IS, Wijnen JT, van der Klift HM, Plug RJ, GriffioenG, Fodde R. 1997. Molecular analysis of the APC gene in 105Dutch kindreds with familial adenomatous polyposis: 67 germ-line mutations identified by DGGE, PTT, and southern analy-sis. Hum Mutat 9:7–16.
Yagi OK, Akiyama Y, Ohkura Y, Ban S, Endo M, Saitoh K, YuasaY. 1997. Analyses of the APC and TGF-beta type II receptorgenes, and microsatellite instability in mucosal colorectal carci-nomas. Jpn J Cancer Res 88:718–724.
MUTATION SPECTRA IN WNT GENES 759
Genes, Chromosomes & Cancer DOI 10.1002/gcc




















